Настоящее изобретение относится к новым циклоспоринам, их использованию в качестве лекарственных препаратов и к содержащим их фармацевтическим композициям, а также к способам их получения.
Циклоспорины включают класс структурно различных, циклических, поли-N-метилированных ундекапептидов, обычно обладающих фармакологическими, в частности, иммуносупрессорными, противовоспалительными или противопаразитическими активностями, или активностью реверсии или повышения сопротивляемости, например, к опухолям или другой лекарственной терапии, в частности, мультилекарственной резистентностью. Первым выделенным циклоспорином был природный фунгицидный метаболит Циклоспорин (Cyclosporine), известный также как циклоспорин A, и коммерчески доступный под торговой маркой SANDIMMUNR или SANDIMMUNER. Циклоспорин является циклоспорином формулы A:
где MeBmt является N-метил-(4R)-4-бут-2E-ен-1-ил-4-метил-(L) треониновым остатком формулы B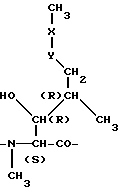
где -x-y- является -CH=CH- (транс).
После первоначального открытия циклоспорина был выделен и идентифицирован широкий круг природных циклоспоринов, а также были получены полностью синтетическими или полусинтетическими способами или микробиологическими способами с применением модифицированных культур множество дополнительных неприродных циклоспоринов. Таким образом, класс циклоспоринов в настоящее время является обширным и включает, например, природные циклоспорины A - Z 1) Traber и др.; Helv. Chim. Acta, 60, 1247 - 1255 (1977); 2) Traber и др.; Helv. Chim. Acta, 65, 1655 - 1667 (1982); 3) Kobel и др.; Eupor. J. Applied Microbiology and Biotechnology, 14, 273 - 240, 1982 и von Wartburg и др., Progress in Allergy, 38, 28-45, 1986, а также различные неприродные производные циклоспоринов и искусственные или синтетические циклоспорины, включая дигидроциклоспорины, в которых фрагмент -x-y- остатка -MeBmt- (Формула B выше) насыщают до получения -x-y- равного -CH2-CH2-, производные циклоспоринов, в которых 3'-0-атом остатка -MeBmt- ацилируют или далее замещают по α- углеродному атому саркозилового остатка в положении 3; и циклоспорины, в которых различные аминокислоты связаны в определенной пептидной последовательности. Например, используют полностью синтетический способ получения циклоспоринов, разработанный R. Wenger (1) Traber и др., 2) Traber и др., 3) Kobel и др. , указанные ранее); патенты US N 4108985, 4220641, 4288431, 4554351, 4396542 и 4798823, EP 34567A, EP 56782A, EP 300784A и EP 300785A, WO 86/02080 и GB 2206119A и 2207678A; Wenger 1, Transpl. Proc., 15, Suppl. 1: 2230 (1983); Wenger 2, Angew. Chem. Int. Ed. 24, 77 (1985) и Wenger 3, Progress in the Chemistry of Organic Natural products, 50, 123 (1986).
Таким образом, класс, включающий циклоспорины, весьма обширен и включает, например [Thr]2-, [Val]2-, [Nva]2 и [Nva]2-[Nva]5-циклоспорин (также известные как циклоспорины C, D, G и M соответственно). [3-0-ацетил-Mebmt] 1циклоспорин (известный также как ацетат циклоспорина A), [дигидро-MeBmt] 1-[Val] 2-циклоспорин (также известный как дигидроциклоспорин D), [(D)Ser] 8-циклоспорин, [Melle] 11-циклоспорин, [(D)MeVal]11-циклоспорин (известный также как циклоспорин H), [MeAla]6-циклоспорин, [(D)Pro]3-циклоспорин и т.д.
В соответствии с обычной номенклатурой для циклоспоринов их определяют в данном изобретении по отношению к структуре циклоспорина, т.е. циклоспорина A. Это делается, во-первых, указанием тех остатков в молекуле, которые отличаются от остатков, присутствующих в циклоспорине, и затем применяя термин "Циклоспорин" для характеристики остальных остатков, которые идентичны остаткам, присутствующим в циклоспорине. Так, [дигидроMeBmt]1-[Val] 2-циклоспорин является циклоспорином, содержащим последовательность, представленную в формуле A, но где остаток -MeBmt- в положении 1 заменен на дигидроMeBmt- (остаток формулы B, приведенной выше, где -x-y- является -CH2CG2-), а α Abu- во втором положении заменен на -Val-.
Кроме того, аминокислотные остатки, которые обозначаются сокращенно, например, -Ala-, MeVal-, - α Abu- и т.д., в соответствии с обычной практикой следует считать, как имеющие (L)-конфигурацию, если нет других указаний, например, как в случае "-(D)Ala-". Сокращение для остатка, в котором предшествует "Me", как например, в случае "-MeLeu-", представляет α -N-метилированные остатки. Отдельные остатки циклоспориновой молекулы пронумерованы, как принято, по часовой стрелке, начиная с остатка -MeBmt-, -дигидро-MeBmt- или остатка, соответствующего остатку в положении 1. Ту же самую цифровую последовательность используют и в описании изобретения и в формуле изобретения.
Настоящее изобретение относится к новым циклоспоринам, наиболее пригодным для местного нанесения, например, при лечении заболеваний легких или болезненных состояний, связанных с легкими.
Более конкретно, настоящее изобретение относится к производным циклоспорина формулы I:
где R означает водород, (C1 - C3)алкил, (C1 - C3)алкокси, галоидзамещенный (C1 - C3)алкил, гидроксизамещенный (C1 - C3)алкил, (C1 - C2)алкиламино или ди(C1 - C2)алкиламино;
X означает кислород;
-x-y- представляет собой -CH=CH- (транс) или -CH2-CH2-;
Q является -(D)Ala-, -(D)Ser-, [0-(2-гидроксиэтил)(D)Ser]- или [0-(2-ацилоксиэтил)(D)(Ser] -, в котором ацильный остаток является физиологически гидролизуемым и приемлемым.
Предпочтительными являются производные циклоспорина формулы I, в которых
R означает метокси;
X означает кислород;
-x-y- является -CH=CH-(транс);
Q является -(D)Ala-.
Особенно предпочтительными являются производные циклоспорина формулы I, в которых
X означает кислород;
-x-y- является -CH=CH- (транс);
Q является -(D)Ala-;
R означает водород, метил, этил, этокси, трифторметил, диметиламино, гидроксиметил, [S]α- гидроксиэтил, [R]α- гидроксиэтил
или в которых
X означает кислород;
-x-y- является -CH2=CH2-;
Q является -(D)Ala-;
R означает метил,
или
X означает кислород;
-x-y- является -CH=CH- (транс);
Q представляет остаток формулы III:
и R в формуле I и R' в формуле III - оба являются метилом и оба - метокси.
Предпочтительными алкильными группами R являются метил и этил. Предпочтительными алкокси R являются метокси, этокси. Галоидзамещенный означает хлор-, бром-, фтор- или иодзамещенный. Галоидзамещенные группы R могут быть моно-, ди- или поли-галоидзамещенными. Подходящей галоидзамещенной(C1 - C3)алкильной группой в качестве R является трифторметил. Подходящими гидроксизамещенными (C1 - C3)алкильными группами в качестве R являются гидроксиметил и 1-гидроксиэтил. Подходящими моно- и ди-алкиламиногруппами в качестве R являются метиламино, диметиламино и метилэтиламино, причем диметиламиногруппа является наиболее предпочтительной.
В одном из вариантов настоящего изобретения R является водородом, (C1 - C2)алкилом, (C1 - C2)алкокси, трифторметилом, гидроксизамещенным (C1 - C2)алкилом, моно- или ди-(C1 - C2алкил)-амино, особенно водородом, (C1 - C2)алкилом, (C1 - C2)алкокси, трифторметилом, гидроксизамещенным(C1 - C2)алкилом или диметиламиногруппой. Предпочтительными значениями R являются водород, (C1 - C2)алкил, (C1 - C2)алкокси и наиболее предпочтительно метилом или метокси. Наиболее предпочтительным значением R является (C1 - C2)алкокси, особенно метокси.
Термин "физиологически гидролизуемый и приемлемый", используемый в определении Q, определяет ацильные остатки, которые могут быть отщеплены в физиологических условиях до получения кислоты, которая сама по себе является физиологически переносимой во вводимых дозах. Подходящие ацильные остатки включают бензоил и салицил, а также остатки формулы R'-CX'-, где R' имеет указанные ранее значения для R, исключая амино и моно- и ди-(C1 - C2алкил)-амино, а X' является кислородом или серой.
Предпочтительно, чтобы Q было -(D)Ala- или группой III, как указано ранее. Особенно предпочтительно, чтобы Q было -(D)Ala-.
Циклоспорины настоящего изобретения, в которых R и/или R' является асимметричной группой, например, такой, в которой R является α- гидроксиэтилом, демонстрируют оптический изомеризм. В этом случае отдельные изомеры можно получить обычным способом, например, синтезом из оптически активных исходных материалов (как, например, в случае примерно 10 и 11, приводимых далее), или разделяя полученные вначале изомерные смеси, например, используя хроматографическую методику разделения хиральных соединений. В тех случаях, когда существуют такие изомеры, следует учитывать, что настоящее изобретение охватывает как отдельные изомерные формы, например, S- и R-энантиомеры, а также их смеси, например, рацемические и диастереомерные смеси, если нет других указаний. Однако, вообще, для фармацевтического использования в соответствии со способом настоящего изобретения, применение отдельных энантиомеров в чистой или практически чистой форме, например, содержащих вплоть до 95% или более чистого отдельного энантиомера, окажется предпочтительным.
В настоящем изобретении предложен также способ получения циклоспорина формулы I, который заключается во взаимодействии производного формулы IV:
где значения "-x-y-" и "Q" соответствуют ранее указанным, с соединением формулы V:
где Z означает отщепляемую группу, R и X имеют ранее указанные значения, при этом, если R представляет собой защищенный по гидроксигруппе гидроксизамещенный (C1 - C3)алкил, удаляют защиту, а если в полученном при этом соединении Q является [0-(2-гидроксиэтил)(D)Ser], в случае необходимости осуществляют ацилирование этого соединения ацилирующим агентом.
Подходящие защитные группы включают трет.-бутилсилил. Удаление защиты можно осуществить обычными способами, например, используя трет-бутиламмонийфторид в соответствии с процедурой, описанной Corey и др. J. Am. Chem. Soc., 94, 6190 (1972).
В циклоспориновых исходных материалах формулы IV 8'-гидроксигруппы остатков в положении 1 являются более реакционноспособными, чем гидроксильные группы, которые могут присутствовать в остатке в положении 8, например, когда Q является -(D)Ser-. Согласно способу можно соответственно вводить группу R-C-(= X)-, преимущественно, по остатку в положении 1. Например, используя 1 эквивалент соединения формулы V. После этого может следовать стадия введения отличной группы R'-C(=X)- в остаток в положении 8. Для этого используют соединение формулы V'R'-C(=X)-Z, где X' означает кислород и R' имеет значение, определенное выше для формулы III. Z является отщепляемой группой.
Если необходимо ввести группу R'-C-(=X)- в положение 8, которая идентична группе R-C(= X)- в положении 1, указанные реакции можно вести одновременно, используя два эквивалента соединения формулы V или V'.
Подходящие соединения V и V' для использования в способе включают ацилгалогениды, например, такие, в которых Z является хлором или бромом, и -ангидриды, например, такие, в которых Z = R-CX-O- или R'-CX'-O. Реакцию удобно вести в присутствии кислотного связующего агента, например, 4-диметиламинопиридина при температуре от приблизительно -20oC до +60oC.
Исходные соединения формул V и V' хорошо известны или могут быть получены аналогично известным соединениям. Гидроксизамещенные ацилгалогены в O-трет. -бутилсилилзащищенной форме описаны, например, в Chem. Abstr. 109-149284 и Bishofsberger и др. J. Org. Chem., 53, 3457 (1988).
Циклоспориновые исходные соединения формулы IV могут быть получены в следующей последовательности реакций. В этой последовательности реакций только остаток в положении 1 молекулы циклоспорина представлен подробно. Остальная часть молекулы обозначена как "(rs2-11)" и представляет собой указанную для формулы I последовательность остатков, а именно следующую последовательность: - α Abu-Sar-MeLeu-Val-Ala-Q-MeLeu-MeVal-; x-y имеют значения, данные для формулы I.
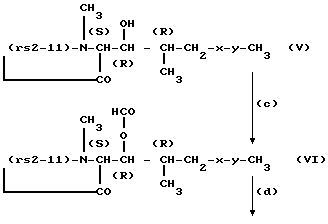
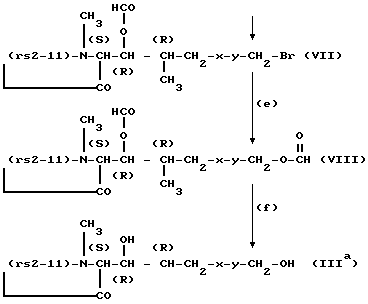
Стадии (c) - (f) удобно вести в соответствии с общими методиками, описываемыми далее в примере 1, или им аналогичными способами. Если остаток в положении 8-(rs2-11), т.е. остаток Q является -(D)Ser- или -(0-2-гидроксиэтил)(D)Ser), то его свободная гидроксильная группа также будет претерпевать формилирование на стадии (c) и последующее деформилирование на стадии (f).
Циклоспориновые исходные материалы формулы V известны или могут быть получены аналогично известным циклоспоринам. Так, если (rs2-11) представляет указанную последовательность, где Q является -(D)Ala-, указанные циклоспорины являются циклоспоринами или дигидроциклоспоринами A, C, D и G. Соответствующие циклоспорины, в которых Q является -(D)Ser- или (-0-ацил(D)Ser)-, описаны, например, в EP 56782A и в патенте GB 2155936A. Соответствующие циклоспорины, в которых Q является -(0-(2-гидроксиэтил)(D)Ser)- или -(0)2-ацилоксиэтил)(D)Ser)- описаны, например, в EP 414632 (заявка N PO 810567.9).
Циклоспорин формулы IV, где -x-y- является -CH=CH- (транс), а Q является (D)Ala-, известен также как метаболит циклоспорина A. Этот метаболит известен специалистам как M17.
Нижеследющие примеры иллюстрируют процесс получения циклоспоринов настоящего изобретения.
Пример 1
Получение [(8'-метоксикарбонилокси)MeBmt]1-циклоспорина (формулы I, где R = CH3O-, X = O, -x-y- = -CH=CH (транс), Q = -(D)Ala-).
0,33 мл метилхлорформиата добавляют к раствору 4,9 г [(8'-гидрокси)MeBmt] 1-циклоспорина и 1,9 г 4-диметиламинопиридина в 50 мл диметилформамида в безводных условиях. Реакционную смесь перемешивают при комнатной температуре в течение 18 часов. Растворитель удаляют в вакууме. Остаток помещают в этилацетат, промывают водной винной кислотой и водным Na2CO3. Растворитель удаляют при пониженном давлении, а остаток очищают на хроматографической колонке с силикагелем, используя этилацетат, насыщенный водой, до получения указанного в заглавии соединения: [α]
Получение [(3'-0-формил)MeBmt]1-циклоспорина формулы VI (процесс стадии c).
15 мл ацетилформиата добавляют к раствору 9,6 г циклоспорина (циклоспорин A) и 3,9 г 4-диметиламинопиридина в 100 мл ацетона в течение 30 минут при комнатной температуре. Реакционную смесь перемешивают в течение 20 часов при комнатной температуре, и растворитель отгоняют при пониженном давлении. Остаток помещают в этилацетат, промывают водной Na2SO4. Органический растворитель удаляют при пониженном давлении. Остаток кристаллизуют из кипящего гексана до получения указанного в заглавии соединения.
tпл = 195 - 197oC.
Получение [(8'-бром-3'-0-формил)MeBmt] 1-циклоспорина формулы VII (процесс стадии d).
75 г продуктам стадии (c), 11,7 г N-бромсукцинимида и 1 г азоизобутиронитрила в суспензии в 750 мл CCl4 нагревают при кипячении с обратным холодильником в течение 2 часов. Остаток фильтруют, промывают водным бикарбонатом натрия, водной винной кислотой и рассолом, а органический слой сушат над сульфатом натрия. Растворитель удаляют при пониженном давлении до получения указанного в заглавии соединения, которое вводят в дальнейшую реакцию без дополнительной очистки.
Получение [(3'-0-формил-8'-формилокси)MeBmt]1- циклоспорина формулы VIII (процесс стадии e).
86 г продукта стадии (d), 1 г NaI и 27 г тетраэтиламмонийформиата в 750 мл метилэтилкетона нагревают при кипячении с обратным холодильником в течение 4 часов. Растворитель удаляют при пониженном давлении, остаток помещают в этилацетат и промывают водным карбонатом натрия, водной винной кислотой и рассолом. Органический слой сушат над сульфатом натрия, а растворитель удаляют при пониженном давлении до получения указанного в заглавии соединения, которое вводят в следующую реакцию без дополнительной очистки.
Получение [(8'-гидрокси)MeBmt] 1-циклоспорина M17 формулы IIIa (процесс стадии f).
71 г продукта со стадии (e) перемешивают в течение 18 часов при комнатной температуре в 0,8М этанольном метиламине. Растворитель удаляют при пониженном давлении, а остаток хроматографируют на силикагеле, используя этилацетат насыщенной H2O. Растворитель удаляют при пониженном давлении, получая указанное в заглавии соединение в виде белой пены Rf = 0,31 (этилацетат насыщенный H2O; силикагель).
Нижеследующие циклоспорины формулы I, указанной ранее, в которых остаток A представлен формулой Ia, где R и -x-y- имеют значения, приведенные в таблице 1 (см. в конце описания), а Q является -(D)Ala-, можно получить аналогично примеру 1.
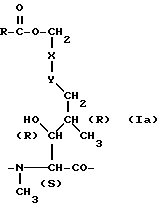
Нижеследующие циклоспорины формулы I, где A является остатком вышеприведенной формулы Ia, где R имеет значения, указанные в таблице 2, а -x-y- является -CH=CH- (транс), а Q является остатком формулы IIIa: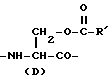
где R' имеет значения, приведенные далее в таблице 2 (см. в конце описания), можно также получить аналогичным образом, но используя 2 эквивалента нужного ацилирующего агента.
Циклоспорины настоящего изобретения обладают потенциальной иммуносуппрессорной и противовоспалительной активностью. В частности, они ингибируют спровоцированную антигеном воспалительную клеточную инфильтрацию, например, в дыхательных путях. In vivo эта активность проявляется после местного введения, например, после местного введения в воздушные пути за счет вдыхания. Было обнаружено, что циклоспорины настоящего изобретения напротив обладают существенно пониженной или практически не обладают активностью, например, противовоспалительной или иммуносуппрессорной активностью in vivo при систематическом приеме, например, после орального введения.
Иммуносуппрессорные и противовоспалительные свойства циклоспоринов настоящего изобретения можно продемонстрировать на стандартных тестовых моделях in vitro и in vivo, например, следующим образом:
1. Иммуносуппрессорная активность (in vitro)
1.1. Смешанная лимфоцитная реакция Мурина.
Примерно 0,5 • 106 лимфоцитов из селезенки самки (8-10) недель мышей штамма BaI b/c инкубируют в течение 5 дней в 0,2 мл среды для клеточного роста с приблизительно 0,5 • 106 лимфоцитов из селезенки самки (8-10 недель) мыши штамма CBA. Тестовое вещество добавляют в среду в различных концентрациях. Активность оценивают по способности подавлять пролиферацию, связанную с ДНК синтезом, что определяют по связыванию радиомеченного тимидина.
Циклоспорины по способу настоящего изобретения ингибируют связывание тимидина при концентрациях порядка от 0,005 до 0,025 мкг/мл.
1.2 Тест Мишель-Даттона
Приблизительно 107 лимфоцитов из селезенки самки мыши штамма OFI культивируют совместно с примерно 3 • 107 эритроцитов овцы в течение 3 дней. Тестовое вещество добавляют к инкубируемой среде в различных концентрациях. Лимфоциты собирают и помещают на агар со свежими эритроцитами овцы в качестве антигена. Сенсибилизированные лимфоциты выделяют антитела, которые покрывают эритроциты, и лизис происходит с образованием бляшек в присутствии добавки. Активность оценивают по снижению числа образующихся бляшек, т.е. клеток, продуцирующих антитела. Циклоспорины настоящего изобретения снижают количество клеток, образующих бляшки при концентрациях порядка от 0,03 до 0,05 мкг/мл.
2. Влияние на вызванную аллергенами легочную эозинофилию (in vivo)
Самцов гималайских пятнистых морских свинок (300 г, BRL) сенсибилизируют овальбумином (ОА) путем внутрибрюшинной инъекции 1 мл суспензии ОА (10 мкг) с Al(OH)3 (100 мг) и вакциной B-пертуссиса (0,25 мл) в физиологическом растворе (0,9% мас./объем). Для оральных исследований процедуру повторяют 1 раз спустя 2 недели, и животных используют еще через неделю. Для ингаляционных исследований процедуру повторяют 2 раза с интервалами в 3 недели, и животных используют спустя неделю после последней инъекции.
Иммунизацию проводят, используя физиологический раствор ОА, распыляемый в опытную камеру. Тестовых животных подвергают воздействию ОА только ингаляцией через нос в течение 60 минут. Для оральных исследований раствор ОА используют в концентрации 0,05%. Для ингаляционных исследований используют ОА раствор в концентрации 0,01%.
Тестовое вещество вводят (a) орально и (b) путем ингаляции. Для оральных исследований тестовое вещество вводят перорально (р.о) в оливковом масле 1 раз в день в течение 3 дней или в порошке с метилцеллюлозой один раз до ОА иммунизации. На третий день тестовые животные получают тестовое вещество за полтора часа и спустя 6 часов после ОА иммунизации. Для ингаляционных исследований тестовое вещество тонко измельчают для введения тестовым животным в жидкой пасте только через нос. Введение путем ингаляции осуществляют за 15 минут до ОА иммунизации.
Эффективность вводимых тестовых соединений определяют бронхоальвеолярным лаважем (BAL) и счетом клеток. Для этой цели животных умерщвляют Na-пентобарбитоном (100 мг/кг внутрибрюшинно) и извлекают трахею и канюлируют. Затем последовательно вводят в легкие 5 последовательных аликвот по 10 мл Ca2+ и Mg2+ свободного сбалансированного солевого раствора Хенка (HBSS), содержащего альбумин бычьей сыворотки (BSA, 0,3%), EDTA (10 мМ) и HEPES (10 мМ), и немедленно отсасывают, осторожно сжимая легочную ткань. Полное количество клеток в собранных элюатах определяют, используя автоматический клеточный счетчик. Жидкость лаважа центрифугируют при 200 об/мин в течение 10 минут, а клеточный осадок повторно суспендируют в 1 мл дополнительного HBSS. 10 мкл клеточной суспензии добавляют к 190 мкл раствора Турка (1:20 разбавление). Проводят дифференциальный счет клеток на мазках, подкрашенных Chiff-Quick. Клетки идентифицируют и считают, погружая в масло (х1000). Учитывают минимально 500 клеток на мазок и оценивают полную популяцию каждого типа клеток.
У необработанных животных ОА заражение вызывает повышение содержания клеток всех типов в BAL жидкости спустя 24 часа после заражения. Предварительное введение циклоспорина по способу настоящего изобретения путем ингаляции в дозах порядка от 1,0 до 15,0 мг/кг снижает содержание эозинофилов в BAL в зависимости от дозы по сравнению с необработанными контрольными опытами. Количество клеток других лейкоцитов (макрофагов, нейтрофилов) также снижается. Напротив, повторное оральное введение циклоспорина настоящего изобретения не оказывает существенного влияния на количество клеток по сравнению с необработанным контролем.
Циклоспорины настоящего изобретения соответственно пригодны для лечения заболеваний или болезненных состояний, реагирующих на или требующих местной противовоспалительной, иммуносуппрессорной или родственной терапии, например, путем местного введения для лечения таких заболеваний или болезненных состояний глаз, носовой полости, ротовой полости, кожи, прямой кишки или, особенно, дыхательных путей или легких. В частности, цефалоспорины настоящего изобретения позволяют осуществлять местную противовоспалительную, иммуносуппрессорную или родственную терапию при одновременном снижении или исключении нежелательных побочных эффектов, например, общей системной иммуносуппрессии.
Циклоспорины настоящего изобретения особенно пригодны для лечения заболеваний или болезненных состояний дыхательных путей или легких, в частности, воспалительных заболеваний или обструктивных заболеваний дыхательных путей. Они особенно пригодны для лечения заболеваний или болезненных состояний дыхательных путей, или легочных заболеваний, связанных с или характеризующихся воспалительной клеточной инфильтрацией или другими воспалительными явлениями, сопровождающими воспаления клеток, например, накопление эозинофилов и/или нейтрофилов. Они особенно пригодны для лечения астмы.
Циклоспорины настоящего изобретения пригодны для лечения астмы любого типа и происхождения, включая как эндогенную, так и экзогенную астму. Они пригодны для лечения атопической или неатопической астмы, включая астму аллергическую, бронхиальную, астму, вызванную нагрузками, астму, связанную с местом пребывания, астму, вызванную бактериальными инфекциями, и другие астмы неаллергического происхождения. Лечение астмы также включает и лечение "стридорного детского синдрома", т.е. лечение детей младше 4-5 лет, с симптомами стридора, в частности, по ночам, с диагнозом "стридорные дети", относящихся к категории постоянных пациентов, а в последнее время более корректно отнесенных к разряду предрасположенных к астме или больных астмой на ранних стадиях. Циклоспорины настоящего изобретения, в частности, полезны при лечении астмы у пациентов, астматический статус которых зависит от стероидов или устойчив по отношению к стероидам.
Циклоспорины настоящего изобретения пригодны также для лечения бронхитов или для лечения хронических или острых осложнений верхних дыхательных путей, с ними связанных. Циклоспорины настоящего изобретения можно использовать для лечения бронхитов любого типа или происхождения, включая, например, острые бронхиты, арахидические бронхиты, катарральные бронхиты, хронические бронхиты, крупозные бронхиты, фтиноидные бронхиты и т.п.
Циклоспорины настоящего изобретения, кроме того, полезны при лечении болезней легких (например, воспалений, обычно связанных с местом, заболеваний легких, часто сопровождающихся воспалениями дыхательных путей, как хронических, так и острых, и вызванных частым вдыханием разного рода пыли), любого типа и происхождения, включая, например, алюминоз, антракоз, асбестоз, бериллоз, халикоз, филоз, сидероз, силикоз, табакоз и, особенно, биссиноз.
Циклоспорины настоящего изобретения можно также использовать для лечения заболеваний дыхательных путей, связанных с эозинофилами (например, включая болезненную эозинофильную инфильтрацию легочных тканей), включая гиперэозинофилию и ее действие на дыхательные пути и/или легкие, а также, например, связанные с эозинофилами заболевания дыхательных путей, сопровождающиеся синдромом Лефлера, эозинофильную пневмонию, паразитическое (в частности, метазоновое) заражение (включая тропическую эозинофилию), бронхолегочный аспергиллез, полиартритные уплотнения (включая синдром Чург-Штраусса), эозинофильную гранулему и связанные с эозинофилами заболевания дыхательных путей, обусловленные реакцией на лекарственные препараты.
Под формулировкой "лечение", как она использована относительно лечения заболеваний дыхательных путей или легких, и, в частности, астмы, следует понимать охватывающую как симптоматический, так и профилактический тип лечения, т.е. немедленное лечение, например, острых воспалений (симптоматическое лечение), так и предварительное лечение для предотвращения, улучшения или ограничения длительной симптоматологии (профилактическое лечение). Термин "лечение", как он использован в настоящем описании и формуле изобретения, относительно таких заболеваний следует понимать, соответственно, как включающий как симптоматическое, так и профилактическое лечение, например, в случае астмы - симптоматическое лечение для снятия острого воспалительного приступа и профилактическое лечение для ограничения продолжающегося воспалительного состояния и для уменьшения дальнейших связанных с ними обострений.
Циклоспорины настоящего изобретения можно также использовать для лечения любых заболеваний или болезненных состояний дыхательных путей или легких, требующих иммуносуппрессорной терапии, например, для лечения автоиммунных заболеваний или легких (например, для лечения саркоидоза, альвеолита или хронических гиперчувствительных пневмоний) или для поддержания аллогенного легочного трансплантата, например, после трансплантации легких или сердца.
Как было указано ранее, для вышеперечисленных целей циклоспорины настоящего изобретения нужно вводить местно в дыхательные пути, например, ингаляцией. Как было указано ранее, обладая потенциальной эффективностью при местном введении, циклоспорины настоящего изобретения не обладают или демонстрируют относительно пониженную системную активность, например, после орального приема. Таким образом, циклоспорины настоящего изобретения обеспечивают средства для лечения заболеваний и болезненных состояний легких, как было указано ранее, позволяя при этом избежать нежелательных системных побочных эффектов, например, сопутствующих непроизвольному проглатыванию лекарственных препаратов при лечении ингаляцией. (По проведенным оценкам во время курса ингаляции при необходимых при этом манипуляциях вплоть до 90% или более лекарственного препарата обычно проглатывается, а не ингалируется).
Путем предложения циклоспоринов, которые активны при местном введении, т.е. эффективны при ингаляции, но не активны систематически, настоящее изобретение открывает доступ к лечению циклоспоринами для тех пациентов, для которых в противном случае такое лечение было бы исключено, например, из-за риска систематических, особенно иммуносуппрессорных, побочных эффектов.
Циклоспорины настоящего изобретения пригодны также для лечения других заболеваний или болезненных состояний, в частности, заболеваний или болезненных состояний с аутоиммунной или воспалительной компонентной, и для которых может практиковаться местная терапия, например, лечение заболеваний или болезненных состояний глаз, например, конъюнктивитов, кератоконъюнктивитов, весенних конъюнктивитов и поддержания роговичных трансплантатов, заболеваний, связанных с носом, включая аллергические риниты, заболеваний и болезненных состояний кожи, включая псориаз, атопические терматиты, пемфигус и контактные дерматиты, а также заболеваний толстой кишки, например, болезни Кронина и язвенных колитов.
Для указанных ранее целей циклоспорины настоящего изобретения можно использовать в любых дозовых формах, пригодных для местного введения в нужное место. Так, для лечения заболеваний дыхательных путей или легких циклоспорины настоящего изобретения можно вводить вдыханием или ингаляцией из соответствующего распределительного устройства.
Для этой цели циклоспорины настоящего изобретения можно использовать в любом тонко измельченном или тонкодиспергированном виде, пригодном для введения в дыхательные пути или легкие, например, в виде тонкоизмельченных сухих частиц или в виде дисперсии или раствора в любом подходящем (т.е. возможном для введения в дыхательные пути) твердом или жидком носителе. Для введения в виде сухих частиц, циклоспорины настоящего изобретения можно, например, использовать как они есть, т.е. в микроизмельченной форме без дополнительных материалов, в разбавленном виде с дополнительными материалами - тонкоизмельченными инертными твердыми носителями или разбавителями (например, глюкозой, лактозой, маннитолом, сорбитолом, рибозой, маннозой или ксилозой), в виде частиц с покрытием или в любой другой форме, известной специалистам для введения в дыхательные пути тонко измельченных твердых веществ.
Введение в легкие можно осуществить, используя любую подходящую систему, известную специалистам для введения лекарственных препаратов в сухом или жидком виде путем ингаляции, например, атомизатор, распылитель, ингалятор для сухого порошка или подобное приспособление. Предпочтительно устройство с мерным приспособлением, с помощью которого можно вводить заранее определенное количество циклоспорина при каждом акте. Такие приспособления известны специалистам.
Для введения в нос циклоспорины настоящего изобретения удобно вводить в жидкой форме из соответствующего устройства для закапывания в нос. Подходящие формы для местного введения для лечения заболеваний или болезненных состояний, связанных с кожей, включают, например, кремы, гели, мази, пасты, катаплазмы, пластыри, трансдермальные бляшки и т.п. Композиции для дермального применения обычно содержат соответствующие агенты, способствующие проникновению сквозь кожу, например, известный специалистам азон. Формы, подходящие для офтальмологического применения, включают лосьоны, тинктуры, гели, мази и офтальмологические вкладыши, также известные специалистам. Для ректального введения, например, для местной терапии прямой кишки, циклоспорины настоящего изобретения можно вводить в виде суппозиториев или клизм, в частности в растворах, например, растворах растительного масла или в подобных масляных системах для использования в виде удерживаемых клизм.
Соответственно, настоящее изобретение обеспечивает:
A. Способ лечения заболеваний или болезненных состояний, требующих противовоспалительной, иммуносуппрессорной или родственной терапии нуждающегося в ней пациента, причем способ включает местное введение эффективного количества циклоспорина настоящего изобретения.
B. Циклоспорины настоящего изобретения предназначены для использования в составе лекарственных средств, например, для лечения воспалительной, иммуносуппрессорной или родственной терапии, например, для применения в способе A, описанном выше, особенно для местного введения в дыхательные пути.
Способ, описанный в пункте A, применим, в частности, для лечения заболеваний или болезненных состояний глаз, носа, горла, ротовой полости, кожи, толстой кишки или, особенно, дыхательных путей или легких. Он применим к любым болезням или болезненным состояниям, перечисленным выше, в частности, к любым заболеваниям или болезненным состояниям дыхательных путей или легких, требующим противовоспалительной или родственной терапии, особенно любым заболеваниям или болезненным состояниям дыхательных путей или легких, отличающихся воспалительной клеточной инфильтрацией и, особенно, для лечения астмы.
Далее объектом настоящего изобретения является:
C. Фармацевтическая композиция для местного введения, т.е. пригодная для местного введения в форме, содержащей циклоспорин настоящего изобретения в качестве иммуносуппрессорного или противовоспалительного агента вместе с фармацевтически приемлемым разбавителем или носителем, или циклоспорин настоящего изобретения в форме, или в средстве, или в приспособлении, обеспечивающем или облегчающем местное введение.
Фармацевтически приемлемые разбавители или носители, указанные ранее, представляют собой разбавители или носители, приемлемые для местного введения в необходимую зону лечения, например, разбавители или носители, приемлемые для местного введения в дыхательные пути, на кожу, через нос, в глаз или ректально. Формы для местного введения, например, облегчающие или обеспечивающие местное введение, включают, например, препараты сухих порошков активного ингредиента (например, циклоспорина настоящего изобретения) в практически чистом виде, например, такие, которые используются специалистами для любого ингаляционного устройства для введения порошка. Средства или приспособления, обеспечивающие или облегчающие местное введение, включают, в частности, устройства для ингаляции, а также контейнеры и т.п., из которых активные ингредиенты можно подавать в форме, пригодной для местного введения. Предпочтительными вариантами, указанными в пункте C, будут такие, которые позволяют обеспечить местное введение в дыхательные пути или легкие, например, путем ингаляции.
Дозы циклоспоринов настоящего изобретения, используемые на практике в способе настоящего изобретения, естественно, будут зависеть от места применения, конкретного состояния пациента, тяжести заболевания нуждающегося в лечении пациента (с точки зрения веса, возраста и т.д.), а также в зависимости от ожидаемого эффекта. Вообще, для лечения заболеваний или болезненных состояний дыхательных путей или легких, например, для использования при лечении воспалений или осложнений после заболеваний дыхательных путей, например, астмы, циклоспорины настоящего изобретения следует вводить местно в дыхательные пути или легкие, т.е. с помощью ингаляции, в дозах порядка от 20 до 400 мг/день, например, от 50 или 100 до 300, или например, от 200 до 300 мг/день. Дозы следует вводить через мерную систему в течение ряда последовательных вдохов от 1 до 5 при каждом приеме, причем прием может повторяться от одного до четырех раз в день. Таким образом, дозы при каждом введении будут порядка от около 5 до 100 мг и, что более удобно, от 12,5 или 25 до 100 мг, т.е. в мерном подающем устройстве будет подаваться, например, от 1 до 25 мг за один раз.
Для лечения заболеваний глаз и носа циклоспорины настоящего изобретения обычно вводят в виде соответствующих композиций, например, глазных капель, гелей, жидкостей или т.п., или капель в нос, спрея в нос или т.п., содержащих от около 0,05 до около 10%, и особенно от около 0,05 до около 5%, более предпочтительно от около 0,1 до около 2,5% по массе циклоспорина, в приемлемом для введения в глаза или нос разбавителе или носителе для нанесения на поверхность глаз или носа в количестве от около 0,05 до около 0,2 мл композиции, например, от около 0,05 до около 0,1 мл композиции один раз или от двух до трех раз в день.
Для лечения заболеваний или болезненных состояний толстой кишки обычно подходящая дневная доза циклоспорина настоящего изобретения составляет величину порядка от около 0,5 до около 15,0, предпочтительно от около 2,5 до около 10,0 мг/кг и обычно вводится как удерживаемая клизма, вводимая один раз или в половинной дозе дважды в день. Таким образом, каждая доза должна содержать от около 17,5 до около 1000, и, предпочтительно, от около 35 до около 700, более предпочтительно, от около 87,5 до около 550 мг циклоспорина настоящего изобретения наряду с соответствующим ректально применимым разбавителем или носителем его. Подходящие концентрации циклоспорина для применения в таких удерживаемых клизмах составляют величину порядка от около 0,5 до около 12,0, и, предпочтительно, от около 1,0 до около 10,0, и более предпочтительно, от около 2,0 до около 7,0 мг/мл.
Для нанесения на кожу для лечения заболеваний или болезненных состояний кожи циклоспорины настоящего изобретения обычно вводят в соответствующем, т. е. приемлемом для кожи виде, содержащем от примерно 1 до 10 мас.% циклоспорина вместе в приемлемым для кожи разбавителем или носителем его. Такие композиции удобно наносить на конкретное место, подлежащее лечению, в количестве приблизительно от 0,005 до 0,05 г/см2, 1, 2 или 3 раза в день.
Предпочтительным циклоспорином настоящего изобретения является продукт примера 1, а именно [(8'-метокси-карбонилокси)MeBmt]1- циклоспорин. Конкретные результаты для этого циклоспорина в серии тестов, проведенных по методикам, описанным в п.п. 1.1, 1.2 и 1.3, а также 2, приведены ранее, указаны в таблице 3.



Описываются новые производные циклоспорина общей формулы I  ,
,
R - водород, C1-C3-алкил, C1-C3-алкокси, галоидзамещенный C1-C3-алкил, гидроксизамещенный C1-C3-алкил, (C1-C2)алкиламино или ди (C1-C2)алкиламино;
X - кислород;
x-y представляет собой -CH=CH- (транс или -CH2-CH2);
Q является -(D)Ala, -(D)Ser, -[0-2-гидроксиэтил (D)Ser]- или -[0-(2-ацилоксиэтил)(D)Ser]-,
где каждый из ацильных остатков является физиологически гидролизуемым и приемлемым.
Описывается способ вышеуказанных соединений и фармацевтическая композиция для местного введения, используемая в качестве иммуносупрессорного или противовоспалительного агента с фармацевтически приемлемыми разбавителями или носителями на базе соединений формулы I. 3 с. и 6 з.п. ф-лы, 3 табл.

где R - водород, (C1-C3)алкил, (C1-C3)алкокси, галоидзамещенный (C1-C3)алкил, гидроксизамещенный (C1-C3)алкил, (C1-C3)алкиламино или ди(C1-C2)алкиламино;
X - кислород;
-x-y- представляют собой -CH=CH-(транс) или -CH2-CH2-;
Q является -(D)Ala-, -(D)Ser-, -[0-(2-гидроксиэтил)(D)Ser]- или -[0-(2-ацилоксиэтил)(D)Ser]-, в котором ацильный остаток является физиологически гидролизуемым и приемлемым.
Q - -(D)Ala-; R - метил, или X - кислород; -x-y- является -CH=CH-(транс); Q остаток формулы III
и R в формуле I и R' в формуле III оба являются метилом или оба - метокси.
где значения -x-y- и Q соответствуют указанным в п.1 формулы изобретения,
с соединением формулы V
в котором Z - отщепляемая группа,
R и X имеют значения, указанные в п.1,
при этом, если R - защищенный по гидроксигруппе гидроксизамещенный (C1-C3)алкил, удаляют защиту, а, если в полученном при этом соединении Q является [0-(2-гидроксиэтил)(D)Ser] , в случае необходимости, осуществляют ацилирование этого соединения ацилирующим агентом.
| Способ получения пептидов | 1976 |
|
SU639446A3 |
| DE 3840354 A1, 1989 | |||
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИНДОЛ-5-ОЛА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2013 |
|
RU2674249C2 |
| УСТРОЙСТВО ДЛЯ ИЗМЕРЕНИЯ И РЕ1 /ЛИРОВАНИЯ ТЕМПЕРАТУРЫ | 0 |
|
SU300784A1 |
| US 5079341 A, 07.01.92 | |||
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1988, ч.11, с.175 - 176. | |||

