Настоящее изобретение относится к специфическим глюкозаминовым дисахаридам, в частности к 2-N- и/или 2'-N- ацилированным глюкозаминовым дисахаридам, где по крайней мере одна из ацильных групп разветвлена, и к соединениям, содержащим эти дисахариды. Далее, настоящее изобретение относится к способам получения этих дисахаридов из исходного продукта, содержащего радикал липида А липополисахаридов, исходный материал которых подвергается специфической щелочной обработке. Изобретение относится также к фармацевтическим композициям, содержащим эти дисахариды в качестве активного ингредиента, и, наконец, к применению этих дисахаридов в лечении и профилактике.
Липополисахариды представляют эндотоксины микроорганизмов, таких как грам-отрицательные бактерии, и включают полисахаридный компонент и липидный компонент. Этот липидный компонент, также называемый липидом А, определяет эндотоксические свойства липополисахаридов (Rietschel Е. Th. et al.in Immunobiology, Volume 186, pages 169-190 [1993]).
В US-A-4 912 094 описываются модифицированные липополисахариды, которые проявляют менее выраженные эндотоксические свойства, тогда как их антигенные, и иммуностимулирующие свойства сохраняются. Эти модифицированные липополисахариды 3-O-деацилированы и могут быть переведены в 3-O-деацилированные дисахариды путем кислотного гидролиза. Из этих соединений монофосфорил 3-O-деацилированный дисахарид является менее токсичным, чем дифосфорил 3-O-деацилированный дисахарид.
Настоящее изобретение касается дисахаридов, которые являются 3-O-деацилированными и 3'-O-деацилированными или содержат в 3-O- положении и/или в 3'-O-положении короткую O-присоединенную алкильную или ацильную группу, и содержат по крайней мере N-связанную разветвленную ацильную группу во 2-положении, 2'-положении, или в обоих: 2-положении и 2'- положении. Эти соединения проявляют, по-прежнему, пониженную эндотоксичность, хотя сохраняют биологическую активность (такую как иммуномодуляция) и проявляют противораковую активность.
Хотя синтетические 3-O- и 3'-O-деацилированные глюкозаминовые дисахариды, содержащие N-присоединенную ацильную группу во 2- и 2' -положении (соединение 307, Takeda, Н. et al. in CRC Critical Reviews in Microbiology, Volume 16, pages 477-523 [1989]; и соединение LA-19-PP, Rietschel et al. [1993]) проявляли некоторую иммунобиологическую активность в определениях in vitro, неожиданным было то, что эти специфические глюкозаминовые дисахариды проявляют сочетание пониженной эндотоксичности и сохраненной биологической активности, но эти активности значительно более слабые, чем у контрольных бактериальных образцов липида А. Они также лишены типичной эндотоксической активности.
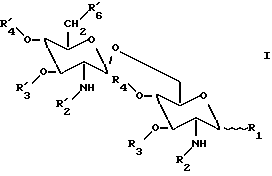
Соответственно, настоящее изобретение касается β(1→6) глюкозаминовых дисахаридов, имеющих общую формулу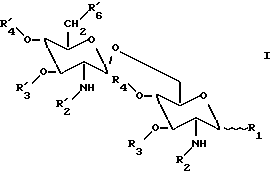
где
R1 представляет гидроксильную группу, дигидроксифосфоноилоксигруппу или ее заряженные формы, (C1-C5) ацилокси группу; (C1-C5) алкилокси группу, или группу X;
R2 и R'2 каждая является ацильной группой или группой Y при условии, что, по крайней мере, R2 или R'2 являются группой Y;
R3 и R'3 каждая представляют водород, (C1-C3) алкильную группу, или (C1-C3) ацильную группу;
R4 представляет водород, (C1-C3) алкильную группу; или (C1-C3) ацильную группу;
R'4 представляет водород, (C1-C5) ацильную группу, (C1-C5) алкильную группу, диметоксифосфоноильную группу, или фосфоногруппу или ее заряженные формы; и
R'6 представляет водород, гидроксильная группа, дигидроксифосфоноилоксигруппу, гидроксисульфонилоксигруппу, их заряженные формы, или группу Z;
где группа X выбрана из группы, содержащей карбокси (C1-C5) алкоксигруппу; -O-CH-[(CH2)m COOH][(CH2)nCOOH] группу,
где m = 0-5 и n = 0-5; фосфоно (C1-C5) алкильную группу; диметоксифосфоноилоксигруппу; гидроксисульфонилоксигруппу; гидроксисульфонил (C1-C5) алкильную группу; и заряженные формы группы X;
где группа Y выбрана из группы, содержащей ацилоксиацильную группу, ациламиноацильную группу, ацилтиоацильную группу, (C1-C24) алкилоксиацильную группу, (C1-C24) алкиламиноацильную группу, (C1-C24) алкилтиоацильную группу; и
где группа Z выбрана из группы, содержащей (C1-C24) алкилоксигруппу; (C1-C24) ацилоксигруппу; 3-деокси-D-манно-2- октулосоновую кислоту (KDO); (KDO)n, где n = 1-10; полисахаридную боковую цепь, такая как боковая цепь натурального липополисахарида; компонент ядра, такой как компонент, происходящий из натурального липополисахарида; и амино- (C1-C8) алкилкарбоксильную группу; и их соли.
Данные глюкозаминовые дисахариды проявляют гораздо более низкую эндотоксичность, определяемую а исследовании лизата амебоцита limulus (LAL), чем липополисахариды (LPS, ЛПС), например, от E.coli, липид A и модифицированный липид A в соответствии с US-A-4912094. Более того данные глюкозаминовые дисахариды индуцируют реакционноспособные промежуточные продукты оксида азота и цитокины, такие как интерлейкин 1-альфа (ILI-alpha, ИЛ1-альфа), IL-6, фактор некроза опухоли (TNF, ФНО) и простагландин (PGE, ПГЕ).
Кроме того, данные дисахариды проявляют противоопухолевую активность, такую как в случае перитонеального карциноматоза.
И наконец, острая токсичность данных дисахаридов крайне низка. После внутривенного введения 100 мг дисахарида на кг массы тела смертности у мышей Swiss зарегистрировано не было.
Настоящее изобретение относится также к способу получения данных глюкозаминовых дисахаридов с использованием в качестве исходного продукта биологического образца, а именно, некоторого исходного продукта, содержащего радикал липида A полисахаридов из микроорганизмов, таких как грамотрицательные бактерии. Согласно изобретению этот исходный продукт подвергают, по крайней мере, обработке щелочью так, чтобы удалить O-присоединенные ацильные и/или O-присоединенные оксиацильные группы сахаров. Если приемлемо, обработанный щелочью исходный продукт может быть подвергнут дальнейшим обработкам для удаления полисахарида и ядерного компонента (путем обработки кислотой) и для замещения и обмена заместителей в 1-положении, 2-положении, 3-положении, 4-положении, 2'-положении, 3'-положении, 4'-положении, 6'- положении.
Однако согласно изобретению глюкозаминовые дисахариды могут быть получены путем синтеза, исходя из соответствующего глюкозаминового дисахарида, и введения целевых заместителей во 2- и/или 2'-положении.
Благодаря крайне низкой эндотоксичности в сочетании с описанной выше биологической активность эти дисахариды по изобретению представляют собой наилучший активный ингредиент фармацевтической композиции. Такая фармацевтическая композиция и дисахариды как таковые могут использоваться в качестве иммуномодулирующего агента, противоопухолевого агента и как компонент вакцины.
Глюкозаминовый дисахарид по изобретению (aβ(1→6)D- глюкозаминовый димер) характеризуется тем, что каждый глюкозамин содержит в 3- и 3'-положении гидроксильную группу или короткую O- присоединенную алкильную или ацильную группу, не существенно изменяющую эндотоксичность и/или биологическую активность, и, кроме того, по крайней мере одну N-присоединенную разветвленную ацильную группу во 2- или 2'-положении или в обоих 2- и 2'- положениях. Оставшееся 2'- или 2-положение ацилировано. Вероятно, наличие двух гидрофобных цепей во 2-положении и 2'-положении, по крайней мере одна из которых находится в виде разветвленной ацильной группы, придает дисахариду сочетание крайне низкой эндотоксичности и сохраненной биологической активности.
Разветвленная ацильная группа, которая здесь обычно обозначается как группа Y, выбрана из групп, содержащих ацилоксиацильную группу, ациламиноацильную группу, ацилтиоацильную группу, (C1-C24)алкилоксиацильную группу, (C1-C24)алкиламиноацильную группу и (C1-C24)алкилтиоацильную группу.
В случае ацилоксиацильной группы две ацильные группы связаны через атом кислорода, в случае ациламиноацильной группы через NH-группу и в случае ацилтиоацильной группы через атом серы. Другие члены группы Y, (C1-C24) алкилоксиацильная группа, (C1-C24)алкилтиоацильная группа и (C1-C24)алкиламиноацильная группа, могут быть получены, исходя из соответствующей гидроксижирной кислоты.
Преимущественно, группа Y представляет собой N-присрединенную ацильную группу, разветвляющуюся в ее 3-положении, например, 3-ацилоксиацильная группа, 3-ациламиноацильная группа, 3-ацилтиоацильная группа. То же относится и к указанным выше (C1-C24) алкильным эквивалентам.
Преимущественно, члены группы Y содержат один или два ацильных радикала, предпочтительно выбранных из остатков жирных кислот, остатков гидроксижирных кислот и остатков оксожирных кислот. Когда ацилоксиацильная группа является, преимущественно, 3-ацилоксиацильной группой, такие ацильные радикалы содержат остаток 3-гидроксижирной кислоты или, для эфирно-присоединенной группы, остаток 3-оксожирной кислоты. Типичными примерами ацилоксиацильной группы являются 3-гидрокси (C4-C24) ацилы жирных кислот, которые эфирно-связаны по 3-гидроксиположению с (C1-C24) карбоновой кислотой. Ацилоксиацильная группа, предпочтительно, является 3-гидрокси(C8-C18)ацилом жирной кислоты, который связан эфирной связью по 3-гидроксиположению с (C10-C18) жирной кислотой. Такие ацилоксиацильные группы представлены в липид A-компоненте грамотрицательных бактерий, таких как Escherichia coli, Haemophilus influenzae, Campylobacter jejuni, Rhodocyclus gelatinosus, Chromobacterium violaceum, Neisseria meningitidis, Salmonella minnesota.
В первой группе глюкозаминовых дисахаридов по изобретению ацилоксиацильная группа является N-присоединенным 3-гидроксиC14-ацилом жирной кислоты, эфирно-связанным по 3- гидроксиположению с C12жирной кислотой с данной ацилоксиацильной группой по 2'-положению. В другом выбранном глюкозаминовом дисахариде по изобретению ацилоксиацильная группа является N-присоединенным 3-гидроксиC14-ацилом жирной кислоты, эфирно-связанным по 3-гидроксиположению с C14-жирной кислотой, и ацилоксиацильная группа находится предпочтительно во 2'- положении.
В другом выбранном глюкозаминовом дисахариде по изобретению ацилоксиацильная группа является N-присоединенным 3- гидроксиC14-ацилом жирной кислоты, эфирно-связанным по 3- гидроксиположению с C12-жирной кислотой с данной ацилоксиацильной группой во 2-положении. В другом выбранном глюкозаминовом дисахариде по изобретению ацилоксиацильная группа является N-присоединенным 3-гидроксиC14-ацилом жирной кислоты, эфирно-связанным по 3-гидроксиположению с C12-жирной кислотой с ацилоксиацильной группой как во 2-положении, так и во 2'- положении.
Когда группа Y содержит хиральный центр, изобретение охватывает все R- и S-энантиомеры и любую рацемическую смесь.
Другой N-присоединенный заместитель может быть ацильной группой или также ацилоксиацильной группой. Согласно второй группе дисахаридов по изобретению ацильная группа является 3- гидрокси (C4-C24) жирной кислотой, предпочтительно 3-гидрокси(C10-C18) жирной кислотой. В выбранных дисахаридах по изобретению ацильная группа является 3-гидроксиC14-жирной кислотой во 2-положении или во 2' -положении.
Однако N-присоединенный заместитель может являться также ацилоксиацильной группой, определенной выше, и содержащей N-присоединеннный 3-гидрокси(C14-C24)ацил жирной кислоты, который эфирно-связан по 3- гидроксиположению с (C1-C20)карбоновой кислотой, предпочтительно, предпочтительно 3-гидрокси(C8-C18) жирной кислоты, эфирно-связанный по 3-гидроксиположению с (C10-C18) -жирной кислотой. Более предпочтителен дисахарид, где R2 является N-присоединеннным 3-гидрокси C14-ацилом жирной кислоты, эфирно-связанным по 3-гидроксиположению с C12-жирной кислотой или C16-жирной кислотой, и где R2 является N-присоединеннным 3-гидроксиC14-ацилом жирной кислоты, эфирно-связанным по 3-гидроксиположению с C12-жирной кислотой и C14-жирной кислотой.
Замечено что в группе Y ацильные группы и/или ацильная или алкильная группа могут быть связаны между собой.
В данной спецификации термин "остаток жирной кислоты" означает: значительно гидрофобная цепь из C2-C30 атомов, которая может быть неразветвленной, разветвленной, насыщенной, моно- или полиненасыщенной, с включенными одним или более гетероатомами, такими как азот, кислород, сера, и которая может быть замещена одним или более заместителями, такими как гидроксил, оксо, ацилокси, алкокси, амино, нитро, циано, галогено, сульфгидрил, при условии, что биологическая активность затрагивается не существенно. Пример замещенного остатка жирной кислоты (содержащего амидно-присоединенный заместитель) описан Onozuka, К. et al. in Int.J.Immunopharmac, Volume 15, pages 657-664 [1993].
Заместитель R1 может быть (C1-C5)ацилоксигруппой или (C1-C5)алкилоксигруппой, тогда как R4 может быть (C1-C5) ацильной группой или (C1-C5) алкильной группой, при условии, что свойства глюкозаминовых дисахаридов затрагиваются не существенно. Кроме того, R1 может быть гидроксильной группой, a R4 может быть водородом. Предпочтительно, R1 и R4, каждая, могут быть фосфоросодержащей группой. В частности, такая группа в 1-положении и 4'-положении может влиять на биологическую активность, такую как различная стимуляция цитокинов (смотри Takada, D.C. and Ryan,J., CRC Press, Volume I, pages 107-134 [1992], в особенности page 123).
Выбранные дисахариды по изобретению содержат дигидрокси-фосфоноилоксигруппу по 1-положению и фосфоногруппу по 4-положению, причем группа в 1-положении находится предпочтительно в α конфигурации.
Заместитель R1 может быть также представлен группой X. Группа X обычно обладает отрицательным зарядом при физиологическом pH. Группа X может быть кapбoкси(C1-C5)алкилоксигруппой. Группа X может быть также дикарбоновой кислотой формулы -O-CH-[(CH2)mCOOH] [(CH2)nCOOH], где m = 0-10 и n = 0-10, как, например, m и n=0, m и n=1, и m=1 и n=3. Заместитель в виде дикарбоновой кислоты по 1-положению, где m и n=1, описан Onozuka, К. et al. (1993). Вместо дикарбоновой кислоты группа X может представлена фосфоно(C1-C5)алкильной группой, такой как фосфонометильная группа или фофсфоноэтильная группа.
Замещенная группа X может также иметь вид сульфатной группы или гидроксисульфонил(C1-C5)алкильной группы, как, например, гидроксисульфонилметильная группа.
Заместитель R3 и R'3 могут быть короткой алкильной или ацильной группой, которая не влияет существенно на эндотоксичность и/или биологическую активность глюкозаминовых дисахаридов по изобретению. Примерами являются (C1-C3)алкильная группа и (C1-C3)ацильная группа. Предпочтительно, заместители R3 и R'3 оба являются водородами, что означает, что 3-положение и 3'-положение не ацилированы.
Заместитель R4 по 4-положению может быть (C1-C3) алкильной группой или (C1-C3) ацильной группой, значение которых было описано выше. 4-O-Ацилированный дисахарид может быть синтезирован с использованием способа, описанного Kusumoto S. et al., ACS Symposium Series, Volume 231, pages 237-254, (1993). Однако по 4-положению предпочтительна гидроксильная группа (R4=H).
Заместитель R6 может быть водородом, гидроксильной группой, дигидроксифосфоноилоксигруппой, дигидроксисульфонилоксигруппой и их заряженными формами.
В целях улучшения водорастворимости глюкозаминовых дисахаридов по изобретению заместитель по 6'-положению может иметь выраженный гидрофильный характер, обусловленный группой Z. Группой Z может быть 3-деокси-D-манно-2-октулосоновая кислота (КДО) или несколько молекул КДО, таких как присутствуют во внутреннем ядре натуральных полисахаридов, вплотную примыкающих к липид A-компоненту.
Группа Z может быть также полной или частичной полисахаридной цепью, такой как боковая цепь из натуральных липополисахаридов или ядерный компонент натуральных липополисахаридов.
Группа Z также может быть амино(C1-C8)алкилкарбоксильной группой.
Водорастворимость дисахаридов по изобретению с одной стороны определяется наличием заряженных групп, гидрофильным характером заместителя по 6'-положению. С другой стороны, водорастворимость может быть также улучшена, если дисахарид находится в виде соли, такой как соль, содержащая один или более катионов щелочных металлов и/или ионов аммония, образующий пару с, например, дигидроксифосфоноилоксигруппами, карбоксильными группами, фосфоногруппами, гидроксисульфонилоксигруппами и гидроксисульфонилалкильными группами, если таковые присутствуют.
Отмечается, что любая алкильная или ацильная цепь или радикал может быть неразветвленной, разветвленной, насыщенной, моно- или полиненасыщенной, с включением одного или более гетероатомов, таких как азот, кислород, сера, и которая может быть замещена одним или более заместителями, такими как гидроксил, оксо, ацилокси, алкокси, амино, нитро, циано, галогено, сульфгидрил, при условии, что биологическая активность не затрагивается существенно.
Глюкозаминовые дисахариды по изобретению могут быть получены из исходного продукта, содержащего радикал липида A липополисахаридов, которые присутствуют в микроорганизмах, таких как грамотрицательные бактерии. Данные липополисахариды присутствуют, например, в фракции, содержащей поверхностную структуру данных микроорганизмов и в липополисахаридах из нее. Выбранными грамотрицательными бактериями, используемыми в качестве источника исходного продукта, является Escherichia coli и Haemophilus influenzae. Однако имеющиеся в продаже ЛПС или липид A могут быть использованы в качестве исходного продукта.
Избирательное деацилирование по 3-положению и по 3'-положению проводится с использованием обработки щелочью. Условия щелочной обработки выбираются такими, чтобы оба глюкозамина 3- гидроксидеацилировались. Обработка щелочью может проводиться с использованием гидроксидов, карбонатов и фосфатов, таких как гидроксид натрия или карбонат калия. Показательными органическими щелочными агентами являются алкиламины, такие как диэтиламин и триэтиламин. Обработка щелочью, как правило, проводится в водной и органической среде. pH обычно находится в пределах 10-14, как, например, 11-13, в практических условиях pH бывает, например, 12.2. Обработка щелочью, как правило, проводится при температуре между температурой окружающей среды и 70oC, такой как 37oC. Временной период зависит от типа исходного продукта. Если исходить из микроорганизмов, период времени колеблется между 1 часом и 10 днями, как, например, 8 часов и 5 дней, но, как правило, в пределах 8-40 часов. Если исходить из липополисахаридов или липида A, период времени может быть 0,2-10 часов, как, например, 1-5 часов. На практике период времени примерно равен от 1.5 до 3 часов.
Когда исходный продукт содержит в 6'-положении ядерный компонент, который надо удалить, исходный материал подвергают обработке кислотой для удаления ядерного компонента. Данная обработка кислотой может проводиться до или после описанной выше обработки щелочью. Обработка кислотой проводится при pH 1-5, предпочтительно, при pH в пределах 2.5-4.5, как правило, при pH выше чем 3, и ниже чем 4.5, как, например, при 3.5. При pH 1 и ниже глюкозаминовый дисахарид дефосфорилируется, что приводит к монофосфорилированной форме. Кислоты, которые можно использовать, - минеральные и органические кислоты, такие как соляная кислота и ледяная уксусная кислота. Период времени обработки кислотой от примерно 30 минут до 5 часов. Как, например, 1-2 часа. Во время обработки кислотой температуру повышают до примерно 70-100oC, как, например, 80-100oC, на практике 95oC. Впоследствии температуру снижают до температуры окружающей среды.
Глюкозаминовые дисахариды по изобретению могут также быть получены, исходя из соответствующего де-, моно- или дифосфорилированного глюкозаминового димера путем присоединения ацилоксиацильной группы, ациламиноацильной группы и/или ацилтиоацильной группы по обоим 2-положению или 2'-положению.
После частичного деацилирования данных глюкозаминовых дисахаридов по изобретению и разделения продуктов получаемые глюкозаминовые дисахариды имеют в своем составе разветвленную ацильную группу по 2-положению или по 2'-положению.
Глюкозаминовые дисахариды по изобретению могут быть применены в фармацевтической композиции или лекарстве и использованы как иммуномодулирующий агент для ингибирования, стимулирования или индуцирования толеризации продукции реакционноспособных промежуточных форм оксида азота и цитокинов в зависимости от частоты применения и от дозировки, как противоопухолевый агент, например, Т-клеточный реактивации, как компонент вакцины, как конкурент за места связывания эндотоксина и как модулятор интерлейкинов. Благодаря крайне низкой эндотоксичности данные дисахариды почти или значительно свободны от побочных эффектов.
Дисахариды по настоящему изобретению могут применяться систематически или ограниченно с использованием внутривенной инъекции, подкожной инъекции, интраперитонеальной инъекции, внутримышечной инъекции и т.п. Дозировка варьируется в зависимости от пациента - животного или человека, возраста, массы тела, симптомов или заболевания, подлежащего излечению, желаемого терапевтического эффекта путем введения, периода лечения и т.п. Удовлетворительные эффекты будут получены при дозировке от 0.001 до 500 мг на кг массы тела, вводимых одной или более дозами в день или в форме для длительного высвобождения.
Фармацевтическая композиция может содержать фармацевтически приемлемый носитель или разбавитель для, например, неперорального введения водных или неводных растворов, суспензий и эмульсий. Водные растворы или суспензии могут содержать дистиллированную воду или физиологический раствор. Неводные растворы могут включать пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, спирты. Композиция может содержать другие добавки, такие как консерванты, увлажняющие агенты, эмульсифицирующие агенты, диспергирующие агенты и т.п.
Для более полного понимания настоящего изобретения дана отсылка к следующим примерам, которые приведены здесь лишь в целях иллюстрации и не предназначаются для ограничения рамок настоящего изобретения.
Пример 1
Escherichia coli I-1147 (депонированную в CNCM 3 октября 1991 г под номером I-1147) культивировали в культуральной среде, композиция которой описана в таблице 1.
Раствор-концентрат олигометаллов: 2,5 г FeCl2•H2O, 0,25 г CoCl2•6H2О, 0,25 г NaMoO4•7H2O, 0,25 г Мn3SO4•4H2O, 0,25 г ZnSO4•7H2O, 0,25 г NiSO4•7H2O, 0,05 г H3BO4, 0,05 г CuSO4, затем добавить 1,0 л H2O, перемешать и добавить 1,1 мл H2SO4 (85%).
pH культуральной среды доводили, используя 5 н. NaOH, 5% аммиак или 25% HCI. Escherichia coli I-1147 культивировали при аэрировании и перемешивании (500 об/мин), 37oC и при pH 6,9.
Содержимое ферментера после этого инактивировали, используя температурную обработку (105oC в течение 2 минут).
Инактивированное содержимое ферментера подвергали ультрафильтрации (отсечение 1000 kD) и оставшиеся бактерии отмыли, используя 0,6% водный раствор NaCl. Отмытую суспензию бактерий концентрировали путем ультрафильтрации. Выход биомассы составлял 764 г сухого веса.
Биомассу разбавляли до 7,0 г/л и подвергали щелочной обработке путем добавления 345 мл 10,77 N NaOH и инкубации при 37oC в течение 40 часов (pH 12,2).
Щелочной экстракт подвергали первой ультрафильтрации (отсечение 1000 kD), и фильтрат - второй ультрафильтрации (10 kD). Задержанный остаток подвергали обработке кислотой.
Задержанный остаток разбавляли 7,0 л воды и подкисляли при помощи 370 мл ледяной уксусной кислоты (конечный pH 3,52). Смесь прогревали при 95oC в течение 120 минут при перемешивании. После этого кислую суспензию охлаждали до 25oC. Осадок отделяли путем центрифугирования (4000 X g в течение 50 минут). Осадок вновь суспендировали в воде (3,7 л) и подвергали экстракции пропан-2-олом (4,3 л) и через 60 минут при 25oC добавляли 252 мл триэтиламина (pH 9,0) и продолжали перемешивание в течение 24 часов.
Супернатант собирали путем центрифугирования (4000 X g, 25oC в течение 50 минут) и осадок вновь экстрагировали 90% пропан-2- олом. Супернатанты объединяли и подвергали хроматографии с обращенной фазой (Waters No. 10001, Preparative C8, 
Альтернативно, обработанный кислотой экстракт подвергали ультрафильтрации и задержанный остаток (>1000 kD) концентрировали и диализовали против 5 объемов воды. Диализированный остаток разводили 9 объемами пропан-2-ола и устанавливали pH 9 с помощью триэтиламина (TEA). Экстракцию проводили при перемешивании в течение 2 часов.
Супернатант удаляли, как описано выше, и осадок вновь экстрагировали пропан-2-олом. Супернатанты объединяли и подвергали вакуумному концентрированию (40oC, 12 Topp), и, в конце, подвергали хроматографии с обращенной фазой C18 Prep Sep Pak (Waters N. 10001).
Каждый из двух супернатантов разводили двумя объемами воды, и смешивали с 5 мМ тетрабутиламмоний фосфатом (TBAP) и наносили на колонку, содержащую 50 г обращенной фазы C18 Prep Sep Pak (Waters N. 10001, препаративная C18,  прекондиционированную 250 мл CH3CN:H2O 1:1, (по объему) + 5 мМ TBAP. Колонку промывали 60% CH3CN:H2O 1:1, (по объему) + 5 мМ TBAP и 40% пропан-2-олом: H2O 9:1, (по объему) + 5 мМ TBAP. Дисахариды по изобретению элюировались во фракции при 30% CH3CN:H2O 1:1, (по объему) + 5 мМ TBAP и 70% пропан-2-ол:H2O 9:1, (по объему) + 5 мМ TBAP.
прекондиционированную 250 мл CH3CN:H2O 1:1, (по объему) + 5 мМ TBAP. Колонку промывали 60% CH3CN:H2O 1:1, (по объему) + 5 мМ TBAP и 40% пропан-2-олом: H2O 9:1, (по объему) + 5 мМ TBAP. Дисахариды по изобретению элюировались во фракции при 30% CH3CN:H2O 1:1, (по объему) + 5 мМ TBAP и 70% пропан-2-ол:H2O 9:1, (по объему) + 5 мМ TBAP.
Дисахаридную фракцию, полученную при хроматографии с обращенной фазой, разбавляли водой 1:1, (по объему) + 25 мМ TBAP и наносили на препаративную ЖХВР колонку (Millipore- Waters Bondapak C18 300  15М, 300 мм X 47 мм). Дисахаридная фракция по изобретению элюировалась 67% пропан-2-олом:H2O 9:1, (по объему) + 25 мМ TBAP и 33% CH3CN:H2O 1:1, (по объему) + 25 мМ TBAP. Эта фракция содержала 55 мг дисахарида A по изобретению.
15М, 300 мм X 47 мм). Дисахаридная фракция по изобретению элюировалась 67% пропан-2-олом:H2O 9:1, (по объему) + 25 мМ TBAP и 33% CH3CN:H2O 1:1, (по объему) + 25 мМ TBAP. Эта фракция содержала 55 мг дисахарида A по изобретению.
Обессоливание дисахарида
Соль удаляли из аликвот фракции дисахарида A следующим образом. Колонка Sep Pak Vac C18 Plus (silica C18, 0,6 мл, Waters N. 20515) была последовательно кондиционирована введениями 5 мл CHCl3 - CH3OH 2:1, (по объему), 5 мл CH3CN и 5 мл CH3CN:H2O 1:1, (по объему). Образец был нанесен на колонку после разведения фракции ЖХВР тремя объемами H2O, с доведением общего объема разведенного образца до 6 мл. TBAP затем был смыт 10 мл CH3CN:H2O 1:1, (по объему) + 10 мл мМ HCl, и последующими 10 мл CH3CN. Чистый дисахарид A затем снимали 5 мл CHCl3-CH3OH 2:1, (по объему).
Фракцию высушивали выпариванием в вакууме (12 Topp) при 35oC. Обессоленный дисахарид A растворяли в H2O:TEA 1000:1, (по объему) для биологических или биохимических определений, или в хлороформ:метаноле 2:1, (по объему) для FAB-MS.
Получение в виде натриевой соли
Колонка, содержащая 10 г обращенной фазы C18Prep Sep Pak (Waters N. 10001, препаративная C18,125  ), была последовательно прекондиционирована 50 мл CH3CN и 50 мл CH3CN:H2O 1:1, (по объему).
), была последовательно прекондиционирована 50 мл CH3CN и 50 мл CH3CN:H2O 1:1, (по объему).
Образец (фракция ЖХВР) был нанесен на колонку после разведения в 1 объеме H2O. После связывания колонку промыли 100 мл CH3CN:H2O 1:1, (по объему) + 5 мл TBAP. Дисахарид снимали 50 мл пропан-2-ола:H2O 9:1, (по объему) + 5 мл TBAP.
Полученную фракцию очищали следующим образом. Колонка, содержащая 20 мл быстро текущей Q-Sepharose (Pharmacia 17-0510-01), была последовательно уравновешена 30 мл NaOH 1М и промыта водой для нейтрализации, затем 30 мл HCl 1М и промыта водой для нейтрализации.
Образец наносили непосредственно на колонку. После связывания несвязавшийся материал смывали 200 мл H2O и 100 мл пропан-2-ола:H2O 9:1, (по объему). Дисахарид элюировали 100 мл NaCl 0,9%:изопропанолом 1:1, (по объему).
Окончательная очистка была проведена следующим образом. Колонку, содержащую 10 г обращенной фазы C18 Prep Sep Pak, последовательно прекондиционированную 50 мл CH3CN, 50 мл CHCl3:CH3OH 2:1, (по объему), 50 мл CH3CN и 50 мл 50% CH3CN:H2O 1:1, (по объему). Образец наносили на колонку после разведения 1 объемом воды. После связывания колонку промывали последовательно 200 мл H2O, 200 мл пропан-2-ола:H2O 9:1, (по объему) и 50 мл CHCl3CN. Дисахарид элюировали 50 мл CHCl3-CH3OH
2:1, (по объему). Фракцию высушивали упариванием в вакууме (12 Topp) при 35oC.
Натриевая соль была легко растворима в воде (до 100 мг/мл).
Пример 2
Haemophilus influenzae (полученный из Национальной Коллекции Типов Культур - National Collection of Type Cultures (АТСС 9795)) культивировали в культуральной среде, состав которой описан в таблице 2.
Культуральная среда была дополнена гемином (10 мг/л) и NADH (4 мг/л). pH доводили до 7,0 +/- 0,3, используя 5 н. NaOH или 25% HCl. После начала установления стационарной фазы культивирование прерывали и содержимое ферментера инактивировали температурной обработкой (100oC в течение 100 секунд). Инактивированную культуру центрифугировали и отделенную биомассу разбавляли 0,6% водным раствором NaCl (приблизительно 60 г/л). Щелочную обработку проводили путем добавления 10 н. NaOH до конечной концентрации 0,2 н. NaOH. Обработку проводили при 37oC в течение 5 дней при непрерывном перемешивании.
Обработанный щелочью лизат непосредственно подвергали кислотной обработке после подкисления до pH 3,5 ледяной уксусной кислотой. Смесь прогревали при 95oC в течение 120 минут, после чего охлаждали до комнатной температуры.
Осадок центрифугировали (10 000 X g, 30 минут при 4oC), a супернатант отбрасывали.
Осадок вновь суспендировали в CH3CN:H2O 1:1, (по объему) и доводили pH до 9, применяя TEA. После центрифугирования (15 000 X g, 10 минут) супернатант доводили до 5 мМ TBAP. Супернатант наносили на колонку Sep Pak Vak Cis (10 г silica 018, 35 мл., Waters N. 43345), кондиционированную 50 мл CH3CN: H2O 1:1, (по объему). Фракцию, содержащую дисахарид B по изобретению, элюировали 50 мл пропан-2-олом:H2O 9:1, (по объему) + 5 мМ TBAP.
Эту фракцию концентрировали выпариванием (35oC, 12 Topp) приблизительно до 2 мл. Фракцию центрифугировали (15 000 X g, 5 минут) и супернатант наносили на полупрепаративную C18 колонку ЖХВР (Macherey-Nagel N.715806, 250 мм х 10 мм O, Nucleosil 300-7C18). Фракцию, содержащую дисахарид B, по изобретению, элюировали во фракцию, содержащую 28% CH3CN:H2O 1:1, (по объему) + 25 мМ TBAP и 72% пропан-2-ол:H2O 9:1, (по объему) + 25 мМ TBAP.
Дисахарид B обессоливали, используя метод, подобный приведенному в примере 1.
Пример 3
Липополисахарид Escherichia coli 0111: D4 (Sigma, продукт No. L3024) подвергали щелочной обработке в присутствие 0,2 М NaOH при 37 С в течение 1,5 часов. Раствор нейтрализовали 1 М фосфорной кислотой.
400 мкл обработанного щелочью раствора LPS концентрировали путем ультрафильтрации (Millipore Ultrafree-MC, UFC3 LGC OO, отсечение 10 kD).
Задержанный остаток (> 10 kD) разводили в 400 мкл H2O и подвергали кислотной обработке путем доведения ледяной уксусной кислотой до 0,2 М концентрации уксусной кислоты. Подкисленный раствор нагревали при 95oC в течение 120 минут. После охлаждения до 25oC осадок осаждали центрифугированием (15 000 X g, 10 минут), а супернатант отбрасывали. Осадок разводили в 20 мкл H2O:TEA 1000:1, (по объему) и этот раствор наносили на аналитическую C18 колонку ЖХВР (Supelco N. 58985, Supelcosil LC-18, 3 мкм, 150 мм х 4,6 мм О). Дисахаридную фракцию по изобретению элюировали во фракцию, содержащую 42% CH3CN: H2O 1: 1, (по объему) + 5 мМ TBAP и 58% пропан-2-ол:H2O 9:1, (по объему) + 5 мМ TBAP.
Пример 4
Раствор 2 мг/мл Липида A из Escherichia coli F-583 (Sigma, продукт N L5399) готовили на смеси H2O:TEA 1000:1 (по объему) и этот раствор подвергали щелочной обработке, используя 2 М NaOH при 37oC в течение 2,5 часов. Раствор нейтрализовали, используя 1 М фосфорную кислоту.
Нейтрализованный раствор наносили на аналитическую колонку ЖХВР (Supelco N 58958, Supelcosil LC- 18, 3 мкм, 150 мм x 4,6 мм). Дисахариды по изобретению элюировали 42% CH3CN:Н2O 1:1, (по объему) + 5 мМ TBAP и 58% пропан-2-ол:Н2O 9:1 (по объему) + 5 мМ TBAP.
Пример 5
2-Амино-2-деокси-6-O-(2-амино-2-деокси-4-O-фосфоно- -D-глюкопиранозил)- α-D-глюкопиранозилдигидрофосфат [Holst et al. Eur. J. Biochem. 214 (1993) 695-701] обрабатывали в метаноле с метоксидом натрия (точно 4,0 мол. эквив.) и затем (R)- 3-додеканоилокситетрадекановым ангидридом (2,2 мол. эквив.) [полученный взаимодействием (R)-3-додеканоилокситетрадекановой кислоты с DDC (0,5 мол. эквив.) в безводном дихлорметане, см. Charon et al. J. Chem. Soc. Perkin Trans. 1. (1984) 2291-2295]. Через 12 часов при комнатной температуре добавляли воду (2-кр. объем по отношению к метанолу) и смесь экстрагировали диэтиловым эфиром (для удаления 3-додеканоилокситетрадекановой кислоты). Водную фазу концентрировали и сырой дисахарид C по изобретению подвергали ЖХВР с обращенной фазой. Продукт растворяли в H2O:TEA 1000:1 (по объему) и добавляли тетрабутиламмоний (TBAP)до концентрации 5 мМ. Этот раствор затем наносили на препаративную колонку ЖХВР (Millipore-Waters Bondapak C18 300  15М, 300 мм х 47 мм). Дисахарид C элюировали градиентом CH3CN:H2O 1:1, (по объему) + 25 мМ TBAP (раствор A) и пропан-2-ола:H2O 9:1 (по объему) +25 мМ TBAP (раствор B) (50%A / 50%B - 0%A-:100%В при 1%/мин, скорость 80 мл/мин.). Обессоливание проводили следующим образом. Фракцию ЖХВР, содержащую дисахарид C, разводили водой, затем наносили на колонку C18-Sep Pak Vak Plus (Waters) [C18 силикагель с обращенной фазой, последовательно прекондиционированной CHCl3:CH3ОН 2:1 (по объему), CH3CN, CH3CN:H2O 1:1, (по объему)]. TBAP удаляли путем последовательного промывания CH3CN:H2O 1:1, (по объему), 10 мМ HCl и CH3CN. Чистый дисахарид C элюировали CHCl3:CH3OH 2:1, (по объему).
15М, 300 мм х 47 мм). Дисахарид C элюировали градиентом CH3CN:H2O 1:1, (по объему) + 25 мМ TBAP (раствор A) и пропан-2-ола:H2O 9:1 (по объему) +25 мМ TBAP (раствор B) (50%A / 50%B - 0%A-:100%В при 1%/мин, скорость 80 мл/мин.). Обессоливание проводили следующим образом. Фракцию ЖХВР, содержащую дисахарид C, разводили водой, затем наносили на колонку C18-Sep Pak Vak Plus (Waters) [C18 силикагель с обращенной фазой, последовательно прекондиционированной CHCl3:CH3ОН 2:1 (по объему), CH3CN, CH3CN:H2O 1:1, (по объему)]. TBAP удаляли путем последовательного промывания CH3CN:H2O 1:1, (по объему), 10 мМ HCl и CH3CN. Чистый дисахарид C элюировали CHCl3:CH3OH 2:1, (по объему).
Пример 6
Водную фазу, содержащую дисахарид C, полученный по примеру 5 (до очистки), обрабатывали водным раствором гидроокиси натрия (точно 1,0 моль. эквив. ; концентрация, приводящая к начальному значению pH 12,5); через 24 часа при комнатной температуре смесь доводили до pH 6,5-7 и наносили на препаративную колонку ЖХВР (Millipore-Waters Bondapak C18 300  15М, 300 мм х 47 мм). Дисахариды A и D элюировали градиентом CH3CN:H2O 1:1, (по объему) + 25 мМ TBAP (раствор A) и пропан-2-ол:H2O 9:1, (по объему) + 25 мМ TBAP (раствор B) (75%A: 25%B - 0%A: 100%B при 1%/мин, скорость тока 80 мл/мин). Фракции ЖХВР, содержащие дисахариды A и D обессоливали, как описано для дисахарида C в примере 5.
15М, 300 мм х 47 мм). Дисахариды A и D элюировали градиентом CH3CN:H2O 1:1, (по объему) + 25 мМ TBAP (раствор A) и пропан-2-ол:H2O 9:1, (по объему) + 25 мМ TBAP (раствор B) (75%A: 25%B - 0%A: 100%B при 1%/мин, скорость тока 80 мл/мин). Фракции ЖХВР, содержащие дисахариды A и D обессоливали, как описано для дисахарида C в примере 5.
Пример для сравнения (не по изобретению)
Раствор липида A 10 мг/мл из Escherichia coli F-583 (продукт Sigma N L5399) получали в смеси H2O:триэтиламин в соотношении 1000:1, (по объему) и затем подвергали щелочной обработке 0,2 М NaOH при 37oC в течение 20 минут. Этот период времени был достаточен только для 3-O деацилирования липида A (Myers et al. in Cellular and Molecular Aspects of Endotoxin Reactions, pages 145-156 [1990], Elsevier Science Publishers).
Раствор нейтрализовывали ортофосфорной кислотой. Для биологических определений его разводили в 0,1% TEA/0,9% NaCI и использовали без дальнейшей очистки.
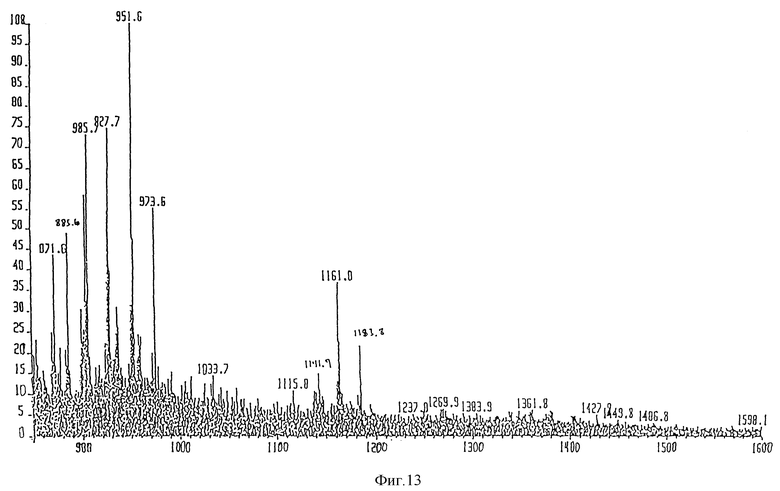
Для FAB-MS образец этого обработанного щелочью липида А очищали при помощи ЖХВР с обращенной фазой (Supelco N 58985, Supelcosil LC18, 3 мкм, 15 мм х 4,6 мм). Главный пик, элюирующийся при 18% CH3CN:H2O 1:1, (по объему) + 5 мМ TBAP и 82% пропан-2-ол:H2O 9:1, (по объему) + 5 мМ TBAP, обессоливали в условиях, описанных в примере 1. Анализ FAB-MS дал молекулярный ион 1570,1 единиц массы (расчетный 1569,1 единиц массы).
Физико-химические характеристики дисахаридов по изобретению
Дисахариды A и B, полученные в примерах 1 и 2 были подвергнуты физико-химически охарактеризованы.
Глюкозамин определяли после кислотного гидролиза (4 М HCl, 16 часов, 100oC, атмосфера аргона) и получения производных, с использованием фенилизотиоцианата и последующего количественного анализа при помощи ЖХВР (см. Anumula, K. R. et al, Analiti-cal Biochemistry, Volume 179, pages 113-122 [1991]).
Общее содержание жирных кислот определяли после кислотного гидролиза (4 М HCl, 4 часа, 100oC), путем метилирования с использованием BF3 в присутствии метанола, и количественного определения методом газовой хроматографии (колонка OV-1, Hewlett Packard) (см. Miller, L., Gas-Liquid Chromatography of Cellular Fatty Acids as a Bacterial Identification Aid, Gas Chromatography Application Note, pages 228-237 [1984]).
Эфирно-присоединенные жирные кислоты определяли при помощи газовой хроматографии после обработки NaOCH3 (см. Rietschel, Е.Т. et al, European Jounal of Biochemistry, Volume 28, pages 166-173 [1972]).
Фосфат определяли по методу Эймса (см. Ames, B.N., Methods in Enzimology, Volume 8, page 115-118 [1966]).
3-Деокси-D-манно-2-октулосоновую кислоту (KDO) определяли по методу Karkhakis, Y. D. et al. (Analytical Biochemistry, Volume 58, pages 595-601 [1978]).
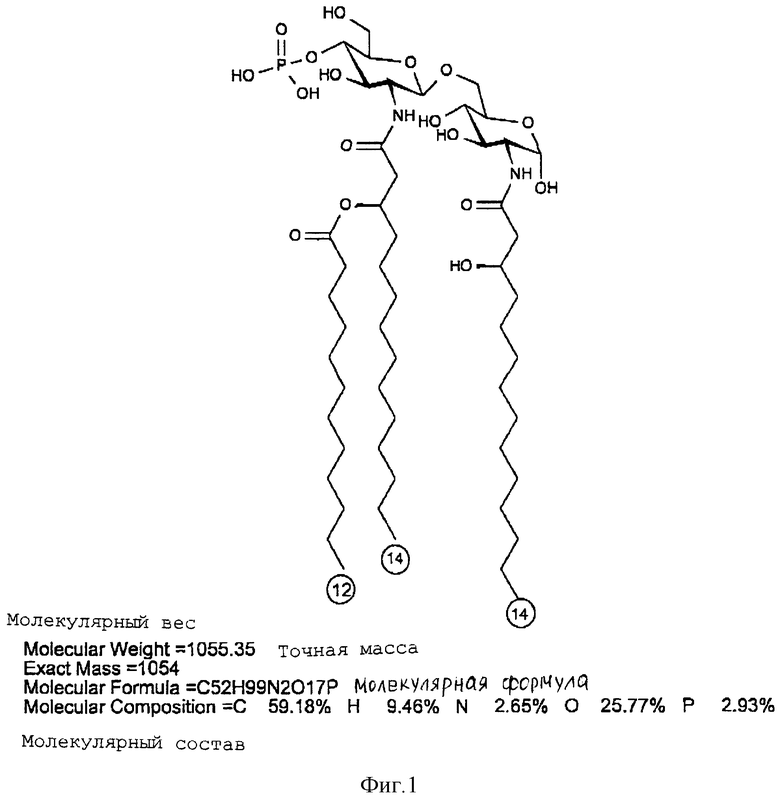
Раствор дисахарида A содержал 2,1 мкмоль/мл фосфата, 1,9 мкмоль/мл глюкозамина, 1,0 мкмоль/мл 012:0 жирной кислоты и 2,2 мкмоль/мл 3OH-C14:o жирной кислоты. Только C12:o жирная кислота была определена после высвобождения эфирно-присоединенных остатков жирной кислоты, свидетельствуя, что остатки 3OН-C14:o жирных кислот были связаны амидной связью. KDO не была обнаружена (< 1 моля на 10 молей дисахарида A).
Таким образом, дисахарид содержит на моль 2 моль фосфата, 2 моль глюкозамина, 2 моль 3OH-C14:o жирной кислоты и 1 моль C12:o жирной кислоты.
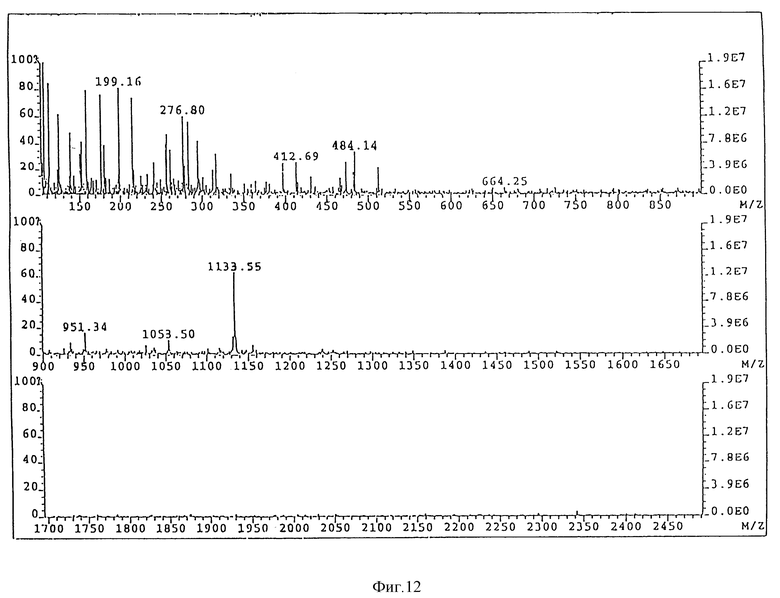
Масс-спектроскопия бомбардировкой быстрыми атомами (FAB-MS), в отрицательном режиме образца в CHCl3:CH3OH 1:1, (по объему), концентрация 1 мг/мл. VG ZAB-2SE масс-спектрометр, настроенный на Vасс 8 kV был использован для получения спектра при 30 kV и тока эмиссии 1 мкА. Спектрометр был калиброван, используя иодид цезия. FAB-MS спектр представлен на фиг. 1. Дисахарид A давал молекулярный пик на 1133,55 единицах массы (расчетная масса 1133,3). Другие пики предполагают расщепление продукта при анализе. Пик 1053,5 представляет потерю фосфатной группы, а 951,3 - потерю C12 жирной кислоты.
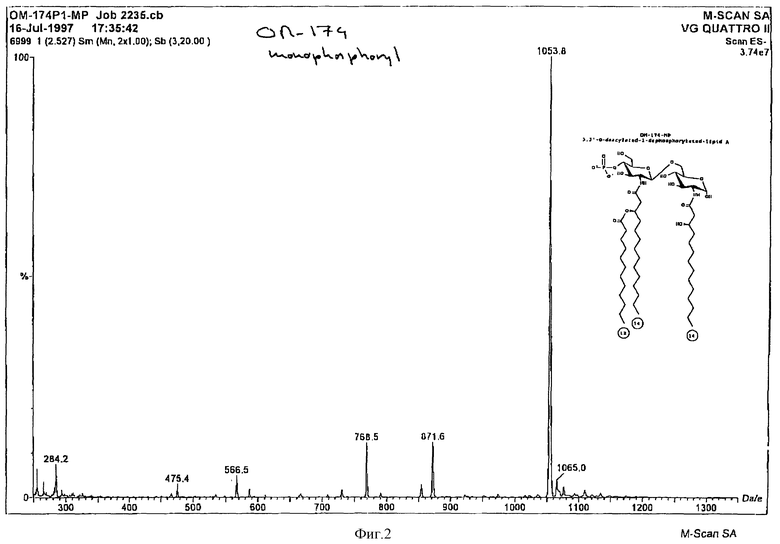
FAB-MS спектр дисахарида B представлен на фиг. 2 и проявляет молекулярный пик на 1161,8 единицах массы (расчетная 1161,3). Пик на 1183,8 единицах массы представляет добавление натрия. Пик на 951,6 представляет потерю C14 жирной кислоты. Пик на 973,6 единицах массы представляет фрагмент пика 951,6 вместе с ионом натрия.
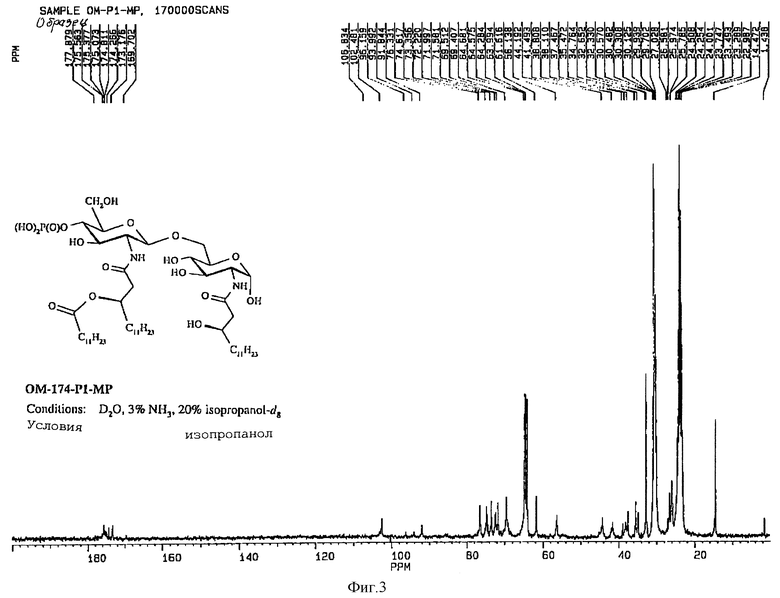
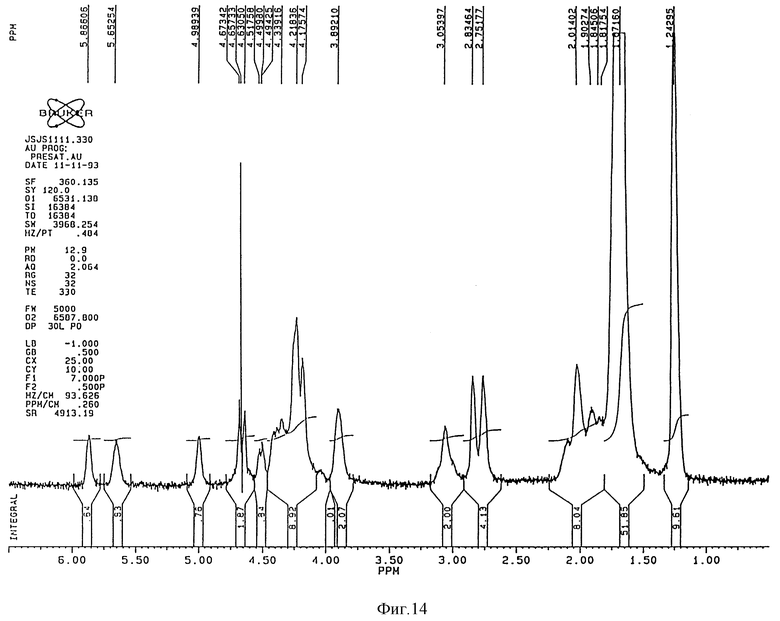
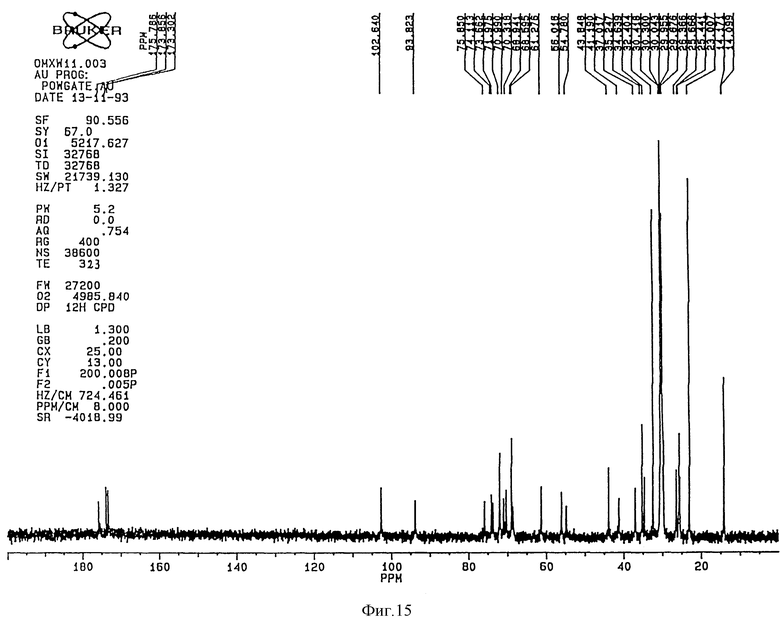
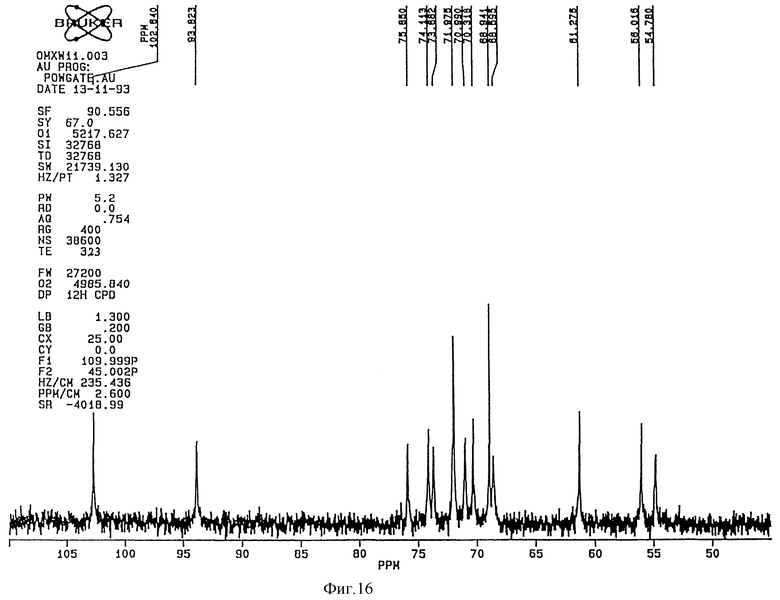
1H-ЯМР-спектр (Bruker 360 Mhz) дисахарида A (натриевая соль в D2O) представлен на фиг. 3 и его 13C-NMR-спектр (Bruker 90 Mhz) представлен на фиг. 4 и 5 (расширенная шкала).
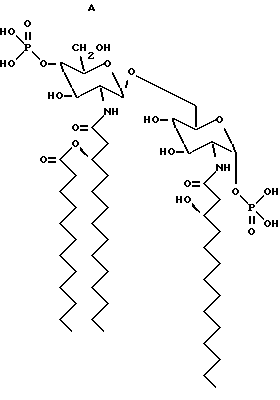
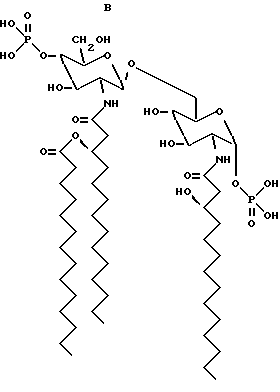
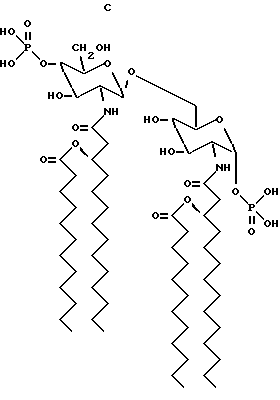
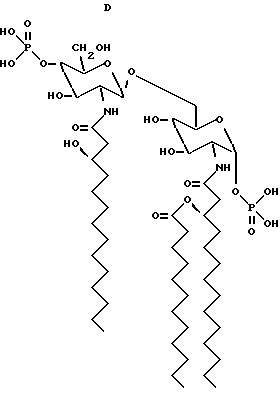
Структурные формулы следующих β- D-глюкозамин-(1-6 -α- -D- глюкозаминдисахаридов:
дисахарид A (2-Деокси-6-O-[2-деокси-2-[(R)-3- додеканоилокситетрадеканоиламино] -4-O-фосфоно -β- D-глюкопиранозил]-2- [(R)-3-гидрокситетрадеканоиламино] -α-D- глюкопиранозилдигидрофосфат);
дисахарид B (2-Деокси-6-O-[2-деокси-2-[(R)-3- тетрадеканоилокситетрадеканоиламино] -4-O-фосфоно -β-D- глюкопиранозил]-2-[(R)-3-гидрокситетрадеканоиламино] -α-D- глюкопиранозилдигидрофосфат);
дисахарид C (2-Деокси-6-O-[2-деокси-2-[(R)-3- додеканоилокситетрадеканоиламино] -4-O-фocфoнo -β-D- глюкопиранозил]-2-[(R)-3-додеканоилокситетрадеканоиламино] α -D-глюкопиранозилдигидрофосфат);
дисахарид D (2-Деокси-6-O-[2-деокси-2-[(R)-3- гидрокситетрадеканоиламино] -4-O-фосфоно -β-D- глюкопиранозил]- 2-[(R)-3-додеканоилокситетрадеканоиламино] - α-D- глюкопиранозилдигидрофосфат); описаны далее.
Эндотоксичность и биологическая активность дисахаридов по изобретению
Эндотоксичность и биологическую активность дисахарида A (пример 1) и дисахарида B (пример 2) определяли и сравнивали с липополисахаридом, выделенным из Escherichia coli 0111: 84 (Sigma, продукт N L3024), липида A (Sigma, продукт N L5399; использованного в качестве исходного продукта в примере 4), и 3-O- деацилированного липида A, полученного из липида A Escherichia coli F583 (полученного как в примере для сравнения), но не очищенного ЖХВР.
1. Эндотоксичность
Эндотоксичность была определена в Limulus amoebocyte лизатном (LAL) тесте. Этот тест основан на наблюдении, что эндотоксин индуцирует коагуляцию гемолимфы Limulus poliphemus.
В гельфикационном тесте были смешаны серийные разведения исследуемых соединений с LAL 1:1, (по объему) (Haemachem Inc., чувствительность LAL 0.06 единиц эндотоксина/мл) и смесь оставили инкубироваться на один час при 37oC. Образование геля регистрировали затем путем измерения оптической плотности при 405 нм. Последнее разведение, в котором образовывался гель определяли путем переворачивания реакционной микроплаты. Активность эндотоксина в образцах была определена путем сравнения с разведениями стандарта липополисахарида (1 единица эндотоксина = 0,1 нг LPS).
Эндотоксичность была определена также в хромогенном тесте, в котором активацию протеазы в LAL под действием LPS измеряли, используя хромоген (Ac-Ile-Glu-Ala-Arg-pNA; Bio Whittaker Kit N 50-650U). Образование цвета (высвобождение pNA (p-нитроанилина)) измеряли при 405 нм.
Образцы предварительно инкубировали при 37oC в течение 10 минут и затем добавляли содержащий хромоген LAL. Было определено время, требующееся для достижения оптической плотности 0,2 при 405 нм. Активность эндотоксина расчитывали в сравнении с контрольной кривой, полученной для стандарта LPS.
Результаты представлены в таблице 3 в виде: нг LPS на нг продукта.
Таким образом, из таблицы 3 видно, что дисахариды А и В по изобретению проявляют наименьшую эндотоксичность, в частности, в сравнении с 3-O-деацилированным липидом A, в соответствии с US-A-4 912 094.
2. Биологическая активность, индуцируемая у макрофагов C57BL/6 мышей, in vitro
Костный мозг был отобран из верхней части бедра, бедренной кости и большой берцовой кости самцов мышей C57BL/6, возрастом шесть недель. После гомогенизации костномозговой суспензии в модифицированной среде Дульбекко и центрифугирования осадок вновь суспендировали в модифицированной среде Дульбекко и клетки культивировали при концентрации 4•105 клеток в мл в той же среде, дополненной 30% супернатантом L-929 фибробластов (обычный источник для колоние-стимулирующего фактора 1 [CSF-1]) и 20% лошадиной сывороткой. Через 7 дней зрелые фибробласты собрали и вновь суспендировали в модифицированной среде Дульбекко, содержащей 5% телячью эмбриональную сыворотку в концентрации 7•106 клеток в мл. Эта клеточная суспензия была смешана 1:1, (по объему) с образцами, разбавленными той же средой, и использована для биологических исследований, проводимых на микроплатах с 70 000 клеток/ячейку (инкубация при 37oC в течение 22 часов, при 100% влажности и 8% CO2).
Образование окиси азота (NO)
Окись азота (NO) продуцируется макрофагами в ответ на бактериальную инфекцию и, в особенности, на LPS. NO, по-видимому, обладает цитостатическими и цитотоксическими свойствами. NO чрезвычайно реакционноспособна и быстро подвергается окислению в нитрит и нитрат.
Образование нитрита определяли, используя Griess тест (добавление 1:1, (по объему) N-(1-нафтил)этилендиамингидрохлорида [1 г/л на воде] и p-аминобензенсульфонамида [10 г/л на 5% H3PO4]). Концентрацию нитрита в супернатантах активированных макрофагов рассчитывали в сравнении со стандартами NaNO2.
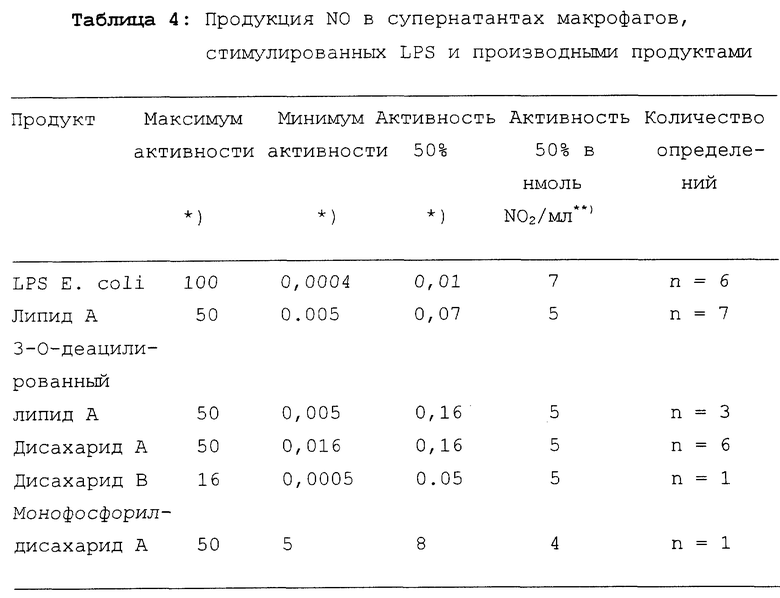
Результаты представлены в таблице 4.
LPS индуцирует наивысшую продукцию NO. Липид A, 3-O- деацилированный липид A, дисахарид A и дисахарид B индуцируют продукцию NO того же порядка.
Таким образом, дисахариды A и B индуцируют продукцию NO в макрофагах так же эффективно, как и липид A и 3-O-деацилированный липид A.
Продукция интерлейкина-1 (IL-1)
IL-1α продуцируется многими клетками, включая макрофаги, при стимулировании LPS. Некоторые данные об активностях IL-1 включают активацию Т-клеток, индукцию экспрессии IL-2 рецептора и экспресии гена цитокина в Т-клетках, ко-стимуляцию пролиферации B-клеток и секрецию Ig, увеличение IL-2 и IFN-индуцированной активации NK-опосредованной цитотоксичности, индукцию острой фазы синтеза белка и индукцию повышения температуры.
Концентрацию IL-1α в супернатантах макрофагов измеряли путем ELISA-теста (Kit GENZIME, Intertest-I).
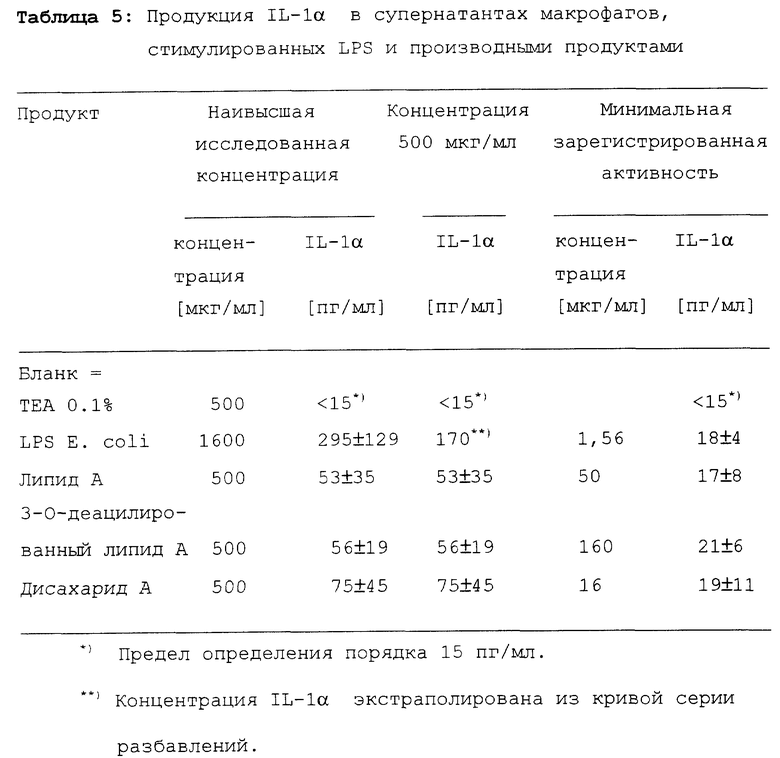
Результаты обобщены в таблице 5.
Определить максимальную продукцию IL-1 α, не представлялось возможности, поскольку продукция все еще возрастала при использовании наивысших концентраций.
Индукция продукции IL-1 α при концентрации 500 мкг/мл существенно не различалась для липида A, 3-O-деацилированного липида A и дисахарида A. LPS индуцирует продукцию IL-1 более сильно.
Продукция IL-1α под действием дисахарида A, по крайней мере, индуцируется так же сильно, как и липидом A и 3-O- деацилированным липидом A.
Продукция интерлейкина-6 (IL-6)
IL-6 продуцируется активированными моноцитами и макрофагами, T- и B- лимфоцитами. IL-6 индуцирует, среди прочего, пролиферацию определенных типов клеток, ингибирование роста определенных клеточных линий меланом, дифференцировку B-лимфоцитов и стимуляцию секреции IgG, дифференцировку цитотоксических T-клеток и слабую антивирусную активность. Концентрацию IL-6 в супернатантах макрофагов определяли с помощью ELISA теста (Kit ENDOGEN, EM-IL- 6).
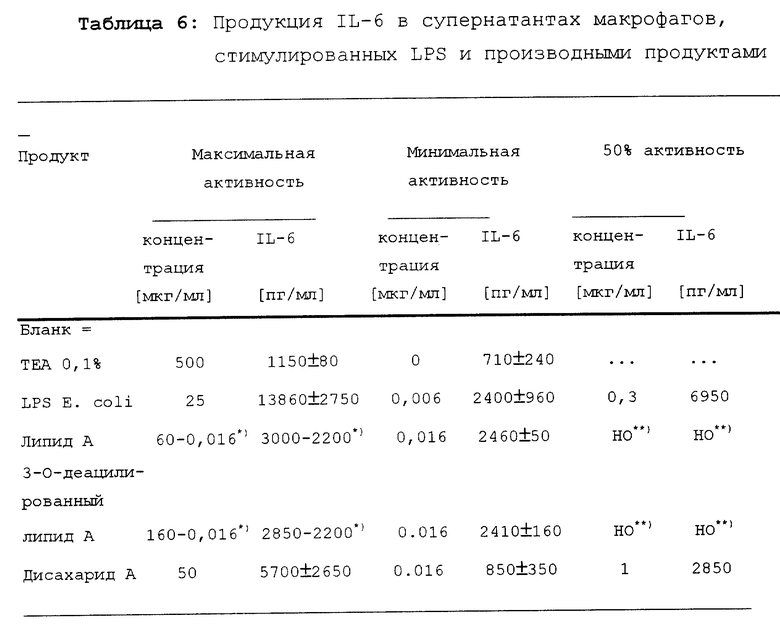
Результаты обобщены в таблице 6.
Стимуляция секреции IL-6 под действием дисахаридом A существенно ниже, чем под действием LPS. Однако дисахарид A индуцирует продукцию IL-6 в макрофагах более сильно, чем липид A и 3-O-деацилированный липид A.
Продукция фактора некроза опухолей альфа (TNF -α);
TNF-α продуцируется главным образом макрофагами и моноцитами, стимулированными LPS. Активностями, индуцируемыми TNF-α являются антивирусная активность, цитолиз и цитостазис определенных типов клеток, рост определенных клеточных линии, экспрессия антигена, такого как главный комплекс тканевой совместимости I и II класса, некроз индуцированной метилхолантреном саркомы, активация полиморфноядерных лейкоцитов (PMN), активация остеокластов и резорбция кости. TNF-α является также главным медиатором при токсическом шоке и сепсисе.
Концентрацию TNF-α супернатантах макрофагов определяли с помощью ELISA теста (Kit GENZYME, Factor-test mTNF).
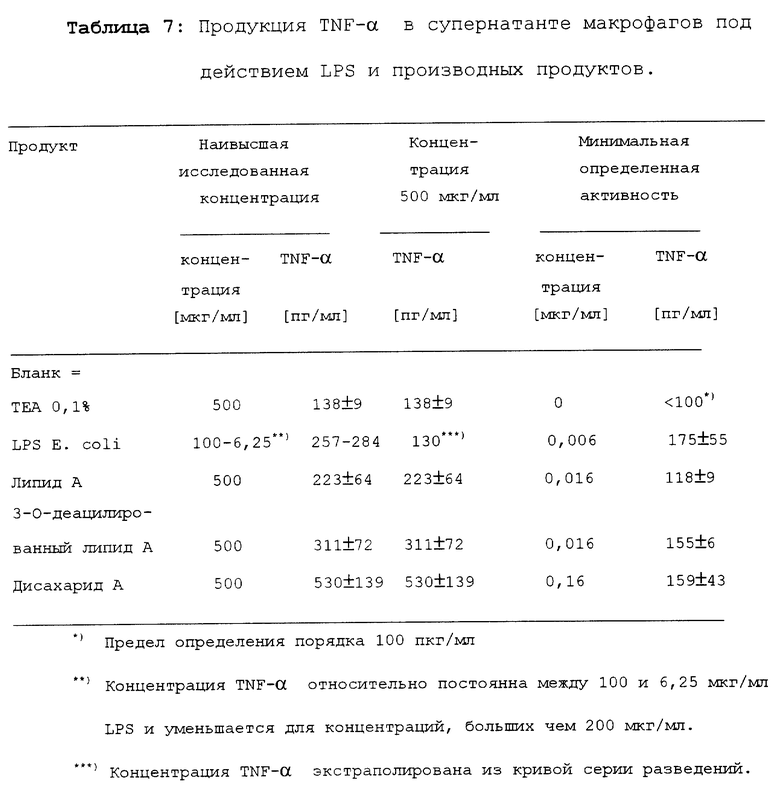
Результаты представлены в таблице 7.
Определить максимальную продукцию TNF-α не представлялось возможным, поскольку продукция продолжала возрастать при наиболее высоких исследуемых концентрациях.
В отличие от других исследований, продукции TNF-α, индуцированная LPS, была ниже, чем для дисахарида A. Дисахарид A индуцировал TNF-α так же или более сильно, чем липид A или 3-O-деацилированный липид А.
Продукция простагландина E2 (PGE2)
PGE1 и PGE2 являются основными метаболитами арахидновой кислоты, синтезируемыми макрофагами при стимуляции LPS, TNF-α, или IL-1. Простагландины E проявляют иммуномодулирующию активность на T- и B-лимфоцитах. Они, по-видимому, индуцируют стимуляцию Th2 и ингибирование Th1 субпопуляций T-лимфоцитов и переключают изотип иммуноглобулинов.
Концентрацию PGE2 в супернатанте макфрофагов определяли с помощью RIA-теста (Kit PAESEL + LORET, 36-104-6001 Prostaglandin E2 3H-RIA Kit).
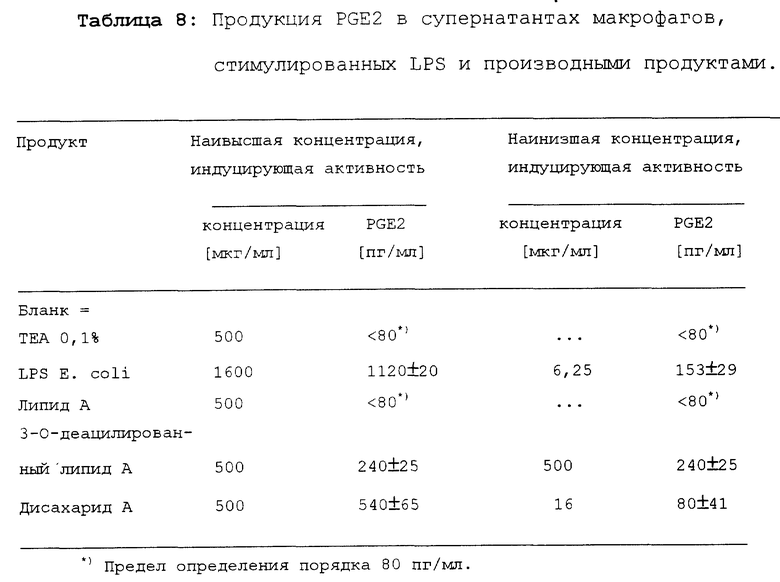
Результаты представлены в таблице 8.
Определить максимальную продукцию PGE2 не представлялось возможным, поскольку продукция продолжала возрастать при использовании наивысших концентраций. Стимуляция продукции PGE2 под действием дисахарида A значительно ниже, чем под действием LPS. Однако дисахарид A более активен, чем липид A и 3-O-деацилированный липид A. Липид A не индуцировал продукцию PGE2, а 3-O-деацилированный липид A индуцировал только в наивысших использованных концентрациях.
Заключение
Дисахариды по изобретению проявляют активность in vitro и индуцируют продукцию NO, IL-1α, IL-6, TNF-α и PGE2. Дисахариды по изобретению так же активны, или даже более активны, чем липид A, но проявляют существенно пониженную эндотоксичность, что определено с помощью LAL теста. Более низкая активность липида A и 3-О-деацилированного липида A может быть связана с различием в чистоте. Дисахариды A и B очищали при помощи ЖХВР, липид A является коммерческим биологическим препаратом (Sigma L-5399), а 3-O-деацилированный липид A получали в соответствии с US-A-4 912 054, исходя из коммерческого препарата липида A. Единственным продуктом с большей активностью является LPS, который индуцирует, обычно, более высокий ответ и требует меньшей концентрации для индукции значительного сигнала.
3. Биологическая активность in vivo
Биологическую активность дисахарида A in vivo исследовали по противоопухолевой активности в случае перитонеального карциноматоза, вызываемого у BDIX крыс. Pro b клетки, полученные согласно методу Martin (Martin, F. et al, International Journal of Cancer, Volume 32, pages 623-627 [1983]) были инъецированы внутрибрюшинно крысам (106 клеток на крысу). Через 10 дней в мезентерии в молочных сосках появляются многочисленные твердые узлы, проникающие в брюшную полость (см. Lagadec, P. et al. Invasion and Metastasis, Volume 7, pages 83-95 [1987]). Геморрагический асцит появился через 4-5 недель и все крысы погибли через 8-12 недель.
Иммунотерапию начали через 14 дней после инъекции опухолевых Pro b клеток. Лечение заключалось во внутрибрюшинных инъекциях дисахарида A: дозы 0,1; 0,3 и 0,8 мг/кг веса тела. Дисахарид A разводили в водном растворе 0,9% NaCl и 0,1% триетиламина. Крысы получили пять инъекций один раз каждые 3,5 дня. Контрольной группе вводили водный раствор. Обе группы включали по 10 крыс.
Через 6 недель после инъекции опухолевых клеток была взята аутопсия. Слепым методом была оценена степень перитонеального карциноматоза и крыс классифицировали в порядке развития карциноматоза.
Классификация размеров узелков следующая:
класс 0: нет видимых опухолевых узелков;
класс 1: редкие узелки размером менее чем 0,2 см;
класс 2: узелков размером до 0,5 см слишком много для подсчета;
класс 3: опухоли размером до 1 см;
класс 4: опухолевая полость полностью инвазирована опухолями, размер несколько см.
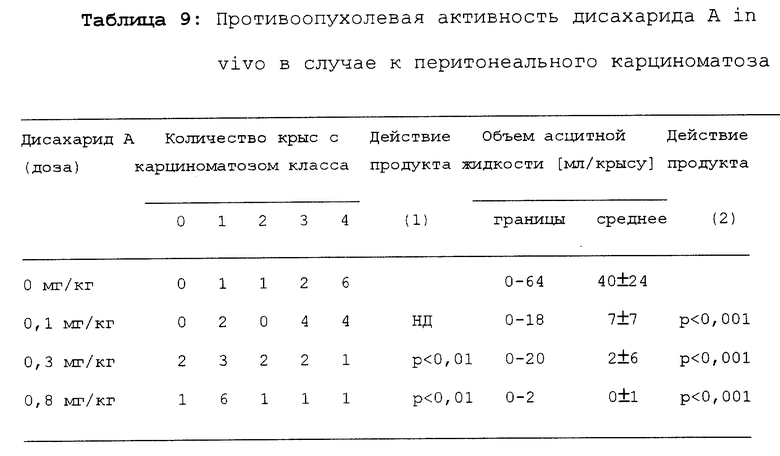
Результаты представлены в таблице 9.
Статистическая значимость противоопухолевой активности была подсчитана согласно Krukal-Wallis тесту (1) или (2), используя вариантный анализ.
Очевидно, дисахарид A проявляет дозо-зависимое противоопухолевое действие.
4. Острая токсичность
Дисахарид A вводили в хвостовую вену самцов и самок NMRI мышей (возрастом 6-7 недель). Дозы вплоть до 100 мг/кг не вызывали никакой смертности.
Пример 7
Исходным продуктом является липополисахарид из Pseudomonas aeruginosa (Sigma, продукт N L7018). Структура липида A уже известна (см. Kulshin et al. in Eur. J. Biochem. 198 (1991) 697-704). В отличие от липида A из E. coil, преобладающие виды содержат ацилоксиацильные остатки по обеим аминогрууппам основной цепи глюкозаминдифосфата.
Более того, по положению 3' находится 3-гидроксидекановая кислота, но такой ацильный остаток жирной кислоты присутствует в положении 3 лишь у незначительной фракции.
Удаление этого ацильного остатка жирной кислоты по положению 3' могло бы привести к аналогу дисахарида С. Дальнейший гидролиз приведет к потере этерифицированного ацильного остатка жирной кислоты при ацилоксиацильной группе как по положению 2, так и 2'. Данные структуры аналогичны дисахаридам C и D.
Липополисахарид от Ps.aeruginosa (Sigma L7018) растворяли в 0.1 М ацетате натрия с pH 4.0 до 5 мг/мл и нагревали в течение 120 мин при 100oC. После охлаждения добавляли 0.5 объема пропан-2-ола, после чего добавляли фосфат тетрабутиламмония (TBAP) так, чтобы конечная концентрация была 25 мМ. Добавляли триэтиламин (TEA) до pH 9.0 (приблизительно, рH-бумажки). Смесь была добавлена к C18Sep-Pak (Waters) с рециркуляцией (10 прогонов). Sep-Pak был промыт 10 мл 5 мМ TBAP в ацетонитриле:H2O 1:1 (по объему), а затем 10 мл ацетонитрила. Адсорбированные вещества были отмыты 4 мл хлороформ:метанол 2: 1 (по объему, %).
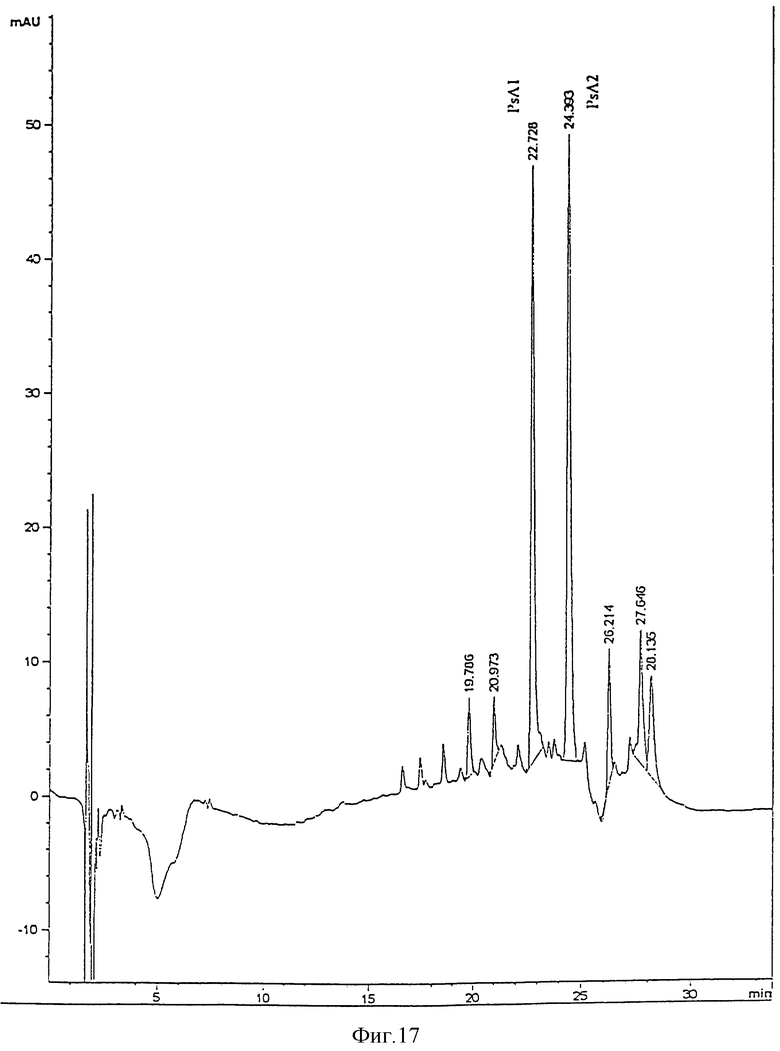
Два главных пика, PsA1 и PsA2 (фиг. 6), очищали с помощью ЖХВР и жирные кислоты анализировали. Композиция жирных кислот соответствует молекулам, описанным Kulshin et al. (таблица ниже). PsA1 менее гидрофобен, чем PsA2, так как он вымывается из колонки с меньшим временем удерживания (Rt). Это соответствует молекуле с двумя 2OH-C12:O кислотным остаткам. PsA2 имеет и 2OH-C12:O, и C12:O.
Пик - Идентифицированная жирная кислота
Ps1 - 3OH-C10:O - 2OH-C12:O - 3OH-C12:O
PsA2 - 3OH-C10:O - C12:O - 2OH-C12:O - 3OH-C12:O
Растворитель удаляли ротационным упариванием, а остаток вновь растворяли в 0.2% TEA на воде.
Гидроксид натрия добавляли к раствору липида A Ps.aeruginosa до концентрации 0.2 М, и раствор инкубировали при комнатной температуре в течение 60 мин. Затем раствор нейтрализовали (8.5%) ортофосфорной кислотой. Его затем помещали в систему ЖХВР с обращенной фазой (HP1050 c Supelco LC18, 3 мкм обращенно-фазовая колонка, с предколонкой), уравновешенной в 75% раствора A (5 мМ TBAP) в ацетонитрил:вода 1:1 (по объему)), 25% раствора B (5 мМ TBAP в пропан-2-ол:вода 9:1 (по объему) и промытой градиентом от 2 % раствора В/мин до 100% B. Пики регистрировали по поглощению при 210 нм. Главные пики собирали (см. фиг. 7). Их разбавляли 2 объемами воды и наносили на картриджи C18 Sep Pak, уравновешенных раствором A. Sep Pak и промывали 10 мл 0.45% хлорида натрия в пропан-2-оле:H2O 1:3 (по объему), 10 мл воды и 10 мл ацетонитрила. Адсорбированные вещества отмывали 4 мл хлороформ:метанол 2:1 (по объему), а растворители удаляли в атмосфере азота. Фракции вновь растворяли в 100 мкл воды.
Содержание ацилов жирных кислот во фракциях анализировалось методом газовой хроматографии после гидролиза в 4 М HCl, 100oC, 4 ч. Высвобожденные жирные кислоты превращали в метильные эфиры по Miller (Miller, Gas Chromatography application note 228-37 [Hewlett Packard]) и анализировали на Hewlett Packard 5890 газовой хроматограмме с колонками со сплавленным кварцем (Supelco 2-4026) с контролем по стандартным метильным эфирам жирных кислот (Supelco).
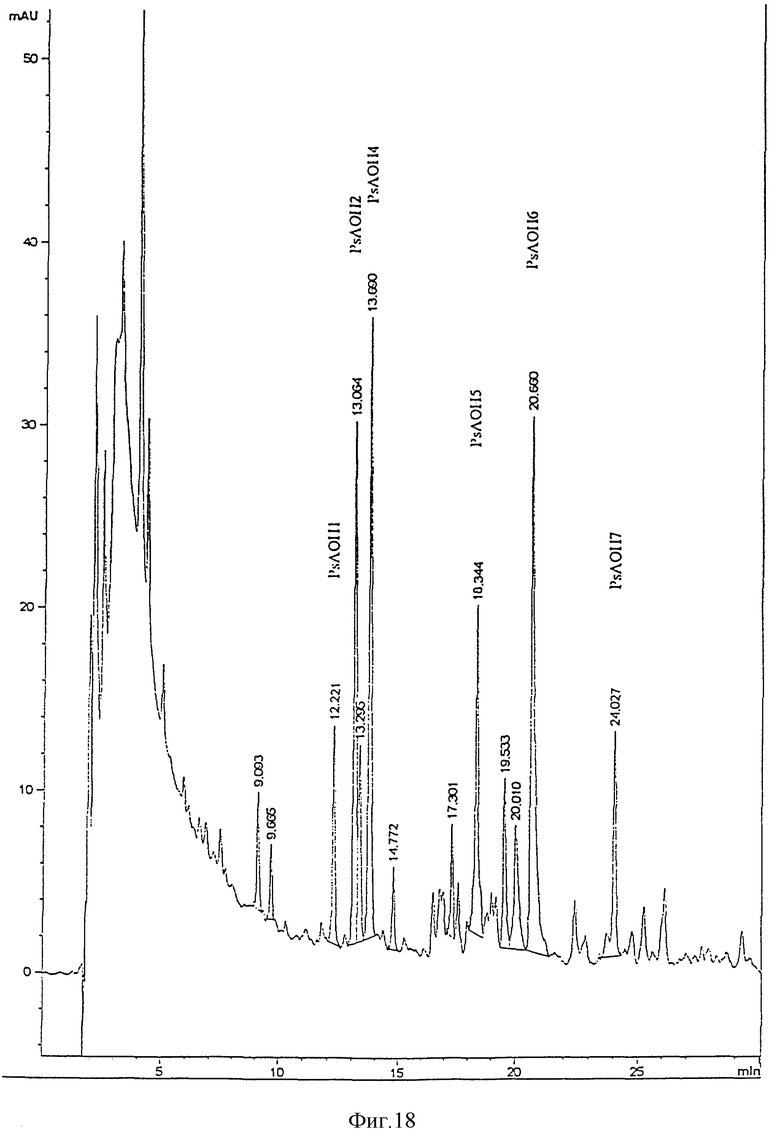
После гидролиза экстракта липида A, на обращенно-фазовой ЖХВР наблюдается множество пиков. Из ЖХВР были отобраны главные пики (PsAOH 1, 2, 4 и 6). Жирные кислоты, идентифицированные в каждой фракции, приведены ниже.
Пик - Идентифицированная жирная кислота
PsAOH1 - 3OH-C12:O - 2OH-C12:O
PsAOH2 - 3OH-C12:O - 2OH-C12:O
PsAOH4 - 3OH-C12:O - C12:O
PsAOH6 - 3OH-C12:O - 2OH-C12:O - C12:O
Структурные формулы данных дисахаридов следующие:
PsAOH1: 2-Деокси-6-O-[2-деокси-2-[(R)-3-[(5)-2- гидроксидодеканоилокси] додеканоиламино] -4-O-фосфоно -β-D- глюкопиранозил]-2-[(R)-3-гидроксидодеканоиламино -α-D- глюкопиранозилдигидрофосфат;
PsAOH2 2-Деокси-6-O-[2-деокси-2-[(R)-3- гидроксидодеканоиламино]-4-O-фосфоно -β-D- глюкопиранозил]-2-[(R)-3-[(R)-3-[(S)-2-гидроксидодеканоилокси] додеканоиламино]-α-D-глюкопиранозилдигидрофосфат;
PsAOH4 2-Деокси-6-O-[2-деокси-2-[(R)-3- додеканоилоксидодеканоиламино] -4-O-фосфоно -β-D- глюкопиранозил]-2-[(R)-3-гидpoкcидoдeкaнoилaминo]- -α-D- глюкопиранозилдигидрофосфат;
PsAOH6 2-Деокси-6-O-[2-деокси-2-[(R)-3- додеканоилоксидодеканоиламино] -4-O-фосфона- -β-D- глюкопиранозил] -2-[(R)-3-[[(S)-2- гидроксидодеканоилокси]додеканоиламино] -α-D- глюкопиранозилдигидрофосфат;
формулы изобретения ниже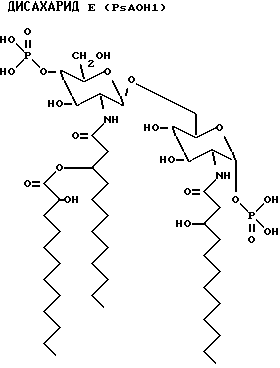
Фракции также анализировались с помощью электровспрыскивателей масс-спектроскопии (ES-MS) в отрицательном режиме. Использовался прибор VG Biotech Bio-Q с тройным четырехполюсным анализатором. От 2 до 4 мкл каждого образца разбавляли 10 мкл раствора ацетонитрил: вода: 25% раствор аммиака, 50: 50: 1 (по объему). 10 мкл затем впрыскивали непосредственно в источник масс- спектрометра. Раствор ацетонитрил:вода 25% раствор аммиака, 50:50:1 (по объему), 7 мкл/мин использовался в качестве элюанта. Для анализа дробления основных ионов, ионы, исходящие из первого четырехполюсника, подвергались разложению, активированному столкновением во втором четырехполюснике, с использованием аргона в качестве газа столкновения. Дочерние ионы обнаруживали в третьем четырехполюснике.
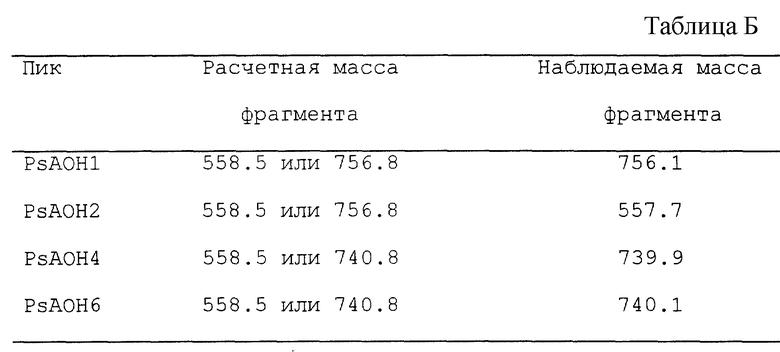
Масса, расчетная для каждого из пиков, и масса, наблюдаемая с помощью ES-MS, даны в таблице A
Налицо очень хорошее соответствие в каждом случае.
В случае PsAOH1 и PsAOH2 массы идентичны. Это свидетельствует о том, что они представляют собой две изоформы молекулы, известно, что липиды A расщепляются при определенных аналитических условиях в масс-спектрометрии (Kulshin, 1991; и Cotter et al. , Biomed. End. Mass Spectrom. 14(1987), 591-598). Производятся ионы, которые представляют собой невосстанавливающую половину молекулы с увеличением на 102 единицы массы. Таким образом, имея два MS, образующих тандем, основной ион в первом MS можно расщепить, а "дочерние" ионы обнаружить во втором MS. Это исключает вероятность того, что наблюдаемые вторичные ионы являются загрязненными; они должны происходить из исходного иона путем расщепления. Масса дочерних ионов, ожидаемых для каждой фракции, и массы, наблюдаемые на ES-MS, показаны в таблице Б.
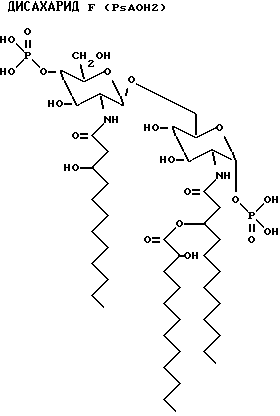
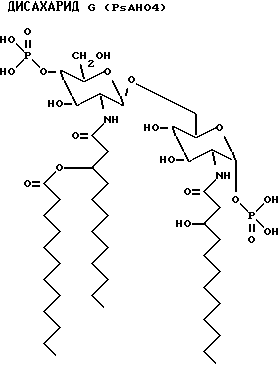
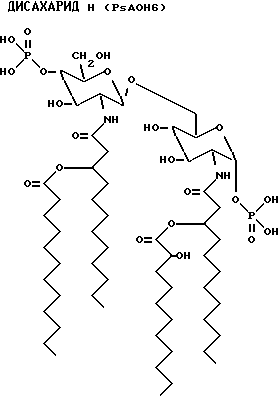
Данные наблюдения ясно идентифицируют структуру дисахаридов E, F, G и Н.
Для внутреннего сравнения дисахарид A также проанализирован с помощью ES-MS. Расчетная масса была 768.9, а наблюдаемая масса фрагмента 768.
Биологическая активность фракций была исследована по стимуляции продукции нитрита перитонеальными макрофагами мыши, как описано выше.
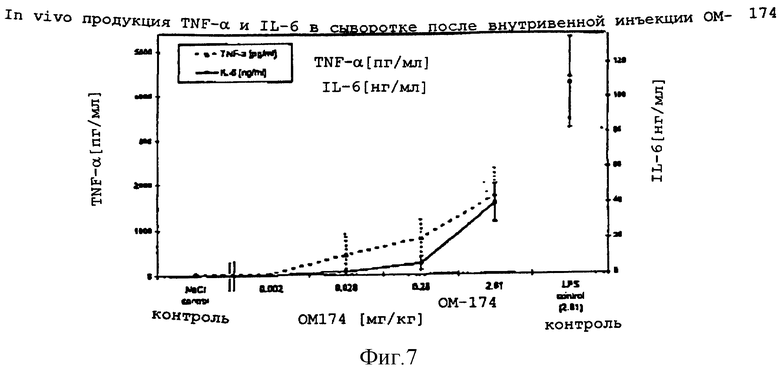
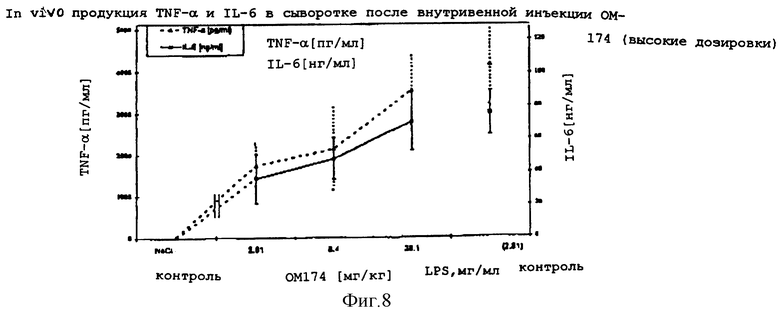
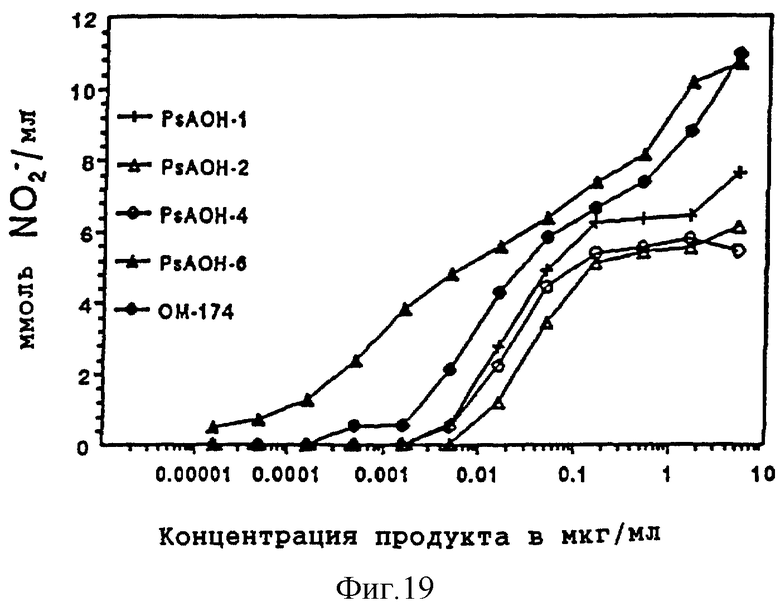
Количество каждого аналога в растворе исходного продукта определяли по поглощению на ЖХВР с контролем в виде таковой дисахарида A. Фракции проявляют активности того же порядка, что и дисахарид A (фиг. 8). Дисахарид H, который имеет две ацилоксиацильные группы и не имеет никаких других ацильных остатков жирных кислот, наиболее активен. Положение ацилоксиацила, 2 в сравнении с 2', лишь незначительно влияет на активность.
В целях исключения возможности того, что активность возникла благодаря загрязнению образцов другими веществами, такими как ЛПС, дисахариды T, F, G и H повторно очищали на обращенно-фазовой ЖХВР, и регионы базовой линии ЖХВР непосредственно до и непосредственно после пика также отбирались и обрабатывались таким же образом, как и фракции, содержащие пики продукта. Исследовалась активность пиковых фракций и фракций базовой линии. Пики, содержащие дисахариды E, F, G и H, проявляли такую же активность, какая наблюдалась в первом исследовании. Пустые образцы, представляющие собой районы профиля ЖХВР непосредственно до и непосредственно после пика аналога липида A, были неактивны. Стимуляция продукции нитрита, таким образом, специфически связана с аналогами липида А.
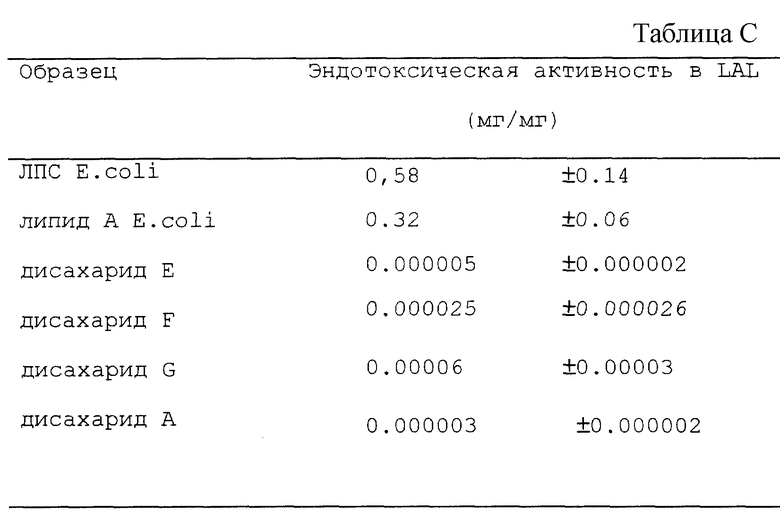
Эндотоксичность дисахаридов E, F и G определяли, используя хромогенное исследование LAL (смотри выше). Однако, вместо использования 1 мг/мл бычьего сывороточного альбумина, использовали 0.1 мг/мл. Результаты (n=4 или 6) получены в двух сериях экспериментов и показаны в таблице С.
Монофосфорил ОМ-174 (ОМ-174-МР)
Получение
ОМ-174 (607 мг) растворяли в 38.4 мл воды для инъекций. В растворе создавали 0.1 М концентрацию HCl с помощью 4,2 мл HCl, после чего раствор нагревали при 95oC в течение 30 минут. После этого раствор охлаждали до комнатной температуры. Добавляли изопропанол (42,6 мл) и с помощью триэтиламина устанавливали pH равным 9.
ЖХВР: очистку осуществляли с помощью обращенно-фазовой ЖХВР на Bondapack C18 (Waters, колонка 47х300 мм). Вкратце, такую операцию проводили разбавлением концентрированного раствора 2000 мл смеси вола: изопропанол, в объемном соотношении 9:1, после чего создавали 50 мМ концентрацию (NH4)HCO3, и смесь пропускали через ЖХВР колонку. Очистку проводили с использованием следующей системы растворителей:
Подвижная фаза A = вода:изопропанол, 9:1, об./об., с установленной 50 мМ концентрацией (NH4)HCO3
Подвижная фаза B = вода:изопропанол, 2:8, об./об., с установленной 50 мМ концентрацией (NH4)HCO3
Градиент: от 40% B до 74% B в течение 10 минут; затем в течение 0.1 мин от 74% B до 78% B; затем в изократном режиме при 78% B в течение 30 минут, при объемной скорости 40 мл/мин. Детекцию проводили при длине волны 210 нм. Фракцию ОМ-174-MP объемом 425 мл элюировали в промежутке времени между 20 и 28 минутами.
Солевой обмен: аммониевую соль ОМ-174-МР адсорбировали на анионообменной смоле (Q-Sepharose-для быстрого потока, четвертичный аммоний, Pharmacia), промывали 1000 мл смеси вода: изопропанол, в объемном соотношении 1:1 и элюировали 1900 мл смеси вода:изопропанол в присутствии 10 г NaCI, 1:1 (по объему) в виде натриевой соли ОМ-174-МР.
Окончательная ЖХВР очистка: Q-Sepharose фракцию разбавляли 1 объемом воды и адсорбировали на обращенно-фазной ЖХВР колонке, Bondapack C18 (Waters, колонка 40х200 мм), которую прекондиционировали в смеси вода: изопропанол, 3: 1, (по объему). Элюирование осуществляли в использованием следующей системы растворителей:
Подвижная фаза A = вода
Подвижная фаза B = изопропанол
Градиент: 10% B в течение 12 мин, затем от 10% B до 100% B в течение 1 минуты; затем изократно при 100% B в течение 20 минут, при объемной скорости 100 мл/мин. Детекцию проводили при длине волны 210 нм. Фракцию ОМ-174-МР объемом 142 мл элюировали в промежутке времени между 25,9 и 28 мин.
Изопропанольную фракцию выпаривали на Rotavapor и высушенный материал (примерно 0,4 г) повторно растворяли в 100 мл бидистиллированной воды, содержащей 0,1% триэтиламина.
Приготовление клеток для испытания пролиферации ствольных клеток костного мозга или продукции оксида азота макрофагами:
Собирали кости, включая тазовую, бедренную и большеберцовую, из задних ног самцов мышей C57/B16 в возрасте 6 недель. Костный мозг экстрагировали инъекцией среды через просвет кости после отрезания ее верхушки. Клетки собирали, гомогенизировали и центрифугировали в течение 5 мин при 200 хг. Затем их однократно промывали и ресуспендировали в свежей среде (Dulbecco' s Hank MEM).
Стволовые клетки костного мозга использовали непосредственно для тестирования пролиферации или подвергали дополнительной дифференцировке до превращения в зрелые макрофаги, которые стимулировали к индуцированию продукции оксида азота.
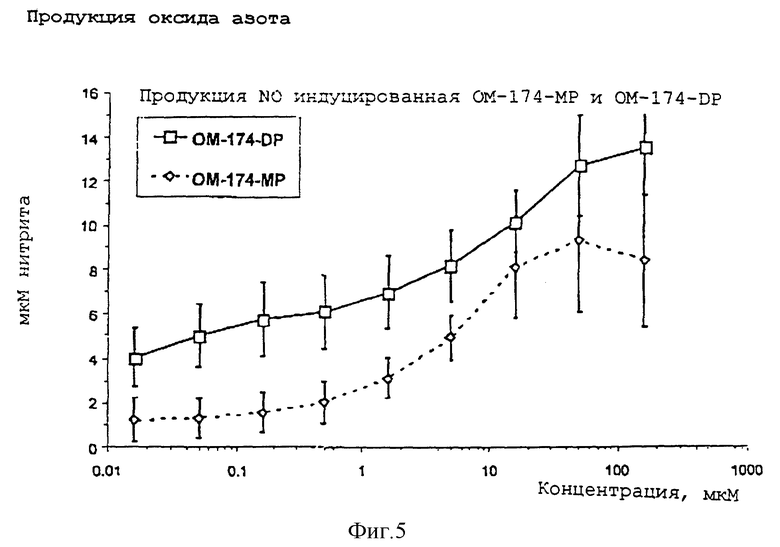
Продукция оксида азота
Дифференцировку стволовых клеток в макрофаги индуцировали культивированием 4•105 стволовых клеток в течение 8 дней, в среде, дополненной 20% лошадиной сыворотки и 30% L929 супернатантной кондиционированной среды. Супернатант L929 представляет собой среду, обогащенную M-CSF, которую получали в чашах Петри культивацией до слияния линии клеток мышиного фибробласта.
Через 8 дней инкубации большая часть клеток (>95%) превращалась в макрофаги присоединенные к чашам Петри, причем некоторые из клеток флоттировали в среде. Супернатанты сливали и после инкубации на холоду клетки могли быть разъединены пипетированием. Клетки центрифугировали, промывали и устанавливали концентрацию 7•105 макрофагов на мл DH среды, дополненной 5% фетальной телячьей сывороткой.
Испытуемые образцы, OM-174-DP (дифосфорил) или ОМ-174-МР (монофосфорил) серийно разбавляли до конечного объема 100 мкл. 100 мкл суспензии макрофагов распределяли по лункам микропланшета, содержащего разбавленный продукт, подлежащий тестированию. Такие продукты инкубировали в течение 22 часов, при 37oC в присутствии 8%CO2. К концу инкубации клеточные супернатанты тестировали на их содержание в нитрите. Нитрит представляет собой продукт реакции оксида азота с водой. Его определяют по колориметрической реакции Griess. Нитрит реагирует с сульфаниламидом в растворе кислого дигидрохлорида N-1(1(-нафтил)-этилендиамина с образованием азо соединения, способного к спектрофотометрическому мониторингу при длине волны 560 нм.
Пролиферация стволовых клеток костного мозга
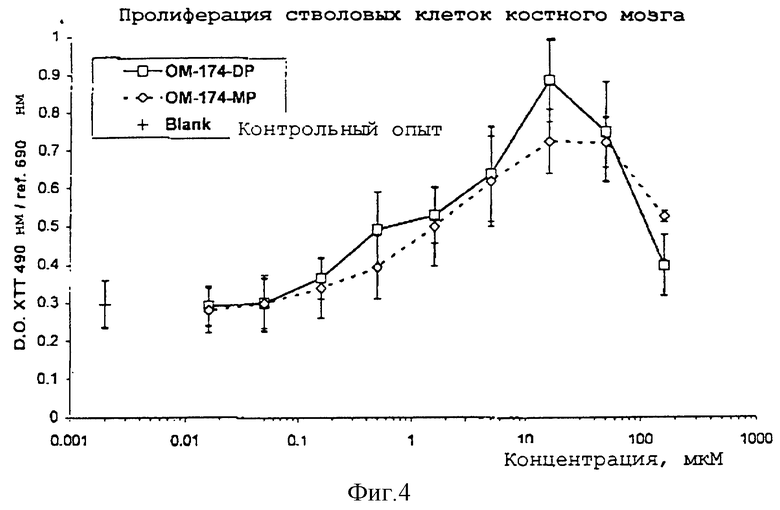
Пролиферацию стволовых клеток костного мозга, индуцированную ОМ-174-DP и ОМ-174-МР, измеряли количественным определением окисления тетразолиевых солей ХТТ под действием митохондриальных дегидрогеназ с образованием растворимого формазанового красителя. Увеличение числа живых клеток приводит к увеличению общего количества митохондриальных дегидрогеназ и непосредственно коррелирует с количеством образовавшегося оранжевого формазона.
Концентрацию стволовых клеток поддерживали на значении 5•105 клеток/мл и 100 мкл такой суспензии инкубировали в течение 8 дней в присутствии 100 мкл серийно разбавленных продуктов. В конце периода инкубации в каждую лунку добавляли по 50 мкл раствора, содержащего 1 мг/мл ХТТ (3'-[1-фениламино-карбонил)-3,4- тетразолий] бис(4-метокси-6-нитро)бензол сульфокислота). Еще через 8 часов инкубации в присутствии раствора ХТТ определяли образование растворимого формазонового красителя. Микропланшет считывали при длине волны 490 нм относительно стандарта при 690 нм и полученные результаты выражали, как среднее±стандартное отклонение от трех лунок с одинаковыми условиями.
Определение эндотоксина в ОМ-174 с помощью хромогенного Limulus теста
Эндотоксин в ОМ-174 определяли количественным, кинетическим анализом, основанным на реакционной способности грам- отрицательного эндотоксина в отношении лизата амебоцитов Limulus (LAL). Этот тест основан на активации, под действием липополисахарида или аналогичных структур, энзимного каскада, обнаруженного в LAL. Начальную скорость активации определяли по концентрации присутствующего эндотоксина. Активированная протеаза катализирует гидролиз - бесцветного субстрата, приводящий к выделению n-нитроанилина, концентрацию которого определяли O.D. считыванием при 405 нм.
Энзимную реакцию осуществляли при 37oC и образование хромогена измеряли, как функцию времени. В конечной точке кинетического метода определяли время, требующееся для достижения значения O.D., равного 0.2 единицы.
Обоснованность дозировки подтверждали выделением положительного контрольного продукта в количестве 0,5EU/мл , а концентрацию в образце определяли сравнением со стандартной кривой.
I EU (эндотоксиновая единица) составляет, примерно, 0,1 нг E.coli 055:B5 стандарта.
Стволовые клетки костного мозга (5•103 на лунку) инкубировали с серийным разбавлением OM-174-DP (дифосфорил) и ОМ-174-МР (монофосфорил), причем интервал разбавления составлял 160 - 0.016 мкМ. Через 8 дней инкубации добавляли 50 мкМ ХТТ реагента и инкубацию продолжали еще в течение 8 часов. Окисление ХТТ, соответствующее количеству митохондрия в живых клетках, измеряли считыванием микропланшета при 490 нм относительно стандарта при 690 нм. Каждое значение представляет собой среднее±стандартное отклонение в четырех независимых экспериментах.
Макрофаги (7•104 на лунку) инкубировали с серийным разбавлением OM-174-DP (дифосфорил) или ОМ-174-МР (монофосфорил), причем интервал разбавления составлял 160-0.016 мкМ. Через 22 часа стимуляции супернатанты собирали и анализировали на продукцию нитрита, с использованием колориметрического теста Griess. Полученные результаты выражали в мкМ нитрита. В каждом случае вычитали нижние (нестимулированные) значения. Каждое значение индуцированной действием OM-174-DP или ОМ-174-МР продукции нитрита представляло собой среднее±стандартное отклонение в 4 независимых экспериментах.
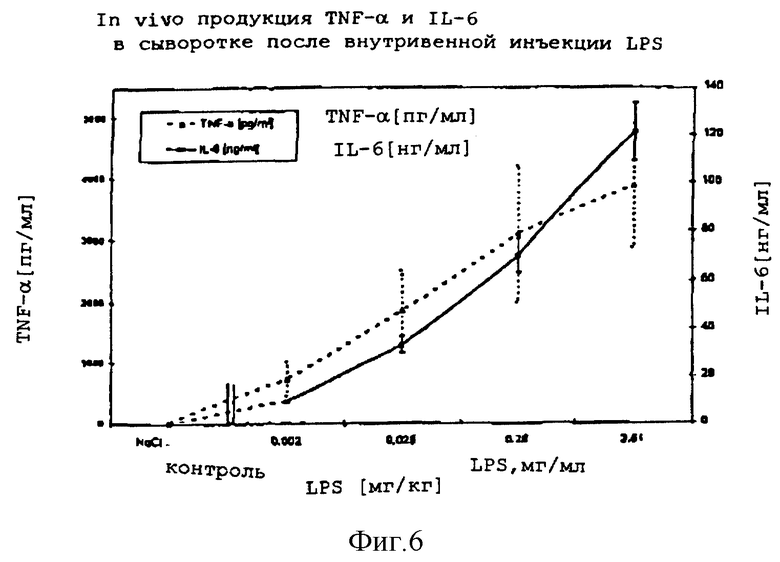
4.1.3.2 Сравнение in vivo продукции TNF- α и IL-6 в сыворотке у крови мышей после внутривенного вливания ОМ-174, относительно действия LPS.
Испытательная лаборатория: Д-р T.V.Pham, проф. д-р J.Mauel Institute of Biochemistry, University of Lausanne 1066 Epalinges/Switzerland Отчет N PA 400
Цель: Сравнение мощности действия ОМ-174 с активностью LPS после внутривенной инъекции мышам, с целью продукции TNF- α и IL-6 в сыворотке крови.
Животные: Мыши. Белые шведские самцы OFI, в возрасте 8 недель, 30-35 г, IOPS, IFFA CREDO, France.
Материалы и методы: Животным делали единичную внутривенную инъекцию 200 мкл ОМ-174, LPS или физиологического раствора (12 мышей на группу). Клинические признаки и смертность наблюдали в течение двух часов после инъекции испытуемых - веществ. Мышей умерщвляли и обескровливали через два часа после внутривенной инъекции. Крови давали коагулироваться при комнатной температуре (2 часа) и подвергали центрифугированию (5 мин, 13000 об/мин, Microfuge). Сыворотку удаляли, разделяли на аликвоты и хранили при -20oC до проведения измерений цитокина.
Измерение TNF- α в сыворотке осуществляли анализом ELISA с помощью набора Factor-Test-Xтм мышиный TNF- α Elisa от GENZYME, а сывороточный IL-6 определяли с помощью набора ЕМ-IL-6 мышиный IL-6 ELISA от ENDOGEN Lnc. Все сыворотки испытывали проведением дублирующих экспериментов.
Дозировки: Испытуемое вещество: ОМ-174:0,0002, 0,028, 0,28, 2,8, 8,4, 28,1 мг/кг в физиологическом растворе. Сравнение: Липополисахарид (E.coli 0111: В4 Sigma):0,002; 0,028; 0,28, 2,8 мг/кг в физиологическом растворе
Положительный контроль: Липополисахарид (E.coli 0111: B4, Sigma): 2,8 мг/кг в физиологическом растворе
Отрицательный контроль: физиологический раствор (0.9% NaCl в воде для инъекций)
Результаты: Внутривенная инъекция ОМ-174 и LPS индуцировала in vivo доза-зависимую продукцию TNF- α и IL-6. Оба цитокина детектировали в сыворотке через 2 часа после внутривенной инъекции. Сывороточный IL-6 был обнаружен в более высокой концентрации, чем TNF- α. Для достижения одинакового уровня содержания TNF- α и IL-6 требовались значительно более высокие дозировки ОМ-174 (десятикратные), чем соответствующие дозировки LPS.
См. фиг. 1 - 19.
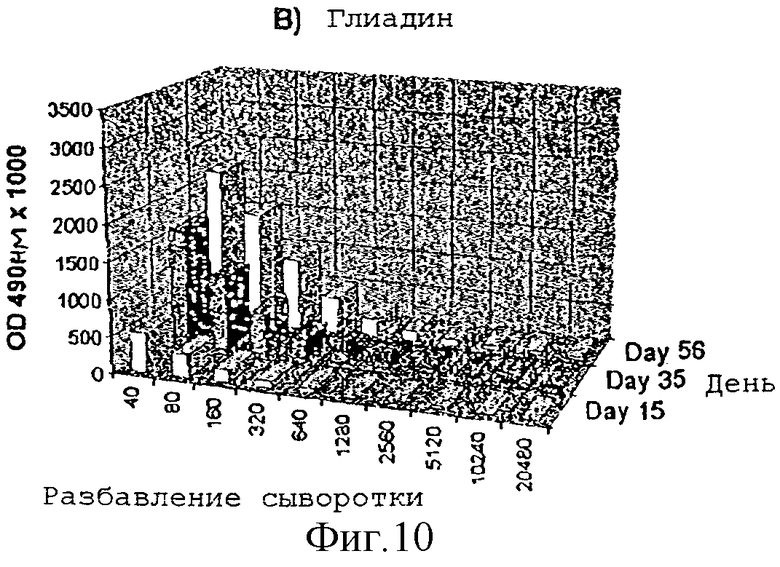
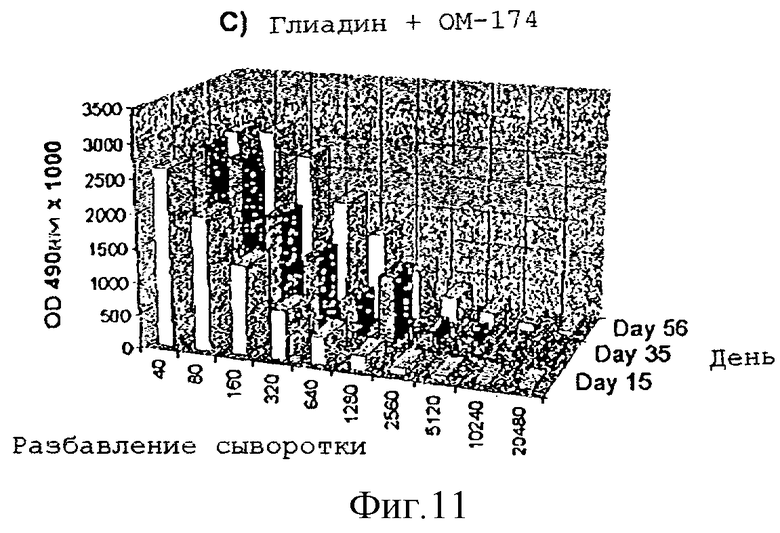
Усиление гуморального иммунного ответа на глиадин
Адьювантную активность дисахарида A в отношении гуморального иммунного ответа на действие слабого антигенного протеина, глиадина, исследовали на мышах. Группы из 4 мышей Balb/с внутрибрюшинно инъектировали в 1, 22 и 44 дни следующими веществами:
1) физиологическим раствором
2) 50 мкг глиадина
3) 50 мкг глиадина в смеси с 50 мкг дисахарида A
Образцы сыворотки отбирали на 15-, 35- и 56-й день и с помощью анализа ELISA измеряли титры антител.






Изобретение относится к β(1→6)глюкозаминовым дисахаридам, имеющим общую формулу I, где R1, R2, R3, R4, R2', R3', R4', R6' имеют значения, указанные в формуле изобретения, а также к способу их получения и к фармацевтической композиции, включающей в качестве активного ингредиента эти дисахариды. Соединения по изобретению могут быть использованы при лечении или профилактике, как иммуномодулирующие, противоопухолевые агенты и компоненты вакцины. 3 c. и 30 з.п.ф-лы, 19 ил., 13 табл.
где R1 представляет гидроксильную группу, дигидроксифосфоноилоксигруппу или ее заряженные формы, (C1 - C5)ацилоксигруппу, (C1 - C5)алкилоксигруппу или группу Х;
R2 и R'2 каждый являются ацильной группой или группой Y при условии, что, по крайней мере, R2 и R'2 являются группой Y;
R3 и R'3 каждый представляет водород, (C1 - C3)алкильную группу или (C1 - C3)ацильную группу;
R4 представляет водород, (C1 - C3)алкильную группу или (C1 - C3)ацильную группу;
R'4 представляет водород, (C1 - C5)ацильную группу, (C1 - C5)алкильную группу или фосфоногруппу или ее заряженные формы;
R'6 представляет водород, гидроксильную группу или группу Z;
где группа Х выбрана из группы, включающей карбокси (C1 - C4)алкоксигруппу; -O-CH-[(CH2)mCOOH][(CH2)nCOOH] группу, где m = 0 - 5, n = 0 - 5; и заряженные формы группы Х;
где группа Y выбрана из группы, включающей ацилоксиацильную группу, (C1 - C24)алкилоксиацильную группу;
где группа Z выбрана из группы, включающей 3-дезокси-D-манно-2-октулосоновую кислоту (KDO); (KDO)n, где n = 1 - 10; полисахаридную цепь, такую, как боковая цепь из натурального полисахарида; компонент ядра, такой, как компонент, происходящий из натурального липополисахарида;
или их соли.
| Производные хитоолигосахаридов, обладающие иммуномодулирующей и противоопухолевой активностью | 1988 |
|
SU1652319A1 |
| Г-. . .' • ' . | 0 |
|
SU192296A1 |
| US 4912094 A, 1990. | |||

