Перекрестные ссылки к связанным заявкам Настоящая заявка имеет приоритет по продолженной заявке US 60/394487, поданной 08.07.02 г. Указанная продолженная заявка относится к US No. 10/137730, поданной 30.04.02 г., которая является частичным продолжением US No. 10/043089, поданной 08.01.2002 г., которая является частичным продолжением US No. 09/905106, поданной 12.07.2001 г., которая является частичным продолжением US No. 09/439839, поданной 12.11.1999 г., в настоящее время патент US 6303347, который является частичным продолжением US No. 08/853826, поданной 08.05.1997 г., в настоящее время патент US 6113918. Настоящая заявка также относится к US 09/074720, поданной 07.05.1998 г., к US 6355257, которая также является частичным продолжением US No. 853826. Эта заявка также имеет приоритет по заявке US 60/438585, поданной 06.05.2003 г. Все указанные патенты и ссылки включены в настоящую заявку как ссылки, во всей их полноте.
Область техники, к которой относится изобретение
Настоящее изобретение относится к процессам получения аминоалкилглюкозаминидфосфата (AGP) и дисахаридных соединений. Такие соединения раскрыты как иммуноэффекторы, адъюванты для вакцин и тому подобное, и, кроме того, они могут обладать своими собственными терапевтическими и/или профилактическими свойствами. Далее настоящее изобретение относится к процессам получения гликозилгалогенидных соединений, которые могут служить промежуточными соединениями в синтезе AGP's соединений, дисахаридов и родственных по структуре молекул.
Уровень техники
Аминоалкилглюкозаминидфосфаты описаны в ряде патентов, заявках на патент и в журнальных статьях. Такие соединения, как правило, имеют пять или шесть ацильных групп в структуре молекулы, вместе с "агликоном" (азотосодержащей частью), которая может быть циклической или ациклической. AGP's, имеющие ациклические агликоновые группы, раскрыты, например, в US 6113918; 6303347 и 6355257. AGP's, имеющие циклические агликоновые группы, раскрыты, например, в WO 02/012258.
Вышеупомянутые документы описывают получение AGP соединений двумя альтернативными способами. В одном из способов защищенный 3-O-ацилоксиацилированный гликозилгалогенид, содержащий фосфонатную боковую цепь, конденсируют с аминоалканолом или аминоалкантиолом того типа, который описан в патентах. Продукт реакции затем селективно ацилируется, чтобы обеспечить введение дополнительных ацильных групп, как описано, а защитные группы удаляют. Во втором из способов и фосфонатную боковую цепь, и жирнокислотные группы вводят после реакции конденсации. Дополнительная информация о способе получения AGP соединений содержится у Johnson et al., Bioorg. Med. Chem. Lett. 9: 2273 (1999).
Дисахариды, которые могут быть получены с помощью способов, описанных в настоящей заявке, включают компоненты хорошо известного иммуностимулятора монофосфориллипида А (содержащегося, например, в MPL® иммуностимуляторе (Corixa Corp.). Другие дисахариды, которые могут быть получены, раскрыты, например, в РСТ WO 01/90129 и US 6013640; 4987237; 4912094; 4436727 и 4436728. В US 6103640 дисахарид получают путем конденсации N-ацилоксиацилированного или N-защищенного гликозильного акцепторного звена с защищенным и/или 3-O-ацилоксиацилированным гликозильным донорным звеном. Защитные группы представляют собой различные бензильные (Bn) и 2,2,2-трихлорэтоксикарбонильные (Troc) группы. Гликозильный акцептор и донорные звенья формируют отдельно, используя серии шагов по защите и снятию защиты с заместителя, начиная с известных исходных продуктов бензил- и 2-(триметилсилил)этил-2-амино-2-дезокси-4,6-O-изопропилиден-β-D-глюкопиранозидов, соответственно.
Гликозилгалогениды используют во многих процессах в области химии сахаров, для введения гликозидного остатка в молекулу, обычно как часть многостадийного синтеза. Они представляют собой полезные промежуточные соединения для введения большого количества групп, обычно реакцией взаимодействия с нуклеофилами, особенно с такими нуклеофилами, как кислород, сера и азот. Это пригодно для обеспечения процесса получения AGP и дисахаридных соединений, используя гликозилгалогенид в качестве исходного продукта.
Описаны различные пути получения гликозилгалогенидов. Как правило, они включают галоидирование существующего гликозида (который может содержать обычные защитные группы на реакционоспособных остатках, таких как аминогруппа или гидроксил).
В патенте US 6299897, например, этиловый эфир того гликозида, который рассматривается (в этом случае, например, N-ацетилнейраминовой кислоты), реагирует с ацетилхлоридом с образованием соответствующего гликозилхлорида. В US 5843463 гликозилхлорид получают путем взаимодействия рассматриваемого гликозида (3-O-аллил-5-О-бензил-1,2-O-метоксибензилиден-альфа-D-рибофуранозы) с триметилсилил-хлоридом. Реакцию проводят путем перемешивания двух реагентов или путем растворения гликозида в триметилсилилхлориде.
US 4613590 раскрывает способ получения гликозилхлорида при обработке гликозида тетрахлоридом титана. В Org. Lett. 2:2713 (2000), Sugiyama et al. гликозилхлориды получают при взаимодействии тиогликозидов с хлоридом хлорсульфония.
У Kovac, Carbohydr. Res. 245: 219 (2993) получают гликозилхлорид при взаимодействии гликозида с дихлорметилметиловым эфиром и хлоридом цинка. Takeo et al., Carbohydr. Res. 245: 81 (1993) получает гликозилхлорид при взаимодействии с хлором. Magnusson et al., 55: 3181 (1990) получает гликозилхлорид при взаимодействии 2-(триметилсилил)этилгликозида с 1,1-дихлорметилметиловым эфиром в присутствии каталитического количества хлорида цинка.
Раскрытие изобретения
Настоящее изобретение относится к группе относительно новых процессов получения аминоалкилглюкозаминидфосфатов и дисахаридов, вместе с промежуточными процессами и соединениями.
В первом аспекте изобретение относится к процессам получения аминоалкилглюкозаминидных соединений (AGP).
Во втором аспекте изобретение относится к процессам получения гликозилгалогенидов, который включает взаимодействие силилгликозида с дигалогенметилалкиловым эфиром в присутствии хлорида цинка, бромида цинка, трифторида бора или подобных кислот Льюиса. Эта стадия также включает первую из двухстадийного процесса стадию удаления аномерной защитной силильной группы с силилгликозида путем его первого взаимодействия с получением гликозилгалогенида, который затем взаимодействует с солью серебра в присутствии воды для получения гемиацеталя.
В другом аспекте настоящее изобретение включает процесс, в котором вначале получают гликозилгалогенид, как указано выше, с последующим взаимодействием гликозилгалогенида с моносахаридом в присутствии соли серебра, с образованием дисахарида.
Другой аспект настоящего изобретения включает процесс получения дисахарида путем взаимодействия моносахарида с силилгликозидом.
Еще один аспект настоящего изобретения включает процесс силилирования дисахарида и необязательного последующего добавления фосфоновой боковой цепи к дисахариду.
Еще один аспект настоящего изобретения включает процесс получения триацилированного дисахарида из дисахарида.
Еще один аспект изобретения представляет собой процесс удаления защитной ацетильной группы с дисахарида.
Еще дальнейший аспект настоящего изобретения включает процесс получения фосфорилированного дисахарида путем (а) селективной защиты 6'-гидроксильного заместителя дисахарида и b) добавления фосфоновой боковой цепи к дисахариду по 5'-положению.
Еще один аспект настоящего изобретения включает процесс одновременного удаления всех основанных на силиле защитных групп с дисахарида, имеющего многочисленные, основанные на силиле защитные группы.
Другие аспекты настоящего изобретения включают другие новые процессы и новые промежуточные соединения, и/или они будут очевидны из последующего описания.
Осуществление изобретения
Определения: как используют в настоящей заявке
Термин "гликозид" относится к тетрагидропирановому кольцу, несущему заместитель по 1-положению (то есть на одном из атомов углерода, смежном с атомом кислорода в кольце), который представляет собой гидроксигруппу, необязательно замещенную алкоксигруппу или тризамещенную силилоксигруппу. Гликозиды могут также иметь заместители по другим положениям, обычно защищенные или незащищенные гидроксильные или аминогруппы.
Термин "силилгликозид" относится к гликозиду, в котором группа, присоединенная по 1-положению, представляет собой тризамещенную силилоксигруппу, такую как триметилсилилокси, трет-бутилдиметилсилилокси или трет-бутилдифенилсилилоксигруппу. Силильный компонент этой группы имеет формулу RaRbRcSi, где Ra, Rb и Rc независимо выбраны из группы, состоящей из C1-С6алкила, С3-С6циклоалкила и необязательно замещенного фенила. Предпочтительно один из Ra, Rb и Rc группы является большим, чем метил; относительно затрудненные группы, такие как трет-бутил, фенил и изопропил, являются предпочтительными. Включенными в силильные компоненты являются арилдиалкилсилильные, диарилалкилсилильные и триарилсилильные группы. Типичными примерами являются триизопропилсилильные, трифенилсилильные, трет-бутилдиметилсилильные (TBS) и трет-бутилдифенилсилильные (TBDPS) группы. Силильный компонент силилгликозида является более предпочтительным в виде TBS или TBDPS группы.
Силилгликозид может обычно быть представлен формулой (II)

где R20 представляет собой тризамещенную силильную группу, предпочтительно TBS или TBDPS и W, X, Y и Z независимо представляют собой Н, необязательно защищенные гидрокси, необязательно защищенные амино или необязательно замещенные алкильные группы. Обычно Z представляет собой необязательно защищенную гидроксиметильную группу.
Термин "дигалогенметилалкиловый эфир" относится к соединению, несущему алкоксигруппу и два атома галогена на единственном атоме углерода. Типичными примерами являются дихлорметилметиловый эфир (CHCl2ОСН3), дихлорметилэтиловый эфир (CHCl2OC2H5), дибромметилметиловый эфир (CHBr2OCH3), 1,1-дихлорэтилэтиловый эфир (СН3CCl2ОС2Н5) и им подобные. Дихлорметилметиловый эфир является предпочтительным в способах настоящего изобретения.
Термин "гликозилгалогенид" относится к 2-галогентетрагидропирановому соединению, например, 2-хлортетрагидропирану или 2-бромтетрагидропирану. Предпочтительными галогенами являются фторид, хлорид и бромид, с хлоридом являющимся более предпочтительным. Кроме того, гликозилгалогениды, используемые в способах настоящего изобретения, могут иметь другие заместители, аналогично тем, которые представлены в формуле (II), приведенной выше.
Гликозилгалогениды могут обычно быть представлены формулой (III):
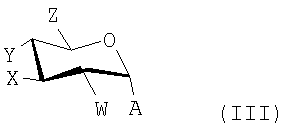
где W, X, Y и Z имеют те же значения, которые описаны выше для формулы (II), и А представляет собой Cl, Br или F.
Термин "алифатический" означает прямую или разветвленную цепь или неароматический циклический, углеводородный радикал или их комбинацию, который может быть полностью насыщенным или моно- или полиненасыщенным и может включать ди- и поливалентные радикалы, имеющие определенное число атомов углерода (то есть С1-С10означает от одного до десяти атомов углерода). Примеры насыщенных ациклических алифатических групп (также определенных как "алкильные" группы) включают, но без ограничения, группы, такие как метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор.-бутил, гомологи и изомеры, например, н-пентил, н-гексил, н-гептил, н-октил и им подобные. Ненасыщенная алифатическая группа представляет собой одну из групп, имеющую одну или более двойных или тройных связей. Примеры ненасыщенных ациклических алифатических групп включают, но без ограничения, винил, 2-пропенил, изопропенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-петадиенил, 3-(1,4-петадиенил), этинил, 1- и 3-пропинил, 3-бутинил и высшие гомологи и изомеры. Примеры циклических алифатических групп включают циклопропил, циклобутил, циклопентил, циклогексил, циклопентенил, циклогексенил, циклогексадиенил и им подобные.
Двухвалентные алифатические группы включают насыщенные и ненасыщенные группы, подобные тем, которые упомянуты выше, например, метилен, -СН2-; этилен, -CH2CH2-; н-бутилен, -СН2СН2СН2СН2-; и ненасыщенные группы, такие как -СН=СН-, -СН=СН-СН2СН2- и им подобные.
Термины "оксиалифатический", "аминоалифатический" и "тиоалифатический" используют в их обычном смысле, и они имеют отношение к алифатическим группам, присоединенным к остатку молекулы через атом кислорода, аминогруппу или атом серы, соответственно. Термины "алкокси", "тиоалкокси" и "аминоалкил" имеют отношение к таким группам, содержащим насыщенные ациклические алифатические остатки.
Термин "гетероалифатический," сам по себе или в комбинации с другим термином, означает, если не обусловлено иное, группу, аналогичную алифатической группе, то есть с насыщенной или ненасыщенной прямой или разветвленной цепью или циклический радикал или их комбинацию, содержащую постоянное число атомов углерода и, кроме того, включающую, по крайней мере, один гетероатом, выбранный из группы, состоящей из О, N, Si и S и где атом азота и атом серы могут необязательно быть окислены и азотный гетероатом может быть необязательно кватернизован. Гетероатом(ы) О, N и S и Si могут быть помещены по любому внутреннему положению гетероалифатической группы или по положению, по которому указанная группа присоединена к остатку молекулы. Примеры включают, но без ограничения, -СН2-СН2-O-СН3, -СН2-СН2-NH-СН3, -СН2-СН2-N(СН3)-СН3, -СН2-S-СН2-СН3, -CH2-CH2, -S(O)-СН3, -СН2-СН2-S(O)2-СН3, -СН=СН-O-СН3, -Si(СН3)3 и -CH2-СН=N-ОСН3.
Алифатические группы могут замещенными или незамещенными. Заместители включают различные группы, выбранные из: -OR',=O,=NR',=N-OR', -NR'R'', -SR', -галогена, -SiR'R''R''', -OC(O)R', -C(O)R', -CO2R', -CONR'R'', -OC(O)NR'R", -NR''C(O)R', -NR'-C(O)NR''R''', -NR''C(O)2R', -NR-C(NRR'R'')=NR''', -NR'C(NR'R'')=NR''', -NR-C(NR'R'')=NR''', -S(O)R', -S(O)2R', -S(O)2NR'R'', -NRSO2R', -CN и -NO2 в числе, находящемся в интервале от 0 до (2m'+1), где m' представляет собой общее число атомов углерода в таком радикале. R', R'' и R''' каждый независимо может быть водородом, необязательно замещенным алкилом, арилом, необязательно замещенным 1-3 галогенами, необязательно замещенным алкокси, необязательно замещенным тиоалкокси или необязательно замещенными арил-(С1-С4)алкильными группами. Когда соединение изобретения включает более чем одну R группу, например, каждая из R групп является независимо выбранной, как каждая из R', R'' и R''' групп, когда присутствует более чем одна из этих групп. Когда R' и R'' присоединены к одному и тому же атому азота, тогда они могут быть объединены с атомом азота, чтобы образовать 5-, 6- или 7-членное кольцо. Например, -NR'R'' означает включение 1-пирролидинила и 4-морфолинила.
Термин "ароматический" или "арил" относится к типичным замещенным или незамещенным неалифатическим углеводородным группам из этого класса, то есть, полиненасыщенным, обычно ароматическим с углеводородным заместителем, которые могут быть в виде единственного кольца или нескольких колец (вплоть до трех колец), которые конденсируются вместе или связаны ковалентно, такие как фенил, нафтил и им подобные.
Термин "арилалкил" относится к алкильным группам, замещенным одной или большим количеством арильных групп; например, бензил, фенэтил, трифенилметил и им подобные.
Термин "ацил" относится к группе, производной от органической кислоты при удалении гидроксильной группы. Ацильные соединения могут, как правило, быть алифатическими, ароматическими или гетероциклическими по природе. "Алифатический ацил" относится к таким группам, производным от насыщенных или ненасыщенных алифатических кислот, и включает группы, такие как ацетил, пропионил, бутирил, гексаноил, деканоил, додеканоил, тетрадеканоил и им подобные. При определении ацильных групп через содержание в них атомов углерода отнесение происходит к содержанию атомов углерода в целой группе. Соответственно, ацетил представляет собой С2 ацильную группу; пропионил представляет собой С3 ацильную группу, тетрадеканоил представляет собой С14 ацильную группу и так далее.
Термин "алканоилоксикарбонил" относится к группам, имеющим насыщенную или ненасыщенную алифатическую группу или арилалкиловую группу, такую как бензильная, связанную через атом кислорода с карбонильной группой, то есть группа, имеющая общую формулу Alk.-OC(O)-, в которой Alk. обозначает алифатическую или арилалкильную группы, как определено выше.
Термин "алканоилоксиацил" относится к насыщенной или ненасыщенной ацильной группе, замещенной на обозначенном положении алифатической группой Al.С(O)O-, в которой Al. обозначает ацикличную насыщенную или ненасыщенную алифатическую группу. В целом алканоилоксигруппа предпочтительно имеет от 2 до 24 атомов углерода, более предпочтительно от 6 до 14 атомов углерода. Ацильный остаток алканоилоксиацильной группы содержит от 6 до 14 атомов углерода. Типичная группа из этой серии представляет собой 3-(н-алканоилокси)ацильную группу, где ацильная группа представляет собой тетрадеканоил, и алканоилоксигруппу, содержащую от 2 до 20, предпочтительно от 6 до 14, атомов углерода включительно. Подобным образом термин "алканоил" относится к группе Al.С(O)-, где Al. является тем, как определено выше.
Термин "защитная группа" относится к любому большому числу групп, используемых для замещения одного или обоих атомов водорода реакционноспособной группы, такой как гидрокси, амино или тиольная группа, для того, чтобы блокировать, предотвращать или ослаблять реакционноспособность группы. Примеры защитных групп (и перечень, обычно используемые сокращения для них) могут быть найдены у Т. W. Green и Р.G. Puts, "Protective Grups in Organic Chemistry" (Wiley), Beaucage и Iyer, Tetrahedron 48:2223 (1992) и Harrison et al., Compendium of Synthetic Organic Methods, vols. 1-8 (Wiley).
Представители аминозащитных групп включают те, которые образуют карбамат или амид с атомом азота, так же как те группы, которые вместе сведены в публикации Green и Puts, как "специальные -NH защитные группы". Типичные примеры аминозащитных групп включают ацетильные (Ас), трифторацетильные, бензилоксикарбонильные (Cbz), трет.-бутоксикарбонильные (Boc), аллилоксикарбонильные (Аос), 9-флуренилметилоксикарбонильные (Fmoc), нитро-версатрилоксикарбонильные (Nvoc), необязательно замещенные фталоилом, и им подобные.
Типичные гидроксизащитные группы включают те, где гидроксигруппа является или ацилированной, или алкилированной, такие, для которых используют образование простых или сложных эфиров, например, ацетильные, бензильные, тритильные, алкильные, тетрагидропиранильные, аллильные и тризамещенные силильные группы.
Выбор защитной группы для данного соединения, цели или установление условий находится в пределах квалификации специалиста в данной области, и его делают, чтобы защитить, вообще или селективно реакционноспособную рассматриваемую группу в преобладающих условиях (наличие других реакционноспособных соединений, рН, температуры и так далее). Защитные группы, которые могут быть использованы в настоящем изобретении и которые упомянуты в нем, включают фталоильные, ацетильные (Ас), бензильные (Bn), 2,2,2-трихлорэтоксикарбонильные (Troc), трет-бутилдиметилсилильные (TBS), трет-бутилдифенилсилильные (TBDPS) и 2,2,2-трихлор-1,1-диметилэтилхлорформильные (ТСВОС) группы. Как известно среднему специалисту, некоторые защитные группы или типы групп могут быть более пригодны, чем другие, для использования с частными соединениями или в конкретной ситуации и преимущество получают от этих соответствий в развитии способов, которые включают соединения с реакционноспособными группами, такими как гидрокси и/или аминогруппы. Таким образом, как будет замечено ниже, реакционная схема может быть развернута для получения или взаимодействия определенных соединений, в которых общая или селективная защита или снятие защиты (удаление защитных групп) выполнены в некоторых точках. Например, чтобы селективно ввести в реакцию гидроксильную группу в соединение, которое также содержит аминогруппу или, наоборот, группу, которую не желательно вводить в реакцию на этой стадии, может быть осуществлено блокирование защитной группой, которая не удаляется в условиях реакции (например, не может гидролизоваться основанием, если реакция должна быть проведена в основных условиях, в то время как группа, которую надо ввести в реакцию, может быть защищена группой, которая гидролизуется основанием, так указанная группа становится не блокированной и, таким образом, реакционноспособной). Аналогично, как будет замечено ниже, чтобы селективно ввести в реакцию группу, например, гидроксильную группу, расположенную в одном положении в молекуле, ее можно защитить другой защитной группой, отличной от других гидроксилов в молекуле. Как используют в настоящем описании, обозначение "PG" относится к защитным группам, которые формируют сложные эфиры, простые эфиры или карбонаты с гидроксильньми группами (то есть с атомом кислорода гидроксилыгой группы) или которые формируют амиды или карбаматы с аминогруппами (то есть с атомом азота аминогруппы). Обозначение "PG'" используют в настоящем описании, чтобы отнести к нему необязательно замещенные фталоильные группы, например, фталоил или тетрахлорфталоил и которые, как показано, могут быть использованы для защиты аминогруппы. Однако в любом случае выбор конкретных защитных групп, использованных или продемонстрированных в процессах, описанных в настоящем описании, ни в коем случае не предназначен, чтобы ограничить область изобретения.
Основные продукты
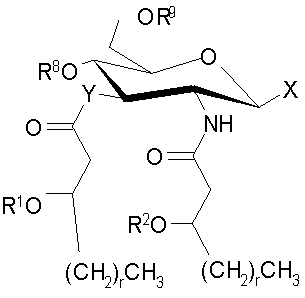
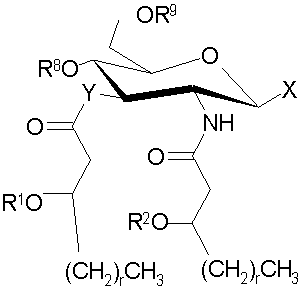
Основные продукты, полученные при использовании процессов и промежуточных соединений настоящего изобретения, содержат группу соединений, которые включают и AGP соединения, которые представляют собой моносахариды и дисахариды до некоторой степени аналогичной структуры. Как правило, продукты могут быть описаны с помощью формул (I) и (Ia-с):
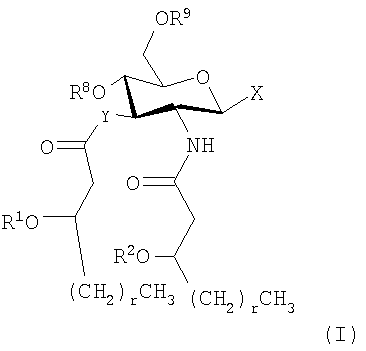
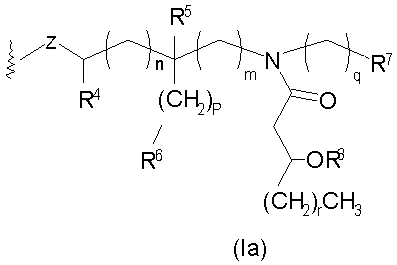
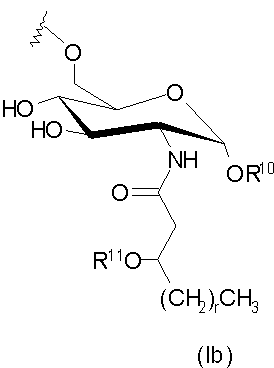
и их фармацевтически приемлемые соли и производные, где Y представляет собой -О- или -NH-; R1 и R2 каждый независимо является выбранным из насыщенных и ненасыщенных (C2-C24) алифатических ацильных групп; R8 представляет собой -Н или -РО3R11R12, где R13 и R12 каждый независимо является -Н или (C1-C4) алифатическими группами; R9 представляет собой -Н, -СН3 или -РО3R13R14, где R13 и R14 каждый является независимо выбранным из -Н и (C1-C4) алифатических групп и где, по крайней мере, один из R8 и R9 является фосфорсодержащей группой, но R8 и R9 оба не являются фосфорсодержащими группами; и Х представляет собой группу, выбранную из формул:
 and
and 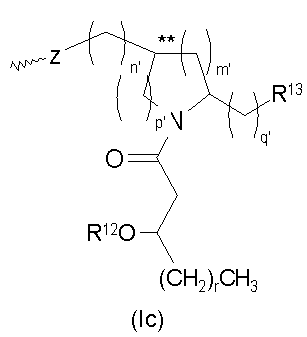
где индексы n, m, p, q, n', т', р' и q' каждый независимо является целым числом от 0 до 6, при условии, что сумма р' и т' является целым числом от 0 до 6; индекс r независимо является целым числом от 0 до 14 и может быть одинаковым или разичным; R3, R11 и R12 независимо являются насыщенными или ненасыщенными алифатическими (C2-C24) ацильными группами; и когда Х имеет формулу (1а) или (1с), один из R, R2, R3, R11 и R12 необязательно является водородом; R4 и R' независимо выбраны из Н и метила; R6 и R7 независимо выбраны из Н, ОН, (C1-C4)оксиалифатических групп, -РО3H2, -ОРО3Н2, -SO3Н, -OSO3H, -NR15R16, -SR15, -CN, -NO2, -СНО, -CO2R15, -CONR15R16, -PO3R15R16, -OPO3R15R16, -SO3R15 и -OSO3R15, где R15 и R16 каждый является независимо выбранным из Н и (C1-C4)алифатических групп; R10 выбран из Н, СН3, -РО3Н2, ω-фосфоноокси(С2-С24)алкила и ω-карбокси(С1-С24)алкила; R13 независимо выбран из Н, ОН, (C1-C4)оксиалифатических групп, -PO3R17R18, -ОРО3R17R18, -SO3R17, -OSO3R17, -NR17R18, -SR17, -CN, -NO2, -СНО, -CO2R17 и -CONR17R18, где R17 и R18 каждый является независимо выбранным из Н и (C1-C4)алифатических групп; и Z представляет собой -O- или -S-.
Процессы и промежуточные соединения
Один из процессов настоящего изобретения относится к получению гликозилгалогенидов, который включает взаимодействие O-силилгликозида с дигалогенметилалкиловым эфиром в присутствии хлорида цинка, бромида цинка, трифторида бора или аналогичной кислоты Льюиса. Более конкретно, в этом процессе гликозилгалогенид образуется при взаимодействии силилгликозида формулы (II):
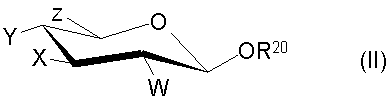
где R20 представляет собой тризамещенную силильную группу, имеющую формулу R3RbRcSi, в которой R3, Rb и Rc независимо выбраны из группы, состоящей из C1-С6алкила, С3-С6 циклоалкила и необязательно замещенного фенила, предпочтительно TBS или TBDPS и W, X, Y и Z независимо представляют собой Н, необязательно защищенную гидроксильную, необязательно защищенную амино и необязательно замещенную алкильную группы, с дигалогенметилалкиловым эфиром, предпочтительно дихлорметилметиловьм эфиром, в присутствии хлорида цинка, бромида цинка, трифторида бора или аналогичной кислотой Льюиса. Кислоту Льюиса используют в приблизительно в стехиометрическом количестве по отношению к силилгликозиду. Реакцию получения гликозилгалогенида проводят при температуре от около -30°С до около 50°С, предпочтительно от около 0°С до около 30°С и в присутствии растворителя, такого как хлороформ, дихлорметан, дихлорэтан, или подобных растворителей, которые являются инертными в условиях, требуемых для осуществления реакции. Температура реакции выбрана, чтобы дать возможность реагентам существенно раствориться и предотвратить выкипание дигалогенметилалкилового эфира. Выход желаемого продукта гликозилгалогенида в основном имеет значение от около 50 до около 95%. Выбор таких растворителей осуществляют на основании знаний среднего специалиста в данной области. Силилгликозиды получают обычно в защищенном виде, что известно среднему специалисту в данной области. Тем не менее, конкретные силилгликозиды, такие как конкретные триацетилированные силилгликозиды и их производные, могут быть получены через новые промежуточные соединения, описанные ниже, что является аспектом изобретения.
Средним специалистом в данной области будет оценено, что гликозилгалогениды могут существовать в виде изомеров, если другие заместители находятся на гликозилгалогенидном кольце. Изобретение включает получение индивидуальных изомеров, так же как смесей обоих изомеров. Условия осуществления реакций большинства нуклеофилов с гликозилгалогенидами хорошо известны среднему специалисту в данной области.
Полученные таким образом гликозилгалогениды обычно имеют формулу (III)
где А представляет собой Cl, Br или F и W, X, Y и Z имеют те же значения, которые описаны выше.
В одном предпочтительном воплощении силилгликозид и полученные продукты замещены по 3-положению (заместитель X) с помощью алифатической ацильной группы, предпочтительно алканоилоксиацильной группы, более предпочтительно 3-н-алканоилоксиацильной группы и более предпочтительно 3-алканоилокситетрадеканоильной группы, в которых алифатическая или алканоильная группа содержит от 2 до 24, предпочтительно от 2 до 18 и более предпочтительно от 6 до 14, атомов углерода и защитные группы в данном соединении являются предпочтительно Тгос группами или подобными алканоилоксикарбонильными группами. В таких воплощениях соединения имеют общую формулу (IV) или (V):
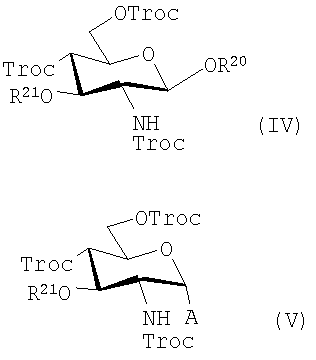
где А представляет собой Cl, Br или F; R20 представляет собой тризамещенную силильную группу и R21 представляет собой алифатическую ацильную группу, предпочтительно 3-н-алканоилокситетрадеканоильную группу. Следует заметить, что в этих и в последующих формулах защитные группы более точно идентифицированы с целью иллюстрации и/или ясности. Однако, как известно среднему специалисту в данной области, другие защитные группы, как в общем определено выше для "PG", могут быть использованы в качестве подходящих групп.Таким образом, например, более обобщенно эти соединения могут быть представлены формулой
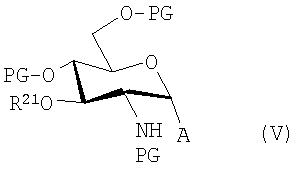
где PG представляет собой защитные группы, которые образуют простой эфир, сложный эфир или карбонат с атомом кислорода или которые образуют амид или карбамат с атомом азота, соответственно.
В другом предпочтительном воплощении силилгликозид имеет гидроксильную группу по 4-положению, замещенную фосфатноэфирной группой, такой как диалкилфосфонильная или диарилфосфонильная группа, R21 представляет собой алканоилоксиацильную группу, предпочтительно 3-н-алканоилокситетрадеканоильную группу, и защитные группы являются предпочтительно "ТСВОС" группами, полученными из 2,2,2-трихлор-1,1-диметилэтилхлорформиата, или подобную алканоилоксикарбонильную защитную группу, такую как Troc; то есть силилгликозид может иметь конкретную формулу (VI):
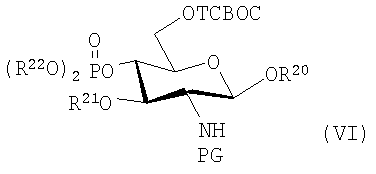
где R20 представляет собой тризамещенную силильную группу, предпочтительно TBS или TBDPS; R21 представляет собой алифатическую ацильную, предпочтительно алканоилоксиацильную группу; и R22 представляет собой алкил, арил или арилалкил, или может иметь более общую формулу, которая позволяет использовать другие подходящие защитные группы и гликозилгалогениды соответственно, имеющие более конкретную формулу (VII):
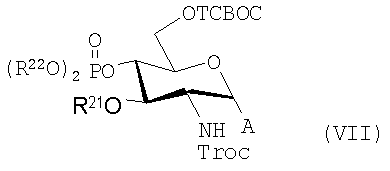
где R21 и R22 имеют те же значения, которые описаны выше, и А представляет собой Cl, Br или F.
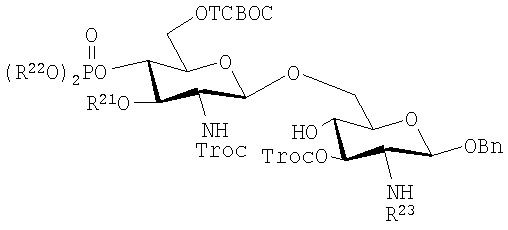
В одном аспекте изобретения гликозилхлориды, полученные таким образом взаимодействуют с моносахаридом, предпочтительно в присутствии соли серебра, для получения дисахарида с помощью этого двухстадийного способа. Моносахариды, которые могут быть использованы как реагенты в этом способе, включают, например, те, которые имеют формулы (i)-(iii):
(i):

(ii):
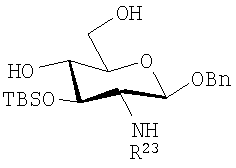
или (iii):

где R23 представляет собой алифатическую ацильную группу, предпочтительно 3-н-алканоилокситетрадеканоильную группу, как описано выше.
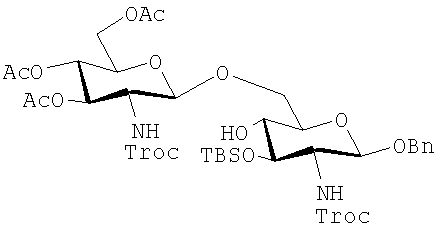
Дисахариды, которые могут быть получены с помощью таких способов, включают те, которые имеют формулы (iA)-(ivA):
(iA):
(iiA):
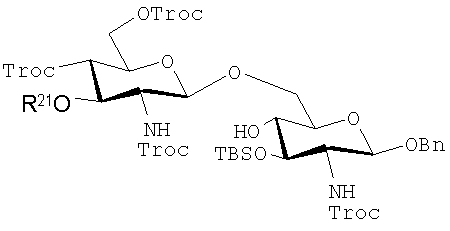
(iiiA):
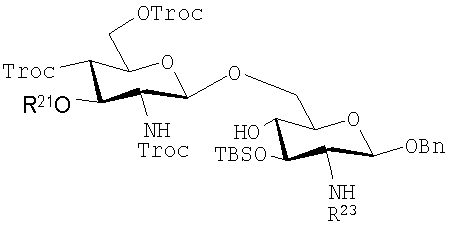
(ivA):
где R21, R22 и R23 имеют те же значения, которые описаны выше, и указанные защитные группы являются обычно теми, которые могут быть использованы.
Реакции, приводящие к синтезу продуктов (iA)-(ivA) в соответствии с настоящим изобретением, обычно проводят при температуре от около -30°С до около 30°С, в хлорсодержащем или другом растворителе в присутствии серебряного катализатора, такого как трифторметансульфонат (трифлат) серебра, и в безводных условиях, с или без других добавок, таких как молекулярные сита или буферные агенты, такие как тетраметилмочевина.
В другом аспекте изобретения силилгликозиды формулы (II) конденсируют непосредственно с моносахаридом, не через процесс образования гликозилгалогенида. Полученный продукт является снова дисахаридом, имеющим заместитель в соответствии с исходными продуктами. Такой процесс обычно проводят при температуре от около -78°С до около 50°С в присутствии подходящего катализатора кислоты Льюиса, такого как триметилсилилтрифлат эфирата трифторида бора с или без дополнительной сушки или буферных агентов. В другом аспекте настоящего изобретения защитные группы могут в действительности быть удалены с силилгликозида, имеющего такие группы, путем его взаимодействия с дигалогеналкиловым эфиром, для получения гликозилгалогенида и последующего взаимодействия гликозилгалогенида с солью серебра, такой как оксид серебра или карбонат серебра, в присутствии воды для получения соответствующего гемиацеталя.
Дисахариды, полученные с помощью любого из процессов, могут быть подвергнуты дополнительно силилированию гидроксильной группы по 4-положению восстановленного сахара силилирующей группой, такой как TBS, в присутствии имидазола и N,N-диметилформамида для получения 3,4-бис-силилированного соединения. Присоединение фосфатной группы по 4-положению невосстановленного сахара затем осуществляют с помощью цепочки стадий, включающей (1) снятие 4,6 защитных групп (обычно ацетата или Troc), (2) N-снятия защиты/ацилирования, (3) селективной защиты сначала 6-положения с помощью группы, такой как ТСВОС, и (4) взаимодействия 6-защищенного дисахарида с фосфонилирующим агентом, таким как фосфорамидитным реагентом, например, дибензилдиизопропилфосфорамидит [обеспечение дибензилфосфононовой боковой цепи] или хлорфосфат, такой как бис(2,2,2-трихлорэтил)хлорфосфат [обеспечение бис(2,2,2-трихлорэтил)фосфоновой боковой цепи] или дифенилхлорфосфат [обеспечение дифенилфосфононовой боковой цепи].
Изобретение аналогично включает также способы получения триацилированных дисахаридов, таких как те, которые имеют формулу (viii):
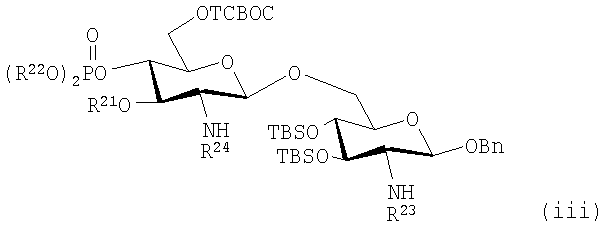
где R21, R23 и R24 представляют собой алифатические ацильные, предпочтительно алканоилоксиацильные группы и R22 является необязательно замещенной алкильной, арильной или арилалкильной группой, путем селективной защиты С-6 гидроксильной группы соответствующего дисахарида с помощью 2,2,2-трихлор-1,1-диметилэтилхлорформиата в присутствии третичного амина, такого как пиридин. Предпочтительно R21, R23 и R24 представляют собой (R)-3-гексадеканоилокситетрадеканоил, (R)-3-октадеканоилокситетрадеканоил и (R)-3-тетрадеканоилокситетрадеканоил соответственно, но они могут быть одинаковыми или различными в зависимости от желаемых замещений и природы используемого моносахаридного донора на стадии гликозилирования.
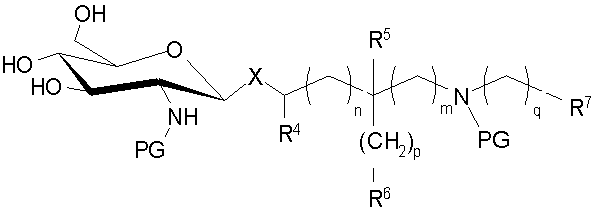
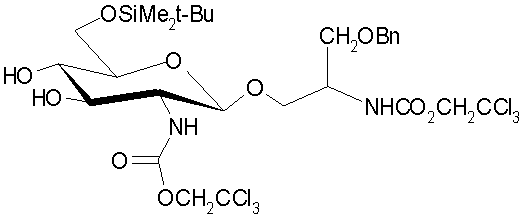
Настоящее изобретение также относится к процессам получения аминоалкильных и циклических аминоалкильных глюкозаминидных соединений (AGP), которые представляют собой соединения формулы (I), в который Х представляет собой (Ia) или (Ic), в которых жирную кислоту и фосфатные группы вводят на основной AGP остов после первоначальной стадии гликозилирования (конденсации). Эти процессы включают применение новых гликозидных триольных промежуточных соединений, которые могут быть селективно защищены по 6-положению сахара, до введения сложного эфирных- и амидсвязанных ацилоксиацильных остатков.
Один из предпочтительных по изобретению способов поучения AGP соединений показан на схеме 1, представленной ниже. Схема 1 раскрывет способ получения конкретных соединений формулы (Ia), но предназначена, чтобы служить только в качестве примера указанного аспекта изобретения, тот же самый или подобной способ может быть использован для получения других соединений типовой формулы (Ia), так же как соединения формулы (Ic).
В указанном процессе введение алифатического ацила, например (R)-3-н-алканоилокситетрадеканоила и фосфатных групп в глюкозаминовые и агликоновые компоненты также выполняют после реакции конденсации, но, в отличие от способа, раскрытого в предшествующем уровне техники, 3-гидроксильную группу селективно этерифицируют с помощью алкановой кислоты, замещенной алифатической ацильной группой, предпочтительно (R)-3-н-алканоилоксиалкановой кислоты, в присутствии незащищенной/нефосфорилированной 4-гидроксильной группы, с блокированием 6-положения. Это достигают путем защиты 6-гидроксильной группы сахарного остатка устойчивой защитной группой по месту временной защиты 4,6-гидроксильных положений ацетонидом. Предпочтительно β-гликозид 8 или соответствующеее бис-Troc производное 9 де-O-ацетилируют с помощью подходящего основания, что дает промежуточный триол 10, который селективно защищают по 6-положению с помощью затрудненной силильной группы, такой как трет-бутилдиметилсилил (TBS) в обычных условиях известных среднему специалисту в данной области, что дает силилзащищенное промежуточное соединение 12. Промежуточный триол 10 представляет собой новое соединение. 3-O-Ацилирование 12 с помощью (R)-3-н-алканоилокситетрадекановой кислоты, например, с последующим снятием защиты/ацилирования сахара и агликоновых аминогрупп, одновременно (PG=Troc) или последовательно (PG=Aoc) используя либо цинк (PG=Troc) или цинк и Pd(0) (PG=Aoc) на стадии снятия защиты и (R)-3-н-алканоилокситетрадекановой кислоты на стадии ацилирования, обеспечивает гексаацилированное промежуточное соединение 13. Пентаацилированные соединения, то есть в которых одна из ацильных групп R1, R2, R3, R11 или R12 является водородом, могут быть получены путем использования различных защитных групп для двух аминогрупп так, что одна или другая может быть селективно ацилирована, например используя Аос группу для первой и Troc группу для другой.
Фосфорилирование 4-гидроксильной группы выполняют с помощью методик, известных среднему специалисту в данной области, используя, предпочтительно, или дибензил, или ди-трет-бутилзащищенный хлорфосфат, или фосфорамидитный реагент, что дает фосфотриэфир 14. Фосфатные, силильные и любые остающиеся защитные группы в 14 затем расщепляют при мягких кислотных условиях или с помощью других соответствующих методик, что дает соединения формулы (Ia). Важно заметить, что порядок, по которому вводят фосфат и N-связанные (R)-3-н-алканоилокситетрадеканоильные группы в 14, может быть обращен с помощью соответствующего выбора ортогональных фосфат- и аминозащитных групп.
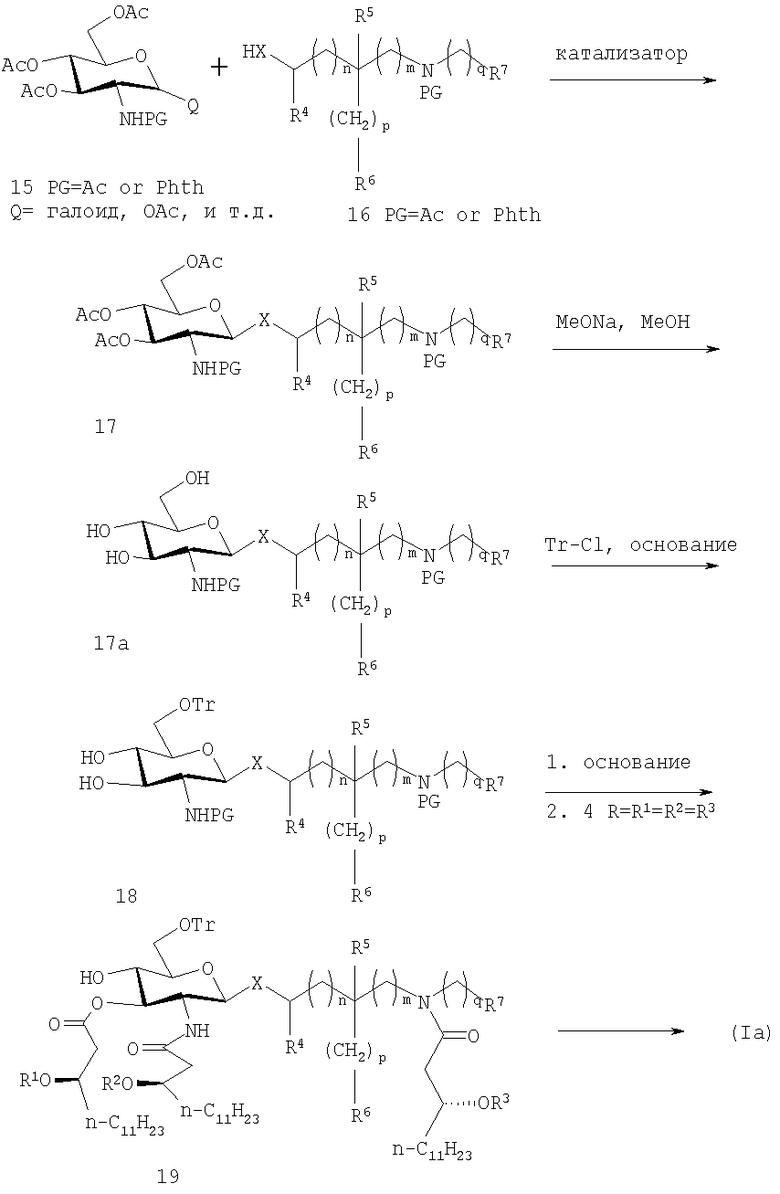
Вариант способа, продемонстрированный на схеме 1, показан на схеме 2 и включает использование коммерчески доступного гликозильного донора, такого как 15, обладающего или ацетильными, или фталимидными азотзащитными группами и либо аномерными ацетокси или галоидной группой. С другой стороны, эта схема представляет способы изобретения для получения соединений формулы (Ia) или (Ic).
На схеме 2 гликозильный донор 15 конденсируют с аналогичным N-защищенным акцепторным компонентом 16 в присутствии подходящего катализатора, что дает β-гликозид 17. Так как N-ацетил и фталимидные группы обычно требуют сильных основных условий для снятия защиты, как правило, необходимо использование устойчивой в основных условиях эфир-связанной защитной группы, такой как трифенилметил (тритил, Tr) по 6-положению. Соответственно, де-O-ацетилирование 17 в обычных условиях, с последующим предпочтительно селективным тритилированием по 6-положению дает диол 18. Вызванное основанием расщепление N-ацетильной или фталимидной группы сопровождаемое одновременньм или последовательным N- и О- ацилированием полученного промежуточного диаминодиола с помощью (R)-3-н-алканоилокситетрадекановой кислоты в присутствии подходящего реагента(ов) конденсации дает гексаацилированное производное 19. Промежуточный диаминодиол образуется путем обработки соединения 18 основанием, имеющим формулу:
Фосфорилирование соединения 19 хлорфосфатом или фосфорамидитным реагентом, как изображено на схеме 1, с последующим снятием защиты в мягких кислотных условиях или с помощью других подходящих методик дает соединения формулы (Ia).
СХЕМА 1
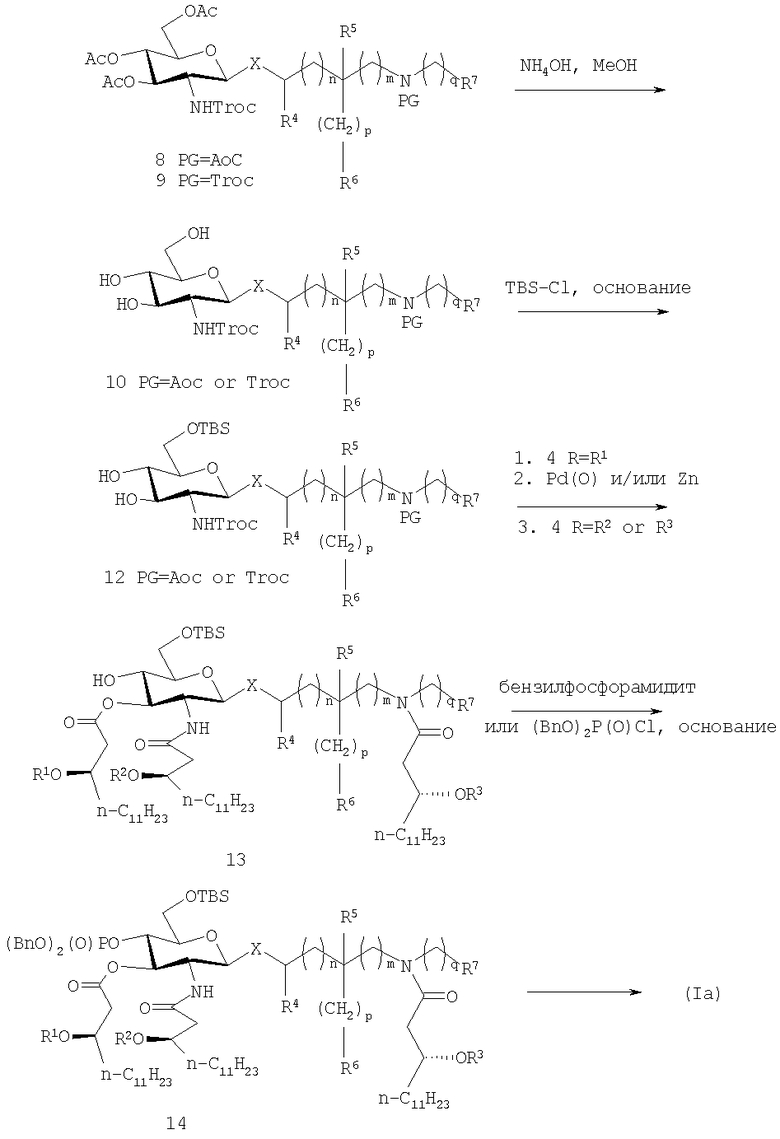
СХЕМА 2
В указанных выше схемах 1 и 2 различные группы R1-R7, n, p и q имеют значения, как определено выше.
Изобретение, кроме того, иллюстрируется с помощью следующих примеров. Эти примеры представлены исключительно как иллюстративные для изобретения и ни в коем случае не ограничивают его определение или границы.
Пример 1: Получение хлорида 2-дезокси-4-O-дифенилфосфоно-3-O-[(R)-3-тетрадеканоилокситетрадеканоил]-6-O-(2,2,2-трихлор-1,1-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтоксикарбониламино)-α-D-глюкопиранозила
Этот процесс изображен на схеме А, приведенной ниже, и включает новые промежуточные соединения (iv), (vi) и (vii), которые представляют собой аспекты настоящего изобретения.
Схема А

(а) Получение трет-бутилдифенилсилил-2-дезокси-4,6-O-изопропилиден-2-(2,2,2-трихлорэтоксикарбониламино)-α-D-глюкопиранозида (Схема А, соединение vi).
(1) 2,2,2-Трихлорэтоксикарбонилхлорид (200 г, 0,944 моль) добавляют порциями в раствор гидрохлорида D-глюкозамина (v, 200 г, 0,927 моль) и NaHCO3 (200 г, 2,4 моль) в воде (4 л) в 3-горлой круглодонной колбе объемом 10 л и полученную смесь механически перемешивают в течение ночи при комнатной температуре. Осадок белого цвета, который образуется, собирают путем фильтрования, используя фриттовую воронку, объемом, 2 л, промывают эфиром (2 л) и сушат под высоким вакуумом в течение 3 часов, что дает 297 г (90%) 2-дезокси-2-(2,2,2-трихлорэтоксикарбонилмино)-D-глюкозу в виде твердого вещества белого цвета (мол. вес 354,57).
(2) Раствор 2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-D-глюкозы (297 г, 0,838 моль), полученной выше в (1), в смеси пиридина (1 л, 12,4 моль) и уксусного ангидрида (1 л, 10,6 моль) в круглодонной колбе объемом 10 л механически перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют под пониженным давлении, что дает масло, которое отгоняют азеотропо с толуолом (2×1 л) и сушат под высоким вакуумом в течение ночи, что дает 438 г (˜100%; 90% из v) тетраацетата в виде сиропа (мол. вес 522,71, ТСХ (EtOAc) Rf 0,75).
(3) Тетраацетат, полученный выше в (2) (438 г, 0,838 моль), растворяют в EtOAc (4 л) и переносят в 3-горлую круглодонную колбу объемом 10 л, обрабатывают морфолином (200 мл, 2,29 моль) и механически перемешивают в течение 8 часов при комнатной температуре. Завершение реакции определяют с помощью ТСХ (50% смесь EtOAc/гексан). Добавляют 3 N водный раствор HCl (2 л) и полученную смесь перемешивают в течение 30 минут. Смесь переносят в отдельную колбу объемом 6 л и слои разделяют. Органическую фазу промывают насыщенным водным раствором NaCl (1 л), сушат (Na2SO4) и концентрируют, что дает 373 г (93%, 84% из v) производного со снятой защитой по 1-O-положению (гемиацеталь) в виде пены белого цвета (мол. вес 480,67; ТСХ (50% смесь EtOAc/гексан) Rf 0,22).
(4) Раствор гемиацеталя, полученного выше в (3) (373 г, 0,776 моль), и имидазола (132 г, 1,94 моль) в N,N-диметилформамиде (ДМФА, 430 мл, 1,8 М) обрабатывают трет-бутилхлордифенилсиланом (242 мл, 0,931 моль) и перемешивают в течение 48 часов при комнатной температуре. Завершение реакции подтверждают с помощью ТСХ (50% смесь EtOAc/гексан). Реакционную смесь распределяют между этиловым эфиром (4 л) и водой (1 л) в отдельной колбе объемом 6 л и слои разделяют. Слой эфира промывают водой (1 л), сушат (Na2SO4) и концентрируют, что дает окрашенное в бронзовый цвет масло, которое кристаллизуют из смеси EtOAc-гексан (˜1:2 об./об.) тремя порциями, что обеспечивает 474 г (85%, 71% из v) трет-бутилдифенилсилилгликозида iv в виде твердого вещества белого цвета (мол. вес 719,08; ТСХ (50% смесь EtOAc/гексан) Rf 0,44).
(5) Раствор силилгликозида, полученный выше в (4) (474 г, 0,659 моль), в МеОН (2 л) в 3-горлой круглодонной колбе объемом 3 л обрабатывают гидроксидом аммония (300 мл, 4,5 моль) (появляется некоторое осаждение) и перемешивают при комнатной температуре в течение ночи, а затем обрабатывают вторьм остатком гидроксида аммония (50 мл, 0,75 моль) и снова перемешивают в течение ночи. Завершение реакции определяют с помощью ТСХ (EtOAc). Реакционную смесь концентрируют и полученный остаток растворяют в EtOAc (500 мл), помещают на подушку из силикагеля (1 кг) во фриттовой стеклянной воронке объемом 3 л и элюируют 50% смесью EtOAc-гексан (5 л) и EtOAc (7 л). Фракции, содержащие продукт, концентрируют в круглодонной колбе объемом 3 л, что дает 329 г (84%, 60% из v) триола (мол. вес 592,97, ТСХ (EtOAc) Rf 0,35).
(6) Суспензию триола, полученного выше в (5) (329 г, 0,555 моль), в 2,2-диметоксипропане (1,5 л) в круглодонной колбе объемом 3 л обрабатывают камфорсульфоновой кислотой (6,4 г, 0,028 моль) и перемешивают с помощью магнитной мешалки при комнатной температуре в течение ночи, получая раствор светло-желтого цвета. Добавляют твердый NaHCO3 (4,6 г, 0,055 моль) и полученную смесь перемешивают в течение 2 часов при комнатной температуре, а затем концентрируют досуха. Полученный сырой продукт растворяют в дихлорметане (1,2 л), разделяют на две равные порции и помещают на силикагель (1 кг, предварительно смоченный 30% смесью EtOAc/гексан) в две индивидуальные фриттовые стеклянные воронки объемом 3 л и элюируют 30% смесью EtOAc/гексан (10 л) и 50% смесью EtOAc/гексан (8 л). Фракции, содержащие очищенный продукт, объединяют и концентрируют, что дает соединение vi в виде аморфного вещества. Указанный продукт может быть в дальнейшем очищен кристаллизацией из гексана, если необходимо.
Молекулярная формула: С28Н36Cl3NO7Si
Молекулярный вес: 633,04
Теоретический выход: 587 г (в расчете на v)
Полученный выход: 306 г (87%, 52% из v)
ТСХ: Rf 0,60 (EtOAc)
(b) Получение трет-бутилдифенилсилил-2-дезокси-4-O-дифенилфосфоно-3-O-[(R)-3-тетрадеканоилокситетрадеканоил]-6-O-(2,2,2-трихлор-1,1-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтоксикарбониламино)-α-D-глюкопиранозида (Схема А, соединение vii).
(1) Раствор соединения vi (141 г, 0,223 моль) в CH2Cl2 (1 л) в круглодонной колбе объемом 2 л обрабатывают 3-(R)-(тетрадеканоилокси)тетрадекановой кислотой (101,7 г, 0,224 моль), DCC (55 г, в виде расплава, 0,267 моль) и 4-пирролидинопиридином (3,3 г, 0,022 моль) и перемешивают при комнатной температуре в течение ночи. Завершение реакции определяют с помощью ТСХ (20% смесь EtOAc/гексан). Реакционную смесь фильтруют, концентрируют до приблизительно половины объема, разделяют на две равные порции и помещают на силикагель (1 кг, предварительно смоченный 2,5% смесью EtOAc/гексан) в две индивидуальные фриттовые стеклянные воронки объемом 3 л. Градиентное элюирование с 2,5%, 5% и 10% смеси EtOAc/гексан (8 л каждая) и концентрирование фракции, содержащей продукт в круглодонной колбе объемом 3 л, дает 220 г (92%) эфира (мол. вес 1069,72, ТСХ (20% смесь EtOAc/гексан) Rf 0,53).
(2) Эфир, полученный выше в (1) (218 г, 0,204 моль), суспендируют в 90% водный раствор АсОН (1 л) в круглодонной колбе объемом 3 л, перемешивают (на роторном испарителе) при температуре 70°С в течение 2,5 часов, получая раствор молочного вида. Завершение реакции определяют с помощью ТСХ (20% смесь EtOAc/гексан). Реакционную смесь концентрируют и остаток АсОН удаляют азеотропно с толуолом (2×500 мл). Полученный сырой продукт растворяют в 10% смеси EtOAc/гексан (400 мл), разделяют на две равные порции и помещают на силикагель (1 кг) в две индивидуальные фриттовые стеклянные воронки объемом 3 л. Градиентное элюирование с 10% смесью EtOAc/гексан (10 л) и 15%, 20% и 30% смесью EtOAc/гексан (5 л каждая) и концентрирование фракции, содержащей продукт, дает 193 г (92%, 85% из vi) диола (мол. вес 1029,66, ТСХ (20% EtOAc) Rf 0,10), содержащего небольшое количество (<5% с помощью ТСХ) 6-O-ацетила как побочного продукта (Rf 0,25). (Примечание: 6-ацетатный побочный продукт легко разделяют с помощью радиальной компрессионной хроматографии как производное 4-дифенилфосфатное, полученное ниже на стадии (3).)
(3) Магнитной мешалкой перемешивают раствор диола, полученный выше в (2) (193 г, 0,187 моль) в CH2Cl2 (1 л) при температуре 0°С, обрабатывают пиридином (18,2 мл, 0,225 моль), а затем 1,1-диметил-2,2,2-трихлорэтилхлорформиатом (49,5 г, 0,206 моль). Развитие реакции контролируют с помощью ТСХ (20% смесь EtOAc/гексан). Когда реакцию доводят до конца, как показывает ТСХ (обычно 30-60 минут, но может быть необходимо более продолжительное время реакции), последовательно добавляют триэтиламин (55 мл, 0,39 моль), 4-пирролидинопиридин (13,9 г, 0,094 моль) и дифенилхлорфосфат (58,2 мл, 0,281 моль) и полученную смесь перемешивают при комнатной температуре в течение ночи. Завершение реакции определяют с помощью ТСХ (20% смесь EtOAc/гексан). Реакционную смесь концентрируют досуха и полученный остаток распределяют между EtOAc (1,5 л) и 1,2 N водным раствором HCl (2 л) в отдельной колбе объемом 6 л и слои разделяют. Слой EtOAc промывают водой (2 л), сушат (Na2SO4) и концентрируют. Полученный остаток растворяют в 10% смеси EtOAc/гексан (500 мл) и очищают с помощью градиентного элюирования на системе Biotage 150 Hi (150 л колонка) с 10% смесью EtOAc/гексан (50 л), собирая 950 мл фракции. Фракции, содержащие соединение vii, объединяют и концентрируют.
Молекулярная формула: C70H98Cl6NO15Psi
Молекулярный вес: 1465,30
Теоретический выход: 326,8 г (в расчете на vi)
Полученный выход: 211 г (77%, 65% из vi)
ТСХ: Rf 0,47 (20% смесь EtOAc/гексан)
(с) Получение хлорида 2-дезокси-4-O-дифенилфосфоно-3-O-[(R)-3-тетрадеканоилок-ситетрадеканоил]-6-O-(2,2,2-трихлор-α-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтокси-карбониламино)-α-D-глюкопиранозила (Схема А, соединение vii).
Раствор соединения vii (192 г, 0,131 моль) в CHCl3 (2 л) при температуре 0°С в круглодонной колбе объемом 5 л обрабатывают, вводя по каплям из делительной воронки а,а-дихлорметилметиловый эфир (78 мл, 0,87 моль), а затем - ZnCl2 (1,0 М в эфире, 100 мл, 0,1 моль). Холодную баню удаляют и полученную смесь перемешивают при комнатной температуре в течение ночи. Завершение реакции определяют с помощью ТСХ (20% смесь EtOAc/гексан). Реакционную смесь обрабатывают холодным насыщенным водным раствором NaHCO3 (1 л), перемешивают в течение часа и слои разделяют в отдельной колбе объемом 6 л. Органический слой сушат (MgSO4) и концентрируют. Полученный остаток очищают на системе Biotage 150 Hi (150 л колонка), элюируя 10% смесью EtOAc/гексан (80 л, 950 мл фракции). Фракции, содержащие чистый продукт, объединяют и концентрируют.
Молекулярная формула: C54H79Cl7NO14P
Молекулярный вес: 1245,36
Теоретический выход: 163,2 г
Полученный выход: 141 г (86%)
ТСХ: Rf 0,42 (20% смесь EtOAc/гексан)
Пример 2 - Получение триэтиламмонийной соли (N-[(R)-3-деканоилокси-тетрадеканоил]-O-[2-дезокси-4-O-фосфоно-2-[(R)-3-деканоилокситетрадеканоиламино]-3-O-[(R)-3-деканоилокситетрадеканоил]-β-D-глюкопиранозил]-L-серина [соединение формулы (Ia), в котором R1=R2=R3=H-C9H19CO, Z=Y=O, n=m=р=q=0, r=10, R4=R5=R7=R9=Н, R6=CO2Н, R8=РО3Н2)], а именно
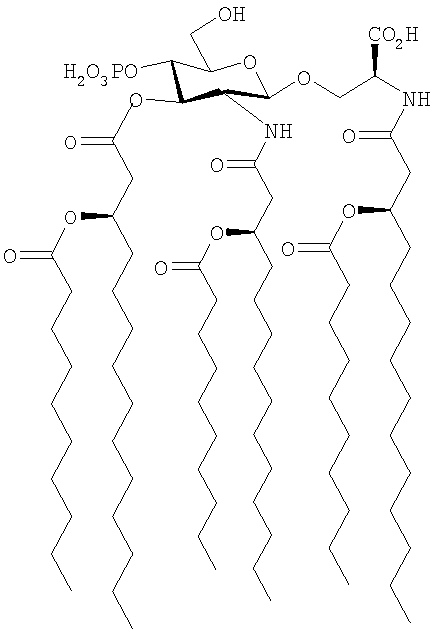
В этом примере используют способ, показанный на схеме 1.
(1) Раствор 1,3,4,6-тетра-O-ацетил-2-дезокси-2-(2,2,2-трихлорэтоксикарбони-ламино)-β-D-глюкопиранозида (5,33 г, 10,2 ммоль) и бензил N-(2,2,2-трихлорэтоксикарбонил)-L-серина (4,16 г, 11,2 ммоль) в безводном СН2Cl3 (15 мл) обрабатывают по каплям эфиратом трифторида бора (2,59 мл, 20,4 ммоль), а затем перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь гасят насыщенным водным раствором NaHCO3 (20 мл) и слои разделяют. Водный слой экстрагируют CHCl3 (2×10 мл) и объединенные органические слои промывают H2O (10 мл), сушат (Na2SO4), а затем концентрируют в вакууме. Флеш хроматография на силикагеле (градиентное элюирование, 20-50% смесь AcOEt/гексан) дает 7,42 г (87%) бензиловый эфир N-(2,2,2-трихлорэтоксикарбонил)-O-[3,4,6-тетра-O-ацетил-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозил]-L-серина в виде твердого вещества белого цвета (соединение 9; Х=О, n=m=р=q=0, r=10, R4=R5=R7=Н, R6=CO2Bn).
(2) Раствор соединения, полученного выше в (1) (408 мг, 0,49 ммоль), в тетрагидрофуране (ТГФ; 20 мл) гидрируют в присутствии 10% палладия на угле (30 мг) при комнатной температуре и атмосферном давлении в течение 3 часов. Реакционную смесь фильтруют через целит и фильтрат концентрируют в вакууме. Флеш хроматография на силикагеле с 2% смесью МеОН-CHCl3, а затем с 10% смесью МеОН-CHCl3 дает 347 мг (98%) N-(2,2,2-трихлорэтоксикарбонил)-O-[3,4,6-тетра-O-ацетил-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозил]-L-серин в виде твердого вещества белого цвета (соединение 9; Х=О, n=m=р=q=0, r=10, R4=R5=R7=Н, R6=СО2Н)
(3) Раствор соединения, полученного выше в (2) (998 мг, 1,34 ммоль) в метаноле (15,5 мл), обрабатывают гидроксидом аммония (0,21 мл, 5,37 ммоль) при комнатной температуре в течение 16 часов, а затем дополнительно гидроксидом аммония (0,21 мл, 5,37 ммоль) в течение 24 часов. Реакционную смесь концентрируют в вакууме, что дает твердое вещество белого цвета. Суспензию твердого вещества белого цвета в CH2Cl2 (33,5 мл) обрабатывают бензилбромидом (0,80 мл, 6,7 ммоль), бромидом тетрабутиламмония (432 мг, 1,34 ммоль) и насыщенным раствором NaHCO3 (33,5 мл) и полученную двухфазную смесь энергично перемешивают при комнатной температуре в течение 24 часов и слои разделяют. Водный слой экстрагируют CHCl3 (2×15 мл) и объединенные органические слои промывают Н2О (10 мл), сушат (Na2SO4) и концентрируют в вакууме. Полученный остаток растворяют в безводном пиридине (10 мл), обрабатывают трет-бутилдиметилсилилхлоридом (242 мг, 1,61 ммоль) и перемешивают при комнатной температуре в течение 1,5 часов. Реакционную смесь обрабатывают дополнительно трет-бутилдиметилсилилхлоридом (242 мг, 1,61 ммоль) и перемешивают в течение 1,5 часов. Реакционную смесь распределяют между CHCl3 (10 мл) и Н2О (10 мл). Водный слой экстрагируют CHCl3 (2×15 мл) и объединенные органические слои промывают Н2О (15 мл), сушат (Na2SO4) и концентрируют в вакууме. Флеш хроматография на силикагеле, используя градиентное элюирование (1,0→1,25% СН3ОН/CHCl3), дает 724 мг (66%) бензилового эфира N-(2,2,2-трихлорэтоксикарбонил)-O-[6-O-трет-бутилдиметилсилил-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозил]-L-серина в виде твердого вещества белого цвета (соединение 12 PG=Troc, X=0, n=m=p=q=0, r=10, R4=R5=R7=Н, R6=CO2H).
(4) Раствор соединения, полученного выше в (3) (892 мг, 1,09 ммоль), в безводном СН2Cl2 (10,5 мл) обрабатывают (R)-3-деканоилокситетрадекановой кислотой (476 мг, 1,20 ммоль), метиодидом 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC-MeI; 355 мг, 1,20 ммоль) и 4-пирролидинопиридином (8 мг, 0,054 ммоль) при температуре 0°С в течение часа. Реакционную смесь обрабатывают дополнительно (R)-3-деканоилокситетрадекановой кислотой (60 мг) и EDC-MeI (60 мг) при температуре 0°С, перемешивают в течение 30 минут и концентрируют в вакууме. Флеш хроматография на силикагеле со смесью 1:6 AcOEt-гексан дает 1,10 г (85%) бензиловый эфир N-(2,2,2-трихлорэтоксикарбонил)-O-[6-O-трет-бутилдиметилсилил-3-O-[(R)-3-деканоилокситетрадеканоил]-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозил]-L-серин в виде бесцветного масла.
(5) Раствор соединения, полученного выше в (4) (1,162 г, 0,967 ммоль), в 20% водном растворе ТГФ (16 мл) обрабатывают порошком цинка (632 мг, 9,67 ммоль) и уксусной кислотой (0,12 мл, 2,13 ммоль), а затем перемешивают в течение часа при комнатной температуре. Реакционную смесь фильтруют через целит и фильтрат концентрируют в вакууме. Полученное твердое вещество грязно-белого цвета растворяют в CHCl3 (15 мл) и промывают последовательно порциями, в объеме 15 мл каждая, 0,1М HCl, насыщенным водным раствором NaHCO3 и Н2О. Органический слой сушат (Na2SO4) и концентрируют в вакууме и полученный остаток сушат в течение ночи под высоким вакуумом. Раствор остатка в безводном СН2Cl2 (9,5 мл) обрабатывают (R)-3-деканоилокситетрадекановой кислотой (848 мг, 2,13 ммоль) и EDC.MeI (632 мг, 2,13 ммоль) и перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь концентрируют в вакууме и полученный остаток очищают с помощью флеш хроматографии на силикагеле (градиентное элюирование; 20→25% AcOEt/гексан), что дает 1,03 г (66%) бензиловый эфир N-[(R)-3-деканоилокситетрадеканоил]-O-[6-O-трет-бутилдиметилсилил-2-дезокси-2-[(R)-3-деканоилокситетрадеканоиламино]-3-O-[(R)-3-деканоилокситетрадеканоил]-β-D-глюкопиранозил]-L-серина в виде стекловидного твердого вещества, (соединение 13 R1=R2=R3=n-C9H19CO, Х=O, n=m=р=q=0, r=10, R4=R5=R7=Н, R6=CO2Bn).
(6) Раствор соединения, полученного выше в (5) (112 мг, 0,069 ммоль), в безводном дихлорметане (1 мл) в атмосфере аргона обрабатывают диизопропилдибензилфосфорамидитом (39 мкл, 0,12 ммоль) и тетразолом (12 мг, 0,173 ммоль) и перемешивают при комнатной температуре в течение часа. Реакционную смесь охлаждают до температуры 0°С и обрабатывают мета-хлорпербензойной кислотой (мета-СРВА; 33 мг, 0,193 ммоль) в течение 30 минут. Реакционную смесь гасят с помощью дополнительного количества насыщенного водного раствора NaHCO3 (5 мл) и перемешивают при комнатной температуре в течение 15 минут. Водный слой экстрагируют хлороформом (3×5 мл) и объединенные органические слои промывают водой (5 мл), сушат (Na2SO4) и концентрируют в вакууме. Флеш хроматография 25% смеси AcOEt-гексан дает частично очищенный продукт, который повторно хроматографируют на силикагеле 20% смесью AcOEt-гексан, что дает 122 мг (93%) бензилового эфира N-[(R)-3-деканоилокситетрадеканоил]-O-[6-O-трет-бутилдиметилсилил-2-дезокси-4-O-дифенилфосфоно-2-[(R)-3-деканоилокситетрадеканоиламино]-3-О-[(R)-3-деканоилокситетрадеканоил]-β-D-глюкопиранозил]-L-серина в виде бесцветного масла.
(7) Раствор соединения, полученного выше в (6) (232 мг, 0,124 ммоль), в безводном ТГФ (10 мл) гидрируют в присутствии 20% гидроксида палладия на угле (46 мг) при комнатной температуре и атмосферном давлении в течение 36 часов. Реакционную смесь фильтруют через целит и фильтрат концентрируют под вакуумом. Полученное масло (181 мг) растворяют в CH2Cl2 (2,5 мл) и обрабатывают трифторуксусной кислотой (29 мкл) и перемешивают в атмосфере аргона при комнатной температуре в течение 18 часов. Реакционную смесь концентрируют и совместно выпаривают с гексаном (2х5 мл). Флеш хроматография на силикагеле со смесью хлороформ-метанол-вода-триэтиламин (градиентное элюирование; 87:12:0,5:0,5→77:22,5:0,5:0,5) дает 102 мг (55%) триэтиламмонийной соли N-[(R)-3-деканоилокситетрадеканоил]-O-[2-дезокси-4-O-фосфоно-2-[(R)-3-деканоилокситетрадеканоиламино]-3-O-[(R)-3-деканоилокситетрадеканоил]-β-D-глюкопиранозил]-L-серина (RC-527) в виде бесцветного твердого вещества.
Пример 3 - Получение бис(триэтил)аммонийной соли (S)-2-[(R)-3-гексаноилокситетрадеканоиламино]-3-фосфонооксипропил 2-дезокси-4-O-фосфоно-3-O-[(R)-3-гексаноилокситетрадеканоил]-2-[(R)-3-гексаноилокситетрадеканоиламино]-β-D-глюкопиранозида [соединение формулы (I), в котором Х представляет собой (Ia), а именно R1=R2=R3=n-C5H11CO, Z=Y=O, n=m=р=q=0, r=10, R4=R5=R7=R9=Н, R6=СН2ОРО3Н2, R8=РО3Н2], а именно:
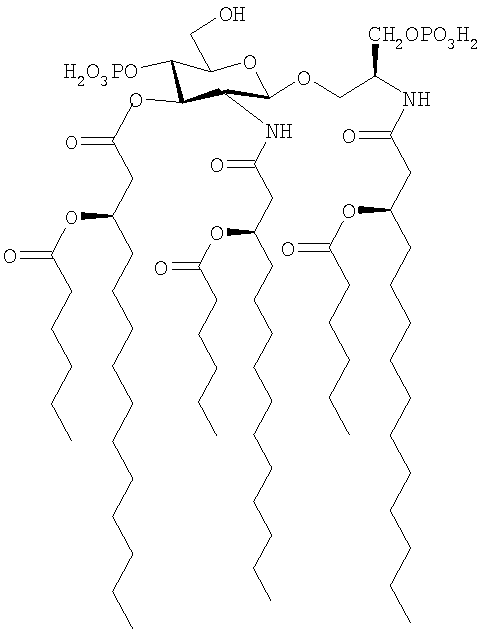
В этом примере используют способ, показанный на схеме 1.
(1) В соответствии с аналогичной методикой, описанной для примера 2-(3), 1,3,4,6-тетра-O-ацетил-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозид (0,62 г, 1,18 ммоль) и (S)-2-(2,2,2-трисхлорэтоксикарбониламино)-3-бензилокси-1-пропанол (0,46 г, 1,30 ммоль) конденсируют в присутствии эфирата трифторида бора (0,3 мл, 2,4 ммоль), что дает (R)-2-(2,2,2-трифторэтоксикарбониламино-3-бензилокси-1-пропил-2-дезокси-3,4,6-тетра-O-ацетил-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозид в виде твердого вещества светло-желтого цвета (соединение 9; Х=О, n=m=р=q=0, r=10, R4=R5=R7=Н, R6=СН2OBn). Раствор этого соединения в метаноле (15 мл) обрабатывают гидроксидом аммония (0,21 мл, 5,37 ммоль) при комнатной температуре в течение 19 часов, а затем дополнительно гидроксидом аммония (0,20 мл, 5,1 ммоль) в течение 25 часов. Реакционную смесь концентрируют в вакууме, что дает твердое вещество белого цвета. Флеш хроматография на силикагеле (градиентное элюирование 5→6% СН3ОН/CHCl3) дает 0,57 г (63%) 3-бензилокси-(R)-2-(2,2,2-трихлорэтоксикарбониламино)пропил 2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозида в виде стекловидного твердого вещества.
(2) Раствор соединения, полученного выше в (2) (0,57 г, 0,83 ммоль), в безводном пиридине (8,5 мл) обрабатывают трет-бутилдиметилсилилхлоридом (0,15 г, 0,99 ммоль) и перемешивают при комнатной температуре в течение 1,5 часов. Дополнительно добавляют трет-бутилдиметилсилилхлорид (0,15 г, 0,99 ммоль) и через еще 1,5 часа реакционную смесь распределяют между CHCl3 (10 мл) и Н2O (10 мл) и слои разделяют. Водный слой экстрагируют CHCl3 (2×10 мл) и объединенные органические слои промывают H2O (10 мл), сушат (Na2SO4) и концентрируют в вакууме. Флеш хроматография на силикагеле (градиентное элюирование; 80:1→60:1 CHCl3/СН3ОН) дает 0,65 г (98%) 3-бензилокси-(R)-2-(2,2,2-трихлорэтоксикарбониламино)пропил 6-O-трет-бутилдиметилсилил-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозида в виде твердого вещества белого цвета.
(3) В соответствии с аналогичной методикой, описанной для примера 2-(4), соединение, полученное выше в (2) (0,47 г, 0,59 ммоль), ацилируют (R)-3-гексаноилокситетрадекановой кислотой (0,22 г, 0,64 ммоль) в присутствии EDC-MeI (0,21 г, 0,70 ммоль) и 4-пирролидинопиридина (4 мг, 0,03 ммоль), что дает 0,58 г (88%) 3-бензилокси-(R)-2-(2,2,2-трихлорэтоксикарбониламино)пропил-6-О-трет-бутилдиметилсилил-3-O-[(R)-3-гексаноилокситетрадеканоил]-2-дезокси-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозида в виде бесцветного масла.
(4) В соответствии с аналогичной методикой, описанной для примера 2-(5), с соединения, полученного выше в (3) (0,58 г, 0,51 ммоль), снимают защиту с помощью цинка (0,34 г, 5,14 ммоль) и ацилируют (R)-3-гексаноилокситетрадекановой кислотой (0,39 г, 1,13 ммоль) в присутствии EDC-MeI (0,34 г, 1,13 ммоль), что дает 0,41 г (56%) 3-бензилокси-(R)-2-[(R)-3-гексаноилокситетрадеканоиламино]пропил-6-O-трет-бутилдиметилсилил-3-O-[(R)-3-гексаноилокситетрадеканоил]2-дезокси-2-[(R)-3-гексаноилокситетрадеканоиламино]-β-D-глюкопиранозида в виде бесцветного масла (соединение 13 R1=R2=R3=n-С3Н11СО, Х=О, n=m=р=q=0, r=10, R4=R5=R7=Н, R6=CH2OBn).
(5) Раствор соединения, полученного выше в (4) (0,41 г, 0,29 ммоль), в ТГФ (18 мл) гидрируют в присутствии хлорида палладия (0,04 г) при комнатной температуре и атмосферном давлении в течение 17 часов. Реакционную смесь фильтруют через целит и фильтрат концентрируют в вакууме. Флеш хроматография на силикагеле (градиентное элюирование; 1:2→1:8 этилацетат/гептан) обеспечивает 0,3 г (77%) 3-гидрокси-(R)-2-[(R)-3-гексаноилокситетрадеканоиламино]пропил-6-O-трет-бутилдиметилсилил-3-O-[(R)-3-гексаноилокситетрадеканоил]-2-дезокси-2-[(R)-3-гексаноилокситетрадеканоиламино]-β-D-глюкопиранозида в виде бесцветного масла (соединение 13 R1=R2=R3=n-C5H11CO, X=О, n=m=р=q=0, r=10, R4=R5=R7=Н, R6=CH2OH).
(6) В соответствии с аналогичной методикой, описанной для примера 2-(6), соединение, полученное выше в (5) (0,30 г, 0,22 ммоль), фосфорилируют дибензилдиизопропилфосфорамидитом (0,25 мл, 0,75 ммоль), тетразолом (0,08 г, 1,11 ммоль) и мета-СРВА (0,33 г, 1,95 ммоль), что дает 0,30 г (73%) 3-дибензилфосфоноокси-(R)-2-[(R)-3-гексаноилокситетрадеканоиламино]пропил 4-дибензилфосфоно-6-O-трет-бутилдиметилсилил-3-O-[(R)-3-гексаноилокситетрадеканоил]-2-дезокси-2-[(R)-3-гексаноилокситетрадеканоиламино]-β-D-глюкопиранозида в виде бесцветного масла.
(7) Раствор соединения, полученного выше в (6) (302 мг, 0,16 ммоль), в безводном ТГФ (13 мл) гидрируют в присутствии 20% хлорида палладия на угле (60 мг) при комнатной температуре и атмосферном давлении в течение 27 часов. Реакционную смесь фильтруют через целит и фильтрат концентрируют в вакууме. Раствор полученного масла (226 мг) в СН2Cl2 (3,5 мл) обрабатывают трифторуксусной кислотой (0,04 мл, 0,49 ммоль) и перемешивают в атмосфере аргона при комнатной температуре в течение 16 часов. Реакционную смесь концентрируют и совместно выпаривают с гексаном (2×5 мл) и полученный остаток сушат под высоким вакуумом, что дает сырой продукт (226 мг). Часть сырого продукта (102 мг) растворяют в смеси 1:2 CHCl3/СН3ОН (9 мл), помещают на DEAE- колонку с целлюлозой (15 г, большой скоростью (потока), Sigma) и элюируют смесью 2:3:1 CHCl3:СН3ОН:Н2O, используя градиент соли от 0 до 0,1 M NH2OAc. Фракции, содержащие очищенный продукт, объединяют, промывают 0,1 N водным раствором HCl и концентрируют в вакууме. Полученный остаток лиофилизуют из 1% водного раствора триэтиламина (отсутствие пирогена), что дает 82 мг (81%) бис(триэтил)аммонийную соль (S)-2-[(R)-3-гексаноилокситетрадеканоиламино]-3-фосфонооксипропил 2-дезокси-4-O-фосфоно-3-O-[(R)-3-гексаноилокситетрадеканоил]-2-[(R)-3-гексаноилокситетрадеканоиламино]-β-D-глюкопиранозида в виде порошка белого цвета: положительный FAB-MS вычислено для [М+Na]+ 1407,8534, найдено 1407,8689; 1Н ЯМР (CDCl3/CD3OD): δ (част. на млн) 5,23-5,16 (м, 4Н), 4,67 (д, 1Н), 4,38 (дд, 1Н), 4,19-3,83 (м, 7Н), 3,49 (м, 2Н), 3,06 (м, 12Н), 2,64-2,23, (м, 12Н), 1,58-1,56 (м, 12Н), 1,23 (м, 94 Н), 0.88-0,87 (м, 18Н). 13С ЯМР (CDCl3/CD3OD): δ (част. на млн) 173,7, 173,3, 173,2, 170,3, 170,1, 100.0, 74,6, 74,0, 70,9, 70,8, 70,3, 66,6, 63,5, 60,4, 54,2, 45,8, 41,1, 40,7, 39,3, 34,4, 34,3, 31,9, 31,3, 29,7, 29,4, 25,3, 24,7, 22,7, 22,3, 14,1, 13,9, 8,5.
Все публикации и патентные заявки, приведенные в настоящем описании, включены в изобретение как ссылки так, как будто каждая индивидуальная публикация или описание к патенту определено и индивидуально обозначено как ссылка в настоящую заявку.
Хотя упомянутое выше изобретение было детально описано только с помощью иллюстраций и примеров для ясного понимания, среднему специалисту в данной области в свете изучения настоящего изобретения будет очевидно, что дополнительно могут быть сделаны некоторые изменения и модификации, не отступая от духа или области прилагаемой формулы изобретения.
Изобретение относится к усовершенствованному способу получения соединения аминоалкилглюкозаминид 4-фосфата формулы
где Х представляет собой 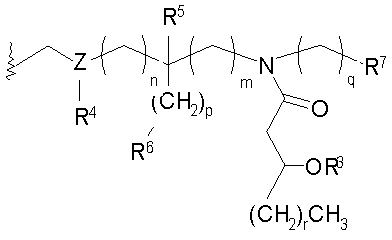 ;
;
Y представляет собой -О- или -NH-; R1, R2 и R3 каждый является независимо выбранным из водорода и насыщенных и ненасыщенных (C2-C24)алифатических ацильных групп; R8 представляет собой -Н или -РО3R11R11a, где R11 и R11a каждый независимо является -Н или (C1-C4)алифатическими группами; R9 представляет собой -Н, -СН3 или -PO3R13aR14, где Rl3a и R14 каждый является независимо выбранным из -Н и (C1-C4) алифатических групп и где индексы n, m, p, q каждый независимо является целым числом от 0 до 6, а г независимо является целым числом от 2 до 10; R4 и R5 независимо выбраны из Н и метила; R6 и R7 независимо выбраны из Н, ОН, (C1-C4)оксиалифатических групп -РО3Н2, -ОРО3Н2, -SO3Н, -OSO3Н, -NR15R16, -SR15, -CN, -NO2, -СНО, -CO2R15, -CONR15R16, -РО3R15Р16, -OPO3R15R16, -SO3Р15 и -OSO3Р15, где R15 и R16 каждый является независимо выбранным из Н и (C1-C4)алифатических групп, где алифатические группы необязательно замещены арилом; и Z представляет собой -О- или -S-; при условии, что, по крайней мере, один из R8 и R9 представляет из себя группу, содержащую фосфор, но R8 и R9 не могут быть одновременно группой, содержащей фосфор, включающий: (а) селективное 6-O-силилирование производного 2-амино-2-дезокси-β-D-глюкопиранозы формулы:
где X представляет собой О или S; и PG независимо представляет защитную группу, которая образует сложный эфир, простой эфир или карбонат с атомом кислорода гидроксильной группы или которая образует амид или карбамат с атомом азота аминогруппы соответственно; с помощью тризамещенного хлорсилана RaRbRcSi-Cl, где Ra, Rb и Rc независимо выбраны из группы, состоящей из C1-С6алкила, С3-С6циклоалкила и необязательно замещенного фенила, в присутствии третичного амина, что дает 6-силилированное производное; (b) селективное ацилирование 4-ОН положения полученного 6-O-силилированного производного с помощью (R)-3-алканоилоксиалкановой кислоты или гидроксизащищенной (R)-3-гидроксиалкановой кислоты в присутствии карбодиимидного реагента и каталитического количества 4-диметиламинопиридина или 4-пирролидинопиридина, что дает 4-0-ацилированное производное; (с) селективное удаление азотных защитных групп, последовательно или одновременно и N,N-диацилирование полученного диамина с помощью (R)-3-алканоилоксиалкановой кислоты или гидроксизащищенной (R)-3-гидроксиалкановой кислоты в присутствии реагента пептидной конденсации; (d) введение защитной фосфатной группы по 3-положению с помощью хлорфосфата или реагента фосфорамидита, что дает фосфотриэфир; и (е) одновременное или последовательное снятие защиты фосфатных, силильных и оставшихся защитных групп. 10 з.п. ф-лы.
где Х представляет собой 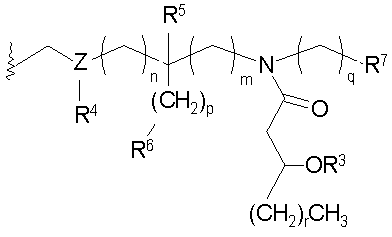 ;
;
Y представляет собой -О- или -NH-;
R1, R2 и R3, каждый является независимо выбранным из водорода и насыщенных и ненасыщенных (С2-С24)алифатических ацильных групп;
R8 представляет собой -Н или -РО3R11R11a, где R11 и R11a, каждый независимо является -Н или (C1-C4)алифатическими группами;
R9 представляет собой -Н, -СН3 или -PO3R13aR14, где R13a и R14, каждый является независимо выбранным из -Н и (C1-C4)алифатических групп; и
где индексы n, m, p, q каждый независимо является целым числом от 0 до 6, а г независимо является целым числом от 2 до 10;
R4 и R5 независимо выбраны из Н и метила;
R6 и R7 независимо выбраны из Н, ОН, (C1-C4)оксиалифатических групп, -РО3Н2, -ОРО3Н2, -SO3Н, -OSO3Н, -NR15R16, -SR15, -CN, -NO2, -СНО, -CO2R15, -CONR15R16, -РО3R15R16, -ОРО3R15R16, -SO3R15 и -OSO3R15, где R15 и R16, каждый является независимо выбранным из Н и (C1-C4)алифатических групп, где алифатические группы необязательно замещены арилом; и
Z представляет собой -О- или -S-;
при условии, что, по крайней мере, один из R8 и R9 представляет из себя группу, содержащую фосфор, но R8 и R9 не могут быть одновременно группой, содержащей фосфор,
включающий
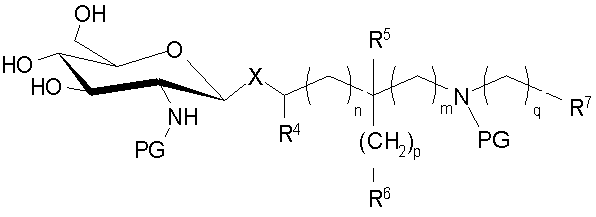
(а) селективное 6-O-силилирование производного 2-амино-2-дезокси-β-D-глюкопиранозы формулы
где Х представляет собой О или S; и PG независимо представляет защитную группу, которая образует сложный эфир, простой эфир или карбонат с атомом кислорода гидроксильной группы или которая образует амид или карбамат с атомом азота аминогруппы соответственно;
с помощью тризамещенного хлорсилана RaRbRcSi-Cl, где Ra, Rb и Rc независимо выбраны из группы, состоящей из C1-С6алкила, С3-С6циклоалкила и необязательно замещенного фенила, в присутствии третичного амина, что дает 6-силилированное производное;
(b) селективное ацилирование 4-ОН положения полученного 6-0-силилированного производного с помощью (R)-3-алканоилоксиалкановой кислоты или гидроксизащищенной (R)-3-гидроксиалкановой кислоты в присутствии карбодиимидного реагента и каталитического количества 4-диметиламинопиридина или 4-пирролидинопиридина, что дает 4-O-ацилированное производное;
(c) селективное удаление азотных защитных групп, последовательно или одновременно и N,N-диацилирование полученного диамина с помощью (R)-3-алканоилоксиалкановой кислоты или гидроксизащищенной (R)-3-гидроксиалкановой кислоты в присутствии реагента пептидной конденсации;
(d) введение защитной фосфатной группы по 3-положению с помощью хлорфосфата или реагента фосфорамидита, что дает фосфотриэфир; и
(e) одновременное или последовательное снятие защиты фосфатных, силильных и оставшихся защитных групп.
 ,
,
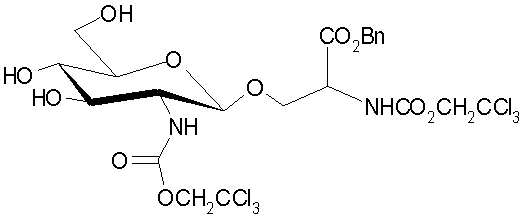 ,
,
или
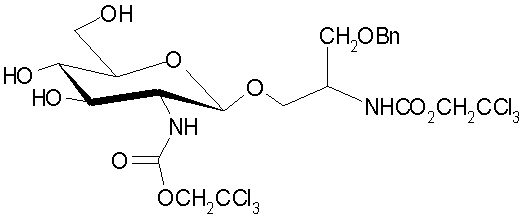 ,
,
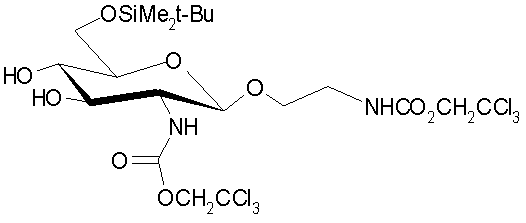
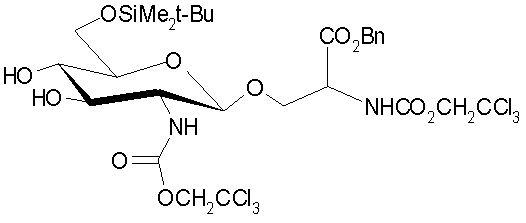
| Gross, H.; "Dichlormethyl-methylether als Reagens fur die organische Synthese und Kohlenhydratchemie"; 1978, Z | |||
| Chemie, vol.18, No.6, pp.201-210 | |||
| Magnusson, G | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |
| Org | |||
| Chem., vol.55, pp.3181-3185 | |||
| Pozsgay, Vince et al.; "Synthesis of Kojidextrins and | |||

