УРОВЕНЬ ТЕХНИКИ
1. ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новому способу получения рацемического небиволола, его энантиомерных форм и к новым соединениям, полученным данным способом
2. ОПИСАНИЕ ПРЕДШЕСТВУЮЩЕГО УРОВНЯ ТЕХНИКИ
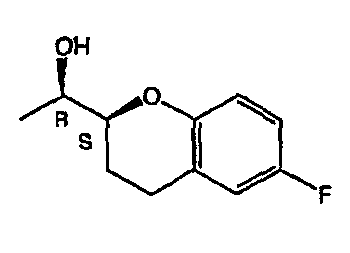
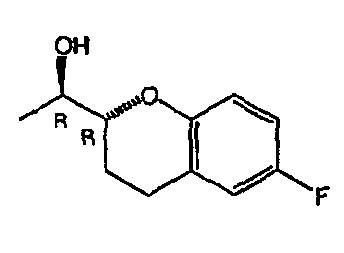
Небиволол (см. фигуру 1А, на которой показан d-Небиволол, химическое название [2R*[R*[R*(S*)]]]-α,α'-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2H-1-бензопиран-2-метанол] или альтернативно [2R*[R*[R*(S*)]]]-α,α'-[иминобис(метилен)]бис[6-фторхроман-2-метанол], и фигуру 1В, на которой показан рацемический небиволол, который представляет собой смесь l- и d-небиволола) известен как адренергический бета-антагонист, антигипертензивное средство, ингибитор агрегации тромбоцитов и сосудорасширяющее средство.
Небиволол вводят в виде таблеток (например, доза гидрохлорида небиволола 5,45 мг эквивалентна 5 мг небиволола), которые содержат небиволол в виде рацемической смеси энантиомеров: SRRR-небиволола (правовращающего d-Небиволола) и RSSS-небиволола (левовращающего l-Небиволола).
Небиволол содержит четыре асимметрических центра, и поэтому теоретически возможно существование 16 стереоизомеров. Однако из-за особенностей структуры и конфигурации стереоизомеров (например, аксиальная симметрия) могут образовываться только 10 стереоизомеров (6 диастереомеров: 4 dl-формы и 2 мезо-формы) (таблица 1).
Нестереоселективное получение этих стереоизомеров в общих чертах описано в патенте США № 4654362 Van Lommen et al. (Janssen Pharmaceutica N. V.) (и его аналог EP 0145067). Стереоселективный синтез [2R,αS,2'S,α'S]-изомера α,α'-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2H-1-бензопиран-2-метанола] описан в патенте США № 6545040 (Janssen Pharmaceutica N. V.) (и его аналоге EP 0334429).
Методика разделения диастереомерной смеси, состоящей из(±)-[2R*[1S*,5S*(S*)]]+[2R*[1S*[5R*(S*)]]]-α,α'-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2H-1-бензопиран-2-метанолов] фракционной кристаллизацией соответствующих гидрохлоридных солей описана в патенте США № 5759580 (Janssen Pharmaceutica N. V.) (и его аналоге EP 0744946). Гидрохлорид небиволола может быть получен только с очень низким выходом 6,6%.
Публикация PCT-патентной заявки WO 2004/041805 (Egis Gyogyszergyar RT.) описывает новый способ получения рацемического [2S[2R*[R[R*]]]] и [2R[2S*[S[S*]]]]-(±)α,α'-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2H-1-бензопиран-2-метанола] и его чистых [2S[2R*[R[R*]]]] и [2R[2S*[S[S*]]]] энантиомеров.
Альтернативные и энантиоселективные синтезы d-небиволола описаны в статьях (J. Am. Chem. Soc. 1998, 120, 8340-8347 и Tetrahedron 56, 2000, 6339-6344).
Стереоизомеры небиволола
общая формула изомеров небиволола
Стереоизомер 1
Стереоизомер 2 d-небиволол
Стереоизомер 3
мезо-форма 1
Стереоизомер 4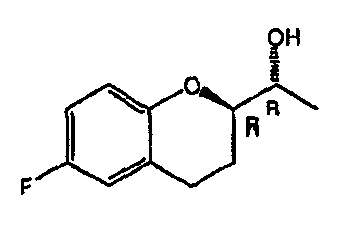
Стереоизомер 2 d-небиволол
Стереоизомер 5
Стереоизомер 6
Стереоизомер 7
мезо-форма 2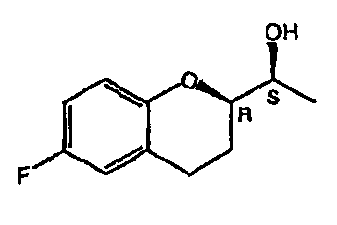
Стереоизомер 3
мезо-форма 1
Стереоизомер 6
Стереоизомер 8
Стереоизомер 9 l-небиволол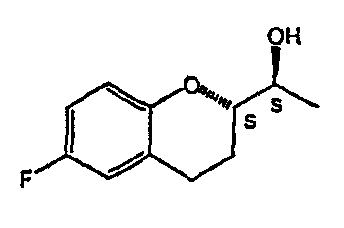
Стереоизомер 4
Стереоизомер 7
мезо-форма 2
Стереоизомер 9 l-небиволол
Стереоизомер 10
Способы получения небиволола, описанные в вышеупомянутых ссылках, кратко обобщены ниже.
а. Патент США № 4654362 (и его аналог EP 0145067 U.S) (Janssen Pharmaceutica N. V.)
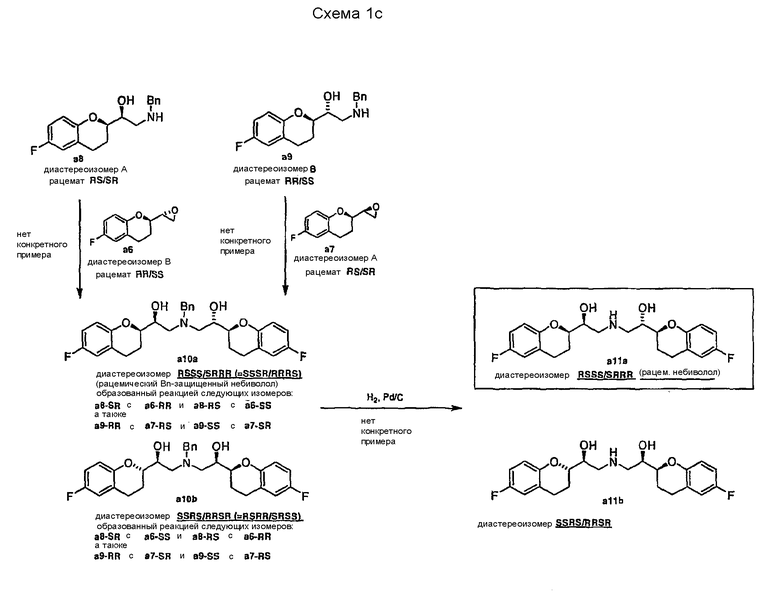
Синтетический путь нестереоселективного получения небиволола описан, исходя из 6-фтор-4-oксo-4H-1-бензопиран-2-карбоновой кислоты a1 (Схема 1a).
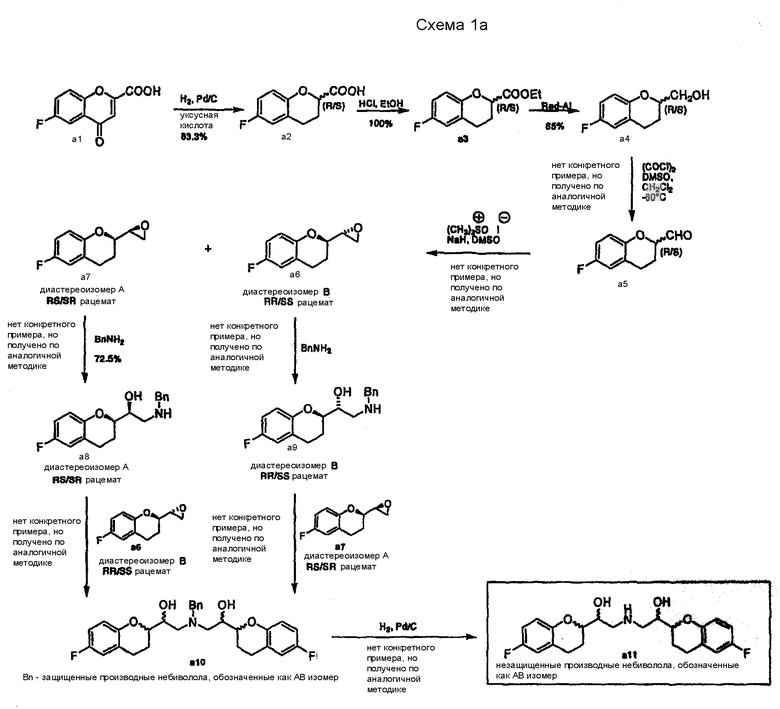
Для получения небиволола по Схеме 1а в патенте США № 4654362 и его аналоге EP 0145067 содержатся подробные примеры синтеза только компонентов a1, a2, a3, a4 и a8. Все другие примеры представляют собой аналоги методик, которые описывают получение родственных производных (например, производных без фторного заместителя в ароматическом кольце). Общая стратегия получения небиволола или его соответствующих производных основана на синтезе 2-оксиранилхроманов (a6 и a7) в качестве ключевых интермедиатов для конечных стадий сочетания. Вследствие наличия двух асимметрических атомов углерода, эти соединения могут образовываться из рацемических альдегидов a5 в виде двух диастереомерных рацематов ("А формы" a7 = RS/SR и "В формы"a6 = SS/RR), которые можно разделять с помощью хроматографии. В рассмотренной ссылке для желательных промежуточных продуктов не приводятся описания методик обработки, кристаллизации и очистки или разделения стереоизомеров, выходы и т.п.
Рацематы a6 или a7 могут быть превращены в соответствующие бензилированные аминоспирты а8 и а9 взаимодействием с бензиламином. Защищенная бензилом АВ смесь небивололов а10 может быть получена взаимодействием рацемата a8 (RS/SR) с рацематом эпоксида a6 (RR/SS) или взаимодействием рацемата a9 (RR/SS) с рацематом эпоксида a7 (RS/SR). Защитная группа может быть удалена на последней стадии каталитическим гидрированием, которое приводит к образованию АВ смеси небивололов a11.
Схема 1b показывает дополнительные способы синтеза аналогичных 2-хроманилальдегидов (a14) и 2-оксиранилхроманов (a16) в качестве ключевых интермедиатов для синтеза производных небиволола, содержащих различные заместители в ароматической группе.
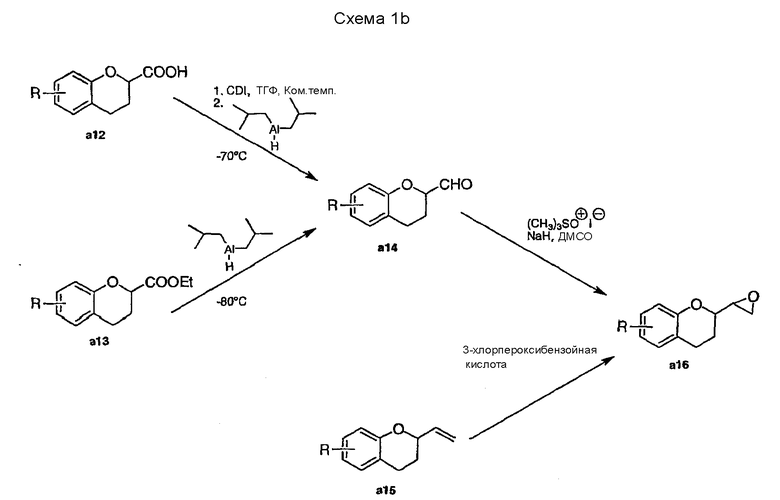
Альдегид a14 может быть получен низкотемпературным восстановлением имидазолида a12 или таким же восстановлением сложного эфира a13. Альдегид a14 затем превращают в 2-оксиранилхроманы a16 взаимодействием с гидридом натрия и йодидом триметилсульфоксония в диметилсульфоксиде по реакции, аналогичной описанной выше. Еще одной возможностью синтеза 2-оксиранилхроманов a16 является окисление 2-винилхромана a15 3-хлорпероксибензойной кислотой (источник 2-винилхромана a15 не указан в этих патентах, но согласно EP 0334429 (см. также ниже) соединение a14 можно превратить в соединение а15 реакцией Виттига).
На схеме 1c показано, что диастереомерные смеси, состоящие из желательных и нежелательных диастереомеров (т.e. RSSS/SRRR и RSRR/SRSS), могут быть получены, способом, представленны на схеме 1a.
Стратегия синтеза, описанная в патенте США № 4654362 и его аналоге EP 0145067, имеет следующие недостатки:
1. Синтез альдегидов a6 и a14 требует очень низких температур и поэтому требует специального оборудования, что усложняет и удорожает способ.
2. Альдегид а5 представляет собой очень нестабильное соединение, как указано в публикации РСТ-заявки WO 2004/041805.
3. Синтез a6/a7 из a5 может быть опасным, так как известно, что использование гидрида натрия в растворителях, подобных ДМСО, диметилформамиду (ДМФА), диметилацетамиду (DMA) и DMI, может вызвать экзотермическую реакцию и, следовательно, может вызвать неконтролируемую реакцию (см. UK Chemical Reaction Hazards Forum: "Sodium Hydride/DMF process stopped").
4. Соединения a6 и a7 были охарактеризованы как маслообразные вещества (см. публикацию РСТ-заявки WO 2004/041805). Так как получение в соответствии с описанной методикой приводит к образованию диастереомерной смеси a6 и a7, может потребоваться хроматографическая очистка, которая коммерчески нерентабельна.
5. Соединения а10 и а11 могут быть получены реакцией рацемического интермедиата а8 ("изомер A") с рацематом a6 ("изомер B") или альтернативно реакцией рацемического интермедиата a9 ("изомер B") с рацематом а7 ("изомер A") с последующим снятием защиты. В патенте США № 4654362 и его аналоге EP 0145067 не содержится четко сформулированного описания того, являются ли индивидуальными изомерами или смесью изомеров соединения а10 и а11 (охарактеризованы только как "AB" изомерная форма). Не представлено никаких соображений по разделению таких смесей. Очевидно, что такие методики могут приводить к образованию диастереомерных смесей, состоящих из желательного RSSS/SRRR диастереомера и нежелательного RSRR/SRSS диастереомера (схема 1c; ср. также таблицу 1, демонстрирующую комбинацию различных фрагментов, которая дает все возможные диастереомеры). Кроме того, из предшествующего уровня техники известно (см. WO 2004/041805), что рацемический небиволол, полученный способом, описанным в патенте США № 4654362 (и его аналоге EP 0145067) (схемы 1a и 1c) в виде диастереомерного рацемата с SRSS/RSRR конфигурацией не удалось успешно разделить фракционной кристаллизацией; и
6. Потери дорогостоящих веществ из-за образования нежелательных изомеров небиволола, особенно во время последних стадий способа.
b. Публикация европейкой патентной заявки EP 0334429 и патент США № 6545040 (Xhonneux et al., Janssen Pharmaceutica N.V.)
Аналогичная стратегия синтеза небиволола описана в EP 0334429 и патенте США № 6545040, но отличие состоит в том, что l-небиволол получают энантиоселективным синтезом, в котором в качестве ключевых интермедиатов используют энантиочистые фрагменты b6 и b11 (Схема 2).
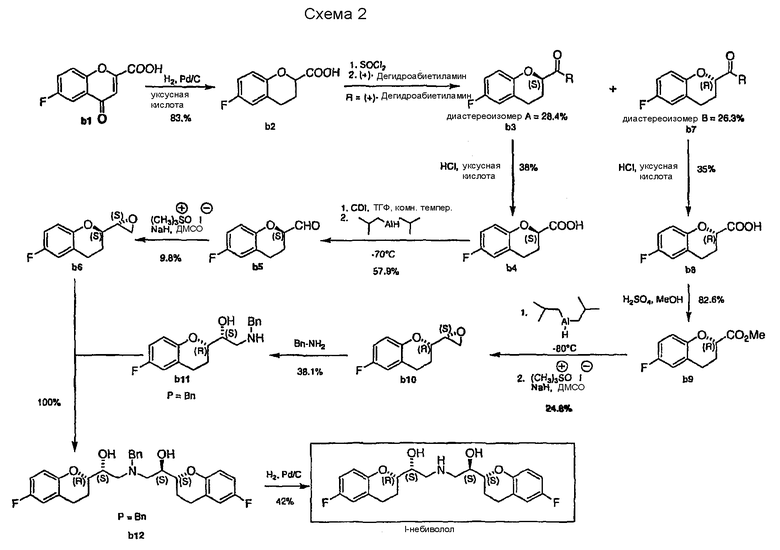
Для этой методики необходимо разделить рацемическую 6-фторхроман-2-илкарбоновую кислоту b2 путем образования диастереомерного амида b3 с (+)-дегидроабиетиламином с последующей фракционной кристаллизацией диастереомеров и гидролизом амидов. Следующие стадии синтеза фрагментов b6 и b11 осуществляют конвергентными путями, используя (S)-форму и (R)-форму 6-фторхроман-2-илкарбоновых кислот b4 и b8. Сначала (S)-6- фторхроман-2-илкарбоновую кислоту b4 превращают в альдегид b5 по методике, уже упомянутой в схеме 1b. Затем можно получить эпоксид b6 взаимодействием b5 с гидридом натрия и йодидом триметилсульфоксония в диметилсульфоксиде. В случае второго пути (R)-6-фторхроман-2-илкарбоновую кислоту b8 сначала этерифицируют с образованием b9. Эпоксид b10 синтезируют по методике “без выделения” восстановлением b9 в соответствующий альдегид с последующей реакцией с гидридом натрия и йодидом триметилсульфоксония в диметилсульфоксиде. Кольцо эпоксида b10 раскрывают реакцией замещения бензиламином, которая дает второй ключевой фрагмент b11, последний затем подвергают взаимодействию с эпоксидом b6 и получают защищенный бензилом l-небиволол b12. Последняя стадия снятия защиты каталитическим гидрированием b12 дает l-небиволол.
Стратегия, описанная в EP 0334429 и патенте США № 6545040, имеет следующие недостатки:
1. Стадии получения соединений b5 из b4 и b10 из b9 требуют очень низких температур для восстановления диизобутилалюминийгидридом (DIBAH), что более усложняет и удорожает способ из-за потребности в специальном холодильном оборудовании.
2. Стадии получения соединений b6 из b5 и b10 из b9 могут представлять угрозу безопасности, так как известно, что использование гидрида натрия в растворителях, подобных ДМСО, ДМФА, DMA и DMI, может привести к экзотермической реакции и, следовательно, может вызвать неконтролируемую реакцию (см. UK Chemical Reaction Hazards Forum: "Sodium Hydride/DMF process stopped").
3. Соединения b5, b6, b9 и b10 представляют собой маслообразные вещества и поэтому их трудно очищать; в вероятном случае, когда соединения b6 и b10 загрязнены нежелательными диастереомерами, может потребоваться их разделение колоночной хроматографией, которая не является коммерчески рентабельным методом.
4. Низкие выходы, особенно выходы на стадиях получения соединений b2-b3-b4, b2-b7-b8 и b5-b6, b9-b10, в результате приводят к очень низкому общему выходу (меньше 0,5%) для синтеза l-небиволола, что делает рассматриваемую методику нерентабельной.
5. Так как получается только l-небиволол, а для получения небиволола требуется рацемическая смесь, необходимы дополнительные стадии для синтеза соответствующей d-формы (т.e., d-небиволола); и
6. При взаимодействии интермедиата b2 образуются диастереомеры b3 и b7, которые затем приходится разделять и обрабатывать по отдельности, получая b6 и b11, затем объединять для получения b12, следовательно, для способа требуется множество дополнительных стадий.
с. Публикация РСТ-патентной заявки WO 2004/041805 (Trinka et al., EGIS GYOGYSZERGYAR RT)
WO 2004/041805 описывает энантиоселективный синтез d- и l-небиволола (см. Схемы 3a-c).
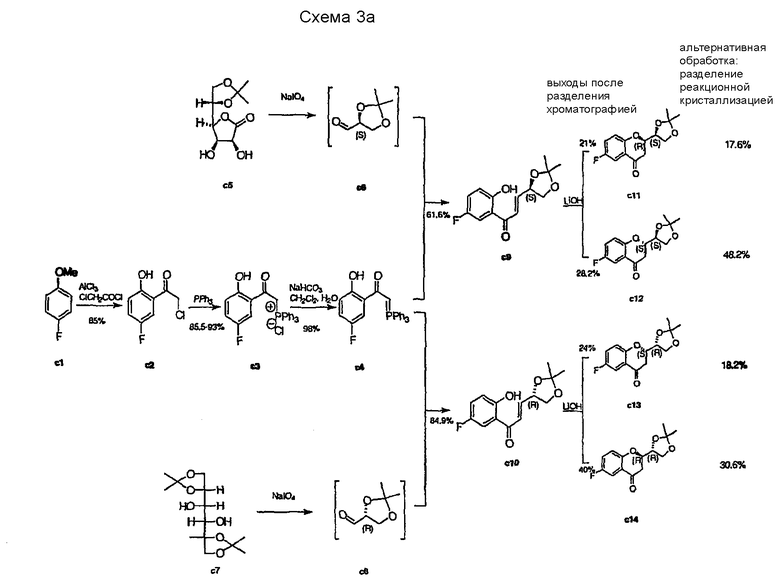
Стратегия этого пути основана на синтезе и разделении защищенных изопропилиденовой группой изомеров (1`,2`-дигидроксиэтил)-6-фторхроман-4-она с11, c12, c13, с14 (Схема 3a). Указанные соединения синтезируют, начиная с ацилирования 4-фторанизола с1 хлорацетилхлоридом по Фриделю-Крафтсу c образованием хлорацетилпроизводного c2, которое далее трансформируется под действием трифенилфосфина с последующей обработкой слабым основанием в стабильное фосфанилиденовое производное c4. Соединение c4 затем подвергают взаимодействию или с защищенными глицериновыми альдегидами c6 (полученными из витамина C) с получением с11 и c12; или с c8 (полученным из D-маннита) с получением c13 и с14 после циклизации.
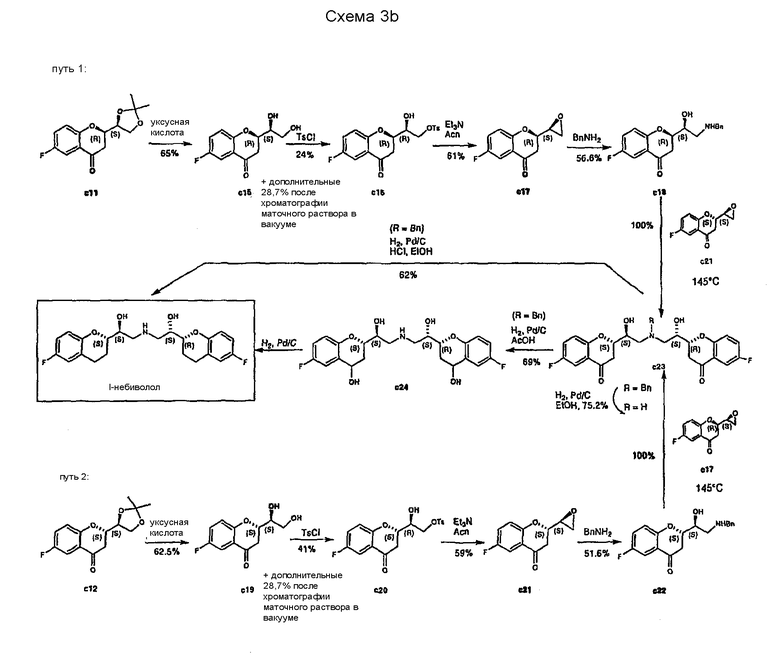
Каждый из этих изомеров был далее трансформирован четырьмя путями и аналогичным образом (Схемы 3b и 3c), в результате чего по путям 1 и 2 получают l-небиволол, используя с11 и c12 в качестве исходных соединений (Схема 3b).
Энантиомерный d-небиволол получают аналогичным образом, причем в качестве исходных веществ используют S,R-изомер c13 и R,R-изомер с14 (1`,2`-дигидроксиэтил)-6-фторхроман-4-она, защищенного изопропилиденовой группой (пути 3 и 4, Схема 3c).
Типичная последовательность реакций для каждого пути начинается со снятия защиты с11 (путь 1, Схема 3b), с12 (путь 2, Схема 3b), с13 (путь 3, Схема 3c), с14 (путь 4, Схема 3c) и получения соответствующих диолов с15, c19, c25, с29. Селективное тозилирование первичной спиртовой группы дает соединения c16, c20, c26, c30, которые можно затем превратить в эпоксиды c17, c21, c27, c31 обработкой основанием. После превращения этих эпоксидов действием бензиламина в c18, c22, c28, c32 и замещения требуемыми эпоксидами (c18+c21, c22+c17, c28+c31, c32+c27) образуются дикетопроизводные с23 и с33, защищенные бензильной группой. Снятие защиты и восстановление карбонильных групп можно проводить в одном реакторе каталитическим гидрированием и получать или l-небиволол, или d-небиволол.
Рацемический небиволол получают, приготавливая смесь (1:1) интермедиатов c23 и c33 перед проведением последней стадии гидрирования (выход 52%).
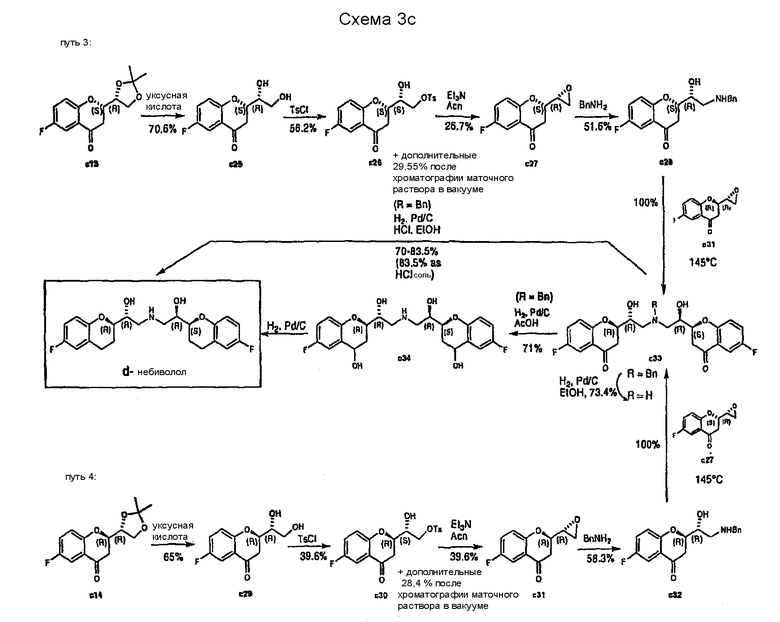
Стратегия, описанная в WO 2004/041805, имеет следующие недостатки:
1. Хотя стратегия основана на использовании всех стереоизомеров для синтеза или l-небиволола или d-небиволола, основным недостатком является то, что для получения рацемической смеси необходимо осуществить 30 стадий, используя все интермедиаты, что делает такой синтез длительным и нерентабельным; и
2. Стадии получения соединений c23 из cl8, c23 из c22, c33 из c28 и c33 из c32 проводят без применения растворителя при 145°С (преимущественно после плавления реагента). Такая методика не подходит для использования в больших масштабах.
d. Johannes et al., J. Am. Chem. Soc. (1998), 120, 8340-8347
В статье Johannes c соавторами описано энантиоселективное получение d-небиволола (Схема 4).
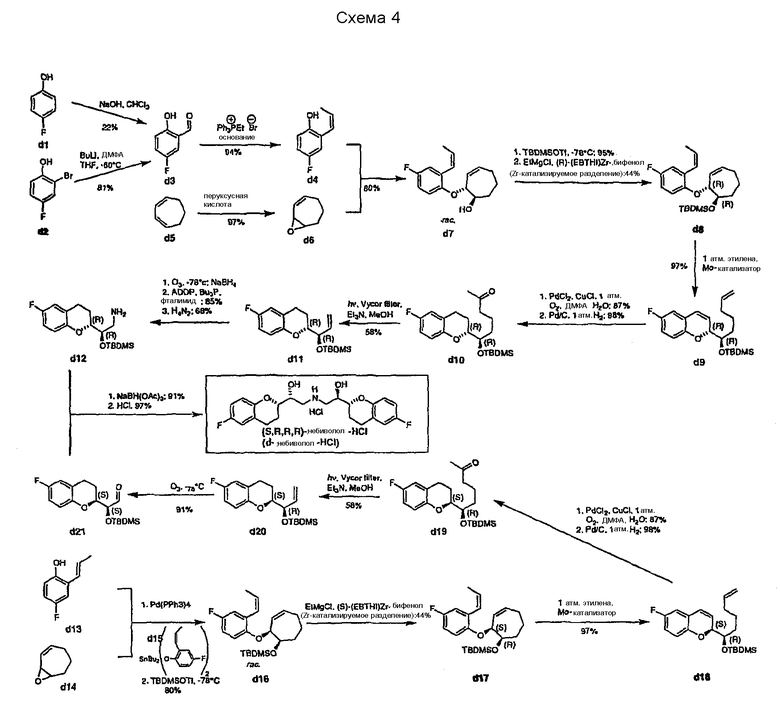
Стратегия основана на синтезах хиральных фрагментов хромана d12 (R,R-конфигурации) и d21 (S,S-конфигурации) в качестве ключевых интермедиатов в конвергентных путях, которые в результате соединяются и дают после снятия защиты d-небиволол. Существенно важной стадией в синтезе упомянутых хиральных хроманов является катализируемое Zr кинетическое разделение рацемических интермедиатов d7 и d16.
В соответствии с первым путем исходным веществом для получения фрагмента хромана d12 является салициловый альдегид d3, который синтезируют или формилированием соединения d1, или реакцией литиилированного соединения d2 с ДМФА при -60°С. Затем эпоксид аллильного циклогептена, который можно получать эпоксидированием циклогептадиена, подвергают взаимодействию с альдегидом d4, получая рацемическое соединение d7 путем региоселективного и стереоселективного нуклеофильного раскрытия цикла эпоксида d8. Защита гидроксильной группы d7 с использованием TBSOT с последующей обработкой 5 экв. EtMgCl и 10 мол.% (R)-(EBTHI)Zr-бифенола дает d8 с выходом 44% и >98%ee. Мо-катализируемая реакция метатезиза в атмосфере этилена с последующим окислением концевой двойной связи по Вакеру (Wacker) и последующим каталитическим гидрированием дает d10 с общим выходом 83%. Чтобы синтезировать d11 из d10 необходимо фотохимическое расщепление Норриша типа II. Следующая последовательность трех стадий: озонолитическое расщепление, реакция Мицунобу, в которой используют трибутилфосфин и фталид, и последующий гидразинолиз, для удаления фталимидильной группы дает интермедиат d12. Второй путь начинается синтезом рацемата d16 с цис-конфигурацией, который затем разделяют в присутствии циркониевого катализатора (S)-(EBTHI)Zr-бифенола. Соединение d17 превращают в соединение d18 Мо-катализируемой реакцией метатезиса. Окисление по Вакеру (Wacker) концевой двойной связи и последующее каталитическое гидрирование дают интермедиат d19, который далее фотохимическим расщеплением Норриша типа II и озонолизом превращают в альдегид d21. D-Небиволол получают затем восстановительным аминированием соединений d12 и d21 с последующим удалением силилэфирных защитных групп.
Стратегия, описанная в статье Johannes с соавторами, имеет следующие недостатки:
1. Получение альдегида d3 происходит или с низким выходом реакцией формилирования d1, использующей хлороформ в присутствии основания, или требует низкой температуры для литиилирования и формилирования производного d2. Кроме того, работа с н-BuLi в промышленном масштабе требует особых мер предосторожности.
2. Стадии получения производных d8 из d7 и d16 и d17 из d13/d14 также требуют низкой температуры (-78°С) для силилирования. Кроме того, необходима трудная стадия разделения, которая использует особый коммерчески недоступный Zr-катализатор.
3. Стадии получения соединений d10-d11 и d19-d20 требуют специального оборудования для проведения фотохимической реакции (расщепление по Норришу, тип 2).
4. Стадия получения соединения d12 из d11 требует низкой температуры (-78°C) и специального оборудования для проведения озонолиза.
5. Для синтеза одного энантиомера небиволола (d-формы) необходимо 16-20 стадий, но требуется рацемическая смесь; поэтому нужны дополнительные стадии, чтобы синтезировать соответствующую l-форму (т.e. l-небиволол).
e. Chandrasekhar et al., Tetrahedron (2000), 56, 6339-6344
В статье Chandrasekhar с соавторами описана еще одна методика энантиоселективного синтеза d-небиволола (см. схему 5).
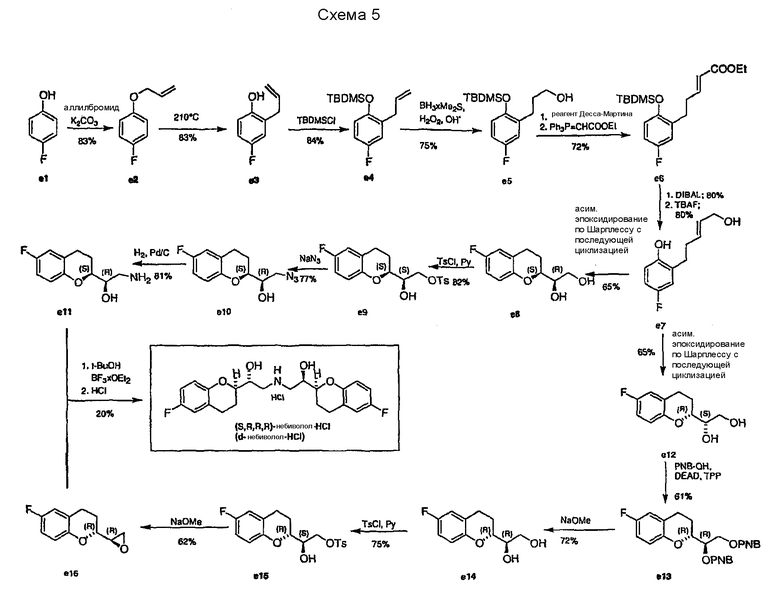
Основой для упомянутой энантиоселективной стратегии является асимметрическое однореакторное эпоксидирование аллилового спирта e7 по Шарплессу, в котором используют (-)-DET и (+)-DET, для получения обоих энантиомерных диолов e8 и e12 после стадии циклизации.
Исходным соединением является 4-фторфенол e1, который сначала превращают в простой аллиловый эфир e2. Перегруппировка Кляйзена при 210°С с последующей защитой фенольной группы (e3) TBDMS-Cl приводит к образованию интермедиата e4. Первичный спирт e5 получают гидроборированием и последующей окислительной обработкой H2O2. Продукт e5 превращают в α,β-ненасыщенный сложный эфир e6 однореакторным окислением периоданом Десса-Мартина и олефинированием по Виттигу. Затем соединение e6 восстанавливают DIBAL-H в аллиловый спирт e7. На этой стадии путь разделяется на два пути, каждый из которых начинается с асимметрического эпоксидирования по Шарплессу и циклизации в одном реакторе. Согласно первому пути диол e8 может быть получен с выходом 65% действием (-)-DET. Селективное тозилирование первичного спирта e8 и замещение e9 азидом с последующим каталитическим восстановлением е10 дает аминоспирт е11. По второму пути синтезируют диол e12, почти аналогично диолу e8, за исключением того, что используют (+)-DET для эпоксидирования по Шарплессу, получая соответствующее энантиомерное соединение. Инверсия на атоме углерода C2 в условиях реакции Мицунобу действием п-нитробензойной кислоты приводит к образованию соединения e13, защищенного двумя PNB группами. После удаления защитных групп может быть получен диастереомерный диол e14. Селективное тозилирование e14 и обработка полученного e15 основанием дает эпоксид е16. Синтез гидрохлорида d-небиволола может быть завершен реакцией сочетания аминоспирта е11 с эпоксидом е16 с последующей трансформацией в гидрохлоридную соль.
Стратегия, описанная в статье Chandrasekhar с соавторами, имеет следующие недостатки:
1. Стадия получения соединения e3 из e2 требует высокой температуры для перегруппировки Кляйзена, которая практически недостижима в промышленных масштабах.
2. Для синтеза только одного энантиомера небиволола требуется до 16 стадий, но необходима рацемическая смесь.
3. Последняя стадия сочетания дает d-небиволол с низким выходом (20%).
4. Асимметрическое эпоксидирование по Шарплессу, как известно, не приводит к энантиочистым продуктам. Поэтому вероятны загрязнения другими стереоизомерами. Для определения таких возможных загрязнений недостаточно описанного способа измерения оптической чистоты, как уже упоминалось в WO 2004/041805.
5. Почти все интермедиаты очищают колоночной хроматографией, так как большинство интермедиатов представляют собой маслообразные соединения.
Таким образом, многочисленные стадии (более 13), низкий выход, использование необычных катализаторов, условий реакции, специального оборудования и колоночной хроматографии для очистки преимущественно маслообразных интермедиатов делают имеющиеся стратегии и большинство стадий слишком трудоемкими и экономически неприемлемыми для промышленного способа.
Несмотря на вышеописанные работы, существует потребность в новом, эффективном и практически осуществимом в промышленных масштабах способе получения рацемического небиволола с увеличенным общим выходом.
Все цитируемые здесь ссылки включены в данную заявку во всей полноте путем ссылки.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым соединениям и интермедиатам, а также к способам, которые можно использовать непосредственно для селективного синтеза небиволола или рацемического ([2S*[R*[R[R*]]]]- и ([2R*[S*[S[S*]]]]-(±)-α,α`- [иминобис(метилен)]бис[6-фторхроман-2-метанола] формулы (I)
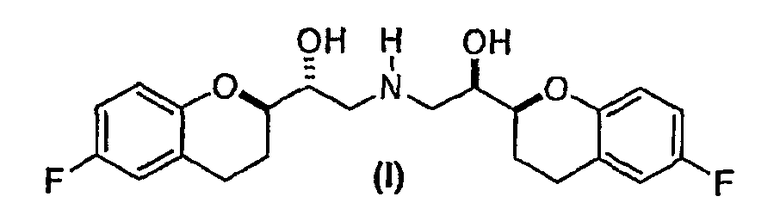
и его чистых ([2S[2R*[R[R*]]]]- и ([2R*[S*[S[S*]]]]-энантиомерных форм и фармацевтически приемлемых солей.
Соответственно способ получения рацемического [2S*[R*[R*[R*]]]] и [2R*[S*[S*[S*]]]]-(±)α,α`-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2H-1-бензопиран-2-метанола] и его фармацевтически приемлемых солей включает
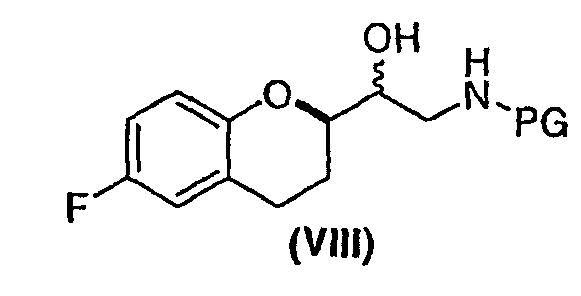
(a) предоставление соединения формулы (VIII)
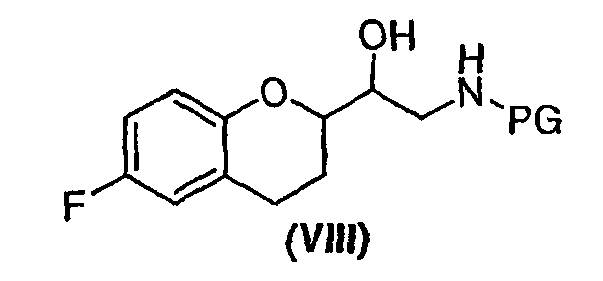
в виде диастереомерно чистого соединения, содержащего по меньшей мере 95% RS/SR конфигурации или RR/SS конфигурации, где PG представляет собой водород или аминозащитную группу, причем аминозащитная группа представляет собой по меньшей мере одну из аллильной группы или арил-C1алкильной группы;
(b) предоставление рацемического соединения формулы (V)
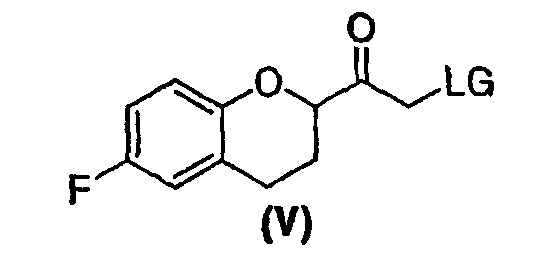
где LG выбрана из группы, состоящей из хлора, брома, йода, алкилсульфонилокси и арилсульфонилокси;
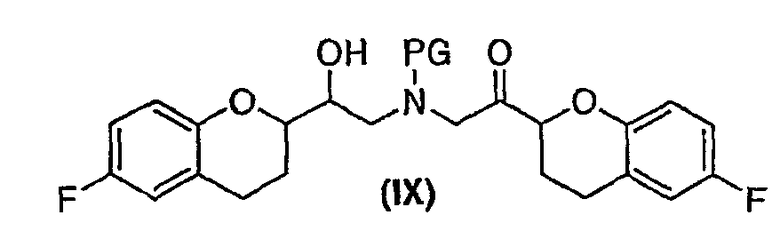
(c) N-алкилирование соединения формулы (VIII) соединением формулы (V), где указанное N-алкилирование проводят в инертном органическом растворителе в присутствии основания и необязательно в присутствии катализатора, с получением соединения формулы (IX),
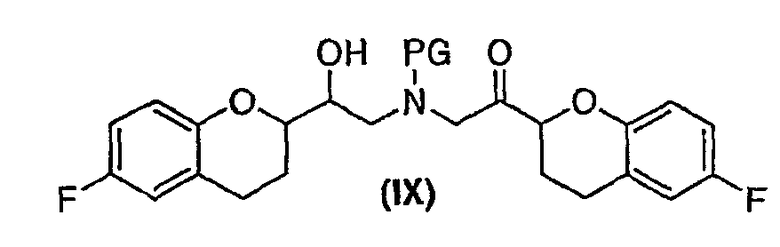
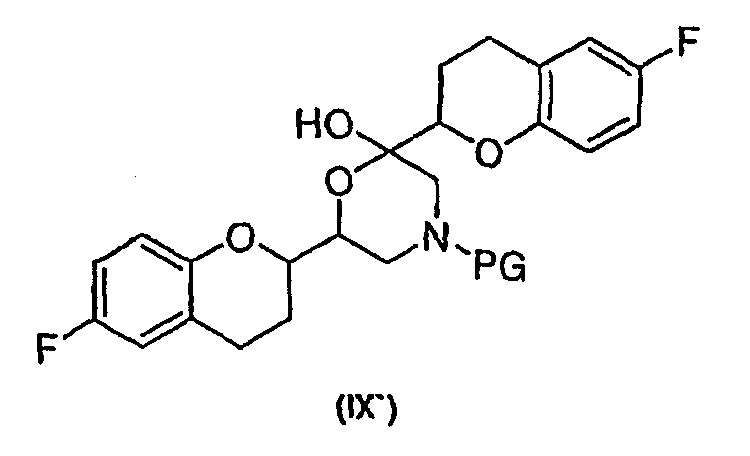
соединения формулы (IX'), которое представляет собой циклическую полукетальную форму соединения формулы (IX),
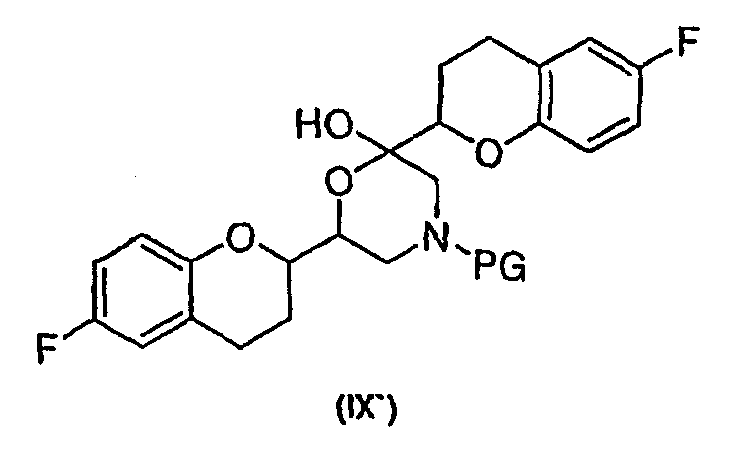
или их смеси, где соединение формулы (IX) и соединение формулы (IX') представляют собой смеси диастереомеров;
(d) разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') по меньшей мере одним из (d1) или (d2) способов, где
(d1) разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') фракционной кристаллизацией после образования соли или после модификации с получением по существу чистых диастереомеров формулы (IX) или формулы (IX'), содержащих по меньшей мере 50% RSS/SRR или RRS/SSR конфигурации;
(d2) разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') с получением по существу чистых диастереомеров формулы (IX) или формулы (IX'), содержащих по меньшей мере 50% RSS/SRR или RRS/SSR конфигурации, на стадии одновременной эпимеризации-кристаллизации, где стадия эпимеризации-кристаллизации включает:
(1) эпимеризацию соединения формулы (IX) или формулы (IX') RSR/SRS конфигурации с получением смеси диастереомеров формулы (IX) или формулы (IX') RSS/SRR конфигурации и RSR/SRS конфигурации или
эпимеризацию соединения формулы (IX) или (IX') RRR/SSS конфигурации с получением смеси диастереомеров формулы (IX) или формулы (IX') RRS/SSR конфигурации и RRR/SSS конфигурации, при условии, что указанную эпимеризацию проводят в присутствии основания и органического растворителя, где смесь необязательно охлаждают, используя градиент температуры и где диастереомеры RSS/SRR конфигурации или RRS/SSR конфигурации в смеси получают по меньшей мере в двукратном избытке относительно диастереомеров RSR/SRS конфигурации и RRR/SSS конфигурации; и
(2) кристаллизацию по существу чистых диастереомеров формулы (IX) или формулы (IX') с RSS/SRR конфигурацией или RRS/SSR конфигурацией по меньшей мере в двукратном избытке относительно диастереомеров RSR/SRS конфигурации и RRR/SSS конфигурации;
разделение смеси фракционной кристаллизацией необязательно после образования соли или после модификации с получением по существу чистых диастереомеров формулы (IX) или формулы (IX') с RSS/SRR или RRS/SSR конфигурацией;
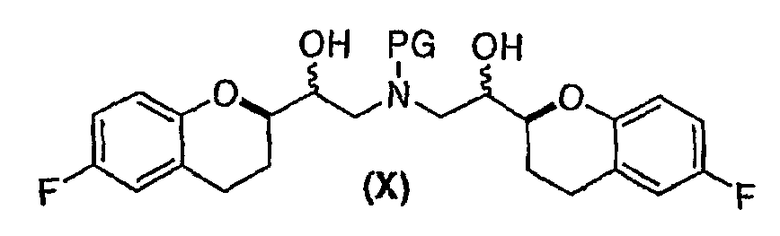
(e) восстановление по существу чистых диастереомеров формулы (IX) или формулы (IX') с RSS/SRR или RRS/SSR конфигурацией с получением соединения формулы (X)
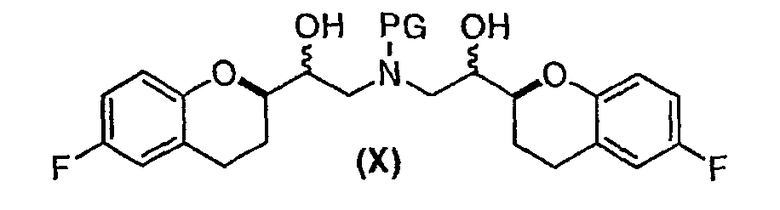
в виде RSSS/SRRR диастереомерной смеси, в которой отношение RSSS/SRRR диастереомерной конфигурации к SRSR или RRSS диастереомерной конфигурации составляет по меньшей мере 1;
(f) снятие защиты соединения формулы (X), при условии, что PG не является H (а если РG представляет собой H, тогда отменяют стадию снятия защиты) с получением соединения формулы (I) или его фармацевтически приемлемых солей; и
(g) удаление соединения формулы (I) или его фармацевтически приемлемых солей RSRS или RRSS диастереомерной конфигурации, если они имеются, перекристаллизацией или суспендированием с получением рацемического [2S*[R*[R*[R*]]]] и [2R*[S*[S*[S*]]]]-(±)[α,α'-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2H-1-бензопиран-2-метанол] или его фармацевтически приемлемые соли.
Дополнительно предоставляется рацемический ([2S*[R*[R*[R*]]]]- и ([2R*[S*[S*[S*]]]]-(±)-α,α`-[имино-бис(метилен)]бис[6-фторхроман-2-метанол] соединения формулы (I), полученное способом, описанным выше.
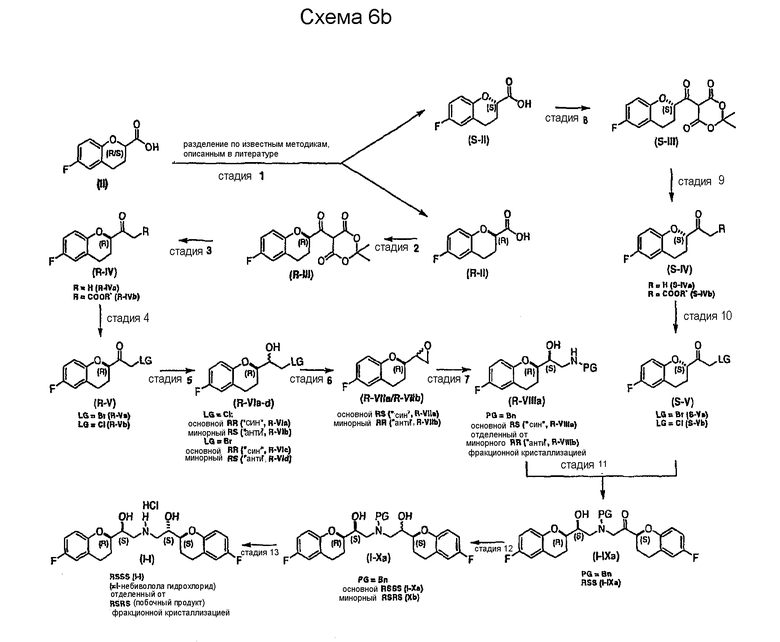
Предпочтительный вариант осуществления способа показан на схеме 6а
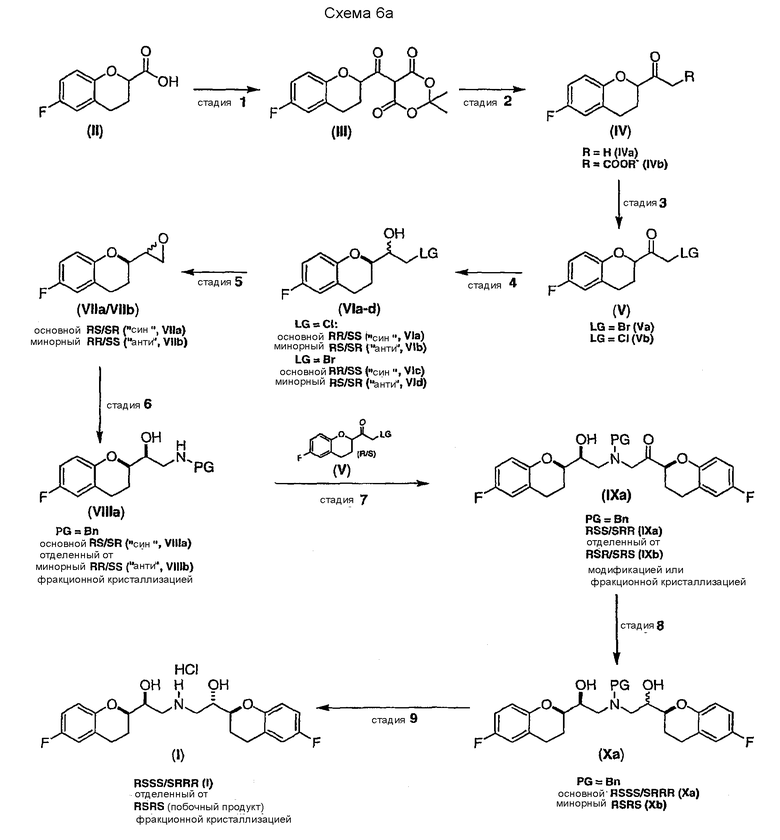
Исходным соединением является 6-фторхроман-2-карбоновая кислота II, которую превращают соответствующими трансформациями (стадии 1, 2 и 3) в соединение V, содержащее подходящую уходящую группу (LG). Селективное восстановление соединения V с последующим образованием эпоксида и замещением защищенным амином дает соединение VIIIa после фракционной кристаллизации. В этом случае порядок трансформаций может быть изменен без необходимости образования эпоксида. Реакция сочетания соединения VIIIa с соединением V дает диастереомерную смесь соединений IXa и Xb, после чего соединение IXa селективно выделяют и почти селективно восстанавливают в смесь соединений Xa (основной продукт) и Xb (минорный продукт). Полученную смесь затем подвергают снятию защиты и после образования соли с HCl рацемический гидрохлорид небиволола I селективно кристаллизуется. Общий выход составляет 8%, однако дополнительные количества соединения V, используемые на стадии 7, не учитываются.
В предпочтительном варианте осуществления изобретения защитная группа представляет собой бензильную группу. В некоторых вариантах осуществления изобретения уходящая группа представляет собой хлор или бром.
В некоторых вариантах осуществления способа на стадии (b) соединение формулы (V) получают в количестве от около 1,0 до около 1,5 эквивалента.
В некоторых вариантах осуществления способа на стадии (c) органический растворитель представляет собой полярный апротонный растворитель, выбранный из группы, состоящей из ДМФА, DMA и NMP.
В некоторых вариантах осуществления способа на стадии (c) основание представляет собой по меньшей мере один из третичных аминов, карбонатов щелочных металлов или гидрокарбонатов щелочных металлов. Предпочтительным основанием является гидрокарбонат натрия. Предпочтительно использовать от около 1,5 до около 2,5 эквивалента основания.
В некоторых вариантах осуществления способа на стадии (c) катализатор представляет собой по меньшей мере один из бромидов щелочных металлов, йодидов щелочных металлов, бромидов тетраалкиламмония или йодидов тетраалкиламмония. Предпочтительным катализатором является бромид натрия. Предпочтительно используют от около 0,1 до около 0,25 эквивалента катализатора и наиболее предпочтительно использовать 0,15 эквивалента катализаторов.
В некоторых вариантах осуществления изобретения на стадии (c) указанное N-алкилирование проводят при температуре от около комнатной до около 80°С.
В некоторых вариантах осуществления на стадии (d) фракционную кристаллизацию проводят в растворителе. Предпочтительным растворителем является ацетонитрил. Предпочтительно использовать свободный амин для фракционной кристаллизации. В некоторых вариантах осуществления используют от около 0,4/n до около 0,6/n эквивалента силилирующего реагента и n обозначает число переносимых силильных групп, приходящихся на силилирующий реагент. Предпочтительно, чтобы силилирующий реагент представлял собой по меньшей мере один из триметилсилилхлорида, HMDS или BSU.
В некоторых вариантах осуществления изобретения на стадии (d1) силилирующий реагент используют для модификации до проведения фракционной кристаллизации из растворителя. Модификацию проводят предпочтительно в присутствии от около 1,0 до около 2,0 эквивалента основания. Предпочтительным основанием является имидазол.
В некоторых вариантах осуществления на стадии (d1) упомянутое разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') проводят в ацетонитриле, метил-трет-бутиловом эфире (MTBE), циклогексане или их смесях.
В некоторых вариантах осуществления способа стадию (d2) проводят для соединения формулы (IX) или (IX`) RSR/SRS конфигурации. Конфигурацию RSS/SRR в смеси получают предпочтительно при примерно девятикратном избытке RSR/SRS.
В некоторых вариантах осуществления способа на стадии (d2) смесь охлаждают при градиенте температуры от около 70°C до около 20°C. В предпочтительном варианте осуществления изобретения градиент температуры составляет от 70° до 40°С.
В некоторых вариантах осуществления способа на стадии (d2) упомянутую эпимеризацию проводят в присутствии по меньшей мере 0,1 эквивалента основания. В некоторых вариантах осуществления упомянутую эпимеризацию проводят в присутствии по меньшей мере 0,25 эквивалентов основания.
В некоторых вариантах осуществления на стадии (d2) основание представляет собой соединение, выбранное из группы, состоящей из алкоксида, амидина, гуанидина и фосфазена. Предпочтительным основанием является амидин. В предпочтительном варианте осуществления изобретения основание представляет собой диазабициклоундецен.
В некоторых вариантах осуществления на стадии (d2) содержание воды, если она имеется, не может превышать 1,0%. В некоторых вариантах осуществления на стадии (d2) содержание воды, если она имеется, не может превышать 0,1%.
В некоторых вариантах осуществления на стадии (e), упомянутое восстановление проводят для соединения формулы (IX) или соединения формулы (IX') RSS/SRR конфигурации в растворителе действием боргидрида щелочного металла, боргидрида тетрабутиламмония, селектрида (SELECTRIDE) щелочного металла или боргидрида цинка необязательно в присутствии кислоты Льюиса. В некоторых вариантах осуществления кислота Льюиса представляет собой по меньшей мере одно вещество из Ti(OАлкил)4, ZnCl2, галогенида щелочного металла или галогенида щелочноземельного металла. Предпочтительно, чтобы растворитель представлял собой по меньшей мере один из эфира, спирта или галогенированного углеводорода. В некоторых вариантах осуществления изобретения упомянутое восстановление проводят при температурах от около -20°C до около комнатной.
В некоторых вариантах осуществления на стадии (f) упомянутое снятие защиты проводят каталитическим гидрированием.
В некоторых вариантах осуществления на стадии (g) упомянутую очистку соединения формулы (I) осуществляют суспендированием его гидрохлоридной соли в растворителе. Суспендирование проводят предпочтительно в метаноле в качестве растворителя.
В некоторых вариантах осуществления изобретения упомянутое предоставление соединения формулы (VIII) включает:
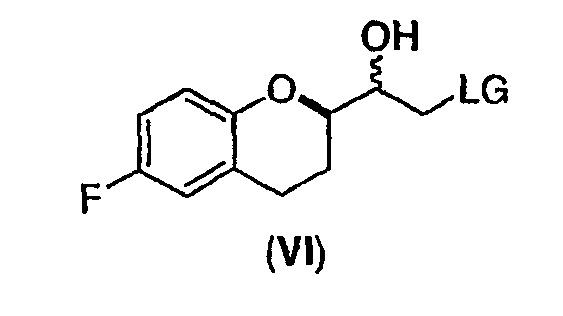
(i) восстановление рацемического соединения формулы (V) в растворителе и необязательно в присутствии кислоты Льюиса, где LG представляет собой бром или хлор, с образованием диастереомерной смеси соединений формулы (VI)
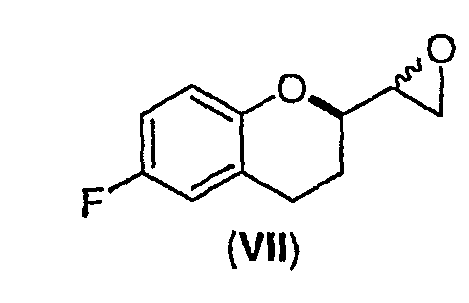
(ii) образование смеси диастереомеров соединения формулы (VII)
 ;
;
(iii) взаимодействие диастереомеров соединения формулы (VII) с NH2PG с образованием смеси диастереомеров соединения формулы (VIII)
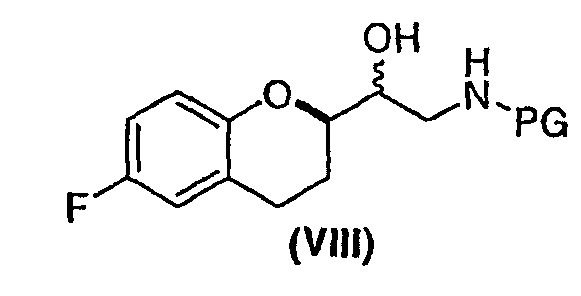
и
(iv) разделение диастереомеров соединения формулы (VIII) смеси диастереомеров фракционной кристаллизацией необязательно после образования соли. В предпочтительном варианте осуществления изобретения PG представляет собой бензильную группу.
В некоторых вариантах осуществления изобретения выделяют по меньшей мере один из диастереомеров соединения формулы VIII, имеющий RR/SS или RS/SR конфигурацию.
Еще один аспект настоящего изобретения относится к способу рециркуляции нежелательных диастереомеров, получаемых во время способа, который уменьшает затраты и делает способ получения небиволола более эффективным. Конкретно во время селективного получения соединений VIIIa и Xa нежелательные диастереомеры образуются в качестве минорных продуктов. Поэтому рециркуляция отходов представляет собой экономическое и экологическое преимущество по сравнению с предшествующими способами получения небиволола.
Алкилирование соединения VIIIa соединением V также образует диастереомерную смесь (смесь IXa и IXb, 1:1), где стадия рециркуляции делает возможной трансформацию нежелательной формы IXb или путем селективного расщепления с образованием соединения VIIIa, или проведением подходящей стадии эпимеризации в смесь соединений IXa и IXb с последующим селективным выделением желательного диастереомера IXa.
В некоторых вариантах осуществления способа проводят рециркуляцию соединения формулы (VIII) RR/SS конфигурации, который включает:
предоставление соединения формулы (VIII) с защитной группой RR/SS конфигурации; и
инверсию конфигурации спирта с образованием соединения формулы VIII SR/RS конфигурации.
В некоторых вариантах осуществления изобретения на стадии i) восстановитель выбирают из боргидрида щелочного металла, боргидрида тетраалкиламмония, боргидрида цинка, триацетоксиборгидрида щелочного металла, SUPERHYDRIDE, RED-AL, селектрида (SELECTRIDE) щелочного металла или координационных боргидридов. В некоторых вариантах осуществления изобретения на стадии i) упомянутое восстановление проводят в условиях реакции Меервейна-Понндорфа-Верлея. В некоторых вариантах осуществления на стадии i) упомянутое восстановление проводят каталитическим гидрированием. В некоторых вариантах осуществления на стадии i) кислота Льюиса представляет собой кислоту, выбранную из группы, состоящей из хлоридов щелочных или щелочноземельных металлов, хлорида цинка, алкоксида титана (IV) и триалкоксида алюминия. В некоторых вариантах осуществления на стадии i) упомянутое восстановление проводят в условиях, которые приводят к образованию соединения формулы (VI) RR/SS изомера в избытке. В некоторых вариантах осуществления на стадии i) упомянутое восстановление проводят при температуре от около -78°C и до примерно комнатной. Предпочтительно проводить упомянутое восстановление при температуре от -20°C и до комнатной. В некоторых вариантах осуществления на стадии i) растворитель представляет собой растворитель, выбранный из группы, состоящей из спиртов, простых эфиров, галогенированных углеводородов и ароматических растворителей.
В некоторых вариантах осуществления на стадии ii) упомянутое образование смеси диастереомеров соединения формулы (VII) проводят в растворителе и в присутствии основания. Растворитель предпочтительно представляет собой спирт и основание представляет собой алкоголят щелочного металла. Используют предпочтительно 1,0-2,0 эквивалента основания.
В некоторых вариантах осуществления на стадии ii) упомянутое образование смеси диастереомеров соединения формулы (VII) проводят при температурах от 0°С до 40°С.
В некоторых вариантах осуществления на стадии iv) фракционную кристаллизацию проводят в толуоле, ацетонитриле, C1-C3-спирте, простом эфире или их смесях. Предпочтительным C1-C3-спиртом является 2-пропанол, а простой эфир представляет собой по меньшей мере один из диизопропилового эфира или MTBE.
В некоторых вариантах осуществления изобретения упомянутое предоставление рацемического соединения формулы (V) включает:
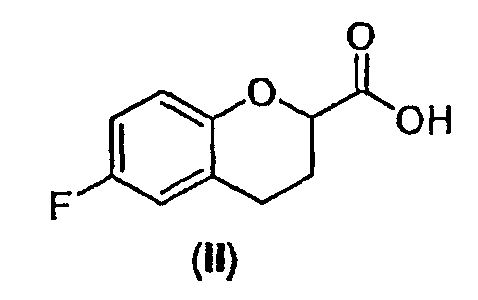
(1) превращение соединения формулы (II)
в активированное производное кислоты;
(2) взаимодействие активированного производного кислоты с кислотой Мелдрума в присутствии основания с образованием соединения формулы (III)
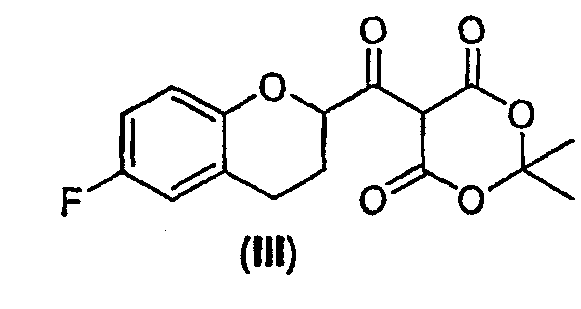 ;
;
(3) превращение соединения формулы (III) в соединение формулы (IV)
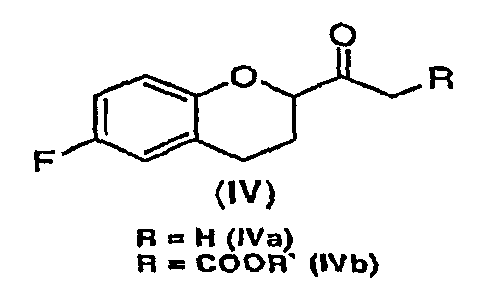
где R представляет собой водород или COOR` и где R` представляет собой C1-C6алкил или арил-C1алкил; и
(4) галогенирование соединения формулы (IV) и необязательно проведение гидролиза и декарбоксилирования с образованием соединения формулы (V).
В некоторых вариантах осуществления изобретения на стадии (1) карбоновую кислоту превращают в соответствующий хлорангидрид кислоты.
В некоторых вариантах осуществления изобретения на стадии (2) основание представляет собой третичный амин. В некоторых вариантах осуществления используют 1-3 эквивалента кислот Мелдрума. В некоторых вариантах осуществления на стадии (2) температура реакции составляет от около -10°C до около +30°C.
В некоторых вариантах осуществления на стадии (3) соединение формулы (III) гидролизуют в смеси органической кислоты и воды и получают соединение формулы (IV), где R представляет собой H. В предпочтительном варианте осуществления изобретения органическая кислота представляет собой уксусную кислоту и гидролиз проводят при температуре кипения. В некоторых вариантах осуществления на стадии (3) соединение формулы (IV), содержащее R в виде СOOR' и R` в виде C1-C6 алкила или арил-C1алкила, получают алкоголизом соединения формулы (III). Алкоголиз проводят предпочтительно этанолом и трет-бутанолом. Алкоголиз проводят предпочтительно при температурах от около 70°C до около 80°C.
В некоторых вариантах осуществления изобретения на стадии (3) растворитель представляет собой по меньшей мере один из спирта или толуола.
В некоторых вариантах осуществления изобретения на стадии (4) перед проведением галогенирования соединение формулы (IV), в котором R представляет собой H, превращают силилированием в соответствующий силиленоловый эфир, содержащий концевую двойную связь. В некоторых вариантах осуществления изобретения силилирование осуществляют путем кинетически контролируемого депротонирования диизопропиламидом лития (LDA) с последующим силилированием при температуре от около -78°C до около -40°C. Предпочтительно проводить силилирование при температуре от -78°C до -70°C. В предпочтительном варианте осуществления изобретения силилирующим агентом является TMSCl.
В некоторых вариантах осуществления изобретения на стадии (4) после превращения в простой силиленоловый эфир проводят галогенирование, используя бромирующий реагент. Предпочтительным бромирующим реагентом является N-бромсукцинимид.
В некоторых вариантах осуществления изобретения на стадии (4) соединение формулы (IV), где R представляет собой COOR`, сначала галогенируют, а затем превращают в соединение формулы (V) гидролизом эфира с последующим декарбоксилированием. Предпочтительно проводить галогенирование в присутствии катализатора. В некоторых вариантах осуществления в качестве галогенирующих реагентов используют от около 1,0 до около 1,5 эквивалента N-бромсукцинимида, N-хлорсукцинимида или SO2Cl2. В некоторых вариантах осуществления в качестве катализатора используют около 0,2-0,4 эквивалента Mg(ClO4)2.
В некоторых вариантах осуществления изобретения на стадии (4) упомянутое галогенирование проводят при температурах от 0°С до примерно комнатной.
В некоторых вариантах осуществления изобретения на стадии (4) после упомянутого галогенирования проводят гидролиз сложного эфира с последующим декарбоксилированием в водном растворе органической кислоты. Органическая кислота представляет собой предпочтительно по меньшей мере одну из трифторуксусной кислоты, муравьиной кислоты и уксусной кислоты. Предпочтительно проводить гидролиз и декарбоксилирование при температурах от около 75°С до около 90°С.
В некоторых вариантах осуществления способ дополнительно включает рециркуляцию соединения формулы (IX) или (IX′) RSR/SRS или RRR/SSS конфигурации, где упомянутая рециркуляция включает:
эпимеризацию соединения формулы (IX) или (IX') RSR/SRS или RRR/SSS конфигурации, с получением смеси диастереомеров формулы (IX) или формулы (IX') RSS/SRR конфигурации, содержащей диастереомеры RSR/SRS конфигурации, или смеси диастереомеров формулы (IX) или формулы (IX') RRS/SSR конфигурации, содержащей диастереомеры RRR/SSS конфигурации, и
разделение смеси фракционной кристаллизацией после образования соли или после модификации, приводящее к по существу чистым диастереомерам формулы (IX) или формулы (IX'), имеющим RSS/SRR или RRS/SSR конфигурацию.
В некоторых вариантах осуществления способ дополнительно включает рециркуляцию соединения формулы (IX) или (IX') RSR/SRS или RRR/SSS конфигурации, где упомянутая рециркуляция включает:
расщепление соединения формулы (IX) или (IX') RSR/SRS или RRR/SSS конфигурации, которое дает смесь, содержащую диастереомеры формулы (VIII) RS/SR или RR/SS конфигурации; и
разделение RS/SR или RR/SS конфигурации диастереомеров формулы (VIII).
Дополнительным аспектом является соединение формулы (III)
Дополнительным аспектом является соединение формулы (IV)
где R представляет собой водород или COOR' и R` представляет собой С1-С6алкил или арил-С1алкил.
Дополнительным аспектом является соединение формулы (V)
где LG представляет собой бром или хлор.
Дополнительным аспектом является соединение формулы (VI)
где LG представляет собой бром или хлор.
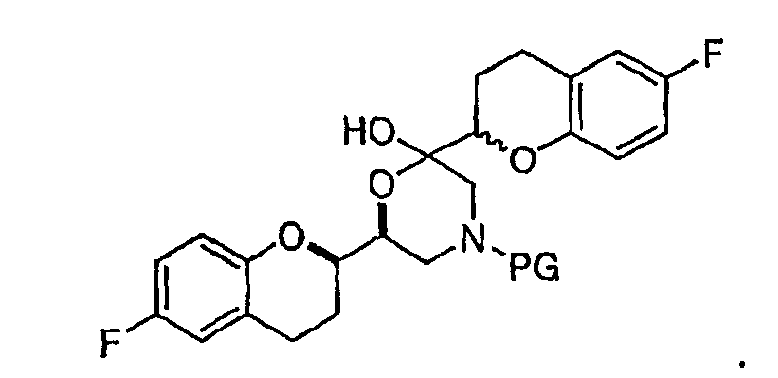
Дополнительным аспектом является соединение формулы (IX)
или его циклическая полукетальная форма формулы (IX'),
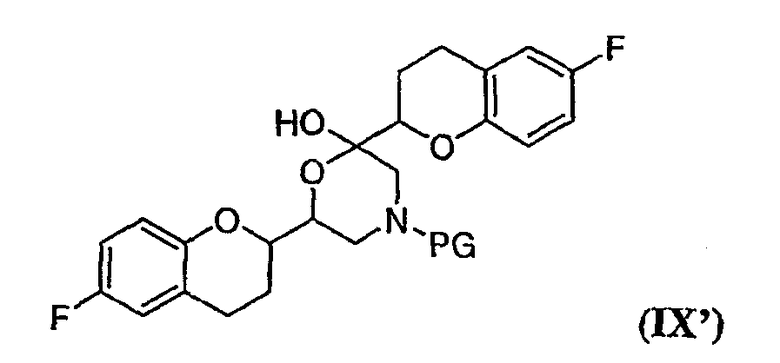
где PG представляет собой защитную группу, выбранную из водорода, аллила и арил-С1алкила.
Дополнительным аспектом является соединение формулы (IX) с RSS/SRR конфигурацией
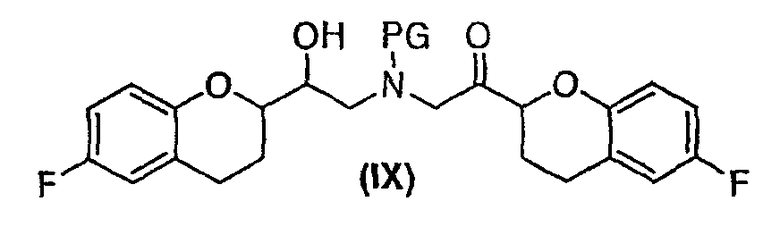
или его циклическая полукетальная форма формулы (IX')
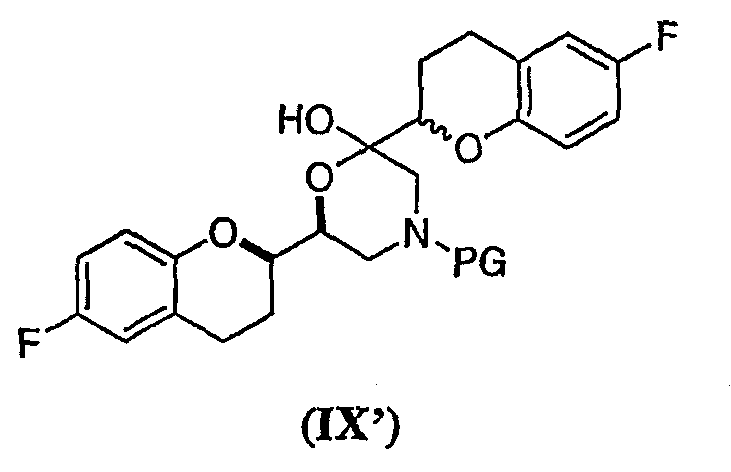
где PG представляет собой бензильную группу.
Аспектом изобретения является также способ получения рацемического [2S*[R*[R*[R*]]]] и [2R*[S*[S*[S*]]]]-(±)α,α`-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2H-1-бензопиран-2-метанола] и его фармацевтически приемлемых солей, включающий: предоставление соединения формулы (IX); и восстановление соединения формулы (IX) с получением соединения формулы (X), содержащего не более 50% стереоизомера, имеющего RSRS конфигурацию. Указанный способ дополнительно включает получение соединения (VIII) и соединения формулы (V).
Согласно настоящему изобретению можно также получать энантиомерно чистый небиволол (l-небиволол или d-небиволол), например, после разделения соединения II (см. Схемы 6b и 6c). Каждый из энантиомеров (S-II и R-II) может быть трансформирован теми же способами, которые описаны для соответствующего рацемического соединения II (ср. схему 6a) с получением первых ключевых интермедиатов S-V и R-V. Гидрохлорид l-небиволола (схема 6b) можно затем получать синтезом R-VIIIa, исходя из R-V, с последующей реакцией сочетания с S-V (стадия 11), селективным восстановлением (стадия 12), снятием защиты и образованием соли (стадия 13). Гидрохлорид d-небиволола может быть получен таким же образом, за исключением того, что в отличие от синтеза l- небиволола, энантиомерный интермедиат S-VIIIa получают из S-V и затем вводят в реакцию сочетания с R-V (схема 6c, стадия 11). Селективное восстановление (стадия 12) с последующим снятием защиты и образованием соли дает гидрохлорид d-небиволола.
Для специалиста в данной области техники очевидно, что различные изменения и модификации рассмотренных путей могут быть использованы для энантиоселективных синтезов l- или d-небиволола. Поэтому применение интермедиатов для энантиоселективного синтеза l- или d-небиволола, получаемых по схемам 6b и 6c, не ограничивается описанными путями. Например, эпоксиды R-VIIa, R-VIIb, S-VIIa и S-VIIb (полученные из ключевых интермедиатов R-V или S-V) можно также использовать непосредственно для синтеза каждого энантиомера небиволола, например, по пути, описанному на схеме 2. От восстановителя зависит, получаются ли эпоксиды R-VIIa, R-VIIb, S-VIIa и S-VIIb в качестве основного или минорного соединения.
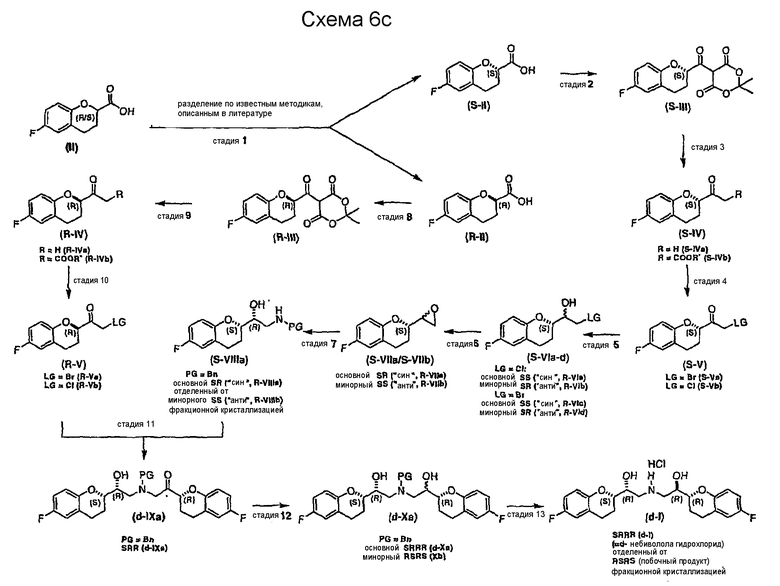
Способ получения соединения формулы (I) в виде рацемической смеси или энантиомерно чистой форме и его фармацевтически приемлемых солей включает:
(a) разделение соединения формулы (II)
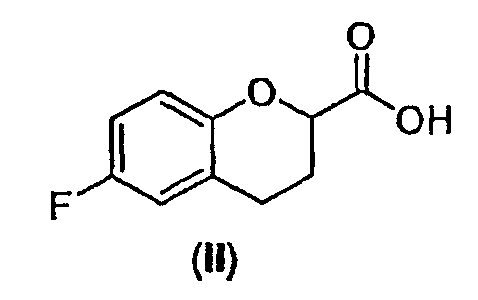
с получением соединения формулы (II) S конфигурации и R конфигурации;
(b) превращение соединения формулы (II) S конфигурации в соединение формулы (V) S конфигурации,
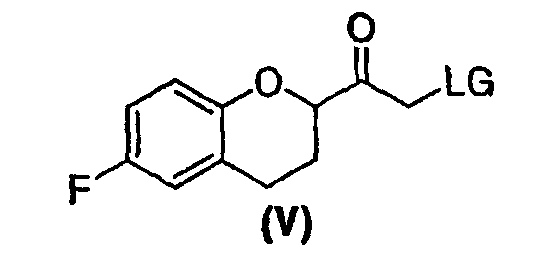
где LG представляет собой заместитель, выбранный из группы, состоящей из хлора, брома, йода, алкилсульфонилокси и арилсульфонилокси, через образование соединения формулы (III) S конфигурации и соединения формулы (IV) S конфигурации;
(c) превращение соединения формулы (II) R конфигурации в соединение формулы (V) R конфигурации через образование соединения формулы (III) R конфигурации и соединения формулы (IV) R конфигурации;
(d) предоставление соединения формулы (VIII)
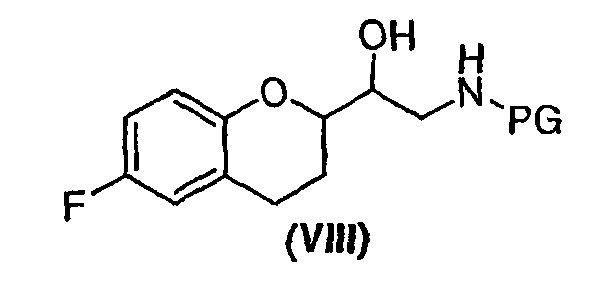
где PG представляет собой водород или аминозащитную группу, где аминозащитная группа представляет собой по меньшей мере одну из аллильной группы или арил-С1алкильной группы и где соединение формулы (VIII) представляет собой энантиомерное соединение с RS или SR конфигурацией;
(e) проведение N-алкилирования (i) соединения формулы (VIII) RS конфигурации соединением формулы (V) S конфигурации или (ii) соединения (VIII) SR конфигурации соединением формулы (V) R конфигурации, при условии, что упомянутое N-алкилирование проводят в инертном органическом растворителе в присутствии основания и необязательно в присутствии катализатора с получением RSS или SRR энантиомерной формы соединения формулы (IX)
или RSS или SRR энантиомерной формы соединения (IX`), которое представляет собой циклическую полукетальную форму соединения формулы (IX)
(f) восстановление по меньшей мере одной из RSS или SRR энантиомерных форм соединения формулы (IX) или формулы (IX') с получением по меньшей мере одной RSSS или SRRR энантиомерной формы соединения формулы (X)
(g) снятие защиты по меньшей мере с одной из RSSS или SRRR энантиомерных форм соединения формулы (X), при условии, что PG не является H (а если PG представляет собой Н, тогда указанную стадию снятия защиты отменяют), чтобы получить соединение формулы (I) или его фармацевтически приемлемые соли; и
(h) удаление соединения формулы (I) или его фармацевтически приемлемых солей RSRS диастереомерной конфигурации, если они имеются, в качестве побочного продукта, перекристаллизацией или суспендированием с получением по меньшей мере одного из ([2S*[R*[R*[R*]]]]- и ([2R*[S*[S*[S*]]]]-энантиомеров соединения формулы (I) и его фармацевтически приемлемых солей; и
(i) необязательно объединение ([2S*[R*[R*[R*]]]]- и ([2R*[S*[S*[S*]]]]- энантиомеров соединения формулы (I) и его фармацевтически приемлемых солей для образования рацемического ([2S*[R*[R*[R*]]]]- и ([2R*[S*[S*[S*]]]]-(±)α,α`-[имино-бис(метилен)]бис[6-фторхроман-2-метанола] формулы (I) и его фармацевтически приемлемых солей.
Еще одним аспектом изобретения является способ получения рацемического [2S*[R*[R*[R*]]]]- и [2R*[S*[S*[S*]]]]-(±)α,α`-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2Н-1-бензопиран-2-метанола] и его фармацевтически приемлемых солей, включающий:
(a) предоставление соединения формулы (VIII) в виде диастереомера с RR/SS конфигурацией, где PG представляет собой водород или аминозащитную группу, где аминозащитная группа представляет собой по меньшей мере одну из аллильной группы или арил-C1 алкильной группы;
b) предоставление рацемического соединения формулы (V), где LG представляет собой заместитель, выбранный из группы, состоящей из хлора, брома, йода, алкилсульфонилокси и арилсульфонилокси;
(c) N-алкилирование соединения формулы (VIII) соединением формулы (V), где упомянутое N-алкилирование проводят в инертном органическом растворителе в присутствии основания и необязательно в присутствии катализатора и получают соединение формулы (IX), соединение формулы (IX'), которое является циклической полукетальной формой соединения формулы (IX), или их смесь, где соединение формулы (IX) и соединение формулы (IX') представляют собой смеси диастереомеров, имеющих RRR/SSS и RRS/SSR конфигурацию;
(d) разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') фракционной кристаллизацией после образования соли или после модификации с получением по существу чистых диастереомеров формулы (IX) или формулы (IX'), имеющих по меньшей мере 50% RRR/SSS или RRS/SSR конфигурации;
(e) восстановление по существу чистых диастереомеров формулы (IX) или формулы (IX') RRS/SSR конфигурации, которое дает соединение формулы (X) в виде RSSS/SRRR диастереомерной смеси, в которой отношение RSSS/SRRR диастереомерной конфигурации к SRSR или RRSS диастереомерной конфигурации, составляет по меньшей мере 1;
(f) снятие защиты соединения формулы (X), при условии, что PG не является H с получением соединения формулы (I) или его фармацевтически приемлемых солей; и
(g) удаление соединения формулы (I) или его фармацевтически приемлемых солей RSRS диастереомерной конфигурации, если имеются, перекристаллизацией или суспендированием, приводящее к образованию рацемического [2S[2R*[R[R*]]]] и [2R[2S*[S[S*]]]]-(±)α,α`-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2H-1-бензопиран-2-метанола] или его фармацевтически приемлемых солей.
В некоторых вариантах осуществления изобретения способ дополнительно включает расщепление соединения формулы (IX) или соединения формулы (IX') RRR/SSS конфигурации с образованием соединения формулы (VIII) в виде диастереомера RR/SS конфигурации.
В одном варианте способ дополнительно включает эпимеризацию соединения формулы (IX) или соединения формулы (IX') RRR/SSS конфигурации с образованием упомянутых смесей диастереомеров соединения формулы (IX) или соединения формулы (IX') RRR/SSS и RRS/SSR конфигурации. В еще одном варианте вышеупомянутого варианта способ дополнительно включает расщепление соединения формулы (IX) или соединения формулы (IX') RRR/SSS конфигурации с образованием соединения формулы (VIII) в виде диастереомера RR/SS конфигурации.
Предоставляется также способ получения соединения формулы (VIII)
способ включает
(i) предоставление рацемического соединения формулы (V)
;
(i) восстановление рацемического соединения формулы (V) в растворителе и необязательно в присутствии кислоты Льюиса, где LG представляет собой бром или хлор, с получением диастереомерной смеси соединения формулы (VI)
 ;
;
(ii) образование смеси диастереомеров соединения формулы (VII)
 ;
;
(iii) взаимодействие диастереомеров соединения формулы (VII) с NH2PG, где PG представляет собой водород или аминозащитную группу и где аминозащитная группа представляет собой по меньшей мере одну из аллильной группы или арил-C1 алкильной группы, которое дает соединение формулы (VIII) в виде смеси диастереомеров; и
(iv) необязательно разделение диастереомеров соединения формулы (VIII) из смеси диастереомеров фракционной кристаллизацией.
Предоставляется также способ получения рацемического [2S*[R*[R*[R*]]]] и [2R*[S*[S*[S*]]]]-(±)α,α`-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2H-1-бензопиран-2-метанола] и его фармацевтически приемлемых солей, включающий:
(a) предоставление соединения формулы (IX), соединения формулы (IX′), которое представляет собой циклическую полукетальную форму соединения формулы (IX) или их смеси, где соединение формулы (IX) и соединение формулы (IX') представляют собой смеси диастереомеров;
(b) разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') по меньшей мере одним из (b1) или (b2) способов, где
(b1) разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') фракционной кристаллизацией после образования соли или после модификации с получением по существу чистых диастереомеров формулы (IX) или формулы (IX'), содержащих по меньшей мере на 50% RSS/SRR или RRS/SSR конфигурации; (b2) разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') с получением по существу чистых диастереомеров формулы (IX) или формулы (IX'), содержащих по меньшей мере на 50% RSS/SRR или RRS/SSR конфигурации на стадии одновременной эпимеризации-кристаллизации, где стадия эпимеризации-кристаллизации включает:
(1) эпимеризацию соединения формулы (IX) или (IX') RSR/SRS конфигурации с получением смеси диастереомеров формулы (IX) или формулы (IX') RSS/SRR конфигурации и RSR/SRS конфигурации или
эпимеризацию соединения формулы (IX) или (IX') RRR/SSS конфигурации, с получением смеси диастереомеров формулы (IX) или формулы (IX') RRS/SSR конфигурации и RRR/SSS конфигурации, при условии, что указанную эпимеризацию проводят в присутствии основания и органического растворителя, где смесь необязательно охлаждают, используя градиент температуры и где RSS/SRR конфигурацию или RRS/SSR конфигурацию в смеси получают по меньшей мере в двухкратном избытке относительно RSR/SRS конфигурации и RRR/SSS конфигурации; и
(2) кристаллизацию по существу чистых диастереомеров формулы (IX) или формулы (IX'), имеющих RSS/SRR конфигурацию или RRS/SSR конфигурацию по меньшей мере в двухкратном избытке относительно RSR/SRS конфигурации и RRR/SSS конфигурации;
разделение смеси фракционной кристаллизацией необязательно после образования соли или после модификации с получением по существу чистых диастереомеров формулы (IX) или формулы (IX') RSS/SRR или RRS/SSR конфигурации;
(c) восстановление по существу чистых диастереомеров формулы (IX) или формулы (IX') RSS/SRR или RRS/SSR конфигурации с получением соединения формулы (X) в виде RSSS/SRRR диастереомерной смеси с отношением RSSS/SRRR диастереомерной конфигурации к SRSR или RRSS диастереомерной конфигурации, которое составляет по меньшей мере 1;
(d) снятие защиты соединения формулы (X) при условии, что PG не является H (а если PG представляет собой H, тогда отменяют стадию снятия защиты) с получением соединения формулы (I)
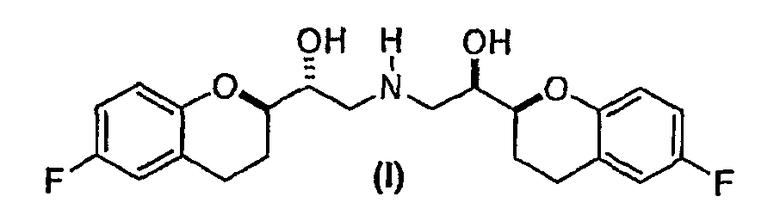
или фармацевтически приемлемых солей; и
(e) удаление соединения формулы (I) RSRS или RRSS диастереомерной конфигурации или его фармацевтически приемлемых солей, если имеются, путем перекристаллизации или суспендирования с получением рацемического [2S*[R*[R*[R*]]]] и [2R*[S*[S*[S*]]]]-(±)α,α`-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2H-1-бензопиран-2-метанол] или его фармацевтически приемлемых солей.
Новые соединения, полученные авторами настоящего изобретения, включают следующие
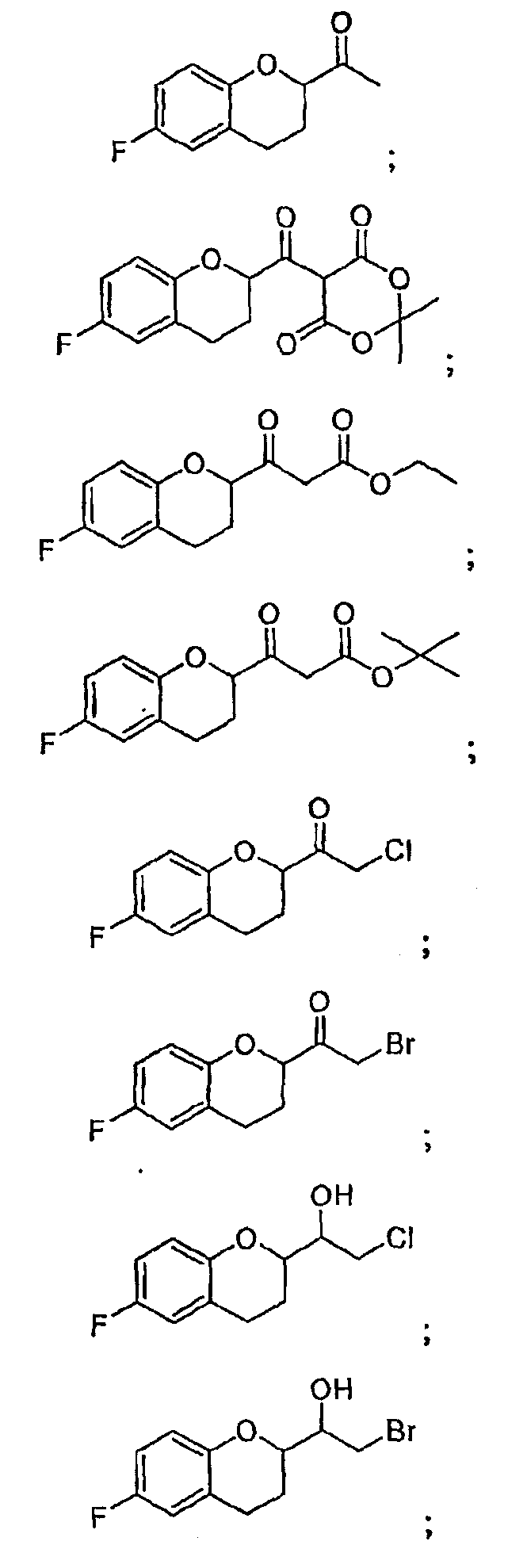
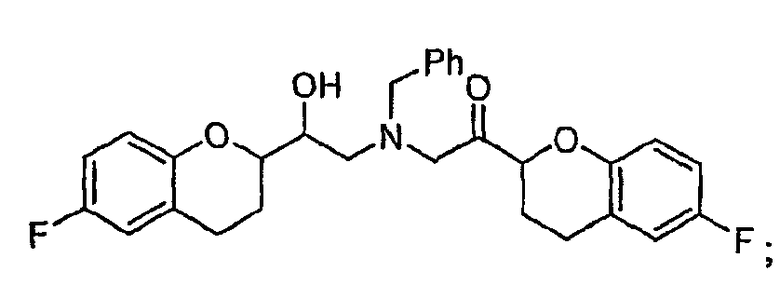 и
и
КРАТКОЕ ОПИСАНИЕ НЕСКОЛЬКИХ ВИДОВ ЧЕРТЕЖЕЙ
Изобретение будет описано вместе с нижеследующими чертежами, в которых подобные номера позиций обозначают подобные элементы и где:
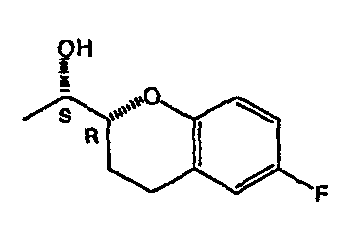
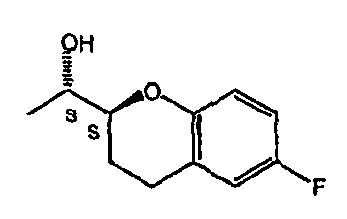
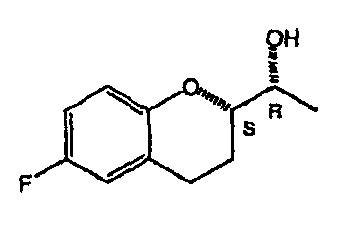
На фигуре 1A изображена структурная формула d-небиволола, на фигуре 1В изображена структурная формула рацемического небиволола.
Фигура 2 представляет собой схематическое изображение молекулы соединения VIIIa с пронумерованными атомами (50% вероятность эллипсоидов; даны произвольные параметры смещения атомов Н для разъяснения).
Фигура 3 представляет собой изображение 13C-ЯМР спектра соединения IXa в циклической полукетальной форме.
Фигура 4 представляет собой изображение 13C-ЯМР спектра соединения IXb в циклической полукетальной форме.
Фигура 5 представляет собой схематическое изображение молекулы соединения IXb в циклической полукетальной форме.
Фигура 6 представляет собой схему, демонстрирующую способ получения рацемического небиволола и его фармацевтически приемлемых солей.
Фигура 7 представляет собой схему, демонстрирующую методику одновременной эпимеризации-кристаллизации по изобретению.
Фигура 8 представляет собой схему, демонстрирующую предпочтительный пример методики одновременной эпимеризации-кристаллизации по изобретению, где защитной группой является бензильная группа.
Фигура 9 представляет собой схему, демонстрирующую способ получения рацемического небиволола и его фармацевтически приемлемых солей, в которой стадия 7b представляет одновременную эпимеризацию-кристаллизацию по изобретению.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым соединениям и способам синтеза рацемического небиволола и его фармацевтически приемлемых солей, а также энантиомерно чистого небиволола и его фармацевтически приемлемых солей. Настоящее изобретение стимулировано желанием создать более эффективный способ, содержащий меньшее количество стадий реакции, чтобы избежать стадий разделения энантиомеров до получения рацемической смеси. Один пример разделения энантиомеров описан в WO 2004/041805 (Схема 3b). Так как все энантиомеры разделяют во время более ранних стадий способа, для получения рацемического небиволола необходимо до 30 стадий в четырех конвергентных путях. Следовательно, способ получения рацемического небиволола, базирующийся на такой стратегии, является сложным и неэффективным. Настоящее изобретение предлагает решение, которое делает возможным селективное получение интермедиатов, как показано на схеме 6a, где каждый из интермедиатов получают в виде рацемической смеси без предшествующего разделения энантиомеров, которые образуются во время осуществления способа.
Кроме того, настоящее изобретение дает возможность получения энантиомерно чистого небиволола, например после разделения выбранных рацемических соединений, например соединения II (Схемы 6b и 6c).
Изобретение также относится к способу получения рацемического небиволола, в котором образование нежелательных диастереомеров (например SRSS/RSRR) на конечных стадиях сводится к минимуму за счет содействия очистке и увеличения эффективности. Авторы настоящего изобретения с удивлением обнаружили, что для эффективного получения небиволола можно с выгодой использовать различия в растворимости других диастереомерных производных небиволола в виде их солей с HCl. Конкретно, авторы изобретения наблюдали, что мезо-форма рацемического небиволола (в виде HCl-соли) конфигурации RSRS обладает повышенной растворимостью по сравнению с гидрохлоридом небиволола, тогда как вторая мезо-форма конфигурации RRSS имеет растворимость, сравнимую с растворимостью гидрохлорида небиволола. Растворимость HCl-солей в MeOH составляет 1,5% для небиволола, 1,0% для RRSS-мезо-формы и более 15% для RSRS-мезо-формы. В предпочтительном варианте осуществления настоящего изобретения обеспечивается селективное получение рацемического небиволола, который может содержать только выбранный диастереомер (например RSRS-мезо-форму) в качестве возможной примеси, которую можно легко удалить простой перекристаллизацией благодаря более высокой растворимости. Следовательно, с помощью такой стратегии синтеза удается избежать трудной и неэффективной (с низким выходом) очистки конечного продукта, так как предотвращается образование плохо растворимых диастереомеров небиволола (SRRS/RSSR, RRSS, SRSS/RSRR и RRRR/SSSS).
Еще один аспект настоящего изобретения относится к диастереоселективному синтезу интермедиата VIIIa, содержащего предпочтительную син-конфигурацию. Указанное соединение является полезным интермедиатом в вышеописанной синтетической стратегии селективного получения рацемического небиволола, так как он может образовывать в качестве загрязняющей примеси только диастереомер RSRS конфигурации, который может быть легко удален на конечных стадиях (схема 6a, стадии 8 и 9).
Еще один аспект настоящего изобретения относится к эффективному способу селективного восстановления соединения IXa, в котором образование нежелательного изомера RSRS сводится к минимуму.
Авторы настоящего изобретения обнаружили, что эффективность способа дополнительно увеличивается способами рециркуляции для повторного использования нежелательных диастереомеров, которые могут получаться в ходе синтеза интермедиатов.
Способ получения рацемического небиволола (как показано на схеме 6a) теперь будет описан подробно.
Исходным веществом для настоящего способа является соединение II, рацемическая кислота, которую можно получать различными путями согласно способам, описанным в патенте США № 5171865 (см. также патент-аналог EP 0331078) и патенте США № 4985574 (см. также патент-аналог EP 0264586).
Стадия 1 включает получение (±)-5-[6-фторхроман-2-карбонил]-2,2-диметил[1,3]диоксан-4,6-диона (соединения III) из соединения II, как показано на схеме 7.
Сначала рацемическую кислоту II превращают в активированное производное кислоты, которое затем реагирует с кислотой Мелдрума в органическом растворителе и дает соответствующий ацилированный мелдрумат III, который представляет собой новое соединение и полезный интермедиат для синтеза небиволола.
Ацилирование кислотой Мелдрума можно проводить по методике, аналогичной традиционной, например методике, описанной в (J. Org. Chem. 43(10), 1978, 2087).
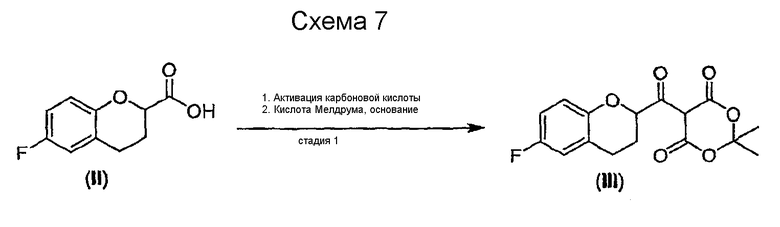
Карбоксильная группа может быть активирована общепринятым методом, например превращением в галогенангидрид с помощью PHaI5, PHaI3, SOHaI2, (COHaI)2, в виде ангидрида карбоновой кислоты, активированного сложного эфира и т.п. Активация с получением галогенангидрида предпочтительна, а хлорангидрид является наиболее предпочтительным галогенангидридом, который получают, используя 1-5 эквивалентов SOСl2, предпочтительно 1-3 эквивалента в присутствии каталитических количеств ДМФА. Упомянутую реакцию можно проводить без применения какого-либо растворителя или в растворителе, таком, например, как бензол, алкил- или галогензамещенный бензол, галогенированные углеводороды и им подобные. Алкилзамещенные или галогензамещенные бензолы являются предпочтительными растворителями, а толуол является наиболее предпочтительным растворителем.
Температура реакции может изменяться в интервале от комнатной до температуры кипения растворителя. В предпочтительном варианте осуществления изобретения температурный интервал составляет от 60°С до 90°С в толуоле в качестве растворителя. Ацилхлорид может быть получен почти с количественным выходом выпариванием растворителя вместе с избытком хлорирующего реагента.
Ацилирование кислоты Мелдрума можно проводить в том же растворителе, который используют для активации карбоновой кислоты. Галогенированные углеводороды представляют собой предпочтительные растворители, а метиленхлорид наиболее предпочтителен. Обычно кислоту Мелдрума используют при молярном соотношении 1-3 моля на моль соединения II, предпочтительно 1-1,5 моля на моль соединения II. Реакцию проводят в присутствии органического или неорганического основания, предпочтительно в присутствии органического основания, такого, например, как третичный амин, и наиболее предпочтительно в присутствии пиридина. В некоторых вариантах осуществления изобретения в качестве основания используют 1-5 эквивалентов, предпочтительно 1,5-3 эквивалента пиридина. Температура реакции может варьироваться в интервале от -10°C (но не ниже температуры плавления чистого растворителя, например, бензола) до около +30°C, предпочтительно в интервале от 0°С до комнатной температуры. Реакция, которую проводят в указанном интервале температур (0°С-комн.температура), обычно завершается за 2 часа.
В конце реакции смесь гидролизуют и экстрагируют водой или разбавленным водным раствором неорганической кислоты, предпочтительно 5%-10% водным раствором хлористоводородной кислоты.
После разделения слоев органический растворитель выпаривают и остаток можно использовать непосредственно на следующей стадии или можно очистить перекристаллизацией или суспендированием в органическом растворителе. Предпочтительно очищать остаток суспендированием в эфире, а наиболее предпочтительно - в метил-трет-бутиловом эфире (MTBE) или диизопропиловом эфире.
Если требуется получение энантиомерно чистого небиволола, его можно получить разделением соединения II, с помощью, например (+)-дегидроабиетиламина, как описано в патенте США № 6545040.
Стадия 2 включает получение соединения IV, схема 6a, где R представляет собой H, тогда получают (±)-1-(6-фторхроман-2-ил)-этанон (соединение IVa, схема 8, путь A), или COOR', где R' представляет собой алкил или замещенный алкил, тогда получают алкиловый эфир (±)-3-(6-фторхроман-2-ил)-3-оксопропионовой кислоты (соединение IVb, схема 9, путь B).
Ацилированные кислоты Мелдрума являются подходящими интермедиатами для получения соответствующего метилкетона после гидролиза и декарбоксилирования или для получения соответствующих бета-кетокислот после алкоголиза и декарбоксилирования. Реакции можно проводить аналогично общепринятой методике, например описанной в (J. Org. Chem. 43(10), 1978, 2087 и Synth. Commun., 10, 1980, 221).
Путь А
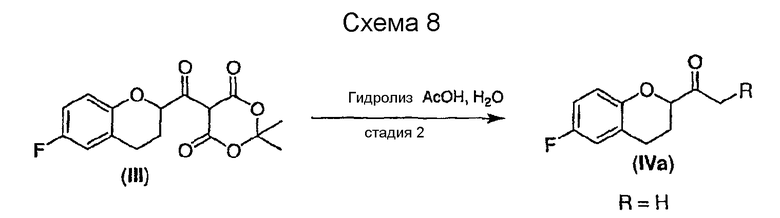
Гидролиз и декарбоксилирование соединения III с образованием соединения IVa (схема 8) в качестве нового соединения и полезного интермедиата для синтеза небиволола, можно проводить в водном растворе кислоты при температуре кипения. Можно использовать неорганическую или органическую кислоту, уксусная кислота является предпочтительной. Воду можно использовать в избытке; предпочтительны равные по объему количества воды и уксусной кислоты. Соединение можно использовать непосредственно в виде сырого продукта или его можно дополнительно очищать колоночной хроматографией.
Путь B
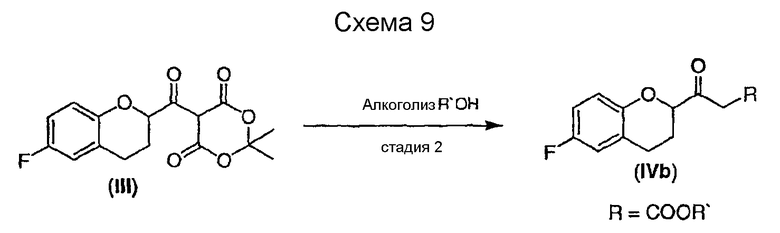
Реакция соединения III со спиртами дает соответствующий бета-кетоэфир IVb в качестве нового соединения и полезного интермедиата для синтеза небиволола, в котором R' представляет собой алкил или замещенный алкил. Этот алкоголиз можно проводить первичными, вторичными или третичными спиртами, предпочтительно первичными и третичными спиртами, и наиболее предпочтительно этанолом или трет-бутанолом. В качестве растворителя можно использовать сам спирт или инертный ароматический растворитель. Предпочтительным растворителем для синтеза соответствующего этилового эфира является этанол, а толуол является предпочтительным растворителем для синтеза трет-бутилового эфира. Tемпература реакции может изменяться от температуры кипения низкокипящих спиртов до температуры кипения толуола или температуры кипения соответствующего азеотропа толуол/спирт. Предпочтительные температуры для получения бета-кетоэтилового эфира, а также для бета-кето-трет-бутилового эфира находятся в интервале от около 70°С до около 80°С. После завершения реакции реакционную смесь можно обрабатывать обычным образом, например, экстракцией, а сырой продукт можно или использовать непосредственно на следующей стадии, или очищать колоночной хроматографией.
Стадия 3 включает получение соединения V из соединения IV, например (±)-2-бром-1-(6-фторхроман-2-ил)этанона (соединение Va) и (±)-2-хлор-1-(6-фторхроман-2-ил)этанона (соединения Vb) (схемы 10 и 11 a-c).
Соединение IV, полученное на стадии 2, можно использовать для синтеза соединения V, содержащего подходящую уходящую группу (LG). Неограничивающие примеры подходящих уходящих групп включают замещенные и незамещенные алкильные и арильные производные сульфоновой кислоты и атомы галогенов. В предпочтительном варианте осуществления изобретения уходящие группы представляют собой атомы галогенов, а наиболее предпочтительными уходящими группами являются бром (соединение Va) и хлор (соединение Vb).
Соединения Va и Vb можно получить по пути A схемы 10 или по пути B схем 11а-с, как описано ниже.
Путь А:
Синтез бромкетона (соединение Va) прямым бромированием метилкетона (соединение IVa) бромом или NBS приводит в большинстве случаев к конкурентному бромированию ароматического кольца. Однако после предварительного превращения метилкетона IVa в соответствующий силиленоловый эфир, содержащий концевую двойную связь, возможно селективное получение соединения Va (см. схему 10).
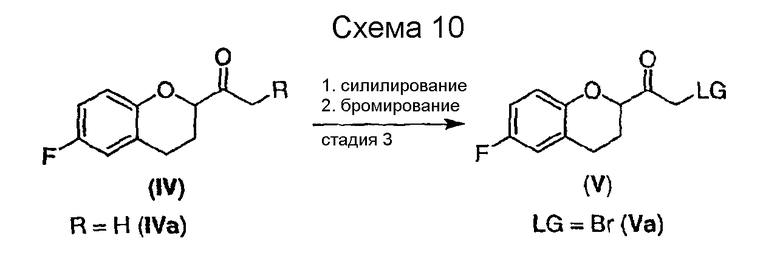
По общей методике силиленоловый эфир может быть получен кинетически контролируемым депротонированием действием сильного основания с последующим силилированием. Примеры растворителя включают простые эфиры или смеси простых эфиров с такими растворителями, в которых обычно доступны растворы сильных оснований. В предпочтительном варианте получения силиленолового эфира в качестве основания используют диизопропиламид лития (LDA), в качестве силилирующего агента используют триметилсилилхлорид (TMSCl), а тетрагидрофуран (TГФ) используют в качестве простого эфира. Реакция начинается при -78°C прибавлением соединения IVa к смеси 1-1,5 эквивалента LDA и 1-2 эквивалентов TMSCl. Предпочтительны 1,2 эквивалента LDA и 1,6 эквивалента TMSCl. Затем реакционной смеси дают нагреться до комнатной температуры, смесь сначала экстрагируют и затем концентрируют. Бромирование можно проводить в подходящем растворителе N-бромсукцинимидом (NBS), 1,3-дибром-5,5-диметилгидантоином или пербромид гидробромида пиридина при температуре от 0°С до комнатной. Подходящие растворители включают, например, галогенированные углеводороды, предпочтительно метиленхлорид. После завершения реакции смесь экстрагируют, а продукт можно очищать колоночной хроматографией или перекристаллизацией. Так как при неселективном бромировании наблюдается образование побочных продуктов, что затрудняет очистку, был разработан более селективный и эффективный способ получения соединения V (см. путь B, Схема 11a).
Путь B:
Путь B представляет собой альтернативный путь получения галогенированных соединений Va и Vb, который является более селективным, чем путь А (см. схему 11a).
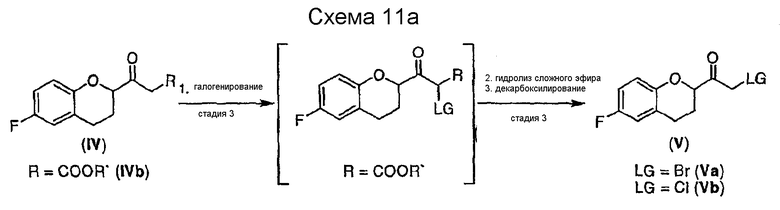
Преимуществом пути В является возможность проведения реакции при более высоких, более удобных температурах последовательным галогенированием бета-кетоэфира IVb с последующим гидролизом и декарбоксилированием. Галогенирование можно проводить подходящим галогенирующим реагентом с катализатором или без катализатора. Типичные галогенирующие реагенты для получения соответствующих бромидов или хлоридов включают, например NBS, NCS, и SO2Cl2. Неограничивающий пример катализатора включает Mg(ClO4)2. Подходящие растворители для рассматриваемой реакции включают ацетонитрил, сложные эфиры или галогенированные углеводороды; ацетонитрил, этилацетат и метиленхлорид предпочтительны. В некоторых вариантах осуществления можно использовать 1,0-1,5 эквивалента галогенирующего реагента и 0,3 эквивалента катализатора. Реакция происходит при температуре от 0°С до комнатной до полной конверсии в течение 3-4 часов. Более высоких температур для галогенирования следует избегать из-за возможных побочных реакций, а при более низких температурах может увеличиться продолжительность реакции. Последующий гидролиз эфира и декарбоксилирование с образованием соединения V можно проводить в водных или неводных растворах кислот при более высоких температурах. Можно использовать органические и неорганические кислоты.
Соединение IVb в виде этилового или трет-бутилового эфира может быть использовано в качестве исходного вещества, причем трет-бутиловый эфир является предпочтительным. В случае использования этилового эфира гидролиз и декарбоксилирование соответствующего галогенированного бета-кетоэфира можно предпочтительно проводить водным раствором трифторуксусной кислоты. Когда используют форму трет-бутилового эфира гидролиз и декарбоксилирование соответствующего галогенированного бета-кетоэфира проводят предпочтительно в смеси муравьиной кислоты и уксусной кислоты предпочтительно в присутствии воды. Tемпература для реакции гидролиза эфира и декарбоксилирования находится в интервале от около 60°С до около 100°С, предпочтительно 75-90°С. Очистку соединения V можно проводить так же, как описано выше для соединения Va. Поскольку соединение Vb обладает большей стабильностью при хранении, чем соединение Va, соединение Vb является предпочтительным.
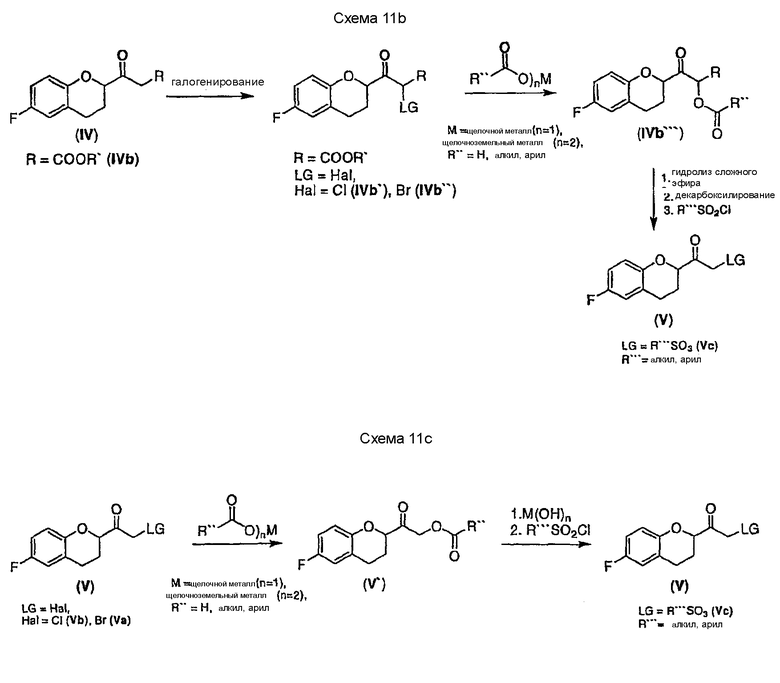
Соединение V, содержащее в качестве уходящих групп замещенные и незамещенные производные алкил- и арилсульфоновых кислот, могут быть получены, например, трансформацией соединения IVb` или Ivb'' (LG = галоген) солями карбоновых кислот в соединение Ivb''' с последующим гидролизом сложного эфира, затем декарбоксилированием и сульфонилированием под действием соответствующих хлорангидридов алкил- или арилсульфоновых кислот (см. схему 11b). Aльтернативно, соединение Vc можно получить аналогичной трансформацией, начиная непосредственно с галогенкетонов Va или Vb (см. Схема 11c)
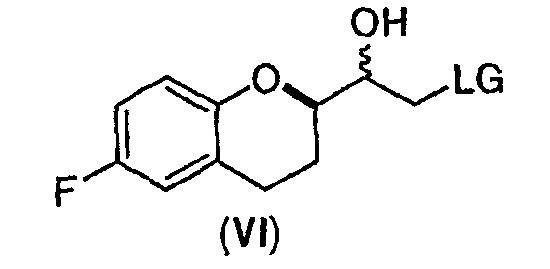
Стадия 4 включает получение соединения VI как показано ниже на схеме 12. Неограничивающие примеры соединения VI включают (±)-2-хлор-1-[6-фтор-(2R*)-хроман-2-ил]-(1R*)-этан-1-oл (интермедиат VIа), (±)-2-хлор-1-[6-фтор-(2R*)-хроман-2-ил]-(1S*)-этан-1-oл (интермедиат VIb), (±)-2-бром-1-[6-фтор-(2R*)-хроман-2-ил]-(1R*)-этан-1-ол (интермедиат VIc) и (±)-2- бром-1-[6-фтор-(2R*)-хроман-2-ил]-(1S*)-этан-1-oл (интермедиат VId).
Целый ряд восстановителей может быть использован для получения галогенметиловых спиртов (соединение VI) из рацемических галогенметилкетонов (соединение V) (см. схему 12). Вообще могут образовываться два рацемических диастереомера, имеющих син(RR/SS) или анти(RS/SR) конфигурации. Относительно стратегии синтеза небиволола, как показано на схеме 6(a), предпочтительны способы восстановления, которые приводят к образованию избытка галогенметилового спирта, имеющего син(RR/SS) конфигурацию. Удивительно, но имеется совсем немного исследований по селективному восстановлению галогенметилкетонов, содержащих алкоксизамещенный хиральный центр в альфа-положении (например, Tetrahedron Letters, 40 (1999), 2863-2864). До настоящего изобретения не было понятно влияние алкоксизамещенного хирального центра в альфа-положении галогенметилкетонов на образование нового хирального центра в альфа-положении, а диастереоселективность для таких реакций точно не установлена.
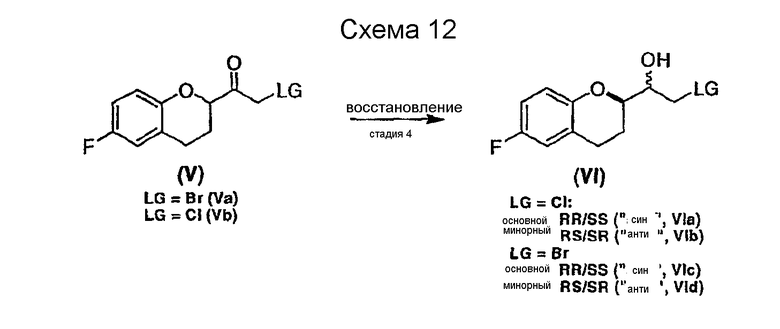
Обычно не имеется ограничений по использованию восстановителей, например боргидридных и алюминийгидридных восстановителей, а также реагентов, которые применимы для восстановления по Меервейну-Пондорфу-Верлею. Неограничивающие примеры восстановителей включают LiBH4, NaBH4, KBH4 N(nBu)4BH4, Zn(BH4)2, NaH(Oac)3, Superhydride®, Red-Al, Li-селектрид (Li-Selectride), BH3хSMe2 или им подобные. В случае каталитического гидрирования подходящими катализаторами являются катализаторы, которые не дают побочных реакций с галогенирорванными соединениями (например, катализаторы, описанные и цитированные в WO 03/064357). Восстановление можно проводить в отсутствие или в присутствие кислоты Льюиса, такой, например, как MgCl2, CaCl2, BaCl2, ZnCl2 Al(Oалкил)3, Ti(Oалкил)4 BF3хOEt2 и т.п. Подходящие растворители включают простые эфиры, спирты, галогенированные углеводороды, галогенированные или алкилированные ароматические растворители и им подобные, за исключением того, что галогенированные растворители не подходят для реакций каталитического восстановления. Предпочтительные галогенметилкетоны соединения V содержат хлор или бром в качестве заместителя "LG". Восстановление удобно проводить при температурах от около -78°C и до примерно комнатной, предпочтительно при температуре от -20°С до комнатной. В таблице 2 показаны репрезентативные результаты восстановления хлорметилкетона Vb (LG = Cl).
RR/SS//RS/SR
(11.1)
(1,0)
(1,4)
(1,4)
(1/1)
(1,4)
(1/2)
(1,4)
(2/1)
(1.1)
(1,3)
(1,1)
(1/1)
(1,1)
(1/1)
(1,1)
(1/1)
(2)
(2/1)
(1,1)
(1/1)
(1,1)
(1/1)
(1,1)
(1,1)
(1,1)
iPrOH (7.0)
BINAPHTHOL
iPrOH (5,0)
Ацетон (2,0)
Ацетон (2,0)
(1/1)
D/L-фенетиловый спирт (2,0)
D/L-фенетиловый спирт (2,0)
sBuOH (7,0)
(0,5)
Циклогексанол
(7,0)
Циклогексанол
(7,0)
Циклогексанол
(7,0)
3-пентанол
(7,0)
9-гидроксифлуорен
(3,4)
9-дифенилкарбинол
В таблице 2 неполная конверсия отмечена символом «*».
Отношение диастереомерных конфигураций RR/SS к RS/SR составляет от около 0,3 до около 2, предпочтительно 1,1-2, наиболее предпочтительно 1,2 - 2.
Обнаружено, что наилучшее отношение диастереомеров (соединение VIa, cин-конфигурация RR/SS) диастереоселективные реакции восстановления дают при температурах выше -20°С, когда восстановление проводят, например NaBH4 в MeOH или EtOH в присутствии катализатора, например ZnCl2 (0,1-2,0 эквивалента). Образованию диастереомера, имеющего антиконфигурацию (соединение VIb, RS/SR), способствует восстановление по Меервейну-Пондорфу-Верлею. В этом случае отношение RS/SR к RR/SS составляет 9.
После почти полной конверсии реакцию можно подвергать обработке, аналогичной описанной на предшествующем уровне техники, концентрированием реакционной смеси и растворением остатка в растворителе, не смешивающимся с водой, предпочтительно в толуоле или МТВЕ, затем последовательным промыванием водным раствором кислоты, предпочтительно 2N раствором HCl, с последующим промыванием водой и/или щелочным раствором, предпочтительно раствором NaHCO3. Смесь диастереомерных продуктов можно очищать колоночной хроматографией или использовать непосредственно для следующей стадии.
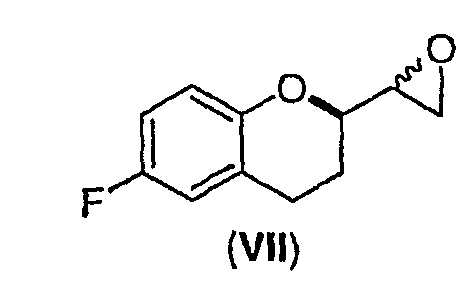
Обнаружено, что в отличие от восстановления соединения Vb, восстановление соединения Va происходит с частичной циклизацией в соответствующие эпоксиды (соединения VIIa и VIIb, см. ниже).
Стадия 5 включает в себя получение соединения VII из соединения VI, как показано на схеме 13. Неограничивающие примеры соединения VII включают (±)-6-фтор-[(2R*)-оксиран-2-ил]-(2S*)-хроман (соединение VIIa) и (±)-6-фтор-[(2R*)-оксиран-2-ил]-(2R*)хроман (соединение VIIb).
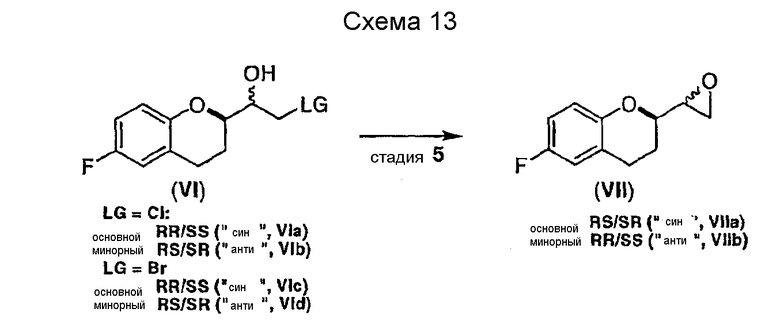
Образование эпоксидов соединения VII из галогенметиловых спиртов соединения VI удобно проводить в растворителях, таких, например, как простые эфиры или спирты, с основанием, таким, например, как гидроксиды щелочных и щелочноземельных металлов, алкоксиды щелочных и щелочноземельных металлов, карбонаты щелочных и щелочноземельных металлов, третичные амины или гидриды щелочных металлов. Применение алкоксидов щелочных металлов в спиртах, используемых в качестве растворителя, является предпочтительным, а наиболее предпочтительным является применение метоксида натрия в метаноле в качестве растворителя. Tемпература для этой реакции может варьироваться в интервале от около 0°C до около 40°C, предпочтительно от 15°С до 25°С. Приблизительно 1,0-2,0 эквивалента основания можно использовать, предпочтительно 1,1 эквивалента. После завершения реакции избыток основания обычно нейтрализуют, прибавляя кислоту, предпочтительно уксусную кислоту. Смесь затем концентрируют и остаток растворяют в подходящем растворителе, например, в эфире или галогенированном углеводороде. Промывание полученного раствора полунасыщенным водным раствором хлорида натрия и концентрирование органического слоя дает эпоксиды соединения VII. Если реакцию проводят, используя смесь диастереомерных галогенметиловых спиртов формулы VI, тогда образуется смесь соответствующих диастереомерных эпоксидов VII. Диастереомерную смесь эпоксидов можно использовать непосредственно на следующей стадии или разделить колоночной хроматографией. В предпочтительном варианте осуществления настоящего способа смесь используют на следующей стадии без разделения диастереоизомеров.
Стадия 6 включает получение соединения VIII из соединения VII, как показано на схеме 14, и разделение диастереомеров. Неограничивающие примеры соединения VIII включают (±)-2-бензиламино-1-[6-фтор-(2R*)-хроман-2-ил]-(1S*)-этан-1-ол (соединение VIIIa) и (±)-2-бензиламино-1-[6-фтор-(2R*)-хроман-2-ил]-(1R*)-этан-1-ол (соединение VIIIb).
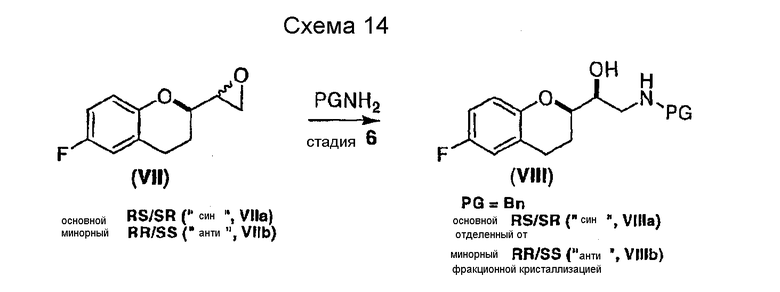
Примеры подходящих защитных групп (PG) включают водород или подходящую аминозащитную группу. Предпочтительными являются защитные группы, которые позволяют провести последующую стадию алкилирования (стадия 7) и могут быть легко удалены на конечной стадии простым способом снятия защиты. Следовательно, предпочтительная PG представляет собой аллильную группу, замещенную или незамещенную арилметильную группу. Если PG представляет собой бензильную группу, получение соединения VIII можно проводить таким же образом, как описано в EP 0145067, но без предварительного разделения диастереомерной смеси соединений VIIa и VIIb. Авторы настоящего изобретения обнаружили, что хроматографическое разделение маслообразных соединений VIIa и VIIb не является необходимым в настоящем способе и что диастереомерную смесь можно разделить фракционной кристаллизацией после превращения в VIIIа и VIIIb. Следовательно, в предпочтительном варианте осуществления изобретения смесь VIIa и VIIb подвергают взаимодействию с бензиламином с образованием соответствующей диастереомерной смеси соединений VIIIа и VIIIb, которую далее разделяют фракционной кристаллизацией. Так как соединения VIIIа и VIIIb обладают основными свойствами, фракционную кристаллизацию можно проводить не только со свободным амином, но также и с соответствующей солью. Соединение VIIIа, а также соединение VIIIb представляют собой интермедиаты, применимые для получения небиволола и, следовательно, способ может быть использован для селективного получения этих двух изомеров в промышленных масштабах.
В отношении данной стратегии получения небиволола, наиболее предпочтительным является селективное получение и выделение соединения VIIIа.
В типичном варианте эквимолярную или обогащенную диастереомерную смесь соединений VIIа (син) и VIIb (анти) с отношением син/анти, равным 1 или больше, полученную на стадиях 4 и 5, обрабатывают избытком бензиламина (≥ 3 эквивалентов) в C1-C3спирте в качестве растворителя при температурах от около комнатной до около 50°С. В предпочтительном варианте осуществления изобретения реакцию проводят при 40°С с 3 эквивалентами бензиламина в 2-пропаноле.
После полной конверсии реакционную смесь охлаждают, чтобы вызвать кристаллизацию. Было показано, что растворитель, предпочтительный для реакции, подходит и для фракционной кристаллизации. Дополнительные порции можно получить путем дополнительной кристаллизации концентрированных маточных растворов из того же спирта или из смеси этого спирта с простыми эфирами, предпочтительно с диизопропиловым эфиром. Дополнительное увеличение диастереомерного отношения касательно соединения VIIIа можно получить путем перекристаллизации или суспендирования в C1-C3спиртах, простых эфирах, толуоле, ацетонитриле или их смесях.
Исходя из диастереомерной смеси соединений VIIa и VIIb с отношением диастереомеров около 57/43, по вышеупомянутой методике можно получить соединение VIIIа, содержащее 5% или меньше соединения VIIIb.
Относительная конфигурация предпочтительного соединения VIIIa (см. фигуру 2) подтверждена данными рентгеноструктурного анализа на единичном кристалле, как показано ниже в таблице 3.
Кристаллографические данные для соединения VIIIа
Молекулярная масса [г-1·мол]
Цвет кристалла, внешний вид
Размеры кристалла [мм]
Температура [К]
Кристаллическая система
пространственная группа
Z
Отражения для определения ячейки
2θ-интервал для определения ячейки [°]
301,6
бесцветные призмы
0,15×0,20×0,25
160(1)
моноклиническая
Р21/с (#14)
4
4489
4-60
Стадия 7 включает в себя получение соединения IX из соединений VIII и V и разделение диастереомеров соединения IX. Неограничивающие примеры соединения IX включают (±)-2-{бензил-[2-(6-фтор-(2R*)-хроман-2-ил)-(2S*)-гидроксиэтил]амино}-1-(6-фтор-(2S*)-хроман-2-ил)этанон (соединение IXa) (схема 15) и (±)-2-{бензил-[2-(6-фтор-(2R*)-хроман-2-ил)-(2S*)-гидроксиэтил]амино}-1-(6-фтор-(2R*)-хроман-2-ил)этанон (соединение IXb) (см. Схема 17).
Реакция предпочтительного рацемического диастереомера соединения VIIIа с рацемическим соединением V, содержащим соответствующую реакционноспособную уходящую группу (LG), дает диастереомерную смесь новых соединений IXa и IXb. Полученную смесь можно разделять колоночной хроматографией или фракционной кристаллизацией для получения требуемого соединения IXa в качестве подходящего интермедиата для синтеза рацемического небиволола. Так как соединения IXa и IXb обладают основными свойствами, фракционную кристаллизацию можно проводить с использованием свободного амина или соответствующей соли.
Как описано выше на стадии 3, подходящие уходящие группы (LG) соединения V включают галоген, алкилсульфонилоксигруппы, замещенные или незамещенные арилсульфонилоксигруппы или им подобные. Предпочтительными уходящими группами являются галогены и наиболее предпочтительными уходящими группами являются бром (соединение Va) и хлор (соединение Vb). Защитные группы (PG) описаны при рассмотрении стадии 6; наиболее предпочтительной защитной группой является бензильная группа.
Реакции алкилирования удобно проводить в подходящем инертном органическом растворителе или в смеси таких растворителей и в присутствии основания и соответствующего количества катализатора. Типичные растворители включают замещенные или галогенированные углеводороды, такие как метиленхлорид, дихлорэтан и т.п.; простые эфиры, например ТГФ, диоксан, диметоксиэтан, и полярные апротонные растворители, например ДМФА, DMA, N-метилпирролидон (NMP), диметилсульфоксид (ДМСО) и им подобные. Предпочтительными растворителями являются полярные апротонные растворители. Можно использовать 1,0-1,5 эквивалента соединения V, предпочтительно 1,1 эквивалента. Типичные основания включают третичные амины, например триэтиламин, пиридин, карбонаты щелочных металлов, гидрокарбонаты щелочных металлов или гидрид натрия. Предпочтительными основаниями являются гидрокарбонаты щелочных металлов, а наиболее предпочтительным основанием является гидрокарбонат натрия. Можно использовать 1,5-2,5 эквивалента основания, предпочтительно 2,0 эквивалента. Типичные катализаторы для ускорения реакции включают галогениды щелочных металлов или галогениды тетраалкиламмония. Если уходящей группой является хлорид, тогда предпочтительными катализаторами являются бромиды и йодиды, а наиболее предпочтительными - бромид натрия и йодид натрия. Можно использовать по меньшей мере 0,1 эквивалента катализатора, предпочтительно - 0,15 эквивалента. В предпочтительном варианте осуществления изобретения реакцию можно проводить при температурах от около комнатной до около 80°С. Более низкие температуры могут увеличить время реакции, а более высокие температуры могут вызвать побочные реакции. После завершения реакции смесь можно обрабатывать методом экстракции, известным в данной области техники. Выпаривание растворителя после экстракции и кристаллизации диастереомерной смеси с помощью подходящего антирастворителя дает диастереомерные соединения IXa и IXb, которые можно выделять колоночной хроматографией или фракционной кристаллизацией из подходящего растворителя. Так как соединения IXa и IXb обладают основными свойствами, фракционную кристаллизацию можно проводить со смесью свободных аминов или соответствующих солей. Разделение смеси соединений IXa и IXb можно эффективно осуществлять фракционной кристаллизацией свободных аминов из ацетонитрила в качестве растворителя.
Подходящая модификация соединений IXa и IXb может также быть применима для разделения диастереомерной смеси. Авторы настоящего изобретения с удивлением обнаружили, что диастереомер IXb можно селективно силилировать в присутствии диастереомера IXa. Из-за более высокой растворимости силилированного соединения IXb по сравнению с несилилированными соединениями эффективность фракционной кристаллизации можно значительно увеличить. Обычно силилирование можно проводить в органических растворителях или смеси растворителей, таких как простые эфиры, сложные эфиры, галогенированные углеводороды, ароматические растворители (например, толуол, хлорбензол и т.п.), полярные апротонные растворители (например, ДМФА, ДМСО), силилирующим реагентом, если необходимо, в присутствии основания. Предпочтительные органические растворители включают ацетонитрил, TГФ, MTBE и их смеси. Если основания необходимы, тогда можно использовать амины, например триэтиламин, пиридин, имидазол и т.п., карбонаты щелочных металлов или гидрокарбонаты щелочных металлов. Амины являются предпочтительными органическими растворителями, а наиболее предпочтительным является имидазол. Можно использовать 1,0-2,0 эквивалента основания, предпочтительно 1,5 эквивалента. В качестве силилирующего реагента можно использовать TMSCl, HMDS, BSU и т.п., но TMSCl предпочтителен. Для успешного разделения диастереомеров важно использовать силилирующий реагент при молярных отношениях 0,40/n - 0,60/n относительно общего количества обоих диастереомеров, где n обозначает общее число переносимых силильных групп, приходящихся на силилирующий реагент. Меньшее число эквивалентов может привести к недостаточному разделению, а большее число эквивалентов может в результате привести к уменьшению выхода. Реакцию обычно проводят в интервале температур от около 0°С до около комнатной. Более низкие температуры могут увеличить время реакции, а более высокие температуры могут привести к недостаточной селективности.
Что касается данной стратегии получения небиволола, предпочтительным является выделение соединения IXa из смеси, состоящей из IXa и IXb, фракционной кристаллизацией или селективной модификацией IXb с последующей кристаллизацией IXa. Tермином «по существу чистые диастереомеры», используемый здесь, обозначают диастереомеры по меньшей мере 50%-ной чистоты, предпочтительно 80%-ной чистоты и более предпочтительно 95%-ной чистоты.
Данные ЯМР-спектров, записанных для выделенных диастереомеров, указывают, что оба изомера циклизуются в полукетальную форму. 13C ЯМР спектры показывают, что вместо пиков карбонильной группы, в спектрах имеются новые пики при 94,616 и 94,707, соответственно, которые указывают на атом углерода циклической полукетальной формы (см. схемы 16 и 17 и фигуры 3 и 4).

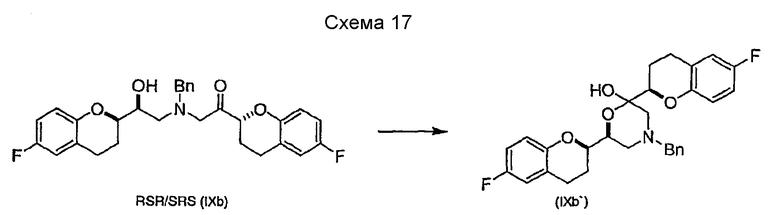
Относительная конфигурация предпочтительного соединения IХb` подтверждена данными рентгеноструктурного анализа на единичном кристалле, как показано ниже в таблице 4.
Кристаллографические данные соединения IХb`
Стадия 8 включает получение соединения X, как показано на схеме 18. Неограничивающие примеры соединения X включают (±)-[2R*[R*[R*(S*)]]]-α,α`-[[(фенилметил)имино]бис(метилен)]-бис[6-фторхроман-2-метанол] (интермедиат Xa) и (±)-[2R*[S*[R*(S*)]]]-α,α`-[[(фенилметил)имино]бис(метилен)]бис[6-фторхроман-2-метанол] (соединение Xb). Целый ряд восстановителей может быть использован для восстановления соединения IXa, при этом два рацемических диастереомера могут образовываться RSSS/SRRR конфигурации (соединение Xa) или RSRS конфигурации (соединение Xb). Относительно стратегии синтеза небиволола, как изображено на схемах 6(a) и 18, предпочтительны способы восстановления, которые приводят к образованию избытка соединения Xa. Имеется совсем немного исследований по селективному восстановлению хиральных 1-гидрокси-5-кетосоединений, регулируемому стереогенными центрами в удаленных положениях цепи (расстояние 4 или более атомных центров; Tetrahedron Letters, 35 (1994), 4891-4894), Tetrahedron Letters 40 (1999) 593-596, J. Org. Chem. 63 (1998) 7964-7981). В отличие от соединений, описанных в указанных работах, соединение IXa coдержит три асимметрических центра, особенно в положениях 1-2, а также в положениях 1-5 и 1-6, которые могут регулировать диастереоселективность при восстановлении кетогруппы.
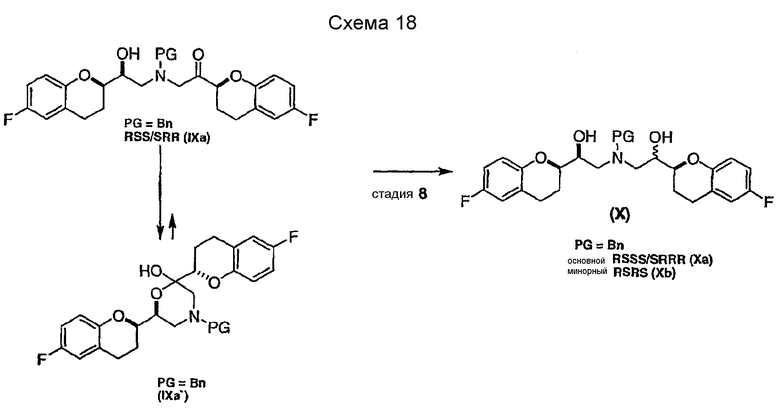
PG может представлять собой водород или аминозащитную группу, как определено выше. Предпочтительными являются те же защитные группы, которые были уже описаны для стадий 6a-c и 7.
В принципе нет ограничений по использованию боргидридных или алюминийгидридных восстановителей, которые могут быть выбраны, например из LiBH4, NaBH4, KBH4 N(nBu)4BH4, Zn(BH4)2, NaH(Oac)3, Superhydride® (триэтилборгидрид натрия), Red-Al® (натрий-бис(2-метоксиэтокси)алюминийгидрид), Li-Selectride® (три-втор-бутилборгидрид, Li, K), BH3хSMe2 или им подобных, или таких реагентов, которые применимы для восстановления по Меервейну-Пондорфу-Верлею. Однако следует учитывать, что реакции восстановления по Меервейну-Пондорфу-Верлею обратимы (Окисление по Оппенауэру). Так как соединение Xa содержит две вторичные спиртовые группы, которые могут находиться в равновесии окисление/восстановление с восстановителем, может образовываться смесь трех диастереомеров (бензилированного небиволола (RSSS/SRRR) и двух их мезо-форм (RSRS и RRSS)).
Чтобы не допустить таких побочных реакций, можно защитить гидроксильную группу соединения IXa перед проведением восстановления по Меервейну-Пондорфу-Верлею. Еще одна возможность состоит в непрерывной отгонке кетона (например, ацетона, если изопропанол используют в качестве донора гидрида).
Каталитическое гидрирование соединения IXa может быть дополнительной возможностью, но если PG представляет собой защитную группу, нестабильную при восстановлении (например, бензил), тогда следует принимать во внимание снятие защиты.
Реакции восстановления можно проводить в отсутствие или в присутствии кислот Льюиса, выбранных из MgCl2, CaCl2, BaCl2, ZnCl2 Al(Oалкил)3, Ti(Oалкил)4 BF3хOEt2 и т.п. Подходящими растворителями являются простые эфиры, спирты, галогенированные углеводороды или им подобные, за исключением того, что галогенированные растворители не подходят для реакций каталитического восстановления. Восстановление удобно проводить при температурах от -20°C до комнатной. Хотя при более низких температурах может повышаться селективность, время реакции будет увеличиваться, а более высокие температуры могут привести к уменьшению селективности. В таблице 5 представлены типичные результаты селективного восстановления соединения IXa.
RSSS/SRRR
//RSRS
Diglyme (3)
В таблице 2 неполная конверсия отмечена символом «*».
Отношение диастереомерных конфигураций RSSS/SRRR к RSRS составляет от около 1 до около 20, предпочтительно 2-20, наиболее предпочтительно 4 - 20.
После полной конверсии реакционную смесь можно обрабатывать стандартным образом. Диастереомерную смесь продуктов можно разделять колоночной хроматографией или фракционной кристаллизацией. Так как соединения Xa и Xb обладают основными свойствами, образование соли перед проведением фракционной кристаллизации является дополнительной возможностью. Смесь диастереомерных продуктов можно также использовать на следующей стадии в виде сырого продукта без дополнительной очистки.
Стадия 9 включает получение гидрохлорида (±)-[2R*[R*[R*(S*)]]]-α,α`-[иминобис(метилен)]бис[6-фторхроман-2-метанола] (соединения I) и отделение от побочного продукта гидрохлорида (±)-[2R*[S*[R*(S*)]]]-α,α`-[иминобис-(метилен)]бис[6-фторхроман-2-метанола], как показано на схеме 19.
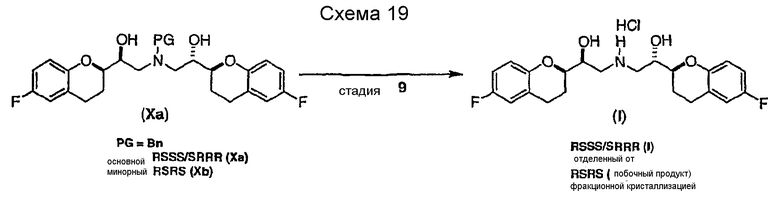
Конечные стадии получения рацемического гидрохлорида небиволола включают снятие защиты, образование соли и очистку фракционной кристаллизацией для удаления побочных продуктов, главным образом нежелательного диастереомера, имеющего RSRS конфигурацию.
PG может представлять собой такие же группы, какие уже были описаны выше, и если PG является группой, отличающейся от водорода, снятие защиты можно проводить по известным методикам. Так как бензильные группы являются наиболее предпочтительными, снятие защиты можно проводить каталитическим гидрированием. Если соединение Xa содержит соединение Xb в качестве побочного продукта, тогда очистку можно осуществить фракционной кристаллизацией. Так как соединения Xa и Xb после снятия защиты обладают основными свойствами, фракционную кристаллизацию можно проводить после образования соответствующей соли. Было установлено, что смесь, состоящую из небиволола и его RSRS диастереомера, можно легко разделить фракционной кристаллизацией после образования соли с HCl или любой другой фармацевтически приемлемой соли.
Соединение I можно превратить в его фармацевтически приемлемую нетоксичную кислотно-аддитивную соль, которая образуется в результате обработки соответствующими кислотами, такими как, например, неорганические кислоты, такие как галогеноводородная кислота, например, хлористоводородная, бромистоводородная и т.п., серная кислота, азотная кислота, фосфорная кислота и им подобные; или органические кислоты, такие, например, как уксусная, пропановая, гидроксиуксусная, 2-гидроксипропановая, 2-оксопропановая, этандиовая, пропандиовая, бутандиовая, (Z)-2-бутендиовая, (E)-2-бутендиовая, 2-гидроксибутандиовая, 2,3-дигидроксибутандиовая, 2-гидрокси-1,2,3-пропантрикарбоновая, метансульфоновая, этансульфоновая, бензолсульфоновая, 4-метилбензолсульфоновая, циклогексансульфаминовая, 2-гидроксибензойная, 4-амино-2-гидроксибензойная и подобные кислоты.
Образование соли с HCl является предпочтительным для фракционной кристаллизации, обеспечивая фармацевтически приемлемую гидрохлоридную соль небиволола. Фракционную кристаллизацию обычно можно проводить в подходящих растворителях, в которых небиволол растворим хуже, чем его RSRS диастереомер. В качестве растворителей для фракционной кристаллизации предпочтительны спирты, а метанол является наиболее предпочтительным растворителем.
СТАДИИ РЕЦИРКУЛЯЦИИ ДИАСТЕРЕОМЕРОВ
Настоящее изобретение также включает стадии рециркуляции диастереомеров, образующихся в ходе осуществления способа.
I. Возможности рециркуляции интермедиатов, образующихся на стадии 5 и стадии 6
Неограничивающий пример рециркуляции иллюстрируется схемой 20, где в качестве уходящей группы используют Cl.
Стадию рециркуляции можно проводить, используя, например реакцию Мицунобу для инверсии вторичной спиртовой группы для рециркуляции нежелательного диастереомера VIIIb, образующегося на стадии 6. Однако может потребоваться подходящая защитная группа (PG`) для азотсодержащей группы. Специалисту в данной области понятно, что примеры введения подходящих защитных групп PG` включают образование соответствующих карбаматов, например, под действием алкилхлорформиатов, или образование соответствующих амидов действием хлорангидридов или ангидридов карбоновых кислот. Защитную группу можно ввести после разделения диастереомерной смеси фракционной кристаллизацией с последующим выделением нежелательного диастереомера из маточных растворов.
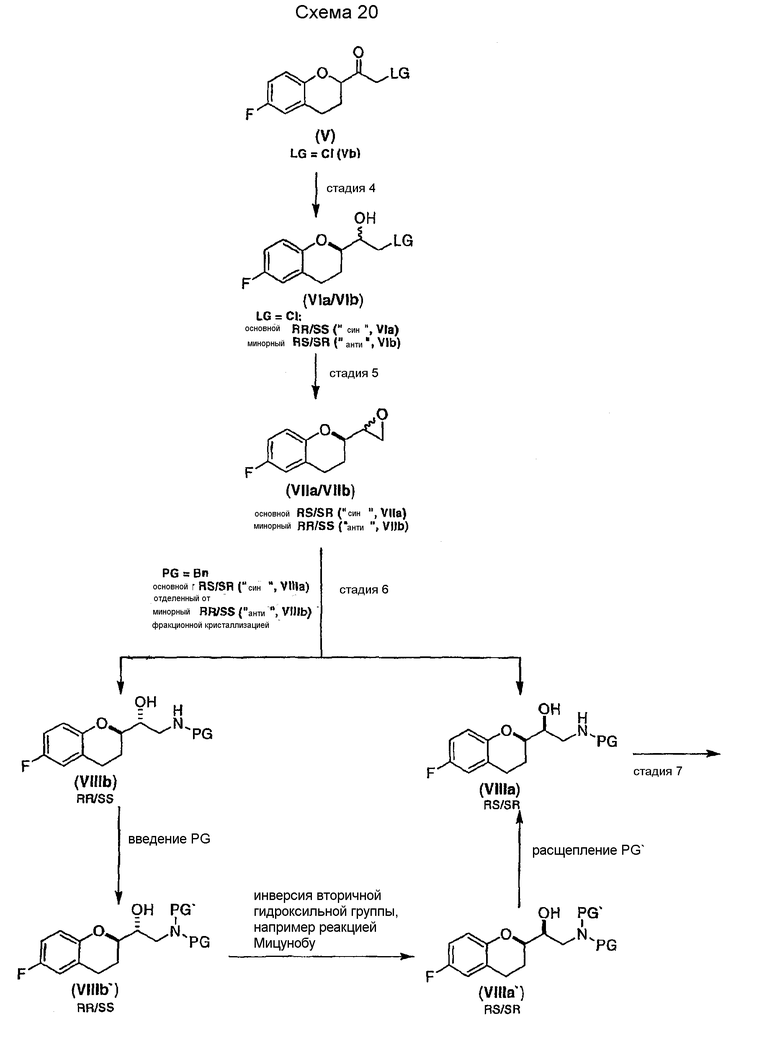
II. Возможность рециркуляции соединений, образующихся на стадии 7
На стадии 7 образуется смесь двух диастереомеров (соединения IXa и IXb), которые разделяют фракционной кристаллизацией. Имеется две возможности рециркуляции нежелательного диастереомера IXb (схема 21, таблицы 6, 7, 8). Первая возможность состоит в эпимеризации нежелательного диастереомера IXb опять с образованием смеси, состоящей из IXa и IXb, которую можно разделять вышеописанным способом.
Исследования эпимеризации в различных растворителях, содержащих 10% 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU), начиная от отношения IXa/IXb около 23/77.
IXa/IXb
IXa+IXb/VIIIa
Смеси анализируют ВЭЖХ через 40 ч. Результаты, представленные в таблице 6, показывают, что расщепление соединений IXa/IXb в метаноле с образованием соединения VIIIa происходит быстрее, чем эпимеризация. Во всех других растворителях через 40 ч эпимеризация давала смесь диастереомеров IXa и IXb (почти 1/1), тогда как тенденция к расщеплению с образованием соединения VIIIa явно подавляется особенно в таких растворителях, как ДМФА и толуол. Результаты дополнительных исследований эпимеризации в ДМФА и толуоле показаны в таблицах 7 и 8.
Исследования эпимеризации в ДМФА, содержащем 5% 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU) при 30°С, начиная от отношения IXa/IXb около 23/77. Смеси анализировали ВЭЖХ.
IXa/IXb
IXa+IXb/VIIIa
Эпимеризация в толуоле, содержащем 5% 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU) при 30°С, начиная от отношения IXa/IXb около 23/77. Смеси анализировали ВЭЖХ.
IXa/IXb
IXa+IXb/VIIIa
Вторая возможность рециркуляции включает расщепление нежелательного диастереомера IXb с образованием смеси соединения VIIIa и некоторых побочных продуктов. Расщепление можно осуществить таутомерным превращением аминокетона в енаминную форму с последующим гидролизом известными способами. Соединение VIIIa можно потом выделить и повторно ввести в процесс, как показано на схеме 21.
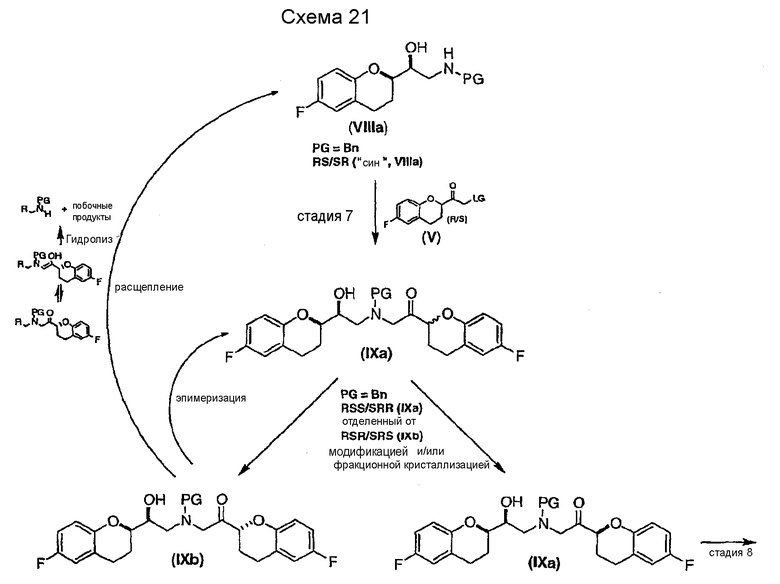
III. Aльтернативный синтез небиволола, использующий диастереомер VIIIb
Хотя в соответствии с вышеописанной стратегией синтеза предпочтительным является получение диастереомера VIIIa в качестве интермедиата, нежелательный диастереомер VIIIb можно также использовать в качестве интермедиата для получения рацемического небиволола, как показано на схеме 22.
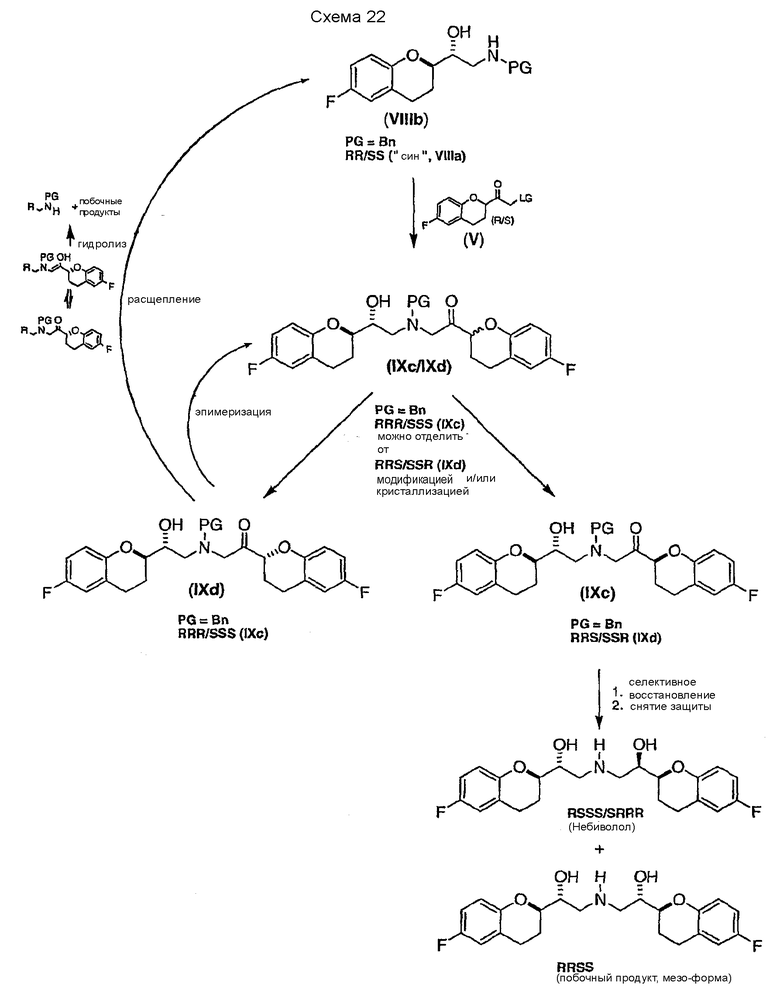
Возможны другие аналогичные способы рециркуляции. Например, в отличие от пути, в котором используют предпочтительное соединение VIIIa, использование соединения VIIIb дает после стадии алкилирования диастереомерную смесь соединений IXc и IXd. Восстановление соединения IXd после снятия защиты дает небиволол и вторую мезо-форму, имеющую RRSS конфигурацию (в отличие от предпочтительного пути, который приводит к образованию мезо-формы, имеющей RSRS конфигурацию). Как было указано выше, мезо-форма, имеющая RSRS конфигурацию, обладает более высокой растворимостью, чем вторая мезо-форма, имеющая RRSS конфигурацию, и поэтому небиволол, загрязненный RSRS диастереомером (по предпочтительному пути), можно легко очистить перекристаллизацией. В случае загрязнения небиволола мезо-формой RRSS конфигурации очистку тоже можно проводить перекристаллизацией, но из-за того, что растворимости RRSS диастереомера и небиволола близки, очистка намного труднее и приходится принимать во внимание снижение выхода.
IV. Рециркуляция мезо-форм небиволола (RSRS и RRSS)
Обе мезо-формы небиволола, получаемые по вышеописанным способам, могут быть превращены непосредственно в небиволол после подходящей защиты (например, циклическим карбаматом, циклической силильной группой и т.п.), последующей инверсии вторичной спиртовой группы (например, реакцией Мицунобу) и снятия защиты, как показано на схеме 23.
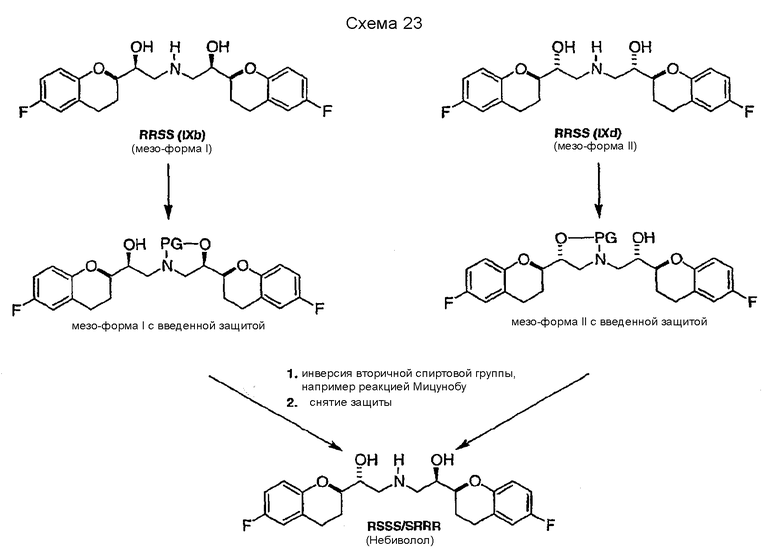
Реакции для защиты, снятия защиты и инверсии можно осуществлять способами, известными в данной области техники.
Хотя рассмотренные способы уменьшают затраты и повышают эффективность путем рециркуляции нежелательного диастереомера, они все же требуют дополнительных стадий для рециркуляции и выделения требуемых соединений. Чтобы уменьшить затраты и воздействие на окружающую среду было сделано дополнительное усовершенствование способа получения соединений IXa/IXa`, как описано ниже.
Кроме того, обнаружено, что в некоторых условиях дополнительное обогащение, способствующее образованию IXa, может быть достигнуто эпимеризацией эквимолярной смеси IXa и IXb, которое приводит к более эффективному способу, чем методы рециркуляции, описанные выше. Такое увеличение отношения желательного диастереомера к нежелательному диастереомеру в смеси приводит к повышению выхода и объемного выхода и способствует выделению IXa фракционной кристаллизацией. И наконец, такой способ также уменьшает отходы и сокращает время получения.
Таким образом, изобретение относится к новым способам и условиям для увеличения отношения диастереомеров IXa и IXb на стадии 7 (фигура 6) от около 1:1 до 9:1 перед проведением выделения IXa (см. фигуры 7-9).
Увеличение диастереомерного отношения можно получить с помощью методики одновременной эпимеризации-кристаллизации, включающей эпимеризацию смеси, состоящей из соединений IXa и IXb в соотношении примерно 1:1, в присутствии подходящего основания и в органическом растворителе, в котором растворимость соединения IXa немного хуже, чем растворимость соединения IXb. Фигура 7 показывает одновременную эпимеризацию-кристаллизацию, которая представляет собой динамический процесс повышения диастереомерного отношения и получения желательного диастереомера (соединения IX показаны в ациклических формах).
Защитная группа (PG) может быть любой соответствующей аминозащитной группой. PG предпочтительно выбирают из аллильной группы или замещенной или незамещенной арилметильной группы; наиболее предпочтительной (PG) является бензильная группа(Bn). Подходящие основания для эпимеризации могут быть выбраны из алкоксидов, амидинов, гуанидинов или фосфазенов, предпочтительными основаниями являются амидины, такие, например, как диазабициклоундецен (DBU), диазабициклононен (DBN), а наиболее предпочтительным основанием является DBU. Основание можно использовать в количестве по меньшей мере 0,05 эквивалента и 0,25 эквивалента предпочтительно. Эпимеризацию предпочтительно проводят в интервале температур от 20°С до 70°С и наиболее предпочтительно в интервале от 40°С до 70°С. Более низкие температуры можно также использовать, но они увеличивают время реакции, а более высокие температуры могут привести к высокой скорости деградации. Эпимеризацию можно также проводить в изотермических условиях, но предпочтительным является медленное охлаждение смеси в указанном интервале температур. Органический растворитель может быть выбран из растворителей, в которых может образовываться суспензия в указанном температурном интервале для проведения одновременной кристаллизации-эпимеризации по изобретению. Подходящие растворители обычно выбирают из апротонных растворителей, например ароматических растворителей, простых эфиров, сложных эфиров, амидов или нитрилов или протонных растворителей, таких, например, как спирты. Кислые растворители, которые инактивируют основание путем образования соли, предпочительно исключать. Предпочтительными являются апротонные растворители и наиболее предпочтительным растворителем является ацетонитрил. Так как присутствие воды может вызвать деградацию соединений IXa и IXb, предпочтительно, кроме того, использовать растворители, в которых содержание воды меньше 1,0% и наиболее предпочтительно - меньше 0,1%.
Реакцию удобно проводить при медленном охлаждении суспензии, содержащей эквимолярные количества соединений IXa и IXb в присутствии основания. Соединение IXa, в котором количество нежелательного диастереомера составляет менее 1%, можно выделить с выходом до 74% и только в одну стадию без необходимости в нескольких кристаллизациях, селективных модификациях или дополнительных стадиях рециркуляции, как описано выше.
Таким образом, одновременная эпимеризация-кристаллизация включает одновременную (a) эпимеризацию смеси, состоящую из соединений IXa и IXb в присутствии подходящего основания и в органическом растворителе и (b) способствует кристаллизации IXa из смеси. Необходимо, чтобы во время эпимеризации соединение IXa удалялось из равновесной системы одновременной кристаллизацией. Исследования эпимеризации проводили в растворе, а не в суспензии.
Методика одновременной эпимеризации-кристаллизации для синтеза соединения IXa превосходит вышеописанные способы кристаллизации и рециркуляции, так как способом селективной модификации получают IXa с выходом только 41% по сравнению с выходом 74% по способу одновременной эпимеризации-кристаллизации. Чтобы получить такой же выход другими методами, необходимо осуществить несколько стадий рециркуляции, что требует больше времени, реагентов и дает больше отходов. Кроме того, небольшие количества соединений IXa/IXb, которые теряются в новом способе во время выделения IXa, можно легко извлечь из маточного раствора и повторно использовать для следующего опыта по эпимеризации. Извлечение потерянного вещества и использование его в следующем опыте дополнительно увеличивает эффективность способа одновременной эпимеризации-кристаллизации.
Методика одновременной эпимеризации-кристаллизации теперь будет описана с использованием фигуры 8 в качестве примера. На фигуре 8 показана эпимеризация смеси соединений IXa и IXb, где PG представляет собой бензильную группу (Bn). Соединения IXa и IXb показаны в форме с открытой цепью (ациклической форме). В растворе имеет место равновесие ациклических форм IXa и IXb с соответствующими циклическими формами IХa' и IХb'. Для эпимеризации требуется ациклическая форма, которая содержит кислый протон на хиральном центре в альфа-положении к карбонильной группе.
Суспензия смеси, состоящей из соединений IXa и IXb в почти эквимолярном отношении, нагревают в ацетонитриле до внутренней температуры 70°С. После охлаждения до 60°С прибавляют DBU (0,25 экв.) и проводят эпимеризацию, охлаждая смесь до 40°С, используя градиент температуры, описанный в таблице 9.
Реакцию прекращают прибавлением уксусной кислоты (0,25 экв.) и смесь охлаждают до 25°С. Сырой продукт выделяют фильтрованием и дополнительно очищают суспендированием в ацетонитриле. Фильтрование проводят после окончания одновременной эпимеризации-кристаллизации для выделения IXa. Образование суспензии в ацетонитриле представляет собой только дополнительную стадию очистки для удаления оставшихся прримесей и небольших количеств нежелательного диастереомера. В таблице 10 показана успешная эпимеризация, контролируемая постоянным технологическим контролем (IPC) (ВЭЖХ).
IXa/IXb
Так как разложению IXa/IXb во время эпимеризации способствует H2O, предпочтительно, чтобы исходное вещество, а также ацетонитрил были почти безводными (таблица 11). Смесь IXa/IXb инкубируют при 50°С в присутствии 0,25 экв. DBU и различных количеств H2O. Количество оставшихся IXa/IXb определяют периодически методом ВЭЖХ. Результаты показаны в таблице 11.
Следует избегать температур выше 70°С в течение более 30 мин, так как IXa/IXb не совсем стабильны при температуре выше 70°С. Разложение и эпимеризацию контролируют, когда проводят перемешивание взвеси IXa в ACN в течение 21 ч при соответствующей температуре, как показано в таблице 12.
(IXa->IXb)
1) Так как скорость разложения IXa и IXb может быть разной, неизвестно, вызвано ли изменение отношения более быстрым разложением IXa, а не эпимеризацией.
На фигуре 9 показан способ получения рацемического небиволола и его фармацевтически приемлемых солей, где стадия 7b изображает стадию одновременной эпимеризации-кристаллизации по изобретению.
Параметры стадии одновременной эпимеризации-кристаллизации по изобретению выбирают, базируясь на понимании того, что растворимость соединения IXa ниже растворимости соединения IXb. Таким образом, обнаружено, что IXa может кристаллизоваться и поэтому удаляться из равновесия, хотя в растворе равновесие обоих диастереомеров может регенерироваться эпимеризацией нежелательного диастереомера IXb в присутствии основания. Равновесие исследовалось при различных температурах в присутствии некоторого основания в некотором растворителе при некоторых скоростях охлаждения. Охлаждение необходимо, чтобы завершить кристаллизацию и оптимизировать выход. Так как охлаждение уменьшает растворимость обоих диастереомеров, создают условия для дополнительной кристаллизации, благоприятствующие желательному диастереомеру. Так как эпимеризация происходит медленнее при более низких температурах, равновесие также регулируется медленнее. Получают сбалансированное отношение между скоростью эпимеризации для регулировки равновесия в растворе и охлаждением смеси, способствующим кристаллизации IXa. Следовательно, градиен температуры (как показано в таблице 9 и ниже в примерах 19 и 20) необходимо сохранять сбалансированное отношение между скоростью эпимеризации и стадиями кристаллизации. Так как более высокие температуры могут также вызывать деградацию соединений IXa и IXb, которому способствует присутствие воды, условия для эпимеризации кристаллизации приходится искать, когда деградация сведена к минимуму, чтобы не допустить уменьшения выхода.
Изобретение будет проиллюстрировано более подробно ссылками на нижеследующие примеры, но следует понимать, что настоящее изобретение не считается ограниченным ими.
ПРИМЕРЫ
Пример 1
Стадия 1. Получение (+)-5-[6-фторхроман-2-карбонил]-2,2-диметил[1,3]диоксан-4,6-диона (соединения III).
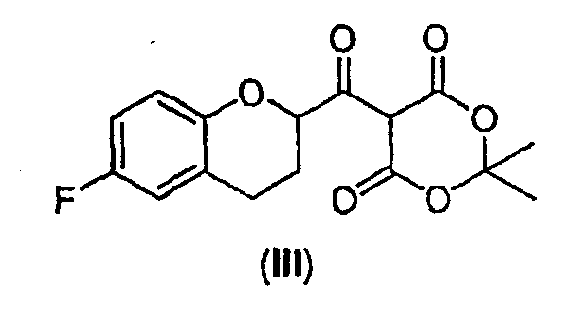
В атмосфере азота тионилхлорид (109,21 г, 918 ммоль) прибавляют при 20-25°С к суспензии 6-фторхроман-2-карбоновой кислоты (90,00 г, 459 ммоль) и ДМФА (1,68 г, 23 ммоль) в толуоле (635 мл). Затем суспензию нагревают до внутренней температуры 60-70°С и получают прозрачный желтый раствор с одновременным выделением газа. При этой температуре реакцию завершают за 70 мин, после чего смесь концентрируют в вакууме (температура бани 45-50°C, давление ≤35 мбар) и получают хлорангидрид хроман-2-карбоновой кислоты в виде желтого масла (112,65 г). Сырой продукт растворяют в метиленхлориде (65 мл) и медленно прибавляют в атмосфере азота к раствору кислоты Мелдрума (70,90 г, 482 ммоль) и пиридина (72,62 г, 918 ммоль) в метиленхлориде (261 мл) при внутренней температуре 0-10°С. Реакционной смеси дают возможность нагреться до 20-25°С в течение 50 мин и перемешивают при этой температуре дополнительно 30 мин. Затем к образовавшейся коричневой суспензии прибавляют метиленхлорид (325 мл) и воду (325 мл). Двухфазную смесь перемешивают в течение 5 мин, разделяют и органический слой затем экстрагируют водой дважды (по 200 мл каждый раз), затем 2N водным раствором HCl (250 мл) и наконец водой (250 мл). После сушки над Na2SO4 органический слой фильтруют и концентрируют в вакууме (≤50 мбар), получая коричневое вязкое масло (170,76 г), которое кристаллизуется через 10 мин при комнатной температуре. Твердое вещество суспендируют в диизопропиловом эфире (500 мл) при 20-25°С в течение 2 ч. После фильтрования суспензии влажный продукт промывают диизопропиловым эфиром (70 мл) и сушат в вакууме (13 ч при 40°С), получая твердое вещество цвета желтой охры (выход: 114,71 г, ВЭЖХ-чистота: 96,98%).
Пример 2
Стадия 2, путь A. Получение (±)-1-(6-фторхроман-2-ил)этанона (соединения IVa).
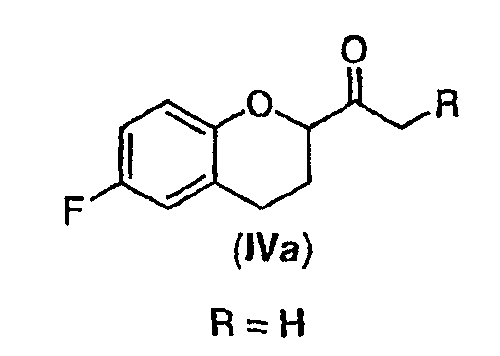
Воспроизводят пример 1 с 16 г 6-фторхроман-2-карбоновой кислоты и остаток, полученный после обработки и выпаривания метиленхлорида, используют непосредственно для стадии 2, пути A. Смесь полученного таким образом сырого продукта (соединения III) с водой (40 мл) и уксусной кислотой (40 мл) нагревают в течение 70 мин при кипячении с обратным холодильником и затем охлаждают до комнатной температуры. Реакционную смесь экстрагируют метиленхлоридом (40 мл) и органический слой два раза промывают 1 N водным раствором NaOH (каждый раз 20 мл). После сушки над MgSO4 органический слой фильтруют и упаривают. Остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь этилацетат/циклогексан (1/3 по объему). Собирают вторую фракцию, выпаривают растворитель и получают продукт в виде желтого масла (выход: 11,89 г, ВЭЖХ-чистота: 98,76%).
Пример 3
Стадия 2, путь В. Получение этилового эфира (±)-3-(6-фторхроман-2-ил)-3-оксопропионовой кислоты (соединения IVb в виде этилового эфира)
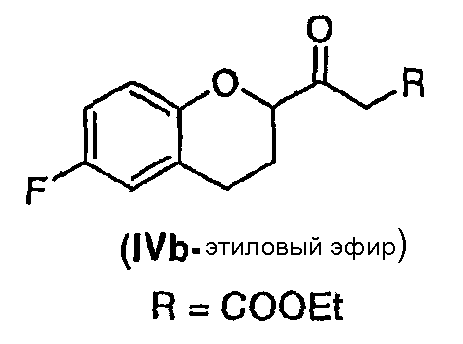
Воспроизводят пример 1 с 16 г 6-фторхроман-2-карбоновой кислоты и остаток, полученный после обработки и выпаривания метиленхлорида, используют непосредственно для стадии 2, путь В. Суспензию этого сырого продукта (соединения III) в этаноле (150 мл) нагревают при кипячении с обратным холодильником в течение 75 мин, в результате получают прозрачный раствор. После охлаждения раствора до комнатной температуры и упаривания остаток распределяют между метиленхлоридом (80 мл) и водой (80 мл). Фазы разделяют и органический слой экстрагируют 1 N водным раствором NaOH (40 мл). Раствор в метиленхлориде сушат над MgSO4, фильтруют и упаривают. Остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь этилацетат/циклогексан (1/4 по объему). Собирают первую фракцию, выпаривают растворитель и получают продукт в виде желто-коричневого масла (выход: 11,89 г, ВЭЖХ-чистота: 92,45%).
Пример 4
Стадия 2, путь В. Получение трет-бутилового эфира (±)-3-(6-фторхроман-2-ил)-3-оксопропионовой кислоты (соединения IVb в виде трет-бутилового эфира)
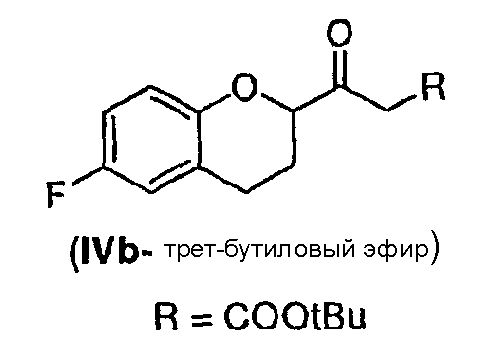
трет-Бутанол (83,90 г) прибавляют при комнатной температуре к суспензии соединения III (94,00 г) (полученного способом, описанным в примере 1) в толуоле (280 мл). Суспензию нагревают до внутренней температуры 70-80°С, в результате получают прозрачный раствор при одновременном выделении газа. Реакцию завершают за 80 мин. Смесь охлаждают до комнатной температуры и экстрагируют последовательно раствором NaHCO3 (235 мл) и насыщенным раствором NaCl. Органический слой сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая продукт в виде оранжево-коричневого масла (выход: 95,79 г, ВЭЖХ-чистота: 97,20%; сырой продукт содержал небольшие количества толуола). Сырой продукт используют на следующей стадии без дополнительной очистки.
Пример 5
Стадия 3, путь А. Получение (±)-2-бром-1-(6-фторхроман-2-ил)-этанона (соединения Va).
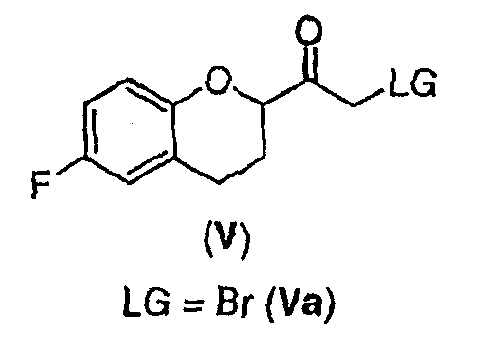
TMSCl (3,2 мл) прибавляют к раствору 2 M LDA (9,0 мл) в 20 мл ТГФ при -78°С в течение 10 мин. Затем прибавляют раствор соединения IVa (3,0 г) (полученного способом, описанным выше в примере 2) в ТГФ (3 мл), через 10 мин реакционной смеси дают нагреться до комнатной температуры в течение 40 мин. Выпавшее белое твердое вещество и осадок распределяют между циклогексаном (100 мл) и холодным 10% раствором NaHCO3 (60 мл). Водный слой разбавляют водой (20 мл) и отделяют. Органический слой экстрагируют дважды 10% раствором NaHCO3 (каждый раз 30 мл), сушат над Na2SO4, фильтруют и концентрируют. Остаток растворяют в метиленхлориде (15 мл) и охлаждают до внутренней температуры 0-5°C. К полученной смеси прибавляют суспензию NBS (2,94 г) в метиленхлориде (10 мл). После перемешивания в течение 1,5-2 ч при указанной температуре реакционную смесь выливают в 10% раствор NaHCO3 (15 мл), органический слой отделяют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле, используя смесь этилацетат/циклогексан (1/5 по объему) в качестве элюента. Собирают первую фракцию, упаривают растворитель и получают смесь продуктов, состоящую из соединения Va (78,1% по данным ВЭЖХ) и соответствующего побочного продукта (VaBP1),
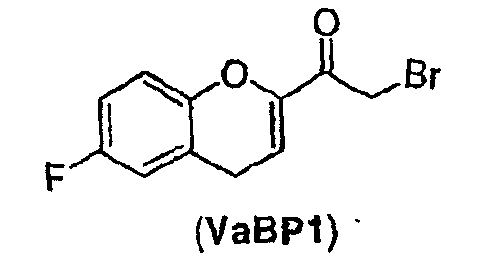
который образуется неселективным бромированием с последующим элиминированием в виде желто-коричневого масла (выход: 2,17 г, так как соединение Va, по-видимому, менее стабильно, чем соединение Vb, его следует хранить предпочтительно в темноте при температуре -20°С).
Пример 6
Стадия 3, путь B: Получение (±)-2-бром-1-(6-фторхроман-2-ил)этанона (соединения Va).
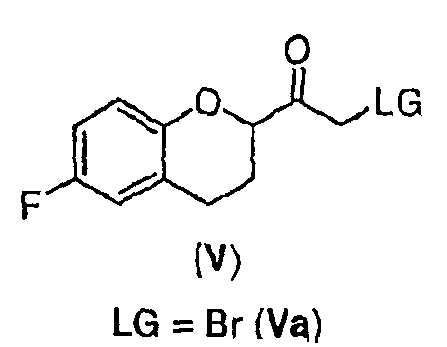
N-Бромсукцинимид (NBS) (5,04 г) прибавляют порциями при 5-10°C к раствору трет-бутилового эфира соединения IVb (10,0 г) (полученного способом, описанным в примере 4) и Mg(ClО4)2 (2,32 г) в этилацетате (100 мл). В дальнейшем реакцию контролируют ВЭЖХ. После прибавления смесь перемешивают при 5-10°C в течение 45 мин. После того как остается 16% аддукта, прибавляют дополнительное количество NBS (1,0 г). Смесь перемешивают в течение 20 мин при 5-10°C, затем дают согреться до комнатной температуры и перемешивают в течение 20 мин при этой температуре. Осадок отфильтровывают и маточный раствор концентрируют в вакууме, получая трет-бутиловый эфир 2-бром-3-(6-фторхроман-2-ил)-3-оксопропионовой кислоты в виде красного масла (15,3 г). Для проведения гидролиза и декарбоксилирования красное масло растворяют в уксусной кислоте (42 мл) и муравьиной кислоте (49 мл) и смесь нагревают до внутренней температуры 80-85°С, при этом наблюдается выделение газа. Через 60 мин реакцию завершают и смесь концентрируют в вакууме, получая коричневое масло. Масло затем растворяют в этилацетате (50 мл) и н-гексане (50 мл) и раствор экстрагируют последовательно дважды полунасыщенным раствором NaCl (каждый раз 20 мл) и насыщенным раствором NaHCO3 (20 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют в вакууме, получая масло янтарного цвета, которое растворяют в циклогексане. Кристаллизацию вызывают с помощью затравки при комнатной температуре. Через 45 мин суспенсию фильтруют, получая после сушки соединение Va (2,98 г, бежевое твердое вещество). Дополнительные количества соединения Va (1,9 г) получают из маточного раствора после перемешивания при 6-7°C в течение 1,5 ч, отфильтровывания и сушки (общий выход: 4,88 г, ВЭЖХ-чистота: 98,5%; так как соединение Va, по-видимому, менее стабильно, чем соединение Vb, его следует хранить предпочтительно в темноте при температуре -20°С).
Пример 7
Стадия 3, путь B: Получение (±)-2-хлор-1-(6-фторхроман-2-ил)этанона (соединения Vb)
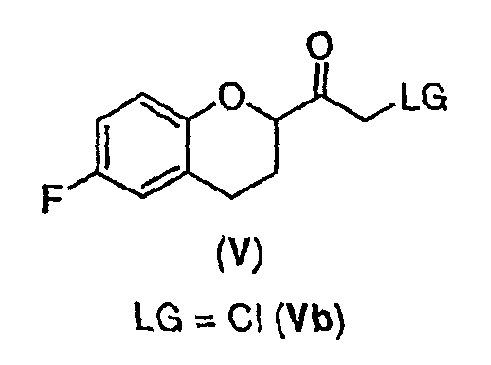
При комнатной температуре Mg(СlO4)2 (21,40 г) медленно прибавляют к раствору трет-бутилового эфира соединение IVb (105,59 г) (полученного способом, описанным в примере 4) в этилацетате (800 мл). Затем порциями прибавляют NCS (41,80 г) при 20-25°С в течение 3,5-4 ч и в дальнейшем реакцию контролируют ВЭЖХ. После завершения прибавления желтую суспензию перемешивают в течение 30 мин при 20-25°С и затем фильтруют. Слой на фильтре промывают этилацетатом (100 мл) и объединенные фильтраты экстрагируют последовательно насыщенным раствором NaCl (150 мл) и водой (150 мл), затем концентрируют в вакууме (60 мбар) и получают коричневое масло (116,82 г). Для проведения гидролиза и декарбоксилирования коричневое масло растворяют в смеси уксусной кислоты (420 мл), муравьиной кислоты (390 мл) и воды (80 мл). Раствор нагревают до внутренней температуры 80-90°С, при этом наблюдается выделение газа. После завершения реакции (в течение 2 ч) раствор концентрируют в вакууме (<30 мбар), получая темно-оранжевое масло (83,25 г), которое растворяют в этилацетате (400 мл). Полученный раствор последовательно экстрагируют полунасыщенным раствором NaCl (300 мл), насыщенным раствором NaHCO3 (300 мл) и водой (100 мл). После сушки над Na2SO4 суспензию фильтруют и концентрируют, получая сначала красное масло (80,00 г), которое медленно кристаллизуется при комнатной температуре. Для дальнейшей очистки сырой продукт (77,0 г) растворяют в изопропаноле (240 мл) при внутренней температуре 45-50°С. В раствор помещают затравку, чтобы вызвать кристаллизацию, и охлаждают до 0-5°С. После 75 мин перемешивания при 0-5°C суспензию фильтруют и слой на фильтре промывают холодным изопропанолом (40 мл). Влажный продукт сушат в вакууме при 35-40°С, получая желтоватое твердое вещество (57,13 г, ВЭЖХ-чистота: 99,00%).
Пример 8
Стадии 1-3. Получение (±)-2-хлор-1-(6-фторхроман-2-ил)этанона (соединения Vb) из 6-фторхроман-2-карбоновой кислоты (II)
Cмесь 6-фторхроман-2-карбоновой кислоты (114,4 г, анализ = 99%, 577 ммоль), тионилхлорида (83,15 г, 692 ммоль) и ДМФА (2,18 г, 30 ммоль) в толуоле (471 г) медленно нагревают в атмосфере азота до внутренней температуры 70-80°C (при внутренней температуре 57°С начинается выделение газа). Когда реакция завершается (в течение 40 мин при 78°С, ВЭЖХ анализ показывает 98,6% соответствующего хлорида кислоты), 208 г растворителя отгоняют в вакууме (давление: 220 (начальное) - 155 (конечное) мбар, внутренняя температура: 73 (начальная) - 69 (конечная)°С, температура паров: 39 (начальная) - 63 (конечная)°С). Во вторую колбу загружают кислоту Мелдрума (89,1 г, 606 ммоль), пиридин (89 мл, 1,11 моль) и метиленхлорид (375 мл). Затем смесь охлаждают до внутренней температуры 0-5°С, медленно прибавляют вышеупомянутый приготовленный ранее раствор хлорида 6-фторхроман-2-карбоновой кислоты в толуоле при внутренней температуре 0-5°С. Реакционной смеси дают нагреться до 20°С в течение 80 мин (в ходе реакции ВЭЖХ показывает 92,6% продукта). К смеси медленно прибавляют трет-бутанол (81,0 г, 1,08 моль) и смесь медленно (в течение 4 часов) нагревают до внутренней температуры 70-80°С при одновременной отгонке растворителя и выделении газа. Во время нагревания (после 75 мин и при внутренней температуре 56°C) прибавляют дополнительное количество трет-бутанола (75 г, 1,00 моль). Перегонка и выделение газа прекращаются, когда внутренняя температура достигает 70-80°С (к этому времени отгоняется 370 г растворителя). Когда ВЭЖХ-анализ процесса показывает завершение реакции, смесь охлаждают до 20°С и прибавляют раствор серной кислоты (41,8 г) в воде (200 мл). Органический слой отделяют, дважды экстрагируют насыщенным раствором NaHCO3 (каждый раз 200 мл), затем концентрируют в вакууме до приблизительно 60% объема (давление: 370-150 мбар; отмечается, что во время перегонки вода должна быть удалена полностью) и разбавляют при комнатной температуре этилацетатом (450 мл). После прибавления Na3PO4 (91,5 г), медленно (в течение 3 ч) прибавляют сульфурилхлорид (53 мл) при внутренней температуре 10-20°С и продолжают перемешивание до тех пор, пока ВЭЖХ анализ процесса не покажет завершение реакции (приблизительно 1 ч). Смесь экстрагируют дважды водой (каждый раз 150 мл) и перегоняют в вакууме (давление: 150-170 мбар) до тех пор, пока не будет получено 305 г дистиллята. Затем прибавляют уксусную кислоту (400 мл) и смесь опять перегоняют в вакууме (давление: 30-40 мбар) до тех пор, пока не будет получено дополнительно 292 г дистиллята. Прибавляют концентрированную хлористоводородную кислоту (84 мл) и смесь перемешивают при внутренней температуре 40-50°С до завершения реакций (гидролиза и декарбоксилирования) (4 ч, контроль ВЭЖХ). После отгонки еще 100 г растворителя в вакууме (давление: 200-40 мбар; удаление оставшихся толуола и трет-бутанола) эмульсию разбавляют уксусной кислотой (70 мл) при внутренней температуре 20°С и получают раствор. Затем прибавляют воду (20 мл) и кристаллы (затравку), чтобы вызвать кристаллизацию. Когда начинается кристаллизация медленно прибавляют дополнительную воду (230 мл). Суспензию перемешивают при комнатной температуре (15 ч), затем фильтруют и слой на фильтре промывают смесью уксусной кислоты и воды (об/об = 1/1, 100 мл). Влажный продукт сушат в вакууме при 40°С, получая твердое вещество цвета охры (общий выход 101,84 г, чистота (ВЭЖХ): 98,9%).
Пример 9
Стадия 4. Получение (±)-2-хлор-1-(6-фтор-(2R*)-хроман-2-ил)-(1R*)-этан-1-ола (соединения VIa) и (±)-2-хлор-1-(6-фтор-(2S*)-хроман-2-ил)-(1R*)-этан-1-ола (соединения VIb)
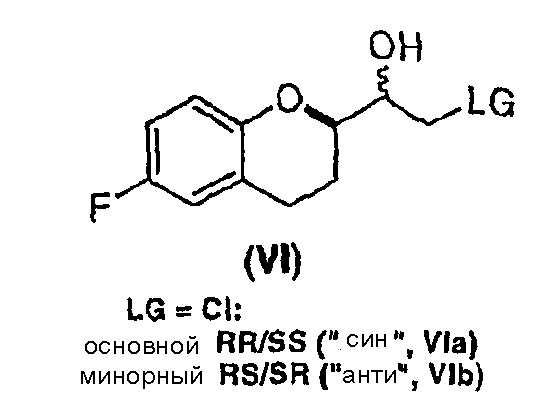
Соединение Vb (33,74 г) прибавляют при 0-5°C к раствору ZnCl2 (40,3 г) в метаноле (470 мл) и смесь перемешивают до полного растворения всех твердых веществ (1 ч). Раствор охлаждают до -10°С и прибавляют порциями NaBH4 в течение 35 мин. После завершения реакции, контролируемой ВЭЖХ, смесь концентрируют до объема около 150 мл и затем разбавляют толуолом (400 мл). Органический слой экстрагируют последовательно два раза 1,0 N раствором HCl (каждый раз 200 мл) и насыщенным раствором NaHCO3 (100 мл). После сушки над MgSO4 суспензию фильтруют и растворитель упаривают в вакууме, получая коричневатое масло (35,28 г, отношение VIa/VIb = 61/39; сырая смесь продуктов содержит небольшие количества толуола). Сырой продукт используют на следующей стадии без дополнительной очистки.
Пример 10
Стадия 5. Получение (±)-6-фтор-[(2R*)-оксиран-2-ил]-(2S*)-хромана (соединение VIIa) и (+)-6-фтор-[(2R*)-оксиран-2-ил]-(2R*)-хромана (соединения VIIb)
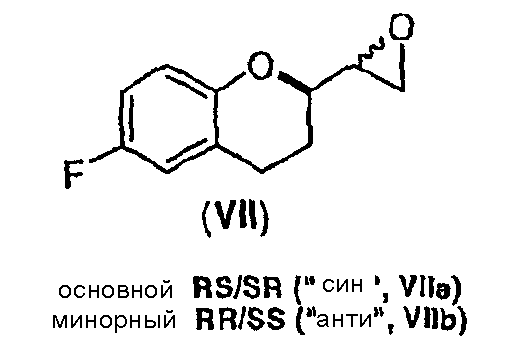
Метанольный раствор NaOMe (30%, 30,9 г) прибавляют при 20-25°C к раствору смеси соединений VIa и VIb (37,9 г, отношение VIa/VIb = 61/39) в метаноле (380 мл). Реакцию контролируют ВЭЖХ и после перемешивания в течение 3,5 ч при 20-25°C прибавляют дополнительное количество метанольного раствора NaOMe (30%, 1,4 г). После завершения реакции (в течение 3,5 ч) смесь нейтрализуют прибавлением уксусной кислоты и затем концентрируют в вакууме. Остаток распределяют между метиленхлоридом (300 мл) и полунасыщенным раствором NaCl (200 мл). Фазы разделяют и органический слой сушат над MgSO4. После фильтрования фильтрат концентрируют, получая коричневатое масло (32,1 г, отношение VIIa/VIIb = 61/39). Сырой продукт используют непосредственно на следующей стадии.
Пример 11
Стадия 6. Получение (±)-2-бензиламино-1-(6-фтор-(2R*)-хроман-2-ил)-(1S*)-этан-1-ола (соединения VIIIа) и (±)-2-бензиламино-1-(6-фтор-(2R*)-хроман-2-ил)-(1R*)-этан-1-ола (соединения VIIIb) и разделение соединений VIIIa и VIIIb
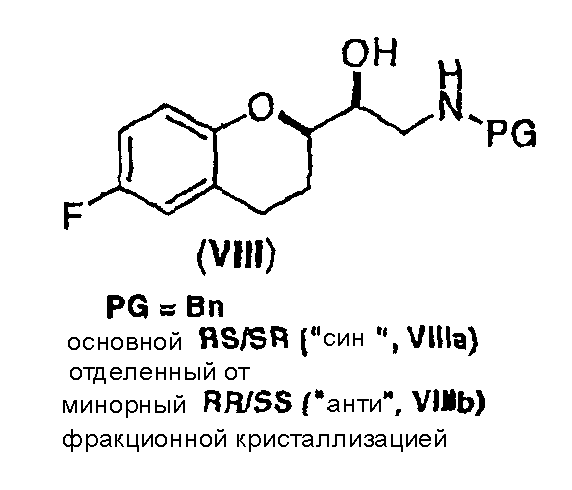
Cмесь соединений VIIа и VIIb (отношение: 57/43) медленно прибавляют (в течение 1,5 ч) при внутренней температуре 40°С к раствору бензиламина (5,4 г) в 2-пропаноле (30 мл). После завершения реакции, контролируемой ВЭЖХ, раствор охлаждают до комнатной температуры и прибавляют вещество-затравку. Затем диастереомеры VIIIa и VIIIb разделяют фракционной кристаллизацией. Суспензию перемешивают при комнатной температуре в течение 1 ч и фильтруют, получая после сушки в вакууме бесцветное твердое вещество (1,01 г). Маточный раствор концентрируют до тех пор, пока не останется 25 г остатка. Затем концентрированную смесь нагревают до 60°C и охлаждают в течение 3 ч до 0-5°C. Дополнительную порцию продукта получают после фильтрования и сушки влажного продукта в вакууме (0,3 г). Маточный раствор концентрируют до тех пор, пока не останется 15 г остатка, и прибавляют диизопропиловый эфир (15 г). Третью порцию получают после фильтрования и сушки влажного продукта в вакууме (0,33 г). Вторую и третью порции перекристаллизовывают из 2-пропанола (3,7 г) и после фильтрования влажный продукт (0,6 г) растворяют с первой порцией в 2-пропаноле (10 г) при кипячении с обратным холодильником. Смесь охлаждают до 0-5°C и затем фильтруют. Влажный продукт сушат в вакууме, получая бесцветное твердое вещество (выход: 1,1 г, отношение VIIIa/VIIIb = 96/4).
Диастереомер VIIIb может быть получен, например, из маточного раствора после концентрирования досуха с последующей экстрактивной обработкой и кристаллизацией.
Пример 12
Стадии 4-6. Получение (±)-2-бензиламино-1-[6-фтор-(2R*)-хроман-2-ил]-(1S*)-этан-1-ола (соединения VIIIа) и (±)-2-бензиламино-1-[6-фтор-(2R*)-хроман-2-ил]-(1R*)-этан-1-ола (соединения VIIIb) из (±)-2-хлор-1-(6-фторхроман-2-ил)этанона (соединения Vb) и разделение соединений VIIIa и VIIIb
Соединение Vb (76,31 г, 324 ммоль) и ZnCl2 (22,53 г, 162 ммоль) растворяют в этаноле (648 мл) при внутренней температуре 20-30°C. Полученный раствор затем охлаждают до внутренней температуры -15°C - (-20)°С и медленно прибавляют раствор NaBH4 (12,77 г, 324 ммоль) и NaOMe (4 мл 30% раствора MeOH) в MeOH (136 мл). Во время прибавления поддерживают внутреннюю температуру от -20 до -10°C и реакцию контролируют ВЭЖХ. После завершения реакции смеси дают возможность нагреться до 0-5°C и прибавляют хлористоводородную кислоту (160 мл 2N раствора HCl). Смеси дают возможность нагреться до 20-25°C и перемешивают при такой температуре 30 минут. Растворитель почти полностью удаляют в вакууме, получая коричневую суспензию (191,3 г). Этот остаток распределяют между хлористоводородной кислотой (160 мл 2N раствора HCl) и MTBE (450 мл). Органический слой отделяют, экстрагируют хлористоводородной кислотой (30 мл 2N водного раствора HCl), затем два раза водой (каждый раз 250 мл) и концентрируют в вакууме, получая коричневое масло (79,77 г, отношение VIa/VIb = 63,5/36,5). После растворения масла в 2-пропаноле прибавляют раствор NaOMe в MeOH (64,18 г, концентрация: 30%) при внутренней температуре 20-25°С. Реакцию контролируют ВЭЖХ. После завершения реакции смесь охлаждают до 0-5°С и нейтрализуют прибавлением уксусной кислоты (1,9 мл). Суспензию фильтруют через целит и слой на фильтре промывают 2-пропанолом (25 мл). Фильтрат концентрируют в вакууме, получая полуконцентрированный мутный коричневатый раствор (115,97 г). Полученную смесь опять фильтруют и слой на фильтре промывают 2-пропанолом (25 мл), получая прозрачный коричневый раствор, который затем медленно прибавляют (в течение 3 ч) к раствору бензиламина (105,2 г, 972 ммоль) в 2-пропаноле (352 мл) при внутренней температуре 33-38°C. Реакцию контролируют ВЭЖХ и прибавляют затравку, чтобы вызвать кристаллизацию продукта во время реакции. После завершения прибавления смесь перемешивают в течение 3,5 ч при 25-30°C, затем охлаждают до 0-5°С и перемешивают при указанной температуре в течение 1,5 ч. Суспензию фильтруют и слой на фильтре промывают предварительно охлажденным (0-5°C) 2-пропанолом (46 мл). Влажный продукт сушат в вакууме при 50-55°C, получая твердое вещество слабо-бежевой окраски (42,23 г, отношение VIIIa/VIIIb = 92/8). Сырой продукт растворяют в ацетонитриле (294 мл) нагреванием при кипячении с обратным холодильником. Раствор медленно охлаждают 0-5°C (6-7 ч), фильтруют и слой на фильтре промывают ацетонитрилом (38 мл). Влажный продукт сушат в вакууме при 50-55°C, получая белое твердое вещество (общий выход: 38,2 г, отношение VIIIa/VIIIb = 98,8/1,2, чистота (ВЭЖХ) продукта VIIIa: 98,62%).
Пример 13
Стадия 7. Получение (±)-2-{бензил[2-(6-фтор-(2R*)-хроман-2-ил)-(2S*)гидроксиэтил]амино}-1-(6-фтор-(2S*)-хроман-2-ил)этанона (соединения IXa), (±)-4-бензил-2-[6-фтор-(2R*)-хроман-2-ил]-(6S*)-[6-фтор-(2S*)-хроман-2-ил]морфолин-2-ола (соединения IXa`), (±)-2-{бензил[2-(6-фтор-(2R*)-хроман-2-ил)-(2S*)-гидроксиэтил]амино}-1-(6-фтор-(2R*)-хроман-2-ил)этанона (соединения IXb) и (±)-4-бензил-(6S*)-2,6-бис-[6-фтор-(2R*)-хроман-2-ил]-морфолин-(2S*)-ола (соединения IXb`)и разделение диастереомеров
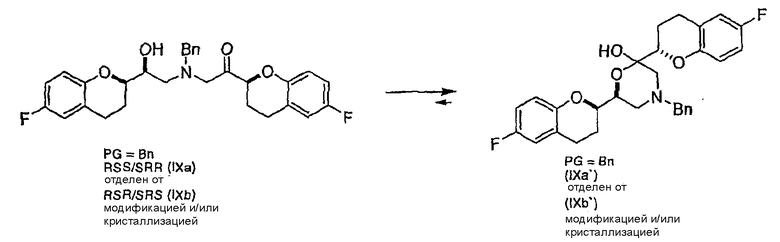
Соединение V (14,62 г) прибавляют при внутренней температуре 40°C к суспензии соединения VIIIa (17,5 г), NaHCO3 (9,6 г) и NaBr (0,9 г) в ДМФА (70 мл). Когда реакция завершается (за 3-3,5 ч, контроль ВЭЖХ), суспензию охлаждают до комнатной температуры и разбавляют MTBE (400 мл) и водой (200 мл). Затем фазы разделяют, органический слой экстрагируют водой (100 мл) и объединенные водные слои повторно экстрагируют MTBE (100 мл). После упаривания объединенных органических слоев оставшееся масло янтарного цвета (35,0 г) растворяют в диизопропиловом эфире (400 мл) и вносят затравку. Суспензию сначала перемешивают при комнатной температуре в течение 1,5 ч и затем при 0-5°С в течение 0,5 ч. После фильтрования влажный продукт сушат в вакууме, получая светло-желтое твердое вещество (выход: 23,95 г, ВЭЖХ-чистота 97,5%, отношение IXa/IXa` к IXb/IXb` = 52/48).
Маточный раствор концентрируют до 56 г, затем охлаждают до 0-5°C и вносят затравку. Вторую порцию получают после фильтрования и сушки (выход: 2,62 г, ВЭЖХ-чистота 92,6%, отношение IXa/IXa` к IXb/IXb` = 43/57).
Разделение диастереомеров проводят фракционной кристаллизацией из ацетонитрила.
Диастереомерную смесь соединений IXa/IXa` и IXb/IXb` (отношение: 55/45, 2,31 г) растворяют в ацетонитриле при внутренней температуре 70°C. В светло-желтый раствор вносят затравку, охлаждают до комнатной температуры (в течение 2-3 ч) и перемешивают при указанной температуре в течение 1,5-2 ч. Фильтрованием суспензии и сушкой влажного продукта получают первую порцию (выход: 0,26 г, отношение IXa/IXa` к IXb/IXb` = 95/5).
Маточный раствор концентрируют до 30 г и перемешивают при комнатной температуре после внесения затравки. Фильтрованием суспензии получают влажный продукт (1,12 г), который перекристаллизовывают из ацетонитрила (11,2 г). После сушки влажного продукта в вакууме получают вторую порцию (0,50 г, отношение IXa/IXa` к IXb/IXb`=62/38). Эту порцию перекристаллизовывают из ацетонитрила (10 мл) и получают влажный продукт (0,57 г), который опять растворяют в ацетонитриле (8 мл) при нагревании. Раствор охлаждают до комнатной температуры и вносят затравку. После фильтрования суспензии и сушки влажного продукта получают третью порцию (выход: 0,16 г, отношение IXa/IXa` к IXb/IXb` = 98/2).
Разделение диастереомеров также проводят селективным силилированием и фракционной кристаллизацией.
Методика A.
Имидазол (0,417 г) прибавляют при 0-5°C к суспензии соединений IX (2,0 г, полученной по вышеописанной методике, отношение IXa/IXa` к IXb/IXb` = 52/48) в смеси ацетонитрила (13,5 мл) и ТГФ (1,5 мл). Затем медленно прибавляют TMSCl (0,228 мг) при указанной температуре в течение 3,5-4 ч и при контроле ВЭЖХ. После завершения прибавления смесь концентрируют в вакууме до 8-10 мл и затем прибавляют ацетонитрил (5 мл). Перемешивание суспензии при 0-5°C в течение 1-1,5 ч с последующим фильтрованием дает белый влажный продукт (1,31 г), который сушат в вакууме (выход: 0,82 г, отношение IXa/IXa` к IXb/IXb` = > 98/2).
Методика B.
Имидазол (0,21 г) прибавляют при 0-5°C к суспензии соединений IX (1,0 г, полученной по вышеописанной методике, отношение IXa/IXa` к IXb/IXb` = 52/48) в MTBE (10 мл). Затем медленно прибавляют TMSCl (0,115 мг) при указанной температуре в течение 3,5-4 ч и при контроле ВЭЖХ. Реакцию завершают прибавлением 4 капель TMSCl. Затем суспензию фильтруют и влажный продукт (0,87 г) сушат в вакууме, получая белый сырой продукт (0,51 г, отношение IXa/IXa` к IXb/IXb` = 98/2, продукт содержит гидрохлорид имидазола). Для удаления гидрохлорида имидазола сырой продукт суспендируют при комнатной температуре в смеси ацетонитрила и воды (3,0 мл, 4/1 по объему) в течение 2,5-3 ч. Фильтрование суспензии и сушка влажного продукта (0,65 г) в вакууме дают белое твердое вещество (выход: 0,31 г, отношение IXa/IXa` к IXb/IXb` = 98/2).
Пример 14
Стадия 7. Получение (±)-2-{бензил[2-(6-фтор-(2R*)-хроман-2-ил)-(2S*)-гидроксиэтил]амино}-1-(6-фтор-(2S*)-хроман-2-ил)этанона (соединения IXa), (±)-4-бензил-2-[6-фтор-(2R*)-хроман-2-ил]-(6S*)-[6-фтор-(2S*)-хроман-2-ил]морфолин-2-ола (соединения IXa`), (±)-2-{бензил[2-(6-фтор-(2R*)-хроман-2-ил)-(2S*)-гидроксиэтил]амино}-1-(6-фтор-(2R*)-хроман-2-ил)этанона (соединения IXb) и (±)-4-бензил-(6S*)-2,6-бис-[6-фтор-(2R*)-хроман-2-ил]-морфолин-(2S*)-ола (соединения IXb`)и разделение диастереомеров
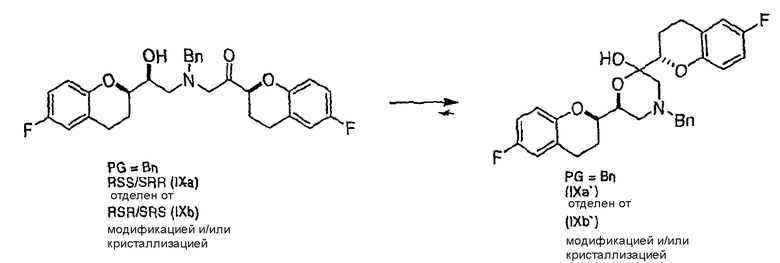
Смесь соединения VIIIa (49,0 г, 162,6 ммоль, полученного по методике примера 12), соединения Vb (42,5 г, 178,9 ммоль, полученного по методике примера 8), NaBr (1,68 г, 16,3 ммоль) и NaHCO3 (20,5 г, 243,9 ммоль) в ДМФА (200 мл) нагревают до внутренней температуры 60-65°C до тех пор, пока ВЭЖХ анализ процесса не покажет почти полную конверсию (приблизительно 1 ч). Затем суспензию фильтруют и слой на фильтре промывают ДМФА (70 мл). К фильтрату прибавляют воду при 50°С (15 мл) и вносят кристаллы-затравку, чтобы вызвать кристаллизацию. Затем продукт осторожно осаждают медленным прибавлением воды (в течение 4 ч) при 50°С. В конечном счете осаждение завершают прибавлением воды (25 мл) при 50°С. Суспензию охлаждают до 20-25°C и фильтруют. Влажный продукт промывают 2-пропанолом (100 мл), сушат в вакууме при 50°С и получают белое твердое вещество (выход: 70,15 г, чистота (ВЭЖХ): 99,1%, отношение IXa/IXa` к IXb/IXb` = 52/48). К суспензии полученного твердого вещества (70,0 г) и имидазола (14,6 г, 214 ммоль) в ацетонитриле (385 мл) медленно прибавляют (1,75 мл/ч) TMSCl (7,56 г, 68,2 ммоль) при внутренней температуре -10-(-15)°C. Затем суспензию перемешивают в течение 2 ч при -5-0°C, контролируя ВЭЖХ. Реакцию завершают прибавлением TMSCl (1,34 г, 12,3 ммоль). Суспензию фильтруют и влажный продукт сушат в вакууме при 40°С, получая белое твердое вещество (66,45 г, отношение IXa/IXa` к IXb/IXb` = 92/8). Полученный продукт суспендируют в смеси циклогексана (285 мл) и MTBE (95 мл) при внутренней температуре 60°C в течение 10 минут. Затем суспензию охлаждают до 25°С и фильтруют, влажный продукт промывают циклогексаном (50 мл) и опять суспендируют в циклогексане (350 мл). Суспензию перемешивают при 60-65°С в течение 20 минут, затем охлаждают до 25°С и фильтруют. Влажный продукт промывают циклогексаном (50 мл) и сушат в вакууме при 40°С, получая белое твердое вещество (общий выход: 28,83 г, отношение IXa/IXa` к IXb/IXb` = 98,6/1,4).
Пример 15
Стадия 8. Получение (±)-[2R*[R*[R*(S*)]]]-α,α`-[[(фенилметил)имино]бис(метилен)]бис[6-фторхроман-2-метанол] (соединения Xa) и (±)-[2R*[S*[R*(S*)]]]-α,α`-[[(фенилметил)имино]бис(метилен)]бис[6-фторхроман-2-метанол] (соединения Xb)
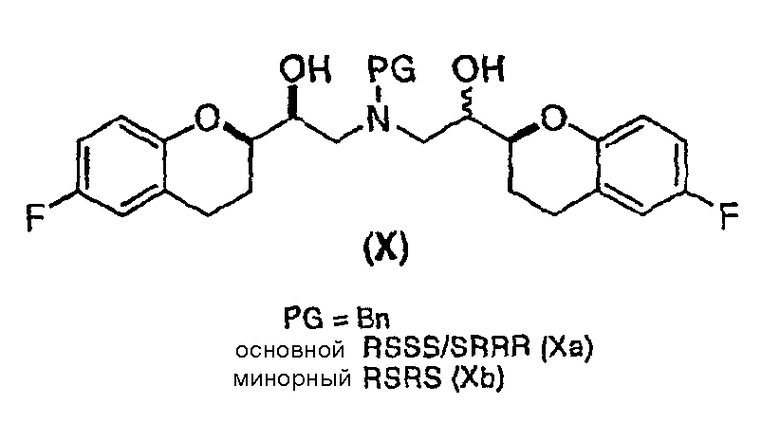
Раствор соединения IXa/IXa` (0,40 г, содержащий 2% IXb) в ТГФ (8,0 мл) охлаждают до внутренней температуры -10 - (-15)°C. К охлажденному раствору прибавляют Ti(OiPr)4 (0,485 мг), затем - LiBH4 (18 мг). После перемешивания при -10-(-15)°C в течение 1 ч и при 0-5°C в течение 1,5-2 ч реакционную смесь выливают в смесь метиленхлорида (10 мл) и насыщенного раствора NaHCO3 (10 мл). Суспензию фильтруют через целит и фазы разделяют. После сушки над MgSO4 органический слой концентрируют и получают бесцветную пену (418 мг отношение Xa/Xb = 78/22). Сырой продукт используют на следующей стадии без дополнительной очистки.
Пример 16
Стадия 8. Получение (±)-[2R*[R*[R*(S*)]]]-α,α`-[[(фенилметил)имино]бис-(метилен)]бис[6-фторхроман-2-метанол] (соединения Xa)
К раствору соединения IXa/IXa`(28,0 г, полученному как в примере 14) и Ti(OiPr)4 (32,9 г, 113,5 ммоль) в DME (280 мл) прибавляют KBH4 (3,15 г, 56,73 ммоль) при внутренней температуре 0°C. После перемешивания при указанной температуре в течение 21 ч (контроль ВЭЖХ) смеси дают нагреться до комнатной температуры и медленно прибавляют хлористоводородную кислоту (280 мл, 10% водный раствор). Суспензию перемешивают в течение 2,5 ч. Суспензию фильтруют и влажный продукт промывают сначала смесью DME (25 мл) и хлористоводородной кислоты (25 мл, 2N водный раствор), затем хлористоводородной кислотой (50 мл, 2N водный раствор) и два раза водой (каждый раз 50 мл). Влажный продукт суспендируют в этаноле (120 мл) и нагревают до 50°С. Затем прибавляют водный раствор NaOH (8,3 г, 30%), получая сначала прозрачный раствор, и смесь нагревают до 60°С. После начала кристаллизации прибавляют воду (33 мл) и суспензию охлаждают до комнатной температуры. Суспензию фильтруют и влажный продукт промывают смесью EtOH/вода (20 мл, об/об = 3/1). Далее влажный продукт растворяют в EtOH (160 мл) нагреванием до 70-75°C и затем охлаждают до 65°C. Прибавляют воду (40 мл) и кристаллы для затравки, смесь охлаждают до комнатной температуры и перемешивают при такой температуре в течение ночи. После фильтрования влажный продукт промывают смесью EtOH/воды (30 мл, об/об = 3/1), сушат в вакууме при 50°С и получают белое твердое вещество (выход: 21,66 г, ВЭЖХ-чистота: 99,85%).
Пример 17
Стадия 9. Получение гидрохлорида (±)-[2R*[R*[R*(S*)]]]-α,α`-[иминобис(метилен)]бис[6-фторхроман-2-метанола] (соединения I) и отделение от побочного продукта гидрохлорида (±)-[2R*[S*[R*(S*)]]]-α,α`-[иминобис(метилен)]бис[6-фторхроман-2-метанола]
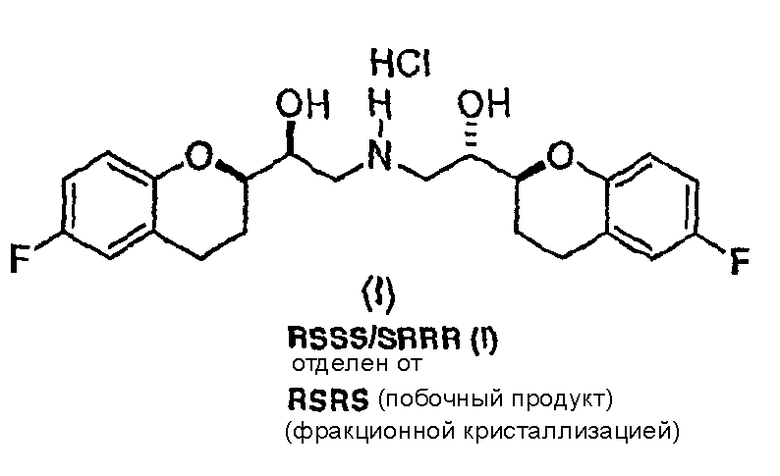
Соединения Xa и Xb (418 мг, отношение Xa/Xb = 78/22, получены по методике примера 11) растворяют в смеси EtOH, содержащего 14% HCl (0,665 г), и MeOH (10 мл). Полученную смесь гидрируют при нормальном давлении и при комнатной температуре с катализатором 5% палладием на угле (100 мг). После завершения реакции (в течение 3 ч) смесь разбавляют MeOH (25 мл), нагревают до 40°С и затем фильтруют через целит. Слой на фильтре промывают горячим MeOH (40°С, 30 мл), объединенные фильтраты концентрируют в вакууме до количества 7-8 г. Полученную суспензию фильтруют, получая бесцветное твердое вещество после сушки влажного продукта (выход: 0,17 г, отношение соединение I/побочный продукт = 95,5/4,5). Маточный раствор концентрируют и остаток растворяют в 2,0 мл MeOH. Суспензию перемешивают при комнатной температуре в течение 0,5 ч, затем фильтруют, получая дополнительную порцию (выход: 28 мг) после сушки влажного продукта в вакууме. Обе порции перекристаллизовывают из MeOH (2,0 мл) и после сушки получают бесцветное твердое вещество (выход: 0,161 г, отношение соединение I/побочный продукт = 98/2).
Пример 18
Стадия 9. Получение гидрохлорида (±)-[2R*[R*[R*(S*)]]]-α,α`-[иминобис-(метилен)]бис[6-фторхроман-2-метанола] (соединения I)
Смесь соединения Xa (21,0 г, 42,3 ммоль, полученного по методике примера 16) и катализатора 5% палладия на угле (1,35 г) в уксусной кислоте (150 мл) гидрируют при нормальном давлении и при внутренней температуре 40°С. После того как ВЭЖХ-анализ показывает полное снятие защиты, суспензию фильтруют через целит. Фильтрат охлаждают до 20°С и прибавляют концентрированную водную хлористоводородную кислоту (4,59 г, 46,5 ммоль). После фильтрования влажный продукт промывают сначала уксусной кислотой (10 мл), затем этанолом (20 мл) и сушат в вакууме, получая белое твердое вещество (выход: 18,05 г, ВЭЖХ-чистота: 99,7 %).
Пример 19
Получение (±)-2-{бензил[2-(6-фтор-(2R*)-хроман-2-ил)-(2S*)гидроксиэтил]амино}-1-(6-фтор-(2S*)-хроман-2-ил)этанона (соединения IXa) ⇔ (±)-4-бензил-2-[6-фтор-(2R*)-хроман-2-ил]-(6S*)-[6-фтор-(2S*)-хроман-2-ил]морфолин-2-ола (соединения IXa`)
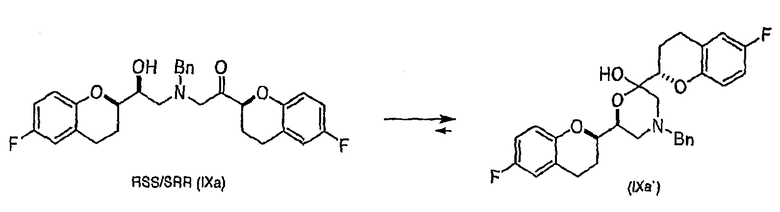
В стеклянный реактор с двойной рубашкой объемом 1 л загружают смесь соединений IXa/IXb (100 г, 202,6 ммоль, отношение IXa/IXb = 50,8/49,2) и ацетонитрил (311 г, вода 0,01% KF). Бесцветную взвесь нагревают до внутренней температуры (IT) = 70°C и перемешивают при такой температуре в течение 13 минут. После медленного охлаждения смеси до 60°С (30 минут) прибавляют DBU (7,79 г, 50,7 ммоль) и реакционную смесь охлаждают со следующим градиентом:
Затем взвесь перемешивают в течение 1 ч при 50°С и отношение IXa/IXb контролируют ВЭЖХ (результат: IXa/IXb = 66,3/33,7). Перемешивание продолжают при 50°С до тех пор, пока отношение IXa/IXb не станет больше 75/25 (контроль ВЭЖХ). В данном случае взвесь перемешивают в течение 4 ч при 50°С (отношение IXa/IXb = 75,1/24,9). Смесь затем охлаждают до 45°С и перемешивают при этой температуре до тех пор, пока отношение IXa/IXb не станет больше 84/16 (контроль ВЭЖХ). Взвесь перемешивают в течение 2 ч при 45°С (отношение IXa/IXb = 85/15). Смесь затем охлаждают до 40°С и перемешивают при этой температуре до тех пор, пока отношение IXa/IXb не станет больше 89/11 (контроль ВЭЖХ), но не дольше 8 ч (в таком случае реакционную смесь следует обрабатывать таким же образом, как описано ниже). В этом случае взвесь перемешивают в течение 5 ч при 40°С (отношение IXa/IXb = 89,2/10,8). Реакционную смесь нейтрализуют уксусной кислотой (3,051 г, 50,7 ммоль) и затем охлаждают до 25°С. После перемешивания в течение 2 ч при 25°С суспензию отфильтровывают и слой на фильтре промывают четыре раза ацетонитрилом (каждый раз 31 г) (отношение IXa/IXb в слое на фильтре после промывания = 99,06/0,94). Маточный раствор янтарного цвета дополнительно обрабатывают (см. ниже, извлечение, ML1). Влажный продукт суспендируют в ацетонитриле (249 г) и взвесь нагревают до 70°С. После суспендирования при этой температуре в течение 1-10 минут смесь охлаждают до 20°С (в течение 2 ч 45 мин) и перемешивают 1,5 ч при 20°С. После фильтрования влажный продукт промывают дважды ацетонитрилом (каждый раз 31 г) и сушат в вакууме при 50°С, получая бесцветное твердое вещество (выход: 74,09 г, 73,4%, ВЭЖХ-чистота: 99,33%, отношение IXa/IXb = 99,37/0,63, анализ = 99,1%). Маточный раствор из взвеси (ML2) используют для стадии извлечения (см. ниже).
Пример 20
Извлечение исходного вещества из маточного раствора, полученного в примере 19.
Воду (117 г) прибавляют к маточному раствору (ML1, 350,02 г) при IT = 24°C и в полученную смесь вносят затравку IXa/IXb (0,05 г). Кристаллизация начинается через несколько минут. После перемешивания в течение 1,5 ч при 20°C - 25°C, взвесь бежевого цвета охлаждают до 0°C-5°C и перемешивают при указанной температуре в течение 3,5 ч. Взвесь отфильтровывают и влажный продукт промывают смесью ацетонитрила (31 г) и воды (10 г). Маточный раствор янтарного цвета удаляют и влажный продукт суспендируют при 20°С-25°С во втором маточном растворе (ML2, 279 г, см. выше). Затем прибавляют воду (93 г) и смесь перемешивают при 0°С-(-5°C) в течение 1,5 ч. После фильтрования взвеси влажный продукт промывают смесью ацетонитрила (31 г) и воды (10 г) и сушат в вакууме при 50°C, получая не совсем белое твердое вещество (выход: 7,74 г, 7,6%, ВЭЖХ-чистота: 99,49%, сумма обоих диастереомеров), отношение IXa/IXb = 39,1/60,9, анализ = 98,2% (сумма обоих диастереомеров)).
Хотя изобретение описано подробно и со ссылкой на его конкретные примеры, для специалиста в данной области техники очевидно, что могут быть сделаны различные изменения и модификации, не выходящие за пределы существа и объема изобретения.
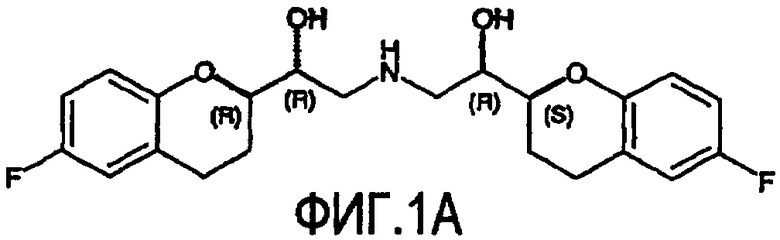
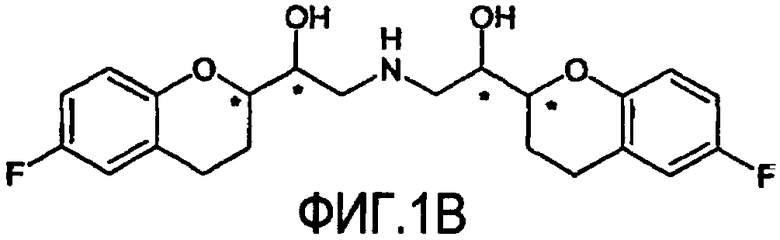
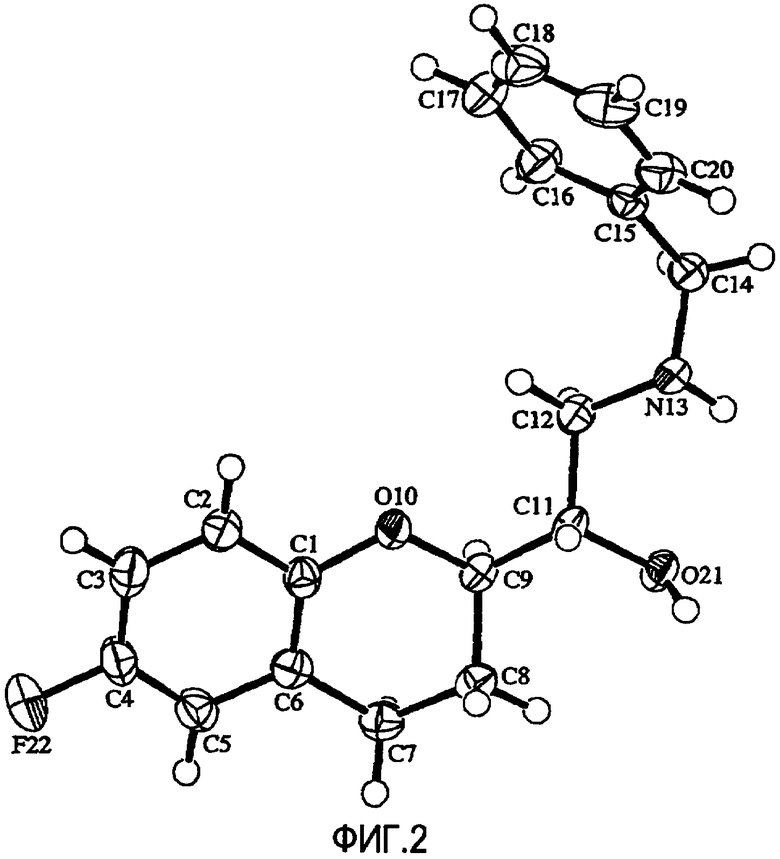
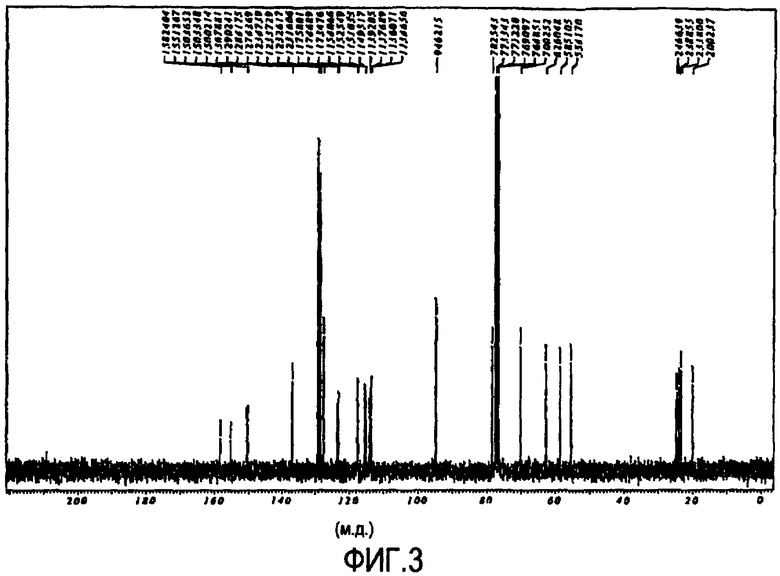
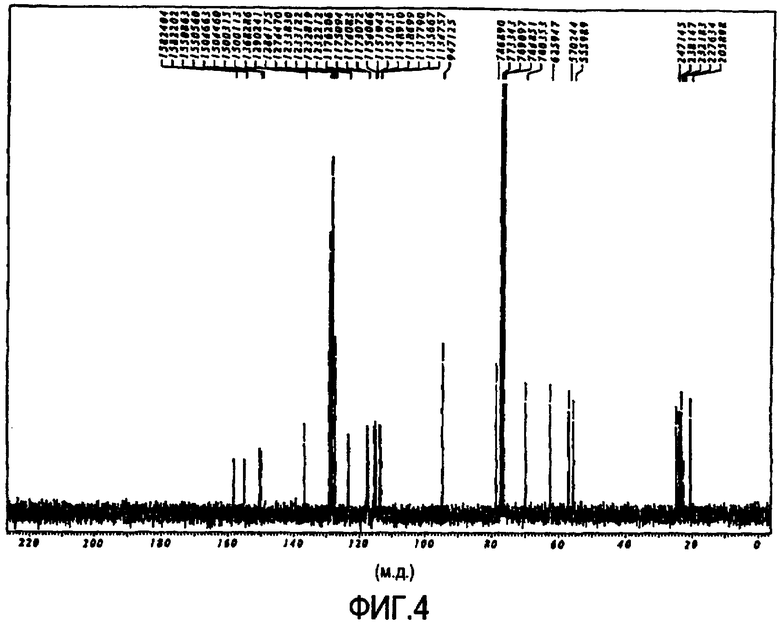
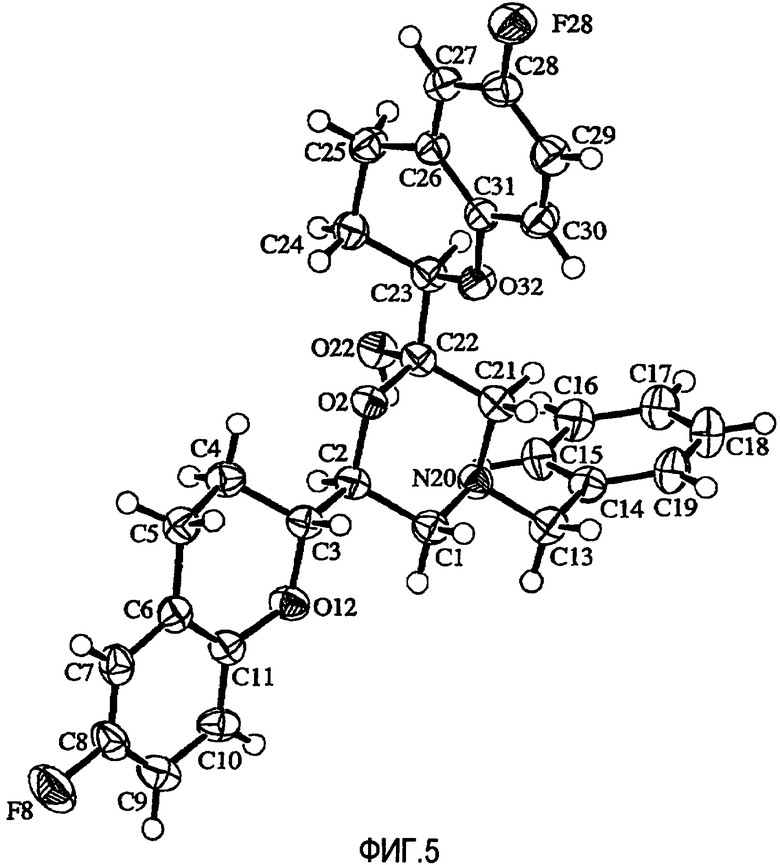
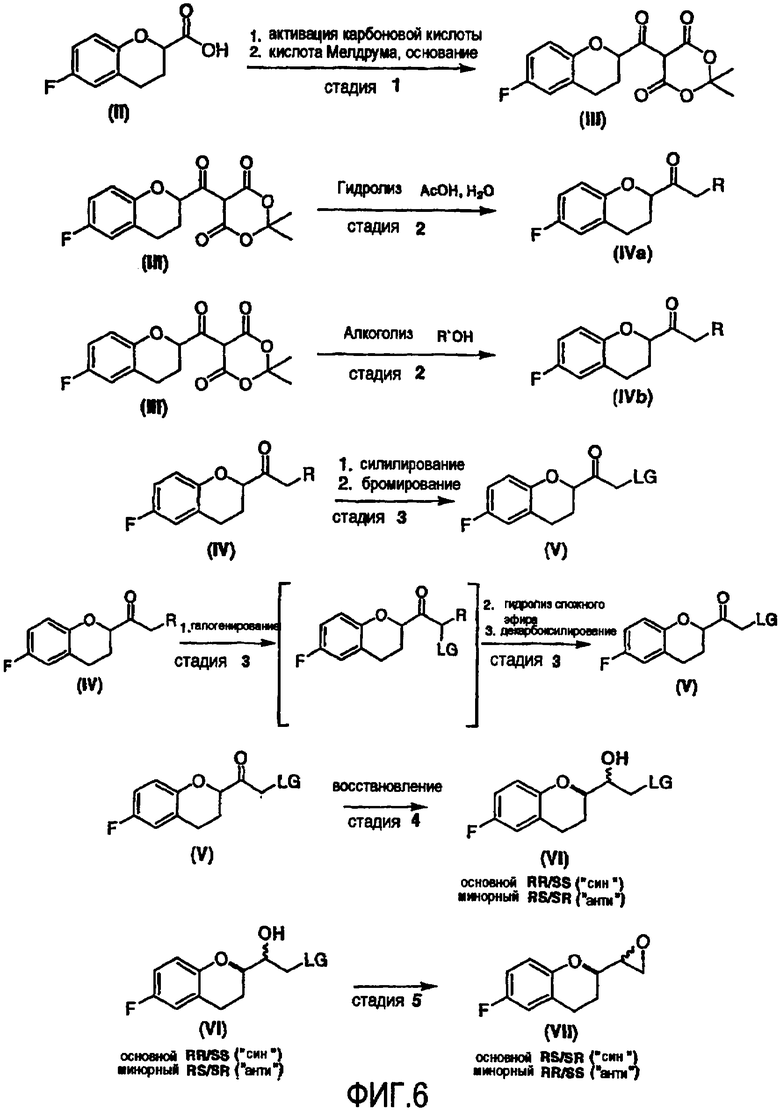
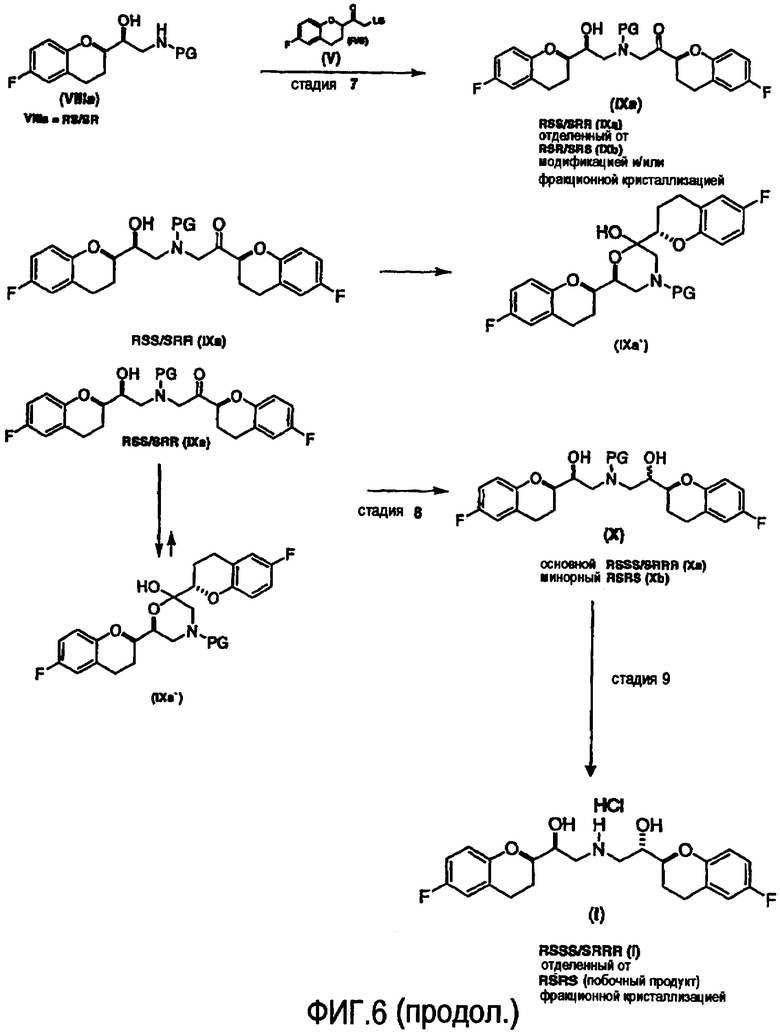
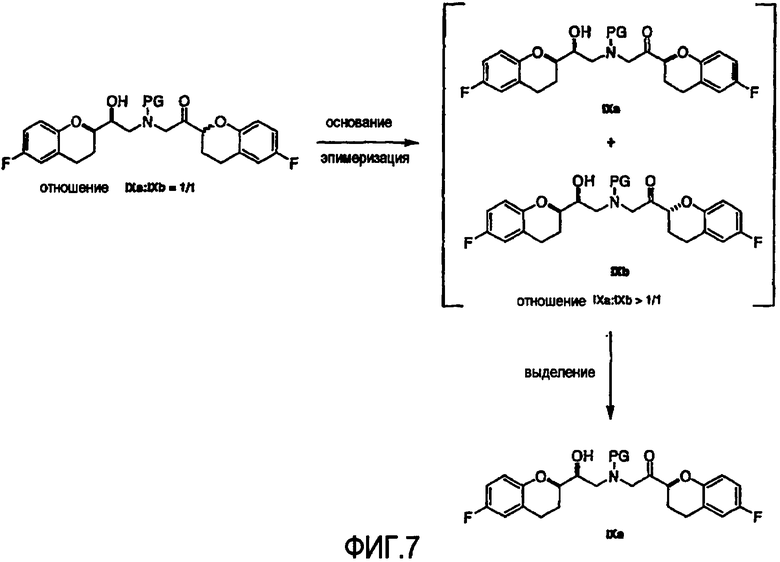
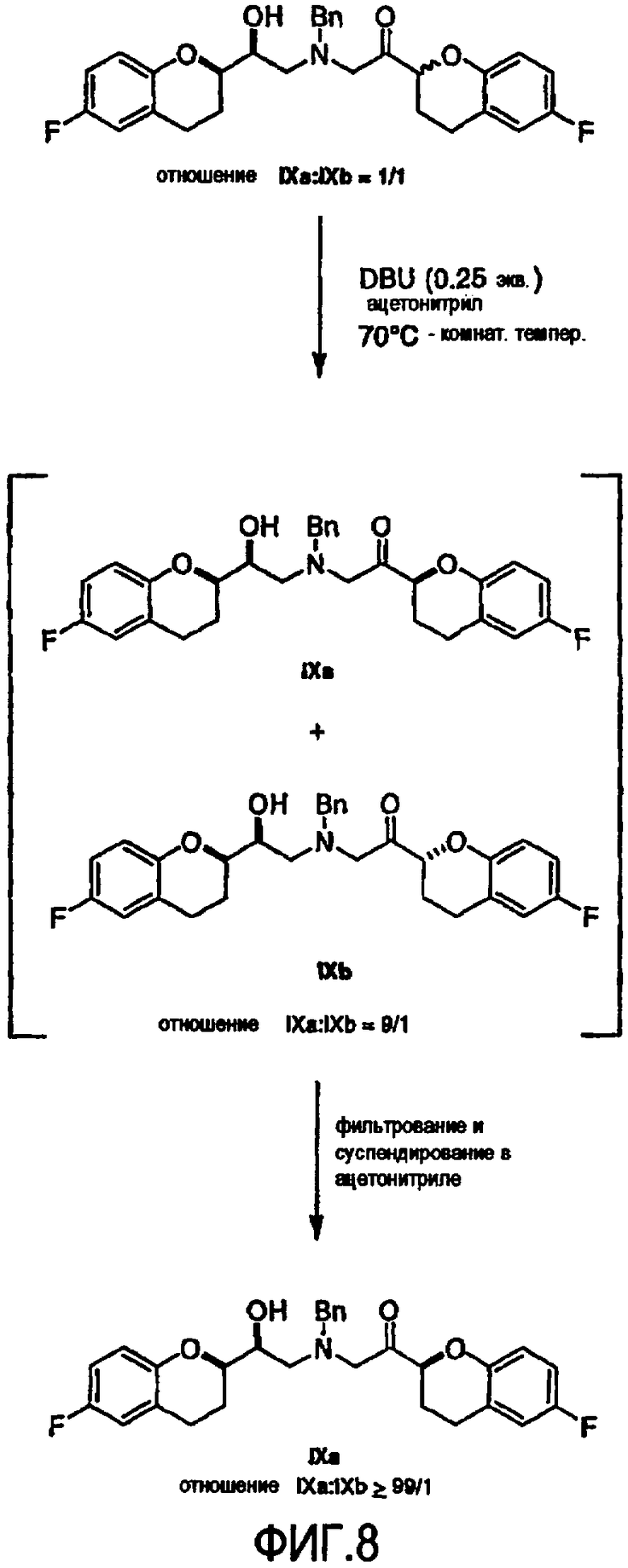
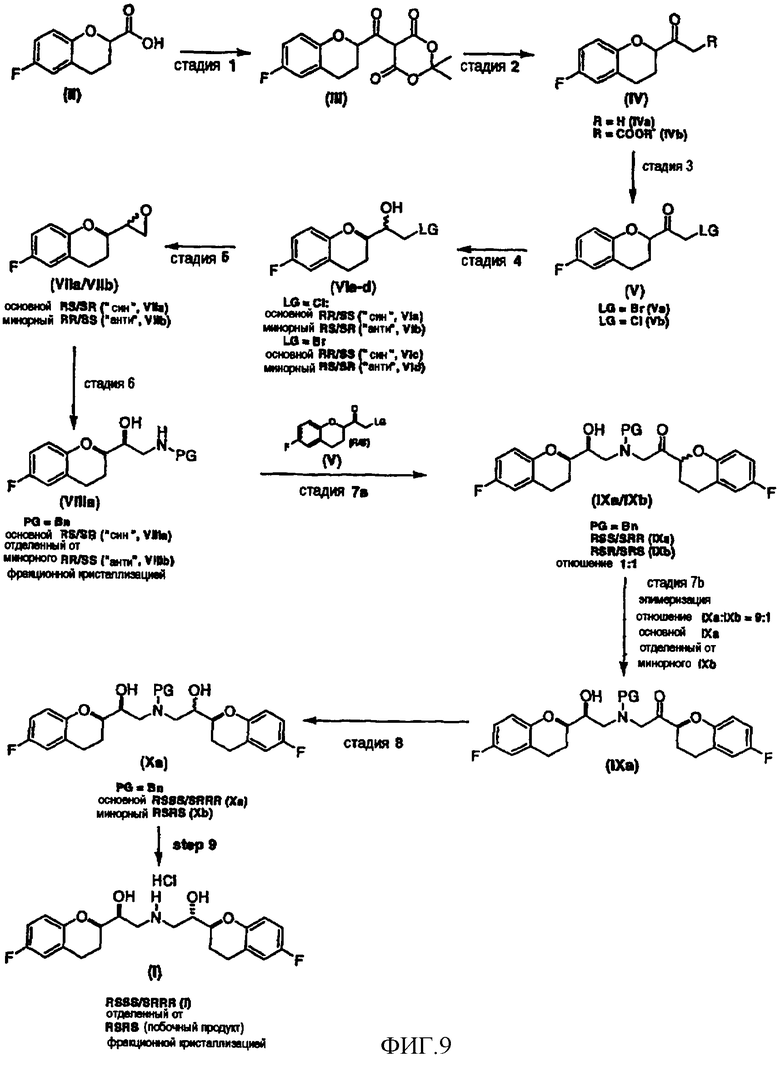
Изобретение относится к новому способу получения рацемического[2S*[R*[R*[R*]]]] и [2R*[S*[S*[S*]]]]-(±)α,α'-[иминобис(метилен)] бис [6-фтор-3,4-дигидро-2Н-1-бензопиран-2-метанола] формулы (I) (небиволола) и его фармацевтически приемлемых солей:
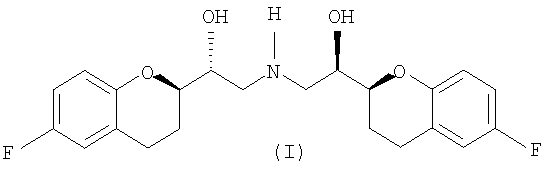
включающим стадии, указанные в описании, а также к промежуточным соединениям и способам их получения. 7 н. и 99 з.п. ф-лы, 12 табл., 9 ил.
1. Способ получения рацемического [2S*[R*[R*[R*]]]] и [2R*[S*[S*[S*]]]]-(±)α,α'-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2Н-1-бензопиран-2-метанола] и его фармацевтически приемлемых солей, включающий (а) предоставление соединения формулы (VIII)
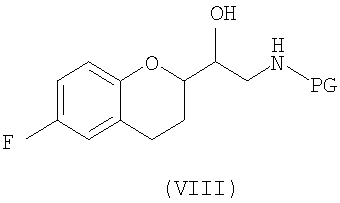
в виде диастереомерно чистого соединения, содержащего по меньшей мере 95% диастереомера RS/SR конфигурации или RR/SS конфигурации, где PG представляет собой водород или аминозащитную группу, причем аминозащитная группа представляет собой по меньшей мере одну аллильную группу или арил-С1алкильную группу;
(b) предоставление рацемического соединения формулы (V)
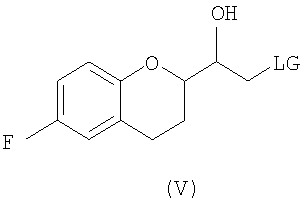
где LG выбран из группы, состоящей из хлора, брома, йода, алкилсульфонилокси и арилсульфонилокси;
(с) N-алкилирование соединения формулы (VIII) соединением формулы (V), где указанное N-алкилирование проводят в инертном органическом растворителе в присутствии основания и необязательно в присутствии катализатора, с получением соединения формулы (IX)
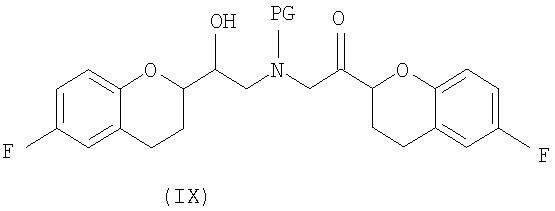 ,
,
соединения формулы (IX'), которое представляет собой циклическую полукетальную форму соединения формулы (IX)
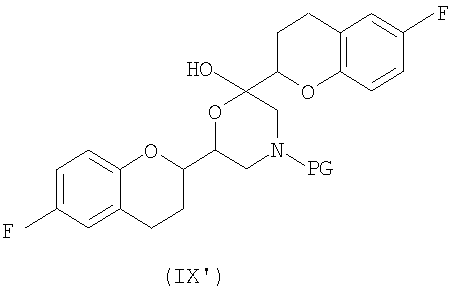
или их смеси, где соединение формулы (IX) и соединение формулы (IX') представляют собой смеси диастереомеров;
(d) разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') по меньшей мере одним из способов (dl) или (d2), где (d1) разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') фракционной кристаллизацией после образования соли или после модификации с получением, по существу, чистых диастереомеров формулы (IX) или формулы (IX), содержащих по меньшей мере 50% RSS/SRR или RRS/SSR конфигурации;
(d2) разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') с получением, по существу, чистых диастереомеров формулы (IX) или формулы (IX'), содержащих по меньшей мере 50% RSS/SRR или RRS/SSR конфигурации, на стадии одновременной эпимеризации-кристаллизации, где стадия эпимеризации-кристаллизации включает:
(1) эпимеризацию соединения формулы (IX) или (IX') RSR/SRS конфигурации с получением смеси диастереомеров формулы (IX) или формулы (IX') RSS/SRR конфигурации и RSR/SRS конфигурации или
эпимеризацию соединения формулы (IX) или (IX') RRR/SSS конфигурации с получением смеси диастереомеров формулы (IX) или формулы (IX') RRS/SSR конфигурации и RRR/SSS конфигурации, при условии, что указанную эпимеризацию проводят в присутствии основания и органического растворителя, где смесь необязательно охлаждают, используя градиент температуры, и диастереомеры RSS/SRR конфигурации или RRS/SSR конфигурации в смеси получают по меньшей мере в двукратном избытке относительно диастереомеров RSR/SRS конфигурации и RRR/SSS конфигурации; и
(2) кристаллизацию, по существу, чистых диастереомеров формулы (IX) или формулы (IX') RSS/SRR конфигурации или RRS/SSR конфигурации по меньшей мере в двукратном избытке относительно диастереомеров RSR/SRS конфигурации и RRR/SSS конфигурации;
разделение смеси фракционной кристаллизацией необязательно после образования соли или после модификации с получением по существу чистых диастереомеров формулы (IX) или формулы (IX') с RSS/SRR или RRS/SSR конфигурацией;
(е) восстановление, по существу, чистых диастереомеров формулы (IX) или формулы (IX') с RSS/SRR или RRS/SSR конфигурацией, с получением соединения формулы (X)
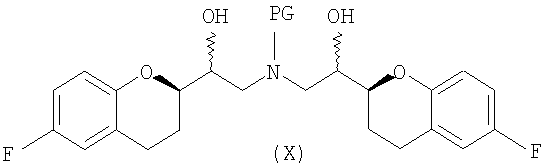
в виде диастереомерной смеси RSSS/SRRR, в которой отношение RSSS/SRRR диастереомерной конфигурации к SRSR или RRSS диастереомерной конфигурации составляет по меньшей мере 1;
(f) снятие защиты с соединения формулы (X), при условии, что PG не является Н (а если PG представляет собой Н, тогда стадию снятия защиты исключают) с получением соединения формулы (I)
или его фармацевтически приемлемых солей; и
(g) удаление соединения формулы (I) или его фармацевтически приемлемых солей RSRS или RRSS диастереомерной конфигурации (если они имеются) перекристаллизацией или суспендированием, с получением рацемического [2S*[R*[R*[R*]]]] и [2R*[S*[S*[S*]]]]-(±)[α,α'-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2Н-1-бензопиран-2-метанола] или его фармацевтически приемлемых солей.
2. Способ по п.1, где защитная группа представляет собой бензильную группу.
3. Способ по п.1, где уходящая группа представляет собой хлор или бром.
4. Способ по п.1, где на стадии (с) органический растворитель представляет собой полярный апротонный растворитель, выбранный из группы, состоящей из диметилформамида, диметилацетамида и N-метилпирролидона.
5. Способ по п,1, где на стадии (с) основание является по меньшей мере одним из третичных аминов, карбонатов щелочных металлов или гидрокарбонатов щелочных металлов.
6. Способ по п.5, где основание представляет собой гидрокарбонат натрия.
7. Способ по п.6, где используют от около 1,5 до около 2,5 эквивалента основания.
8. Способ по п.1, где на стадии (с) катализатор представляет собой по меньшей мере один из бромидов щелочного металла, йодидов щелочного металла, бромидов тетраалкиламмония или йодидов тетраалкиламмония.
9. Способ по п.8, где катализатор представляет собой бромид натрия.
10. Способ по п.9, где используют от около 0,1 до около 0,25 эквивалента катализатора.
11. Способ по п.10, где используют 0,15 эквивалента катализатора.
12. Способ по п.1, где на стадии (с) указанное N-алкилирование проводят при температуре приблизительно от комнатной до около 80°С.
13. Способ по п.1, где на стадии (b) соединение формулы (V) используют в количестве от около 1,0 до около 1,5 эквивалента.
14. Способ по п.1, где на стадии (d1) фракционную кристаллизацию проводят в растворителе.
15. Способ по п.14, где на стадии (d1) используют свободный амин для фракционной кристаллизации.
16. Способ по п.14, где растворитель представляет собой ацетонитрил.
17. Способ по п.1, где на стадии (d1) используют силилирующий реагент для модификации перед фракционной кристаллизацией из растворителя.
18. Способ по п.17, где силилирующий реагент представляет собой по меньшей мере один из триметилсилилхлорида, гексаметилдисилазана или бис-триметилсилилмочевины.
19. Способ по п.17, где используют от около 0,4/n до около 0,6/n эквивалентов силилирующего реагента и где n обозначает число переносимых силильных групп, приходящихся на силилирующий реагент.
20. Способ по п.17, где модификацию проводят в присутствии от около 1,0 до около 2,0 эквивалента основания.
21. Способ по п.20, где основание представляет собой имидазол.
22. Способ по п.17, где указанное разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') проводят в ацетонитриле, метил-трет-бутиловом эфире, циклогексане или их смесях.
23. Способ по п.1, где на стадии (е), указанное восстановление проводят для соединения формулы (IX) или соединения формулы (IX') RSS/SRR конфигурации в растворителе с помощью боргидрида щелочного металла, боргидрида тетрабутиламмония, три-втор-бутилборгидрида щелочного металла или боргидрида цинка необязательно в присутствии кислоты Льюиса.
24. Способ по п.23, где кислота Льюиса представляет собой одно из соединений: Тi(Oалкил)4, ZnCl2, галогенид щелочного металла или галогенид щелочноземельного металла.
25. Способ по п.23, где растворитель представляет собой по меньшей мере один из простого эфира, спирта или галогенированного углеводорода.
26. Способ по п.23, где указанное восстановление проводят в интервале температур от около -20°С до около комнатной.
27. Способ по п.1, где на стадии (f) указанное снятие защиты проводят каталитическим гидрированием.
28. Способ по п.1, где на стадии (g) указанную очистку соединения формулы (I) осуществляют суспендированием его гидрохлорида в растворителе.
29. Способ по п.28, где суспендирование проводят в метаноле в качестве растворителя.
30. Способ по п.1, где указанное предоставление соединения формулы (VIII) включает:
(i) восстановление рацемического соединения формулы (V) в растворителе и необязательно в присутствии кислоты Льюиса, где LG представляет собой бром или хлор, с получением смеси диастереомеров соединения формулы (VI)
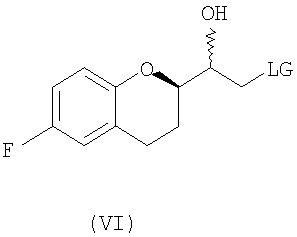
(ii) образование смеси диастереомеров соединения формулы (VII)
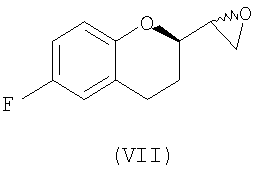
(iii) реакцию диастереомеров соединения формулы (VII) с NH2PG, с получением смеси диастереомеров соединения формулы (VIII)
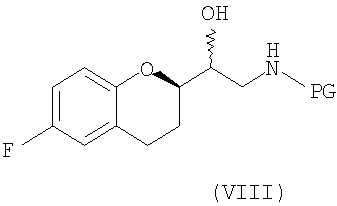 и
и
(iv) выделение отдельных диастереомеров соединения формулы (VIII) из смеси диастереомеров соединения формулы (VIII) фракционной кристаллизацией необязательно после образования соли.
31. Способ по п.30, где выделяют по меньшей мере один из диастереомеров соединения формулы VIII с RR/SS или RS/SR конфигурацией.
32. Способ по п.31, дополнительно включающий рециркуляцию соединения формулы (VIII) RR/SS конфигурации, где указанная рециркуляция включает:
предоставление соединения формулы (VIII) RR/SS конфигурации с защитной группой; и
инверсию конфигурации спирта с получением соединения формулы (VIII) SR/RS конфигурации.
33. Способ по п.30, где на стадии i) восстанавливающий агент выбирают из боргидрида щелочного металла, боргидрида тетраалкиламмония, боргидрида цинка, триацетоксиборгидрида щелочного металла, триэтилборгидрида натрия, бис(2-метоксиэтокси)алюминийгидрида натрия, три-втор-бутилборгидрида щелочного металла или координационных боргидридов.
34. Способ по п.30, где на стадии i) проводят восстановление в условиях Меервейна-Пондорфа-Верлея.
35. Способ по п.30, где на стадии i) указанное восстановление проводят путем каталитического гидрирования.
36. Способ по п.30, где на стадии i) кислоту Льюиса выбирают из группы, состоящей из хлоридов щелочных или щелочноземельных металлов, хлорида цинка, алкоксида титана (IV) и триалкоксида алюминия.
37. Способ по п.30, где на стадии i) указанное восстановление осуществляют в условиях, которые приводят к избытку RR/SS изомера соединения формулы (VI).
38. Способ по п.30, где на стадии i) указанное восстановление проводят в интервале температур от около -78°С до около комнатной.
39. Способ по п.38, где указанное восстановление проводят при температуре от -20°С до комнатной.
40. Способ по п.30, где на стадии i) растворитель выбран из группы, состоящей из спиртов, простых эфиров, галогенированных углеводородов и ароматических растворителей.
41. Способ по п.30, где PG представляет собой бензильную группу.
42. Способ по п.30, где на стадии ii) указанное образование смеси диастереомеров соединения формулы (VII) проводят в растворителе и в присутствии основания.
43. Способ по п.42, где растворитель представляет собой спирт, а основание представляет собой алкоголят щелочного металла.
44. Способ по п.43, где используют 1,0-2,0 эквивалента основания.
45. Способ по п.30, где на стадии ii) указанное образование смеси диастереомеров соединения формулы (VII) проводят при температурах от 0 до 40°С.
46. Способ по п.30, где на стадии iv) фракционную кристаллизацию проводят в толуоле, ацетонитриле, С1-С3-спирте, простом эфире или их смесях.
47. Способ по п.46, где С1-С3-спирт представляет собой 2-пропанол, а простой эфир представляет собой по меньшей мере один из диизопропилового эфира или метил-трет-бутилового эфира.
48. Способ по п.1, где указанное предоставление рацемического соединения формулы (V) включает:
(1) трансформацию соединения формулы (II)
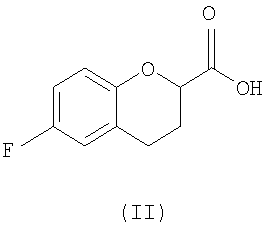
в активированное производное кислоты;
(2) взаимодействие активированного производного кислоты с кислотой Мелдрума в присутствии основания с получением соединения формулы (III)
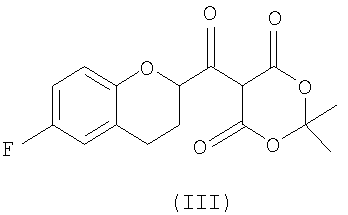
(3) превращение соединения формулы (III) в соединение формулы (IV)
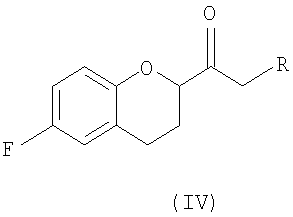
где R представляет собой водород или COOR' и где R' представляет собой C1-С6алкил или арил-С1алкил; и
(4) галогенирование соединения формулы (IV) и необязательно проведение гидролиза и декарбоксилирования с получением соединения формулы (V).
49. Способ по п.48, где на стадии (1) карбоновая кислота превращается в соответствующий хлорангидрид.
50. Способ по п.48, где на стадии (2) основание представляет собой третичный амин.
51. Способ по п.48, где на стадии (2) используют 1-3 эквивалента кислоты Мелдрума.
52. Способ по п.48, где на стадии (2) температура реакции составляет от около -10°С до около +30°С.
53. Способ по п.48, где на стадии (3) соединение формулы
(III) гидролизуют в смеси органической кислоты и воды, получая соединение формулы (IV), где R представляет собой Н.
54. Способ по п.53, где органическая кислота представляет собой уксусную кислоту и гидролиз проводят при температуре кипения.
55. Способ по п.48, где на стадии (3) соединение формулы
(IV), в котором R представляет собой COOR' и R' представляет собой C1-С6алкил или арил-С1алкил, получают алкоголизом соединения формулы (III).
56. Способ по п.55, где алкоголиз проводят этанолом и трет-бутанолом.
57. Способ по п.48, где на стадии (3) растворитель представляет собой по меньшей мере один из спирта или толуола.
58. Способ по п.48, где на стадии (3) алкоголиз проводят при температурах от около 70°С до около 80°С.
59. Способ по п.48, где на стадии (4) до проведения галогенирования соединение формулы (IV), где R представляет собой Н, превращают путем силилирования в соответствующий силиленоловый эфир, содержащий концевую двойную связь.
60. Способ по п.59, где силилирование осуществляют через кинетически контролируемое депротонирование, используя литийдиизопропиламид, с последующим силилированием при температуре от около -78°С до около - 40°С.
61. Способ по п.60, где силилирование осуществляют при температуре от -78°С до -70°С.
62. Способ по п.60, где в качестве силилирующего реагента используют триметилсилилхлорид.
63. Способ по п.48, где на стадии (4) после превращения в силиленоловый эфир галогенирование проводят с помощью бромирующего реагента.
64. Способ по п.63, где бромирующий реагент представляет собой N-бромсукцинимид.
65. Способ по п.48, где на стадии (4) соединение формулы (IV), в котором R представляет собой COOR', сначала галогенируют, а затем превращают в соединение формулы (V) гидролизом сложного эфира с последующим декарбоксилированием.
66. Способ по п.65, где галогенирование проводят в присутствии катализатора.
67. Способ по п.66, где от около 1,0 до около 1,5 эквивалента N-бромсукцинимида, N-хлорсукцинимида или SO2Cl2 используют в качестве галогенирующих реагентов.
68. Способ по п.67, где от около 0,2 до около 0,4 эквивалента Mg(ClO4)2 используют в качестве катализатора.
69. Способ по п.48, где на стадии (4) указанное галогенирование проводят при температурах от 0°С и до около комнатной.
70. Способ по п.48, где на стадии (4) после указанного галогенирования проводят гидролиз сложного эфира с последующим декарбоксилированием в водном растворе органической кислоты.
71. Способ по п.70, где органическая кислота представляет собой по меньшей мере одну из трифторуксусной кислоты, муравьиной кислоты и уксусной кислоты.
72. Способ по п.71, где гидролиз и декарбоксилирование проводят при температурах от около 75°С до около 90°С.
73. Способ по п.1, дополнительно включающий рециркуляцию соединения формулы (IX) или (IX') RSR/SRS или RRR/SSS конфигурации, где указанная рециркуляция включает:
эпимеризацию соединения формулы (IX) или (IX') RSR/SRS или RRR/SSS конфигурации с получением смеси диастереомеров формулы (IX) или формулы (IX') RSS/SRR конфигурации, содержащей диастереомеры RSR/SRS конфигурации, или смеси диастереомеров формулы (IX) или формулы (IX') RRS/SSR конфигурации, содержащей диастереоизомеры RRR/SSS конфигурации, и
разделение смеси фракционной кристаллизацией после образования соли или после модификации с получением, по существу, чистых диастереомеров формулы (IX) или формулы (IX'), имеющих RSS/SRR или RRS/SSR конфигурацию.
74. Способ по п.1, дополнительно включающий рециркуляцию соединения формулы (IX) или (IX') RSR/SRS или RRR/SSS конфигурации, где указанная рециркуляция включает:
расщепление соединения формулы (IX) или (IX') RSR/SRS или RRR/SSS конфигурации, с образованием смеси диастереомеров формулы (VIII) RS/SR или RR/SS конфигурации; и
разделение диастереомеров формулы (VIII) RS/SR или RR/SS конфигурации.
75. Соединение, выбранное из следующей группы:
(а) соединение формулы (III)
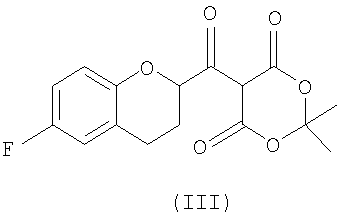
(b) соединение формулы (IV)
где R представляет собой COOR' и где R' представляет собой С1-С6алкил или арил-С1алкил,
(с) соединение формулы (V)
где LG представляет собой бром или хлор,
d) соединение формулы (IX)
или его циклическая полукетальная форма формулы (IX')
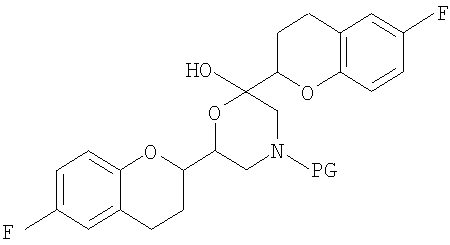
где PG представляет собой защитную группу, выбранную из водорода, аллила и арил-С1алкила,
(е) соединение формулы (IX), имеющее RSS/SRR конфигурацию,
или его соответствующая циклическая полукетальная форма формулы (IX')
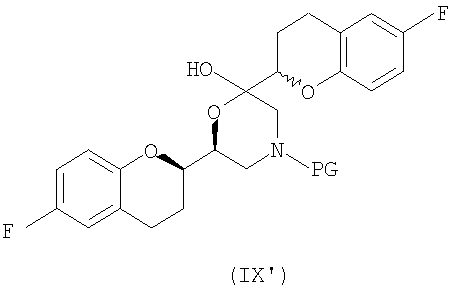
где PG представляет собой бензильную группу, или
f) соединение, имеющее формулу, изображенную ниже
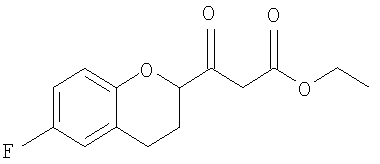 .
.
76. Соединение по п.75, имеющее формулу (IV)
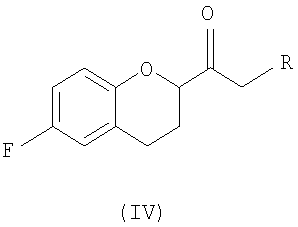
где R представляет собой COOR' и где R' представляет собой С1-С6алкил или арил-С1алкил.
77. Соединение по п.75, имеющее формулу (V)
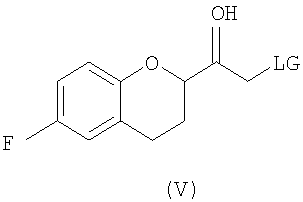
где LG представляет собой бром или хлор.
78. Соединение по п.75, имеющее формулу (IX)
или его циклическая полукетальная форма формулы (IX')
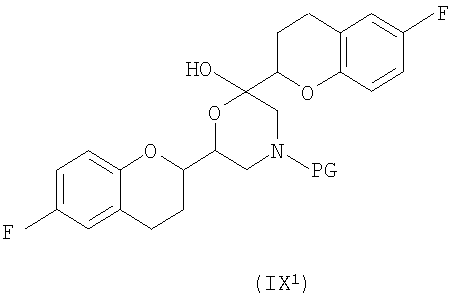
где PG представляет собой защитную группу, выбранную из водорода, аллила и арил-С1алкила.
79. Соединение по п.75, имеющее формулу (IX), имеющее RSS/SRR конфигурацию,
или его соответствующая циклическая полукетальная форма формулы (IX')
где PG представляет собой бензильную группу.
80. Соединение по п.75, имеющее формулу, изображенную ниже
81. Соединение по п.75, имеющее формулу (III)
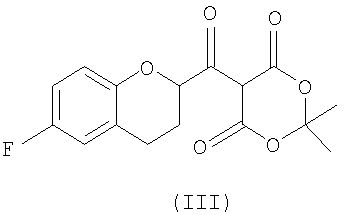
82. Способ получения рацемического [2S*[R*[R*[R*]]]] и [2R*[S*[S*[S*]]]]-(±)α,α'-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2Н-1-бензопиран-2-метанола] и его фармацевтически приемлемых солей, включающий:
предоставление соединения формулы (IX)
его соли или его производного;
разделение стереоизомеров соединения, соли или производного формулы (IX) фракционной кристаллизацией с получением, по существу, чистых их диастереомеров, которые содержат по меньшей мере 50% RSS/SRR или RRS/SSR конфигурации; и
восстановление, по существу, чистых диастереомеров с получением соединения формулы (X),
содержащего самое большее 50% стереоизомера с RSRS конфигурацией.
83. Способ по п.82, в котором соединение формулы (IX) получают путем N-алкилирования соединения формулы (VIII) соединением формулы (V), как определено в п.1.
84. Способ получения соединения формулы (I) и его фармацевтически приемлемых солей,
включающий:
(а) разделение соединения формулы (II)
с получением соединения формулы (II) с S-конфигурацией и R-конфигурацией;
(b) превращение соединения формулы (II) с S-конфигурацией в соединение формулы (V) с S-конфигурацией,
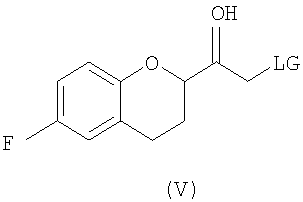
где LG представляет собой заместитель, выбранный из группы, состоящей из хлора, брома, йода, алкилсульфонилокси и арилсульфонилокси, через образование соединения формулы (III) с S-конфигурацией и соединения формулы (IV) с S-конфигурацией;
(c) превращение соединения формулы (II) с R-конфигурацией в соединение формулы (V) с R-конфигурацией через образование соединения формулы (III) с R-конфигурацией и соединения формулы (IV) с R-конфигурацией;
(d) предоставление соединения формулы (VIII),
где PG представляет собой водород или аминозащитную группу, где аминозащитная группа представляет собой по меньшей мере одну из аллильной группы или арил-С1алкильной группы и где соединение формулы (VIII) является энантиомерным соединением с RS или SR конфигурацией;
(е) проведение N-алкилирования (i) соединения формулы (VIII) с RS конфигурацией соединением формулы (V) с S-конфигурацией или (ii) соединения формулы (VIII) с SR конфигурацией соединением формулы (V) с R-конфигурацией при условии, что указанное N-алкилирование проводят в инертном органическом растворителе в присутствии основания и необязательно в присутствии катализатора, с получением RSS или SRR энантиомерной формы соединения формулы (IX)
или RSS или SRR энантиомерной формы соединения формулы (IX'), которое представляет собой циклическую полукетальную форму соединения формулы (IX)
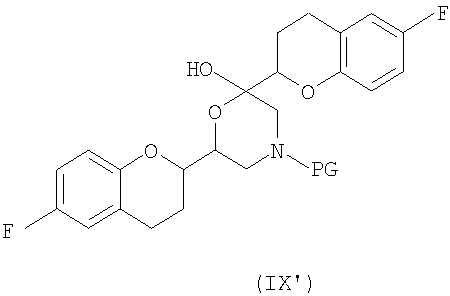
(f) восстановление по меньшей мере одной из RSS или SRR энантиомерных форм соединения формулы (IX) или формулы (IX') с получением по меньшей мере одной RSSS или SRRR энантиомерной формы соединения формулы (X);
(g) снятие защиты по меньшей мере с одной из RSSS или SRRR энантиомерных форм соединения формулы (X), при условии, что PG не является Н (а если PG представляет собой Н, тогда отменяют указанную стадию снятия защиты), с получением соединения формулы
(I) или его фармацевтически приемлемых солей; и
(h) удаление соединения формулы (I) или его фармацевтически приемлемых солей RSRS диастереомерной конфигурации, если они присутствуют в качестве побочного продукта, кристаллизацией или суспендированием с получением по меньшей мере одного из ([2S*[R*[R*[R*]]]]- и ([2R*[S*[S*[S*]]]]-энантиомеров соединения формулы (I) и их фармацевтически приемлемых солей; и
(i) необязательно объединение ([2S*[R*[R*[R*]]]]- и ([2R*[S*[S*[S*]]]]-энантиомеров соединения формулы (I) и их фармацевтически приемлемых солей с образованием рацемического ([2S*[R*[R*[R*]]]]- и ([2R*[S*[S*[S*]]]]-(±)α,α'-[имино-бис(метилен)]бис[6-фторхроман-2-метанола] формулы (I) и его фармацевтически приемлемых солей.
85. Способ получения рацемического [2S*[R*[R*[R*]]]]- и [2R*[S*[S*[S*]]]]-(±)-α,α'-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2Н-1-бензопиран-2-метанола] и его фармацевтически приемлемых солей, включающий:
(а) предоставление соединения формулы (VIII)
в виде диастереомера, имеющего RR/SS конфигурацию, где PG представляет собой водород или аминозащитную группу, где аминозащитная группа представляет собой по меньшей мере одну из аллильной группы или арил-С1алкильной группы;
(b) предоставление рацемического соединения формулы (V),
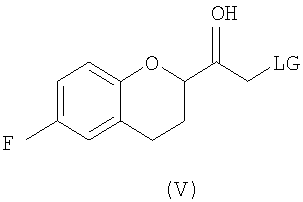
где LG представляет собой заместитель, выбранный из группы, состоящей из хлора, брома, йода, алкилсульфонилокси и арилсульфонилокси;
(с) N-алкилирование соединения формулы (VIII) соединением формулы (V), где указанное N-алкилирование проводят в инертном органическом растворителе в присутствии основания и необязательно в присутствии катализатора, с получением соединения формулы (IX)
соединения формулы (IX'), которое представляет собой циклическую полукетальную форму соединения формулы (IX),
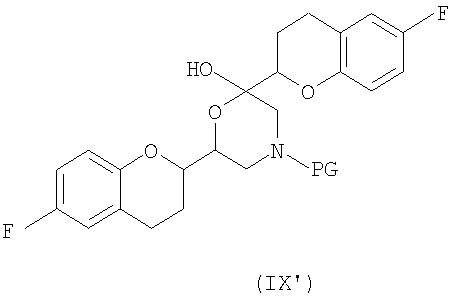
или их смеси, где соединение формулы (IX) и соединение формулы (IX') представляют собой смеси диастереомеров, имеющих RRR/SSS и RRS/SSR конфигурацию;
(а) разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') фракционной кристаллизацией после образования соли или после модификации с получением, по существу, чистых диастереомеров формулы (IX) или формулы (IX'), содержащих по меньшей мере 50% стереоизомера RRR/SSS или RRS/SSR конфигурации;
(е) восстановление, по существу, чистых диастереомеров формулы (IX) или формулы (IX'), имеющих RRS/SSR конфигурацию с получением соединения формулы (X)
в виде RSSS/SRRR диастереомерной смеси, в которой отношение RSSS/SRRR диастереомерной конфигурации к SRSR или RRSS диастереомерной конфигурации составляет по меньшей мере 1;
(f) снятие защиты соединения формулы (X), при условии, что PG не является Н, с получением соединения формулы (I)
или его фармацевтически приемлемых солей; и
(g) удаление соединения формулы (I) или его фармацевтически приемлемых солей, если они имеются, RSRS диастереомерной конфигурации перекристаллизацией или суспендированием, с получением рацемического [2S[2R*[R[R*]]]] и [2R[2S*[S[S*]]]]-(±)α,α'-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2Н-1-бензопиран-2-метанола] или его фармацевтически приемлемых солей.
86. Способ по п.85, дополнительно включающий:
эпимеризацию соединения формулы (IX) или соединения формулы (IX') RRR/SSS конфигурации с получением смеси диастереомеров соединения формулы (IX) или соединения формулы (IX'), имеющих RRR/SSS и RRS/SSR конфигурацию.
87. Способ по п.85, дополнительно включающий расщепление соединения формулы (IX) или соединения формулы (IX') RRR/SSS конфигурации с образованием соединения формулы (VIII) в виде диастереомера, имеющего RR/SS конфигурацию.
88. Способ по п.86, дополнительно включающий расщепление соединения формулы (IX) или соединения формулы (IX') RRR/SSS конфигурации с образованием соединения формулы (VIII) в виде диастереомера, имеющего RR/SS конфигурацию.
89. Способ получения RS/SR или RR/SS диастереомеров соединения (VIII),
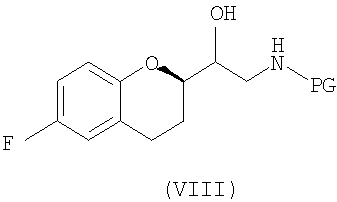
включающий:
(i) предоставление рацемического соединения формулы (V)
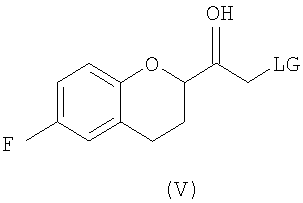
(ii) восстановление рацемического соединения формулы (V) в растворителе и необязательно в присутствии кислоты Льюиса, где LG представляет собой бром или хлор, с получением диастереомерной смеси соединения формулы (VI)
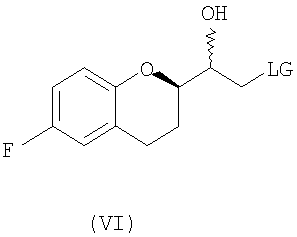
(iii) образование смеси диастереомеров соединения формулы (VII)
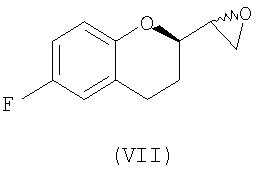
(iv) взаимодействие диастереомеров соединения формулы (VII) с NH2PG, где PG представляет собой водород или аминозащитную группу, где аминозащитная группа представляет собой по меньшей мере одну из аллильной группы или арил-С1алкильной группы с получением соединения формулы (VIII) в виде смеси диастереомеров; и (v) выделение RS/SR или RR/SS диастереомеров соединения формулы (VIII) из смеси диастереомеров фракционной кристаллизацией.
90. Способ по п.1, где стадию (d2) проводят для соединения формулы (IX) или (IX1) с RSR/SRS конфигурацией.
91. Способ по п.90, где диастереомер RSS/SRR конфигурации в смеси получают примерно в девятикратном избытке по отношению к диастереомеру RSR/SRS конфигурации.
92. Способ по п.1, где на стадии (d2) смесь охлаждают, используя градиент температуры от около 70°С до около 20°С.
93. Способ по п.92, где градиент температуры составляет от около 70°С до около 40°С.
94. Способ по п.1, где на стадии (d2) органический растворитель представляет собой ацетонитрил.
95. Способ по п.1, где на стадии (d2) указанную эпимеризацию проводят в присутствии по меньшей мере 0,1 эквивалента основания.
96. Способ по п.1, где указанную эпимеризацию проводят в присутствии по меньшей мере 0,25 эквивалента основания.
97. Способ по п.1, где на стадии (d2) основание представляет собой соединение, выбранное из группы, состоящей из алкоксида, амидина, гуанидина и фосфазена.
98. Способ по п.97, где основание представляет собой амидин.
99. Способ по п.98, где основание представляет собой диазабициклоундецен.
100. Способ по п.1, где на стадии (d2) содержание воды, если она присутствует, не может превышать 1,0%.
101. Способ по п.1, где на стадии (d2) содержание воды, если она присутствует, не может превышать 0,1%.
102. Способ получения рацемического [2S*[R*[R*[R*]]]] и [2R*[S*[S*[S*]]]]-(±)α,α'-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2Н-1-бензопиран-2-метанола] и его фармацевтически приемлемых солей, включающий:
(а) предоставление соединения формулы (IX),
соединения формулы (IX'), которое представляет собой циклическую полукетальную форму соединения формулы (IX),
или их смеси, где соединение формулы (IX) и соединение формулы (IX') представляют собой смеси диастереомеров;
(b) разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') по меньшей мере по одному из (b1) или (b2), где
(b1) разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') фракционной кристаллизацией после образования соли или после модификации с получением, по существу, чистых диастереомеров формулы (IX) или формулы (IX'), содержащих по меньшей мере 50% RSS/SRR или RRS/SSR конфигурации;
(b2) разделение диастереомеров соединения формулы (IX) или соединения формулы (IX') с получением, по существу, чистых диастереомеров формулы (IX) или формулы (IX'), содержащих по меньшей мере 50% RSS/SRR или RRS/SSR конфигурации на стадии одновременной эпимеризации-кристаллизации, где стадия эпимеризации-кристаллизации включает:
(1) эпимеризацию соединения формулы (IX) или (IX') RSR/SRS конфигурации с получением смеси диастереомеров формулы (IX) или формулы (IX') RSS/SRR конфигурации и RSR/SRS конфигурации, или
эпимеризацию соединения формулы (IX) или (IX') RRR/SSS конфигурации с получением смеси диастереомеров формулы (IX) или формулы (IX') RRS/SSR конфигурации и RRR/SSS конфигурации при условии, что указанную эпимеризацию проводят в присутствии основания и органического растворителя, где смесь необязательно охлаждают с градиентом температуры и где диастереомеры RSS/SRR конфигурации или RRS/SSR конфигурации в смеси получают по меньшей мере в двукратном избытке относительно диастереомеров RSR/SRS конфигурации и RRR/SSS конфигурации; и
(2) кристаллизацию, по существу, чистых диастереомеров формулы (IX) или формулы (IX') RSS/SRR конфигурации или RRS/SSR конфигурации по меньшей мере в двукратном избытке относительно диастереомеров RSR/SRS конфигурации и RRR/SSS конфигурации;
разделение смеси фракционной кристаллизацией необязательно после образования соли или после модификации с получением, по существу, чистых диастереомеров формулы (IX) или формулы (IX'), имеющих RSS/SRR или RRS/SSR конфигурацию;
(с) восстановление, по существу, чистых диастереомеров формулы (IX) или формулы (IX'), имеющих RSS/SRR или RRS/SSR конфигурацию с получением соединения формулы (X)
в виде RSSS/SRRR диастереомерной смеси, в которой отношение RSSS/SRRR диастереомерной конфигурации к SRSR или RRSS диастереомерной конфигурации составляет по меньшей мере 1;
(d) снятие защиты соединения формулы (X) при условии, что PG не является Н, (а если PG представляет собой Н, тогда отменяют указанную стадию снятия защиты), с получением соединения формулы (I)
или его фармацевтически приемлемых солей; и (е) удаление соединения формулы (I) или его фармацевтически приемлемых солей RSRS или RRSS диастереомерной конфигурации, если они имеются, перекристаллизацией или суспендированием, с получением рацемического [2S*[R*[R*[R*]]]] и [2R*[S*[S*[S*]]]]-(±)α,α'-[иминобис(метилен)]бис[6-фтор-3,4-дигидро-2Н-1-бензопиран-2-метанола] или его фармацевтически приемлемых солей.
103. Способ по п.102, где стадию (b) проводят для соединения формулы (IX) или (IX') RSR/SRS конфигурации.
104. Способ по п.103, где диастереомер RSS/SRR конфигурации в смеси получают в примерно девятикратном избытке по отношению к диастереомеру RSR/SRS конфигурации.
105. Способ по п.102, где на стадии (b) смесь охлаждают при градиенте температуры от около 70°С до около 20°С.
106. Способ по п.105, где градиент температуры составляет от 70 до 40°С.
| WO 2004041805 A1, 21.04.2004 | |||
| ЭЛЕМЕНТ ГЛУШИТЕЛЯ ШУМА | 0 |
|
SU334429A1 |
| Магнитный логический элемент | 1961 |
|
SU145067A1 |
| WO 9932476 A1, 01.07.1999 | |||
| STN of the Web БД REGISTRY RN 620176-90-1 | |||
| УСТАНОВКА ДЛЯ ПРИГОТОВЛЕНИЯ ФОРМОВОЧНЫХ И СТЕРЖНЕВЫХ СМЕСЕЙ | 1965 |
|
SU223277A1 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ МОРФОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ СНИЖЕНИЯ КОЛИЧЕСТВА ТАХИКИНИНОВ ПРИ ЛЕЧЕНИИ ИЛИ ПРОФИЛАКТИКЕ ФИЗИОЛОГИЧЕСКИХ СОСТОЯНИЙ, АССОЦИИРУЕМЫХ С ИЗБЫТКОМ ТАХИКИНИНОВ | 1994 |
|
RU2131426C1 |

