Настоящее изобретение относится к новым производным индола со сродством к рецептору 5-НТ6 и содержащим эти производные фармацевтическим композициям, способам их применения в качестве терапевтических средств и способам их получения.
Действие нейромодулятора 5-гидрокситриптамина (5-НТ) как главного модулирующего нейромодулятора в мозге медиируется через рецепторы ряда семейств, названных 5-НТ1, 5-НТ2, 5-НТ3, 5-НТ4, 5-НТ5, 5-НТ6 и 5-НТ7. На основании высокого уровня mRNA рецептора 5-НТ6 в мозге было констатировано, что рецептор 5-НТ6 может играть роль в патологии и лечении расстройств центральной нервной системы. Так, в частности, селективные лиганды рецептора 5-НТ6 были идентифицированы как потенциально полезные при лечении некоторых расстройств ЦНС, таких как болезнь Паркинсона, болезнь Хантингтона, страх, депрессия, маниакальная депрессия, психозы, эпилепсия, обсессивные компульсивные расстройства, мигрень, болезнь Альцгеймера (ослабление познавательной памяти), расстройства сна, расстройства питания, такие как анорексия и булемия, приступы паники, гиперфункция дефицита внимания (ADHD), дефицит внимания (ADD), синдром отмены вследствие злоупотребления сильнодействующими средствами, такими как кокаин, этанол, никотин и бензодиазепины, шизофрения, а также расстройства, связанные со спинно-мозговой травмой и/или повреждением головы, в частности при гидроцефалии. Полагают также, что такие соединения могут быть использованы и при лечении некоторых желудочно-кишечных расстройств (GI), таких как функциональное расстройство кишечника (см., например, B.L.Roth и др. J. Pharmacol. Exp. Ther, 1994, 268, с.1403-14120; D.R.Sibley и др., Mol. Pharmacol., 1993, 43, 320-327; A.J.Sleight и др., Neurotransmission, 1995 11, 1-5, и А.J.Sleight и др. Serotonin ID Research Alert, 1997, 2(3), 115-8).
Более того, была описана способность антагониста 5-НТ6 и 5-НТ6 антисмысловых олигонуклеотидов уменьшать у крыс потребление пищи (Br.J.Pharmac., 1999, Suppl 126, с.66, и J.Psychopharmacol Suppl. A64, 1997, c.255).
Объектом настоящего изобретения являются соединения общей формулы I
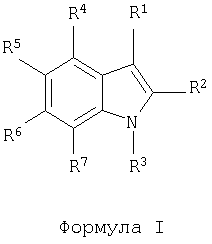
в которой R1 обозначает -S(O)0-2-A, -C(O)-A или -(СН2)0-1-А, где А обозначает арил или гетероарил, необязательно замещенный одной или несколькими группами, выбранными из гидрокси-, циано-, низшего алкила, соответственно (низш.)алкила, (низш.)алкокси, тиоалкила, гало, галоалкила, гидроксиалкила, нитро-, алкоксикарбонила, алкилкарбонила, алкилсульфонила, галоалкилсульфонила, амино-, алкиламино-, диалкиламиногрупп, алкиламинокарбонила, алкилкарбониламиногруппы, алкиламиносульфонила и алкилсульфониламиногруппы;
R2 имеет значения, выбранные из водородного атома, С1-6-алкила, С1-6-алкокси и C1-6-алкилтио;
R3 имеет значения, выбранные из водородного атома и C1-6-алкила;
R4 имеет значения, выбранные из атома водорода или галогена, С1-6-алкила, C1-6-алкокси, C1-6-алкилтио, галоалкила, цианогруппы и алкилкарбонила;
один из R5, R6 и R7 обозначает группу общей формулы В, в которой W обозначает группу -СН- или атом азота, а каждый из R8, R9 и R10 имеет значения, независимо выбранные из водородного атома, C1-10-алкила и бензила, или R8 и R9 совместно могут образовывать С3-С4алкилен,
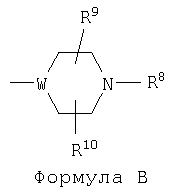
а значения каждого из остальных независимо выбирают из атомов водорода и галогена, C1-6-алкила, С1-6-алкокси, C1-6-алкилтио, галоалкила, цианогруппы и алкилкарбонила,
или их отдельные изомеры, рацемические или нерацемические смеси изомеров, пролекарства или фармацевтически приемлемые соли или сольваты.
Другим объектом изобретения являются фармацевтические композиции, содержащие терапевтически эффективное количество по меньшей мере одного соединения формулы I или отдельных изомеров, рацемических или нерацемических смесей изомеров или их фармацевтически приемлемых солей или сольватов, в смесях с по меньшей мере одним приемлемым носителем.
Еще одним объектом изобретения является соединение формулы I или отдельные изомеры, рацемические или нерацемические смеси изомеров или их фармацевтически приемлемые соли или сольваты, предназначенные для применения в качестве лекарственного средства.
Тем не менее другим объектом настоящего изобретения является применение соединения формулы I при приготовлении лекарственного средства для лечения или профилактики болезненных состояний, которые облегчают агонисты 5-НТ6, в частности болезненных состояний, включающих болезнь Альцгеймера, расстройства центральной нервной системы, такие, например, как психозы, шизофрения, маниакальные депрессии, неврологические расстройства, болезнь Паркинсона, амиотрофический латеральный склероз и болезнь Хантингтона. К другим болезненным состояниям, которые облегчают 5-НТ6 агонисты, а следовательно, соединения формулы I, относятся желудочно-кишечные заболевания, включая слизистый колит (IBS) и ожирение.
Другим объектом изобретения является способ, который включает:
I) реакцию соединения, отвечающего общей формуле 4

в которой Р представляет собой защитную группу, а R2, R4, R9 и R10 имеют указанные выше значения,
с соединением общей формулы (А-S)2, в которой А обозначает арил или гетероарил,
II) окисление атома серы соединения общей формулы (А-S)2,
III) необязательное алкилирование атома азота индоловой группы,
IV) удаление защитной группы у атома азота пиперазинового или пиперидинового кольца с получением соединения общей формулы I
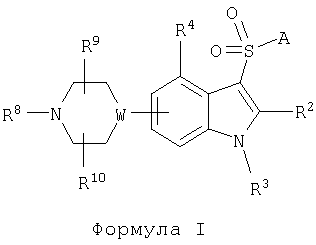
в которой R8 обозначает водородный атом, a A, R2, R3, R4, R9 и R10 имеют указанные выше значения, и
V) необязательное алкилирование атома азота пиперазинового или пиперидинового кольца с получением соединения общей формулы I, в которой R8 обозначает С1-10-алкил, a A, R2, R3, R4, R9 и R10 имеют указанные выше значения.
По другому варианту объектом изобретения является, кроме того, способ, который включает:
I) реакцию соединения, отвечающего общей формуле 4
в которой Р представляет собой защитную группу, а R2, R4, R9 и R10 имеют указанные выше значения,
с соединением общей формулы (А-S)2, в которой А обозначает арил или гетероарил,
II) необязательное алкилирование атома азота индоловой группы,
III) удаление защитной группы с получением соединения общей формулы I

в которой R8 обозначает водородный атом, a A, R2, R3, R4, R9 и R10 имеют указанные выше значения, и
IV) необязательное алкилирование атома азота пиперазиновой или пиперидиновой группы с получением соединения общей формулы I, в которой R8 обозначает C1-10-алкил, a A, R2, R3, R4, R9 и R10 имеют значения, указанные в кратком изложении сущности изобретения.
По другому варианту объектом изобретения является, кроме того, способ, который включает:
I) реакцию 1-гало-2-нитробензола с галометансульфонилбензолом,
II) аминирование продукта, полученного на стадии I), 1-алкилпиперазином,
III) восстановление нитрогруппы продукта, полученного на стадии II),
IV) присоединение ортоформиата к продукту, полученному на стадии III), и
V) циклизацию продукта, полученного на стадии IV), с получением соединений формулы 18а,
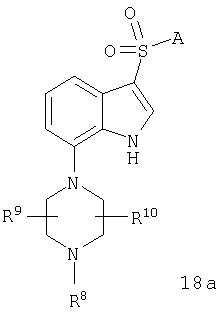
в которой R8 обозначает C1-10-алкил, а А, R9 и R10 имеют указанные выше значения.
Используемые в данной заявке, включая настоящее описание и формулу изобретения, следующие термины во всех случаях, если не указано иное, имеют приведенные ниже значения. Необходимо отметить, что во всех случаях, если из контекста не вполне очевиден иной смысл, использованные в настоящем описании и формуле изобретения в единственном числе понятия распространяются также на их варианты, приведенные во множественном числе.
Понятие "алкил" означает одновалентный линейный или разветвленный насыщенный углеводородный радикал, состоящий во всех случаях, если не указано иное, только из атомов углерода и водорода, содержащий от одного до двенадцати углеродных атомов включительно. Примеры алкильных радикалов включают, хотя ими их список не ограничен, метил, этил, пропил, изопропил, изобутил, втор-бутил, трет-бутил, пентил, н-гексил, октил, додецил и т.п. Понятие "алкил" также означает циклический радикал или сочетание линейного или разветвленного и циклического насыщенного углеводородного радикала, состоящего во всех случаях, если не указано иное, только из атомов углерода и водорода, содержащего от одного до двенадцати углеродных атомов включительно. Примеры таких алкильных радикалов включают, хотя ими их список не ограничен, циклопропил, циклопропилметил, циклогексил, циклопропилэтил и т.п.
Понятие "(низш.)алкил" означает одновалентный линейный или разветвленный насыщенный углеводородный радикал, состоящий во всех случаях, если не указано иное, только из атомов углерода и водорода, содержащий от одного до шести углеродных атомов включительно. Примеры (низш.)алкильных радикалов включают, хотя ими их список не ограничен, метил, этил, пропил, изопропил, втор-бутил, трет-бутил, н-бутил, н-пентил, н-гексил и т.п.
Понятие "алкилен" означает двухвалентный линейный или разветвленный насыщенный углеводородный радикал, состоящий во всех случаях, если не указано иное, только из атомов углерода и водорода, содержащий от одного до шести углеродных атомов включительно. Примеры алкиленовых радикалов включают, хотя ими их список не ограничен, метилен, этилен, пропилен, 2-метилпропилен, бутилен, 2-этилбутилен и т.п.
Понятие "алкокси" означает радикал -O-R, у которого R обозначает (низш.)алкильный радикал, как он представлен в настоящем описании. Примеры алкоксирадикалов включают, хотя ими их список не ограничен, метокси, этокси, изопропокси и т.п.
Понятие "алкилтио" или "алкилсульфанил" означает радикал -SR, у которого R обозначает (низш.)алкильный радикал, как он представлен в настоящем описании. Примеры алкилтиорадикалов включают, хотя ими их список не ограничен, метилтио, бутилтио и т.п.
Понятие "алкилсульфонил" означает радикал -SO2R, у которого R обозначает (низш.)алкильный радикал, как он представлен в настоящем описании. Примеры алкилсульфонильных радикалов включают, хотя ими их список не ограничен, метилсульфонил, этилсульфонил и т.п.
Понятие "арил" означает одновалентный циклический ароматический углеводородный радикал, состоящий из одного или нескольких конденсированных колец, из которых по меньшей мере одно кольцо по своей природе является ароматическим, который во всех случаях, если не указано иное, может быть необязательно замещенным гидроксилом, цианогруппой, (низш.)алкилом, (низш.)алкокси, тиоалкилом, гало, галоалкилом, гидроксиалкилом, нитрогруппой, алкоксикарбонилом, амино-, алкиламино-, диалкиламиногруппой, аминокарбонилом, карбониламиногруппой, аминосульфонилом, сульфониламиногруппой и/или трифторметилом. Примеры арильных радикалов включают, хотя ими их список не ограничен, фенил, нафтил, дифенил, инданил, антрахинолил и т.п. Примеры замещенных арильных радикалов включают, хотя ими их список не ограничен, фторфенил, хлорфенил, дихлорфенил, трифторметилфенил, толил и т.п.
Понятие "гетероарил" означает одновалентный ароматический карбоциклический радикал, содержащий одно или несколько колец, включающих в кольце один, два или три гетероатома (выбранных из атомов азота, кислорода или серы), который во всех случаях, если не указано иное, может быть необязательно замещенным гидроксилом, цианогруппой, (низш.)алкилом, (низш.)алкокси, тиоалкилом, гало, галоалкилом, гидроксиалкилом, нитрогруппой, алкоксикарбонилом, амино-, алкиламино-, диалкиламиногруппой, аминокарбонилом, карбониламиногруппой, аминосульфонилом, сульфониламиногруппой и/или трифторметилом. Примеры гетероарильных радикалов включают, хотя ими их список не ограничен, имидазолил, оксазолил, тиазолил, пиразинил, тиофенил, фуранил, пиранил, пиридинил, хинолинил, изохинолинил, бензофурил, бензотиофенил, бензотиопиранил, бензимидазолил, бензооксазолил, бензотиазолил, бензопиранил, индазолил, индолил, изоиндолил, нафтиридинил и т.п.
Понятие "уходящая группа" используют в качестве названия группы в том значении, которое обычно связано с ним в химии органического синтеза, т.е. для обозначения атома или группы, способной замещаться в условиях алкилирования. Примеры уходящих групп включают, хотя ими их список не ограничен, атом галогена, алкил- и арилсульфонилокси, такой как метансульфонилокси, этансульфонилокси, тиометил, бензолсульфонилокси, тозилокси и тиенилокси, дигалофосфиноилокси, необязательно замещенный бензилокси, изопропилокси, ацилокси и т.п.
Понятие "аминозащитная группа" означает защитную группу, которая относится к тем органическим группам, которые предназначены для защиты атома азота от нежелательной реакции во время осуществления методов синтеза; оно охватывает, хотя ими их список не ограничен, бензил (Bnz), бензилоксикарбонил (карбобензилокси, БЗК), п-метоксибензилоксикарбонил, п-нитробензилоксикарбонил, трет-бутоксикарбонил (БОК), трифторацетил и т.п. В предпочтительном варианте благодаря относительной легкости удаления, например, кислотами в случае БОК, в частности трифторуксусной кислотой или соляной кислотой в этилацетате, или каталитической гидрогенизацией в случае БЗК, в качестве аминозащитной группы используют либо БОК, либо БЗК.
Понятия "необязательный" и "необязательно" означают, что описываемый далее случай или обстоятельства могут возникать или не возникать и что описание включает варианты, когда такие случаи и обстоятельства возникают, и варианты, когда они не возникают. Так, например, "необязательно связанный" означает, что связь может содержаться или отсутствовать и что описание охватывает одинарные, двойные и тройные связи.
Понятие "защитная группа" или "защищающая группа" означает группу, которая селективно блокирует один реакционноспособный участок в содержащем много функциональных групп соединении, вследствие чего химическое взаимодействие можно проводить селективно по другому, незащищенному реакционноспособному участку; его используют в качестве названия группы в том значении, которое обычно связано с ним в химии органического синтеза. Осуществление некоторых способов по настоящему изобретению гарантируется блокированием защитной группой содержащегося в реагентах реакционноспособного атома кислорода. Приемлемые защитные группы для спиртовых или фенольных гидроксильных групп, которые могут быть последовательно и селективно удалены, включает группы, защищенные в форме ацетатов, галоалкилкарбонатов, простых бензиловых эфиров, простых алкилсилиловых эфиров, простых гетероциклических эфиров и метиловых или других простых алифатических эфиров и т.п. Защитные или блокирующие группы для карбоксильных групп аналогичны тем, которые описаны для гидроксильных групп, предпочтительно остатки трет-бутиловых, бензиловых или метиловых сложных эфиров.
Понятие "инертный органический растворитель" или "инертный растворитель" означает растворитель, который в реакционных условиях, описанных в сочетании с ним, инертен; это понятие охватывает, например, бензол, толуол, ацетонитрил, тетрагидрофуран, N,N-диметилформамид, хлороформ, метиленхлорид или дихлорметан, дихлорэтан, диэтиловый эфир, этилацетат, ацетон, метилэтилкетон, метанол, этанол, пропанол, изопропанол, трет-бутанол, диоксан, пиридин и т.п. Во всех случаях, если не указано иное, растворители, используемые в реакциях по настоящему изобретению, являются инертными растворителями.
Понятием "фармацевтически приемлемый" обозначают то, что может быть использовано при приготовлении фармацевтической композиции, которая в общем безопасна, нетоксична и ни в биологическом, ни в каком-либо другом смысле не является нежелательной и включает то, что является приемлемым для применения в ветеринарии, а также с фармацевтическими целями для человека.
Понятием "фармацевтически приемлемые соли" соединения обозначают соли, которые фармацевтически приемлемы, как это представлено в настоящем описании, и которые обладают целевым фармакологическим действием исходного соединения. Такие соли включают следующие соединения:
(1) кислотно-аддитивные соли, образованные неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., и образованные органическими кислотами, такими как уксусная кислота, бензолсульфоновая кислота, бензойная кислота, камфорсульфоновая кислота, лимонная кислота, этансульфоновая кислота, фумаровая кислота, глюкогептоновая кислота, глюконовая кислота, глутаминовая кислота, гликолевая кислота, гидроксинафтойная кислота, 2-гидроксиэтансульфоновая кислота, молочная кислота, малеиновая кислота, яблочная кислота, малоновая кислота, миндальная кислота, метансульфоновая кислота, муконовая кислота, 2-нафталинсульфоновая кислота, пропионовая кислота, салициловая кислота, янтарная кислота, винная кислота, п-толуолсульфоновая кислота, триметилуксусная кислота, трифторуксусная кислота и т.п.;
(2) соли, образующиеся когда протон кислоты, находящийся в исходном соединении, либо замещается ионом металла, например ионом щелочного металла, ионом щелочноземельного металла или алюминиевым ионом, либо образует координационную связь с органическим или неорганическим основанием. Приемлемые органические основания включают диэтаноламин, этаноламин, N-метилглюкамин, триэтаноламин, трометамин и т.п.; приемлемые неорганические основания включают гидроксид алюминия, гидроксид кальция, гидроксид калия, карбонат натрия и гидроксид натрия.
Предпочтительные фармацевтически приемлемые соли представляют собой соли, полученные из уксусной кислоты, трифторуксусной кислоты, соляной кислоты, серной кислоты, метансульфоновой кислоты, малеиновой кислоты, фосфорной кислоты, винной кислоты, лимонной кислоты, натрия, калия, кальция, цинка и магния.
Следует иметь в виду, что все ссылки на фармацевтически приемлемые соли охватывают связанные с растворителями формы (сольваты) и кристаллические формы (полиморфы), как они представлены в настоящем описании, той же кислотно-аддитивной соли.
Понятие "сольваты" означает связанные с растворителями формы, которые содержат либо стехиометрические, либо нестехиометрические количества растворителя. Некоторые соединения в кристаллическом твердом состоянии проявляют тенденцию к захвату в фиксированном молярном соотношении молекул растворителя, образуя таким образом сольват. Если растворителем является вода, то образующийся сольват представляет собой гидрат, а когда растворителем является спирт, образующийся сольват представляет собой алкоголят. Гидраты образуются сочетанием одной или нескольких молекул воды с одним из веществ, в которых вода удерживается в ее молекулярном состоянии, как Н2О, причем такое сочетание способно образовывать один или несколько гидратов.
Понятием "пролекарство" или "пролекарственное средство" обозначают фармакологически неактивную форму соединения, в которой оно должно in vivo подвергаться метаболизму, например, под действием биологических жидкостей или ферментов пациента после введения в его организм, превращаясь в фармакологически действующую форму соединения и производя целевой фармакологический эффект. Пролекарства соединения формулы I получают модификацией одной или нескольких функциональных групп, содержащихся в соединении формулы I, таким путем, чтобы модифицирующая группа (группы) могла быть удалена in vivo с высвобождением исходного соединения. Пролекарства включают соединения формулы I, где гидрокси, амино-, сульфгидрильная, карбоксильная или карбонильная группа в соединении формулы I связана с любой группой, которая может быть in vivo удалена с восстановлением свободной соответственно гидроксильной, амино-, сульфгидрильной, карбоксильной или карбонильной группы. Примеры пролекарств включают, хотя ими их список не ограничен, сложные эфиры (например, ацетатные, диалкиламиноацетатные, формиатные, фосфатные, сульфатные и бензоатные производные) и карбаматы гидроксильных функциональных групп (например, N,N-диметилкарбонил), эфиры карбоксильных функциональных групп (например, этиловые эфиры, морфолиноэтанольные эфиры), N-ациловые производные (например, N-ацетил), N-основания Манниха, Шиффовы основания и енаминоны аминовых функциональных групп, оксимы, ацетали, кетали и енольные эфиры кетоновых и альдегидных функциональных групп в соединениях формулы I и т.п.
Пролекарство может подвергнуться метаболизму до всасывания, во время всасывания, после всасывания или в определенном месте. Хотя метаболизм многих соединений происходит главным образом в печени, метаболизм в разной степени может происходить в почти всех других тканях и органах, в частности в легком. Соединения в пролекарственных формах можно использовать, например, для повышения биологической доступности, повышения переносимости пациентом, в частности, посредством маскировки или ослабления неприятных характеристик, таких как ощущение горечи или раздражение желудочно-кишечного тракта, изменения растворимости, в частности для внутривенного применения, с целью обеспечить долговременное или непрерывное высвобождение или доставку, с целью упростить приготовление или обеспечить доставку этого соединения к конкретному месту. В настоящем описании ссылка на соединение охватывает пролекарственные формы соединения. Пролекарства описаны в The Organic Chemistry of Drug Design and Drug Action, Richard B. Silverman, Academic Press, San Diego, 1992. Chapter 8: "Prodrugs and Drug delivery Systems" c.352-401; Design of Prodrugs под редакцией H.Bundgaard, Elsevier Science, Amsterdam, 1985; Design of Biopharmaceutical Properties through Prodrugs and Analogs под редакцией E.B.Roche, American Pharmaceutical Association, Washington, 1977; Drug Delivery Systems под редакцией R.L.Juliano, Oxford Univ. Press, Oxford, 1980.
Понятие "пациент" распространяется на млекопитающих и немлекопитающих. Под млекопитающим подразумевается любой представитель класса млекопитающих, включая, хотя ими их список не ограничен, людей; приматы, кроме человека, такие как шимпанзе и другие обезьяны и виды обезьян; сельскохозяйственные животные, такие как крупный рогатый скот, лошади, овцы, козы и свиньи; домашние животные, такие как кролики, собаки, кошки; лабораторные животные, включая грызунов, такие как крысы, мыши, морские свинки и т.п. Примеры немлекопитающих включают, хотя ими их список не ограничен, птиц и т.п. Понятие "пациент" не указывает конкретный возраст или пол.
Понятие "терапевтически эффективное количество" означает количество соединения, которого, когда оно введено в организм пациента для лечения болезненного состояния, достаточно для эффекта лечения при таком болезненном состоянии. "Терапевтически эффективное количество" обычно варьируют в зависимости от соединения, болезненного состояния, которое необходимо устранить лечением, серьезности заболевания, от которого лечат, возраста и относительного состояния здоровья пациента, пути и формы введения в организм, мнения соответствующего лечащего врача или практикующего ветеринара и других факторов.
Под понятием "болезненное состояние" подразумевают любое заболевание, состояние, симптом или показание.
В данной заявке следующие аббревиатуры использованы в следующих значениях:
Номенклатура, используемая в настоящей заявке, основана в общем на компьютеризированной системе AUTONOM™ том 4.0, Beilstein Institute, разработанной для данного поколения системной номенклатуры ИЮПАК. Однако поскольку жесткая привязка к рекомендациям ИЮПАК приводила бы к существенному изменению названий при замене всего лишь одного заместителя, форма наименований соединений сохраняет соответствие номенклатуре базовой структуры молекулы.
Так, например, соединение формулы I, в которой каждый из R2, R3, R4, R5 и R6 обозначает водородный атом, R1 обозначает фенилсульфонил, а R7 обозначает пиперазинил, называют 3-бензолсульфонил-7-пиперазин-1-ил-1H-индолом.
Хотя выше приведено самое широкое определение настоящего изобретения, предпочтительны некоторые следующие соединения формулы I или их отдельные изомеры, рацемические или нерацемические смеси изомеров или фармацевтически приемлемые соли или сольваты, у которых
R1 обозначает группу -S(O)0-2-A, в которой А обозначает арил или гетероарил. Другими предпочтительными соединениями являются те, у которых предпочтительным значением R1 является группа -S(O)0-2-A, в которой А обозначает фенил, необязательно замещенный одним или несколькими заместителями, выбранными из С1-6-алкила, С1-6-алкокси, атома галогена и галоалкила. Другими предпочтительными соединениями являются те, у которых предпочтительным значением R1 является группа -S(O)2-A, в которой А обозначает фенил, необязательно замещенный одним или несколькими заместителями, выбранными из C1-6-алкила, C1-6-алкокси, атома галогена и галоалкила. Другими предпочтительными соединениями являются те, у которых предпочтительным значением R1 является группа -S-A, в которой А обозначает фенил, необязательно замещенный одним или несколькими заместителями, выбранными из С1-6-алкила, С1-6-алкокси, атома галогена и галоалкила.
В другом предпочтительном варианте предпочтительным значением R1 является группа -S(O)0-2-A, в которой А обозначает гетероарил, более предпочтительным значением R1 является группа -S(O)2-A, в которой А обозначает пиридинил или бензотиазолил.
R2 и R3 каждый в предпочтительном варианте обозначает водородный атом или C1-6-алкил.
R4 в предпочтительном варианте обозначает водородный атом.
R8 в предпочтительном варианте обозначает водородный атом или C1-10-алкил, а каждый из R9 и R10 в предпочтительном варианте обозначает водородный атом.
В предпочтительном варианте R7 обозначает пиперазинильную группу общей формулы В, в которой W обозначает атом азота, а каждый из R5 и R6 обозначает водородный атом.
В другом предпочтительном варианте R5 обозначает пиперазинильную группу общей формулы В, в которой W обозначает атом азота, а каждый из R5 и R6 обозначает водородный атом.
Типичные особенно предпочтительные соединения или отдельные изомеры, рацемические или нерацемические смеси изомеров или их фармацевтически приемлемые соли или сольваты включают:
3-бензолсульфонил-7-пиперазин-1-ил-1H-индол,
3-бензолсульфонил-1-метил-7-пиперазин-1-ил-1H-индол,
3-бензолсульфонил-2-метил-7-пиперазин-1-ил-1H-индол,
3-(4-хлорбензолсульфонил)-2-метил-7-пиперазин-1-ил-1H-индол,
3-(4-метоксибензолсульфонил)-2-метил-7-пиперазин-1-ил-1H-индол,
3-бензолсульфонил-5-пиперазин-1-ил-1H-индол,
7-пиперазин-1-ил-3-(пиридин-4-сульфонил)-1H-индол,
7-пиперазин-1-ил-3-(пиридин-2-сульфонил)-1H-индол,
1-метил-7-пиперазин-1-ил-3-(пиридин-2-сульфонил)-1H-индол,
3-бензолсульфонил-7-(4-метилпиперазин-1-ил)-1H-индол,
3-бензолсульфонил-1-метил-5-пиперазин-1-ил-1H-индол,
3-(3,4-дихлорбензолсульфонил)-7-пиперазин-1-ил-1Н-индол,
2-(7-пиперазин-1-ил-1H-индол-3-сульфонил)бензотиазол,
3-(4-фторбензолсульфонил)-2-метил-7-пиперазин-1-ил-1H-индол,
3-(4-фторбензолсульфонил)-7-пиперазин-1-ил-1H-индол,
3-бензолсульфонил-7-пиперидин-4-ил-1H-индол,
7-пиперазин-1-ил-3-(толуол-4-сульфонил)-1H-индол,
3-(3,5-дихлорбензолсульфонил)-7-пиперазин-1-ил-1H-индол,
3-(3-хлорбензолсульфонил)-7-пиперазин-1-ил-1H-индол,
3-(2-хлорбензолсульфонил)-7-пиперазин-1-ил-1H-индол,
7-пиперазин-1-ил-3-(2-трифторметилбензолсульфонил)-1H-индол,
1-метил-7-пиперазин-1-ил-3-(2-трифторметилбензолсульфонил)-1H-индол,
3-(4-фторбензолсульфонил)-1-метил-7-пиперазин-1-ил-1H-индол,
1-метил-7-пиперазин-1-ил-3-(пиридин-2-сульфонил)-1H-индол,
1-метил-7-пиперазин-1-ил-3-(3-трифторметилбензолсульфонил)-1H-индол,
3-(2-хлорбензолсульфонил)-1-метил-7-пиперазин-1-ил-1H-индол,
3-(3-хлорбензолсульфонил)-1-метил-7-пиперазин-1-ил-1H-индол,
3-бензолсульфонил-1-метил-7-(4-метилпиперазин-1-ил)-1H-индол,
3-(2,3-дихлорфенилсульфанил)-5-пиперазин-1-ил-1H-индол,
3-(2,3-дихлорбензолсульфонил)-5-пиперазин-1-ил-1H-индол,
3-(2,3-дихлорбензолсульфонил)-1-метил-5-пиперазин-1-ил-1H-индол,
1-метил-5-пиперазин-1-ил-3-(3-трифторметилбензолсульфонил)-1H-индол,
5-пиперазин-1-ил-3-(4-трифторметилбензолсульфонил)-1H-индол,
3-(4-хлорбензолсульфонил)-5-пиперазин-1-ил-1H-индол,
3-(3,5-дихлорбензолсульфонил)-5-пиперазин-1-ил-1H-индол,
3-(3,5-дихлорбензолсульфонил)-1-метил-5-пиперазин-1-ил-1H-индол,
3-фенилсульфанил-5-пиперазин-1-ил-1H-индол,
3-(2-хлорбензолсульфонил)-5-пиперазин-1-ил-1H-индол,
3-(4-фторбензолсульфонил)-5-пиперазин-1-ил-1H-индол,
3-(4-фторбензолсульфонил)-1-метил-5-пиперазин-1-ил-1H-индол,
3-(2-хлорбензолсульфонил)-1-метил-5-пиперазин-1-ил-1H-индол,
3-(3,4-дихлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1H-индол,
3-(2-хлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1H-индол,
3-(3-хлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1H-индол,
3-(2,4-дихлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1H-индол,
3-(3,5-дихлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1H-индол,
7-(4-метилпиперазин-1-ил)-3-(2-трифторметилбензолсульфонил)-1H-индол и
3-фенилсульфанил-7-пиперазин-1-ил-1H-индол.
Было показано, что соединения формулы I обладают хорошим сродством к рецептору 5-НТ6. Предпочтительные соединения проявляют значения pKi>8,0.
Соединения по настоящему изобретению могут быть получены по методам, представленным на иллюстративных схемах реакций синтеза, приведенных и описанных ниже.
Исходные материалы и реагенты, используемые при получении этих соединений, в общем либо доступны у промышленных поставщиков, таких как фирма Aldrich Chemical Co., либо получают по методам, специалистам в данной области техники известным, т.е. осуществлением следующих методов, приведенных в такой литературе, как Fieser and Fieser's Reagents for Organic Synthesis; Wiley & Sons: New York, 1991, тома 1-15; Rodd's Chemistry of Carbon Compounds. Elsevier Science Publishers, 1989, тома 1-5 и Supplementals; Organic Reactions, Wiley & Sons: New York, 1991, тома 1-40. Следующие схемы реакций синтеза являются лишь иллюстрациями некоторых методов, в соответствии с которыми могут быть синтезированы соединения по настоящему изобретению, поэтому в такие схемы можно вносить различные модификации, которые обычно предполагаются любым специалистом в данной области техники, когда он обращается к описанию, содержащемуся в данной заявке.
Исходные материалы и промежуточные продукты, представленные на схемах реакций синтеза, при необходимости можно выделять и очищать с использованием обычных методов, включая, хотя ими их список не ограничен, фильтрование, дистилляцию, кристаллизацию, хроматографию и т.п. Такие материалы могут быть охарактеризованы с использованием обычных средств, включая физические константы и данные спектрального анализа.
Во всех случаях, если не указано иное, представляемые в настоящем описании реакции в предпочтительном варианте проводят под атмосферным давлением в температурном интервале от примерно -78 до примерно 150°С, более предпочтительно от примерно 0 до примерно 125°С, а наиболее предпочтительно при примерно комнатной температуре (или температуре окружающей среды), например при приблизительно 20°С.
Схема А
На схеме А представлены методы получения пиперазинилиндолов.
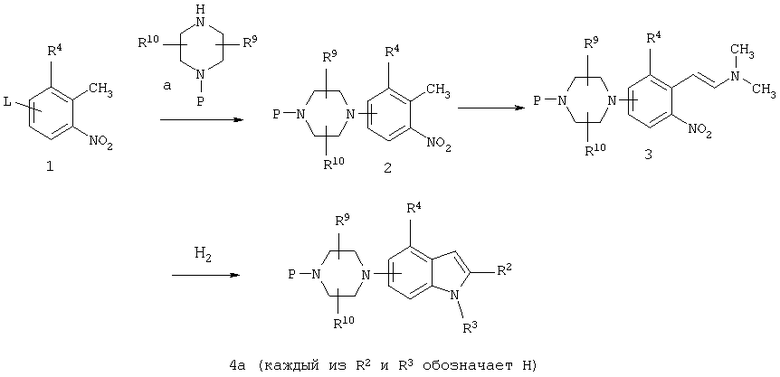
орто-Нитротолуолы, замещенные в фенильном кольце защищенной пиперазиниловой группой, можно превращать в 2-незамещенные индолы (4a, R2 и R3 обозначают Н, Р обозначает защитную группу) посредством синтеза Leimgruber-Batcho так, как изложено в Organic Synthesis, общий том 7, с.34, и в Heterocycles, 22, 195 (1984). Проводят реакцию конденсации орто-нитротолуола (2) с N,N-диметилформамиддиметилом или диэтилацеталем, трет-бутоксибис(диметиламино)метаном и т.п. в приемлемом растворителе, таком как ДМФ, необязательно в присутствии добавленного амина, такого как пирролидин, с получением диалкиламинонитростирола (3). Восстановление нитростирола до индола (4а, R2 и R3 обозначают Н) можно осуществлять по самым разнообразным методам, таким как каталитическая гидрогенизация, гидрогенизация с переносом и с помощью химических восстановителей, таких как трихлорид титана, металлические железо и цинк.
Защищенные в 1-м положении 4-пиперазинилнитротолуольные предшественники для синтеза индола по Batcho-Leimgruber могут быть получены нуклеофильным замещением нитробензола с уходящей группой, такой как гало и трифторметансульфонилокси, с использованием соответствующим образом защищенного в 1-м положении пиперазина общей формулы а, в которой Р обозначает защитную группу, как это изложено в Synthesis, 1145 (1990), для замещения 2-трифторметансульфонилоксинитробензола 1-бензилпиперазином. Другие приемлемые защитные группы включают N,N-трет-бутоксикарбонильную (БОК), карбобензилокси (БЗК), карбэтокси, ацетильную, бензоильную и формильную группы.
По другому варианту пиперазин можно использовать в процессе замещения так, как изложено в J.Med.Chem., 42, 4794 (1999), для получения 1-(2-нитрофенил)пиперазина, а полученный нитрофенилпиперазин может быть защищен по обычным методам, таким как обработка ди-трет-бутилдикарбонатом, с получением производного с БОК-группой.
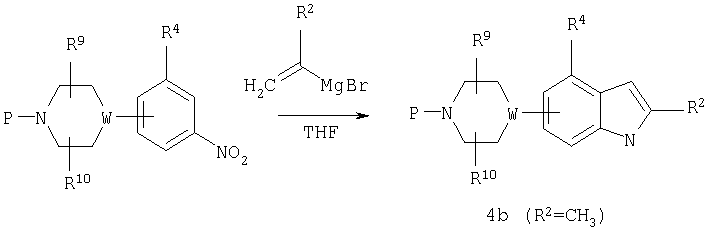
Пиперазинилиндолы или пиперидинилиндолы могут быть также получены синтезом индола по Bartoli, который описан в Tetrahedron Letters, 30, 2129 (1989). Соответствующим образом защищенный 2-пиперазинил или 2-пиперидинилнитробензол обрабатывают винильным реактивом Гриньяра в ТГФ с получением индола (4b) в одну стадию. По такому пути могут быть получены 2-алкилзамещенные индолы, которые недоступны посредством синтеза Leimgruber-Batcho.
Пиперазинилиндолы могут быть также получены из соответствующего аминоиндола реакцией аминогруппы с бисхлорэтиламином или соответствующим образом защищенным его производным, как это хорошо известно в данной области техники. Так, например, получение 7-(1-пиперазинил)индола обработкой 7-аминоиндола бисхлорэтиламином описано в WO 94/15919.
Пиперазинилиндолы могут быть также получены из орто-нитрофенилацетонитрилов так, как изложено для случая получения 7-(4-карбэтоксипиперазин-1-ил)индола в заявке GB 2097790.
Пиперазинилиндолы могут быть также получены из соответствующего броминдола катализируемой палладием реакцией сочетания с соответствующим образом защищенным пиперазином, таким как содержащий БОК-группу пиперазин (реакция Buchwald).
Пиперидинилиндолы могут быть получены превращением соответствующим образом защищенного галоиндола в литийсодержащее производное с последующей реакцией конденсации с соответствующим образом защищенным 4-пиперидоном и последующими дегидратацией и восстановлением такого олефина.
Схема В
На схеме В представлены методы получения соединения формулы I, в которой R1, R2, R3, R4, R5, R6, R7, R8, R9, R10 и А имеют значения, указанные в кратком изложении сущности изобретения.
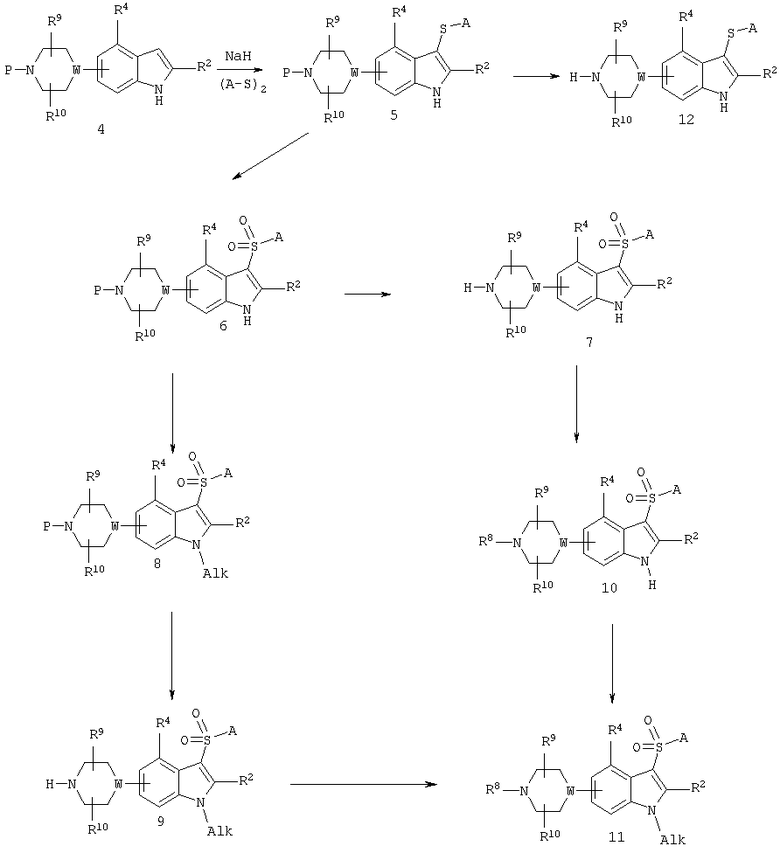
3-Арил- или 3-гетероарилсульфанилиндолы общей формулы (5), в которой Р обозначает защитную группу, a W обозначает -СН- или N, получают реакцией натриевой соли индола с арил или гетероарилдисульфидом, как это изложено в Synthesis 480 (1988). Арил- или гетероарилдисульфиды технически доступны или могут быть легко получены окислением соответствующего арил- или гетероарилтиола с помощью средства, хорошо известного специалистам в данной области техники.
Соединения формулы (12) могут быть получены удалением защитной группы (Р) у пиперазина или пиперидина общей формулы (5) в обычных условиях.
Окисление арил- или гетероарилсульфанильной группы до арил- или гетероарилсульфонильной группы может быть осуществлено подходящим окислителем, таким как пероксимоносульфат калия (Oxone™), 3-хлорпероксибензойная кислота, перуксусная кислота, пероксид водорода, озон и т.п. Необходимо иметь в виду, что при применении некоторых из этих окислителей возможно также протекание окисления пиперазинилового атома азота с образованием N-оксида. В этих случаях обратное восстановление N-оксида до исходного пиперазина общей формулы (6) может быть осуществлено подходящим восстановителем, таким как водород (каталитическая гидрогенизация), трифенилфосфин и т.п.
Удаление защитной группы (Р) у пиперазина или пиперидина общей формулы (6) осуществляют в обычных условиях, например отщеплением БОК-группы обработкой сильной кислотой, такой как трифторуксусная, в приемлемом растворителе, таком как дихлорметан, или соляная кислота, в приемлемом растворителе, таком как вода, этанол и этилацетат, с получением соединения общей формулы (7).
Замещенные в положении 1 производные общей формулы (8), в которой Alk представляет собой алкильную группу, получают алкилированием натриевой соли исходного индола общей формулы (6) подходящим алкилирующим агентом, таким как алкилгалогенид или сульфонат, в приемлемом растворителе, таком как тетрагидрофуран и ДМФ, с получением соединения общей формулы (9).
Реакцией сочетания соединений общей формулы (7) или (9) с карбоксиальдегидом в условиях восстановительного аминирования могут быть получены соединения соответственно (10) или (11), у которых R8 обозначает C1-10-алкильную группу.
Схема С
На схеме С представлен альтернативный метод получения соединения формулы I, в которой R1 обозначает -SO2-A, a R8 обозначает C1-10-алкил, каждый из R2, R3, R4, R5, R6 обозначает водородный атом, а А имеет значения, указанные в кратком изложении сущности изобретения.
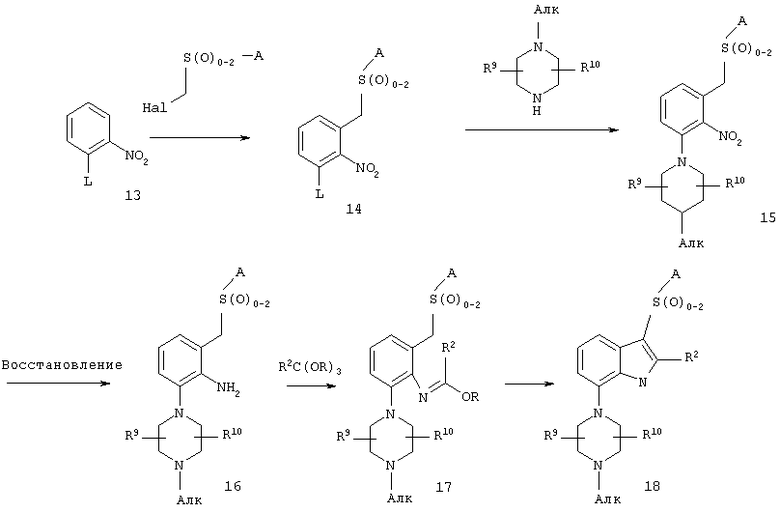
Нитробензолы формулы 14 могут быть получены из нитробензолов формулы 13 с использованием галометанового производного, такого как галометансульфонилбензол, галометансульфинилбензол и галометансульфанилбензол, в присутствии сильного основания, такого как трет-бутоксид калия, гидроксид натрия, гидроксид лития и метоксид натрия, в приемлемом растворителе, таком как ТГФ. В результате нуклеофильного замещения нитробензола формулы 14 с уходящей группой L, такой как галогруппа, с использованием 1-алкилпиперазина можно получить нитробензольное соединение формулы 15. Восстановлением нитрогруппы по самым разнообразным методам, хорошо известным в данной области техники, таким как каталитическая гидрогенизация, предпочтительно в присутствии катализатора Pearlman (гидроксид палладия), в приемлемом растворителе, таком как ТГФ, можно получить амин формулы 16. Добавлением сложного орто-эфира общей формулы R2С(OR)3, в которой R обозначает алкильную группу, а R2 обозначает водородный атом или алкил, в присутствии кислоты, такой как п-толуолсульфоновая кислота, с последующей циклизацией можно получить индол формулы 18, в которой R2 обозначает водородный атом или алкил.
Соединения по изобретению обладают селективным сродством к рецептору 5-НТ6, и поэтому предполагают, что они полезны при лечении некоторых расстройств ЦНС, таких как болезнь Паркинсона, болезнь Хантингтона, страх, депрессия, маниакальная депрессия, психозы, эпилепсия, обсессивные компульсивные расстройства, мигрень, болезнь Альцгеймера (ослабление познавательной памяти), расстройства сна, расстройства питания, такие как анорексия и булемия, приступы паники, гиперфункция дефицита внимания (ADHD), дефицит внимания (ADD), синдром отмены при злоупотреблении сильнодействующими средствами, такими как кокаин, этанол, никотин и бензодиазепины, шизофрения, а также расстройства, связанные со спинно-мозговой травмой и/или повреждением головы, в частности при гидроцефалии. Полагают также, что такие соединения эффективны и при лечении некоторых (GI) расстройств (желудочно-кишечных), таких как функциональное расстройство кишечника и слизистый колит (IBS), также как и при лечении ожирения.
Фармакологию соединений по настоящему изобретению определяли, используя известные методы. Методы in vitro для определения сродства тестируемых соединений к рецептору 5-НТ6 по связыванию радиолиганда и функциональные эксперименты описаны в примере 15.
Объем настоящего изобретения охватывает фармацевтические композиции, включающие по меньшей мере одно соединение по настоящему изобретению или отдельный изомер, рацемическую или нерацемическую смесь изомеров или его фармацевтически приемлемую соль или сольват совместно с по меньшей мере одним фармацевтически приемлемым носителем и необязательными другими терапевтическими и/или профилактическими компонентами.
В общем соединения по настоящему изобретению вводят в организм в терапевтически эффективном количестве по любому из общепринятых методов введения средств, которые служат достижению аналогичных эффектов. В зависимости от многочисленных факторов, таких как серьезность заболевания, от которого необходимо лечение, возраст и относительное состояние здоровья пациента, действие используемого соединения, путь и форма введения в организм, показание, в соответствии с которым предусмотрено введение, выбор и опыт лечащего врача, приемлемая доза, как правило, находится в интервале от 1 до 500 мг ежедневно, предпочтительно от 1 до 100 мг ежедневно, а наиболее предпочтительно от 1 до 30 мг ежедневно. Терапевтически эффективное количество соединений по настоящему изобретению при данном конкретном заболевании без чрезмерного количества экспериментов в состоянии установить обычный специалист в области лечения таких заболеваний, опираясь на собственные познания и исходя из описания настоящей заявки.
Соединения по настоящему изобретению обычно вводят в виде фармацевтических композиций, включая композиции, приемлемые для применения перорально (включая трансбуккальный и подъязычный методы), ректально, интраназально, локально, через легкие, вагинально и парентерально (включая внутримышечный, внутриартериальный, подоболочечный, подкожный и внутривенный методы), или в препаративной форме, приемлемой для введения ингаляционно или инсуффляционно. Предпочтительным методом введения является пероральный, в котором используют удобную схему ежедневного приема, в которую можно вносить коррективы в соответствии с серьезностью заболевания.
Соединению или соединениям по настоящему изобретению совместно с одним или несколькими обычными адъювантами, носителями или разбавителями можно придавать форму фармацевтических композиций и их доз на один прием. Фармацевтические композиции и одноразовые дозированные формы могут включать обычные компоненты в обычных пропорциях вместе с дополнительными действующими соединениями или действующими началами или без них, и в такой одноразовой дозированной форме могут содержать любое приемлемое эффективное количество действующего компонента, соответствующее предусмотренному диапазону ежедневно вводимых доз. Такие фармацевтические композиции можно применять в виде твердых препаратов, таких как таблетки и наполненные капсулы, полутвердых препаратов, порошков, препаратов с высвобождением в постоянной концентрации или жидкостей, таких как растворы, суспензии, эмульсии, эликсиры и наполненные капсулы для перорального введения, в форме суппозиториев для ректального или вагинального введения или в форме стерильных растворов для инъекций при парентеральном введении. Следовательно, типичным примером приемлемых препаративных форм в дозах на один прием служат композиции, содержащие примерно 1 миллиграмм действующего вещества или, если брать шире, от примерно 0,01 до примерно 100 миллиграммов на таблетку.
На основе соединений по настоящему изобретению можно готовить самые разнообразные дозированные формы для перорального введения. В качестве действующего компонента фармацевтические композиции и дозированные формы могут включать соединение или соединения по настоящему изобретению или их фармацевтически приемлемые соли. Фармацевтически приемлемые носители могут находиться либо в твердой, либо в жидкой форме. Твердые препаративные формы включают порошки, таблетки, пилюли, капсулы, крахмальные облатки, суппозитории и диспергируемые гранулы. Твердый носитель может включать одно или несколько веществ, которые могут также выполнять функции разбавителей, корригентов, солюбилизаторов, смазывающих добавок, суспендирующих веществ, связующих веществ, консервантов, добавок, способствующих механическому разрушению таблеток, или инкапсулирующего материала. В порошках носитель находится в виде тонкоизмельченного твердого вещества, которое представляет собой смесь с тонкоизмельченным действующим компонентом. Для изготовления таблеток действующий компонент обычно смешивают с носителем, обладающим необходимой связующей способностью, в приемлемых пропорциях и прессуют с приданием целевых формы и размера. Предпочтительные порошки и таблетки содержат от примерно 1 до примерно 70 процентов действующего вещества. Приемлемые носители включают, хотя ими их список не ограничен, карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатину, трагакант, метилцеллюлозу, натрийкарбоксиметилцеллюлозу, низкоплавкий воск, масло какао и т.п. Понятие "препарат" использовано как охватывающее сочетание действующего вещества с инкапсулирующим материалом как носителем, образующим капсулу, в которой действующий компонент совместно с другими носителями или без них окружен носителем, который связан с ним. Подобным же образом этим понятием охватываются крахмальные облатки и лепешки. Таблетки, порошки, капсулы, пилюли, крахмальные облатки и лепешки могут находиться в твердой форме, приемлемой для перорального введения.
Другие формы, приемлемые для перорального введения, включают препараты в жидком виде, к которым относятся эмульсии, сиропы, эликсиры, водные растворы, водные суспензии, и препараты в твердом виде, которые предназначены для перевода незадолго до применения в форму жидких препаратов. Эмульсии могут быть приготовлены в виде растворов, например, в водных пропиленгликолевых растворах или могут содержать эмульгаторы, например такие как лецитин, моноолеат сорбитана и гуммиарабик. Водные растворы можно готовить растворением действующего компонента в воде и добавлением приемлемых красителей, корригентов, стабилизаторов и загустителей. Водные суспензии можно готовить диспергированием тонкоизмельченного действующего компонента в воде вместе с вязким материалом, таким как природные и синтетические камеди, смолы, метилцеллюлоза, натрийкарбоксиметилцеллюлоза и другие хорошо известные суспендирующие агенты. Препараты в жидкой форме включают растворы, суспензии и эмульсии, они могут содержать, в дополнение к действующему компоненту, красители, корригенты, стабилизаторы, буферные добавки, искусственные и природные подслащивающие вещества, диспергаторы, загустители, солюбилизирующие агенты и т.п.
С использованием соединений по настоящему изобретению можно готовить препараты для парентерального введения (например, путем инъекций, в частности для инъекции ударной дозы вещества или постоянного вливания), они могут содержаться в лекарственном препарате в дозах на один прием в виде ампул, предварительно наполненных шприцев, контейнеров малого объема для вливания и контейнеров для нескольких доз с добавленным консервантом. Композициям можно придавать такие формы, как суспензии, растворы и эмульсии в маслянистых или водных наполнителях, например растворы в водном полиэтиленгликоле. Примеры маслянистых или неводных носителей, разбавителей, растворителей или наполнителей включают пропиленгликоль, полиэтиленгликоль, растительные масла (в частности, оливковое масло) и органические сложные эфиры для инъекций (в частности, этилолеат); они могут содержать используемые для приготовления препаратов добавки, такие как консерванты, смачивающие вещества, эмульгаторы, суспендирующие агенты, стабилизаторы и/или диспергаторы. По другому варианту действующий компонент может находиться в виде порошка, приготовленного выделением в асептических условиях стерильного твердого вещества или лиофилизацией из раствора, и перед использованием его совмещают с приемлемым наполнителем, в частности со стерильной апирогенной водой.
Для местного введения в эпидермис на основе соединений по настоящему изобретению можно готовить мази, кремы, лосьоны или пластыри для введения через кожу. Мази и кремы можно готовить, например, с использованием водной или маслянистой основы с добавлением приемлемых загущающих веществ и/или гелеобразователей. Лосьоны можно готовить с использованием водной или маслянистой основы, обычно они содержат также один или несколько эмульгаторов, стабилизирующих агентов, диспергаторов, суспендирующих добавок, загущающих веществ и красителей. Препараты, приемлемые для местного введения в полость рта, включают лепешки, содержащие действующие вещества в обработанной корригентами основе, обычно в сахарозе и гуммиарабике или трагаканте; пастилки, содержащие действующий компонент в инертной основе, такой как желатина, глицерин или сахароза и гуммиарабик; жидкости для полоскания рта, содержащие действующее вещество в подходящем жидком носителе.
С использованием соединений по настоящему изобретению можно готовить препараты для введения в виде суппозиториев. Низкоплавкий воск, такой как смесь глицеридов жирных кислот, или масло какао вначале плавят и гомогенно диспергируют, например, перемешиванием действующий компонент. Далее расплавленную гомогенную смесь разливают по формам подходящего размера, дают остыть и затвердеть.
С использованием соединений по настоящему изобретению можно готовить препараты для вагинального введения. Приемлемыми можно считать вагинальные суппозитории, тампоны, кремы, гели, пасты, губки или спреи, содержащие, помимо действующего вещества, такие носители, которые известны в данной области техники.
С использованием соединений по настоящему изобретению можно готовить препараты для интраназального введения. Растворы и суспензии вводят непосредственно в носовую полость с помощью обычных средств, например капельницы, пипетки или распылительного устройства. Препараты могут быть приготовлены в дозированной форме на один или несколько приемов. В этом последнем случае при наличии капельницы или пипетки может быть обеспечено введение пациенту соответствующего, предопределенного объема раствора или суспензии. В случае спрея этого можно добиться, например, с помощью атомизационного распылителя с насосом-дозатором.
С использованием соединений по настоящему изобретению можно готовить препараты для введения в форме аэрозоля, в частности для введения в дыхательные пути, включая интраназальное введение. При этом вещество обычно характеризуется малым размером частиц, в частности примерно 5 микрометров или меньше. Такие размеры частиц могут быть достигнуты с помощью средств, известных в данной области техники, например обработкой в микронной коллоидной мельнице. Действующее вещество находится в упаковке под давлением приемлемого пропеллента, такого как хлорфторуглерод (ХФУ), например дихлордифторметан, трихлорфторметан или дихлортетрафторметан, диоксид углерода или другой приемлемый газ. Аэрозольный препарат обычно может также включать поверхностно-активное вещество, такое как лецитин. Дозу лекарственного средства можно регулировать с помощью дозирующего клапана. В другом варианте с использованием действующих компонентов могут быть приготовлены сухие порошки, например порошкообразная смесь такого соединения с приемлемой порошкообразной основой, такой как лактоза, крахмал, производные крахмала, такие как гидроксипропилметилцеллюлоза, и поливинилпирролидон (ПВП). В носовой полости порошкообразный носитель обычно образует гель. Порошкообразную композицию можно готовить в форме дозы на один прием, например капсул или облаток, в частности желатиновых и пузырьковых упаковок, из которых порошки можно вводить с помощью ингалятора.
При необходимости препараты могут быть снабжены покрытием для тонкой кишки, приспособленном для постоянного или регулируемого высвобождения в организм действующего компонента. Так, например, на основе соединений по настоящему изобретению могут быть изготовлены приспособления для чрезкожного или подкожного введения лекарственного средства. Эти системы доставки обладают преимуществом, когда требуется постоянное высвобождение соединения и когда решающее значение имеет согласие пациента с режимом лечения. Соединения в системах чрезкожной доставки часто закрепляют на обладающей адгезией к коже твердой подложке. Представляющее интерес соединение можно также совмещать с усиливающей проникновение внутрь добавкой, например с продуктом Azone (1-додецилазациклогептан-2-он). Системы доставки с постоянным высвобождением внедряют подкожно, внутрь подкожного слоя хирургическим путем или инъекцией. Эти подкожные имплантанты инкапсулируют такое соединение в липидорастворимой мембране, например, из силиконового каучука или в биоразлагаемом полимере, в частности в полимере молочной кислоты.
В предпочтительном варианте фармацевтические композиции готовят в формах доз на один прием. В такой форме препарат разделен на унифицированные дозы, содержащие соответствующие количества действующего компонента. Унифицированная дозированная форма может представлять собой упакованный препарат, причем такая упаковка содержит раздельные количества препарата, в частности упакованные таблетки, капсулы, порошки в пузырьках или ампулы. К тому же лекарственный препарат в дозах на один прием может представлять собой капсулу, таблетку, крахмальную облатку или лепешку или он может включать соответствующее число доз в любой из таких упакованных форм.
Другие подходящие фармацевтические носители и приготовленные с их применением препараты описаны в работе Remington: The Science and Practice of Pharmacy, 1995, под редакцией E.W.Martin, Mack Publishing Company, издание 19-е, Истон, шт. Пенсильвания. Типичные фармацевтические композиции, содержащие соединения по настоящему изобретению, описаны в примерах с 8 по 14.
ПРИМЕРЫ
Следующие ниже композиции и примеры приведены с целью дать возможность специалистам в данной области техники более четко понять сущность настоящего изобретения. Их не следует рассматривать как ограничивающие объем изобретения, а как приведенные лишь с целью его иллюстрации.
Пример получения 1
трет-Бутиловый эфир 4-(1H-индол-7-ил)пиперазин-1-карбоновой кислоты

Стадия 1:
3-метил-2-нитрофениловый эфир трифторметансульфоновой кислоты

Раствор 3-метил-2-нитрофенола (15,3 г, 100 ммолей) в 200 мл дихлорметана и 42 мл триэтиламина охлаждали до -30°С и медленно добавляли трифторметансульфоновый ангидрид (21 мл, 125 ммолей). После 15 мин смесь промывали насыщенным водным бикарбонатом натрия и рассолом, сушили и выпаривали до остатка в виде темного масла. Хроматографией на силикагеле (10% этилацетата/гексан) в виде бесцветного масла получали 26,7 г 3-метил-2-нитрофенилового эфира трифторметансульфоновой кислоты.
Стадия 2:
трет-бутиловый эфир 4-(3-метил-2-нитрофенил)пиперазин-1-карбоновой кислоты
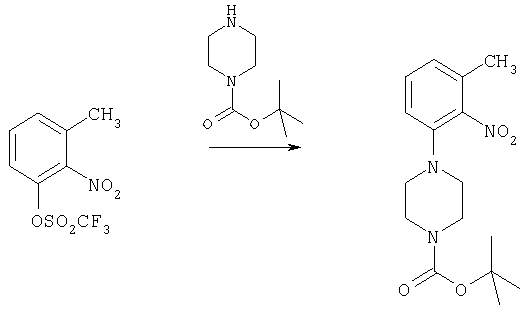
3-Метил-2-нитрофениловый эфир трифторметансульфоновой кислоты (26,7 г, 93,6 ммоля), 1-трет-бутоксикарбонилпиперазин (17 г, 91,3 ммоля) и триэтиламин (14 мл, 100 ммолей) в 250 мл ацетонитрила кипятили с обратным холодильником в течение 25 ч. Смесь концентрировали под вакуумом, разбавляли водой и экстрагировали диэтиловым эфиром. Эфирный экстракт промывали водным гидроксидом аммония и рассолом, сушили и выпаривали. В результате очистки хроматографией на силикагеле (10% этилацетата/гексан) в виде золотистого твердого вещества получали 11 г трет-бутилового эфира 4-(3-метил-2-нитрофенил)пиперазин-1-карбоновой кислоты, tпл 101-102°С.
Стадия 3:
трет-бутиловый эфир 4-(1H-индол-7-ил)пиперазин-1-карбоновой кислоты

Раствор трет-бутилового эфира 4-(3-метил-2-нитрофенил)пиперазин-1-карбоновой кислоты (10 г, 31 ммоль), N,N-диметилформамиддиметилацеталя (13,2 мл, 100 ммолей) и пирролидина (4 мл, 50 ммолей) в 45 мл ДМФ кипятили с обратным холодильником в течение 20 ч. Смесь концентрировали под вакуумом и остаток разделяли между этилацетатом и водой. Этилацетатный слой сушили и выпаривали. Остаток гидрировали в 200 мл ТГФ, содержавшего 2,5 г 10%-ного Pd-C, под давлением 50 фунтов/кв.дюйм в течение 6 ч. Смесь фильтровали, концентрировали под вакуумом и разделяли между этилацетатом и водным HCl. Этилацетатный слой промывали рассолом, сушили и выпаривали. Хроматографией на силикагеле (10% этилацетата/гексан) в виде белого твердого вещества получали 2,5 г трет-бутилового эфира 4-(1H-индол-7-ил)пиперазин-1-карбоновой кислоты (19), tпл 150-151°С.
Пример получения 2
трет-Бутиловый эфир 4-(1H-индол-5-ил)пиперазин-1-карбоновой кислоты
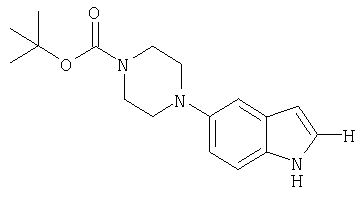
Стадия 1:
трет-бутиловый эфир 4-(3-метил-4-нитрофенил)пиперазин-1-карбоновой кислоты

Смесь 5-фтор-2-нитротолуола (2 мл, 16,4 ммоля), 1-трет-бутоксикарбонилпиперазина (3,35 г, 18 ммолей) и карбоната калия (2,76 г, 20 ммолей) в 7 мл ДМСО перемешивали и выдерживали при 100°С в течение 2 ч. Добавляли воды (40 мл) и гексана (30 мл), ярко-желтый осадок собирали, промывали водой и гексаном и сушили под вакуумом с получением 4,9 г трет-бутилового эфира 4-(3-метил-4-нитрофенил)пиперазин-1-карбоновой кислоты, tпл 145-146°С.
Стадия 2:
трет-бутиловый эфир 4-(1H-индол-5-ил)пиперазин-1-карбоновой кислоты

Раствор трет-бутилового эфира 4-(3-метил-4-нитрофенил)пиперазин-1-карбоновой кислоты (4,3 г, 13,5 ммоля), N,N-диметилформамиддиметилацеталя (2,12 мл, 16 ммолей) и пирролидина (1,3 мл, 16 ммолей) в 15 мл ДМФ выдерживали при 110°С в течение 3 ч. Дополнительно добавляли N,N-диметилформамиддиметилацеталя (0,7 мл, 5 ммолей) и пирролидина (0,42 мл, 5 ммолей) и выдержку при 110°С продолжали в течение 15 ч. Смесь концентрировали под вакуумом, растворяли в 50 мл этанола и медленно вводили в горячий (непосредственно ниже температуры кипения) раствор 50 мл этанола, содержавший 2 мл воды, 1 г 10%-ного Pd-C и формиат аммония (4,4 г, 70 ммолей). После того как добавление завершали, смесь кипятили с обратным холодильником в течение 10 мин. Смесь фильтровали, концентрировали под вакуумом и разделяли между диэтиловым эфиром и водным карбонатом натрия. Эфирный слой промывали водой и рассолом, сушили (сульфат магния) и выпаривали. Хроматографией на силикагеле (20% этилацетата/гексан) в виде густого масла получали 3 г трет-бутилового эфира 4-(1H-индол-5-ил)пиперазин-1-карбоновой кислоты (20).
Альтернативный путь получения трет-бутилового эфира 4-(1H-индол-5-ил)пиперазин-1-карбоновой кислоты (20)

Смесь 5-бром-1-триизопропилсиланил-1H-индола (9 г, 25,5 ммоля), 1-трет-бутоксикарбонилпиперазина (5 г, 27 ммолей), ацетата палладия (0,3 г, 1,3 ммоля), три-трет-бутилфосфина (0,263 г, 1,3 ммоля) и трет-бутоксида натрия (3,65 г, 38 ммолей) в 75 мл ксилола выдерживали в течение 2 ч при 110°С. Реакционную смесь разбавляли 200 мл 50% этилового эфира/гексана и смесь фильтровали через слой силикагеля. Использовали достаточное количество 10% этилацетата/гексана для элюирования трет-бутилового эфира 4-(1-триизопропилсиланил-1H-индол-5-ил)пиперазин-1-карбоновой кислоты (11,7 г), который концентрированием элюата под пониженным давлением выделяли в виде светло-коричневого вязкого сиропа.
В раствор трет-бутилового эфира 4-(1-триизопропилсиланил-1H-индол-5-ил)пиперазин-1-карбоновой кислоты (11,7 г, 25 ммолей) в 150 мл ТГФ добавляли 26 мл 1,0 М тетрабутиламмонийфторида в ТГФ. После 2 ч выдержки при комнатной температуре раствор концентрировали под пониженным давлением и остаток разделяли между 100 мл этилового эфира и 20 мл насыщенного раствора бикарбоната натрия. Органическую фазу промывали 20 мл воды, 10 мл насыщенного раствора хлорида натрия, сушили (сульфат магния) и концентрировали под пониженным давлением. Остаток подвергали хроматографии в колонке с силикагелем, элюируя 20% этилацетатом/гексаном, с получением 7 г трет-бутилового эфира 4-(1H-индол-5-ил)пиперазин-1-карбоновой кислоты (20).
Пример получения 3
трет-Бутиловый эфир 4-(2-метил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты

Изопропенилмагнийбромид (234 мл 0,5 М раствора в ТГФ, 117 ммолей) медленно вводили в охлажденный до -40°С раствор трет-бутилового эфира 4-(4-нитрофенил)пиперазин-1-карбоновой кислоты (12 г, 39 ммолей) в 200 мл ТГФ и образовавшуюся смесь перемешивали при -40°С в течение 20 мин. Добавляли насыщенного водного аммонийхлорида и смесь экстрагировали этилацетатом. Экстракт промывали водой и рассолом, сушили (сульфат натрия) и выпаривали. Хроматографией на силикагеле (20% этилацетата/гексан) в виде масла получали 4,8 г трет-бутилового эфира 4-(2-метил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты (21), масс-спектр: М+ 315.
Пример получения 4
трет-Бутиловый эфир 4-(1H-индол-7-ил)пиперидин-1-карбоновой кислоты
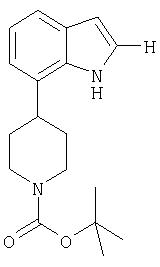
Стадия 1:
трет-бутиловый эфир 4-гидрокси-4-(1H-индол-7-ил)пиперидин-1-карбоновой кислоты
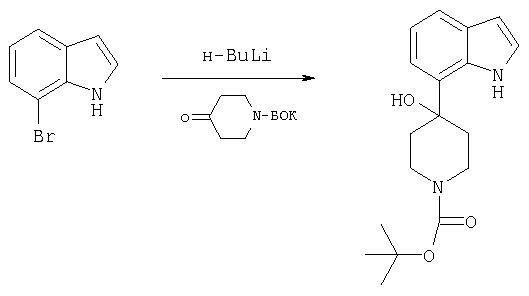
В охлажденный до -78°С раствор 7-броминдола (400 мг, 2,04 ммоля) в ТГФ (20 мл) в аргоновой атмосфере по каплям вводили н-бутиллитий (3,1 мл, 2,0 М, 6,1 ммоля). Реакционную смесь перемешивали при -78°С в течение 15 мин, затем нагревали до 5°С и выдерживали в течение 30 мин. Реакционную смесь вновь охлаждали до -78°С и по каплям добавляли трет-бутилового эфира 4-оксопиперидин-1-карбоновой кислоты (813 мг, 4,08 ммоля) в ТГФ (5 мл). Реакционной смеси давали достичь комнатной температуры и перемешивали в течение 1 ч. Реакцию в реакционной смеси гасили водой (15 мл) и экстрагировали EtOAc (3 порции по 20 мл). Объединенные ацетатные слои промывали рассолом, сушили над MgSO4 и фильтровали. Фильтрат концентрировали под вакуумом и хроматографировали на диоксиде кремния, элюируя 20%-ным ацетоном в гексанах, с получением в виде белой пены трет-бутилового эфира 4-гидрокси-4-(1H-индол-7-ил)пиперидин-1-карбоновой кислоты (520 мг).
Стадия 2:
трет-бутиловый эфир 4-(1H-индол-7-ил)-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты
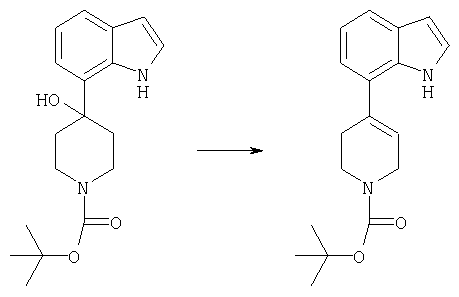
В раствор трет-бутилового эфира 4-гидрокси-4-(1H-индол-7-ил)пиперидин-1-карбоновой кислоты (117 мг, 0,37 ммоля) в пиридине (5 мл) одной порцией вводили POCl3 (70 мкл, 0,74 ммоля). Реакционную смесь перемешивали в течение 24 ч, редакцию гасили медленным добавлением воды (10 мл) и экстрагировали EtOAc (3 порции по 10 мл). Объединенные ацетатные слои промывали рассолом, сушили (MgSO4) и концентрировали с получением в виде светло-желтого масла трет-бутилового эфира 4-(1H-индол-7-ил)-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (100 мг).
Стадия 3:
трет-бутиловый эфир 4-(1H-индол-7-ил)пиперидин-1-карбоновой кислоты

В раствор трет-бутилового эфира 4-(1H-индол-7-ил)-3,6-дигидро-2Н-пиридин-1-карбоновой кислоты (100 мг) в EtOH (20 мл) вводили 10%-ный Pd-C (20 мг). Реакционную смесь помещали на 24 ч на вибратор Парра под давлением H2 55 фунтов/кв.дюйм. Реакционную смесь фильтровали через продукт Celite™ и концентрировали с получением в виде прозрачного масла трет-бутилового эфира 4-(1H-индол-7-ил)пиперидин-1-карбоновой кислоты (22) (78 мг).
ПРИМЕР 1
3-Бензолсульфонил-7-пиперазин-1-ил-1H-индол

Стадия 1:
трет-бутиловый эфир 4-(3-фенилсульфанил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты

Гидрид натрия (0,4 г 60%-ной дисперсии в минеральном масле, 10 ммолей) вводили в охлажденный льдом раствор трет-бутилового эфира 4-(1H-индол-7-ил)пиперазин-1-карбоновой кислоты (19), полученного так, как изложено в примере получения 1 (2,3 г, 7,6 ммоля), в 20 мл ДМФ и образовавшуюся смесь перемешивали в течение 10 мин. Добавляли фенилдисульфида (1,85 г, 8,5 ммоля) и раствор перемешивали при комнатной температуре в течение 16 ч. Добавляли воды и смесь экстрагировали этилацетатом. Экстракт промывали рассолом, сушили и выпаривали с получением в виде белого твердого вещества 2,9 г трет-бутилового эфира 4-(3-фенилсульфанил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты, tпл 165-166°С.
Стадия 2:
трет-бутиловый эфир 4-(3-бензолсульфонил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты
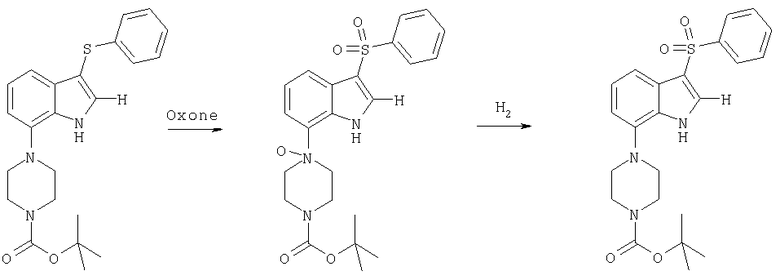
Oxone™ (9,2 г, 15 ммолей) в 40 мл воды вводили в перемешиваемый раствор трет-бутилового эфира 4-(3-фенилсульфанил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты (2,9 г) в 50 мл метанола и 10 мл ТГФ. Смесь перемешивали в течение 2 ч, разбавляли водой и экстрагировали этилацетатом. Этилацетатный слой сушили (сульфат натрия) и выпаривали с получением в виде твердого остатка сульфон-N-оксида. Его растворяли в 50 мл метанола и гидрировали под атмосферным давлением над 0,3 г 10%-ного Pd-C в течение 12 ч. Добавляли дихлорметана и смесь фильтровали и выпаривали. Растиранием в порошок остатка с диэтиловым эфиром в виде белого твердого вещества получали 2,2 г трет-бутилового эфира 4-(3-бензолсульфонил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты, tпл 168-169°С.
Стадия 3:
3-бензолсульфонил-7-пиперазин-1-ил-1H-индолгидрохлорид

трет-Бутиловый эфир 4-(3-бензолсульфонил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты (1 г, 2,2 ммоля) растворяли в 5 мл дихлорметана и добавляли 6 мл трифторуксусной кислоты. По истечении 10 мин смесь концентрировали под вакуумом, растворяли в воде и промывали диэтиловым эфиром. Водный слой подщелачивали гидроксидом аммония, экстрагировали дихлорметаном и экстракт сушили (сульфат натрия) и выпаривали с получением сырого свободного основания. Гидрохлоридную соль кристаллизовали из этанола с получением 550 мг 3-бензолсульфонил-7-пиперазин-1-ил-1H-индолгидрохлорида (101), tпл 278-280°C.
Аналогичным образом заменой на стадии 1 фенилдисульфида соответствующими замещенными фенилдисульфидами получали следующие соединения:
7-пиперазин-1-ил-3-(толуол-4-сульфонил)-1H-индол (102), tпл 285-287°C;
3-(3,4-дихлорбензолсульфонил)-7-пиперазин-1-ил-1H-индол (103), tпл 290°С;
3-(4-фторбензолсульфонил)-7-пиперазин-1-ил-1H-индол (104), tпл 247-249°С;
3-(3,5-дихлорбензолсульфонил)-7-пиперазин-1-ил-1H-индол (105), tпл 290°С;
3-(2,4-дихлорбензолсульфонил)-7-пиперазин-1-ил-1H-индол (106), tпл 300°С;
3-(3-хлорбензолсульфонил)-7-пиперазин-1-ил-1H-индол (107), tпл 295°C;
3-(2-хлорбензолсульфонил)-7-пиперазин-1-ил-1H-индол (108), tпл 280-282°С;
7-пиперазин-1-ил-3-(2-трифторметилбензолсульфонил)-1H-индол (109), tпл 186-187°C;
3-(3-хлорбензолсульфонил)-5-пиперазин-1-ил-1H-индол (110), tпл 192-197°С и
3-(3,4-дихлорбензолсульфонил)-5-пиперазин-1-ил-1H-индол (111), tпл >300°С.
Аналогичным образом заменой на стадии 1 фенилдисульфида соответствующими гетероарилдисульфидами получали следующие соединения:
7-пиперазин-1-ил-3-(пиридин-4-сульфонил)-1H-индол (112), tпл 207-208°С;
7-пиперазин-1-ил-3-(пиридин-2-сульфонил)-1H-индол (113), tпл 198-199°C;
2-(7-пиперазин-1-ил-1H-индол-3-сульфонил)бензотиазол (114), tпл 295°C и
6-пиперазин-1-ил-3-(пиридин-2-сульфонил)-1H-индол (115), tпл 246,7-247,2°С.
Пример 23
3-Бензолсульфонил-5-пиперазин-1-ил-1H-индол

Стадия 1:
трет-бутиловый эфир 4-(3-фенилсульфанил-1H-индол-5-ил)пиперазин-1-карбоновой кислоты
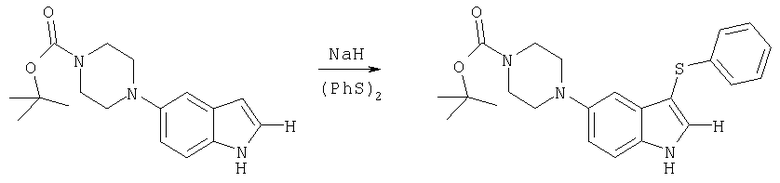
Гидрид натрия (0,29 г, 12 ммолей) вводили в охлажденный льдом раствор трет-бутилового эфира 4-(1H-индол-5-ил)пиперазин-1-карбоновой кислоты (20), полученного так, как изложено в примере получения 2 (3 г, 10 ммолей), в 30 мл ДМФ и образовавшуюся смесь перемешивали в течение 10 мин. Добавляли фенилдисульфида (2,6 г, 12 ммолей) и раствор перемешивали при комнатной температуре в течение 16 ч. Добавляли воды и смесь экстрагировали этиловым эфиром. Экстракт промывали рассолом, сушили и выпаривали с получением в виде желтовато-коричневого кристаллического твердого вещества 3,83 г трет-бутилового эфира 4-(3-фенилсульфанил-1H-индол-5-ил)пиперазин-1-карбоновой кислоты, tпл 174°C.
Стадия 2:
трет-бутиловый эфир 4-(3-бензолсульфонил-1H-индол-5-ил)пиперазин-1-карбоновой кислоты

Раствор трет-бутилового эфира 4-(3-фенилсульфанил-1H-индол-5-ил)пиперазин-1-карбоновой кислоты (2,57 г, 6,28 ммоля) в дихлорметане (50 мл) охлаждали во льду и обрабатывали 70%-ной м-хлорпербензойной кислотой (4,5 г). Охлаждающую баню удаляли и перемешивание при комнатной температуре продолжали в течение 3 ч. Далее смесь концентрировали досуха, остаток обрабатывали этиловым эфиром (50 мл), тщательно перемешивали в течение 10 мин, фильтровали, фильтровальный пирог тщательно промывали этиловым эфиром и сушили на воздухе с получением в виде бежевого твердого вещества соли сульфон-N-оксид-м-хлорбензойной кислоты. Ее растворяли в 45 мл ДМФ, обрабатывали 0,24 г катализатора Pearlman и гидрировали под атмосферным давлением в течение 4 ч. Далее смесь фильтровали для выделения катализатора, фильтрат концентрировали досуха, остаток растворяли в 450 мл этилацетата/хлороформа (4:1), промывали 1,5 М карбонатом натрия, а затем водой, сушили, фильтровали и частично концентрировали до ˜50 мл. После разбавления смеси 75 мл этилового эфира твердый материал отфильтровывали, промывали этиловым эфиром и сушили с получением в виде розовато-бежевого твердого вещества 2,4 г трет-бутилового эфира 4-(3-бензолсульфонил-1H-индол-5-ил)пиперазин-1-карбоновой кислоты, tпл 238-241°С (с разложением).
Стадия 3:
3-бензолсульфонил-5-пиперазин-1-ил-1H-индолгидрохлорид
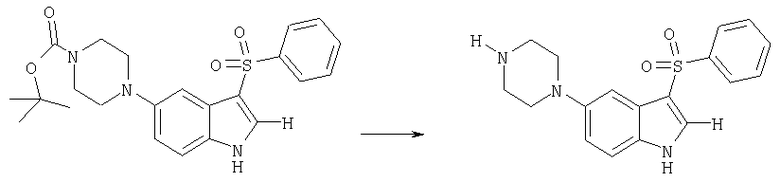
трет-Бутиловый эфир 4-(3-бензолсульфонил-1H-индол-5-ил)пиперазин-1-карбоновой кислоты (0,5 г, 1,13 ммоля) растворяли в 5 мл этанола и добавляли 3 мл концентрированной соляной кислоты. Смесь выдерживали при слабом кипении в течение 2-3 мин, охлаждали до комнатной температуры, подщелачивали гидроксидом аммония и разбавляли 20 мл воды. После того как смеси давали постоять в течение ночи, твердый материал отфильтровывали, промывали водой и сушили с получением в виде желтовато-коричневого кристаллического твердого вещества 0,37 г 3-бензолсульфонил-5-пиперазин-1-ил-1H-индола (201), tпл 254-264°C (с разложением).
Аналогичным образом в соответствии с методом, описанным выше, но с заменой фенилдисульфида соответствующими замещенными фенилдисульфидами, получали следующие соединения:
3-(2,3-дихлорбензолсульфонил)-5-пиперазин-1-ил-1H-индол (202), tпл >300°C;
5-пиперазин-1-ил-3-(4-трифторметилбензолсульфонил)-1H-индол (203), tпл 274,9-280,9°С;
3-(4-хлорбензолсульфонил)-5-пиперазин-1-ил-1H-индол (204), tпл 282-286,4°С;
3-(3,5-дихлорбензолсульфонил)-5-пиперазин-1-ил-1H-индол (205), tпл >300°С;
3-(2-хлорбензолсульфонил)-5-пиперазин-1-ил-1Н-индол (206), tпл >300°C;
3-(4-фторбензолсульфонил)-5-пиперазин-1-ил-1H-индол (207), tпл 289-291°С и
3-(3-хлорбензолсульфонил)-7-пиперидин-4-ил-1H-индол (208), tпл 272,3-272,9°С.
Пример 3
3-Бензолсульфонил-7-пиперидин-4-ил-1H-индол

Стадия 1:
трет-бутиловый эфир 4-(3-фенилсульфанил-1H-индол-7-ил)пиперидин-1-карбоновой кислоты

В раствор трет-бутилового эфира 4-(1H-индол-7-ил)пиперидин-1-карбоновой кислоты (22), полученного так, как изложено в настоящем описании в примере получения 4 (220 мг, 0,73 ммоля), в ДМФ (15 мл) одной порцией добавляли NaH (95%-ный, 25 мг, 0,95 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и добавляли фенилдисульфид (185 мг, 0,88 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 24 ч и выливали в воду (50 мл). Водный слой экстрагировали EtOAc (3 порции по 20 мл) и объединенные органические слои промывали рассолом, сушили (MgSO4) и концентрировали под вакуумом. Экспресс-хроматографией, элюируя 20%-ным ацетоном в гексанах, в виде желтовато-коричневого твердого вещества получали трет-бутиловый эфир 4-(3-фенилсульфанил-1H-индол-7-ил)пиперидин-1-карбоновой кислоты (200 мг).
Стадия 2:
трет-бутиловый эфир 4-(3-бензолсульфонил-1H-индол-7-ил)пиперидин-1-карбоновой кислоты
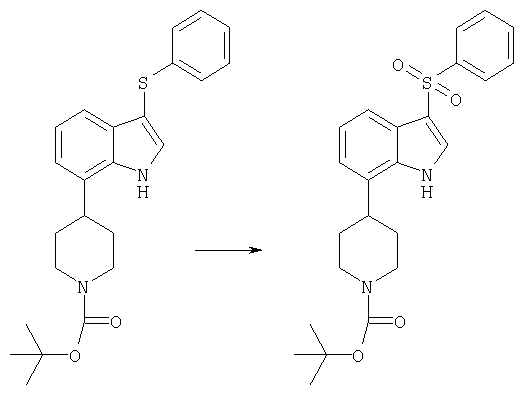
В холодный как лед раствор трет-бутилового эфира 4-(3-фенилсульфанил-1H-индол-7-ил)пиперидин-1-карбоновой кислоты (190 мг, 0,48 ммоля) в метиленхлориде (20 мл) в азотной атмосфере отдельными порциями вводили м-хлорпербензойную кислоту (166 мг, 0,96 ммоля). Реакционную смесь, полностью подготовленную после 2 ч выдержки при 0°С, разбавляли метиленхлоридом (30 мл) и промывали водой (15 мл). Органическую фазу промывали 5%-ным КОН (15 мл) и концентрировали с получением трет-бутилового эфира 4-(3-бензолсульфонил-1H-индол-7-ил)пиперидин-1-карбоновой кислоты (156 мг).
Стадия 3:
3-бензолсульфонил-7-пиперидин-4-ил-1H-индол

В раствор трет-бутилового эфира 4-(3-бензолсульфонил-1H-индол-7-ил)пиперидин-1-карбоновой кислоты (200 мг, 0,45 ммоля) в метиленхлориде (20 мл) в азотной атмосфере добавляли ТФК (5 мл). Спустя 30 мин реакционную смесь концентрировали под вакуумом и разделяли между 10%-ным водным КОН (30 мл) и EtOAc (30 мл). Ацетатный слой сушили (MgSO4) и концентрировали с получением продукта в виде светло-коричневого масла. Растиранием в порошок с диэтиловым эфиром в виде желтовато-коричневого твердого вещества получали 3-бензолсульфонил-7-пиперидин-4-ил-1H-индол (301) (113 мг), tпл 186-189,5°С.
Пример 4
3-Бензолсульфонил-1-метил-7-пиперазин-1-ил-1H-индол

Стадия 1:
трет-бутиловый эфир 4-(3-бензолсульфонил-1-метил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты

Гидрид натрия (0,12 г 60%-ной дисперсии в минеральном масле, 3 ммоля) с охлаждением льдом вводили в раствор трет-бутилового эфира 4-(3-бензолсульфонил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты (1 г, 2,3 ммоля) в 20 мл ДМФ. После перемешивания в течение 15 мин при комнатной температуре смесь обрабатывали метилиодидом (0,17 мл, 3 ммоля). Добавляли воды и смесь экстрагировали этилацетатом. Экстракт промывали водой и рассолом, сушили (сульфат натрия) и выпаривали. Хроматографией на силикагеле (20% этилацетата/гексан) в виде пены получали 0,9 г трет-бутилового эфира 4-(3-бензолсульфонил-1-метил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты, масс-спектр: М+ 455.
Стадия 2:
3-бензолсульфонил-1-метил-7-пиперазин-1-ил-1H-индол

Удалением защитной группы у трет-бутилового эфира 4-(3-бензолсульфонил-1-метил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты так, как изложено в описании стадии 3 примера 1, в виде белого твердого вещества получали свободное основание 3-бензолсульфонил-1-метил-7-пиперазин-1-ил-1H-индолгидрохлорида (401), tпл 249-250°C, tпл гидрохлоридной соли составляла 293-295°С.
Аналогичным образом в соответствии с методом, описанным выше, но с заменой на стадии 1 трет-бутилового эфира 4-(3-бензолсульфонил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты соответствующими индоловыми производными, получали следующие соединения:
1-метил-7-пиперазин-1-ил-3-(пиридин-2-сульфонил)-1H-индол (402), tпл 297-298°С;
3-бензолсульфонил-1-метил-5-пиперазин-1-ил-1H-индол (403), tпл 239-240°С (с разложением);
1-метил-7-пиперазин-1-ил-3-(2-трифторметилбензолсульфонил)-1H-индол (404), tпл 295°С;
3-(4-фторбензолсульфонил)-1-метил-7-пиперазин-1-ил-1H-индол (405), tпл 300°С;
1-метил-7-пиперазин-1-ил-3-(3-трифторметилбензолсульфонил)-1H-индол (406), tпл 279-280°C;
3-(2-хлорбензолсульфонил)-1-метил-7-пиперазин-1-ил-1H-индол (407), tпл 295-297°С;
3-(3-хлорбензолсульфонил)-1-метил-7-пиперазин-1-ил-1H-индол (408), tпл 300°C;
3-(2,3-дихлорбензолсульфонил)-1-метил-5-пиперазин-1-ил-1H-индол (409), tпл 198-203°C;
1-метил-5-пиперазин-1-ил-3-(3-трифторметилбензолсульфонил)-1H-индол (410), tпл 235-240°C;
3-(3,5-дихлорбензолсульфонил)-1-метил-5-пиперазин-1-ил-1H-индол (411), tпл 282-284,5°С;
1-метил-7-пиперазин-1-ил-3-(пиридин-2-сульфонил)-1H-индол (412), tпл 297-298°С;
3-(4-фторбензолсульфонил)-1-метил-5-пиперазин-1-ил-1H-индол (413), tпл 195-207°С;
3-(2-хлорбензолсульфонил)-1-метил-5-пиперазин-1-ил-1H-индол (414), tпл 249,6-253°С;
-(3-хлорбензолсульфонил)-1-метил-5-пиперазин-1-ил-1H-индол (415), tпл 185,5-188,5°С и
1-метил-5-пиперазин-1-ил-3-(пиридин-2-сульфонил)-1H-индол (416), tпл 256,8-257,5°С.
Аналогичным образом в соответствии с методом, описанным выше, но с заменой на стадии 1 метилиодида изопропилиодидом, получали следующее соединение:
3-бензолсульфонил-1-изопропил-5-пиперазин-1-ил-1H-индол (417), М+Н =384.
Пример 5
3-Бензолсульфонил-7-(4-метилпиперазин-1-ил)-1H-индол

Раствор 3-бензолсульфонил-7-пиперазин-1-ил-1H-индола (101) (500 мг, 1,46 ммоля) и 1 мл 37%-ного водного формальдегида в 25 мл этанола гидрировали под атмосферным давлением в присутствии 250 мг 10%-ного Pd-C в течение 30 мин. Смесь фильтровали, разбавляли водой и экстрагировали этилацетатом. Этилацетатный слой промывали водой и рассолом, сушили и выпаривали с получением сырого свободного основания 3-бензолсульфонил-7-(4-метилпиперазин-1-ил)-1H-индола.
Гидрохлоридную соль кристаллизовали из этанола/диэтилового эфира с получением 290 мг продукта (501), tпл 160-162°С.
Аналогичным образом в соответствии с методом, описанным выше, но с заменой 3-бензолсульфонил-7-пиперазин-1-ил-1H-индола соответствующими индолами, получали следующие соединения:
3-бензолсульфонил-1-метил-7-(4-метилпиперазин-1-ил)-1H-индол (502), tпл 260-262°С;
3-(3,4-дихлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1H-индол (503), tпл 196-203°C;
3-(2-хлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1H-индол (504), tпл 168,5-175,9°С;
3-(3-хлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1H-индол (505), tпл 163-171°С;
3-(2,4-дихлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1H-индол (506),
tпл 199-203°C;
3-(3,5-дихлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1H-индол (507), tпл 237,1-241,5°С;
7-(4-метилпиперазин-1-ил)-3-(2-трифторметилбензолсульфонил)-1H-индол (508), tпл 237,1-241,5°С;
3-бензолсульфонил-5-(4-метилпиперазин-1-ил)-1H-индол (509), М+Н =356, и
3-(4-фторбензолсульфонил)-7-(1-метилпиперидин-4-ил)-1H-индол (510), tпл 178-182°С.
Аналогичным образом в соответствии с методом, описанным выше, но с заменой формальдегида соответствующими альдегидами, получали следующие соединения:
3-бензолсульфонил-7-(4-циклопропилметилпиперазин-1-ил)-1H-индол (511), tпл 280-282°С;
3-бензолсульфонил-7-(4-пропилпиперазин-1-ил)-1H-индол (512), tпл 290-295°С, и
3-бензолсульфонил-7-(4-этилпиперазин-1-ил)-1H-индол (513), tпл 274-275°С.
Альтернативный путь получения 3-бензолсульфонил-7-(4-метилпиперазин-1-ил)-1H-индола (501)
Стадия 1:
1-бензолсульфонилметил-3-хлор-2-нитробензол

100 г 1-хлор-2-нитробензола (0,52 моля) растворяли в 450 мл ТГФ, содержавшего 86 г хлорметансульфонилбензола (0,52 моля). Смесь вводили в 12%-ный (1 М) раствор трет-бутоксида калия (трет-BuOK) в ТГФ (1,1 кг, 2,2 экв.), который охлаждали до -48°С и механически перемешивали. Скорость введения регулировали таким образом, чтобы температура массы раствора ТГФ/трет-BuOK не превышала -40°С. После добавления смесь (темно-пурпурную) перемешивали в течение еще 3 ч при температуре от -45 до -40°С до тех пор, пока ТСХ анализ (гексаны/этилацетат в соотношении 4:1) не свидетельствовал о затрате всего исходного материала. Реакцию в смеси гасили 200 мл уксусной кислоты и нагревали после гашения до -15°C. Затем реакцию медленно гасили с использованием в общем 2,4 л воды. Смесь перемешивали в течение дополнительных 18 ч, фильтровали и промывали 1,5 л воды в виде 500-миллилитровых аликвот. Фильтровальный пирог промывали гексанами и сушили в вакуумном сушильном шкафу при 45-50°С в токе азота с получением 136 г 1-бензолсульфонилметил-3-хлор-2-нитробензола, tпл 141-142°С.
Стадия 2:
1-(3-бензолсульфонилметил-2-нитрофенил)-4-метилпиперазин

10 г 1-бензолсульфонилметил-3-хлор-2-нитробензола в азотной атмосфере суспендировали в 25 мл N-метилпиперазина и перемешивали. Смесь выдерживали при 80°С (температура масляной бани) в течение 14-часового периода. Реакцию гасили при 80°С 125 мл воды и густую суспензию фильтровали, промывали 100 мл воды и 100 мл гексанов и сушили при 45°С под вакуумом в токе азота в течение 4 ч с получением 11,7 г 1-(3-бензолсульфонилметил-2-нитрофенил)-4-метилпиперазина с tпл 180-182°С.
Стадия 3:
2-бензолсульфонилметил-6-(4-метилпиперазин-1-ил)фениламин

Раствор 1-(3-бензолсульфонилметил-2-нитрофенил)-4-метилпиперазина (25 г) в тетрагидрофуране (500 мл), приготовленный нагреванием до 45°С, добавляли в предварительно гидрированную суспензию катализатора Pearlman (20% Pd(OH)2/C, 0,5 г) в тетрагидрофуране (20 мл). Перемешивание продолжали в атмосфере водорода при 45°С до завершения реакции (примерно 29 ч). После охлаждения катализатор отфильтровывали через солка-флок (SolkaflocТМ) (10 г) и промывали тетрагидрофураном (50 мл). Фильтрат концентрировали под вакуумом с получением в виде пены 24,9 г 2-бензолсульфонилметил-6-(4-метилпиперазин-1-ил)фениламина.
Стадия 4:
3-бензолсульфонил-7-(4-метилпиперазин-1-ил)-1H-индол
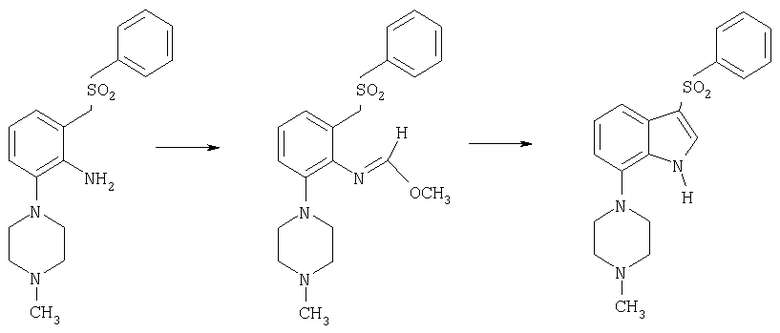
В раствор 2,3 г 2-бензолсульфонилметил-6-(4-метилпиперазин-1-ил)фениламина в 20 мл триметилортоформиата вводили п-толуолсульфоновую кислоту (120 мг). Смесь кипятили с обратным холодильником в азотной атмосфере в течение 2 ч. Смесь охлаждали до 60°С и добавляли одну чешуйку КОН. Вновь смесь кипятили с обратным холодильником в течение 1,5 ч. Греющую масляную баню отключали и смесь перемешивали в азотной атмосфере в течение ночи. Смесь обрабатывали 50 мл насыщенного раствора аммонийхлорида и 100 мл EtOAc. От прозрачного водного раствора отделяли пурпурный органический слой, промывали водой (45 мл) и выпаривали до получения вещества в виде пены. Это вещество в виде пены растворяли в 80 мл EtOH крепостью 200. После растворения кристаллизовалось свободное основание. Образовавшуюся смесь кипятили с обратным холодильником до повторного растворения твердого вещества, а после удаления источника нагрева в эту смесь добавляли 5 мл насыщенного раствора HCl в этаноле. Смесь охлаждали с перемешиванием до комнатной температуры и вносили затравку для гарантии немедленной кристаллизации. Смесь концентрировали до примерно 40 мл и фильтровали. Собранный при этом твердый материал промывали примерно 3 мл EtOH и сушили при 50°С в вакуумном сушильном шкафу с получением 1,9 г 3-бензолсульфонил-7-(4-метилпиперазин-1-ил)-1H-индола (501) с tпл 160-162°С.
ПРИМЕР 6
3-Бензолсульфонил-2-метил-7-пиперазин-1-ил-1H-индол
Стадия 1:
трет-бутиловый эфир 4-(2-метил-3-фенилсульфанил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты
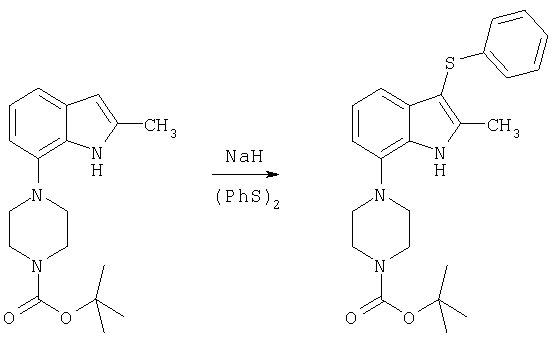
Гидрид натрия (0,08 г 60%-ной дисперсии в минеральном масле, 1,9 ммоля) вводили в охлажденный льдом раствор трет-бутилового эфира 4-(2-метил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты (21), полученного так, как изложено в примере получения 3 (0,4 г, 1,3 ммоля), в 5 мл ДМФ и образовавшуюся смесь перемешивали в течение 10 мин. Добавляли фенилдисульфид (0,3 г, 1,4 ммоля) и раствор перемешивали при комнатной температуре в течение 4 ч. Добавляли воды и смесь экстрагировали этилацетатом. Экстракт промывали рассолом, сушили и выпаривали с получением в виде масла 0,5 г трет-бутилового эфира 4-(2-метил-3-фенилсульфанил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты, М+=409.
Стадия 2:
трет-бутиловый эфир 4-(3-бензолсульфонил-2-метил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты

Oxone™ (1,5 г, 2,4 ммоля) в 20 мл воды вводили в перемешиваемый раствор трет-бутилового эфира 4-(2-метил-3-фенилсульфанил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты (0,5 г, 1,2 ммоля) в 25 мл метанола. Смесь перемешивали в течение 2 ч, разбавляли водой и экстрагировали этилацетатом. Этилацетатный экстракт сушили (сульфат натрия) и выпаривали с получением в виде твердого остатка сульфон-N-оксида. Этот продукт растворяли в 50 мл метанола и гидрировали под атмосферным давлением в течение 12 ч над 0,3 г 10%-ного Pd-C. Добавляли дихлорметан и смесь фильтровали и выпаривали. Растиранием в порошок остатка с диэтиловым эфиром в виде белого твердого вещества получали 0,25 г трет-бутилового эфира 4-(3-бензолсульфонил-2-метил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты, tпл 168-169°С.
Стадия 3:
3-бензолсульфонил-2-метил-7-пиперазин-1-ил-1H-индолгидрохлорид

трет-Бутиловый эфир 4-(3-бензолсульфонил-2-метил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты (0,25 г) растворяли в 2 мл дихлорметана и добавляли 2 мл трифторуксусной кислоты. По истечении 10 мин смесь концентрировали под вакуумом, растворяли в воде и промывали диэтиловым эфиром. Водный слой подщелачивали гидроксидом аммония, экстрагировали дихлорметаном и экстракт сушили (сульфат натрия), выпаривали с получением сырого свободного основания, которое кристаллизовали из дихлорметана/диэтилового эфира с получением 35 мг 3-бензолсульфонил-2-метил-7-пиперазин-1-ил-1H-индола (601), tпл 188-190°С.
Аналогичным образом в соответствии с методом, описанным выше, но с заменой фенилдисульфида на стадии 1 соответствующими замещенными фенилдисульфидами, получали следующие соединения:
3-(4-хлорбензолсульфонил)-2-метил-7-пиперазин-1-ил-1H-индол (602), tпл 118-120°С;
3-(4-фторбензолсульфонил)-2-метил-7-пиперазин-1-ил-1Н-индол (603), tпл 232-236°С, и
3-(4-метоксибензолсульфонил)-2-метил-7-пиперазин-1-ил-1Н-индол (604), tпл 182-184°C.
Пример 7
3-Фенилсульфанил-7-пиперазин-1-ил-1H-индол
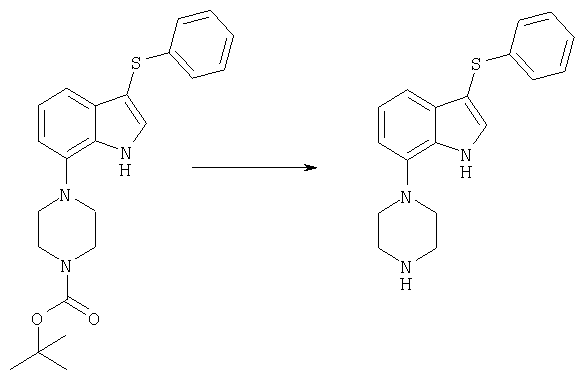
Трифторуксусную кислоту (5 мл) вводили в раствор трет-бутилового эфира 4-(3-фенилсульфанил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты (700 мг, 1,7 ммоля), полученного так, как изложено в примере 1, стадия 1, в 4 мл дихлорметана. По истечении 15 мин раствор разбавляли водой, подщелачивали гидроксидом аммония и экстрагировали дихлорметаном. Дихлорметановый экстракт промывали рассолом и выпаривали с получением в виде твердого вещества 3-фенилсульфанил-7-пиперазин-1-ил-1H-индола (701). Из этанола/HCl кристаллизовали гидрохлоридную соль с получением 500 мг белого твердого вещества, tпл 300°С.
Аналогичным образом с заменой трет-бутилового эфира 4-(3-фенилсульфанил-1H-индол-7-ил)пиперазин-1-карбоновой кислоты соответствующими сульфаниловыми производными, полученными так, как изложено в настоящем описании в других примерах, получали следующие соединения:
3-(2,3-дихлорфенилсульфанил)-5-пиперазин-1-ил-1H-индол (702), tпл 255,1-255,5°С;
3-фенилсульфанил-5-пиперазин-1-ил-1H-индол (703), tпл 246-247, и
3-(2,6-дихлорфенилсульфанил)-5-пиперазин-1-ил-1H-индол (704), М+Н =378.
Пример 8
Композиция для перорального введения
Эти компоненты смешивают и распределяют по капсулам вместимостью примерно по 100 мг; одна капсула заключает в себе приблизительно целую ежедневную дозу.
Пример 9
Композиция для перорального введения
Эти компоненты объединяют и гранулируют с использованием растворителя, такого как метанол. Далее композицию сушат и с помощью соответствующей таблетировочной машины из нее формуют таблетки (содержащие примерно по 20 мг действующего вещества).
Пример 10
Композиция для перорального введения
Эти компоненты смешивают с получением суспензии для перорального введения.
Пример 11
Композиция (IV) для парентерального введения
Действующий компонент растворяют в части воды для инъекций. Далее с перемешиванием добавляют хлорид натрия в нужном для придания раствору изотоничности количестве. Оставшейся водой для инъекций доводят массу раствора до нужной, фильтруют через 0,2-микрометрический мембранный фильтр и упаковывают в стерильных условиях.
Пример 12
Композиция для суппозиториев
Эти компоненты совместно плавят, перемешивают на паровой бане и выливают в формы, вмещающие в общей сложности по 2,5 г.
Пример 13
Композиция для местного применения
Все компоненты, за исключением воды, совмещают и при перемешивании нагревают до примерно 60°С. Далее при примерно 60°С и интенсивном перемешивании добавляют достаточное количество воды для эмульгирования компонентов, а затем воду добавляют в количестве, необходимом для достижения массы примерно 100 г.
Пример 14
Композиция для интраназального спрея
В качестве аэрозольных композиций для интраназального введения готовят несколько водных суспензий, содержащих примерно от 0,025 до 0,5% действующего соединения. Эти композиции необязательно включают такие неактивные компоненты, как, например, микрокристаллическая целлюлоза, натрийкарбоксиметилцеллюлоза, декстрин и т.п. Для регулирования рН можно добавлять соляную кислоту. Композиции для интраназального спрея можно дозировать с помощью дозировочного насоса интраназального спрея, который за один импульс, как правило, подает примерно от 50 до 100 мкл композиции. Типичная схема применения включает от 2 до 4 впрсыскиваний каждые 4-12 ч.
Пример 15
Изучение связывания радиолиганда
Связывающую активность соединений по настоящему изобретению in vitro определяли следующим образом.
Сродство лигандов определяли в 2 повторах с использованием конкурентного метода по связыванию [3H]LSD с клеточными мембранами, выделенными из клеток НЕК293, которые стабильно экспрессируют рекомбинантный рецептор 5-НТ6 человека.
Все определения осуществляли в буферном растворе, содержавшем 50 мМ Трис-HCl, 10 мМ MgSO4, 0,5 мМ ЭДТК, 1 мМ аскорбиновую кислоту, при рН 7,4 и 37°С, в реакционном объеме 250 мкл. Пробирки, содержавшие [3H]LSD (5 нМ), конкурентный лиганд и мембраны, инкубировали встряхиванием в течение 60 мин при 37°С на водяной бане, фильтровали в планшет фирмы Packard GF-B (предварительно пропитанный 0,3% ПЭИ (полиэтиленимин)), используя харвастер для 96-луночного планшета фирмы Packard, и 3 раза промывали охлажденным льдом 50 мМ Трис-HCl. Связанный [3H]LSD определяли по числу радиоактивных импульсов в минуту с использованием счетчика фирмы Packard TopCount.
Замещение [3H] LSD из сайтов связывания определяли количественно, подставляя данные "концентрация-связывание" в 4-параметрическое логическое уравнение
где Hill обозначает коэффициент Хилла, [ligand] обозначает концентрацию конкурирующего радиолиганда, a IC50 обозначает концентрацию радиолиганда, соответствующую половине максимально связанного радиолиганда. Интервал специфического связывания является разницей между Bmax и базальными параметрами.
Соединения формулы I тестировали согласно протоколу примера 15 и обнаружили, что они являлись селективными антагонистами 5-НТ6.
Хотя настоящее изобретение описано со ссылкой на конкретные варианты его выполнения, специалистам в данной области техники следует иметь в виду, что в них можно вносить самые разнообразные изменения и их заменять эквивалентами, не выходя из объема изобретения. Кроме того, для достижения соответствия конкретных ситуации, материала, рассматриваемой композиции, способа и стадии или стадий способа целевой сущности и объему настоящего изобретения можно прибегнуть ко многим модификациям. Все эти модификации необходимо рассматривать как охватываемые объемом прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| 2,4-ЗАМЕЩЕННЫЕ ИНДОЛЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ К 5-ГТ6 РЕЦЕПТОРУ | 2003 |
|
RU2326112C2 |
| 2,7-ЗАМЕЩЕННЫЕ ИНДОЛЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АНТАГОНИСТИЧЕСКУЮ АКТИВНОСТЬ ПО ОТНОШЕНИЮ К 5-ГТ6 РЕЦЕПТОРУ | 2003 |
|
RU2327685C2 |
| ПРОИЗВОДНЫЕ ПРОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КАТЕПСИНА | 2010 |
|
RU2535479C2 |
| 4-ПИПЕРАЗИНИЛБЕНЗОЛСУЛЬФОНИЛИНДОЛЫ, ХАРАКТЕРИЗУЮЩИЕСЯ СРОДСТВОМ К РЕЦЕПТОРУ 5-НТ6 | 2003 |
|
RU2324693C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ЦИКЛОПЕНТАНА | 2010 |
|
RU2572555C2 |
| 2,5- ЗАМЕЩЕННЫЕ ТЕТРАГИДРОИЗОХИНОЛИНЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ МОДУЛЯЦИИ 5-НТ6 | 2004 |
|
RU2327689C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 1-ОКСА-2,8-ДИАЗАСПИРО[4.5]ДЕЦ-2-ЕНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО, ОБЛАДАЮЩЕЕ АНАЛЬГЕТИЧЕСКИМ ДЕЙСТВИЕМ | 2002 |
|
RU2296128C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2005 |
|
RU2382770C2 |
| СОЕДИНЕНИЯ, ЯВЛЯЮЩИЕСЯ АКТИВНЫМИ ПО ОТНОШЕНИЮ К РЕЦЕПТОРАМ, АКТИВИРУЕМЫМ ПРОЛИФЕРАТОРОМ ПЕРОКСИСОМ | 2004 |
|
RU2356889C2 |
| АРИЛСУЛЬФОНИЛЬНЫЕ ПРОИЗВОДНЫЕ, ОБЛАДАЮЩИЕ СРОДСТВОМ К РЕЦЕПТОРУ 5-HT, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2268884C2 |
Изобретение относится к новым производным индола общей формулы I

где R1 обозначает -S(O)0-2-A, где А обозначает фенил, необязательно замещенный одной или несколькими группами, выбранными из (низш.)алкила, (низш.)алкокси, гало, (низш.)галоалкила, (низш.)алкилсульфонила или (низш.)галоалкилсульфонила, или означает нафтил, пиридинил, бензотиазолил;
R2 обозначает водород, C1-6-алкил;
R3 обозначает водород, С1-6-алкил;
R4 обозначает водород;
один из R5, R6 или R7 обозначает группу общей формулы В, в которой W обозначает группу -СН- или атом азота,
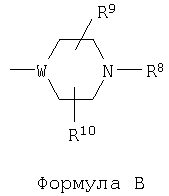
а другие независимо обозначают водород;
R8 обозначает водород, С1-10-алкил;
R9 и R10 независимо обозначают водород;
или их фармацевтически приемлемым солям.
Соединения формулы I обладают сродством к рецептору 5-НТ6, что позволяет использовать их в фармацевтической композиции. 2 н. и 18 з.п. ф-лы.
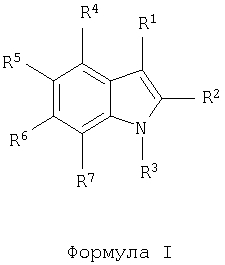
в которой R1 обозначает -S(O)0-2-A,
где А обозначает фенил, необязательно замещенный одной или несколькими группами, выбранными из (низш.)алкила, (низш.)алкокси, гало, (низш.)галоалкила, (низш.)алкилсульфонила или (низш.)галоалкилсульфонила,
или означает нафтил, пиридинил, бензотиазолил;
R2 обозначает водород, C1-6-алкил;
R3 обозначает водород, C1-6-алкил;
R4 обозначает водород;
один из R5, R6 или R7 обозначает группу общей формулы В, в которой W обозначает группу -СН- или атом азота,

а другие независимо обозначают водород;
R8 обозначает водород, С1-10-алкил;
R9 и R10 независимо обозначают водород;
или их фармацевтически приемлемые соли.
3-бензолсульфонил-7-пиперазин-1-ил-1Н-индол,
3-бензолсульфонил-1-метил-7-пиперазин-1-ил-1Н-индол,
3-бензолсульфонил-2-метил-7-пиперазин-1-ил-1Н-индол,
3-(4-хлорбензолсульфонил)-2-метил-7-пиперазин-1-ил-1Н-индол,
3-(4-метоксибензолсульфонил)-2-метил-7-пиперазин-1-ил-1Н-индол,
7-пиперазин-1-ил-3-(пиридин-4-сульфонил)-1Н-индол,
7-пиперазин-1-ил-3-(пиридин-2-сульфонил)-1Н-индол,
1-метил-7-пиперазин-1-ил-3-(пиридин-2-сульфонил)-1Н-индол,
3-бензолсульфонил-7-(4-метилпиперазин-1-ил)-1Н-индол,
3-(3,4-дихлорбензолсульфонил)-7-пиперазин-1-ил-1Н-индол,
2-(7-пиперазин-1-ил-1Н-индол-3-сульфонил)бензотиазол,
3-(4-фторбензолсульфонил)-2-метил-7-пиперазин-1-ил-1Н-индол,
3-(4-фторбензолсульфонил)-7-пиперазин-1-ил-1Н-индол,
3-бензолсульфонил-7-пиперидин-4-ил-1Н-индол,
7-пиперазин-1-ил-3-(толуол-4-сульфонил)-1Н-индол,
3-(3,5-дихлорбензолсульфонил)-7-пиперазин-1-ил-1Н-индол,
3-(3-хлорбензолсульфонил)-7-пиперазин-1-ил-1Н-индол,
3-(2-хлорбензолсульфонил)-7-пиперазин-1-ил-1Н-индол,
7-пиперазин-1-ил-3-(2-трифторметилбензолсульфонил)-1Н-индол,
1-метил-7-пиперазин-1-ил-3-(2-трифторметилбензолсульфонил)-1Н-индол,
3-(4-фторбензолсульфонил)-1-метил-7-пиперазин-1-ил-1Н-индол,
1-метил-7-пиперазин-1-ил-3-(пиридин-2-сульфонил)-1Н-индол,
1-метил-7-пиперазин-1-ил-3-(3-трифторметилбензолсульфонил)-1Н-индол,
3-(2-хлорбензолсульфонил)-1-метил-7-пиперазин-1-ил-1Н-индол,
3-(3-хлорбензолсульфонил)-1-метил-7-пиперазин-1-ил-1Н-индол,
3-бензолсульфонил-1-метил-7-(4-метилпиперазин-1-ил)-1Н-индол,
3-(3,4-дихлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1Н-индол,
3-(2-хлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1Н-индол,
3-(3-хлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1Н-индол,
3-(2,4-дихлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1Н-индол,
3-(3,5-дихлорбензолсульфонил)-7-(4-метилпиперазин-1-ил)-1Н-индол,
7-(4-метилпиперазин-1-ил)-3-(2-трифторметилбензолсульфонил)-1Н-индол,
3-фенилсульфанил-7-пиперазин-1-ил-1Н-индол
или его фармацевтически приемлемая соль.
3-бензолсульфонил-5-пиперазин-1-ил-1Н-индол,
3-бензолсульфонил-1-метил-5-пиперазин-1-ил-1Н-индол,
3-(2,3-дихлорфенилсульфонил)-5-пиперазин-1-ил-1Н-индол,
3-(2,3-дихлорбензолсульфонил)-5-пиперазин-1-ил-1Н-индол,
3-(2,3-дихлорбензолсульфонил)-1-метил-5-пиперазин-1-ил-1Н-индол,
1-метил-5-пиперазин-1-ил-3-(3-трифторметилбензолсульфонил)-1Н-индол,
5-пиперазин-1-ил-3-(4-трифторметилбензолсульфонил)-1Н-индол,
3-(4-хлорбензолсульфонил)-5-пиперазин-1-ил-1Н-индол,
3-(3,5-дихлорбензолсульфонил)-5-пиперазин-1-ил-1Н-индол,
3-(3,5-дихлорбензолсульфонил)-1-метил-5-пиперазин-1-ил-1Н-индол,
3-фенилсульфанил-5-пиперазин-1-ил-1Н-индол,
3-(2-хлорбензолсульфонил)-5-пиперазин-1-ил-1Н-индол,
3-(4-фторбензолсульфонил)-5-пиперазин-1-ил-1Н-индол,
3-(4-фторбензолсульфонил)-1-метил-5-пиперазин-1-ил-1Н-индол,
3-(2-хлорбензолсульфонил)-1-метил-5-пиперазин-1-ил-1Н-индол,
или его фармацевтически приемлемая соль.
| СПОСОБ УЛЬТРАЗВУКОВОГО ОБЕССЕРИВАНИЯ ИСКОПАЕМЫХ ТОПЛИВ В ПРИСУТСТВИИ ДИАЛКИЛОВЫХ ЭФИРОВ | 2003 |
|
RU2287551C2 |
| Bioorganic and Medicinal Chemistry Letters | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| WO 00/63203 А1, 26.10.2000 | |||
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| ПРОИЗВОДНЫЕ ИНДОЛА, ЗАМЕЩЕННЫЕ ИНДОЛЫ И СПОСОБ ИНГИБИРОВАНИЯ 5HT РЕЦЕПТОРА СЕРОТОНИНА | 1992 |
|
RU2126399C1 |

