Область техники, к которой относится изобретение
Изобретение относится к соединениям, активным в отношении метаботропных глутаматных рецепторов и которые могут быть использованы для лечения неврологических и психических заболеваний и нарушений.
Уровень техники
Последние достижения в изучении нейрофизиологических функций метаботропных глутаматных рецепторов показали, что эти рецепторы являются перспективными мишенями для лекарственных средств при терапии острых и хронических неврологических и психических заболеваний и нарушений. Однако при реализации этой идеи основной проблемой является разработка соединений, избирательных к подвиду метаботропных глутаматных рецепторов.
Глутамат является главным стимулирующим нейромедиатором центральной нервной системы (ЦНС) млекопитающих. Глутамат оказывает воздействие на центральные нейроны посредством связывания с рецепторами клеточной поверхности и их активации. Эти рецепторы подразделяются на два главных класса, ионотропные и метаботропные глутаматные рецепторы, на основе структурных характеристик рецепторных белков, с помощью которых рецептор передает сигнал в клетку, и совокупности их фармакологических параметров.
Метаботропные глутаматные рецепторы (mGluR) это опосредованные G-белками рецепторы, которые после связывания с глутаматом активируют различные внутриклеточные системы вторичных мессенджеров (посредников). Активация mGluR в интактных нейронах млекопитающих индуцирует один или более следующих ответов: активацию фосфолипазы С, усиление гидролиза фосфоинозитидов (ФИ), высвобождение внутриклеточного кальция, активацию фосфолипазы D, активацию или ингибирование аденилциклазы, усиление или подавление образования циклоаденозинмонофосфата (цАМФ), активацию гуанилилциклазы, усиление образования циклогуанозинмонофосфата (цГМФ), активацию фосфолипазы A2, усиление высвобождения арахидоновой кислоты, а также усиление или подавление активности потенциали лиганд-управляемых ионных каналов. См. статьи Schoepp и соавт., Trends Pharmacol. Sci., 14:13 (1993); Schoepp, Neurochem. Int., 24:439 (1994), Pin и соавт., Neuropharmacology, 34:1 (1995).
Методом молекулярного клонирования идентифицировано восемь различных подвидов mGluR, получивших названия mGluR1-mGluR8. См., например, в статьях Nakanishi, Neuron, 13:1031 (1994); Pin и соавт., Neuropharmacology, 34:1 (1995); Knopfel и соавт., J. Med. Chem., 38:1417 (1995). Образование ряда других рецепторов происходит путем экспрессии альтернативно сплайсированных форм некоторых подвидов mGluR. См. в статьях Pin и соавт.; PNAS, 89:10331 (1992); Minakami и соавт., BBRC, 199:1136 (1994); Joly и соавт., J. Neurosci., 15:3970 (1995).
Подвиды метаботропного глутаматного рецептора подразделяются на три группы, группу I, группу II и группу III mGluR, на основе гомологии аминокислотной последовательности, систем вторичного мессенджера, используемых рецепторами, и их фармакологических характеристиках. См. в статьях Nakanishi, Neuron, 13:1031 (1994); Pin и соавт., Neuropharmacology, 34:1 (1995); Knopfel и соавт., J. Med. Chem., 38:1417 (1995).
Группа I mGluR включает mGluRI, mGluR5 и их альтернативно сплайсированные варианты. Связывание агонистов с этими рецепторами вызывает активацию фосфолипазы С и последующую мобилизацию внутриклеточного кальция. Для демонстрации этих эффектов выполнены электрофизиологические измерения, например, на овоцитах Xenopus, которые экспрессируют рекомбинантные рецепторы mGluRI. См., например, в статьях Masu и соавт., Nature, 349:760 (1991); Pin и соавт., PNAS, 89:10331 (1992). Аналогичные результаты получены на овоцитах, экспрессирующих рекомбинантные рецепторы mGluR5. См. в статьях Abe и соавт., J. Biol. Chem., 267:13361 (1992); Minakami и соавт., BBRC, 199:1136 (1994); Joly и соавт., J. Neurosci., 15:3970 (1995). В другом эксперименте, активация агонистом рекомбинантных рецепторов mGluR 1, экспрессированных в клетках яичника китайского хомячка (СНО), стимулирует гидролиз ФИ, образование цАМФ и высвобождение арахидоновой кислоты, что было определено измерениями с помощью стандартных биохимических испытаний. См. в статье Aramori и соавт., Neuron, 8:757 (1992).
Для сравнения, активация рецепторов mGluR5, экспрессированных в клетках СНО, стимулирует гидролиз ФИ и последующее изменение потоков внутриклеточного кальция, но при этом не наблюдается стимуляции образования цАМФ или высвобождения арахидоновой кислоты. См. в статье Abe и соавт., J. Biol. Chem., 267:13361 (1992). Однако, активация рецепторов mGluR5, экспрессированных в клетках LLC-PK1, приводит к гидролизу ФИ и образованию повышенной концентрации цАМФ. См. в статье Joly и соавт., J. Neurosci., 15:3970 (1995). Эффективность действия агониста рецептора mGluR группы I изменяется следующим образом: квисквалат > глутамат = иботенат > (28,1′ S,2′S)-2-карбоксициклопропилглицин (L-CCG-I) > (18,3R)-1-аминоциклопентан-1,3-дикарбоновая кислота (АЦПД). Квисквалат действует относительно избирательно на рецепторы группы I по сравнению с mGluR групп II и III, но, кроме того, является сильным активатором ионотропных рецепторов АМФ кислоты. См. в статьях Pin и соавт., Neuropharmacology, 34:1 (1995); Knopfel и соавт., J. Med. Chem., 38:1417 (1995).
Отсутствие агонистов и антагонистов к подвидам mGluR препятствует изучению физиологической роли отдельных mGluR, в связи с этим в настоящее время необходимо исследовать опосредованные этими рецепторами патофизиологические процессы, действующие на ЦНС. Однако исследования с использованием имеющихся неспецифических агонистов и антагонистов дают некоторое общее представление о рецепторах mGluR группы I в сравнении с рецепторами mGluR групп II и III.
Попытки выяснения физиологических функций mGluR группы I свидетельствуют о том, что активация этих рецепторов вызывает возбуждение нейронов. Различного рода исследования показали, что АЦПД может вызывать постсинаптическое возбуждение после воздействия на нейроны в гиппокампе, коре головного мозга, мозжечке и таламусе, а также в других отделах мозга. Факты свидетельствуют о том, что это возбуждение происходит благодаря прямой активации постсинаптических mGluR, но кроме того, предполагают, что имеет место активация предсинаптических mGluR, что приводит к повышенному выбросу нейромедиатора. См. в статьях Baskys, Trends Pharmacol. Sci., 15:92 (1992); Schoepp, Neurochem. Int., 24:439 (1994); Pin и соавт., Neuropharmacology, 34:1 (1995).
В фармакологических экспериментах рецепторы mGluR группы I используются в качестве медиаторов этого механизма возбуждения. Действие АЦПД может быть воспроизведено при низких концентрациях квисквалата в присутствии антагонистов iGluR. См. в статьях Ни и соавт., Brain Res., 568:339 (1991); Green и соавт., Eur. J. Pharmacol., 226:279 (1992). Известно, что два производных фенилглицина, (S)-3-гидроксифенилглицин ((S)-ЗГФГ) и (S)-3,5-дигидроксифенилглицин ((S)-ДГФГ), активируют mGluR 1 и, таким образом, вызывают возбуждение. См. в статье Watkins и соавт., Trends Pharmacol. Sci., 15:33 (1994). Кроме того, возбуждение может быть блокировано (S)-4-карбоксифенилглицином ((S)-4КФГ), (S)-4-карбокси-3-гидроксифенилглициноом ((S)-4КЗГФГ) и (+)-α-метил-4-карбоксифенилглицином ((+)-4МКФГ), то есть соединениями, известными как антагонисты mGluRI. См. в статьях Eaton и соавт., Eur. J. Pharmacol., 244:195 (1993); Watkins и соавт., Trends Pharmacol., Sci. 15:33 (1994).
Метаботропные глутаматные рецепторы принимают участие в ряде нормальных процессов ЦНС млекопитающих. Показано, что активация mGluR необходима для индукции долговременного потенциирования гиппокампа и долговременного торможения мозжечка. См. в статьях Bashirn соавт., Nature, 363:347 (1993); Bortolotto и соавт., Nature, 368:740 (1994); Aiba и соавт., Cell, 79:365 (1994); Aiba и соавт., Cell, 79:377 (1994). Установлена также роль активации mGluR в проявлении болевых ощущений и в аналгезии. См. в статье Meller и соавт., Neuroreport, 4:879 (1993). Кроме того, предполагается, что активация mGluR выполняет модуляторную функцию во многих других нормальных процессах, включая синаптическую передачу сигнала, развитие нейронов, апоптотическую гибель нейронов, синаптическую пластичность, пространственное обучение, обонятельную память, центральную регуляцию сердечной активности, пробуждение от сна, регуляцию двигательного центра и регуляцию вестибулярно-зрительного рефлекса. См.,например, в статьях Nakanishi, Neuron, 13:1031 (1994); Pin и соавт., Neuropharmacology, 34:1 (1995); Knopfel и соавт., J. Med. Chem., 38:1417 (1995).
Предполагается также, что метаботропные глутаматные рецепторы принимают участие во многих патофизиологических процессах и патологических состояниях, затрагивающих ЦНС. Такие состояния включают инсульт, травмы головы, гипоксические и ишемические повреждения, гипогликемию, эпилепсию и нейродегенеративные заболевания, такие как болезнь Альцгеймера. См. в статьях Schoepp и соавт., Trends Pharmacol. Sci., 14:13 (1993); Cunningham и соавт., Life Sci., 54:135 (1994); Hollman и соавт., Ann. Rev. Neurosci., 17:31 (1994); Pin и соавт., Neuropharmacology, 34:1 (1995); Knopfel и соавт., J. Med. Chem., 38:1417 (1995). Полагают, что многие патологические процессы при этих состояниях происходят из-за возбуждения нейронов ЦНС, вызванных избытком глутамата. Поскольку считается, что mGluR группы I усиливает возбуждение нейронов, опосредованное глутаматом, через постсинаптические механизмы и усиленный предсинаптический выброс глутамата, их активация, вероятно, вносит определенный вклад в развитие патологических состояний. Соответственно, избирательные антагонисты mGluR группы I могут быть эффективны в терапии, прежде всего, в качестве нейропротективных агентов, аналгетиков или противосудорожных средств.
Предварительные исследования по оценке терапевтических характеристик известных агонистов и антагонистов mGluR привели к кажущимся противоречивым результатам. Например, было установлено, что нанесение АЦПД на нейроны гиппокампа приводит к эпилептическому припадку и повреждению нейронов (см. в статьях Sacaan и соавт., Neurosci. Lett., 139:77 (1992); Lipparti и соавт., Life Sci., 52:85 (1993)). Однако другие исследования свидетельствуют о том, что АЦПД ингибирует эпилептоформную активность, а также может проявлять нейропротективные свойства. См. в статьях Taschenberger и соавт., Neuroreport, 3:629 (1992); Sheardown, Neuroreport, 3:916 (1992); Koh и соавт., Ргос. Natl. Acad. Sci. USA, 88:9431 (1991); Chiamulera и соавт., Eur. J. Pharmacol., 216:335 (1992); Siliprandi и соавт., Eur. J. Pharmacol., 219:173 (1992); Pizzi и соавт., J. Neurochem., 61:683 (1993).
Вероятно, эти противоречивые результаты связаны с недостаточной избирательностью АЦПД, который вызывает активацию нескольких различных подвидов mGluR. Работы, в которых обнаружено повреждение нейронов, по-видимому, свидетельствуют о том, что происходит активация mGluR группы I, что в свою очередь приводит к усилению нежелательного переноса возбуждающего нервного импульса. Работы, в которых установлено нейропротективное действие, свидетельствуют о том, что происходит активация mGluR группы II и/или III, ингибирование предсинаптического выброса глутамата и снижение переноса возбуждающего нервного импульса.
Такая интерпретация результатов согласуется с наблюдением, что (S)-4КЗГФГ, антагонист mGluR группы I и агонист mGluR группы II, защищает мышей линии DBA/2 от аудиогенных эпилептических припадков, в то время как агонисты DCG-IV и L-CCG-1, селективные в отношении mGluR группы II, защищают нейроны от токсичного действия, индуцированного NMDA и КА. См. в статьях Thomsen и соавт., J. Neurochem., 62:2492(1994); Вгипо и соавт., Eur. J. Pharmacol., 256:109(1994); Pizzi и соавт., J. Neurochem., 61:683(1993).
Из вышесказанного очевидно, что известные в настоящее время агонисты и антагонисты mGluR имеют ограниченное значение ввиду недостаточной эффективности и селективности действия. Кроме того, большинство известных соединений являются аминокислотами или их производными, обладающими ограниченной биодоступностью, что препятствует исследованиям in vivo по оценке физиологической роли mGluR, фармакологии и терапевтических свойств таких соединений. Соединения, которые избирательно ингибируют активацию подвидов метаботропных глутаматных рецепторов группы I, могут найти применение при терапии неврологических нарушений и заболеваний, таких как старческое слабоумие, болезнь Паркинсона, болезнь Альцгеймера, хорея Гентингтона, боли, мигрень, эпилепсия, травма головы, гипоксические или ишемические повреждения, психические нарушения, такие как шизофрения, депрессия и тревога, офтальмологические нарушения, такие как различные ретинопатии, например, диабетическая ретинопатия, глаукома, и неврологические нарушения слуховой природы, такие как шум в ушах, и невропатические болевые состояния, включая невропатические заболевания, такие как диабетическая невропатия, невропатии, вызванные химиотерапией, пост-герпетическая невралгия и невралгия тройничного нерва.
Соответственно, существует необходимость в эффективных агонистах и антагонистах mGluR, обладающих высокой избирательностью в отношении подвидов mGluR, прежде всего, в отношении подвида рецепторов группы I.
Сущность изобретения
Объектом настоящего изобретения являются соединения, активные в отношении метаботропных глутаматных рецепторов, обладающие высокой эффективностью и избирательностью к отдельным подвидам метаботропного глутаматного рецептора, и разработка способов получения таких соединений.
Другим объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение, которое обладает высокой эффективностью и избирательностью в отношении отдельных подвидов метаботропного глутаматного рецептора, и разработка способов получения таких фармацевтических композиций.
Еще одним объектом настоящего изобретения является способ ингибирования активации mGluR группы 1 и ингибирования неврологических заболеваний, вызванных стимулирующей активацией mGluR группы I, прежде всего, mGluR5.
Еще одним объектом изобретения является способ лечения заболеваний, вызванных стимулирующей активацией рецептора mGluR группы I, прежде всего, mGluR5.
Для достижения этих и других объектов и целей настоящее изобретение включает в себя эффективные антагонисты mGluR группы I, прежде всего mGluR5. Эти антагонисты могут быть представлены формулой I
A1-G-Ar2,
где Ar1 означает необязательно замещенный гетероароматический остаток, а Ar2 означает необязательно замещенный бензольный цикл. G означает группу, которая связана с Ar1 и Ar2 не только ковалентными связями, и обеспечивает формирование правильной пространственной ориентации Ar1 и Ar2, но также может сама по себе взаимодействовать с белком, тем самым вызывая связывание с рецептором.
В одном из вариантов воплощения изобретения, G выбирают из группы, включающей в себя -NH-, -S-, -O-, -СО-, -CONH-, -CONHCH-, -СН2CONH-, -CNHNH-, -CNHNHCH2-, -С=NO-СН2-, -CH2NCH2-, -CH2CH2NH-, -NHCH2CO-, -NHCH2CHOH-, -NHCNHNH-, -NHCONH-, циклопентан, циклопентадиен, фуран, тиофуран, пирролидин, пиррол, 2-имидазолин, 3-имидазолин, 4-имидазолин, имидазол, пиразолин, пиразолидин, имидазолидин, оксазол, 2-оксазол, тиазол, изоксазол, изотиазол, 1Н-1,2,4-триазол, 1Н-1,2,3-триазол, 1,2,4-оксатиазол, 1,3,4-оксатиазол, 1,4,2-диоксазол, 1,4,2-оксатиазол, 1,2,4-оксадиазол, 1,2,4-тиадиазол, 1,2,5-оксадиазол, 1,2,5-тиадиазол, 1,3,4-оксадиазол, 1,3,4-тиадиазол, 1Н-тетразол, циклогексан, пиперидин, тетрагидропиридин, 1,4-дигидропиридин, пиридин, бензол, тетрагидропиран, 3,4-дигидро-2Н-пиран, 2Н-пиран, 4Н-пиран, тетрагидротиопиран, 3,4-дигидро-2Н-тиопиран, 2Н-тиопиран, 4Н-тиопиран, морфолин, тиоморфолин, пиперазин, пиридазин, пиримидин, пиразин, 1,2,4-триазин, 1,2,3-триазин, 1,3,5-триазин и 1,2,4,5-тетразин.
В другом варианте воплощения изобретения Ar1 выбирают из группы, включающей в себя фенил, бензил, нафтил, флуоренил, антренил, инденил, фенантренил и бензонафтенил, а Ar2 выбирают из группы, включающей в себя тиазолил, фурил, пиранил, 2Н-пирролил, тиенил, пирролил, имидазолил, пиразолил, пиридил, пиразинил, пиримидинил, пиридазинил, бензотиазолил, бензимидазолил, 3Н-индолил, индолил, индазолил, пуринил, хинолизинил, изохинолил, хинолил, фтализинил, нафтиридинил, хиназолинил, циннолинил, изотиазолил, хиноксалинил, индолизинил, изоиндолил, бензотиенил, бензофуранил, изобензофуранил и хроменил.
В еще одном варианте воплощения изобретения, соединения по настоящему изобретению имеют формулу II
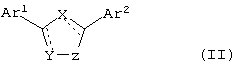
где X, Y и Z независимо выбирают из группы, состоящей из N, О, S, С и СО, причем по крайней мере один из X, Y и Z означает гетероатом,
Ar1 и Ar2 независимо выбирают из группы, включающей в себя гетероциклический или конденсированный гетероциклический остаток, содержащий от 1 до 4 гетероатомов, выбранных из группы, состоящей из N, О и S, и ароматический остаток, выбранный из группы, состоящей из фенила, бензила, 1-нафтила, 2-нафтила, флуоренила, антренила, инденила, фенантренила и бензонафтенила,
причем остатки Ar1 и Ar2 необязательно замещены одним или более заместителями, выбранными из группы, состоящей из -F, -Cl, -Br, -I, -OR, -SR, -SOR, -SO2R, -SO2NRR', -OCOR, -OCONRR', -NRCOR', -NRCO2R', -CN, -NO2, -CO2R, -CONRR', -C(O)R, -CH(OR)R', -CH2(OR). -R и -A-(CH2)n-NRR', где R или R' выбирают из группы, включающей в себя Н, CF3, С1-С10алкил, циклоалкил, алкиларил, алкилгетероарил, гетероциклоалкил, арил, и где R и R' могут быть объединены с образованием цикла, а A означает СН2, О, NH, S, SO, SO2, и n равно 1, 2, 3 или 4. Гетероциклический или конденсированный гетероциклический остаток предпочтительно выбирают из группы, состоящей из хинолила, хиназолила, хиноксалила, 2-пиримидила, 4-пиримидила, 5-пиримидила, 2-пиридила, 3-пиридила, 4-пиридила и пиразила.
В предпочтительном варианте воплощения изобретения соединение выбирают из группы, включающей в себя
3-(2-пиридил)-5-(3,5-дихлорфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-хлорфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-метоксифенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(2-хлорфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-[3-(трифторметил)фенил]-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-метилфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(1-нафтил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-[3-(трифторметокси)фенил]-1,2,4-оксадиазол,
3-(2-пиридил)-5-(2,3-дифторфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(2,5-дифторфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3,5-дифторфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-цианофенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3,5-диметоксифенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(2,3-дихлорфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-хлор-5-цианофенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-фтор-5-цианофенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-хлор-5-фторфенил)-1,2,4-оксадиазол,
3-(5-хлорпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол,
3-(5-фторпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол,
3-(5-фторпирид-2-ил)-5-(3-циано-5-фторфенил)-1,2,4-оксадиазол,
3-(3-фторпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол,
3-(5-фторпирид-2-ил)-5-(3,5-диметоксифенил)-1,2,4-оксадиазол,
3-(5-метоксипирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол,
3-(2-хинолинил)-5-(3-цианофенил)-1,2,4-оксадиазол,
3-(3-хлор-5-трифторметилпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(5-хлор-2-метоксифенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(2-хлор-5-метилтиофенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(2-бром-5-метоксифенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(2,5,6-трифторфенил)-1,2,4-оксадиазол,
2-(3-хлорфенил)-4-(пиридин-2-ил)-1,3-оксазол и
3-(2-пиридил)-5-(2,5,6-трифторфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-нитрофенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-бромфенил)-1,2,4-оксадиазол и
их фармацевтически приемлемые соли.
В другом варианте осуществления изобретения соединение выбирают из группы, включающей в себя
2-(3,5-дихлорфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-хлорфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-метоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2-хлорфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-трифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-метилфенил)-4-(2-пиридил)-1,3-оксазол,
2-( 1 -нафтил)-4-(2-пиридил)-1,3-оксазол,
2-(3-трифторметоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2,3-дифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(2,5-дифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3,5-дифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-цианофенил)-4-(2-пиридил)-1,3-оксазол,
2-(3,5-диметоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2,3-дихлорфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-хлор-5-цианофенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-фтор-5-цианофенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-хлор-5-фторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-цианофенил)-4-(5-хлорпирид-2-ил)-1,3-оксазол,
2-(3-цианофенил)-4-(5-фторпирид-2-ил)-1,3-оксазол,
2-(3-циано-5-фторфенил)-4-(5-фторпирид-2-ил)-1,3-оксазол,
2-(3-цианофенил)-4-(3-фторпирид-2-ил)-1,3-оксазол,
2-(3,5-диметоксифенил)-4-(5-фторпирид-2-ил)-1,3-оксазол,
2-(3-цианофенил)-4-(5-метоксипирид-2-ил)-1,3-оксазол,
2-(3-цианофенил)-4-(2-хинолинил)-1,3-оксазол,
2-(3-цианофенил)-4-(3-хлор-5-трифторметилпирид-2-ил)-1,3-оксазол,
2-(5-хлор-2-метоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2-хлор-5-метилтиофенил)-4-(2-пиридил)-1,3-оксазол,
2-(2-бром-5-метоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2,5,6-трифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-хлорфенил)-4-(пиридин-2-ил)-1,3-оксазол,
2-(3-нитрофенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-бромфенил)-4-(2-пиридил)-1,3-оксазол и
их фармацевтически приемлемые соли.
Еще один вариант воплощения изобретения относится к фармацевтической композиции, содержащей вышеописанное соединение формулы I и формулы II в смеси с фармацевтически приемлемым разбавителем или наполнителем.
Еще один вариант воплощения изобретения относится к способу получения вышеописанного соединения. Более подробно, соединения по изобретению в общем виде могут быть получены путем образования фрагмента G между двумя соединениями-предшественниками, содержащими соответствующие остатки Ar1 и Ar2. Если связующая группа содержит 1,2,4-оксадиазол, с помощью известных способов может быть получен гетероцикл, например, по реакции между амидоксимом и хлорангидридом кислоты или по реакции амидоксима с ацилимидазолом. Иллюстрация этого превращения представлена ниже в Примерах 3-6.
Амидоксимы могут быть получены с помощью известных способов по реакции нитрила, замещенного Ar1, с гидроксиламином. Иллюстрация такого превращения приведена ниже в Примере 1.
В большинстве случаев соединения-предшественники, хлорангидриды карбоновой кислоты (карбонилхлориды), содержащие Ar2, доступны или могут быть получены с использованием известных методов органической химии. Например, карбоновые кислоты могут быть превращены в хлорангидриды, например, по реакции с тионилхлоридом или оксалилхлоридом.
Если связующая группа содержит 1,3-оксазол, соединения получают по методике, аналогичной описанной в статье Kelly и соавт., J. Org. Chem., 61, 4623-4633 (1996). 3,5-Дизамещенные-1,3-оксазолы получают при взаимодействии галогенкетона с карбоксамидом при кипячении в толуоле с обратным холодильником в течение 3 сут. Полученную смесь охлаждают до комнатной температуры, растворитель удаляют, а остаток подвергают очистке.
Еще один вариант воплощения изобретения относится к способу ингибирования активации рецептора mGluR группы I, а именно mGluR5, включающему обработку клеток, содержащих упомянутые рецепторы mGluR группы I, эффективным количеством соединения, описанного выше.
Другим объектом изобретения является разработка способа ингибирования повреждения нейронов, вызванных возбуждающей активацией рецептора mGtuR группы I, включающего обработку нейронов эффективным количеством соединения, описанного выше.
В соответствии с еще одним воплощением изобретения представлен способ лечения заболевания или нарушения, ассоциированного с глутамат-индуцированными повреждениями нейронов, или способ лечения заболевания или нарушения, ассоциированного с активацией mGlu группы I или подвергающегося терапевтическому действию вместе с антагонистом mGlu группы I, причем способ включает введение пациенту, страдающему упомянутым заболеванием или нарушением, эффективного количества композиции, описанной выше, при этом упомянутое заболевание или нарушение выбирают из группы, включающей в себя старческое слабоумие, болезнь Паркинсона, болезнь Альцгеймера, хорею Гентингтона, боли, мигрень, эпилепсию, травмы головы, гипоксические или ишемические повреждения, психические нарушения, такие как шизофрения, депрессия, тревога, диабетическая ретинопатия, глаукома, шум в ушах, диабетическая невропатия, невропатии, индуцированные химиотерапией, постгерпетическая невралгия и невралгия тройничного нерва.
Другие объекты, признаки и преимущества настоящего изобретения представляются очевидными из следующего подробного описания изобретения. Однако следует понимать, что данное подробное описание и специфические примеры, несмотря на определение предпочтительных вариантов воплощения изобретения, представлены только для иллюстрации, так как для специалиста в данной области техники при прочтении данного подробного описания представляются очевидными возможные различные изменения и модификации в пределах объема и сущности изобретения.
Перечень фигур чертежей
На чертеже в качестве иллюстрации представлены соединения, полученные согласно изобретению.
Сведения, подтверждающие возможность осуществления
изобретения
В настоящем изобретении представлены соединения, являющиеся сильнодействующими и селективными антагонистами mGluR5. Соединения, рассматриваемые в изобретении, могут быть представлены общей формулой
Ar1-G-Ar2 (I),
где Ar1 означает необязательно замещенный гетероциклический остаток и Ar2 означает необязательно замещенный карбоциклический остаток. G означает группу, которая не только ковалентно связана с остатками Ar1 и Ar2 и облегчает формирование правильной пространственной ориентации остатков Ar1 и Ar2, но также может сама по себе взаимодействовать с белком, делая тем самым возможным связывание с рецептором.
Структура остатков Ar1 и Ar2
Остаток Ar1 в основном определен как гетероциклический остаток, а остаток Ar2 в основном определен как карбоциклический остаток. Остатки Ar1 и Ar2 могут означать моноциклические или конденсированные бициклические группы. Ar2 предпочтительно определен как арильный или алкарильный остаток. Ar1 предпочтительно определен как гетероциклический, гетероарильный или гетероарилалкильный остаток. Кольцевые системы, определенные как Ar1, могут содержать вплоть до четырех гетероатомов, которые независимо выбирают из группы, включающей в себя N, S и О. Если Ar1 является гетероарильным кольцом или кольцевой системой, то он предпочтительно содержит один или два гетероатома. По крайней мере один из гетероатомов предпочтительно является азотом (N). Гетероциклический или конденсированный гетероциклический остаток предпочтительно выбирают из группы, включающей в себя хинолил, хиназолил, хиноксалил, 2-пиримидил, 4-пиримидил, 5-пиримидил, 2-пиридил, 3-пиридил, 4-пиридил и пиразил.
Моноциклические группы Ar1 включают в себя, без ограничения перечисленным, тиазолил, фурил, пиранил, 2Н-пирролил, тиенил, пирролил, имидазоил, пиразоил, пиридил, пиразинил, пиримидинил и пиридазинил. Моноциклические группы Ar2 включают в себя, без ограничения перечисленным, фенил и бензил. Конденсированная бициклическая группа Ar2 включает в себя, без ограничения перечисленным, нафтил, флуоренил, антренил, инденил, фенантренил и бензонафтенил. Конденсированные бициклические группы Ar1 включают в себя, без ограничения перечисленным, бензотиазолил, бензимидазолил, 3Н-индолил, индолил, индазолил, пуринил, хинолизинил, изохинолил, хинолил, фтализинил, нафтиридинил, хиназолинил, циннолинил, изотиазолил, хиноксалинил, индолизинил, изоиндолил, бензотиенил, бензофуранил, изобензофуранил и хроменил. Ar1 предпочтительно является 2-пиридильным остатком. Ar2 предпочтительно является замещенным фенильным остатком.
Остатки Ar1 и Ar2 необязательно могут быть независимо замещены по меньшей мере одним остатком, выбранным из группы, включающей в себя галоген, С1-С3 алкил, С1-С3O-алкил, -OН, -OCF3, -COOR, -COR, -SOR, -SO2NRR′, -NRR′, -CN, -CF3, -CO-NRR′, -A-(CH2)n-NRR′, где А означает С, О, N, SO, SO2, a R и R′ независимо выбирают из группы, включающей в себя С1-С3алкил, Н, циклоалкил, гетероциклоалкил, арил, и n равно 1, 2, 3 или 4.
Структура остатка G
Остаток G в основном составлен из 1-14 атомов. G независимо может быть выбран из группы атомов: С, Н, N, О и S.
Таким образом, остаток G может являться нециклическим остатком. К некоторым примерам таких остатков, например, относятся -NH(амин), -S-(тиоэфир), -O(простой эфир), -СО(кетон), -CONH(амид), -CONHCH2-, -СН2CONH-, -CNHNH(амидин), -CNHNHCH2-, -C=NO-CH2(метоксим), -CH2NHCH2-, -СН2СН2NH-, -NHCH2CO-, -NHCH2CHOH-, -NHCNHNH-(гуанидин) и -NHCONH(мочевина).
Конфигурация атомов в остатке G может образовать пятичленное кольцо. К некоторым примерам таких остатков, например, относятся циклопентан, циклопентадиен, фуран, тиофуран, пирролидин, пиррол, 2-имидазолин, 3-имидазолин, 4-имидазолин, имидазол, пиразолин, пиразолидин, имидазолидин, оксазол, 2-оксазол, тиазол, изоксазол, изотиазол, 1Н-1,2,4-триазол, 1Н-1,2,3-триазол, 1,2,4-оксатиазол, 1,3,4-оксатиазол, 1,4,2-диоксазол, 1,4,2-оксатиазол, 1,2,4-оксадиазол, 1,2,4-тиадиазол, 1,2,5-оксадиазол, 1,2,5-тиадиазол, 1,3,4-оксадиазол, 1,3,4-тиадиазол и 1Н-тетразол. Наиболее предпочтительным является 1,2,4-оксадиазол.
Конфигурация атомов в остатке G может также образовать шестичленное кольцо. К некоторым примерам таких остатков, например, относятся циклогексан, пиперидин, тетрагидропиридин, 1,4-дигидропиридин, пиридин, бензол, тетрагидропиран, 3,4-дигидро-2Н-пиран, 2H-пиран, 4Н-пиран, тетрагидротиопиран, 3,4-дигидро-2Н-тиопиран, 2Н-тиопиран, 4Н-тиопиран, морфолин, тиоморфолин, пиперазин, пиридазин, пиримидин, пиразин, 1,2,4-триазин, 1,2,3-триазин, 1,3,5-триазин и 1,2,4,5-тетразин.
Конфигурация атомов в остатке G может также образовать пяти- или шестичленное кольцо, содержащее одну или более карбонильных групп. К некоторым примерам таких остатков, например, относятся 2-азетидинон, 1,2-диазетидин-3-он, циклопентанон, 2-циклопентенон, 2-пирролидинон, 3-пирролин-2-он, сукцинимид, малеимид, 3-пиразолидинон, 2-имидазолидон, 4-имидазолин-2-он, 2Н-имидазол-2-он, 4-имидазолинон, З-пиразолин-5-он, гидантоин, 1Н-имидазол-2,5-дион, 2-оксазолин-4-он, 2-оксазолидинон, 3-оксазолин-5-он, 3(2Н)-изоксазолон, 2,4-оксазолидиндион, 1,2,4-триазолин-3,5-дион, 2,4-дигидро-3Н-1,2,4-триазол-3-он, 2Н-пиран-2-он, 2(1H)-пиридон, 2(1Н)-пиразинон, 4(3Н)-пиримидон, 3,4-дигидропиримидин-4-он,глутаримид, 4,6-(1H,5Н)-пиримидиндион, 1,3,5-триазин-2(1Н)-он и циануровая кислота.
В предпочтительном варианте воплощения изобретения остаток G включает гетероциклическую пятичленную кольцевую систему. G предпочтительно является оксазоловым или 1,2,4-оксадиазоловым кольцом.
Остаток G может иметь одну или две возможные ориентации относительно групп Ar1 и Ar2. Таким образом, предпочтительными соединениями по изобретению, например, являются следующие конфигурации: 4-(Ar1)-2-(Ar2)-оксазол или 3-(Ar1)-5-(Ar2)-1,2,4-оксадиазол.
В другом варианте воплощения изобретения соединения по настоящему изобретению могут быть представлены общей формулой II:
где X, Y и Z независимо выбирают из группы, состоящей из N, О, S, С и СО, причем по крайней мере один из X, Y и Z является гетероатомом;
Ar1 и Ar2 независимо выбраны из группы, состоящей из гетероциклического или конденсированного гетероциклического остатка, содержащего от 1 до 4 гетероатомов, выбранных из группы, состоящей из N, О и S, а ароматический остаток выбран из группы, включающей в себя фенил, бензил, 1-нафтил, 2-нафтил, флуоренил, антренил, инденил, фенантренил и бензонафтенил, причем остатки Ar1 и Ar2 необязательно замещены по меньшей мере одним остатком, выбранным из группы, состоящей из -F, -Cl, -Br, -I, -OR, -SR, -SOR, -SO2R, -SO2NRR′, -OCOR, -OCONRR′, -NRCOR′, -NRCO2R′, -CN, -NO2, -CO2R, -CONRR′, -C(O)R, -CH(OR)R′, -CH2(OR), -R и -A-(CH2)n-NRR′, причем R или R′ выбраны из группы, включающей в себя Н, CF3, C1-С10алкил, циклоалкил, алкиларил, алкилгетероарил, гетероциклоалкил, арил, где R и R′ могут быть объединены с образованием кольца, а A определен как CH2, О, NH, S, SO, SO2 и n равно 1, 2, 3 или 4.
В предпочтительном варианте воплощения изобретения соединение
выбирают из группы, включающей в себя
3-(2-пиридил)-5-(3,5-дихлорфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-хлорфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-метоксифенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(2-хлорфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-[3-(трифторметил)фенил]-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-метилфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(1-нафтил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-[3-(трифторметокси)фенил]-1,2,4-оксадиазол,
3-(2-пиридил)-5-(2,3-дифторфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(2,5-дифторфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3,5-дифторфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-цианофенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3,5-диметоксифенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(2,3-дихлорфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-хлор-5-цианофенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-фтор-5-цианофенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-хлор-5-фторфенил)-1,2,4-оксадиазол,
3-(5-хлорпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол,
3-(5-фторпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол,
3-(5-фторпирид-2-ил)-5-(3-циано-5-фторфенил)-1,2,4-оксадиазол, 3-(3-фторпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол,
3-(5-фторпирид-2-ил)-5-(3,5-диметоксифенил)-1,2,4-оксадиазол,
3-(5-метоксипирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол,
3-(2-хинолинил)-5-(3-цианофенил)-1,2,4-оксадиазол,
3-(3-хлор-5-трифторметилпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(5-хлор-2-метоксифенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(2-хлор-5-метилтиофенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(2-бром-5-метоксифенил)-1,2,4-оксадиазол, 3-(2-пиридил)-5-(2,5,6-трифторфенил)-1,2,4-оксадиазол,
2-(3-хлорфенил)-4-(пиридин-2-ил)-1,3-оксазол,
3-(2-пиридил)-5-(3-нитрофенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3-бромфенил)-1,2,4-оксадиазол и
их фармацевтически приемлемые соли.
В другом варианте воплощения изобретения соединение выбирают из группы, включающей в себя
2-(3,5-дихлорфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-хлорфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-метоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2-хлорфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-трифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-метилфенил)-4-(2-пиридил)-1,3-оксазол,
2-(1-нафтил)-4-(2-пиридил)-1,3-оксазол,
2-(3-трифторметоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2,3-дифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(2,5-дифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3,5-дифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-цианофенил)-4-(2-пиридил)-1,3-оксазол,
2-(3,5-диметоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2,3-дихлорфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-хлор-5-цианофенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-фтор-5-цианофенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-хлор-5-фторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-цианофенил)-4-(5-хлорпирид-2-ил)-1,3-оксазол,
2-(3-цианофенил)-4-(5-фторпирид-2-ил)-1,3-оксазол,
2-(3-циано-5-фторфенил)-4-(5-фторпирид-2-ил)-1,3-оксазол,
2-(3-цианофенил)-4-(3-фторпирид-2-ил)-1,3-оксазол,
2-(3,5-диметоксифенил)-4-(5-фторпирид-2-ил)-1,3-оксазол,
2-(3-цианофенил)-4-(5-метоксипирид-2-ил)-1,3-оксазол,
2-(3-цианофенил)-4-(2-хинолинил)-1,3-оксазол,
2-(3-цианофенил)-4-(3-хлор-5-трифторметилпирид-2-ил)-1,3-оксазол,
2-(5-хлор-2-метоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2-хлор-5-метилтиофенил)-4-(2-пиридил)-1,3-оксазол,
2-(2-бром-5-метоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2,5,6-трифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-хлорфенил)-4-(пиридин-2-ил)-1,3-оксазол,
2-(3-нитрофенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-бромфенил)-4-(2-пиридил)-1,3-оксазол и
их фармацевтически приемлемые соли.
Получение антагонистов mGluR группы I
Многие исходные материалы для получения соединений по настоящему изобретению можно приобрести на фирмах, таких как Aldrich Chemical Company (Milwaukee, Wl). В то же время, соединения по настоящему изобретению легко получить из имеющихся предшественников с использованием прямых превращений, известных в данной области техники.
Для специалистов в данной области представляется очевидным, что антагонисты mGluR группы 1 могут быть получены с помощью хорошо известной методологии с использованием широко распространенных методов органической химии. Подходящие реакции описаны в стандартных учебниках по органической химии. Например, см. учебник March, Advanced Organic Chemistry (Органическая химия), 2-е изд., McGraw Hill (1977).
Более подробно, соединения по настоящему изобретению могут быть получены путем образования остатка G из двух исходных соединений, содержащих подходящие остатки Ar1 и Ar2. Если связующая группа содержит 1,2,4-оксадиазол, то гетероцикл может быть сформирован с использованием хорошо известного способа, такого как реакция между амидоксимом и хлорангидридом кислоты, или с использованием реакции между амидоксимом и ацилимидазолом. Иллюстрация таких превращений представлена ниже в Примерах 3-6.
Амидоксимы могут быть получены с использованием хорошо известных методов путем взаимодействия нитрила, замещенного Ar1, с гидроксиламином. Иллюстрация такого превращения представлена ниже в Примере 1.
В большинстве случаев предшественник Ar2 в виде хлорангидридов легко доступен или может быть получен с использованием простых методов органической химии. Например, карбоновые кислоты могут быть превращены в соответствующие хлорангидриды с помощью взаимодействия, например, с тионилхлоридом или оксалилхлоридом.
В случае если связующая группа содержит 1,3-оксазол, соединения могут быть получены по аналогичному методу, описанному в статье Kelly и соавт., J. Org. Chem., т.61 (1996), стр.4623-4633. Таким образом, 3,5-дизамещенные 1,3-оксазолы получают при смешивании галогенкетона с карбоксамидом при кипячении в толуоле с обратным холодильником в течение 3 сут. Полученную смесь охлаждают до комнатной температуры, растворитель удаляют и остаток подвергают очистке.
Исследование соединений для определения активности антагонистов mGluR группы 1
Фармакологические свойства соединений по настоящему изобретению могут быть исследованы с использованием стандартных методов анализа функциональной активности. Примеры методов определения глутаматных рецепторов хорошо известны в данной области техники, например, см. в статьях Aramori и соавт., Neuron, 8:757 (1992); Tanabe и соавт., Neuron, 8:169 (1992); Miller и соавт., J. Neuroscience, 15:6103 (1995); Balazs и соавт., J. Neurochemistry, 69:151 (1997). Методология, описанная в указанных публикациях, включена в текст данного описания в качестве ссылки.
В основном, соединения по настоящему изобретению могут быть исследованы с помощью анализа, в котором измеряют мобилизацию внутриклеточного кальция, [Са2+]; в клетках, экспрессирующих mGluR5, который может связывать соединения. Хорошо известная клеточная линия, которая подходит для этой цели, описана в статье Miller и соавт., J. Neuroscience, 15:6103 (1995), содержание которой включено в текст описания в качестве ссылки. Показано, что выдерживание астроцитов крысы с основным фактором роста фибробластов факторами роста, EPG, или с α-трансформирующим фактором роста, приводит к значительному увеличению экспрессии белка и функциональной активности эндогенного mGluR5 (Miller и соавт., J. Neuroscience, 15(9):6103-6109 (1995)).
В кратком изложении, первичные культуры астроцитов получают из 3-5-дневных детенышей крыс Spraque-Dawley с использованием модификации по методу Miller и соавт. и наносят на флаконы, покрытые поли-1-лизином, в среде DMEM (среда Игла, модифицированная Дульбеко), содержащей эмбриональную сыворотку коров (FCS). В случае измерений в кюветах, культуры инициируют факторами роста во флаконах в течение 3-5 дней, затем клетки собирают и проводят измерение мобилизации [Са2+]1, как описано ранее (Nemeth и соавт., 1998).
Для анализа с использованием флуоресцентного ридера для планшет (FLIPR) клетки высеивают на 96-луночные планшеты с прозрачным дном и черными стенками, покрытые поли-D-лизином, и определение мобилизации [Са2+]i проводят через 3 сут после инициирования факторами роста.
Эксперименты с использованием FLIPR проводят с помощью лазера, установленного на 0,800 Вт, со скоростью затвора камеры прибора с зарядовой связью (CCD) 0,4 с. Каждый FLIPR-эксперимент инициируют с помощью 180 мкл буферного раствора, который присутствует в каждой лунке клеточного планшета. После каждого добавления соединения измеряют флуоресцентный сигнал 50 раз с интервалом 1 с с последующими 3 измерениями с интервалом 5 с. В качестве ответа принимают высоту пика ответного сигнала в течение измерения.
Величины ЕС50 и IC50 определяют с использованием калибровочных кривых, полученных для 8 концентраций и рассчитанных по результатам двух параллельных опытов. Калибровочные кривые для агониста получают путем сканирования всех ответов для выявления максимального ответа на планшете. Подавление антагонистом активности введенного агониста нормализуют по среднему ответу на введение агониста в 14 контрольных лунках одного и того же планшета. Подробная методика тестирования соединений по настоящему изобретению представлена ниже в Примере 10.
Получение фармакологических композиций, содержащих антагонисты mGluR, и их использование для лечения неврологических нарушений
Соединения по настоящему изобретению используются для лечения неврологических нарушений или заболеваний. В то время как эти соединения в основном будут использованы для лечения пациентов (человека), они также могут быть использованы в ветеринарной медицине для лечения сходных или аналогичных заболеваний.
При использовании соединений по настоящему изобретению в области терапии и/или диагностики они могут быть включены в составы для различных форм введения, включая системное и местное или локальное введение. Методы и составы в основном можно найти в книге Remington's Pharmaceutical Sciences (Фармацевтика Ремингтона), 18-е изд., Mack Publishing Co. (1990).
Соединения по настоящему изобретению эффективны в широком диапазоне дозировок. Например, при лечении взрослых пациентов могут быть использованы суточные дозы от приблизительно 0,01 до приблизительно 1000 мг для взрослых весом 60-70 кг, предпочтительно от приблизительно 0,5 до приблизительно 100 мг для взрослых весом 60-70 кг. Наиболее предпочтительная суточная доза составляет от приблизительно 2 мг до приблизительно 70 мг для взрослых весом 60-70 кг. Точная доза зависит от способа введения, формы вводимого соединения, от субъекта, нуждающегося в лечении, массы тела субъекта, нуждающегося в лечении, от опыта и заключения практикующего врача.
Фармацевтически приемлемыми солями в основном являются соли, известные для специалистов в данной области техники, причем соли могут включать в себя, например, без ограничения перечисленным, ацетат, бензолсульфонат, бензилат, бензоат, бикарбонат, битартрат, бромид, эдетат кальция, камзилат, карбонат, цитрат, эдетат, эдизилат, эстолат, эзилат, фумарат, глуцептат, глюконат, глутамат, гликолиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, иодид, изотионат, лактат, лактобионат, малат, малеат, манделат, мезилат, мукат, напсилат, нитрат, памоат (эмбонат), пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, субацетат, сукцинат, сульфат, таннат, тартрат или теоклат. Другие фармацевтически приемлемые соли можно найти в книге Remington's Pharmaceutical Sciences (Фармацевтика Ремингтона), 18-е изд., Mark Publishing Co. (1990).
Предпочтительные фармацевтически приемлемые соли включают, например, ацетат, бензоат, бромид, карбонат, цитрат, глюконат, гидробромид, гидрохлорид, малеат, мезилат, напзилат, памоат, (эмбонат), фосфат, салицилат, сукцинат, сульфат или тартрат.
В зависимости от специфических условий лечения такие агенты могут быть включены в составы жидких или твердых лекарственных форм и введены системным или местным способом. Агенты могут быть доставлены, например, в пролонгированной форме или форме с непрерывным высвобождением, которые известны специалистам в данной области техники. Методы формирования составов и введения можно найти в книге Remington's Pharmaceutical Sciences (Фармацевтика Ремингтона), 18-е изд., Mack Publishing Co. (1990). Подходящие способы введения могут включать оральный, щечный, подъязычный, ректальный, чрескожный, вагинальный, чресслизистый, назальный или кишечный способы; парентеральную доставку, включая внутримышечную, подкожную, интрамедуллярную инъекции, а также наряду с прочими, подоболочечную, прямую внутрижелудочковую, внутривенную, внутрибрюшинную, интраназальную или внутриглазную инъекции.
В случае инъекций агенты по настоящему изобретению могут быть получены в виде водных растворов, предпочтительно в физиологически совместимых буферных растворах, таких как раствор Хенка, раствор Ринджера или физиологический солевой буферный раствор. В случае чресслизистого введения в композиции могут быть использованы проникающие агенты в соответствии с барьером, через который необходимо проникнуть. Такие проникающие агенты в основном известны в данной области техники.
Использование фармацевтически приемлемых носителей для получения композиций, содержащих соединения, которые описаны в данном изобретении, в подходящих для системного введения дозах, находятся в пределах объема настоящего изобретения. При правильном выборе носителя и подходящего метода изготовления композиций по настоящему изобретению, прежде всего, таких как составы в виде растворов, композиции могут быть введены парентеральным способом, таким как внутривенная инъекция. Соединения могут быть включены в готовые композиции в подходящих для орального введения дозах с использованием фармацевтически приемлемых носителей, известных в данной области техники. Такие носители позволяют получить составы, содержащие соединения по настоящему изобретению, в виде таблеток, пилюль, капсул, жидкостей, гелей, сиропов, взвесей, суспензий и т.п. для орального приема внутрь пациентом, подлежащим лечению.
Фармацевтические композиции, подходящие для использования по настоящему изобретению, включают композиции, в которых активные компоненты содержатся в эффективном количестве для достижения предназначенной цели. Определение эффективных количеств зависит от специалиста в данной области техники, прежде всего, с учетом подробного описания, представленного в данном контексте.
Кроме активных компонентов, такие композиции могут содержать подходящие, фармацевтически приемлемые носители, включающие наполнители и вспомогательные вещества, облегчающие переработку активных соединений в препараты, которые могут быть использованы в фармацевтических целях. Препараты, предназначенные для орального введения, могут быть в форме таблеток, драже, капсул или растворов.
Фармацевтические препараты для орального введения могут быть получены путем смешивания активных соединений с твердыми наполнителями, необязательно путем измельчения полученной смеси, и переработки смеси гранул при необходимости после добавления подходящих вспомогательных веществ для изготовления таблеток или ядер драже. Подходящими наполнителями, прежде всего, являются наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит; препараты целлюлозы, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрий карбоксиметилцеллюлоза (КМЦ) и/или поливинилпирролидон (ПВП, повидон). При необходимости могут быть добавлены дезинтегрирующие агенты, такие как поперечно сшитый поливинилпирролидон, агар или альгиновая кислота или ее соли, такие как альгинат натрия.
Ядра драже покрыты подходящими покрытиями. Для этих целей могут быть использованы концентрированные растворы сахаров, которые по выбору могут содержать аравийскую камедь, тальк, поливинилпирролидон, карбополь-гель, полиэтиленгликоль (ПЭГ) и/или диоксид титана, растворы лаков и подходящие органические растворители или смеси растворителей. В покрытия для таблеток или драже могут быть добавлены красители или пигменты для идентификации или характеристики различных комбинаций доз активных соединений.
Фармацевтические препараты для орального введения включают штампованные капсулы из желатина, а также мягкие запаянные капсулы из желатина и пластификатора, такого как глицерин или сорбит. Штампованные капсулы могут содержать активные компоненты в смеси с наполнителем, таким как лактоза, связующими, такими как крахмалы, и/или замасливателем, таким как тальк или стеарат магния, а также необязательно стабилизаторы. В случае мягких капсул активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как растительные масла, жидкий парафин или жидкие полиэтиленгликоли (ПЭГ). Кроме того, могут быть добавлены стабилизаторы.
Для более глубокого понимания настоящего изобретения ниже представлены следующие примеры, которые даны для иллюстрации и не предназначены для ограничения объема изобретения.
Примеры
Основные экспериментальные методы
Данные масс-спектрометрии и капиллярной газовой хроматографии получают с использованием газового хроматографа Hewlett-Packard (HP) серии 5890 II, соединенным с масс-селективным детектором серии HP 5971 [капиллярная колонка сверхвысокого разрешения Ultra-2 (поперечно сшитый 5% РПМе силикон); длина 25 м; внутренний диаметр 0,20 мм; скорость потока гелия 60 мл/мин; температура инжектора 250°С; температурная программа 20°С/мин от 125 до 325°С в течение 10 мин, затем постоянная температура 325°С в течение 6 мин]. Тонкослойную хроматографию (ТСХ) проводят с использованием пластинок с силикагелем Analtech Uniplate HF TLC с размером гранул 250 мкм. Для проявления соединений на пластинках ТСХ используют УФ свет, иногда в сочетании с реагентами в аэрозольной форме, содержащими нингидрин и реагенты Драгендорфа (Sigma Chemical Co.). Для синтеза в основном используют реагенты производства Aldrich Chemical Co. (Milwaukee, Wl), Sigma Chemical Co. (Saint Louis, МО), Fluka Chemical Corp. (Milwaukee, Wl), Fisher Scientific (Pittsburgh, PA), TCI America (Portland, OR) или Lancaster Synthesis (Windham, NH).
Пример 1. Синтез производных амидоксима
Пирид-2-иламидоксим
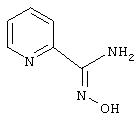
Используя методику, описанную в статье Shine и соавт., J. Heterocyclic Chem., 26:125-128 (1989), гидроксид гидроксиламина (7,65 г, 110 ммоль) в этаноле (100 мл) обрабатывают раствором гидроксида натрия (11 мл, 10 н., 110 ммоль). При этом быстро образуется осадок и реакционную смесь перемешивают при температуре окружающей среды в течение 30 мин. Неорганический осадок отфильтровывают и промывают этанолом (100 мл). Фильтрат и этанольный промывные растворы объединяют и обрабатывают 2-цианопиридином (10,4 г, 100 ммоль). Реакционную смесь кипятят с обратным холодильником в течение 20 ч. Затем летучие растворители удаляют в вакууме и получают 13,3 г (97%) пирид-2-иламидоксима.
3-Метоксибензамидоксим
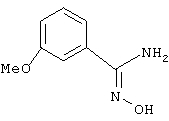
Используя основную методику синтеза амидоксимов, из гидрохлорида гидроксиламина (7,65 г, 110 ммоль), гидроксида натрия (11 мл, 10 н., 110 ммоль) и 3-метоксибензилнитрила (12,2 мл, 100 ммоль) получают 9,9 г (60%) 3-метоксибензамидоксима.
5-Хлорпирид-2-иламидоксим
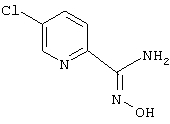
Смесь 2,5-дихлорпиридина (1,48 г, 10 ммоль), цианида цинка (705 мг, 6 ммоль), цинка (пыль, 29 мг, 0,45 ммоль), [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), комплекс с дихлорметаном (1:1) (0,18 г, 0,22 ммоль) в N,N-диметилформамиде (10 мл) кипятят с обратным холодильником в течение 5 ч. После охлаждения реакционную смесь разбавляют этилацетатом и экстрагируют водой и солевым раствором. После хроматографии на силикагеле получают 735 мг (53%) 2-циано-5-хлорпиридина.
Используя основную методику синтеза амидоксимов, 2-циано-5-хлорпиридин (735 мг, 5,3 ммоль), раствор гидрохлорида гидроксиламина (1,2 мл, 5 М, 6 ммоль) в этаноле (7 мл) и гидроксид натрия (0,61 мл, 10 н., 6,1 ммоль), кипятят с обратным холодильником в течение 24 ч. С помощью стандартной обработки получают 707 мг (77%) 5-хлорпирид-2-иламидоксима.
5-Фторпирид-2-иламидоксим
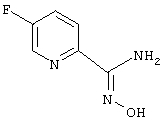
Смесь 2-циано-5-хлорпиридина (1 г, 7,22 ммоль) и фторида калия (1,26
г, 21,68 ммоль) в 1-метил-2-пирролидиноне (25 мл) кипятят с обратным холодильником в течение 18 ч. После охлаждения реакционную смесь разбавляют этилацетатом и экстрагируют водой и солевым раствором. Затем органические растворители удаляют в вакууме. После хроматографии на силикагеле получают 425 мг (48%) 2-циано-5-фторпиридина.
Используя основную методику синтеза амидоксимов, 2-циано-5-фторпиридин (425 мг, 3,48 ммоль), раствор гидрохлорида гидроксиламина (0,79 мл, 5 М, 3,95 ммоль) в этаноле (5 мл) и гидроксид натрия (0,398 мл, 10 н., 3,98 ммоль), кипятят с обратным холодильником в течение 24 ч. С помощью стандартной обработки получают 330 мг (61%) 5-фторпирид-2-иламидоксима.
5-Метоксипирид-2-иламидоксим
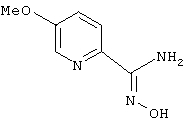
Раствор 2-циано-5-фторпиридина (0,65 г, 5,3 ммоль) в метоксиде натрия (1,83 мл, 25 мас.%-ный раствор в метаноле, 7,95 ммоль) перемешивают при 0°С в течение 1,5 ч и при температуре окружающей среды в течение 2 ч. Затем реакционную смесь разбавляют этилацетатом и промывают водой и солевым раствором. После удаления растворителя в вакууме получают 304 мг (43%) 2-циано-5-метоксипиридина.
Используя основную методику синтеза амидоксимов, 2-циано-5-метоксипиридин (270 мг, 2,01 ммоль), раствор гидрохлорида гидроксиламина (0,457 мл, 5 М, 2,28 ммоль) в этаноле (4 мл) и гидроксид натрия (0,230 мл, 10 н., 2,30 ммоль), кипятят с обратным холодильником в течение 24 ч. С помощью стандартной обработки получают 79 мг (24%) 5-метоксипирид-2-иламидоксима.
3-Фторпирид-2-иламидоксим
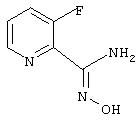
Смесь 2,3-дихлорпиридина (1,48 г, 10 ммоль), цианида цинка (705 мг, 6 ммоль), цинка (пыль, 29 мг, 0,45 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), комплекс с дихлорметаном (1:1) (0,18 г, 0,22 ммоль) в N,N-диметилформамиде (10 мл) кипятят с обратным холодильником в течение 5 ч. После охлаждения реакционную смесь разбавляют этилацетатом и экстрагируют водой и солевым раствором. После удаления растворителя и хроматографии на силикагеле получают 1,05 г (76%) 2-циано-3-хлорпиридина.
Раствор 2-циано-3-хлорпиридина (1 г, 7,22 ммоль) в 1-метил-2-пирролидиноне (25 мл) обрабатывают фторидом калия (1,26 г, 21,68 ммоль) и кипятят с обратным холодильником в течение 18 ч. После охлаждения реакционную смесь разбавляют этилацетатом и экстрагируют водой и солевым раствором. После хроматографии на силикагеле получают 442 мг (50%) 2-циано-3-фторпиридина.
Используя основную методику синтеза амидоксимов, 2-циано-3-фторпиридин (442 мг, 3,62 ммоль), раствор гидрохлорида гидроксиламина (0,82 мл, 5 М, 4,1 ммоль) в этаноле (5 мл) и гидроксид натрия (0,415 мл, 10 н., 4,15 ммоль), кипятят с обратным холодильником в течение 24 ч. С помощью стандартной обработки получают 368 мг (66%) 3-фторпирид-2-иламидоксима.
Хинол-2-иламидоксим
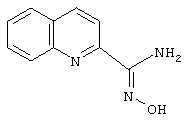
Используя основную методику синтеза амидоксимов, 2-хинолинкарбонитрил (1,02 г, 6,6 ммоль), раствор гидрохлорида гидроксиламина (1,44 мл, 5 н., 7,2 ммоль) в этаноле (10 мл) и гидроксид натрия (0,72 мл, 10 н., 7,2 ммоль), кипятят с обратным холодильником в течение 18 ч. С помощью стандартной обработки получают 990 мг (80%) хинол-2-иламидоксима.
Пример 2. Синтез производных карбоновых кислот
3-хлор-5-цианобензойная кислота
Смесь метилового эфира 3,5-дихлорбензойной кислоты (14,66 г, 71,5 ммоль), цианида цинка (5,04 г, 42,9 ммоль), цинка (пыль, 0,21 г, 3,21 ммоль), [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), комплекс с дихлорметаном (1:1) (1,3 г, 1,57 ммоль) в N,N-диметилформамиде (70 мл) кипятят с обратным холодильником в течение 5 ч. После охлаждения реакционную смесь разбавляют этилацетатом и экстрагируют водой и солевым раствором. После хроматографии на силикагеле получают 2,34 г (17%) метилового эфира 2-хлор-5-цианобензойной кислоты.
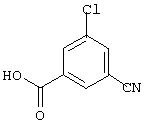
Промежуточный эфир обрабатывают раствором гидроксида натрия (7,5 мл, 4 н., 30 ммоль) в метаноле (50 мл) и перемешивают при температуре окружающей среды в течение 18 ч. Растворитель удаляют в вакууме и остаток растворяют в этилацетате. Органический раствор промывают 5%-ной HCI и солевым раствором. После удаления растворителя получают 1,8 г (83%) 3-хлор-5-цианобензойной кислоты.
3-хлор-5-фторбензойная кислота
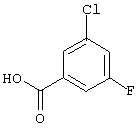
Смесь 1-бром-3-хлор-5-фторбензола (25,0 г, 120 ммоль), цианида цинка (8,45 г, 72 ммоль), цинка (пыль, 235 г, 3,6 ммоль), [1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладия (II), комплекс с дихлорметаном (1:1) (1,5 г, 1,8 ммоль) в N,N-диметилформамиде (70 мл) кипятят с обратным холодильником в течение 1 ч. После охлаждения реакционную смесь разбавляют этилацетатом и экстрагируют водой и солевым раствором. После хроматографии на силикагеле получают 15,9 г (85%) 3-хлор-5-фторбензонитрила.
Промежуточный нитрил обрабатывают раствором гидроксида натрия (100 мл, 10 н., 1 моль) в 100 мл воды и кипятят с обратным холодильником в течение 2 ч. Затем раствор охлаждают и подкисляют концентрированной соляной кислотой. После экстракции дихлорметаном и упаривания растворителя получают 15,14 г (85%) 3-хлор-5-фторбензойной кислоты.
3-фтор-5-цианобензойная кислота
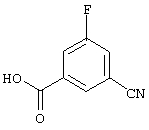
3-хлор-5-фторбензойную кислоту (13,74 г, 78,7 ммоль) обрабатывают 50 мл тионилхлорида и кипятят с обратным холодильником в течение 2 ч. Избыток тионилхлорида удаляют в вакууме и остаток обрабатывают 100 мл сухого метанола, при этом получают 13,6 г (92%) метилового эфира 3-хлор-5-фторбензойной кислоты.
Смесь метилового эфира 3-хлор-5-фторбензойной кислоты, цианида цинка (8,46 г, 72,3 ммоль), цинка (пыль, 235 г, 3,6 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II), комплекс с дихлорметаном (1:1) (1,5 г, 1,8 ммоль) в N, N-диметилформамиде (70 мл) кипятят с обратным холодильником в течение 1 ч. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют этилацетатом. Органический раствор экстрагируют водой и солевым раствором, концентрируют в вакууме и получают неочищенный метиловый эфир 3-хлор-5-цианобензойной кислоты.
Неочищенный метиловый эфир 3-хлор-5-цианобензойной кислоты обрабатывают раствором гидроксида натрия (45 мл, 4 н., 180 ммоль) в метаноле (350 мл) при температуре окружающей среды в течение 4 ч. Растворитель удаляют в вакууме и остаток растворяют в этилацетате. Органический раствор промывают 5%-ной HCl и солевым раствором. После хроматографии на силикагеле получают 7,0 г (54%) 3-фтор-5-цианобензойной кислоты.
Пример 3. Синтез 3,5-дизамещенных 1,2,4-оксадиазолов из хлорангидридов кислот
В основном используют модифицированную методику, описанную в статье Shine и соавт., J. Heterocyclic Chem., 26:125-128 (1989). В типичном случае 3,5-дизамещенные 1,2,4-оксадиазолы получают при добавлении ацилхлорида в раствор амидоксима в пиридине с последующим кипячением реакционной смеси с обратным холодильником или нагреванием реакционной смеси в закрытой пробирке. Оксадиазолы выделяют осаждением холодной водой и фильтрованием или экстракцией органическим растворителем. При необходимости оксидиазолы очищают хроматографией или перекристаллизацией.
3-(2-Пиридил)-5-(3,5-дихлорфенил)-1,2,4-оксадиазол (NPS 64982)
(404) В2
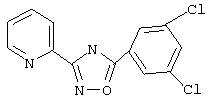
Смесь 3,5-дихлорбензоилхлорида (2,1 г, 10 ммоль) и пирид-2-иламидоксима (1,37 г, 10 ммоль) в пиридине (5 мл) нагревают в закрытой пробирке при 190°С в течение 2 ч. Затем реакционную смесь добавляют в ледяную воду для осаждения оксадиазола. Твердое вещество собирают фильтрованием, промывают водой и затем перекристаллизовывают из этанола, при этом получают 2,1 г (72%) 3-(2-пиридил)-5-(3,5-дихлорфенил)-1,2,4-оксадиазола: т.пл. 162-166°С; ГХ/МС(El) m/z (отн.инт.) 291 (М+, 38), 293 (25), 261 (1), 173 (6), 145 (13), 120 (100), 90 (20), 78 (28), 51 (15).
3-(2-Пиридил)-5-(3-хлорфенил)-1,2,4-оксадиазол (NPS 64983) (405)
В3
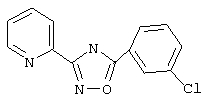
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-хлорбензоилхлорид (127 мкл, 1 ммоль) и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) кипятят с обратным холодильником в течение 4 ч. После стандартной обработки получают 156 мг (61%) 3-(2-пиридил)-5-(3-хлофенил)-1,2,4-оксадиазола: т.пл. 136-140°С; ГХ/МС (El) m/z (отн.инт.) 257 (M+, 64), 259 (21), 227 (3), 120 (100), 111 (22), 90 (24), 78 (32), 75 (26), 51 (20).
3-(2-Пиридил)-5-(3-метоксифенил)-1,2,4-оксадиазол (В1)
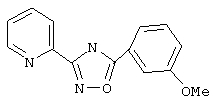
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-анизоилхлорид (151 мкл, 1 ммоль) и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) кипятят с обратным холодильником в течение 4 ч. После стандартной обработки получают 200 мг (79%) 3-(2-пиридил)-5-(3-метоксифенил)-1,2,4-оксадиазола: т.пл. 96-99°С; ГХ/МС (EI) m/z (отн.инт.) 253 (М+, 100), 223 (3), 179 (3), 135 (74), 133 (90), 92 (27), 78 (29), 77 (32), 64 (23), 63 (23).
3-(2-Пиридил)-5-(2-хлорфенил)-1,2,4-оксадиазол (В5)
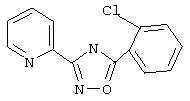
Используя основной метод синтеза 1,2,4-оксадиазолов, 2-хлорбензоилхлорид (127 мкл, 1 ммоль) и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) кипятят с обратным холодильником в течение 4 ч. После стандартной обработки получают 157 мг (61%) 3-(2-пиридил)-5-(2-хлорфенил)-1,2,4-оксадиазола: т.пл. 93-94°С; ГХ/МС (El) m/z (отн.инт.) 257 (М+ 76), 259 (26), 227 (4), 139 (11), 120 (100), 111 (21), 90 (27), 78 (35), 75 (29), 51 (21).
3-(2-Пиридил)-5-[3-(трифторметил)фенил]-1,2,4-оксадиазол (В6)
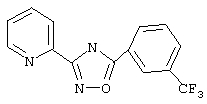
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-(трифторметил)бензоилхлорид (151 мкл, 1 ммоль) и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) кипятят с обратным холодильником в течение 16 ч. После стандартной обработки получают 233 мг (80%) 3-(2-пиридил)-5-[3-(трифторметил)фенил]-1,2,4-оксадиазола: т.пл. 116-118°С; ГХ/МС (EI) m/z (отн.инт.) 291 (М+, 81), 272 (7), 173 (6), 145 (25), 120 (100), 90 (20), 78 (23), 51 (11).
3-(2-Пиридил)-5-(3-фторфенил)-1,2,4-оксадиазол (В7)
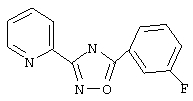
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-фторбензоилхлорид (122 мкл, 1 ммоль) и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) кипятят с обратным холодильником в течение 16 ч.
После стандартной обработки получают 176 мг (73%) 3-(2-пиридил)-5-(3-фторфенил]-1,2,4-оксадиазола: т.пл. 88-98°С; ГХ/МС (El) m/z (отн.инт.) 241 (M+, 95), 211 (5), 120 (100), 107 (13), 95 (30), 90 (21), 78 (27), 75 (19), 51 (15).
3-(2-Пиридил)-5-(3-метилфенил)-1,2,4-оксадиазол (В9)
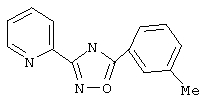
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-метилфенилхлорид (264 мкл, 2 ммоль) и пирид-2-иламидоксим (274 мг, 2 ммоль) в пиридине (1 мл) нагревают в закрытой пробирке при 200°С в течение 2 ч. После стандартной обработки получают 387 мг (82%) 3-(2-пиридил)-5-(3-метилфенил)-1,2,4-оксадиазола: т.пл. 127-128°С; ГХ/МС (EI) m/z (отн. инт.) 237 (M+, 100), 222 (2), 207 (8), 120 (68), 117 (24), 91 (29), 90 (29), 78 (32), 65 (26), 51 (23).
3-(2-Пиридил)-5-(1-нафтил)-1,2,4-оксадиазол (В10)

Используя основной метод синтеза 1,2,4-оксадиазолов, 1-нафтоилхлорид (150 мкл, 1 ммоль) и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают в закрытой пробирке при 200°С в течение 3 ч. После стандартной обработки получают 50 мг (18%) 3-(2-пиридил)-5-(1-нафтил)-1,2,4-оксадиазола; т.пл. 132-136°С; ГХ/МС (EI) m/z (отн. инт.) 273 (M+, 75), 195 (5), 169 (88), 153 (100), 139 (12), 127 (66), 126 (29), 105 (23), 78 (14), 51 (14).
3-(2-Пиридил)-5-[3-(трифторметокси)фенил]-1,2,4-оксадиазол (В11)
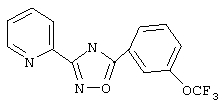
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-(трифторметокси)бензоилхлорид (220 мг, 1 ммоль) и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают в закрытой пробирке при 200°С в течение 3 ч. После стандартной обработки получают 175 мг (57%) 3-(2-пиридил)-5-[3-(трифторметокси)фенил]-1,2,4-оксадиазола; т.пл.86-88°С; ГХ/МС (El) m/z (отн. инт.) 307 (M+ 73), 277 (3), 222 (3), 189 (6), 161 (5), 120 (100), 78 (21), 69(17), 51 (10).
3-(2-Пиридил)-5-(2,3-дифторфенил)-1,2,4-оксадиазол (В16)
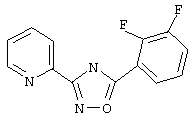
Используя основной метод синтеза 1,2,4-оксадиазолов, 2,3-дифторбензоилхлорид (124 мкл, 1 ммоль) и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают в закрытой пробирке при 100°С в течение 16 ч. После стандартной обработки получают 158 мг (61%) 3-(2-пиридил)-5-(2,3-дифторфенил)-1,2,4-оксадиазола; т.пл. 120-121 °С; ГХ/МС(El) m/z (отн. инт.) 259 (M+, 97), 229(5), 228(4), 141 (11), 120 (100), 113 (26), 90 (27), 78 (34), 51 (17).
3-(2-Пиридил)-5-(2,5-дифторфенил)-1,2,4-оксадиазол (В17)
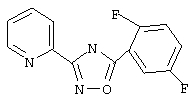
Используя основной метод синтеза 1,2,4-оксадиазолов, 2,5-дифторбензоилхлорид(124 мкл, 1 ммоль) и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают при 100°С в течение 16 ч. После стандартной обработки получают 3-(2-пиридил)-5-(2,5-дифторфенил)-1,2,4-оксадиазол; т.пл.120-126°С; ГХ/МС (El) m/z (отн. инт.) 259 (M+, 91), 229 (5), 228 (4), 141 (13), 120 (100), 113 (25), 90 (23), 78 (27), 51 (14).
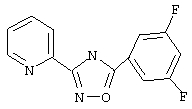
3-(2-Пиридил)-5-(3,5-дифторфенил)-1,2,4-оксадиазол (В18)
Используя основной метод синтеза 1,2,4-оксадиазолов, 3,5-дифторбензоилхлорид (1,25 мл, 10 ммоль) и пирид-2-иламидоксим (1,37 г, 10 ммоль) в пиридине (5 мл) нагревают в закрытой пробирке при 200°С в течение 4 ч. После стандартной обработки получают 1,2 г (46%) 3-(2-пиридил)-5-(3,5-дифторфенил)-1,2,4-оксадиазола; т.пл. 115-119°С; ГХ/МС(El) m/z (отн. инт.) 259 (M+, 100), 229 (4), 228 (5), 141 (9), 125 (13), 113 (30), 90 (19), 78 (27), 63 (23), 51 (15).
3-(2-Пиридил)-5-(3-цианофенил)-1,2,4-оксадиазол (В21)

Используя основной метод синтеза 1,2,4-оксадиазолов, 3-цианобензоилхлорид (165 мг, 1 ммоль) и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают при 100°С в течение 72 ч. После стандартной обработки получают 158 мг (64%) 3-(2-пиридил)-5-(3-цианофенил)-1,2,4-оксадиазола; т.пл.148-149°С; ГХ/МС (El) m/z (отн. инт.) 248 (M+, 85), 218 (5), 130 (6), 120 (100), 114 (9), 102 (28), 90 (26), 78 (37), 75 (19), 51 (30).
3-(2-Пиридил)-5-(3,5-диметоксифенил)-1,2,4-оксадиазол (В23)
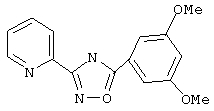
Используя основной метод синтеза 1,2,4-оксадиазолов, 3,5-диметоксибензоилхлорид (200 мг, 1 ммоль) и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают при 100°С в течение 72 ч. После стандартной обработки получают 210 мг (74%) 3-(2-пиридил)-5-(3,5-диметоксифенил)-1,2,4-оксадиазола; т.пл.145-148°С; ГХ/МС (El) m/z (отн. инт.) 283 (M+, 100), 253 (3), 165 (69), 163 (19), 137 (36), 122 (33), 107 (17), 90 (10), 78(25), 63(19), 51 (19).
3-(2-Пиридил)-5-(2,3-дихлорфенил)-1,2,4-оксадиазол (В25)
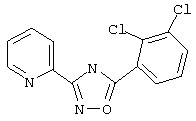
Используя основной метод синтеза 1,2,4-оксадиазолов, 2,3-дихлорбензоилхлорид (209 мг, 1 ммоль) и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают при 100°С в течение 48 ч. После стандартной обработки получают 236 мг (81%) 3-(2-пиридил)-5-(2,3-дихлорфенил)-1,2,4-оксадиазола; т.пл.128-133°С; ГХ/МС (El) m/z (отн. инт.) 291 (M+, 66), 293 (43), 256 (6), 173 (10), 145 (11), 120 (100), 90 (19), 78 (27), 51 (14).
3-(2-Пиридил)-5-(3-хлор-5-цианофенил)-1,2,4-оксадиазол (В26)
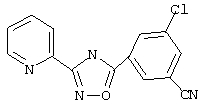
3-Хлор-5-цианобензойную кислоту (0,82 г, 4,97 ммоль) обрабатывают раствором оксалилхлорида (10 мл, 2,5 М в дихлорметане, 25 ммоль) и каталитическим количеством N,N-диметилформамида. Реакционную смесь перемешивают при температуре окружающей среды в течение 2,5 ч. Избыток оксалилхлорида удаляют в вакууме и при этом получают 3-хлор-5-цианобензоилхлорид.
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-хлор-5-цианобензоилхлорид и пирид-2-иламидоксим (682 мг, 5 ммоль, 1 экв.) в пиридине (5 мл) нагревают в закрытой пробирке при 175°С в течение 4 ч. После стандартной обработки и перекристаллизации из 2-пропанола получают 250 мг (19%) 3-(2-пиридил)-5-(3-хлор-5-цианофенил)-1,2,4-оксадиазола; ГХ/МС (EI) m/z (отн. инт.) 282 (M+, 100), 283 (18), 284 (34), 251 (4), 136 (10), 120 (53), 100 (10), 78 (15), 51 (6).
3-(2-Пиридил)-5-(3-фтор-5-цианофенил)-1,2,4-оксадиазол (В27)
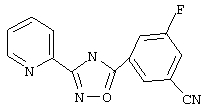
3-Фтор-5-цианобензойную кислоту (2,5 г, 15,14 ммоль) обрабатывают раствором оксалилхлорида (30 мл, 2,5 М в дихлорметане, 75 ммоль) и каталитическим количеством N,N-диметилформамида. Реакционную смесь перемешивают при температуре окружающей среды в течение 2,5 ч. Избыток оксалилхлорида удаляют в вакууме и при этом получают 3-фтор-5-цианобензоилхлорид.
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-фтор-5-цианобензоилхлорид и пирид-2-иламидоксим (2,076 г, 15,15 ммоль, 1 экв.) в пиридине (5 мл) нагревают в закрытой пробирке при 175°С в течение 4 ч. После стандартной обработки и перекристаллизации из 2-пропанола получают 1,5 г (37%) 3-(2-пиридил)-5-(3-фтор-5-цианофенил)-1,2,4-оксадиазола; ГХ/МС (El) m/z (отн. инт.) 266 (M+, 81), 267 (13), 235 (5), 132 (12), 120 (100), 100 (18), 90 (18), 78 (35), 51 (20).
3-(2-Пиридил)-5-(3-хлор-5-фторфенил)-1,2,4-оксадиазол (В28)
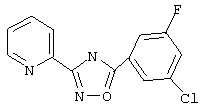
3-Хлор-5-фторбензойную кислоту (400 мг, 2,3 ммоль) обрабатывают раствором оксалилхлорида (4,6 мл, 2,5 М в дихлорметане, 11,5 ммоль) и каталитическим количеством N,N-диметилформамида. Реакционную смесь перемешивают при температуре окружающей среды в течение 2,5 ч. Избыток оксалилхлорида удаляют в вакууме и при этом получают 3-хлор-5-фторбензоилхлорид.
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-хлор-5-фторбензоилхлорид и пирид-2-иламидоксим (314 мг, 2,3 ммоль, 1 экв.) в пиридине (5 мл) нагревают в закрытой пробирке при 175°С в течение 4 ч. После стандартной обработки и перекристаллизации из 2-пропанола получают 250 мг (39%) 3-(2-пиридил)-5-(3-хлор-5-фторфенил)-1,2,4-оксадиазола; ГХ/МС (El) m/z (отн. инт.) 275 (M+, 89), 276 (14), 277 (29), 129 (26), 120 (100), 109 (7), 90 (20), 78 (31), 51 (14).
3-(5-Хлорпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол (В29)
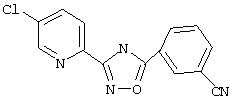
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-цианобензоилхлорид (675 мг, 4 ммоль) и 5-хлорпирид-2-иламидоксим (686 мг, 4 ммоль) в пиридине (5 мл) нагревают в закрытой пробирке при 175°С в течение 4 ч. После стандартной обработки и перекристаллизации из 2-пропанола получают 357 мг (32%) 3-(5-хлорпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазола; ГХ/МС (EI) m/z (отн. инт.) 282 (M+, 85), 283 (14), 284 (27), 156 (31), 154 (100), 112 (19), 102 (30), 76 (28), 64 (13).
3-(5-Фторпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол (В30)
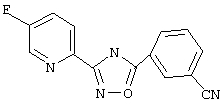
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-цианобензоилхлорид (0,534 г, 3,2 ммоль) и 5-фторпирид-2-иламидоксим (0,5 г, 3,2 ммоль) в пиридине (5 мл) нагревают в закрытой пробирке при 175°С в течение 4 ч. После стандартной обработки и перекристаллизации из 2-пропанола получают 370 мг (43%) 3-(5-фторпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазола; ГХ/МС (El) m/z (отн. инт.) 266 (M+, 100), 267 (10), 138 (80), 114 (8), 102 (19), 96 (22), 76 (17), 57 (8).
3-(5-Фторпирид-2-ил)-5-(3-циано-5-фторфенил)-1,2,4-оксадиазол (В31)
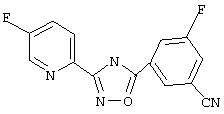
3-Фтор-5-цианобензойную кислоту (1,0 г, 6 ммоль) обрабатывают раствором оксалилхлорида (12 мл, 2,5 М в дихлорметане, 30 ммоль) и каталитическим количеством N,N-диметилформамида. Реакционную смесь перемешивают при температуре окружающей среды в течение 2,5 ч. Избыток оксалилхлорида удаляют в вакууме и при этом получают 3-фтор-5-цианобензоилхлорид.
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-фтор-5-цианобензоилхлорид (1,1 г, 6 ммоль) и 5-фторпирид-2-иламидоксим (0,93 г, 6 ммоль) в пиридине (5 мл) нагревают в закрытой пробирке при 175°С в течение 4 ч. После стандартной обработки и перекристаллизации из 2-пропанола получают 0,41 г (24%) 3-(5-фторпирид-2-ил)-5-(3-циано-5-фторфенил)-1,2,4-оксадиазола; ГХ/МС (EI) m/z (отн. инт.) 284 (M+, 100), 285 (16), 253 (2), 138 (99), 120 (23), 108 (16), 96 (25), 82 (15), 57 (11).
3-(3-Фторпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол (В32)
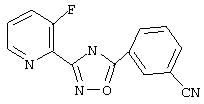
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-цианобензоилхлорид (107 мг, 0,64 ммоль) и З-фторпирид-2-иламидоксим (0,1 г, 0,64 ммоль) в пиридине (5 мл) нагревают в закрытой пробирке при 175°С в течение 4 ч. После стандартной обработки, хроматографии на силикагеле и перекристаллизации из 2-пропанола получают 32 мг (19%) 3-(3-фторпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазола; ГХ/МС (El) m/z (отн. инт.) 266 (M+, 75), 267(12), 138(100), 114(11), 102(19), 96(17), 76(16), 57(5), 51 (5).
3-(5-Фторпирид-2-ил)-5-(3,5-диметоксифенил)-1,2,4-оксадиазол (B33)
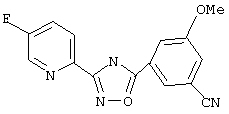
Используя основной метод синтеза 1,2,4-оксадиазолов, 3,5-диметоксибензоилхлорид (0,10 г, 0,5 ммоль) и 5-фторпирид-2-иламидоксим (78 мг, 0,5 ммоль) в пиридине (3 мл) нагревают в закрытой пробирке при 175°С в течение 4 ч. После стандартной обработки, хроматографии на силикагеле и перекристаллизации из 2-пропанола получают 94 мг (62%) 3-(5-фторпирид-2-ил)-5-(3,5-диметоксифенил)-1,2,4-оксадиазола; ГХ/МС (EI) m/z (отн. инт.) 301 (M+, 100), 302 (17), 165(41), 137(23), 122 (27), 96 (15), 77 (11), 63 (12).
3-(5-Метоксипирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол (В34)
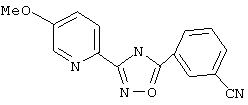
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-цианобензоилхлорид (79 мг, 0,47 ммоль) и 5-метоксипирид-2-иламидоксим (79 мг, 0,47 ммоль) в пиридине (2,5 мл) нагревают в закрытой пробирке при 175°С в течение 4 ч. После стандартной обработки, хроматографии на силикагеле и перекристаллизации из 2-пропанола получают 59 мг (45%) 3-(5-метоксипирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазола; ГХ/МС (El) m/z (отн. инт.) 278 (M+,100), 279 (16), 150 (56), 128 (7), 107 (21), 102 (17), 80 (12), 64 (5).
3-(2-Хинолинил)-5-(3-цианофенил)-1,2,4-оксадиазол (В35)
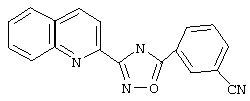
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-цианобензоилхлорид (68 мг, 0,41 ммоль) и хинол-2-иламидоксим (75,9 мг, 0,405 ммоль) в пиридине (0,5 мл) нагревают в закрытой пробирке при 165°С в течение 22 ч. После стандартной обработки, перекристаллизации из этанола и твердофазной экстракции получают 23,7 мг (20%) 3-(2-хинолинил)-5-(3-цианофенил)-1,2,4-оксадиазола. 1H (CDCl3), δ (часть/млн): 8,62 (s, 1Н), 8,54 (d, 1Н), 8,36 (d, 2H), 8.28 (d, 1Н), 7,90 (d, 2H), 7,80 (t, 1H), 7,72 (t, 1H), 7,64 (t, 1H).
3-(3-Хлор-5-трифторметилпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол (В36)
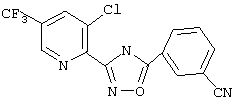
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-цианобензоилхлорид (66 мг, 0,40 ммоль) и 3-хлор-5-трифторметилпирид-2-иламидоксим (96,5 мг, 0,403 ммоль) в пиридине (0,5 мл) нагревают в закрытой пробирке при 165°С в течение 22 ч. После стандартной обработки и твердофазной экстракции получают 45,9 мг (33%) 3-(3-хлор-5-трифторметилпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазола. 1H ЯМР (CDCl3), δ (часть/млн): 8,99 (s, 1Н), 8,57 (s, 1Н), 8,49 (d, 1H), 8,19 (s, 1H), 7,92 (d, 1H), 7,72 (t, 1H).
3-(2-Пиридил)-5-(5-хлор-2-метоксифенил)-1,2,4-оксадиазол (В37)
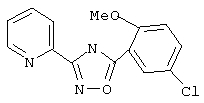
5-Хлор-О-анисовую кислоту (187 мг, 1 ммоль) обрабатывают раствором оксалилхлорида (1,5 мл, 2 М в дихлорметане, 3 ммоль) и каталитическим количеством N, N-диметилформамида. Реакционную смесь перемешивают при температуре окружающей среды в течение 2 ч. Избыток оксалилхлорида удаляют в вакууме и при этом получают 5-хлор-2-метоксибензоилхлорид.
Используя основной метод синтеза 1,2,4-оксадиазолов, 5-хлор-2-метоксибензоилхлоридипирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают при 115°С в течение 17 ч. После стандартной обработки и хроматографии на силикагеле получают 49 мг (17%) 3-(2-пиридил)-5-(5-хлор-2-метоксифенил)-1,2,4-оксадиазола. 1H ЯМР (CDCl3), δ (часть/млн): 4,00 (s, 3Н), 7,03 (d, J=8,9 Гц, 1Н), 7,42-7,47 (m, 1H), 7,50 (dd, J=8,9 Гц, 2,8 Гц, 1Н), 7,87 (ddd, J=1,4 Гц, 7,4 Гц, 8,2 Гц, 1H), 8,22 (d, J=8,2 Гц, 1H), 8,28 (d, J=2,4 Гц, 1H), 8,84 (m, 1H).
3-(2-Пиридил)-5-(2,3-диметоксифенил)-1,2,4-оксадиазол (В38)
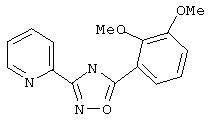
2,3-Диметоксибензойную кислоту (182 мг, 1 ммоль) обрабатывают раствором оксалилхлорида (1,5 мл, 2 М в дихлорметане, 3 ммоль) и каталитическим количеством N,N-диметилформамида. Реакционную смесь перемешивают при температуре окружающей среды в течение 2 ч. Избыток оксалилхлорида удаляют в вакууме и при этом получают 2,3-диметоксибензоилхлорид.
Используя основной метод синтеза 1,2,4-оксадиазолов, 2,3-диметоксибензоилхлорид и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают при 115°С в течение 17 ч. После стандартной обработки и хроматографии на силикагеле получают 120 мг (42%) 3-(2-пиридил)-5-(2,3-диметоксифенил)-1,2,4-оксадиазола.
3-(2-Пиридил)-5-(2-хлор-5-метилтиофенил)-1,2,4-оксадиазол (В39)
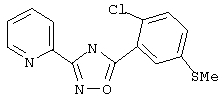
2-Хлор-5-метилтиобензойную кислоту (182 мг, 1 ммоль) обрабатывают раствором оксалилхлорида (1,5 мл, 2 М в дихлорметане, 3 ммоль) и каталитическим количеством N,N-диметилформамида. Реакционную смесь перемешивают при температуре окружающей среды в течение 2 ч. Избыток оксалилхлорида удаляют в вакууме и при этом получают 2-хлор-5-метилтиобензоилхлорид.
Используя основной метод синтеза 1,2,4-оксадиазолов, 2-хлор-5-метилтиобензоилхлорид и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают при 115°С в течение 17 ч. После стандартной обработки и хроматографии на силикагеле получают 250 мг (82%) 3-(2-пиридил)-5-(2-хлор-5-метилтиофенил)-1,2,4-оксадиазола. 1H ЯМР (CDCl3), δ (часть/млн): 7,37 (dd, J=2,4 Гц, 8,2 Гц, 1 Н), 7,40-7,50 (m, 2H), 7,89 (add, J=1,4 Гц, 7,4 Гц, 8,2 Гц, 1Н), 8,05 (d, J=2,4 Гц, 1H), 8,23 (dd, J=2,2 Гц, 8,0 Гц, 1Н), 8,85 (m, 1H).
3-(2-Пиридил)-5-(3-феноксифенил)-1,2,4-оксадиазол (В40)
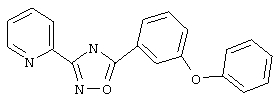
3-Феноксибензойную кислоту (214 мг, 1,0 ммоль) обрабатывают раствором оксалилхлорида (1,5 мл, 2 М в дихлорметане, 3 ммоль) и каталитическим количеством N,N-диметилформамида. Реакционную смесь перемешивают в течение ночи при температуре окружающей среды. Избыток оксалилхлорида удаляют в вакууме и при этом получают 3-феноксибензоилхлорид.
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-феноксибензоилхлорид и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают в закрытом сосуде при 110°С в течение ночи. После стандартной обработки получают 118 мг (37%) 3-(2-пиридил)-5-(3-феноксифенил)-1,2,4-оксадиазола в виде твердого вещества белого цвета.
3-(2-Пиридил)-5-(3-бензоилфенил)-1,2,4-оксадиазол (В41)
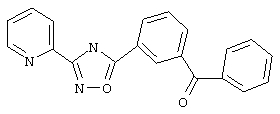
3-Бензоилбензойную кислоту (226 мг, 1 ммоль) в дихлорметане (1,5 мл) обрабатывают раствором оксалилхлорида (1,5 мл, 2 М в дихлорметане, 3 ммоль) и каталитическим количеством N,N-диметилформамида. Реакционную смесь перемешивают при температуре окружающей среды в течение ночи. Избыток оксалилхлорида удаляют в вакууме и при этом получают 3-бензоилбензоилхлорид.
Используя основной метод синтеза 1,2,4-оксадиазолов, 3-бензоилбензоилхлорид и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают в закрытом сосуде при 110°С в течение ночи. После стандартной обработки и фильтрования через силикагель (в дихлорметане) получают 200мг (61 %) 3-(2-пиридил)-5-(3-бензоилфенил)-1,2,4-оксадиазола в виде твердого вещества белого цвета. 1Н ЯМР (CDCl3), δ (часть/млн): 8,85 (d, 1H),8,68(m, 1H),8,53(dd, 1H), 8,23 (d, 1H),8,07(m, 1H), 7,88 (m, 3H), 7,70 (m, 2H), 7,49 (m, 3H).
3-(2-Пиридил)-5-(2-бром-5-метоксифенил)-1,2,4-оксадиазол (В42)
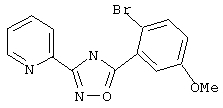
2-Бром-5-метоксибензойную кислоту (231 мг, 1 ммоль) в дихлорметане (1,5 мл) обрабатывают раствором оксалилхлорида (1,5 мл, 2 М в дихлорметане, 3 ммоль) и каталитическим количеством N,N-диметилформамида. Реакционную смесь перемешивают при температуре окружающей среды в течение ночи. Избыток оксалилхлорида удаляют в вакууме и при этом получают 2-бром-5-метоксибензоилхлорид.
Используя основной метод синтеза 1,2,4-оксадиазолов, 2-бром-5-метоксибензоилхлоридипирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают в закрытом сосуде при 110°С в течение ночи. После стандартной обработки и фильтрования через силикагель (в дихлорметане) получают 147 мг (44%) 3-(2-пиридил)-5-(2-бром-5-метоксифенил)-1,2,4-оксадиазола. 1H ЯМР (CDCl3), δ (часть/млн): 8,85 (d, 1 Н), 8,24 (d, 1 Н), 7,89 (m, 1Н), 7,65 (m. 2Н), 7,47 (m, 1Н), 6,99 (m, 1H), 3,89 (s, 3H).
3-(2-Пиридил)-5-(2-хлор-5-(трифторметил)фенил)-1,2,4-оксадиазол (В43)
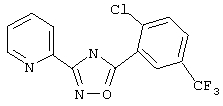
2-Хлор-5-(трифторметил)бензойную кислоту (224 мг, 1 ммоль) в дихлорметане (1,5 мл) обрабатывают раствором оксалилхлорида (1,5 мл, 2 М в дихлорметане, 3 ммоль) и каталитическим количеством N,N-диметилформамида. Реакционную смесь перемешивают при температуре окружающей среды в течение ночи. Избыток оксалилхлорида удаляют в вакууме и при этом получают 2-хлор-5-(трифторметил)бензоилхлорид.
Используя основной метод синтеза 1,2,4-оксадиазолов, 2-хлор-5-(трифторметил)бензоилхлорид и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают в закрытом сосуде при 110°С в течение ночи. После стандартной обработки и фильтрования через силикагель (в дихлорметане) получают 136 мг (42%) 3-(2-пиридил)-5-(2-хлор-5-(трифторметил)фенил)-1,2,4-оксадиазола в виде твердого вещества бежевого цвета. 1H ЯМР (CDCl3), δ (часть/млн): 8,87 (d, 1Н), 8,56 (s, 1H), 8,25 (d, 1H), 7,89 (m, 1H), 7,78 (m, 2H), 7,50 (m, 1H).
3-(2-Пиридил)-5-(3,4,5-трифторфенил)-1,2,4-оксадиазол (В44)
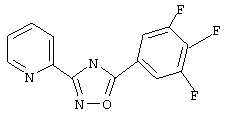
3,4,5-Трифторбензойную кислоту (0,176 г, 1,0 ммоль) в дихлорметане (1,5 мл) обрабатывают раствором оксалилхлорида (1,5 мл, 2 М в дихлорметане, 3 ммоль) и каталитическим количеством N,N-диметилформамида. Реакционную смесь перемешивают при температуре окружающей среды в течение ночи. Избыток оксалилхлорида удаляют в вакууме и при этом получают 3,4,5-трифторбензоилхлорид.
Используя основной метод синтеза 1,2,4-оксадиазолов, 3,4,5-трифторбензоилхлорид и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают в закрытом сосуде при 110°С в течение ночи. После стандартной обработки и хроматографии на силикагеле (10-30% этилацетата в гексане) получают 15 мг (5%) 3-(2-пиридил)-5-(3,4,5-трифторфенил)-1,2,4-оксадиазола в виде твердого вещества белого цвета.
3-(2-Пиридил)-5-(2,5,6-трифторфенил)-1,2,4-оксадиазол (В45)
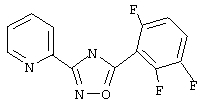
2,5,6-Трифторбензойную кислоту (176 мг, 1 ммоль) обрабатывают раствором оксалилхлорида (1,5 мл, 2 М в дихлорметане, 3 ммоль) и каталитическим количеством N,N-диметилформамида. Реакционную смесь перемешивают при температуре окружающей среды в течение 16 ч. Избыток оксалилхлорида удаляют в вакууме и при этом получают 2,5,6-трифторбензоилхлорид.
Раствор промежуточного 2,5,6-трифторбензоилхлорида и пирид-2-иламидоксима (137 мг, 1 ммоль) в дихлорметане перемешивают при температуре окружающей среды в течение 0,5 ч. После хроматографии на силикагеле получают 151 мг (51%) N-[(2,5,6-трифторбензоил)окси]пиридин-2-карбоксимидамид.
Раствор N-[(2,5,6-трифторбензоил)окси]пиридин-2-карбоксимидамида (50 мг, 0,169 ммоль) в пиридине (0,3 мл) нагревают при 115°С в течение 17 ч. После стандартной обработки и хроматографии на силикагеле получают 9,5 мг (20%) 3-(2-пиридил)-5-(2,5,6-трифторфенил)-1,2,4-оксадиазола.
Пример 4: Синтез 3,5-дизамещенных-1,2,4-оксадиазолов из ацилимидазолов
3-(3-Метоксифенил)-5-(2-пиридил)-1,2,4-оксадиазол (В8)
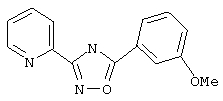
Используя модифицированный метод Shine и соавт., J. Heterocyclic Chem., 26:125-128 (1989), раствор пиколиновой кислоты (123 мг, 1 ммоль) в пиридине (1 мл) обрабатывают 1,1 '-карбонилдиимидазолом (162 мг, 1 ммоль) и реакционную смесь перемешивают при температуре окружающей среды до прекращения выделения диоксида углерода (30 мин). Затем промежуточный ацилимидазол обрабатывают 3-метоксибензамидоксимом (166 мг, 1 ммоль) и реакционную смесь нагревают с обратным холодильником в течение 1 ч. Затем для осаждения оксадиазола к реакционной смеси добавляют ледяную воду. Твердое вещество собирают фильтрованием, промывают водой и сушат, при этом получают 80 мг (32%) 3-(3-метоксифенил)-5-(2-пиридил)-1,2,4-оксадиазола; т.пл.90-94°С; ГХ/МС (El) m/z (отн. инт.) 253 {M+, 100), 254 (17), 179 (2), 175 (2), 149 (77), 133 (33), 119 (4), 106 (29), 78 (45), 51 (18).
Пример 5: Синтез 3,5-дизамещенных-1,2,4-оксадиазолов из сложных эфиров
3-(Пирид-2-ил)-5-(2-гидроксифенил)-1,2,4-оксадиазол (В46)
Используя метод Korbonits и соавт., J. Chem. Soc. Perkin Trans., (1982) t.i стр. 759-766, смесь этилсалицилата (200 мг, 1,2 ммоль), пирид-2-иламидоксима (82,5 мг, 0,6 ммоль) и 21%-ного этоксида натрия (19,4 мл, 6 ммоль) в этаноле (12 мл) нагревают с обратным холодильником в течение 16 ч. После охлаждения реакционную смесь разбавляют дихлорметаном (50 мл) и промывают водой и насыщенным гидрокарбонатом натрия. Органический слой сушат над сульфатом натрия и концентрируют в вакууме. При перекристаллизации из диэтилового эфира получают 15 мг (5%) 3-(пирид-2-ил)-5-(2-гидроксифенил)-1,2,4-оксадиазола.
3-(2-Пиридил)-5-(5-хлор-2-гидроксифенил)-1,2,4-оксадиазол (В47)
Аналогичным способом, метиловый эфир 5-хлор-2-гидроксибензойной кислоты (372 мг, 2 ммоль), пирид-2-иламидоксим (137 мг, 1 ммоль) и 21%-ного этоксида натрия (32,4 мл, 10 ммоль) в этаноле (20 мл) нагревают с обратным холодильником в течение 16 ч. После стандартной обработки и перекристаллизации из диэтилового эфира получают 14,2 мг (5%) 3-(2-пиридил)-5-(5-хлор-2-гидроксифенил)-1,2,4-оксадиазола.
Пример 6: Синтез 3,5-дизамещенных-1,2,4-оксадиазолов из ангидридов изатовой кислоты
3-(2-Пиридил)-5-(2-аминофенил)-1,2,4-оксадиазол (В48)
Используя модифицированный метод Nagahara и соавт., Chem. Pharm.
Bull., 23:3178-3183 (1975), смесь ангидрида изатовой кислоты (163 мг, 1 ммоль), пирид-2-иламидоксима (137 мг, 1 ммоль) в пиридине (1 мл) нагревают при 115°С в течение 17 ч. После охлаждения реакционную смесь разбавляют 50 мл дихлорметана и промывают водой и насыщенным гидрокарбонатом натрия. Органический слой сушат над сульфатом натрия, фильтруют через силикагель и концентрируют в вакууме. При перекристаллизации из диэтилового эфира получают 45,6 мг (19%) 3-(2-пиридил)-5-(2-аминофенил)-1,2,4-оксадиазола.
3-(2-Пиридил)-5-(2-аминофенил)-1,2,4-оксадиазол (В49)
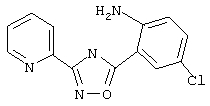
Аналогичным способом, ангидрид 5-хлоризатовой кислоты (197 мг, 1 ммоль) и пирид-2-иламидоксим (137 мг, 1 ммоль) в пиридине (1 мл) нагревают при 115°С в течение 17 ч. После обработки получают 138 мг (51%) 3-(2-пиридил)-5-(2-аминофенил)-1,2,4-оксадиазола.
Пример 7: Синтез 2,4-дизамещенных 1,3-оксазолов
2-[3-хлорфенил]-4-[пиридин-2-ил]-1,3-оксазол (В50)
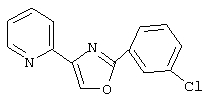
Используя метод Kelly и соавт., J. Org. Chem., 61:4623-4633 (1996), раствор 2-бромацетилпиридина (120 мг, 0,6 ммоль) в толуоле (5 мл) обрабатывают 3-хлорбензамидом (300 мг, 1,9 ммоль) и смесь нагревают в закрытом сосуде с обратным холодильником в течение 60 ч. Затем смесь охлаждают и растворитель удаляют в вакууме. После хроматографии на силикагеле с использованием градиента от гексана до этилацетата получают 38 мг (9%) 2-[3-хлорфенил]-4-[пиридин-2-ил]-1,3-оксазола в виде твердого вещества бледно-желтого цвета. 1H ЯМР (CDCl3), δ (часть/млн): 8,62 (d, 1H), 8,35 (s, 1H), 8,15 (m, 1H), 8,00 (m, 2H), 7,80 (td, 1H), 7,42 (m, 2H), 7,23 (m, 1H).
2-[3-Бромфенил]-4-[пиридин-2-ил]-1,3-оксазол (В51)
Аналогичным способом, 2-бромацетилпиридин (500 мг, 2,5 ммоль) и 3-хлорбензамид (1,2 г, 6 ммоль) в толуоле (10 мл) нагревают в закрытом сосуде с обратным холодильником в течение 60 ч. После обработки и хроматографии на силикагеле с использованием градиента от гексана до этилацетата получают 50 мг (7%) 2-[3-бромфенил]-4-[пиридин-2-ил]-1,3-оксазола в виде твердого вещества белого цвета. 1H ЯМР (CDCl3), δ (часть/млн): 8,60 (d, 1H), 8,34 (s, 1H), 8,30 (t, 1H), 8,00 (m, 2H), 7,80 (td, 1H), 7,60 (dd, 1H), 7,35 (t, 1H), 7,23 (m, 1H).
2-[3-Цианофенил]-4-[пиридин-2-ил]-1,3-оксазол (В52)
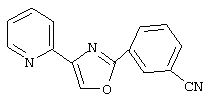
Смесь 2-[3-бромфенил]-4-[пиридин-2-ил]-1,3-оксазола (23 мг, 0,076 ммоль) и цианида цинка (112 мг, 0,96 ммоль) в N, N-диметилформамиде (2 мл) обрабатывают Pd(PPh3)4 (74 мг, 0,064 ммоль) и нагревают при 80°С в течение ночи. После стандартной обработки и хроматографии получают 6 мг (32%) 2-[3-цианофенил]-4-[пиридин-2-ил]-1,3-оксазола в виде твердого вещества белого цвета. 1Н ЯМР (CDCl3), δ (часть/млн): 8,61 (d, 1H), 8,45 (s, 1Н), 8,38 (s, 1H), 8,36 (m, 1H), 8,00 (d, 1H), 7,80 (m, 2H), 7,61 (t, 1H), 7,23 (m, 1H).
Пример 8: Синтез 3,5-дизамещенных 1,2-оксазолов
5-[3-Гидроксифенил]-3-[пиридин-2-ил]-1,2-оксазол (В53)
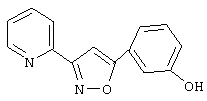
Перемешиваемый раствор пиридин-2-карбогидроксимоилхлорида (300 мг, 1,9 ммоль) и 3-гидроксифенилацетилена (760 мг, 6,4 ммоль) в смеси (1:1) ТГФ/CH2Cl2 (10 мл) при 0°С обрабатывают триэтиламином (2 мл, 1,45 г, 15 ммоль). Полученную смесь нагревают при комнатной температуре в течение ночи. Растворитель удаляют в вакууме. Остаток растворяют в дихлорметане, промывают солевым раствором и сушат над безводным сульфатом натрия. После удаления растворителя в вакууме и растирания с 10%-ным этилацетатом в гексане получают 200 мг (44%) 5-[3-гидроксифенил]-3-[пиридин-2-ил]-1,2-оксазола в виде твердого вещества бежевого цвета. 5-[3-Цианофенил]-3-[пиридин-2-ил]-1,2-оксазол (В54)
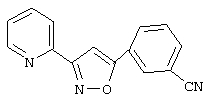
Смесь 5-[3-трифторметансульфонилфенил]-3-[пиридин-2-ил]-1,2-оксазола (98 мг, 0,26 ммоль), KCN (230 мг, 4 ммоль), NiBr2(PPh3)2 (52,4 мг, 0,07 ммоль) и PPh3 (42 мг, 0,16 ммоль) в ацетонитриле (1 мл) обрабатывают порошкообразным цинком (20 мг, 0,3 ммоль) и полученную смесь нагревают при 60°С в течение ночи. После хроматографии на силикагеле с использованием градиента от гексана до этилацетата получают 15 мг (23%) 5-[3-цианофенил]-3-[пиридин-2-ил]-1,2-оксазола в виде твердого вещества белого цвета.
Пример 9: Синтез 3,5-дизамещенных 1,2,4-триазолов 3-хлорбензгидразид
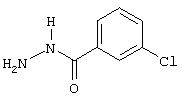
Смесь 3-хлорбензойной кислоты (0,5 г, 3,19 ммоль), 1,3-дициклогексилкарбодиимида (0,72 г, 3,51 ммоль), 4-диметиламинопиридина (0,04 г, 0,32 ммоль) в этаноле перемешивают при температуре окружающей среды в течение 1,5 ч. Твердое вещество белого цвета отфильтровывают и фильтрат разбавляют дихлорметаном (100 мл). Органический раствор промывают 1н. гидросульфатом натрия (100 мл), насыщенным бикарбонатом натрия (100 мл), водой (100 мл) и солевым раствором (100 мл). Органическую фазу сушат над безводным сульфатом магния и фильтруют. Фильтрат концентрируют в вакууме. Неочищенный остаток растворяют в этаноле (15 мл) и обрабатывают моногидратом гидразина (0,46 мл, 9,58 ммоль). Полученный прозрачный раствор перемешивают при температуре окружающей среды в течение ночи. Затем реакционную смесь концентрируют досуха в вакууме. После хроматографии остатка на силикагеле с использованием 3%-ного метанола в дихлорметане получают 0,29 г (53%) 3-хлорбензгидразида в виде твердого вещества белого цвета.
3-(2-Пиридил)-5-(3-хлорфенил)-1,2,4-триазол (В55)
Используя метод Browne и соавт., Aust. J. Chem., 28:2543-2546 (1975), раствор 2-цианопиридина (0,1 мл, 1,00 ммоль) в метаноле (5 мл) обрабатывают металлическим натрием (6,9 мг, 0,30 ммоль) и перемешивают при температуре окружающей среды в течение 1 ч. После этого добавляют раствор 3-хлорбензгидразида (0,17 г, 1,0 ммоль) в метаноле (5 мл) и полученный раствор нагревают с обратным холодильником в течение 3 ч. Реакционную смесь концентрируют в вакууме, и полученное твердое вещество желтого цвета (100 мг) растворяют в толуоле (2 мл). Смесь нагревают при 175°С в течение 3 ч и затем перемешивают при температуре окружающей среды в течение ночи. После упаривания растворителя в вакууме и хроматографии на силикагеле с использованием 1%-ного метанола в дихлорметане получают 29 мг (11%) 3-(2-пиридил)-5-(3-хлорфенил)-1,2,4-триазола в виде твердого вещества грязно-белого цвета.
3-(2-Пиридил)-5-(3-иодфенил)-1,2,4-триазол (В56)
Аналогичным способом, из 2-цианопиридина (0,15 мл, 1,53 ммоль), металлического натрия (10,5 мг, 0,46 ммоль) и 3-иодбензгидразида (0,40 г, 1,53 ммоль) получают после стандартной обработки и хроматографии 210 мг (40%) 3-(2-пиридил)-5-(3-иодфенил)-1,2,4-триазола в виде твердого вещества белого цвета.
Пример 10. Исследование активности антагонистов рецептора группы 1
Скрининг с использованием астроцитов
Первичные культуры астроцитов получают из детенышей крыс возрастом 3-5 дней согласно модифицированному методу Миллера (Miller и соавт., J. Neuroscience, 15(9): 6103-6109 (1995)). Первичные культуры наносят на покрытые поли-1-лизином флаконы в среде Игла, модифицированной Дульбеко (DMEM) и содержащей FCS. Через 6 сут клеточные культуры встряхивают в течение ночи при 280 об/мин, затем переносят в среду для астроцитов, содержащую факторы роста, которые стимулируют экспрессию mGluR5 (Miller и соавт., 1995). Для анализа в кюветах, культуры клеток стимулируют факторами роста во флаконах в течение 3-5 сут, затем клетки собирают и готовят для измерений мобилизации [Са2+]i, как описано ранее (Nemeth и соавт., 1998).
Для FLIPR-анализа клетки высеивают на покрытые поли-О-лизином 96-луночные планшеты с прозрачным дном и черными стенками, определение мобилизации [Са2+] проводят через 3 сут после стимулирования факторами роста. На клеточные культуры в 96-луночном планшете наносят 4 мкМ раствор флуоресцентного индикатора кальция fluo-3 в форме ацетоксиметилового эфира (Molecular Probes, Eugene, Oregon) в 0,01%-ном Pluronic. Все анализы проводят в буферном растворе, содержащем 127 мМ NaCI, 5 мМ KCI, 2 мМ MgCl2, 0,7 мМ NaH2РО4, 2 мМ CaCl2, 0,422 мг/мл NaHCO3, 2,4 мг/мл HEPES, 1,8 мг/мл глюкозы и 1 мг/мл БСА, фракция IV (рН 7,4).
Эксперименты с использованием FLIPR проводят с помощью лазера, установленного на 0,800 Вт, со скоростью затвора камеры прибора с зарядовой связью (CCD) 0,4 с. Каждый FLIPR-эксперимент инициируют с помощью 180 мкл буферного раствора, который присутствует в каждой лунке клеточного планшета. Добавляют по 20 мкл из планшета с антагонистом, затем по 50 мкл из планшета с агонистом. После каждого добавления измеряют флуоресцентный сигнал 50 раз с интервалом 1 с с последующими 3 измерениями с интервалом 5 с. В качестве ответа принимают высоту пика ответного сигнала в течение измерения.
Величины EC50 и IC50 определяют с использованием калибровочных кривых, полученных для 8 концентраций и рассчитанных по результатам двух параллельных опытов. Калибровочные кривые для агониста получают путем сканирования всех ответов для выявления максимального ответа на планшете. Подавление антагонистом активности введенного агониста нормализуют по среднему ответу на введение агониста в 14 контрольных лунках одного и того же планшета.
Скрининг с использованием CaR/mGluRSd
Клетки НЕК 293, экспрессирующие химерный рецептор CaR/mGluR5d (клональная клеточная линия hCaR/hmGluR5d_hek6), наносят за 24 ч до анализа при плотности 100000 клеток на лунку 96-луночного планшета, покрытого коллагеном I, с прозрачным дном (Becton Dickenson) в среде DMEM с добавкой 10% FCS (Hyclone).
В день анализа культуральную среду отсасывают из лунок планшета и в каждую лунку добавляют 80 мкл буферного раствора для анализа (20 мМ HEPES, 146 мМ NaCI, 5 мМ KCI, 1 мМ MgCl2, 1 мМ CaCl2, 1 мг/мл БСА, 1 мг/мл глюкозы, рН 7,4) с добавлением 6 мкМ Са2+-чувствительного красителя Fluo-3 AM (Molecular Probes) и 0,025% Pluronic (Molecular Probes). Затем для более эффективного взаимодействия клеток с Fluo-3 планшет инкубируют в темноте при комнатной температуре в течение 1 ч. При завершении инкубирования внеклеточный Fluo-3 удаляют путем промывки планшета буферным раствором для анализа. Перед началом анализа в каждую лунку снова добавляют буферный раствор для анализа (конечный объем = 160 мкл).
Планшет помещают в загрузочный робот прибора FLIPR (Molecular Devices) с лазером, установленным на 0,8 Вт. Через 10 с после инициирования анализа в каждую лунку, содержащую 160 мкл буферного раствора для анализа, добавляют 40 мкл буферного раствора, содержащего 62,5 мкМ тестируемого соединения и 2% ДМСО, при этом конечная концентрация тестируемого соединения составляет 12 мкМ и концентрация ДМСО 0,4%. Через 75 с после инициирования анализа к 200 мкл раствора, присутствующего в каждой лунке, добавляют 50 мкл буферного раствора для анализа, содержащего 6 мМ CaCl2, при этом конечная концентрация Са2+ составляет 2,0 мМ, а конечная концентрация тестируемого соединения составляет 10 нМ. Для определения активации и/или ингибирования рецептора в ходе анализа регистрируют относительную интенсивность флуоресценции (Л возбуждения = 488 нм, Л испускания =510 нм) через соответствующие промежутки времени.
Для иллюстрации: для 1,2,4-оксадиазола, описанного выше и обозначенного как "В21" (см. Пример 3), величина 1650 по отношению к CaR/mGluR5d составляет 43 нМ, а величина IC50 по отношению к природному рецептору mGluR5d составляет 121 нМ. Показано, что соответствующий 1,3-оксазол, обозначенный как "В52" (см. Пример 7), обладает равной активностью по отношению к химерному CaR/mGluR5d, то есть величина 1650 составляет 45 нМ, но проявляет повышенную активность по отношению к природному рецептору mGluR5d, то есть величина IC50 составляет 74 нМ.
Таким образом, настоящее изобретение описано в широком диапазоне и проиллюстрировано на примере соответствующих вариантов воплощения, представленных выше. Для специалистов в данной области техники представляется очевидным, что возможны различные модификации настоящего изобретения, не выходя за пределы его объема и сущности.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛИГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МЕТАБОТРОПНОГО РЕЦЕПТОРА ГЛУТАМАТА | 2005 |
|
RU2381226C2 |
| СОЕДИНЕНИЯ ХИНОЛИНА, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ, ЯВЛЯЮЩИХСЯ ОТВЕТОМ НА МОДУЛЯЦИЮ РЕЦЕПТОРА 5-HT СЕРОТОНИНА | 2008 |
|
RU2483068C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ АРИЛСУЛЬФОНЫ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, КОТОРЫЕ ОТВЕЧАЮТ НА МОДУЛИРОВАНИЕ 5HT РЕЦЕПТОРОВ | 2007 |
|
RU2451012C2 |
| ЗАМЕЩЕННЫЕ ПИРИМИДИНЫ, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ИЛИ N-ОКСИДЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2113437C1 |
| СПОСОБ СИНТЕЗА ПРОИЗВОДНЫХ ИМИДАЗОЛАМИНОКИСЛОТ И РОДСТВЕННЫХ СОЕДИНЕНИЙ | 2005 |
|
RU2444516C2 |
| ПИРРОЛИДИНИЛАЛКИЛАМИДНЫЕ ПРОИЗВОДНЫЕ, ИХ ПОЛУЧЕНИЕ И ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛИГАНДОВ РЕЦЕПТОРА CCR3 | 2009 |
|
RU2514824C2 |
| ПРОИЗВОДНЫЕ ДИАЗАГОМОАДАМАНТАНА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2549551C2 |
| ПРОИЗВОДНЫЕ (3-МЕТИЛПИРРОЛИДИН-3-ИЛ)-МЕТИЛ ПИРИДИНИЛОВОГО ЭФИРА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА NK-3 | 2011 |
|
RU2625798C2 |
| ПРОИЗВОДНЫЕ (ИНДОЛ-3-ИЛ)ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ В КАЧЕСТВЕ АГОНИСТОВ КАННАБИНОИДНЫХ РЕЦЕПТОРОВ СВ1 | 2005 |
|
RU2376302C2 |
| ПРОИЗВОДНЫЕ НИТРОКАТЕХОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ СОМТ | 2006 |
|
RU2441001C2 |
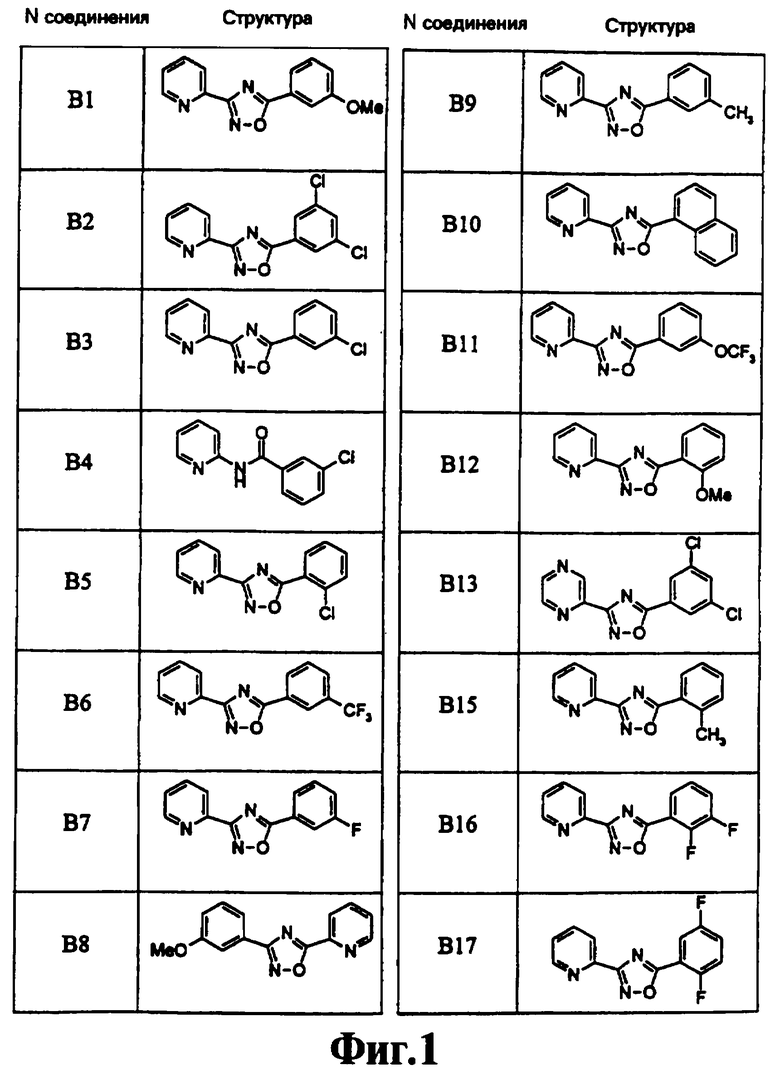
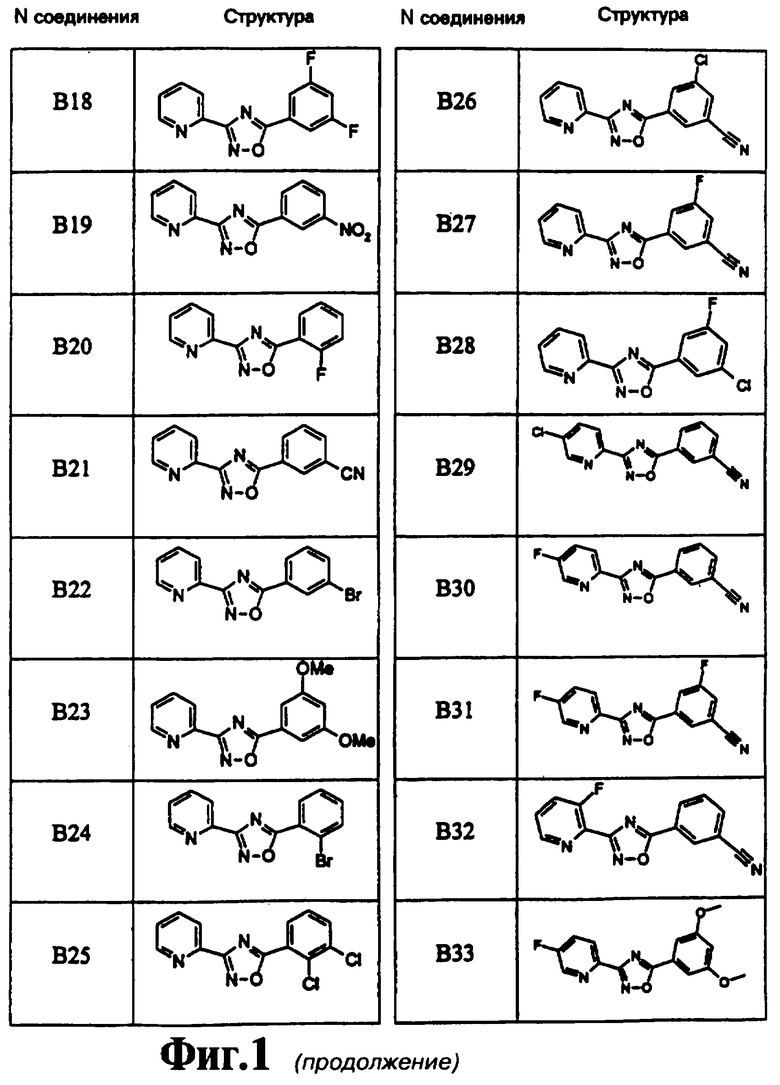
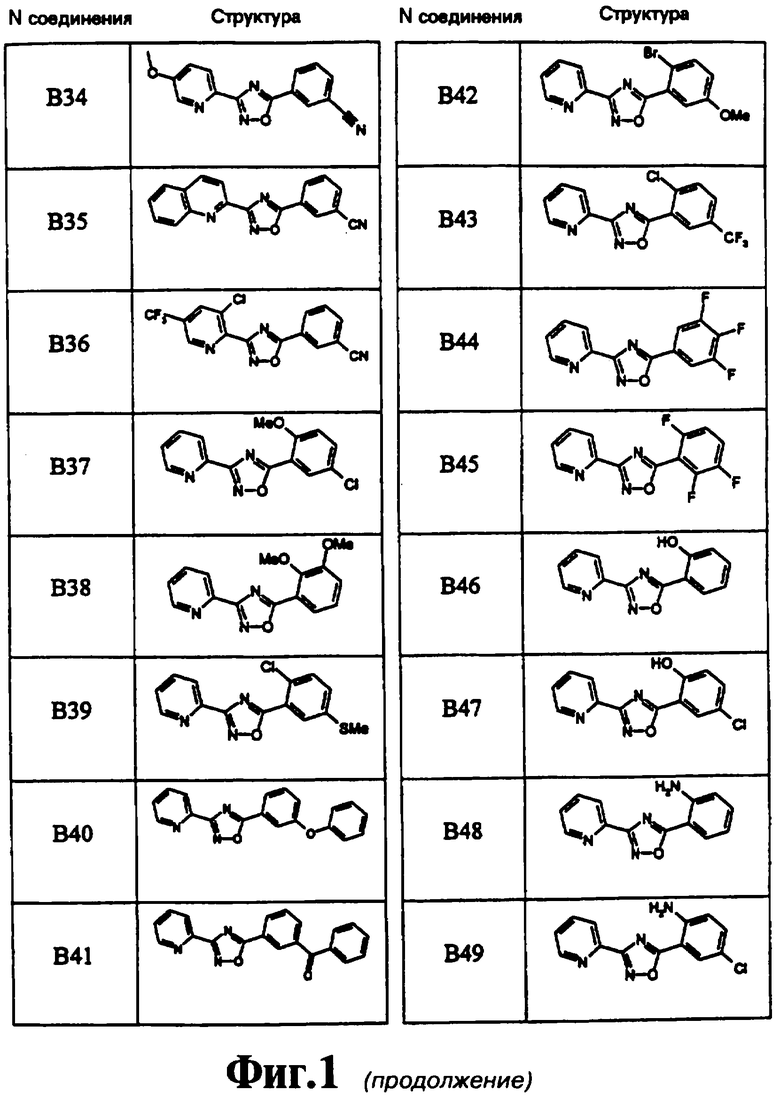
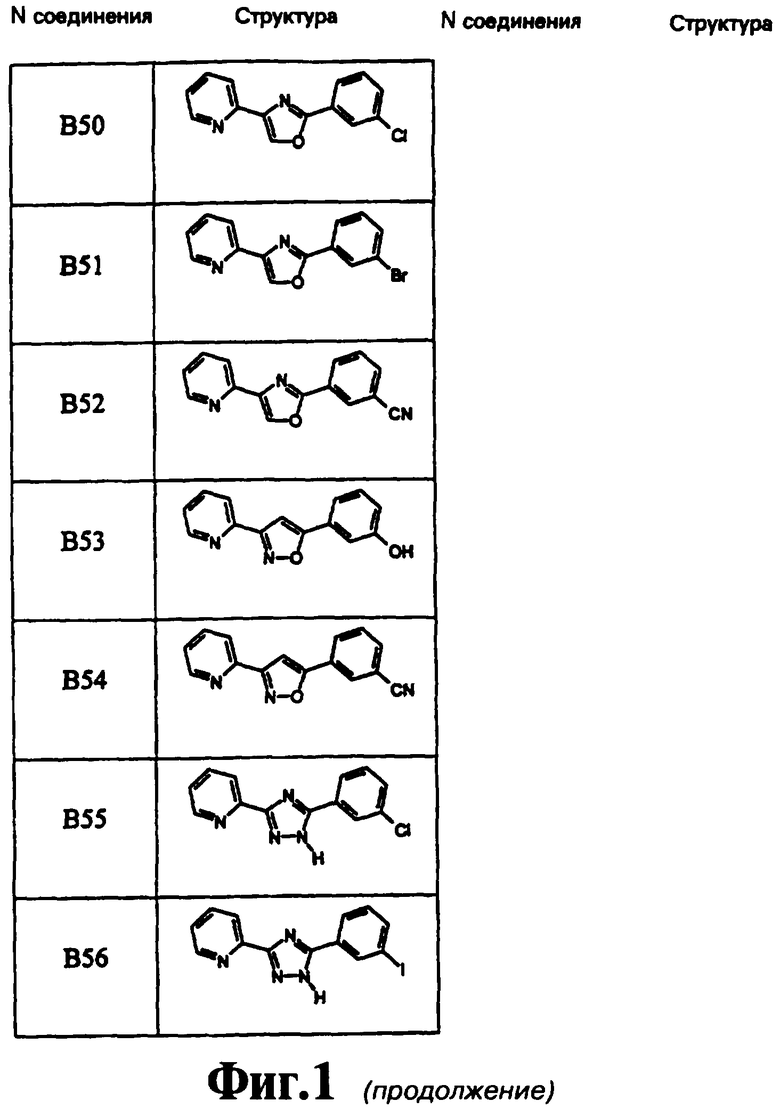
Изобретение относится к новым соединениям формулы II
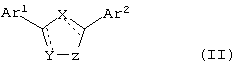
где X, Y представляют собой N, Z представляет собой О; Ar1 представляет собой 2-пиридил; Ar2 представляет собой фенил; причем по крайней мере один из остатков Ar1 и Ar2 замещен по меньшей мере одним остатком, выбранным из группы, состоящей из -F, -CI, -Br, -I, -SR, -CN, -C(O)R, -CH(OR)R', -CH2(OR), -CF3, C1-С10-алкила и арила, где R или R' выбраны из группы, включающей в себя Н, -CF3, C1-С10-алкил и арил, где R или R' могут быть объединены с образованием кольца, и при условии, что соединение не является 3-(2-пиридил)-5-(2-хлорфенил)-1,2,4-оксадиазолом или 3-(2-пиридил)-5-[3-(трифторметил)фенил]-1,2,4-оксадиазолом; или к новым соединениям, перечисленным в формуле изобретения, а также относится к фармацевтической композиции, обладающей активностью в отношении метаботропных глутаматных рецепторов на их основе, и к способу модулирования метаботропных глутаматных рецепторов. 3 н. и 11 з.п. ф-лы, 1 ил.
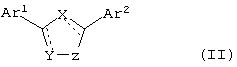
где X, Y представляют собой N;
Z представляет собой О;
Ar1 представляет собой 2-пиридил;
Ar2 представляет собой фенил;
причем по крайней мере один из остатков Ar1 и Ar2 замещен по меньшей мере одним остатком, выбранным из группы, состоящей из -F, -CI, -Br, -I, -SR, -CN, -C(O)R, -CH(OR)R', -CH2(OR), -CF3, C1-С10-алкила и арила,
где R или R' выбраны из группы, включающей в себя Н, -CF3, C1-С10-алкил и арил, где R или R' могут быть объединены с образованием кольца, и
при условии, что соединение не является 3-(2-пиридил)-5-(2- хлорфенил)-1,2,4-оксадиазолом или 3-(2-пиридил)-5-[3-(трифторметил)фенил]-1,2,4-оксадиазолом;
или из следующих соединений:
3-(2-пиридил)-5-(3-(трифторметокси)фенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(2,3-дифторфенил)-1,2,4-оксадиазол,
3-(2-пиридил)-5-(3,5-диметоксифенил)-1,2,4-оксадиазол,
3-(5-фторпирид-2-ил)-5-(3,5-диметоксифенил)-1,2,4-оксадиазол,
3-(5-метоксипирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазол,
2-(3-хлорфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3,5-дихлорфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-метоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2-хлорфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-трифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-метилфенил)-4-(2-пиридил)-1,3-оксазол,
2-(1-нафтил)-4-(2-пиридил)-1,3-оксазол,
2-(3-трифторметоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2,3-дифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(2,5-дифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3,5-дифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-цианофенил)-4-(2-пиридил)-1,3-оксазол,
2-(3,5-диметоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2,3-дихлорфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-хлор-5-цианофенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-фтор-5-цианофенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-хлор-5-фторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-цианофенил)-4-(5-хлорпирид-2-ил)-1,3-оксазол,
2-(3-цианофенил)-4-(5-фторпирид-2-ил)-1,3-оксазол,
2-(3-циано-5-фторфенил)-4-(5-фторпирид-2-ил)-1,3-оксазол,
2-(3-цианофенил)-4-(3-фторпирид-2-ил)-1,3-оксазол,
2-(3,5-диметоксифенил)-4-(5-фторпирид-2-ил)-1,3-оксазол,
2-(3-цианофенил)-4-(5-метоксипирид-2-ил)-1,3-оксазол,
2-(3-цианофенил)-4-(2-хинолинил)-1,3-оксазол,
2-(3-цианофенил)-4-(3-хлор-5-трифторметилпирид-2-ил)-1,3-оксазол,
2-(5-хлор-2-метоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2-хлор-5-метилтиофенил)-4-(2-пиридил)-1,3-оксазол,
2-(2-бром-5-метоксифенил)-4-(2-пиридил)-1,3-оксазол,
2-(2,5,6-трифторфенил)-4-(2-пиридил)-1,3-оксазол,
2-(3-бромфенил)-4-(2-пиридил)-1,3-оксазол
или их фармацевтически приемлемые соли.
3-(2-пиридил)-5-(3,5-дихлорфенил)-1,2,4-оксадиазола,
3-(2-пиридил)-5-(3-хлорфенил)-1,2,4-оксадиазола,
3-(2-пиридил)-5-(3-метилфенил)-1,2,4-оксадиазола,
3-(2-пиридил)-5-(2,5-дифторфенил)-1,2,4-оксадиазола,
3-(2-пиридил)-5-(3,5-дифторфенил)-1,2,4-оксадиазола,
3-(2-пиридил)-5-(3-цианофенил)-1,2,4-оксадиазола,
3-(2-пиридил)-5-(2,3-дихлорфенил)-1,2,4-оксадиазола,
3-(2-пиридил)-5-(3-хлор-5-цианофенил)-1,2,4-оксадиазола,
3-(2-пиридил)-5-(3-фтор-5-цианофенил)-1,2,4-оксадиазола,
3-(2-пиридил)-5-(3-хлор-5-фторфенил)-1,2,4-оксадиазола,
3-(5-хлорпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазола,
3-(5-фторпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазола,
3-(5-фторпирид-2-ил)-5-(3-циано-5-фторфенил)-1,2,4-оксадиазола,
3-(3-фторпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазола,
3-(3-хлор-5-трифторметилпирид-2-ил)-5-(3-цианофенил)-1,2,4-оксадиазола,
3-(2-пиридил)-5-(5-хлор-2-метоксифенил)-1,2,4-оксадиазола,
3-(2-пиридил)-5-(2-хлор-5-метилтиофенил)-1,2,4-оксадиазола,
3-(2-пиридил)-5-(2,5,6-трифторфенил)-1,2,4-оксадиазола,
3-(2-пиридил)-5-(3-бромфенил)-1,2,4-оксадиазола и их фармацевтически приемлемые соли.
| US 3647809 А, 07.03.1972 | |||
| US 4022901 A, 10.05.1977 | |||
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Helene PERRIER et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Biorganic & Medicinal Chemistry Letters | |||
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| Т.Ross Kelly and Fengrui Lang | |||
| Total Synthesis of Dimethyl Sulfomycinamate | |||
| Tetrahedron Letters | |||
| Топка с качающимися колосниковыми элементами | 1921 |
|
SU1995A1 |

