Настоящее изобретение предлагает процесс химического ацилирования и, в частности, процесс безводного ацилирования, для получения антибиотика, цефепимгидрохлорид гидрата, который также известен как 7-/2-(2-аминотиазол-4-ил)-2-(Z)-метоксииминоацета- мидо/-3-/ (1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилат. Настоящее изобретение также предлагает устойчивую, кристаллическую соль син-изомера промежуточного тиазолила и способ ее получения, который может быть использован при получении полезных противобактериальных веществ широкого диапазона действия.
Известен целый ряд цефалоспорин-антибиотиков, которые содержат 2-(2-аминотиазол-4-ил)-(Z)-2-метоксииминоуксусную кислоту в боковой цепи, которая присоединена к 7-аминогруппе цефалоспорановой кислоты посредством выполнения хорошо известных методик ацилирования. Во многих случаях необходимо защитить амино-часть и активировать карбоновую кислоту боковой цепи в качестве составляющей методику ацилирования. Следовательно, в литературе излагается целый ряд аминозащитных групп для 2-аминогруппы кольца тиазола и целый ряд активирующих групп для карбоновой кислоты. Предметом многочисленных публикаций все еще остается поиск более совершенных защитных групп и активирующих групп для получения искомого антибиотика с точки зрения стоимости и токсичности, ассоциируемых с некоторыми активирующими группами. Поэтому все еще существует необходимость при получении пригодных антибиотиков широкого круга действий в простой, устойчивой, кристаллической, экономичной нетоксичной боковой цепи, обладающей искомым геометрическим (Z)-изомером, который может быть легко присоединен к 7-аминогруппе ядра цефалоспорина. Ниже приведены некоторые характерные представители группы боковой цепи тиазола.
В патенте США N 4203899 на имя Очиаи и др. выданном 20 мая 1980 года, раскрываются соединения формулы
R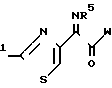 где R1 представляет собой амино, защищенный амино, гидроксил или защищенный гидроксил, R5 представляет собой гидрокси или защищенный гидроксил и W представляет собой гидроксил, С1-С4 алкокси, галоген или ОМ, где М обозначает щелочной металл.
где R1 представляет собой амино, защищенный амино, гидроксил или защищенный гидроксил, R5 представляет собой гидрокси или защищенный гидроксил и W представляет собой гидроксил, С1-С4 алкокси, галоген или ОМ, где М обозначает щелочной металл.
В заявке на патент Великобритании N GВ-2144424, опубликованной 6 марта 1985 года, раскрывает получение ряда пиридиниевых производных цефалоспорина при помощи различных методов, включая использование соединения формулы
R4HN или его соли, где R1 обозначает атом водорода или галогена, R2обозначает атом водорода или радикал С1-С6-алкила и R4 обозначает атом водорода или аминозащитную группу, или с активированным производным данного соединения.
или его соли, где R1 обозначает атом водорода или галогена, R2обозначает атом водорода или радикал С1-С6-алкила и R4 обозначает атом водорода или аминозащитную группу, или с активированным производным данного соединения.
В заявке на Европатент N ЕР-160546, опубликованной 6 ноября 1985 года, также раскрывается получение ряда цефалоспориновых соединений различными способами, включая использование замещенных оксииминотиазолилсоединений уксусной кислоты формулы
R8HN или их реакционноспособных производных, где R8 обозначает атом водорода или защитную группу для аминогруппы. Пригодными примерами таких реакционноспособных производных являются смешанные ангидриды кислот, ангидриды кислоты, галоидангидриды, активные сложные эфиры, активные амиды и азиды кислоты.
или их реакционноспособных производных, где R8 обозначает атом водорода или защитную группу для аминогруппы. Пригодными примерами таких реакционноспособных производных являются смешанные ангидриды кислот, ангидриды кислоты, галоидангидриды, активные сложные эфиры, активные амиды и азиды кислоты.
В патенте США N 4385181 на имя Фарге и др. выданном 24 мая 1983 года, раскрываются сложные тиолоэфиры формулы
R′NH где R' обозначает атом водорода или защитный радикал, Ro обозначает атом водорода, алкильный, винильный, цианометильный или защитный радикал и R обозначает алкил, L-2-амино-2-карбоксиэтил, фенил или целый ряд различных гетероциклических радикалов, приведенных в столбцах 4-8, а также их син- и анти-изомеры и их смеси.
где R' обозначает атом водорода или защитный радикал, Ro обозначает атом водорода, алкильный, винильный, цианометильный или защитный радикал и R обозначает алкил, L-2-амино-2-карбоксиэтил, фенил или целый ряд различных гетероциклических радикалов, приведенных в столбцах 4-8, а также их син- и анти-изомеры и их смеси.
Кроме вышеприведенных отсылок, существует целый ряд отсылок, которые раскрывают различные защитные группы для 2-аминозаместителя, а также целый ряд активирующих групп/уходящих групп части карбоновой кислоты, которые могут быть использованы при ацилировании 7-аминосоединения цефалоспорина.
Однако наиболее точной отсылкой является патент Чехословакии N 238950, опубликованный 16 марта 1987 года /Кемикал Эбстрактс, Том 110, стр. 544 (1989)/, который раскрывает соединение настоящего изобретения, имеющего формулу
NH · HCl где соединение имеет син-конфигурацию. Единственным свидетельством, присутствующим в патенте в отношении продукта, является содержание хлора, равное от 99% до 100,5% от теоретического значения.
· HCl где соединение имеет син-конфигурацию. Единственным свидетельством, присутствующим в патенте в отношении продукта, является содержание хлора, равное от 99% до 100,5% от теоретического значения.
В связи с попытками разработать новые синтетические методы получения антибиотиков заявитель, как и другие изобретатели в данной области техники, почувствовал необходимость в новых простых, удобных, экономичных, кристаллических, устойчивых, нетоксичных исходных веществах для использования при производстве антибиотиков. Первоначальные попытки получить и использовать хлорангидрид 2-(2-аминотиазол-ил)-2-метоксииминоуксусной кислоты без применения защитных групп не имели успех. Однако заявитель нашел, что соединение предлагаемого изобретения может быть получено при специально определенных реакционных условиях. Это открытие было в дальнейшем подтверждено заявителем, когда он смог воспроизвести доктрину вышеприведенного патента Чехословакии. Искомый син-изомер хлористоводородной соли хлорангидрида, которая необходима для получения искомого антибиотика, нельзя повторить. Кроме того, дополнительные эксперименты подтвердили, что доктрина противопоставленного патента не может привести к получению хлористоводородной соли искомого син-изомера хлорангидрида, по существу свободного от син-изомера и имеющего описываемый в данной работе спектр протонного ядерного магнитного резонанса (1Н ЯМР).
Антибиотик цефепим широкого спектра действия раскрыт Абураки и др. в патенте США N 4406899, выданном 27 сентября 1983 года, и его получение описано в двух Схемах Реакции, в которых реагенты и продукты нуждаются в использовании блокирующих и деблокирующих группах. В приведенной примерами реакционной схеме продукт нуждается в стадии хроматографической очистки с целью разделения смеси Δ2 и Δ3 изомеров, и поэтому полученный таким образом продукт цефепим продуцируется в форме цвиттериона. Однако форма цвиттериона соединения цефепим является неустойчивой при комнатной и повышенной температурах.
Мюррей А. Каплан и др. в патенте США N 4910301, выданном 20 марта 1990 года, раскрывают устойчивые кристаллические соли цефепима в форме сухого порошка, имеющего превосходную устойчивость при комнатной и намного повышенных температурах по сравнению с формой цвиттериона соединений цефепим, раскрытого Абураки и др. в патенте США N 4406899.
В патенте США N 4868294, выданном 19 сентября 1990 года на имя Брундиджа и др. раскрывается получение солей 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоновой кислоты, по существу свободных от Δ2-изомера, а также их использование в методе водного ацилирования с целью получения антибиотика цефепим в виде соли серной кислоты (сульфата).
В патенте США N 4754031, выданном 28 июня 1988 года на имя Ангербауэра и др. раскрывается способ получения нескольких антибиотиков на основе цефалоспорина, включая цефепим в форме цвиттериона. Хотя данный способ не предусматривает использование защитных групп, в нем применяют ангидрид для активации в реакции водного ацилирования, которая предусматривает стадии хроматографической очистки с получением цвиттерион-формы цефепима.
В патенте США N 4943631, выданном 24 июля 1990 года на имя Брайен Е.Лукер, описывается усовершенствованный способ получения антибиотика цефепим в виде иодистоводородной соли. Способ обеспечивает контроль за образованием нежелательного Δ2-изомера путем использования промежуточного сульфоксида цефалоспорина. Однако, описываемый в патенте Способ остается дорогостоящим и неэффективным, поскольку в нем используются две дополнительные стадии и продолжается применение защитных групп, которые требуют проведения методик блокирования и деблокирования. Кроме того, способ предполагает использование колоночной хроматографии в качестве метода очистки, которая является непрактичной в промышленном масштабе.
Получение кристаллической соли серной кислоты (сульфата) в цвиттериона цефепима, которые описаны в литературе, предполагает использование по существу того же процесса водного ацилирования и различных блокирующих и деблокирующих групп и активных сложных эфиров. Во всех случаях предпочтительная кристаллическая форма гидрата дигидрохлорида цефепима должна быть получена через очищенную форму цвиттериона цефепима. Таким образом существует необходимость в разработке простой, непосредственной и эффективной методики ацилирования, которая обеспечила бы устранение реакционных стадий с целью добавления и отщепления защитных групп, стереохимических контролирующих стадий и хроматографических методик и, что более важно, методики ацилирования, которая привела бы к получению гидрата дигидрохлорида антибиотика цефепим, по существу свободного от анти-изомера и Δ2-изомера.
Настоящее изобретение предлагает процесс химического ацилирования и, в частности, процесс безводного ацилирования для получения антибиотика, гидрата дигидрохлорида цефепима, по существу свободного от анти-изомера и Δ2-изомера. Настоящее изобретение также предлагает устойчивый, кристаллический синизомер хлоргидрата 2-(2-аминтииазол-4-ил)-2-метоксиимино-ацетилхлорида, по существу свободного от анти-изомера, который используют в процессе ацилирования для получения широкоспектрового антибиотика цефепим.
Фиг.1 показывает спектр протонного ядерного магнитного резонанса хлоргидрата син-2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхлорида Примера 10 в уксусной кислоте-d4 (100 МГц).
Фиг.2 показывает спектр протонного ядерного магнитного резонанса продукта примеpа 12 в уксусной кислоте d4 (100 МГц).
Фиг.3 показывает спектр протонного ядерного магнитного резонанса продукта Примера 13 в уксусной кислоте d4 (100 МГц).
Фиг.4 показывает спектр протонного ядерного магнитного резонанса продукта Примера 14 в уксусной кислоте d4 (100 МГц).
Настоящее изобретение предлагает безводный способ ацилирования для N-ацилирования 7-амино-3-/(1-метил-1-пирролидино)метил/цеф-3-ем-4-карбоксилата с син-изомером хлоргидрата 2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхлорида, который по существу свободен от анти-изомера, с получением температурно-устойчивого гидрата дигидрохлорида цефепима, по существу свободного от анти-изомера и Δ2-изомера и представленного формулой Y, в которой Z обозначает 1 или 2. H2N
 · 2HCl·zH2O
· 2HCl·zH2O
Преимущества предлагаемого способа безводного ацилирования становятся очевидными и могут быть оценены специалистами в данной области, когда все преимущества объединены и рассмотрены в целом. Элиминирование формальных амино- и карбоксильных защитных групп и соответствующее элиминирование дополнительных химических стадий, необходимых для блокировки и деблокировки, позволяют получить очевидное преимущество в общей эффективности процесса и стоимости материалов перед ранее известными процессами. Предлагаемый способ дополнительно обеспечивает и поддерживает проведение контроля стереохимической конфигурации метоксиимино- изомера и Δ3 двойной связи ядра цефалоспорина без необходимости в разделении нежелательных побочных продуктов цефалоспорина и без необходимости в использовании стереохимических контролирующих сульфоксидпроизводных, таких, которые описаны в патенте США N 4043631. Другим преимуществом настоящего изобретения является получение и использование незащищенной кристаллической хлористоводородной соли син-изомера 2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхлорида формулы III, которая устраняет необычные и иногда сложные органические уходящие группы, описанные в литературе. Использование простого хлорид-иона в качестве уходящей группы устраняет необходимость использования потенциальных токсичных уходящих групп, таких как 2-меркаптобензотиазол. Другим преимуществом предпочтительного варианта настоящего процесса безводного ацилирования является получение искомого температурно-устойчивого кристаллического гидрата дигидрохлорида цефепима непосредственно из реакционной смеси способа ацилирования без необходимости в получении и выделении соли серной кислоты или цвиттериона цефепима. Данный способ также позволяет получить водорастворимый кристаллический моногидрат цефепим дигидрохлорида (или дигидрат), по существу свободный от анти-изомера и Δ2-изомера, с высоким выходом, непосредственно из водной фазы двухфазного раствора.
Настоящее изобретение также предлагает устойчивый кристаллический син-изомер хлоргидрата 2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхлорида, который по существу свободен от антиизомера и представлен формулой III:
H2N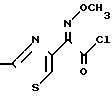 · HCl (III)
· HCl (III)
В результате того, что соединение III является по существу свободным от анти-изомера, оно конвертируется в цефалоспорины широкого круга действия, которые сами по себе по существу свободны от анти-изомера, причем нет необходимости в хроматографическом разделении син- и анти-изомеров. В результате повышенной устойчивости соединение III может быть выделено и сохранено и, когда необходимо, соединение III может быть превращено в конечные продукты в другом растворителе, который благоприятен для получения искомого антибиотика, по существу свободного от Δ2-изомера. Дополнительное преимущество промежуточного соединения формулы III заключается в том, что оно не требует блокировки (защиты) аминогруппы перед ацилированием или деблокировки (освобождения от защиты) аминогруппы после ацилирования, что тем самым обеспечивает эффективность способа. Другим преимуществом хлорангидрата формулы III является его использование в процессе ацилирования с получением цефалоспоринов широкого спектра действия. В противоположность другим методам, например тем, которые описаны в патенте США N 4406899 на имя Абураки и др. промежуточное соединение формулы III имеет хлорид-ион в виде простой и нетоксичной уходящей группы без какой-либо предосторожности в отщеплении ее от искомого антибиотика, что имеет место со многими другими уходящими группами, известными специалистами. Кроме того, некоторые промежуточные соединения, которые, как известно, содержат другие уходящие группы, трудно получить, в то время как другие промежуточные соединения, содержащие такие уходящие группы, как 2-меркаптобензотиазол, являются токсичными /Кемикал Эбстрактс, 1989, Том III (3), стр. 19243/.
Схема Реакции I
Син-изомер хлоргидрата хлоргидрида формулы III можно получить из син-изомера кислоты формулы I, как отмечается в Схеме Реакции I. Кислоту формулы I вначале превращают в соответствующую хлористоводородную соль формулы II известными методами, а затем, если необходимо, выделять в виде безводного кристаллического соединения формулы II. Образование хлористоводородной соли преимущественно осуществляют с помощью, по меньшей мере, одного молярного эквивалента газообразного хлороводорода в инертном органическом растворителе, таком как толуол, ацетонитрил, дихлорметан, ацетон, бензол, ксилол, циклогексан, гексаны, диоксан или простой диэтиловый эфир, при температуре в диапазоне приблизительно от -10оС до 50оС. Предпочтительно, реакцию осуществляют в толуоле, дихлорметане или ацетонитриле, и полученный таким образом хлоргидрат формулы II может быть выделен или использован 
 . Когда реакцию осуществляют в ацетонитриле, полученный хлоргидрат формулы II, как правило, сохраняет слегка привязанный растворитель. Поэтому благоприятно использовать хлоргидрат кислоты формулы II из ацетонитрила в должный период времени в следующей стадии, с тем чтобы избежать сольватное вытеснение под действием атмосферной влаги. Наиболее предпочтительно, если реакцию осуществляют в толуоле или дихлорметане при температуре от 0оС до комнатной температуры.
. Когда реакцию осуществляют в ацетонитриле, полученный хлоргидрат формулы II, как правило, сохраняет слегка привязанный растворитель. Поэтому благоприятно использовать хлоргидрат кислоты формулы II из ацетонитрила в должный период времени в следующей стадии, с тем чтобы избежать сольватное вытеснение под действием атмосферной влаги. Наиболее предпочтительно, если реакцию осуществляют в толуоле или дихлорметане при температуре от 0оС до комнатной температуры.
Кислую соль формулы II затем благоприятно обработать хлорирующим агентом и, наиболее предпочтительно, оксалилхлоридом в сочетании с диметилформамидом для получения устойчивого кристаллического син-изомера соединения III. Как показано в данном описании, использование других известных хлорирующих агентов может привести к изомеризации с производством нежелательного анти-изомера или смесей син- и анти-изомеров. Кроме того, такие хлорирующие реагенты, как пятихлористый фосфор, могут привести к хлорированию 5-положения на кольце тиазола, что затем создаст нежелательную примесь в антибиотике. Заявитель обнаружил, что, кроме получения хлоргидрата кислоты формулы II, правильный выбор хлорирующего реагента и реакционных условий, таких как растворитель и температура, являются решающими факторами в способе получения син-изомера соединения III, которое по существу свободно от анти-изомера.
Методы хлорирования, обычно используемые для активации кислот, хорошо известны в данной области техники. Пятихлористый фосфор, являющийся наиболее широко используемым хлорирующим реагентом, не подходит для хлорирования соединения II, поскольку он также приводит к изомеризации метоксииминогруппы с получением нежелательного анти-изомера соединения III. Это ясно продемонстрировано в Примерах 12, 13, 14 и 16, которые приведены ниже. Другим известным методом хлорирования является использование оксалилхлорида в сочетании с диметилформамидом. Однако заявитель обнаружил, что метод оксалилхлорида, в котором диметилформамид используют в качестве катализатора, не приводит к получению значительных количеств искомого син-изомера соединения III. Это также ясно продемонстрировано в Примере 15 ниже. В результате обширных исследований заявитель установил, что использование диметилформамида в количестве меньшем, нежели эквимолярное количество относительно количества оксалилхлорида, имеет отрицательное воздействие на производство искомого син-изомера хлоргидрата хлорангидрида формулы III. Наиболее предпочтительно, если молярное количество диметилформамида превышает молярное количество оксалилхлорида. Заявитель также обнаружил, что использование избыточных молярных количеств диметилформамида также оказывает отрицательное влияние как на реакцию, так и на устойчивость искомого продукта. Таким образом заявитель открыл способ управления нестабильностью реакционных веществ как в отношении избыточного хлорид-иона, производимого оксалилхлоридом, так и в отношении диметилформамида, каждый из которых имеет решающее значение при производстве устойчивого кристаллического син-изомера соединения III, которое по существу свободно от анти-изомера. В том случае, когда превращение соединения формулы II в соединение формулы III не завершено, остается небольшое количество син-изомера кислоты формулы II в выделенном продукте соединения III. Присутствие некоторого количества непрореагировавшего соединения II в продукте соединения III не оказывает воздействие на последующую реакцию ацилирования с точки зрения успешного производства искомого антибиотика, который по существу свободен от антиизомера указанного антибиотика.
Заявитель также обнаружил, что решающим также являются температура и реакционный растворитель хлорирования. Предпочтительно, если реакцию осуществляют в инертном органическом растворителе, таком как дихлорметан, хлороформ или ацетонитрил, при температуре менее, чем -10оС. Наиболее предпочтительно, если реакцию преимущественно осуществляют в дихлорметане при температуре приблизительно от -15оС до -40оС.
Использование син-изомера хлоргидрата хлорангидрида формулы III для получения полезных антибиотиков широкого спектра действия при помощи общей реакции ацилирования приведено в Схеме Реакции 2. Более конкретно, Схема Реакции 2 иллюстрирует использование хлорангидрида формулы III для получения широкоспектрового антибиотика цефепим, который по существу свободен от анти-изомера и Δ2-изомера. Кроме того, хлорангидридгидрохлорид формулы III можно использовать для получения цефалоспориновых антибиотиков, имеющих син-изомер 2-(2-аминотиазол-4-ил)-2-метоксииминоацетила, присоединенный к 7-аминогруппе ядра цефалоспорина, например, цефодизим, цефменоксим, цефатоксим, цефпиром, цефподоксим, цефхином, цефтерам, цефтиофур, цефетамет и цефузонам.
Кроме того, с целью подтверждения того, что хлоргидрат хлорангидрида в соответствии с известными способами находится в форме анти-изомера, а не в форме искомого син-изомера, заявитель заменил продукт известных способов, например, полученный в Примере 14, на син-изомер соединения формулы III в реакциях ацилирования, приведенных в Схеме Реакции 2 и в Примере 4. Полученный цефалоспориновый продукт, который был изготовлен в соответствии с описанием Примеров 17, 18 и 23, сравнивали с антибиотиком цефепим, полученным в соответствии с настоящим изобретением. Как можно видеть из сравнения в Примере 19, анти-цефепим, полученный известными способами, отличается от пригодного широкоспектрового син-цефепима, полученного в соответствии с настоящим изобретением.
Определенный в описании и в формуле изобретения термин "по существу свободен" означает, что соединение содержит менее, чем приблизительно 5% нежелательного изомера. Предпочтительно, если соединение содержит менее, чем приблизительно 1% нежелательно изомера.
В соответствии со способом настоящего изобретения дигидрохлорид-гидрат широкоспектрового антибиотика цефепим, который по существу свободен от анти-изомера и Δ2-изомера, получают N-ацилированием соединения формулы IV с син-изомером хлоргидрата формулы III, как показано в Схеме Реакции 2.

7-Амино-3-/(1-метил-1-пирролидино)метил/цеф-3-ем-4-карбоксилатную соль, которая по существу свободна от Δ2-изомера и представлена формулой IV, где НХ обозначает НСl, HI или H2SO4, может быть получена общими методами, описанными С.П.Брундиджем и др. в патенте США N 4868294.
Промежуточное соединение цефалоспорина формулы IV; где НХ предпочтительно обозначает НI, может быть силилировано в инертном органическом растворителе с образованием раствора in situ растворимого силилированного производного, представленного формулой VI, где R и R1каждый самостоятельно обозначает Н или силильную группу, или же их смесь. В одном исследовании IH ЯМР определено, что R1 главным образом силилирован, а R обозначает главным образом водород. Специалист должен понять, что вследствие постоянного изменения уравновешивания различных видов в растворе трудно идентифицировать специфическое соединение формулы VI, полученное in situ в реакционной смеси в любое конкретное время. Однако важно прибавить достаточное количество силилирующего реагента и, если необходимо, основания с тем, чтобы солюбилизировать промежуточное соединение цефалоспорина формулы IV перед тем, как безводный раствор обработан соединением формулы III. Силилирующие реагенты, которые могут быть использованы, хорошо известны специалистам в данной области техники и включают, например, триметилхлорсилан, триметилиодосилан, гексаметилдисилазан трет-бутилдиметилхлорсилан, триметилсилилацетамид, бис-(триметилсилил)мочевину и тому подобное. Предпочтительно, в процессе ацилирования можно использовать триметилхлорсилан или смесь триметилхлорсилана и гексаметилдисилазана.
Хотя при взаимодействии требуется по меньшей мере один молярный эквивалент силилирующего реагента и по меньшей мере один молярный эквивалент основания, на практике обнаружено, что благоприятно использовать около двух молярных эквивалентов силилирующего реагента и около двух молярных эквивалентов, или меньше, основания, с тем чтобы получить раствор промежуточного соединения формулы VI. Однако, когда силилирующими реагентом является смесь триметилхлорсилана и гексаметилдисилазана, предпочтительно не добавлять основание для получения растворимого производного формулы VI. Как оценит специалист в данной области, гексаметилдисилазан в качестве силилирующего реагента продуцирует побочный продукт основания, который достаточен для нейтрализации некоторого количества продуцируемой кислоты. Поскольку избыточное количество основания оказывает отрицательное воздействие на производство искомого антибиотика, по существу свободного от Δ2-изомера, смесь триметилхлорсилана и гексаметилдисилазана наиболее предпочтительна в соответствии с настоящим изобретением. Количество Δ3-Δ2 изомеризации в ядре цефалоспорина является чувствительным к используемым реакционным условиям при получении растворимого силилированного производного формулы VI и при его N-ацилирования с помощью хлоргидрата хлорангидрида формулы III, которое приводит к получению соединения формулы V. Степень изомеризации зависит от таких факторов, как количество и порядок прибавления основания, растворитель и температура, используемые в данном способе. Наиболее важно, что основные условия в результате использования избыточного основания или в результате прибавления основания перед прибавлением силилирующего реагента или хлоргидрата хлорангидрида формулы III, увеличивают изомеризацию двойной связи цефема от Δ3 к Δ2. Поэтому наиболее предпочтительно поддерживать неосновные реакционные условия в течение процесса безводного ацилирования.
Пригодными основаниями, которые можно использовать в предлагаемом способе, являются неорганические и органические основания, являющиеся пригодными акцепторами кислоты, например NaHCO3, KHCO3, Na2CO3, K2CO3, аммиак, первичный амин, вторичный амин, третичный амин и так далее. Наиболее предпочтительно использовать такие органические основания, как, например, 1,8-диазабицикло-/5.4.0/ундец-7-ен, N-метилморфолин, 2,6-лутидин, 2-метил-6-этилпиридин, N,N-диметиланилин, N,N-диэтиланилин, триэтиламин, диизопропилэтиламин и так далее. Предпочтительно, и если необходимо, в реакциях силилирования и N-ацилирования можно использовать N-метилморфолин или триэтиламин, или их смесь. Наиболее предпочтительно при N-ацилировании соединения формулы VI с соединением формулы III использовать N-метилморфолин или триэтиламин.
Растворимое силилированное производное формулы VI, которое получают in situ, затем обрабатывают хлорангидридом-хлоргидратом формулы III и, предпочтительно, с одним молярным эквивалентом и, наиболее предпочтительно, с незначительным избытком гидрохлорида хлорангидрида формулы III с последующим прибавлением эквимолярного количества и, предпочтительно менее, чем эквимолярное количество, основания относительно количества соединения формулы III, получая смесь, содержащую искомый антибиотик. Предпочтительно, если количество хлоргидрата хлорангидрида формулы III и основания прибавляют медленное по порциям. Хотя прибавлением реагентов можно осуществить сразу за один прием, если температура и неосновные условия реакции можно контролировать, благоприятно прибавлять реагенты в две или три порции с тем, чтобы гарантировать полное взаимодействие реагентов.
Пригодные растворители, которые можно использовать в предлагаемом способе, представляют собой инертные органические растворители, в которых силилированное производное формулы VI является растворимым и в которых изомеризация двойной связи Δ3 минимизирована, например, толуол, тетрагидрофуран, ацетон, ацетонитрил, дихлорметан, хлороформ, диметилацетамид и так далее, или их смеси. Наиболее предпочтительно использовать в данном способе ацетонитрил или дихлорметан. Способ настоящего изобретения можно осуществлять при температуре в диапазоне от -60 до +50оС, предпочтительно около -40оС комнатная температура. Получение силилированного соединения формулы VI преимущественно проводят при температуре в диапазоне приблизительно от -10оС до комнатной температуры, тогда как N-ацилирование преимущественно осуществляют при температуре от -40 до 0оС приблизительно.
Когда N-ацилирование растворимого силилированного соединения формулы VI завершено, что подтверждается известными методами определения, например, тонкослойной хроматографией, жидкостной хроматографией высокого давления и спектроскопическими методами, затем, в соответствии с предпочтительным вариантом способа настоящего изобретения, в реакционную смесь прибавляют воду в количестве, достаточном для растворения реакционной смеси, если желательно, видимых твердых частиц и получения двухфазного раствора органической фазы и воды. Количество подлежащей прибавлению в реакционную смесь воды определяют на основе выбора и количества инертного органического растворителя, используемого в способе, и оно должно быть достаточным для того, чтобы обеспечить или вызвать разделение фаз. После завершения фазового разделения органическую фазу благоприятно отделить и декантировать с получением водного богатого раствора, содержащего искомый антибиотик. Водный богатый раствор затем обрабатывают достаточным количеством кислоты или ее растворимой нетоксичной соли, как например, хлористоводородная кислота, хлористый натрий, хлористый аммоний, хлористый калий, серная кислота, сульфат натрия, сульфат калия, сульфат аммония, фосфорная кислота, фосфат натрия, фосфат калия, фосфат аммония, азотная кислота, нитрат натрия, нитрат калия и так далее, с получением достаточного количества противоаниона с тем, чтобы гарантировать кристаллизацию искомого цефепима в виде соли, по выбору разбавленной соответствующим, смешивающимся с водой органическим растворителем, таким как метилэтилкетон, ацетон, изопропанол, бутанол и так далее, с тем, чтобы индуцировать или завершить кристаллизацию. Предпочтительно, водный богатый раствор обрабатывают достаточным количеством серной кислоты с тем, чтобы кристаллизовать соль серной кислоты (сульфат) цефепима, который по существу свободен от анти-изомера и Δ2-изомера. Сульфат цефепима затем можно превратить в предпочтительный моногидрат дигидрохлорида кристаллического цефепима способом, описанным Капланом и др. в патенте США N 4910301. Сульфат цефепима, получаемый в соответствии с настоящим способом, может быть нейтрализован основанием и, предпочтительно, слабоосновной ионообменной смолой, известной в данной области и, предпочтительно, коммерчески доступной, как например, Amberlite IA-2, Dowex WGR Bio-Rad AG3-X4A, Amberlite IRA 93, Amberlite IRA 35 и так далее, с получением водного или водно-органического раствора, содержащего цвиттерион-форму цефепима. Раствор затем обрабатывают достаточным количеством хлористоводородной кислоты и, по выбору, смешивающимся с водой органическим растворителем с тем, чтобы индуцировать кристаллизацию предпочтительно кристаллического дигидрохлоридгидрата цефепима. Наиболее предпочтительно, если водный богатый раствор, полученный в результате безводного ацилирования в соответствии с настоящим способом, обработан достаточным количеством хлористоводородной кислоты, с тем чтобы индуцировать и гарантировать кристаллизацию указанного антибиотика, дигидрохлорид гидрата цефепима, когда прибавлен смешивающийся с водой органический растворитель, такой как ацетон. Количество смешивающегося с водой органи- ческого растворителя должно быть достаточным для получения полной кристаллизации указанного антибиотика и, преимущественно, в количестве около 2-9 объемов водной фазы, с тем чтобы получить температурно-устойчивый моногидрат или дигидрат кристаллического дигидрохлорида цефепима, который по существу свободен от анти-изомера и Δ2-изомера.
Когда необходимо получить только дигидрохлорид-моногидрат цефепима, богатый водный раствор в результате безводного ацилирования преимущественно обрабатывают достаточным количеством хлористоводородной кислоты и разбавляют подходящим количеством смешивающегося с водой органического растворителя, как описано в данной работе, с тем, чтобы гарантировать кристаллизацию желаемой моногидратной формы. Альтернативно, когда необходимо получить устойчивый дигидрохлорид-дигидрат цефепима, богатый водный раствор преимущественно обрабатывают более высокой эквивалентной концентрацией хлористоводородной кислоты и количеством смешивающегося с водой органического растворителя с тем, чтобы удержать кристаллизацию на точке помутнения перед прибавлением органического растворителя (дополнительного количества) для завершения кристаллизации. Однако следует понять, что в том случае, если стадия выделения из водного богатого раствора способа не контролируется тщательно, возможно, что может быть получена смесь кристаллического дигидрохлорид-моногидрата и дигидрата цефепима. В любом случае приготовление только одного из желаемых гидратов можно осуществить из любого гидрата или смеси гидратов с последующим проведением методики перекристаллизации, описанной ниже.
Кристаллический дигидрохлорид-моногидрат цефепима, полученный в соответствии с настоящим изобретением, может быть использован для получения устойчивого кристаллического дигидрохлорид-дигидрата цефепима при регулируемых концентрациях растворителя и хлористоводородной кислоты, а также периода времени у точки помутнения (начальной кристаллизации), как описано ниже. Альтернативно, кристаллический дигидрохлорид-дигидрат, полученный предлагаемым способом, также можно использовать для получения устойчивого кристаллического дигидрохлорид-моногидрата цефепима путем перекристаллизации при отличных контролируемых условиях, как описано ниже. Таким образом способ настоящего изобретения может быть применен для получения либо моногидрата, либо дигидрата указанного антибиотика.
В противоположность лабильному дигидрохлорид-дигидрату цефепима, описанному в патенте США N 4910301, который легко теряет второй моль воды, полученный в соответствии с предлагаемым изобретением кристаллический дигидрохлорид-дигидрат цефепима, как обнаружено, имеет хорошо выраженную кристаллическую структуру, которая сохраняет второй моль воды. Обнаружено, что новая кристаллическая дигидративная форма (игольчатые кристаллы) являются удивительно устойчивой, а морфология ее кристаллов не изменяется при различных условиях, например, в воздухе при температуре 70оС в течение более, чем двух месяцев, в вакууме с P2O5 при температуре 50оС в течение 48 ч, при печной сушке при температуре 70оС в течение 96 ч, и в условиях высокой и низкой относительной влажности. Кристаллический дигидрат проявляет характерные пики поглощения в инфракрасной области спектра при 3574 см-1 и 3432 см-1, как отмечается при диффузной отражательной инфракрасной спектроскопии с фурье-преобразованием с применением КBr и 13 мм испытательской чашки и с использованием спектрометра Nicolet 20SX. Эта температурно- и влагоустойчивая кристаллическая дигидратная форма цефепима также характеризуется порошковой рентгенограммой, приведенной в табл.1.
В табл.1 "d" относится к межплоскостным расстояниям и "I/Io" относится к относительным интенсивностям в процентах. Порошковую рентгенограмму собирали с помощью рентгеновского дифрактометра Rigaku Geigerflex и длины волны излучения никель-отфильтрованной меди (Ка), равной 1,5425  .
.
Таким образом, вариант настоящего изобретения предлагает способ получения антибиотика, цефепим-дигидрохлорид-гидрата, который по существу свободен от анти-изомера и Δ2-изомера, предусматривающий взаимодействие силилированного производного 7-амино-3-/(1-метил-1-пирролидино)метил/цеф-3-ем-4-карбоксилата с син-изомером хлоргидрата 2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхлорида, по существу свободного от анти-изомера, в инертном органическом растворителе.
Предпочтительный вариант настоящего изобретения кроме того предусматривает получение син-изомера хлоргидрата 2(2-амино-тиазол-4-ил)-2-метоксииминоацетил- хлорида, по существу свободного от анти-изомера, путем взаимодействия безводной кислой хлористоводородной соли син-изомера 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты со смесью, содержащей по меньше мере один молярный эквивалент оксалилхлорида и по меньшей мере один молярный эквивалент (до незначительного избытка) диметилформамида (относительно указанного оксалилхлорида), в инертном органическом растворителе при температуре менее, чем -10оС.
Другой предпочтительный вариант настоящего изобретения предусматривает получение силилированного производного путем взаимодействия 7-амино-3-/(1-метил-1-пирролидино)метил/цеф-3-ем-4-карбокс- илатной соли в инертном органическом растворителе с силилирующим реагентом.
Более предпочтительный вариант настоящего изобретения предлагает способ получения антибиотика, цефепим-дигидрохлорид-гидрата, который по существу свободен от анти-изомера и Δ2-изомера, предусматривающий взаимодействие силилирующего производного 7-амино-3-/(1-метил-1-пирролидино)метил/цеф-3-ем-4-кар-боксилата с син-изомером хлоргидрата 2-(2-аминотиазол-4-ил)-2-метоксииминоацетил- хлорида в инертном органическом растворителе с последующим прибавлением достаточного количества воды в реакционную смесь для получения органического водного двухфазного раствора, и с последующим прибавлением достаточного количества кислоты или ее растворимой нетоксичной соли и, по выбору, смешивающегося с водой органического растворителя в отделенный водный раствор.
Наиболее предпочтительный вариант настоящего изобретения предлагает способ получения антибиотиков, цефепим-дигидрохлорид-моногидрата и цефепим-дигидрохлорид-дигидрата, непосредственно из богатого водного раствора предлагаемого способа безводного ацилирования.
Пригодность цефепима (соединения V) показана в работе Абураки и др. патент США N 4406899. Устойчивая дигидратная форма цефепима, продуцируемая с использованием настоящего способа, проявляет антибиотические свойства вышеупомянутого цефепима по патенту США N 4406899 и находит применение в качестве антибиотика аналогичным образом.
Следует понять, что описание изобретения и примеры являются лишь иллюстративными и не должны истолковываться как ограничивающие объем изобретения.
П р и м е р 1. Хлористоводородная соль син-2-(2-аминотиазол-4-ил)-2-метоксиими-ноуксусной кислоты. Суспензию (25 г, 124,25 ммоль) 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты в толуоле (250 мл) насыщают газом HCl при температуре 20-28оС. HCl вводят вглубь в две аликвоты 8,1 г (222,2 ммоль) и 4,8 г (131,7 ммоль) при 30 минутном перемешивании между аликвотными прибавлениями. Через 1 ч при температуре 20оС продукт собирают фильтрацией в атмосфере азота, промывают толуолом (50 мл) и гексаном (250 мл) и сушат при температуре 20-25оС в вакууме с получением 28,68 г (97%) указанного в заголовке соединения.
П р и м е р 2. Хлоргидрат син-2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхло-рида. К раствору (0,77 мл, 10 ммоль) диметилформамида в дихлорметане (40 мл) при температуре 5оС прибавляют (0,89 мл, 10 ммоль) 98% оксалилхлорид в дихлорметане (4,1 мл). Покапельное прибавление поддерживает температуру на уровне 4-5оС. К полученной суспензии, охлажденной до температуры -27оС, прибавляют (2,37 г,10 ммоль) хлоргидрата 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты, полученного в Примере 1. Суспензию перемешивают в течение 2,5 часа при температуре -25оС. Фильтрация в атмосфере азота и промывка дихлорметаном (50 мл) и гексаном (100 мл) позволяют получить 1,78 г (69,5% ) указанного в заголовке кристаллического соединения белого цвета после сушки при температуре 20оС в вакууме. Указанный в заготовке хлорангидрид ацилирует хлоргидрат сложного дифенилметилового эфира 7-аминодезацетокси-цефалоспорановой кислоты в растворе пиридина с получением продукта с одной зоной (тонкослойная хроматография), который согласуется с аутентичным образом (и не отделяется от него) искомого сложного эфира дезацетокси-цефалоспорина.
П р и м е р 3. Хлоргидрат син-2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхло- рида. К раствору 1,55 мл, 20 ммоль) диметилформамида в дихлорметане (80 мл) при температуре 5оС прибавляют (1,78 мл, 20 ммоль) оксалилхлорида со степенью чистоты 98% в дихлорметане (8,2 мл). Время прибавления составляет 5 мин при температуре 5-8оС. Полученную суспензию перемешивают в течение 10 мин при температуре 5оС и затем охлаждают до температуры -30оС. Прибавляют хлоргидрат 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты из Примера 1. Суспензию перемешивают в течение 2,5 ч при температуре от -25 до 30оС. Фильтрация в атмосфере азота и промывка дихлорметаном (75 мл) и гексаном (100 мл) позволяют получить 3,57 г (69,7%) указанного в заголовке кристаллического соединения после сушки при температуре 20оС в вакууме.
Аликвота твердого хлоргидрата хлоргидрида ацилирует хлоргидрат сложного дифенилметилового эфира 7-аминодезацетокси- цефалоспорановой кислоты в растворе пиридина с получением в основном продукта с одной зоной (тонкослойная хроматография): который согласуется с аутентичным образом (и не отделяется от него) искомого сложного эфира дезацетоксицефалоспорина.
П р и м е р 4. Получение 7-/2-(2-аминотиазол-4-ил)-2-(Z)- метоксииминоацетамидо/-3-(1-метил-1-пирролидино)метил/цеф-3-ем-4 -карбоксилата (цефепим).



Моногидроиодид 7-амино-3-/(1-метил-1-пирролидинио)метил/-цеф-3-ем-4-карбо- ксилата (0,85 г, 2,0 ммоль)/ получен в соответствии с методикой, описанной С.П.Брундиджем и др. в патенте США N 4714760/ растворяют в 9 мл смеси ацетон-вода (2:1) с триэтиламином при pH 6,5 и температуре 20оС. Хлоргидрат син-2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида (0,56 г; 2,2 ммоль) /получен в Примере 3/ прибавляют с использованием триэтиламина для регулирования pH на уровне 5-7. Анализ полученного раствора с помощью жидкостной хроматографии высокого давления показывает 58% выход искомого цефалоспорина (цефепима). Подкисление серной кислотой до pH 2,2 позволяет получить 0,63 г указанного в заголовке антибиотика в виде его соли серной кислотой (51% выход активности), как описано Абураки и др. в патенте США N 4406899, выданном 27 сентября 1983 года, и Капланом и др. в патенте США N 4910301, выданном 20 марта 1990 года.
П р и м е р 5. Хлоргидрат син-2-(2-аминотиазол-4-ил) -2-метоксииминоацетилхлорида. К раствору (9,75 мл, 125,9 ммоль) диметилформамида в дихлорметане (450 мл) при температуре 5оС прибавляют по каплям раствор (11,21 мл, 125,9 ммоль) оксалилхлорида (98%) в дихлорметане (15 мл). Прибавлением завершают через 10 мин при температуре 5-7оС. К полученной суспензии, охлажденной до температуры -25оС, прибавляют (28,5 г, 119,9 ммоль) хлористоводородную соль син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты в одну аликвоту. Суспензию перемешивают в течение 3,5 ч при температуре от -25 до -30оС, фильтруют в атмосфере азота, промывают дихлорметаном (100 мл) и гексаном (400 мл) и сушат при температуре 20-25оС в вакууме. Выход указанного в заголовке кpисталлического соединения составляет 30,7 г (72,5%).
Указанный в заголовке хлорангидрид ацилирует хлоргидрат сложного дифенилметилового эфира 7-аминодезацетокси-цефалоспорановой кислоты в растворе пиридина с получением по существу одной зоны (тонкослойная хроматография) искомого сложного эфира дезацетоксицефалоспорина с отсылкой на аутентичный образец.
Указанный в заголовке хлорангидрид (200 мг, 0,8 ммоль) гидролизуют в воде. IH ЯМР выделенного продукта идентичен исходной син-кислоте.
П р и м е р 6. Хлоргидрат син-2-(2-аминотиазол-4-ил)-метоксиимино-ацетилхлор- ида. К раствору (8,13 мл, 105 ммоль) диметилформамида в дихлорметане (350 мл) при температуре 5оС прибавляют по каплям (9,34 мл, 105 ммоль) оксалилхлорид (чистота 98%) в дихлорметане (5 мл). Максимальная температура достигает во время прибавления 7оС. Полученную суспензию перемешивают в течение 10 мин при температуре 5оС и затем охлаждают до -27оС. Хлоргидрат 2-(2-аминотиазол-4-ил)-2-метоксииминоук- сусной кислоты (23,8 г, 100 ммоль) прибавляют в одну аликвоту. Суспензию перешивают в течение 2,5 часа при температуре от -25 до -30оС, фильтруют в атмосфере азота, промывают дихлорметаном (25 мл) и гексаном (125 мл) и сушат при температуре 20оС в вакууме. Выход составляет 21,39 г (83,5%) кристаллического хлоргидрата хлорангидрида.
Аналитически вычислено для C6H7N3O2SCl2:
С 28,14, Н 2,76, N 16,41, S 12,52,
Найдено: С 28,25, Н 2,93, N 16,32, S 12,67.
IH ЯМР (ДМСО-d6) δ: 3,93 (CH3), 7,04 (H5).
П р и м е р 7. Хлористоводородная соль син-2-(2-аминотиазол-4-ил)-2-метоксиими-ноуксусной кислоты. Суспензию (87,0 г, 432,4 ммоль) син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты в толуоле (870 мл) при температуре 22оС насыщает газом с помощью двух аликвот HCl: 17,5 г, 480 ммоль в течение 30 мин и 15,0 г, 410 ммоль в течение 20 мин при 20-минутных периодах перемешивания между аликвотами. Суспензию перемешивают в течение 1,5 ч при температуре 25оС, фильтруют в атмосфере азота, промывают толуолом (100 мл) и гексаном (400 мл) и сушат при температуре 20-25оС в вакууме. Выход указанного в заготовке соединения составляет 100,2 г (97,5%).
Аналитически вычислено для C6H8N3O3SCl:
С 30,32, Н 3,39, N 17,68, S 13,49, Cl 14,92.
Найдено: С 30,51, Н 3,39, N 17,54, S 13,37, Cl 14,90.
П р и м е р 8. Хлоргидрат син-2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхло- рида. К раствору (32,4 мл, 419,7 ммоль) диметилформамида в дихлорметане (400 мл) при температуре 5оС прибавляют (37,4 мл, 419,7 ммоль) по каплям 98% оксалилхлорид. Полученную суспензию охлаждают до температуры -25оС и прибавляют к суспензии (-25оС) (95 г, 399,7 ммоль) хлоргидрата син-2-(2-аминотиазол-4-ил)-2-метоксииминоук-сусной кислоты из Примера 7. Суспензию перемешивают в течение 2,5 ч при температуре от -25 до -28оС, фильтруют в атмосфере азота, промывают дихлорметаном (100 мл) и гексаном (500 мл) и сушат при температуре 20-25оС в вакууме. Выход указанного в заголовке кристаллического соединения составляет 84,3 г (82,3%).
Аналитически вычислено для С6Н7N3O2SCl2:
C 28,14, H 2,76, N 16,41, S 12,52,
Найдено: С 27,90, Н 3,10, N 16,14, S 12,27.
IH ЯМР (ДМСО-d6) δ: 3,95 (CH3), 7,04 (Н5).
П р и м е р 9. Получение 7-/2-(2-аминотиазол-4-ил)-2-(Z)-метоксииминоацетамидо/ -3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата (цефепим). К раствору 240 мл ацетона и 80 мл воды прибавляют 20,0 г гидроиодида 7-амино-3-/(1-метил-1-пирролидинио)метил/-цеф-3-ем-4-карбоксилата (0,047 моль) и перемешивают с отверждением. С использованием автотитратора Radiometer ABU80 конечной точкой, установленной на pH 6,5, и заполненного N-метилморфолином, хлоргидрат син-2-(2- аминотиазол-4-ил)-2-метоксииминоацетилхлорида (20,0 г, 0,0785 моль) /получен в Примере 5/ прибавляют в 4 порции с 5-минутными интервалами, поддерживая pH на уровне 6,5. После прибавления густую суспензию перемешивают в течение 20 мин при комнатной температуре. pH реакционной смеси понижают до pH 2,65 с помощью 21 мл 6 н. раствора H2SO4. Происходит осаждение указанного в заголовке соединения. Суспензию просеивают и перемешивают при комнатной температуре в течение 20 минут. pH суспензию доводят до 1,8 с помощью 16 мл 6 н. раствора H2SO4 и перемешивание продолжают еще 60 минут. Суспензию фильтруют в вакууме и промывают 70 мл смеси вода-ацетон (1:1), а затем 70 мл ацетона, получая 24,09 г (88,5% стехиометрический массовый выход) указанного в заголовке соединения, которое идентично соединению Примера 4 и цефепиму, описанному Абураки и др. в патенте США N 4406899, выданном 27 сентября 1983 года, и Капланом и др. в патенте США N 4910301, выданном 20 марта 1990 года.
П р и м е р 10. Получение хлоргидрата син-2-(2-аминотиазол-4-ил)-2-метоксиими-ноацетилхлорида. К раствору диметилформамида (8,76 мл, 0,113 моль) в дихлорметане (375 мл) при температуре 5оС прибавляют оксалилхлорид (9,64 мл, 0,111 моль) по каплям, поддерживая температуру на уровне 5-6оС. Суспензию перемешивают в течение 10 мин и затем охлаждают до температуpы -25оС. Затем прибавляют хлористоводородную соль син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты (25,0 г) в аликвотах в течение 11 минут в атмосфере сухого азота. Суспензию перемешивают в течение 2,5 часа при температуре -25оС. Продукт фильтруют в атмосфере сухого азота и промывают фильтровальный пирог дихлорметаном (80 мл). Продукт сушат при температуре 20-25оС в вакууме в присутствии Р2O5 с получением 23,88 г (88,6%) указанного в заголовке соединения в виде палево-желтого кристаллического твердого тела.
Аналитически вычислено для C6H7N3O2SCl2:
C 28,14, H 2,76, N 16,41, S 12,52, Cl 27,68
Найдено: C 28,06, H 2,71, N 16,26, S 12,30, Cl 27,23.
Продукт предыдущего эксперимента характеризуется спектром протонного ядерного магнитного резонанса в уксусной кислоте d4 (IН ЯМР), приведенном на Рис.1.
IH ЯМР (СD4CO2D) δ: 4,14 (CH3), 7,10 (H5). Уровень остаточного хлоргидрата кислоты из CH3 (4,11) составляет 5,1% Ничтожный уровень изомерного Н5 виден при 7,67 ppm.
П р и м е р 11. Хлоргидрат син-2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхло- рида. К раствору диметилформамида (17,92 мл, 231,9 ммоль) в дихлорметане (375 мл) при температуре 5оС прибавляют оксалилхлорид (19,76 мл, 220,8 ммоль). Время прибавления составляет 15 мин при температуре 5-6оС. Полученную суспензию перемешивают в течение 10 мин при температуре 5-6оС и затем охлаждают до температуры -25оС. Прибавляют хлоргидрат 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты (25,0 г, 105,2 ммоль). Полученный раствор затравливают указанным в заголовке соединением с получением продуктовой суспезии. Суспензию перемешивают в течение 3,5 ч при температуре -25оС, фильтруют в атмосфере сухого азота, промывают дихлорметаном (150 мл) и сушат при температуре 20-25оС в вакууме. Выход указанного в заголовке кристаллического соединения составляет 9,61 г (35,7%).
Аликвота твердого продукта хлоргидрата хлорангидрида ацилирует хлоргидрат сложного дифенилметилового эфира 7-амино-дезацетоксицефалоспорановой кислоты в растворе пиридина с получением продукта, по существу с одной зоной (тонкослойная хроматография), который согласуется с аутентичным образцом (и не отделяется от него) искомого сложного эфира дезацетоксицефалоспорина.
П р и м е р 12. Получение хлоргидрата 2-(2-аминотиазол-4-ил)-2-метоксиимино- ацетилхлорида. Экспериментальную часть примера 1 в патенте Чехословакии N 238950 повторяют следующим образом:
Пробу син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты (4,0 г), имеющую коэффициент Маккини 0,06% суспендируют в 30 мл бензола при температуре 21оС. Прибавляют одну каплю диметилформамида с последующим прибавлением 5 г порошкообразного пятихлористого фосфора в одну аликвоту. Температура возрастает до 34оС приблизительно через 2 мин, а затем повышается до 40оС через одну минуту, что приводит к получению полного раствора. Раствор охлаждают, и при температуре 36оС образуется осадок. После перемешивания в течение 30 мин температура составляет 22оС. Твердое тело светло-желтого цвета собирают фильтрацией в атмосфере сухого азота и промывают 30 мл бензола и 20 мл гептана. Выход продукта составляет 2,88 г после сушки в вакууме в присутствии P2O5 при температуре 20-25оС в течение 18 ч.
Продукт вышеприведенного эксперимента характеризуется спектром протонного ядерного магнитного резонанса в уксусной кислоте do4 (IH ЯМР), как показано на Рис.2, где Н5 составляет значение при 7,56 ppm и СН3 при 4,34 ppm. Данный спектр согласуется с продуктом указанного в заголовке соединения, имеющего конфигурацию анти-изомера, а не син-изомера, как раскрыто в противопоставленном патенте Чехословакии.
П р и м е р 13. Получение хлоргидрата 2-(2-аминотиазол-4-ил)-2-метоксиимино-ац- етилхлорида. Экспериментальную часть Примера 2 в патенте Чехословакии N 238950 повторяют следующим образом:
Пробу син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты (4,0 г), имеющую коэффициент Маккини 0,06% суспендируют в 20 мл ацетонитрила, который доводят до коэффициента Маккини 0,22% Прибавляют каплю диметилформамида, и температура составляет 20оС. После прибавления 6,0 г порошкообразного пятихлористого фосфора температуру повышают до 40оС и получают полный раствор. Раствор охлаждают до температуры 20оС, и преципитат образуется при температуре 33оС. После перемешивания в течение 30 мин продукт собирают в атмосфере сухого азота и промывают 30 мл бензола и 20 мл гептана. Выход составляет 1,86 г после сушки в течение 18 ч при температуре 20-25оС в вакууме в присутствии P2O5.
Продукт вышеприведенного эксперимента характеризуется спектром пpотонного ядерного магнитного резонанса в уксусной кислоте d4 (IH ЯМР), как показано на Фиг.3, где Н5 составляет значение при 7,56 ppm, а СН3 при 4,31 ppm. IH ЯМР-спектр согласуется с продуктом указанного в заголовке соединения, имеющего конфигурацию анти-изомера, а не син-изомера, как раскрывается в противопоставленном патенте Чехословакии.
П р и м е р 14. Получение хлоргидрата 2-(2-аминотиазол-4-ил)-2-метоксиимино- ацетилхлорида. Экспериментальную часть в Примере 3 патента Чехословакии N 238950 повторяют следующим образом:
Концентрированную хлористоводородную кислоту (0,16 мл) прибавляют к 30 мл дихлорметана. После охлаждения до температуры -10оС, прибавляют с приращениями 6,5 г пятихлористого фосфора. После нагревания до температуры 0оС прибавляют в одну аликвоту 4,0 г син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты, имеющей коэффициент Маккини 0,06% Температуру повышают до 2оС. Полный раствор получают через 9 минут при температуре 0оС. Через 40 мин начинает образовываться осадок. Продуктовую суспензию перемешивают в течение 2,8 ч при температуре 2-3оС фильтруют в атмосфере сухого азота, промывают 30 мл бензола и 20 мл гептана и сушат в вакууме при температуре 20-25оС в присутствии P2O5 в течение 18 ч. Выход составляет 3,42 г порошка светло-желтого цвета.
Продукт вышеприведенного эксперимента характеризуется спектром протонного ядерного магнитного резонанса в уксусной кислоте d4 (IH ЯМР), как показано на Фиг.4, где Н4 проявляется при 7,56 ppm, а СН3 при 4,31 ppm. IH ЯМР спектр согласуется с продуктом указанного в заголовке соединения, имеющего конфигурацию анти-изомера, а не син-изомера, как раскрыто в противопоставленном патенте Чехословакии.
П р и м е р 15. Предпринятое получение хлоргидрата 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида.
Общую методику, описанную в Примере 7 патента США N 4203899 для превращения защищенной аминотиазолуксусной кислоты в соответствующий хлорангидрид, применяют в отношении незащищенной аминотиазолуксусной кислоты следующим образом:
Пробу хлористоводородной соли син-2-(2-аминотиазол-4-ил)-2-метоксииминоуксус- ной кислоты (2,38 г, 0,01 моль) суспендируют в 30,5 мл бензола и охлаждают до температуры 20оС. Прибавляют оксалилхлорид (2,09 мл, 0,024 моль) с последующим прибавлением диметилформамида (0,50 мл, 0,0065 моль). Температура возрастает до 22оС при энергичном выделении газа. В течение 20 мин при температуре 20оС происходит облагороживание газом, и суспензию перемешивают при температуре 20 ± 2оС в течение 2 ч. Суспензию концентрируют в вакууме тем, чтобы удалить растворитель, и полученный продукт желтого цвета сушат в вакууме в присутствии P2O5 при температуре 20-25оС в течение 16 ч. Выход составляет 2,59 г.
Продукт в результате вышеприведенного эксперимента характеризуется спектром протонного ядерного магнитного резонанса (IH ЯМР) в уксусной кислоте d4, который показывает Н5 при 7,60 ppm и СН3 при 4,37 ppm. Спектр продукта согласуется с указанным в заголовке соединением, имеющим конфигурацию антиизомера.
П р и м е р 16. Предпринятое получение хлоргидрата 2-(2-аминотиазол-4-ил)2-метоксииминоацетилхлорида.
Общую методику, описанную в Примере 59 патента США N 4203899 для превращения защищенной аминотиазолоуксусной кислоты в соответствующий хлорангидрид, применяют в отношении незащищенной аминотиазолоуксусной кислоты следующим образом:
Пробу хлористоводородной соли син 2-(2-аминотиазол-4-ил)-2-метоксииминоуксус- ной кислоты (2,38 г, 0,01 моль) суспендируют в 25 мл дихлорметана. После охлаждения до температуры 4оС прибавляют 2,08 г (0,01 моль) пятихлористого фосфора. При охлаждении на льду температуру повышают до 6оС, и после охлаждения до 4оС суспензию перемешивают в течение 1 ч. Осадок собирают фильтрацией в атмосфере сухого азота, промывают дихлорметаном (10 мл) и сушат в вакууме при температуре 20-25оС с получением 1,4 г палево-желтого твердого вещества.
Продукт вышеприведенного эксперимента характеризуется спектром протонного ядерного магнитного резонанса (IH ЯМР) в уксусной кислоте d4, который показывает Н5 при 7,61 ppm, а СН3 при 4,34 ppm. Спектр продукта согласуется с указанным в заголовке соединением, имеющим анти-изомер-конфигурацию. Кроме того, продукт загрязнен неконвертируемой кислотой (IH ЯМР, имеющий Н5 при 7,07 ppm и СН3 при 4,06 ppm), что дополнительно подтверждается всплеском исходной кислоты.
П р и м е р 17. Ацилирование соли Н1 7-амино-3-/(1-метил-1-пирролидинио)-мет- ил/-цеф-3-ем-4-карбоксилата с использованием хлоргидрата 2-(2-ами отиазол-4-ил)-2-метоксииминоацетилхлорида (анти- изомерами из Примера 14). К предварительно охлажденному раствору 9 мл ацетона и 3,4 мл воды при температуре 10оС прибавляют Н1 соль 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата (1,13 г, 2,66 ммоль). Хлоргидрат 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлори-да (1,09 г, 4,21 ммоль) /получен в Примере 14/ прибавляют в 5 порций при температуре 0оС вместе с триэтиламином (0,37 мл, 2,66 ммоль) с тем, чтобы поддержать pH на уровне 6,0-7,0. Реакционную смесь перемешивают при окружающей температуре в течение 15 минут. Анализ полученного раствора жидкостной хроматографией высокого давления (градиентная колонка С18, от 2 до 25% ацетонитрила в 0,005М растворе NH4H2PO4) показывает 72,4% площади анти-цефепима на 13,08 мин и полное отсутствие син-цефепима, который, как ожидают, имеет время удерживания около 8,5 мин. Подкисление серной кислоты до pH 1,9 приводит к получению 1,48 г антицефепима в виде соли серной кислоты (сульфата). Идентификацию продукта подтверждают IH ЯМР-спектроскопией (ДМСО-d6), и показано, что он содержит 0,58 моль соли триэтиламина.
П р и м е р 18. Ацилирование Н1 соли 7-амино-3-/(1-метил-1-пирролидинио)-мет- ил/цеф-3-ем-4-карбоксилата с использованием хлоргидрата 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида (антиизомера из Примера 14). К предварительно охлажденному раствору 108 мл ацетона и 40,5 мл воды при температуре 10оС прибавляют Н1 соль 7-амино-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата (13,5 г, 0,0317 моль). pH суспензии доводят до 7,0 с использованием 2,7 мл 14% раствора NH4OH. При температуре 10оС порционно, в течение 60 минут прибавляют хлоргидрат 2-(2-аминотиазол-4-ил)-2-метоксииминоаце- тилхлорида (13,5 г, 0,015 мол) /получен методикой Примера 14/, используя 14% раствор NH4OH (27 мл) для поддержания pH на уровне 6,3-7,0 во время первой половины времени прибавления и pH 6,1-6,6 во время второй половины. Реакционную смесь перемешивают при температуре окружающей среды в течение 30 мин. Реакционную смесь фильтруют для окончательной очистки и промывают 6 мл 2:1 ацетон-вода, после чего в фильтрат медленно прибавляют 6 н. раствор H2SO4 (15 мл) с тем, чтобы отрегулировать pH до уровня 1,87-1,90. После перемешивания в течение одного часа нерастворимые вещества отфильтровывают и лепешку промывают 21 мл 2:1 ацетон/вода, а затем 30 мл ацетона. К фильтрату прибавляют 1 литр ацетона в течение 30 мин и смесь перемешивают при температуре 5-8оС в течение 40 мин. Продукт собирают фильтрацией, дважды промывают 24 мл 4:1 ацетон/вода, 60 мл ацетона и сушат в вакууме с получением 20,64 г (116% стехиометрический массовый выход) анти-цефепима в виде соли серной кислоты (95,4% чистота при жидкостной хроматографии высокого давления). Спектры IH ЯМР согласуются со структурой анти-цефепима, содержащего около 3 молей солей аммония.
П р и м е р 19. Сравнение продукта Примера 9 (син-изомера цефепима) и продукта Примера 17 (анти-изомера цефепима) показывает следующие отличия в физических характеристиках. Жидкостную хроматографию высокого давления изомеров цефепима осуществляют на колонке Water M Bondapack C18(3,9х300 мл) с использованием системы растворителей 1000 мл, воды, содержащей 2,88 г (0,013 моль) натриевой соли гептансульфокислоты, и pH доводят до 4,0 с помощью уксусной кислоты и 100 мл ацетонитрила со скоростью растекания фронта растворителя, равной 2,0 мл/мин. Продукты наблюдают визуально с помощью детектора с переменной длиной волны Water Модели 450, установленного на 254λ, с получением следующих результатов.
Время удерживания, мин
Син-изомер цефепима (пример 9) 10,5
Анти-изомер цефепима (пример 17) 37,8
Спектры протонного ЯМР син- и анти-метоксим-изомеров цефепима в виде двухлористоводородных солей определяют на спектрометре Bruker АМХ-400 F Т ядерного магнитного резонанса с использованием дейтерированного диметилсульфоксида в качестве растворителя. Изложенные химические сдвиги упоминаются в отношении ДМСО при 2,49 ppm. Система счисления, приведенная ниже в формуле и в таблице, дана только для удобства.
Син- и анти цефепим.
NH

* Волнистая связь обозначает син- и анти-метоксиимино-изомеры.
Таблица сравнения химических сдвигов протонного ЯМР (ppm)
Задание Син-цефепим Анти-цефепим
С2-Н2 4.04, 3.65 4.02, 3.65
С6-Н 5.33 5.31
С7-Н 5.88 5.85
С11-Н2 4.60, 4.31 4.59, 4.30
С12-Н4 2.93 2.93
С13-Н4 3.7, 3.4 3.6, 3.3
С14-Н4 2.10 2.10
С18-Н4 6,88 7.57
С20-Н3 3.92 4.05
NH 9.83 9.56
NH2 8.60 8.70
Спектры IH ЯМР двух метоксим-изомеров цефепима, как показано выше, существенно отличаются друг от друга. Тиазоловое кольцо СН(18) син-(Z) метоксим-изомера при 6,88 ppm уменьшается на высоту поля анти-(Е) метоксим-изомера СН(18) при 7,57.
П р и м е р 20. Безводное ацилирование Н1 соли 7-амино-3-/(1-метил-1-пирролидино)метил/цеф-3-ем-карбоксилата с использованием хлоргидрата 2-(2-аминотиазол- 4-ил)-2-метоксииминоацетилхлорида (син-изомера). В атмосфере азота Н1 соль 7-амино-3-/(1-метил-1-пирролидинио)-метил/цеф -3-ем-4-карбоксилата (50 г, 0,1176 моль) охлаждают до температуры -20оС в 500 мл ацетонитрила. Прибавляют триметилхлорсилан (39 мл, 2,5 эквивалента) и триэтиламин (38 мл, 2,3 эквивалента), поддерживая температуру на уровне ниже -10оС. После перемешивания силиловой смеси в течение 1,5 ч при температуре -10оС хлоргидрат 2-(2-аминотиазол-4-ил)-2-метоксииминоацетил- хлорида (син-изомер, полученный в Примере 5) прибавляют в две аликвоты (15 г, 0,50 эквивалента каждая). Затем 8 мл (0,5 эквивалента) триэтиламина прибавляют с 7,5 г (0,25 эквивалента) хлоргидрата 2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхлори- да (син-изомера). Суспензию перемешивают при температуре -10оС в течение 15 мин, после чего прибавляют 150 мл воды и перемешивание продолжают при температуре окружающей среды до растворения всех твердых веществ. Слой ацетонитрила отделяют от богатого водного слоя, в который прибавляют 6 н. раствор НСl (2,5 эквивалента) с 400 мл ацетона. Раствор затравливают и кристаллизуют в течение 15 минут. Для завершения кристаллизации прибавляют еще 1000 мл ацетона. Суспензию перемешивают в течение одного часа и затем фильтруют, промывают 400 мл ацетона и сушат при температуре около 40оС. Выход продукта составляет 56,51 г (84,1% стехиометрическая масса) цефепим (98,6% чистота, как определено анализом жидкостной хроматографии высокого давления в виде цефепим 2НСl ˙ H2O), который идентичен цефепим. 2НСl ˙ H2O, описанному Капланом и др. в патенте США N 4910301, выданном 20 марта 1990 года.
П р и м е р 21. Безводное ацилирование Н1 соли 7-амино-3-/(1-метил-1-пирролидино)метил/цеф-3-ем-4-карбоксилата с использованием хлоргидрата 2-(2-аминотиазол- 4-ил)-2-метоксиимино-ацетилхлорида (син-изомера). В атмосфере азота Н1 соль 7-амино-3-/(1-метил-1-пирролидино)метил/цеф-3-ем-4-карбоксилата (5,0 г, 0,01176 моль) охлаждают до температуры 0-5оС в 50 мл ацетонитрила. Прибавляют триметилхлорсилан (3,3 мл, 2,2 эквивалента) и N-метилморфолин (2,7 мл, 2,1 эквивалента), поддерживая температуру ≅ 5оС. Силиловую смесь перемешивают в течение 1,5 ч при температуре 0-5оС. 2-(2-Аминотиазол-4-ил)-2-метоксиимино-ацетилхлорид-хлоргидрат (1,5 г, 0,5 эквивалента) прибавляют в две аликвоты, после чего перемешивают в течение 10 мин. Другие две аликвоты хлоргидрата 2-(аминотиазол-4-ил)-2-метоксиимино-ацетилхлорида (1,5 г, 0,5 эквивалента) прибавляют с триэтиламином (0,8 мл, 0,5 эквивалента каждая). Суспензию перемешивают при температуре 0-5оС в течение одного часа, затем прибавляют 15 мл воды и смесь перемешивают при температуре окружающей среды до растворения всех твердых тел. Органический слой отделяют от богатого водного слоя, в водный слой прибавляют 6 н. раствор HCl (5 мл, 2,5 эквивалента) и 60 мл ацетона и перемешивают в течение 15 мин до начала кристаллизации. Для завершения кристаллизации прибавляют 80 мл ацетона. Суспензию перемешивают в течение 1 ч и затем фильтруют, промывают 50 мл ацетона и сушат при температуре около 40оС. Выход моногидрата дигидрохлорида цефепима составляет 5,78 г (86,0% стехиометрической массы).
П р и м е р 22. Безводное ацилирование Н1 соли 7-амино-3-/(1-метил-1-пирролидино)метил/цеф-3-ем-4-карбоксилата с использованием хлоргидрата 2-(2-аминотиазол- 4-ил)-2-метоксиимино-ацетилхлорида (син-изомера). В атмосфере азота Н1 соль 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата (5,0 г, 0,01176 моль) охлаждают до температуры 0-5оС в 50 мл ацетонитрила. Прибавляют триметилхлорсилан (1,34 мл, 0,90 эквивалента) и гексаметилдисилазан (1,8 мл, 0,75 эквивалента), поддерживая температуру ≅ 5оС. Силиловую смесь перемешивают в течение 1 ч при температуре 0-5оС. Затем прибавляют в две аликвоты хлоргидрат 2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхлорида (1,8 г, 0,59 эквивалента каждая) и перемешивание продолжают в течение 10 мин. Затем прибавляют еще две аликвоты хлоргидрата 2-(аминотиазол-4-ил)-2-метоксиимино-ацетил- хлорида (1,5 г, 0,5 эквивалента каждая) с триэтиламином (0,8 мл, 0,5 эквивалента каждая). Суспензию перемешивают при температуре 0-5оС в течение 1,5 ч с последующим прибавлением 15 мл воды и перемешиванием при температуре окружающей среды до растворения всех твердых веществ. Органический слой отделяют от богатого водного, в водный слой прибавляют 6 н. раствор НСl (5 мл, 2,5 эквивалента) и 30 мл ацетона и перемешивание продолжают в течение 10 мин с целью кристаллизации продукта. Для завершения кристаллизации прибавляют еще 110 мл ацетона. Суспензию перемешивают в течение одного часа, затем фильтруют, промывают 75 мл ацетона и сушат пpи температуре около 40оС. Выход моногидрата цефепим-дигидрохлорида составляет 5,45 г (81,1% стехиометрической массы).
П р и м е р 23. Безводное ацилирование Н1 соли 7-амино-3-/(1-метил-1-пирролидино)метил/цеф-3-ем-4-карбоксилата с использованием хлоргидрата 2-(2-аминотиазол- 4-ил)-2-метоксиимино-ацетилхлорида (анти-изомера из Примера 12. В атмосфере азота Н1 соль 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата (2,0 г 4,70 ммоль) охлаждают до температуры -20оС в 20 мл ацетонитрила. Прибавляют триметилхлорсилан (1,26 мл, 2,1 эквивалента) и триэтиламин (1,32 мл, 2,05 эквивалента), поддерживая температуру на уровне или ниже -10оС после перемешивания силиловой смеси в течение 1,5 ч при температуре -10оС прибавляют хлоргидрат 2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхлорида (анти-изомер, полученный в Примере 12) (1,2 г, 1,49 эквивалента) и реакционную смесь перемешивают в течение 1 ч при температуре -10оС.
Продукт вышеприведенной ацилированной смеси сравнивают с аналогичным продуктом смеси безводного ацилирования с использованием хлоргидрата син-2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетил- хлорида, как в примере 20. Основной пик продукта при жидкостной хроматографии высокого давления вышеупомянутой реакции ацилирования не соответствует пику искомого антибиотика цефепим. Пробу продукта в результате вышеприведенной реакции ацилирования выделяют хроматографией, и данные IH ЯМР и жидкостной хроматографии высокого давления относительно данной пробы согласуются с анти-изомером цефепима, как описано в Примере 19.
П р и м е р 24. Безводное ацилирование Н1 соли 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата с использованием хлоргидрата 2-(2-аминотиазол- 4-ил)-2-метоксиимино-ацетилхлорида (син-изомера). В атмосфере азота Н1 соль 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф-3-ем-4-карбоксилата (10,0 г, 0,02252 моль) перемешивают с 100 мл дихлорметана при температуре 20оС. Прибавляют триметилхлорсилан (2,35 мл, 0,82 эквивалента) и гексаметилдисилазан (3,85 мл, 1,62 эквивалента), и температуру силиловой смеси повышают до 25оС и сохраняют в течение 1,5 ч при температуре от 25 до 30оС. Затем силиловую смесь охлаждают до температуры -40оС и в течение 40 мин прибавляют хлоргидрат 2-(2-аминотиазол-4-ил)-2-метоксиимино-ац- етилхлорида (6,04 г, 0,93 эквивалента) при температуре от -40 до 20оС. Прибавляют триэтиламин (1,65 мл, 0,5 эквивалента) и хлоргидрат 2-(2-аминотиазол-4-ил)-2-метоксиимино-ацет- илхлорида (1,21 г, 0,19 эквивалента) и суспензию сохраняют при температуре от -20 до 25оС в течение 65 мин.
Затем суспензию добавляют в воду (50 мл) в течение 10 мин и перемешивают при температуре окружающей среды в течение 1 ч для растворения большинства твердых веществ. Прибавляют целит (броунмиллерит) (0,5 г), смесь фильтруют и лепешку промывают дихлорметаном (10 мл). Органический слой отделяют от богатого водного слоя, в который прибавляют концентрированную серную кислоту (5,9 мл, 0,111 моль) в течение 5 мин при температуре 20-25оС. Затем в богатый водный слой прибавляют в течение 35 мин ацетон (320 мл) с целью кристаллизации продукта. Суспензию сохраняют на 20 мин при температуре окружающей среды и затем охлаждают до 0-5оС в течение одного часа. Суспензию фильтруют, промывают ацетоном (150 мл) и твердое вещество сушат при температуре около 40оС. Выход сульфата цефепима составляет 11,54 г (84,9 стехиометрическая масса).
П р и м е р 25. Безводное ацилирование Н1 соли 7-амино-3-/(1-метил-1-пирролидино)метил/цеф-3-ем-4-карбоксилата с использованием хлоргидрата 2-(2-аминотиазол- 4-ил)-2-метоксиимино-ацетилхлорида (син-изомера). В атмосфере азота Н1 соль 7-амино-3-/(1-метил-1-пирролидинио)метил/цеф- 3-ем-4-карбоксилата (10,0 г, 0,02252 моль) перемешивают с 100 мл дихлорметана при температуре 20оС. Прибавляют триметилхлорсилан (2,35 мл, 0,82 эквивалента) и гексаметилдисилазан (3,85 мл, 1,62 эквивалента), и температуру силиловой смеси повышают до 25оС и сохраняют в течение 1,5 ч при температуре от 25 до 30оС. Силиловую смесь охлаждают до температуры -40оС и в течение 40 мин при температуре от -40 до -20оС прибавляют хлоргидрат 2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхлорида. Прибавляют триэтиламин (1,65 мл, 0,5 эквивалента) и хлоргидрат 2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхлорида (1,21 г, 0,19 эквивалента) и суспензию поддерживают при температуре от -20 до 25оС в течение 45 мин. Суспензию прибавляют в воду (45 мл) в течение 10 мин и перемешивают при температуре окружающей среды в течение одного часа до растворения большинства твердых веществ. Прибавляют броунмиллерит (0,5 г) и смесь фильтруют через броунмиллеритовую (1,0 г) набивку. Органический слой отделяют от богатого водного слоя, который дважды перемешивают с углеродом (1,0 г каждый раз) и фильтруют. Объединенную углеродную лепешку промывают раствором воды (10,5 мл), 12 н. HCl (5 мл, 2,5 эквивалента) и ацетона (20,5 мл). Затем в объединенный фильтрат прибавляют ацетон (320 мл) и промывают в течение 35 мин для кристаллизации продукта. Суспензию сохраняют 30 мин при температуре окружающей среды и затем охлаждают до температуры 0оС в течение 1 ч. Суспензию фильтруют, промывают 80 мл ацетона и твердое тело сушат при температуре около 40оС. Выход полученного моногидрата дигидрохлорида цефепима составляет 10,25 г (76,3% стехиометрическая масса).
П р и м е р 26. Хлористоводородная соль син 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты. Син 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусную кислоту (85,3 г, 424 ммоль) в дихлорметане (570 мл) измельчают под азотом в течение 15 мин в смесителе. Полученную тонкоизмельченную суспензию разбавляют дихлорметаном (100 мл) и переносят под азотом в реактор с рубашкой Buchi объемом 1 л. В реакторе создают повышенное давление с помощью азота (5 фунтов на кв. дюйм 0,352 кг/см2), смесь перемешивают при 375 об/мин и охлаждают до температуры -2оС. В головную часть реактора вводят хлористый водород (15,4 г, 424 ммоль) со скоростью 0,2 г в минуту. Температура повышается до 2оС. Смесь перемешивают еще 30 мин при температуре 0оС, фильтруют и промывают дихлорметаном (350 мл) под азотом. Твердое вещество сушат в вакууме при температуре 45оС в течение 18 ч. Указанное в заголовке соединение имеет вид порошка не совсем белого цвета (110,9 г, неоткорректированный выход 111%).
Аналитически вычислено для С6Н8N3O3SCl:
C 30,32, H 3,39, N 17,68, S 13,49, Cl 14,91.
Найдено: С 29,37, Н 3,17, N 16,34, S 12,70, Cl 16,99.
IH ЯМР (ДМСО-d6) δ: 4,05 (с, 3Н, СН3), 5,9 (с. 15 мол. остаточного СН2Сl2), 7,1 (с, IH, C-5 H). Сигналы видны при 4,18 (с, 3Н, СН3) и 7,7 (с, 1Н, с-5 Н), что соответствует приблизительно 2% анти-изомера.
П р и м е р 27. Хлористоводородная соль син 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты. Син 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусную кислоту (25 г, 124 ммоль) в ацетонитриле (125 мл) под азотом титруют, 1,39 М раствором НСl в ацетонитриле (89,2 мл, 123,9 ммоль) и поддерживают температуру на уровне 10-15оС. Смесь перемешивают в течение 30 мин при температуре 10-15оС, фильтруют и промывают ацетонитрилом (200 мл) под азотом. Твердое тело сушат в вакууме при температуре 45оС в течение 3 часов. Указанное в заголовке соединение представляет собой порошок не совсем белого цвета (29,5 г, неоткорректированный выход 97,4%).
IH ЯМР (CD3OD) δ: 2,05 (c, 13% масс. остаточный ацетонитрил),
4,1 (с, 3Н, СН3), 7,1 (с, 1Н, С-5 Н). Сигналы также видны при 4,2 (с, 3Н, СН3) и 7,8 (с, 1Н, С-5 Н), что соответствует приблизительно 0,5% анти-изомера.
П р и м е р 28. Хлоргидрат син 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхло-рида. Хлоргидрат син 2-(2-аминотиазол-4-ил)-2-метоксииминоуксусной кислоты (56,24 г, 210 ммоль), содержащий приблизительно 11 мас. остаточного ацетонитрила, в дихлорметане (450 мл) измельчают под азотом в течение 3 мин в смесителе, затем охлаждают до температуры -35оС и переносят под азотом в течение 5 мин в хорошо перемешанную суспензию реагента Вильсмайера, также при температуре -35оС. Суспензию реагента Вильсмайера, получают путем прибавления оксалилхлорида (28,2 г 221 ммоль) порционно в раствор диметилформамида (16,89 г, 231 ммоль) в дихлоpметане (330 мл) при температуре 0оС, с последующим охлаждением до температуры -35оС. Во время прибавления реакционная температура повышается до -28оС. После прибавления реакционную смесь затравливают продуктом. Еще через 2,5 ч при температуре от -28 до -35оС смесь фильтруют и фильтровальную лепешку промывают дихлорметаном (200 мл) под азотом. Азот пропускают через лепешку в течение 30 мин, затем твеpдое вещество сушат в вакууме при комнатной температуре в течение 12 ч. Указанное в заголовке соединение получают в виде порошка не совсем белого цвета (42,9 г, выход 72%).
IH ЯМР (CD3OD) δ: 4,06 (с, 3Н, СН3), 7,12 (с, 1Н, С-5 Н). Сигналы видны при 7,18, что соответствует приблизительно 5% кислой хлористоводородной соли, и при 7,80 (с, С-5 Н), что соответствует приблизительно 0,5% анти-изомера. После дериватизации диэтиламином в ацетонитриле анализ жидкостной хроматографии высокого давления показывает указанное в заголовке соединение (син-изомер в качестве диэтиламидпроизводного) со временем удерживания 9,6 мин, кислую хлористоводородную соль со временем удерживания 2,8 мин и анти-изомер (в качестве диэтиламинпроизводного) со временем удерживания 16,4 мин. Отношение син-изомер: кислая хлористоводородная соль: анти-изомер составляет 90:5:<1.
П р и м е р 29. Хлоргидрат син-2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхло- рида.
Син-2-(аминотиазол-4-ил)-2-метокси- имино-уксусную кислоту (84,7 г, 421 ммоль) в дихлорметане (570 мл) измельчают под азотом в течение 20 мин в смесителе. Полученную тонкоизмельченную суспензию разбавляют дихлорметаном (100 мл) и переносят под азотом в реактор с рубашкой Buchi объемом 1 л. В реакторе создают с помощью азота повышенное давление (5 фунтов на кв. дюйм 0,352 кг/см2) и смесь перемешивают при 375 об/мин и охлаждают до температуры -2оС. В головную часть реактора вводят хлористый водород (15,3 г, 421 ммоль) со скоростью 0,2 г в минуту. Температура повышается до 2оС. Смесь перемешивают в течение 30 мин при температуре 0оС, измельчают в течение 3 мин в смесителе, затем охлаждают до температуры -35оС и переносят под азотом в течение 5 мин в хорошо перемешанную суспензию реагента Вильсмайера, также при температуре -35оС. Суспензию реагента Вильсмайера получают путем прибавления оксалилхлорида (56,1 г, 439 ммоль) порционно в раствор диметилформамида (33,8 г, 462 ммоль) в дихлорметане (880 мл) при температуре 0оС с последующим охлаждением до температуры -35оС. Во время прибавления температура реакции возрастает до -28оС. После прибавления реакционную смесь затравливают продуктом. Еще через 2,5 ч при температуре от -28 до -35оС смесь фильтруют и фильтровальную лепешку промывают дихлорметаном (350 мл) под азотом. Азот пропускают через лепешку в течение 30 мин, после чего твердое вещество сушат в вакууме при комнатной температуре в течение 12 ч. Указанное в заголовке соединение получают в виде порошка не совсем белого цвета (95,2 г, 89% неоткорректированный выход). Аналитически вычислено для C6H7N3O2SCl2:
C 28,14, H 2,76, N 16,41, S 12,52, Cl 27,68.
Найдено: C 28,11, H 2,62, N 16,20, S 12,22, Cl 26,74.
IH ЯМР (CD3OD) δ: 4,06 (c, 3H, CH3), 7,12 Эс, 1Н, С-5, Н).
Сигналы видны также при 7,18 (С, С-5 Н), что соответствует приблизительно 4% кислой хлористоводородной соли, и при 7,80 (с, С-5 Н), что соответствует приблизительно 2% анти-изомера. После дериватизации диэтиламином в ацетонитриле анализ с помощью жидкостной хроматографии высокого давления показывает указанное в заголовке соединение (в качестве его диэтиламид-производного) со временем удерживания 9,6 мин, исходную кислоту со временем удерживания 2,8 мин и анти-изомер (в качестве его диэтиламид-производного) со временем удерживания 16,4 мин. Отношение син-изомер: исходная кислота анти-изомер составляет 90:4:2.
П р и м е р 30. Превращение моногидрата дигидрохлорида цефепима в дигидрат дигидрохлорида цефепима. Моногидрат цефепим-дигидрохлорида (300 г, степень чистоты 99,9% при жидкостной хроматографии высокого давления, коэффициент Маккини 3,8%) растворяют в деионизированной воде (1200 мл). Прибавляют 6 н. раствор хлористоводородной кислоты (132 мл, 1,5 эквивалента). Раствор фильтруют и промывают деионизированной водой (300 мл).
К отфильтрованному раствору прибавляют ацетон (1500 мл). Затем в течение 20 мин прибавляют по каплям дополнительное количество ацетона (4000 мл). Раствор сохраняют при температуре помутнения до образования тяжелых кристаллов дигидрата (игольчатые кристаллы при микроскопическом анализе; затравка по выбору при температуре помутнения). В течение 25 мин прибавляют дополнительно ацетон (8000 мл). Густую суспензию перемешивают при температуре 25оС в течение 1 ч.
Форму кристаллов подтверждают в качестве дигидратной (игольчатые кристаллы) микроскопическим анализом, сравнивая кристаллы с аутентичным образом. Суспензию фильтруют и промывают ацетоном (2х1500 мл). Лепешку сушат при температуре 40оС в вакууме в течение 15 ч. Выход дигидрата цефепим-дигидрохлорида составляет 305,10 г (98,6%) степень чистоты 99,0% при жидкостной хроматографии высокого давления, коэффициент Маккини 6,5%
П р и м е р 31. Превращение дигидрата цефепим-дигидрохлорида в моногидрат цефепим-дигидрохлорида. Дигидрат цефепим-дигидрохлорида (15,0 г, степень чистоты 99,2% при жидкостной хроматографии высокого давления, коэффициент Маккини 6,4% ) растворяют в деионизированной воде (75 мл). Прибавляют 6 н. раствор хлористоводородной кислоты (0,9 мл, 0,2 эквивалента). Раствор фильтруют через фильтр с размерами сита 0,45 мкм. К отфильтрованному раствору в течение 20 мин прибавляют по каплям ацетон (200 мл) с получением мутного раствора (необязательное внесение затравки в этот момент). Не удерживая в данной точке, прибавляют еще по каплям ацетон (400 мл) в течение 40 мин. Суспензию охлаждают в ванне со льдом при температуре 0-5оС в течение одного часа. Форму кристаллов подтверждают в качестве моногидрата микроскопическим анализом, сравнивая кристаллы с аутентичным образцом. Суспензию фильтруют и промывают ацетоном (2х60 мл). Лепешку сушат при температуре 40оС в вакууме в течение 15 ч. Выход моногидрата цефепим-дигидрохлорида составляет 13,28 г (91,8%), и структуру кристаллов подтверждают аналогичной, как описано Капланом и др. в патенте США N 4910301.
П р и м е р 32. Получение гидратов 7-/2-(2-аминотиазол-4-ил)-2-(Z)-метоксиимино-ацетамидо/-3- (1-метил-1-пирролидинио)метил/цеф-3-ем-4- карбоксилат-дигидрохлорида. В инертной атмосфере при комнатной температуре гидроиодид 7-амино-3-(1-метил-1-пирролидинио)метил/цеф-3-ем-4карбоксилата (14,67 г, 0,0345 моль) суспендируют в дихлорметане (150 мл). В суспензию прибавляют триметилхлорсилан (4,7 мл) и гексаметилдисилазан (7,7 мл) и смесь нагревают до температуры 25-30оС в течение 1,5 ч. Затем реакционную смесь охлаждают до температуры около -50оС, и в три приема в течение 35 минут прибавляют хлоргидрат син-2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхлорида (7,24 г, 0,0283 моль), тогда как температуру постепенно повышают до -30оС. Прибавляют триэтиламин (1,47 мл) и 1,78 г (0,0069 моль) хлоргидрата син-2-(2-аминотиазол-4-ил)-2-метоксиимино-ацетилхлорида, и ацилирование продолжают при температуре от -20 до -25оС приблизительно один час. По завершении реакции (как отмечает анализ жидкостной хроматографии высокого давления) смесь нагревают до температуры -5оС, после чего прибавляют 56 мл воды и 10 мл диметилацетамида. Реакционную суспензию перемешивают при температуре 25оС до растворения твердых веществ. Фазы разделяют и водную фазу окончательно фильтруют. Водную фазу обесцвечивают активированным углем (3 г), фильтруют и фильтрат разделяют на две равные порции.
Метод А
Одну порцию богатого водного фильтрата подкисляют 12 н. раствором хлористоводородной кислоты (11,7 мл, 0,14 моль), Суспензию затравливают дигидратом цефепим-дигидрохлорида (0,5 г) и суспензию нагревают при температуре 40оС в течение одного часа, после чего сохраняют при комнатной температуре в течение ночи. Смесь разбавляют ацетоном (126 мл), перемешивают при комнатной температуре в течение 0,5 ч, затем охлаждают при комнатной температуре 0-5оС в течение одного часа. Продукт собирают фильтрацией, промывают ацетоном и сушат в вакууме при температуре 45оС. Дигидратную форму дигидрохлорида указанного в заголовке соединения (игольчатые кристаллы) получают со степенью чистоты 96,3% (9,11 г, 87,6 стехиометрический массовый выход). Содержание воды при анализе Карла Фишера составляет 6,3% а анализ инфракрасной спектроскопией с Фурье-преобразованием (диффузионный коэффициент отражения с КВr) показывает пики спектральной поглощательной способности при 3574 см-1 и 3432 см-1.
Метод В.
Вторую порцию богатого водного фильтрата подкисляют 6 н. раствором хлористоводородной кислоты (15 мл, 0,09 моль), разбавляют ацетоном (280 мл) в течение 20 мин и затем охлаждают до температуры 0-5оС в течение одного часа. Суспензию фильтруют, промывают ацетоном и сушат в вакууме при температуре 45оС. Моногидратную форму дигидрохлорида (гранулированные кристаллы) указанного в заголовке соединения получают со степенью чистоты 95,5% (8,38 г, 83,1% стехиометрический массовый выход). Содержание воды при анализе Карла Фишера составляет 3,9%
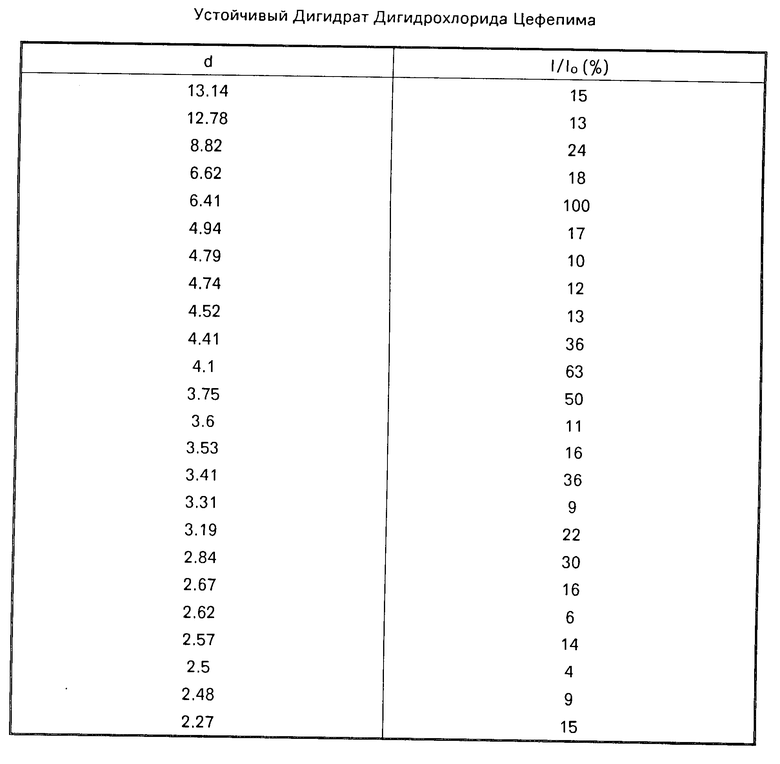
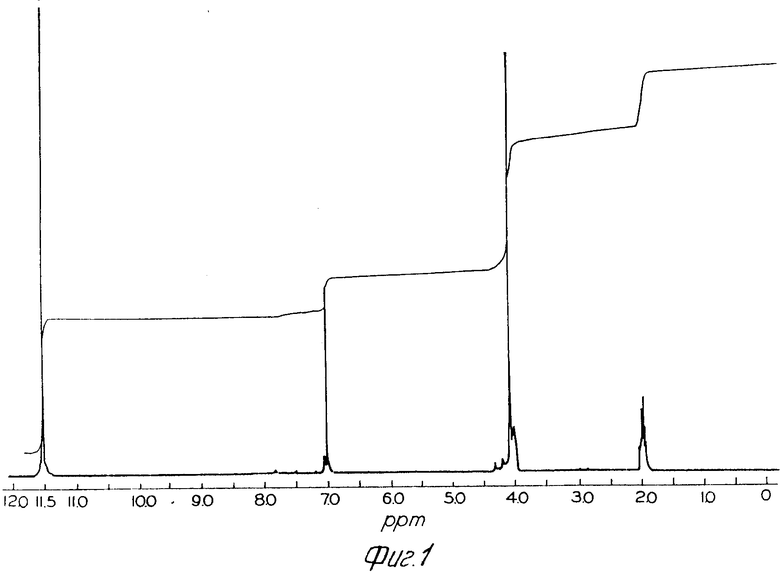
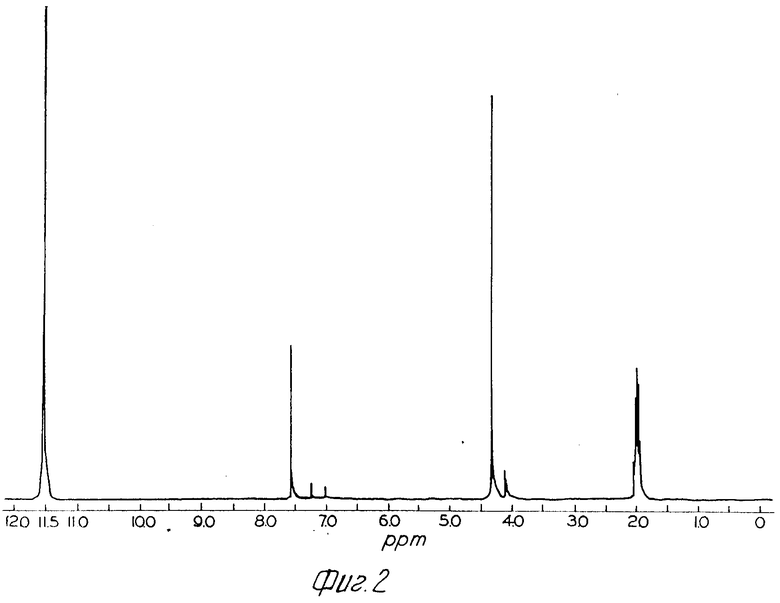


Сущность изобретения: по существу свободное от дельта (2)-изомера силилированное производное формулы 1, где R и R′ каждый представляет собой атом водорода или силильную защитную группу, при условии, что R и R′ одновременно не являются атомами водорода, Xx остаток хлористоводородной, йодистоводородной или серной кислоты, подвергают взаимодействию с син-изомером гидрохлорида 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида по существу свободным от анти-изомера в инертном органическом растворителе в присутствии основания с последующим выделением из полученной реакционной смеси целевого продукта в виде его кристаллического моно- или дигидрата дигидрохлорида. Структура соединения формулы 1:  18 з.п.ф-лы, 4 ил. 1 табл.
18 з.п.ф-лы, 4 ил. 1 табл.

где R и R1 каждый водород или силильная защитная группа при условии, что R и R1 одновременно не водород;
X- остаток хлористоводородной, йодистоводородной или серной кислоты,
подвергают взаимодействию с син-изомером гидрохлорида 2-(2-аминотиазол-4-ил)-2-метоксииминоацетилхлорида, по существу свободным от антиизомера, в инертном органическом растворителе, в присутствии основания с последующим выделением из полученной реакционной смеси целевого продукта в виде его кристаллического моно-или дигидрата дигидрохлорида.
метоксииминоацетамидо] -3-[(1- метил-1- пирролидинио) метил]-цеф-3- ем-4-карбоксилат.
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Патент США N 4754031, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
