Перекрестная ссылка на смежную заявку
Настоящая заявка претендует на приоритет предварительной заявки США, серийный №62/205,121, поданной 14 августа 2016 г., которая полностью включена в настоящий документ посредством ссылки, включая любые фигуры, таблицы и рисунки.
Предшествующий уровень техники
Конъюгаты антитело-лекарственное средство (ADC, Antibody drug conjugate), быстро растущий класс таргетных терапевтических средств, представляет новый перспективный подход в направлении улучшения как селективности, так и цитотоксической активности противораковых лекарственных средств. Примером лекарственного средства ADC, одобренного для терапевтического применения в США, является брентуксимаб ведотин (ADCETRIS®), представляющий собой химерное антитело к CD30, конъюгированное с монометилауристатином Е, применяемый в лечении анапластической крупноклеточной лимфомы и лимфомы Ходжкина.
Один из традиционных способов, применяемых в дизайне конъюгатов антитело-лекарственное средство (ADC), включает связывание молекул лекарственного средства с тиольными группами цепей антитела посредством линкерной группировки. Свободные тиольные группы получают в результате разрыва цистеиновых межцепьевых дисульфидных связей антитела посредством реакции восстановления. Типичное антитело содержит 4 межцепьевые дисульфидные связи (2 между тяжелыми цепями и 2 между тяжелыми и легкими цепями). Эти межцепьевые дисульфиды могут быть селективно восстановлены дитиотрейтолом, трис(2-карбоксиэтил)фосфином или другими слабыми восстанавливающими агентами с получением в результате 8 реакционноспособных сульфгидрильных групп для конъюгации. Этот способ позволяет присоединить к определенному антителу до восьми молекул лекарственного средства.
За счет факта разрыва по меньшей мере двух дисульфидных связей ADC, сконструированные с использованием этого принципа, нестабильны после поступления в кровообращение, и поэтому период полувыведения этих ADC будет сокращен. В результате недавних разработок дизайна и синтеза ADC адаптирован другой подход, а именно основанный на ковалентном соединении двух тиольных групп посредством агента сочетания, в результате чего устанавливаются мостики между двумя тяжелыми цепями и между тяжелыми и легкими цепями определенного антитела. В настоящее время усилия по освоению такого подхода главным образом сосредоточены на разработке дизайна структуры агента сочетания, не только имеющего функциональные группы для образования ковалентной мостиковой связи между двумя тиольными группами, но также охватывающего необходимые компоненты, способствующие специфическим видам биологической активности.
В более ранних исследованиях в этой области для взаимодействия с двумя тиольными группами, образовавшимися в результате разрыва дисульфидной связи, использовали бис-малеимиды. Ковалентное связывание между малеимидами и тиолами представляет собой классическую реакцию алкенового преобразования. Реакции образования мостиковой связи между тиолами, исследованные в более недавнее время, также основаны на этом принципе, где в иллюстративные реакции вовлечены малеимиды, бис-малеимиды и малеимиды с галогеновыми заместителями. К настоящему времени, однако, линкерные композиции, образующие тиольные мостиковые связи, ограничены только соединениями на основе малеимида и часто не предназначены для применения в ADC для таргетной терапии опухолей. Например, описанные ранее способы ковалентного связывания тиолов, включающие использование подобных соединений на основе малеимида, не предназначены для этих областей применения в сочетании с активными агентами, такими как нацеленные на опухоль молекулы лекарственных средств, белки или полипептиды (см., например, публикацию заявки на патент РСТ № WO 2013132268). В других способах, в которых конкретно раскрыты применения ADC в направленной доставке терапевтического препарата в область опухоли, где используют соединения на основе малеимида, не используют механизм ковалентной мостиковой связи с тиолами, в котором один линкер одновременно взаимодействует с двумя тиольными группами, как предложено в настоящем изобретении (см., например, публикацию заявки на патент Китая CN 103933575).
Краткое описание изобретения
В настоящем изобретении предложены новые и обладающие преимуществом линкеры для связывания антитела с другим соединением. В дополнение к линкерам, структурный синтез и применение которых описаны в настоящем документе, в заявленном изобретении также предложены конъюгаты антитела с активным агентом и области их применения, например, область применения в конъюгатах антитело-лекарственное средство.
В некоторых воплощениях изобретения предложен линкер-активный агент, имеющий структуру формулы I:
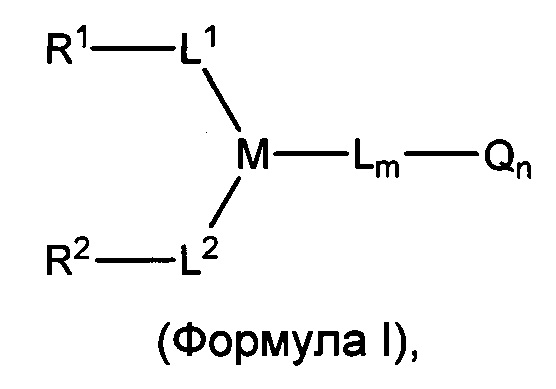
или его фармацевтически приемлемая соль,
где:
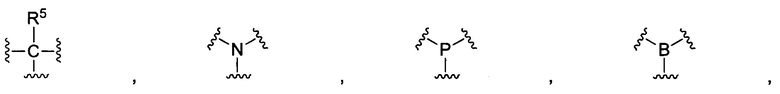
R1 и R2 представляют собой функциональные группы, которые могут взаимодействовать с тиолами;
L1, L2, L представляют собой линкер или могут отсутствовать;
М представляет собой линкерную группу;
Q представляет собой активный агент или может отсутствовать;
m представляет собой целое число от 0 до 6;
n представляет собой целое число от 0 до 8.
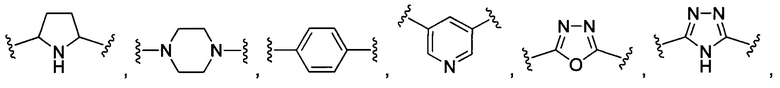
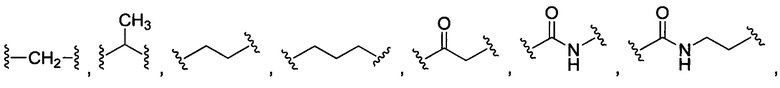
В некоторых воплощениях изобретения R1 и R2 независимо выбраны из 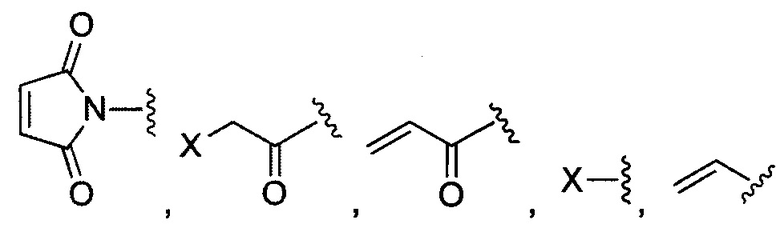 и их производных.
и их производных.
В некоторых воплощениях изобретения предложен конъюгат нацеливающей группировки с линкером-активным агентом, имеющий структуру формулы II:
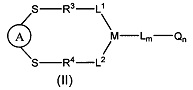
или его фармацевтически приемлемая соль,
А представляет собой нацеливающую группировку;
L1, L2, L представляют собой линкер или отсутствуют;
М представляет собой линкерную группу;
Q представляет собой активный агент или отсутствует;
m представляет собой целое число, выбранное из чисел от 0 до 6;
n представляет собой целое число, выбранное из чисел от 0 до 8;
каждый из R3 и R4 независимо выбран из:
 их производных или отсутствует.
их производных или отсутствует.
При использовании в настоящем документе ссылка на отсутствие означает, что объект не присутствует.
Краткое описание графических материалов
На Фиг. 1 схематично показана ковалентная мостиковая связь между тиолами, образованная в результате взаимодействия линкера с двумя соседними тиольными группами разорванной или восстановленной дисульфидной связи антитела.
На Фиг. 2 представлено изображение электрофореза в ДСН-ПААГ иллюстративного ADC с антителом к Her-2, содержащего моноклональное антитело к Her-2, конъюгированное с цитотоксином, полученного с использованием Mc-VC-PAB-MMAE.
На Фиг. 3 показана хроматограмма гидрофобной интерактивной хроматографии (ГИХ)-высокоэффективной жидкостной хроматографии (ВЭЖХ) MSL-C31.
На Фиг. 4 показаны результаты электрофореза MSL-C75 в ДСН-ПААГ в восстанавливающих условиях.
На Фиг. 5 показана хроматограмма ГИХ-ВЭЖХ MSL-C75.
На Фиг. 6 показан результат электрофореза в ДСН-ПААГ в восстанавливающих условиях для а) чистого антитела, b) CD59-C78.
На Фиг. 7 показана хроматограмма ГИХ-ВЭЖХ CD59-C78.
Подробное описание изобретения
Следующее ниже подробное описание приведено со ссылкой на сопроводительные графические материалы, на которых изображены не имеющие ограничительного характера и неисчерпывающие воплощения изобретения. Эти воплощения описаны достаточно подробно, чтобы обеспечить специалистам в данной области техники возможность осуществления изобретения на практике, и понятно, что можно использовать другие воплощения и выполнить другие изменения без отклонения от сущности или объема изобретения. Поэтому следующее ниже подробное описание не следует рассматривать в ограничительном смысле, и объем изобретения определен только прилагаемой формулой изобретения. Все патенты, заявки на патенты, предварительные заявки и публикации, относящиеся к настоящему документу или цитируемые в нем, полностью включены в него путем ссылки, включая все фигуры и таблицы, в той степени, в которой они явным образом не соответствуют положениям данного описания.
В настоящем изобретении предложены новые и обладающие преимуществом линкерные молекулы, способные к ковалентному связыванию с двумя свободными тиолами антитела посредством одного конца линкера и присоединению к активному агенту на другом конце линкера. В настоящем документе также предложены конъюгаты антитела с активным агентом, включающие, например, конъюгаты антитело-лекарственное средство (ADC). В некоторых воплощениях изобретения предложенные в настоящем документе ADC включают в себя противораковое лекарственное средство в качестве активного агента.
Определения
Ниже приведены используемые в настоящем документе сокращения распространенных органических соединений:
Термин «активный агент», используемый в соответствии с настоящим изобретением, включает любое природное или синтетическое вещество, оказывающее физиологическое действие при введении животному. Активный агент можно применять в соответствии с изобретением для лечения, например, теплокровных животных, в частности млекопитающих, включая людей, ветеринарных животных и сельскохозяйственных животных. Активный агент может действовать на желаемую мишень или может быть визуализирован на ней внутри или на поверхности тела животного, включая опухолевую ткань.
Неограничивающими примерами «активных агентов» являются лекарственные средства, действующие на синаптические области или области нейроэффекторных соединений; общие и локальные анестетики; снотворные и седативные средства; лекарственные средства для лечения расстройств психики, таких как депрессия и шизофрения; противоэпилептические и противосудорожные средства; лекарственные средства для лечения болезни Паркинсона и Гентингтона, возрастных изменений и болезни Альцгеймера; антагонисты возбуждающих аминокислот, нейротрофические факторы и нейрорегенеративные агенты; трофические факторы; лекарственные средства, нацеленные на лечение травмы центральной нервной системы (ЦНС) или инсульта; лекарственные средства для лечения бактериальных, вирусных и/или микробных инфекций, таких как грипп, ВИЧ, герпес, ветряная оспа и т.п.; антацидные и противовоспалительные лекарственные средства; химиотерапевтические агенты для лечения паразитарных инфекций и заболеваний, вызванных микроорганизмами; иммуносупрессивные средства; противораковые лекарственные средства; гормоны и антагонисты гормонов; тяжелые металлы и антагонисты тяжелых металлов; антагонисты неметаллических токсических агентов; цитостатические агенты; агенты визуализации и другие диагностические вещества; иммуноактивные и иммунореактивные агенты; трансмиттеры и соответствующие агонисты рецепторов и антагонисты рецепторов; ингибиторы транспорта; антибиотики; спазмолитики; антигистаминные средства; средства против тошноты; релаксанты; стимуляторы; смысловые и антисмысловые олигонуклеотиды; церебральные сосудорасширяющие средства; психотропные средства; противоманиакальные средства; сосудорасширяющие и сосудосуживающие средства; гипотензивные средства; лекарственные средства для лечения мигрени; гипергликемические и гипогликемические средства; минералы и нутритивные средства; средства против ожирения; анаболики; антигистаминные средства и их смеси.
«Антитело», также известное как иммуноглобулин, представляет собой крупный белок Y-образной формы, используемый иммунной системой для идентификации и нейтрализации чужеродных элементов, таких как бактерии и вирусы. Антитело имеет четыре полипептидные цепи, две идентичные тяжелые цепи и две идентичные легкие цепи, соединенные цистеиновыми дисульфидными связями.
«Моноклональное антитело» представляет собой моноспецифическое антитело, где все молекулы антитела идентичны, поскольку они вырабатываются идентичными иммунными клетками, которые все представляют собой клоны уникальной родительской клетки. Моноклональные антитела могут быть получены путем слияния клеток миеломы с клетками селезенки от мыши (или В-лимфоцитами от кролика), которая была иммунизирована желаемым антигеном, с последующей очисткой полученных в результате гибридом такими методами, как аффинная очистка. Рекомбинантные моноклональные антитела могут быть вероятнее получены в вирусах или дрожжевых клетках, чем у мышей, методами, включающими методы клонирования репертуара или фагового дисплея/дрожжевого дисплея, клонирования сегментов генов иммуноглобулинов с созданием библиотек антител с незначительно различающимися аминокислотными последовательностями, из которых могут быть получены антитела желаемой специфичности. Полученные в результате антитела можно производить в широком масштабе путем ферментации.
Химерные или гуманизированные антитела представляют собой антитела, содержащие комбинацию исходных (обычно мышиных) и человеческих последовательностей ДНК, используемых в процессе рекомбинации, например, таких, в которых ДНК мыши, кодирующая связывающий участок моноклонального антитела, объединена с продуцирующей антитело ДНК человека с образованием частично мышиного, частично человеческого моноклонального антитела. Полностью гуманизированные антитела получают с помощью трансгенных мышей (сконструированных методами генной инженерии для выработки человеческих антител) или библиотек фагового дисплея.
«Линкер» представляет собой группировку с двумя активными концами, одним для связывания или ассоциации иным путем биологической группировки или ее фрагмента, такой как антитело (или его фрагмент), а другим - для конъюгации с активными агентом, таким как цитотоксин.
«Цитотоксин» представляет собой соединение, которое в присутствии клетки, такой как раковая клетка, является токсичным или индуцирует ключевые функциональные изменения в этой клетке.
При использовании в настоящем документе «алкил» относится к прямоцепочечной или разветвленной углеводородной цепи, которая является полностью насыщенной (т.е. не содержит двойные или тройные связи). Алкильная группа может иметь от 1 до 9 атомов углерода (везде, где он встречается в данном документе, числовой диапазон, такой как «от 1 до 9», относится к каждому целому числу в данном диапазоне; например, «от 1 до 9 атомов углерода» означает, что алкильная группа может содержать 1 атом углерода, 2 атома углерода, 3 атома углерода и т.д., вплоть до 9 атомов углерода включительно, хотя настоящее определение также охватывает случаи, когда термин «алкил» встречается без обозначения числового диапазона). Алкильная группа может также представлять собой алкил среднего размера, имеющий от 1 до 9 атомов углерода. Типичные алкильные группы включают, но никоим образом не ограничены ими, метил, этил, пропил, изопропил, бутил, изобутил, третичный бутил, пентил, гексил и т.п.
При использовании в настоящем документе «алкенил» относится к прямоцепочечной или разветвленной углеводородной цепи, содержащей одну или более двойных связей. Алкенильная группа может иметь от 2 до 9 атомов углерода, хотя настоящее определение также охватывает случаи, где термин «алкенил» встречается без обозначения числового диапазона. Алкенильная группа может также представлять собой алкенил среднего размера, имеющий от 2 до 9 атомов углерода. Алкенильная группа может также представлять собой низший алкенил, имеющий от 2 до 4 атомов углерода. Алкенильная группа может быть обозначена как «С2-4 алкенил» или подобными обозначениями. Только в качестве примера «С2-4 алкенил» указывает на наличие от двух до четырех атомов углерода в алкенильной цепи, т.е. алкенильная цепь выбрана из этенила, пропен-1-ила, пропен-2-ила, пропен-3-ила, бутен-1-ила, бутен-2-ила, бутен-3-ила, бутен-4-ила, 1-метил-пропен-1-ила, 2-метил-пропен-1-ила, 1-этил-этен-1-ила, 2-метил-пропен-3-ила, бута-1,3-диенила, бута-1,2-диенила и бута-1,2-диен-4-ила. Типичные алкенильные группы включают, но никоим образом не ограничены ими, этенил, пропенил, бутенил, пентенил и гексенил и т.п.
При использовании в настоящем документе «алкинил» относится к прямоцепочечной или разветвленной углеводородной цепи, содержащей одну или более тройных связей. Алкинильная группа может иметь от 2 до 9 атомов углерода, хотя настоящее определение также охватывает случаи, где термин «алкинил» встречается без обозначения числового диапазона. Алкинильная группа может также представлять собой алкинил среднего размера, имеющий от 2 до 9 атомов углерода. Алкинильная группа может также представлять собой низший алкинил, имеющий от 2 до 4 атомов углерода. Алкинильная группа может быть обозначена как «С2-4 алкинил» или подобными обозначениями. Только в качестве примера «С2-4 алкинил» указывает на наличие от двух до четырех атомов углерода в алкинильной цепи, т.е. алкинильная цепь выбрана из этинила, пропин-1-ила, пропин-2-ила, бутин-1-ила, бутин-3-ила, бутин-4-ила и 2-бутинила. Типичные алкинильные группы включают, но никоим образом не ограничены ими, этинил, пропинил, бутинил, пентинил и гексинил и т.п.
Термин «ароматический» относится к кольцу или системе колец, имеющих конъюгированную π-электронную систему, и включает как карбоциклические ароматические (например, фенил), так и гетероциклические ароматические группы (например, пиридин). Этот термин включает моноциклические или полициклические группы с конденсированными кольцами (т.е. кольцами, имеющими общие соседние пары атомов) при условии, что полная кольцевая система является ароматической.
При использовании в настоящем документе «циклоалкил» означает полностью насыщенное карбоциклическое кольцо или систему колец. Примеры включают без ограничений циклопропил, циклобутил, циклопентил и циклогексил.
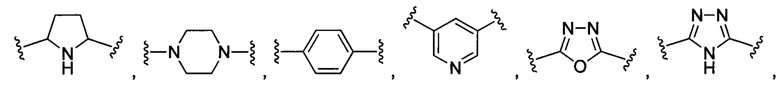
При использовании в настоящем документе «гетероарил» относится к ароматическому кольцу или системе колец (т.е. к двум или более конденсированных колец, имеющих два общих соседних атома), которые содержат один или более гетероатомов, то есть элементов, отличающихся от атомов углерода, включающих без ограничений атом азота, кислорода и серы в кольцевой структуре. Когда гетероарил представляет собой систему колец, каждое кольцо в системе является ароматическим. Гетероарильная группа может иметь 5-18 атомов-членов кольца (т.е. число атомов, составляющих кольцевую структуру, включая атомы углерода и гетероатомы), хотя настоящее определение также охватывает термин «гетероарил» без указания числового диапазона. Примеры гетероарильных колец включают без ограничений фурил, тиенил, фталазинил, пирролил, оксазолил, тиазолил, имидазолил, пиразолил, изоксазолил, изотиазолил, триазолил, тиадиазолил, пиридинил, пиридазинил, пиримидинил, пиразинил, триазинил, хинолинил, изохинолинил, бензимидазолил, бензоксазолил, бензотиазолил, индолил, изоиндолил и бензотиенил.
При использовании в настоящем документе гетероциклил означает неароматическое циклическое кольцо или систему колец, содержащие по меньшей мере один гетероатом в кольцевой структуре. Гетероциклилы могут быть соединены вместе в конденсированной, мостиковой или спиро-соединенной системе. Гетероциклилы могут иметь любую степень насыщенности при условии, что по меньшей мере одно кольцо в системе колец является неароматическим. Гетероатом(-ы) может(-гут) присутствовать либо в неароматическом, либо в ароматическом кольце в системе колец. Гетероциклильная группа может иметь от 3 до 20 атомов-членов кольца (т.е. число атомов, составляющих кольцевую структуру, включая атомы углерода и гетероатомы), хотя настоящее определение также охватывает термин «гетероциклил» без указания числового диапазона. Гетероциклильная группа может также представлять собой гетероциклил среднего размера, имеющий от 3 до 10 членов кольца. Гетероциклильная группа может также представлять собой гетероциклил, имеющий от 3 до 6 членов кольца. Примеры гетероциклильных колец включают без ограничений азепинил, акридинил, карбазолил, циннолинил, диоксоланил, имидазолинил, имидазолидинил, морфолинил, оксиранил, оксепанил, тиепанил, пиперидинил, пиперазинил, диоксопиперазинил, пирролидинил, пирролидонил, пирролидинонил, 4-пиперидонил, пиразолинил, пиразолидинил, 1,3-диоксинил, 1,3-диоксанил, 1,4-диоксинил, 1,4-диоксанил, 1,3-оксатионил, 1,4-оксатиинил, 1,4-оксатианил, 2Н-1,2-оксазинил, триоксанил, гексагидро-1,3,5-триазинил, 1,3-диоксолил, 1,3-диоксоланил, 1,3-дитиолил, 1,3-дитиоланил, изоксазолинил, изоксазолидинил, оксазолинил, оксазолидинил, оксазолидинонил, тиазолинил, тиазолидинил, 1,3-оксатиоланил, индолинил, изоиндолинил, тетрагидрофуранил, тетрагидропиранил, тетрагидротиофенил, тетрагидротиопиранил, тетрагидро-1,4-тиазинил, тиаморфолинил, дигидробензофуранил, бензимидазолидинил и тетрагидрохинолин.
(Гетероциклил)алкил представляет собой гетероциклильную группу, соединенную в качестве заместителя посредством алкиленовой группы. Примеры включают без ограничений имидазолинилметил ииндолинилэтил.
Линкер/активный агент
В одном аспекте в настоящем изобретении предложен линкер-активный агент, который может быть представлен формулой I:
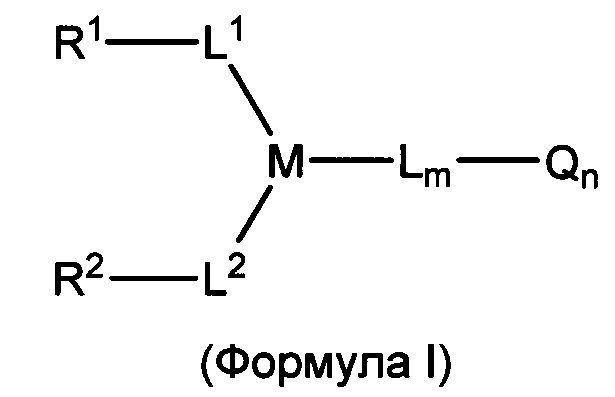
или его фармацевтически приемлемая соль,
где:
L1, L2, L представляют собой линкер или отсутствуют;
М представляет собой линкерную группу;
Q представляет собой активный агент или отсутствует;
m представляет собой целое число, выбранное из чисел от 0 до 6;
n представляет собой целое число, выбранное из чисел от 0 до 8;
каждый из R1 и R2 представляют собой функциональную группу, которая может взаимодействовать с тиолом.
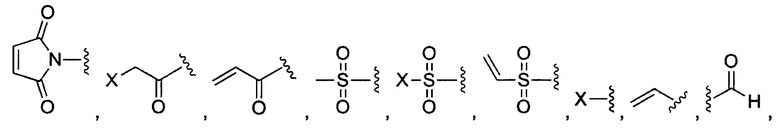
В некоторых воплощениях изобретения каждая из двух функциональных групп, взаимодействующих с тиолом, может независимо содержать малеимид, атом галогена, галоген-замещенные функциональные группы, алканаль, алканон, сульфонил-алкен, силан, изоцианат или норборнен, но обе они не могут представлять собой малеимид, и обе не могут представлять собой атом галогена.
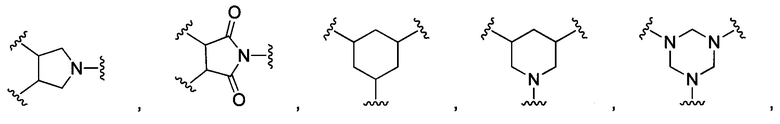
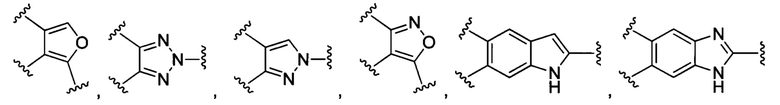
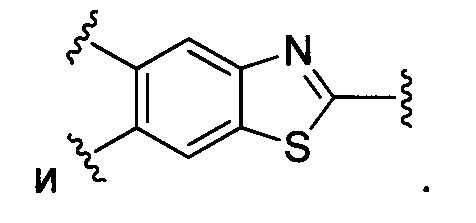
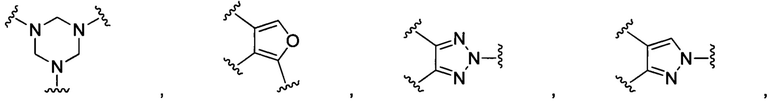
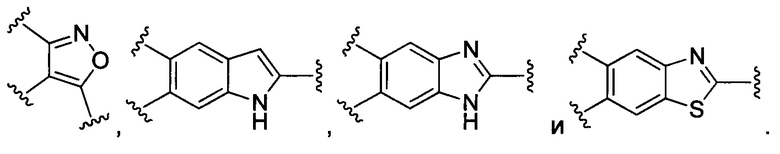
В конкретных воплощениях изобретения каждый из R1 и R2 независимо выбран из:
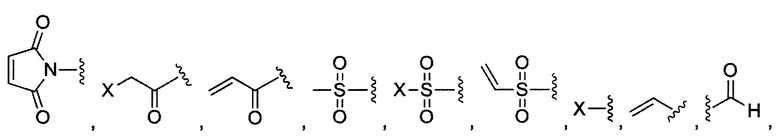
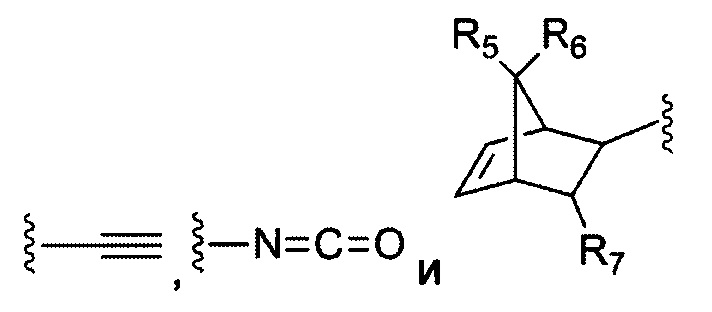
и их производных;
где в данном воплощении изобретения R5, R6 и R7 независимо выбраны из Н, атома галогена, С1-С6 алкилов, С2-С6 алкенилов, С2-С6 алкинилов, ароматических групп, гетероарилов, С3-С9 циклоалкилов, С3-С9 гетероциклила, полиэтиленгликоля, NO2, CN, SCN, OR8, SR9, NR10R11, C(=O)R12, C(=O)OR8, C(=O)NR10R11, C(=S)OR8, C(=S)NR10R11, C(=S)SR9, NR10(C=O)R12, NR10(C=S)NR10R11, O(C=O)NR10R11, SO2R9, S(=O)2OR8 и их комбинаций, где R8, R9, R10, R11, R12 независимо выбраны из H, С1-С6 алкила, С2-С6 алкенилов, С2-С6 алкинилов и их комбинаций.
X представляет собой атом галогена; в некоторых воплощениях изобретения X может представлять собой F, Cl, Br или I;
R1 и R2 могут быть одинаковыми или разными;
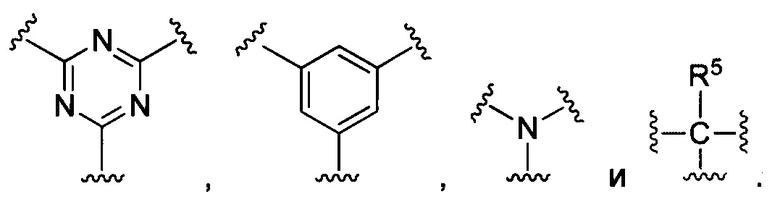
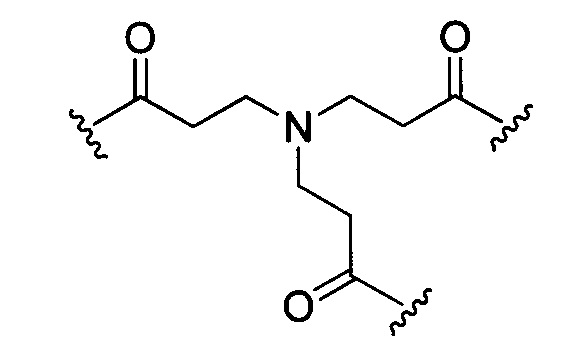
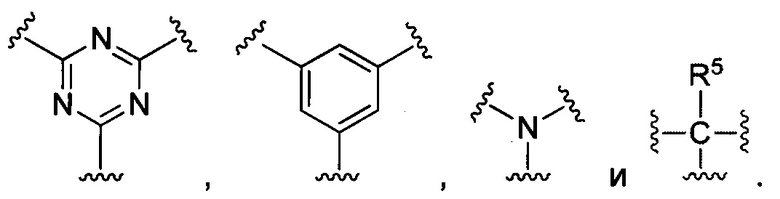
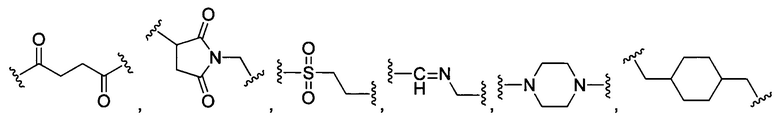
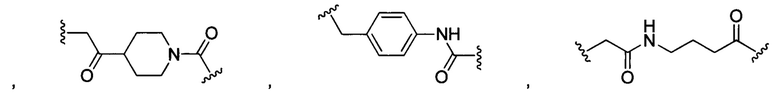
В некоторых воплощениях изобретения М выбран, например, из C-R5, N, В, Р, Si-R5, ароматических групп, гетероарилов, циклоалкилов и (гетероциклил)алкилов, R5 выбран из Н, C1-C6 алкилов, С2-С6 алкенилов и С2-С6 алкинилов.
В других воплощениях изобретения М выбран из группы, состоящей из
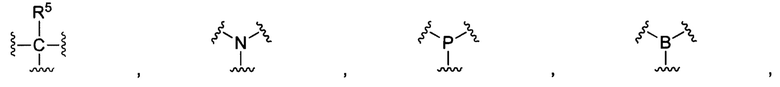
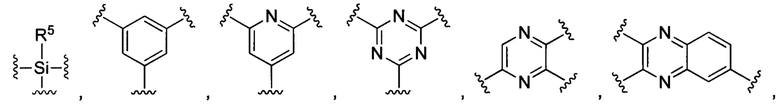
В некоторых воплощениях изобретения М выбран из 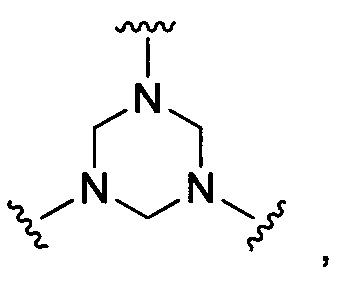
В некоторых воплощениях изобретения Q представляет собой активный агент, выбранный из группы, состоящей из агентов, связывающих тубулин, алкилирующих агентов ДНК, интеркаляторов ДНК, ингибиторов ферментов, иммуномодуляторов, пептидов и нуклеотидов.
В некоторых воплощениях изобретения Q выбран из группы, состоящей из майтанзиноидов, ауристатинов, калихеамицинов, доксорубицинов, дуокармицинов и пирролобензодиазепинов (PBD).
В некоторых воплощениях изобретения Q выбран из ММАЕ, MMAF, димера PBD, DM1 и DM4. Активный агент может представлять собой, например, любой из агентов, раскрытых в WO 2013085925 А1, которая полностью включена в настоящий документ посредством ссылки. «Лекарственное средство» и «активный агент» используют в настоящем документе взаимозаменяемо.
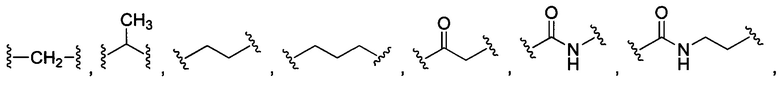
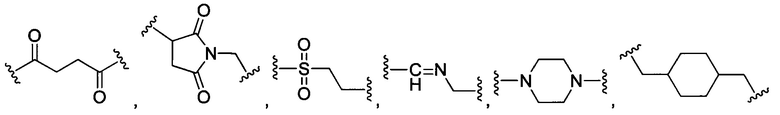
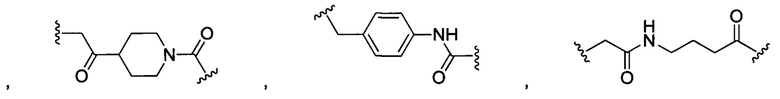
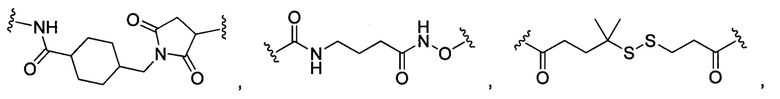
В некоторых воплощениях изобретения каждая из внутренних линкерных группировок L1, L2 независимо выбрана из С1-С6 алкилов, С2-С6 алкенилов, С2-С6 алкинилов, ароматических групп, гетероарилов, С3-С9 циклоалкилов, С3-С9 гетероциклила, полиэтиленгликоля, О, S, NR6, С(=O), С(=O)O, C(=O)NR6, C=NR6, C(=S)O, C(=S)NR6, C(=S)S, NR6(C=O), NR6(C=S)NR7, O(C=O)NR6, S(=O)2 и любой их комбинации;
где в данном воплощении изобретения R6 и R7 независимо выбраны из Н, C1-C6 алкилов, С2-С6 алкенилов и С2-С6 алкинилов.
В других воплощениях изобретения каждый из L1, L2 независимо выбран из:
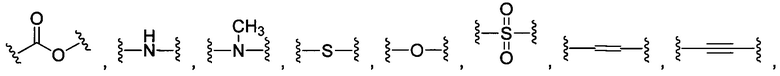
 или отсутствует.
или отсутствует.
В некоторых воплощениях изобретения каждый из L1, L2 независимо выбран из: 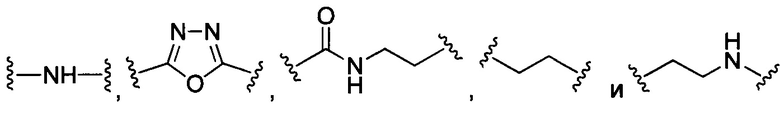 или отсутствует.
или отсутствует.
L представляет собой линкер, соединяющий активный агент Q с линкерной группой М. В некоторых воплощениях изобретения L включает нерасщепляемое звено, в других воплощениях изобретения L включает расщепляемое звено. Конъюгат линкер-активный агент может иметь один линкер, два линкера, три линкера, четыре линкера, пять линкеров или шесть линкеров.
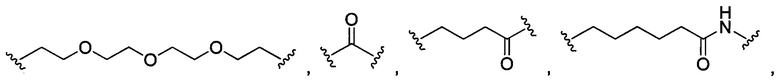
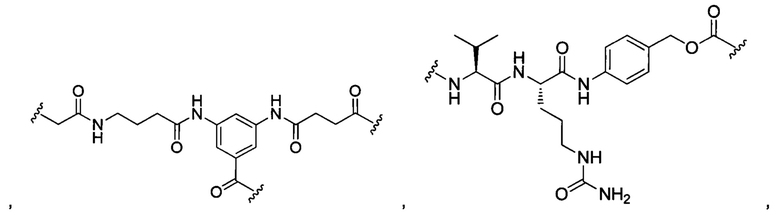
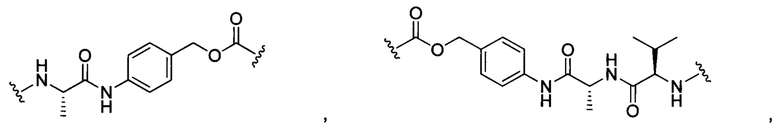
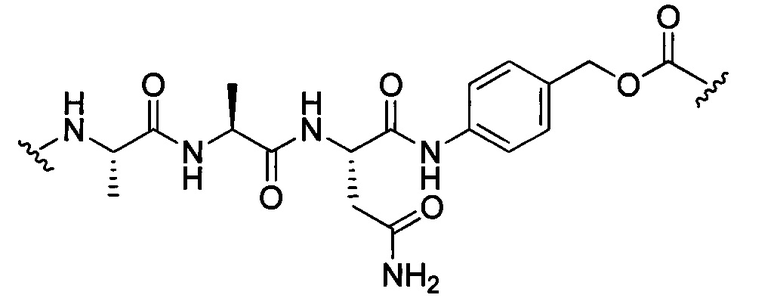
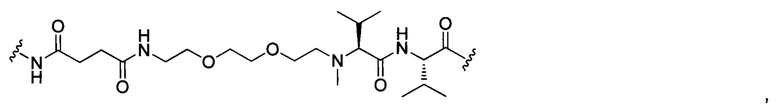
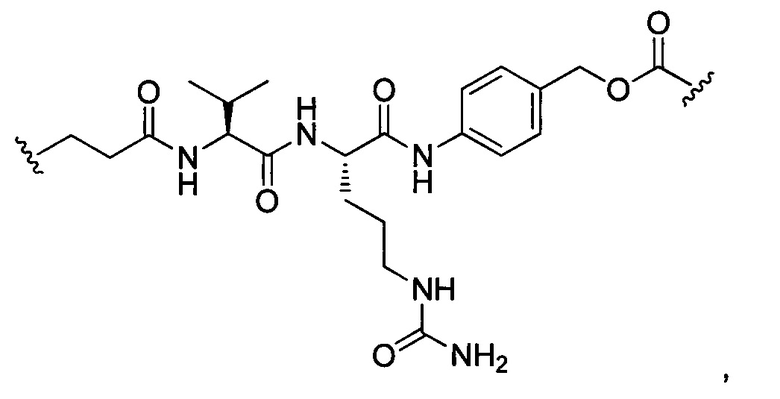
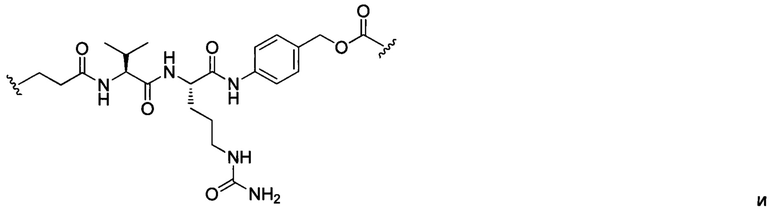
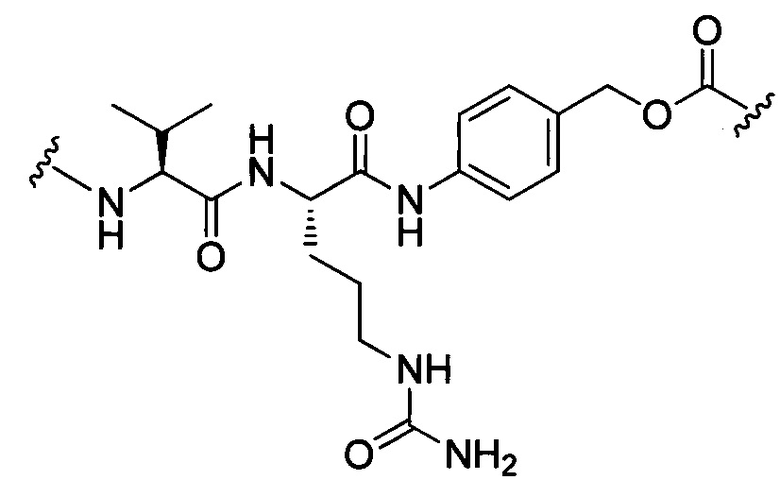
В некоторых воплощениях изобретения L независимо выбран из C1-C9 алкилов, С2-С9 алкенилов, С2-С9 алкинилов, ароматических групп, гетероарилов, С3-С9 циклоалкилов, С3-С9 гетероциклила, полиэтиленгликоля, О, S, NR8, С(=O), С(=O)O, C(=O)NR8, C=NR8, C(=S)O, C(=S)NR8, C(=S)S, NR8(C=O), NR8(C=S)NR9, O(C=O)NR8, S(=O)2, Val-Cit-PAB, Val-Ala-PAB, Val-Lys(Ac)-PAB, Phe-Lys-PAB, Phe-Lys(Ac)-PAB, D-Val-Leu-Lys, Gly-Gly-Arg, Ala-Ala-Asn-PAB, Ala-PAB, PAB и любой их комбинации.
В данном воплощении изобретения R8 и R9 независимо выбраны из Н, C1-C6 алкилов, С2-С6 алкенилов и С2-С6 алкинилов.
В некоторых воплощениях изобретения L отсутствует. Когда L отсутствует, активный агент Q соединен непосредственно с М.
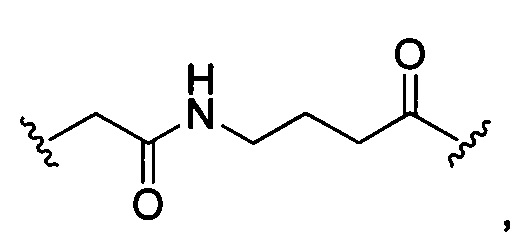
В некоторых воплощениях изобретения L независимо выбран из группы, состоящей из:


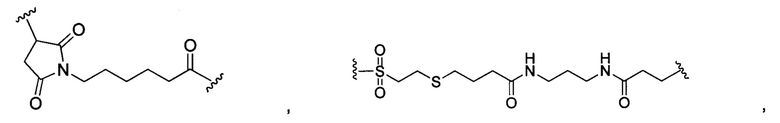
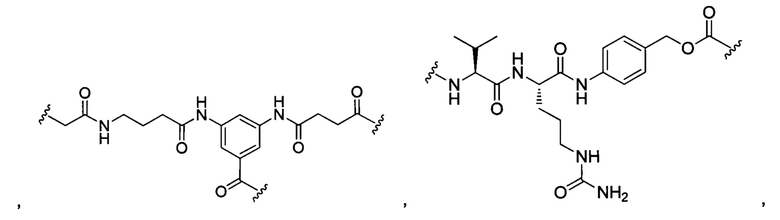
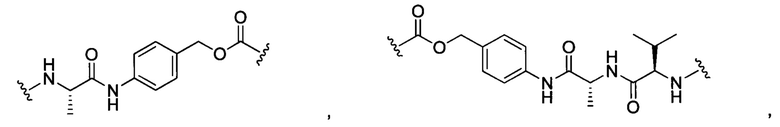
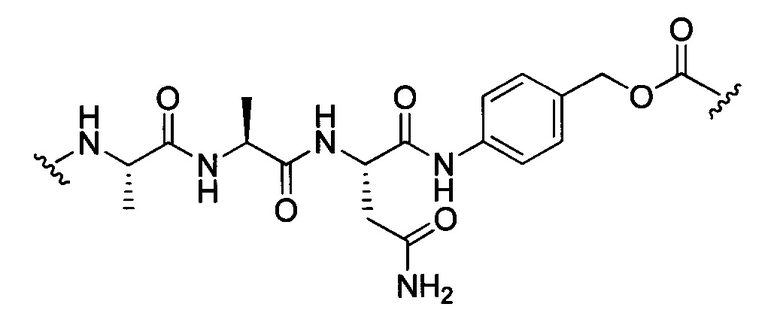
 или отсутствует.
или отсутствует.
В некоторых воплощениях изобретения L независимо выбран из следующих групп:
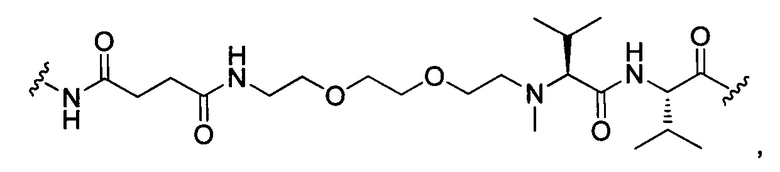
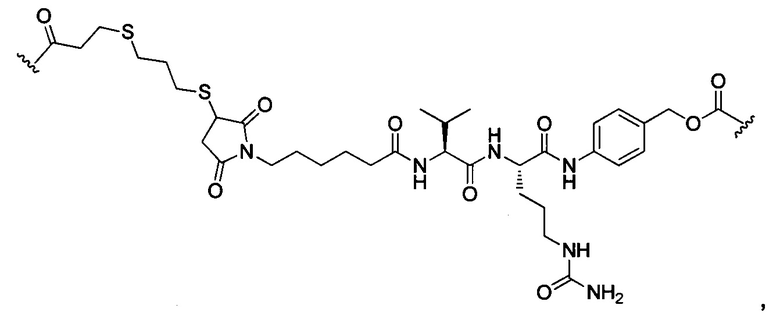
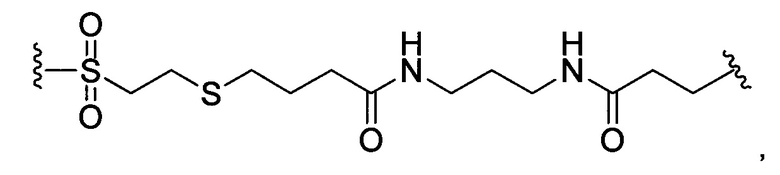
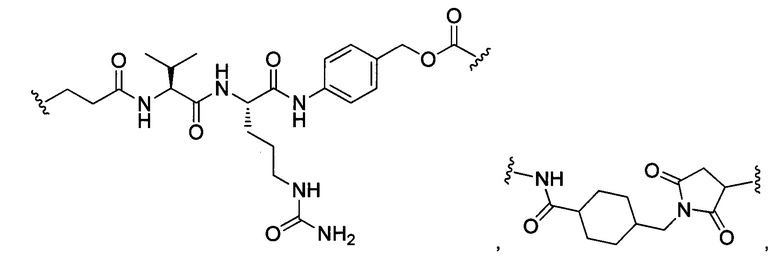
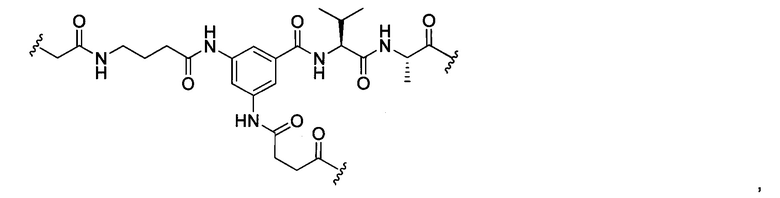
 или отсутствует.
или отсутствует.
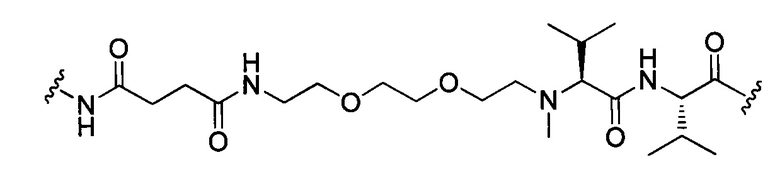
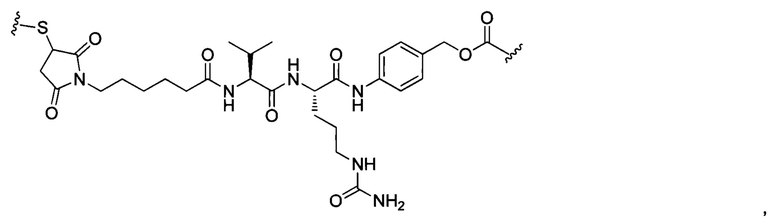
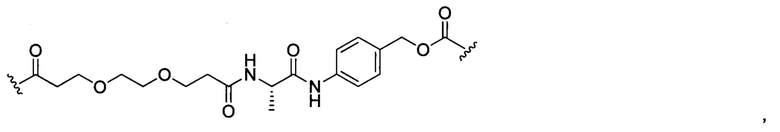
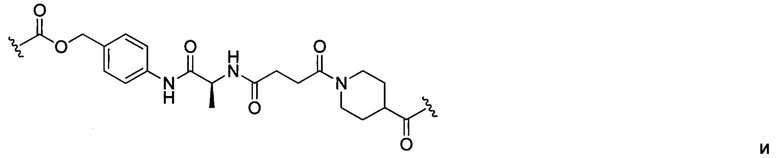
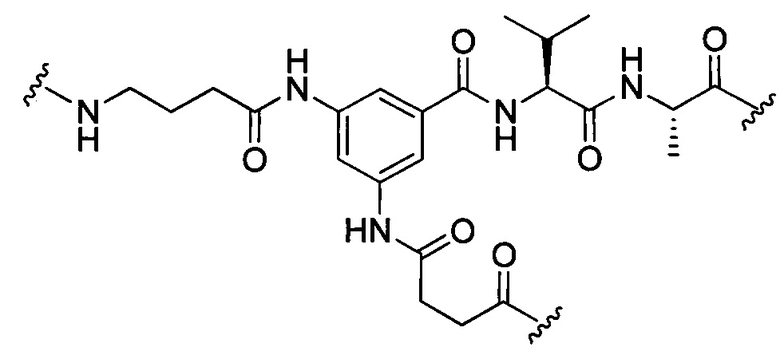
В некоторых воплощениях изобретения конъюгат линкер-активный агент имеет следующую структуру формулы I:
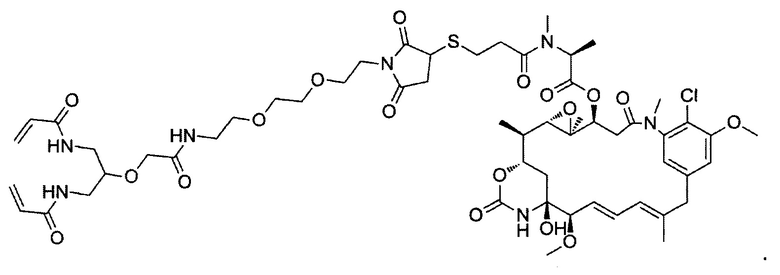
В некоторых воплощениях изобретения конъюгат линкер-активный агент имеет следующую структуру формулы I:
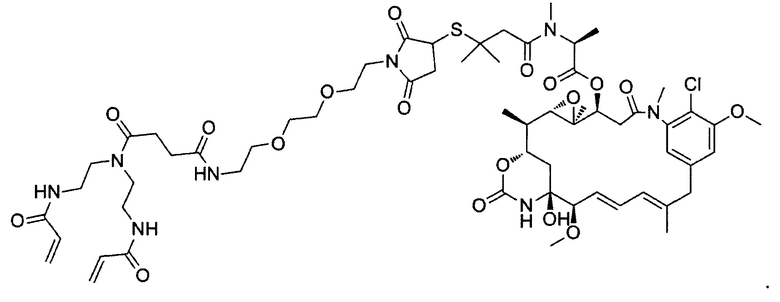
В некоторых воплощениях изобретения конъюгат линкер-активный агент имеет следующую структуру формулы I:
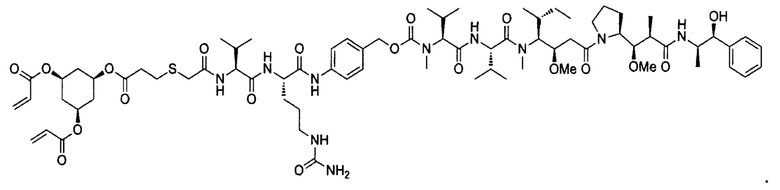
В некоторых воплощениях изобретения конъюгат линкер-активный агент имеет следующую структуру формулы I:
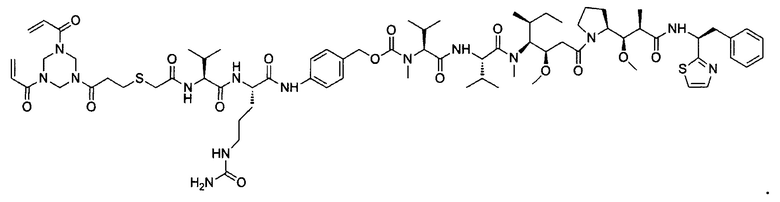
В некоторых воплощениях изобретения конъюгат линкер-активный агент имеет следующую структуру формулы I:
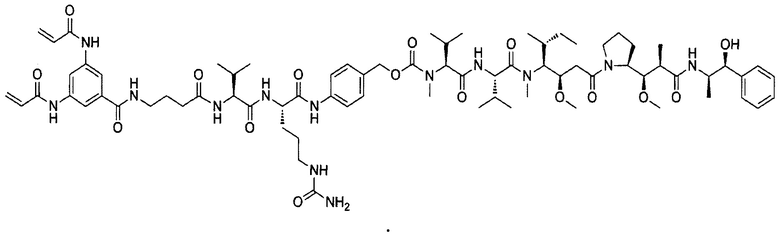
В некоторых воплощениях изобретения конъюгат линкер-активный агент имеет следующую структуру формулы I:
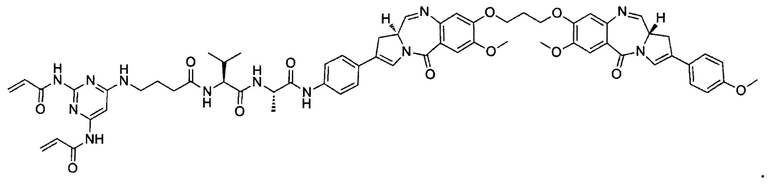
В некоторых воплощениях изобретения конъюгат линкер-активный агент имеет следующую структуру формулы I:
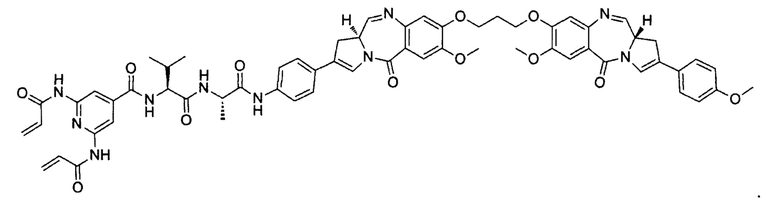
В некоторых воплощениях изобретения конъюгат линкер-активный агент имеет следующую структуру формулы I:
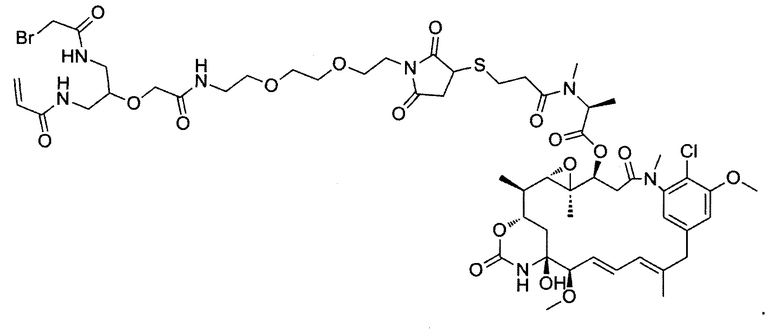
В некоторых воплощениях изобретения конъюгат линкер-активный агент имеет следующую структуру формулы I:
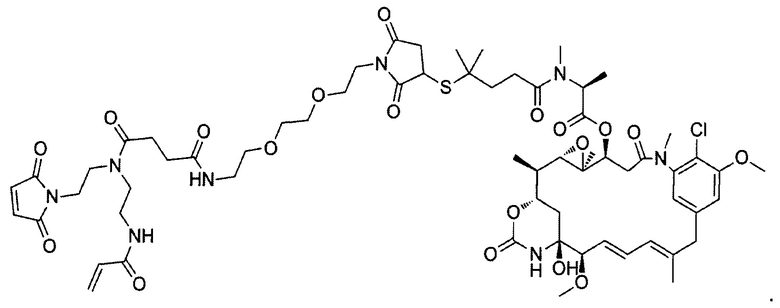
В некоторых воплощениях изобретения конъюгат линкер-активный агент имеет следующую структуру формулы I:
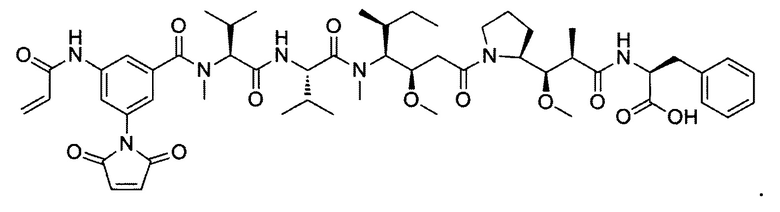
В некоторых воплощениях изобретения конъюгат линкер-активный агент имеет следующую структуру формулы I:
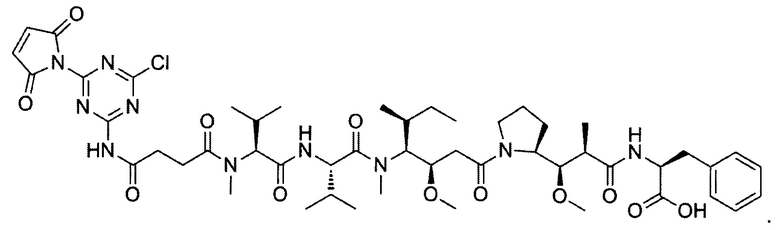
В некоторых воплощениях изобретения конъюгат линкер-активный агент имеет следующую структуру формулы I:
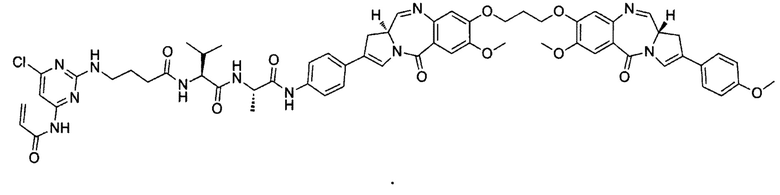
В другом аспекте в настоящем изобретении предложен конъюгат антитело-лекарственное средство, имеющий следующую формулу II:
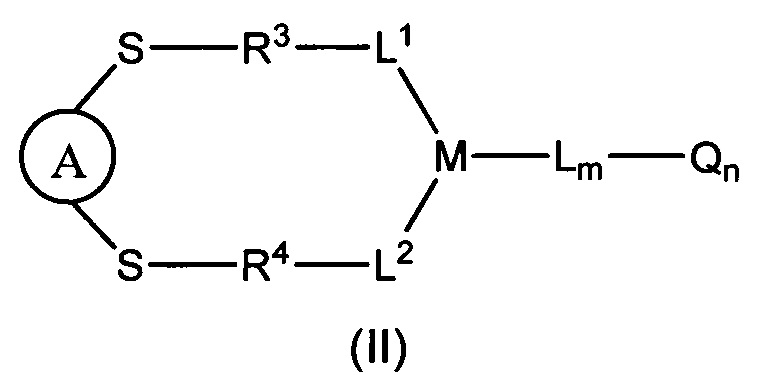
или его фармацевтически приемлемая соль,
А представляет собой нацеливающую группировку;
S представляет собой атом серы тиольной группы нацеливающей группировки;
L1, L2, L представляют собой линкер или отсутствуют;
М представляет собой линкерную группу;
Q представляет собой активный агент или отсутствует;
m представляет собой целое число, выбранное из чисел от 0 до 6;
n представляет собой целое число, выбранное из чисел от 0 до 8; и
каждый из R3 и R4 образован в результате взаимодействия между S и функциональной группой, которая может взаимодействовать с тиолами.
В некоторых воплощениях изобретения функциональные группы, взаимодействующие с тиолом, могут независимо содержать малеимид, атом галогена, галоген-замещенные функциональные группы, алканаль, алканон, сульфонил-алкен, силан, изоцианат или норборнен, но обе они не могут представлять собой малеимид, и обе не могут представлять собой атом галогена.
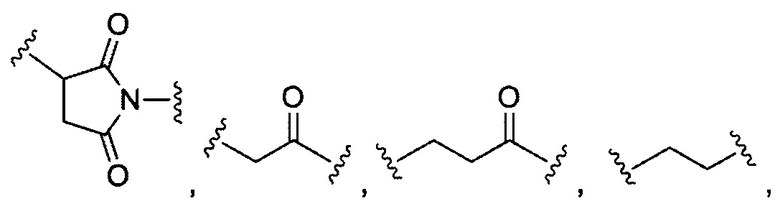
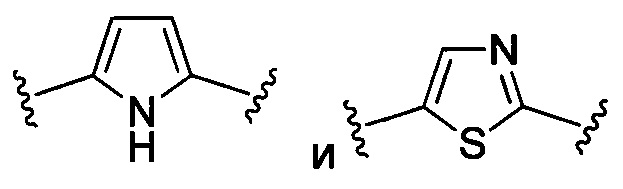
В некоторых воплощениях изобретения каждый из R3 и R4 образуется независимо в результате взаимодействия между тиольной группой и группой, выбранной из
 и их производных.
и их производных.
где в данном воплощении изобретения R5, R6 и R7 независимо выбраны из Н, атом галогена, C1-C6 алкилов, С2-С6 алкенилов, С2-С6 алкинилов, ароматических групп, гетероарилов, С3-С9 циклоалкилы, С3-С9 гетероциклил, полиэтиленгликоль, NO2, CN, SCN, OR8, SR9, NR10R11, C(=O)R12, C(=O)OR8, C(=O)NR10R11, C(=S)OR8, C(=S)NR10R11, C(=S)SR9, NR10(C=O)R12, NR10(C=S)NR10R11, O(C=O)NR10R11, SO2R9, S(=O)2OR8 и их комбинации, где R8, R9, R10, R11, R12 независимо выбраны из H, С1-С6 алкила, С2-С6 алкенилов, С2-С6 алкинилов и их комбинаций.
В некоторых воплощениях изобретения R3 и R4 имеют 10 или менее, предпочтительно 5 или менее атомов углерода, являются циклическими и/или содержат кетоновую группу.
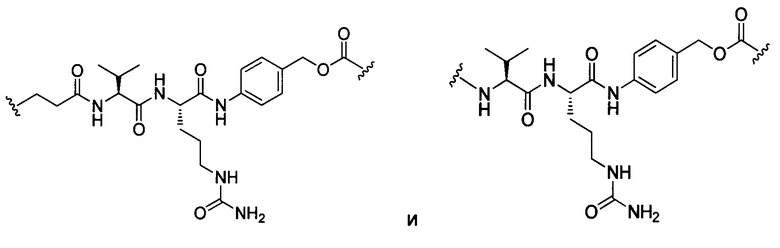
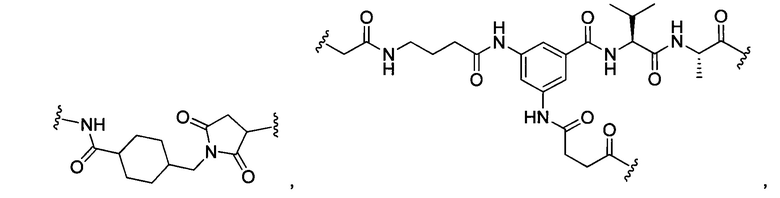
В конкретных воплощениях изобретения каждый из R3 и R4 независимо выбран из: 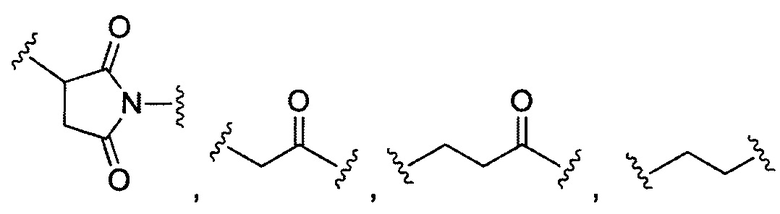 и их производных. В одном воплощении изобретения R3 и R4 способны связывать 2 сульфгидрильные группы и образовывать легкие-тяжелые цепи антитела более стабильно.
и их производных. В одном воплощении изобретения R3 и R4 способны связывать 2 сульфгидрильные группы и образовывать легкие-тяжелые цепи антитела более стабильно.
Когда R3 или R4 отсутствует, L1 или L2 конъюгированы непосредственно с нацеливающей группировкой.
R3 и R4 могут быть одинаковыми или разными.
Только R3 представляет собой 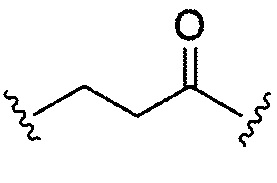 , R3 и R4 могут быть одинаковыми.
, R3 и R4 могут быть одинаковыми.
В некоторых воплощениях изобретения М выбран, например, из C-R5, N, В, Р, Si-R5, ароматических групп, гетероарилов, циклоалкилов и (гетероциклил)алкилов, R5 выбран из Н, С1-С6 алкилов, С2-С6 алкенилов и С2-С6 алкинилов. В других воплощениях изобретения М выбран из группы, состоящей из
В некоторых воплощениях изобретения М выбран из 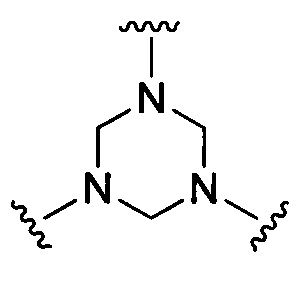 ,
, 
В некоторых воплощениях изобретения Q представляет собой активный агент, выбранный из группы, состоящей из агентов, связывающих тубулин, алкилирующих агентов ДНК, интеркаляторов ДНК, ингибиторов ферментов, иммуномодуляторов, пептидов и нуклеотидов. «Лекарственное средство» и «активный агент» используют в настоящем документе взаимозаменяемо.
В некоторых воплощениях изобретения Q выбран из группы, состоящей из майтанзиноидов, ауристатинов, калихеамицинов, доксорубицинов, дуокармицинов и PBD.
В некоторых воплощениях изобретения Q выбран из ММАЕ, MMAF, PBD, DM1 и DM4. Активный агент может представлять собой, например, любой из агентов, раскрытых в WO 2013085925 А1, которая полностью включена в настоящий документ посредством ссылки.
В некоторых воплощениях изобретения каждая из внутренних сшивающих группировок L1, L2 независимо выбрана из С1-С6 алкилов, С2-С6 алкенилов, С2-С6 алкинилов, ароматических групп, гетероарилов, С3-С9 циклоалкилов, С3-С9 гетероциклила, полиэтиленгликоля, О, S, NR6, С(=O), С(=O)O, C(=O)NR6, C=NR6, C(=S)O, C(=S)NR6, C(=S)S, NR6(C=O), NR6(C=S)NR7, O(C=O)NR6, S(=O)2 и любой их комбинации;
где в данном воплощении изобретения R6 и R7 независимо выбраны из Н, С1-С6 алкилов, С2-С6 алкенилов и С2-С6 алкинилов.
В других воплощениях изобретения каждый из L1 и L2 независимо выбран из:
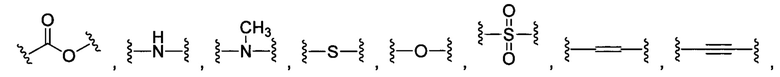
 или отсутствует.
или отсутствует.
В некоторых воплощениях изобретения каждый из L1 и L2 независимо выбран из: 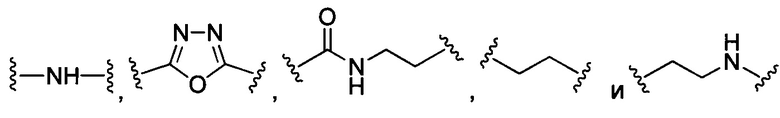 или отсутствует.
или отсутствует.
L представляет собой линкер, соединяющий лекарственное средство Q с линкерной группой М. В некоторых воплощениях изобретения L включает нерасщепляемое звено, в других воплощениях изобретения L включает расщепляемое звено. Конъюгат антитело-лекарственное средство может иметь один линкер, два линкера, три линкера, четыре линкера, пять линкеров или шесть линкеров.
В некоторых воплощениях изобретения L независимо выбран из С1-С9 алкилов, С2-С9 алкенилов, С2-С9 алкинилов, ароматических групп, гетероарилов, С3-С9 циклоалкилов, С3-С9 гетероциклила, полиэтиленгликоля, О, S, NR8, С(=O), С(=O)O, C(=O)NR8, C=NR8, C(=S)O, C(=S)NR8, C(=S)S, NR8(C=O), NR8(C=S)NR9, O(C=O)NR8, S(=O)2, Val-Cit-PAB, Val-Ala-PAB, Val-Lys(Ac)-PAB, Phe-Lys-PAB, Phe-Lys(Ac)-PAB, D-Val-Leu-Lys, Gly-Gly-Arg, Ala-Ala-Asn-PAB, Ala-PAB, PAB и любой их комбинации;
где в данном воплощении изобретения R8 и R9 независимо выбраны из Н, C1-C6 алкилов, С2-С6 алкенилов и С2-С6 алкинилов.
В некоторых воплощениях изобретения L отсутствует. Когда L отсутствует, активный агент Q соединен непосредственно с М.
В некоторых воплощениях изобретения L независимо выбран из группы, состоящей из:

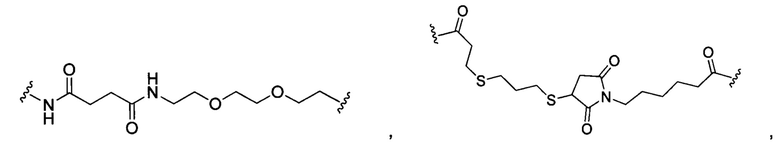
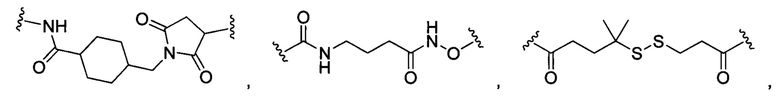
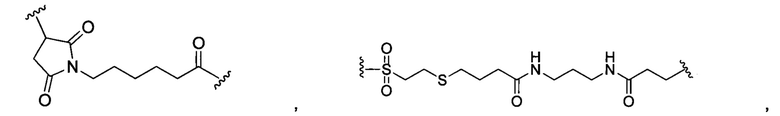
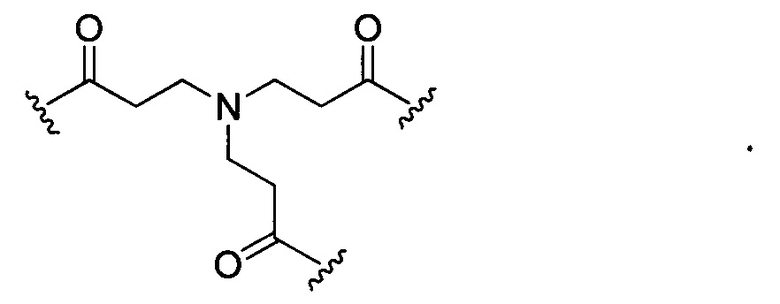
или отсутствует.
В некоторых воплощениях изобретения L независимо выбран из следующих групп: 

или отсутствует.
А представляет собой нацеливающую группировку. В некоторых воплощениях изобретения А представляет собой антитело; фрагмент, имитатор или вариант антитела; белковый лиганд; белковый каркас; пептид или низкомолекулярный лиганд. Фрагмент антитела может представлять собой, например, Fab, Fab', (Fab')2 или scFv, Fv.
Примеры опухолеассоциированных антигенов, которые могут быть нацеленными в соответствии с заявленным изобретением, включают без ограничений опухолеассоциированные антигены (1)-(36), перечисленные ниже. Приведено название, альтернативное название и номер доступа по GenBank каждого из приведенных примеров антигена. Нуклеиново-кислотные и белковые последовательности, соответствующие этим примерам опухолеассоциированных антигенов, можно найти в публично доступных базах данных, таких как GenBank.
(1) BMPR1B (рецептор типа 1В костного морфогенетического белка, номер доступа GenBank NM_001203);
(2) Е16 (LAT1, SLC7A5, номер доступа GenBank bandit _00: 3486);
(3) STEAPI (эпителиальный антиген простаты с шестью трансмембранными доменами, номер доступа GenBank NM_012449);
(4) 0772Р (СА125, MUC16, номер доступа GenBank AF361486);
(5) MPF (MPF, MSLN, SMR, фактор стимуляции мегакариоцитов, мезотелин, номер доступа GenBank NM_005823);
(6) Napi3b (NAP1-3B, NPTIIb, SLC34A2, член (фосфат натрия) член 2 семейства 34 растворимых носителей, натрийзависимый переносчик фосфатов 3b II типа, номер доступа GenBank NM_006424);
(7) Sema5b (FLJ10372, KIAA1445, Mm.42015, SEMA5B, SEMAG, семафорин 5b Hlog, сема-домен, трансмембранный домен (TM) и короткий цитоплазматический домен с семью тромбоспондиновыми повторами (I типа и II типа), (семафорин) 5b, номер доступа GenBank АВ040878);
(8) ген PSCA hlg (2700050C12Rik, C530008016Rik, RIKEN CDNA2700050C12, RIKENcDNA2700050C12, номер доступа GenBank AY358628);
(9) ETBR (рецептор эндотелина В, номер доступа GenBank AY275463);
(10) MSG783 (RNF124, псевдобелок FLJ20315, номер доступа GenBank NM_017763);
(11) STEAP2 (HGNC_8639, IPCA-1, PCANAP I, STAMP I, STEAP2, STMP, гены I, связанные с раком простаты, белок I, ассоциированный с раком простаты, эпителиальный антиген 2 простаты с шестью трансмембранными доменами, белок простаты с шестью трансмембранными доменами, номер доступа GenBank AF455138);
(12) TrpM4 (BR22450, FLJ20041, TRPM4, TRPM4B, катионный канал, действующий по механизму транзиторного рецепторного потенциала, подсемейство М, член 4, номер доступа GenBank NM_017636);
(13) CRIPTO (CR, CRI, CRGF, CRIPTO, TDGFI, фактор роста тератомы, номер доступа GenBank NP_003203 или NM_003212);
(14) CD21 (CR2 (рецептор комплемента типа 2) или C3DR (C3d/рецептор вируса ЕВ) или Hs.73792, номер доступа GenBank М26004);
(15) CD79b (CD79B, CD79 β, IGb (иммуноглобулин, родственный β), В29, номер доступа GenBank NM_000626);
(16) FcRH2 (IFGP4, IRTA4, SPAPIA (содержащие заякоренные домены SH2 белков фосфатаз Ia), SPAP1B, SPAP 1С, номер доступа GenBank NM_030764);
(17) HER2 (ErbB2, номер доступа GenBank MI1730);
(18) NCA (СЕАСАМ6, номер доступа GenBank М18728);
(19) MDP (DPEPI, номер доступа GenBank ВС017023);
(20) IL20Ra (IL20Ra, ZCYT0R7, номер доступа GenBank AF184971);
(21) Бревикан (BCAN, ВЕНАВ, номер доступа GenBank AF229053);
(22) EphB2R (DRT, ERK, Hek5, ЕРНТ3, Tyro5, номер доступа GenBank NM_004442);
(23) ASLG659 (B7h, номер доступа GenBank AX092328);
(24) PSCA (прежнее название антиген стволовой клетки простаты, номер доступа GenBank AJ297436);
(25) GEDA (номер доступа GenBank AY260763);
(26) BAFF-R (рецептор фактора активации В-лимфоцитов, рецептор 3 BLyS, BR3, номер доступа GenBank AF116456);
(27) CD22 (рецептор изоформы CD22-B рецептора В-лимфоцитов, номер доступа GenBank AK026467);
(28) CD79a (CD79A, CD79a, иммуноглобулин, родственный α, способный к ковалентным взаимодействиям с Ig (CD79B) и образованию комплекса с молекулами IgM на поверхности трансдуцированных В-лимфоцитов, в который вовлечен сигнальный специфический белок В-лимфоцитов, номер доступа GenBank NP_001774.1);
(29) CXCR5 (рецептор 1 лимфомы Беркитта, сопряженный с G-белком рецепторный хемокин, активируемый CXCL13, играет роль в миграции лимфоцитов и гуморальной защите, возможно, влияет на инфекцию ВИЧ-2, СПИД, лимфому, миелому и лейкоз, номер доступа GenBank NP_001701.1);
(30) HLA-D0B (бета-субъединица молекулы главного комплекса гистосовместимости (ГКГ) II (антиген Ia), который связывает пептиды и презентирует их Т-лимфоцитам CD4+ в пейеровых бляшках, номер доступа GenBank NP_002111.1);
(31) Р2Х5 (Р2Х ионный канал 5, гейтируемый лигандом пуринергического рецептора, внеклеточный АТФ-гейтируемый ионный канал, может быть вовлечен в синаптическую передачу и нейрогенез, его дефект может лежать в основе патофизиологии идиопатической нестабильности детрузора, номер доступа GenBank NP_002552.2);
(32) CD72 (антиген CD72 дифференцировки В-лимфоцитов, Lyb-2, номер доступа GenBank NP_001773.I);
(33) LY64 (лимфоцитарный антиген 64 (RP105), семейство мембранных белков с лейцин-богатым повтором I типа (LRR), регулирует активацию В-лимфоцитов, апоптоз и утрату функций, может быть вовлечен в повышение активности у пациентов с системной красной волчанкой, номер доступа GenBank NP_005573.1);
(34) FcRHI (белок, подобный Fc 1 рецептору, рецептор предполагаемого Fc домена иммуноглобулина, содержащий Ig-подобные домены С2 типа и домены ITAM, может играть роль в дифференцировке В-лимфоцитов, номер доступа GenBank NP_443170.1);
(35) IRTA2 (рецептор 2, ассоциированный с транслокацией, суперсемейства иммуноглобулинов, предполагаемый иммунный рецептор, может играть роль в развитии В-лимфоцитов и в развитии лимфомы; генетические нарушения, вызванные транслокацией, встречаются при некоторых злокачественных В-лимфоцитарных опухолях, номер доступа GenBank NP_112571.1);
(36) TENB2 (предполагаемый трансмембранный протеогликан, и относящийся к факторам роста EGF/семейству херегулина и фоллистатина, номер доступа GenBank AF179274).
В некоторых воплощениях изобретения А представляет собой гуманизированное моноклональное антитело, нацеленное на опухолеассоциированный антиген. Гуманизированное антитело может представлять собой без ограничений: трастузумаб (антитело к HER2), пертузумаб (антитело к HER2), ритуксимаб (антитело к CD20), алемтузумаб (антитело к CD52), бевацизумаб (антитело к фактору роста эндотелия сосудов (VEGF)), адалимумаб (антитело к фактору некроза опухоли (ФНО) альфа), цетуксимаб (антитело к рецептору эпидермального фактора роста (EGFR)), аматуксимаб (антитело к мезотелину), блинатумомаб (антитело к CD19), брентуксимаб (антитело к CD30), сертолизумаб пегол (антитело к ФНО альфа), панитумумаб (антитело к EGFR), нимотузумаб (антитело к EGFR), гемтузумаб (антитело к CD33), голимумаб (антитело к ФНО альфа), ибритумомаб (антитело к CD20), инфликсимаб (антитело к ФНО альфа), ипилимумаб (антитело к CTLA-4), офатумумаб (антитело к CD20) и тозитумомаб (антитело к CD20).
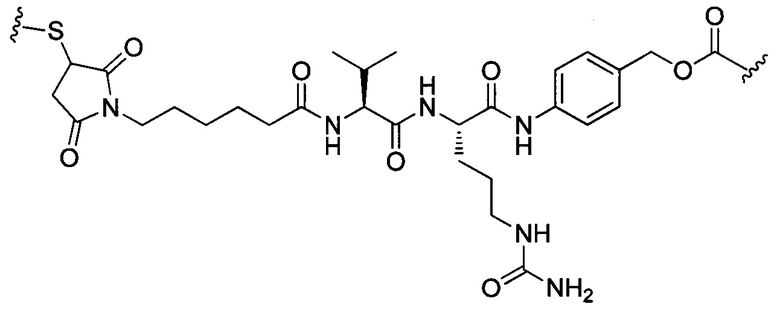
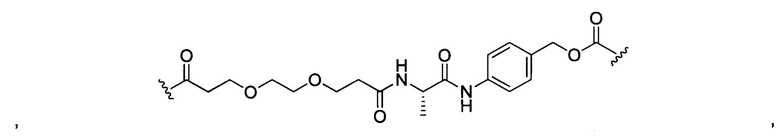
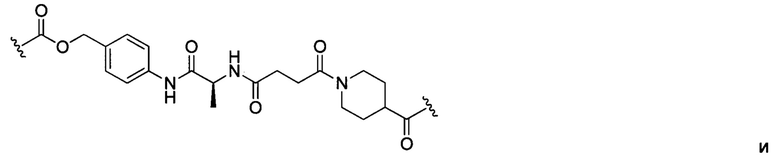
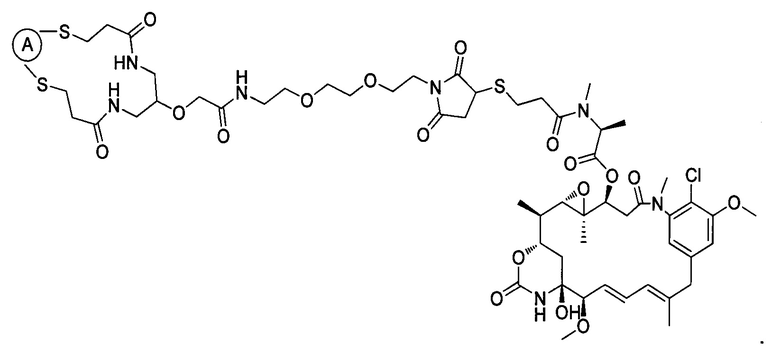
В некоторых воплощениях изобретения конъюгат антитело-лекарственное средство имеет следующую структуру формулы II:
В некоторых воплощениях изобретения конъюгат антитело-лекарственное средство имеет следующую структуру формулы II:
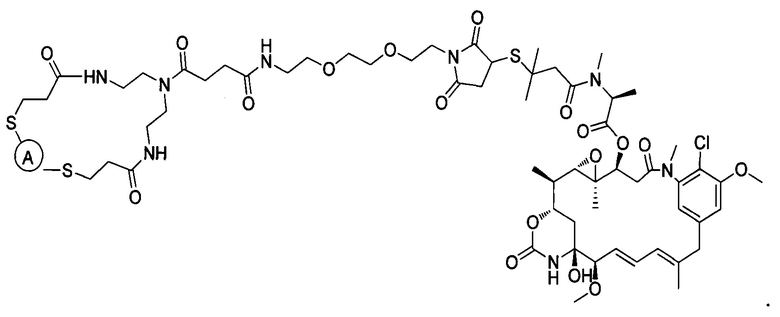
В некоторых воплощениях изобретения конъюгат антитело-лекарственное средство имеет следующую структуру формулы II:
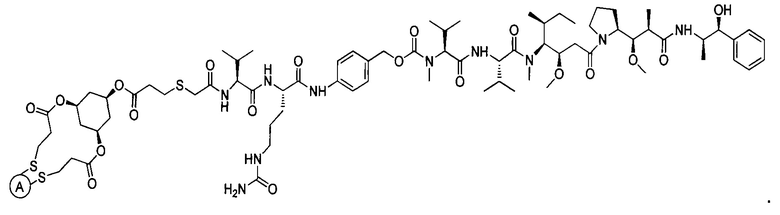
В некоторых воплощениях изобретения конъюгат антитело-лекарственное средство имеет следующую структуру формулы II:
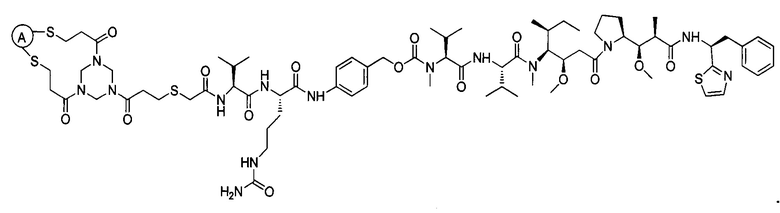
В некоторых воплощениях изобретения конъюгат антитело-лекарственное средство имеет следующую структуру формулы II:
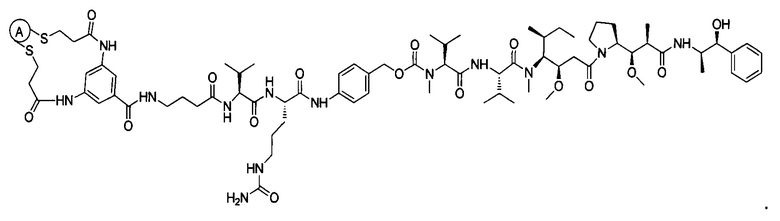
В некоторых воплощениях изобретения конъюгат антитело-лекарственное средство имеет следующую структуру формулы II:
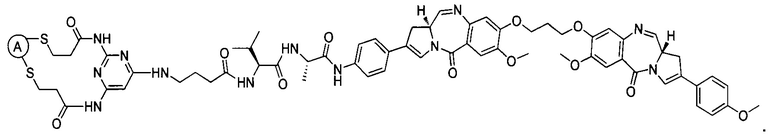
В некоторых воплощениях изобретения конъюгат антитело-лекарственное средство имеет следующую структуру формулы II:
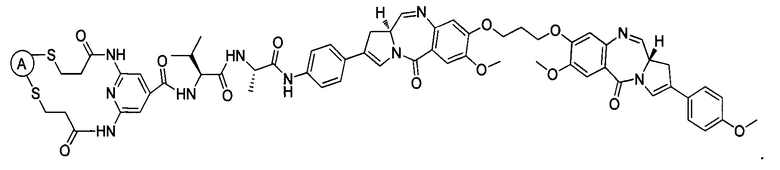
В некоторых воплощениях изобретения конъюгат антитело-лекарственное средство имеет следующую структуру формулы II:
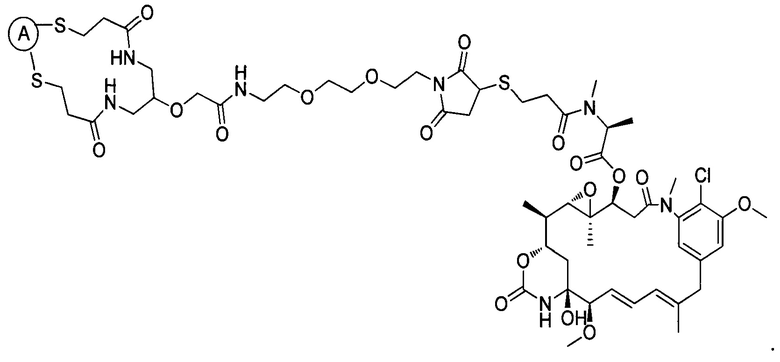
В некоторых воплощениях изобретения конъюгат антитело-лекарственное средство имеет следующую структуру формулы II:
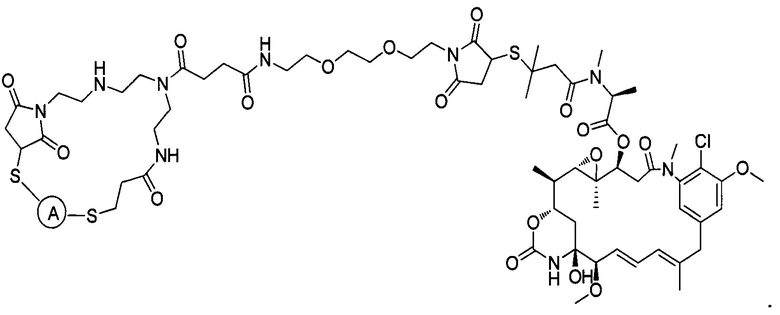
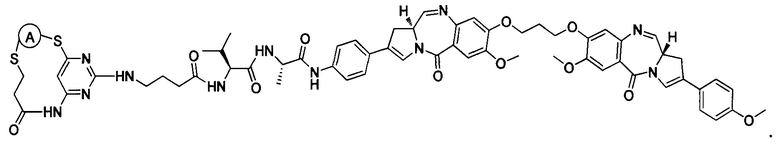
В некоторых воплощениях изобретения конъюгат антитело-лекарственное средство имеет следующую структуру формулы II:
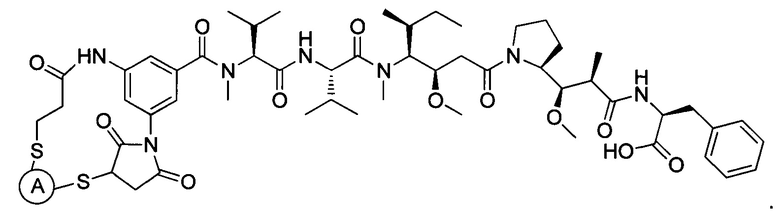
В некоторых воплощениях изобретения конъюгат антитело-лекарственное средство имеет следующую структуру формулы II:
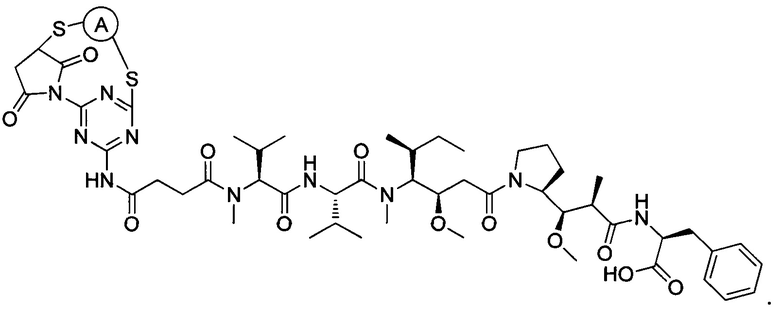
В некоторых воплощениях изобретения конъюгат антитело-лекарственное средство имеет следующую структуру формулы II:
В некоторых воплощениях изобретения А включает без ограничений трастузумаб.
Области терапевтического применения
В некоторых воплощениях изобретения конъюгаты заявленного изобретения обеспечивают лечение злокачественных опухолей, включающих без ограничения карциному, лимфому, бластому, саркому и лейкоз или лимфоидные злокачественные опухоли. Более конкретные примеры таких злокачественных опухолей включают плоскоклеточный рак (например, эпителиальный плоскоклеточный рак), рак легкого, включающий мелкоклеточный рак легкого, немелкоклеточный рак легкого, аденокарциному легкого и плоскоклеточную карциному легкого, рак брюшины, печеночно-клеточный рак, рак желудочно-кишечного тракта или желудка, включая желудочно-кишечный рак, рак поджелудочной железы, глиобластому, рак шейки матки, рак яичника, рак полости рта, рак печени, рак мочевого пузыря, рак мочевыводящих путей, гепатому, рак молочной железы, включающий, например, HER2-положительный рак молочной железы, рак ободочной кишки, рак прямой кишки, колоректальный рак, карциному эндометрия или матки, карциному слюнной железы, рак почки или почечно-клеточный рак, рак предстательной железы, рак вульвы, рак щитовидной железы, карциному печени, карциному анальной области, карциному полового члена, меланому, множественную миелому и В-клеточную лимфому, рак головного мозга, злокачественные опухоли органов головы и шеи и ассоциированные метастазы.
Конъюгаты заявленного изобретения можно также применять для доставки активных агентов для лечения ряда других состояний, включающих без ограничений воспалительные расстройства, аутоиммунные расстройства, расстройства нервной системы и сердечно-сосудистые расстройства.
Области диагностического применения
В других воплощениях изобретения конъюгат заявленного изобретения содержит в качестве активного агента обнаружимое химическое соединение. Обнаружимое химическое соединение может представлять собой, например, агент визуализации. Агент может представлять собой синтетический или натуральный препарат и способен отражать или испускать свет при длине волны, при которой его можно обнаружить при помощи приемлемого оборудования для визуализации и/или диагностики, такого как микроскоп. Неограничивающие примеры приемлемых агентов визуализации включают органические красители, пищевые красители и флуоресцентные красители.
Таким образом, в одном воплощении заявленного изобретения предложен способ диагностики, который включает приведение в контакт исследуемого образца, подозреваемого на наличие интересующего анализируемого вещества, с конъюгатом антитело-лекарственное средство заявленного изобретения, в котором нацеливающая группировка селективно связывается с интересующим анализируемым веществом, a Q представляет собой обнаружимое химическое соединение и/или химическое соединение, с которым будет связываться одно или более дополнительных химических соединений, при этом сами указанные дополнительные химические соединения представляют собой обнаружимые химические соединения, и где указанный способ дополнительно включает анализ исследуемого образца для определения присутствия обнаружимого химического соединения, которое будет указывать на наличие интересующего анализируемого вещества. В одном воплощении изобретения анализируемое вещество представляет собой HER2 или другой антиген, ассоциированный с раком. Этот способ может быть выполнен либо in vivo, либо in vitro.
Биологическая активность конъюгатов
Оценку эффективности и токсичности конъюгатов заявленного изобретения можно проводить известными способами. Например, токсикологические свойства конкретного соединения или подгруппы соединений, имеющих определенные общие химические группировки, могут быть установлены путем определения токсичности in vitro в отношении линии клеток, такой как линия клеток млекопитающего и предпочтительно человека. На основании результатов таких исследований часто можно предсказать токсичность у животных, таких как млекопитающие, или более конкретно у людей. Альтернативно токсичность конкретных соединений можно определить известными способами в биологической модели, такой как мыши, крысы, кролики, собаки или обезьяны.
Эффективность конкретного соединения может быть установлена признанными в данной области техники способами, такими как способы in vitro, биологические модели или клинические исследования с участием людей. Признанные в данной области техники способы in vitro существуют почти для каждого класса состояния, включая состояния, разрешаемые посредством соединений, раскрытых в настоящем документе, включающие злокачественную опухоль, сердечно-сосудистое заболевание, различные нарушения иммунной системы и инфекционные заболевания. Аналогичным образом для установления эффективности химических соединений для лечения таких состояний можно использовать приемлемые биологические модели. При выборе модели для определения эффективности обычный специалист в данной области техники может руководствоваться уровнем техники по выбору соответствующей модели, дозы, пути и схемы введения.
Количественное определение комплексов, предложенных в настоящем документе, можно проводить на основании аффинности связывания и специфичности к желаемой мишени любым из способов, традиционно используемых для количественного определения антител. Количественное определение эффективности комплексов в качестве противораковых средств можно проводить любым из способов, традиционно используемых для количественного определения цитостатических/цитотоксических агентов, таких как анализы активности на клеточных культурах, ксенотрансплантатах и т.п.
Используя преимущества настоящего описания, обычный специалист в данной области техники не будет испытывать затруднений с учетом своих навыков и имеющейся литературы при определении приемлемых методов количественного определения; при определении на основании результатов этих анализов приемлемых доз для испытания в качестве противораковых средств с участием людей и при определении на основании результатов этих испытаний приемлемых доз для применения для лечения злокачественных опухолей у людей.
Лекарственные формы и введение
Композиции заявленного изобретения в дополнение к описанным в настоящем документе комплексам содержат физиологически приемлемый носитель и/или разбавитель, позволяющий транспортировать комплексы к мишени в организме животного после введения. Носитель и/или разбавитель может, как правило, представлять собой любую приемлемую среду, с помощью которой можно достичь желаемой цели, и которая не влияет на способность конъюгатов к направлению к желаемой мишени и к транспортировке активного агента к этой мишени для получения желаемого эффекта. В частности, носитель и/или разбавитель не должен приводить к ухудшению фармакологической активности активного агента и способности комплекса к направлению к желаемой мишени внутри или на поверхности тела животного. Предпочтительно указанный носитель и/или разбавитель выбран/выбраны из воды, физиологически приемлемых водных растворов, содержащих соли, и/или буферных растворов и любого другого раствора, приемлемого для введения животному. Такие носители и разбавители хорошо известны специалисту в данной области техники и могут представлять собой, например, дистиллированную воду, деионизированную воду, чистую или ультрачистую воду, солевой раствор, фосфатно-солевой буферный раствор (ФСБ), растворы, содержащие обычные буферные агенты, совместимые с другими компонентами системы доставки лекарственных средств, и т.д.
Композиции можно вводить субъекту способами, включающими без ограничений: (а) введение посредством пероральных путей, включающее введение в капсуле, таблетке, грануле, спрее, сиропе или других таких формах; (b) введение посредством не пероральных путей, включающее введение в виде водной суспензии, масляного препарата и т.п. или в виде капельной клизмы, суппозитория, бальзама, мази и т.п.; введение посредством инъекции подкожно, интраперитонеально, внутривенно, внутримышечно, внутрикожно и т.п.; а также (с) введения местным путем; (d) введение ректальным путем или (е) введение вагинальным путем, в зависимости от того, что считают приемлемым специалисты в данной области техники для приведения соединения по настоящему воплощению изобретения в контакт с живой тканью; и (f) введение посредством лекарственных форм контролируемого высвобождения, депо-препаратов и систем доставки с помощью инфузионных насосов.
В качестве дополнительных примеров таких способов введения и в качестве дополнительного описания способов введения в настоящем документе раскрыты различные способы введения раскрытых соединений и фармацевтических композиций, включая способы введения посредством внутриглазных, интраназальных и внутрисуставных путей.
В конкретных воплощениях изобретения комплексы по данному изобретению можно включать в лекарственные формы в виде растворов для внутривенного введения или в виде лиофилизированных концентратов для восстановления для приготовления внутривенных растворов (для восстановления, например, физиологическим раствором, 5% раствором декстрозы или подобными изотоническими растворами). Как правило, их будут вводить путем внутривенной инъекции или инфузии. Обычный специалист в области изготовления фармацевтических лекарственных форм, в частности лекарственной формы противораковых антител, не будет испытывать затруднений при разработке подходящих лекарственных форм с учетом своих навыков и имеющейся литературы.
Примеры
Ниже следуют примеры, которые иллюстрируют методики для осуществления изобретения на практике. Эти примеры не следует рассматривать как имеющие ограничительный характер.
ПРИМЕР I синтез конъюгатов линкер-активный агент
ПРИМЕР I-1 синтез соединения 9
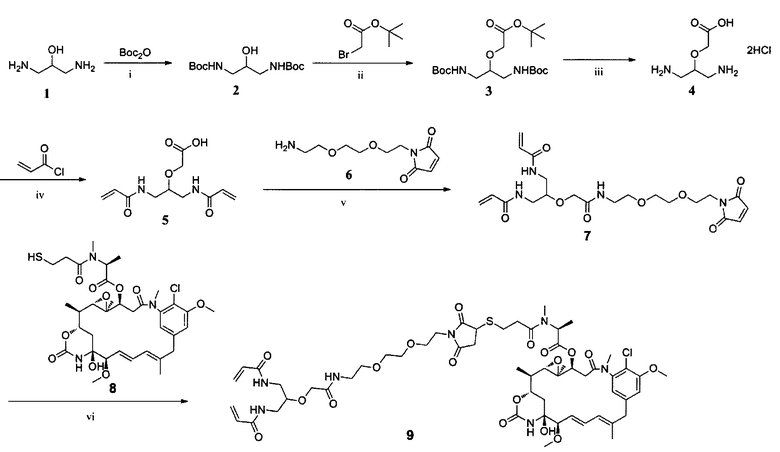
i) Et3N, МеОН, К. Т., 2 ч; ii) NaH, ТГФ, К. Т., 5 ч; iii) Хлористо-водородная кислота, 1,4-диоксан, К. Т., 16 ч; iv) NaHCO3, Н2О/ТГФ, К. Т., 3 ч; v) HOBt, DIC, DIPEA, ДМФ, К. Т., 12 ч; vi) Et3N, CH2Cl2, К. Т., 2 ч.
Синтез соединения 2
Раствор 1,3-диамино-2-пропанола (3,15 г, 0,035 моль) и Et3N (4,85 мл, 0,035 моль) в метаноле (120 мл) нагревали до 45°С. К раствору медленно добавляли по каплям (Вос)2O (17,05 г, 0,078 моль) в метаноле (80 мл). Реакционный раствор перемешивали при 45°С в течение 30 мин. После дополнительного перемешивания при комнатной температуре в течение 1,5 ч растворитель удаляли при пониженном давлении. Неочищенный продукт экстрагировали диэтиловым эфиром (200 мл×3) и высушивали над сульфатом натрия с получением соединения 2 (9,94 г, 97,8%) в виде белого порошка. ЖХ-МС m/z (электрораспыление (ЭР)+), 291,19 (М+Н)+.
Синтез соединения 3
трет-Бутилбромацетат (5,41 мл, 33,5 ммоль) добавляли к раствору соединения 2 (3,89 г, 13,4 ммоль) в безводном ТГФ (40 мл) при комнатной температуре. Впоследствии к этому раствору добавляли гидрид натрия (60% дисперсия в минеральном масле, 2,42 г, 60,5 ммоль). Смесь фильтровали через 5 часов. Фильтрат выпаривали, и остаток очищали колоночной хроматографией (петролейный эфир : этилацетат составляет от 10:1 до 5:1) с получением продукта 3 (3,96 г, 73,1%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 405,26 (М+Н)+.
Синтез соединения 4
5 мл хлористо-водородной кислоты добавляли к перемешанному раствору соединения 3 (1,0 г, 2,5 ммоль) в 1,4-диоксане (10 мл) при комнатной температуре. Реакция была завершена через 16 часов. Затем растворитель удаляли с получением белого неочищенного продукта (384,2 мг, 69,8%). Продукт 4 мог быть использован в следующей стадии без очистки. ЖХ-МС m/z (ЭР+), 221,05 (М+Н)+.
Синтез соединения 5
Акрилоилхлорид (267 мкл, 3,3 ммоль) добавляли к раствору соединения 4 (242,1 мг, 1,1 ммоль) в смеси насыщенного раствора бикарбоната натрия/ТГФ (об./об. составляет 1:1, 20 мл) при 0°С. Полученную в результате смесь энергично перемешивали при 0°С. Через 10 минут раствор оставляли для подогрева до комнатной температуры и проводили реакцию в течение 3 часов. Смесь подкисляли хлористоводородной кислотой до достижения рН<4. Смесь экстрагировали этилацетатом (50 мл×2). Органические слои объединяли и промывали насыщенным раствором хлорида натрия (40 мл), высушивали над безводным сульфатом магния, фильтровали и удаляли растворитель. Твердый остаток очищали препаративной ВЭЖХ с получением продукта 5 в виде серого порошка (196,0 мг, 69,6%). ЖХ-МС m/z (ЭР+), 257,12 (М+Н)+.
Синтез соединения 7
К перемешанному раствору соединения 5 (25,6 мг, 0,10 ммоль) и DIPEA (16,5 мкл, 0,10 ммоль) в безводном ДМФ (6 мл) при 0°С добавляли HOBt (14,9 мг, 0,11 ммоль) и DIC (13,9 мг, 0,11 ммоль). Через 15 минут к раствору добавляли соединение 6 (20,5 мг, 0,09 ммоль). Реакционную смесь оставляли для подогрева до комнатной температуры и перемешивали в течение ночи. Смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (20 мл×3). Объединенные органические слои высушивали над безводным сульфатом магния, и растворитель удаляли в вакууме. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир : этилацетат составляет 3:1) с получением соединения 7 в виде белого твердого вещества (24,3 мг, 62,6%). ЖХ-МС m/z (ЭР+), 467,22 (М+Н)+.
Синтез соединения 9
Соединение 7 (140,1 мг, 0,3 ммоль) растворяли в 5 мл CH2Cl2 и охлаждали до 0°С в ледяной бане. К полученной в результате смеси добавляли Et3N (1 мг, 0,01 ммоль) и соединение 8 (73,7 мг, 0,1 ммоль) и оставляли для перемешивания на 30 минут. Раствор оставляли для подогрева до комнатной температуры и перемешивали еще в течение 1,5 часов. Растворитель удаляли при пониженном давлении, и твердый остаток очищали препаративной ВЭЖХ с получением соединения 9 в виде белого порошка (90 мг, 74,8%). ЖХ-МС m/z (ЭР+), 291,19 (М+Н)+.
ПРИМЕР I-2 синтез соединения 17
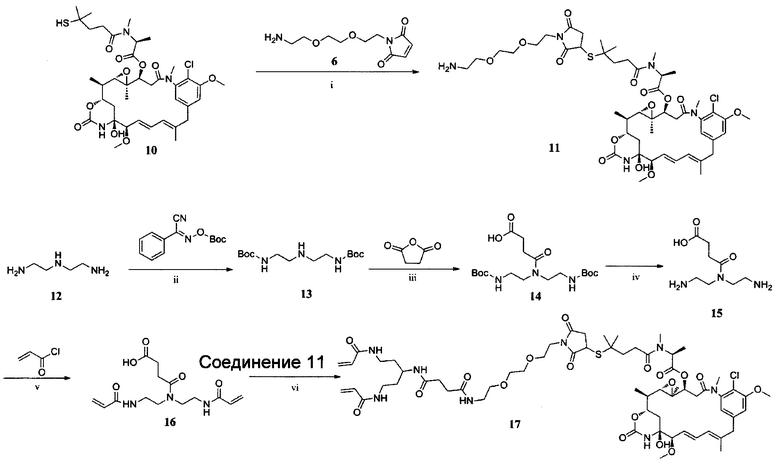
i) Et3N, CH2Cl2, К. Т., 5 ч; ii) Et3N, ТГФ, К. Т., 4 ч; iii) CH2Cl2, К. Т., 12 ч; iv) ТФУ, CH2Cl2, К. Т., 3 ч; V) K2CO3, H2O/EtOAc, К. Т., 5 ч; vi) HOBt, DIC, DIPEA, ДМФ, К. Т., 24 ч.
Синтез соединения 11
Соединение 10 (263,3 мг, 1,15 ммоль) растворяли в CH2Cl2 (15 мл), к полученной в результате смеси добавляли Et3N (5,9 мкл, 0,04 ммоль) и соединение 6 (300,1 мг, 0,39 ммоль) при 0°С. Раствор оставляли для подогрева до комнатной температуры и выдерживали в течение 5 часов. Растворитель удаляли в вакууме, и неочищенный продукт очищали препаративной ВЭЖХ с получением соединения 11 (193,2 мг, 49,7%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 994,43 (М+Н)+.
Синтез соединения 13
К смеси соединения 12 (0,45 г, 4,2 моль) и Et3N (8 мл, 0,06 моль) в ТГФ (15 мл) добавляли по каплям раствор
2-(трет-бутоксикарбонилоксиимино)-2-фенилацетонитрила (2,1 г, 8,3 моль) в ТГФ (30 мл) при 0°С. После полного добавления раствор оставляли для подогрева до комнатной температуры и оставляли для перемешивания в течение 4 часов. Реакционную смесь концентрировали до масла при пониженном давлении и добавляли CH2Cl2 (50 мл). Смесь промывали гидроксидом натрия (5%, 30 мл) и раствором хлорида натрия (30 мл). Органический слой высушивали над сульфатом натрия и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (силикагель, МеОН: CH2Cl2 составляет 1:10, об./об.) с получением соединения 13 в виде желтого масла (803,1 мг, 60,7%). ЖХ-МС m/z (ЭР+), 304,22 (М+Н)+.
Синтез соединения 14
Ангидрид янтарной кислоты (265,2 мг, 2,65 ммоль) добавляли к раствору соединения 13 (800,1 мг, 2,65 ммоль) в CH2Cl2 (10 мл). Смесь перемешивали при комнатной температуре в течение ночи, а затем концентрировали до масла при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией (силикагель, МеОН : CH2Cl2 составляет 1:8, об./об.) с получением соединение 14 (506,5 мг, 47,6%) в виде серого масла. ЖХ-МС m/z (ЭР+), 404,24 (М+Н)+.
Синтез соединения 15
Соединение 14 (503,5 мг, 1,25 ммоль) растворяли в CH2Cl2 (10 мл) вместе с трифторуксусной кислотой (2 мл). Раствор оставляли для взаимодействия на 3 часа при комнатной температуре. Растворитель удаляли при пониженном давлении с получением соединения 15 в виде серого масла (190,2 мг, 75,1%) без дополнительной очистки. ЖХ-МС m/z (ЭР+), 204,13 (М+Н)+.
Синтез соединения 16
К смеси карбоната калия (68,3 мг, 0,5 ммоль) в воде (5 мл) и соединения 15 (67,2 мг, 0,33 ммоль) в этилацетате (10 мл) добавляли по каплям раствор акрилоилхлорида (53,6 мкл, 0,67 ммоль) в этилацетате (10 мл) при 0°С. Полученную в результате смесь перемешивали при 0°С в ледяной бане в течение 10 минут, а затем оставляли для взаимодействия при комнатной температуре на 5 часов. Смесь подкисляли хлористоводородной кислотой до достижения рН<5. Смесь экстрагировали этилацетатом (20 мл×2). Органические слои объединяли и промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом магния, фильтровали и удаляли растворитель. Твердый остаток очищали колоночной хроматографией (МеОН : CH2Cl2 составляет 10:1) с получением продукта 16 в виде желтого масла (63,1 мг, 61,7%). ЖХ-МС m/z (ЭР+) 312,15 (М+Н)+.
Синтез соединения 17
Соединение 16 (22,2 мг, 0,07 ммоль), HOBt (9,7 мг, 0,07 ммоль) и DIC (11 мкл, 0,07 ммоль) растворяли в 8 мл ДМФ и охлаждали до 0°С в ледяной бане. Впоследствии к смеси добавляли соединение 11 (59,1 мг, 0,06 ммоль) и DIPEA (12,4 мкл, 0,07 ммоль). Раствор оставляли для подогрева до комнатной температуры и оставляли для взаимодействия на 24 часов. Полученную в результате смеси концентрировали и очищали препаративной ВЭЖХ с получением соединения 17 (11,3 мг, 39,5%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 1287,56 (М+Н)+.
ПРИМЕР I-3 синтез соединения 25
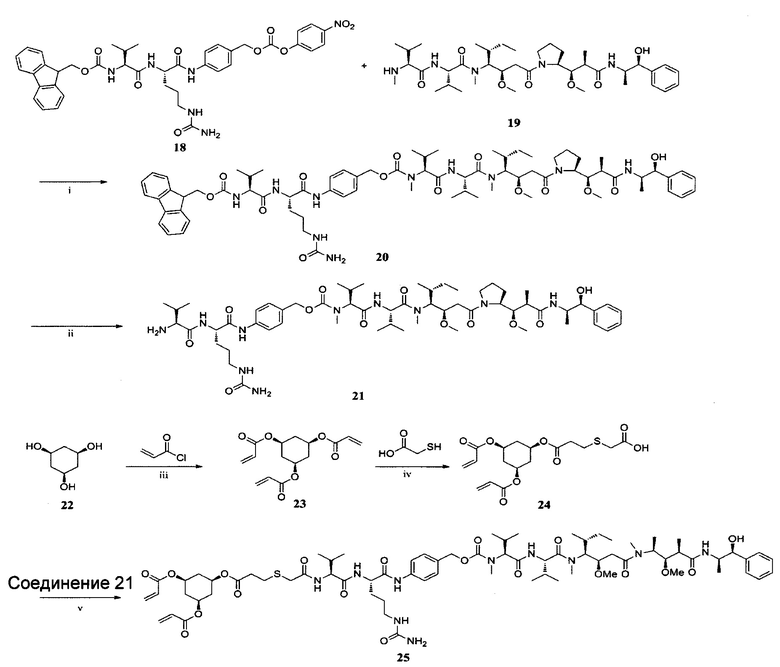
i) HOBt, DIPEA, пиридин, ДМФ, К. Т., 32 ч; ii) DEA, ДМФ, К. Т., 2 ч; iii) Et3N, CH2Cl2, К. Т., 4 ч; iv) Et3N, CH2Cl2, К. Т., 12 ч; v) EDCl, HOBt, DIPEA, CH2Cl2, К. Т., 24 ч. Синтез соединения 20
К перемешанному раствору 18 (191,6 мг, 0,25 ммоль) в безводном ДМФ (15 мл) при 0°С в атмосфере азота добавляли HOBt (33,8 мг, 0,25 ммоль) и соединение 19 (197,3 мг, 0,26 ммоль). Через 15 минут добавляли пиридин (3 мл) и DIPEA (52,4 мкл, 0,30 ммоль), и реакцию проводили в течение 30 минут при 0°С. Затем реакционную смесь оставляли для подогрева до комнатной температуры и перемешивали в течение 3 часов. Смесь дополняли DIPEA (52,4 мкл, 0,30 ммоль) и проводили реакцию в течение 32 часов. После завершения реакции растворитель удаляли при пониженном давлении. Остаток очищали колоночной хроматографией (МеОН : CH2Cl2 составляет 20:1) с получением соединения 20 (126,4 мг, 37,5%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 1344,77 (М+Н)+.
Синтез соединения 21
Диэтиламин (4 мл, 38,8 ммоль) добавляли к раствору соединения 20 (126,4 мг, 0,1 ммоль) в ДМФ (8 мл) при комнатной температуре. После перемешивания в течение 2 часов растворитель удаляли при пониженном давлении с получением неочищенного продукта 21(104,1 мг, 99%). Продукт мог быть использован в следующей стадии без очистки. ЖХ-МС m/z (ЭР+), 1123,72 (М+Н)+.
Синтез соединения 23
Раствор акрилоилхлорида (7,31 мл, 90,00 ммоль) в безводном CH2Cl2 (50 мл) добавляли по каплям к смеси соединения 22 (2,64 г, 20,00 ммоль) и Et3N (16,60 мл; 120,00 ммоль) в безводном CH2Cl2 (60 мл) при 0°С в атмосфере аргона. Раствор оставляли для подогрева до комнатной температуры и проводили реакцию в течение 4 часов. Затем смесь обрабатывали насыщенным раствором бикарбоната натрия (150 мл) и раствором хлорида натрия (150 мл) и экстрагировали CH2Cl2 (150 мл×3). Объединенные органические слои высушивали над безводным сульфатом магния, и растворитель удаляли в вакууме. Неочищенный продукт очищали колоночной хроматографией (петролейный эфир : этилацетат составляет 10:1) с получением соединения 23 в виде белого твердого вещества (4,88 г, 83,0%). ЖХ-МС m/z (ЭР+), 295,10 (М+Н)+.
Синтез соединения 24
К раствору соединения 23 (444,1 мг, 1,5 ммоль) и Et3N (0,23 мл, 1,66 ммоль) в CH2Cl2 (30 мл) добавляли по каплям меркаптоуксусную кислоту (0,23 мл, 3,32 ммоль) в CH2Cl2 (5 мл) в ледяной бане. После полного добавления раствор оставляли для подогрева до комнатной температуры и оставляли для перемешивания в течение ночи. Растворитель удаляли в вакууме, и остаток очищали колоночной хроматографией (МеОН : CH2Cl2 составляет 60:1-30:1) с получением соединения 24 в виде желтого масла (1,72 г, 36,87%). ЖХ-МС m/z (ES), 385,10 (M-H)-.
Синтез соединения 25
Соединение 24 (22,2 мг, 0,057 ммоль), HOBt (9,3 мг, 0,069 ммоль) и EDCl (16,5 мг, 0,086 ммоль) растворяли в 5,0 мл CH2Cl2. Впоследствии к смеси добавляли соединение 21 (64,5 мг, 0,063 ммоль) и DIPEA (20,3 мкл, 0,104 ммоль). Раствор оставляли для подогрева до комнатной температуры и оставляли для взаимодействия на 24 часа. Полученный в результате раствор разделяли препаративной ВЭЖХ с получением соединение 25 в виде белого твердого вещества (25,3 мг, 29,7%). ЖХ-МС m/z (ES+), 1479,80 (М+Н)+.
ПРИМЕР I-4 синтез соединения 31
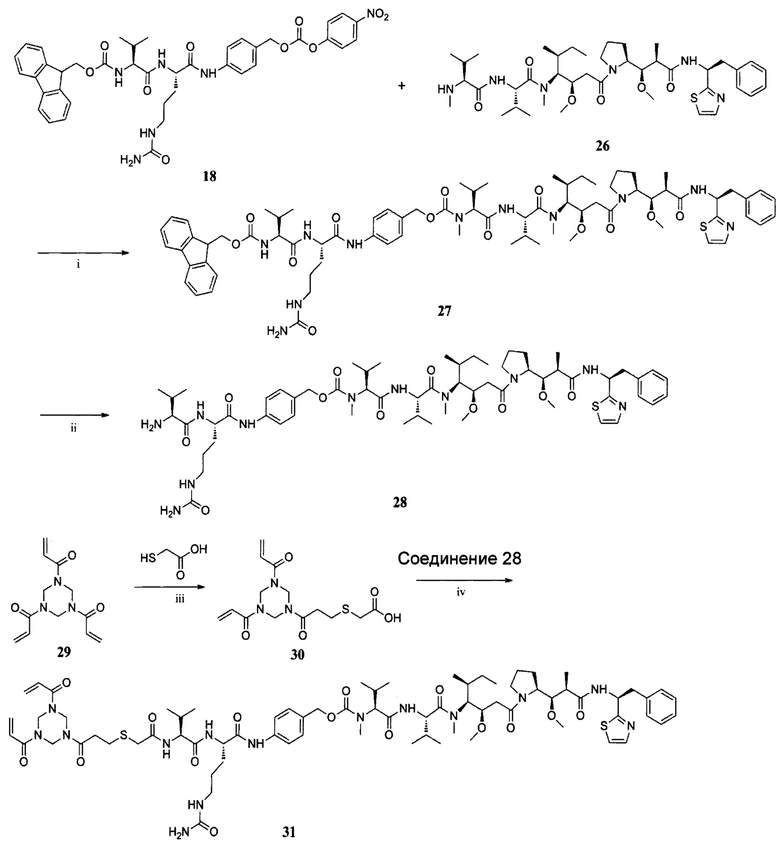
i) HOBt, DIPEA, пиридин, ДМФ, К. Т., 24 ч; ii) DEA, ДМФ, К. Т., 2,5 ч; iii) Et3N, CH2Cl2, К. Т., 12 ч; iv) DIC, HOBt, DIPEA, ДМФ, К. Т., 24 ч.
Синтез соединения 27
К раствору соединения 18 (194,2 мг, 0,25 ммоль) в безводном ДМФ (15 мл) при 0°С в атмосфере азота добавляли HOBt (34,2 мг, 0,25 ммоль) и соединение 26 (200,3 мг, 0,28 ммоль). После перемешивания в течение 15 минут добавляли пиридин (3 мл) и DIPEA (53,1 мкл, 0,32 ммоль), и оставляли для взаимодействия на 30 минут при 0°С. Затем реакционную смесь оставляли для подогрева до комнатной температуры на 3 часа. Смесь дополняли DIPEA (53,1 мкл, 0,32 ммоль) и проводили реакцию еще в течение 24 часов. После завершения реакции смесь концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией с получением соединения 27 (126,2 мг, 37,0%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 1398,75 (М+Н)+.
Синтез соединения 28
Диэтиламин (4 мл) добавляли к раствору соединения 27 (126,2 мг, 0,09 ммоль) в ДМФ (8 мл) при комнатной температуре. После перемешивания в течение 2,5 часов растворитель удаляли при пониженном давлении с получением неочищенного продукта 28 (73,3 мг, 68,9%), который можно использовать без дополнительной очистки. ЖХ-МС m/z (ЭР+), 1176,68 (М+Н)+.
Синтез соединения 30
К раствору соединения 29 (1,87 г, 7,5 ммоль) и Et3N (104 мкл, 0,75 ммоль) в CH2Cl2 (40 мл) добавляли по каплям меркаптоуксусную кислоту (103,9 мкл, 1,5 ммоль) в CH2Cl2 (10 мл) при 0°С. После полного добавления раствор оставляли для подогрева до комнатной температуры и оставляли для перемешивания в течение ночи. Растворитель удаляли в вакууме, и остаток очищали колоночной хроматографией с получением соединения 30 в виде белого твердого вещества (1,72 г, 36,87%). ЖХ-МС m/z (ЭР-), 342,11(М-Н)\
Синтез соединения 31
Соединение 30 (22,2 мг, 0,065 ммоль), HOBt (8,8 мг, 0,065 ммоль) и DIC (11 мкл, 0,065 ммоль) добавляли к ДМФ (8 мл) и охлаждали до 0°С в ледяной бане. К раствору добавляли соединение 28 (63,7 мг, 0,054 ммоль) и DIPEA (12,4 мкл, 0,054 ммоль). Смесь оставляли для подогрева до комнатной температуры и оставляли для взаимодействия на 24 часов. Остаток очищали колоночной хроматографией (МеОН : CH2Cl2 составляет от 60:1 до 30:1) с получением соединения 31 в виде белого твердого вещества (11,3 мг, 39,5%). ЖХ-МС m/z (ЭР+), 1499,78 (М+Н)+.
ПРИМЕР I-5 синтез соединения 36
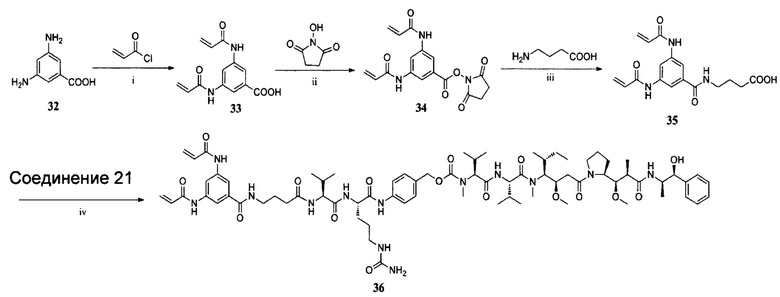
i) K2CO3, EtOAc/H2O, К. Т., 2 ч; ii) EDCl, ДМФ, К. Т., 12 ч; iii) DIPEA, ДМФ, К. Т., 12 ч; iv) EDCl, HOBt, DIPEA, ДМФ, К. Т., 24 ч.
Синтез соединения 33
2,4-Диаминобензойную кислоту 32 (501,8 мг, 3,3 ммоль) диспергировали в этилацетате (20 мл) и добавляли раствор карбоната калия (9,0 г, 66 ммоль) в воде (20 мл). К смеси осторожно добавляли акрилоилхлорид (1 мл, 13 ммоль), в результате чего образовался светло-коричневый осадок. Затем реакционную смесь оставляли до достижения комнатной температуры, в результате чего осадок растворялся. Через 2 часа реакция была завершена. Органический слой отбрасывали, и водный слой подкисляли 5% хлористоводородной кислотой с образованием остатка при рН<4. Остаток фильтровали, промывали петролейным эфиром и разбавляли этилацетатом. Затем органический слой высушивали над безводным сульфатом магния, фильтровали и удаляли в вакууме. Твердый остаток суспендировали в воде, фильтровали и тщательно промывали диэтиловым эфиром с получением продукта 33 (670,2 мг, 78%) в виде серого порошка. ЖХ-МС m/z (ЭР+), 261,09 (М+Н)+.
Синтез соединения 34
Соединение 33 (260,1 мг, 1,0 ммоль) и EDCl (210,9 мг, 1,1 ммоль), N-гидроксисукцинимид (126,6 мг, 1,1 ммоль) растворяли в ДМФ (14 мл). Раствор перемешивали в течение ночи при комнатной температуре. После завершения реакции растворитель удаляли при пониженном давлении. Твердый остаток очищали колоночной хроматографией с CH2Cl2/MeOH (10:1) в качестве элюента с получением продукта 34 в виде светло-коричневого твердого вещества (273 мг, 76,4%). ЖХ-МС m/z (ЭР+), 358,11 (М+Н)+.
Синтез соединения 35
К раствору 4-аминомасляной кислоты (82,4 мг, 0,8 ммоль) и DIPEA (489 мкл, 2,8 ммоль) в ДМФ (10 мл) и добавляли раствор соединения 34 (260,7 мг, 0,73 ммоль) в ДМФ (10 мл) при 0°С в ледяной бане. Смесь перемешивали в течение 10 минут при 0°С, а затем перемешивали при комнатной температуре в течение ночи. Растворитель удаляли в вакууме после того, как реакция была завершена. Твердый остаток очищали колоночной хроматографией (CH2Cl2: МеОН составляет 10:1, об./об.) с получением соединения 35 (192,3 мг, 76,2%). ЖХ-МС m/z (ЭР+), 346,14 (М+Н)+.
Синтез соединения 36
HOBt (14,9 мг, 0,11 ммоль), EDCl (21,1 мг, 0,11 ммоль) и DIPEA (19,2 мкл, 0,11 ммоль) добавляли к перемешанному раствору 35 (34,5 мг, 0,1 ммоль) в безводном ДМФ (10 мл) при 0°С. Через 15 минут добавляли соединение 21 (112,3 мг, 0,1 ммоль). Реакционную смесь оставляли для подогрева до комнатной температуры и перемешивали в течение 24 часов. К смеси добавляли воду (15 мл) и экстрагировали этилацетатом (20 мл×3). Объединенные органические слои высушивали над безводным сульфатом магния, фильтровали и удаляли в вакууме. Неочищенный продукт очищали колоночной хроматографией (CH2Cl2 : МеОН составляет 10:1, об./об.) с получением соединения 36 в виде белого твердого вещества (56,5 мг, 39%). ЖХ-МС m/z (ЭР+), 1450,84 (М+Н)+.
ПРИМЕР I-6 синтез соединения 43
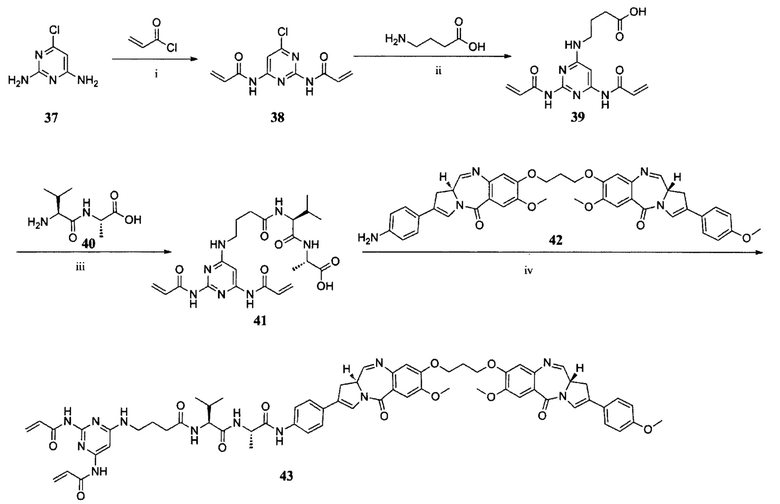
i) DMAP, CH2Cl2, К. Т., 1 ч; ii) NaOH, EtOH/H2O, 80°C, 2 ч; iii) EDCl, HOBt, DIPEA, ДМФ, К. Т., 18 ч; (iv) EEDQ, CH2Cl2, МеОН, К. Т., 18 ч.
Синтез соединения 38
В круглодонную колбу емкостью 100 мл помещали раствор соединения 37 (1,05 г, 6,9 ммоль) и DMAP (1,94 г, 15,9 ммоль) в CH2Cl2 (20 мл) добавляли по каплям акрилоилхлорид (1,38 г, 15,2 ммоль) при перемешивании при 0°С. Реакционную смесь перемешивали в течение 1 часа при комнатной температуре. Реакционную смесь гасили добавлением воды (50 мл) и экстрагировали CH2Cl2 (20 мл×3). Органическую фазу объединяли, промывали насыщенным раствором бикарбоната натрия (30 мл) и раствором хлорида натрия (30 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 38 (1,51 г, 85,8%) в виде светло-желтого полутвердого вещества. ЖХ-МС m/z (ЭР+), 253,04 (М+Н)+.
Синтез соединения 39
Соединение 38 (1,56 г, 5,94 ммоль) и 4-аминобутановую кислоту (673,0 мг, 6,53 ммоль) растворяли в EtOH (15 мл) вместе с гидроксидом натрия (713,3 мг, 17,82 ммоль) в воде (15 мл). Реакционную смесь перемешивали в течение 2 часов при 80°С. После охлаждения до комнатной температуры растворитель концентрировали при пониженном давлении. Смесь подкисляли до рН менее 7 хлористоводородной кислотой (0,5 н.). Твердое вещество собирали фильтрованием, промывали водой (10 мл×2) и высушивали с получением соединения 39 (490,2 мг, 25,8%) в виде беловатого твердого вещества. ЖХ-МС m/z (ЭР+), 320,03 (М+Н)+.
Синтез соединения 41
Соединение 39 (230,1 мг, 0,72 ммоль), HOBt (116,7 мг, 0,86 ммоль), EDCl (151,8 мг, 0,79 ммоль) и DIPEA (279,2 мг, 2,2 ммоль) добавляли к ДМФ (20 мл) и перемешивали при 0°С в ледяной бане. Через 1 час к раствору добавляли соединение 40 (149,1 мг, 0,79 ммоль). Смесь оставляли для подогрева до комнатной температуры и оставляли для взаимодействия на 18 часов. Реакционную смесь разбавляли раствором хлорида натрия (50 мл), экстрагировали этилацетатом (20 мл×3). Органические слои объединяли и высушивали безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией (МеОН : CH2Cl2 составляет 200/1 ~10/1, об./об.) с получением соединения 41 в виде желтого твердого вещества (97,3 мг, 27,6%). ЖХ-МС m/z (ЭР+), 490,23 (М+Н)+.
Синтез соединения 43
Раствор соединения 41 (90,1 мг, 0,18 ммоль), EEDQ (54,6 мг, 0,22 ммоль) в смеси безводного CH2Cl2 (7 мл) и безводного МеОН (0,07 мл) перемешивали в течение 1 часа при комнатной температуре. Впоследствии добавляли соединение 42 (133,5 мг, 0,184 ммоль). Реакционную смесь перемешивали дополнительно в течение 18 часов. Реакционную смесь концентрировали в вакууме и очищали препаративной ВЭЖХ с получением соединения 43 (12,3 мг, 5,6%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 1197,51 (М+Н)+.
ПРИМЕР I-7 синтез соединения 49
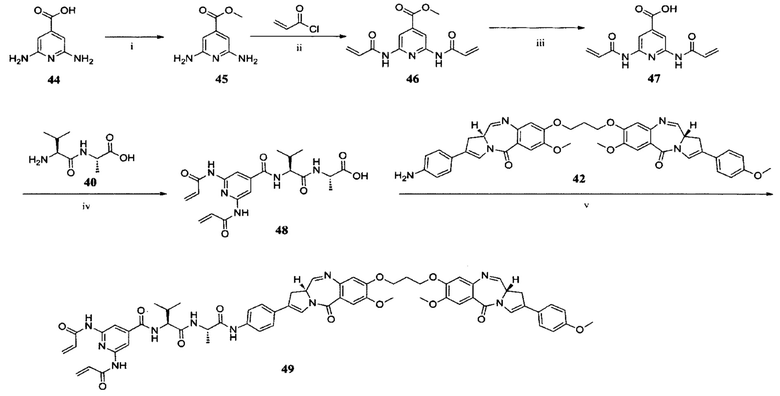
i) HCl (г), МеОН, 0°C, 2 ч; ii) пиридин, 100°C, 2 ч; iii) NaOH, ТГФ/H2O, К. Т., 4 ч; iv) EDCl, HOBt, DIPEA, ДМФ, К. Т., 18 ч; v) EEDQ, CH2Cl2/MeOH, К. Т., 18 ч.
Синтез соединения 45
В раствор соединения 44 (1,13 г, 6,53 ммоль) в метаноле (30 мл) вводили хлористоводородную кислоту (газ) при перемешивании при 0°С. Реакционную смесь перемешивали в течение 2 часов при 0°С. Смесь концентрировали, и остаток растворяли в воде (50 мл). Смесь доводили до рН 8 насыщенным раствором бикарбоната натрия, экстрагировали этилацетатом (20 мл×3). Органический слой объединяли и высушивали безводным сульфатом натрия, фильтровали. Фильтрат концентрировали с получением соединения 45 (710,1 мг, 65,1%) в виде желтого твердого вещества. ЖХ-МС m/z (ЭР+), 168,07 (М+Н)+.
Синтез соединения 46
Акрилоилхлорид (947,1 мг, 10,47 ммоль) добавляли по каплям к раствору соединения 45 (700,5 мг, 4,19 ммоль) в пиридине (20 мл) при перемешивании при 0°С в ледяной бане. Реакционную смесь перемешивали в течение 2 часов при 100°С. После охлаждения до комнатной температуры реакционную смесь концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (МеОН : CH2Cl2 составляет 200/1 ~10/1, об./об.) с получением соединения 46 (520,4 мг, 45,2%) в виде желтого твердого вещества. ЖХ-МС m/z (ЭР+), 276,09 (М+Н)+.
Синтез соединения 47
Смесь соединения 46 (510,2 мг, 1,85 ммоль), гидроксида натрия (370,6 мг, 9,26 ммоль), ТГФ (10 мл) и воды (10 мл) перемешивали в течение 4 часов при комнатной температуре. После завершения реакции смесь концентрировали и доводили рН до 7 хлористоводородной кислотой (1 н). Соединение 47 (360,2 мг, 74,1%) собирали фильтрованием в виде беловатого твердого вещества. ЖХ-МС m/z (ЭР+), 262,07 (М+Н)+.
Синтез соединения 48
Соединение 47 (120,5 мг, 0,5 ммоль), HOBt (4,7 мг, 0,6 ммоль), EDCl (96,8 мг, 0,5 ммоль) и DIPEA (178,1 мг, 1,4 ммоль) добавляли к ДМФ (10 мл) и перемешивали при 0°С в ледяной бане. Через 1 час к раствору добавляли соединение 40 (95,1 мг, 0,5 ммоль). Смесь оставляли для подогрева до комнатной температуры и оставляли для взаимодействия на 18 часов. Реакционную смесь разбавляли раствором хлорида натрия (20 мл), экстрагировали этилацетатом (10 мл×3). Органические слои объединяли и высушивали безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией (МеОН : CH2Cl2 составляет 100/1~20/1, об./об.) с получением соединения 41 в виде беловатого твердого вещества (71 мг, 35,8%). ЖХ-МС m/z (ЭР+), 432,07 (М+Н)+.
Синтез соединения 49
Соединение 48 (60,2 мг, 0,14 ммоль), EEDQ (41,3 мг, 0,17 ммоль) растворяли в смеси безводного CH2Cl2 (6 мл) и безводного МеОН (0,06 мл) и оставляли для перемешивания в течение 1 часа при комнатной температуре. К раствору добавляли соединение 42 (100,9 мг, 0,14 ммоль). Реакционную смесь перемешивали дополнительно в течение 18 часов. Реакционную смесь концентрировали при пониженном давлении и очищали препаративной ВЭЖХ с получением соединения 49 (12,4 мг, 7,61%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 1139,45 (М+Н)+.
ПРИМЕР I-8 синтез соединения 55
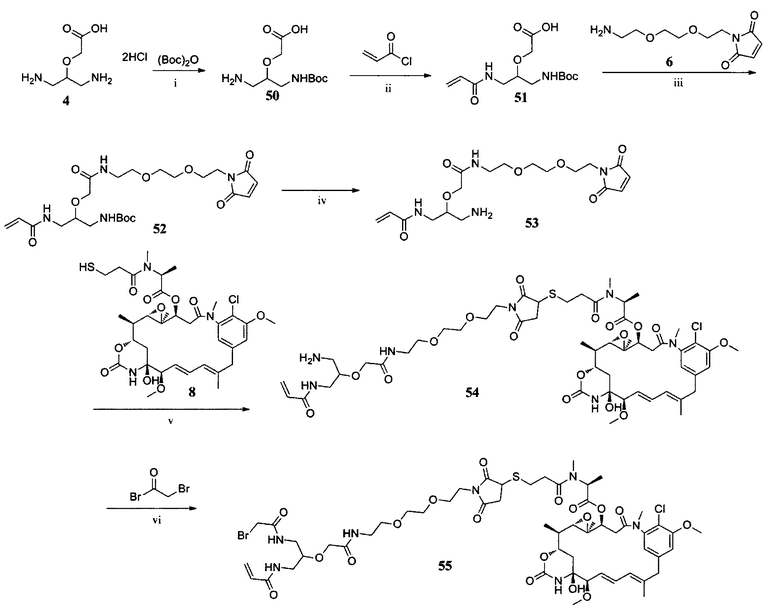
i) Et3N, МеОН, К. Т., 3 ч; ii) Na2CO3, ТГФ/H2O, К. Т., 3 ч; iii) HOBt, DIC, DIPEA, ДМФ, К. Т., 12 ч; iv) ТФУ, CH2Cl2, К. Т., 3 ч; v) Et3N, CH2Cl2, К. Т., 2 ч; vi)
Синтез соединения 50
Et3N (332,7 мкл, 2,4 ммоль) добавляли к раствору соединения 4 (440,0 мг, 2 ммоль) в МеОН (10 мл) при 45°С. Затем добавляли раствор (Вос)2O (436,2 мг, 2 ммоль) в МеОН (10 мл). Полученную в результате смесь энергично перемешивали при 45°С в течение 30 минут с последующим перемешиванием при комнатной температуре в течение 2 часов. Затем растворитель удаляли, остаток промывали хлористоводородной кислотой (5%, 10 мл), насыщенным раствором хлорида натрия (15 мл) и экстрагировали этилацетатом (10 мл×3). Органический слой высушивали над безводным сульфатом магния, фильтровали и удаляли в вакууме. Твердый остаток перекристаллизовывали смесью петролейный эфир/этилацетат (10 мл, об.:об. составляет 2:1,) с получением соединения 50 (375,4 мг, 75,6%) в виде белого порошка. ЖХ-МС m/z (ЭР+), 249,15 (М+Н)+.
Синтез соединения 51
Акрилоилхлорид (121 мкл, 1,5 ммоль) добавляли к раствору соединения 50 (248,0 мг, 1,0 ммоль) в смеси насыщенного раствора бикарбоната натрия/ТГФ (об./об. составляет 1:1, 20 мл) при 0°С. Полученную в результате смесь энергично перемешивали при 0°С в течение 10 мин. Раствор оставляли для подогрева до комнатной температуры и проводили реакцию в течение 3 часов. Смесь подкисляли хлористоводородной кислотой до достижения рН менее 4. Смесь экстрагировали этилацетатом (20 мл×3). Органические слои объединяли и промывали насыщенным раствором хлорида натрия (30 мл), высушивали над безводным сульфатом магния, фильтровали и удаляли растворитель. Твердый остаток очищали препаративной ВЭЖХ с получением продукта 51 (176,1 мг, 58,3%) в виде серого масла. ЖХ-МС m/z (ЭР+), 303,16 (М+Н)+.
Синтез соединения 52
Соединение 51 (30,2 мг, 0,1 ммоль), HOBt (14,7 мг, 0,11 ммоль) и DIC (13,9 мг, 0,11 ммоль) растворяли в безводном ДМФ (6 мл) и охлаждали до 0°С в ледяной бане. Через 15 мин к смеси добавляли соединение 6 (20,5 мг, 0,09 ммоль) и DIPEA (19,2 мкл, 0,11 ммоль). Раствор оставляли для подогрева до комнатной температуры и перемешивали в течение ночи. Смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (20 мл×3). Объединенные органические слои высушивали над безводным сульфатом магния, и растворитель удаляли в вакууме. Неочищенный продукт очищали колоночной хроматографией (CH2Cl2 : МеОН составляет 20:1, об./об.) с получением соединения 52 в виде белого твердого вещества (28,2 мг, 60,7%). ЖХ-МС m/z (ЭР+), 513,26 (М+Н)+.
Синтез соединения 53
Раствор соединения 52 (28,2 мг, 0,06 ммоль) в трифторуксусной кислоте (1,0 мл) в CH2Cl2 (5 мл) перемешивали в течение 3 часов при комнатной температуре. Растворитель удаляли при пониженном давлении с получением соединения 53 (20,4 мг, 88,2%) в виде серого твердого вещества, которое можно было использовать в следующей стадии без дополнительной очистки. ЖХ-МС m/z (ЭР+), 413,21 (М+Н)+.
Синтез соединения 54
Соединение 53 (41,2 мг, 0,1 ммоль) растворяли в 10 мл ДМФ и охлаждали до 0°С в ледяной бане. Et3N (6,9 мкл, 0,05 ммоль) и соединение 8 (73,7 мг, 0,1 ммоль) добавляли по каплям к раствору и оставляли для перемешивания в течение 30 минут. Раствор оставляли для подогрева до комнатной температуры и перемешивали еще в течение 5 часов. Смесь концентрировали при пониженном давлении, и остаток очищали препаративной ВЭЖХ с получением соединения 54 (73 мг, 63,6%) в виде белого порошка. ЖХ-МС m/z (ЭР+), 1148,47 (М+Н)+.
Синтез соединения 55
Бромацетилбромид (150,0 мг, 0,75 ммоль) добавляли к раствору соединения 54 (573,7 мг, 0,5 ммоль) в ДМФ (10 мл) при 0°С. Полученную в результате смесь перемешивали при 0°С в течение 10 минут, а затем оставляли для перемешивания при комнатной температуре еще на 3 часа. К смеси добавляли воду (20 мл) и экстрагировали этилацетатом (20 мл×3). Объединенные органические слои концентрировали, и остаток очищали препаративной ВЭЖХ с получением соединения 55 (421 мг, 66,4%) в виде желтого порошка. ЖХ-МС m/z (ЭР+), 1270,38 (М+Н)+.
ПРИМЕР I-9 синтез соединения 59
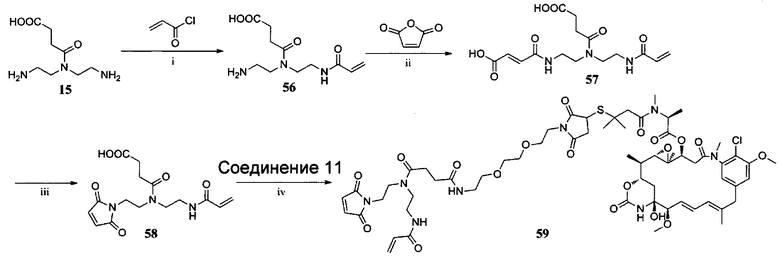
i) K2CO3, H2O/EtOAc, К. Т., 5 ч; ii) CHCl3, кипячение с обратным холодильником, 12 ч; iii) Ac2O, NaAc, 100°С, 2 ч; iv) HOBt, DIC, DIPEA, ДМФ, К. Т., 24 ч.
Синтез соединения 56
К смеси карбоната калия (51,2 мг, 0,37 ммоль) в воде (5 мл) и соединения 15 (50,0 мг, 0,25 ммоль) в этилацетате (10 мл) медленно добавляли по каплям раствор акрилоилхлорида (16 мкл, 0,2 ммоль) в этилацетате (8 мл) при 0°С. После завершения добавления полученную в результате смесь перемешивали при 0°С в ледяной бане дополнительно в течение 10 минут. Затем смесь оставляли для подогрева до комнатной температуры и проводили реакцию в течение 5 часов. Смесь подкисляли хлористоводородной кислотой до достижения рН менее 5. Смесь экстрагировали этилацетатом (15 мл×3). Объединенные органические слои промывали насыщенным раствором хлорида натрия (15 мл), высушивали над безводным сульфатом магния, фильтровали и удаляли растворитель. Твердый остаток очищали колоночной хроматографией (МеОН : CH2Cl2 составляет 8:1) с получением продукта 56 (26,2 мг, 40,1%) в виде серого масла. ЖХ-МС m/z (ЭР+), 258,14 (М+Н)+.
Синтез соединения 57
Соединение 56 (26,0 мг, 0,10 ммоль) перемешивали в CHCl3(10 мл), к смеси добавляли малеиновый ангидрид (28,1 мг, 0,11 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 12 часов. После охлаждения до комнатной температуры, выпавший в осадок твердый продукт фильтровали и промывали CHCl3 (30 мл×3) с получением соединения 57 (20,1 мг, 59,1%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 356,14 (М+Н)+.
Синтез соединения 58
Раствор соединения 57 (20,1 мг, 0,054 ммоль) и ацетата натрия (6,3 мг, 0,047 ммоль) в уксусном ангидриде (10 мл) нагревали до 100°С в течение 2 часов. Как только реакция была завершена, реакционный раствор медленно наливали в колотый лед при перемешивании и добавляли ледяную воду. Твердое вещество выпадало в осадок после 1 часа перемешивания. Твердый остаток фильтровали и промывали ледяной водой (15 мл×3) с получением соединения 58 (9,1 мг, 46,1%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 338,13 (М+Н)+.
Синтез соединения 59
Соединение 58 (8,1 мг, 0,02 ммоль), HOBt (3,25 мг, 0,02 ммоль) и DIC (3,7 мкл, 0,02 ммоль) растворяли в 5 мл ДМФ и охлаждали до 0°С в ледяной бане. Впоследствии к смеси добавляли соединение 11 (20,1 мг, 0,02 ммоль) и DIPEA (4,1 мкл, 0,02 ммоль). Раствор оставляли для подогрева до комнатной температуры и оставляли для взаимодействия на 24 часов. Полученную в результате смесь концентрировали и очищали препаративной ВЭЖХ с получением соединения 59 (9,3 мг, 34,9%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 1313,53 (М+Н)+.
ПРИМЕР I-10 синтез соединения 64
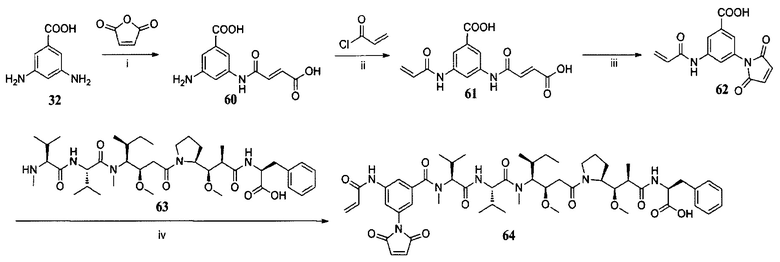
i) ДМФ, 80°C, 6 ч; ii) K2CO3, EtOAc/H2O, К. Т., 5 ч; iii) Ac2O, NaAc, 90°С, 3 ч; iv) HOBt, DIC, DIPEA, ДМФ, К. Т., 24 ч.
Синтез соединения 60
К раствору соединения 32 (140,0 мг, 0,9 ммоль) в ДМФ (6 мл) добавляли по каплям малеиновый ангидрид (89,8 мг, 0,9 ммоль). Смесь перемешивали при 60°С в течение 6 часов, а затем концентрировали до масла при пониженном давлении. Соединение 60 (белое твердое вещество, 209,1 мг, 90,7%) было получено путем перекристаллизации из этилацетата (5 мл). ЖХ-МС m/z (ЭР+), (М+Н)+ 251,06.
Синтез соединения 61
К смеси карбоната калия (201,3 мг, 1,4 ммоль) в воде (15 мл) и соединения 60 (209,1 мг, 0,9 ммоль) в этилацетате (20 мл) добавляли по каплям раствор акрилоилхлорида (90 мкл, 1,1 ммоль) в этилацетате (10 мл) при 0°С. После полного добавления смесь оставляли для подогрева до комнатной температуры и проводили реакцию в течение 5 часов. Смесь подкисляли хлористоводородной кислотой до достижения рН менее 5. Смесь экстрагировали этилацетатом (20 мл×2). Органические слои объединяли и промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом магния и фильтровали. Растворитель удаляли в вакууме с получением соединения 61 (201,0 мг, 82,7%) в виде серого твердого вещества, которое можно было использовать в следующей стадии без дополнительной очистки. ЖХ-МС m/z (ЭР+), (М+Н)+ 304,07.
Синтез соединения 62
К раствору неочищенного соединения 61 (201,0 мг, 0,7 ммоль) в уксусном ангидриде (30 мл) добавляли ацетат натрия (60,5 мг, 0,7 ммоль). Смесь перемешивали при 90°С в течение 3 часов. Реакционную смесь наливали в ледяную воду и перемешивали в течение 30 мин и экстрагировали этилацетатом (20 мл×3). Объединенные органические слои концентрировали и очищали препаративной ВЭЖХ с получением соединения 62 (92,2 мг, 48,6%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), (М+Н)+287,06.
Синтез соединения 64
Соединение 62 (57,0 мг, 0,2 ммоль), HOBt (27,2 мг, 0,2 ммоль), DIC (29,3 мкл, 0,2 ммоль) и DIPEA (0,2 ммоль, 33,1 мкл) добавляли к ДМФ (6 мл). Через 30 мин добавляли соединение 63 (109,8 мг, 0,15 ммоль) и добавляли при 0°С. Затем смесь оставляли для подогрева при комнатной температуры на 24 часа. Раствор концентрировали и очищали препаративной ВЭЖХ с получением соединения 64 (94,8 мг, 47,6%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 1000,53 (М+Н)+.
ПРИМЕР I-11 синтез соединения 69
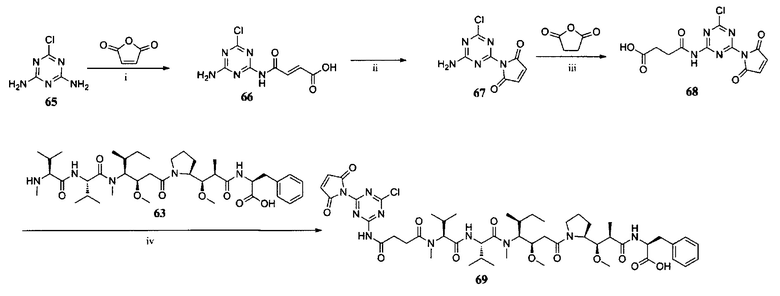
i) ДМФ, кипячение с обратным холодильником, 12 ч; iii) Ac2O, NaAc, кипячение с обратным холодильником, 6 ч; iii) ДМФ, 80°С, 12 ч; iv) HOBt, DIC, DIPEA, NMP, К. Т., 36 ч.
Синтез соединения 66
К раствору соединения 65 (145,0 мг, 1,0 ммоль) в ДМФ (6 мл) медленно добавляли по каплям малеиновый ангидрид (89,8 мг, 0,9 ммоль). Смесь нагревали с обратным холодильником в течение 12 часов. После охлаждения до комнатной температуры выпавший в осадок твердый продукт фильтровали и промывали CH2Cl2 (20 мл×3) с получением соединения 66 (110,2 мг, 45,1%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 244,02 (М+Н)+.
Синтез соединения 67
Смесь соединения 66 (110,2 мг, 0,5 ммоль) и ацетата натрия (14,2 мг, 0,2 ммоль) в уксусном ангидриде (10 мл) нагревали с обратным холодильником в течение 6 часов. Как только реакция была завершена, реакционную смесь медленно наливали в колотый лед при перемешивании и добавляли ледяную воду. Твердый остаток выпадал в осадок после 1 часа перемешивания. Твердый остаток фильтровали и промывали ледяной водой (10 мл×3) с получением соединения 67 (87,3 мг, 85,7%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 226,01 (М+Н)+.
Синтез соединения 68
Ангидрид янтарной кислоты (38,8 мг, 0,4 ммоль) добавляли к смеси соединения 67 (87,3 мг, 0,4 ммоль) в ДМФ (6 мл). Смесь перемешивали при 80°С в течение ночи, а затем концентрировали до масла при пониженном давлении. Неочищенный продукт очищали препаративной ВЭЖХ с получением соединения 68 (45,0 мг, 35,7%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 326,02 (М+Н)+.
Синтез соединения 69
Соединение 68 (45,0 мг, 0,14 ммоль), HOBt (19,0 мг, 0,14 ммоль), DIC (20,5 мкл, 0,14 ммоль) и DIPEA (23,2 мкл, 0,14 ммоль) добавляли к ДМФ (10 мл) и перемешивали при 0°С в ледяной бане в течение 1 часа. К раствору добавляли соединение 63 (76,9 мг, 0,11 ммоль). Смесь оставляли для подогрева до комнатной температуры и оставляли для взаимодействия на 36 часов. Реакционную смесь очищали препаративной ВЭЖХ с получением соединения 69 (64,3 мг, 44,3%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 1039,49 (М+Н)+.
ПРИМЕР I-12 синтез соединения 74
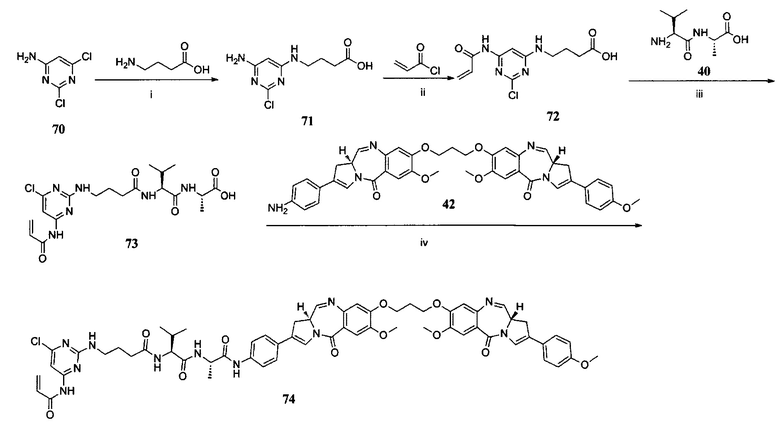
i) Et3N, н-бутанол, 90°С, 18 ч, ii) Et3N, CH2Cl2, К. Т., 18 ч, iii) EDCl, HOBt, DIPEA, ДМФ, К. Т., 18 ч, iv) EEDQ, CH2Cl2, МеОН, К. Т., 18 ч.
Синтез соединения 71
Соединение 69 (1,0 г, 6,10 ммоль) и 4-аминобутановую кислоту (691,9 мг, 6,71 ммоль) и триэтиламин (3,09 г, 30,5 ммоль) растворяли в н-бутаноле (30 мл). Реакционную смесь перемешивали в течение 18 часов при 90°С. После охлаждения до комнатной температуры растворитель концентрировали при пониженном давлении. Остаток разбавляли водой (100 мл) и экстрагировали этилацетатом (30 мл×3). Органическую фазу объединяли и высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией (МеОН : CH2Cl2 составляет 200/1 ~10/1, об./об.) с получением соединения 71 в виде желтого твердого вещества (482,3 мг, 34,3%). ЖХ-МС m/z (ЭР+), 231,06 (М+Н)+.
Синтез соединения 72
Акрилоилхлорид (947,2 мг, 10,47 ммоль) добавляли по каплям к раствору соединения 71 (450,5 мг, 1,95 ммоль) и ТЭА (590,8 мг, 5,85 ммоль) в CH2Cl2 (20 мл) при перемешивании при 0°С в ледяной бане. Реакционную смесь перемешивали в течение 18 часов при комнатной температуре. Реакционную смесь гасили добавлением воды (50 мл) и экстрагировали CH2Cl2 (20 мл×3). Органический слой объединяли, промывали раствором хлорида натрия (30 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали с получением соединения 72 (255,4 мг, 46%) в виде желтого твердого вещества. ЖХ-МС m/z (ЭР+), 285,07 (М+Н)+.
Синтез соединения 73
Соединение 3 (155,7 мг, 0,547 ммоль), HOBt (88,7 мг, 0,656 ммоль), EDCl (115,4 мг, 0,602 ммоль) и DIPEA (212,1 мг, 1,64 ммоль) добавляли к ДМФ (10 мл) и перемешивали при 0°С в ледяной бане в течение 1 часа. К раствору добавляли соединение 40 (113,3 мг, 0,602 ммоль). Смесь оставляли для подогрева до комнатной температуры и оставляли для взаимодействия на 18 часов. Реакционную смесь разбавляли раствором хлорида натрия (30 мл), экстрагировали CH2Cl2 (10 мл×3). Органический слой объединяли и высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией (МеОН : CH2Cl2 составляет 200/1-10/1, об./об.) с получением соединения 73 в виде желтого твердого вещества (90,2 мг, 36,2%). ЖХ-МС m/z (ЭР+), 455,17 (М+Н)+.
Синтез соединения 74
Раствор соединения 73 (85,4 мг, 0,19 ммоль), EEDQ (55,4 мг, 0,23 ммоль) в смеси безводного CH2Cl2 (7 мл) и безводного МеОН (0,07 мл) перемешивали в течение 1 часа при комнатной температуре. Впоследствии добавляли соединение 42 (135,7 мг, 0,19 ммоль). Реакционную смесь перемешивали дополнительно в течение 18 часов. Реакционную смесь концентрировали в вакууме и очищали препаративной ВЭЖХ с получением соединения 74 (19,1 мг, 6,7%) в виде белого твердого вещества. ЖХ-МС m/z (ЭР+), 1162,45 (М+Н)+.
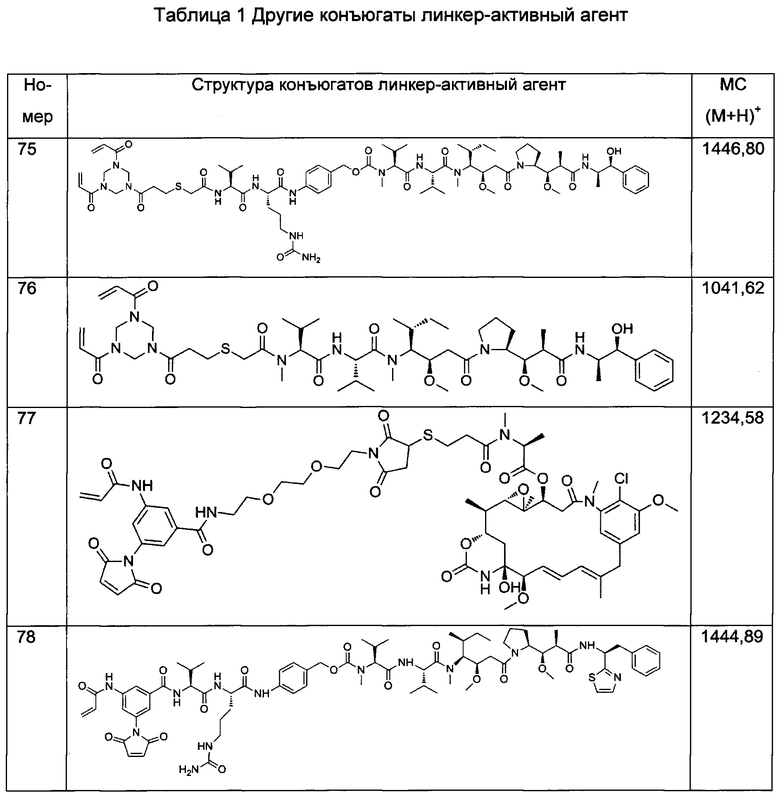
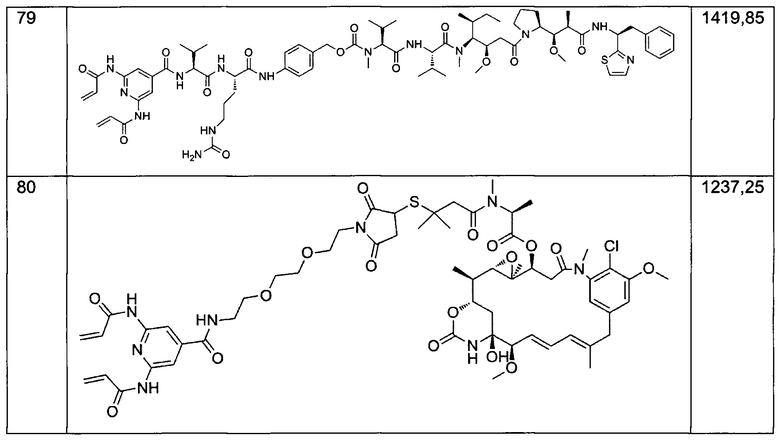
ПРИМЕР II получение конъюгатов антитело-лекарственное средство
Общая методика конъюгации
Диэтилентриаминпентауксусную кислоту (DTPA) (1 мМ) и 3,0-5,0 экв. трис-(2-карбоксиэтил)фосфина (ТСЕР) (10 мМ) добавляли к антителу (10 мг/мл) в пробирке для ПЦР и перемешивали в течение 1 часа для восстановления дисульфидных связей на антителе. К реакционной смеси добавляли различные количества конъюгата линкер-активный агент и перемешивали встряхиванием в течение ночи с помощью вихревой мешалки. Все реакции проводили при комнатной температуре. Полученные в результате продукты анализировали в гель-электрофорезе (ДСН-ПААГ) и ГИХ (гидрофобной интерактивной хроматографии)-ВЭЖХ.
Конъюгат антитело-лекарственное средство анализировали методом электрофореза в ДСН-ПААГ в восстанавливающих условиях: исследуемые образцы, смешанные с буферным раствором, наносили на 8%-15% акриламидные гели и делали фотоснимки гелей с помощью системы визуализации UVP BioDoc-it.
Полосы (больше или равно 50 кДа) были видны на дорожках 2 и 3, что позволяет предположить, что конъюгация (Н, L) посредством мостиковых связей с тиолами была эффективной. Полосы при 75 кДа представляют фрагмент, состоящий из тяжелой и легкой цепи, связанных одной парой мостиковых тиолов (LH). Две тяжелые цепи были связаны одной или двумя парами мостиковых тиолов, которые образовывали полосы при 100 кДа (НН). Полосы при 125 кДа представляют собой фрагмент, в котором только одна мостиковая связь между тяжелой и легкой цепью не образовалась (LHH). Наконец, полосы при 150 кДа выявляют образование полного мостикового антитела (LHHL).
Конъюгат антитело-лекарственное средство анализировали ГИХ-ВЭЖХ в следующих условиях: колонка ВЭЖХ: колонка TSK-GEL бутил-NPR 4,6-35 мм; буфер А: 75% фосфат натрия (20 мМ, рН 7,0), 25% изопропанол (об./об.); буфер В: фосфат натрия (20 мМ, рН 7,0), сульфат аммония (1,5 мМ); скорость потока: 1,0 мл/мин.
Оценка конъюгатов антитело-лекарственное средство in vitro
В первый день линию опухолевых клеток из линий клеток рака яичника человека, такую как OVCAR3, высевали в количестве 5000 клеток в 100 мкл культуральной среды в 96-луночный культуральный планшет (Cellstar). Клетки инкубировали в CO2 инкубаторе с насыщением влагой при 37°С в течение ночи. На второй день готовили серийные разведения двух ADC MSL-31 и MSL-75, конъюгированных посредством ковалентных тиольных связей, и добавляли в лунки 96-луночного планшета, содержащие 100 мкл на лунку клеток OVCAR3. Первоначальная концентрация конъюгата составляла 100000 нг/мл, и его разводили до 0,001 нг/мл. Клетки OVCAR3 с добавлением ADC инкубировали при 37°С в течение 72 часов. Впоследствии жизнеспособность опухолевых клеток, обработанных ADC, конъюгированным посредством ковалентных тиольных связей, измеряли на считывающем устройстве для планшетов (BIO-RAD iMark SpectraMax L от компании Molecular device), используя набор реагентов для определения жизнеспособности клеток Cell Counting Kit-8 (DOJINDO, CK04) в соответствии с протоколом производителя. Значение 50% ингибирования роста клеток IC50 рассчитывали с помощью программного обеспечения для подбора кривой Graphpad.
ПРИМЕР II-1 получение msl-c31
DTPA (1 мМ) и 5,0 экв. ТСЕР (10 мМ) добавляли к антителу к MSL (10 мг/мл) в пробирке для ПЦР. Смесь перемешивали встряхиванием на вихревой мешалке в течение 1 часа для восстановления дисульфидных связей на антителе. Впоследствии к реакционным смесям добавляли 25% ДМСО. Соединение 31 (5,0 экв., 10 мМ) добавляли к реакционным смесям и перемешивали встряхиванием на вихревой мешалке в течение ночи. Все реакции проводили при комнатной температуре. После завершения реакции смесь загружали в центрифужную пробирку для ультрафильтрации Millipore емкостью 4 мл и промывали три раза ФСБ, затем собирали реакционные смеси. На Фиг. 2 показано, что продукты (дорожка справа) ковалентной конъюгации по меньшей мере одной пары сульфгидрильных групп будут мигрировать медленнее, чем тяжелая (Н) или легкая (L) цепь отдельно (показано на дорожке слева). В результате ковалентной конъюгации тяжелой и легкой цепей образовывалась полоса 75 кДа (HL); в результате ковалентной конъюгации двух тяжелых цепей образовывалась полоса 100 кДа (НН). На хроматограмме ГИХ-ВЭЖХ наблюдали 3 пика, представляющих долю лекарственного средства на антитело (DAR, drug antibody ratio) 3, 4 и 5 соответственно. DAR полученного в результате MSL-C31 составляла 3,60 на основании результатов ГИХ-ВЭЖХ (Фиг. 3).
На Фиг. 2 показан результат электрофореза в ДСН-ПААГ в восстанавливающих условиях для а) чистого антитела, b) MSL-C31, продукта ковалентной конъюгации с тиолом.
На Фиг. 3 показана хроматограмма ГИХ-ВЭЖХ MSL-C31.
ПРИМЕР II-2 получение msl-c75
DTPA (1 мМ) и 5,0 экв. ТСЕР (10 мМ) добавляли к антителу к MSL (10 мг/мл) в пробирке для ПЦР. Смесь перемешивали встряхиванием на вихревой мешалке в течение 1 часа для восстановления дисульфидных связей на антителе. Впоследствии к реакционным смесям добавляли 25% ДМСО. 5,0 экв. соединения 75 (5,0 экв., 10 мМ) добавляли к реакционным смесям и перемешивали встряхиванием на вихревой мешалке в течение ночи. Все реакции проводили при комнатной температуре. После завершения реакции смесь загружали в центрифужную пробирку для ультрафильтрации Millipore емкостью 4 мл и промывали три раза ФСБ, затем собирали реакционные смеси. Образцы подвергали анализу методом электрофореза. Результаты электрофореза в ДСН-ПААГ в восстанавливающих условиях представлены на Фиг. 4. На дорожке слева нанесено чистое антитело, а на дорожке справа нанесен ковалентный конъюгат MSL-C75. DAR полученного в результате MSL-C75 составляла 3,87 на основании результатов ГИХ-ВЭЖХ.
На Фиг. 4 показаны результаты электрофореза MSL-C75 в ДСН-ПААГ в восстанавливающих условиях.
На Фиг. 5 показана хроматограмма ГИХ-ВЭЖХ MSL-C75.
В таблице 2 приведены значения IC50 двух образцов: ADC-C31 и ADC-C75, протестированных на клетках рака яичника человека.

ПРИМЕР II-3 получение cd59-c78
DTPA (1 мМ) и 5,0 экв. ТСЕР (10 мМ) добавляли к антителу к CD59 (10 мг/мл) в пробирке для ПЦР. Смесь перемешивали встряхиванием на вихревой мешалке в течение 1 часа для восстановления дисульфидных связей на антителе. Впоследствии к реакционным смесям добавляли 25% ДМСО. Соединение 78 (5,0 экв., 10 мМ) добавляли к реакционным смесям и перемешивали встряхиванием на вихревой мешалке в течение ночи. Все реакции проводили при комнатной температуре. После завершения реакции смесь загружали в центрифужную пробирку для ультрафильтрации Millipore емкостью 4 мл и промывали три раза ФСБ. DAR полученного в результате CD59-C78 составляла 3,66 на основании результатов ГИХ-ВЭЖХ.
На Фиг. 6 показан результат электрофореза в ДСН-ПААГ в восстанавливающих условиях для а) очищенного антитела, b) CD59-C78. На Фиг. 7 показана хроматограмма ГИХ-ВЭЖХ CD59-C78.
ПРИМЕР II-4 получение конъюгата her2-c78
DTPA (1 мМ) и 5,0 экв. ТСЕР (10 мМ) добавляли к антителу к Her2 (10 мг/мл) в пробирке для ПЦР. Смесь перемешивали встряхиванием на вихревой мешалке в течение 1 часа для восстановления дисульфидных связей на антителе. Впоследствии к реакционным смесям добавляли 25% ДМСО. Соединение 75 (5,0 экв., 10 мМ) добавляли к реакционным смесям и перемешивали встряхиванием на вихревой мешалке в течение ночи. Все реакции проводили при комнатной температуре. После завершения реакции смесь загружали в центрифужную пробирку для ультрафильтрации Millipore емкостью 4 мл и промывали три раза ФСБ.
Исследование стабильности конъюгата HER2-C78
Для определения степени распада ADC ковалентной тиольной конъюгации по сравнению со стандартной тиольной конъюгацией для исследования стабильности использовали три образца ADC, приготовленных с помощью описанных выше методик твердофазного иммуносорбентного анализа (ELISA). ELISA выполняли по следующему плану: сначала внеклеточный домен (ECD) Her2 (100 нг) наносили в виде покрытия в каждую лунку 96-луночного планшета; затем периферическую кровь человека, обработанную конъюгатом антитело к Her2-Mc-VC-PAB-MMAE (полученным стандартной тиольной конъюгацией), конъюгатом антитело к Her2-Di-Mc-VC-PAB-MMAE (полученным конъюгацией посредством би-малеимида-тиола) и конъюгатом антитело к Her2-PY-QJ2-ADC (Her2-С78, полученным ковалентной тиольной конъюгацией по данному изобретению) инкубировали в лунке с нанесенным покрытием, чтобы дать возможность связывания с ECD Her-2. Наконец, количество ADC к her2, связанного с антигеном Her2-ECD, измеряют на основании связывания с антителом к IgG человека, конъюгированным со стрептавидином/пероксидазой хрена, которая впоследствии катализирует преобразование хромогенного субстрата орто-фенилендиамина (OPD) в окрашенный продукт. Результаты показывают, что в течение 3 дней инкубации с образцами периферической крови степень распада ADC в результате стандартной тиольной конъюгации составляла 26,7%, для 2го ADC с бималеимид-тиольной конъюгацией она составляла 18,9%, а для 3го ADC по данному изобретению она составляла 5,1%. Степень распада ADC с ковалентным линкером по данному изобретению является самой низкой среди трех ADC, как показано ниже в таблице 3.
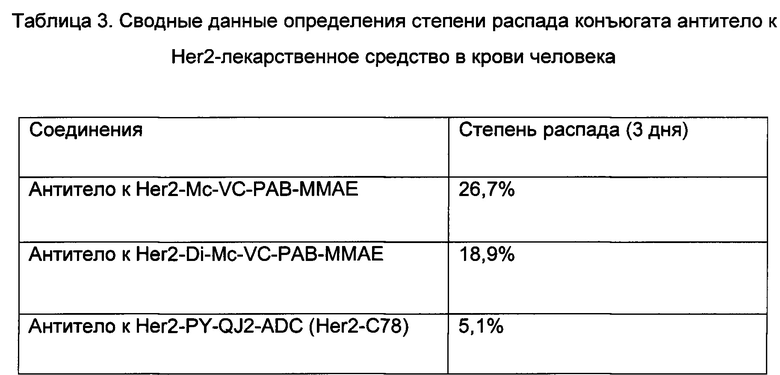
Примечания: Антитело к Her2-Mc-VC-PAB-MMAE было получено традиционной тиольной конъюгацией (ссылка 1) и антитело к Her2-Di-Mc-VC-PAB-MMAE было получено ковалентной конъюгацией с использованием структуры и методики, описанных в заявке на патент (ссылка 2). Значения DAR обоих конъюгатов были сопоставимы со значениями для Her2-С78.
ПРИМЕР III - Активность конъюгатов антитело-лекарственное средство
Конъюгаты антитело-лекарственное средство заявленного изобретения в одном воплощении изобретения можно использовать для специфического нацеливания на опухолевые клетки и связывания с опухолеассоциированными антигенами на поверхности этих опухолевых клеток. Комплекс ADC-антиген будет проникать в опухолевую клетку посредством эндоцитоза и высвобождает нагрузку после слияния эндосомы комплекса ADC-антиген с лизосомами. Высвобождаемая нагрузка ингибирует ключевые функции опухолевой клетки и/или уничтожать опухолевые клетки. Если нагрузки высвобождаются на поверхности клеток или вблизи опухолевых клеток, опухолевая клетка может быть также уничтожена посредством обходного пути.
Таким образом, в изобретении предложены различные комплексы белок-лекарственное средство, которые ингибируют рост опухолевых клеток и/или их дифференцировку. Эти соединения, содержащие различные виды активных нагрузок, можно применять как для лечения новообразований, аутоиммунных расстройств и воспаления, так и для различения опухолевых или стволовых клеток.
ПРИМЕР III-1 - Активность против опухолей
В одном воплощении заявленного изобретения в конъюгатах антитело-лекарственное средство (ADC) объединены свойства уникального нацеливания mAb со способностью к уничтожению раковых клеток цитотоксическими лекарственными средствами, что позволяет ADC быть активными противоопухолевыми агентами. Кроме того, использование ковалентной тиольной конъюгации в соответствии с заявленным изобретением повышает стабильность ADC, ограничивая разложение ADC в кровообращении, за счет чего снижается их токсичность. В данном примере представлены данные, показывающие лечение рака человека с применением ADC, полученных, как описано выше, у бестимусных мышей с имплантированными клетками рака молочной железы человека (MDA-MB-231).
Чтобы определить биологическую функцию различных ADC с ковалентными или стандартными конъюгатами, mAb к Her-2 конъюгировали посредством тиола путем стандартной конъюгации или ковалентной тиольной конъюгации с тем же линкером и цитотоксином. Бестимусным мышам имплантировали линию клеток рака человека (MDA-MB-231) и вводили ADC (0, 2,5, 5,0 и 10 мг/кг) посредством интраперитонеальной инъекции после достижения объема опухоли около 100 кубических миллиметров. Объемы опухоли контролировали раз в 3 дня, и по окончании 21-дневного периода животных умерщвляли, и опухоли вырезали и взвешивали.
В таблице 4 ниже приведено сравнение эффективности стандартных ADC с ADC, полученными коавлентной тиольной конъюгацией, введенных бестимусным мышам, которым были имплантированы раковые клетки молочной железы человека.
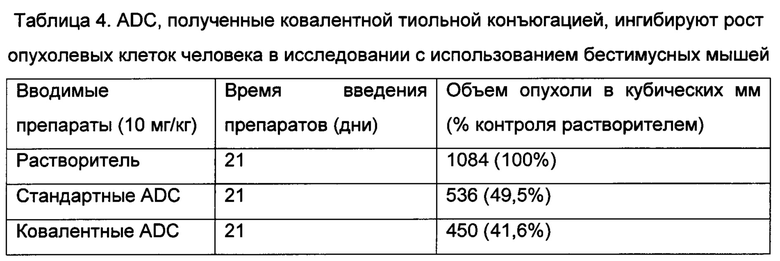
Ссылки
1. Yao Xuejing, Jing Jiang, Xin Wang, Changjiang Huang Dong Li, Kuan Xie, Qiaoyu Xu, Hongwen Li, Zhuanglin Li, Liguang Lou and Jianmin Fang, 2015. A novel humanized anti-her2 antibody conjugated with MMAE exerts potent anti-tumor activity, Breast Cancer Research & Treatment, 153(1): 123-133.
2. An Deqiang et al., 2014 07 23  CN 103933575 A
CN 103933575 A
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛИНКЕР ДЛЯ КОНЪЮГАТОВ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2792201C2 |
| ПЕПТИДОМИМЕТИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ КОНЪЮГАТЫ АНТИТЕЛ С ЛЕКАРСТВЕННЫМИ СРЕДСТВАМИ | 2014 |
|
RU2689388C1 |
| МОНОМЕТИЛВАЛИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБНЫЕ ОБРАЗОВЫВАТЬ КОНЪЮГАТЫ С ЛИГАНДАМИ | 2004 |
|
RU2448117C2 |
| ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ КОНЪЮГАТА АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ SN38, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2022 |
|
RU2840789C2 |
| Бифункциональные цитотоксические агенты | 2015 |
|
RU2669807C2 |
| АНТИ-МЕЗОТЕЛИН АНТИТЕЛО И ЕГО КОНЪЮГАТ С ЛЕКАРСТВЕННЫМИ СРЕДСТВАМИ | 2019 |
|
RU2747995C1 |
| Модулирующие стабильность линкеры для использования с конъюгатами антитело-лекарственное средство | 2015 |
|
RU2680238C2 |
| ХЕМОСЕЛЕКТИВНАЯ ТИОЛ-КОНЪЮГАЦИЯ С АЛКЕН- ИЛИ АЛКИН-ФОСФОНОТИОЛАТАМИ И -ФОСФОНАТАМИ | 2019 |
|
RU2825582C2 |
| ПАРА-АМИНОБЕНЗИЛЬНЫЕ ЛИНКЕРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КОНЪЮГАТАХ | 2021 |
|
RU2839675C1 |
| ЦИТОТОКСИЧЕСКИЕ ПЕПТИДЫ И ИХ КОНЪЮГАТЫ АНТИТЕЛО-ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2012 |
|
RU2586885C2 |
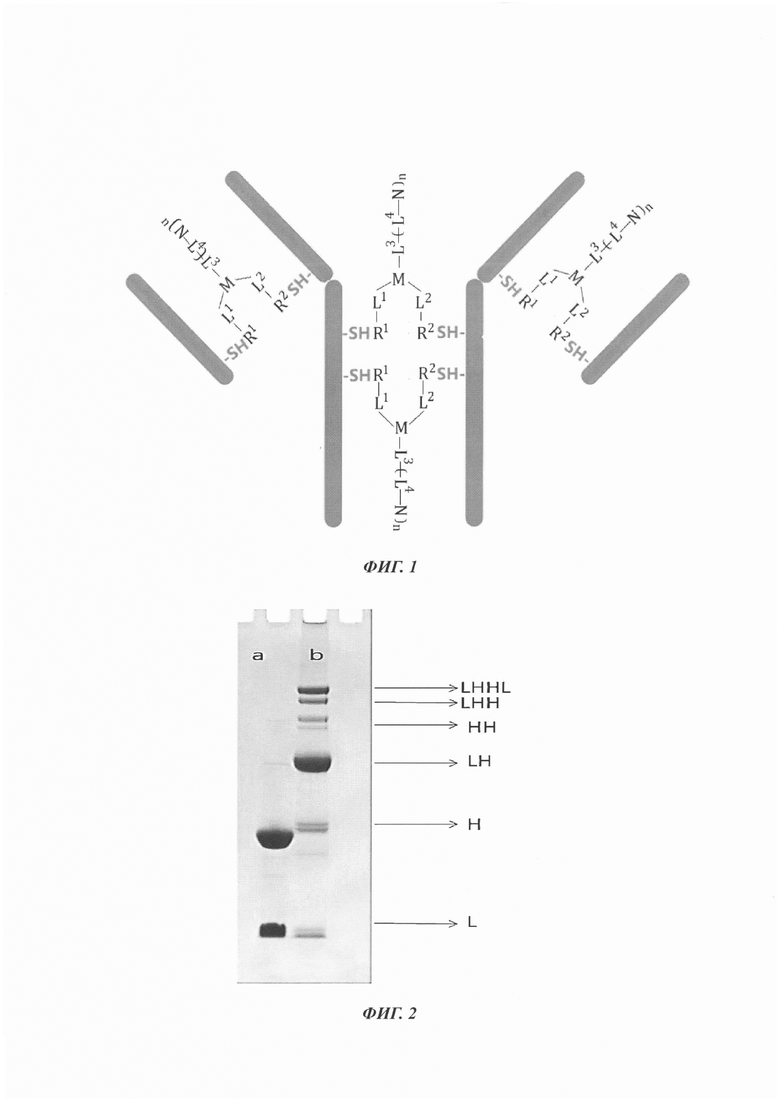
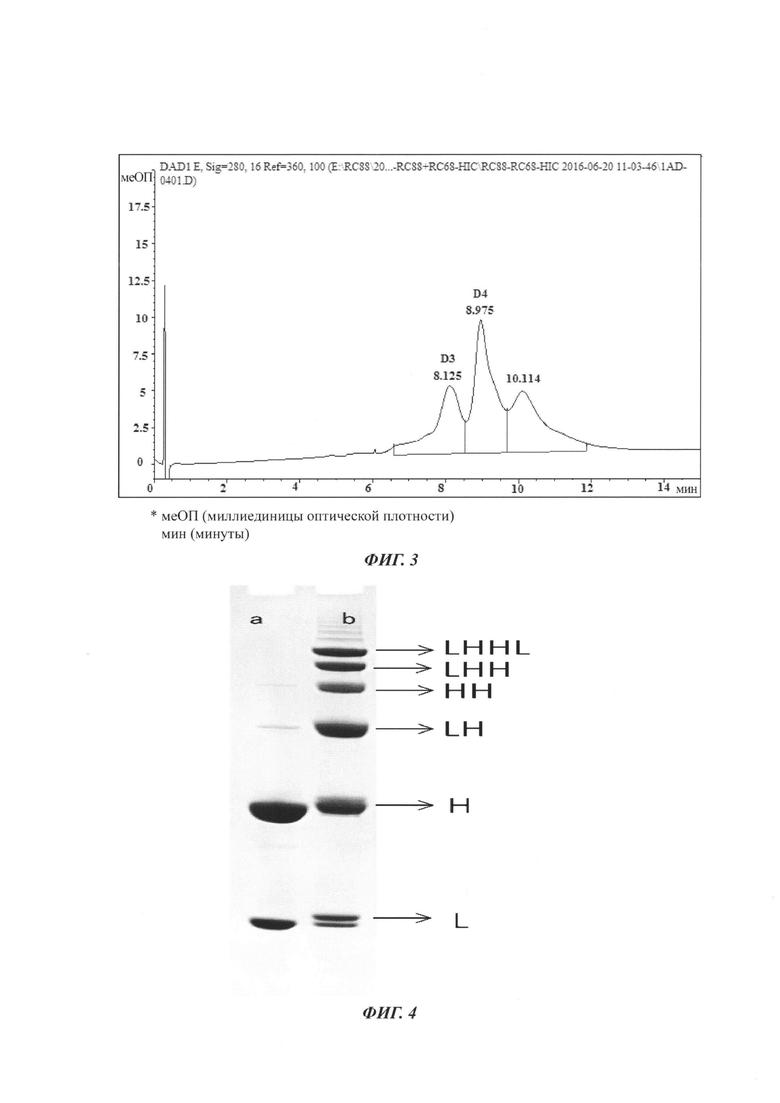
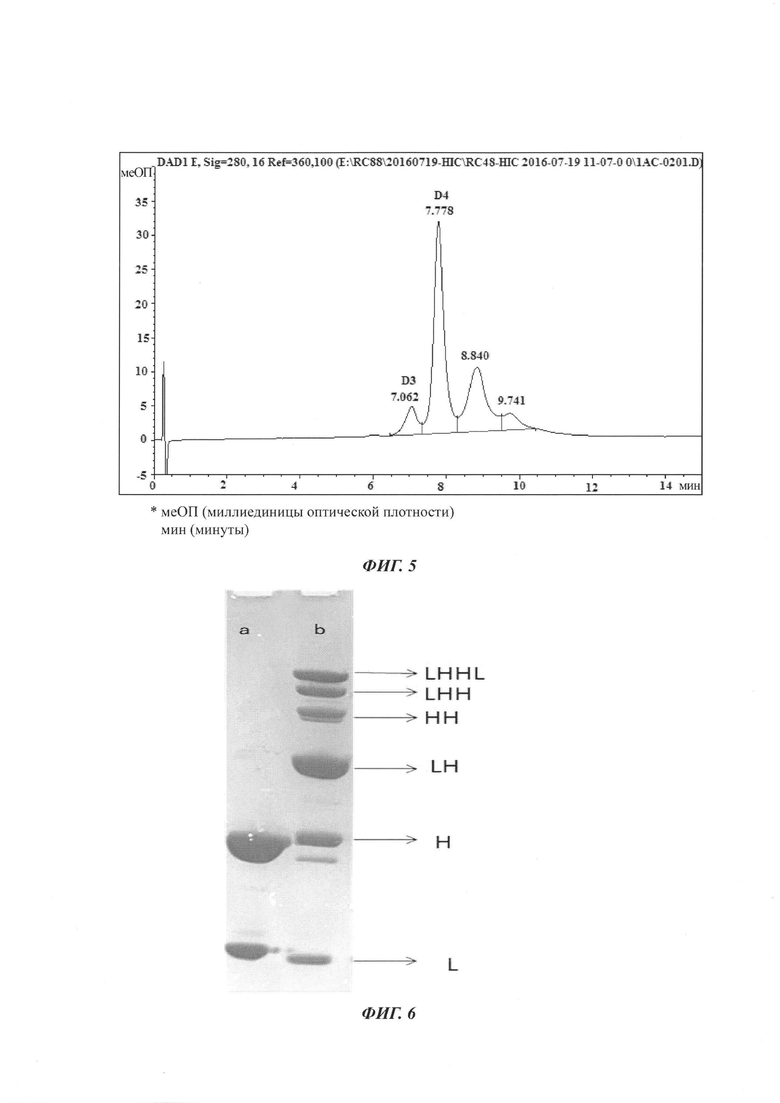
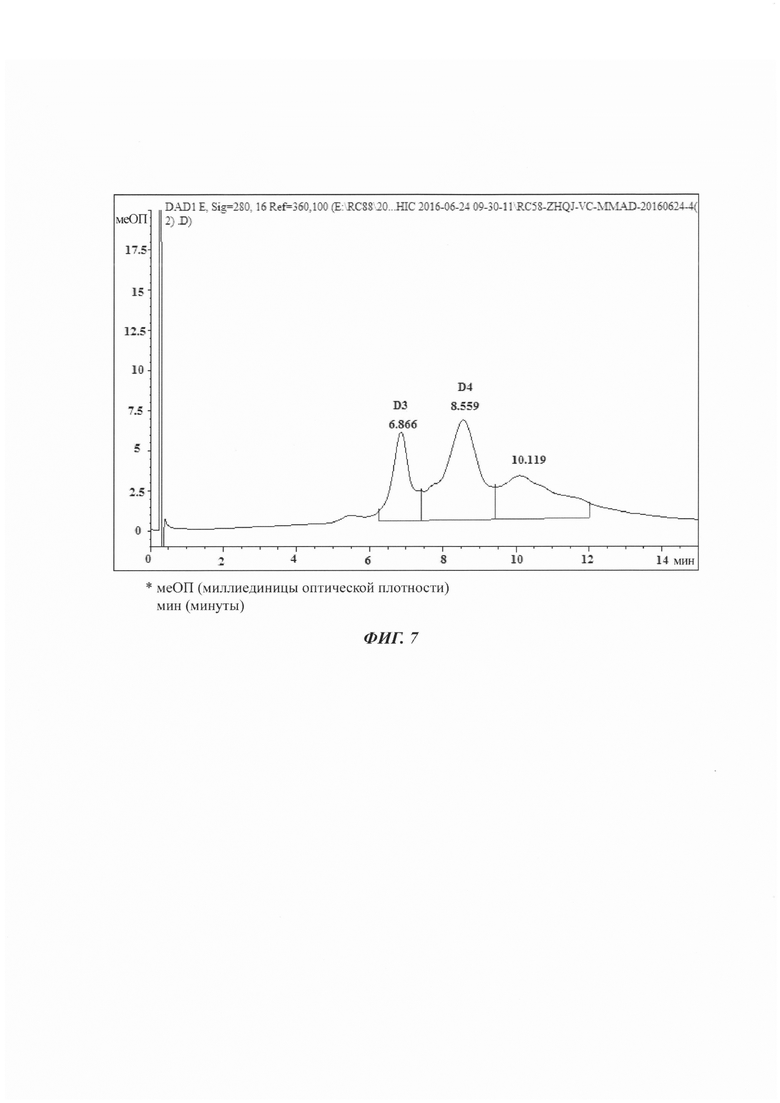
Группа изобретений относится к медицине и касается линкер-активного агента для получения конъюгата антитело-лекарственное средство, содержащего соединение формулы I или его фармацевтически приемлемую соль. Группа изобретений также касается конъюгата нацеливающая группировка-лекарственное средство для лечения HER2-положительного рака у субъекта; способа лечения HER2-положительного рака у субъекта, включающего введение субъекту, нуждающемуся в таком лечении, конъюгата нацеливающая группировка-лекарственное средство. Группа изобретений обеспечивает снижение скорости разложения противоракового коньюгата по сравнению с известными коньюгатами. 3 н. и 10 з.п. ф-лы, 3 пр., 7 ил., 4 табл.
1. Линкер-активный агент для получения конъюгата антитело-лекарственное средство, содержащий
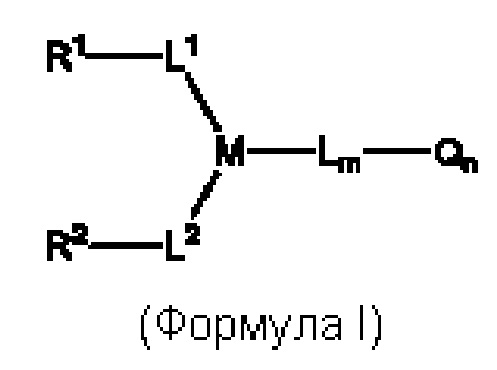
или его фармацевтически приемлемую соль,
где R1 и R2 представляют собой функциональные группы, которые могут взаимодействовать с тиолами;
L1, L2, L представляют собой линкер или могут отсутствовать;
М представляет собой линкерную группу;
Q представляет собой активный агент;
m представляет собой целое число от 0 до 6;
n представляет собой целое число от 0 до 8, при этом n не представляет собой 0;
каждый R1 и R2 содержит по меньшей мере одну группу, выбранную из группы, состоящей из малеимида, атома галогена, галоген-замещенных функциональных групп, алканаля, алканона, сульфонил-алкена, силана, изоцианата, 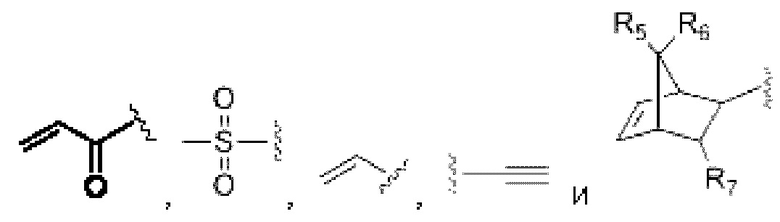
и их производных, где R5, R6 и R7 независимо выбраны из группы, состоящей из Н, атома галогена, С1-С6 алкилов, С2-С6 алкенилов, С2-С6 алкинилов, ароматических групп, гетероарилов, С3-С9 циклоалкилов, C3-C9 гетероциклила, полиэтиленгликоля, NO2, CN, SCN, OR8, SR9, NR10R11, C(=O)R12, C(=O)OR8, C(=O)NR10R11, C(=S)OR8, C(=S)NR10R11, C(=S)SR9, NR10(C=O)R12, NR10(C=S)NR10R11, O(C=O)NR10R11, SO2R9, S(=O)2OR8 и их комбинаций, где R8, R9, R10, R11, R12 независимо выбраны из группы, состоящей из Н, C1-C6 алкила, C1-C6 алкенилов, C2-C6 алкинилов и их комбинаций;
и R1 и R2 оба не могут быть малеимидом, и R1 и R2 оба не могут быть атомом галогена;
М выбран из группы, состоящей из C-R5, N, В, Р, Si-R5, ароматических групп, гетероарилов, циклоалкилов и (гетероциклил)алкилов, где R5 выбран из группы, состоящей из Н, C1-C6 алкилов, C2-C6 алкенилов и С2-С6 алкинилов;
L1 и L2 независимо выбраны из группы, состоящей из C1-C6 алкилов, С2-С6 алкенилов, С2-С6 алкинилов, ароматических групп, гетероарилов, С3-С9 циклоалкилов, C3-C9 гетероциклилов, полиэтиленгликоля, О, S, NR6, С(=O), С(=O)O, C(=O)NR6, C=NR6, C(=S)O, C(=S)NR6, C(=S)S, NR6(C=O), NR6(C=S)NR7, O(С=O)NR6, S(=O)2 и их комбинаций, и R6 и R7 независимо выбраны из группы, состоящей из Н, C1-C6 алкилов, C2-C6 алкенилов и C2-C6 алкинилов;
L содержит нерасщепляемое звено и/или расщепляемое звено, и L содержит C1-C9 алкил, C2-C9 алкенил, C2-C9 алкинил, ароматическую группу, гетероарил, С3-С9 циклоалкил, С3-С9 гетероциклил, полиэтиленгликоль, О, S, NR8, С(=O), С(=O)O, C(=O)NR8, C=NR8, C(=S)O, C(=S)NR8, C(=S)S, NR8(C=O), NR8(C=S)NR9, O(С=O)NR8, S(=O)2, Val-Cit-PAB, Val-Ala-PAB, Val-Lys(Ac)-PAB, Phe-Lys-PAB, Phe-Lys(Ac)-PAB, D-Val-Leu-Lys, Gly-Gly-Arg, Ala-Ala-Asn-PAB, Ala-PAB, РАВ или их комбинацию, где R8 и R9 независимо выбраны из группы, состоящей из Н, C1-C6 алкилов, О2-С6 алкенилов и С2-С6 алкинилов; и
Q выбран из группы, состоящей из агентов, связывающих тубулины, алкилирующих агентов ДНК, интеркаляторов ДНК, ингибиторов ферментов, иммуномодуляторов, пептидов и нуклеотидов.
2. Линкер-активный агент по п. 1, где R1 и R2 независимо выбраны из группы, состоящей из 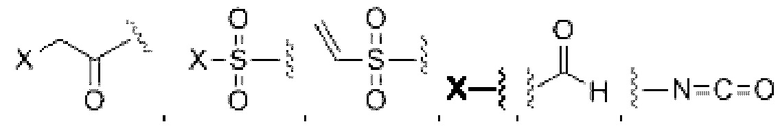
и норборнена и их производных, и X выбран из группы, состоящий из F, Cl, Br и I.
3. Линкер-активный агент по п. 1 или 2, где R1 и R2 являются разными.
4. Линкер-активный агент по п. 1 или 2, где Q выбран из группы, состоящей из майтанзиноидов, ауристатинов, калихеамицинов, доксорубицинов, дуокармицинов и пирролобензодиазепинов.
5. Линкер-активный агент по п. 1 или 2, где структура формулы I представляет собой любую, выбранную из группы, состоящей из:
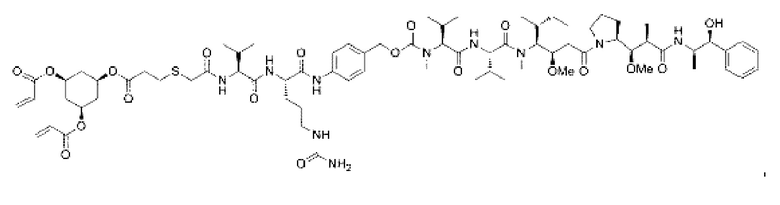
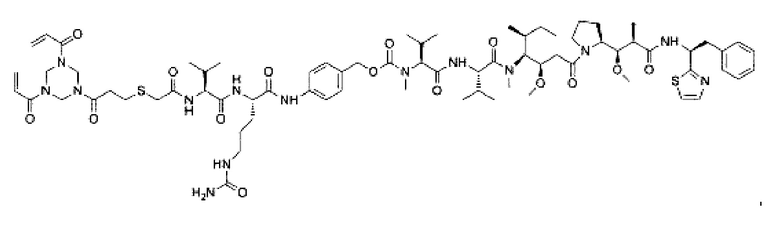
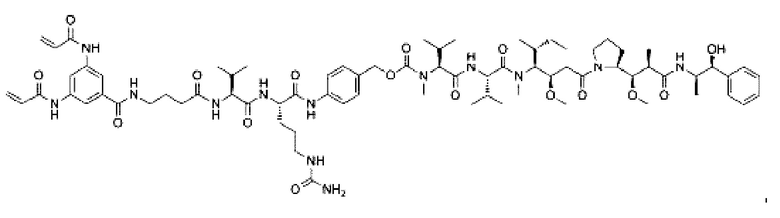
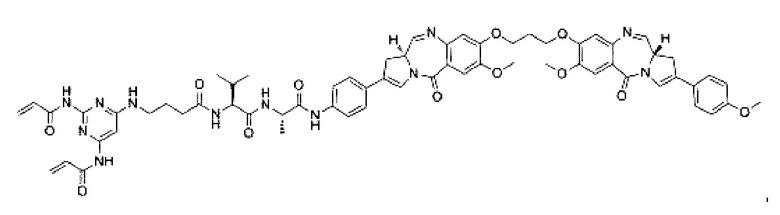
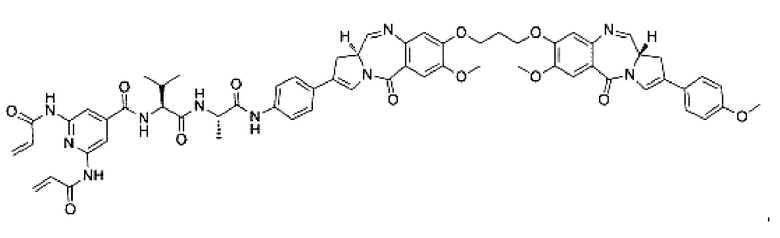
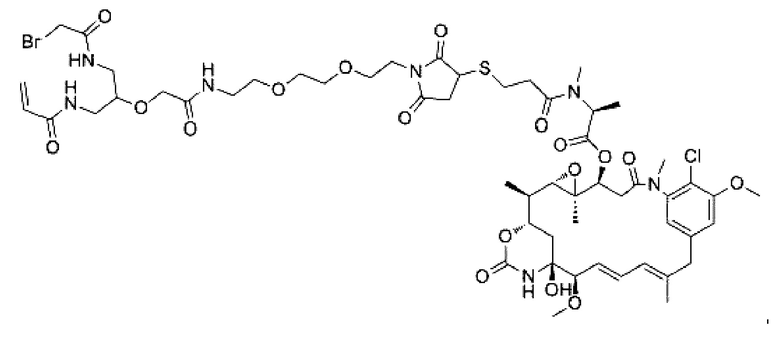
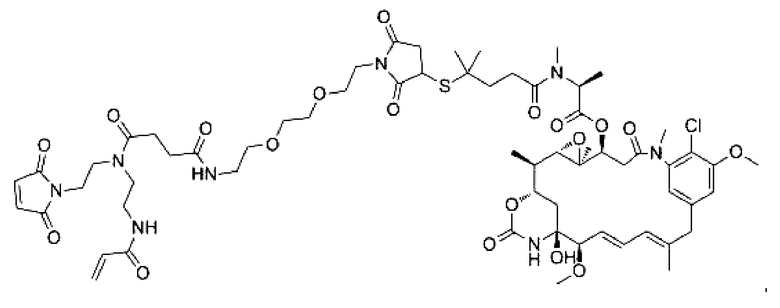
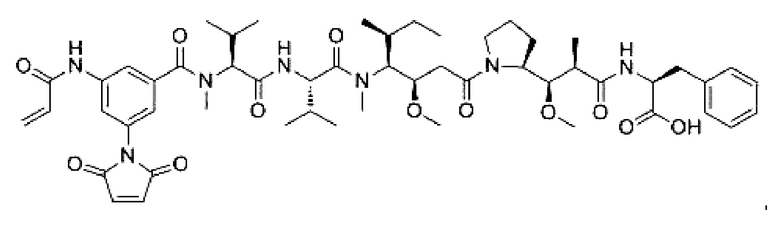
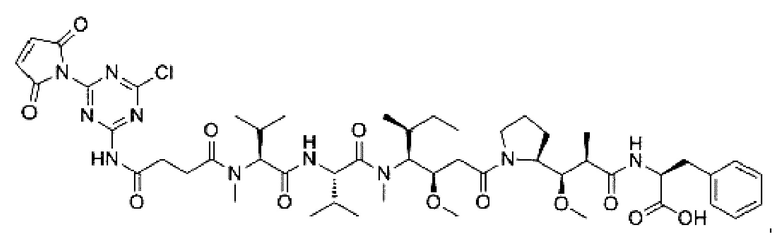
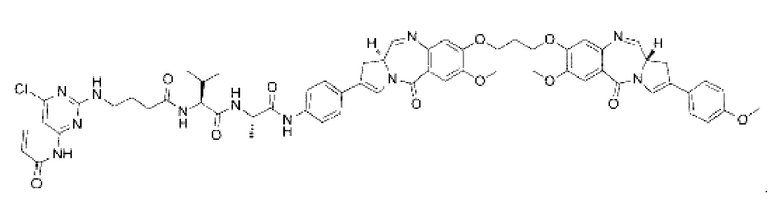
6. Конъюгат нацеливающая группировка-лекарственное средство для лечения HER2-положительного рака у субъекта, имеющий следующую формулу:
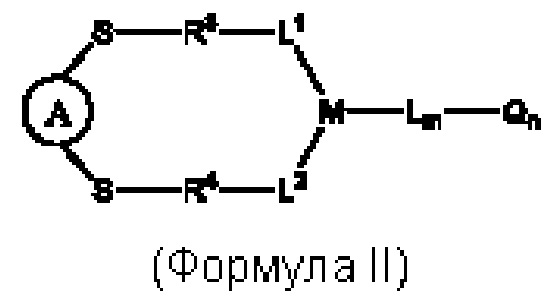
или его фармацевтически приемлемая соль,
где L1, L2, L представляют собой линкер или могут отсутствовать;
М представляет собой линкерную группу;
Q представляет собой активный агент;
m представляет собой целое число от 0 до 6;
n представляет собой целое число от 0 до 8, при этом n не представляет собой 0;
А представляет собой нацеливающую группировку, где А содержит моноклональное антитело, фрагмент антитела, белковый лиганд, белковый каркас, пептид или низкомолекулярный лиганд, где фрагмент антитела выбран из группы, состоящей из Fab, Fab', (Fab')2, scFv и Fv;
S представляет собой атом серы тиольной группы нацеливающей группировки; и
каждый из R3 и R4 образован в результате взаимодействия между S и функциональной группой, которая может взаимодействовать с тиолами, и R3 и R4 являются разными, и где каждый R3 и R4 образуется независимо в результате взаимодействия между тиольной группой и группой, выбранной из группы, состоящей из
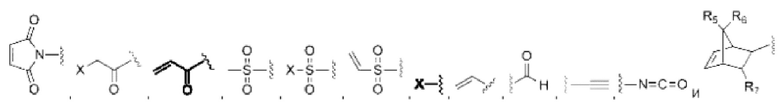
и их производных, где
R5, R6 и R7 независимо выбраны из группы, состоящей из Н, атома галогена, C1-C6 алкилов, С2-С6 алкенилов, C2-С6 алкинилов, ароматических групп, гетероарилов, С3-С9 циклоалкилов, С3-С9 гетероциклила, полиэтиленгликоля, NO2, CN, SCN, OR8, SR9, NR10R11, C(=O)R12, C(=O)OR8, C(=O)NR10R11, C(=S)OR8, C(=S)NR10R11, C(=S)SR9, NR10(C=O)R12, NR10(C=S)NR10R11, O(C=O)NR10R11, SO2R9, S(=O)2OR8 и их комбинаций, где R8, R9, R10, R11, R12 независимо выбраны из группы, состоящей из Н, C1-С6 алкила, С2-С6 алкенилов, С2-С6 алкинилов и их комбинаций, и X выбран из группы, состоящей из F, Cl, Br и I;
М выбран из группы, состоящей из C-R5, N, В, Р, Si-R5, ароматических групп, гетероарилов, циклоалкилов и (гетероциклил)алкилов, и где R5 выбран из группы, состоящей из Н, C1-С6 алкилов, C2-C6 алкенилов и С2-С6 алкинилов;
L1 и L2 независимо выбраны из группы, состоящей из C1-С6 алкилов, С2-С6 алкенилов, С2-С6 алкинилов, ароматических групп, гетероарилов, С3-С9 циклоалкилов, С3-С9 гетероциклилов, полиэтиленгликоля, О, S, NR6, С(=O), С(=O)O, C(=O)NR6, C=NR6, C(=S)O, C(=S)NR6, C(=S)S, NR6(C=O), NR6(C=S)NR7, O(C=O)NR6, S(=O)2 и их комбинаций, и R6 и R7 независимо выбраны из группы, состоящей из Н, C1-С6 алкилов, С2-С6 алкенилов и С2-С6 алкинилов;
L содержит нерасщепляемое звено и/или расщепляемое звено, и L содержит C1-C9 алкил, С2-С9 алкенил, С2-С9 алкинил, ароматическую группу, гетероарил, С3-С9 циклоалкил, С3-С9 гетероциклил, полиэтиленгликоль, О, S, NR8, С(=O), С(=O)O, C(=O)NR8, C=NR8, C(=S)O, C(=S)NR8, C(=S)S, NR8(O=O), NR8(С=S)NR9, O(C=O)NR8, S(=O)2, Val-Cit-PAB, Val-Ala-PAB, Val-Lys(Ac)-PAB, Phe-Lys-PAB, Phe-Lys(Ac)-PAB, D-Val-Leu-Lys, Gly-Gly-Arg, Ala-Ala-Asn-PAB, Ala-PAB, РАВ или их комбинацию, где R8 и R9 независимо выбраны из группы, состоящей из Н, C1-C6 алкилов, С2-С6 алкенилов и С2-С6 алкинилов; и
Q выбран из группы, состоящей из агентов, связывающих тубулины, алкилирующих агентов ДНК, интеркаляторов ДНК, ингибиторов ферментов, иммуномодуляторов, пептидов и нуклеотидов.
7. Конъюгат нацеливающая группировка-лекарственное средство по п. 6, где каждый из R3 и R4 независимо выбран из группы, состоящей из: 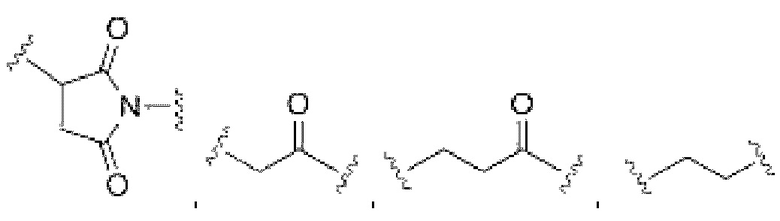 и их производных.
и их производных.
8. Конъюгат нацеливающая группировка-лекарственное средство по п. 6 или 7, где структура формулы II представляет собой любую, выбранную из группы, состоящей из:
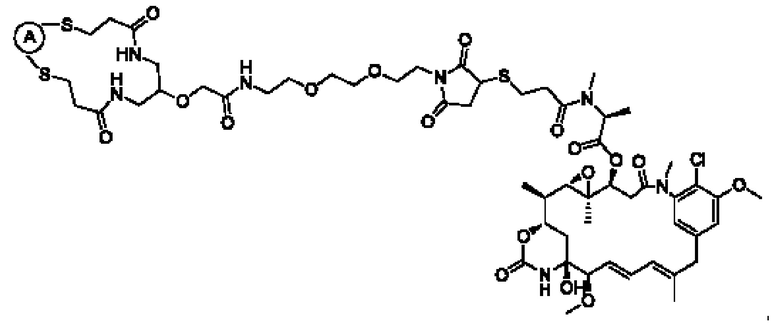
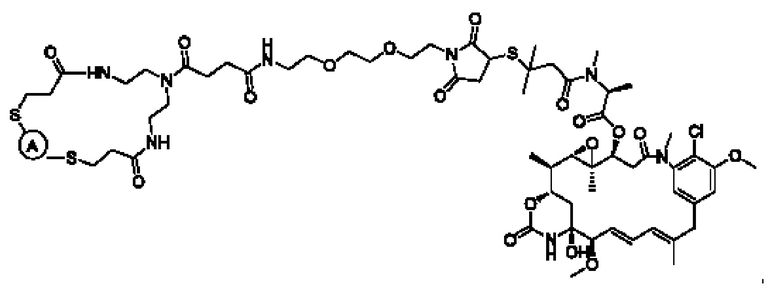
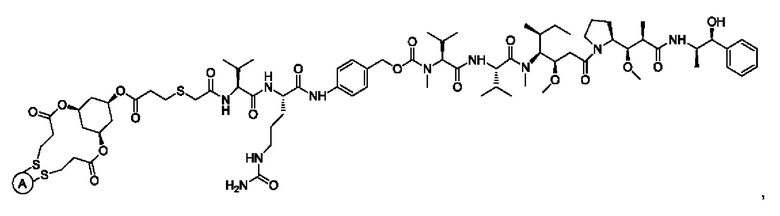
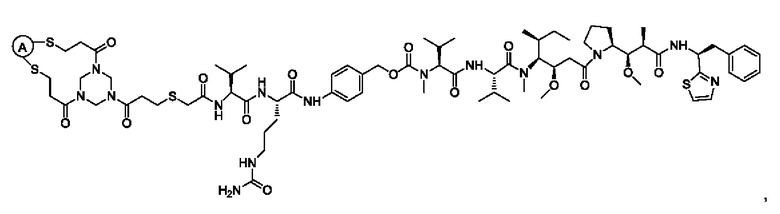
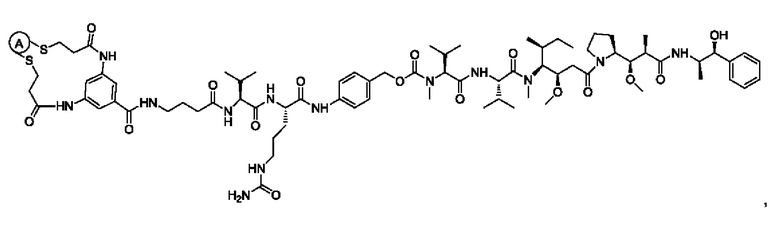
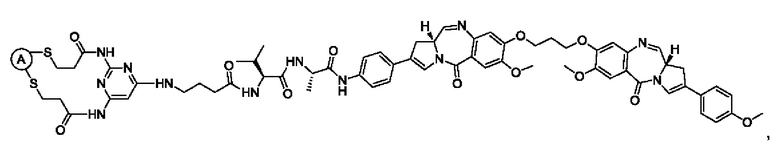
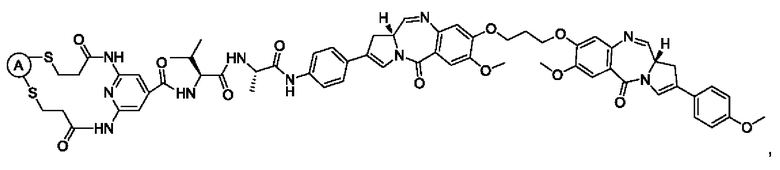
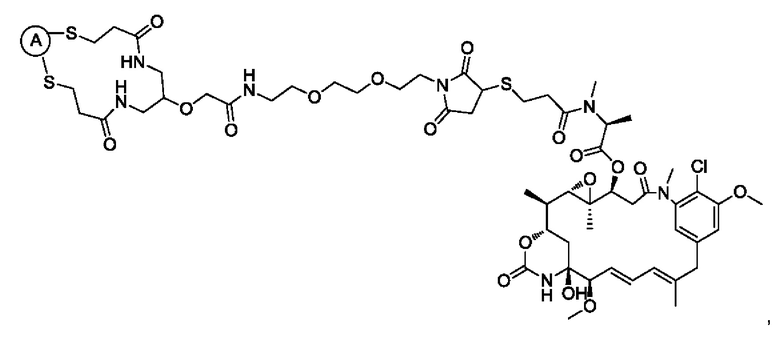
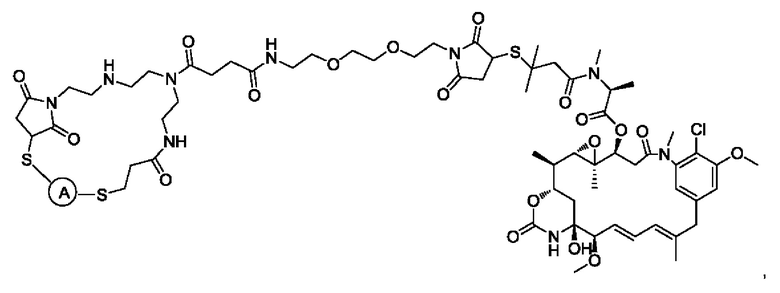
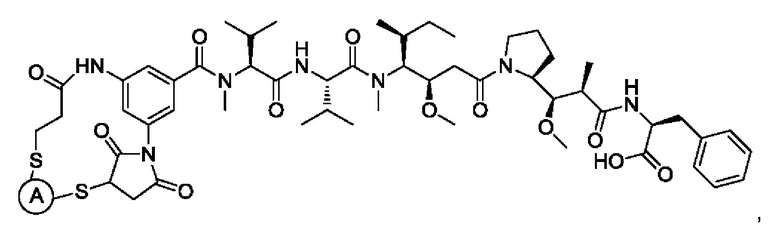
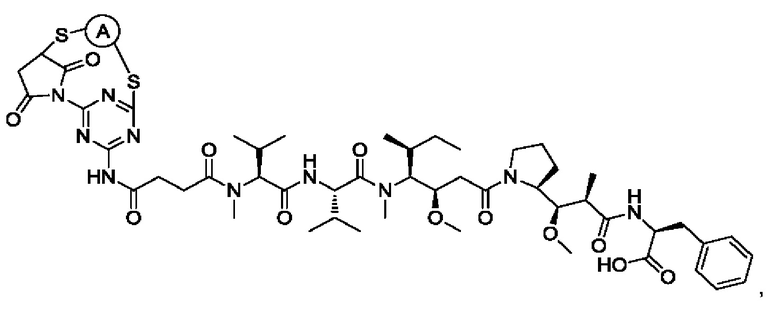
9. Способ лечения патологического состояния у субъекта, включающий введение субъекту, нуждающемуся в таком лечении, конъюгата нацеливающая группировка-лекарственное средство по любому из пп. 6-8, где указанное патологическое состояние представляет собой HER2-положительный рак.
10. Способ по п. 9, где А представляет собой антитело или его фрагмент, связывающиеся с HER2, и указанный фрагмент выбран из группы, состоящей из Fab, Fab', (Fab')2, scFv и Fv.
11. Способ по п. 9 или 10, где Q представляет собой цитотоксин.
12. Способ по п. 9 или 10, где субъект представляет собой человека.
13. Способ по п. 9 или 10, где рак представляет собой рак молочной железы.

