Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения промежуточных продуктов для хинолоновых антибиотиков. Способ настоящего изобретения основан на неожиданном результате, состоящем в том, что побочные продукты, сложные эфиры 3-(2-алкоксифенил)-2-енамино-3-оксопропионовой кислоты, могут быть превращены далее и являются подходящими промежуточными продуктами для конечных хинолоновых препаратов.
Предпосылки изобретения
Хинолоновые и хинолониловые антибиотики представляют собой класс современных сильнодействующих противомикробных средств широкого спектра действия, и соответствующие исследования были направлены на разработку данного нового класса противомикробных средств, особенно средств, эффективных против грамположительных патогенов, в частности, против Enterococcus faecium, устойчивого к ванкомицину. Данные хинолоновые и хинолониловые антибиотики и их получение хорошо описаны в публикациях, цитированных ниже.
Однако некоторые типы аналогов хинолоновых антибиотиков имеют такие заместители в системе хинолонового цикла, которые делают синтез подобных аналогов трудным, непрямым и осуществляемым с низким выходом. Поэтому существует насущная потребность в способе получения промежуточных продуктов для хинолоновых и хинолониловых антибиотиков, который описан в данном изобретении, причем указанный способ включает ряд прямых и недорогих реакций, дающих высокий выход и обеспечивающих потребителя эффективными, доступными антибиотиками.
Сущность изобретения
Настоящее изобретение представляет собой неожиданное открытие, состоящее в том, что промежуточные продукты для хинолоновых антибиотиков и соответствующие формуле:
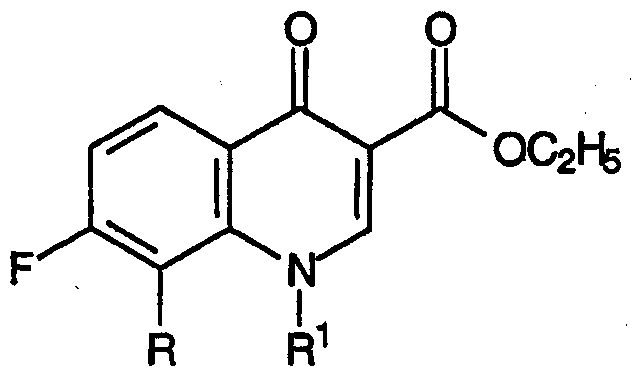
где R представляет собой С1-С2алкил, С1-С2фторалкил, С2-С4алкенил, метокси, хлор или бром; R1 представляет собой заместитель, выбранный из группы, содержащей С1-С2алкил, С2-С3алкенил, С3-С5циклоалкил и фенил, каждый из которых может быть замещен одним или несколькими атомами фтора;
могут быть получены взаимодействием смеси промежуточных предшественников хинолоновых антибиотиков, содержащей 2-этоксизамещенный промежуточный продукт, соответствующий формуле:

с силилирующим агентом, используемым в качестве катализатора реакции циклизации.
В общем, настоящее изобретение относится к способу получения промежуточных продуктов для хинолоновых антибиотиков и соответствующих формуле:
где R и R1 принимают значения, определенные выше; причем указанный способ включает следующие стадии:
a) взаимодействие ацетофенона, соответствующего формуле:
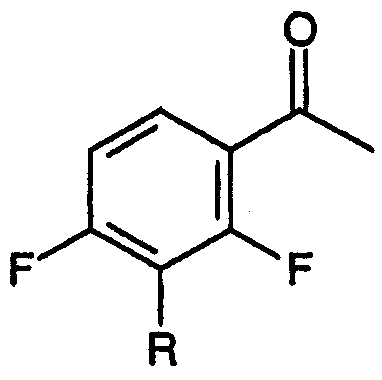
с диэтилкарбонатом в присутствии основания, приводящее к образованию сложных 4-фтор-β-кетоэфиров, соответствующих формуле:
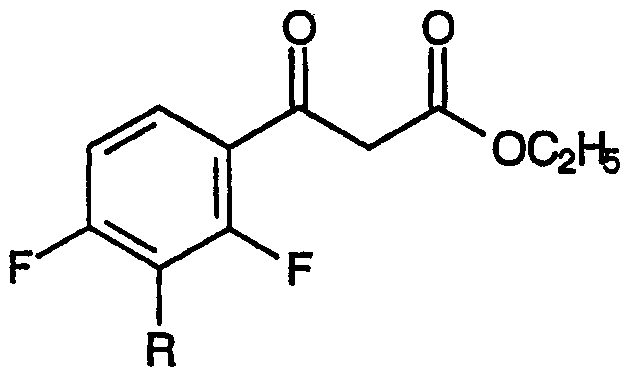
и
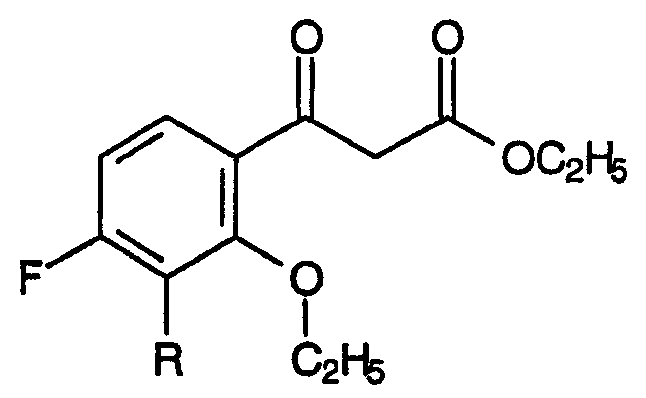
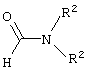
b) взаимодействие указанной смеси с амином, способным подвергаться реакции Кневенагеля и соответствующим формуле:

где R2 представляет собой С1-С4 нормальный или разветвленный алкил, фенил или их комбинацию; Х представляет собой альдегидную группу или эквивалент альдегидной группы,
приводящее к образованию смеси енаминовых промежуточных продуктов, соответствующих формуле:
и
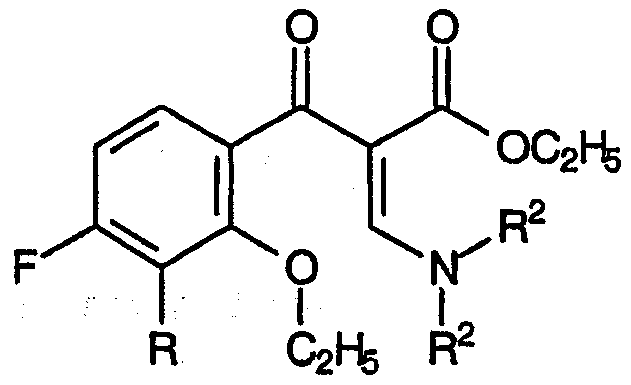
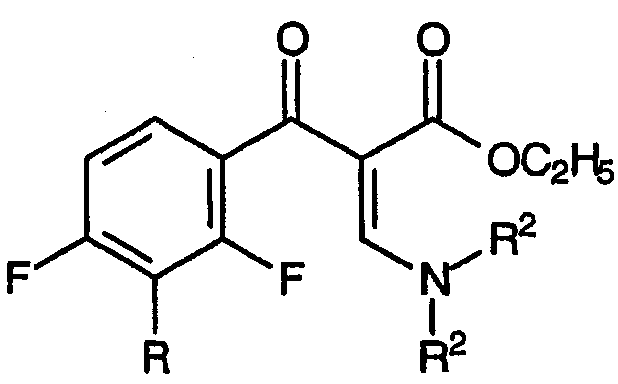
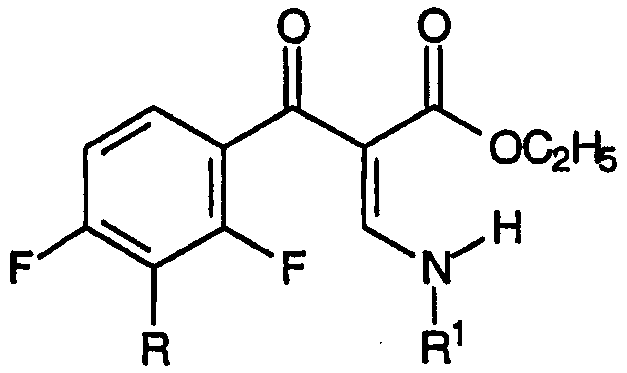
с) взаимодействие смеси указанных енаминовых промежуточных продуктов с амином, соответствующим формуле:
R1-NH2
приводящее к образованию смеси промежуточных продуктов для хинолонов, соответствующих формуле:
и
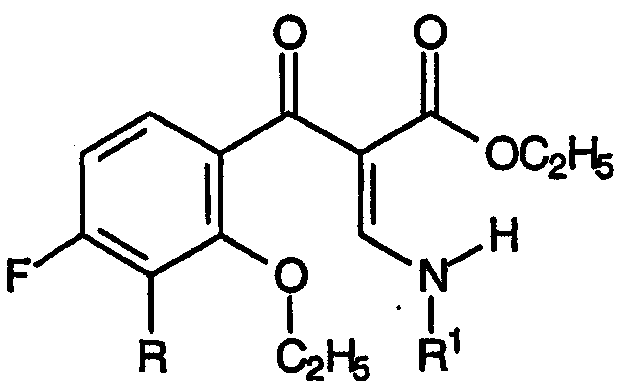
и
d) циклизацию указанной смеси промежуточных продуктов для хинолинов в присутствии силилирующего агента, приводящую к образованию промежуточных продуктов для хинолиновых антибиотиков согласно настоящему изобретению.
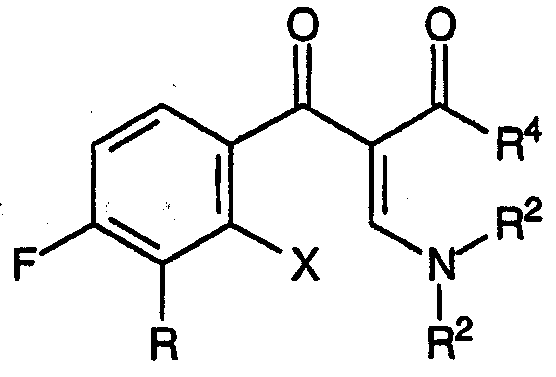
Настоящее изобретение, кроме того, относится к новым соединениям, соответствующим формуле:
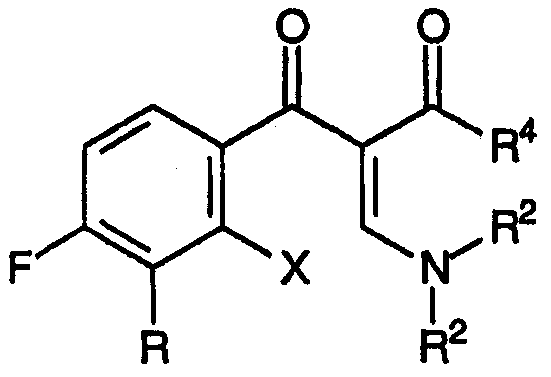
и формуле:
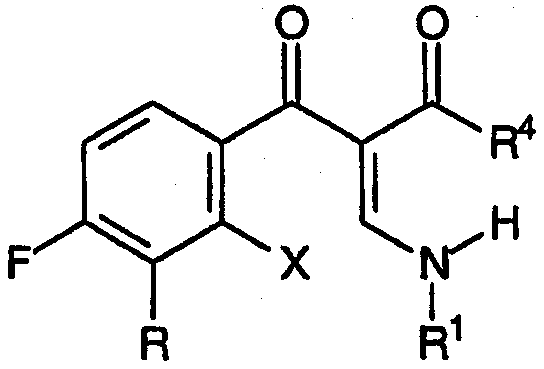
где R, R1 и R4 принимают значения, определенные выше, Х представляет собой подходящую уходящую группу, причем указанные соединения подходят для применения в качестве промежуточных продуктов при получении хинолоновых антибиотиков согласно настоящему изобретению.
Данные и другие аспекты, особенности и преимущества станут очевидными для обычных специалистов в данной области после ознакомления с подробным описанием и прилагаемой формулой изобретения. Все проценты, отношения и пропорции, приведенные в данном описании, являются массовыми, если не оговорено особо. Все температуры даны в градусах Цельсия (°С), если не оговорено особо. Все цитируемые документы находятся в соответствующей части, включенные в данное описание посредством ссылок; цитирование любого документа не следует истолковывать как признание того, что он относится к известному уровню техники для данного изобретения.
Подробное описание изобретения
Настоящее изобретение представляет собой неожиданное открытие, состоящее в том, что 1-низший алкил-8-замещенные хинолоны, в частности, 1-циклопропил и 1-этил-8-метоксихинолонкарбоновые кислоты формулы:
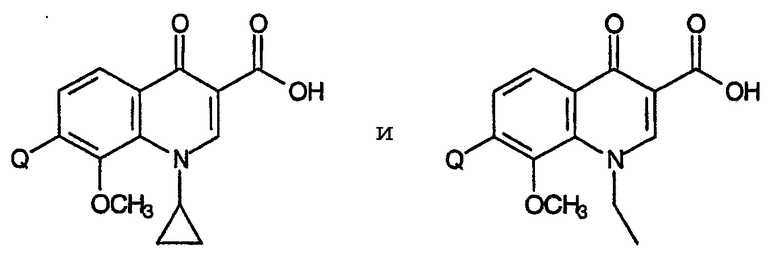
где Q представляет собой аминозамещенный пирролидин или пиперидин, в частности, группы, соответствующие формулам:
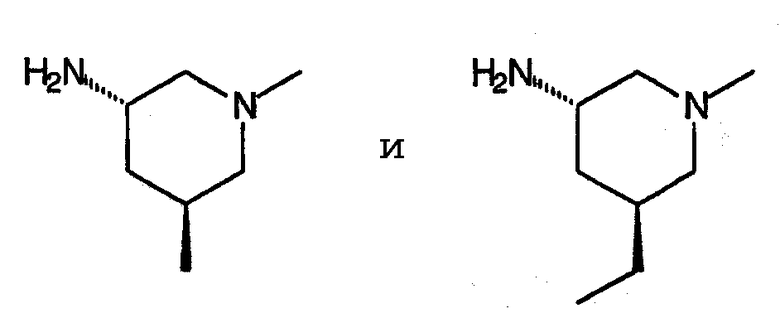
проявляют высокую эффективность против грамположительных бактерий.
В результате данного неожиданного открытия неизбежно требуется высокий выход и соответствующий недорогой синтез антибиотиков указанного класса соединений. Кроме того, требуется способ, пригодный для широкого спектра аналогов, включающих данный класс антибиотиков.
Исследование хинолоновых антибиотиков подобного типа детально описано в патентах: U.S. 6329391 B1 Ledoussal et al., выданном 11 декабря 2001 и U.S. 6387928 B1 Ledoussal et al., выданном 14 мая 2002, включенных в данное описание посредством ссылок. Описанный в них способ включал условия реакций и методики, которые препятствовали нежелательному отщеплению атома фтора из положения 2 в ходе получения предшественников хинолоновых антибиотиков, один из которых имеет формулу:
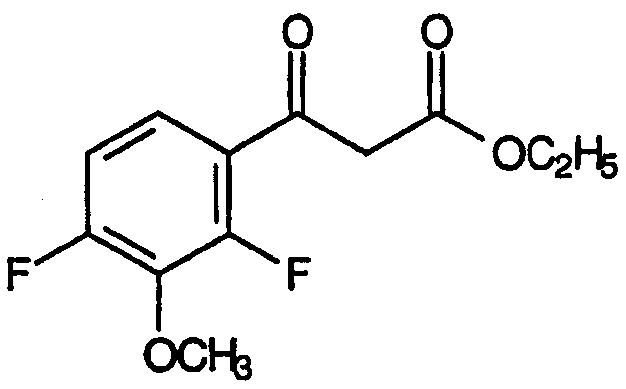
В целях изыскания недорогого способа получения хинолоновых антибиотиков с высоким выходом авторы настоящего изобретения попытались провести циклизацию промежуточных продуктов сложных 2-енамино-β-кетоэфиров - предшественников хинолоновых антибиотиков, соответствующих общей формуле:
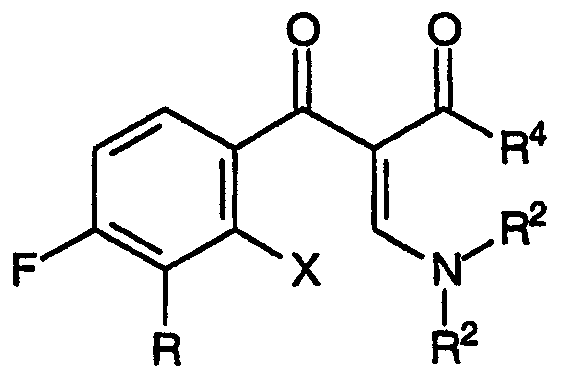
где Х представляет собой уходящую группу и R4 представляет собой группу, совместимую с условиями реакции замыкания кольца. Подход к данным промежуточным продуктам для хинолоновых антибиотиков авторы начали с взаимодействия 3-замещенных-2,4-дифторбензофенонов с диэтилкарбонатом, ожидая получить соответствующий сложный β-кетоэфир. Однако анализ продуктов, полученных по реакции диэтилкарбоната с 2,4-дифтор-3-метоксибензофеноном, указал на смесь продуктов, приведенных ниже,
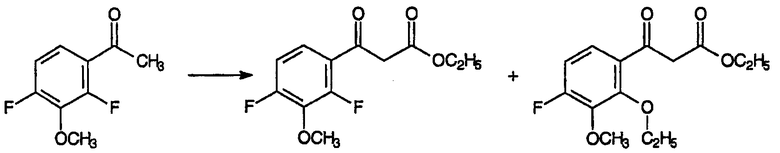
где значительное количество атомов фтора в положении 2 было заменено этоксигруппой в результате нежелательной побочной реакции.
Неожиданно было установлено, что присутствующая примесь, соответствующая общей формуле:
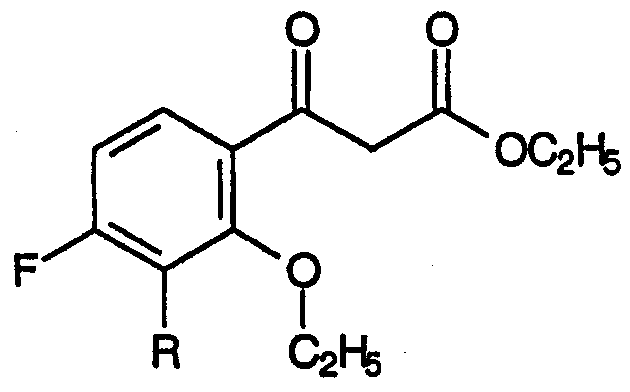
которая образуется в значительных количествах как побочный продукт взаимодействия 3-замещенных-2,4-дифторбензофенонов с диэтилкарбонатом, может быть подвергнута успешной циклизации, вместе с циклизацией соответствующего 2-фтор-аддукта, которая приводит к желаемому промежуточному продукту для хинолонового антибиотика, с высоким выходом и в мягких условиях. Также неожиданно было установлено, что заместитель R может дополнительно включать С1-С2алкил, С1-С2фторалкил, С2-С4алкенил, хлор, бром и метокси.
Промежуточные продукты способа
Способ настоящего изобретения, описанный ниже, основан на использовании новых соединений в качестве промежуточных продуктов. Данные промежуточные продукты относятся к двум категориям. Первая категория относится к сложным β-кетоэфирам енаминов, которые образуются взаимодействием вторичного амина и эфира 3-арил-3-оксопропионовой кислоты и соответствуют формуле:
где R представляет собой С1-С2алкил, С1-С2фторалкил, С2-С4алкенил, метокси, хлор или бром; R2 представляет собой С1-С4 нормальный или разветвленный алкил, фенил или группу, включающую одновременно указанные остатки; R4 представляет собой группу, совместимую с последующими условиями реакции замыкания кольца. Неограничивающим примером группы, подходящей для R4, является алкоксигруппа, которая приводит к образованию сложного метилового эфира. Сложные эфиры являются самой удобной формой, так как антибиотики, которые образуются из промежуточных продуктов, описанных в данном описании, включают 3-карбоксихинолон.
Первый аспект настоящего изобретения относится к новым соединениям, для которых R представляет собой метокси. Вариант осуществления данного аспекта R соответствует формуле:

в то время как другой вариант осуществления соответствует формуле:

Второй аспект относится к заместителям R, представляющим собой низший алкил или фторзамещенный низший алкил, последовательные варианты которых включают -СН3, -СН2F, -CHF2, и -CF3.
Третий аспект относится к заместителям R, представляющим собой атомы хлора или брома. Вариант осуществления данного аспекта R соответствует формуле:
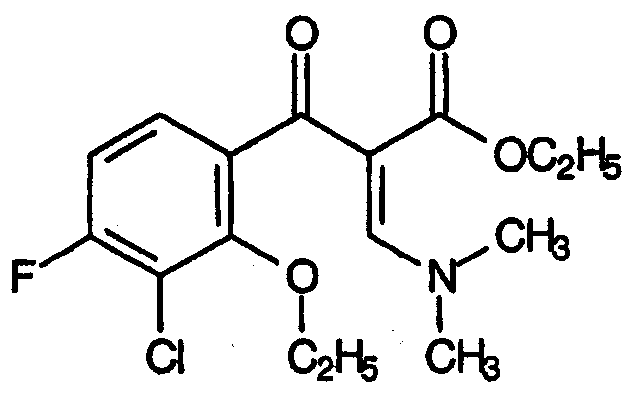
Четвертый аспект относится к заместителям R, представляющим собой олефин, в частности -СН2СН=СН2.
Вторая категория относится к сложным β-кетоэфирам енаминов, которые образуются взаимодействием вторичного амина и эфира 3-арил-3-оксопропионовой кислоты, и соответствуют формуле:
где R представляет собой С1-С2алкил, С1-С2фторалкил, С2-С4алкенил, метокси, хлор или бром; R2 представляет собой С1-С4 нормальный или разветвленный алкил, фенил или их комбинацию; R4 представляет собой группу, совместимую с последующими условиями реакции замыкания кольца.
Первый аспект относится к R, представляющему собой метокси. Вариант осуществления данного аспекта R соответствует формуле:
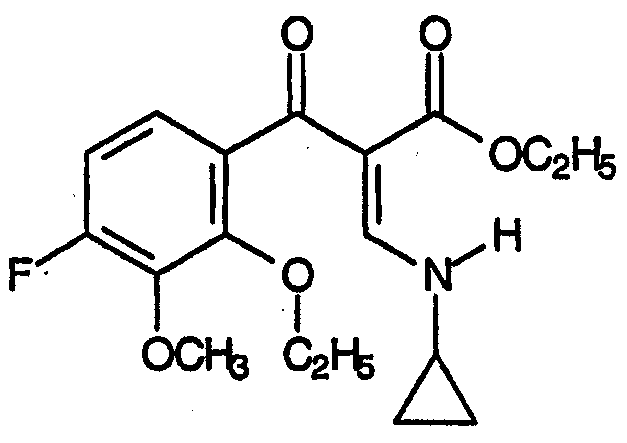
Второй аспект относится к заместителям R, представляющим собой низший алкил или фторзамещенный низший алкил, последовательные варианты которых включают -СН3, -СН2F, -CHF2 и -CF3.
Третий аспект относится к заместителям R, представляющим собой атомы хлора или брома. Вариант осуществления данного аспекта R соответствует формуле:
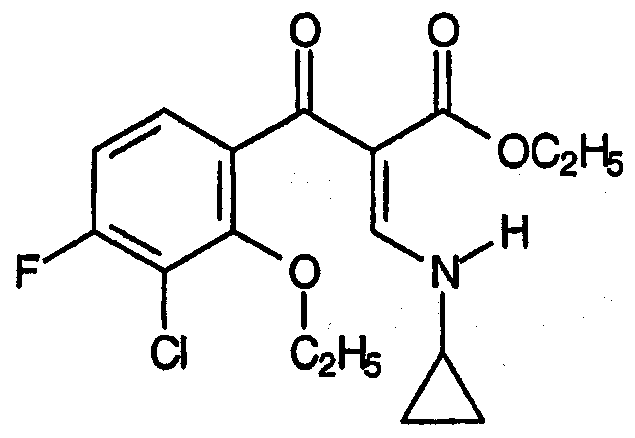
Четвертый аспект относится к заместителям R, представляющим собой олефин, в частности, -СН2СН=СН2.
Способ
Способ настоящего изобретения относится к получению промежуточных продуктов для хинолоновых антибиотиков и соответствующих формуле:

где R представляет собой С1-С2алкил, С1-С2фторалкил, С2-С4алкенил, метокси, хлор или бром; R1 представляет собой заместитель, выбранный из группы, содержащей С1-С2алкил, С2-С3алкенил, С3-С5 циклоалкил и фенил, каждый из которых может быть замещен одним или несколькими атомами фтора.
Стадия (а)
Стадия (а) настоящего способа относится к взаимодействию 3-замещенного-2,4-дифторбензофенона, соответствующего формуле:
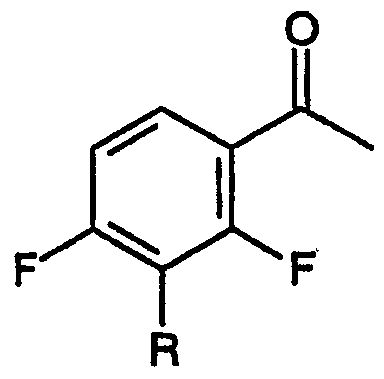
с диэтилкарбонатом в присутствии основания, приводящему к образованию смеси метилового эфира 3-[2,4-дифтор-3-R-замещенной]-3-оксопропионовой кислоты:
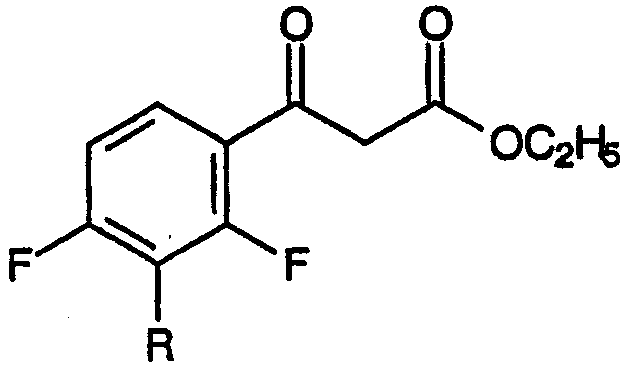
и метилового эфира 3-[2-этокси-3-R-замещенной-4-фтор]-3-оксопропионовой кислоты:
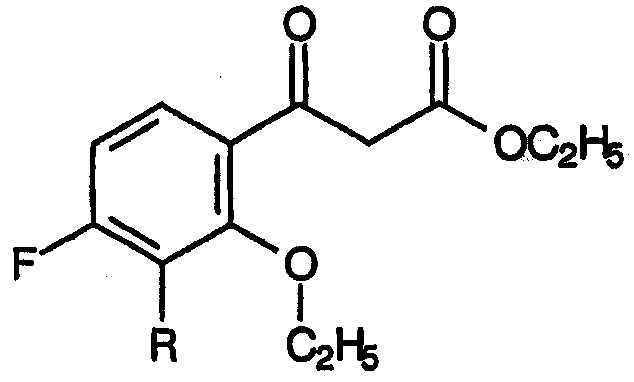
Согласно стехиометрии для стадии (а) требуется, по меньшей мере, один эквивалент диэтилкарбоната на эквивалент бензофенона. Однако с других точек зрения, на стадии (а) используют избыточные количества диэтилкарбоната свыше одного моля эквивалента.
Согласно стехиометрии для стадии (а) требуется, по меньшей мере, один эквивалент основания. Подходящие основания включают гидриды металлов, например, LiH, NaH, KH, CaH2 и т.п. При одном подходе, касающемся выбора основания, используют суспензию NaH в минеральном масле, которая может быть применена удобно и безопасно как в крупномасштабных, так и в мелкомасштабных синтезах. Другие основания включают неорганические основания, в частности Na2CO3, NaHCO3 и K2CO3, или органические основания, в частности бутиллитий и литийдиизопропиламид.
Один из аспектов настоящего изобретения относится к применению избытка диэтилкарбоната. Первый подход к стадии (а), где применяется избыток диэтилкарбоната, представляет собой также и сопутствующее применение избытка основания. Пример стадии (а), относящийся к данному аспекту, но без ограничения только им, включает взаимодействие одного моля 2,4-дифтор-3-R-замещенного бензофенона, 2,2 молей основания и 2,4 молей диэтилкарбоната.
Синтетик может применить апротонный растворитель на стадии (а) как для растворения реагентов, так и для создания среды с эффективным переносом тепла. Примеры растворителей, но без ограничения только ими, включают метиленхлорид, дихлорметан, гексаметилфосфотриамид, тетрагидрофуран, бензол, толуол, алканы, в частности гексан, или смеси растворителей. В большинстве случаев для проведения реакции требуется применение безводных растворителей, однако синтетик может применить избыток основания для связывания любой остаточной или образующейся в процессе реакции воды, которая присутствует. Один из аспектов настоящего изобретения относится к применению толуола в качестве растворителя, который в синтезах, проводимых без выделения промежуточных продуктов, может быть использован для обезвоживания реакционного продукта азеотропной отгонкой.
Температуру, при которой проводят стадию (а), устанавливают на основе нескольких факторов, включающих реакционную способность исходного бензофенона, выбор основания и температуру замерзания/кипения любого примененного растворителя. Один пример настоящего изобретения, имеющий отношение к температуре проведения стадии (а), описан ниже в примере 1, где диэтилкарбонат медленно добавляют к суспензии основания (NaH) в толуоле при 90°С с последующим добавлением ацетофенона при 90-95°С. Однако в зависимости от выбора реагентов и масштаба проведения реакции синтетик может предпочесть проведение реакции при более низких температурах.
Один из аспектов относится к первоначальному депротонированию бензофенона действием алкиллитиевого реагента на холоде и последующему нагреванию раствора перед добавлением диэтилкарбоната.
Стадия (b)
Стадия (b) настоящего изобретения относится к взаимодействию смеси, полученной на стадии (а), с аддуктом, который способен подвергаться реакции Кневенагеля или ей подобной реакции, приводящему к образованию смеси N,N-дизамещенных енаминов, соответствующих формуле:
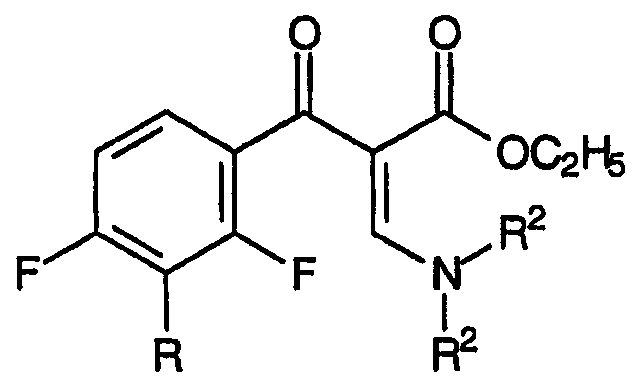
и формуле:
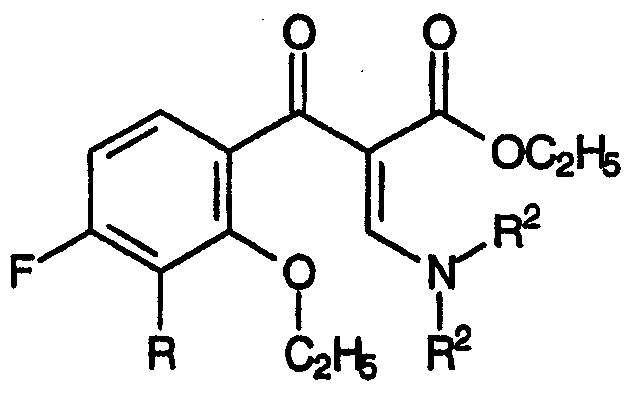
Аддукт, который подвергают взаимодействию со смесью β-кетоэфиров, образовавшихся на стадии (а), соответствует формуле:
где R2 представляет собой С1-С4 нормальный или разветвленный алкил, фенил или их комбинацию; Х представляет собой альдегидную группу или эквивалент альдегидной группы. В приведенном в данном описании определении Х подразумевается, что указанный аддукт может быть альдегидом, соответствующим формуле:
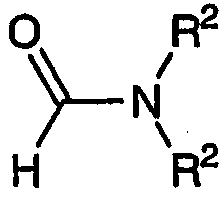
или соединением, эквивалентным альдегиду, например, диметилацеталем, соответствующим формуле:
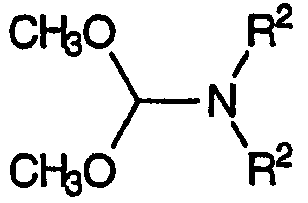
В зависимости от условий проведения стадии (b) синтетик может выбрать любой тип аддукта, способный образовывать требуемую смесь енаминов.
Согласно стехиометрии для стадии (b) требуется, по меньшей мере, один эквивалент альдегида или аддукта, эквивалентного альдегиду, во всех формах на эквивалент бензофенона. Однако с других точек зрения, на стадии (b) используют избыточные количества аддукта свыше одного моля эквивалента.
Один из аспектов настоящего изобретения относится к применению избытка аддукта, который представляет собой соединение, эквивалентное альдегиду. Неограничивающий пример стадии (b), относящийся к данному аспекту, но без ограничения только им, включает взаимодействие одного моля смеси сложных β-кетоэфиров с 1,5 молями производных диметилацеталя, соответствующих формуле:
например, взаимодействие, приведенное в общих чертах ниже в примере 1.
Обычно смесь сложных β-кетоэфиров и аддукт соединяют вместе при комнатной температуре и нагревают. В случае ацетальсодержащих альдегидных эквивалентов реакция может быть доведена до завершения отгонкой выделяющегося спирта, затем альдегидный карбонил реагирует со смесью сложных β-кетоэфиров.
Синтетик может применить любой растворитель на стадии (b), который не взаимодействует с реагентами в условиях осуществления стадии, для растворения реагентов и создания среды с эффективным переносом тепла. Неограничивающие примеры растворителей, включают метиленхлорид, дихлорметан, гексаметилфосфотриамид, тетрагидрофуран, бензол, толуол, алканы, в частности гексан, или смеси растворителей. Однако спиртовые растворители, особенно в присутствии соединений, эквивалентных альдегидам, могут сильно ограничивать реакционную способность аддукта, и поэтому их применять не следует. Один из аспектов настоящего изобретения относится к применению толуола в качестве растворителя, который в синтезах, проводимых без выделения промежуточных продуктов, может быть использован для обезвоживания реакционного продукта азеотропной отгонкой.
Температуру, при которой проводят стадию (b), устанавливают на основе нескольких факторов, включающих реакционную способность аддукта, представляющего собой альдегид или соединение, эквивалентное альдегиду, и температуру замерзания/кипения любого примененного растворителя. Пример 1, приведенный ниже, представляет собой пример температурного режима, при котором выполняют все ступени стадии (b).
Стадия (с)
Стадия (с) настоящего изобретения относится к взаимодействию смеси N,N-дизамещенных енаминов, полученных на стадии (b), с первичным амином, соответствующим формуле:
R1-NH2
приводящему к образованию смеси промежуточных продуктов синтеза хинолонов, соответствующих формуле:
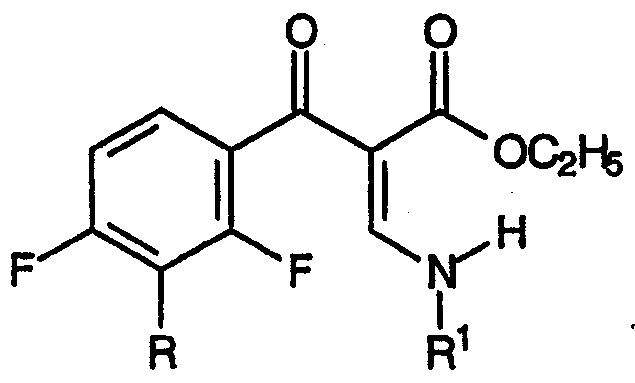
и формуле:
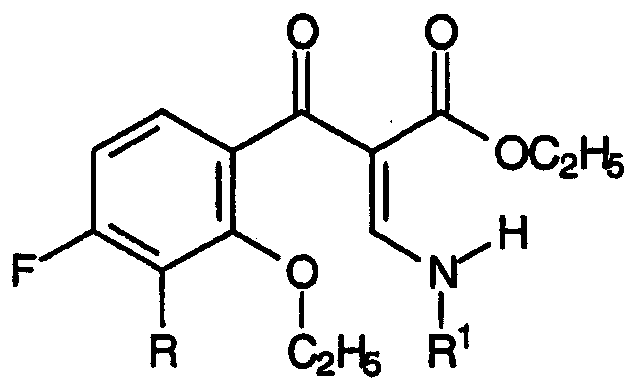
где R1 представляет собой заместитель, выбранный из группы, содержащей С1-С2алкил, С2-С3алкенил, С3-С5циклоалкил и фенил, каждый из которых может быть замещен одним или несколькими атомами фтора.
Согласно стехиометрии для стадии (с) требуется, по меньшей мере, один эквивалент амина на общее количество енамина, присутствующего в смеси. Однако с других точек зрения, на стадии (с) используют избыточные количества амина свыше одного моля эквивалента.
Синтетик может применить апротонный растворитель на стадии (с) как для растворения реагентов, так и для создания среды с эффективным переносом тепла. Примеры растворителей, но без ограничения только ими, включают метиленхлорид, дихлорметан, гексаметилфосфотриамид, тетрагидрофуран, бензол, толуол, алканы, в частности гексан или смеси растворителей. В большинстве случаев для проведения реакции требуется применение безводных растворителей, однако синтетик может применить избыток основания для связывания любой остаточной или образующейся в процессе реакции воды, которая присутствует. Один из аспектов настоящего изобретения относится к применению толуола в качестве растворителя, который в синтезах, проводимых без выделения промежуточных продуктов, может быть использован для обезвоживания реакционного продукта азеотропной отгонкой.
Обычно амин может быть добавлен в реакционную смесь в виде раствора в подходящем растворителе или непосредственно, если амин представляет собой жидкость. Стадия (с) может быть проведена при любой температуре, достаточной для завершения реакции. В одном из вариантов осуществления, которое подтверждено в примере 1, приведенным ниже, реакцию проводят при комнатной температуре.
Один из аспектов настоящего изобретения относится к применению избытка амина. Первый подход к стадии (с), где применяют избыток амина, включает обработку до нейтрального продукта, которая состоит в обработке реакционного раствора кислотой.
Гибкость настоящего способа подтверждается тем, что синтетик выборочно может соединить стадии (b) и (с) таким образом, что смесь, полученная на стадии (b), не выделяют, а вводят на стадию (с). Подобная возможность предоставляет синтетику преимущество при больших серийных производствах промежуточных продуктов для хинолоновых антибиотиков, если только определены параметры реакции.
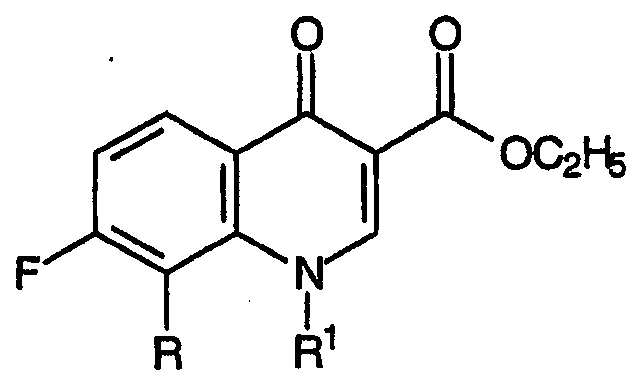
Стадия (d)
Стадия (d) настоящего способа относится к циклизации смеси промежуточных продуктов для хинолинов, полученных на стадии (с), в присутствии силилирующего агента, приводящей к образованию промежуточных продуктов для хинолиновых антибиотиков и соответствующих формуле:
Под термином "силилирующий агент" в данном описании подразумевается любое "кремнийорганическое" соединение или любое кремнийсодержащее соединение, которое обычно используют в реакциях силилирования, в которых атом водорода или азота, или кислорода замещают силильной группой. Примеры силильных групп, но без ограничения только ими, включают триметилсилил и фенилдиметилсилил. Примеры силилирующих агентов, но без ограничения только ими, включают: хлортриметилсилан,
N,O-бис(триметилсилил)ацетамид,
N,O-бис(триметилсилил)трифторацетамид,
бис(триметилсилил)мочевину, гексаметилтрисилазан,
N-метил-N-триметилсилилтрифторацетамид, 1-триметилсилилимидазол,
триметилсилилтрифторметансульфонат, трет-бутилдиметилхлорсилан,
1-(трет-бутилдиметилсилил)имидазол,
N-трет-бутилдиметилсилил-N-метилтрифторацетамид,
трет-бутилдиметилсилилтрифторметансульфонат,
трет-бутилфенилхлорсилан, трет-бутилметоксифенилбромсилан,
диметилфенилхлорсилан, триэтилхлорсилан,
триметилсилилтрифторметансульфонат и трифенилхлорсилан.
Применение силилирующих агентов описано у Cooper B., в "Silylation in Organic Synthesis", Proc. Biochem. 9, (1980), включенной в данное описание посредством ссылки. Кроме того, применение силилирующих агентов для получения хинолонов описано в: US 5801242 Randall et al., выданном 1 сентября 1998 и US 5703231 Randall et al., выданном 30 декабря 1997, причем оба источника включены в данное описание посредством ссылок.
Не желая ограничиваться какой-либо теорией, можно предположить, что согласно стехиометрии на стадии (d) требуется, по меньшей мере, один эквивалент силилирующего агента на каждый моль енамина в смеси, полученной на стадии (с). Однако с других точек зрения, на стадии (d) используют избыточные количества силилирующего агента свыше одного моля эквивалента.
Синтетик может применить апротонный растворитель на стадии (d) как для растворения реагентов, так и для создания среды с эффективным переносом тепла. Примеры растворителей, но без ограничения только ими, включают метиленхлорид, дихлорметан, гексаметилфосфотриамид, тетрагидрофуран, бензол, толуол, алканы, в частности гексан, или смеси растворителей. В большинстве случаев для проведения реакции требуется применение безводных растворителей, однако, синтетик может применить избыток основания для связывания любой остаточной или образующейся в процессе реакции воды, которая присутствует. Один из аспектов настоящего изобретения относится к применению толуола в качестве растворителя, который в синтезах, проводимых без выделения промежуточных продуктов, может быть использован для обезвоживания реакционного продукта азеотропной отгонкой.
Обычно силилирующие агенты представляют собой жидкости, которые могут быть добавлены непосредственно к реакционному раствору смеси енаминов, полученных на стадии (с), или силилирующие агенты представляют собой низкоплавкие твердые вещества, которые могут быть добавлены в виде раствора в подходящем растворителе. В одном из вариантов осуществления, которое подтверждено в примере 1, приведенном ниже, силилирующий агент добавляют при комнатной температуре, затем реакционную смесь нагревают в подходящем растворителе с обратным холодильником до завершения реакции.
Гибкость настоящего способа допускает проведение полунепрерывного процесса, где промежуточные продукты смеси не нужно выделять из реакционной среды и очищать. Удобство состоит также в том, что стадии настоящего способа могут быть доступно проконтролированы на завершение реакции тонкослойной хроматографией (ТСХ) или любой подходящей ВЭЖХ-системой.
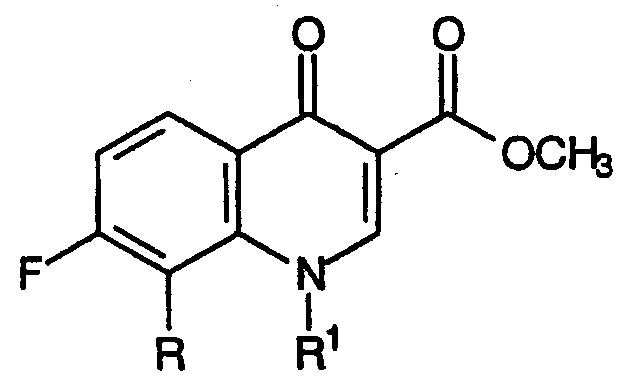
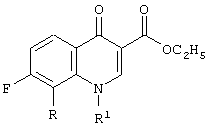
Первая категория промежуточных продуктов для хинолоновых антибиотиков, которые можно получить способом настоящего изобретения, представляют собой метиловые эфиры 3-карбоксихинолона, соответствующие формуле:
Следующая схема реакции и пример 1 иллюстрируют настоящий способ получения соединений категории один, где R для данного примера представляет собой метокси и R1 представляет собой циклопропил.
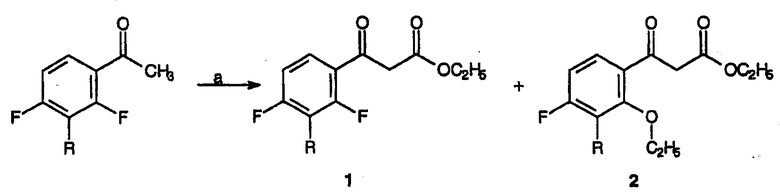
Реагенты и условия:(а) (EtO)2CO, NaH, толуол, 90-95°С, 1,5 час.
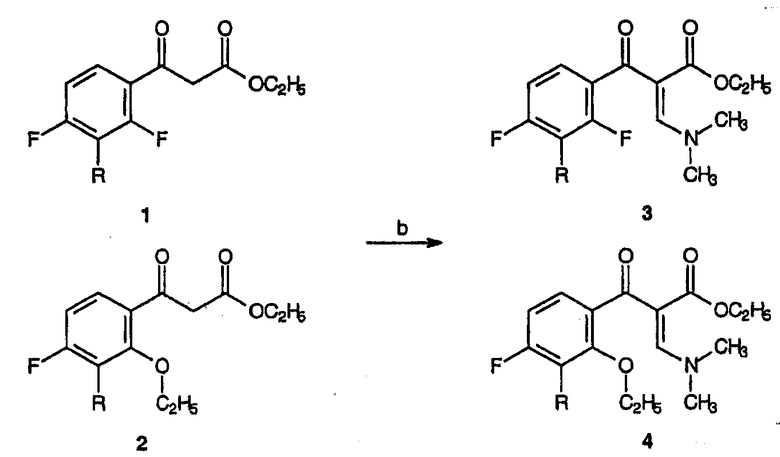
Реагенты и условия: (b) C(OCH3)2N(CH3)2, толуол, кипячение с обратным холодильником, 2 час.
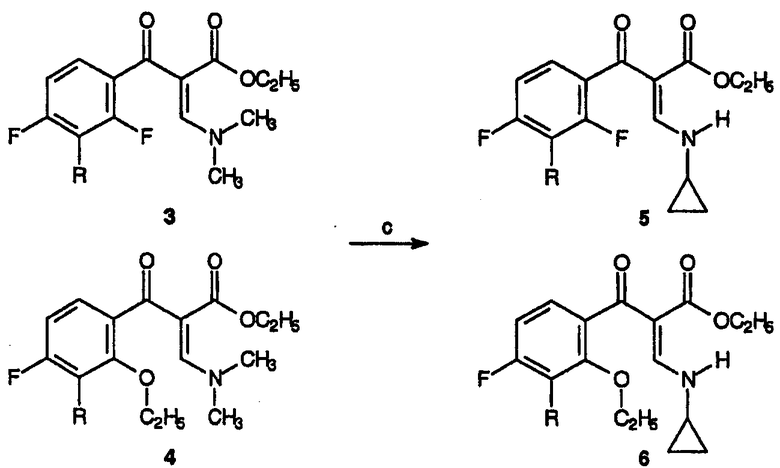
Реагенты и условия: (с) циклопропиламин, толуол, кипячение с обратным холодильником, 1 час.
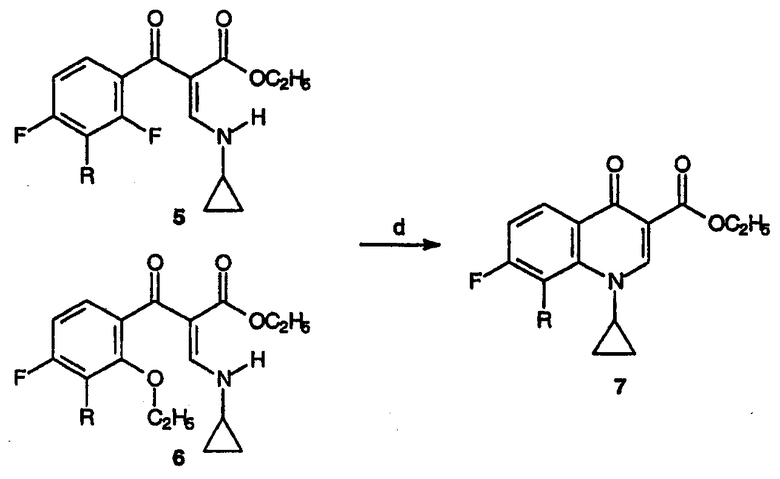
Реагенты и условия: (d) N,O-бис(триметилсилил)ацетамид, толуол, кипячение с обратным холодильником, 1 час.
Пример 1
Этиловый эфир 7-Фтор-8-метоксихинолона (7)
Получение смеси этилового эфира 3-(2,4-дифтор-3-метоксифенил)-3-оксопропионовой кислоты и этилового эфира 3-(2-этокси-4-фтор-3-метоксифенил)-3-оксопропионовой кислоты (1 и 2)
В реакционный сосуд помещают толуол (2087 мл) и масляную дисперсию NaH, содержащую 60% активного основания (264 г, 6,6 моль). К суспензии NaH при 90°С медленно за 1 час добавляют диэтилкарбонат (850,5 г, 7,2 моль). 2,4-Дифтор-3-метоксиацетофенон (558 г, 3 моль) растворяют в достаточном количестве толуола для образования гомогенного раствора (около 2 л) и полученный раствор осторожно добавляют в реакционный сосуд, поддерживая температуру реакции в интервале от 90 до 95°С. После прекращения выделения газообразного водорода реакционную смесь перемешивают дополнительно 30 минут, после чего реакционную смесь охлаждают до 20°С и гасят добавлением 10% мас./мас. водного раствора H2SO4 (3822 г). Слои разделяют и растворитель концентрируют в вакууме (при 40°С, <100 мбар), органическую фазу обезвоживают азеотропной отгонкой с толуолом. Полученную смесь соединений 1 и 2 используют без дальнейшей очистки.
Получение смеси этилового эфира 2-(2,4-дифтор-3-метоксибензоил)-3-диметиламиноакриловой кислоты и этилового эфира 2-(2-этокси-3-метокси-4-фторбензоил)-3-диметиламиноакриловой кислоты (3 и 4)
В реакционный сосуд помещают смесь этилового эфира 3-(2,4-дифтор-3-метоксифенил)-3-оксопропионовой кислоты и этилового эфира 3-(2-этокси-4-фтор-3-метоксифенил)-3-оксопропионовой кислоты, 1 и 2, (850 г, ˜3 моль) и толуол (3850 мл). Примерно за 15 минут добавляют диметилацеталь диметилформамида (536,3 г, 4,5 моль), затем реакционную смесь нагревают до примерно 90°С с отгонкой образовавшегося метанола. Затем раствор кипятят с обратным холодильником около 2 часов. На данной стадии последующая реакция может быть проведена без выделения соединений 3 и 4. Продукты выделяют концентрированием реакционного раствора в вакууме, остаток переводят в метиленхлорид и экстрагируют полученный органический слой водой. Повторное концентрирование в вакууме приводит к смеси 3 и 4.
Получение смеси этилового эфира 3-циклопропиламино-2-(2,4-дифтор-3-метоксибензоил)акриловой кислоты и этилового эфира 3-циклопропиламино-2-(2-этокси-3-метокси-4-фторбензоил)акриловой кислоты (5 и 6)
Реакционную смесь, полученную выше, т.е. смесь соединений 3 и 4, взятую в момент после завершения 2-часового кипячения раствора с обратным холодильником, охлаждают до комнатной температуры и добавляют толуол (2087 мл). Затем добавляют циклопропиламин (205,6 г, 3,6 моль) и перемешивание продолжают при комнатной температуре до завершения реакции по данным ТСХ (около 30 минут). Далее реакционную смесь гасят добавлением 10%-ного водного раствора H2SO4 (2940 г, 3 моль). Органический слой концентрируют в вакууме до полного исчезновения присутствующей воды. Полученный раствор соединений 5 и 6 в толуоле может быть введен в реакцию циклизации без дальнейшей очистки.
Получение этилового эфира 1-циклопропил-7-фтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (7)
В реакционный сосуд помещают смесь этилового эфира 3-циклопропиламино-2-(2,4-дифтор-3-метоксибензоил)акриловой кислоты и этилового эфира 3-циклопропиламино-2-(2-этокси-3-метокси-4-фторбензоил)акриловой кислоты, 5 и 6, (1050 г, ˜3 моль) и толуол (4270 мл). Затем добавляют N,O-бис(триметилсилил)ацетамид (610,3 г, 3 моль) и реакционную смесь кипятят с обратным холодильником 30 минут. Дополнительно в реакционную смесь вносят N,O-бис(триметилсилил)ацетамид (183 г, 0,9 моль) и продолжают ее нагревание в течение дополнительных 30 минут. Реакционную смесь охлаждают и растворитель удаляют в вакууме (при 40°С, 100 мбар). Раствор охлаждают на бане со льдом и образовавшееся твердое вещество отделяют фильтрованием, промывают дважды дистиллированной водой, получая 757 г (выход 82%) желаемого продукта.
Хотя были проиллюстрированы и описаны частные варианты осуществления настоящего изобретения, специалистам в данной области будет очевидно, что могут быть реализованы другие различные варианты и модификации, не изменяющие сущности и объема изобретения, поэтому предполагается охватить в прилагаемой формуле изобретения все такие варианты и модификации, которые входят в объем данного изобретения.
Настоящее изобретение относится к способу получения промежуточного продукта для хинолонового антибиотика. Описывается способ получения промежуточного продукта для хинолонового антибиотика, соответствующего формуле:
где R представляет собой С1-С2алкил, C1-С2фторалкил, С2-С4алкенил, метокси, хлор или бром; R1 представляет собой заместитель, выбранный из группы, содержащей С1-С2алкил, С2-С3алкенил, С3-С5циклоалкил и фенил, каждый из которых может быть замещен одним или несколькими атомами фтора; причем указанный способ включает стадию циклизации смеси предшественников хинолонов
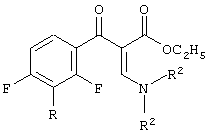 и
и 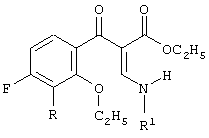
включающий 2-этоксизамещенный промежуточный продукт, соответствующий формуле:
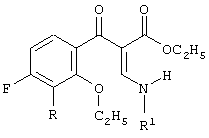
в присутствии силилирующего агента. Технический результат - способ настоящего изобретения основан на неожиданном результате, состоящем в том, что побочные продукты, сложные эфиры 3-(2-алкоксифенил)-2-енамино-3-оксопропионовой кислоты, могут быть превращены далее и являются подходящими промежуточными продуктами для конечных хинолоновых препаратов. 2 н. и 23 з.п. ф-лы.
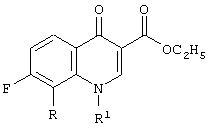
где R представляет собой С1-С2алкил, С1-С2фторалкил, С2-С4алкенил, метокси, хлор или бром;
R1 представляет собой заместитель, выбранный из группы, содержащей С1-С2алкил, С2-С3алкенил, С3-С5циклоалкил и фенил, каждый из которых может быть замещен одним или несколькими атомами фтора;
причем указанный способ включает стадию циклизации смеси предшественников хинолонов
 и
и 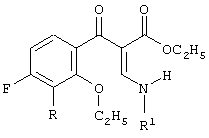
включающий 2-этоксизамещенный промежуточный продукт, соответствующий формуле
в присутствии силилирующего агента.
хлортриметилсилан,
N,O-бис(триметилсилил)ацетамид,
N,O-бис(триметилсилил)трифторацетамид,
бис(триметилсилил)мочевину, гексаметилтрисилазан,
N-метил-N-триметилсилилтрифторацетамид, 1-триметилсилилимидазол,
триметилсилилтрифторметансульфонат, трет-бутилдиметилхлорсилан,
1-(трет-бутилдиметилсилил)имидазол,
N-трет-бутилдиметилсилил-N-метилтрифторацетамид,
трет-бутилдиметилсилилтрифторметансульфонат,
трет-бутилфенилхлорсилан,трет-бутилметоксифенилбромсилан,
диметилфенилхлорсилан, триэтилхлорсилан
и трифенилхлорсилан.
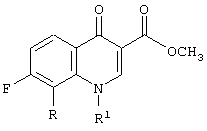
где R представляет собой С1-С2алкил, С1-С2фторалкил, С2-C4алкенил, метокси, хлор или бром;
R1 представляет собой заместитель, выбранный из группы, содержащей С1-С2алкил, С2-С3алкенил, С3-С5циклоалкил и фенил, каждый из которых может быть замещен одним или несколькими атомами фтора,
причем указанный способ включает следующие стадии:
а) взаимодействие ацетофенона, соответствующего формуле
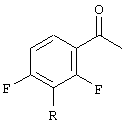
с диэтилкарбонатом в присутствии основания, приводящее к образованию смеси сложных 4-фтор-β-кетоэфиров, соответствующих формуле
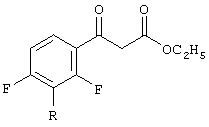
и
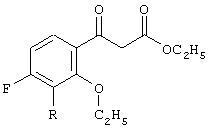
b) взаимодействие указанной смеси с аддуктом реакции Кневенагеля, соответствующим формуле
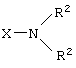
где R2 представляет собой С1-С4 нормальный или разветвленный алкил, фенил или их комбинацию;
Х представляет собой альдегидную группу или эквивалент альдегидной группы,
приводящее к образованию смеси иминных промежуточных продуктов, соответствующих формуле
и

с) взаимодействие смеси указанных иминных промежуточных продуктов с амином, соответствующим формуле

приводящее к образованию смеси промежуточных продуктов синтеза хинолонов, соответствующих формуле
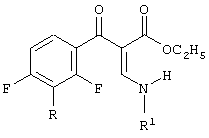
и
d) циклизацию указанной смеси промежуточных продуктов синтеза хинолинов в присутствии силилирующего агента, приводящую к образованию указанных промежуточных продуктов для хинолиновых антибиотиков и соответствующих формуле
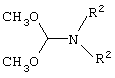
где R2 представляет собой метил, этил и их смесь.
хлортриметилсилан,
N,O-бис(триметилсилил)ацетамид,
N,O-бис(триметилсилил)трифторацетамид,
бис(триметилсилил)мочевину, гексаметилтрисилазан,
N-метил-N-триметилсилилтрифторацетамид, 1-триметилсилилимидазол,
триметилсилилтрифторметансульфонат, трет-бутилдиметилхлорсилан,
1-(трет-бутилдиметилсилил)имидазол,
N-трет-бутилдиметилсилил-N-метилтрифторацетамид,
трет-бутилдиметилсилилтрифторметансульфонат,
трет-бутилфенилхлорсилан, трет-бутилметоксифенилбромсилан,
диметилфенилхлорсилан,триэтилхлорсилан
и трифенилхлорсилан.
| КАНТОВАТЕЛЬ ДЛЯ СБОРКИ И СВАРКИ КРУПНОГАБАРИТНЫХИЗДЕЛИЙ | 0 |
|
SU248113A1 |
| US 5703231 A, 30.12.1997 | |||
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Способ получения 1-циклопропил-7-хлор-6-фтор-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты | 1988 |
|
SU1660582A3 |

