Настоящее изобретение относится к пролекарству, представляющему циклический ундекапептид и его применению в качестве лечебного продукта, в частности, предназначенного для лечения патологических состояний глаза.
Циклоспорины представляют собой в структурном отношении особую группу циклических пептидов, общим для которых является тот факт, что они состоят из цепочки из одиннадцати аминокислот, некоторые из которых являются атипичными либо за счет их D-конфигурации, либо за счет сложной химической структуры их боковой цепи, либо за счет того, что их аминогруппа алкилирована.
К настоящему времени из грибов выделено примерно тридцать циклоспоринов, и полусинтетическим путем или полным синтезом получено много циклических ундекапептидов, подобных этим натуральным продуктам. В число этих аналогов циклических ундекапептидов входят пептолиды или депсипептиды, т.е. циклические полипептиды, содержащие в их цепи также сложноэфирные связи.
В дальнейшем в настоящем описании, если не указано иначе, термин "циклоспорин" предназначен для обозначения как циклических ундекапептидов, полученных из естественных источников, так и их аналогов, полученных полусинтезом или полным синтезом, включая пептолиды, полученные из естественных источников или их аналоги, полученные полусинтезом или полным синтезом.
Первый член данного семейства циклоспоринов был выделен и идентифицирован как циклоспорин А. Пептидная цепь, составляющая его ундекапептидное кольцо, является следующей:
-MeBmt-Abu-Sar-MeLeu-Val-MeLeu-Ala-(D)Ala-MeLeu-MeLeu-MeVal-
Его более полная химическая структура является следующей:
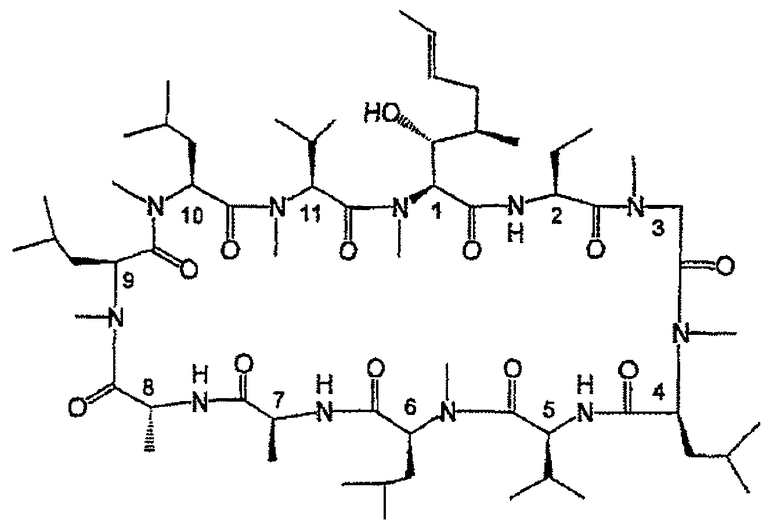
(циклоспорин А)
Среди атипичных аминокислот, которые включает данный циклический ундекапептид в 1-м положении, особо следует упомянуть N-метил-(4R)-4-((Е)-2-бутенил)-4-метил-L-треонин, обозначенный MeBmt.
Данная аминокислота является специфической для циклоспоринов, и допускается, чтобы этиленовая группа была необязательно восстановленнной. Он имеет аминогруппу, которая метилирована. Кроме того, гидроксильная группа, которую она включает, очень примечательна в том смысле, что она является единственной группой во всем данном циклическом ундекапептиде, которая способна химически модифицироваться. Также можно отметить, что она находится в значительной степени затрудненном пространственном окружении, что делает любой подход реагента очень затруднительным.
Данные циклические ундекапептиды, происходят ли они из естественных источников или получены синтетически, проявляют широкий спектр видов биологической активности, среди которых наиболее хорошо известно иммуносупрессорное, противовоспалительное или антипаразитарное действие или активность, которая делает возможным бороться со злокачественными опухолями или уменьшать их резистентность к другим видам лечения. Было установлено, что некоторые из этих циклических ундекапептидов обладают обещающим противовирусным действием, в частности, при лечении СПИДа путем подавления репликации вируса иммунодефицита человека типа 1 (ВИЧ-1).
В этом отношении в заявке на патент WO 00/01715, поданной заявителями настоящей заявки, описан определенный ряд полусинтетических циклических ундекапептидов, обладающих структурой, близкой к таковой для циклоспорина А, но в которых изменена природа аминокислот в 4-м положении, или в 3-м и 4-м положениях относительно аминокислоты MeBmt.
Недавние разработки в области фармакологии сделали возможным надеяться, что иммуносупрессорный эффект циклоспоринов, в частности циклоспорина А, чье действие является обратимым и немиелотоксичным и для которого отмечены незначительные побочные эффекты, можно с успехом использовать, в частности, в области офтальмологии для местного лечения, в частности, поверхностных патологических состояний глаза и окружающих его придатков.
Данные патологические состояния включают сухой кератоконъюнктивит, также называемый сухим кератитом глаз, синдром Шегрена, формы аллергического кератоконъюнктивита, в частности, устойчивые к действию кортикостероидов, конъюнктивит со слизью и синехией, герпетический стромальный кератит, связанный с иммунной системой лимбический кератит и кератит Тигесона, и профилактику отторжения трансплантата роговицы, и в качестве вспомогательного лечения при хирургических операциях с целью удаления.
Циклические ундекапептиды семейства циклоспоринов по своей природе являются очень гидрофобными соединениями, что тем более снижает их растворимость в воде. Данное свойство связано с природой боковой цепи большинства аминокислот, входящих в их состав, и также с тем фактором, что аминогруппа некоторых аминокислот метилирована, что ограничивает возможность образования межмолекулярных водородных связей между циклическим ундекапептидом и, например, водной солюбилизирующей средой.
В результате для внутривенного (в/в) введения данных циклоспоринов требуется разработка очень сложных фармацевтических композиций в основном в виде эмульсий, которые в некоторых случаях обладают низкой стабильностью и требуют осторожного обращения с ними и которые являются причиной проявления нежелательных побочных эффектов.
В качестве примера, один из препаратов для в/в инфузии циклоспорина А, промышленно выпускаемый под торговым названием сандиммун, представляет микроэмульсию, которую получают при использовании в качестве наполнителя полиоксиэтилинированного касторового масла, известного под торговым названием кремофор. Данный препарат хранится в виде концентрата и должен быть разбавлен непосредственно перед введением.
Из-за применения данного касторового масла, которое, как известно, солюбилизирует некоторые компоненты синтетических веществ, изготовитель рекомендует использовать при работе с данным препаратом только материалы из стекла или синтетических материалов, отвечающих "стандартам Европейской фармакопеи для резервуаров, предназначенных для крови", все эти материалы не должны содержать силиконовое масло и жиры.
Кроме того, врачей предупреждают о том, что данное касторовое масло способно вызывать анафилактоидные реакции, и в результате им дают рекомендации о том, что прибегать к внутривенному введению следует только в тех случаях, когда невозможно пероральное применение.
Разработка обещающих применений циклоспоринов местным введением в области офтальмологии развивается медленно также из-за трудностей в создании подходящих фармацевтических композиций, в частности, которые проявляют хорошую местную переносимость и не вызывают затуманенного зрения в результате присутствия вязких агентов.
Так, в качестве примера, недавно в журнале J. Fr. Opthalmol., 2001, 24(5), 527, Robert et al. привели в своем обзоре все технические трудности, которые необходимо преодолеть, в результате липофильной природы фармацевтических композиций для местного применения циклоспорина А в офтальмологии и все проблемы местной переносимости, которые возникают при использовании таких композиций.
Одним из выводов, к которому можно прийти в результате данного обзора, является то, что к настоящему времени отсутствуют композиции в виде раствора для промывания глаз, который можно применять местно для лечения патологических состояний глаза и окружающих его придатков. Это заключение можно распространить на применение циклоспорина для местного лечения состояний слизистых мембран или состояний кожи.
Следовательно, по-прежнему имеется потребность в том, чтобы сделать циклоспорины доступными для врачей, независимо от того, происходят ли они из естественного источника или являются синтетическими, или производные этих циклоспоринов, которые можно было бы легко вводить пациенту, в частности, местно или внутривенно, избегая при этом применения сложных фармацевтических композиций, которые обладают низкой стабильностью и с которыми трудно обращаться, и которые приводят к проявлению нежелательных побочных эффектов.
Эта потребность является тем более насущной, если данные циклоспорины, естественного или синтетического происхождения, должны применяться местно на глаз или окружающие его придатки.
Одной из возможностей, доступных для специалиста при решении проблемы создания гидрофобной, фармакологически активной молекулы, ассимилируемой в физиологической среде, является ее химическая модификация в целях придания ей гидрофильных свойств.
Для того чтобы избежать изменения фармакологических свойств такой фармакологически активной молекулы, данная химическая модификация может заключаться в получении предшественника, по возможности, неактивного предшественника, данной фармакологически активной молекулы, который при введении под влиянием физиологических условий, имеющихся в организме, будет модифицироваться химически или под действием ферментов с образованием фармакологически активной молекулы, если возможно, либо в месте проявления ее фармакологического действия, либо в крови, которая будет доставлять данную фармакологически активную молекулу, образовавшуюся таким образом, к месту ее действия, соответствуя понятию "пролекарство". В дальнейшем в данном описании предшественник указанной фармакологически активной молекулы называется "пролекарство".
Уже известна практика химической модификации структуры циклоспорина А в целях придания получаемому продукту гидрофильных свойств.
Так, Rothbard et al. описали в заявке на патент WO 01/13957 способ улучшения введения фармакологически активных молекул и облегчения их проникновения через дерму и эпителиальные мембраны, состоящий из обратимого введения в данные молекулы боковой цепи, состоящей из фрагментов цепи полиаргинина. Среди этих фармакологически активных молекул находятся молекулы гидрофобной природы такие, как циклоспорин А.
Однако подобные конъюгаты требуют очень осторожного обращения с ними и их хранения, как указывается в цитируемой заявке, из-за того, что фармакологически активная молекула образуется, если рН среды более 7. Кроме того, при образовании данной фармакологически активной молекулы фрагменты цепи полиаргинина высвобождаются в организме. Поскольку известно, что они обладают токсичностью и раздражающими свойствами, данные полиаргинины будут оказывать раздражающее действие, и таким образом, применение подобных конъюгатов циклоспоринов в области офтальмологии делается невозможным.
Crooks et al. в заявке на патент WO 00/67801 описали получение пролекарств противовоспалительных средств таких, как флурбипрофен, для их местного применения при контакте с глазом, одновременно избегая при этом проявления местного раздражающего действия. Они достигли этого для определенного ряда продуктов введением оксигенированных или полиоксигенированных цепей.
С другой стороны, при попытке подвергнуть циклоспорин А таким же химическим модификациям они преуспели только в получении продуктов, которые описывают как стабильные, другими словами, которые не расщепляются с образованием циклоспорина А при контакте с сывороткой человека или в фосфатном буфере при рН 7,4.
Следовательно, задача настоящего изобретения состоит в том, чтобы сделать доступными для врачей пролекарства циклических ундекапептидов семейства циклоспоринов, которые, во-первых, можно вводить в физиологической среде без необходимости создания сложных фармацевтических композиций и, во-вторых, можно было бы хранить и затем обращаться и вводить, в частности, не беспокоясь о рН окружающей среды.
Также задача настоящего изобретения состоит в том, чтобы сделать доступными для врачей пролекарства циклических ундекапептидов семейства циклоспоринов, которые можно было бы применять местно вообще и на поверхность глаза или на слизистые оболочки, в частности, и которые затем могли бы образовать в течение подходящего времени полужизни фармакологически активный циклический ундекапептид без проявления местного раздражающего действия.
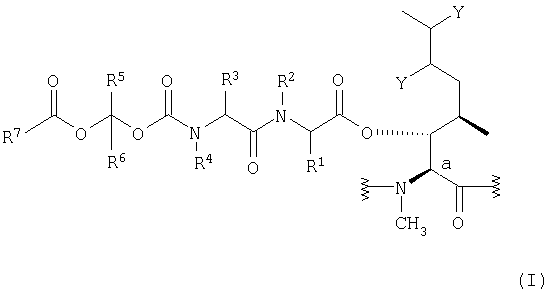
Для осуществления этого настоящее изобретение относится к пролекарству, представляющему циклический ундекапептид, в котором пептидная цепь включает, по меньшей мере, один аминокислотный остаток общей формулы (I) ниже:
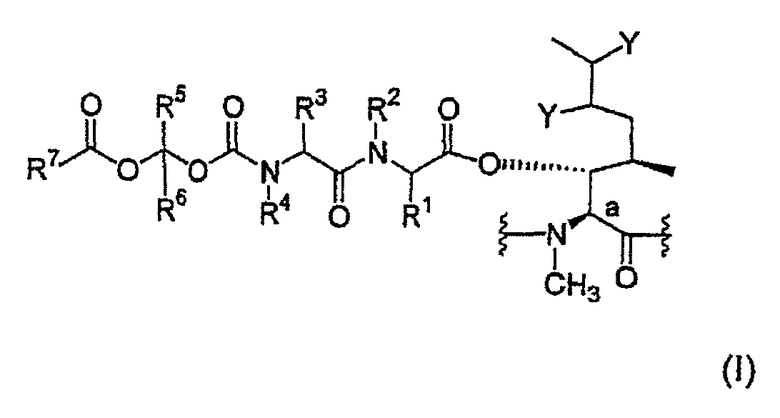
в которой:
- атом углерода Са образует одну из связей в кольце ундекапептида;
- каждый из заместителей Y представляет атом водорода или вместе они образуют связь;
- заместители R1 и R3, независимо друг от друга, представляют атом водорода, аралкил, алкарил, гетероалкил, гетероцикл, алкилгетероцикл, гетероциклоалкил или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода, причем указанные группы необязательно замещены, по меньшей мере, одной группой, выбранной из -COOH, -CONHR8, -NHC=NH(NH2), -NHC=NR8(NH2), -NH2, -NHR8, -NR8 2, -N+R8 3, -OH, -OPO(OR8)2, -OPO(OH)(OR8), -OPO(OH)2, -OSO(OR8)2, -OSO(OH)(OR8), -OSO(OH)2 и различных солевых форм этих групп, где каждый из заместителей R8, независимо друг от друга, представляет нормальный или разветвленный алкил, содержащий 1-6 атомов углерода;
- заместители R2 и R4, независимо друг от друга, представляют атом водорода, алкарил или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода;
- заместители R5 и R6, независимо друг от друга, представляют атом водорода, аралкил, или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода; и
- заместитель R7 представляет аралкил, алкарил, гетероалкил, гетероцикл, алкилгетероцикл, гетероциклоалкил или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода, причем указанные группы необязательно замещены, по меньшей мере, одной группой, выбранной из -COOH, -CONHR8, -NHC=NH(NH2), -NHC=NR8(NH2), -NH2, -NHR8, -NR8 2, -N+R8 3, -OH, -OPO(OR8)2, -OPO(OH)(OR8), -OPO(OH)2, -OSO(OR8)2, -OSO(OH)(OR8), -OSO(OH)2 и различных солевых форм этих групп, где каждый из заместителей R8 имеет значения, определенные выше.
Когда два заместителя Y образуют связь, то указанный аминокислотный остаток общей формулы (I) является производным остатка N-метил-(4R)-4-((Е)-2-бутенил)-4-метил-L-треонина, в котором гидроксильная группа треонина соответствующим образом этерифицирована, и фармакологически активная молекула, которая образуется при расщеплении пролекарства в организме, будет представлять циклический ундекапептид, в котором пептидная цепь содержит, по меньшей мере, один остаток N-метил-(4R)-4-((Е)-2-бутенил)-4-метил-L-треонина (MeBmt).
Аналогично, когда каждый из двух заместителей Y представляет атом водорода, указанный аминокислотный остаток общей формулы (I) является производным остатка N-метил-(4R)-4-бутил-4-метил-L-треонина, в котором гидроксильная группа треонина соответствующим образом этерифицирована, и фармакологически активная молекула, которая образуется при расщеплении пролекарства в организме, будет представлять собой циклический ундекапептид, в котором пептидная цепь содержит, по меньшей мере, один остаток N-метил-(4R)-4-бутил-4-метил-L-треонина (Dh-MeBmt).
Предпочтительно в общей формуле (I), определяющей указанный аминокислотный остаток, по меньшей мере, один из заместителей R1 и R3 представляет аралкил, алкарил, гетероалкил, гетероцикл, алкилгетероцикл, гетероциклоалкил или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода, где указанные группы необязательно замещены, по меньшей мере, одной группой, выбранной из -COOH, -CONHR8, -NHC=NH(NH2), -NHC=NR8(NH2), -NH2, -NHR8, -NR8 2, -N+R8 3, -OH, -OPO(OR8)2, -OPO(OH)(OR8), -OPO(OH)2, -OSO(OR8)2, -OSO(OH)(OR8), -OSO(OH)2 и различных солевых форм данных групп, где каждый из заместителей R8 имеет значения, определенные выше. Данные группы, известны как полярные по своей природе, значительно повышают гидрофильные свойства указанного пролекарства.
Более предпочтительно, когда вышеуказанные аралкил, алкарил, гетероалкил, гетероцикл, алкилгетероцикл, гетероциклоалкил или алкил замещены, по меньшей мере, одной из групп, выбранных из -NR8 2, -N+R8 3, -OPO(OH)2 или различных солевых форм этих групп, где каждый из заместителей R8 имеет значения, определенные выше.
Более предпочтительно, когда один из указанных заместителей R1 и R3 представляет нормальный алкил, содержащий 1-6 атомов углерода, замещенный, по меньшей мере, одной из групп, выбранных из -NR8 2, -N+R8 3, -OPO(OH)2 или различных солевых форм данных групп, где каждый из заместителей R8 имеет значения, определенные выше.
Когда указанные заместители R1 и R3 представляют нормальный алкил, содержащий 1-6 атомов углерода, замещенный, по меньшей мере, одной из групп, выбранных из -NR8 2, -N+R8 3, -OPO(OH)2 или различных солевых форм данных групп, где каждый из заместителей R8 имеет значения, определенные выше, тогда соответствующие аминокислотные остатки предпочтительно являются производными:
- либо остатков серина, гомосерина, треонина, аллотреонина, N-метилсерина, N-метилтреонина или N-метилгомосерина, в любой одной из (D) или (L) конфигураций, предпочтительно (L) конфигурации, и в которых гидроксильная группа соответствующим образом функционализирована так, что боковая цепь данных аминокислотных остатков включает полярные и/или солюбилизирующие группы;
- либо остатков лизина, орнитина, аргинина, N-дельта-метиларгинина, N-альфа-метиларгинина или N-метиллизина в любой одной из (D) или (L) конфигураций, предпочтительно (L) конфигурации, и в которых соответственно амино- или иминогруппа соответствующим образом функционализирована так, что боковая цепь данных аминокислотных остатков включает полярные и/или солюбилизирующие группы.
Когда заместители R1, R2 и/или R3 и R4, образующие пары (R1, R2) и/или (R3, R4), представляют алкил, содержащий 1-6 атомов углерода, то они могут образовать внутри каждой пары алкиленовую цепь, которая образует с атомом углерода и атомом азота, с которыми они связаны, кольцо. Предпочтительно они представляют боковую цепь остатка пролина.
Когда заместители R1 и R3, независимо друг от друга, представляют атом водорода, аралкил, алкарил, гетероалкил, гетероцикл, алкилгетероцикл, гетероциклоалкил или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода, но указанные группы не замещены, по меньшей мере, одной группой, выбранной из -COOH, -CONHR8, -NHC=NH(NH2), -NHC=NR8(NH2), -NH2, -NHR8, -NR8 2, -N+R8 3, -OH, -OPO(OR8)2, -OPO(OH)(OR8), -OPO(OH)2, -OSO(OR8)2, -OSO(OH)(OR8), -OSO(OH)2 и различных солевых форм данных групп, тогда они предпочтительно представляют боковые цепи аминокислотных остатков в (D) или (L) конфигурациях, предпочтительно (L) конфигурации, или остатков указанных аминокислот в защищенных и/или активированных формах, необязательно имеющих алкилированную аминогруппу, которые, как правило, являются промышленно выпускаемыми. Более предпочтительно, когда аминокислотные остатки выбраны из 20 аминокислот, обычно называемых натуральными (природными) аминокислотами.
Также является предпочтительным, когда в общей формуле (I) заместители R5 и R6 не могут одновременно представлять атом водорода. Также предпочтительно, когда, по меньшей мере, один из указанных заместителей R5 и R6 представляет нормальный или разветвленный алкил, содержащий 1-6 атомов углерода, а заместитель R7 представляет аралкил или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода.
Более предпочтительно, когда заместители R5 и R6, независимо друг от друга, представляют атом водорода или метил.
Предпочтительно, указанное пролекарство представляет циклический ундекапептид, в котором пептидная цепь включает один аминокислотный остаток общей формулы (I) и таким образом образует кольцо ундекапептида с линейной последовательностью из 10 аминокислот общей формулы (II) ниже:
-T-U-V-W-MeLeu-Ala-X-MeLeu-Z-MeVal- (II)
в которой:
- Т выбран из аминокислот Ala, Abu, Nval, Val и Thr;
- U выбран из аминокислот Sar, (D)MeSer, (D)MeAla и (D)MeSer(OCOR9), где R9 представляет атом водорода, алкарил или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода;
- V представляет аминокислоту общей формулы (N-R10)aa, где аа выбрана из аминокислот Val, Leu, Ile, Thr, Phe, Tyr и Thr, и R10 представляет нормальный или разветвленный алкил, содержащий 1-6 атомов углерода;
- W выбран из аминокислот Val, Nval и Leu;
- Х выбран из аминокислот (D)Ala, (D)Ser, (D)Hiv, (D)Val и (D)Thr при (D)Hiv, представляющем остаток D-2-гидроксиизовалериановой кислоты; и- Z выбран из аминокислот Leu и MeLeu.
Таким образом, если в указанном аминокислотном остатке общей формулы (I) каждый из двух заместителей Y представляет атом водорода, то фармакологически активная молекула, которая будет образовываться во время расщепления пролекарства в организме, будет представлять циклический ундекапептид семейства циклоспоринов, в котором пептидная цепь содержит остаток N-метил-(4R)-4-бутил-4-метил-L-треонина (Dh-MeBmt).
Аналогичным образом, если в указанном аминокислотном остатке общей формулы (I) два заместителя Y вместе образуют связь, то фармакологически активная молекула, которая будет образовываться во время расщепления пролекарства в организме, будет представлять циклический ундекапептид семейства циклоспоринов, в котором пептидная цепь содержит остаток N-метил-(4R)-4-((Е)-2-бутенил)-4-метил-L-треонина (MeBmt).
Предпочтительно, чтобы данные циклические ундекапептиды соответствовали циклоспоринам, которые уже описаны в литературе как имеющие фармакологические свойства и которые все содержат в их пептидной цепи либо остаток N-метил-(4R)-4-((Е)-2-бутенил)-4-метил-L-треонина (MeBmt), либо остаток N-метил-(4R)-4-бутил-4-метил-L-треонина (Dh-MeBmt).
Более предпочтительно, когда линейная последовательность из десяти остальных аминокислотных остатков, образующая с указанным аминокислотным остатком общей формулы (I) указанный циклический ундекапептид, была выбрана из следующих последовательностей формул (III)-(XIV):
-Abu-Sar-MeLeu-Val-MeLeu-Ala-(D)Ala-MeLeu-MeLeu-MeVal- (III);
-Abu-(D)MeAla-EtVal-Val-MeLeu-Ala-(D)Ala-MeLeu-MeLeu-MeVal- (IV);
-Thr-Sar-MeLeu-Val-MeLeu-Ala-(D)Ala-MeLeu-MeLeu-MeVal- (V);
-Val-Sar-MeLeu-Val-MeLeu-Ala-(D)Ala-MeLeu-MeLeu-MeVal- (VI);
-Nval-Sar-MeLeu-Val-MeLeu-Ala-(D)Ala-MeLeu-MeLeu-MeVal- (VII);
-Val-(D)MeAla-MeLeu-Val-MeLeu-Ala-(D)Ala-MeLeu-MeLeu-MeVal- (VIII);
-Val-Sar-MeLeu-Val-MeLeu-Ala-(D)Val-MeLeu-Leu-MeVal- (IX);
-Val-Sar-MeLeu-Val-MeLeu-Ala-(D)Thr-MeLeu-Leu-MeVal- (X);
-Abu-(D)MeSer(OAc)-MeLeu-Val-MeLeu-Ala-(D)Ala-MeLeu-Leu-MeVal- (XI);
-Abu-Sar-MeLeu-Val-MeLeu-Ala-(D)Ser-MeLeu-MeLeu-MeVal- (XII);
-Thr-Sar-MeLeu-Leu-MeLeu-Ala-(D)-Hiv-MeLeu-Leu-MeVal- (XIII); и -Abu-Sar-MeLeu-Val-MeLeu-Ala-(D)Val-MeLeu-Leu-MeVal- (XIV).
Фармакологически активная молекула, которая будет образовываться во время расщепления пролекарства в организме, будет соответственно представлять один из следующих циклоспоринов с остатком, в зависимости от ситуации, являющийся производным треонина с бутениловой (MeBmt) или бутиловой (Dh-MeBmt) цепью:
циклоспорин А (CsA); (D)MeAla3EtVal4CsA (WO 00/01715); циклоспорин С (CsC); циклоспорин D (CsD); циклоспорин G (CsG); (D)MeAla3CsD; (D)Val8Csl; (D)Thr8Csl; (D)MeSer(Oac)3CsT; (D)Ser8CsA (Progress in Medicinal Chemistry, Vol. 25, ed. Ellis and West, Elsevier Science Publ., Biomedical Division, 1998, pp. 1-33); Thr2Leu5(D)Hiv8Leu10CsC (The Journal of Biological Chemistry, 1991, 266(24), 15570); (D)Val8Leu10CsA; где циклоспорины А, C, D, G, I и Т описаны в Progress in the Chemistry of Organic Natural Products, 1986, 50, 124, и остальные циклоспорины получены аналогично способу, описанному в Helvetica Chimica Acta, 1984, 67, 502.
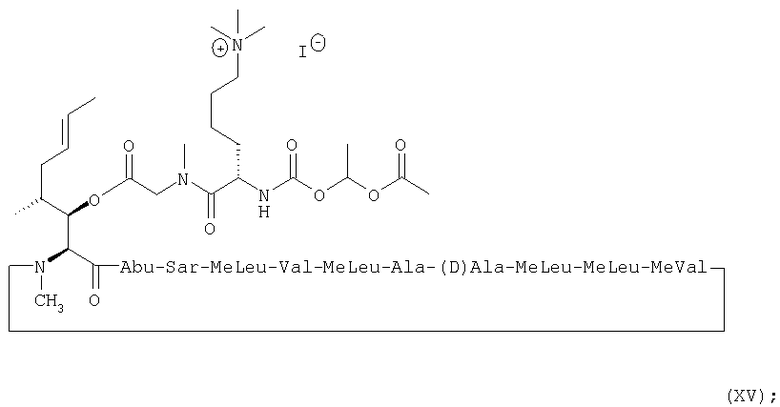
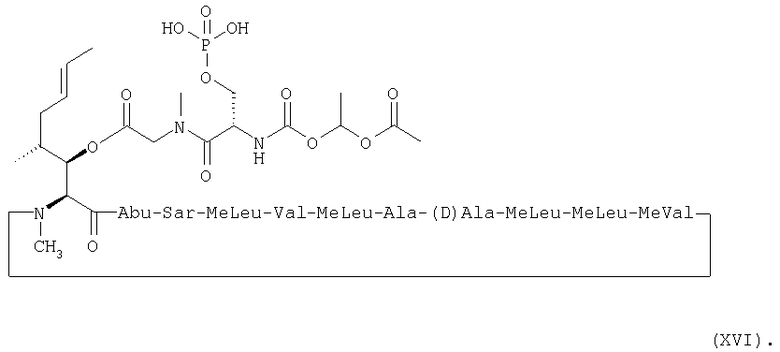
Более предпочтительно, пролекарства по настоящему изобретению имеют, соответственно, формулы (XV) и (XVI) ниже:
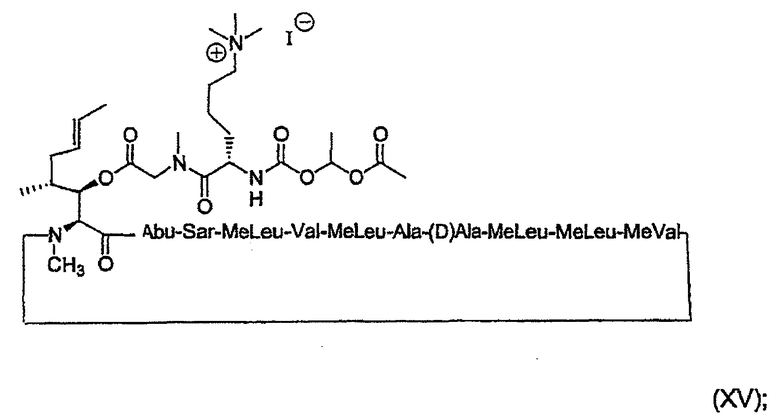
и
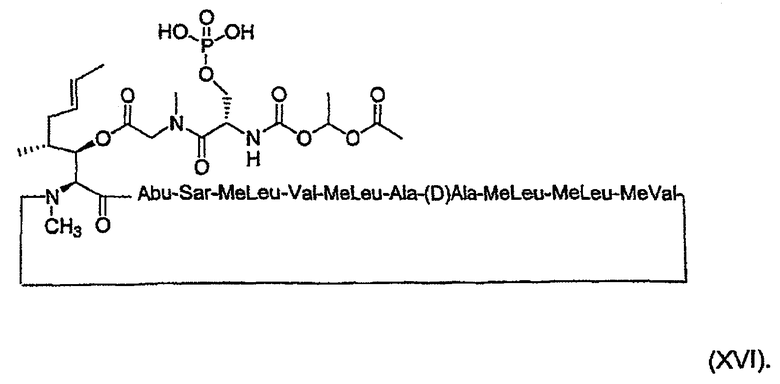
Пролекарства по настоящему изобретению можно получить используемыми способами химического синтеза, хорошо известными специалистам в области химии пептидов, и особенно в области химии циклоспоринов.
В результате соответствующего выбора различных заместителей, определяющих аминокислотный остаток общей формулы (I), было установлено, что пролекарства по настоящему изобретению обладают, в особенности значительно лучшими гидрофильными свойствами по сравнению с фармакологически активной молекулой, образующейся во время расщепления указанного пролекарства. В качестве примера, растворимость некоторых пролекарств по настоящему изобретению, которые образуют циклоспорин А после расщепления, по меньшей мере, в 3000 раз выше, чем у циклоспорина А.
Следовательно, пролекарства по настоящему изобретению легко включать в состав водной фармацевтической композиции.
Также примечательно, что, как было установлено, пролекарства по настоящему изобретению не чувствительны к значениям рН, обычно имеющимся при данном типе применения, когда они находятся в водном растворе.
Кроме того, пролекарства по настоящему изобретению полностью осуществляют свою роль, образуя в течение периода полужизни, подходящего для терапевтического применения, фармакологически активную молекулу при контакте с ферментами, находящимися в биологических жидкостях.
Также настоящее изобретение относится к применению пролекарства, как описано выше, в качестве лечебного средства.
Подобное лечебное средство предпочтительно применяют для лечения патологических состояний или физиологических состояний, для которых заранее требуется применение циклоспорина, в частности всех патологических состояний, для которых необходимо применение циклоспорина А, местно или системно при внутривенном введении.
Такое лечебное средство, в частности, предназначено для обеспечения длительной приживаемости аллотрансплантатов органов таких, как почки, сердце, печень, поджелудочная железа, легкие, тонкий кишечник или костный мозг. Также он может быть предназначен для подавления репликации вируса иммунодефицита человека типа (ВИЧ-1).
При таких применениях дозировка пролекарства по настоящему изобретению при системном введении с помощью внутривенной инъекции является таковой, чтобы концентрация циклоспорина, образующегося во время расщепления, например, циклоспорина А, соответствовала обычно рекомендуемым терапевтическим концентрациям.
Более предпочтительно, если такое лечебное средство применяют в области офтальмологии и предназначено, в частности, для лечения патологических состояний глаза и окружающих его придатков.
Данными патологическими состояниями являются, в числе прочего, сухой кератоконъюнктивит, также называемый сухим кератитом глаз, синдром Шегрена, формы аллергического кератоконъюнктивита, в частности, устойчивые к действию кортикостероидов, конъюнктивит со слизью и синехией, герпетический стромальный кератит, связанный с иммунной системой лимбический кератит и кератит Тигесона, и профилактику отторжения трансплантата роговицы, и в качестве вспомогательного лечения при хирургических операциях с целью удаления. Более предпочтительно лечебный продукт по настоящему изобретению используется для лечения сухого кератоконъюнктивита.
При таких применениях дозировка пролекарства по настоящему изобретению является такой, чтобы концентрация в слезной жидкости циклоспорина, образующегося во время расщепления пролекарства, например, циклоспорина А, была выше 0,5 мкг/л при местном применении.
Лечебное средство по настоящему изобретению можно применять местно, в частности, для местного лечения состояний слизистых мембран или состояний кожи, или парентерально, в частности, внутривенно. Его также можно вводить перорально с целью повышения биодоступности циклоспорина, например циклоспорина А.
Когда лечебное средство по настоящему изобретению вводят парентерально, то подходящие фармацевтические препараты могут представлять собой стерильные, концентрированные водные растворы или порошки для инъекционных препаратов.
Предпочтительно лечебное средство по настоящему изобретению вводят внутривенно. Подходящие фармацевтические препараты для подобного введения представляют собой водные растворы для инъекции или инфузии, которые хорошо известны специалистам в данной области.
Более предпочтительно лечебное средство по настоящему изобретению применяют местно. Подходящими фармацевтическими препаратами для такого введения, в частности, для применения в области офтальмологии, являются растворы для промывания и лечения глаз в виде водных стерильных растворов, офтальмологических мазей, офтальмологических гелей и офтальмологических вставок. Настоящее изобретение, а также его преимущественные свойства, подробно представлены, однако не являются ограниченными, в примерах и с помощью чертежей, где :
- на фиг.1 представлена кривая кинетики превращения в условиях in vitro пролекарства по изобретению в результате гидролиза эстеразами и кривая кинетики появления циклоспорина А;
- на фиг.2а представлена концентрация циклоспорина А в крови после в/в введения циклоспорина А в виде масляного раствора крысам;
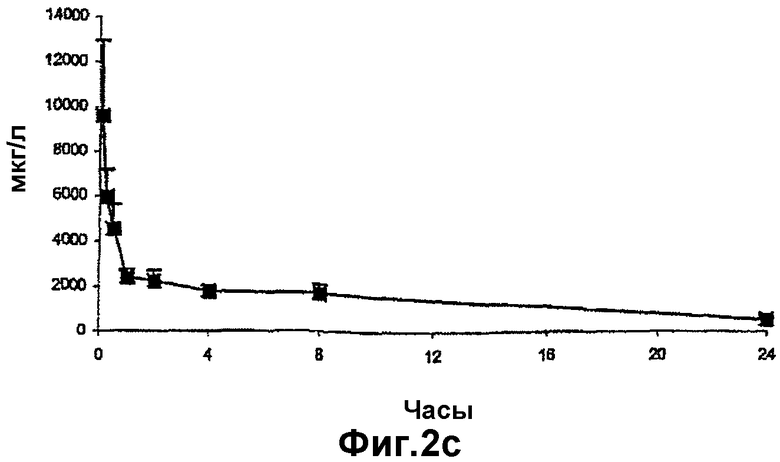
- на фиг.2b и 2с представлена концентрация циклоспорина А в крови после в/в введения водного раствора соответственно двух пролекарств по изобретению крысам; и
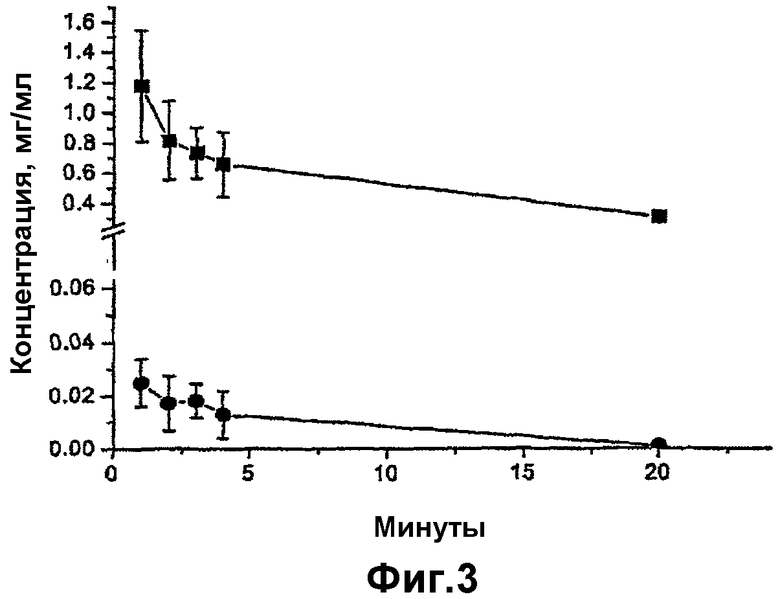
- на фиг.3 соответственно представлена концентрация во времени циклоспорина А и пролекарства по изобретению в слезной жидкости кроликов.
В номенклатуре, использованной в примерах, для описания полученных продуктов остаток циклоспорина А сокращенно обозначен CsA, остаток противоположного фрагмента, связанного с единственной функциональной группой данного циклического ундекапептида, а именно гидроксильной группой аминокислоты, имеющей 1-е положение, обозначается как N-метил-(4R)-4-((Е)-2-бутенил)-4-метил-L-треонин (MeBmt). В структурных химических формулах промежуточных продуктов, полученных из циклоспорина А, будет представлен только аминокислотный остаток в 1-м положении с соответствующей боковой цепью.
Пример 1
Получение циклического ундекапептида формулы (XV)
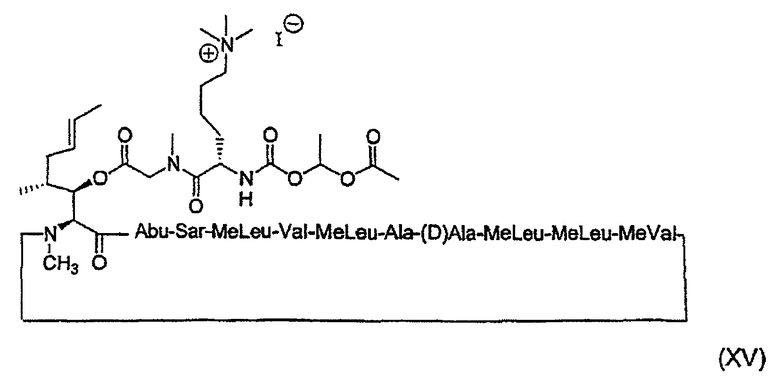
1. Получение MeBmt(O-Sar-Lys((Nε +Me3)-COOCH(CH3)OCOCH3))1-CsA (XV)
1.1. Получение α-ацетоксиэтил-пара-нитрофенилкарбоната (2)
1.1.1. Получение α-хлорэтил-пара-нитрофенилкарбоната (1)
2,6 мл (23,7 ммоль, 1,1 экв.) α-хлорэтилхлорформиата добавляют при 0°С к раствору 3 г (21,6 ммоль, 1 экв.) п-нитрофенола и 1,7 мл (21,7 ммоль, 1 экв.) пиридина в 108 мл хлороформа. Реакционную смесь перемешивают в течение 30 минут при 0°С и затем в течение 16 часов при температуре окружающей среды. Реакционную смесь экстрагируют водой, 0,5%-ным раствором NaOH и затем водой. Органическую фазу высушивают над Na2SO4, фильтруют и упаривают при пониженном давлении с получением желтого масла, которое после кристаллизации из гексана дает чистое белое твердое вещество (5,8 г).
1H-ЯМР (400 МГц, CDCl3); δ: 8,32 (д, 2H), 7,44 (д, 2H), 6,52 (кв, 1H), 1,95 (с, 3H).
1.1.2. Получение α-ацетоксиэтил-пара-нитрофенилкарбоната (2)
7,8 г (24,4 ммоль, 1,5 экв.) ацетата ртути добавляют к раствору 4 г (16,3 ммоль, 1 экв.) соединения (1), растворенного в 100 мл уксусной кислоты. Реакционную смесь перемешивают в течение 1 суток при температуре окружающей среды и затем добавляют еще 1 г (3,13 ммоль, 0,2 экв.) ацетата ртути. После перемешивания еще в течение 1 суток при температуре окружающей среды реакцию заканчивают. Уксусную кислоту выпаривают в глубоком вакууме и остаток переводят в эфир. Органическую фазу экстрагируют раствором соли и затем сушат над Na2SO4, фильтруют и упаривают при пониженном давлении с получением желтого масла. Сырой продукт хроматографируют на силикагеле с получением бесцветного масла (4,4 г).
1H-ЯМР (400 МГц, CDCl3); δ: 8,31 (д, 2H), 7,43 (д, 2H), 6,87 (кв, 1H), 2,16 (с, 3H), 1,64 (с, 3H).
1.2. Получение α-ацетоксиэтоксикарбониллизина (Nε(Fmoc)) (5)
1.2.1. Получение бензилового сложного эфира α-ацетоксиэтоксикарбониллизина (Nε(Z)) (3)
500 мг (1,23 ммоль, 1 экв.) H-Lys(Z)Obn·HCl суспендируют в 2,5 мл диоксана. 231 мкл (1,35 ммоль, 1,1 экв.), N,N-диизопропилэтиламина (DIPEA) и 396 мг (1,47 ммоль, 1,2 экв.) соединения (2) добавляют при температуре окружающей среды. После перемешивания в течение 1 суток при температуре окружающей среды реакцию заканчивают. Диоксан выпаривают при пониженном давлении и остаток переводят в 20 мл этилацетата. Органическую фазу экстрагируют трижды 6% раствором лимонной кислоты (20 мл), насыщенным раствором NaHCO3 (20 мл) и насыщенным раствором NaCl (20 мл), высушивают над безводным Na2SO4, фильтруют и растворитель выпаривают при пониженном давлении. Полученный сырой продукт хроматографируют на силикагеле с получением светлого прозрачного масла (576 мг).
ESI-MC m/z: 501,34 [M+H+], 518,28 (M+H2O+H+].
1.2.2. Получение α-ацетоксиэтоксикарбониллизина (4)
50 мг палладия на активированном древесном угле добавляют к раствору 509 мг (1,02 ммоль) соединения (3) в 10 мл этанола. После перемешивания в течение 3 часов при температуре окружающей среды в атмосфере водорода реакцию заканчивают. Реакционную смесь фильтруют через целит и фильтрат упаривают при пониженном давлении с получением сырого вещества в виде коричневатых кристаллов, которое непосредственно используют на следующей стадии (254 мг).
ES-MC m/z: 276,87 [M+H+].
1.2.3. Получение α-ацетоксиэтоксикарбониллизина (Nε(Fmoc)) (5)
1,11 мл N,N-дизиопропилэтиламина (DIPEA) (6,53 ммоль, 1,7 экв.) и 1,555 г (4,61 ммоль, 1,2 экв.) Fmoc-O-Suc добавляют к раствору 1,062 (3,84 ммоль, 1 экв.) соединения (4), растворенного в 38 мл диоксана. После перемешивания в течение 1 часа при температуре окружающей среды реакцию заканчивают. Диоксан выпаривают при пониженном давлении и остаток переводят в 20 мл EtOAc. Органическую фазу промывают один раз 6% раствором лимонной кислоты (20 мл) и насыщенным раствором NaCl (20 мл), высушивают над безводным Na2SO4, фильтруют и растворитель выпаривают при пониженном давлении. Полученный сырой продукт хроматографируют на силикагеле с получением белой пены (1,026 г).
ES-MC m/z: 499,37 [M+H+], 516,29 [M+H2O+H+].
1.3. Получение MeBmt(O-Sar-Lys((Nε +Me3)-COOCH(CH3)OCOCH3))1-CsA.I- (XV)
1.3.1. Получение MeBmt(O-COCH2Br)1-CsA (6)
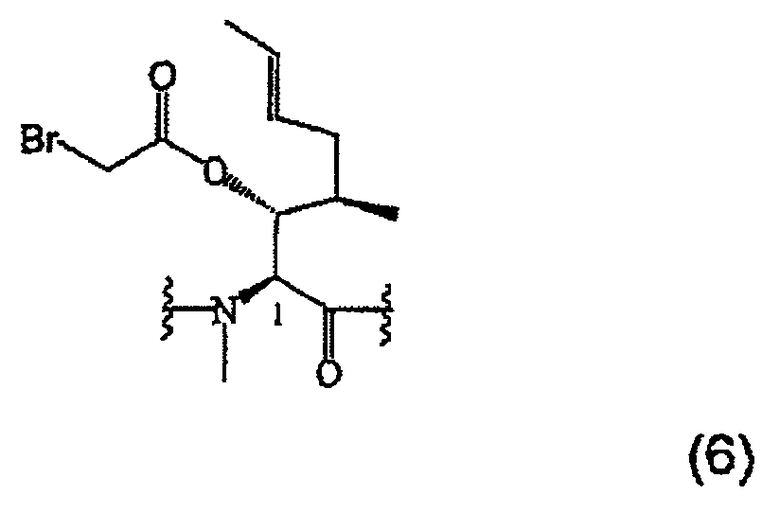
4 г (3,33 ммоль, 1 экв.) сухого CsA растворяют в атмосфере аргона в 66 мл (0,76 моль) бромацетилбромида. 2 г (16,64 ммоль, 5 экв.) диметиламинопиридина добавляют небольшими порциями и реакционную смесь перемешивают при температуре окружающей среды в течение 40 минут. Затем реакцию заканчивают. Реакционную смесь осторожно и при энергичном перемешивании выливают в смесь гидрокарбоната (77 г, 0,91 моль), воды (500 мл) и размолотого льда. Возможное добавление еще нескольких порций NaHCO3 делает возможным довести рН раствора до 7-8. Отделенную водную фазу дважды экстрагируют дихлорметаном, и объединенные органические фазы трижды экстрагируют насыщенным раствором NaHCO3 и насыщенным раствором NaCl, высушивают над безводным Na2SO4, фильтруют и растворитель выпаривают при пониженном давлении. Полученный сырой продукт хроматографируют на силикагеле с получением белой пены (3,2 г).
ESI-MC m/z: 622,6 [M+2H+], 673,9 [M+Na++H+].
1.3.2. Получение MeBmt(O-Sar-Н)1-CsA (7)
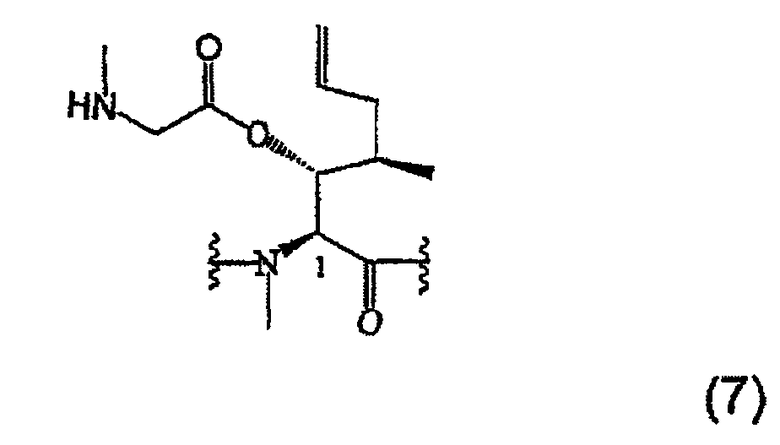
300 мг (0,23 ммоль, 1 экв.) соединения (6) растворяют в 2,3 мл этанола. 95 мкл (0,68 ммоль, 3 экв.) триэтиламина (TEA) и 31 мг (0,45 ммоль, 2 экв.) хлорида метиламмония добавляют при температуре окружающей среды. После перемешивания в течение 3 суток при температуре окружающей среды рН доводят до 12 добавлением ТЕА и добавлением 15 мг (0,23 ммоль, 1 экв.) хлорида метиламмония, реакцию заканчивают через 1 часа. Этанол выпаривают при пониженном давлении и остаток переводят в этилацетат. Органическую фазу экстрагируют водой и насыщенным раствором NaCl, высушивают над безводным Na2SO4, фильтруют и растворитель выпаривают при пониженном давлении. Полученный сырой продукт хроматографируют на силикагеле с получением белой пены (200 мг).
ESI-MC m/z: 1273,7 [M+H+].
1.3.3. Получение MeBmt(O-Sar-Lys(Nε(FMOC-COOCH(CH3)-OCOCH3))1-CsA (8)
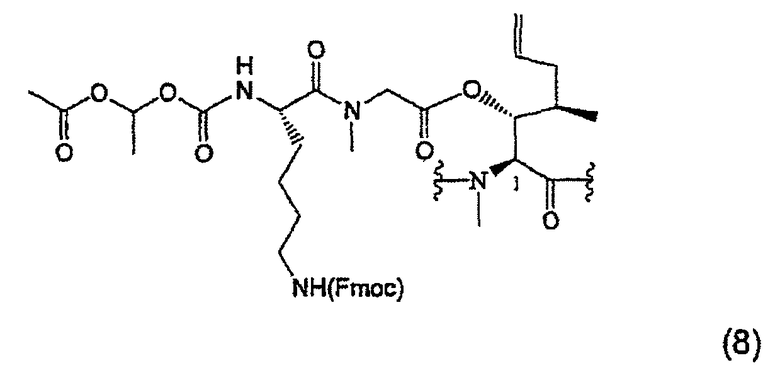
79 мг (0,06 ммоль, 1 экв.) соединения (7) растворяют в 0,5 мл дихлорметана (DCM) в атмосфере аргона. 31,6 мкл (0,18 ммоль, 3 экв.) DIPEA, 35 мг (0,09 ммоль, 1,5 экв.) гексафторфосфата О-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (HATU) и 40 мг (0,08 ммоль, 1,3 экв.) соединения (5), растворенного в 0,8 мл DCM, добавляют последовательно в атмосфере аргона. После перемешивания в течение 3 часов при температуре окружающей среды реакцию заканчивают. DCM выпаривают при пониженном давлении и остаток переводят в 20 мл EtOAc. Органическую фазу промывают один раз 6% раствором лимонной кислоты (20 мл), насыщенным раствором NaHCO3 (20 мл) и насыщенным раствором NaCl (20 мл), высушивают над безводным Na2SO4, фильтруют и растворитель выпаривают при пониженном давлении. Сырой продукт хроматографируют на силикагеле с получением белой пены (59 мг).
ESI-MC m/z: 1754,36 [M+H+], 877,86 [M+2H+].
1.3.4. Получение MeBmt(O-Sar-Lys(Nα-COOCH(CH3)OCOCH3))1-CsA (9)
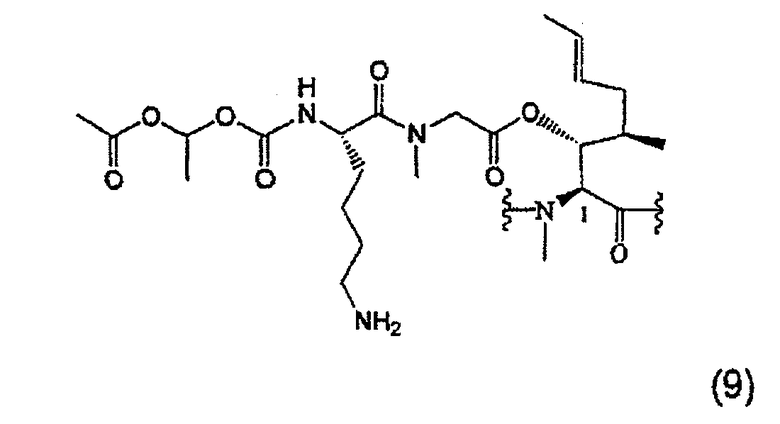
90 мкл (0,86 ммоль, 10 экв.) диэтиламина добавляют к раствору 150 мг (0,09 ммоль, 1 экв.) соединения (8), растворенного в 900 мкл ацетонитрила. После перемешивания в течение 3 часов при температуре окружающей среды реакцию заканчивают. Растворитель выпаривают при пониженном давлении и сырой продукт хроматографируют на силикагеле с получением белой пены (52 мг).
ESI-MC m/z: 1532,79 [M+H+], 766,79 [M+2H+].
1.3.5. Получение MeBmt(O-Sar-Lys(Nε +Me3)-COOCH(CH3)-OCOCH3))1-CsA.I- (XV)
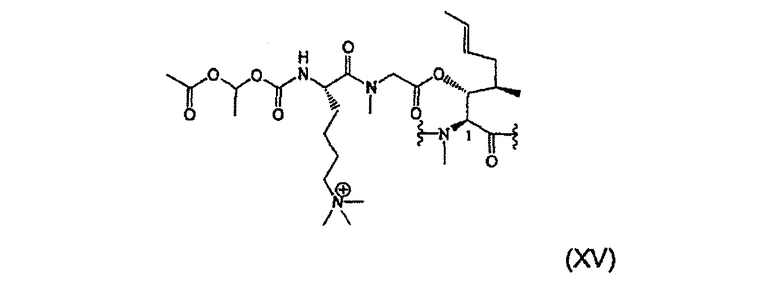
49 мг (0,03 ммоль, 1 экв.) соединения (9) растворяют в 640 мкл безводного DCM и затем добавляют 30 мкл (0,48 ммоль, 15 экв.) MeI с последующим добавлением 14 мкл (0,08 ммоль, 2,5 экв.) DIPEA. После перемешивания в течение 1 часа при температуре окружающей среды реакцию заканчивают. DCM выпаривают при пониженном давлении и сырой продукт очищают полупрепаративной ВЭЖХ для выделения чистого вещества в виде лиофилизата (30 мг).
ESI-MC m/z: 1574,37 [M+H+], 787,83 [M+2H+].
Пример 2
Получение циклического ундекапептида формулы (XVI)
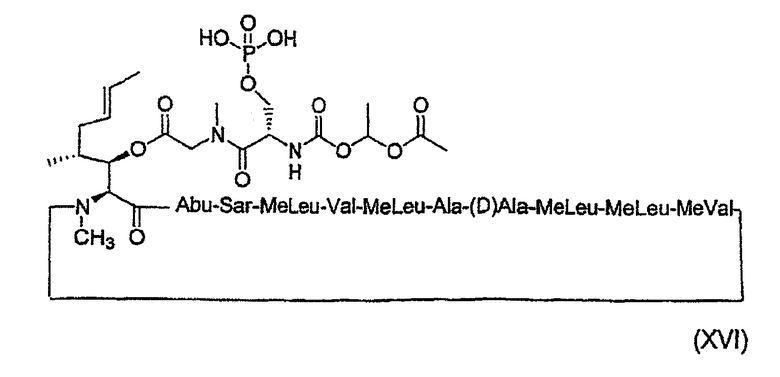
1. Получение MeBmt(O-Sar-Ser(OPO(OH2))-COOCH(CH3)OCOCH3))1-CsA (XVI)
1.1. Получение α-ацетоксиэтоксикарбонилсерина (11)
1.1.1. Получение бензилового сложного эфира α-ацетоксиэтоксикарбонилсерина (10)
1,4 г (6,04 ммоль, 1 экв.) H-Ser-Obn·HCl) суспендируют в 12 мл диоксана. 1,14 мл (6,64 ммоль, 1,1 экв.) DIEA и 2,1 г (7,85 ммоль, 1,3 экв.) соединения (2), полученного в примере 1, добавляют при температуре окружающей среды. После перемешивания в течение ночи при температуре окружающей среды реакцию заканчивают. Диоксан выпаривают при пониженном давлении и остаток переводят в этилацетат. Органическую фазу трижды экстрагируют 6%-ным раствором лимонной кислоты, насыщенным раствором NaHCO3 и насыщенным раствором NaCl, высушивают над безводным Na2SO4, фильтруют и растворитель выпаривают при пониженном давлении. Полученный сырой продукт хроматографируют на силикагеле с получением белой пены (1,7 г).
1Н-ЯМР (400 МГц, CDCl3); δ: 7,34-7,41 (м, 5H), 6,79-6,85 (м, 1H), 5,72-5,82 (м, 1H), 5,24 (с, 2H), 4,47 (м, 1H), 3,94-4,05 (м, 2H), 2,06 и 2,08 (с, 3H), 1,49 и 1,50 (д, 3H).
1.1.2. Получение α-ацетоксиэтоксикарбонилсерина (11)
40 мг палладия на активированном древесном угле добавляют к раствору 400 мг (1,23 ммоль) соединения (10) в 13 мл этанола. После перемешивания в течение 4 часов при температуре окружающей среды в атмосфере водорода реакцию заканчивают. Реакционную смесь фильтруют через целит и фильтрат упаривают при пониженном давлении с получением сырого вещества в виде прозрачного осадка, который непосредственно используют на последующей стадии (328 мг).
1Н-ЯМР (400 МГц, CDCl3); δ: 6,3-6,8 (м, 1H), 5,8-6,3 (м, 1H), 4,3-4,4 (м, 1H), 3,7-4,1 (м, 2H), 2,08 (с), 1,49 и 1,50 (д, 3H).
1.2. Получение MeBmt(O-Sar-Ser(OPO(OH)2)-COOCH(CH3)-OCOCH3)1-CsA (XVI)
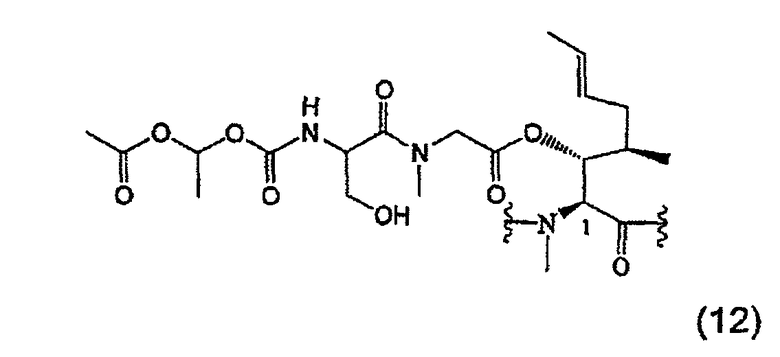
170 мг (0,13 ммоль) соединения (7), полученного в примере 1, растворяют в 3 мл дихлорметана в атмосфере аргона. Последовательно в атмосфере аргона добавляют 92 мкл (0,53 ммоль, 4 экв.) DIEA, 51 мг (0,26 ммоль, 2 экв.) HATU и 60 мг соединения (11) (0,25 ммоль, 2 экв.). После перемешивания в течение 5 часов при температуре окружающей среды реакцию заканчивают. Дихлорметан выпаривают при пониженном давлении и остаток переводят в этилацетат. Органическую фазу трижды экстрагируют 6%-ным раствором лимонной кислоты, насыщенным раствором NaHCO3 и насыщенным раствором NaCl, высушивают над безводным Na2SO4, фильтруют и растворитель выпаривают при пониженном давлении. Полученный сырой продукт хроматографируют на силикагеле с получением белой пены (143 мг).
ESI-MC m/z: 1513,22 [M+Na+], 1508,34 [M+H2O+H+], 1491,36 [M+H+], 746,36 [M+2H+].
1.2.2. Получение MeBmt(O-Sar-Ser(OPO(Oall)2)-COOCH(CH3)-OCOCH3)1-CsA (13)
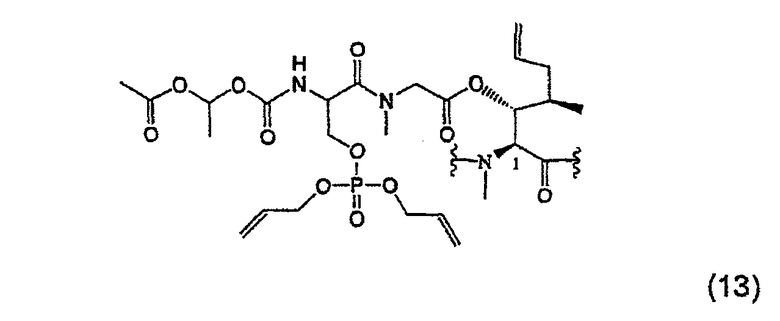
130 мг (0,09 ммоль, 1 экв.) соединения (12) растворяют в 880 мкл безводного СН2Cl2. Затем добавляют 20 мг (0,27 ммоль, 3 экв.) 1Н-тетразола с последующим добавлением 52 мкл (0,17 ммоль, 2 экв.) (AllO)2PN(iPr)2. После перемешивания в течение 4 часов при температуре окружающей среды реакционную смесь охлаждают до -60°С, добавляют 44 мг (0,17 ммоль, 2 экв.) м-хлорпербензойной кислоты и продолжают перемешивание при -60°С в течение 30 минут, при 0°С - в течение 15 минут и при температуре окружающей среды - в течение 45 минут. 0,5 мл 10%-ного раствора Na2S2O5 вносят в реакционную среду при 0°С для разрушения избытка окислителя и затем проводят экстракцию дихлорметаном. Органическую фазу промывают 10%-ным раствором Na2S2O5 и затем дихлорметан выпаривают при пониженном давлении. Остаток переводят в метил-трет-бутиловый эфир и данную органическую фазу экстрагируют 6% раствором лимонной кислоты и насыщенным раствором NaCl, высушивают над безводным Na2SO4, фильтруют и растворитель выпаривают при пониженном давлении. Полученный продукт хроматографируют на силикагеле с получением белой пены (98 мг).
ESI-MC m/z: 1651,38 [M+H+], 826,35 [M+2H+].
1.2.3. Получение MeBmt(O-Sar-Ser(OPO(OH)2)-COOCH(CH3)-OCOCH3)1-CsA (XVI)
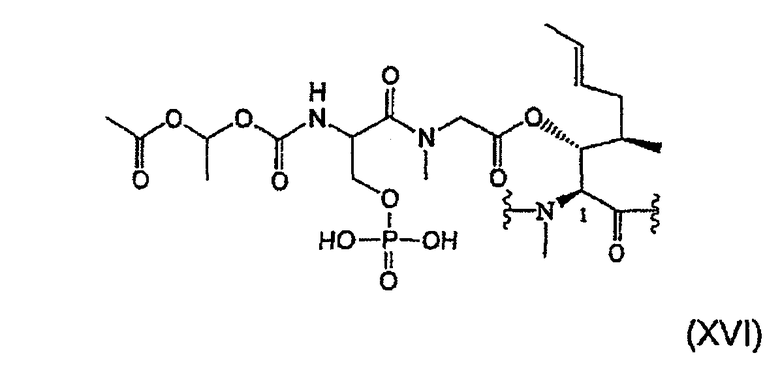
206 мкл (1,55 ммоль, 8 экв.) Me3SiN3 и 1,12 г (0,97 ммоль, 5 экв.) (PPh3)4Pdo в атмосфере аргона при температуре окружающей среды добавляют к раствору 184 мг (0,58 ммоль, 3 экв.) Bu4N+F-H2O в 2 мл CH2Cl2. После перемешивания при температуре окружающей среды в течение 10 минут добавляют 320 мг (0,19 ммоль, 1 экв.) соединения (13) и реакционную смесь перемешивают при температуре окружающей среды в течение 30 минут. Затем реакцию заканчивают. Реакционную смесь гидролизуют добавлением 6%-ного раствора лимонной кислоты и дихлорметан выпаривают при пониженном давлении. Остаток переводят в этилацетат и полученную органическую фазу трижды экстрагируют 6%-ным раствором лимонной кислоты и насыщенным раствором NaCl, высушивают над безводным Na2SO4, фильтруют и растворитель выпаривают при пониженном давлении. Полученный сырой продукт хроматографируют на картридже Sep-Pack® и затем подвергают препаративной ВЭЖХ для выделения чистого соединения в виде лиофилизата (342 мг).
ESI-MC m/z: 1593,32 [M+Na+]; 1571,81 [M+H+].
Пример 3
Физико-химические свойства циклических ундекапептидов формул (XV) и (XVI)
1. Растворимость в воде циклических ундекапептидов формул (XV) и (XVI)
Растворимость в воде определяли визуально при температуре лабораторной комнаты непосредственным растворением взвешенного количества циклического ундекапептида в 67 мМ фосфатом буфере типа Соренсена. Значения представлены в таблице 1.
Для сведения: описано, что циклоспорин А обладает максимальной растворимостью в воде, равной 33 мкг/мл, при температуре 20°С и рН 7, что соответствует максимальной концентрации 0,027 мМ.
2. Химическая и ферментативная стабильность циклических ундекапептидов формул (XV) и (XVI)
Первичное определение химической стабильности циклического ундекапептида формулы (XV) в течение времени проводили в сначала в изотоническом растворе маннита, а затем в фосфатном буфере (PBS) при рН 7 и температуре 4, 20 и 37°С.
Процентное содержание обнаруженного циклоспорина А представлено в таблице 2 ниже.
Как можно видеть из данных таблицы, циклический пептид был стабилен в растворе маннита в течение, по меньшей мере, 90 суток при температуре 4°С.
Второе определение стабильности как химической, так и ферментативной, проводили с двумя циклическими ундекапептидами формул (XV) и (XVI), растворенными в 50 мМ буфере Hepes при рН 7,4, при 37°С в присутствии и в отсутствие эстераз. Во время инкубации при 37°С отбирали аликвотные порции объемом 40 мкл в соответствующие периоды времени и анализировали ВЭЖХ и ESI-MC.
В отсутствие фермента в течение более 3 суток наблюдали химическую стабильность для обоих циклических ундекапептидов.
Результаты, полученные во время гидролиза в присутствии эстераз, представлены на фиг.1. Кривая кинетики превращения циклического ундекапептида (XVI) представлена значками в виде ромбов в то время, как кривая кинетики появления циклоспорина А представлена значками в виде квадратов. Как можно отметить из представленных на фигуре данных, в присутствии эстераз циклический ундекапептид (XVI) быстро разрушается с образованием циклоспорина А. Аналогичное наблюдение было сделано для циклического ундекапептида (XV).
Циклические ундекапептиды (XV) и (XVI) инкубировали в бычьей сыворотке при 37°С. Определяли периоды полупревращения циклических ундекапептидов в циклоспорин А, которые составляли соответственно 3,66 и 3,50 чача.
Пример 4
Применение внутривенного введения в виде водного раствора
Изучение фармакокинетики циклических ундекапептидов формул (XV) и (XVI)
Для того чтобы сделать доступным для врачей средство, которое представляет альтернативу фармацевтическим композициям циклоспорина А, обычно используемого в виде микроэмульсии в полиоксиэтиленированном касторовом масле, которая обладает низкой стабильностью, с ней относительно трудно обращаться, и она вызывает побочные эффекты, пролекарства по настоящему изобретению в виде обычных водных растворов оценивали на возможность внутривенного применения.
Так, два циклических ундекапептида (XV) и (XVI) в растворе фосфатного буфера вводили внутривенно крысам в дозе, экв.ивалентной 10 мг/кг циклоспорина А.
В качестве стандарта использовали пробу инъекционного раствора промышленно доступного циклоспорина под торговым названием сандиммун после соответствующего разведения.
Через регулярные интервалы времени отбирали пробы крови и затем подвергали анализу на содержание циклоспорина А.
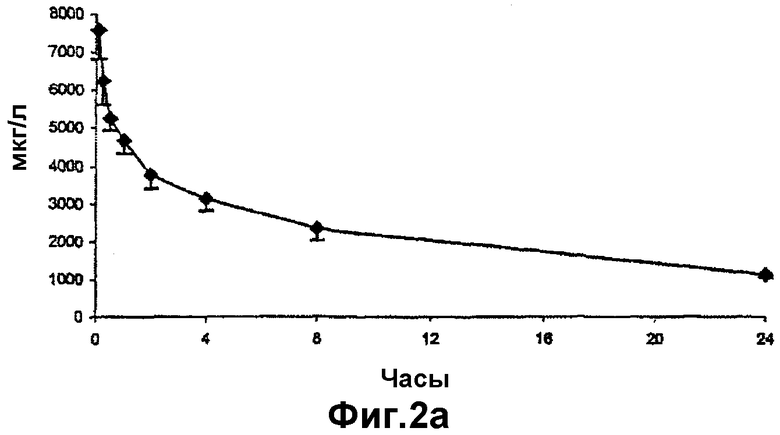
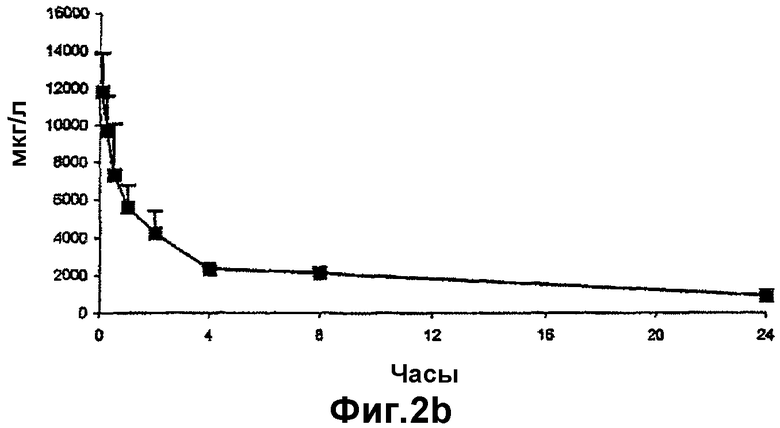
Концентрации циклоспорина А в крови после в/в введения циклоспорина А представлены на фиг.2а. Концентрации циклоспорина А в крови, полученные после в/в введения водного раствора двух пролекарств (XV) и (XVI), представлены на фиг.2b и 2с соответственно. Данные, полученные из кривых на фиг.2а, 2b и 2с, представлены в таблице 3 ниже.
В данной таблице сокращения имеют следующие значения:
- AUC: площадь под кривой концентрации;
- CL: клиренс;
- MRT: среднее время удерживания;
- Vss: объем распределения в стационарной фазе;
- Т1/21: первоначальный период полураспада;
- Т1/22: конечный период полураспада.
Как можно отметить из данных фиг.2 и таблицы 3, фармакокинетические параметры, установленные в контрольном опыте с циклоспорином А, были сравнимы с имеющимися в литературе. Площадь под кривой концентрации для циклоспорина А, образовавшегося во время расщепления циклического ундекапептида (XV), была сравнима с таковой для циклоспорина А в контрольном опыте в то время, как данный показатель для циклоспорина А, образовавшегося в результате расщепления циклического ундекапептида (XVI), был ниже на 25% по сравнению с таковым для циклоспорина А в контрольном опыте.
Два циклических ундекапептида (XV) и (XVI) показывали одинаковые профили образования циклоспорина А. Данные профили сходны с таковым для циклоспорина А при его введении в виде фармацевтической формы на основе полиоксиэтилинированного касторового масла.
Исходя из этих данных можно заключить, что пролекарства по настоящему изобретению дают эквивалентный профиль образования циклоспорина А при следующих важных преимуществах по сравнению с имеющимися фармацевтическими композициями циклоспорина А:
- простота применения при простом растворении в воде; и
- отсутствие необходимости использовать наполнители, которые являются токсичными; и
- отсутствие необходимости использовать специфические вещества при работе с ними.
Пример 5
Применение для местного введения в глаз в виде водного раствора
Изучение фармакокинетики циклического ундекапептида формулы (XV)
Для того, чтобы сделать доступным для врача средство для местного применения в глазу на основе циклоспорина А, не вызывающее раздражения или неприятных ощущений, или расплывчатого зрения, оценивали пролекарства по настоящему изобретению в виде обычных водных растворов.
1. Приготовление растворов
Готовили изотонические водные растворы циклического ундекапептида (XV), содержащие 5% маннита и с рН 7,0. Концентрация циклоспорина А была равна 1% (масс./об.). Растворы стерилизовали пропусканием их через фильтры из нитроцеллюлозы с размером пор 0,22 мкм. Готовили стандартную композицию циклоспорина А в виде 1% раствора на оливковом масле.
2. Определение переносимости циклического ундекапептида формулы (XV)
Определяли переносимость для глаз двумя методами, а именно тестом Драйза в модификации и с использованием софокусного лазерного сканирующего офтальмоскопа.
2.1. Модифицированный тест Драйза (тест по острой переносимости)
Данное исследование проводили на шести самцах-альбиносах кроликов. Каждому животному в один глаз закапывали 50 мкл раствора, описанного выше, другой глаз не обрабатывали, и он служил контролем.
Клиническую оценку возможного раздражающего действия проводили визуально посредством оценки истечений из глаза, наличия хемоза конъюнктивы и покраснения конъюнктивы согласно классификации, представленной в таблице 4.
У каждого животного исследовали возможное раздражающее действие по показателям, представленным выше, в определенные периоды времени в течение 48 часов после закапывания и рассчитывали общий индекс раздражающего действия (Iirr) по общей сумме установленных показателей. Результаты представлены в таблице 4 в п.2.3. ниже.
2.2. Софокусный лазерный сканирующий офтальмоскоп (тест для оценки подострой токсичности в течение 4-дневного периода введения)
Данный тест проводили на том же виде животных, как описано ранее. 25 мкл раствора, описанного выше, закапывали на роговицу правого глаза три раза в день в течение четырех суток, и затем один раз на четвертые сутки непосредственно перед обследованием. После последнего закапывания кроликов усыпляли введением кетамина гидрохлорида и ксилазина. В глаз вносили 25 мкл (всего) раствора натриевой соли флуоресцеина с концентрацией 0,5% в отношении масса/объем для избирательного мечения, возможно, поврежденных поверхностей глаза. Затем глаз промывали в течение одной минуты физиологическим раствором при 37°С.
Наконец, глаз обследовали с помощью софокусного лазерного сканирующего офтальмоскопа по методу, описанному Furrer et al., J. Ocular Pharmacol., 1997, 13, 559. Офтальмоскоп был соединен с системой анализа изображений для получения изображения в трех направлениях и для оценки поврежденных поверхностей.
Степень переносимости оценивали в зависимости от процента участков поражения роговицы и по следующим показателям:
- от 0 до 25%: хорошая переносимость;
- от 25 до 40%: приемлемая переносимость;
- от 40 до 60%: низкая переносимость;
- выше 60%: неприемлемая переносимость.
Следует отметить, и это в целом является общепризнанным, что процент участков поражения, меньший или равный 5%, соответствует обычному показателю гибели клеток в организме, не подвергавшемуся какому-либо воздействию.
Полученные результаты представлены в таблице 5 в п.2.3. ниже.
2.3. Результаты оценки переносимости для глаз циклического ундекапептида формулы (XV)
Из данных таблицы 5 следует, что в тесте Драйза для циклического ундекапептида формулы (XV) был получен общий индекс раздражающего действия, равный 1,8, и что 7% роговицы подвергалось повреждению при введении данного продукта. Два этих результата свидетельствует об очень хорошей переносимости циклического ундекапептида, эта переносимость оказывается явно улучшенной по сравнению с циклоспорином А при введении последнего в оливковом масле.
По очевидным причинам на животных не оценивали субъективное улучшение качества зрения при использовании водного раствора по сравнению с масляным раствором.
3. Стабильность циклического ундекапептида формулы (XV)
Пробы растворов, описанных выше, хранили соответственно при 4 и 20°С. Регулярно в течение 3 месяцев проводили анализ методом ВЭЖХ. Было установлено, что данные пробы обладают хорошей стабильностью при хранении в таких условиях.
4. Кинетика превращения циклического ундекапептида формулы (XV) в условиях ex vivo
Данный тест по кинетике превращения проводили при инкубировании при 37°С 25 мкл пробы раствора, описанного выше, при мягком перемешивании с 8 мкл свежей слезной жидкости кроликов. Через 1, 2, 3 и 30 минут отбирали пробы объемом 2 мкл и затем анализировали методом ВЭЖХ.
Полученные результаты показывают, что циклический ундекапептид формулы (XV) играет свою роль пролекарства с первой минуты контакта со слезной жидкостью, образуя циклоспорин А. Через 3 минуты превращению подвергалось 3% пролекарства, и затем через 30 минут - 4,7%.
5. Кинетика превращения циклического ундекапептида формулы (XV) в условиях in vivo
Данный тест по кинетике превращения проводили при закапывании 25 мкл пробы раствора, описанного выше, в правый глаз самцов-альбиносов кроликов (массой 4 кг). Через 1, 2, 3, 4 и 20 минут отбирали пробы слезной жидкости и затем анализировали методом ВЭЖХ.
Полученные результаты представлены на фиг.3. Как можно отметить, результаты данного теста подтверждают результаты, уже полученные в опыте в условиях ex vivo. Циклический ундекапептид формулы (XV) (квадраты) явно выполняет роль пролекарства с первой минуты контакта со слезной жидкостью кроликов, выделяя циклоспорин А (кружки), и это выделение продолжается в течение последующих 20 минут. Через 1 минуту концентрация циклоспорина А в слезной жидкости равнялась 0,025 мг/мл.
6. Выводы
На основании данных результатов можно заключить, что пролекарства по настоящему изобретению предоставляют следующие преимущества при местном применении для глаз:
- простота приготовления фармацевтической композиции, такой как жидкость для глаз, простым растворением в водном растворе без необходимости применения масляных адъювантов;
- хорошая острая переносимость и очень хорошая подострая переносимость, которые выше по сравнению с установленными для фармацевтической композиции циклоспорина в масляной форме;
- хорошая стабильность; и
- время полужизни, подходящее для применения в офтальмологии.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЦИКЛОСПОРИНЫ | 1991 |
|
RU2085589C1 |
| НЕИММУНОСУПРЕССОРНЫЙ ЦИКЛОСПОРИН ДЛЯ ЛЕЧЕНИЯ ВРОЖДЕННОЙ МИОПАТИИ УЛЬРИХА | 2008 |
|
RU2462262C2 |
| ПРИМЕНЕНИЕ МОДИФИЦИРОВАННЫХ ЦИКЛОСПОРИНОВ | 2007 |
|
RU2463071C2 |
| ПРИМЕНЕНИЕ МОДИФИЦИРОВАННЫХ ЦИКЛОСПОРИНОВ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ВЫЗВАННЫХ HCV | 2004 |
|
RU2389501C2 |
| НОВЫЕ ЦИКЛОСПОРИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ И ПРЕДУПРЕЖДЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2011 |
|
RU2601742C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОСПОРИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМКОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2131883C1 |
| СРЕДСТВО ДЛЯ РОСТА ВОЛОС, СОДЕРЖАЩЕЕ ПРОИЗВОДНЫЕ ЦИКЛОСПОРИНА ПО ПОЛОЖЕНИЮ 3 | 2002 |
|
RU2291682C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОСПОРИНА ДЛЯ ЛЕЧЕНИЯ ГЛАЗНЫХ И КОЖНЫХ ЗАБОЛЕВАНИЙ И СОСТОЯНИЙ | 2009 |
|
RU2521358C2 |
| КОВАЛЕНТНЫЕ ЛИНКЕРЫ В КОНЪЮГАТАХ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2698727C1 |
| СПОСОБ СИНТЕЗА ЦИКЛИЧЕСКИХ ДЕПСИПЕПТИДОВ | 2018 |
|
RU2817013C1 |
Настоящее изобретение относится к фармакологии, в частности, к пролекарству, представляющему собой циклический ундекапептид, в котором его пептидная цепь включает, по меньшей мере, один аминокислотный остаток общей формулы (I), в которой: атом углерода Сa образует одну из связей в кольце ундекапептида; каждый из заместителей Y представляет атом водорода или вместе они образуют связь; заместители R1 и R3, независимо друг от друга, представляют атом водорода, группу аралкил, алкарил, гетероалкил, гетероцикл, алкилгетероцикл, гетероциклоалкил или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода, где указанные группы необязательно замещены, по меньшей мере, одной группой, выбранной из -COOH, -CONHR8, -NHC=NH(NH2), -NHC=NR8(NH2), -NH2, -NHR8, -NR8 2, -N+R8 3, -OH, -OPO(OR8)2, -OPO(OH)(OR8), -OPO(OH)2, -OSO(OR8)2, -OSO(OH)(OR8), -OSO(OH)2 и различных солевых форм этих групп, где каждый из заместителей R8, независимо друг от друга, представляет нормальный или разветвленный алкил, содержащий 1-6 атомов углерода; заместители R2 и R4, независимо друг от друга, представляют атом водорода, группу алкарил или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода; заместители R5 и R6, независимо друг от друга, представляют атом водорода, группу аралкил или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода; и заместитель R7 представляет группу аралкил алкарил, гетероалкил, гетероцикл, алкилгетероцикл, гетероциклоалкил, или нормальный или разветвленный алкил, а также к применению указанного пролекарства для получения лекарственного средства для лечения патологических состояний слизистых мембран. Изобретение обеспечивает повышение доступности лекарства при его использовании. 6 и 11 з.п. ф-лы, 5 табл., 3 ил.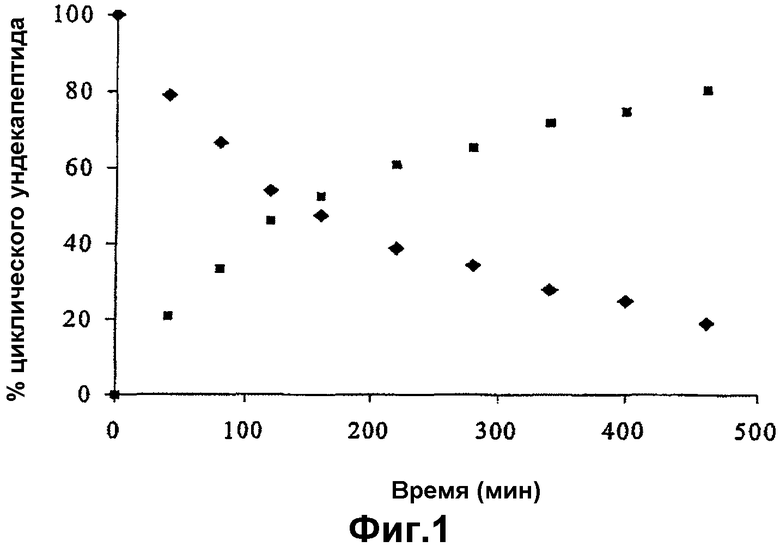
в которой атом углерода Сa образует одну из связей в кольце ундекапептида;
каждый из заместителей Y представляет атом водорода, или вместе они образуют связь;
заместители R1 и R3, независимо друг от друга, представляют атом водорода, группу аралкил, алкарил, гетероалкил, гетероцикл, алкилгетероцикл, гетероциклоалкил или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода, где указанные группы необязательно замещены, по меньшей мере, одной группой, выбранной из -СООН, -CONHR8, -NHC=NH(NH2), -NHC=NR8(NH2), -NH2, -NHR8, -NR8 2, -N+R8 3, -OH, -OPO(OR8)2, -OPO(OH)(OR8), -OPO(OH)2, -OSO(OR8)2, -OSO(OH)(OR8), -OSO(OH)2 и различных солевых форм этих групп, где каждый из заместителей R8, независимо друг от друга, представляет нормальный или разветвленный алкил, содержащий 1-6 атомов углерода;
заместители R2 и R4, независимо друг от друга, представляют атом водорода, группу алкарил или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода;
заместители R5 и R6, независимо друг от друга, представляют атом водорода, группу аралкил или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода; и
заместитель R7 представляет группу аралкил, алкарил, гетероалкил, гетероцикл, алкилгетероцикл, гетероциклоалкил или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода, где указанные группы необязательно замещены, по меньшей мере, одной группой, выбранной из -СООН, -CONHR8, -NHC=NH(NH2), -NHC=NR8(NH2), -NH2, -NHR8, -NR8 2, -N+R8 3, -ОН, -OPO(OR8)2, -OPO(OH)(OR8), -OPO(OH)2, -OSO(OR8)2, -OSO(OH)(OR8), -OSO(OH)2 и различных солевых форм этих групп, где каждый из заместителей R8 имеет значения, определенные выше.
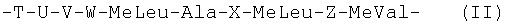
в которой Т выбран из аминокислот Ala, Abu, Nval, Val и Thr;
U выбран из аминокислот Sar, (D)MeSer, (D)MeAla и (D)MeSer(OCOR9), причем R9 представляет атом водорода, алкарил, или нормальный или разветвленный алкил, содержащий 1-6 атомов углерода;
V представляет аминокислоту общей формулы (N-R10)aa, где аа выбрана из аминокислот Val, Leu, Ile, Thr, Phe, Tyr и Thr, и R10 представляет нормальный или разветвленный алкил, содержащий 1-6 атомов углерода;
W выбран из аминокислот Val, Nval и Leu;
Х выбран из аминокислот (D)Ala, (D)Ser, (D)Hiv, (D)Val и (D)Thr; и
Z выбран из аминокислот Leu и MeLeu.
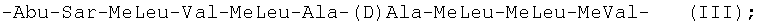
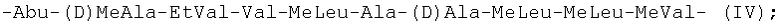
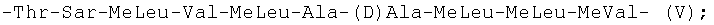
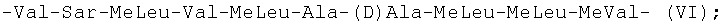
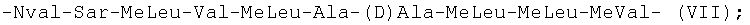
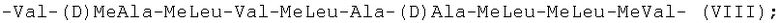
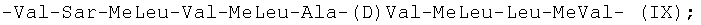
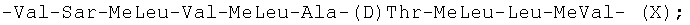
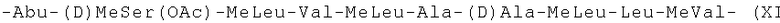
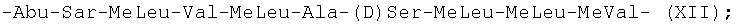
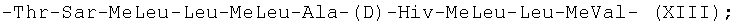
и
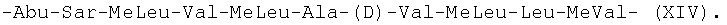
и
| Установка для приготовления асфальтобетонных смесей | 1946 |
|
SU67801A1 |
| ЦИКЛОСПОРИНЫ | 1991 |
|
RU2085589C1 |
| ПРОИЗВОДНЫЕ ЦИКЛОСПОРИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМКОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2131883C1 |
| Автоматический пистолет | 1927 |
|
SU8033A1 |
| US 5079341 А, 07.01.1992 | |||
| US 5156960 А, 06.04.1999. | |||

