Система врожденного иммунитета координирует воспалительный ответ к патогенам через систему, которая отличает "собственное" от "чужого" через рецепторы, распознающие классы молекул, синтезируемых исключительно микробами. Эти классы иногда обозначают как связанные с патогеном молекулярные профили (РАМР), и к ним относятся, к примеру, липополисахариды (LPS), пептидогликаны, липотейхоевые кислоты и бактериальные липопротеиды (BLP).
LPS - это распространенный компонент наружной клеточной стенки грамотрицательных бактерий, который распознается системой врожденного иммунитета. Хотя химическая структура LPS уже известна, однако молекулярный механизм распознавания LPS белками и/или клетками сыворотки еще только начинает проясняться. В ряде недавних публикаций семейство рецепторов, называемых рецепторами типа Toll (TLR), было связано с сильным врожденным иммунным ответом на LPS и другие микробные компоненты. TLR - это мембранные белки, имеющие один трансмембранный домен. Цитоплазматические домены состоят примерно из 200 аминокислот и проявляют сходство с цитоплазматическим доменом рецептора IL-1. Внеклеточные домены сравнительно большие (550-980 аминокислот) и могут содержать множественные сайты связывания лигандов.
Важное значение TLR в иммунном ответе к LPS было продемонстрировано конкретно, по меньшей мере, для двух рецепторов типа Toll-Tlr2 и Тlr4. Так, опыты по трансфекции эмбриональных почечных клеток показали, что Tlr2 человека достаточен для придания реактивности к LPS (Yang et al., Nature 395: 284-288 (1998); Kirschning et al., J. Exp, Med. 11: 2019-97 (1998)). По-видимому, для сильного ответа к LPS требуется LPS-связывающий белок (LBP) и CD 14, который связывает LPS с высоким сродством. Прямое связывание LPS с Tlr2 наблюдалось с относительно низким сродством, что предполагает, что связыванию LPS с Tlr2 и/или активации in vivo могут способствовать вспомогательные белки.
Важное значение Тlr4 в иммунном ответе к LPS было продемонстрировано в связи с позиционным клонированием у линий мутантных мышей lps. Были идентифицированы два мутантные аллеля гена lps: полудоминантный аллель у мышей линии С3Н/HeJ и второй, рецессивный аллель у мышей линий C57BL/10ScN и C57BL/10ScCr. Мыши, гомозиготные по мутантным аллелям гена lps, восприимчивы к заражению грамотрицательными бактериями, но устойчивы к вызванному LPS септическому шоку. Локус Ips из этих мышей клонировали, и было показано, что эти мутации связаны с изменением гена Тlr4 в обоих случаях (Portorak et al., Science 383: 2085-2088 (1998); Qureshi et al., J. Exp. Med. 4: 615-625 (1999)). В этих работах сделан вывод, что Тlr4 необходим для ответа к LPS.
Биологически активной эндотоксической частью структуры LPS является липид А - фосфорилированный, множественно ацилированный жирными кислотами дисахарид глюкозамина, который служит для закрепления всей структуры в наружной мембране грамотрицательных бактерий. Ранее мы сообщали, что токсические эффекты липида А можно уменьшить путем избирательной химической модификации липида А с образованием соединений монофосфориллипида А (иммуностимулятор MPL, Corixa Corporation, Seattle, WA). Способы получения и применения иммуностимулятора MPL и близких по структуре соединений как адъювантов для вакцин и других применений уже описаны (например, см. Patent US № 4436727; 4877611; 4866034 и 4912094; 4987237; Johnson et al., J. Med. Chem. 42: 4640-4649 (1999); Ulrich and Myers, in Vaccine Design: The Subunit and Adjuvant Approach, Powell and Newman, eds., Plenum: New York, 495-524, 1995). В частности, эти и другие работы показали, что иммуностимулятор MPL и родственные соединения обладают значительной адъювантной активностью в усилении гуморального и/или клеточного иммунитета к антигенам при их применении в составе вакцин вместе с белковыми и углеводными антигенами.
Молекулы синтетических моно- и дисахаридов, сходных по структуре с иммуностимулятором MPL и именуемых аминоалкилглюкозаминидфосфатами (АГФ) описаны, к примеру, в Patent US № 6113918; Patent US № 6303347 и WO 98/50399, опубликованном 12 октября 1998 г. Эти соединения в значительной мере сохраняют свойства адъювантов при включении их в состав вакцинных композиций и обладают близким или лучшим профилем токсичности по сравнению с монофосфориллипидом А. Описано применение этих соединений в сочетании с антигенами в составе вакцин (Patent US № 6113918) и в отсутствие антигена как монотерапия (WO 01/90129, опубликовано 29 ноября 2001 г.).
Циклические аминоалкилглюкозаминидфосфаты (циклические АГФ) описаны в РСТ Patent Application № PCT/US01/24284. Эти циклические АГФ являются эффективными иммуноэффекторными молекулами, усиливающими гуморальные и клеточные ответы к антигенам вакцин. В применении к настоящему изобретению термин "циклический АГФ" означает такой азациклоалкил- или (азациклоалкил)алкилглюкозаминидфосфат, в котором 2-дезокси-2-амино-β-D-глюкопираноза (глюкозамин) соединена гликозидной связью с азациклоалкильной или (азациклоалкил)алкильной группировкой (агликоном).
Настоящее изобретение обеспечивает монотерапию, которая составлена и вводится в отсутствие экзогенных антигенов для профилактического и/или терапевтического лечения заболеваний и состояний растений и животных, таких как инфекционные заболевания, аутоиммунные заболевания и аллергии. Монотерапия настоящего изобретения включает один или несколько АГФ. Эти и другие аспекты изобретения станут понятными при обращении к следующему подробному описанию и прилагаемым рисункам.
Раскрытие изобретения
В одном из аспектов настоящее изобретение обеспечивает способы лечения, облегчения или существенного предупреждения заболеваний или состояний у животных путем введения эффективного количества соединения формулы I:
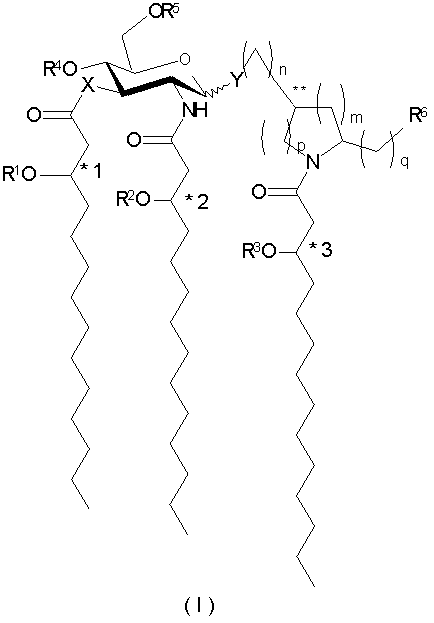
и его фармацевтически приемлемых солей, где Х означает -О- или -NH-, Y означает -О- или -S-; a R1, R2 и R3 независимо друг от друга представляют собой (С2-С20)ацильные группы, включая насыщенные, ненасыщенные и разветвленные ацильные группы; R4 представляет собой -Н или -РО3R7R8, где R7 и R8 независимо друг от друга представлены Н или (С1-С4)алифатической группой; R5 - это -Н, -СН3 или -РО3R9R10, где R9 и R10 независимо друг от друга выбраны из - Н или (С1-С4)алифатических групп; R6 независимо выбран из числа Н, ОН, (С1-С4)оксиалифатических групп, -РО3R11R12, -ОРО3R11R12, -SO3R11, -OSO3R11, -NR11R12, -SR11, -CN, -NO2, -CHO, -CO2R11 и -CONR11R12, где R11 и R12 независимо друг от друга выбраны из Н или (C1-С4)алифатических групп; при условии, что одна из групп R4 и R5 содержит фосфор и что, когда R4 представлен -РО3R7R8, то R5 не является -РО3R9R10, при этом *1-3 и ** обозначают хиральные центры; причем индексы n, m, p и q независимо друг от друга означают целые числа от 0 до 6 при условии, что сумма p и m составляет от 0 до 6.
В некоторых воплощениях соединения настоящего изобретения содержат -О- при Х и Y, R4 означает РО3R7R8, R5 и R6 представлены Н, а индексы n, m, p и q - целые числа от 0 до 3. В более предпочтительном воплощении R7 и R8 представлены - Н. В одном из воплощений n=1, m=2, а p и q=0. В других воплощениях R1, R2 и R3 представлены (С6-С14)-, (C6-C12)- или (С6-С8)ацильными группами, а в предпочтительном воплощении (C6-С12)ацильными группами. В следующем воплощении *1-3 находятся в R-конфигурации, Y находится в экваториальном положении, а ** находятся в S-конфигурации.
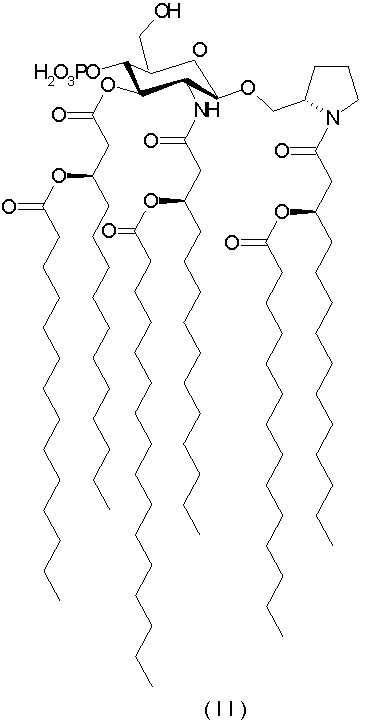
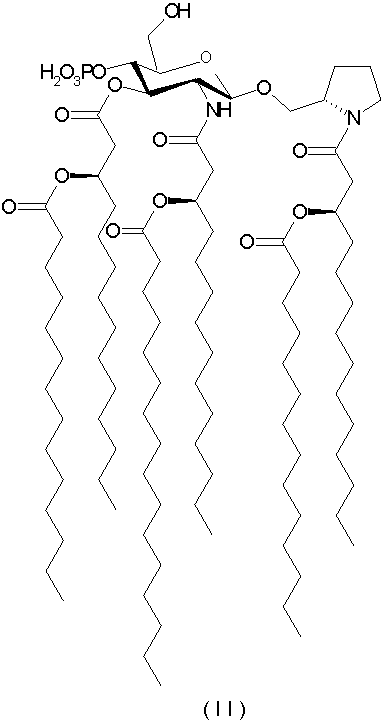
К иллюстративным воплощениям относятся N-[(R)-3-тетрадеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-тетрадеканоилокситетрадеканоиламино]-3-O-[(R)-3-тетрадеканоилокситетрадеканоил]-β-D-глюкопиранозид и его фармацевтически приемлемые соли (формула II),
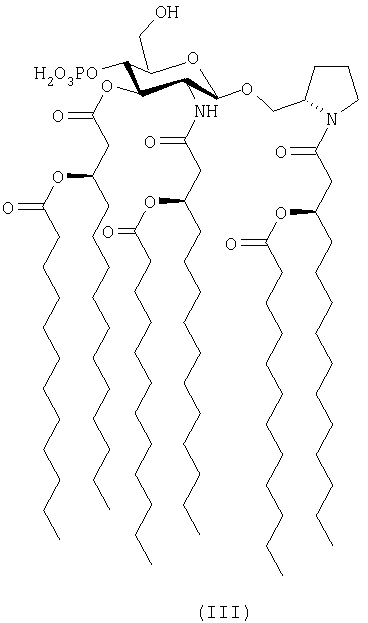
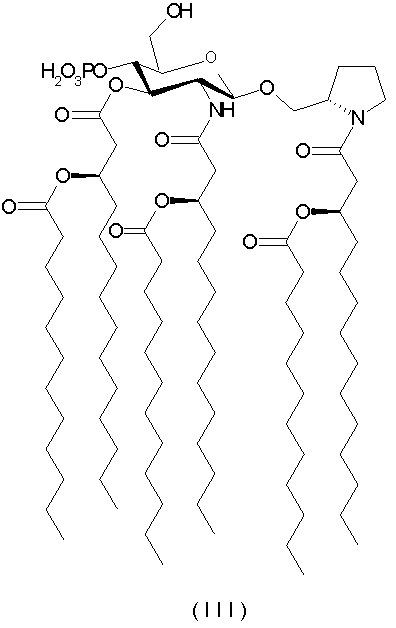
[N-(R)-3-додеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-O-[(R)-3-додеканоилокситетрадеканоил]-β-D-глюкопиранозид и его фармацевтически приемлемые соли (формула III) и
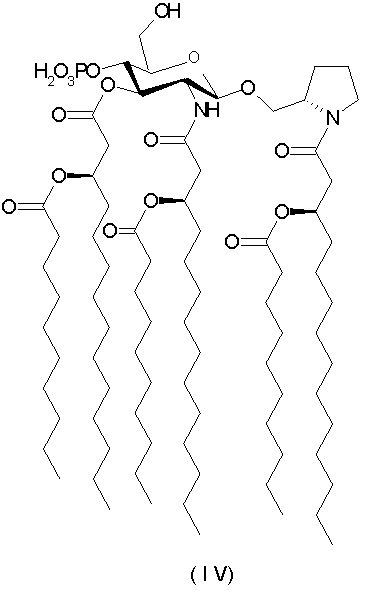
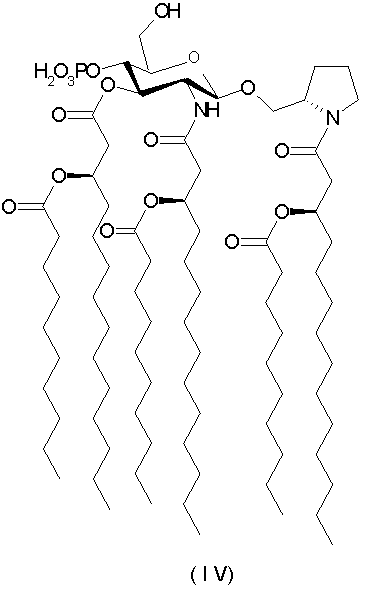
[N-(R)-3-деканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-деканоилокситетрадеканоиламино]-3-O-[(R)-3-деканоилокситетрадеканоил]-β-D-глюкопиранозид и его фармацевтически приемлемые соли (формула IV).
В определенных иллюстративных аспектах изобретения вышеуказанные способы применяются для лечения, облегчения или существенного предупреждения инфекционных заболеваний, аутоиммунных заболеваний и аллергий.
В других аспектах настоящее изобретение обеспечивает фармацевтические композиции, включающие одно или несколько вышеописанных соединений в подходящем наполнителе, которые составлены и/или вводятся в отсутствие экзогенных антигенов.
Осуществление изобретения
Предпочтительные профилактические и терапевтические применения
Настоящее изобретение в широком смысле касается профилактических и терапевтических способов лечения определенных заболеваний и других медицинских показаний путем введения одного или нескольких описанных нами соединений или фармацевтической композиции, включающей одно или несколько таких соединений. Хотя применение некоторых циклических соединений АГФ в качестве адъювантов в сочетании с экзогенно вводимыми антигенами в составе вакцин уже было описано, как и их использование в некоторых других применениях, однако в настоящем изобретении представлены новые терапевтические способы, в которых эти соединения предпочтительно применяются в монотерапевтических применениях, то есть без введения экзогенного антигена.
В одном из аспектов настоящего изобретения предусматриваются способы лечения, облегчения и/или существенного предупреждения инфекционных заболеваний у эукариот, особенно у животных, предпочтительно у человека. Учитывая важное значение опосредованного TLR сигнального пути во врожденном иммунном ответе на микробное заражение, способность к избирательному стимулированию таких путей с минимальной токсичностью является мощным подходом к возможным способам профилактического и/или терапевтического лечения против целого ряда возбудителей инфекций.
Описанные здесь способы применимы практически против любого типа возбудителей, включая бактерии, вирусы, паразиты и грибы. Например, изобретение применимо к профилактическому и/или терапевтическому лечению бактериальных инфекций, вызываемых различными видами Pseudomonas, Escherichia, Klebsiella, Enterobacter, Proteus, Serratia, Candida, стафилококками, стрептококками, Chlamydia, Mycoplasma, Bacillus и многими другими. Иллюстративные вирусные заболевания, которые можно лечить в соответствии с изобретением, включают заболевания, вызываемые, к примеру, вирусом гриппа, аденовирусами, вирусами парагриппа, риновирусами, респираторно-синцитиальными вирусами (RSV), герпесвирусами, цитомегаловирусами, вирусами гепатита, например гепатита В и С, и др. Примеры грибов включают, к примеру, Aspergillus, Candida albicans, Cryptococcus neoformans, Coccidioides immitus и др.
В одном иллюстративном воплощении изобретение предусматривает способы лечения пациентов, в частности иммунокомпрометированных пациентов, которые уже приобрели инфекцию или подвергаются риску развития инфекций, таких как внутрибольничные бактериальные или вирусные инфекции. Около 2 миллионов из 40 миллионов лиц, поступающих в больницы каждый год, приобретают внутрибольничные инфекции во время пребывания в больнице и около 1% из них, то есть около 400000 пациентов, приобретают внутрибольничные пневмонии, причем более 7000 из них умирают. Это делает нозокомиальные пневмонии ведущей причиной смерти среди внутрибольничных инфекций. Таким образом, данное воплощение восполняет значительную потребность в эффективном профилактическом подходе к лечению внутрибольничных инфекций.
В одном связанном с этим воплощении настоящее изобретение предусматривает способы профилактического лечения иммунокомпрометированных пациентов, например ВИЧ-положительных пациентов, которые уже приобрели пневмонию или подвергаются риску развития пневмонии либо вследствие оппортунистической инфекции, либо из-за реактивации подавленной или латентной инфекции. В 1992 г. только в США зарегистрировано около 20000 случаев заражения Pneumocystis carinii у больных СПИД. Кроме того, 60-70% больных СПИД заражаются Р. carinii в тот или иной момент во время болезни. Таким образом, настоящее изобретение в данном воплощении предусматривает эффективные способы профилактики для этой подвергающейся риску популяции.
В другом связанном с этим воплощении способы настоящего изобретения применяются для лечения других популяций пациентов либо иммунокомпрометированных, либо/и подвергающихся риску приобретения инфекционных заболеваний, в том числе, к примеру, больных кистозным фиброзом, хронической обструкцией дыхательных путей и других иммунокомпроментированных пациентов и/или помещенных в больницу.
В следующем аспекте изобретения описанные нами соединения применяются в способах лечения, облегчения или существенного предупреждения аллергических заболеваний и состояний, таких как синусит, хронический риносинусит, астма, диффузный нейродерматит и псориаз. Такой подход основывается, по меньшей мере, частично на способности этих соединений активировать выработку у клеток-мишеней тех цитокинов, которые могут конкурировать со стереотипическими цитокиновыми ответами аллергического типа, характеризующимися продукцией IL-4 или гиперчувствительностью к действию IL-4. Применение некоторых из соединений, описанных в данном изобретении, приводит к экспрессии IFN-γ и IL-12 антиген-презентирующими и процессирующими клетками, а также и другими клетками, что ведет к понижающей регуляции (downregulation) цитокинов, связанных с аллергическими реакциями, таких как IL-4, 5, 6, 10 и 13.
В следующем аспекте изобретения соединения применяются в способах лечения аутоиммунных заболеваний и состояний. Соединения для этого воплощения, как правило, выбирают из числа тех, что способны быть антагонизистами, ингибировать или иным образом отрицательно модулировать один или несколько рецепторов типа Toll, в частности Тlr2 и/или Тlr4, таким образом, что аутоиммунная реакция, связанная с данным состоянием, уменьшается или предотвращается. В качестве иллюстрации способы данного воплощения могут применяться при лечении таких заболеваний, как хроническое воспаление кишечника, ревматоидный артрит, хронический артрит, множественный склероз и псориаз.
Не желая ограничивать себя какой-либо одной теорией, можно полагать, что эффективность вышеописанных профилактических и терапевтических применений основывается, по меньшей мере, частично на участии этих соединений в модуляции активности рецепторов типа Toll. В частности, предполагается, что Тlr2, Тlr4 и прочие рецепторы типа Toll подвергаются специфической активации, конкурентному ингибированию или иному воздействию нетоксических производных и миметиков LPS, раскрытых в настоящем изобретении. Соответственно, способы изобретения обеспечивают мощный и избирательный подход к модулированию механизмов врожденного иммунитета у животных, не вызывая токсических эффектов, зачастую связанных с природными бактериальными компонентами, которые обычно стимулируют эти механизмы.
Иллюстративные соединения циклических АГФ
К иллюстративным соединениям, используемым в вышеизложенных профилактических и терапевтических применениях, относятся соединения формулы I:
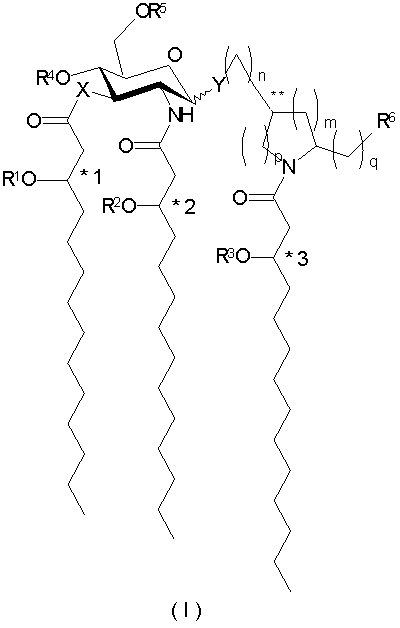
и их фармацевтически приемлемые соли, где Х означает -О- или -NH-, Y означает -О- или -S-; a R1, R2 и R3 независимо друг от друга представляют собой (С2-С20)ацильные группы, включая насыщенные, ненасыщенные и разветвленные ацильные группы; R4 - это -Н или -РО3R7R8, где R7 и R8 независимо друг от друга представлены Н или (C1-С4)алифатической группой; R5 - это -Н, -СН3 или -РО3R9R10, где R9 и R10 независимо друг от друга выбраны из -Н и (С1-С4)алифатических групп; R6 независимо выбран из числа Н, ОН, (С1-С4)оксиалифатических групп, -PO3R11R12, -ОРО3R11R12, -SO3R11, -OSO3R11, -NR11R12, -SR11, -CN, -NO2, -CHO, -CO2R11 и -CONR11R12, где R11 и R12 независимо друг от друга выбраны из Н или (С1-С4)алифатических групп; при условии, что одна из групп R4 и R5 содержит фосфор и что, когда R4 представлен -РО3R7R8, то R5 не является -РО3R9R10, при этом *1-3 и ** обозначают хиральные центры; причем индексы n, m, p и q независимо друг от друга означают целые числа от 0 до 6 при условии, что сумма p и m составляет от 0 до 6.
Хотя гексопиранозид в формуле I представлен в глюкоконфигурации, другие гликозиды также входят в объем изобретения. Например, гликопиранозиды, в том числе и другие гексопиранозиды (алло-, альтро-, манно-, гуло-, идо-, галакто-, тало-), входят в объем изобретения.
В вышеприведенной общей формуле 3'-стереогенные центры, по которым происходит присоединение нормальных жирнокислотных остатков и которые обозначаются как *1, *2 и *3, находятся в R- или S-конфигурации, предпочтительно в R-конфигурации. Абсолютная стереохимия атомов углерода в циклическом агликоне, к которому присоединяются R6 и глюкозамин, прямо или опосредованно (обозначается как **), может быть представлена R- или S-конфигурацией. В вышеприведенной общей формуле Y может находиться в экваториальном или аксиальном положении, предпочтительно экваториальном. Все стереоизомеры, энантиомеры, диастереомеры и их смеси рассматриваются как входящие в объем настоящего изобретения.
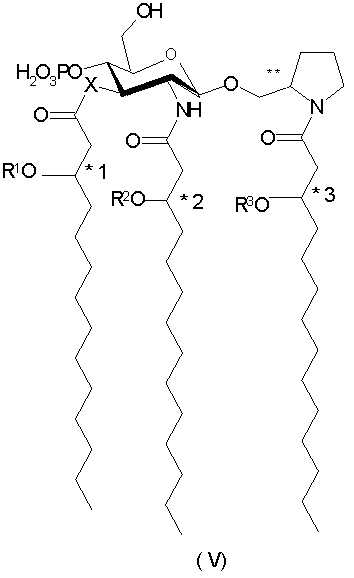
В иллюстративных воплощениях настоящего изобретения Х и Y означают -О-, R4 является фосфоно, R5 и R6 означают Н, а индексы n, m, p и q - целые числа от 0 до 3, более предпочтительно от 0 до 2. В являющемся примером предпочтительном воплощении n=1, m=2, а p и q равны 0. В этом воплощении соединения данного изобретения представлены 2-пирролидинилметил-β-D-глюкозаминид-4-фосфатами общей формулы V:
В другом иллюстративном воплощении настоящего изобретения R1, R2 и R3 в формуле III представляют собой тетрадеканоильные остатки, а 3'-стереогенные центры (*1-3), по которым происходит их присоединение, находятся в R-конфигурации, Y находится в экваториальном положении, а абсолютная стереохимия стереогенного центра пирролидина (**) представлена S-конфигурацией.
К другим показательным воплощениям относятся N-[(R)-3-тетрадеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-тетрадеканоилокситетрадеканоиламино]-3-O-[(R)-3-тетрадеканоилокситетрадеканоил]-β-D-глюкопиранозид и его фармацевтически приемлемые соли (формула II),
[N-(R)-3-додеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-O-[(R)-3-додеканоилокситетрадеканоил]-β-D-глюкопиранозид и его фармацевтически приемлемые соли (формула III) и
[N-(R)-3-деканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-деканоилокситетрадеканоиламино]-3-O-[(R)-3-деканоилокситетрадеканоил]-β-D-глюкопиранозид и его фармацевтически приемлемые соли (формула IV).
Соединения настоящего изобретения могут быть получены методами, изложенными в Johnson et al., Bioorg. Med. Chem. Lett. 9: 2273, 1999, и РСТ/WO 98/50399 и приведенных там ссылках. В общем, методы синтеза, описанные в вышеприведенных источниках, применимы в широком смысле для получения соединений с различными ацильными группами и заместителями. Например, некоторые соединения, применимые в настоящем изобретении, описаны в U.S. Provisional Application № 60/223056 и International Application PCT/USO 1/24284. В общем, для получения этих соединений в широком смысле применимы методы синтеза, описанные в вышеприведенных источниках и в настоящем изобретении, а также другие методы синтеза, уже известные в этой области. Например, при получении соединений с различными ацильными группами и замещениями специалисты в этой области должны понимать, что описанные в приведенных ссылках сходящиеся методы могут быть модифицированы с использованием других ацилирующих реагентов или могут исходить из коммерчески доступных материалов с присоединенными соответствующими ацильными группами.
Термин "ацил" относится к тем группам, которые происходят из органической кислоты при удалении ее гидроксильной части. Соответственно, ацил может означать, к примеру, ацетил, пропионил, бутирил, деканоил и пивалоил.
"(С2-С20)ацил" означает ацильную группу, содержащую от 2 до 20 атомов углерода. Аналогичным образом (С6-С14)-, (С6-С12)-, (C9-C12)- и (С6-С8)ацил означает группы, содержащие от 6 до 14, от 6 до 12, от 9 до 12 и от 6 до 8 атомов углерода соответственно. Термин "ацил" также охватывает ацильные группы, содержащие типичные заместители, такие как гидрокси-, кето- и др.
Термин "алифатический" сам по себе или в составе другого заместителя означает, если не указано иначе, неразветвленную или разветвленную углеводородную цепь либо циклическую углеводородную группу, включая группы, содержащие как циклические элементы, так и цепи, которые могут быть полностью насыщенными либо моно- или полиненасыщенными, содержащими указанное число атомов углерода (например, С1-С4 означает от 1 до 4 атомов углерода). Примеры насыщенных углеводородных радикалов включают такие группы, как метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, циклопропил, циклопропилметил, метилен, этилен и н-бутилен. Ненасыщенная алкильная группа - это группа, содержащая одну или несколько двойных связей и/или тройных связей. Примеры ненасыщенных алкильных групп включают винил, 2-пропенил, кротил, 1-пропинил и 2-бутадиенил.
Термин "оксиалифатический" относится к тем группам, которые содержат алифатическую группу, соединенную с остальной молекулой через атом кислорода.
Каждый из вышеперечисленных терминов (например, "алифатический", "ацил") может включать и замещенные, и незамещенные формы указанных групп. Предпочтительные заместители для каждого типа групп представлены ниже.
Заместителями в алифатических группах может служить целый ряд групп, выбранных из числа -OR', =O, -S, -NR',=N-OR', -NR'R", -SR', -галоген, -SiR'R''R''', -OC(O)R', -C(O)R', -CO2R', -CONR'R'', -OC(O)NR'R'', -NR''C(O)R', -NR'-C(O)NR''R''', -NR''C(O)2R', -NH-C(NH2)=NH, -NR'C(NH2)=NH, -NH-C(NH2)=NR', -S(O)R', -S(O)2R', -S(O)2NR'R'', -CN и NO2, число которых варьирует от 0 до (2m'+1), где m' = это суммарное число атомов углерода в таком радикале. R', R'' и R''' независимо друг от друга означают водород и незамещенные (С1-С4)алифатические группы. Когда R' и R'' присоединены к одному и тому же атому азота, они могут соединяться с атомом азота с образованием 5-, 6- или 7-членного кольца. Например, -NR'R'' может включать 1-пирролидинил и 4 морфолинил. Из вышеизложенного обсуждения заместителей должно быть понятно, что термин "алифатический" может включать и такие группы, как галоалкил (к примеру, -CF3 и -СН2CF3) и им подобные.
Термины "гало" или "галоген" сами по себе или в составе другого заместителя означают, если не указано иначе, атом фтора, хлора, брома или йода. В соединениях с несколькими галогенными заместителями галогены могут быть одинаковыми или разными.
Термин "фармацевтически приемлемые соли" охватывает такие соли активных соединений, которые получают с помощью относительно нетоксичных кислот или оснований, в зависимости от конкретных заместителей, находящихся в соединениях, описанных в настоящем изобретении. В том случае, когда соединения настоящего изобретения содержат сравнительно кислые функциональные группы, их соли с основаниями могут быть получены путем добавления требуемого основания, как в соответствующем инертном растворителе, так и без него. Примеры фармацевтически приемлемых солей, образованных с основаниями, включают соли натрия, калия, кальция, аммония, органических аминов, магния и им подобные. В том случае, когда соединения настоящего изобретения содержат сравнительно основные функциональные группы, их соли с кислотами могут быть получены путем добавления требуемой кислоты, как в соответствующем инертном растворителе, так и без него. Примеры фармацевтически приемлемых солей, образованных с кислотами, включают соли неорганических кислот, таких как соляная, бромистоводородная, азотная, угольная, однозамещенная угольная, фосфорная, однозамещенная фосфорная, двузамещенная фосфорная, серная, однозамещенная серная, иодистоводородная, фосфористая и им подобные кислоты, а также соли относительно нетоксичных органических кислот, таких как уксусная, пропионовая, изомасляная, щавелевая, малеиновая, малоновая, бензойная, янтарная, субериновая, фумаровая, миндальная, фталевая, бензолсульфоновая, пара-толилсульфоновая, лимонная, винная, метансульфоновая и им подобные. Также охватываются соли аминокислот, такие как аргинат и им подобные, и соли таких органических кислот, как глюкуроновая или галактуроновая кислота и им подобные (см., к примеру, Berge S.M. et al., "Pharmaceutical Salts", Journal of Pharmaceutical Science, 1977, 66, 1-19). Определенные соединения настоящего изобретения содержат как основные, так и кислотные функциональные группы, что позволяет их превратить в соли, образованные либо основанием, либо кислотой.
Нейтральные формы соединений могут быть регенерированы путем обработки соли основанием или кислотой и выделения исходного соединения стандартным методом. Исходная форма соединения отличается от разнообразных солевых форм по некоторым физическим свойствам, таким как растворимость в полярных растворителях, но в остальном эти соли эквивалентны исходной форме соединения в целях настоящего изобретения.
Наряду с солевыми формами настоящее изобретение предусматривает соединения, находящиеся в виде пролекарств (предшественников). Пролекарства описанных здесь соединений - это такие соединения, которые легко подвергаются химическим изменениям в физиологических условиях с образованием соединений настоящего изобретения. Кроме того, пролекарства могут быть превращены в соединения настоящего изобретения с помощью химических или биохимических методов в условиях ex vivo. Например, пролекарства могут медленно превращаться в соединения настоящего изобретения, если их поместить в трансдермальный пластырь-резервуар вместе с соответствующим ферментом или химическим реагентом.
Некоторые соединения настоящего изобретения могут существовать в несольватированных формах наряду с сольватированными формами, включая гидраты. В общем, сольватированные формы эквивалентны несольватированным формам и предусматривается, что они охватываются рамками настоящего изобретения. Некоторые соединения настоящего изобретения могут существовать во множественных кристаллических или аморфных формах. В общем, все физические формы эквивалентны для применения в соответствии с настоящим изобретением и предусматривается, что они охватываются рамками настоящего изобретения.
Некоторые соединения настоящего изобретения имеют асимметричные атомы углерода (оптические центры) или двойные связи. Предусматривается, что рацематы, диастереоизомеры, геометрические изомеры и индивидуальные изомеры охватываются рамками настоящего изобретения.
Соединения настоящего изобретения также могут содержать атомные изотопы в неестественных пропорциях по одному или нескольким атомам, входящим в состав таких соединений. Например, соединения могут быть помечены радиоактивными изотопами, к примеру такими как тритий (3H), иод-125 (125I) или углерод-14 (14С). Предусматривается, что все изотопные варианты соединений настоящего изобретения, радиоактивные и нерадиоактивные, охватываются рамками настоящего изобретения.
Предпочтительные фармацевтические композиции и их введение
В следующем воплощении настоящее изобретение касается фармацевтических композиций, содержащих одно или несколько соединений, которые составляют и вводят в отсутствие экзогенного антигена, то есть они применяются в монотерапевтических применениях, в сочетании с фармацевтически приемлемыми носителями и наполнителями. Такие фармацевтические композиции применимы для введения в клетки, ткани, животное или растение, как сами по себе, так и в сочетании с одним или несколькими средствами терапии. Во многих таких воплощениях фармацевтические композиции по изобретению содержат одно или несколько соединений, описанных в данном изобретении.
Фраза "фармацевтически приемлемые" относится к молекулам и композициям, не вызывающим аллергических или иных нежелательных реакций при введении человеку. В применении к настоящему изобретению к "носителям" или "наполнителям" относятся всевозможные растворители, дисперсионные среды, носители, покрытия, разбавители, антибактериальные и противогрибковые вещества, изотонические и замедляющие всасывание вещества, буферы, растворы-носители, суспензии, коллоиды и пр. Применение таких сред и веществ для фармацевтически активных веществ хорошо известно в данной области. За исключением тех случаев, когда традиционные среды или вещества несовместимы с активным ингредиентом, их применение в терапевтических композициях предусмотрено.
Примеры носителей для приготовления фармацевтических композиций включают, к примеру, эмульсии масло-в-воде или вода-в-масле, водные композиции, включающие или не включающие органические растворители, пригодные для внутривенного применения, липосомы или содержащие детергент везикулы, микросферы, микрошарики и микросомы, порошки, таблетки, капсулы, свечи, водные суспензии, аэрозоли и другие носители, известные в этой области.
В некоторых воплощениях фармацевтические композиции включают один или несколько буферов (например, нейтральный солевой буфер или фосфатно-солевой буфер), углеводы (например, глюкозу, маннозу, сахарозу или декстраны), маннит, белки, полипептиды или такие аминокислоты, как глицин, антиоксиданты, бактериостатики, хелаторы типа ЭДТА, глютатион, адъюванты (например, гидроокись алюминия), вещества, делающие композицию изотоничной, гипотоничной или слабо гипертоничной относительно крови реципиента, суспендирующие вещества, загустители и/или консерванты.
Для некоторых применений предпочтительны водные композиции, особенно такие, которые включают эффективное количество одного и более поверхностно-активных веществ (детергентов). Например, композиция может иметь вид мицеллярной дисперсии, включающей по меньшей мере один подходящий детергент, к примеру фосфолипидный детергент. Примеры фосфолипидов включают: диацилфосфатидилглицерины, такие как димиристоилфосфатидилглицерин (DMPG), дипальмитоилфосфатидилглицерин (DPPG) и дистеароилфосфатидилглицерин (DSPG); диацилфосфатидилхолины, такие как димиристоилфосфатидилхолин (DMPC), дипальмитоилфосфатидилхолин (DPPC) и дистеароилфосфатидилхолин (DSPC); диацилфосфатидные кислоты, такие как димиристоилфосфатидная кислота (DMPA), дипальмитоилфосфатидная кислота (DPPA) и дистеароилфосфатидная кислота (DSPA); и диацилфосфатидилэтаноламины, такие как димиристоилфосфатидилэтаноламин (DMPE), дипальмитоилфосфатидилэтаноламин (DPPE) и дистеароилфосфатидилэтаноламин (DSPE). Как правило, молярное отношение детергента к моно-/дисахаридам в водном составе составляет от 10:1 до 1:10, более предпочтительно от 5:1 до 1:5, однако в водном составе может применяться любое эффективное количество детергента, наилучшим образом соответствующее поставленным целям.
В применении к настоящему изобретению "эффективное количество" - это такое количество, которое вызывает ответ выше уровня носителя или отрицательного контроля. Как обсуждалось выше, точная дозировка соединений данного изобретения при введении пациентам зависит от способа применения, фармацевтической композиции и пациента.
Соединения и фармацевтические композиции изобретения могут быть составлены практически для любого способа применения, например, инъекции, ингаляции через рот или нос, ректального, вагинального или внутритрахеального введения, приема внутрь, трансдермального или трансмукозального применения и т.д. Таким образом, эффекты, достигаемые при помощи способов и композиций изобретения, к примеру, могут быть системными, местными, тканеспецифичными и т.д., в зависимости от конкретных требований данного применения изобретения.
Иллюстративные композиции могут составляться и вводиться парэнтерально, то есть внутрибрюшинно, подкожно, внутримышечно или внутривенно. Один из показательных примеров носителя для внутривенного применения включает смесь из 10% этанола, 40% пропиленгликоля (Фармакопея США, USP) или полиэтиленгликоля 600, остальное - вода для инъекций USP (ВДИ). К другим предпочтительным носителям относятся 10% этанол USP и ВДИ USP 0,01-0,1% триэтаноламин в ВДИ USP, 0,01-0,2% дипальмитоилдифосфатидилхолин в ВДИ USP и 1-10% сквален или эмульсия из растительного масла в воде для парентерального введения. Фармацевтически приемлемые растворители для парентерального введения обычно выбирают таким образом, чтобы они представляли собой растворы или дисперсии, которые можно фильтровать через фильтр на 0,22 мкм без потери активного ингредиента.
Показательные примеры носителей для подкожного или внутримышечного применения включают фосфатно-солевой буфер (PBS), 5% глюкозу в ВДИ и 0,01-0,1% триэтаноламин в 5% глюкозе или 0,9% NaCl в ВДИ USP, либо разбавленную 1:2 или 1:4 смесь из 10% этанола USP, 40% пропиленгликоля, остальное - приемлемый изотонический раствор типа 5% глюкозы или 0,9% NaCl, или же 0,01-0,2% дипальмитоилдифосфатидилхолин в ВДИ USP и 1-10% сквален либо эмульсия из растительного масла в воде для парентерального введения.
Примеры носителей для применения через слизистые поверхности зависят от конкретного способа, к примеру, перорально, под язык, в нос и т.п. При пероральном введении показательные примеры включают маннит, крахмал, лактозу, стеарат магния, сахарид натрия, целлюлозу, карбонат магния фармацевтической чистоты и др., при этом предпочтителен маннит. При интраназальном введении показательные примеры включают полиэтиленгликоль, фосфолипиды, гликоли и гликолипиды, сахарозу и/или метилцеллюлозу, порошковые суспензии с наполнителем типа лактозы или без нее, консерванты типа бензалконийхлорида, ЭДТА. В особенно предпочтительном воплощении в качестве изотонического водного носителя используется фосфолипид 1,2-дипальмитоил-sn-глицеро-3-фосфохолин (DPPC) в концентрации 0,01-0,2% для интраназального введения соединений данного изобретения в концентрации от 0,1 до 3,0 мг/мл.
При введении путем ингаляции к предпочтительным носителям относятся полиэтиленгликоль или гликоли, DPPC, метилцеллюлоза, порошковые диспергирующие вещества и консерванты, при этом предпочтительны полиэтиленгликоль и DPPC. Во многих случаях предпочтительно, чтобы соединения находились в аэрозольной форме при введении путем ингаляции. Например, можно использовать одноразовое устройство доставки, распылитель, активируемый дыханием порошковый ингалятор, аэрозольный дозатор-ингалятор (MDI) или любое другое из многочисленных распылительных устройств доставки, доступных в этой области. Кроме того, можно использовать фумигационную палатку или прямое введение через интубационную трубку. Внутритрахеальное или носоглоточное введение эффективно при некоторых показаниях.
Специалисты в данной области должны понимать, что предыдущее описание носит скорее иллюстративный, чем всеобъемлющий характер. В самом деле, в данной области известны многие другие методы составления композиций и фармацевтически приемлемые наполнители и растворы носителей, а также разработаны соответствующие схемы дозировки и лечения для применения конкретных композиций данного изобретения в различных режимах лечения.
Соединения можно оценивать в различных форматах анализа, включая описанные в данном изобретении, чтобы идентифицировать и выбрать соединения, обладающие характеристиками, наилучшим образом соответствующими конкретному применению изобретения. Например, можно использовать животные модели для идентификации и оценки профиля выделения цитокинов в системное кровообращение после введения соединения циклического АГФ. Кроме того, существуют различные модели in vitro и in vivo для оценки изменений одного и более аспектов иммунного ответа к различным антигенным компонентам для того, чтобы идентифицировать соединения, наиболее подходящие для выработки конкретного представляющего интерес иммунного ответа. Например, можно обработать соединением клетки-мишени, такие как макрофаги, дендритные клетки или клетки Лангерганса in vitro и измерить продукцию цитокинов. Кроме того, можно использовать матрицы экспрессии генов для идентификации конкретных путей, активируемых или ингибируемых определенным циклическим АГФ.
Следует иметь в виду, что, при желании, описанные в данном изобретении соединения можно вводить в сочетании с другими лекарственными средствами, такими как антимикробные, антивирусные и противогрибковые соединения или препараты, различные препараты на основе ДНК, препараты на основе РНК, препараты на основе полипептидов, и/или с другими иммуноэффекторами. Так, можно включать практически любые другие компоненты при условии, что дополнительные компоненты не вызывают значительных неблагоприятных эффектов при контакте с клетками-мишенями или тканями хозяина. Таким образом, композиции можно вводить вместе с различными другими веществами, необходимыми или желательными для конкретных воплощений изобретения.
В качестве иллюстрации, фармацевтические композиции изобретения могут включать или применяться вместе с ДНК, кодирующей один или несколько терапевтических белков, антисмысловой РНК, рибозимами и т.п. ДНК может находиться в любой из целого ряда систем доставки, известных в данной области, включая системы экспрессии нуклеиновых кислот, бактериальные и вирусные системы экспрессии. В этой области известны многие методы доставки генов, как те, что описаны в Rolland, Crit. Rev. Therap. Drug Carrier Systems 15: 143-198, 1998, и приведенных там ссылках. Подходящие системы экспрессии нуклеиновых кислот содержат необходимые последовательности ДНК для экспрессии у пациента (такие как промотор и сигнал терминации). В предпочтительном воплощении ДНК вводится с помощью вирусной системы экспрессии (например, вируса осповакцины или другого поксвируса, ретровируса или аденовируса), что обычно включает использование непатогенного (дефектного), способного к репликации вируса. Подходящие системы раскрыты, к примеру, в Fisher-Hoch et al., Proc. Natl. Acad. Sci. USA 86: 317-321, 1989; Flexner et al., Ann. N.Y. Acad. Sci. 569: 86-103, 1989; Flexner et al. Vaccine 8: 17-21, 1990; U.S. Patent № 4603112, 4769330 и 5017487; WO 89/01973; U.S. Patent № 4777127; GB 2200651; EP 0345242; WO 91/02805; Berkner, Biotechniques 6: 616-627, 1988; Rosenfeld et al., Science 252: 431-434, 1991; Kolls et al., Proc. Natl. Acad. Sci. USA 91: 215-219, 1994; Kass-Eisler et al., Proc. Natl. Acad. Sci. USA 90: 11498-11502. 1993; Guzman et al., Circulation 88,2838-2848,1993; и Guzman et al., Cir. Res. 73: 1202-1207, 1993. Методы включения ДНК в такие системы экспрессии хорошо известны в данной области.
ДНК может быть и "голой", как описано, к примеру, в Ulmer et al., Science 259: 1745-1749, 1993, и в обзоре Cohen, Science 259: 1691-1692, 1993. Поглощение голой ДНК можно повысить путем нанесения ДНК на биодеградируемые шарики, которые эффективно транспортируются в клетки. Следует иметь в виду, что фармацевтическая композиция по изобретению может включать и полинуклеотид, и белковый компонент.
В композиции данного изобретения можно включать любые из целого ряда дополнительных иммуностимуляторов. Например, такие цитокины, как GM-CSF, интерфероны или интерлейкины для дополнительной модуляции определенного иммунного ответа. Так, в некоторых воплощениях в композиции могут быть включены дополнительные компоненты для усиления индукции высоких уровней цитокинов типа Th-1 (например, IFN-γ, TNF-α, IL-2 и IL-12). В качестве альтернативы или в дополнение к этому могут быть желательны высокие уровни цитокинов типа Th-2 (например, IL-4, IL-5, IL-6 и IL-10) для определенных терапевтических применений. Уровни этих цитокинов можно легко определить стандартными методами. См. обзор по семейству цитокинов: Mossmann and Cofftnan, Ann. Rev. Immunol. 7: 145-173,1989.
Примеры композиций для применения для индукции цитокинов типа Th-1 включают, к примеру, комбинации из CpG-содержащих олигонуклеотидов (в которых динуклеотид CpG не метилирован), как описано, к примеру, в WO 96/02555, WO 99/33488 и U.S. Patent № 6008200 и 5856462. Иммуностимуляторные последовательности ДНК также описаны, к примеру, Sato et al., Science 273: 352, 1996. Другие подходящие иммуностимуляторы включают сапонины, такие как QS21 (Aquila Biopharmaceuticals Inc., Framingham, MA), GPI-100 (Marciani et al., Vaccine 18: 3141, 2000; U.S. Patent No.6,080,725) и родственные производные других сапонинов и их миметики.
Другие примеры иммуностимуляторов, которые можно применять в сочетании с настоящим изобретением, включают Montanide ISA 720 (Seppic, Франция), SAF (Chiron, Калифорния, США), ISCOMS (CSL), MF-59 (Chiron), адъюванты серии SBAS (SBAS-2 или SBAS-4 фирмы Smith-Kline Beecham, Rixensart, Бельгия) и иммуностимулятор Enhanzyn™ (Corixa, Hamilton, MT). Иммуностимуляторы на основе эфиров полиоксиэтилена описаны BW 099/52549A1.
Далее изобретение раскрывается на следующих неограничивающих примерах.
ПРИМЕРЫ
Пример 1. Получение триэтиламмониевой соли N-[(R)-3-тетрадеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-тетрадеканоилокситетрадеканоиламино]-3-O-[(R)-3-тетрадеканоилокситетрадеканоил]-β-D-глюкопиранозида (триэтиламмониевой соли соединения формулы II)
(1a) В раствор 2-дезокси-4-O-дифенилфосфоно-3-O-[(R)-3-тетрадеканоилокси-тетрадеканоил]-6-O-(2,2,2-трихлор-1,1-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтокси-карбониламино)-β-D-глюкопиранозилбромида (1,05 г, 0,81 ммоль) в безводном 1,2-дихлорэтане (10 мл) добавляли молекулярное сито на 4 Å (0,5 г), безводный CaSO4 (2,2 г, 16 ммоль) и N-[(R)-3-тетрадеканоилокситетрадеканоил]-(S)-2-пирролидинметанол (0,40 г, 0,75 ммоль). Эту смесь перемешивали 1 час при комнатной температуре, обрабатывали Hg(CN)2 (1,02 г, 4,05 ммоль) и кипятили с обратным холодильником 16 часов в темноте. Реакционную смесь охлаждали, разбавляли СН2Cl2 и фильтровали. Фильтрат промывали 1 N водным раствором KI, сушили с помощью Na2SO4 и концентрировали. После флэш-хроматографии на силикагеле (градиентная элюция, 15-20% EtOAc/гексан) получили 0,605 г (43%) N-[(R)-3-тетрадеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-дифенилфосфоно-3-O-[(R)-3-тетрадеканоилокситетрадеканоил]-6-O-(2,2,2-трихлор-1,1-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозида в виде аморфного твердого вещества.
(1b) Раствор полученного по предыдущему п.1а соединения (0,50 г, 0,29 ммоль) в АсОН (10 мл) при 60°С обрабатывали цинковой пылью (0,98 г, 15 ммоль) тремя равными порциями на протяжении 1 часа. Реакционную смесь охлаждали, обрабатывали ультразвуком, фильтровали через слой целита и концентрировали. Полученный осадок подвергали распределению между СН2Cl2 и насыщенным водным раствором NaHCO3 и разделяли слои. Органический слой высушивали с помощью Na2SO4 и концентрировали. Раствор полученного неочищенного аминоспирта и (R)-3-тетрадеканоилокситетра-декановой кислоты (0,155 г, 0,34 ммоль) в СН2Cl2 перемешивали вместе с порошком молекулярного сита на 4 Å (0,25 г) в течение 0,5 ч, а затем обрабатывали 2-этокси-1-этоксикарбонил-1,2-дигидрохинолином (0,11 г, 0,44 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 8 ч, фильтровали через целит и концентрировали. После флэш-хроматографии на силикагеле с использованием 50% EtOAc/гексана получили 0,355 г (68%) N-[(R)-3-тетрадеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-дифенилфосфоно-2-[(R)-3-тетрадеканоилокситетра-деканоиламино]-3-O-[(R)-3-тетрадеканоилокситетрадеканоил]-β-D-глюкопиранозида в виде бесцветного сиропа.
(1с) Раствор полученного по предыдущему п.1b соединения (0,300 г, 0,166 ммоль) в смеси из АсОН (1 мл) и тетрагидрофурана (9 мл) подвергали гидрогенизации в присутствии PtO2 (0,15 г) при комнатной температуре и давлении 70 фунтов/кв.дюйм в течение 18 ч. Реакционную смесь разбавляли смесью 2:1 CHCl3-МеОН (50 мл) и кратко обрабатывали ультразвуком. Собирали катализатор и промывали смесью 2:1 CHCl3-МеОН, и объединенные фильтрат и промывные воды концентрировали. После флэш-хроматографии на силикагеле с использованием CHCl3-МеОН-H2О-Et3N (90:10:0,5:0,5) получили частично очищенный продукт, который растворяли в ледяной смеси 2:1 CHCl3-МеОН (30 мл) и промывали ледяным раствором 0,1 N HCl (12 мл). Органическую фазу фильтровали и лиофилизировали из 2% водного раствора Et3N (5 мл, свободный от пирогенов), получая 0,228 г (79%) триэтиламмониевой соли N-[(R)-3-тетрадеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-тетрадеканоилокситетрадеканоиламино]-3-О-[(R)-3-тетрадеканоилокситетрадеканоил]-β-D-глюкопиранозида в виде бесцветного порошка. Т.пл. 67-70°С, ИК-спектр (пленка): 3306, 2955, 2923, 2853, 1736, 1732, 1644, 1548, 1466, 1378, 1245, 1177, 1110,1053, 844 см-1; 1Н-ЯМР (CDCl3-CD3OD): δ 0,88 (m, 18H), 1,0-1,205 (m, H), 2,20-2,70 (m, 12Н), 3,06 (q, 6Н, J=7,2 Гц), 3,3-3,25 (m, H), 4,52 (d, 1H, J=8 Гц), 5,05-5,28 (m, 4Н), 7,44 (d, 1H, J=9 Гц); 13С-ЯМР (CDCl3): δ 173,3, 173,0, 170,3, 169,6, 168,6, 101,8, 100,4, 75,8, 72,5, 72,4, 70,9, 70,8, 70,3, 70,2, 69,9, 69,3, 67,9, 66,6, 56,5, 56,3, 54,5, 47,4, 45,8, 44,6, 41,4, 41,0, 39,7, 39,2, 39,0, 34,5, 34,3, 34,1, 32,0, 29,7, 29,4, 28,1, 27,3, 25,7, 25,3, 25,2, 25,1, 24,0, 22,7, 21,6, 14,1, 8,6.
Анализ - теоретически для С101Н194N3O17P·Н2О: С=68,47, Н=11,15, N=2,37, Р=1,75; фактически: С=68,79, Н=11,00, N=2,24, Р=1,97.
Пример 2. Получение триэтиламмониевой соли N-[(R)-3-додеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-O-[(R)-3-додеканоилокситетрадекано-ил]-β-D-глюкопиранозида (триэтиламмониевой соли соединения III)
(2а) В раствор 2-дезокси-4-O-дифенилфосфоно-3-O-[(R)-3-додеканоилокси-тетрадеканоил]-6-O-(2,2,2-трихлор-1,1-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтокси-карбониламино)-α-D-глюкопиранозилбромида (1,60 г, 1,27 ммоль) в безводном 1,2-дихлорэтане (3,2 мл) добавляли молекулярное сито на 4 Å (0,6 г), безводный CaSO4 (1,0 г, 7,3 ммоль) и N-[(R)-3-додеканоилокситетрадеканоил]-(S)-2-пирролидинметанол (0,58 г, 1,14 ммоль). Эту смесь перемешивали 1 час при комнатной температуре, обрабатывали Hg(CN)2 (0,58 г, 2,3 ммоль) и кипятили с обратным холодильником 6 часов в темноте. Реакционную смесь охлаждали, разбавляли СН2Cl2 и фильтровали через слой целита. Фильтрат промывали 1 N водным раствором KI, сушили с помощью Na2SO4 и концентрировали. После флэш-хроматографии на силикагеле (градиентная элюция, 25→35% EtOAc/ гексан) получили 1,72 г (82%) N-[(R)-3-додеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-дифенилфосфоно-3-O-[(R)-3-додеканоилокситетраде-каноил]-6-O-(2,2,2-трихлор-1,1-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозида в виде бесцветного масла.
(2b) Раствор полученного по предыдущему п.2а соединения (1,58 г, 0,806 ммоль) в АсОН (40 мл) при 60°С обрабатывали цинковой пылью (2,6 г, 40 ммоль) тремя равными порциями на протяжении 1 часа. Реакционную смесь охлаждали, обрабатывали ультразвуком, фильтровали через слой целита и концентрировали. Полученный осадок подвергали распределению между EtOAc и насыщенным водным раствором NaHCO3 и разделяли слои. Органический слой промывали насыщенным раствором соли, высушивали с помощью Na2SO4 и концентрировали, получая 1,3 г белого твердого вещества. Раствор полученного неочищенного аминоспирта и (R)-3-додеканоилокситетрадекановой кислоты (0,45 г, 1,05 ммоль) в СН2Cl2 (20 мл) обрабатывали 2-этокси-1-этоксикарбонил-1,2-дигидрохинолином (0,30 г, 1,21 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 18 ч и концентрировали. После флэш-хроматографии на силикагеле с использованием 40→50% EtOAc/гексана получили 0,89 г (56%) N-[(R)-3-додеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-дифенилфосфоно-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-O-[(R)-3-додеканоилокситетрадеканоил]-β-D-глюкопиранозида в виде белой пены.
(2с) Раствор полученного по предыдущему п.2b соединения (0,75 г, 0,44 ммоль) в смеси из АсОН (4,5 мл) и тетрагидрофурана (45 мл) подвергали гидрогенизации в присутствии PtO2 (0,45 г) при комнатной температуре и давлении 70 фунтов/кв.дюйм в течение 18 ч. Реакционную смесь разбавляли смесью 2:1 CHCl2-МеОН (35 мл) и кратко обрабатывали ультразвуком. Собирали катализатор и промывали смесью 2:1 CHCl3-МеОН, и объединенные фильтрат и промывные воды концентрировали. После флэш-хроматографии на силикагеле с использованием CHCl3-MeOH-H2O-Et3N (градиентная элюция: 96:4:0,3:0,3→90:10:0,5:0,5) получили частично очищенный продукт (0,51 г), который растворяли в ледяной смеси 2:1 CHCl3-МеОН (50 мл) и промывали ледяным водным раствором 0,1 N HCl (20 мл). Органическую фазу фильтровали и концентрировали. Полученный белый воск лиофилизировали из 2% водного раствора Et3N (70 мл, свободный от пирогенов), получая 0,54 г (78%) триэтиламмониевой соли N-[(R)-3-додеканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-О-[(R)-3-додеканоилокситетрадеканоил]-β-D-глюкопиранозида в виде белого порошка. Т.пл. 146-151°С, ИК-спектр (пленка): 3292, 3100, 2958, 2922, 2852, 1739, 1731, 1659, 1651, 1644, 1562, 1555, 1468, 1455, 1433, 1377, 1339, 1310, 1253, 1238, 1183, 1160, 1107, 1080, 1047, 960, 856, 722 см-1; 1H-ЯМР (CDCl3-CD3OD): δ 0,88 (m, 18 Н), 1,0-2,10 (mH), 2,20-2,75 (m, 12Н), 3,04 (q, 6H, J=7,2 Гц), 3,3-4,3 (mH), 4,45 (d, 1H, J=8,5 Гц), 5,0-5,28 (m, 4Н); 13С-ЯМР (CDCl3): δ 173,9, 173,4, 173,2, 170,6, 170,1, 169,2, 101,4, 75,5, 74,0, 70,8, 70,7, 70,2, 68,5, 60,5, 56,6, 53,6, 47,4, 45,6, 40,9, 39,6, 38,8, 34,5, 34,3, 34,2, 34,1, 31,9, 29,7, 29,6, 29,5, 29,4, 29,4, 29,3, 29,2, 27,3, 25,2, 25,0, 23,6, 22,7, 21,6, 14,0, 8,3.
Масс-спектроскопия MALDI: теоретически для [М+Na]+ 1590, 1900, фактически 1590, 1866. Анализ - теоретически для С95Н182O17Р·3Н2O: С=66,20, Н=10,99, N=2,44; фактически: С=66,36, Н=10,69, N=2,15.
Пример 3. Получение триэтиламмониевой соли N-[(R)-3-деканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-деканоилокситетрадеканоиламино]-3-O-1(R)-3-деканоилокситетрадеканоил]-β-D-глюкопиранозида (триэтиламмониевой соли соединения IV)
(3а) В раствор 2-дезокси-4-O-дифенилфосфоно-3-O-[(R)-3-деканоилокситетрадеканоил]-6-O-(2,2,2-трихлор-1,1-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозилбромида (1,70 г, 1,38 ммоль) в безводном 1,2-дихлорэтане (3,5 мл) добавляли молекулярное сито на 4 Å (0,6 г), безводный CaSO4 (1,2 г, 8,8 ммоль) и N-[(R)-3-деканоилокситетрадеканоил]-(S)-2-пирролидинметанол (0,60 г, 1,24 ммоль). Эту смесь перемешивали 1 час при комнатной температуре, обрабатывали Hg(CN)2 (0,63 г, 2,5 ммоль) и кипятили с обратным холодильником 6 часов в темноте. Реакционную смесь охлаждали, разбавляли СН2Cl2 и фильтровали через слой целита. Фильтрат промывали 1 N водным раствором KI, сушили с помощью Na2SO4 и концентрировали. После флеш-хроматографии на силикагеле (градиентная элюция, 25→40% EtOAc/гексан) получили 1,82 г (80%) N-[(R)-3-деканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-дифенилфосфоно-3-O-[(R)-3-деканоилокситетраде-каноил]-6-O-(2,2,2-трихлор-1,1-диметилэтоксикарбонил)-2-(2,2,2-трихлорэтоксикарбониламино)-β-D-глюкопиранозида в виде бесцветного масла.
(3b) Раствор полученного по предыдущему п.3а соединения (1,67 г, 1,02 ммоль) в АсОН (50 мл) при 60°С обрабатывали цинковой пылью (3,33 г, 51 ммоль) тремя равными порциями на протяжении 1 часа. Реакционную смесь охлаждали, обрабатывали ультразвуком, фильтровали через слой целита и концентрировали. Полученный осадок подвергали распределению между EtOAc и насыщенным водным раствором NaHCO3 и разделяли слои. Органический слой промывали насыщенным раствором соли, высушивали с помощью Na2SO4 и концентрировали, получая 1,25 г белого твердого вещества. Раствор полученного неочищенного аминоспирта и (R)-3-деканоилокситетрадекановой кислоты (0,53 г, 1,33 ммоль) в CH2Cl2 (20 мл) обрабатывали 2-этокси-1-этоксикарбонил-1,2-дигидрохинолином (0,38 г, 1,53 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 18 ч и концентрировали. После флэш-хроматографии на силикагеле с использованием 40→50% EtOAc/гексана получили 1,23 г (74%) N-[(R)-3-деканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-дифенилфосфоно-2-[(R)-3-деканоилокситетрадеканоиламино]-3-O-[(R)-3-деканоилокситетрадеканоил]-β-D-глюкопиранозида в виде белой пены.
(3с) Раствор полученного по предыдущему п.3b соединения (1,07 г, 0,654 ммоль) в смеси из АсОН (6,5 мл) и тетрагидрофурана (65 мл) подвергали гидрогенизации в присутствии PtO2 (0,66 г) при комнатной температуре и давлении 70 фунтов/кв.дюйм в течение 18 ч. Реакционную смесь разбавляли смесью 2:1 CHCl3-МеОН (50 мл) и кратко обрабатывали ультразвуком. Собирали катализатор и промывали смесью 2:1 CHCl3-МеОН, и объединенные фильтрат и промывные воды концентрировали. Полученное воскообразное твердое вещество лиофилизировали из 2% водного раствора триэтиламина, получая ˜1 г неочищенной триэтиламмониевой соли в виде белого порошка. После флэш-хроматографии на силикагеле с использованием CHCl3-MeOH-H2O-Et3N (градиентная элюция: 96:4:0,3:0,3→88:12:1:0,6) получили частично очищенный продукт (0,84 г), который растворяли в ледяной смеси 2:1 CHCl3-МеОН (168 мл) и промывали ледяным раствором 0,1 N HCl (67 мл). Органическую фазу фильтровали и концентрировали. Полученный белый воск лиофилизировали из 2% водного раствора Et3N (70 мл, свободный от пирогенов), получая 0,79 г (79%) триэтиламмониевой соли N-[(R)-3-деканоилокситетрадеканоил]-(S)-2-пирролидинилметил-2-дезокси-4-O-фосфоно-2-[(R)-3-деканоилокситетрадеканоиламино]-3-O-[(R)-3-деканоилокситетрадеканоил]-β-D-глюкопиранозида в виде белого порошка. Т.пл. 121-122°С, ИК-спектр (пленка): 3287, 3093, 2961, 2913, 2850, 1745, 1738, 1732, 1716, 1666, 1660, 1651, 1644, 1635, 1565, 1556, 1538, 1470, 1455, 1434, 1416, 1378, 1337, 1311, 1248, 1184, 1104, 1081, 1021, 964, 721 см-1; 1Н-ЯМР (CDCl3-CD3OD): δ 0,88 (m, 18H), 1,0-2,05 (mH), 2,20-2,75 (m, 12Н), 3,04 (q, 6H, J=7,2 Гц), 3,3-4,3 (mH), 4,45 (d, 1H, J=8,5 Гц), 5,0-5,28 (m, 4Н); 13С-ЯМР (CDCl3): δ 173,7, 173,4, 173,2, 170.5, 170,1, 169,1, 101,4, 75,6, 74,0, 70,8, 70,2, 68,7, 60,4, 56,6, 53,8, 47,4, 45,6, 41,0, 39,6, 38,9, 34,5, 34,3, 34,2, 34,1, 31,9, 29,7, 29,6, 29,5, 29,4, 29,4, 29,3, 29,2, 27,3, 25,3, 25,0, 23,7, 22,7, 21,6, 14,1, 8,4.
Масс-спектроскопия MALDI: теоретически для [M+Na]+ 1506,0961, фактически 1506,1008. Анализ - теоретически для C89H170N3O17P: С=67,43, Н=10,81, N=2,65; фактически: С=67,26, Н=10,85, N=2,47.
Пример 4. Проба на введение Listeria monocytogenes мышам
В данном примере представлены опыты по оценке индукции неспецифической устойчивости методом пробы на введение Listeria monocytogenes мышам, которую проводили с использованием соединений, полученных в примерах 1, 2 и 3. Мышам (по 5 в группе) вводили внутривенно 1 мкг циклического АГФ или MPL, растворенного в 0,2% триэтаноламине (ТЭА). Через 2 дня мышам вводили внутривенно ˜105 клеток Listeria monocytogenes серотипа 10403 (исходная культура получена от Jory Baldridge, Washington State University, Pullman, WA). Через 2 дня после введения мышей забивали и определяли количество колониеобразующих единиц (к.о.е.) в селезенках мышей, высевая 10-кратные серийные разведения гомогенатов селезенки на чашки с агаризованным триптическим гидролизатом сои. Рассчитывали степень защиты, вызываемой данным АГФ или MPL, вычитая среднее число бактерий на 1 селезенку (значение log10) в группе мышей, получавших данное соединение, из среднего числа бактерий на 1 селезенку (значение log10) в контрольной группе, которой в качестве симуляции вводили растворитель (0,2% ТЭА) перед проведением пробы на введение L. monocytogenes.
Из всех исследованных соединений наиболее активным было соединение из примера 3, которое индуцировало защиту в степени, сравнимой с MPL (˜0,9 единиц log10). Соединение из примера 2 индуцировало слегка меньшую степень защиты, а соединение из примера 1 было наименее активным (0,7 и 0,2 единиц log10 соответственно).
Пример 5. Защита от летального инфицирования вирусом гриппа путем профилактического введения циклических АГФ
В данном примере представлены опыты по оценке защиты от летальной инфекции вирусом гриппа у мышей, получавших циклические АГФ. Мышам BALB/c (по 10 в группе) вводили в нос 20 мкг соединений из примеров 1, 2 и 3, либо MPL за 48 часов до летального интраназального инфицирования вирусом гриппа А/НК/68 (5 доз LD50). Защитный эффект оценивали по выживаемости, появлению клинических симптомов (взъерошенная шерсть, сгорбленная поза и затрудненное дыхание) и предупреждению потери веса в течение 21 дня после инфекции.
Как и в случае модели с введением Listeria, соединения из примеров 2 и 3 обеспечивали высокую степень защиты по сравнению с контролем-растворителем. Выживаемость составила 60% у мышей, получавших соединение из примера 3, 40% у мышей, получавших соединение из примера 2, и 30% у мышей, получавших MPL. Не выжила ни одна из мышей, получавших соединение 1. Эти данные свидетельствуют, что соединение из примера 3 обеспечивает наибольшую защиту, а за ним следуют соединения из примеров 2 и 1.
Все процитированные публикации и патентные заявки включены в настоящее изобретение путем отсылки во всей полноте. Хотя данное изобретение описано достаточно подробно с привлечением иллюстраций и примеров для облегчения понимания, однако в свете положений данного изобретения специалисты в этой области должны понимать, что в нем могут производиться определенные изменения и модификации, не отклоняющиеся от духа и рамок прилагаемой формулы изобретения.
Изобретение относится к способам облегчения или существенного предупреждения инфекционного заболевания, аутоиммунного заболевания или аллергического состояния у пациента, включающим контактирование пациента с эффективным количеством одного или нескольких соединений формулы (V), и к фармацевтической композиции для лечения или облегчения заболеваний, таких как инфекционные заболевания, аутоиммунные заболевания и аллергии, которая составлена и вводится в отсутствие экзогенного антигена, включающей одно или несколько соединений формулы (V). 3 н. и 49 з.п. ф-лы.
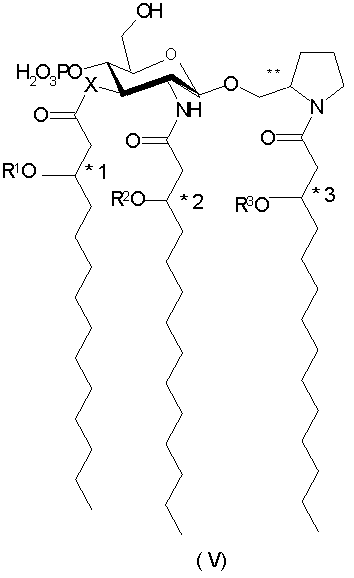
или их фармацевтически приемлемых солей, где Х выбран из группы, состоящей из -O-и -NH-;
R1, R2 и R3 независимо друг от друга выбраны из группы, состоящей из (С2-С20)ацильных групп,
при этом *1, *2, *3 и ** обозначают хиральные центры.
или их фармацевтически приемлемых солей, где Х выбран из группы, состоящей из -O- и -NH-;
R1, R2 и R3 независимо друг от друга выбраны из группы, состоящей из (С2-С20)ацильных групп,
при этом *1, *2, *3 и ** обозначают хиральные центры.
или их фармацевтически приемлемых солей, где Х выбран из группы, состоящей из -O- и -NH-;
R1, R2 и R3 независимо друг от друга выбраны из группы, состоящей из (С2-С20)ацильных групп,
при этом *1, *2, *3 и ** обозначают хиральные центры.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| ЦЕНТРОБЕЖНЫЙ РЕГУЛЯТОР ДАВЛЕНИЯ | 0 |
|
SU194563A1 |
| ГЛЮКОЗАМИНОВЫЕ ДИСАХАРИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2154068C2 |

