Область техники, к которой относится изобретение
Настоящее изобретение, в общем, касается новых иммуноэффекторных соединений, их применения в фармацевтических композициях и способов их получения и применения для профилактической и/или терапевтической вакцинации. В частности, настоящее изобретение касается новых соединений, содержащих 2-дезокси-2-амино-β-D-глюкопиранозу (глюкозамин), соединенную гликозидной связью с циклической аминоалкильной группой (агликоном), и их применения в фармацевтических адъювантных системах.
Уровень техники
Гуморальный иммунитет и клеточный иммунитет представляют собой две главные ветви иммунного ответа у млекопитающих. Гуморальный иммунитет включает образование антител к чужеродным антигенам. Антитела вырабатываются В-лимфоцитами. Клеточный иммунитет включает активацию Т-лимфоцитов, которые либо действуют на инфицированные клетки, несущие чужеродные антигены, либо стимулируют другие клетки, которые действуют на инфицированные клетки. Обе ветви иммунной системы млекопитающих важны в борьбе с болезнью. Гуморальный иммунитет представляет собой основную линию защиты против бактериальных патогенов. В случае вирусных заболеваний индукция цитотоксических Т-лимфоцитов (ЦТЛ) оказывается решающей для защитного иммунитета. Таким образом, эффективная вакцина предпочтительно стимулирует обе ветви иммунной системы для защиты от болезни.
Вакцины презентируют чужеродные антигены возбудителей болезни хозяину с тем, чтобы у хозяина возникал защитный иммунный ответ. Часто антигены вакцины представлены убитыми или аттенюированными формами микробов, вызывающих заболевание. Присутствие существенных компонентов и антигенов в таких убитых или аттенюированных вакцинах привело к направлению значительных усилий на очистку компонентов вакцин, включая разработку четко определенных синтетических антигенов с использованием химических и рекомбинантных методов. Очистка и упрощение микробных вакцин привели, однако, к сопутствующему снижению активности. Низкомолекулярные синтетические антигены, хотя и лишенные потенциально вредных примесей, часто сами по себе недостаточно иммуногенны. Такие наблюдения привели исследователей к добавлению стимуляторов иммунной системы, известных как адъюванты, в вакцинные композиции для усиления активности компонентов вакцин.
Иммунные адъюванты - это соединения, которые при введении индивидууму или при тестировании in vitro усиливают иммунный ответ на антиген у субъекта, которому данный антиген введен, или усиливают определенные активности клеток иммунной системы. Был получен и протестирован целый ряд соединений, обладающих адъювантной активностью в различной степени (см., к примеру, Shimizu et al. 1985, Bulusu et al. 1992, Ikeda et al. 1993, Shimizu et al. 1994, Miyajima et al. 1996). Однако эти и другие разработанные ранее адъювантные системы зачастую проявляют токсические свойства, нестабильны и/или обладают неприемлемо низким иммуностимулирующим действием.
В настоящее время единственным адъювантом, который разрешен для применения на людях в Соединенных Штатах, являются квасцы - группа солей алюминия (к примеру, гидроокись алюминия, фосфат алюминия), в которые заключают антигены вакцин. Носители в виде частиц типа квасцов, как сообщалось, способствуют захвату, процессингу и презентации растворимых антигенов макрофагами. Однако квасцы не лишены побочных эффектов и, к сожалению, ограничиваются только гуморальным иммунитетом (антитела).
Открытие и разработка эффективных адъювантных систем необходимы для улучшения эффективности и безопасности существующих и будущих вакцин. Таким образом, существует постоянная потребность в новых и усовершенствованных адъювантных системах, особенно таких, которые бы усиливали обе эффекторные ветви иммунной системы, что будет способствовать разработке следующего поколения синтетических вакцин. Настоящее изобретение удовлетворяет этим и другим потребностям.
Раскрытие изобретения
Соединения настоящего изобретения представляют собой иммуноэффекторные молекулы, которые усиливают гуморальные и клеточные иммунные ответы на антигены вакцин. Эти соединения, в общем, можно описать как принадлежащие к классу циклических соединений АГФ, где АГФ означает аминоалкилглюкозаминидфосфаты. Термин "циклический АГФ" означает такой азациклоалкил- или (азациклоалкил)алкилглюкозаминидфосфат, в котором 2-дезокси-2-амино-β-D-глюкопираноза (глюкозамин) соединена гликозидной связью с азациклоалкильной или (азациклоалкил)алкильной группировкой (агликоном).
Соединения данного изобретения охватывают 2-дезокси-2-амино-β-D-глюкопиранозу (глюкозамин), соединенную гликозидной связью с азациклоалкильной или (азациклоалкил)алкильной группировкой (агликоном). Эти соединения фосфорилированы в положении 4 или 6 кольца глюкозамина и ацилированы алканоилокситетрадеканоиловыми остатками по азоту агликона и в положении 2 и 3 кольца глюкозамина. Соединения данного изобретения и их фармацевтически приемлемые соли в общем описываются формулой I:
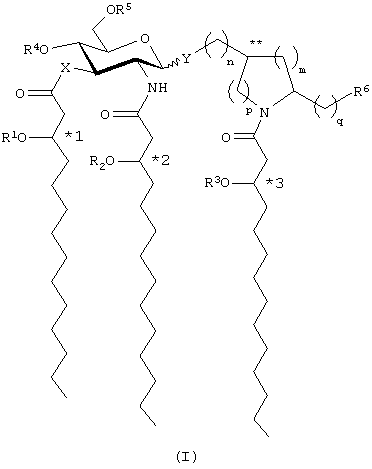
где Х означает -О-или -NH-, Y означает -О-или -S-; a R1, R2 и R3 независимо друг от друга представляют собой (С9-С14)ацильные группы, включая насыщенные, ненасыщенные и разветвленные ацильные группы; R4 - это -Н или -РО3R7R8, где R7 и R8 независимо друг от друга представлены Н или (С1-С4)алифатической группой; R5 - это -Н, -СН3 или -РО3R9R10, где R9 и R10 независимо друг от друга выбраны из -Н или (C1-С4)алифатических групп; R6 независимо выбран из числа Н, ОН, (С1-С4)оксиалифатических групп, -РО3R11R12, -ОРО3R11R12, -SO3R11, -OSO3R11, -NR11R12, -SR11, -CN, -NO2, -СНО, -CO2R11 и -CONR11R12, где R11 и R12 независимо друг от друга выбраны из Н или (С1-С4)алифатической группой; при условии, что одна из групп R4 и R5 содержит фосфор, а когда R4 представлен -РО3R7R8, то R5 не является -РО3R9R10, при этом *1-3 и ** обозначают хиральные центры; причем индексы n, m, р и q независимо друг от друга означают целое число от 0 до 6 при условии, что сумма р и m составляет от 0 до 6.
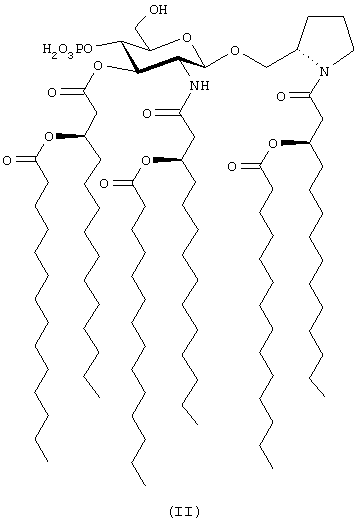
В некоторых воплощениях соединений настоящего изобретения Х и Y представлены кислородом, R4 означает РО3R7R8, R5 и R6 представлены Н, а индексы n, m, р и q - целые числа от 0 до 3. В более предпочтительном воплощении R7 и R8 представлены - Н. В еще более предпочтительном воплощении n=1, m=2, а подстрочные индексы р и q=0. В еще более предпочтительном воплощении R1, R3 и R3 представлены (С9-С13)ацильными группами, более предпочтительно (С10-С12) ванильными группами. В еще более предпочтительном воплощении *1-3 находятся в R-конфигурации, Y находится в экваториальном положении, а ** находятся в s-конфигурации. Особенно предпочтительны N-[(R)-3-тетрадеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-тетрадеканоилокситетрадеканоиламино]-3-O-[(R)-3-тетрадеканоилокситетрадеканоил]-β-D-глюкопиранозид (формула II),
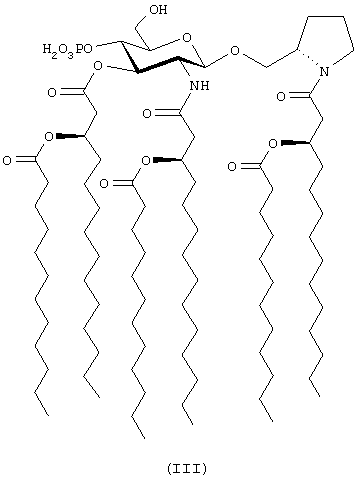
[N-(R)-3-додеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-D-фосфоно-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-О-[(R)-3-додеканоилокситетрадеканоил]-β-D-глюкопиранозид (формула III),
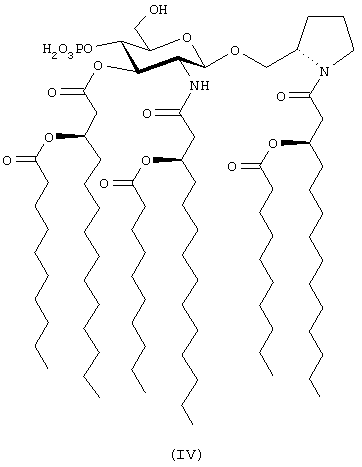
[N-(R)-3-деканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-деканоилокситетрадеканоиламино]-3-О-[(R)-3-деканоилокситетрадеканоил]-β-D-глюкопиранозид (формула IV)
и их фармацевтически приемлемые соли.
Настоящее изобретение также предусматривает фармацевтические композиции, содержащие соединения представленной выше общей формулы и конкретных формул. Фармацевтические композиции могут комбинироваться с различными антигенами и различными лекарственными составами, известными в этой области.
Соединения настоящего изобретения также полезны при различных способах индукции иммунного ответа у индивида. Такой способ заключается во введении индивиду терапевтически эффективного количества одного или нескольких соединений настоящего изобретения, предпочтительно в виде фармацевтической композиции, содержащей также и фармацевтически приемлемый носитель.
Настоящее изобретение также охватывает способы лечения млекопитающих, страдающих от или подверженных инфекции патогеном, раковому или аутоиммунному заболеванию. Такой способ заключается во введении млекопитающему терапевтически эффективного количества одного или нескольких соединений настоящего изобретения, предпочтительно в виде фармацевтической композиции, содержащей также и фармацевтически приемлемый носитель.
Кроме того, настоящее изобретение включает и способ лечения заболеваний или состояний, улучшающихся при образовании оксида азота у пациента. Способ заключается в обработке пациента эффективным количеством соединения или соединений настоящего изобретения либо эффективным количеством композиции, содержащей одно или несколько соединений настоящего изобретения и фармацевтически приемлемый носитель. В некоторых воплощениях соединения настоящего изобретения могут быть введены за 48 часов до, непосредственно перед или во время ишемии.
Осуществление изобретения
Определения
Термин "ацил" относится к тем группам, которые происходят из органической кислоты при удалении ее гидроксильной части. Соответственно, ацил может означать, к примеру, ацетил, пропионил, бутирил, деканоил и пивалоил. Например, "(С9-С14)ацил" означает ацильную группу, содержащую от 9 до 14 атомов углерода.
Термин "алифатический" сам по себе или в составе другого заместителя означает, если не указано иначе, неразветвленную или разветвленную цепь либо циклическую углеводородную группу, включая группы, содержащие как циклические элементы, так и цепи, которые могут быть полностью насыщенными либо моно- или полиненасыщенными, содержащими указанное число атомов углерода (например, C1-C4 означает от 1 до 4 атомов углерода). Примеры насыщенных углеводородных радикалов включают такие группы, как метил, этил, н-пропил, изопропил, н-бутил, t-бутил, изобутил, втор-бутил, циклопропил, циклопропилметил, метилен, этилен и н-бутилен. Ненасыщенная алкильная группа - это группа, содержащая одну или несколько двойных связей и/или тройных связей. Примеры ненасыщенных алифатических групп включают винил, 2-пропенил, кротил, 2-бутадиенил, 1-пропинил и 3-пропинил.
Термин "оксиалифатический" относится к тем группам, которые содержат алифатическую группу, соединенную с остальной частью молекулы через атом кислорода.
Каждый из вышеперечисленных терминов (например, "алкил", "ацил") может включать и замещенные, и незамещенные формы указанных групп.
Заместителями в алифатических группах может служить целый ряд групп, выбранных из числа -OR', -О,=S, -NR', -N-OR', -NR'R'', -SR', -галоген, -SiR'R''R''', -OC(O)R', -C(O)R', -CO2R', -CONR'R'', -OC(O)NR'R'', -NR''C(O)R', -NR'C(O) NR''R''', -NRC(O)2R', -NH-C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R'', -CN и NO2, число которых варьирует от 0 до (2m'+1), где m' = это суммарное число атомов углерода в таком радикале. R', R'' и R''' независимо друг от друга означают водород и незамещенные (С1-С4)алифатические группы. Из вышеизложенного обсуждения замещений должно быть понятно, что термин "алкил" может включать и такие группы, как галоалкил (к примеру, -CF3 и -СН2CF3) и им подобные.
Термин "гало" или "галоген" сам по себе или в составе другого заместителя означает, если не указано иначе, атом фтора, хлора, брома или иода. В соединениях с несколькими галогенными заместителями галогены могут быть одинаковыми или разными.
Термин "фармацевтически приемлемые соли" охватывает такие соли активных соединений, которые получают с помощью относительно нетоксичных кислот или оснований, в зависимости от конкретных заместителей, находящихся в соединениях, описанных в настоящем изобретении. В том случае, когда соединения настоящего изобретения содержат сравнительно кислые функциональные группы, их соли с основаниями могут быть получены путем добавления требуемого основания как в соответствующем инертном растворителе, так и без него. Примеры фармацевтически приемлемых солей, образованных с основаниями, включают соли натрия, калия, кальция, аммония, органических аминов, магния и им подобные. В том случае, когда соединения настоящего изобретения содержат сравнительно основные функциональные группы, их соли с кислотами могут быть получены путем добавления требуемой кислоты как в соответствующем инертном растворителе, так и без него. Примеры фармацевтически приемлемых солей, образованных с кислотами, включают соли неорганических кислот, таких как соляная, бромистоводородная, азотная, угольная, однозамещенная угольная, фосфорная, однозамещенная фосфорная, двузамещенная фосфорная, серная, однозамещенная серная, иодистоводородная, фосфористая и им подобные кислоты, а также соли относительно нетоксичных органических кислот, таких как уксусная, пропионовая, изомасляная, щавелевая, малеиновая, малоновая, бензойная, янтарная, субериновая, фумаровая, миндальная, фталевая, бензолсульфоновая, пара-толилсульфоновая, лимонная, винная, метансульфоновая и им подобные. Также охватываются соли аминокислот, такие как аргинат и им подобные, и соли таких органических кислот, как глюкуроновая или галактуроновая кислота и им подобные (см., к примеру, Berge S.M. et al., "Pharmaceutical Salts", Journal of Pharmaceutical Science, 1977, 66, 1-19). Определенные соединения настоящего изобретения содержат как основные, так и кислотные функциональные группы, что позволяет их превратить в соли, образованные либо основанием, либо кислотой.
Нейтральные формы соединений могут быть регенерированы путем обработки соли основанием или кислотой и выделения исходного соединения стандартным методом. Исходная форма соединения отличается от разнообразных солевых форм по некоторым физическим свойствам, таким как растворимость в полярных растворителях, но в остальном эти соли эквивалентны исходной форме соединения в целях настоящего изобретения.
Наряду с солевыми формами настоящее изобретение предусматривает соединения, находящиеся в виде пролекарств (предшественников). Пролекарства описанных здесь соединений - это такие соединения, которые легко подвергаются химическим изменениям в физиологических условиях с образованием соединений настоящего изобретения. Кроме того, Пролекарства могут быть превращены в соединения настоящего изобретения с помощью химических или биохимических методов в условиях ex vivo. Например, Пролекарства могут медленно превращаться в соединения настоящего изобретения, если их поместить в трансдермальный пластырь-резервуар вместе с соответствующим ферментом или химическим реагентом.
Некоторые соединения настоящего изобретения могут существовать в несольватированных формах наряду с сольватированными формами, включая гидраты. В общем, сольватированные формы эквивалентны несольватированным формам и предусматривается, что они охватываются рамками настоящего изобретения. Некоторые соединения настоящего изобретения могут существовать во множественных кристаллических или аморфных формах. В общем, все физические формы эквивалентны для применения в соответствии с настоящим изобретением и предусматривается, что они охватываются рамками настоящего изобретения.
Некоторые соединения настоящего изобретения имеют асимметричные атомы углерода (оптические центры) или двойные связи. Предусматривается, что рацематы, диастереоизомеры, геометрические изомеры и индивидуальные изомеры охватываются рамками настоящего изобретения.
Соединения настоящего изобретения также могут содержать атомные изотопы в неестественных пропорциях по одному или нескольким атомам, входящим в состав таких соединений. Например, соединения могут быть радиоактивно помечены радиоактивными изотопами, к примеру, такими как тритий (3H) иод-125 (125I) или углерод-14 (14С). Предусматривается, что все изотопные варианты соединений настоящего изобретения, радиоактивные и нерадиоактивные, охватываются рамками настоящего изобретения.
Введение
В стремлении улучшить безопасность вакцин их производители избегают убитых вакцин из целых клеток и производят рекомбинантные или субъединичные вакцины. При получении этих более безопасных вакцин устраняются посторонние компоненты бактерий и вирусов, а остаются минимальные структуры или эпитопы, считающиеся необходимыми для защитного иммунитета. Безопасность этих вакцин улучшается благодаря устранению посторонних компонентов бактерий и вирусов, которые зачастую оказываются токсичными и пирогенными. Однако именно эти компоненты, вызывающие токсичность, обеспечивают неспецифическую иммуностимуляцию, которая делает вакцины из целых клеток столь эффективными. Без дополнительной иммуностимуляции минимальные структуры и эпитопы, образующие рекомбинантные и субъединичные вакцины, зачастую обладают слабой иммуногенностью.
Молекула дисахарида, происходящего из LPS Salmonella minnesota R595, - это иммуностимулятор MPL (Corixa Corp.), обладающий иммуностимулирующими свойствами. Иммуностимулятор MPL, или монофосфориллипид А, является структурным производным липида А (или LPS) и обладает лучшим терапевтическим индексом, чем липид А (см. структуру монофосфориллипида А в U.S. Patent 4,987,237 и описание получения монофосфориллипида А в U.S. Patent Nos.4,436,727 и 4,436,728). К другим полезным иммуностимуляторам относится 3-де-O-ацилированный монофосфориллипид А (3D-MPL), который описан в U.S. Patent No.4,912,094. Это соединение можно безопасно вводить человеку в дозах по меньшей мере вплоть до 20 мкг/кг, хотя у некоторых пациентов может отмечаться повышенная температура, симптомы гриппа, учащенный пульс и умеренное повышение кровяного давления при дозах ≥10 мкг/кг. Опыты на культурах клеток и животных подтверждают, что иммуностимулятор MPL все-таки сохраняет некоторую иммуностимулирующую активность исходного LPS в том, что сохраняется пирогенность и способность к индукции воспалительных цитокинов типа TNF и IL-8, хотя и при более высоких дозах. Таким образом, потребность в эффективных адъювантах для вакцин хорошо осознана. В идеальном случае такие адъюванты должны усиливать защитный иммунный ответ, но не вызывать нежелательной токсичности и пирогенности.
В стремлении получить иммуностимулятор, обладающий низкой пирогенностью, были получены синтетические молекулы, имеющие структурное сходство с иммуностимулятором MPL. Эти новые молекулы, которые собирательно именуют аминоалкилглюкозаминидфосфатами (АГФ), состоят из ацилированной молекулы глюкозы, соединенной с ацилированной аминоалкильной группой (Johnson et al. (1999) Bioorg. Med. Chem. Lett. 9:2273-2278; PCT/WO 98/50399 и приведенные в них ссылки). Каждая молекула имеет 6 жирнокислотных остатков, что считается оптимальным числом для максимальной активности адъюванта. Замещение различных химических группировок в аминоалкильных структурах АГФ было задумано с целью оптимизации стабильности и растворимости. Таким образом, в общих чертах АГФ можно разделить на несколько семейств на основании структуры аминоалкильных групп.После первоначальной биологической оценки стало очевидно, что аминоалкильные группировки могут сильно влиять на пирогенные свойства АГФ (см. U.S. Patent Application Serial No.09/074,720 filed May 7, 1998, и U.S. Patent Nos.6,113,918 и 6,303,347). В ходе первоначального процесса скрининга синтетических адъювантных соединений были получены данные по пирогенности на кроликах. Было отмечено, что некоторые из этих соединений не вызывают повышения температуры при внутривенном введении в дозе 10 мкг/кг. В общем, именно эти соединения не индуцировали воспалительные цитокины TNF-α или IL-1β в заметной степени при анализе индукции цитокинов ex vivo на мононуклеарных клетках периферической крови человека. Далее мы сообщим об изучении адъювантных свойств одного из классов АГФ, вызывающих минимальную активность при тестировании на пирогенность у кроликов и при анализе индукции цитокинов ех vivo.
Соединения и композиции
В настоящем изобретении представлены соединения, описываемые общей формулой I:
и их фармацевтически приемлемые соли, где Х означает -О-или -NH-, Y означает -O-или -S-; a R1, R2 и R3 независимо друг от друга представляют собой (С9-С14)анильные группы, включая насыщенные, ненасыщенные и разветвленные ацильные группы; R4 - это
- Н или -РО3R7R8, где R7 и R8 независимо друг от друга представлены H или (C1-С4)алифатической группой; R5 - это -Н, -СНз или -РО3R9R10, где R9 и R10 независимо друг от друга выбраны из -Н или (С1-С4)алифатических групп; R6 независимо выбран из числа Н, ОН, (С1-С4)оксиалифатических групп, -РО3R11R12, -ОРО3R11R12, -SO3R11, -OSO3R11, -NR11R12, -SR11, -CN, -NO2, -CHO, -CO2R11 и -CONR11R12, где R11 и R12 независимо друг от друга представлены Н или (С1-С4)алифатической группой; при условии, что одна из групп R4 и R5 содержит фосфор, а когда R4 представлен -РО3R9R8, то R5 не является -РО3R9R10, при этом *1-3 и ** обозначают хиральные центры;
причем n, m, р и q независимо друг от друга являются целыми числами от 0 до 6 при условии, что сумма р и m составляет от 0 до 6.
Хотя гексопиранозид в формуле I представлен в глюко-конфигурации, другие гликозиды также входят в объем изобретения. Например, гликопиранозиды, в том числе и другие гексопиранозиды (алло-, альтро-, манно-, гуло-, идо-, галакто-, тало-), входят в объем изобретения.
В вышеприведенной общей формуле 3'-стереогенные центры, по которым происходит присоединение нормальных жирнокислотных остатков и которые обозначаются как *1, *2 и *3, находятся в R- или S-конфигурации, предпочтительно в R-конфигурации. Абсолютная стереохимия атомов углерода в циклическом агликоне, к которому присоединяются R6 и глюкозамин прямо или опосредованно (обозначается как **), может быть представлена R- или S-конфигурацией. В вышеприведенной общей формуле Y может находиться в экваториальном или аксиальном положении, предпочтительно экваториальном. Все стереоизомеры, энантиомеры, диастереомеры и их смеси рассматриваются как входящие в объем настоящего изобретения.
В предпочтительных воплощениях настоящего изобретения Х и Y означают -О-, R4 является фосфоно, R5 и R6 означают Н, а n, m, р и q - целые числа от 0 до 3, более предпочтительно от 0 до 2. Наиболее предпочтительно n=1, m=2, а р и q равны 0. В этом предпочтительном воплощении соединения данного изобретения представлены 2-пирролидинилметил-β-D-глюкозаминид-4-фосфатами общей формулы V:
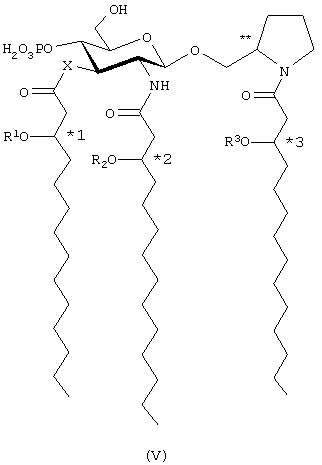
В предпочтительном воплощении настоящего изобретения 3'-стереогенные центры (*1-3), по которым происходит их присоединение, находятся в R-конфигурации, Y находится в экваториальном положении, а абсолютная стереохимия стереогенного центра пирролидина (**) представлена S- конфигурацией.
Особенно предпочтительными воплощениями являются N-[(R)-3-тетрадеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-тетрадеканоилокситетрадеканоиламино]-3-О-[(R)-3-тетрадеканоилокситетрадеканоил]-β-D-глюкопиранозид и его фармацевтически приемлемые соли формулы II,
[N-(R)-3-додеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-О-[(R)-3-додеканоилокситетрадеканоил]-β-D-глюкопиранозид и его фармацевтически приемлемые соли формулы III,
[N(R)-3-деканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-деканоилокситетрадеканоиламино]-3-O-[(R)-3-деканоилокситетрадеканоил]-β-D-глюкопиранозид и его фармацевтически приемлемые соли формулы IV.
Получение соединений
Соединения настоящего изобретения могут быть получены методами, изложенными в Johnson et al., Bioorg. Med. Chem. Lett. 9:2273-2278 (1999) и РСТ/WO 98/50399 и приведенных там ссылках. В общем, методы синтеза, описанные в вышеприведенных источниках, применимы в широком смысле к получению соединений с различными ацильными группами и заместителями. Специалисты в этой области должны понимать, что описанные в этих источниках сходящиеся методы могут быть модифицированы с использованием других ацилирующих реагентов или могут исходить из коммерчески доступных материалов, уже имеющих присоединенные соответствующие ацильные группы.
Оценка соединений
Представленные в настоящем изобретении соединения можно оценивать в различных форматах анализа, чтобы выбрать соединение с подходящим фармакофорным профилем. Например, в U.S. Patent No.6,013,640 описаны животные модели, пригодные для оценки кардиопротекторных свойств описанных соединений. В приведенных ниже примерах также представлены методы оценки пирогенности рассматриваемых соединений и другие методы для оценки провоспалительных свойств этих соединений.
Настоящее изобретение также обеспечивает фармацевтические композиции, содержащие представленные в нем соединения в смеси с одним и более фармацевтически приемлемыми носителями. Подходящие носители зависят от подлежащего лечению заболевания вместе со способом применения. Соответственно, обсуждение носителей приводится ниже в сочетании со способами применения.
Фармацевтические композиции и их применение
В одном из воплощений настоящее изобретение обеспечивает фармацевтические композиции, содержащие соединение настоящего изобретения и фармацевтически приемлемый носитель. Соединение находится в терапевтически эффективном количестве, необходимом для достижения требуемого эффекта в смысле лечения болезни или заболевания или достижения биологического проявления. Фармацевтическая композиция может действовать в качестве адъюванта при введении ее вместе с антигеном.
Композиции данного изобретения включают как композиции, составленные для непосредственного введения активных соединений пациентам без разбавления, либо вместе с вакциной или другим активным агентом, либо поодиночке, так и более концентрированные композиции соединений, которые могут быть составлены для последующего разбавления с тем, чтобы избежать перевозки и/или хранения больших количеств разбавителя (воды, физраствора, водных материалов). В общем, фармацевтические композиции данного изобретения, предназначенные для прямого или немедленного введения пациенту (то есть без разбавления), должны содержать одно или несколько соединений в терапевтически эффективном количестве. Такое количество варьирует в зависимости от конкретного терапевтического соединения или соединений и от требуемого терапевтического эффекта. Более концентрированные композиции должны содержать такие количества соединения или соединений изобретения, которые подходят для таких композиций.
При получении фармацевтических композиций фармацевтически приемлемые носители могут находиться в твердом или жидком виде. К препаратам в твердом виде относятся порошки, таблетки, пилюли, капсулы, облатки, свечи и дисперсивные гранулы. Твердый носитель может представлять собой одно или несколько веществ, которые также могут действовать как разбавители, ароматизаторы, связущие вещества, консерванты, дезинтегрирующие вещества или инкапсулирующие материалы.
В порошках носитель представляет собой тонкоизмельченное твердое вещество, которое смешано с тонкоизмельченным активным компонентом. В таблетках активный компонент смешан в соответствующей пропорции с носителем, обладающим необходимыми связывающими свойствами, и подвергнут сжатию до требуемой формы и размера.
Твердые формы композиций также могут быть получены путем распылительной сушки водных составов активных адъювантов (напр., в виде соли) или лиофилизации и измельчения вместе с наполнителями.
К подходящим носителям для твердых композиций данного изобретения относятся, к примеру, карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлоза, натриевая карбоксиметилцеллюлоза, легкоплавкий воск, масло какао и др. Термин "препарат" включает рецептуры активного соединения с инкапсулирующим материалом в качестве носителя, обеспечивающего капсулы, в которых активный компонент, с другими носителями или без них, окружен носителем, который таким образом находится в связи с ним. Аналогично включены и облатки и лепешки. Таблетки, порошки, капсулы, пилюли, облатки и лепешки могут применяться в качестве твердых дозированных форм, пригодных для перорального применения.
Для получения свечей сначала расплавляют легкоплавкий воск типа смеси глицеридов жирных кислот или масла какао и в нем гомогенно диспергируют активный компонент, например, путем перемешивания. Расплавленную гомогенную смесь затем разливают в формы соответствующего размера и охлаждают, при этом она затвердевает.
К жидким формам препаратов относятся растворы, суспензии и эмульсии, например, водные растворы или растворы в смеси вода/пропиленгликоль. Для парентеральных инъекций жидкие препараты можно приготовить в виде раствора в водном растворе полиэтиленгликоля. В определенных воплощениях фармацевтические композиции готовят в виде стабильных эмульсий (напр., эмульсий вода-в-масле или масло-в-воде) или водных составов, предпочтительно включающих одно и более поверхностно-активных веществ (детергентов). В таких эмульсиях могут использоваться подходящие детергенты, хорошо известные в этой области. В одном воплощении композиция имеет вид мицеллярной дисперсии, включающей по меньшей мере один подходящий детергент. К детергентам, пригодным для таких мицеллярных дисперсий, относятся фосфолипиды. Примеры фосфолипидов включают: диацилфосфатидилглицерины, такие как димиристоилфосфатидилглицерин (DMPG), дипальмитоилфосфатидилглицерин (DPPG) и дистеароилфосфатидилглицерин (DSPG); диацилфосфатидилхолины, такие как димиристоилфосфатидилхолин (DMPC), дипальмитоилфосфатидилхолин (DPPC) и дистеароилфосфатидилхолин (DSPC); диацилфосфатидные кислоты, такие как димиристоилфосфатидная кислота (DMPA), дипальмитоилфосфатидная кислота (DPPA) и дистеароилфосфатидная кислота (DSPA); и диацилфосфатидилэтаноламины, такие как димиристоилфосфатидилэтаноламин (DMPE), дипальмитоилфосфатидилэтаноламин (DPPE) и дистеароилфосфатидилэтаноламин (DSPE). Другие примеры включают производные этаноламина (такие как фосфатидилэтаноламин, указанный выше, или кефалин), серина (такие как фосфатидилсерин) и 3'-O-лизилглицерина (такие как 3'-O-лизилфосфатидилглицерин), но не ограничиваются ими.
Водные растворы, пригодные для перорального применения, могут быть получены путем растворения активного компонента в воде и добавления подходящих красителей, ароматизаторов, стабилизаторов и загустителей, какие потребуются. Водные суспензии, пригодные для перорального применения, могут быть получены путем диспергирования тонкоизмельченного активного компонента в воде вместе с вязким материалом, таким как природные или синтетические камеди, смолы, метилцеллюлоза, натриевая карбоксиметилцеллюлоза и другие хорошо известные суспендирующие вещества.
Также включены препараты в твердом виде, которые нужно превратить, незадолго до употребления, в жидкие формы для перорального применения. Такие препараты, наряду с активным компонентом, могут содержать красители, ароматизаторы, стабилизаторы, буферы, искусственные и натуральные подслащивающие вещества, диспергирующие вещества, загустители, солюбилизирующие вещества и др.
Фармацевтические препараты предпочтительно находятся в дозированной форме. В такой форме препарат разделен на единичные дозы, содержащие надлежащее количество активного компонента. Дозированная форма может представлять собой расфасованный препарат, при этом упаковка содержит определенное количество препарата, например, упаковка таблеток, капсул, и порошки во флаконах или ампулах. Также дозированной формой может служить сама капсула, таблетка, облатка или лепешка, либо соответствующее количество их в упакованном виде.
Итак, адъювантные системы по изобретению особенно выгодны при изготовлении и применении вакцин и других иммуностимулирующих композиций для лечения и предупреждения заболеваний, индуцируя активный иммунитет к антигенам у млекопитающих, предпочтительно у человека. Получение вакцин представляет собой хорошо развитую область и общие указания по получению и составлению вакцин легкодоступны из многочисленных источников. Одним из таких примеров является New Trends and Developments in Vaccines, edited by Voller et al., University Park Press, Baltimore, Md., USA, 1978.
В одном из иллюстративных воплощений антиген в вакцинной композиции по изобретению представлен пептидом, полипептидом или его иммуногенной частью. "Иммуногенная часть" в применении к данному изобретению - это часть белка, которая распознается (то есть специфически связывается с) рецептором антигена на поверхности В-клеток и/или Т-клеток. Такие иммуногенные части обычно включают по меньшей мере 5 аминокислотных остатков, более предпочтительно по меньшей мере 10, еще более предпочтительно по меньшей мере 20 аминокислотных остатков антигенного белка или его варианта.
Иммуногенные части антигенных полипептидов в общем можно идентифицировать хорошо известными методами, как те, что изложены в Paul, Fundamental Immunology, 3 rrd ed., 243-247 (Raven Press, 1993) и приведенных там ссылках. Такие методы включают скрининг полипептидов по их способности к реакции с антиген-специфичными антителами, антисыворотками и/или линиями или клонами Т-клеток. В применении к данному изобретению антисыворотки и антитела являются "антиген-специфичными", если они специфически связываются с антигеном (то есть реагируют с белком при иммуноанализе ELISA или другим методом и не реагируют в заметной степени с неродственными белками). Такие антисыворотки и антитела могут быть получены, как описано в данном изобретении, хорошо известными методами. Иммуногенная часть белка - это часть, реагирующая с такими антисыворотками и/или Т-клетками в не меньшей степени, чем реагирует полноразмерный полипептид (напр., при анализе методом ELISA и/или Т-клеточной реакции). Такие иммуногенные части могут реагировать при таком анализе в такой же или большей степени, чем полноразмерный полипептид. Такой скрининг в общем может проводиться методами, хорошо известными в данной области, как те, что описаны в Harlow and Lane, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, 1988. Например, полипептид может быть иммобилизован на твердой подложке и подвергнут реакции с сыворотками пациентов, что ведет к связыванию антител в этих сыворотках с иммобилизованным полипептидом. Затем можно удалить несвязавшуюся сыворотку и провести детектирование связавшихся антител, используя, к примеру, меченный 125I белок А.
Пептидные и полипептидные антигены получают любым из целого ряда хорошо известных методов. Рекомбинантные белки, кодируемые последовательностями ДНК, можно легко получить из изолированных последовательностей ДНК с помощью целого ряда экспрессионных векторов, известных в данной области. Экспрессию можно осуществлять в любых подходящих клетках-хозяевах, трансформированных или трансфицированных экспрессионным вектором, содержащим молекулу ДНК, кодирующую рекомбинантный полипептид. К подходящим клеткам хозяина относятся прокариоты, клетки дрожжей и высших эукариот, такие как клетки млекопитающих и клетки растений. Предпочтительно применяются клетки Е.coli, дрожжевые клетки или клеточные линии млекопитающих, такие как COS или СНО.
Части и другие варианты белковых антигенов, содержащие менее 100 аминокислот, обычно менее 50 аминокислот, можно получить и с помощью синтеза, используя методы, хорошо известные в этой области. Например, такие полипептиды можно синтезировать любым из коммерчески доступных твердофазных методов, таких как метод твердофазного синтеза Memfield, в котором аминокислоты последовательно добавляются к растущей аминокислотной цепочке, см. Merrifield, J. Am. Chem. Soc. 85:2149-2146, 1963. Оборудование для автоматизированного синтеза полипептидов коммерчески доступно от таких поставщиков, как Perkin Elmer/ Applied BioSystems Division (Foster City, CA), и на нем можно работать согласно инструкциям изготовителя.
В некоторых специфических воплощениях полипептидный антиген, используемый в вакцинных композициях изобретения, может представлять собой слитый белок, включающий два или несколько отдельных полипептидов. Партнер по слиянию может, к примеру, способствовать презентации Т-хелперных эпитопов (иммунологический партнер по слиянию), предпочтительно таких, которые распознаются у человека, либо он может способствовать экспрессии белка (усилитель экспрессии) с большим выходом, чем у нативного рекомбинантного белка. Некоторые предпочтительные партнеры по слиянию являются как иммунологическими, так и усиливающими экспрессию партнерами. Можно выбрать и других партнеров для слияния с тем, чтобы повысить растворимость белка или обеспечить его попадание в требуемые внутриклеточные компартменты. К другим партнерам по слиянию относятся аффинные метки, облегчающие очистку белка.
Слитые белки в общем могут быть получены стандартными методами, включая химическое конъюгирование. Предпочтительно слитый белок экспрессируется как рекомбинантный белок, что позволяет увеличить уровень продукции по сравнению с неслитым белком в экспрессионной системе. Вкратце, последовательности ДНК, кодирующие полипептидные компоненты, могут быть собраны отдельно и подвергнуты лигированию в соответствующий экспрессионный вектор. Лидируют 3'-конец последовательности ДНК, кодирующей один полипептидный компонент, вместе с пептидным линкером или без него, с 5'-концом последовательности ДНК, кодирующей второй полипептидный компонент таким образом, чтобы рамки считывания этих последовательностей были в одной фазе. Это делает возможным трансляцию единого слитого белка, сохраняющего биологическую активность обоих полипептидов, входящих в его состав.
Последовательность пептидного линкера может использоваться для того, чтобы развести первый и второй полипептидные компоненты на расстояние, обеспечивающее укладку каждого из полипептидов в свою вторичную и третичную структуру. Последовательность такого пептидного линкера встраивают в слитый белок стандартными методами, хорошо известными в данной области. Подходящие последовательности пептидного линкера можно выбрать на основании следующих факторов: (1) способность принимать гибкую развернутую конформацию, (2) неспособность принимать такую вторичную структуру, которая могла бы взаимодействовать с функциональными эпитопами на первом и втором полипептидах, (3) отсутствие гидрофобных или заряженных остатков, которые могли бы реагировать с функциональными эпитопами полипептидов. Предпочтительно пептидные линкеры содержат остатки Gly, Asn и Ser. Можно использовать и другие нейтральные аминокислоты в последовательности линкера, такие как Thr и Ala. Аминокислотные последовательности, которые можно выгодно использовать в качестве линкеров, включают те, что раскрыты в Maratea et al., Gene 40:39-46, 1985; Murphy et al., Proc. Natl. Acad. Sci. USA 83:8258-8262, 1986; U.S. Patent No.4,935,233 и U.S. Patent No.4,751,180. Линкерная последовательность, в общем, может иметь длину от 1 до 50 аминокислот. Линкерные последовательности не нужны, если первый и второй полипептиды имею;
несущественные N-концевые аминокислотные участки, которые могут использоваться для разделения функциональных доменов и предотвращения стерических помех.
В предпочтительных воплощениях иммунологический партнер по слиянию происходит из белка D, поверхностного белка грамотрицательной бактерии Haemophilus influenza В (WO 91/18926). Предпочтительно производное белка D включает примерно первую треть белка (напр., первые 100-110 аминокислот с N-конца) и может содержать липид. В некоторых предпочтительных воплощениях первые 109 остатков партнера по слиянию липопротеида D включены в N-конец для придания полипептиду дополнительных экзогенных Т-клеточных эпитопов и для увеличения уровня экспрессии в Е.coli (так что они функционируют как усилители экспрессии). Липидный хвост обеспечивает оптимальную презентацию антигена для антигенпрезентирующих клеток. Другие партнеры для слияния включают неструктурный белок NS1 (гемагглютинин) вируса гриппа. Как правило, используют первую 81 аминокислоту с N-конца, хотя можно использовать и другие фрагменты, содержащие Т-хелперные эпитопы.
В другом воплощении иммунологический партнер по слиянию представляет собой белок, известный как LYTA, или его часть (предпочтительно С-концевой участок). LYTA происходит из Streptococcus pneumoniae, которая синтезирует N-ацетил-L-аланинамидазу, известную как амидаза LYTA (кодируется геном LytA, см. Gene 43:265-292, 1986). LYTA является аутолизином, специфически разрушающим определенные связи в остове пептидогликанов. С-концевой участок белка LYTA отвечает за сродство к холину или к некоторым аналогам холина, таким как DEAE. Это свойство используется для разработки плазмид, служащих для экспрессии C-LYTA в Е.coli и полезных для экспрессии слитых белков. Описана очистка гибридных белков, содержащих фрагмент C-LYTA на N-конце (см. Biotechnology 10:795-798, 1992). В предпочтительном воплощении в слитый белок можно встроить повторяющуюся часть LYTA. Повторяющаяся часть находится в С-концевом участке, начиная с остатка 178. Особенно предпочтительная повторяющаяся часть охватывает остатки 188-305.
В следующем воплощении изобретения описанная в нем адъювантная система применяется при получении основанных на ДНК вакцинных композиций. Показательные вакцины этого типа содержат ДНК, кодирующую один или несколько полипептидных антигенов таким образом, что антиген образуется in situ. ДНК может находиться в любой из целого ряда систем доставки, известных в данной области, в том числе систем экспрессии нуклеиновых кислот, бактериальных и вирусных систем экспрессии. В этой области известны многочисленные методы доставки генов, как те, что описаны в Rolland, Crit. Rev. Therap. Drug Carrier Systems 15:143-198, 1998, и в приведенных там ссылках. Подходящие системы экспрессии нуклеиновых кислот содержат необходимые последовательности ДНК для экспрессии у пациента (такие как промотор и сигнал терминации). Бактериальные системы доставки включают введение бактерий (таких как бациллы Кальметта-Черена), экспрессирующих иммуногенную часть полипептида на поверхности клетки или секретирующих данный эпитоп.В одном из предпочтительных воплощений ДНК вводится с помощью вирусной системы экспрессии (напр., вируса осповакцины или другого поксвируса, ретровируса или аденовируса), что обычно включает использование непатогенного (дефектного), способного к репликации вируса. Показательные системы раскрыты, к примеру, в Fisher-Hoch et al., Proc. Natl. Acad. Sci. USA 86:317-321, 1989; Flexner et al., Ann. N.Y. Acad. Sci. 569:86-103, 1989; Flexner et al., Vaccine 8:17-21, 1990; U.S. Patent Nos.4,603,112, 4,769,330 и 5,017,487; WO 89/01973; U.S. Patent No.4,777,127; GB 2,200,651; EP 0,345,242; WO 91/02805; Berkner, Biotechniques 6:616-627, 1988; Rosenfeld et al., Science 252:431-434, 1991; Kolls et al., Proc. Natl. Acad. Sci. USA 91:215-219, 1994; Kass-Eisler et al., Proc. Natl. Acad. Sci. USA 90:11498-11502. 1993; Guzman et al.,Circulation 88, 2838-2848, 1993; и Guzman et al., Cir. Res. 73:1202-1207, 1993. Методы включения ДНК в такие системы экспрессии хорошо известны в данной области.
В качестве альтернативы ДНК может быть "голой", как описано, к примеру, в Ulmer et al., Science 259:1745-1749, 1993, и в обзоре Cohen, Science 259:1691-1692, 1993. Поглощение голой ДНК можно повысить путем нанесения ДНК на биодеградируемые шарики, которые эффективно транспортируются в клетки. Следует иметь в виду, что вакцина при желании может включать как полинуклеотидный, так и полипептидный компонент.
Более того, очевидно, что вакцина может содержать фармацевтически приемлемые соли требуемых полинуклеотидных, полипептидных и/или углеводных антигенов. Например, такие соли могут быть получены из фармацевтически приемлемых нетоксических оснований, в том числе органических оснований (напр., соли первичных, вторичных и третичных аминов и основных аминокислот) и неорганических оснований (напр., соли натрия, калия, лития, аммония, кальция и магния).
Адъювантная система настоящего изобретения проявляет сильные адъювантные эффекты при введении в широком диапазоне доз и широком диапазоне пропорций.
Количество антигена в каждой дозе вакцины обычно выбирают как то количество, которое вызывает защитный иммунитет без значительных неблагоприятных побочных эффектов в типичных вакцинах. Такое количество варьирует в зависимости от того, какой применяется специфический иммуноген и как он представлен. В общем случае, каждая доза должна содержать 1-1000 мкг белка, чаще всего 2-100 мкг, предпочтительно 5-50 мкг. Конечно, вводимая доза может зависеть от возраста, веса, типа сопутствующего лечения, если оно проводится, и природы вводимого антигена.
Иммуногенная активность данного количества вакцинной композиции настоящего изобретения может быть легко установлена, например, измерением повышения титра антител против антигена, используемого в вакцинной композиции (Dalsgaard К. Acta Veterinia Scandinavica 69:1-40, 1978). Другой распространенный метод включает внутрикожное введение мышам CD-1 различных доз вакцинной композиции, а потом взятие образцов сыворотки у этих мышей и тестирование на антитела против иммуногена, напр., методом ELISA. Эти и другие подобные подходы известны специалистам.
Антиген может быть получен и/или выделен практически из любого нужного источника в зависимости от инфекционного заболевания, аутоиммунного заболевания, состояния, ракового заболевания, патогена или болезни, подлежащих лечению данной вакцинной композицией. В качестве примера: антигены могут происходить из вирусных источников, таких как вирус гриппа, вирус лейкемии кошек, вирус иммунодефицита кошек, ВИЧ-1 человека, ВИЧ-2, вирус герпеса 2 типа, цитомегаловирус человека, вирус гепатита А, В, С или Е, респираторно-синцитиальный вирус, вирус папилломы человека, вирусы бешенства, кори или ящура. Показательные антигены также могут происходить из бактериальных источников, таких как сибирская язва, дифтерия, болезнь Лайма, малярия, туберкулез, лейшманиоз, Т.cruzi, Ehrlichia, Candida и др., или из простейших, таких как Babesia bovis или Plasmodium. Как правило, антигены состоят из природных или синтетических аминокислот, напр., в виде пептидов, полипептидов или белков, и могут состоять из полисахаридов либо представлять собой их смеси. Показательные антигены могут быть выделены из природных источников, синтезированы путем твердофазного синтеза или получены методами рекомбинантной ДНК.
В другом воплощении используются опухолевые антигены в вакцинных композициях настоящего изобретения для профилактики и/или лечения рака. Раковые клетки зачастую несут особые антигены на своей поверхности, такие как укороченный фактор роста эпидермиса, фолат-связывающий белок, эпителиальные муцины, меланоферрин, карциноэмбриональный антиген, специфический мембранный антиген простаты, HER2-neu, которые являются кандидатами для применения в лечебных противораковых вакцинах. Поскольку опухолевые антигены являются нормальными или родственны нормальным компонентам организма, иммунная система зачастую не может выработать эффективный иммунный ответ против таких антигенов и уничтожить раковые клетки. Для получения такого ответа можно использовать адъювантные системы данного изобретения. При этом экзогенные белки могут поступать в путь процессинга эндогенных антигенов, что ведет к выработке цитолитических или цитотоксических Т-клеток (ЦТК). Этот эффект адъювантов способствует образованию антиген-специфичных ЦТК, которые разыскивают и уничтожают те раковые клетки, которые несут на своей поверхности опухолевый антиген, использовавшийся для иммунизации. Показательные виды рака, при которых может применяться этот подход, включают рак простаты, толстой кишки, молочной железы, яичников, поджелудочной железы, мозга, головы и шеи, меланомы, лейкемии, лимфомы и др.
В следующем воплощении изобретения адъювантная система настоящего изобретения может применяться одна, то есть без одновременного введения антигена, для стимулирования иммунной системы при лечении хронических инфекционных заболеваний, особенно у пациентов с нарушениями иммунитета. Показательные примеры инфекционных заболеваний, при которых этот подход может применяться для лечения или профилактики, можно найти в U.S. Patent No.5,508,310. Стимулирование иммунной системы таким способом может быть полезным и в качестве профилактической меры для ограничения риска возникновения внутрибольничных и/или послеоперационных инфекций.
В другом воплощении находящийся в вакцинной композиции антиген не является чужеродным антигеном, а напротив, представляет собой собственный антиген, то есть вакцинная композиция направлена против аутоиммунного заболевания, такого как диабет I типа, обычные органоспецифичные аутоиммунные заболевания, неврологические заболевания, ревматические заболевания, псориаз, коллагеноз, аутоиммунные цитопении и другие аутоиммунные заболевания. К таким обычным органоспецифичным аутоиммунным заболеваниям можно отнести тиреоидит (болезни Грейвса и Хашимото), гастрит, адреналит (болезнь Аддисона), оофорит, первичный билиарный цирроз печени, миастению, гонадную недостаточность, гипопаратиреоз, облысение, синдром мальабсорбции, злокачественную анемию, гепатит, заболевания, вызванные образованием антител к рецепторам, и витилиго. К таким неврологическим заболевания можно отнести шизофрению, болезнь Альцгеймера, депрессию, гипопитуитаризм, несахарный диабет, синдром sicca и множественный склероз. К таким ревматическим заболеваниям / коллагенозам можно отнести ревматоидный артрит, системную красную волчанку или обычную волчанку, склеродермию, полимиозит, хроническое воспаление кишечника, дерматомиозит, язвенный колит, болезнь Крона, васкулит, псориатический артрит, эксфолиативный псориатический дерматит, обыкновенная пузырчатка, синдром Шегрена. К прочим аутоиммунным заболеваниям можно отнести аутоиммунный увеоретинит, гломерулонефрит, постинфарктный посткардиотомный синдром, гемосидероз легких, амилоидоз, саркоидоз, афтозный стоматит и другие иммунологические заболевания, приведенные нами и известные в соответствующих областях.
Хотя в вакцинных композициях данного изобретения могут применяться любые подходящие носители, известные в этой области, однако тип носителя обычно варьирует в зависимости от требуемого способа применения. Композиции настоящего изобретения могут быть составлены для любого подходящего способа применения, включая, к примеру, местное, пероральное, внутриносовое, внутривенное, внутричерепное, внутрибрюшинное, внутрикожное, подкожное или внутримышечное введение. При парентеральном введении типа подкожной инъекции носитель зачастую включает воду, физраствор, спирт, жир, воск или буфер. При пероральном введении часто используются вышеуказанные носители либо применяются твердые носители, такие как маннит, лактоза, крахмал, стеарат магния, сахарин натрия, тальк, целлюлоза, глюкоза, сахароза и карбонат магния. Также можно использовать биодеградируемые микросферы (напр., из полилактата-полигликолата) в качестве носителей для композиций данного изобретения. Подходящие биодеградируемые микросферы раскрыты, к примеру, в U.S. Patent Nos.4,897,268; 5,075,109; 5,928,647; 5,811,128; 5,820,883; 5,853,763; 5,814,344 и 5,942,252, содержание которых включено в настоящее изобретение путем отсылки во всей полноте. Также подходят системы носителей на основе модифицированного корового белка вируса гепатита В типа тех, что описаны в WO 99/40934 и приведенных в нем источниках, которые все включены в настоящее изобретение путем отсылки. Можно использовать и носитель, содержащий корпускулярные белковые комплексы типа тех, что описаны в U.S. Patent No.5,928,647, содержание которого включено в настоящее изобретение путем отсылки во всей полноте, причем они способны индуцировать подвергающийся рестрикции по классу I ответ цитотоксических Т-лимфоцитов в организме хозяина.
В одном из показательных воплощений вакцинные композиции наносятся на слизистые оболочки, в частности ротовой полости, предпочтительно под язык, для выработки иммунного ответа. Применение в ротовой полости может быть предпочтительным во многих случаях вместо традиционного парентерального введения вследствие легкости и удобства неинвазивных методов введения. Более того, этот подход также обеспечивает способ выработки мукозального иммунитета, чего бывает трудно добиться при традиционном парентеральном введении и что может обеспечить защиту от переносимых по воздуху патогенов и/или аллергенов. Дополнительным преимуществом применения в ротовой полости является то, что при подъязычном введении вакцины улучшается соблюдение пациентом назначения, особенно при применении в педиатрии, или когда по традиции требуются многочисленные инъекции на протяжении длительного времени, как при лечении аллергии путем десенситизации.
Вакцинные композиции также могут содержать буферы (напр., физраствор с нейтральным буфером, физраствор с фосфатным буфером или фосфатный буфер без физраствора), углеводы (напр., глюкозу, маннозу, сахарозу или декстран), маннит, белки, полипептиды или такие аминокислоты, как глицин, антиоксиданты, бактериостатики, хелаторы типа ЭДТА, глютатион, адъюванты (напр., гидроокись алюминия), растворимые вещества, поддерживающие изотоничность, гипотоничность или легкую гипертоничность относительно крови реципиента, суспендирующие вещества, загустители и/или консерванты. В качестве альтернативы композиции настоящего изобретения могут быть приготовлены в лиофилизованном виде. Композиции также могут быть заключены в липосомы при помощи хорошо известной технологии.
Итак, в одном из воплощений вакцинные композиции представляют собой водные составы, содержащие эффективное количество одного или нескольких поверхностно-активных веществ. Например, композиция может иметь вид мицеллярной дисперсии, включающей по меньшей мере один подходящий детергент, напр., фосфолипидный детергент. Показательные примеры фосфолипидов включают: диацилфосфатидилглицерины, такие как димиристоилфосфатидилглицерин (DMPG), дипальмитоилфосфатидилглицерин (DPPG) и дистеароилфосфатидилглицерин (DSPG); диацилфосфатидилхолины, такие как димиристоилфосфатидилхолин (DMPC), дипальмитоилфосфатидилхолин (DPPC) и дистеароилфосфатидилхолин (DSPC); диацилфосфатидные кислоты, такие как димиристоилфосфатидная кислота (DMPA), дипальмитоилфосфатидная кислота (DPPA) и дистеароилфосфатидная кислота (DSPA); и диацилфосфатидилэтаноламины, такие как димиристоилфосфатидилэтаноламин (DMPE), дипальмитоилфосфатидилэтаноламин (DPPE) и дистеароилфосфатидилэтаноламин (DSPE).
Как правило, молярное отношение детергент: адъювант в водной композиции составляет от 10:1 до 1:10, чаще всего от 5:1 до 1:5, однако, можно использовать любое эффективное количество детергента в водной композиции, которое наилучшим образом подходит для конкретных целей.
В другом воплощении композиция представляет собой эмульсию, к примеру эмульсию вода-в-масле или масло-в-воде. Такие эмульсии в общем хорошо известны в данной области.
Адъювантная система настоящего изобретения может применяться как единственная адъювантная система или, альтернативно, может применяться вместе с другими адъювантами или иммуноэффекторами. Например, такие адъюванты могут включать масляные адъюванты (к примеру, полный и неполный адъювант Фрейнда), липосомы, минеральные соли (к примеру, AlK(SO4)2, AlNa(SO4)2, AlNH4(SO4)2, кремнезем, квасцы, Al(ОН)3, Са3(PO4)2, каолин и углерод), полинуклеотиды (к примеру, кислоты полиIC и полиAU), полимеры (к примеру, неионные блоксополимеры, полифосфазены, цианоакрилаты, сополимеры DL-лактида и гликозида), среди прочего, и некоторые природные вещества (к примеру, липид А и его производные, воск D из Mycobacterium tuberculosis, а также вещества, обнаруженные в Corynebacterium parvum, Bordetella pertussis и у представителей рода Brucella), бычий сывороточный альбумин, дифтерийный токсоид, столбнячный токсоид, эдестин, гемоцианин моллюска морское блюдечко, токсин А псевдомонад, холерогеноид, холерный токсин, коклюшный токсин, вирусные белки и такие белки эукариот как интерфероны, интерлейкины или фактор некроза опухолей. Такие белки могут быть получены из природных или рекомбинантных источников в соответствии с методами, хорошо известными в этой области. При получении из рекомбинантных источников адъювант может содержать фрагмент белка, включающий как минимум иммуностимулирующую часть молекулы. Другие известные иммуностимулирующие макромолекулы, которые могут использоваться в практике изобретения, включают (не ограничиваясь этим) полисахариды, тРНК, неметаболизируемые синтетические полимеры, такие как поливиниламин, полиметакриловая кислота, поливинилпирролидон, смешанные поликонденсаты (сравнительно высокого молекулярного веса) 4,4'-диаминодифенилметан-3,3'-дикарбоновой кислоты и 4-нитро-2-аминобензойной кислоты (см. Sela M., Science 166:1365-1374, 1969) или гликолипиды, липиды или углеводы.
В одном из воплощений адъювантная система предпочтительно предназначается для выработки иммунного ответа преимущественно типа Тh1. Высокие уровни цитокинов типа Тh1 (напр., IFN-γ, TNFα, IL-2 и IL-12) способствуют выработке клеточных иммунных ответов на введенный антиген. Напротив, высокие уровни цитокинов типа Th2 (напр., IL-4, IL-5, IL-6 и IL-10) способствуют выработке гуморальных иммунных ответов. После применения вакцины в соответствии с изобретением у пациента происходит иммунный ответ, включающий ответы типа Тh1 и Th2. В предпочтительном воплощении, в котором ответ представлен преимущественно типом Тh1, уровень цитокинов типа Тh1 будет повышаться в большей степени, чем уровень цитокинов типа Th2. Уровни этих цитокинов можно легко определить стандартными методами. См. обзор по семействам цитокинов: Mosmann and Coffman, Ann. Rev. Immunol. 7:145-173, 1989.
Например, к дополнительным адъювантам для выработки ответа преимущественно типа Тh1 относятся, к примеру, комбинации из монофосфориллипида А типа 3-O-деацилированного монофосфориллипида A (3D-MPL) и соли алюминия. Адъюванты MPL доступны от Corixa Corporation (Seattle, WA; см. U.S. Patent Nos.4,436,727; 4,877,611; 4,866,034 и 4,912,094). CpG-содержашие олигонуклеотиды (в которых динуклеотид CpG не метилирован) также вызывают ответы преимущественно типа Тh1. Такие олигонуклеотиды хорошо известны и описаны, к примеру, в WO 96/02555, WO 99/33488 и U.S. Patent Nos.6,008,200 и 5,856,462. Иммуностимулирующие последовательности ДНК также описаны, к примеру, Sato et al., Science 273:352, 1996. Другие показательные адъюванты, которые можно включать в вакцинные композиции, включают Montanide ISA 720 (Seppic, Франция), SAF (Chiron, Калифорния, США), ISCOMS (CSL), MF-59 (Chiron), адъювант Detox™ (Corixa, Hamilton, MT).
Описанные нами композиции могут применяться в составе рецептур с замедленным высвобождением (то есть таких рецептур, как капсулы, губки или гели, к примеру, состоящие из полисахаридов), которые обеспечивают медленное высвобождение вещества после введения. Такие рецептуры, в общем, можно получить при помощи хорошо известной технологии (к примеру, см. Coombes et al., Vaccine 14:1429-1438, 1996) и применять, к примеру, путем перорального, ректального введения или имплантации под кожу или в другое место. Составы с замедленным высвобождением могут содержать полипептид, полинуклеотид или антитела, диспергированные в матриксе носителя и/или заключенные в резервуар, окруженный мембраной, контролирующей скорость высвобождения. Носители для таких рецептур являются биосовместимыми и также могут быть биодеградируемыми; предпочтительно рецептура обеспечивает относительно постоянный уровень высвобождения активного компонента. К таким носителям относятся микрочастицы из сополимеров лактида и гликолида, полиакрилата, латекса, крахмала, целлюлозы, декстрана и др. Другие носители с замедленным высвобождением включают надмолекулярные биовекторы, состоящие из внутреннего нежидкого гидрофильного слоя (напр., поперечносшитого полисахарида или олигосахарида) и, необязательно, наружного слоя, содержащего амфифильное соединение, например, фосфолипид (см. U.S. Patent No.5,151,254 и заявки РСТ WO 94/20078, WO 94/23701 и WO 96/06638). Количество активного соединения, содержащегося в составе с замедленным высвобождением, варьирует в зависимости от места имплантирования, скорости и ожидаемой продолжительности высвобождения и природы заболевания, подлежащего лечению или профилактике.
Можно использовать любые известные средства доставки в фармацевтических композициях и вакцинах для облегчения выработки антиген-специфичного иммунного ответа, направленного на клетки. Средства доставки включают антиген-презентирующие клетки (АПК), такие как дендритные клетки, макрофаги, В-клетки, моноциты и другие клетки, которые могут служить эффективными АПК. Такие клетки, хотя и необязательно, могут подвергаться генетической модификации для повышения способности к презентации антигена, усиления активации и/или поддержания Т-клеточного ответа, действия на мишень per se и/или иммунологической совместимости с получателем (то есть совпадения по гаплотипу HLA). В общем, АПК можно выделить из целого ряда биологических жидких сред и органов, включая опухоли и окружающие их ткани, и они могут представлять собой аутологические, аллогенные, сингенные или ксеногенные клетки.
В некоторых предпочтительных воплощениях настоящего изобретения в качестве антиген-презентирующих клеток применяются дендритные клетки или их предшественники. Дендритные клетки являются очень сильными АПК (Banchereau and Steinman, Nature 392:245-251, 1998) и они оказались эффективными в качестве физиологических адъювантов для выработки профилактического или лечебного иммунитета против рака (см. Timmerman and Levy, Ann. Rev. Med. 50:507-529, 1999). В общем, дендритные клетки можно идентифицировать по их типичной форме (звездчатые in situ, с заметными цитоплазматическими отростками-дендритами, различимыми in vitro), их способности к захвату, процессингу и презентации антигенов с высокой эффективностью и их способности к активации "наивных" Т-клеток. Конечно, дендритные клетки можно модифицировать таким образом, что они будут экспрессировать специфические поверхностные рецепторы или лиганды, которые обычно не встречаются на дендритных клетках in vivo или ex vivo, и такие модифицированные клетки предусматриваются настоящим изобретением. В качестве альтернативы дендритным клеткам в вакцине можно использовать секретируемые везикулы (экзосомы) из нагруженных антигеном дендритных клеток (см. Zitvogel et al., Nature Med. 4:594-600, 1998).
Дендритные клетки и их предшественники могут быть получены из периферической крови, костного мозга, клеток, инфильтрирующих опухоли или клеток, инфильтрующих окружающие опухоль ткани, лимфатических узлов, селезенки, кожи, пуповинной крови или любой другой подходящей ткани или жидкой среды. Например, дендритные клетки можно получить путем дифференцировки ex vivo, добавляя набор цитокинов типа GM-CSF, IL-4, IL-13 и/или TNFα в культуры моноцитов, полученных из периферической крови. С другой стороны, можно подвергнуть дифференцировке в дендритные клетки СD34-положительные клетки, полученные из периферической крови, пуповинной крови или костного мозга, добавляя в культуральную среду различные комбинации из GM-CSF, IL-3, TNFα, лиганда CD40, LPS, лиганда flt3 и/или других соединений, вызывающих дифференцировку, созревание и пролиферацию дендритных клеток.
Дендритные клетки для удобства разделяют на "незрелые" и "зрелые" клетки, что позволяет простым способом отличать два хорошо изученных фенотипа. Однако не следует полагать, будто эта номенклатура исключает все возможные промежуточные стадии дифференцировки. Незрелые дендритные клетки характеризуют как АПК с высокой способностью к захвату и процессингу антигенов, что коррелирует с высоким уровнем экспрессии рецептора Fcγ и маннозного рецептора. Зрелый фенотип обычно характеризуется снижением экспрессии этих маркеров и высоким уровнем экспрессии ответственных за активацию Т-клеток поверхностных молекул, таких как МНС класса I и класса II, молекулы адгезии (напр., CD54 и CD11) и костимуляторные молекулы (напр., CD40, CD80, CD86 и 4-1 ВВ).
Обычно АПК можно трансфицировать полинуклеотидом, кодирующим полипептид антигена (или часть его или другой вариант) таким образом, что полипептид антигена или его иммуногенная часть будет экспрессироваться на поверхности клетки. Такую трансфекцию можно проводить ех vivo, а затем использовать композицию или вакцину, содержащую трансфицированные клетки, и описанные нами адъюванты в лечебных целях. В качестве альтернативы пациенту можно ввести средство доставки генов, направленное на дендритные или другие антиген-презентирующие клетки, что приведет к трансфекции in vivo. Трансфекция дендритных клеток in vivo и ех vivo, к примеру, обычно проводится любыми методами, известными в этой области, типа тех, что описаны в WO 97/24447, или методом "генной пушки", описанным в Mahvi et al., Immunology and Cell Biology 75:456-460, 1997. Нагрузка антигеном дендритных клеток может осуществляться путем инкубации дендритных клеток или их предшественников с полипептидом, ДНК ("голой" или находящейся в плазмидном векторе) или РНК антигена либо с экспрессирующими антиген рекомбинантными бактериями или вирусами (напр., векторами на основе вируса осповакцины, птичьей оспы, аденовируса или лентивируса). Перед нагрузкой полипептид может быть ковалентно конъюгирован с иммунологическим партнером, обеспечивающим содействие Т-клеток (напр., с молекулой носителя). С другой стороны, дендритные клетки можно проинкубировать с неконъюгированным иммунологическим партнером, отдельно или в присутствии полипептида.
Лечение заболеваний, связанных с оксидом азота
Один из аспектов настоящего изобретения предусматривает способы лечения заболеваний или состояний, опосредованных оксидом азота, в частности ишемии и реперфузионных повреждений. Эти способы включают введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения по настоящему изобретению. Считается, что индукторы транскрипции гена iNOS и синтеза белка iNOS способствуют воспалению и поэтому в некоторой степени "токсичны" или плохо переносятся животными и человеком. Эндотоксин (LPS) и воспалительные цитокины типа IL-1, TNF-α и IFN-γ известны как индукторы iNOS. Все они по сути токсичны и способны вызывать системные воспалительные реакции, синдром дыхательной недостаточности у взрослых, множественную недостаточность органов и сердечно-сосудистую недостаточность при введении животным.
Исследования защитного действия иммуностимулятора MPL на сердце показали, что индукция синтаз оксида азота (iNOS) имеет важное значение в отсроченном кардиопротекторном действии этого соединения. Кроме того, сигнальная система оксида азота (NO), предположительно через конститутивные пулы NOS, играет важную роль в остром кардиопротекторном действии этого соединения. Ввиду остаточной эндотоксиноподобной активности иммуностимулятора MPL не удивительно, что это соединение может оказаться способным к индукции сигнальной системы оксида азота. Более того, предполагается, что сигнальная система оксида азота является тем путем, через который ишемическое прекопдиционирование оказывает защитное действие на сердце. Это наблюдение в сочетании с тем фактом, что доноры оксида азота оказывают защитное действие на сердце, служит дополнительным подтверждением того, что путь NOS/NO опосредует кардиопротекторное действие иммуностимулятора MPL.
Соединения настоящего изобретения применимы в способах лечения заболеваний или состояний, модулируемых или улучшаемых оксидом азота, в частности ишемии и реперфузионных повреждений (см. описание кардиопротекторных свойств аминоалкилглюкозаминидфосфатов и методов анализа кардиопротекторных свойств в патентной заявке США №09/808669 от 14 марта 2001 г.).
ПРИМЕРЫ
Нижеследующие примеры приводятся для иллюстрации, но не для ограничения заявленного изобретения.
Пример 1. Получение триэтиламмониевой соли N-[(R)-3-тетрадеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-тетрадеканоилокситетрадеканоиламино]-3-O-[(R)-3-тетрадеканоилокситетрадеканоил]-β-D-глюкопиранозида (триэтиламмониевой соли соединения формулы II)
(1а) В раствор 2-дезокси-4-O-дифенилфосфопо-3-O-[(R)-3-тетрадеканоилокситетрадеканоил]-6-O-(2,2,2-трихлор-1,1-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозилбромида (1,05 г, 0,81 ммоль) в безводном 1,2-дихлорэтане (10 мл) добавляли молекулярное сито на 4 (0,5 г), безводный CaSO4 (2,2 г, 16 ммоль) и N-[(R)-3-тетрадеканоилокситетрадеканоил]-(S)-2-пирролидинметанол (0,40 г, 0,75 ммоль). Эту смесь перемешивали 1 час при комнатной температуре, обрабатывали Hg(CN)2 (1,02 г, 4,05 ммоль) и кипятили с обратным холодильником 16 часов в темноте. Реакционную смесь охлаждали, разбавляли CH2Cl2 и фильтровали. Фильтрат промывали 1N водным раствором KI, сушили с помощью Na2SO4 и концентрировали. После флэш-хроматографии на силикагеле (градиентная элюция, 15→20% EtOAc/гексан) получили 0,605 г (43%) N-[(R)-3-тетрадеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-О-дифенилфосфоно-3-О-[(R)-3-тетрадеканоилокситетрадеканоил]-6-O-(2,2,2-трихлор-1,1-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозида в виде аморфного вещества.
(1b) Раствор полученного по предыдущему п.1а соединения (0,50 г, 0,29 ммоль) в АсОН (10 мл) при 60°С обрабатывали цинковой пылью (0,98 г, 15 ммоль) тремя равными порциями на протяжении 1 часа. Реакционную смесь охлаждали, обрабатывали ультразвуком, фильтровали через слой целита и концентрировали. Полученный осадок подвергали разделению между CH2Cl2 и насыщенным водным раствором NaHCO3 и разделяли слои. Органический слой высушивали с помощью Na2SO4 и концентрировали. Раствор полученного неочищенного аминоспирта и (R)-3-тетрадеканоилокситетрадекановой кислоты (0,155 г, 0,34 ммоль) в СН3Cl2 перемешивали вместе с порошком молекулярного сита на 4 А (0,25 г) в течение 0,5 ч, а затем обрабатывали 2-этокси-1-этоксикарбонил-1,2-дигидрохинолином (0,11 г, 0,44 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 8 ч, фильтровали через целит и концентрировали. После флэш-хроматографии на силикагеле с использованием 50% EtOAc/гексана получили 0,355 г (68%) N-[(R)-3-тетрадеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-дифенилфосфоно-2-[(R)-3-тетрадеканоилокситетрадеканоиламино]-3-О-[(R)-3-тетрадеканоилокситетрадеканоил]-β-D-глюкопиранозида в виде бесцветного сиропа.
(1 с) Раствор полученного по предыдущему п.1b соединения (0,300 г, 0,166 ммоль) в смеси из АсОН (1 мл) и тетрагидрофурана (9 мл) подвергали гидрогенизации в присутствии PtO2 (0,15 г) при комнатной температуре и давлении 70 фунтов на квадратный дюйм в течение 18 ч. Реакционную смесь разбавляли смесью 2:1 CHCl3-MeOH (50 мл) и кратко обрабатывали ультразвуком. Собирали катализатор, промывали смесью 2:1 CHCl3-МеОН, и объединенные фильтрат и промывные воды концентрировали. После флэш-хроматографии на силикагеле с использованием CHCl3-MeOH-H2O-Et3N (90:10:0,5:0,5) получили частично очищенный продукт, который растворяли в ледяной смеси 2:1 CHCl3-МеОН (30 мл) и промывали ледяным раствором 0,1 N HCl (12 мл). Органическую фазу фильтровали и лиофилизировали из 2% раствора Et3N (5 мл, свободный от пирогенов), получая 0,228 г (79%) триэтиламмониевой соли N-[(R)-3-тетрадеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-тетрадеканоилокситетрадеканоиламино]-3-O-[(R)-3-тетрадеканоилокситетрадеканоил]-β-D-глюкопиранозида в виде бесцветного порошка. Т.пл. 67-70°С, ИК-спектр (пленка): 3306, 2955, 2923, 2853, 1736, 1732, 1644, 1548, 1466, 1378, 1245, 1177, 1110, 1053, 844 см-1; 1Н-ЯМР (CDCl3-CD3OD): δ 0,88 (m, 18H), 1,0-1,205 (mH), 2,20-2,70 (m, 12Н), 3,06 (q, 6H, J=7,2 Гц), 3,3-3,25 (mH), 4,52 (d, 1H, J=8 Гц), 5,05-5,28 (m, 4Н), 7,44 (d, 1Н, J=9 Гц); 13С-ЯМР (CDCl3): δ 173,3, 173,0, 170,3, 169,6, 168,6, 101,8, 100,4, 75,8, 72,5, 72,4, 70,9, 70,8, 70,3, 70,2, 69,9, 69,3, 67,9, 66,6, 56,5, 56,3, 54,5, 47,4, 45,8, 44,6, 41,4, 41,0, 39,7, 39,2, 39,0, 34,5, 34,3, 34,1, 32,0, 29,7, 29,4, 28,1, 27,3, 25,7, 25,3, 25,2, 25,1, 24,0, 22,7, 21,6, 14,1, 8,6.
Анализ: теоретически для C101H194N3O17P·H2O:С=68,47, Н=11,15, N=2,37, Р=1,75; фактически: С=68,79, Н=11,00, N=2,24, Р=1,97.
Пример 2. Получение триэтиламмониевой соли N-[(R)-3-додеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-O-[(R)-3-додеканоилокситетрадеканоил]-β-D-глюкопиранозида (триэтиламмониевой соли соединения III)
(2а) В раствор 2-дезокси-4-O-дифенилфосфоно-3-О-[(R)-3-додеканоилокситетрадеканоил]-6-O-(2,2,2-трихлор-1,1-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтоксикарбониламино)-α-D-глюкопиранозилбромида (1,60 г, 1,27 ммоль) в безводном 1,2-дихлорэтане (3,2 мл) добавляли молекулярное сито на 4 А (0,6 г), безводный CaSO4 (1,0 г, 7,3 ммоль) и N-[(R)-3-додеканоилокситетрадеканоил]-(S)-2-пирролидинметанол (0,58 г, 1,14 ммоль). Эту смесь перемешивали 1 час при комнатной гемпературе, обрабатывали Hg(CN)2 (0,58 г, 2,3 ммоль) и кипятили с обратным холодильником 6 часов в темноте. Реакционную смесь охлаждали, разбавляли СН2Cl2 и фильтровали через слой целита. Фильтрат промывали 1 N годным раствором KI, сушили с помощью Na2SO4 и концентрировали. После флэш-хроматографии на силикагеле (градиентная элюция, 25→35% EtOAc/гексан) получили 1,72 г (82%) N-[(R)-3-додеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-дифенилфосфоно-3-О-[(R)-3-додеканоилокситетрадеканоил]-6-O-(2,2,2-трихлор-1,1-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозида в виде бесцветного масла.
(2b) Раствор полученного по предыдущему п.2а соединения (1,58 г, 0,806 ммоль) в АсОН (40 мл) при 60°С обрабатывали цинковой пылью (2,6 г, 40 ммоль) тремя равными порциями на протяжении 1 часа. Реакционную смесь охлаждали, обрабатывали ультразвуком, фильтровали через слой целита и концентрировали. Полученный осадок подвергали распределению между EtOAc и насыщенным водным раствором NaHCO3 и разделяли слои. Органический слой промывали насыщенным раствором соли, подсушивали с помощью Na2SO4 и концентрировали, получая 1,3 г белого вещества. Раствор полученного неочищенного аминоспирта и (R)-3-додеканоилокситетрадекановой кислоты (0,45 г, 1,05 ммоль) в CH2Cl2 (20 мл) обрабатывали 2-этокси-1-этоксикарбонил-1,2-дигидрохинолином (0,30 г, 1,21 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 18 ч и концентрировали. После флэш-хроматографии на силикагеле с использованием 40→50% EtOAc/гексана получили 0,89 г (56%) N-[(R)-3-додеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-дифенилфосфоно-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-O-[(R)-3-додеканоилокситетрадеканоил]-β-D-глюкопиранозида в виде белой пены.
(2 с) Раствор полученного по предыдущему п.2b соединения (0,75 г, 0,44 ммоль) в смеси из АсОН (4,5 мл) и тетрагидрофурана (45 мл) подвергали гидрогенизации в присутствии PtO2 (0,45 г) при комнатной температуре и давлении 70 фунтов на квадратный дюйм в течение 18 ч. Реакционную смесь разбавляли смесью 2:1 CHCl3-МеОН (35 мл) и кратко обрабатывали ультразвуком. Собирали катализатор, промывали смесью 2:1 CHCl3-МеОН, и объединенные фильтрат и промывные воды концентрировали. После флэш-хроматографии на силикагеле с использованием CHCl3-MeOH-H2O-Et3N (градиентная элюция: 96:4:0,3:0,3→90:10:0,5:0,5) получили частично очищенный продукт (0,51 г), который растворяли в ледяной смеси 2:1 CHCl3-МеОН (50 мл) и промывали ледяным раствором 0,1 N HCl (20 мл). Органическую фазу фильтровали и концентрировали. Полученный белый воск лиофилизировали из 2% раствора Et3N (70 мл, свободный от плрогенов), получая 0,54 г (78%) триэтиламмониевой соли N-[(R)-3-додеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-додеканоилокситет)адеканоиламино]-3-O-[(R)-3-додеканоилокситетрадеканоил]-β-D-глюкопиранозида в виде белого порошка. Т.пл. 146-151°С, ИК-спектр (пленка): 3292, 3100, 2958, 2922, 2852, 1739, 1731, 1659, 1651, 1644, 1562, 1555, 1468, 1455, 1433, 1377, 1339, 1310, 1253, 1238, 1183, 1160, 1107, 1080, 1047, 960, 856, 722 см-1; 1Н-ЯМР (CDCl3-CD3OD): δ 0,88 (m, 18 Н), 1,0-2,10 (mH), 2,20-2,75 (m, 12Н), 3,04 (q, 6H, J=7,2 Гц), 3,3-4,3 (mH), 4,45 (d, 1H, J=8,5 Гц), 5,0-5,28 (m, 4Н); 13С-ЯМР (CDCl3): δ 173,9, 173,4, 173,2, 170,6, 170,1, 169,2, 101,4, 75,5, 74,0, 70,8, 70,7, 70,2, 68,5, 60,5, 56,6, 53,6, 47,4, 45,6, 40,9, 39,6, 38,8, 34,5, 34,3, 34,2, 34,1, 31,9, 29,7, 29,6, 29,5, 29,4, 29,4, 29,3, 29,2, 27,3, 25,2, 25,0, 23,6, 22,7, 21,6, 14,0, 8.3.
Масс-спектроскопия MALDI: теоретически для [M+Na]+ 1590, 1900, фактически 1590, 1866.
Анализ: теоретически для С95Н182O17Р·3H2O:С=66,20, Н=10,99, N=2,44; фактически: С=66,36, Н=10,69, N=2,15.
Пример 3. Получение триэтиламмониевой соли N-[(R)-3-деканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-деканоилокситетрадеканоиламино]-3-O-[(R)-3-деканоилокситетрадеканоил]-β-D-глюкопиранозида (триэтиламмониевой соли соединения IV)
(3а) В раствор 2-дезокси-4-O-дифенилфосфоно-3-O-[(R)-3-деканоилокситетрадеканоил]-6-O-(2,2,2-трихлор-1,1-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтоксикарбониламино)-α-D-глюкопиранозилбромида (1,70 г, 1,38 ммоль) в безводном 1,2-дихлорэтане (3,5 мл) добавляли молекулярное сито на 4 (0,6 г), безводный CaSO4 (1,2 г, 8,8 ммоль) и N-[(R)-3-деканоилокситетрадеканоил]-(8)-2-пирролидинметанол (0,60 г, 1,24 ммоль). Эту смесь перемешивали 1 час при комнатной температуре, обрабатывали Hg(CN)2 (0,63 г, 2,5 ммоль) и кипятили с обратным холодильником 6 часов в темноте. Реакционную смесь охлаждали, разбавляли СН2Cl2 и фильтровали через слой целита. Фильтрат промывали 1 N водным раствором KI, сушили с помощью Na2SO4 и концентрировали. После флэш-хроматографии на силикагеле (градиентная элюция, 25→40% EtOAc/гексан) получили 1,82 г (80%) N-[(R)-3-деканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-дифенилфосфоно-3-O-[(R)-3-деканоилокситетрадеканоил]-6-О-(2,2,2-трихлор-1,1-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозида в виде бесцветного масла.
(3b) Раствор полученного по предыдущему п.3а соединения (1,67 г, 1,02 ммоль) в АсОН (50 мл) при 60°С обрабатывали цинковой пылью (3,33 г, 51 ммоль) тремя равными порциями на протяжении 1 часа. Реакционную смесь охлаждали, обрабатывали ультразвуком, фильтровали через слой целита и концентрировали. Полученный осадок подвергали распределению между EtOAc и насыщенным водным раствором NaHCO3 и разделяли слои. Органический слой промывали насыщенным раствором соли, подсушивали с помощью Na2SO4 и концентрировали, получая 1,25 г белого вещества. Раствор полученного неочищенного аминоспирта и (R)-3-деканоилокситетрадекановой кислоты (0,53 г, 1,33 ммоль) в СН2Cl2 (20 мл) обрабатывали 2-этокси-1-этоксикарбонил-1,2-дигидрохинолином (0,38 г, 1,53 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 18 ч и концентрировали. После флэш-хроматографии на силикагеле с использованием 40→50% EtOAc/гексана получили 1,23 г (74%) N-[(R)-3-деканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-дифенилфосфоно-2-[(R)-3-деканоилокситетрадеканоиламино]-3-O-[(R)-3-деканоилокситетрадеканоил]-β-D-глюкопиранозида в виде белой пены.
(3 с) Раствор полученного по предыдущему п.3b соединения (1,07 г, 0,654 ммоль) в смеси из АсОН (6,5 мл) и тетрагидрофурана (65 мл) подвергали гидрогенизации в присутствии PtO2 (0,66 г) при комнатной температуре и давлении 70 фунтов на квадратный дюйм в течение 18 ч. Реакционную смесь разбавляли смесью 2:1 CHCl3-МеОН (50 мл) и кратко обрабатывали ультразвуком. Собирали катализатор, промывали смесью 2:1 CHCl3-МеОН, и объединенные фильтрат и промывные воды концентрировали. Получали воскообразное твердое вещество, которое лиофилизировали из 2% раствора триэтиламина, получая ˜1 г неочищенной триэтиламиновой соли в виде белого порошка. После флэш-хроматографии на силикагеле с использованием CHCl3-MeOH-H2O-Et3N (градиентная элюция: 96:4:0,3:0,3→88:12:1:0,6) получили частично очищенный продукт (0,84 г), который растворяли в ледяной смеси 2:1 CHCl3-МеОН (168 мл) и промывали ледяным раствором 0,1 N HCl (67 мл). Органическую фазу фильтровали и концентрировали. Полученный белый воск лиофилизировали из 2% водного раствора Et3N (70 мл, свободный от пирогенов), получая 0,79 г (79%) триэтиламмониевой соли N-[(R)-3-деканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-деканоилокситетрадеканоиламино]-3-О-[(R)-3-деканоилокситетрадеканоил]-β-D-глюкопиранозида в виде белого порошка. Т.пл. 121-122°С, ИК-спектр (пленка): 3287, 3093, 2961, 2913, 2850, 1745, 1738, 1732, 1716, 1666, 1660, 1651, 1644, 1635, 1565, 1556, 1538, 1470, 1455, 1434, 1416, 1378, 1337, 1311, 1248, 1184, 1104, 1081, 1021, 964, 721 см-1;
1Н-ЯМР (CDCl3-CD3OD): δ 0,88 (m, 18H), 1,0-2,05 (mH), 2,20-2,75 (m, 12Н), 3,04 (q, 6H, J=7,2 Гц), 3.3-4,3 (mH), 4,45 (d, 1H, J=8,5 Гц), 5,0-5,28 (m, 4Н); 13С-ЯМР (CDCl3): δ 173,7, 173,4, 173,2, 170,5, 170,1, 169,1, 101,4, 75,6, 74,0, 70,8, 70,2, 68,7, 60,4, 56,6, 53,8, 47,4, 45,6, 41,0, 39,6, 38,9, 34,5, 34.3, 34,2, 34,1, 31,9, 29,7, 29,6, 29,5, 29,4, 29,4, 29,3, 29,2, 27,3, 25,3. 25,0, 23,7, 22,7, 21,6, 14,1, 8,4.
Масс-спектроскопия MALDI: теоретически для [M+Na]+ 1506, 0961, фактически 1506, 1008.
Анализ: теоретически для C89H170N3O17P: С=67,43, Н=10,81, N=2,65; фактически: С=67,26, Н=10,85, N=2,47.
ПРИМЕРЫ 4-8
Главной целью Примеров 4-8 является определение того, будет ли соединение формулы II, полученное в Примере 1 (в виде триэтиламмониевой соли) (в дальнейшем именуемое "соединение II"), проявлять минимальную пирогенность и обладать адъювантной активностью при введении его в состав вакцин вместе с антигенами.
Пример 4. Адъювантная активность в отношении HBsAg (поверхностного антигена гепатита В)
Группам мышей BALB/c (Jackson Laboratories, Bar Harbor, Maine) в возрасте 6-8 недель вводили подкожно 2 мкг HBsAg (Laboratorio Pablo Cassara) ±20 мкг адъюванта (иммуностимулятора MPL или соединения II) в день 0 и день 21. Вакцины получали, смешивая содержащие адъювант составы в триэтаноламине (ТЕоА) с рекомбинантным HBsAg. Титры антител к HBsAg определяли методом ELISA в объединенных сыворотках (5 мышей на группу), полученных через 21 день после вторичной вакцинации (табл.1). Неиммунные контроли не подвергались вакцинации.
В сыворотке мышей, получавших соединение II, титры антител к HBsAg были значительно выше, чем в сыворотке контрольных мышей, получавших один лишь антиген (табл.1). Особенно заметным было повышение титров по изотипам IgG2a и IgG2b. Эти титры были эквивалентны титрам в контрольных группах, получавших иммуностимулятор MPL.
Сравнение слабопирогенных адъювантов вместе с HBsAg
Пример 5. Адъювантная активность в отношении белка гемагглютинина в вакцине FluZone против гриппа
Группам мышей BALB/c (Jackson Laboratories, Bar Harbor, Maine) в возрасте 6-8 недель вводили подкожно 0,2 мкг белка гемагглютинина в вакцине FluZone против гриппа (Connaught Laboratories)±20 мкг адъюванта (иммуностимулятора MPL или соединения II) в день 0 и день 14. Титры антител к FluZone определяли методом ELISA на FluZone в объединенных сыворотках из 5 мышей, полученных через 14 дней после вторичной вакцинации (табл.2). Неиммунные контроли не подвергались вакцинации. Использовали разведения сывороток из тестируемых групп, начиная с 1:1600.
Результаты были близки результатам предыдущего примера. Опять у получавших соединение II титры антител были значительно выше, чем в сыворотке контрольных мышей, получавших один лишь антиген (табл.2). Повышение титров также выражалось в возрастании изотипов IgG2a и IgG2b. Эти титры были эквивалентны титрам в контрольных группах, получавших иммуностимулятор MPL.
Сравнение слабопирогенных адъювантов вместе с вакциной против гриппа
Пример 6. Адъювантная активность в отношении HBsAg
Группам мышей BALB/c вводили подкожно 2,0 мкг HBsAg (Rhein Americana и Rhein Biotech)±25 мкг адъюванта (иммуностимулятора MPL или соединения II) в день О и день 21. Титры антител к HBsAg изотипов IgGI и IgG2a определяли методом ELISA в объединенных сыворотках, полученных через 21 день после вторичной вакцинации (табл.3). Неиммунные контроли не подвергались вакцинации. В этом опыте соединение II вызывало повышение титров по сравнению с контрольной группой, получавшей антиген в забуференном фосфатом физрастворе (PBS). RC-553 вызывал повышение титров в равной мере с положительными контролями, получавшими иммуностимулятор MPL (табл.3).
Сравнение слабопирогенных адъювантов вместе с HBsAg
Пример 7. Соединение II повышает активность ЦТЛ иммунизированных мышей в отношении HBsAg
Некоторых мышей из каждой группы в Примере 4 также использовали как доноров клеток селезенки для того, чтобы оценить активность цитотоксических Т-лимфоцитов (ЦТЛ). Вызванный HBsAg специфический лизис оценивали стандартным методом высвобождения 5lCr за 4 часа (Moore et al. (1988) Cell 55:777-785). Суспензии одиночных клеток получали из селезенок мышей через 9 дней после вакцинации. Клетки селезенки обрабатывали NH4Cl в трис-буфере для удаления эритроцитов и ресуспендировали до концентрации 7,5×106 клеток/мл в среде RPMI с 10% телячьей сыворотки с добавлением 5 мМ HEPES, 4 мМ L-глутамина, 0,05 мМ 2-меркаптоэтанола и антибиотиков. К клеткам добавляли синтетический пептид, представляющий известный эпитоп (IPQSLDSWWTSL) ЦТЛ, подвергающихся Ld-рестрикции по МНС класса I, в конечной концентрации 75 нМ. После инкубации в течение 4 дней клетки извлекали и определяли активность ЦТЛ. Измеряли специфическую гибель меченных 5lCr трансфицированных клеток P815S, экспрессирующих эпитоп, подвергающийся L-рестрикции. Клетки-мишени представляют собой трансфицированные клетки линии Р815 (P815S), которые экспрессируют эпитоп ЦТЛ, подвергающийся Ld-рестрикции. Неспецифический лизис составил <10% при соотношении эффектор:мишень = 50:1 относительно клеток мишени Р815 (табл.4). В отличие от антительного ответа, RC-553 вызывал значительное повышение уровня активности ЦТЛ по сравнению с контролем в присутствии одного лишь антигена (табл.4).
Сравнение слабопирогенных адъювантов вместе с HBsAg
Пример 8. Индукция цитокинов соединением II ex vivo
Влияние соединения II на выработку TNF-α и IL-1β измеряли в условиях ех vivo на мононуклеарных клетках периферической крови человека. Иммуностимулятор MPL и соединение II готовили в виде водных растворов в 0,2% ТЕоА/вода для инъекций.
Использовали цельную кровь человека для оценки способности гликолипидов (AGP) индуцировать провоспалительные цитокины. Цельную кровь человека собирали в гепаринизированные пробирки, отбирали 0,45 мл и смешивали с 0,05 мл физраствора с фосфатным буфером (PBS, рН 7,4), содержащего гликолипид (то есть исследуемые соединения). Пробирки инкубировали в течение 4 ч при 37°С на встряхивателе. Затем образцы разбавляли 1,5 мл стерильного PBS и центрифугировали. Супернатанты удаляли и проводили анализ на TNF-α и IL-1β в клетках сэндвич-методом ELISA с помощью наборов Quantikine фирмы R&D Systems для иммуноанализа TNF-α и IL-1β человека.
Соединение II в концентрациях 1, 5 и 10 мкг/мл при анализе не вызывало образования TNF-α, когорое могло бы быть выявлено в условиях данного метода. Напротив, положительный контроль - LPS эффективно стимулировал секрецию TNF-a из клеток в концентрации 1 нг/мл. Иммуностимулятор MPL эффективно индуцировал TNF-α в диапазоне концентраций от 100 до 10000 нг/мл.
Аналогичным образом соединение II (1, 5 и 10 мкг/мл) не вызывало заметного образования IL-1β. Для сравнения эффектов этого соединения уровень IL-lp, индуцированный иммуностимулятором MPL, приняли за 1, тогда относительная индукция цитокинов соединением II составила ≤0,05.
Обсуждение примеров 4-8
Данные этих опытов свидетельствуют о том, что соединение II способно повышать иммунитет к антигенам вакцин. Это соединение повышало титры сыворотки к двум разным антигенам вакцин - антигену гриппа и поверхностному антигену гепатита. Подобно иммуностимулятору MPL, оно вызывало сдвиг профиля антител с ответа, преимущественно представленного изотипом IgG1, на ответ с высоким уровнем антител IgG2a. Наряду с повышением антительного ответа это соединение оказалось хорошим адъювантом в индуцировании активности ЦТЛ.
Замечательной особенностью результатов этого исследования является то, что соединение II оказывает влияние на иммунный ответ, не вызывая заметного образования воспалительных цитокинов TNF-a или IL-1β.Эти цитокины вырабатываются клетками системы врожденного иммунитета как реакция на продукты клеточной стенки бактерий, включая липид А. Поскольку это соединение обладает структурным сходством с липидом А, то можно полагать, что оно также будет стимулировать образование TNF-α или IL-1β, как это делают многие молекулы AGF. Как воспалительные цитокины, TNF-α и IL-1β вызывают запуск каскада других цитокинов, ответственных за активацию фагоцитов и мобилизацию специфического иммунитета. Поначалу IL-1 называли эндогенным пирогеном, так как он вызывает повышение температуры. Таким образом, отсутствие заметного урвня IL-1 после введения соединения II совпадает с явным отсутствием повышения температуры при тестировании на пирогенность на кроликах.
Остается возможным, что в этих опытах данное соединение на самом деле стимулирует секрецию TNF-α и IL-1β на уровне, достаточно высоком для активации специфического иммунитета, но слишком низком для обнаружения при анализе на цитокины ex vivo. Другая возможность состоит в том, что это соединение стимулирует образование не TNF-α и IL-1β, а других цитокинов, которые и вызывают специфический иммунный ответ на совместно введенные антигены вакцины. По-видимому, образуется IFN-y. Этот цитокин, как полагают, отвечает за индукцию сдвига изотипа в сторону антител подкласса IgG2a, а также является активатором ответов ЦТЛ, опосредованных ТН-1. Таким образом, и повышение титра IgG2a, и появление активных популяций ЦТЛ отражает образование IFN-γ.
Пример 9. Стимуляция индуцибельной синтазы оксида азота (iNOS) соединением II
Данный пример иллюстрирует эффекты различных гликолипидов на индукцию iNOS в макрофагах мышей J774. Клеточная линия макрофагов мышей J774 подвержена примированию IFN-γ in vitro и очень чувствительна к последующей стимуляции LPS повышающей регуляции (upregulation) iNOS, которую измеряют методом ELISA с применением стандартного реагента Грейсса. В этом методе клетки J774 в концентрации 1×106 клеток/мл инокулируют в колбу на 30 мл и инкубируют в течение 16-24 ч с добавлением IFN-γ 100 ед./мл. Затем клетки собирают, промывают, ресуспендируют и вносят по 2×105 клеток/лунку в 96-луночный планшет и оставляют для прикрепления к стенкам. В лунки добавляют серийные разведения гликолипидных соединений исследуемой группы и эти культуры инкубируют еще в течение 36-40 ч, после чего отбирают супернатанты культур для анализа на нитрит с помощью реагента Грейсса (Green et al. (1982) Anal. Biochem. 126:131-138). Содержание нитрита тесно коррелирует с активностью iNOS.
Эффективность соединений определяли как концентрацию (нг/мл) гликолипида в культуре, способную вызвать полумаксимальное образование нитрита (ED50). Чем меньше значение ED50, тем сильнее способность к индукции iNOS. Значения ED50 рассчитывали в соответствии с методами, изложенными в Johnson et al. (1999) J. Med. Chem. 42:4640-4649.
Иммуностимулятор MPL проявлял значение ED50 около 2 нг/мл, что приводит к высокому уровню образования нитрита, тогда как соединение II проявляло номинальное значение ED50≥3000 нг/мл.
Очень низкий уровень максимальной активности iNOS, наблюдавшийся в присутствии этого соединения, свидетельствует о том, что оно практически неактивно в данной системе индукции iNOS.
Предусматривается, что описанные в данном изобретении примеры и воплощения приводятся только в целях иллюстрации и что специалистами в данной области будут предложены различные модификации и видоизменения, которые должны включаться в соответствии с духом и сферой действия настоящей заявки и объемом прилагаемой формулы изобретения. Все процитированные публикации, патенты и патентные заявки включены в настоящее изобретение путем отсылки во всей их полноте для всех целей.
Изобретение относится к новым иммуноэффекторным аминоалкилглюкозаминидфосфатным соединениям формулы (V)
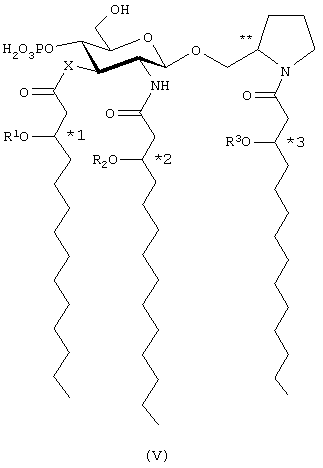 ,
,
или его фармацевтически приемлемым солям, где Х выбран из группы, состоящей из -О- и -NH-; R1, R2 и R3 независимо друг от друга выбраны из группы, состоящей из (С9-С14) ванильных групп. Кроме того, изобретение относится к иммуностимулирующей фармацевтической композиции, включающей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы (V), и к способу индуцирования иммунного ответа у пациента. 3 н. и 16 з.п. ф-лы, 4 табл.
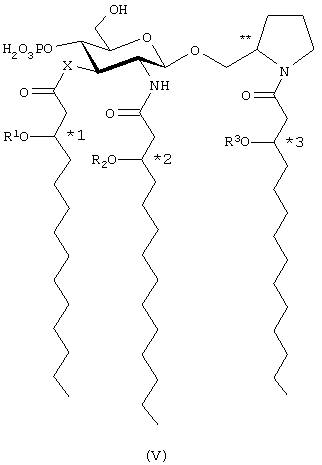
или его фармацевтически приемлемые соли,
где Х выбран из группы, состоящей из -О- и -NH-;
R1, R2 и R3 независимо друг от друга выбраны из группы, состоящей из (C9-С14)ацильных групп.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| D.A | |||
| JOHNSON et al, Synthesis and biological evaluation of a new class vaccine adjuvants: aminoalkyi glucosaminide 4-phosphates (AGPs) | |||
| Bioorganic & Medicinal Chemistry Letters, 1999, vol.9, pp.2273-2278 | |||
| ЦЕНТРОБЕЖНЫЙ РЕГУЛЯТОР ДАВЛЕНИЯ | 0 |
|
SU194563A1 |
| ГЛЮКОЗАМИНОВЫЕ ДИСАХАРИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2154068C2 |

