Настоящее изобретение относится к производным (индол-3-ил)гетероциклических соединений, к содержащим их фармацевтическим композициям и к применению этих производных (индол-3-ил)гетероциклических соединений в терапии, особенно при лечении боли.
Лечение боли часто ограничено побочными эффектами доступных в настоящее время лекарственных средств. При боли, от умеренной до тяжелой, широко применяют опиоиды. Данные средства являются недорогими и эффективными, но обладают серьезными и потенциально опасными для жизни побочными эффектами, в особенности угнетением дыхания и ригидностью мышц. Кроме того, дозы опиоидов, которые можно вводить, ограничены тошнотой, рвотой, запором, зудом и задержкой мочи, что часто приводит к тому, что пациенты предпочтительнее выбирают недостаточно оптимальное устранение боли, чем терпеть эти причиняющие страдание побочные действия. Более того, эти побочные эффекты часто приводят к тому, что пациенты нуждаются в продолжительной госпитализации. Опиоиды представляют собой вызывающие сильное привыкание средства и внесены в список наркотиков во многих областях. Следовательно, существует необходимость в новых болеутоляющих средствах, обладающих улучшенным профилем побочных эффектов по сравнению с используемыми в настоящее время продуктами при равных болеутоляющих дозах.
Накапливаются доказательства, что каннабиноидные агонисты обладают потенциалом в качестве болеутоляющих и противовоспалительных средств. Каннабиноидные рецепторы включают два типа рецепторов: каннабиноидные рецепторы CB1, которые главным образом расположены в центральной нервной системе, но также экспрессируются периферическими нейронами и в меньшей степени в других периферических тканях; и каннабиноидные рецепторы CB2, которые главным образом расположены в клетках иммунной системы (Howlett, A.C. et al.: International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors. Pharmacol. Rev. 54, 161-202, 2002). Хотя рецепторы CB2 вовлечены в модуляцию иммунного и противовоспалительного ответа каннабиноидов, агонисты каннабиноидных рецепторов, главным образом те из них, которые взаимодействуют с рецепторами CB1, были предложены в качестве пригодных для лечения боли (см. Iversen, L. и Chapman, V. Current Opinion in Pharmacology, 2, 50-55, 2002 и приведенные там ссылки).
В патенте США 4939138 (Sterling Drug Inc.) в качестве болеутоляющего средства описан WIN 55,212-2, мезилатная соль (R)-(+)-[2,3-дигидро-5-метил[(морфолинил)метил]пирроло[1,2,3-de]-1,4-бензоксазинил]-(1-нафталенил)метанона. Данное соединение представляет собой прототип аминоалкилиндолов (Eissenstat, M.A. et al., J. Med. Chem. 38, 3094-3105, 1995), которые являются сильными агонистами рецепторов CB1, способными вызывать антиноцицепцию с равной морфину эффективностью на моделях животных острой боли, длительной боли при воспалении и невропатической боли.
Ключевыми структурными признаками аминоалкилиндолов, обладающих каннабимиметическими свойствами (Adam, J. и Cowley, P. Expert Opin. Ther. Patents, 12, 1475-1489, 2002), являются аминоалкильный заместитель в положении 1 индольной группы и, кроме того, объемный заместитель в положении 3 индольного кольца, примером которого является ароильная группа в аминоалкилиндолах, описанных в патенте США 4939138 (Sterling Drug Inc.) или в более поздней заявке WO02060447 (University of Connecticut), или замещенная амидогруппа в соединениях, описанных в WO0158869 (Bristol-Myers Squibb). Недавно в WO0236590 (Amrad Operations PTY Ltd.) в качестве модуляторов каннабиноидных рецепторов и пригодных в качестве болеутоляющих средств были описаны 1-(аминоалкил)индольные производные, имеющие замещенное оксадиазол-5-ильное кольцо в положении 3.
Сохраняется необходимость в каннабиноидных агонистах с улучшенными свойствами, такими как увеличенная растворимость в воде, для применения в качестве терапевтических средств.
В связи с этим, настоящее изобретение относится к производным (индол-3-ил)гетероциклических соединений, имеющим общую формулу I
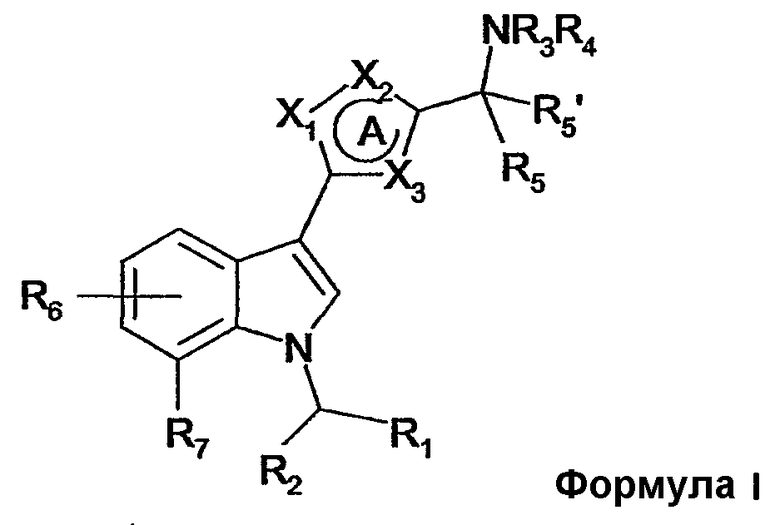
где A представляет собой 5-членное ароматическое гетероциклическое кольцо, где X1, X2 и X3 независимо выбирают из N, O, S и CR;
R представляет собой H или (C1-4)алкил; или
R, когда он присутствует в X2 или X3, может вместе с R3 образовывать 5-8-членное кольцо;
R1 представляет собой 5-8-членное насыщенное карбоциклическое кольцо, необязательно содержащее гетероатом, выбранный из O и S;
R2 представляет собой H, CH3 или CH2-CH3; или
R2 соединен с R7 с образованием 6-членного кольца, необязательно содержащего гетероатом, выбранный из O и S, и где гетероатом связан с положением 7 индольного кольца;
R3 и R4 независимо представляют собой H, (C1-6)алкил или (C3-7)циклоалкил, где указанные алкильные группы необязательно замещены OH, (C1-4)алкилокси, (C1-4)алкилтио, (C1-4)алкилсульфонилом, CN или галогеном; или
R3 вместе с R4 и N, с которым они связаны, образует 4-8-членное кольцо, необязательно содержащее дополнительный гетероатом, выбранный из O и S, и необязательно замещенное OH, (C1-4)алкилом, (C1-4)алкилокси, (C1-4)алкилокси-(C1-4)алкилом или галогеном; или
R3 вместе с R5 образует 4-8-членное кольцо, необязательно содержащее дополнительный гетероатом, выбранный из O и S, и необязательно замещенное OH, (C1-4)алкилом, (C1-4)алкилокси, (C1-4)алкилокси-(C1-4)алкилом или галогеном; или
R3 вместе с R, когда он присутствует в X2 или X3, образует 5-8-членное кольцо;
R5 представляет собой H, (C1-4)алкил; или
R5 вместе с R3 образует 4-8-членное кольцо, необязательно содержащее дополнительный гетероатом, выбранный из O и S, и необязательно замещенное OH, (C1-4)алкилом, (C1-4)алкилокси, (C1-4)алкилокси-(C1-4)алкилом или галогеном;
R5' представляет собой H, (C1-4)алкил;
R6 представляет собой 1-3 замещающие группы, независимо выбранные из H, (C1-4)алкила, (C1-4)алкилокси, CN или галогена;
R7 представляет собой H, (C1-4)алкил (C1-4)алкилокси, CN или галоген; или
R7 соединен с R2 с образованием 6-членного кольца, необязательно содержащего дополнительный гетероатом, выбранный из O и S, и где гетероатом связан с положением 7 индольного кольца; или их фармацевтически приемлемым солям, в качестве агонистов каннабиноидных рецепторов CB1, которые можно применять для лечения боли, например, такой как периоперационная боль, хроническая боль, невропатическая боль, боль при раке и боль и мышечная спастичность при рассеянном склерозе.
Как использовано в определении формулы I, гетероцикл A представляет собой 5-членное ароматическое гетероциклическое кольцо, которое содержит 1-3 гетероатома, выбранных из N, O и S. Это означает, что по меньшей мере один из X1, X2 и X3, используемых для определения гетероцикла A, не может представлять собой CR. Репрезентативными гетероциклами A являются гетероциклы - производные тиофена, фурана, триазола, тиазола, тиадиазола, оксазола, оксадиазола и их изомеров, включающие изотиазол, изотиадиазол, изоксазол и изоксадиазол. Предпочтительные гетероциклы A представляют собой 1,2,4-оксадиазол (X1 представляет собой N, X2 представляет собой O, X3 представляет собой N), 1,2,4-тиадиазол (X1 представляет собой N, X2 представляет собой S, X3 представляет собой N) и тиазол (X1 представляет собой S, X2 представляет собой CR, X3 представляет собой N).
В определении формулы I R, когда он присутствует в X2 или X3, может образовывать с R3 5-8-членное кольцо, таким образом, образуя вместе с кольцом A бициклическую кольцевую систему, содержащую 5-8-членное N-содержащее кольцо, конденсированное с 5-членным ароматическим гетероциклическим кольцом A. Примерами таких конденсированных кольцевых систем являются конденсированные кольцевые системы, производные 5,6-дигидро-4H-пирроло[3,4-d]изоксазола, 4,5,6,7-тетрагидрооксазоло[5,4-c]пиридина, 4,5,6,7-тетрагидротиазоло[5,4-c]пиридина, 5,6,7,8-тетрагидро-4H-изоксазоло[5,4-c]азепина, 5,6-дигидро-4H-пирроло[3,4-d]тиазола и 5,6-дигидро-4H-пирроло[3,4-d]изотиазола.
Как использовано в определении формулы I, термин (C1-4)алкил означает разветвленную или неразветвленную алкильную группу с 1-4 атомами углерода, например бутил, изобутил, трет-бутил, пропил, изопропил, этил и метил.
В термине (C1-4)алкилокси, (C1-4)алкил имеет значение, указанное выше.
Термин галоген означает F, Cl, Br или I.
Как использовано в определении R1 формулы I, термин 5-8-членное насыщенное карбоциклическое кольцо представляет собой циклопентильное, циклогексильное, циклогептильное или циклооктильное кольцо. Такие кольца могут содержать гетероатом, выбранный из O и S, образующий насыщенное гетероциклическое кольцо, такое как тетрагидропиранил, тетрагидрофуранил, тетрагидротиопиранил или тетрагидротиенил. Предпочтительные карбоциклические кольца представляют собой циклогексил и тетрагидропиранил.
В определении формулы I R2 может быть соединен с R7 с образованием 6-членного кольца, необязательно содержащего гетероатом, выбранный из O и S, который связан с положением 7 индольного кольца. В таких (индол-3-ил)замещенных 5-членных гетероциклах по данному изобретению индол-3-ильная группа является частью трициклической конденсированной кольцевой системы, т.е. системы 2,3-дигидропирроло[3,2,1-ij]хинолина (R7 и R2 представляют собой -CH2-CH2-), системы 2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазина (R7 и R2 представляют собой -O-CH2-) или системы 2,3-дигидропирроло[1,2,3-de]-1,4-бензотиазин (R7 и R2 представляют собой -S-CH2-).
В определении формулы I R3 вместе с R4 и N, с которым они связаны, образует 4-8-членное кольцо, необязательно содержащее дополнительный гетероатом, выбранный из O и S. Примерами таких колец являются пирролидин-1-ил, пиперидин-1-ил, азепин-1-ил, морфолин-4-ил и тиоморфолин-4-ил. Предпочтительными являются пирролидин-1-ил, пиперидин-1-ил и морфолин-4-ил.
В определении формулы I R3 вместе с R5 может образовывать 4-8-членное кольцо, необязательно содержащее дополнительный гетероатом, выбранный из O и S. Понятно, что N, с которым связан R3, и атом углерода, с которым связан R5, представляют собой часть 4-8-членного кольца. Примерами таких колец являются пирролидин-2-ил, пиперидин-2-ил, азепин-2-ил, морфолин-3-ил и тиоморфолин-3-ил.
В соответствии с формулой I существует предпочтение для производных (индол-3-ил)гетероциклических соединений, где R2 представляет собой H или где R2 соединен с R7 с образованием 6-членного кольца, необязательно содержащего гетероатом, выбранный из O и S, и где гетероатом соединен с положением 7 индольного кольца.
Кроме того, предпочтительными являются производные (индол-3-ил)гетероциклических соединений по настоящему изобретению, где R, R5, R5' и R6 представляют собой H.
Также предпочтительными являются производные (индол-3-ил)гетероциклических соединений в соответствии с формулой I, где R1 представляет собой циклогексил или тетрагидропиранил.
Особенно предпочтительными являются производные (индол-3-ил)гетероциклических соединений в соответствии с формулой I, где гетероцикл A представляет собой 1,2,4-оксадиазол (X1 представляет собой N, X2 представляет собой O, X3 представляет собой N), 1,2,4-тиадиазол (X1 представляет собой N, X2 представляет собой S, X3 представляет собой N) или тиазол (X1 представляет собой S, X2 представляет собой CR, X3 представляет собой N).
Особенно предпочтительными производными (индол-3-ил)гетероциклических соединений по настоящему изобретению являются:
7-хлор-3-(5-{[N-этил-N-(2-метоксиэтил)амино]метил}-[1,2,4]тиадиазол-3-ил)-1-(тетрагидропиран-4-ил)метил-1-H-индол;
7-хлор-3-{5-[(пирролидин-1-ил)метил]-[1,2,4]тиадиазол-3-ил}-1-(тетрагидропиран-4-ил)метил-1H-индол;
7-хлор-3-(5-{[N-этил-N-(2-гидроксиэтил)амино]метил}-[1,2,4]тиадиазол-3-ил)-1-(тетрагидропиран-4-ил)метил-1H-индол;
7-хлор-3-(4-{[N-(2-гидроксиэтил)-N-изопропиламино]метил}-[1,3]тиазол-2-ил)-1-(тетрагидропиран-4-ил)метил-1H-индол;
7-хлор-3-(4-{[N-этил-N-(2-гидроксиэтил)амино]метил}-[1,3]тиазол-2-ил)-1-(тетрагидропиран-4-ил)метил-1H-индол;
7-хлор-3-(4-{[N-(2-метоксиэтил)-N-метиламино]метил}-[1,3]тиазол-2-ил)-1-(тетрагидропиран-4-ил)метил-1H-индол;
7-хлор-3-{5-[(2,2-диметилпирролидин-1-ил)метил]-[1,2,4]оксадиазол-3-ил}-1-(тетрагидропиран-4-ил)метил-1H-индол,
или их фармацевтически приемлемые соли.
Как правило, производные (индол-3-ил)гетероциклических соединений по настоящему изобретению можно получать способами, известными в области органической химии. Например, производные (индол-3-ил)гетероциклических соединений формулы I можно получать из соединений формулы II, где Y представляет собой уходящую группу, такую как галоген или алкилсульфонатная группа, нуклеофильным замещением уходящей группы амином формулы NHR3R4. Соединения формулы II, где Y представляют собой алкилсульфонатную группу, можно получать из соединений формулы II, где Y представляет собой гидроксигруппу, реакцией с алкилсульфонилгалогенидом в присутствии основания, такого как триэтиламин. Производные (индол-3-ил)гетероциклических соединений формулы I, где R5' представляет собой водород, можно получать из соединений формулы III восстановительным аминированием, с использованием амина формулы NHR3R4 в присутствии восстановителя, такого как триацетоксиборгидрид натрия. В данной области хорошо известно, что соединения формулы II, где Y представляет собой гидроксигруппу и R5' представляет собой водород, можно взаимопревращать с соединениями формулы III окислением и восстановлением с использованием подходящих окислителей или восстановителей, как описано Burke D.S., Danheiser, R.L. в Handbook of Reagents for organic Synthesis: Oxidising and Reducing agents (Wiley: New York, 1999). Подобным образом, соединения формулы II, где Y представляет собой гидроксигруппу и R5 и R5' представляют собой водород, и соединения формулы III, где R5 представляет собой водород, можно получать из соединений формулы IV, где R8 представляет собой водород или (C1-4)алкил, восстановлением с использованием подходящих восстановителей. Соединения формулы II, где Y представляет собой гидроксигруппу и R5' представляет собой (C1-4)алкил, можно получать из соединений формулы III нуклеофильным присоединением с использованием (C1-4)алкилметаллического реагента, такого как алкильный реагент Гриньяра или алкиллитий.
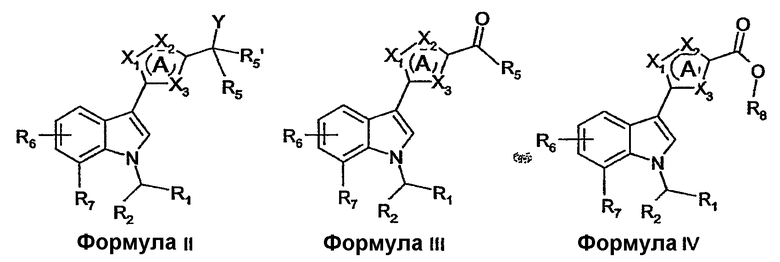
Соединения формулы I, формулы II, формулы III или формулы IV можно получать из соединений формулы V-XII, включительно, с применением хорошо известных в области конструирования гетероциклических колец способов. Такие способы описаны в основной ссылке Katritzky, A.R.: Comprehensive heterocyclic chemistry (First Edition, Pergamon Press, 1984, особенно см. Volume 4, Part 3, Five-membered rings with one oxygen, sulfur or nitrogen atom и Volume 6, Part 4B, Five-membered rings with two or more oxygen, sulfur or nitrogen atoms).
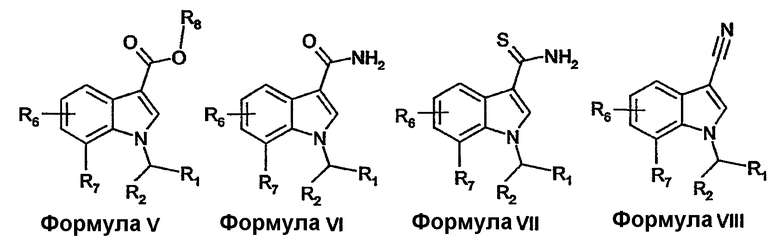
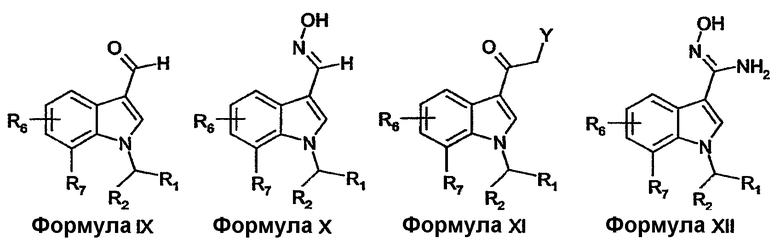
Соединения формулы V-XII, включительно, где R1, R2, R6 и R7 имеют определенные выше значения и R8 представляет собой H или (C1-4)алкил, можно получать описанными в литературе способами или известными специалисту в данной области модификациями описанных в литературе способов.
Например, соединения формулы VI можно получать из соединений формулы V или их активированных производных реакцией с аммиаком в приемлемом растворителе.
Соединения формулы VII можно получать из соединений формулы VI с использованием тионирующих реагентов, таких как пентасульфид фосфора или реагент Лавессона.
Альтернативно, соединения формулы VII можно получать из соединений формулы VIII реакцией с тиоацетамидом в растворителе, таком как диметилформамид.
Соединения формулы VIII можно получать из соединений формулы VI дегидратацией, например, с использованием трифторуксусного ангидрида в присутствии основания, такого как триэтиламин.
Соединения формулы X можно получать из соединений формулы IX реакцией с гидроксиламином в приемлемом растворителе.
Соединения формулы XI, где Y представляет собой NH2, можно получать из соединений формулы V или их активированных производных реакцией с цианид-анионом с образованием оксоацетонитрила с последующим восстановлением нитрила до первичного амина с использованием восстановителя, такого как газообразный водород, в присутствии катализатора, такого как палладий на активированном угле.
Соединения формулы XII можно получать из соединений формулы VIII реакцией с гидроксиламином в приемлемом растворителе.
Соединения формулы V и соединения формулы XI можно получать ацилированием соединений формулы XIII. Например, соединения формулы V, где R8 представляет собой водород, можно получать ацилированием соединений формулы XIII с использованием трифторуксусного ангидрида в растворителе, таком как диметилформамид, с последующим гидролизом с использованием водного гидроксида натрия при повышенной температуре. Соединения формулы XI, где Y представляет собой хлор, можно получать ацилированием соединений формулы XIII с использованием хлорацетилхлорида в присутствии основания, такого как пиридин.
Соединения формулы IX можно получать из соединений формулы XIII формилированием, например, с применением реакции Вильсмейера (для обзора см. Jutz, Adv. Org. Chem. 9, pt. 1, 225-342, 1976).
Альтернативно, соединения формулы V можно получать из соединений формулы XIV с применением способов, описанных Wijngaarden et al. (J. Med. Chem. 36, 3693-3699, 1993) или Hwu et al. (J. Org. Chem. 59, 1577-1582, 1994), или с применением модификаций этих способов.
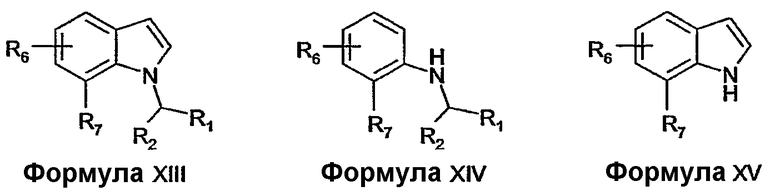
Соединения формулы XIII можно получать описанными в литературе способами или известными специалисту в данной области модификациями описанных в литературе способов. Например, соединения формулы XIII можно получать алкилированием соединений формулы XV посредством обработки основанием, таким как гидрид натрия, с последующей реакцией с алкилирующим агентом R1R2CHY, где Y представляет собой уходящую группу, такую как галоген или алкилсульфонатная группа. Соединения формулы XV можно получить из коммерческих источников, можно получать описанными в литературе способами или известными специалисту в данной области модификациями описанных в литературе способов.
Альтернативно, соединения формулы XIII можно получать из соединений формулы XIV с применением синтеза индолов по Фишеру или его модификаций (Chem. Rev. 69, 227-250, 1969).
Соединения формулы XIV можно получать описанными в литературе способами или известными специалисту в данной области модификациями описанных в литературе способов. Например, соединения формулы XIV, где R2 соединен с R7 с образованием 6-членного карбоциклического кольца, можно получать из соединений формулы XVI восстановлением с использованием восстановителя, такого как боргидрид натрия, в присутствии катализатора, такого как хлорид никеля (II). Соединения формулы XVI можно получать, например, реакцией конденсации, такой как реакция 2-хлорхинолина с реактивом Гриньяра в присутствии катализатора никеля (II).
Соединения формулы XIV, где R2 соединен с R7 с образованием 6-членного кольца, содержащего кислород или серу, можно получить реакцией с соединением формулы XVII, где Z представляет собой OH или SH, с соединением формулы XVIII, где Y представляет собой уходящую группу, с образованием эфира или тиоэфира с последующим восстановлением нитрогруппы до амина и восстановительной циклизацией. Восстановление и циклизацию можно осуществлять, например, с использованием газообразного водорода в присутствии катализатора, такого как палладий на активированном угле.
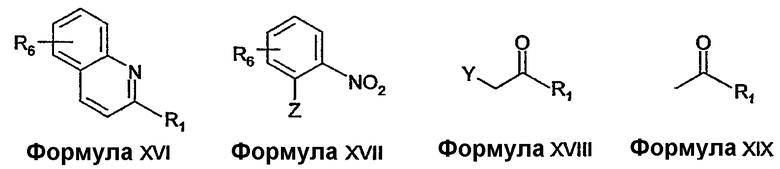
Соединения формулы XVII и соединения формулы XVIII можно получить из коммерческих источников, можно получить описанными в литературе способами или известными специалисту в данной области модификациями описанных в литературе способов. Например, соединения формулы XVIII, где Y представляет собой бром, можно получать из соединений формулы XIX с использованием бромирующего вещества, такого как бром, в растворителе, таком как метанол.
Альтернативно, соединения формулы I, формулы II, формулы III или формулы IV можно получать из соединений формулы XX с использованием переходного металла, катализирующего реакции сочетания, как описано в основной ссылке Hegedus, L.S. Transition Metals in the Synthesis of Complex Organic Molecules (Second Edition, University Science: Sausalito 1999).
Например, соединения формулы III можно получить реакцией соединений формулы XX, где Y1 представляет собой галоген, с соединениями формулы XXI, где Y2 представляет собой бороновую кислоту или сложный эфир бороновой кислоты, с применением реакции Сузуки (Chem. Rev. 95, 2457-2483, 1995) или ее модификации.
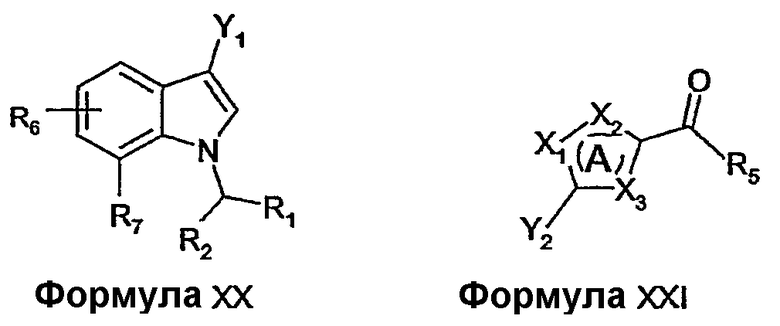
Соединения формулы XX и соединения формулы XXI можно получать из коммерческих источников, можно получить описанными в литературе способами или известными специалисту в данной области модификациями описанных в литературе способов. Например, соединения формулы XX, где Y1 представляет собой бром, можно получить бромированием соединений формулы XIII с использованием брома в растворителе, таком как диметилформамид.
Специалисту в данной области понятно, что азот индола при описанных выше превращениях можно временно защитить с использованием защитной группы, такой как арилсульфонильная группа, с последующим снятием защиты и алкилированием на поздних стадиях синтеза. Кроме того, понятно, что такие защитные группы можно использовать для изменения стабильности промежуточных продуктов и реакционной способности индольного кольца в отношении электрофилов. Подходящие защитные группы описаны Kocienski, P.J.: в Protecting Groups, Thieme, Stuttgart; New York, 1994.
Также специалисту в данной области понятно, что различные производные (индол-3-ил)гетероциклических соединений формулы I можно получать посредством подходящих реакций конверсии функциональных групп, соответствующих определенным заместителям R3-R7. Например, соединения формулы I, где R3 или R4 представляют собой линейные, разветвленные или циклические C1-C6алкильные группы, необязательно замещенные гидроксилом, (C1-4)алкилокси, (C1-4)алкилтио,
(C1-4)алкилсульфонилом или циано, можно получить реакцией соединения формулы I, где R3 или R4 представляет собой водород, с C1-C6алкилгалогенидом или с функционализированным C1-C6алкилгалогенидом в присутствии основания, такого как карбонат калия.
Производные (индол-3-ил)гетероциклических соединений формулы I и их соли могут содержать по меньшей мере один хиральный центр и, следовательно, находиться в виде стереоизомеров, включая энантиомеры и диастереоизомеры. Настоящее изобретение в свой объем включает указанные выше стереоизомеры и каждый отдельный из R и S энантиомеров соединений формулы I и их соли, по существу чистые, т.е. ассоциированные менее чем с 5%, предпочтительно менее чем с 2%, конкретно, менее чем с 1% другого энантиомера, и смеси таких энантиомеров в любых пропорциях, включая рацемические смеси, содержащие по существу равные количества двух энантиомеров.
Способы асимметричного синтеза или хирального разделения, которыми получают стереоизомеры, хорошо известны в данной области, например синтез с хиральной индукцией или исходя из коммерчески доступных хиральных субстратов, или разделение стереоизомеров, например, с использованием хроматографии на хиральной среде, или кристаллизация с хиральным противоионом.
Фармацевтически приемлемые соли можно получать обработкой свободного основания соединения формулы I неорганической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота и серная кислота, или органической кислотой, например, такой как аскорбиновая кислота, лимонная кислота, винная кислота, молочная кислота, малеиновая кислота, малоновая кислота, фумаровая кислота, гликолевая кислота, янтарная кислота, пропионовая кислота, уксусная кислота и метансульфоновая кислота.
Соединения по настоящему изобретению могут находиться как в несольватированной форме, так и в сольватированной форме с фармацевтически приемлемым растворителем, таким как вода, этанол и т.п. В основном, для целей настоящего изобретения сольватированные формы рассматриваются как эквивалентные несольватированным.
Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим производное (индол-3-ил)гетероциклического соединения общей формулы I или его фармацевтически приемлемую соль, в смеси с фармацевтически приемлемыми добавками и, необязательно, другими терапевтическими средствами. Термин "приемлемый” означает сочетаемый с другими ингредиентами композиции и безвредный для его потребителей. Например, композиции включают композиции, приемлемые для перорального, сублингвального, подкожного, внутривенного, эпидурального, интратекального, внутримышечного, трансдермального, легочного, местного или ректального введения и т.п., все в единичных дозированных формах для введения. Предпочтительным путем введения является внутривенный.
Для перорального введения активный ингредиент может находиться в виде отдельных единиц, таких как таблетки, капсулы, порошки, гранулированные формы, растворы, суспензии и т.п. Для парентерального введения фармацевтическая композиция по настоящему изобретению может находиться в контейнере для единичной дозы или многократных доз, например жидкость для инъекций в предопределенных количествах, например в герметичных пузырьках и ампулах, а также может храниться в высушенном вымораживанием состоянии (лиофилизированном), при котором необходимо только добавление стерильного жидкого носителя, например воды, перед применением.
Смешанное с такими фармацевтически приемлемыми добавками, например, как описано в стандартной ссылке Gennaro, A.R. et al., Remington: The Science and Practice of Pharmacy (20th Edition, Lippincott Williams & Wilkins, 2000, особенно см. Part 5: Pharmaceutical Manufacturing), активное средство можно спрессовать в твердые единичные дозы, такие как пилюли, таблетки, или обрабатывать с получением капсул, суппозиториев или пластырей. Посредством фармацевтически приемлемых жидкостей активное средство можно применять как жидкую композицию, например, в качестве инъекционного препарата в виде раствора, суспензии, эмульсии или спрея, например нозального спрея.
Для получения твердых дозированных единиц предусмотрено применение общепринятых добавок, таких как наполнители, красители, полимерные связующие вещества и т.п. В основном можно применять любые фармацевтически приемлемые добавки, которые не препятствуют действию активных соединений. Приемлемые носители, с которыми активное вещество по настоящему изобретению можно вводить в виде твердых композиций, включают производные лактозы, крахмала, целлюлозы и т.п., или их смеси, применяемые в подходящих количествах. Для парентерального введения можно применять водные суспензии, изотонические солевые растворы и стерильные инъецируемые растворы, содержащие фармацевтически приемлемые диспергирующие средства и/или увлажняющие средства, такие как пропиленгликоль или бутиленгликоль.
Кроме того, данное изобретение относится к описанной выше фармацевтической композиции в комбинации с пригодными для указанной композиции упаковочными материалами, где указанные упаковочные материалы включают инструкцию по применению композиции для указанного выше применения.
Обнаружено, что производные (индол-3-ил)гетероциклических соединений по настоящему изобретению являются агонистами рецептора CB1, как установлено в анализе репортера человеческих рецепторов CB1 с использованием клеток CHO. Способы определения связывания рецепторов, а также биологической активности модуляторов каннабиноидных рецепторов in vitro хорошо известны в данной области. В общем, экспрессированный рецептор приводят в контакт с соединением, которое должно быть испытано, и измеряют связывание или стимуляцию или ингибирование функционального ответа.
Для измерения функционального ответа выделенную ДНК, кодирующую ген рецептора CB1, предпочтительно рецептора человека, экспрессируют в подходящих клетках хозяина. Такие клетки могут представлять собой клетки яичника китайского хомяка, но также пригодны другие клетки. Предпочтительно, клетки происходят от млекопитающих.
Способы конструирования экспрессирующих рекомбинантный CB1 клеточных линий хорошо известны в данной области (Sambrook et al., Molecular Cloning: a Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, последнее издание). Экспрессии рецептора достигают посредством экспрессии кодирующей желаемый белок ДНК. Все способы лигирования дополнительных последовательностей и конструирования подходящих экспрессирующих систем к настоящему времени хорошо известны в данной области. Часть или всю ДНК, кодирующую желаемый белок, можно конструировать синтетически с применением стандартных твердофазных способов, предпочтительно с введением сайтов рестрикции для облегчения лигирования. Для кодирующих последовательностей ДНК можно обеспечить приемлемые контрольные элементы для транскрипции и трансляции введенных кодирующих последовательностей. Как хорошо известно, на сегодняшний день доступны системы экспрессии, которые совместимы с широким разнообразием хозяев, включая прокариотических хозяев, таких как бактерии, и эукариотических хозяев, таких как дрожжи, клетки растений, клетки насекомых, клетки млекопитающих, клетки птиц и т.п.
Затем экспрессирующие рецептор клетки приводят в контакт с тестируемым соединением для исследования связывания или стимуляции или ингибирования функционального ответа.
Альтернативно, для измерения связывания соединения можно применять изолированные клеточные мембраны, содержащие экспрессируемый рецептор CB1 (или CB2).
Для измерения связывания можно применять радиоактивно или флуоресцентно меченые соединения. Наиболее широко применяемым радиоактивным каннабиноидным зондом является [3H]CP55940, который обладает приблизительно равной аффинностью для сайтов связывания CB1 и CB2.
Другое исследование включает скрининг соединений-агонистов каннабиноидных CB1 посредством определения ответа вторичных мессенджеров, например, таких как измерение опосредованных рецептором изменений в путях цАМФ или MAP-киназы. Таким образом, такой способ включает экспрессию рецептора CB1 на клеточной поверхности клетки-хозяина и воздействие на клетку тестируемым соединением. Затем измеряют ответ вторичных мессенджеров. Уровень вторичных мессенджеров будет сниженным или повышенным в зависимости от действия тестируемого соединения при связывании с рецептором.
Кроме прямого измерения, например, уровней цАМФ в подвергаемой воздействию клетке, можно использовать клетки, которые, кроме трансфекции кодирующей рецептор ДНК, трансфицированы второй ДНК, кодирующей репортерный ген, экспрессия которого коррелирует с активацией рецептора. В основном экспрессию репортерного гена можно контролировать любым отвечающим элементом, реагирующим на измененные уровни вторичного мессенджера. Приемлемые репортерные гены представляет собой, например, LacZ, щелочную фосфатазу, люциферазу светляка и зеленый флуоресцентный белок. Принципы таких трансактивационных анализов хорошо известны в данной области, например, в Stratowa, Ch, Himmler, A. and Czernilofsky, A. P., Curr. Opin.Biotechnol. 6, 574 (1995). Для выбора активных соединений-агонистов для рецептора CB1 значение EC50 должно составлять <10-5М, предпочтительно <10-7М.
Соединения можно применять в качестве болеутоляющих средств для лечения боли, например, такой как периоперационная боль, хроническая боль, невропатическая боль, боль при раке и боль и спастичность, ассоциированные с рассеянным склерозом.
Также каннабиноидные агонисты по данному изобретению могут быть потенциально пригодными при лечении других нарушений, включая рассеянный склероз, спастичность, воспаление, глаукому, тошноту и рвоту, потерю аппетита, нарушения сна, нарушения дыхания, аллергию, эпилепсию, мигрень, сердечно-сосудистые нарушения, нейродегенеративные нарушения, тревога, травматическое повреждение мозга и удар.
Соединения также можно применять в комбинации с другими лекарственными средствами, например обезболивающими лекарственными средствами, такими как опиоды и нестероидные противовоспалительные лекарственные средства (NSAID), включающие селективные ингибиторы COX-2.
Соединения по данному изобретению можно вводить людям в достаточном количестве и в течение достаточного периода времени для облегчения симптомов. В качестве иллюстрации уровни дозирования для людей могут находиться в диапазоне 0,001-50 мг на 1 кг массы тела, предпочтительно в дозе 0,01-20 мг на кг массы тела.
Изобретение проиллюстрировано следующими примерами.
Общие способы
Если не указано иное, микроволновые реакции проводили с использованием Emrys™ Optimizer (Personal Chemistry). Колоночную флэш-хроматографию проводили на силикагеле. Полупрепаративную высокоэффективную жидкостную хроматографию (полупрепаративная ВЭЖХ) проводили с применением указанных ниже способов:
Способ (i): Agilent CombiHT (SB-C18, 5 мкм) 12 мм ID×100 мм; 5-95% ацетонитрил-вода при 9-минутном градиенте; 25 мл/мин; 0,1% трифторацетатного кислотного буфера; детекция посредством УФ при 254 нм.
Способ (ii): Waters Xterra (RP18, 5 мкм) 30 мм×100 мм; 10-100% ацетонитрил-вода при 25-минутном градиенте; 30 мл/мин; 0,1% трифторацетатного кислотного буфера; детекция посредством УФ при 254 нм.
Константы взаимодействия 1H ЯМР приведены в герцах.
Пример 1
1-(Циклогексил)метил-3-{5-[(диметиламино)метил]-[1,2,4]оксадиазол-3-ил-7-метокси-1H-индол, гидрохлоридная соль
Раствор 7-метоксииндола (45,0 г, 306 ммоль) в диметилформамиде (360 мл) охлаждали до 5°C в атмосфере азота в течение 20 минут и добавляли трифторуксусный ангидрид (60,5 мл, 433 ммоль), поддерживая температуру ниже 10°C. Смесь перемешивали при 5-10°C в течение 2 часов, затем выливали в воду (1600 мл). Полученную суспензию перемешивали в течение 15 минут и осадок 7-метокси-3-[(трифторметил)карбонил]-1H-индола отфильтровывали, промывая водой до нейтральности.
Влажное твердое вещество суспендировали в 4М водном гидроксиде натрия (1700 мл) и нагревали до температуры кипения с обратным холодильником при перемешивании в течение 2 часов. Смесь охлаждали и промывали диэтиловым эфиром (2×400 мл). Затем водную фазу подкисляли до pH 1, используя 5М хлористоводородную кислоту, и полученный чистый осадок отфильтровывали, промывали водой до нейтральности и сушили с получением 7-метокси-1H-индол-3-карбоновой кислоты в виде твердого розового вещества (42,7 г).
К раствору 7-метокси-1H-индол-3-карбоновой кислоты (42,7 г, 224 ммоль) в диметилформамиде (1250 мл) при 10°C в атмосфере азота порциями в течение 20 минут, поддерживая температуру ниже 15°C, добавляли гидрид натрия (60% дисперсия в минеральном масле, 23,0 г, 575 ммоль). Охлаждающую баню удаляли и суспензию перемешивали в течение 90 минут. Добавляли циклогексилметилбромид (64,7 мл, 464 ммоль). Смесь нагревали при 60°C при перемешивании в течение 3 часов. Смесь охлаждали до 10°C и выливали в воду (3600 мл). Эмульсию промывали диэтиловым эфиром (3×500 мл). Водную фазу подкисляли до pH 1, используя 5М хлористоводородную кислоту, и осадок отфильтровывали, промывали водой до нейтральности и сушили с получением 1-(циклогексил)метил-7-метокси-1H-индол-3-карбоновой кислоты (55 г) в виде белого твердого вещества.
К смеси 1-(циклогексил)метил-7-метокси-1H-индол-3-карбоновой кислоты (7,0 г, 24,4 ммоль) и дихлорметана (150 мл) при охлаждении ледяной водой по каплям добавляли оксалилхлорид (12,4 г, 97,4 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 18 часов. Дихлорметан и избыток оксалилхлорида удаляли выпариванием и полученный осадок смешивали с дихлорметаном (150 мл). Газообразный аммиак барботировали в полученную смесь в течение 30 мин при охлаждении на бане лед-вода. Реакционную смесь концентрировали в вакууме, затем полученное твердое вещество последовательно растирали с 0,5M хлористоводородной кислотой, 5% водным карбонатом натрия и водой и сушили при пониженном давлении с получением амида 1-(циклогексил)метил-7-метокси-1H-индол-3-карбоновой кислоты (5,1 г) в виде твердого коричневого вещества.
К смеси амида 1-(циклогексил)метил-7-метокси-1H-индол-3-карбоновой кислоты (4,1 г, 14,3 ммоль), триэтиламина (11,6 г, 115 ммоль) и 1,4-диоксана (250 мл) при охлаждении в ледяной воде по каплям добавляли трифторуксусный ангидрид (12,0 г, 57,1 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 12 часов. Добавляли воду (30 мл) и полученную смесь концентрировали в вакууме. К полученному осадку добавляли воду (300 мл) и смесь экстрагировали дихлорметаном (4×300 мл). Органические слои объединяли, промывали 5% водным гидрокарбонатом натрия и насыщенным раствором соли, сушили над сульфатом магния и концентрировали в вакууме. Полученный осадок очищали колоночной хроматографией, элюируя 10% (об./об.) этилацетатом в н-гептане с получением 1-(циклогексил)метил-7-метокси-1H-индол-3-карбонитрила в виде кристаллического твердого вещества (2,48 г).
К смеси 1-(циклогексил)метил-7-метокси-1H-индол-3-карбонитрила (2,48 г, 9,24 ммоль), триэтиламина (1,41 г, 13,9 ммоль) и этанола (50 мл) добавляли гидрохлорид гидроксиламина (966 мг, 13,9 ммоль), затем полученную смесь перемешивали при кипячении с обратным холодильником в течение 20 часов. После охлаждения до комнатной температуры реакционную смесь концентрировали в вакууме. Полученный осадок смешивали с водой (150 мл), доводили до pH 10 добавлением водного гидроксида натрия и экстрагировали дихлорметаном (4×100 мл). Органические слои объединяли, промывали насыщенным раствором соли, сушили над сульфатом магния и концентрировали в вакууме. Полученный осадок очищали колоночной хроматографией, элюируя 10% (об./об.) ацетоном в дихлорметане, с получением 1-(циклогексил)метил-N-гидрокси-7-метокси-1H-индол-3-карбоксамидина (940 мг).
К суспензии 1-(циклогексил)метил-N-гидрокси-7-метокси-1H-индол-3-карбоксамидина (250 мг, 0,829 ммоль) в тетрагидрофуране (6 мл) в атмосфере азота добавляли молекулярные сита (4 Å, в виде порошка, 200 мг) и смесь перемешивали при комнатной температуре в течение 30 мин. Добавляли гидрид натрия (60% суспензия в масле, 36 мг, 0,900 ммоль), затем полученную смесь перемешивали при 60°C в течение 20 мин. Реакционную смесь охлаждали до комнатной температуры и к смеси добавляли N,N-диметилглицинметиловый эфир (194 мг, 1,66 ммоль). Полученную смесь перемешивали при температуре кипения с обратным холодильником в течение 2 часов и затем концентрировали в вакууме. Полученный осадок смешивали с дихлорметаном (200 мл), промывали 5% водным карбонатом натрия, сушили над сульфатом магния и концентрировали в вакууме. Полученное масло очищали колоночной флэш-хроматографией, элюируя 0,6% (об./об.) метанолом в дихлорметане, с получением масла. Полученное масло растворяли в изопропаноле (3 мл), к раствору добавляли хлористый водород (1М раствор в диэтиловом эфире; 3 мл). Полученную смесь концентрировали в вакууме с получением указанного в заголовке вещества (1:1 гидрохлоридная соль) (66 мг).
1H ЯМР (400 МГц, CD3OD) δ 0,96-1,30 (5H, м), 1,52-1,94 (6H, м), 3,13 (6H, с), 3,97 (3H, с), 4,30 (2H, д, J=6,8), 4,83 (2H, с), 6,81 (1H, д, J=8,0), 7,13 (1H, дд, J=8,0, 8,0), 7,71 (1H, д, J=8,0), 7,85 (1H, с). EsIMS: m/z 369,2 [M+H]+.
Пример 2
Далее, способ примера 1 применяли для получения следующих соединений, используя альтернативные синтетические или коммерчески доступные сложные эфиры аминокислот вместо N,N-диметилглицинметилового эфира.
Способы синтеза промежуточных сложных эфиров аминокислот
Способ А
К смеси пирролидина (171 мг, 2,40 ммоль) и карбоната натрия (254 мг, 2,40 ммоль) в тетрагидрофуране (7 мл) добавляли бензилбромацетат (500 мг, 2,18 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, затем концентрировали в вакууме. Осадок смешивали с водой (200 мл) и экстрагировали дихлорметаном (3×100 мл). Органические слои объединяли, сушили над сульфатом натрия и концентрировали в вакууме. Очистка флэш-хроматографией при элюировании 0-10% (об./об.) метанолом в дихлорметане приводила к получению бензилового эфира пирролидин-1-ил-уксусной кислоты (230 мг, 1,05 ммоль).
Способ B
К смеси (S)-2-метоксиметилпирролидина (268 мкл, 2,17 ммоль), карбоната калия (319 мг, 2,31 ммоль) и иодида натрия (315 мг, 2,10 ммоль) в ацетонитриле (3 мл) добавляли метилбромацетат (199 мкл, 2,10 ммоль). Смесь подвергали микроволновому облучению в течение 5 мин при 160°C, затем распределяли между дихлорметаном и водой. Водный слой экстрагировали дихлорметаном и смешанные органические слои промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали в вакууме. Очистка флэш-хроматографией при элюировании 0-10% (об./об.) метанолом в дихлорметане приводила к получению метилового эфира (S)-(2-метоксиметилпирролидин-1-ил)уксусной кислоты (133 мг, 0,71 ммоль).
Способ C
К раствору серной кислоты (3,5 мл, 65,3 ммоль) в метаноле (45 мл) добавляли D-пролин (10,0 г, 86,9 ммоль). Смесь кипятили с обратным холодильником при перемешивании в течение 18 часов. Затем раствор охлаждали до 0°C и нейтрализовали добавлением водного карбоната калия (2,5М; 10 мл). Добавляли формальдегид (37% раствор в воде; 11 мл, 136 ммоль) и смесь перемешивали при 0°C в течение 15 минут. Добавляли боргидрид натрия (1,6 г, 42,3 ммоль) при 0°C и смесь перемешивали при комнатной температуре в течение 3 часов. Осадок отфильтровывали и фильтрат распределяли между дихлорметаном и водой. Отделенный водный слой доводили до pH 10, используя твердый карбонат натрия, и экстрагировали дихлорметаном. Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме с получением неочищенного метилового эфира (R)-1-метилпирролидин-2-карбоновой кислоты (13,13 г).
Порцию полученного неочищенного продукта (5,0 г) очищали колоночной флэш-хроматографией, элюируя 0-2% (об./об.) метанолом в дихлорметане с получением метилового эфира (R)-1-метилпирролидин-2-карбоновой кислоты (1,30 г).
2A : 1-(Циклогексил)метил-7-метокси-3-{5-[(пирролидин-1-ил)метил]-[1,2,4]оксадиазол-3-ил}-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали, следуя способу примера 1, с использованием бензилового эфира пирролидин-1-илуксусной кислоты, полученного способом A.
1H ЯМР (400 МГц, CD3OD) δ 0,98-1,31 (5H, м), 1,54-1,94 (6H, м), 2,10-2,24 (4H, м), 3,46-3,74 (4H, м), 3,97 (3H, с), 4,30 (2H, д, J=7,2), 4,86 (2H, с), 6,81 (1H, д, J=8,0), 7,14 (1H, дд, J=8,0, 8,0), 7,70 (1H, д, J=8,0), 7,84 (1H, с). EsIMS: m/z 395,2 [M+H]+.
2B: 1-(Циклогексил)метил-3-{5-[(N-этил-N-изопропиламино)метил]-[1,2,4]оксадиазол-3-ил}-7-метокси-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием метилового эфира (N-этил-N-изопропиламино)уксусной кислоты, который получали способом А с использованием метилбромацетата и N-этилизопропиламина. EsIMS: m/z 411,1 [M+H]+.
2C: 1-(Циклогексил)метил-7-метокси-3-(5-{[бис-(2-метоксиэтил)амино]метил}-[1,2,4]оксадиазол-3-ил}-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием метилового эфира [бис-(2-метоксиэтил)амино]уксусной кислоты, который получали способом А с использованием метилбромацетата и бис-(2-метоксиэтил)амина. EsIMS: m/z 457,5 [M+H]+.
2D: 1-(Циклогексил)метил-3-{5-[1-(диметиламино)этил]-[1,2,4]оксадиазол-3-ил}-7-метил-1H-индол, гидрохлоридная соль.
Указанное в заголовке соединение получали с использованием метилового эфира 2-диметиламинопропионовой кислоты, который получали способом А с использованием метил-2-бромпропионата и диметиламина. EsIMS: m/z 383,0 [M+H]+.
2E: (S)-1-(Циклогексил)метил-7-метокси-3-{5-[(2-метоксиметилпирролидин-1-ил)метил]-[1,2,4]оксадиазол-3-ил-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием метилового эфира (S)-(2-метоксиметилпирролидин-1-ил)уксусной кислоты, который получали способом B. EsIMS: m/z 439,3 [M+H]+.
2F : (R)-1-(Циклогексил)метил-7-метокси-3-{5-[(2-метоксиметилпирролидин-1-ил)метил]-[1,2,4]оксадиазол-3-ил}-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием метилового эфира (R)-(2-метоксиметилпирролидин-1-ил)уксусной кислоты, который получали способом B с использованием (R)-2-метоксиметилпирролидина.
EsIMS: m/z 439,1 [M+H]+; [α]D 22 +21,6° (c=0,8 мг/мл в хлороформе).
2G : (R)-1-(Циклогексил)метил-7-метокси-3-[5-(1-метилпирролидин-2-ил)-[1,2,4]оксадиазол-3-ил]-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием метилового эфира (R)-1-метилпирролидин-2-карбоновой кислоты, который получали способом C.
EsIMS: m/z 395,0 [M+H]+ [α]D 22 +50,1° (c=1,70 мг/мл в хлороформе).
2H : (S)-1-(Циклогексил)метил-7-метокси-3-[5-(1-метилпирролидин-2-ил)-[1,2,4]оксадиазол-3-ил]-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием метилового эфира (S)-1-метилпирролидин-2-карбоновой кислоты, который получали способом C, с использованием L-пролина вместо D-пролина. EsIMS: m/z 395,0 [M+H]+ [α]D 22 -51,7 (c=1,35 мг/мл в хлороформе).
2I: 1-(Циклогексил)метил-7-метокси-3-[5-(1-метилпиперидин-2-ил)-[1,2,4]оксадиазол-3-ил]-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали, следуя способу примера 1, с использованием этил-1-метилпипеколината вместо метилового эфира N,N-диметилглицина.
EsIMS: m/z 409,3 [M+H]+.
Пример 3
1-(Циклогексил)метил-3-[(5-аминометил)-[1,2,4]оксадиазол-3-ил]-7-метокси-1H-индол, гидрохлоридная соль
К суспензии 1-(циклогексил)метил-N-гидрокси-7-метокси-1H-индол-3-карбоксамидина (500 мг, 1,66 ммоль) в тетрагидрофуране (10 мл) добавляли молекулярные сита (4 Å, в виде порошка, 300 мг) и полученную смесь перемешивали при комнатной температуре в течение 30 мин. Добавляли гидрид натрия (60% суспензия в масле, 100 мг, 2,55 ммоль) и полученную смесь перемешивали при 65°C в течение 20 мин. Реакционную смесь охлаждали до комнатной температуры и к смеси добавляли N-гидроксисукцинимидный эфир N-Boc-глицина (871 мг, 3,32 ммоль). Полученную смесь нагревали до температуры кипения с обратным холодильником при перемешивании в течение 2 часов, затем охлаждали до комнатной температуры. Добавляли водный гидроксид натрия (4М; 5 мл) и полученную смесь перемешивали в течение 14 часов. Реакционную смесь концентрировали в вакууме, затем полученный осадок смешивали с водой (200 мл). Полученную смесь экстрагировали дихлорметаном (4×200 мл). Органические слои объединяли, промывали насыщенным раствором соли, сушили над сульфатом магния и концентрировали в вакууме. Полученное масло очищали колоночной хроматографией, элюируя 0,4% (об./об.) метанолом в дихлорметане, с получением трет-бутилового эфира ({3-[1-(циклогексил)метил-7-метокси-1H-индол-3-ил]-[1,2,4]оксадиазол-5-ил}метил)карбаминовой кислоты (125 мг).
Смесь трет-бутилового эфира ({3-[1-(циклогексил)метил-7-метокси-1H-индол-3-ил]-[1,2,4]оксадиазол-5-ил}метил)карбаминовой кислоты (110 мг, 0,25 ммоль) и трифторуксусной кислоты (4 мл) перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь осторожно выливали в 5% водный карбонат натрия (200 мл) и полученную смесь экстрагировали дихлорметаном (4×200 мл). Органические слои объединяли, затем промывали насыщенным раствором соли, сушили над сульфатом магния и концентрировали в вакууме. Полученное масло очищали колоночной хроматографией, элюируя 1,5% (об./об.) метанолом в дихлорметане, с получением свободного основания указанного в заголовке соединения в виде желтого масла. Полученное масло растворяли в диэтиловом эфире и затем к раствору добавляли хлористый водород (1М раствор в диэтиловом эфире; 3 мл). Полученную смесь концентрировали в вакууме с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1 (71 мг).
1H ЯМР (400 МГц, CD3OD) δ 0,98-1,29 (5H, м), 1,52-1,78 (5H, м), 1,79-1,94 (1H, м), 3,98 (3H, с), 4,31 (2H, д, J=7,2), 4,55 (2H, с), 6,81 (1H, д, J=7,6), 7,14 (1H, дд, J=7,6, 8,0), 7,72 (1H, д, J=8,0), 7,83 (1H, с). EsIMS: m/z 341,1 [M+H]+.
Пример 4
1-(Циклогексил)метил-3-{5-[(диметиламино)метил]-[1,2,4]оксадиазол-3-ил}-7-фтор-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали, следуя способу примера 1, с использованием 7-фториндола вместо 7-метоксииндола. EsIMS: m/z 357,3 [M+H]+, 247,4.
Пример 5
7-Хлор-1-(циклогексил)метил-3-{5-[(диметиламино)метил]-[1,2,4]оксадиазол-3-ил}-1H-индол, гидрохлоридная соль.
Указанное в заголовке соединение получали, следуя способу примера 1, с использованием 7-хлориндола вместо 7-метоксииндола EsIMS: m/z 375,1, 373,1 [M+H]+.
Пример 6
1-(Циклогексил)метил-3-(5-{[N-изопропил-N-(2-метоксиэтил)амино]метил}-[1,2,4]-триазол-3-ил)-7-метокси-1H-индол, гидрохлоридная соль
Газообразный хлористый водород барботировали в течение 30 минут через охлажденный (0°C) раствор 1-(циклогексил)метил-7-метокси-1H-индол-3-карбонитрила (полученного, как описано в примере 1; 3,15 г, 11,0 ммоль) в метаноле (200 мл). Перед концентрированием полученной смеси давали отстояться в течение 72 часов на две трети в вакууме. Кристаллизацию продукта проводили добавлением диэтилового эфира, твердый продукт собирали фильтрованием с получением метилового эфира 1-(циклогексил)метил-7-метокси-1H-индол-3-карбоксимидной кислоты в виде гидрохлоридной соли (3,82 г).
Метиловый эфир 1-(циклогексил)метил-7-метокси-1H-индол-3-карбоксимидной кислоты, гидрохлоридная соль (0,10 г, 0,297 ммоль), гидрат гидразина (0,289 мл, 5,94 ммоль), хлорид алюминия (39,6 мг, 0,297 ммоль) и толуол (18 мл) смешивали и смесь подвергали микроволновому облучению в течение 60 мин при 120°C. Полученную смесь концентрировали в вакууме, повторно растворяли в толуоле и концентрировали в вакууме еще два раза. Полученный осадок суспендировали в смеси толуола/ацетонитрил (12/1) (19,5 мл) и добавляли хлорацетилхлорид (0,118 мл, 1,49 ммоль), затем смесь подвергали микроволновому облучению в течение 12 мин при 120°C. Полученную смесь концентрировали в вакууме и повторно растворяли в ацетонитриле (3 мл). Смесь отстаивали в течение 72 часов, добавляли N-(2-метоксиэтил)изопропиламин (0,068 мл, 0,446 ммоль), карбонат калия (45,2 мг, 0,327 ммоль) и иодид натрия (44 мг, 0,297 ммоль, затем подвергали микроволновому облучению в течение 5 мин при 160°C и затем концентрировали в вакууме. Полученный осадок очищали колоночной хроматографией, элюируя 2,5%-5% (об./об.) метанолом в дихлорметане, с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1 (46 мг).
1H ЯМР (400 МГц, CD3OD): 0,95-1,12 (2H, м), 1,13-1,24 (3H, м), 1,50 (6H, с(ушир.)), 1,55-1,73 (5H, м), 1,79-1,93 (1H, м), 3,34-3,50 (5H, м), 3,70-3,99 (6H, м), 4,24 (2H, д, J=6,4), 4,67 (2H, c(ушир.)), 6,70 (1H, д, J=7,7), 7,16 (1H, т, J=7,7), 7,95 (1H, д, J=7,7), 8,02 (1H, с(ушир.)); EsIMS: m/z 440,3 [M+H]+.
Пример 7
1-(Циклогексил)метил-3-{5-[(диэтиламино)метил]-[1,2,4]тиадиазол-3-ил}-7-метокси-1H-индол, гидрохлоридная соль
К суспензии амида 1-(циклогексил)метил-7-метокси-1H-индол-3-карбоновой кислоты (полученного из 7-метоксииндола, как описано в примере 1; 4,0 г, 14 ммоль) в тетрагидрофуране (120 мл) добавляли хлоркарбонилсульфенилхлорид (2,4 мл, 28,4 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 15 минут и давали остыть. Раствор и избыточный реагент затем удаляли в вакууме с получением 5-(1-циклогексилметил-7-метокси-1H-индол)-[1,3,4]оксатиазол-2-она (5,2 г, 14,4 ммоль) в виде твердого розового вещества.
К суспензии 5-(1-циклогексилметил-7-метокси-1H-индол)-[1,3,4]оксатиазол-2-она (1,0 г, 2,77 ммоль) в м-ксилоле (15 мл) добавляли этилцианоформиат (2,74 мл, 27,7 ммоль) и реакционную смесь подвергали микроволновому облучению при 160°C в течение 10 мин с использованием Emrys™ Optimizer EXP. Реакцию повторяли три раза при тех же количествах, объединяя и очищая флэш-хроматографией, элюируя 0-50% (об./об.) дихлорметаном в гептане, с получением этилового эфира 3-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-[1,2,4]тиадиазол-5-карбоновой кислоты (4,38 г, 11 ммоль) в виде твердого белого вещества.
К охлажденному раствору (баня лед/метанол) этилового эфира 3-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-[1,2,4]тиадиазол-5-карбоновой кислоты (4,0 г, 10 ммоль) в тетрагидрофуране (80 мл) и метаноле (80 мл) последовательными порциями добавляли боргидрид натрия. Реакционную смесь перемешивали в течение последующих 20 минут и гасили 1М хлористоводородной кислотой (20 мл). Метанол и тетрагидрофуран удаляли в вакууме и добавляли дихлорметан (200 мл) и 2М хлористоводородную кислоту (50 мл). Органические вещества разделяли и промывали насыщенным раствором соли (50 мл), сушили над сульфатом магния и растворитель удаляли в вакууме. Полученный осадок очищали колоночной флэш-хроматографией, элюируя 50% (об./об.) диэтиловым эфиром в гептане, с получением [3-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-[1,2,4]тиадиазол-5-ил]метанола (3,15 г, 8,8 ммоль) в виде твердого светло-розового вещества.
К охлажденному раствору (баня лед/метанол) [3-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-[1,2,4]тиадиазол-5-ил]метанола (2,3 г, 6,4 ммоль) в дихлорметане (150 мл) последовательно добавляли метансульфонилхлорид (0,595 мл, 7,68 ммоль) и триэтиламин (1,16 мл, 8,32 ммоль). Реакционную смесь перемешивали в течение 10 мин и затем выливали в разделительную воронку. Органические вещества промывали 5% водным раствором карбоната натрия (2×100 мл), насыщенным раствором соли (100 мл), сушили над сульфатом магния и растворитель удаляли в вакууме с получением 3-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-[1,2,4]тиадиазол-5-илметилового эфира метансульфоновой кислоты (2,9 г, 6,7 ммоль), которую использовали без дополнительной очистки.
К раствору 3-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-[1,2,4]тиадиазол-5-илметилового эфира метансульфоновой кислоты (93 мг, 0,2 ммоль) в тетрагидрофуране (1 мл) добавляли диэтиламин (0,22 мл, 2,13 ммоль) и реакционную смесь подвергали микроволновому облучению при 150°C в течение 15 минут. Реакционную смесь выливали в разделительную воронку и разбавляли дихлорметаном (40 мл). Объединенную органическую фазу промывали 5% водным раствором карбоната натрия (2×20 мл), насыщенным раствором соли (2×20 мл), сушили над сульфатом магния и растворитель удаляли в вакууме. Полученный осадок очищали колоночной флэш-хроматографией с получением указанного в заголовке соединения (54 мг, 0,13 ммоль) в виде свободного основания. Свободное основание растворяли в дихлорметане и добавляли хлористый водород (2М раствор в диэтиловом эфире; 1,0 мл, 2,0 ммоль). Смесь концентрировали в вакууме с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1.
1H ЯМР (400 МГц, CD3OD): 0,95-1,12 (2H, м), 1,16-1,27 (3H, м), 1,45 (6H, т, J=7), 1,55-1,63 (2H, м), 1,63-1,8 (3H, м), 1,8-1,95 (1H, м), 3,28-3,32 (4H, м), 3,97 (3H, с), 4,3 (2H, д, J=7), 4,96 (2H, с), 6,79 (1H, д, J=8), 7,13 (1H, т, J=8), 7,95 (1H, с), 8,04 (1H, д, J=8); EsIMS: m/z 413,1 [M+H]+.
Пример 8
Далее, способ примера 7 применяли для получения следующих соединений:
8A : 1-(Циклогексил)метил-7-метокси-3-(5-{[бис-(2-метоксиэтил)амино]метил}-[1,2,4]тиадиазол-3-ил)-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием бис-(2-метоксиэтил)амина вместо диэтиламина. EsIMS: m/z 473,1 [M+H]+.
8B : 1-(Циклогексил)метил-7-метокси-3-{5-[(пирролидин-1-ил)метил]-[1,2,4]тиадиазол-3-ил-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием пирролидина вместо диэтиламина. EsIMS: m/z 411,1 [M+H]+, 342,0.
8C : 1-(Циклогексил)метил-7-метокси-3-{5-[(2-метилпиперидин-1-ил)метил]-[1,2,4]тиадиазол-3-ил}-1H-индол, соль трифторуксусной кислоты
Указанное в заголовке соединение получали с использованием 2-метилпиперидина вместо диэтиламина. Свободное основание очищали полупрепаративной ВЭЖХ [способ (i)] с получением указанного в заголовке соединения в виде соли трифторуксусной кислоты, 1:1. EsIMS: m/z 439,3 [M+H]+.
8D : 1-(Циклогексил)метил-3-(5-{[N-(2-гидроксиэтил)-N-метиламино]метил}-[1,2,4]тиадиазол-3-ил)-7-метокси-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием (2-гидроксиэтил)метиламина вместо диэтиламина. Свободное основание очищали полупрепаративной ВЭЖХ [способ (i)] с получением указанного в заголовке соединения в виде соли трифторуксусной кислоты, 1:1. EsIMS: m/z 415,3 [M+H]+, 328,3.
8E : 1-(Циклогексил)метил-7-метокси-3-(5-{[N-(2-метоксиэтил)-N-метиламино]метил[1,2,4]тиадиазол-3-ил)-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием N-(2-метоксиэтил)-N-метиламина вместо диэтиламина. EsIMS: m/z 429,4 [M+H]+.
Пример 9
1-(Циклогексил)метил-3-{5-[1-(диэтиламино)этил]-[1,2,4]тиадиазол-3-ил}-7-метокси-1H-индол, гидрохлоридная соль
К охлажденному раствору (баня сухой лед/ацетон) этилового эфира 3-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-[1,2,4]тиадиазол-5-карбоновой кислоты (500 мг, 1,3 ммоль) в диэтиловом эфире (50 мл) добавляли раствор метилмагнийбромида (0,52 мл, 3М в диэтиловом эфире, 1,56 ммоль) и реакционную смесь перемешивали в течение 15 минут, затем добавляли другую часть раствора метилмагнийбромида (0,25 мл, 3М в диэтиловом эфире, 0,75 ммоль) и реакционную смесь перемешивали в течение 5 минут. Затем реакцию гасили насыщенным водным хлоридом аммония (5 мл) и реакционной смеси давали нагреваться до комнатной температуры. Реакционную смесь выливали в разделительную воронку и органическую фазу промывали водой (20 мл). Затем водный слой промывали диэтиловым эфиром (20 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали и удаляли растворитель в вакууме. Полученный осадок очищали колоночной флэш-хроматографией с получением 1-[3-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-[1,2,4]тиадиазол-5-ил]этанона (170 мг, 0,46 ммоль)в виде твердого желтого вещества.
К раствору 1-[3-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-[1,2,4]тиадиазол-5-ил]этанона (90 мг, 0,24 ммоль) в ацетонитриле (3 мл) добавляли диэтиламин (0,248 мл, 2,4 ммоль) и уксусную кислоту (0,137 мл, 2,4 ммоль) и реакционную смесь перемешивали в течение 30 минут. К реакционной смеси добавляли нанесенный на полимер цианоборгидрид (204 мг, загрузка 2,35 ммольг-1, 0,48 ммоль) и реакционную смесь подвергали микроволновому облучению при 150°C в течение 10 минут. Реакционную смесь фильтровали через колонку 5 г Strata™ SCX giga. Колонку промывали метанолом и затем элюировали 2М аммиаком в метаноле. Раствор аммиака в метаноле выпаривали и полученный осадок очищали колоночной флэш-хроматографией с получением указанного в заголовке соединения (62 мг, 0,145 ммоль) в виде свободного основания. Свободное основание растворяли в дихлорметане (1 мл) и добавляли 2М HCl в диэтиловом эфире (1 мл, 2 ммоль), избыток реагента и растворитель удаляли в вакууме с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1.
1H ЯМР (400 МГц, CD3OD): 1,0-1,2 (2H, м), 1,16-1,26 (3H, м), 1,38-1,5 (6H, м), 1,55-1,78 (5H, м), 1,82-1,94 (4H, м), 3,32-3,68 (4H, м), 3,97 (3H, с), 4,3 (2H, д, J=7,5), 5,36-5,48 (1H, м), 6,8 (1H, д, J=8), 7,14 (1H, т, J=7,5), 7,94 (1H, с), 8,02 (1H, д, J=8); EsIMS: m/z 427,4 [M+H]+, 328,4.
Пример 10
1-(Циклогексил)метил-3-{5-[(диэтиламино)метил]-[1,2,4]тиадиазол-3-ил}-7-фтор-1H-индол, соль трифторуксусной кислоты
Указанное в заголовке соединение получали, следуя способу примера 7, с использованием амида 1-(циклогексил)метил-7-фтор-1H-индол-3-карбоновой кислоты (полученного из 7-фториндола) и диэтиламина. Свободное основание очищали полупрепаративной ВЭЖХ [способ (i)] с получением указанного в заголовке соединения в виде соли трифторуксусной кислоты, 1:1. EsIMS: m/z 401,3 [M+H]+.
Пример 11
7-Хлор-1-(циклогексил)метил-3-{5-[(пирролидин-1-ил)метил]-[1,2,4]тиадиазол-3-ил}-1H-индол, соль трифторуксусной кислоты
Указанное в заголовке соединение получали, следуя способу примера 7, с использованием амида 7-хлор-1-(циклогексил)метил-1H-индол-3-карбоновой кислоты (полученного из 7-хлориндола) и с использованием пирролидина вместо диэтиламина. Свободное основание очищали полупрепаративной ВЭЖХ [способ (i)] с получением указанного в заголовке соединения в виде соли трифторуксусной кислоты, 1:1. EsIMS: m/z 417,3, 415,3 [M+H]+.
Пример 12
1-(Циклогексил)метил-7-этил-3-{5-[(пирролидин-1-ил)метил]-[1,2,4]тиадиазол-3-ил}-1H-индол, соль трифторуксусной кислоты
Указанное в заголовке соединение получали, применяя способ примера 7, с использованием амида 1-(циклогексил)метил-7-этил-1H-индол-3-карбоновой кислоты (полученного из 7-этилиндола) и с использованием пирролидина вместо диэтиламина. Свободное основание очищали полупрепаративной ВЭЖХ [способ (i)] с получением указанного в заголовке соединение в виде соли трифторуксусной кислоты, 1:1. EsIMS: m/z 409,3 [M+H]+.
Пример 13
(R)-3-циклогексил-6-{5-[(диэтиламино)метил]-[1,2,4]тиадиазол-3-ил-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазин, гидрохлоридная соль
К смеси (R)-N-Boc-2-циклогексилэтаноламина (полученного, как описано для (S) энантиомера, Luly et al., J. Org. Chem. 52, 1487-1492, 1987; 29,4 г, 94,5 ммоль) и трифенилфосфина (37,2 г, 141,8 ммоль) в толуоле (150 мл) при 0°C добавляли диизопропилазодикарбоксилат (19,5 мл, 99,2 ммоль). После перемешивания в течение 1 часа к смеси добавляли 2-бромфенол (12,1 мл, 104,0 ммоль) при 0°C. Реакционную смесь перемешивали в течение 2 часов при 0°C и в течение 20 часов при комнатной температуре. Полученную смесь распределяли между дихлорметаном и водой. Водный слой экстрагировали дихлорметаном, объединенные органические слои промывали 2н. раствором гидроксида натрия и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Осадок очищали колоночной флэш-хроматографией, элюируя 0-10% (об./об.) этилацетатом в гептане, с получением (R)-2-(2-трет-бутоксикарбониламино-2-циклогексилэтокси)бромбензола (12,80 г, 32,1 ммоль).
Смесь (R)-2-(2-трет-бутоксикарбониламино-2-циклогексилэтокси)бромбензола (500 мг, 1,26 ммоль), тетракис(трифенилфосфин)палладия (0) (146 мг, 0,126 ммоль) и трет-бутоксида натрия (181 мг 1,88 ммоль) в толуоле (4,0 мл) подвергали микроволновому облучению в течение 10 мин при 120°C. Полученную смесь распределяли между дихлорметаном и водой. Водный слой экстрагировали дихлорметаном и объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Осадок очищали колоночной флэш-хроматографией, элюируя 0-17% (об./об.) этилацетатом в гептане, с получением (R)-4-трет-бутоксикарбонил-3-циклогексил-3,4-дигидро-2H-1,4-бензоксазина (270 мг, 0,85 ммоль). Данную реакцию повторяли 13 раз при тех же количествах с получением указанного выше промежуточного соединения (суммарно 3,98 г, 12,5 ммоль).
Смесь (R)-4-трет-бутоксикарбонил-3-циклогексил-3,4-дигидро-2H-1,4-бензоксазина (3,98 г, 12,5 ммоль), 5н. хлористоводородной кислоты (10 мл) и этанола (10 мл) перемешивали при 70°C в течение 50 мин. Этанол удаляли в вакууме и осадок распределяли между дихлорметаном и 2н. раствором гидроксида натрия. Водный слой экстрагировали дихлорметаном и объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали с получением (R)-3-циклогексил-3,4-дигидро-2H-1,4-бензоксазина (2,72 г, 12,5 ммоль).
(R)-3-циклогексил-3,4-дигидро-2H-1,4-бензоксазин (2,72 г, 12,5 ммоль) растворяли в N,N-диметилформамиде (20 мл) и добавляли раствор нитрита натрия (949 мг, 13,8 ммоль) в воде (3,0 мл) при 0°C. Затем добавляли 5н. хлористоводородную кислоту (6,0 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа, затем распределяли между этилацетатом и водой. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Полученный осадок растворяли в диэтиловом эфире (50 мл) и добавляли литийалюминийгидрид в тетрагидрофуране (1,0М; 9,51 мл, 9,51 ммоль) при 0°C. Реакционную смесь перемешивали при 0°C в течение 1 часа, затем гасили ледяной водой. К смеси добавляли этилацетат, смесь фильтровали через целит и осадок на фильтре промывали этилацетатом. Фильтрат разделяли и водный слой экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Осадок очищали колоночной флэш-хроматографией, элюируя 0-17% (об./об.) этилацетатом в гептане, с получением (R)-4-амино-3-циклогексил-3,4-дигидро-2H-1,4-бензоксазина (1,47 г, 6,33 ммоль).
Этилпируват (882 мг, 7,59 ммоль) добавляли к раствору (R)-4-амино-3-циклогексил-3,4-дигидро-2H-1,4-бензоксазина (1,47 г, 6,33 ммоль) в этаноле (40 мл). Реакционную смесь перемешивали при комнатной температуре в течение 15 мин. К реакционной смеси добавляли серную кислоту (10% об./об. в этаноле; 8,0 мл). Реакционную смесь кипятили с обратным холодильником в течение 2 часов. Смесь охлаждали до комнатной температуры и распределяли между этилацетатом и раствором карбоната натрия. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Осадок очищали колоночной флэш-хроматографией, элюируя 0-10% (об./об.) этилацетатом в гептане, с получением этил-(R)-3-циклогексил-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазин-5-карбоксилата (1,49 г, 4,76 ммоль).
К раствору этил-(R)-3-циклогексил-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазин-5-карбоксилата (1,49 г, 4,76 ммоль) в этаноле (50 мл) добавляли 4н. гидроксид натрия (5,94 мл, 23,8 ммоль). Смесь перемешивали при 70°C в течение 40 мин. Этанол удаляли в вакууме, осадок нейтрализовали 2н. хлористоводородной кислотой и распределяли между дихлорметаном и водой. Водный слой экстрагировали дихлорметаном и объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Осадок растворяли в хинолине (20 мл), затем добавляли порошок меди (453 мг, 7,13 ммоль). Смесь перемешивали при 210°C в течение 1 часа. К смеси добавляли этилацетат и воду при комнатной температуре, смесь фильтровали через целит и осадок на фильтре промывали этилацетатом. Фильтрат подкисляли 5н. хлористоводородной кислотой и разделяли. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали 1н. хлористоводородной кислотой и насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Осадок очищали колоночной флэш-хроматографией, элюируя 0-10% (об./об.) этилацетатом в гептане, с получением (R)-3-циклогексил-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазина (984 мг, 4,08 ммоль).
К раствору (R)-3-циклогексил-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазина (600 мг, 2,49 ммоль) в N,N-диметилформамиде (5,0 мл) при 0°C добавляли трифторуксусный ангидрид (0,311 мл, 2,73 ммоль). Смесь перемешивали при комнатной температуре в течение 5 часов, затем распределяли между дихлорметаном и водой. Водный слой экстрагировали дихлорметаном и объединенные органические слои промывали насыщенным раствором соли, сушили над Na2SO4 и концентрировали. Осадок очищали флэш-хроматографией, элюируя 0-25% (об./об.) этилацетатом в гептане, с получением (R)-3-циклогексил-6-трифторметилкарбонил-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазина (628 мг, 1,86 ммоль).
К раствору (R)-3-циклогексил-6-трифторметилкарбонил-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазина (628 мг, 1,86 ммоль) в 1,4-диоксане (20 мл) добавляли 4н. NaOH (5,0 мл). Смесь кипятили с обратным холодильником в течение 42 часов, затем подкисляли до pH 1, используя 5н. хлористоводородную кислоту, и распределяли между дихлорметаном и водой. Водный слой экстрагировали дихлорметаном и объединенные органические слои промывали насыщенным раствором соли, сушили над Na2SO4 и концентрировали с получением (R)-3-циклогексил-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазин-6-карбоновой кислоты (572 мг).
Указанное в заголовке соединение, следуя способу примера 7, с использованием амида (R)-3-циклогексил-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазин-6-карбоновой кислоты (полученного из (R)-3-циклогексил-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазин-6-карбоновой кислоты) вместо амида 1-(циклогексил)метил-7-метокси-1H-индол-3-карбоновой кислоты. EsIMS: m/z 411,0 [M+H]+; [α]D 22 -30,7° (c=1,50 мг/мл в хлороформе).
Пример 14
Следующие соединения получали, следуя способу примера 7, с использованием амида 7-фтор-1-(тетрагидропиран-4-ил)метил-1H-индол-3-карбоновой кислоты вместо амида 1-(циклогексил)метил-7-метокси-1H-индол-3-карбоновой кислоты.
Амид 7-фтор-1-(тетрагидропиран-4-ил)метил-1H-индол-3-карбоновой кислоты получали, следуя способу примера 1, с использованием 7-фториндола вместо 7-метоксииндола и тетрагидропиран-4-илметилового эфира толуол-4-сульфоновой кислоты вместо циклогексилметилбромида.
Способ синтеза промежуточного тетрагидропиран-4-илметилового эфира толуол-4-сульфоновой кислоты
К смеси тетрагидро-2H-пиран-4-илметанола (20,0 г, 172 ммоль) и пиридина (25,2 мл, 313 ммоль) в дихлорметане (200 мл) порциями добавляли п-толуолсульфонилхлорид (29,8 г, 157 ммоль). Смесь перемешивали при комнатной температуре в течение 17 часов, затем гасили водным раствором хлористоводородной кислоты (2М; 100 мл). Слои разделяли и экстрагировали водный слой дихлорметаном (2×100 мл). Органические слои объединяли и концентрировали в вакууме. Перекристаллизация из смеси дихлорметан:гептан (5:1) приводила к получению тетрагидропиран-4-илметилового эфира толуол-4-сульфоновой кислоты. Затем маточные растворы очищали хроматографией на колонке с силикагелем, элюируя 50% дихлорметаном в н-гептане, с получением дополнительного количества тетрагидропиран-4-илметилового эфира толуол-4-сульфоновой кислоты (суммарный выход 41,6 г, 154 ммоль).
14A: 3-{5-[(Диэтиламино)метил]-[1,2,4]тиадиазол-3-ил}-7-фтор-1-(тетрагидропиран-4-ил)метил-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием диэтиламина. EsIMS: m/z 403,1 [M+H]+.
14B: 7-Фтор-3-{5-[(пирролидин-1-ил)метил]-[1,2,4]тиадиазол-3-ил}-1-(тетрагидропиран-4-ил)метил-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием пирролидина вместо диэтиламина. EsIMS: m/z 401,0 [M+H]+.
14C: 3-{5-[(Диметиламино)метил}-[1,2,4]тиадиазол-3-ил}-7-фтор-1-(тетрагидропиран-4-ил)метил-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием диметиламина вместо диэтиламина. EsIMS: m/z 375,0 [M+H]+.
Пример 15
Следующие соединения получали, следуя способу примера 7, с использованием амида 7-хлор-1-(тетрагидропиран-4-ил)метил-1H-индол-3-карбоновой кислоты вместо амида 1-(циклогексил)метил-7-метокси-1H-индол-3-карбоновой кислоты.
Амид 7-хлор-1-(тетрагидропиран-4-ил)метил-1H-индол-3-карбоновой кислоты получали, следуя способу примера 1, с использованием 7-хлориндола вместо 7-метоксииндола и тетрагидропиран-4-илметилового эфира толуол-4-сульфоновой кислоты вместо циклогексилметилбромида.
15A: 7-Хлор-3-(5-{[N-этил-N-(2-метоксиэтил)амино]метил}-[1,2,4]тиадиазол-3-ил)-1-(тетрагидропиран-4-ил)метил-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием N-этил-N-(2-метоксиэтил)амина вместо диэтиламина. EsIMS: m/z 451,0, 449,0 [M+H]+.
15B: 7-Хлор-3-{5-[(пирролидин-1-ил)метил]-[1,2,4]тиадиазол-3-ил}-1-(тетрагидропиран-4-ил)метил-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием пирролидина вместо диэтиламина. EsIMS: m/z 419,3, 417,3 [M+H]+.
15C: 7-Хлор-3-(5-{[N-этил-N-(2-гидроксиэтил)амино]метил}-[1,2,4]тиадиазол-3-ил)-1-(тетрагидропиран-4-ил)метил-1H-индол
Указанное в заголовке соединение получали с использованием N-этил-N-(2-гидроксиэтил)амина вместо диэтиламина. EsIMS: m/z 437,1, 435,1 [M+H]+.
Пример 16
1-(Циклогексил)метил-7-метокси-3-(4-{[N-(2-метоксиэтил)-N-метиламино]метил}-[1,3]тиазол-2-ил)-1H-индол, гидрохлоридная соль
Смесь амида 1-(циклогексил)метил-7-метокси-1H-индол-3-карбоновой кислоты (полученного из 7-метоксииндола, как описано в примере 1; 5,10 г, 17,8 ммоль), реагента Лавессона (7,92 г, 19,6 ммоль) и толуола (150 мл) перемешивали при комнатной температуре в течение 4 дней. Реакционную смесь концентрировали в вакууме и полученный осадок очищали колоночной хроматографией, элюируя дихлорметаном, с получением амида 1-(циклогексил)метил-7-метокси-1H-индол-3-тиокарбоновой кислоты (3,58 г).
Смесь амида 1-(циклогексил)метил-7-метокси-1H-индол-3-тиокарбоновой кислоты (200 мг, 0,66 ммоль), 1,3-дихлорацетона (126 мг, 0,99 ммоль) и этанола (2,0 мл) перемешивали при 60°C в течение 1 часа. Реакционную смесь концентрировали в вакууме и полученный осадок смешивали с 5% водным карбонатом натрия (100 мл). Полученную смесь экстрагировали дихлорметаном (4×100 мл). Органические слои объединяли, промывали насыщенным раствором соли, сушили над сульфатом магния и концентрировали в вакууме. Полученный осадок очищали колоночной хроматографией, элюируя 25% (об./об.) этилацетатом в н-гептане, с получением 3-[4-(хлорметил)тиазол-2-ил]-1-(циклогексил)метил-7-метокси-1H-индола (200 мг).
Смесь 3-[4-(хлорметил)тиазол-2-ил]-1-(циклогексил)метил-7-метокси-1H-индола (100 мг, 0,27 ммоль), (2-метоксиэтил)метиламина (119 мг, 1,33 ммоль), 1,4-диоксана (2 мл) и ацетонитрила (1 мл) подвергали микроволновому облучению в течение 10 мин при 160°C. Реакционную смесь концентрировали в вакууме и полученный осадок смешивали с водным гидроксидом натрия (1М; 50 мл) и экстрагировали дихлорметаном (4×50 мл). Объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом магния и концентрировали в вакууме. Полученный осадок очищали колоночной хроматографией, элюируя этилацетатом, с получением свободного основания указанного в заголовке соединения в виде масла. Образования гидрохлоридной соли достигали добавлением хлористого водорода (1М раствор в диэтиловом эфире; 3 мл) к раствору свободного основания диэтиловом эфире (15 мл). Смесь концентрировали в вакууме с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1 (95,1 мг).
1H ЯМР (400 МГц, CD3OD) δ 1,00-1,30 (5H, м), 1,55-1,94 (6H, м), 3,00 (3H, с), 3,32-3,66 (5H, м), 3,80 (2H, т, J=5,0), 3,97 (3H, с), 4,29 (2H, д, J=7,2), 4,52 (2H, с), 6,81 (1H, д, J=8,0), 7,16 (1H, дд, J=8,0, 8,0), 7,62 (1H, с), 7,80 (1H, д, J=8,0), 7,85 (1H, с). EsIMS: m/z 428,1 [M+H]+, 339,0.
Пример 17
Далее, способ примера 16 применяли для получения следующих соединений с использованием альтернативных аминов вместо (2-метоксиэтил)метиламина.
17A: 1-(Циклогексил)метил-7-метокси-3-{4-[(морфолин-4-ил)метил]-[1,3]тиазол-2-ил}-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием морфолина вместо (2-метоксиэтил)метиламина. EsIMS: m/z 426,3 [M+H]+, 339,1.
17B: 1-(Циклогексил)метил-3-{4-[(4-гидроксипиперидин-1-ил)метил]-[1,3]тиазол-2-ил}-7-метокси-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием 4-гидроксипиперидина вместо (2-метоксиэтил)метиламина. EsIMS: m/z 440,1 [M+H]+, 399,0.
17C: 1-(Циклогексил)метил-3-(4-{[ N -изопропил-N-(2-метоксиэтил)амино]метил}-[1,3]тиазол-2-ил)-7-метокси-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием N-изопропил-N-(2-метоксиэтил)амина вместо (2-метоксиэтил)метиламина. EsIMS: m/z 456,4 [M+H]+, 339,1.
17D: (S)-1-(Циклогексил)метил-3-{4-[(2-гидроксиметилпирролидин-1-ил)метил]-[1,3]тиазол-2-ил-7-метокси-1H-индол
Указанное в заголовке соединение получали с использованием (S)-(+)-пролинола вместо (2-метоксиэтил)метиламина и выделяли в виде свободного основания. EsIMS: m/z 440,1 [M+H]+, 339,1; [a]D 22 -10,0° (c=0,65 мг/мл в хлороформе).
17E: 1-(Циклогексил)метил-7-метокси-3-{4-[(тиоморфолин-4-ил)метил]-[1,3]тиазол-2-ил}-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием тиоморфолина вместо (2-метоксиэтил)метиламина. EsIMS: m/z 442,0 [M+H]+, 339,0.
Пример 18
1-(Циклогексил)метил-7-метокси-3-{4-[1-(пирролидин-1-ил)этил]-[1,3]тиазол-2-ил}-1H-индол, гидрохлоридная соль
Раствор 1-хлор-2,3-бутандиона (0,717 г, 5,95 ммоль) в этаноле (3 мл) по каплям добавляли к раствору амида 1-(циклогексил)метил-7-метокси-1H-индол-3-тиокарбоновой кислоты (полученного, как в примере 16; 1,20 г, 3,97 ммоль) в этаноле (12 мл) при комнатной температуре и затем полученную смесь перемешивали при комнатной температуре в течение 3 суток. Реакционную смесь концентрировали в вакууме, полученный осадок смешивали с дихлорметаном (50 мл) и последовательно промывали водой и насыщенным раствором соли, сушили над сульфатом магния и концентрировали в вакууме. Полученный осадок очищали колоночной хроматографией, элюируя 33% (об./об.) этилацетатом в н-гептане, с получением 1-{2-[1-(циклогексил)метил-7-метокси-1H-индол-3-ил]тиазол-4-ил}этанона в виде твердого коричневого вещества (1,11 г).
Смесь 1-{2-[1-(циклогексил)метил-7-метокси-1H-индол-3-ил]тиазол-4-ил}этанона (100 мг, 0,27 ммоль), пирролидина (193 мг, 2,71 ммоль), уксусной кислоты (163 мг, 2,71 ммоль) и ацетонитрила (3 мл) перемешивали при комнатной температуре в течение 2 часов. Добавляли крупнопористый метилполистиролцианоборгидрид триэтиламмония (МП-цианоборгидрид, загрузка: 2,35 ммоль/г, 231 мг, 0,543 ммоль) и полученную смесь подвергали микроволновому облучению при 130°C в течение 10 мин. Смолу удаляли фильтрованием, промывая дихлорметаном, и фильтрат концентрировали в вакууме. Полученный осадок смешивали с водным гидроксидом натрия (1М; 100 мл) и экстрагировали дихлорметаном (4×100 мл). Органические слои объединяли, затем промывали насыщенным раствором соли, сушили над сульфатом магния и концентрировали в вакууме. Полученный осадок очищали колоночной хроматографией, элюируя 5% метанолом в этилацетате. Полученное масло растворяли в диэтиловом эфире (10 мл), затем к раствору добавляли HCl (1М раствор в диэтиловом эфире; 3 мл). Полученную смесь концентрировали в вакууме с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1 (30,1 мг).
1H ЯМР (400 МГц, CD3OD) δ 0,98-1,32 (5H, м), 1,54-2,22 (13H, м), 3,22-3,44 (3H, м), 3,66-3,84 (1H, м), 3,97 (3H, с), 4,29 (2H, д, J=7,2), 4,60-4,72 (1H, м), 6,81 (1H, д, J=7,6), 7,15 (1H, дд, J=7,6, 8,0), 7,51 (1H, с), 7,79-7,90 (2H, м). EsIMS: m/z 424,1 [M+H]+, 353,1.
Пример 19
1-(Циклогексил)метил-7-фтор-3-(4-{[N-изопропил-N-(2-метоксиэтил)амино]метил}-[1,3]тиазол-2-ил)-1H-индол, соль трифторуксусной кислоты
Указанное в заголовке соединение получали, следуя способу примера 16, с использованием амида 1-(циклогексил)метил-7-фтор-1H-индол-3-карбоновой кислоты (полученного из 7-фториндола) и N-изопропил-N-(2-метоксиэтил)амина. Свободное основание очищали полупрепаративной ВЭЖХ [способ (i)] с получением указанного в заголовке соединения в виде соли трифторуксусной кислоты, 1:1. EsIMS: m/z 444,3 [M+H]+, 327,3.
Пример 19A
1-(Циклогексил)метил-6-фтор-3-[4-(диэтиламино)метил]-[1,3]тиазол-2-ил)-1H-индол, соль трифторуксусной кислоты
Указанное в заголовке соединение получали, следуя способу примера 16, с использованием амида 1-(циклогексил)метил-6-фтор-1H-индол-3-карбоновой кислоты (полученного из 6-фториндол) и диэтиламина. Свободное основание очищали полупрепаративной ВЭЖХ [способ (i)] с получением указанного в заголовке соединения в виде соли трифторуксусной кислоты, 1:1. EsIMS: m/z 400,1 [M+H]+, 327,1.
Пример 20
7-Хлор-1-(циклогексил)метил-3-(4-{[N-изопропил-N-(2-метоксиэтил)амино]метил}-[1,3]тиазол-2-ил)-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали, следуя способу примера 16, с использованием амида 7-хлор-1-(циклогексил)метил-1H-индол-3-карбоновой кислоты (полученного из 7-хлориндола) и N-изопропил-N-(2-метоксиэтил)амина. EsIMS: m/z 462,3, 460,3 [M+H]+, 343,1.
Пример 21
1-(Циклогексил)метил-7-этил-3-(4-{[N-изопропил-N-(2-метоксиэтил)амино]метил}-[1,3]тиазол-2-ил)-1H-индол, соль трифторуксусной кислоты
Указанное в заголовке соединение получали, следуя способу примера 16, с использованием амида 1-(циклогексил)метил-7-этил-1H-индол-3-карбоновой кислоты (полученного из 7-этилиндол) и N-изопропил-N-(2-метоксиэтил)амина. Свободное основание очищали полупрепаративной ВЭЖХ [способ (i)] с получением указанного в заголовке соединения в виде соли трифторуксусной кислоты, 1:1. EsIMS: m/z 454,5 [M+H]+, 337,3.
Пример 22
Следующие соединения получали, следуя способу примера 16, с использованием амида (R)-3-циклогексил-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазин-6-карбоновой кислоты (полученного из (R)-3-циклогексил-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазин-6-карбоновой кислоты, описанной в примере 13) вместо амида 1-(циклогексил)метил-7-метокси-1H-индол-3-карбоновой кислоты.
22A: (R)-3-циклогексил-6-{4-[(диэтиламино)метил]-[1,3]тиазол-2-ил}-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазин, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием диэтиламина вместо (2-метоксиэтил)метиламина. EsIMS: m/z 410,3 [M+H]+, 337,3; [α]D 22 -37,5° (c=1,30 мг/мл в хлороформе).
22B: (R)-3-циклогексил-6-{4-[(N-этил-N-изопропиламино)метил]-[1,3]тиазол-2-ил}-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазин, соль трифторуксусной кислоты
Указанное в заголовке соединение получали с использованием N-этил-N-изопропиламина вместо (2-метоксиэтил)метиламина. Свободное основание очищали полупрепаративной ВЭЖХ [способ (i)] с получением указанного в заголовке соединения в виде соли трифторуксусной кислоты, 1:1. EsIMS: m/z 424,3 [M+H]+, 337,3; [α]D 22 -27,4° (c=1,25 мг/мл в хлороформе).
22C: (R)-3-циклогексил-6-{4-[(пирролидин-1-ил)метил]-[1,3]тиазол-2-ил}-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазин, соль трифторуксусной кислоты
Указанное в заголовке соединение получали с использованием пирролидина вместо (2-метоксиэтил)метиламина. Свободное основание очищали полупрепаративной ВЭЖХ [способ (i)] с получением указанного в заголовке соединения в виде соли трифторуксусной кислоты, 1:1.
EsIMS: m/z 408,3 [M+H]+, 337,3; [a]D 22 -32,6° (c=2,15 мг/мл в хлороформе).
22D: (R)-3-циклогексил-6-(4-{[N-изопропил-N-(2-метоксиэтил)амино]метил}-[1,3]тиазол-2-ил)-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазин, соль трифторуксусной кислоты
Указанное в заголовке соединение получали с использованием N-изопропил-N-(2-метоксиэтил)амина вместо (2-метоксиэтил)метиламина. Свободное основание очищали полупрепаративной ВЭЖХ [способ (i)] с получением указанного в заголовке соединения в виде соли трифторуксусной кислоты, 1:1. EsIMS: m/z 454,3 [M+H]+, 337,3; [α]D 22 -58,4° (c=2,09 мг/мл в метаноле).
22E: (R)-3-циклогексил-6-(4-{[бис-(2-метоксиэтил)амино]метил}-[1,3]тиазол-2-ил)-2,3-дигидропирроло[1,2,3-de]-1,4-бензоксазин, соль трифторуксусной кислоты
Указанное в заголовке соединение получали с использованием бис-(2-метоксиэтил)амина вместо (2-метоксиэтил)метиламина. Свободное основание очищали полупрепаративной ВЭЖХ [способ (i)] с получением указанного в заголовке соединения в виде соли трифторуксусной кислоты, 1:1. EsIMS: m/z 470,3 [M+H]+, 337,3; [α]D 22 -28,5° (c=1,20 мг/мл в хлороформе).
Пример 23
Следующие соединения получали, следуя способу примера 16, с использованием амида 7-хлор-1-(тетрагидропиран-4-ил)метил-1H-индол-3-карбоновой кислоты вместо амида 1-(циклогексил)метил-7-метокси-1H-индол-3-карбоновой кислоты.
Амид 7-хлор-1-(тетрагидропиран-4-ил)метил-1H-индол-3-карбоновой кислоты получали, следуя способу примера 1, с использованием 7-хлориндола вместо 7-метоксииндола и тетрагидропиран-4-илметилового эфира толуол-4-сульфоновой кислоты (полученного, как описано в примере 14) вместо циклогексилметилбромида.
23A: 7-Хлор-3-{4-[(диэтиламино)метил]-[1,3]тиазол-2-ил}-1-(тетрагидропиран-4-ил)метил-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием диэтиламина. EIMS: m/z 420,0, 418,4 [M+H]+, 347,0, 345,0.
23B: 7-Хлор-3-{4-{{N-(2-гидроксиэтил)-N-изопропиламино]метил}-[1,3]тиазол-2-ил)-1-(тетрагидропиран-4-ил)метил-1H-индол
Указанное в заголовке соединение получали с использованием N-(2-гидроксиэтил)-N-изопропиламина. EIMS: m/z 448,4 [M+H]+, 347,1, 345,1.
23C: 7-Хлор-3-(4-{[N-этил-N-(2-гидроксиэтил)амино]метил}-[1,3]тиазол-2-ил)-1-(тетрагидропиранил-4-ил)метил-1H-индол
Указанное в заголовке соединение получали с использованием N-этил-N-(2-гидроксиэтил)амина. EIMS: m/z 436,3, 434,4 [M+H]+, 347,0, 345,0.
23D : 7-Хлор-3-(4-{[N-(2-метоксиэтил)-N-метиламино]метил}-[1,3]тиазол-2-ил)-1-(тетрагидропиран-4-ил)метил-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали с использованием N-(2-метоксиэтил)-N-метиламина. EIMS: m/z 436,1, 434,1 [M+H]+, 347,0, 345,0.
Пример 24
1-(Циклогексил)метил-3-{4-[(диметиламино)метил]-5-этил-[1,3]тиазол-2-ил}-7-метокси-1H-индол, гидрохлоридная соль
Этоксид натрия (0,68 г, 9,94 ммоль) добавляли порциями к смеси этилдихлорацетата (1,22 мл, 9,94 ммоль) и диэтилового эфира (10 мл) при охлаждении в ледяной воде и полученную смесь перемешивали при 0°C в течение 30 минут. Затем добавляли пропионовой альдегид (0,79 мл, 10,93 ммоль) и реакционной смеси давали медленно нагреваться до комнатной температуры, непрерывно перемешивая в течение 72 часов. Затем реакционную смесь выливали в воду (10 мл) и экстрагировали диэтиловым эфиром (2×15 мл). Органические слои объединяли, сушили над сульфатом магния и концентрировали в вакууме с получением неочищенного этилового эфира 3-хлор-2-оксопентановой кислоты (1,8 г), который использовали на следующей стадии без дополнительной очистки.
Смесь амида 1-(циклогексил)метил-7-метокси-1H-индол-3-тиокарбоновой кислоты (0,227 г, 0,754 ммоль) и неочищенного этилового эфира 3-хлор-2-оксопентановой кислоты (1,34 г, 7,52 ммоль) в диметилформамиде (4 мл) подвергали микроволновому облучению в течение 25 мин при 140°C. Реакционную смесь концентрировали в вакууме и полученный осадок очищали колоночной хроматографией на силикагеле, элюируя 25% ацетоном в гептане, с получением неочищенного этилового эфира 2-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-5-этилтиазол-4-карбоновой кислоты (0,490 г). Полученное вещество использовали на следующей стадии без дополнительной очистки.
К смеси этилового эфира 2-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-5-этилтиазол-4-карбоновой кислоты (490 мг, 1,15 ммоль) и тетрагидрофурана (5 мл) добавляли порциями боргидрид лития (200 мг, 9,09 ммоль) при охлаждении в ледяной воде и полученную смесь перемешивали при 0°C в течение 2 часов. Реакционную смесь гасили водным раствором хлористоводородной кислоты (2М; 2 мл) и водный слой экстрагировали дихлорметаном (2×100 мл). Органические слои объединяли, сушили над сульфатом магния и концентрировали в вакууме с получением неочищенного продукта. Колоночной хроматографией на силикагеле получали неочищенный [2-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-5-этилтиазол-4-ил]метанол (210 мг), элюируя 66% этилацетатом в гептане. Полученное вещество использовали на следующей стадии без дополнительной очистки.
К смеси [2-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-5-этилтиазол-4-ил]метанола (210 мг, 0,547 ммоль), диизопропилэтиламина (150 мкл, 0,91 ммоль) и дихлорметана (5 мл) добавляли при охлаждении в ледяной воде метансульфонилхлорид (90 мкл, 1,16 ммоль) и полученной смеси давали медленно нагреться до комнатной температуры. Перемешивание продолжали в течение 22 часов. Реакционную смесь гасили 5% водным карбонатом натрия (2 мл) и водный слой экстрагировали дихлорметаном (2×10 мл). Органические слои объединяли, сушили над сульфатом магния и концентрировали в вакууме с получением неочищенного продукта. Колоночная флэш-хроматография при элюировании 10% ацетоном в н-гептане приводила к получению 3-(4-хлорметил-5-этилтиазол-2-ил)-1-циклогексилметил-7-метокси-1H-индола (109 мг).
К смеси 3-(4-хлорметил-5-этилтиазол-2-ил)-1-циклогексилметил-7-метокси-1H-индола (38 мг, 0,095 ммоль), карбоната калия (16 мг, 0,113 ммоль) и иодида натрия (14 мг, 0,095 ммоль) в ацетонитриле (2 мл) добавляли диметиламин (2,2М раствор в тетрагидрофуране; 0,50 мл). Смесь подвергали микроволновому облучению в течение 5 мин при 160°C, затем распределяли между дихлорметаном (20 мл) и 5% водным карбонатом натрия (5 мл). Водный слой экстрагировали дихлорметаном (10 мл) и объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме. Полученное масло растворяли в диэтиловом эфире, затем добавляли к раствору хлористый водород (1М раствор в диэтиловом эфире; 3 мл). Полученную смесь концентрировали в вакууме с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1 (40 мг, 0,089 ммоль).
1H ЯМР (400 МГц, CD3OD) δ 1,04-1,15 (2H, м), 1,16-1,30 (3H, м), 1,43 (3H, т, J=7,0), 1,55-1,78 (5H, м), 1,82-1,92 (1H, м), 3,05 (8H, м), 3,99 (3H, с), 4,33 (2H, д, J=7), 4,62 (2H, с), 6,91 (1H, д, J=8,0), 7,29 (1H, т, J=8,0), 7,62 (1H, д, J=8), 8,30 (1H, с). EsIMS: m/z 412,3 [M+H]+, 367,0.
Пример 25
1-(Циклогексил)метил-3-{5-[(диэтиламино)метил]-4-метил[1,3]тиазол-2-ил}-7-метокси-1H-индол, гидрохлоридная соль
К раствору амида 1-(циклогексил)метил-7-метокси-1H-индол-3-тиокарбоновой кислоты (604 мг, 2,00 ммоль) в EtOH (5,0 мл) добавляли этил-2-хлор-3-оксобутаноат (0,332 мл, 2,40 ммоль). Смесь кипятили с обратным холодильником в течение 1 часа. После охлаждения до 0°C осадок собирали фильтрованием с получением 1-циклогексилметил-3-(5-этоксикарбонил-4-метилтиазол-2-ил)-7-метокси-1H-индола (505 мг, 1,22 ммоль).
К раствору 1-циклогексилметил-3-(5-этоксикарбонил-4-метилтиазол-2-ил)-7-метокси-1H-индола (680 мг, 1,65 ммоль) в ТГФ (20 мл) добавляли литийалюминийгидрид (125 мг, 3,30 ммоль) при 0°C. Смесь перемешивали при 0°C в течение 1 часа, затем гасили ледяной водой и экстрагировали дихлорметаном. Объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Осадок очищали флэш-хроматографией, элюируя 25-50% (об./об.) этилацетатом в гептане, затем 10% (об./об.) метанолом в дихлорметане, с получением 1-циклогексилметил-3-(5-гидроксиметил-4-метилтиазол-2-ил)-7-метокси-1H-индола (532 мг, 1,44 ммоль).
К раствору 1-циклогексилметил-3-(5-гидроксиметил-4-метилтиазол-2-ил)-7-метокси-1H-индола (74 мг, 0,20 ммоль) и триэтиламина (26 мг, 0,26 ммоль) в дихлорметане (1,0 мл) добавляли метансульфонилхлорид (28 мг, 0,24 ммоль). Смесь перемешивали при комнатной температуре в течение 40 мин и распределяли между дихлорметаном и 5% водным гидрокарбонатом натрия. Водный слой экстрагировали дихлорметаном, затем объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали с получением неочищенного 1-циклогексилметил-3-(5-метансульфонилоксиметил-4-метилтиазол-2-ил)-7-метокси-1H-индола (65 мг, 0,15 ммоль). Смесь неочищенного 1-циклогексилметил-3-(5-метансульфонилоксиметил-4-метилтиазол-2-ил)-7-метокси-1H-индола (64 мг, 0,14 ммоль), карбоната калия (29 мг, 0,21 ммоль), иодида натрия (31 мг, 0,21 ммоль) и диэтиламина (21 мг, 0,28 ммоль) в ТГФ (1,5 мл) и ацетонитриле (1,5 мл) подвергали микроволновому облучению в течение 5 мин при 160°C. Полученную смесь распределяли между дихлорметаном и водой. Водный слой экстрагировали дихлорметаном и объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом натрия и концентрировали. Осадок очищали флэш-хроматографией, элюируя 50-100% (об./об.) этилацетатом в гептане, с получением 1-циклогексилметил-3-(5-диэтиламиноэтил-4-метилтиазол-2-ил)-7-метокси-1H-индола (27 мг, 0,064 ммоль). Образования гидрохлоридной соли достигали добавлением хлористого водорода (1М раствор в диэтиловом эфире; 1 мл) к раствору свободного основания в диэтиловом эфире (1 мл) и этаноле (2 мл). Растворитель удаляли в вакууме и осадок сушили с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1 (26 мг, 0,056 ммоль).
1H ЯМР (400 МГц, ДМСО-d6) δ 0,95-1,25 (5H, м), 1,29 (6H, т, J=7,2), 1,40-1,52 (2H, м), 1,55-1,70 (3H, м), 1,72-1,84 (1H, м), 3,10-3,25 (4H, м), 3,93 (3H, с), 4,27 (2H, д, J=7,0), 4,56 (2H, д, J=5,0), 6,82 (1H, д, J=7,6), 7,14 (1H, т, J=7,6), 7,72 (1H, д, J=7,6), 8,00 (1H, с), 9,90 (1H, ушир. с); EsIMS: m/z 426,3 [M+H]+, 353,1.
Пример 26
1-(Циклогексил)метил-3-{2-[(диэтиламино)метил]-[1,3]тиазол-4-ил-7-метокси-1H-индол, гидрохлоридная соль
К раствору 7-метоксииндола (5,00 г, 34,0 ммоль) в диметилформамиде (50 мл) в атмосфере азота добавляли гидрид натрия (60% дисперсия в минеральном масле; 1,50 г, 37,4 ммоль). Перед добавлением бромметилциклогексана (5,20 мл, 37,4 ммоль) смесь перемешивали при комнатной температуре в течение 10 минут. Полученную смесь перемешивали при комнатной температуре в течение 42 часов и затем распределяли между этилацетатом (150 мл) и водой (150 мл). Водный слой экстрагировали этилацетатом (150 мл) и объединенные органические слои промывали насыщенным раствором соли (150 мл), сушили над сульфатом натрия и концентрировали в вакууме. Неочищенное промежуточное соединение очищали колоночной флэш-хроматографией, элюируя 0-10% (об./об.) этилацетатом в н-гептане, с получением 1-(циклогексил)метил-7-метокси-1H-индола (7,48 г, 30,7 ммоль).
К перемешиваемому раствору пиридина (2,20 мл, 27,2 ммоль) и 1-(циклогексил)метил-7-метокси-1H-индола (6,60 г, 27,2 ммоль) в толуоле (50 мл) при 55°C в течение 1,5 часов по каплям добавляли хлорацетилхлорид (8,66 мл, 109 ммоль). Полученную смесь нагревали до 55°C в течение следующих 0,5 часа, затем давали охладиться до комнатной температуры. Добавляли воду (60 мл) и метанол (10 мл). Органический слой разделяли и концентрировали при пониженном давлении с получением темно-коричневого осадка. Осадок очищали колоночной хроматографией, элюируя 5% (об./об.) этилацетатом в н-гептане. Полученное твердое вещество повторно перекристаллизовывали из эфира с получением 2-хлор-1-[1-(циклогексил)метил-7-метокси-1H-индол-3-ил]этанона в виде твердого белого вещества. (1,40 г).
2-хлор-1-[1-(циклогексил)метил-7-метокси-1H-индол-3-ил]этанон (0,73 г, 2,30 ммоль) и 2-(трет-бутилкарбонилокси)тиоацетамид (1,21 г, 6,89 ммоль) суспендировали в этаноле (10 мл) и полученную смесь подвергали микроволновому облучению при 150°C в течение 10 мин с использованием Emrys™ Optimizer EXP. Реакционную смесь концентрировали в вакууме и полученный осадок очищали колоночной флэш-хроматографией, элюируя 5% (об./об.) этилацетатом в н-гептане, с получением 1-(циклогексил)метил-7-метокси-3-{2-[(трет-бутилкарбонилокси)метил]тиазол-4-ил}-1H-индола в виде желтого масла (1,01 г).
1-(Циклогексил)метил-7-метокси-3-{2-[(третбутилкарбонилокси)метил]тиазол-4-ил}-1H-индол (0,92 г, 2,10 ммоль) растворяли в метаноле (20 мл) и добавляли 4н. гидроксид натрия (5 мл). Раствор перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали в вакууме и переносили осадок в дихлорметан. Добавляли воду и разделяли органические слои. Водный слой экстрагировали дихлорметаном и объединенные органические слои концентрировали в вакууме с получением 1-(циклогексил)метил-3-[2-(гидроксиметил)тиазол-4-ил]-7-метокси-1H-индола в виде светло-оранжевой пены (0,55 г).
Метансульфонилхлорид (174 мкл, 2,25 ммоль) добавляли к раствору 1-(циклогексил)метил-3-[2-(гидроксиметил)тиазол-4-ил]-7-метокси-1H-индола (0,40 г, 1,12 ммоль) и пиридина (182 мкл, 2,25 ммоль) в дихлорметане (8 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли метансульфонилхлорид (87 мкл, 1,12 ммоль) и непрерывно перемешивали в течение 0,5 часов. Смесь концентрировали в вакууме и полученный оранжевый осадок очищали колоночной флэш-хроматографией, элюируя дихлорметаном, с получением 3-[2-(хлорметил)тиазол-4-ил]-1-(циклогексил)метил-7-метокси-1H-индола в виде желтого масла (0,415 г).
3-[2-(хлорметил)тиазол-4-ил]-1-(циклогексил)метил-7-метокси-1H-индол (0,08 г, 0,214 ммоль) и диэтиламин (221 мкл, 2,14 ммоль) растворяли в ацетонитриле (2 мл). Полученную смесь подвергали микроволновому облучению при 100°C в течение 5 мин. Смесь концентрировали в вакууме и полученный осадок очищали колоночной хроматографией, элюируя 33% (об./об.) этилацетатом в н-гептане. Полученный продукт переносили в диэтиловый эфир и добавляли хлористый водород (1М раствор в диэтиловом эфире; 1 мл). Раствор концентрировали в вакууме и полученное твердое вещество растирали с эфиром, затем сушили с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1 (0,034 г).
1H ЯМР (400 МГц, CD3OD) δ 0,99-1,25 (5H, м), 1,43 (6H, т, J=7,5), 1,56-1,90 (6H, м), 3,35-3,42 (4H, м), 3,96 (3H, с), 4,27 (2H, д, J=7,5), 4,75-4,80 (2H, с, замаскирован пиком H2O), 6,75 (1H, д, J=8,0), 7,08 (1H, дд, J=8,0, 8,0), 7,64-7,68 (3H, м). EsIMS:
m/z 412,1 [M+H]+, 339,0.
Пример 27
1-(Циклогексил)метил-7-метокси-3-{2-[(пирролидин-1-ил)метил]-[1,3]тиазол-4-ил}-1H-индол, гидрохлоридная соль
Указанное в заголовке соединение получали способом примера 26 с использованием пирролидина вместо диэтиламина. EsIMS: m/z 410,3 [M+H]+, 339,1.
Пример 28
1-(Циклогексил)метил-3-{2-[(диметиламино)метил]-[1,3]тиазол-5-ил}-7-метокси-1H-индол, гидрохлоридная соль
К суспензии 1-циклогексилметил-7-метокси-1H-индол-3-карбоновой кислоты (5 г, 17,4 ммоль) в дихлорметане (100 мл) добавляли оксалилхлорид (3,04 мл, 34,8 ммоль) и полученный раствор перемешивали в течение ночи. Затем избыток растворителя и реагента удаляли выпариванием. К полученному осадку добавляли цианид меди (I) (6,2 г, 69,6 ммоль), толуол (200 мл) и ацетонитрил (10 мл) и полученную реакционную смесь кипятили с обратным холодильником в течение 7 часов. Затем добавляли другую порцию цианида меди (I) (1,6 г, 17,9 ммоль) и реакционную смесь кипятили с обратным холодильником в течение ночи. Реакционную смесь охлаждали и фильтровали через слой дикалита, промывали ацетонитрилом и объединенный фильтрат выпаривали с получением твердого красного вещества. Твердое вещество очищали колоночной флэш-хроматографией, элюируя 50% (об./об.) дихлорметаном в гептане, с получением (1-циклогексилметил-7-метокси-1H-индол-3-ил)оксоацетонитрила (4,7 г, 14,7 ммоль).
К раствору (1-циклогексилметил-7-метокси-1H-индол-3-ил)оксоацетонитрила (975 мг, 3,29 ммоль) в уксусной кислоте (40 мл) в атмосфере азота добавляли 10% палладий на активированном угле (90 мг). Реакционную смесь помещали в атмосферу водорода и перемешивали в течение 14 часов. Затем реакционную смесь фильтровали через слой дикалита. Дикалит промывали уксусной кислотой и объединенный фильтрат выпаривали с получением красного масла. Красное масло переносили в дихлорметан (50 мл) и к нему добавляли метилхлороксоацетат (0,393 мл, 4,28 ммоль) с последующим добавлением по каплям N-этилдиизопропиламина (1,7 мл, 9,87 ммоль). Реакционную смесь перемешивали в течение 1 часа и выливали в разделительную воронку. Органические слои последовательно промывали 2М водным раствором хлористоводородной кислоты (50 мл), 5% водным карбонатом натрия (50 мл) и насыщенным раствором соли (50 мл). Органические вещества сушили над сульфатом натрия, фильтровали и растворитель удаляли в вакууме с получением коричневого масла. Масло очищали колоночной флэш-хроматографией с использованием дихлорметана и затем 66% (об./об.) диэтилового эфира в гептане с получением метилового эфира N-[(1-циклогексилметил-7-метокси-1H-индол-3-ил)-2-оксоэтил]оксаламовой кислоты (573 мг, 1,48 ммоль) в виде твердого желто/коричневого вещества.
К раствору метилового эфира N-[(1-циклогексилметил-7-метокси-1H-индол-3-ил)-2-оксоэтил]оксаламовой кислоты (429 мг, 1,11 ммоль) в хлороформе (20 мл) добавляли пентасульфид фосфора (538 мг, 1,21 ммоль) и реакционную смесь нагревали в течение 3,5 часов. Реакционную смесь охлаждали, выливали в разделительную воронку, промывали водой и затем насыщенным раствором соли. Затем органические слои сушили над сульфатом магния, фильтровали и растворитель удаляли в вакууме. Полученное твердое вещество очищали колоночной флэш-хроматографией, элюируя дихлорметаном, с получением метилового эфира 5-(1-циклогексилметил-7-метокси-1H-индол-3-ил)тиазол-2-карбоновой кислоты (418 мг, 1,09 ммоль) в виде твердого коричневого вещества.
К раствору метилового эфира 5-(1-циклогексилметил-7-метокси-1H-индол-3-ил)тиазол-2-карбоновой кислоты (418 мг, 1,09 ммоль) в метаноле (10 мл) и тетрагидрофуране (10 мл) добавляли боргидрид натрия (83 мг, 2,18 ммоль) порциями в течение 2 минут. Затем реакционную смесь перемешивали в течение 1 часа и затем гасили водным раствором хлористоводородной кислоты (1М; 10 мл). Смесь выливали в разделительную воронку, разбавляли дихлорметаном (50 мл) и промывали водой (20 мл). Объединенные органические слои сушили, фильтровали и растворитель удаляли в вакууме с получением желтого масла. Масло очищали колоночной флэш-хроматографией с использованием 50-100% (об./об.) диэтилового эфира в гептане с получением [5-(1-циклогексилметил-7-метокси-1H-индол-3-ил)тиазол-2-ил]метанола (308 мг, 0,86 ммоль) в виде не совсем белой пены.
К раствору 5-(1-циклогексилметил-7-метокси-1H-индол-3-ил)тиазол-2-ил]метанола (308 мг, 0,86 ммоль) в дихлорметане (20 мл) добавляли метансульфонилхлорид (80 мкл, 1,03 ммоль) и затем триэтиламин (0,156 мл, 1,12 ммоль). Реакционную смесь перемешивали в течение 30 минут, затем выливали в разделительную воронку, промывали 5% водным карбонатом натрия, затем насыщенным раствором соли и сушили над сульфатом магния. Растворитель удаляли в вакууме с получением метилового эфира 5-(1-циклогексилметил-7-метокси-1H-индол-3-ил)тиазол-2-илметансульфоновой кислоты (411 мг, 0,94 ммоль). К раствору метилового эфира 5-(1-циклогексилметил-7-метокси-1H-индол-3-ил)тиазол-2-илметансульфоновой кислоты (93 мг, 0,215 ммоль) в тетрагидрофуране (2 мл) добавляли диметиламин (2М в тетрагидрофуране; 1 мл, 2 ммоль) и реакционную смесь подвергали микроволновому облучению при 150°C в течение 15 минут. Реакционную смесь разбавляли дихлорметаном (40 мл) и промывали смесью насыщенного раствора соли и насыщенного бикарбоната натрия в соотношении 1:1 (об./об.), сушили над сульфатом магния, фильтровали и растворитель удаляли в вакууме. Полученное масло очищали колоночной флэш-хроматографией с получением указанного в заголовке соединение (70 мг, 0,18 ммоль) в виде свободного основания. Свободное основание растворяли в дихлорметане (5 мл), добавляли хлористый водород (2М раствор в диэтиловом эфире; 1 мл, 2 ммоль) и растворитель удаляли в вакууме с получением указанного в заголовке соединение в виде гидрохлоридной соли, 1:1.
1H ЯМР (400 МГц, CD3OD): 0,96-1,12 (2H, м), 1,13-1,26 (3H, м), 1,5-1,62 (2H, м), 1,62-1,78 (3H, м), 1,78-1,92 (1H, м), 3,01 (6H, с), 3,96 (3H, с), 4,26 (2H, д, J=5,5), 4,71 (2H, с), 6,78 (1H, д, J=8,0), 7,11 (1H, т, J=8,0), 7,41 (1H, д, J=8,0), 7,52 (1H, с), 8,08 (1H, с); EsIMS: m/z 384,0 [M+H]+, 339,0, 243,1.
Пример 29
1-(Циклогексил)метил-3-{4-[(диэтиламино)метил]-[1,3]оксазол-2-ил-7-метокси-1H-индол, гидрохлоридная соль
К раствору амида 1-циклогексилметил-7-метокси-1H-индол-3-карбоновой кислоты (500 мг, 1,75 ммоль) в толуоле (4 мл) добавляли 1,3-дихлорацетон (333 мг, 2,62 ммоль) и реакционную смесь подвергали микроволновому облучению при 150°C в течение 30 минут. Полученный черный раствор разбавляли дихлорметаном (50 мл) и промывали 5% водным раствором карбоната натрия (5×20 мл), сушили над сульфатом магния, фильтровали и растворитель удаляли в вакууме. Полученное коричневое масло очищали колоночной флэш-хроматографией с использованием 5% (об./об.) ацетона в петролейном эфире 40-60 мл с получением 3-(4-хлорметилоксазол-2-ил)-1-циклогексилметил-7-метокси-1H-индола (510 мг, 1,42 ммоль) в виде твердого белого вещества.
К раствору 3-(4-хлорметилоксазол-2-ил)-1-циклогексилметил-7-метокси-1-индола (100 мг, 0,28 ммоль) в тетрагидрофуране (1 мл) добавляли диэтиламин (0,29 мл, 2,8 ммоль) и реакционную смесь подвергали микроволновому облучению при 150°C в течение 15 минут. Реакционную смесь вносили в разделительную воронку, разбавляли дихлорметаном (40 мл) и промывали 5% водным раствором карбоната натрия (2×25 мл), насыщенным раствором соли (20 мл), сушили над сульфатом магния и растворитель выпаривали в вакууме с получением оранжевого масла. Масло очищали колоночной флэш-хроматографией с использованием 10% (об./об.) метанола в дихлорметане с получением указанного в заголовке соединения (92 мг, 0,23 ммоль) в виде свободного основания. Свободное основание растворяли в дихлорметане и добавляли хлористый водород (1М раствор в диэтиловом эфире; 2 мл, 2 ммоль). Смесь концентрировали в вакууме с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1.
1H ЯМР (400 МГц, CD3OD): 0,97-1,28 (5H, м), 1,44 (6H, т, J=7,0), 1,5-1,8 (5H, м), 1,8-1,95 (1H, м), 3,3-3,5 (4H, м), 3,97 (3H, с), 4,29 (2H, д, J=7,0), 4,38 (2H, с), 6,8 (1H, д, J=8,0), 7,2 (1H, т, J=8,0), 7,8 (1H, с), 7,82 (1H, с), 8,2 (1H, с); EsIMS: m/z 396,0 [M+H]+, 323,4, 295,4, 268,3.
Пример 30
1-(Циклогексил)метил-7-метокси-3-{5-[(пирролидин-1-ил)метил]-[1,3]оксазол-2-ил}-1H-индол, соль трифторуксусной кислоты
Смесь амида 1-циклогексилметил-7-метокси-1H-индол-3-карбоновой кислоты (563 мг, 1,97 ммоль), метилового эфира 2-хлор-3-оксопропионовой кислоты (Gangjee et al., J. Med. Chem. 44, 1993-2003, 2001; 1,48 г, 9,85 ммоль) и диметилацетамида (10 мл) подвергали микроволновому облучению при 90°C в течение 2×5 минут с использованием Emrys™ Optimizer EXP. Реакционную смесь разбавляли дихлорметаном (150 мл), затем промывали 5% водным сульфатом магния (2×100 мл) и насыщенным раствором соли (150 мл). Органические экстракты сушили над сульфатом магния и концентрировали в вакууме. Полученный осадок очищали колоночной флэш-хроматографией, элюируя 25% (об./об.) этилацетатом в гептане, с получением неразделимой смеси метилового эфира 2-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-оксазол-5-карбоновой кислоты и 1-циклогексилметил-7-метокси-3-оксазол-2-ил-1H-индола (соотношение ВЭЖХ 87:13; 0,613 г).
К смеси метилового эфира 2-(1-циклогексилметил-7-метокси-1H-индол-3-ил)оксазол-5-карбоновой кислоты и 1-циклогексилметил-7-метокси-3-оксазол-2-ил-1H-индола (557 мг), растворенных в тетрагидрофуране (10 мл), при охлаждении в системе лед-метанол по каплям добавляли раствор литийалюминийгидрида (1М раствор в диэтиловом эфире; 2,88 мл, 2,88 ммоль). Полученную смесь перемешивали в течение 30 минут при 0°C, затем разбавляли диэтиловым эфиром (40 мл). Затем добавляли избыток декагидрата сульфата натрия и полученную смесь перемешивали при комнатной температуре в течение 18 часов. Смесь фильтровали через слой дикалита и промывали диэтиловым эфиром (100 мл); затем фильтрат концентрировали в вакууме. Полученный осадок очищали колоночной хроматографией, элюируя 50% этилацетатом в н-гептане, с получением [2-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-оксазол-5-ил]метанола в виде твердого желтого вещества (242 мг, 0,71 ммоль).
К раствору [2-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-оксазол-5-ил]метанола (242 мг, 0,71 ммоль), растворенного в дихлорметане (15 мл), при охлаждении в системе лед-метанол по каплям добавляли метансульфонилхлорид (98 мг, 0,85 ммоль), затем по каплям добавляли триэтиламин (93 мг, 0,92 ммоль), затем прекращали охлаждение и смесь перемешивали в течение 16 часов. Затем смесь разбавляли дихлорметаном (30 мл), промывали насыщенным раствором карбоната натрия (30 мл) в гидрофобной пробирке с фриттой. Органические экстракты сушили над сульфатом магния и затем концентрировали в вакууме. Смесь полученного осадка (110 мг, 0,26 ммоль), пирролидина (185 мг, 2,60 ммоль) и тетрагидрофурана (2,5 мл) подвергали микроволновому облучению при 150°C в течение 15 минут. Полученную смесь концентрировали в вакууме и очищали колоночной флэш-хроматографией (2% (об./об.) аммиак в системе метанол/дихлорметан в соотношении 1:49 в качестве элюента) с получением коричневой смолы. Затем смолу очищали полупрепаративной ВЭЖХ [способ (i)] с получением 1-циклогексилметил-7-метокси-3-(5-пирролидин-1-илметилоксазол-2-ил)-1H-индола в виде соли трифторуксусной кислоты (14 мг).
1H ЯМР (400 МГц, CD3OD) δ 0,99-1,27 (5H, м), 1,54-1,78 (5H, м), 1,86 (1H, м), 2,00-2,24 (4H, м), 3,25-3,44 (2H, м), 3,53-3,74 (2H, м), 3,97 (3H, с), 4,29 (2H, д, J=7,0), 4,64 (2H, с), 6,81 (1H, с, J=7,0), 7,15 (1H, т, J=8,0), 7,41 (1H, с), 7,79 (1H, д, J=7,5), 7,82 (1H, с). EsIMS: m/z 394,1 [M+H]+, 323,1.
Пример 31
1-(Циклогексил)метил-3-{5-[(диэтиламино)метил]-4-метил[1,3]оксазол-2-ил}-7-метокси-1H-индол, гидрохлоридная соль
Смесь амида 1-(циклогексил)метил-7-метокси-1H-индол-3-карбоновой кислоты (500 мг, 1,75 ммоль), этил-2-хлорацетоацетата (2,88 г, 17,6 ммоль) и диметилформамида (10 мл) подвергали микроволновому облучению при 185°C в течение 15 минут с использованием Emrys™ Optimizer EXP. Реакционную смесь разбавляли дихлорметаном (100 мл), затем промывали 5% водным сульфатом магния (2×50 мл), водой (50 мл) и насыщенным раствором соли (50 мл). Органические экстракты сушили над сульфатом магния и концентрировали в вакууме. Полученный осадок очищали колоночной хроматографией, элюируя 33% (об./об.) дихлорметаном в н-гептане, с получением неразделяемой смеси этилового эфира 2-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-4-метилоксазол-5-карбоновой кислоты и 1-циклогексилметил-7-метокси-3-(4-метилоксазол-2-ил)-1H-индола (соотношение 78:22 по данным ВЭЖХ; 0,586 г). Данную реакцию повторяли при тех же количествах.
К смеси этилового эфира 2-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-4-метилоксазол-5-карбоновой кислоты и 1-циклогексилметил-7-метокси-3-(4-метилоксазол-2-ил)-1H-индола (1171 мг), растворенных в тетрагидрофуране (20 мл), при охлаждении в системе лед-метанол по каплям добавляли литийалюминийгидрид (1М раствор в диэтиловом эфире; 5,8 мл, 5,8 ммоль). Полученную смесь перемешивали в течение 20 минут при 0°C, затем разбавляли диэтиловым эфиром (40 мл). Добавляли избыток декагидрата сульфата натрия и полученную смесь перемешивали при комнатной температуре в течение 18 часов. Смесь фильтровали через слой дикалита, промывая диэтиловым эфиром (100 мл) и фильтрат концентрировали в вакууме. Осадок очищали колоночной флэш-хроматографией, элюируя 50% (об./об.) этилацетатом в н-гептане, с получением [2-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-4-метилоксазол-5-ил]метанола в виде твердого белого вещества (774 мг). К раствору [2-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-4-метилоксазол-5-ил]метанола (724 мг, 2,04 ммоль) в дихлорметане (40 мл) при -78°C в атмосфере азота по каплям добавляли метансульфонилхлорид (281 мг, 2,45 ммоль). По каплям добавляли триэтиламин (269 мг, 2,66 ммоль) и смеси давали нагреться до комнатной температуры при перемешивании в течение 2 часов. Затем смесь разбавляли дихлорметаном (100 мл), промывали насыщенным раствором карбоната натрия (2×100 мл) и насыщенным раствором соли (100 мл). Органические экстракты сушили над сульфатом магния и затем концентрировали в вакууме. Смесь полученного осадка (100 мг, 0,23 ммоль), диэтиламина (169 мг, 2,30 ммоль) и тетрагидрофурана (2 мл) подвергали микроволновому облучению при 150°C в течение 15 минут. Полученную смесь концентрировали в вакууме и очищали колоночной флэш-хроматографией, элюируя 2% аммиаком в системе метанол/дихлорметан в соотношении 1:49, с получением коричневой смолы. Смолу растворяли в дихлорметане (0,5 мл), затем добавляли хлористый водород (1М раствор в диэтиловом эфире; 0,5 мл) и смесь концентрировали в вакууме с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1 (32 мг).
1H ЯМР (400 МГц, CD3OD) δ 0,85-1,38 (6H, м), 1,46 (6H, т, J=7,5), 1,55-1,78 (4H, м), 1,88 (1H, м), 2,39 (3H, с), 3,34 (4H, q, J=7,6), 3,98 (3H, с), 4,34 (2H, д, J=6,9), 4,65 (2H, с), 6,88 (1H, д, J=8,2), 7,23 (1H, т, J=8,0), 7,74 (1H, д, J=8,0), 8,00 (1H, с). EsIMS: m/z 410,3 [M+H]+, 337,1.
Пример 32
1-(Циклогексил)метил-3-{2-[(диэтиламино)метил]-[1,3]оксазол-5-ил}-7-метокси-1H-индол, гидрохлоридная соль
К раствору (1-циклогексилметил-7-метокси-1H-индол-3-ил)-оксоацетонитрила (полученного, как описано в примере 28; 2,39 г, 8,06 ммоль) в уксусной кислоте (50 мл) в атмосфере азота добавляли 10% палладий на активированном угле (240 мг). Реакционную смесь помещали в атмосферу водорода и перемешивали в течение ночи. Затем реакционную смесь фильтровали через рыхлый слой дикалита. Дикалит промывали уксусной кислотой и объединенный фильтрат выпаривали с получением красного масла. Красное масло переносили в дихлорметан (50 мл), добавляли к нему хлорацетилхлорид (0,77 мл, 9,67 ммоль) и затем по каплям триэтиламин (3,4 мл, 24,2 ммоль). Реакционную смесь перемешивали в течение 30 минут и помещали в разделительную воронку. Органические вещества последовательно промывали 5% водным карбонатом натрия (2×30 мл) и насыщенным раствором соли (30 мл). Органические вещества сушили над сульфатом магния, фильтровали, удаляли растворитель в вакууме с получением красно/коричневого масла. Масло очищали колоночной флэш-хроматографией с использованием 20-100% (об./об.) дихлорметана в гептане и затем 25-50% (об./об.) диэтилового эфира в гептане с получением 2-хлор-N-[2-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-2-оксоэтил]ацетамида (2,32 г, 6,1 ммоль).
К раствору 2-хлор-N-[2-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-2-оксоэтил]ацетамида (200 мг, 0,53 ммоль) в тетрагидрофуране (2 мл) добавляли диэтиламин (0,55 мл, 5,3 ммоль) и реакционную смесь подвергали микроволновому облучению при 150°C в течение 15 минут. Реакционную смесь выливали в разделительную воронку и добавляли дихлорметан (30 мл). Органические вещества последовательно промывали 5% водным карбонатом натрия и насыщенным раствором соли. Органические вещества сушили над сульфатом магния, фильтровали и растворитель удаляли в вакууме с получением твердого коричневого вещества. Твердое коричневого вещество растворяли в тетрагидрофуране (2 мл) и добавляли гидроксид (метоксикарбонилсульфамоил)триэтиламмония, внутреннюю соль (505 мг, 2,12 ммоль). Полученную реакционную смесь подвергали микроволновому облучению при 150°C в течение 15 минут и гасили метанолом (20 мл). Растворитель удаляли в вакууме и осадок очищали флэш-хроматографией с использованием 50% (об./об.) этилацетата в гептане с последующей полупрепаративной ВЭЖХ [способ (ii)] с получением твердого белого вещества. Твердое вещество растворяли в дихлорметане (≈5 мл) и добавляли хлористый водород (1М раствор в диэтиловом эфире; 1 мл). Смесь концентрировали в вакууме с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1 (77 мг, 0,2 ммоль).
1H ЯМР (400 МГц, CD3OD) δ 0,97-1,12 (2H, м), 1,15-1,25 (3H, м), 1,44 (6H, т, J=6,9), 1,52-1,62 (2H, м), 1,62-1,77 (3H, м), 1,77-1,9 (1H, м), 3,36 (4H, кв, J=6,7), 3,95 (3H, с), 4,26 (2H, д,J=7), 4,64 (2H, с), 6,77 (1H, д, J=8), 7,11 (1H, т, J=8), 7,38 (1H, с), 7,41 (1H, д, J=8), 7,56 (1H, с); EsIMS: m/z 396,1 [M+H]+, 323,4, 268,4.
Пример 33
1-(Циклогексил)метил-3-(5-этил-5,6-дигидро-4H-пирроло[3,4-d]изотиазол-3-ил)-7-метокси-1H-индол, гидрохлоридная соль
К суспензии 5-(1-циклогексилметил-7-метокси-1H-индол)-[1,3,4]оксатиазол-2-она (полученного, как описано в примере 7; 100 мг, 0,25 ммоль) в м-ксилоле (0,5 мл) добавляли диэтилацетилендикарбоксилат (0,2 мл, 1,25 ммоль) и реакционную смесь подвергали микроволновому облучению при 200°C в течение 5 минут. Затем реакционную смесь сразу очищали колоночной флэш-хроматографией с использованием 0-100% (об./об.) дихлорметана в гептане с получением диэтилового эфира 3-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-изотиазол-4,5-дикарбоновой кислоты (141 мг, 0,3 ммоль). Данную реакцию повторяли при количестве 1,255 ммоль, все объединяли и очищали колоночной флэш-хроматографией с использованием 50-60% (об./об.) дихлорметана в гептане с получением указанного выше промежуточного соединения (882 мг, 1,87 ммоль).
К охлажденному раствору (баня лед/метанол) диэтилового эфира 3-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-изотиазол-4,5-дикарбоновой кислоты (400 мг, 0,85 ммоль) в тетрагидрофуране (20 мл) добавляли литийалюминийгидрид (1М раствор в ТГФ; 1,91 мл, 1,91 ммоль) и реакционную смесь перемешивали в течение 20 минут. К реакционной смеси добавляли избыток декагидрата сульфата натрия и реакционную смесь энергично перемешивали в течение 1,5 часов. Полученную смесь фильтровали через рыхлый слой дикалита, промывая диэтиловым эфиром. Фильтрат концентрировали в вакууме и полученное масло очищали колоночной флэш-хроматографией с получением 3-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-4-гидроксиметилизотиазол-5-ил]метанола (141 мг, 0,3 ммоль). К раствору 3-(1-циклогексилметил-7-метокси-1H-индол-3-ил)-4-гидроксиметилизотиазол-5-ил]метанола (194 мг, 0,5 ммоль) в дихлорметане (10 мл) добавляли метансульфонилхлорид (0,182 мл, 1,16 ммоль), затем триэтиламин (0,175 мл, 1,26 ммоль) и реакционную смесь перемешивали в течение 45 минут. Добавляли дополнительную порцию метансульфонилхлорида (0,07 мл, 0,44 ммоль) и триэтиламина (0,15 мл, 1 ммоль) и реакционную смесь перемешивали в течение следующих 2 часов. Реакционную смесь помещали в разделительную воронку, промывали 5% водным раствором карбоната натрия, затем насыщенным раствором соли, сушили над сульфатом магния и растворитель выпаривали в вакууме. Полученный осадок очищали колоночной флэш-хроматографией с использованием 33-100% (об./об.) дихлорметана в гептане и затем диэтилового эфира с получением метилового эфира {4-хлорметил-3-[1-(циклогексил)метил-7-метокси-1H-индол-3-ил]изотиазол-5-ил}метансульфоновой кислоты (113 мг, 0,23 ммоль).
К раствору метилового эфира {4-хлорметил-3-[1-(циклогексил)метил-7-метокси-1H-индол-3-ил]изотиазол-5-илметансульфоновой кислоты (90 мг, 0,19 ммоль) в тетрагидрофуране (1 мл) добавляли этиламин (0,186 мл, 0,37 ммоль) и триэтиламин (0,05 мл, 0,37 ммоль) и реакционную смесь подвергали микроволновому облучению при 150°C в течение 15 минут. Реакционную смесь помещали в разделительную воронку, разбавляли дихлорметаном (30 мл) и промывали 5% водным раствором карбоната натрия (2×10 мл), насыщенным раствором соли (10 мл), сушили над сульфатом магния и растворитель удаляли в вакууме. Реакцию повторяли при количестве 0,166 ммоль с получением указанного выше промежуточного соединения. Неочищенные продукты объединяли и очищали колоночной флэш-хроматографией с использованием этилацетата с получением указанного в заголовке соединения (36 мг, 0,09 ммоль) в виде свободного основания. Свободное основание растворяли в дихлорметане и добавляли хлористый водород (2М раствор в диэтиловом эфире; 1 мл, 2 ммоль). Смесь концентрировали в вакууме с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1.
1H ЯМР (400 МГц, CD3OD): 0,97-1,15 (2H, м), 1,15-1,27 (3H, м), 1,47 (3H, т, J=7), 1,52-1,61 (2H, м), 1,62-1,77 (3H, м), 1,81-1,95 (1H, м), 3,63 (2H, кв, J=7), 3,95 (3H, с), 4,28 (2H, д, J=7), 4,62-4,75 (2H, м), 4,95-5,12 (2H, м), 6,77 (1H, д, J=8), 7,09 (1H, т, J=8), 7,43 (1H, с), 8,01 (1H, д, J=8); EsIMS: m/z 396,0 [M+H]+, 353,4, 351,3, 320,3.
Пример 34
1-(Циклогексил)метил-7-метокси-3-{5-[(пирролидин-1-ил)метилизоксазол-3-ил]-1H-индол, соль трифторуксусной кислоты
Оксихлорид фосфора (12 мл, 0,13 моль) медленно добавляли в течение 30 минут к диметилформамиду (30 мл) при -10°C. Раствору давали нагреться до 0°C в течение 1 часа, затем порциями добавляли 1-(циклогексил)метил-7-метокси-1H-индол (полученный, как в примере 26; 3,2 г, 13 ммоль) и раствор перемешивали при комнатной температуре в течение 16 часов. Раствор охлаждали в ледяной бане и разбавляли водой, затем осторожно нейтрализовали бикарбонатом натрия и экстрагировали этилацетатом (3×50 мл). Органические экстракты объединяли и растворитель удаляли в вакууме. Осадок (3,5 г) растворяли в водном растворе гидроксида натрия (5М; 100 мл) и смесь кипятили с обратным холодильником при 100°C в течение следующих 16 часов. Раствор охлаждали и экстрагировали этилацетатом (3×50 мл). Органические экстракты объединяли и растворитель удаляли в вакууме с получением 1-(циклогексил)метил-7-метокси-1H-индол-3-карбальдегида (2,2 г, 8,12 ммоль) в виде твердого белого вещества.
К раствору 1-(циклогексил)метил-7-метокси-1H-индол-3-карбальдегида (780 мг, 2,9 ммоль) в смеси этанола (8 мл) и воды (2 мл) добавляли гидрохлорид гидроксиламина (403 мг, 5,8 ммоль) и ацетат натрия (713 мг, 8,7 ммоль) и раствор перемешивали в течение 64 часов при комнатной температуре. Смесь концентрировали в вакууме, осадок разбавляли водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Продукт перекристаллизовывали из системы диэтиловый эфир/гексан с получением оксима 1-циклогексилметил-7-метокси-1H-индол-3-карбалдегида (380 мг, 1,3 ммоль) в виде желтого порошка.
К раствору оксима 1-циклогексилметил-7-метокси-1H-индол-3-карбальдегида (170 мг, 0,59 ммоль) в дихлорметане (5 мл), охлажденному до 0°C, добавляли N-хлорсукцинимид (119 мг, 0,89 ммоль) и смеси давали нагреться до комнатной температуры при перемешивании в течение 1 часа. Добавляли пропаргилбромид (8 мкл, 0,65 ммоль) и триэтиламин (9 мкл, 0,65 ммоль) и смесь перемешивали при комнатной температуре в течение 16 часов. Растворитель удаляли в вакууме и осадок очищали колоночной флэш-хроматографией, элюируя 60-80% (об./об.) дихлорметаном в н-гептане, с получением 3-(5-бромметилизоксазол-3-ил)-1-циклогексилметил-7-метокси-1H-индола (150 мг, 0,37 ммоль) в виде твердого желтого вещества.
К раствору 3-(5-бромметилизоксазол-3-ил)-1-циклогексилметил-7-метокси-1H-индола (120 мг, 0,31 ммоль) в ацетонитриле (3 мл) добавляли диэтиламин (0,024 мл, 0,34 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов. Смесь фильтровали и растворитель удаляли в вакууме. Осадок очищали полупрепаративной ВЭЖХ [способ (ii)] с получением указанного в заголовке соединения в виде соли трифторуксусной кислоты (20 мг).
1H ЯМР (400 МГц, CD3OD) δ 1,03-1,09 (2H, м), 1,21 (3H, м), 1,57-1,74 (5H, м), 1,83-1,89 (1H, м), 2,14 (4H, м), 3,38-3,60 (4H, м), 3,96 (3H, с), 4,27-4,29 (2H, д, J=6,9), 4,69 (2H, с), 6,78 (1H, д, J=7,5), 7,02 (1H, с), 7,08-7,12 (1H, м), 7,67 (1H, с), 7,67-7,69 (1H, д, J=8,1). EsIMS: m/z 394,1 [M+H]+, 323,4.
Пример 35
1-(Циклогексил)метил-7-метокси-3-(5-{[бис-(2-гидроксиэтил)амино]метил}изоксазол-3-ил)-1H-индол, соль трифторуксусной кислоты
Указанное в заголовке соединение получали способом примера 34 с использованием диэтаноламина вместо пирролидина. EsIMS: m/z 428,4 [M+H]+, 323,4.
Пример 36
1-(Циклогексил)метил-7-фтор-3-{5-[(пирролидин-1-ил)метил]тиофен-2-ил)-1H-индол, гидрохлоридная соль
К охлажденному льдом раствору 7-фториндола (2,0 г, 14,8 ммоль) в диметилформамиде (50 мл) добавляли гидрид натрия (60% дисперсия в минеральном масле; 0,88 г, 22,2 ммоль), смесь перемешивали в течение 15 мин и затем добавляли по каплям бензолсульфонилхлорид (2,26 мл, 17,8 ммоль). Затем смесь перемешивали при комнатной температуре в течение 18 часов. Затем суспензию разбавляли водой (200 мл), экстрагировали трет-бутилметиловым эфиром (3×100 мл) и объединенные органические слои промывали водой (3×100 мл), сушили сульфатом натрия и концентрировали в вакууме. Осадок очищали колоночной флэш-хроматографией, элюируя 20% (об./об.) этилацетатом в изогексане, с получением 1-бензолсульфонил-7-фториндола в виде твердого бесцветного вещества. (3,96 г, 14,4 ммоль). К раствору 1-бензолсульфонил-7-фториндола (2,0 г, 7,27 ммоль) в диметилформамиде (10 мл) по каплям добавляли раствор брома (0,75 мл, 14,55 ммоль) в диметилформамиде (25 мл) в течение 3 мин. Затем смесь перемешивали при комнатной температуре в течение 10 мин и выливали в смесь метабисульфита натрия (2 г), раствора гидроксида аммония (3 мл), воды (100 мл) и дробленого льда (100 г). Полученную суспензию перемешивали до обесцвечивания и экстрагировали трет-бутилметиловым эфиром (2×100 мл). Объединенные органические слои промывали водой (2×100 мл), сушили сульфатом натрия и растворитель удаляли в вакууме с получением 1-бензолсульфонил-3-бром-7-фториндола в виде твердого оранжево-розового вещества (2,35 г, 6,64 ммоль).
Раствор 1-бензолсульфонил-3-бром-7-фториндола (0,5 г, 1,41 ммоль), 5-формил-2-тиофенбороновой кислоты (0,24 г, 1,55 ммоль), дихлорида бис(трифенилфосфин)палладия (II) (0,06 г, 0,08 ммоль) и триэтиламина (0,39 мл, 2,82 ммоль) в этаноле (4 мл) подвергали микроволновому облучению при 150°C в течение 420 с. Полученную суспензию концентрировали при пониженном давлении и осадок пропускали через слой диоксида кремния, элюируя дихлорметаном, с получением неочищенного 5-(1-бензолсульфонил-7-фториндол-3-ил)-тиофен-2-карбоксальдегида в виде твердого коричневого вещества (0,42 г), которое использовали на следующей стадии без дополнительной очистки.
К неочищенному 5-(1-бензолсульфонил-7-фториндол-3-ил)тиофен-2-карбоксальдегиду последовательно добавляли 4 Е молекулярные сита (1 г), пирролидин (0,44 мл, 5,36 ммоль), цианоборгидрид натрия (0,034 г, 0,54 ммоль) и ледяную уксусную кислоту (1 каплю). Затем полученную смесь перемешивали при комнатной температуре в течение 18 часов, фильтровали и осадок на фильтре промывали метанолом (2×30 мл) и дихлорметаном (2×30 мл). Объединенный фильтрат концентрировали в вакууме, растворяли в дихлорметане (20 мл), промывали водным гидроксидом натрия (2М; 15 мл), сушили сульфатом натрия и концентрировали в вакууме. Затем осадок очищали колоночной флэш-хроматографией, элюируя 0-10% (об./об.) метанолом в дихлорметане, с получением 1-бензол-сульфонил-7-фтор-3-(5-пирролидин-1-илметилтиофен-2-ил)индола в виде желтого масла (0,22 г, 0,46 ммоль).
1-бензолсульфонил-7-фтор-3-(5-пирролидин-1-илметилтиофен-2-ил)индол (0,20 г, 0,46 ммоль) и карбонат калия (0,25 г, 1,82 ммоль) обрабатывали смесью метанола и воды (3:1 об./об.; 4 мл) и подвергали микроволновому облучению при 100°C в течение 600 с. Затем полученную суспензию концентрировали при пониженном давлении и распределяли между дихлорметаном (10 мл) и водой (10 мл). Органическую фазу отделяли и водную фазу промывали дихлорметаном (10 мл). Объединенные органические слои сушили сульфатом натрия и концентрировали в вакууме. Осадок очищали колоночной флэш-хроматографией, элюируя 0-10% (об./об.) метанолом в дихлорметане, с получением 7-фтор-3-(5-пирролидин-1-илметилтиофен-2-ил)индола в виде твердого бесцветного вещества (0,10 г, 0,35 ммоль).
К раствору 7-фтор-3-(5-пирролидин-1-илметилтиофен-2-ил)индола (0,10 г, 0,35 ммоль) в диметилформамиде (3 мл) добавляли гидрид натрия (60% дисперсия в минеральном масле; 0,03 г, 0,71 ммоль) и смесь перемешивали при комнатной температуре в течение 15 минут. Добавляли бромметилциклогексан (0,05 мл, 0,39 ммоль) и смесь перемешивали при 60°C в течение 18 часов. Затем суспензию разбавляли водой (30 мл), экстрагировали трет-бутилметиловым эфиром (2×30 мл), объединенные органические слои промывали водой (2×20 мл), сушили сульфатом натрия и выпаривали при пониженном давлении. Осадок очищали флэш-хроматографией, элюируя 5% (об./об.) дихлорметаном в метаноле, с получением указанного в заголовке соединения (свободного основания) в виде бесцветного твердого вещества(0,05 г, 0,13 ммоль). Полученное вещество растворяли в диэтиловом эфире (3 мл) и обрабатывали хлористоводородной кислотой (1М раствор в диэтиловом эфире) с получением посредством выпаривания указанного в заголовке соединения (1:1 с солью хлористоводородной кислоты) в виде бесцветного твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 1,02-1,34 (5H, м), 1,57-1,93 (6H, м), 2,11-2,19 (4H, м), 3,38-3,57 (4H, м), 4,19 (2H, д, J=7,4), 4,65 (2H, с), 6,91-7,36 (4H, м), 7,58 (1H, с), 7,71 (1H, д, J=8,1); EsIMS: m/z 396,9 [M+H]+, 326,0.
Пример 37
1-(Циклогексил)метил-3-{5-[(диэтиламино)метил]-[1,3,4]оксадиазол-2-ил}-7-метокси-1H-индол, гидрохлоридная соль
К суспензии 1-циклогексилметил-7-метокси-1H-индол-3-карбоновой кислоты (1,0 г, 3,48 ммоль) в дихлорметане (20 мл) добавляли оксалилхлорид (0,6 мл, 6,96 ммоль) и реакционную смесь перемешивали в течение 3,5 часов. Растворитель выпаривали при пониженном давлении и полученный осадок повторно растворяли в дихлорметане (20 мл). К раствору добавляли гидразид хлоруксусной кислоты (1,3 г, 8,97 ммоль) и триэтиламин (2,9 мл, 20,9 ммоль) и реакционную смесь перемешивали в течение 4 часов и затем давали отстаиваться в течение ночи. Растворитель выпаривали при пониженном давлении и полученный осадок очищали колоночной флэш-хроматографией, элюируя 50-100% (об./об.) этилацетатом в гептане, с получением N-(2-хлорацетил)гидразида 1-циклогексилметил-7-метокси-1H-индол-3-карбоновой кислоты (397 мг, 1,05 ммоль).
К раствору N'-(2-хлорацетил)гидразида 1-циклогексилметил-7-метокси-1H-индол-3-карбоновой кислоты (250 мг, 0,662 ммоль) в тетрагидрофуране (3 мл) добавляли внутреннюю соль гидроксида (метоксикарбонилсульфамоил)триэтиламмония (315 мг, 1,32 ммоль) и полученную реакционную смесь подвергали микроволновому облучению при 150°C в течение 15 минут. Реакционную смесь гасили метанолом и растворитель выпаривали. Полученный осадок очищали флэш-хроматографией с использованием 33-50% (об./об.) этилацетата в гептане с получением 3-(5-хлорметил[1,3,4]оксадиазол-2-ил)-1-циклогексилметил-7-метокси-1H-индола (169 мг, 0,47 ммоль) в виде твердого желтого вещества. Данную реакцию повторяли при количестве 0,53 ммоль с получением указанного выше промежуточного соединения (суммарно 276 мг, 0,77 ммоль).
К раствору 3-(5-хлорметил[1,3,4]оксадиазол-2-ил)-1-циклогексилметил-7-метокси-1H-индола (92 мг, 0,26 ммоль) в тетрагидрофуране (1 мл) добавляли диэтиламин (0,134 мл, 1,28 ммоль) и реакционную смесь подвергали микроволновому облучению при 150°C в течение 15 минут. Полученную смесь очищали колоночной флэш-хроматографией с получением указанного в заголовке соединения (87 мг, 0,22 ммоль) в виде свободного основания. Свободное основание растворяли в дихлорметане и добавляли хлористый водород (2М раствор в диэтиловом эфире; 1 мл, 2 ммоль). Избыток реагента и растворителя удаляли выпариванием с получением указанного в заголовке соединения (1:1 с солью хлористоводородной кислоты) в виде твердого белого вещества.
1H ЯМР (400 МГц, CD3OD): 0,97-1,12 (2H, м), 1,15-1,26 (3H, м), 1,46 (6H, т, J=7), 1,53-1,63 (2H, м), 1,63-1,78 (3H, м), 1,8-1,95 (1H, м), 3,44 (4H, кв, J=7), 3,98 (3H, с), 4,33 (2H, д, J=7), 4,84 (2H, с), 6,85 (1H, д, J=7), 7,19 (1H, т, J=7,9), 7,73 (1H, д, J=8), 7,94 (1H, с). EsIMS: m/z 397,1 [M+H]+, 324,4, 270,5.
Пример 38
1-(Циклогексил)метил-7-метокси-3-{5-[(пирролидин-1-ил)метил]-[1,3,4]тиадиазол-2-ил}-1H-индол, гидрохлоридная соль
К раствору N'-(2-хлорацетил)гидразида 1-циклогексилметил-7-метокси-1H-индол-3-карбоновой кислоты (полученного, как описано в примере 37; 50 мг, 0,139 ммоль) в тетрагидрофуране (0,5 мл) добавляли пентасульфид фосфора (62 мг, 0,139 ммоль) и реакционную смесь подвергали микроволновому облучению при 150°C в течение 5 минут. Реакцию повторяли два раза при количестве 0,7 ммоль. Объединенную реакционную смесь помещали в разделительную воронку и разбавляли дихлорметаном (60 мл). Органическую фазу промывали 5% водным карбонатом натрия (2×30 мл), насыщенным раствором соли (30 мл), сушили над сульфатом натрия и растворитель удаляли в вакууме. Полученный осадок очищали колоночной флэш-хроматографией с получением 3-(5-хлорметил[1,3,4]тиадиазол-2-ил)-1-циклогексилметил-7-метокси-1H-индола (186 мг, 0,49 ммоль).
К раствору 3-(5-хлорметил[1,3,4]тиадиазол-2-ил)-1-циклогексилметил-7-метокси-1H-индола (93 мг, 0,25 ммоль) в тетрагидрофуране добавляли пирролидин (0,101 мл, 1,235 ммоль) и смесь подвергали микроволновому облучению при 150°C в течение 5 минут. Полученную реакционную смесь очищали колоночной флэш-хроматографией с получением указанного в заголовке соединения (42 мг, 0,1 ммоль) в виде свободного основания. Свободное основание растворяли в дихлорметане и добавляли хлористый водород (1М раствор в диэтиловом эфире; 1 мл, 1 ммоль). Избыток реагента и растворителя удаляли в вакууме с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1.
1H ЯМР (400 МГц, CDCl3): 0,94-1,1 (2H, м), 1,13-1,23 (3H, м), 1,5-1,75 (8H, м), 1,8-1,9 (5H, м), 2,66-2,73 (4H, м), 3,95 (3H, с), 4,12 (2H, с), 4,23 (2H, д, J=7), 6,73 (1H, д, J=7,8), 7,17 (1H, т, J=8), 7,66 (1H, с), 7,77 (1H, д, J=8); EsIMS: m/z 411,1 [M+H]+, 340,0, 324,4, 286,1, 270,5.
Пример 39
7-Хлор-3-{5-[(2,2-диметилпирролидин-1-ил)метил]-[1,2,4]оксадиазол-3-ил-1-(тетрагидропиран-4-ил)метил-1H-индол, гидрохлоридная соль
Способ синтеза 2,2-диметилпирролидинона:
К перемешанному и охлажденному раствору (0°C) NiCl2·6H2О в метаноле (200 мл) порциями добавляли боргидрид натрия (3,36 г, 89 ммоль). Реакционную смесь перемешивали в течение 30 мин и затем добавляли метил-4-метил-4-нитропентаноат в виде раствора в метаноле (100 мл). Температуру реакции поддерживали при 0°C перед добавлением порциями боргидрида натрия (7,86 г, 208 ммоль). Реакционной смеси давали отстаиваться в течение 72 часов перед фильтрованием через слой целита. Полученный осадок промывали метанолом (150 мл) и фильтрат выпаривали досуха. Полученное твердое вещество растирали с дихлорметаном (400 мл) и фильтровали через слой целита. После промывки осадка дихлорметаном (200 мл) фильтрат выпаривали с получением продукта 2,2-диметилпирролидинона в виде светло-зеленой смолы (6,8 г).
Способ синтеза 2,2-диметилпирролидингидрохлорида:
Литийалюминийгидрид (1М раствор в тетрагидрофуране; 120 мл, 120 ммоль) медленно добавляли к перемешиваемому раствору 2,2-диметилпирролидинона (6 г, 53 ммоль) в тетрагидрофуране (150 мл). По окончании добавления реакцию нагревали до температуры кипения с обратным холодильником и перемешивали при кипячении с обратным холодильником в атмосфере аргона в течение 16 часов. Реакционной смеси давали охладиться до 0°C и затем добавляли воду (2,2 мл), 10% раствор гидроксида натрия (2,2 мл) и воду (6,6 мл) с 45 минутными интервалами. Полученную пастообразную смесь разбавляли диэтиловым эфиром (150 мл) и фильтровали через слой целита. Осадок промывали диэтиловым эфиром (250 мл) и фильтрат подкисляли хлористоводородной кислотой (1М раствор в диэтиловом эфире; 63 мл). Полученное твердое желтое вещество отфильтровывали с получением гидрохлорида 2,2-диметилпирролидина (4,8 г).
Способ синтеза этилового эфира (2,2-диметилпирролидин-1-ил)уксусной кислоты:
Этилхлорацетат (0,15 мл, 1,37 ммоль), карбонат калия (416 мг, 1,5 ммоль) и 2,2-диметилпирролидин (0,280 мг, 2,06 ммоль) суспендировали в этаноле (3 мл) и подвергали микроволновому облучению при 120°C в течение 45 мин. Полученную смесь суспендировали в диэтиловом эфире (30 мл), экстрагировали 2М хлористоводородной кислотой (30 мл) и отбирали органический слой. Водный слой обрабатывали небольшим избытком 4 н. раствора гидроксида натрия и экстрагировали диэтиловым эфиром (3×30 мл). Органические слои объединяли, сушили над сульфатом натрия, фильтровали и растворитель удаляли при пониженном давлении с получением этилового эфира (2,2-диметилпирролидин-1-ил)уксусной кислоты (156 мг).
Указанное в заголовке соединение получали, следуя способу примера 1, с использованием 7-хлориндола вместо 7-метоксииндола; тетрагидропиран-4-илметилового эфира толуол-4-сульфоновой кислота (полученного, как описано в примере 14) вместо циклогексилметилбромида и этилового эфира (2,2-диметилпирролидин-1-ил)уксусной кислоты вместо метилового эфира N,N-диметилглицина.
1H ЯМР (400 МГц, CD3OD) δ 1,45 (10H, м), 2,22 (5H, м), 3,35 (2H, м), 3,59 (1H, ушир.с), 3,91 (2H, д, J=11,1), 4,12 (1H, ушир.с), 4,52 (2H, д, J=7,1), 4,71 (1H, ушир.с), 4,95 (1H, ушир.с), 7,23 (1H, т, J=7,6), 7,32 (1H, д, J=7,6), 8,08 (1H, с), 8,13 (1H, д, J=7,6). EsIMS: m/z 429,5 [M+H]+.
Пример 40
1-(Циклогексил)метил-3-{5-[(пирролидин-1-ил)метил]-[1,2,4]тиадиазол-3-ил}-1H-индол-7-карбонитрил, гидрохлоридная соль
Суспензию [3-(7-бром-1-циклогексилметил-1H-индол-3-ил)-[1,2,4]тиадиазол-5-ил]метанола (полученного из 7-броминдола, как описано в примере 7; 2×250 мг, 0,16 ммоль), цианида цинка (II) (2×72 мг, 0,61 ммоль) и тетракис(трифенилфосфин)палладия (0) (2×21 мг, 18,3 моль) в ДМФ (2×4 мл) подвергали микроволновому облучению при 200°C в течение 5 мин с использованием Emrys™ Optimizer EXP. Реакционные смеси объединяли и выливали в разделительную воронку, добавляли к ним дихлорметан (≈50 мл). Органическую фазу последовательно промывали водой (2×20 мл), 1М водным раствором HCl (20 мл) и насыщенным раствором соли (20 мл), сушили над сульфатом магния, фильтровали и растворитель удаляли в вакууме. Полученное масло очищали колоночной флэш-хроматографией, элюируя 50-100% (об./об.) дихлорметаном в гептане и затем диэтиловым эфиром, с получением 1-циклогексилметил-3-(5-гидроксиметил[1,2,4]тиадиазол-3-ил)-1H-индол-7-карбонитрила (416 мг, 1,18 ммоль) в виде светло-желтого масла, которое кристаллизовалось при стоянии.
К раствору 1-циклогексилметил-3-(5-гидроксиметил[1,2,4]тиадиазол-3-ил)-1H-индол-7-карбонитрила (416 мг, 1,18 ммоль) в дихлорметане (40 мл) последовательно добавляли метансульфонилхлорид (0,110 мл, 1,42 ммоль) и триэтиламин (0,214 мл, 1,53 ммоль). Реакционную смесь перемешивали в течение 1 часа и выливали в разделительную воронку. Органическую фазу промывали 2М водным раствором карбоната натрия (20 мл), насыщенным раствором соли (20 мл), сушили над сульфатом магния, фильтровали, растворитель удаляли в вакууме с получением 3-(7-циано-1-циклогексилметил-1H-индол-3-ил)-[1,2,4]тиадиазол-5-илметилового эфира метансульфоновой кислоты (503 мг, 1,17 ммоль), который использовали без дополнительной очистки.
К раствору 3-(7-циано-1-циклогексилметил-1H-индол-3-ил)-[1,2,4]тиадиазол-5-илметилового эфира метансульфоновой кислоты (120 мг, 0,28 ммоль) в дихлорметане (3 мл) добавляли пирролидин (0,12 мл, 1,4 ммоль) и реакционную смесь подвергали микроволновому облучению при 100°C в течение 5 минут. Реакционную смесь сразу очищали колоночной флэш-хроматографией, элюируя дихлорметаном, затем 25-50% (об./об.) этилацетатом в гептане, с получением указанного в заголовке соединения (71 мг, 0,175 ммоль) в виде свободного основания. Свободное основание (35 мг, 0,086 ммоль) растворяли в дихлорметане (2 мл) и добавляли хлористый водород (1М раствор в диэтиловом эфире). Смесь концентрировали в вакууме с получением указанного в заголовке соединения в виде гидрохлоридной соли, 1:1.
1H ЯМР (400 МГц, CD3OD): 1,06-1,36 (7H, м), 1,62-1,80 (5H, м), 1,91-2,07 (1H, м), 2,09-2,34 (2H, ушир. м), 3,35-4,20 (4H, ушир.м), 4,42 (2H, д, J=7), 5,07 (2H, с), 7,37 (1H, т, J=8), 7,69 (1H, дд, J=7, 1), 8,23 (1H, с); 8,84 (1H, дд, J=8,1). EsIMS: m/z 406,4 [M+H]+.
Пример 41
Определение in vitro силы действия и эффективности в отношении человеческого рецептора CB1, экспрессирующегося в клетках CHO
Клетки яичника китайского хомяка (CHO), экспрессирующие человеческий рецептор CB1 и репортерный ген люциферазы, суспендировали в смеси феноловый красный/DMEM без сыворотки/питательная смесь F-12, содержащая пенициллин/стрептомицин (50 ед/50 мкг/мл) и фунгизон (1 мкг/мл), и высевали в 96-луночные планшеты в концентрации 3×104 клеток на лунку (конечный объем 100 мкл). Перед испытанием клетки инкубировали в течение ночи (приблизительно 18 часов при 37°C, 5% CO2/95% воздуха). Тестируемое соединение (10 мМ раствор в диметилсульфоксиде) разводили питательной смесью F12 с получением маточных растворов в диапазоне от 0,11 мМ до 0,11 нМ. Маточные растворы (10 мкл) непосредственно добавляли в соответствующие лунки. Для получения индуцированной агонистами экспрессии люциферазы планшеты инкубировали при 37°C в течение 5 часов. При ослабленном освещении в каждую лунку добавляли субстрат LucLite (Packard; воспроизведенный по инструкции производителя; 100 мкл). Планшеты накрывали верхней крышкой и затем инкубировали при комнатной температуре в течение 5 минут перед подсчетом на устройстве TopCount, Packard (подсчет единичных фотонов, время счета 0,01 минуты, 5-минутная задержка).
Для получения значения EC50 по методу наименьших квадратов по графику зависимости счета в секунду (CPS) от концентрации соединения (М) подбирали кривую наилучшего соответствия. В таблице 1 показаны значения pEC50, полученные для некоторых репрезентативных соединений по настоящему изобретению.

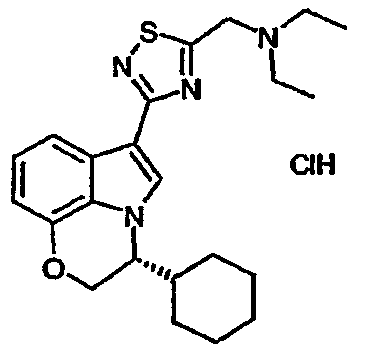
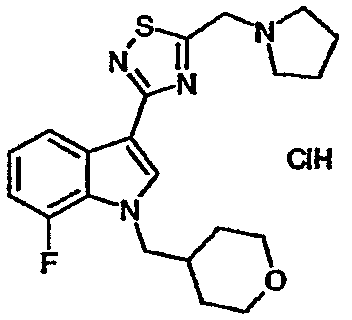
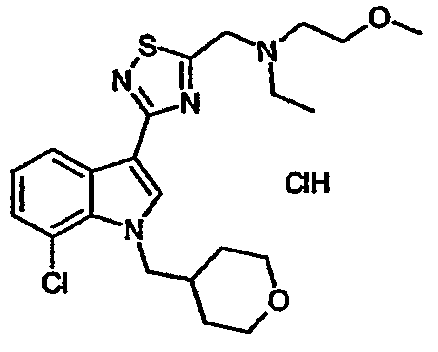
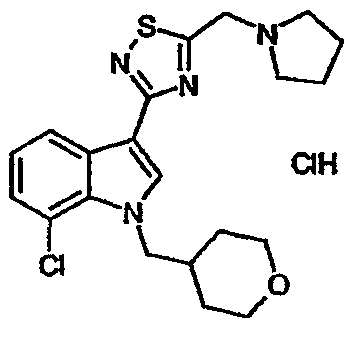
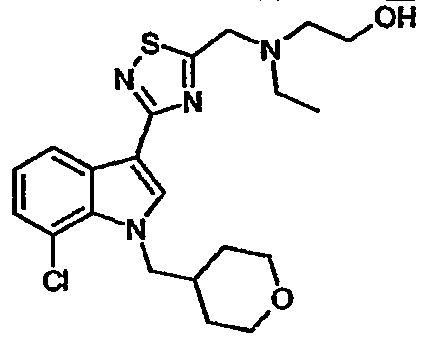
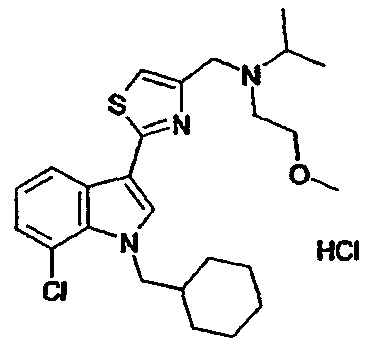
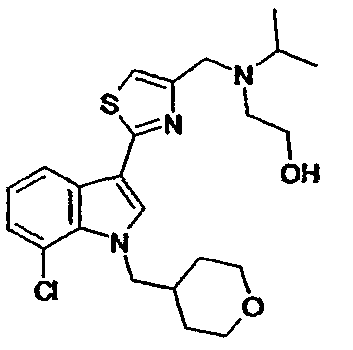
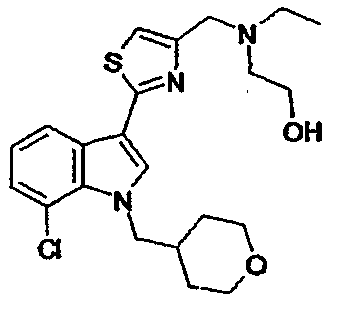
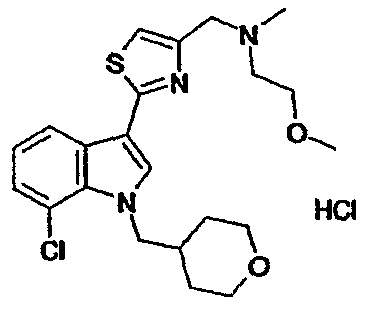

Пример 42
Латентный период отдергивания хвоста у мышей
Мышей обучали спокойно сидеть в устройстве для испытания отдергивания хвоста (Ugo Basile, Италия) в течение времени измерения латентного периода отдергивания хвоста. Хвост подвергали тепловому облучению фокусированным лучом в точке приблизительно 2,5 см от конца хвоста. Латентный период отдергивания хвоста определяли как промежуток времени между применением термического стимула и отдергиванием хвоста. Для предотвращения повреждения тканей применяли 12-секундные интервалы. Четыре группы, по восемь мышей каждая, обрабатывали носителем или одной из трех доз тестируемого соединения, вводимых внутривенно (носитель: 10% Tween-80 в физиологическом растворе; объем введения 10 мл/кг). Латентный период отдергивания хвоста измеряли перед введением тестируемого соединения, а также через регулярные промежутки времени (как правило, через 20, 40 и 60 минут) после введения соединения. ED50 вычисляли при Tmax.
Соединения по примерам 2G, 13, 14B, 15A, 15B, 15C, 20, 23B, 23C, 23D и 39 значительно увеличивали латентный период отдергивания хвоста с ED50<5 мкмоль/кг.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ 1-[(ИНДОЛ-3-ИЛ) КАРБОНИЛ] ПИПЕРАЗИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ И ЕГО ПРИМЕНЕНИЕ | 2003 |
|
RU2318821C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| СОЕДИНЕНИЯ 8-МЕТИЛ-1-ФЕНИЛИМИДАЗО[1, 5-а]ПИРАЗИНА | 2011 |
|
RU2560162C2 |
| МОЛЕКУЛЫ С ОПРЕДЕЛЕННОЙ ПЕСТИЦИДНОЙ АКТИВНОСТЬЮ И ОТНОСЯЩИЕСЯ К НИМ ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ, КОМПОЗИЦИИ И СПОСОБЫ | 2014 |
|
RU2650498C2 |
| Бициклические гетероциклические соединения и их применения в терапии | 2012 |
|
RU2662827C2 |
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2019 |
|
RU2809763C2 |
| ПОЛИГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МЕТАБОТРОПНОГО РЕЦЕПТОРА ГЛУТАМАТА | 2005 |
|
RU2381226C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 4-АМИНОЦИКЛОГЕКСАНА | 2009 |
|
RU2503660C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 4-АМИНОЦИКЛОГЕКСАНА | 2009 |
|
RU2532545C2 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
Изобретение относится к новым производным (индол-3-ил)гетероциклическим соединениям формулы 1:

где А представляет собой 5-членное ароматическое гетероциклическое кольцо, где
Х1, Х2 и Х3 независимо выбирают из N, О, S, CR; R означает Н, (С1-4)алкил; или R, когда он присутствует в Х2 или Х3, может вместе с R3 образовывать 5-8-членное кольцо; R1 означает 5-8-членное насыщенное карбоциклическое кольцо, необязательно содержащее гетероатом О; R2 означает Н; или R2 соединен с R7 с образованием 6-членного кольца, необязательно содержащего гетероатом О, и где указанный гетероатом связан с положением 7 индольного кольца; R3 и R4 независимо означают Н, (С1-6)алкил, необязательно замещенный ОН, (С1-4)алкилокси; или R3 вместе с R4 и N, с которым они связаны, образует 4-8-членное кольцо, необязательно содержащее дополнительный гетероатом, выбранный из О и S, и необязательно замещенное ОН, (С1-4)алкилом, (С1-4)алкилокси или (С1-4)алкилокси-(С1-4)алкилом; или R3 вместе с R5 образует 4-8-членное кольцо, необязательно замещенное ОН, (С1-4)алкилом, (С1-4)алкилокси; или R3 вместе с R, когда присутствуют в Х2 или Х3, образует 5-8-членное кольцо; R5 означает Н; или R5 вместе с R3 образует 4-8-членное кольцо, необязательно замещенное ОН, (С1-4)алкилом, (С1-4)алкилокси; R5′ означает Н; R6 означает один заместитель, выбранный из Н, (С1-4)алкила, (С1-4)алкилокси, галогена; R7 означает Н, (С1-4)алкил, (С1-4)алкилокси, галоген; или R7 соединен с R2 с образованием 6-членного кольца, необязательно содержащего дополнительный гетероатом О, и где гетероатом связан с положением 7 индольного кольца; или его фармацевтически приемлемая соль. Соединения формулы I проявляют активность агонистов к каннабиноидному рецептору СВ1, что позволяет использовать их для лечения боли, например, такой как хроническая боль, невропатическая боль, боль при раке. 4 н. и 6 з.п. ф-лы, 1 табл.
1. Производное (индол-3-ил)гетероциклического соединения общей формулы I
где А представляет собой 5-членное ароматическое гетероциклическое кольцо, где X1, Х2 и Х3 независимо выбирают из N, О, S и CR;
R представляет собой Н или (С1-4)алкил; или
R, когда он присутствует в Х2 или Х3, может вместе с R3 образовывать 5-8-членное кольцо;
R1 представляет собой 5-8-членное насыщенное карбоциклическое кольцо, необязательно содержащее гетероатом О;
R2 представляет собой Н; или
R2 соединен с R7 с образованием 6-членного кольца, необязательно содержащего гетероатом О, и где указанный гетероатом связан с положением 7 индольного кольца;
R3 и R4 независимо представляют собой Н или (С1-6)алкил, необязательно замещенный ОН или (С1-4)алкилокси; или
R3 вместе с R4 и N, с которым они связаны, образует 4-8-членное кольцо, необязательно содержащее дополнительный гетероатом, выбранный из О и S, и необязательно замещенное ОН, (С1-4)алкилом, (С1-4)алкилокси или (С1-4)алкилокси-(С1-4)алкилом; или
R3 вместе с R5 образует 4-8-членное кольцо, необязательно замещенное ОН, (С1-4)алкилом, (С1-4)алкилокси; или
R3 вместе с R, когда присутствуют в Х2 или Х3, образует 5-8-членное кольцо;
R5 представляет собой Н; или
R5 вместе с R3 образует 4-8-членное кольцо, необязательно замещенное ОН, (С1-4)алкилом или (С1-4)алкилокси;
R5′ представляет собой Н;
R6 представляет собой один заместитель, выбранный из Н, (С1-4)алкила, (С1-4)алкилокси или галогена;
R7 представляет собой Н, (С1-4)алкил, (С1-4)алкилокси или галоген; или
R7 соединен с R2 с образованием 6-членного кольца, необязательно содержащего дополнительный гетероатом О, и где гетероатом связан с положением 7 индольного кольца; или его фармацевтически приемлемая соль.
2. Производное (индол-3-ил)гетероциклического соединения по п.1, где R2 представляет собой Н или соединен с R7 с образованием 6-членного кольца, необязательно содержащего гетероатом О, связанный с положением 7 индольного кольца.
3. Производное (индол-3-ил)гетероциклического соединения по п.1 или 2, где R, R5, R5′ и R6 представляют собой Н.
4. Производное (индол-3-ил)гетероциклического соединения по п.1, где R1 представляет собой циклогексил или тетрагидропиранил.
5. Производное (индол-3-ил)гетероциклического соединения по п.4, где гетероцикл А представляет собой 1,2,4-оксадиазол (X1 представляет собой N, Х2 представляет собой О, Х3 представляет собой N), 1,2,4-тиадиазол (X1 представляет собой N, Х2 представляет собой S, Х3 представляет собой N) или тиазол (X1 представляет собой S, Х2 представляет собой CR, Х3 представляет собой N).
6. Производное (индол-3-ил)гетероциклического соединения по п.1, выбранное из:
7-хлор-3-(5-{[N-этил-N-(2-метоксиэтил)амино]метил}-[1,2,4]тиадиазол-3-ил)-1-(тетрагидропиран-4-ил)метил-1-Н-индола;
7-хлор-3-{5-[(пирролидин-1-ил)метил]-[1,2,4]тиадиазол-3-ил}-1-(тетрагидропиран-4-ил)метил-1Н-индола;
7-хлор-3-(5-{[N-этил-N-(2-гидроксиэтил)амино]метил}-[1,2,4]тиадиазол-3-ил)-1-(тетрагидропиран-4-ил)метил-1Н-индола;
7-хлор-3-(4-{[N-(2-гидроксиэтил)-N-изопропиламино]метил}-[1,3]тиазол-2-ил)-1-(тетрагидропиран-4-ил)метил-1Н-индола;
7-хлор-3-(4-{[N-этил-N-(2-гидроксиэтил)амино]метил}-[1,3]тиазол-2-ил)-1-(тетрагидропиран-4-ил)метил-1Н-индола;
7-хлор-3-(4-{[N-(2-метоксиэтил)-N-метиламино]метил}-[1,3]тиазол-2-ил)-1-(тетрагидропиран-4-ил)метил-1Н-индола;
7-хлор-3-{5-[(2,2-диметилпирролидин-1-ил)метил]-[1,2,4]оксадиазол-3-ил}-1-(тетрагидропиран-4-ил)метил-1Н-индола;
или их фармацевтически приемлемых солей.
7. Производное (индол-3-ил)гетероциклического соединения по п.1, проявляющее активность агонистов к каннабиноидному рецептору СВ1.
8. Фармацевтическая композиция, проявляющее активность агонистов к каннабиноидному рецептору СВ1, содержащая производное (индол-3-ил)гетероциклического соединения по любому из пп.1-6 в смеси с фармацевтически приемлемыми добавками.
9. Применение производного (индол-3-ил)гетероциклического соединения формулы I, определенного в п.1, в производстве лекарственного средства для лечения боли.
10. Способ лечения боли, такой как хроническая боль, невропатическая боль и боль при раке, включающий введение нуждающемуся в этом пациенту терапевтически эффективного количества производного (индол-3-ил)гетероциклического соединения по любому из пп.1-6.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| 3-АРИЛКАРБОНИЛ-1-АМИНОАЛКИЛ-1H-ИНДОЛЫ | 1992 |
|
RU2073670C1 |

