Перекрестная ссылка на родственные заявки
Настоящая заявка является заявкой с частичным продолжением одновременно рассматриваемой заявки №08/608776, поданной 29 февраля 1996, которая заявляет приоритет предварительной заявки №60/007071, поданной 26 октября 1995 г.
Ссылка на документы, раскрывающие сущность изобретения
Настоящая заявка включает материал, заключающийся в документе, раскрывающем сущность изобретения №377063, поданном 23 июня 1995; №386504, поданном 11 декабря 1995; №391109, поданном 7 февраля 1996, и №391228, поданном 7 февраля 1996.
Предпосылки создания изобретения
1. Область техники, к которой относится изобретение
Настоящее изобретение относится к способам, композициям и наборам для улучшения биологической доступности фармацевтических агентов, которые плохо абсорбируются из желудочно-кишечного тракта, а также к способам лечения пациентов путем перорального введения указанных агентов. В одном из своих аспектов настоящее изобретение относится к использованию циклоспоринов в целях усиления биологической доступности паклитаксела и родственных таксанов.
2. Описание предшествующего уровня
Многие ценные фармакологически активные соединения не могут достаточно эффективно вводиться перорально из-за их недостаточной системной абсорбции из желудочно-кишечного тракта. Поэтому все такие фармацевтические агенты обычно вводят внутривенно или внутримышечно, что требует вмешательства врача или специального медицинского персонала, и вызывает у пациента значительный дискомфорт, и влечет за собой возможные местные травмы, и даже возможное помещение пациента в стационар для оперативного вмешательства в случае проведения некоторых внутривенных вливаний.
Было сделано предположение, что в некоторых случаях недостаточная биологическая доступность лекарственного средства после его перорального введения является результатом активности связанного с мембраной Р-гликопротеина, который является переносчиком многих лекарственных соединений и который осуществляет энергетически зависимый транспорт или действует как отсасывающий насос для снижения внутриклеточной аккумуляции лекарственного средства путем удаления ксенобиотиков из клетки. Указанный Р-гликопротеин был иденфицирован в нормальных тканях секреторного эндотелия, такого как билиарная выстилка, щеточная каемка почечных проксимальных канальцев и люминальной поверхности тонкого кишечника и васкулярные эндотелиальные клетки, выстилающие гематоэнцефалический барьер, плаценту и яички.
Полагают, что Р-гликопротеин, действующий как откачивающий насос, предупреждает прохождение фармацевтических соединений через клетки слизистой оболочки тонкого кишечника и тем самым абсорбцию этих соединений в большой круг кровообращения. Было показано, что некоторые известные нецитотоксичные фармацевтические агенты ингибируют Р-гликопротеины, включая циклопротеин А (известный также как циклоспорин), верапамил, тамоксифен, хинидин, и фенотиазины, и т.п. Многие из этих исследований были направлены на достижение большей аккумуляции цитотоксических соединений внутри опухолевых клеток. Действительно, были проведены клинические испытания в целях исследования действия циклоспорина на фармакокинетику и токсичность паклитаксела (Fisher et al., Proc. Am. Soc. Clin. Oncol., 13, 143, 1994); доксорубицина (Bartlett et al., J. Clin. Onc. 12, 835-842, 1994); и этопозида (Lum et al., J.Clin. Onc. 10, 1635-42, 1992), которые являются противораковыми агентами и к которым, как известно, у пациентов вырабатывается полирезистентность (резистентность ко многим лекарственным средствам) (MDR). Эти испытания показали, что пациенты, которым внутривенно вводили циклоспорин вместе с противоопухолевыми средствами или до их введения, имели высокие уровни этих лекарственных средств в крови, что, вероятно, обусловлено снижением выведения лекарственного средства из организма; причем эти лекарственные средства обнаруживали ожидаемое токсическое действие при значительно более низких дозах. Полученные результаты свидетельствуют о том, что введение циклоспорина вместе с лекарственными средствами подавляет MDR-действие Р-гликопротеина, способствуя, тем самым, большей аккумуляции терапевтических агентов внутри клеток. Общее обсуждение фармакологических аспектов клинического использования ингибиторов Р-гликопротеина можно найти в работах Lum et al., Drug Resist. Clin. One. Hemat., 9, 319-336 (1995); Schinkel et al., Eur.J.Cancer, 31A: 1295-1298 (1995).
В вышеописанных исследованиях, касающихся использования циклоспорина для повышения уровней фармацевтических агентов в крови, по отношению к которым у пациента вырабатывается Р-гликопротеинопосредованная резистентность, указанные активные агенты и циклоспорин были введены внутривенно. В этих публикациях не было сделано каких-либо предположений относительно того, что циклоспорин или другие соединения, которые, как полагают, способны ингибировать Р-гликопротеиновый "откачивающий насос", могли бы быть введены перорально в целях значительного повышения биологической доступности перорально вводимых противораковых средств и других фармацевтических агентов, плохо абсорбируемых из кишечника в кровоток, но при этом так, чтобы они не продуцировали побочных эффектов с высоким уровнем токсичности. Действительно, в вышеуказанном обзоре 1995 Lum и др. показали, что совместное внутривенное введение MDR-ингибиторов и химиотерапевтических агентов, к которым вырабатывается полирезистентность (MDR), способствует увеличению уровней токсичности и обострению серьезных побочных эффектов у пациента. Schinkel и др. вкратце упоминают о том факте, что клетки слизистой оболочки тонкого кишечника являются обогащенными MDRI и Р-гликопротеинами, и это может влиять на биологическую доступность лекарственных средств, имеющих в качестве субстрата Р-гликопротеин, однако эти авторы не высказывали каких-либо предположений или не подразумевали, что пероральное введение MDR-подавляющих агентов может способствовать повышению биологической доступности пероральных лекарственных средств, обладающих плохой биодоступностью. Кроме того, как и Lum и др., Schinkel и др. предупреждают, что ингибиторы Р-гликопротеина могут резко увеличивать токсичность при проведении химиотерапии, а поэтому они должны применяться с большой осторожностью.
В более ранних публикациях Schinkel и др. показали, что абсорбция ивермецина, перорально вводимого мышам, гомозиготным по разрушению гена MDR-1, была более высокой, чем у нормальных мышей, что свидетельствовало о том, что Р-гликопротеин играет важную роль в снижении биологической доступности этого агента (Cell, 77, 491-502, 1994). Кроме того, это исследование также показало, что проникновение винбластина в различные ткани у мутантных мышей было более высоким.
При этом ни в одном из опубликованных исследований не описаны условия осуществления эффективного перорального введения относительно плохо усвояемых лекарственных средств, так, например, нигде не указаны соответствующие интервалы доз и схемы введения конкретных целевых лекарственных средств и агентов, усиливающих их биологическую доступность, а также нигде не указано, какие именно MDR-ингибирующие агенты являются наиболее подходящими для стимуляции абсорбции каждого из перорально вводимых лекарственных средств или определенного класса лекарственных средств.
Раскрытые ранее способы повышения кишечной абсорбции лекарственных средств, вводимых до настоящего времени лишь парентерально, были направлены, в основном, на использование агентов, усиливающих проницаемость и растворимость, в качестве стимулирующих агентов либо на совместное использование ингибиторов Р-гликопротеина путем внутрипросветной перфузии в тонкий кишечник или путем внутривенного введения, как описано, например, Leu и др. в Cancer Chemother. Pharmacol., 35: 432-436, 1995 (перфузия или инфузия i.v. хинидина подавляет выход из кровотока в полость желудочно-кишечного тракта). Однако все эти методы имеют ряд недостатков. Так, например, агенты, усиливающие растворимость и проницаемость, являются часто непригодными или неэффективными для перорального введения в необходимых дозах и могут неблагоприятным образом влиять на фармакологическую активность целевого лекарственного средства. Парентеральное введение человеку ингибиторов Р-гликопротеина в терапевтических (или близких к тератевтическим) дозах могут вызывать серьезные клинические осложнения. Например, в случае использования хинидина внутривенное введение может вызвать аритмию, вазодилатацию, желудочно-кишечные расстройства и т.п.
В опубликованной (10 августа 1995) РСТ-заявке WO 95/20980, Benet и др. раскрывают способ повышения биологической доступности перорально вводимых гидрофобных фармацевтических соединений. Этот метод заключается в пероральном введении пациенту указанных соединений вместе с биоусилителем, содержащим ингибитор фермента цитохрома Р450 3А или ингибитор Р-гликопротеинопосредованного мембранного транспорта. Однако, по существу, Benet и др. не раскрывают способа идентификации наиболее подходящих агентов, повышающих биологическую доступность специфических "целевых" фармацевтических соединений, и не указывают конкретных доз или схем применения усиливающих или целевых агентов. Действительно, хотя в заявке Benet указано множество потенциальных агентов, усиливающих биологическую доступность (ингибиторов Р450 3А), и целевых лекарственных средств (субстратов для Р450 3А), однако в этой заявке описана лишь экспериментально подтвержденная комбинация кетоконазола, используемого в качестве усиливающего агента, и циклоспорина А, используемого в качестве целевого лекарственного средства.
При описании основных характеристик соединений, которые могут быть использованы в качестве агентов, повышающих биологическую доступность благодаря подавлению транспортной активности Р-гликопротеина, Benet и др. показали, что эти соединения являются гидрофобными и содержат, но необязательно, два копланарных ароматических кольца, положительно заряженную азотную группу или карбонильную группу; причем этот класс соединений охватывает огромное число соединений, большинство из которых не в состоянии обеспечить нужное усиление абсорбции конкретно используемых целевых агентов. Кроме того, классы целевых агентов, раскрываемых Benet и др., включают в подавляющем большинстве фармацевтические агенты, приведенные в Physicians Desk References. Указанные критерии включения не представляют собой ценности для практических врачей, стремящихся найти безопасные, удобные в применении и эффективные способы перорального введения конкретных фармацевтических агентов.
Другим недостатком метода Benet и др. является критерий, применяемый для оценки повышения биологической доступности плохо абсорбируемого лекарственного средства после перорального введения. Benet и др. показали, что любой агент, ингибирующий Р-гликопротеин, присутствующий в кишечнике в данной концентрации и способствующий снижению транспорта через мембрану опосредованного Р-гликопротеином Родамина 123 в мембранных везикулах щеточной каемки или в Р-гликопротеинсодержащих клетках на 10% или более может рассматриваться как агент, увеличивающий биологическую доступность при данной концентрации, и может быть использован для практического осуществления их изобретения. Однако увеличение абсорбции из кишечника почти не абсорбируемого в обычных условиях лекарственного агента на 10% является недостаточным для того, чтобы сделать данный агент терапевтически ценным для использования в любых целях. Действительно, согласно нормативным документам Федерального управления по контролю за качеством пищевых продуктов и медикаментов два фармацевтических препарата, содержащих один и тот же активный ингредиент, но отличающихся по уровню своей биологической доступности на -20%/+25%, считаются все же биологически эквивалентными, поскольку для большинства лекарственных средств различие в концентрации активного ингредиента в крови на -20%/+25% не является клинически значимым. Approved Drug Products with Therapeutics Equivalence Evaluation (Dept. of HHS, 14th ed. 1994). Если в соответствии с нормами FDA два фармацевтических препарата считаются биологически эквивалентными, то лечащие врачи и фармацевты рассматривают эти два фармацевтических препарата взаимозаменяемыми.
Вообще говоря, Benet и др. не дают конкретных указаний относительно того, какими именно соображениями должны руководствоваться специалисты медики и фармацевты для идентификации подходящих комбинаций биоусилителя/целевого лекарственного средства или для разработки конкретных схем или приемов лечения, которые позволили бы добиться нужного терапевтического эффекта после перорального введения целевого агента.
Таким образом, необходимость в разработке безопасного и к тому же эффективного способа повышения системной доступности вводимых перорально лекарственных средств, которые до настоящего времени вводились только парентерально из-за их недостаточной или неадекватной абсорбции при пероральном введении, до сих пор остается актуальной, поскольку такой способ еще не был описан в предшествующих работах.
Краткое описание изобретения
Неожиданно было обнаружено и экспериментально подтверждено, что некоторые агенты, явно ингибирующие транспортную активность Р-гликопротеина, особенно циклоспорины, могут быть использованы для значительного повышения пероральной биологической доступности плохо усвояемых или вообще неусвояемых фармацевтических агентов, например, противораковых лекарственных средств таких, как паклитаксел (известный ранее как таксол), его аналоги и производные, и этопозид.
В одном из своих аспектов настоящее изобретение относится к способу повышения биологической доступности при пероральном введении фармацевтических агентов, которые плохо или вообще не абсорбируются из желудочно-кишечного тракта или кишечника, где указанный способ осуществляют путем предварительного и/или одновременного перорального введения одного или комбинации агентов, о которых известно, что они являются эффективными ингибиторами Р-гликопротеина, действующего как насос-переносчик лекарственного средства. В случае предварительного введения агент или агенты, повышающие биологическую доступность лекарственного средства, должны быть введены в достаточных количествах и незадолго до введения этого лекарственного средства ("целевое лекарственное средство" или "целевой агент") так, чтобы ко времени введения целевого агента в месте его абсорбции оставалось достаточное количество агента, повышающего биологическую доступность, и тем самым обеспечивалось эффективное ингибирование активности Р-гликопротеина или других переносчиков одновременно нескольких лекарственных соединений.
В своем втором аспекте настоящее изобретение относится к композициям и лекарственным формам для перорального введения фармацевтических агентов, которые до настоящего времени применялись только для парентерального введения. В своем третьем аспекте настоящее изобретение относится к введению таких пероральных лекарственных форм или их комбинации пациентам для лечения заболеваний, поддающихся воздействию активных агентов, содержащихся в указанных пероральных формах.
Настоящее изобретение также относится к фармацевтическим наборам, включающим одну или несколько пероральных лекарственных форм, содержащих целевой агент, и одну или несколько пероральных лекарственных форм, содержащих усиливающий агент.
Краткое описание чертежей
Фиг.1 - график, иллюстрирующий уровни паклитаксела в образцах сыворотки, взятых в течение периода 6-8 часов от трех групп крыс, где одной группе вводили внутривенно только паклитаксел, второй группе вводили паклитаксел только перорально; а третьей группе перорально вводили паклитаксел и циклоспорин А (называемый далее циклоспорином или CsA); причем дозы циклоспорина вводили до и непосредственно после введения дозы паклитаксела.
Фиг.2 - график, иллюстрирующий сравнительные уровни паклитаксела в сыворотке, взятой от двух из трех групп крыс, описанных в Фиг.1, а именно от группы, которой перорально вводили только один паклитаксел, и от группы, которой перорально вводили паклитаксел с предварительным или одновременным пероральным введением доз циклоспорина.
Фиг.3 - график, иллюстрирующий уровень паклитаксела в образцах плазмы, взятых за период 24 часа от двух групп крыс, где одной группе (А) перорально вводили циклоспорин за один час до перорального введения комбинации циклоспорина и паклитаксела, а второй группе (Р) перорально вводили один циклоспорин за один час до перорального введения паклитаксела.
Фиг.4 - график, иллюстрирующий уровни паклитаксела в образцах плазмы, взятых от двух групп крыс, где одной группе (G) вводили паклитаксел IV через 3 часа после введения пероральной дозы циклоспорина, а второй группе (Н) вводили только паклитаксел IV.
Фиг.5 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых от трех групп крыс за период 24 часа, где одной группе (Группа А) вводили только радиоактивный меченный паклитаксел IV, второй группе (Группа В) вводили перорально только радиоактивно меченный паклитаксел, а третьей группе (Группа С) вводили перорально радиоактивно меченный паклитаксел вместе с пероральными дозами циклоспорина, которые вводили за 1 час до и сразу после введения дозы паклитаксела.
Фиг.6 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых у отдельных крыс Группы В (описанной для Фиг.5).
Фиг.7 - график, иллюстрирующий уровни радиоактивности, зарегистрированной в образцах цельной крови, взятых у отдельных крыс Группы С (определенной выше в описании Фиг.5).
Фиг.7А - график, иллюстрирующий уровни полной радиоактивности и неизмененного паклитаксела, зарегистрированные в образцах цельной крови, взятых за период 24 часа от группы из 10 крыс, которым был перорально введен радиоактивно меченный паклитаксел (9 мг/кг) и пероральные дозы циклоспорина (5 мг/кг), введенные за 2 часа до и непосредственно после введения дозы паклитаксела.
Фиг.7В - график, иллюстрирующий уровни полной радиоактивности и метаболитов паклитаксела 1, 2 и 3, зарегистрированные в образцах цельной крови, взятых от группы из 10 крыс (определенной выше для Фиг.7А) за период времени 24 часа.
Фиг.8 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых за период 24 часа от трех групп крыс, где одной группе перорально вводили 10 мг/кг верапамила в качестве усиливающего агента, второй группе перорально вводили прогестерон в качестве усиливающего агента, а третьей группе перорально вводили дипиридамол в качестве усиливающего агента; причем каждой из этих групп вводили пероральную дозу того же самого усиливающего агента через один час после введения пероральной дозы радиоактивно меченного паклитаксела.
Фиг.9 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятые за период 24 часа у крыс первой группы, определенной выше для Фиг.8 (которой вводили 10 мг/кг верапамила перорально); у крыс группы, которой перорально вводили лишь один радиоактивно меченный паклитаксел, и у крыс группы, которой перорально вводили циклоспорин за один час и непосредственно после введения пероральной дозы радиоактивно меченного паклитаксела.
Фиг.10 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятые за период 24 часа у крыс второй группы, определенной выше для Фиг.8 (которой вводили перорально прогестерон), у группы крыс, которой вводили перорально один паклитаксел, и у группы крыс, которым перорально вводили циклоспорин за 1 час до введения, а затем сразу после введения радиоактивно меченного паклитаксела.
Фиг.11 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых за период 24 часа у крыс третьей группы, определенной выше для Фиг.8 (которой перорально вводили дипиридамол); у группы крыс, которым перорально вводили лишь один паклитаксел; и у группы крыс, которым перорально вводили циклоспорин за 1 час до введения, а затем сразу после введения перорального радиоактивно меченного паклитаксела.
Фиг.12 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых за период 24 часа от трех групп крыс, где одной группе перорально вводили 100 мг/кг верапамила (Как показано на Фиг.12, крысы, получившие высокую дозу верапамила не выживали более чем примерно 8 часов) в качестве усиливающего агента, второй группе вводили перорально мегестерола ацетат (торговой марки MEGACER, Bristol-Myers Squibb Oncology) в качестве усиливающего агента, а третьей группе перорально вводили кетоконазол в качестве усиливающего агента; причем каждой группе вводили ту же самую пероральную дозу того же самого усиливающего агента через один час после введения пероральной дозы радиоактивно меченного паклитаксела.
Фиг.13 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых за период 24 часа у крыс первой группы, определенной выше для Фиг.12 (которой перорально вводили 100 мг/кг верапамила); у крыс группы, которой перорально вводили лишь один радиоактивно меченный паклитаксел; у крыс группы, которой перорально вводили циклоспорин за один час до введения, а затем сразу после введения пероральной дозы радиоактивно меченного паклитаксела.
Фиг.14 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых за период 24 часа у крыс второй группы, определенной выше для Фиг.12 (которой перорально вводили магестролацетат); у крыс группы, которой перорально вводили лишь один радиоактивно меченный паклитаксел; у крыс группы, которой перорально вводили циклоспорин за один час до введения, а затем сразу после введения пероральной дозы радиоактивно меченного паклитаксела.
Фиг.15 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых за период 24 часа у крыс третьей группы, определенной выше для Фиг.12 (которым перорально вводили кетоконазол); у крыс группы, которой перорально вводили лишь один радиоактивно меченный паклитаксел; у крыс группы, которой перорально вводили циклоспорин за один час до введения, а затем сразу после введения пероральной дозы радиоактивно меченного паклитаксела.
Фиг.16 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых за период 24 часа у крыс первой группы, определенной для Фиг.8 (которой вводили 10 мг/кг верапамила); у крыс первой группы, определенной выше для Фиг.12 (которой вводили 100 мг/кг верапамила); у крыс группы, которой перорально вводили лишь один радиоактивно меченный паклитаксел; у крыс группы, которой перорально вводили циклоспорин за один час до введения и затем сразу после введения перорально дозы радиоактивно меченного паклитаксела.
Фиг.17 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых за период 24 часа у крыс второй группы, определенной выше для Фиг.8 (которой перорально вводили прогестерон); у крыс второй группы, определенной выше для Фиг.12 (которой перорально вводили мегестролацетат); у крыс группы, которой перорально вводили лишь один радиоактивно меченный паклитаксел; у крыс группы, которой перорально вводили циклоспорин за один час до введения, а затем сразу после введения перорально дозы радиоактивно меченного паклитаксела.
Фиг.17А - график, иллюстрирующий сравнение кривой дозовой зависимости, построенный для группы крыс, которым перорально вводили циклоспорин за один час до введения, а затем сразу после введения пероральной дозы радиоактивно меченного паклитаксела, с кривой дозой зависимости, построенной для группы крыс, которым перорально вводили кетоканазол за один час до введения, а затем сразу после введения пероральной дозы радиоактивно меченного паклитаксела.
На Фиг.17В показано сравнение величин AUC0-24, определенных для тех же самых двух групп крыс.
Фиг.18 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых за период времени 24 часа от трех групп крыс, где одной группе вводили внутривенно (IV) только радиоактивно меченный этопозид; второй группе перорально вводили только радиоактивно меченный этопозид; а третьей группе перорально вводили радиоактивно меченный этопозид в сочетании с пероральными дозами циклоспорина, которые вводили до и сразу после введения дозы этопозида; причем на этом графике по оси ординат отложены эквиваленты этопозида (млн.д.) от 0 до 1 в цельной крови.
Фиг.19 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых от трех групп крыс, определенных выше для Фиг.18; где по оси ординат отложены эквиваленты радиоактивно меченного этопозида (млн.д.) от 0 до 0,2 в цельной крови.
Фиг.20 - график, иллюстрирующий процент (%) средней кумулятивной дозы радиоактивности, зарегистрированной в экскрементах и моче, взятых от трех групп крыс за период 168 часов, где одной группе вводили внутривенно только радиоактивно меченный паклитаксел; второй группе перорально вводили только радиоактивно меченный паклитаксел; а третьей группе перорально вводили радиоактивно меченный паклитаксел в сочетании с пероральными дозами циклоспорина до или сразу после введения паклитаксела.
Фиг.21 - гистограмма, иллюстрирующая средние величины (млн.д.) эквивалентов паклитаксела, зарегистрированные в крови и плазме от трех групп крыс, определенных для Фиг.20, через 168 часов (7 дней) после введения паклитаксела.
Фиг.22 - гистограмма, иллюстрирующая средние величины (млн.д.) эквивалентов плакситаксела, зарегистрированные в различных тканях (печени, почках, яичках и костях) для тех групп крыс, определенных выше для Фиг.20, через 168 часов (7 дней) после введения паклитаксела.
Фиг.23 - гистограмма, иллюстрирующая средние величины (млн.д.) эквивалентов паклитаксела, зарегистрированные в различных тканях (мышцах, поджелудочной железе, костях, легких и семенных пузырьках), происходящих от крыс трех групп, определенных выше для Фиг.20, через 168 часов (7 дней) после введения паклитаксела.
Фиг.24 - гистограмма, иллюстрирующая средние величины (млн.д.) эквивалентов паклитаксела, зарегистрированные в различных тканях (головном мозге, сердце, желудочно-кишечном тракте, селезенке и предстательной железе) крыс от трех групп, определенных выше для Фиг.20, через 168 часов (7 дней) после введения паклитаксела.
Фиг.25 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых за период времени 24 часа от трех групп крыс, где одной группе перорально вводили циклоспорин D за час до и сразу после введения пероральной дозы радиоактивно меченного паклитаксела; второй группе перорально вводили циклоспорин G за один час до и сразу после введения пероральной дозы радиоактивно меченного паклитаксела; а третьей группе вводили циклоспорин А за один час до и сразу после введения пероральной дозы радиоактивно меченного паклитаксела.
Фиг.26 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых за период времени 24 часа от трех групп крыс, где одной группе перорально вводили кетоконазол за один час до и сразу после введения пероральной дозы радиоактивно меченного паклитаксела; второй группе перорально вводили комбинированную дозу циклоспорина А и кетоконазола за один час до и сразу после введения пероральной дозы радиоактивно меченного паклитаксела; а третьей группе вводили циклоспорин А за один час и сразу после введения пероральной дозы радиоактивно меченного паклитаксела.
Фиг.27 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых за период времени 24 часа от трех групп крыс, где одной группе перорально вводили каптоприл за два часа до и сразу после введения пероральной дозы радиоактивно меченного паклитаксела; второй группе вводили циклоспорин А за один час до и сразу после введения пероральной дозы радиоактивно меченного паклитаксела; а третьей группе перорально вводили лишь один радиоактивно меченный паклитаксел.
Фиг.28 иллюстрирует профиль радиоактивности в ВЭЖХ-экстракте плазмы от крыс Группы С, определенной выше для Фиг.5.
Фиг.29 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых за период времени 24 часа от четырех групп крыс, где одной группе перорально вводили 10 мг/кг циклоспорина D за один час до и сразу после введения пероральной дозы радиоактивно меченного паклитаксела; второй группе перорально вводили 10 мг/кг циклоспорина F за один час до и сразу после введения пероральной дозы радиоактивно меченного паклитаксела; третьей группе вводили 5 мг/кг циклоспорина D за один час до и сразу после введения пероральной дозы радиоактивно меченного паклитаксела; а четвертой группе вводили 5 мг/кг циклоспорина F за один час до и сразу после введения пероральной дозы радиоактивно меченного паклитаксела.
Фиг.30 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых от трех групп крыс за период времени 24 часа, где одной группе (Группе А) внутривенно вводили только радиоактивно меченный доцетаксел ("Таксотер"); второй группе (Группе В) перорально вводили только радиоактивно меченный доцетаксел; а третьей группе (Группе С) перорально вводили радиоактивно меченный доцетаксел в сочетании с пероральными дозами циклоспорина, вводимого за 1 час до и сразу после введения дозы доцетаксела; причем на этом графике по оси ординат отложены средние величины (мл.д.) эквивалентов доцетаксела.
Фиг.31 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых от трех групп крыс, определенных выше для Фиг.30; причем на этом графике по оси ординат отложены средние величины (мл. д.) эквивалентов доцетаксела от 0 до 2.0.
Фиг.32 - график, иллюстрирующий уровни радиоактивности, зарегистрированные в образцах цельной крови, взятых от трех групп крыс за период времени 24 часа, где одной группе (Группе А) внутривенно вводили лишь один радиоактивно меченный паклитоксел; второй группе (Группа В) перорально вводили лишь один радиоактивно меченный паклитаксел; а третьей группе (Группе С) перорально вводили радиоактивно меченный паклитаксел в сочетании с пероральными дозами CSP, вводимыми до и сразу после введения дозы паклитаксела.
Фиг.33 - график, иллюстрирующий уровни неизмененного радиоактивно меченного паклитаксела в образцах цельной крови, взятых от трех групп крыс, определенных выше для Фиг.32, через 1-24 часа после введения дозы.
Фиг.34 - график, иллюстрирующий уровни неизмененного радиоактивно меченного паклитаксела в образцах цельной крови, взятых через 0-12 часов после введения дозы у крыс Группы А, определенной выше для Фиг.32, и крыс четвертой группы (Группы D), которой вводили внутривенно радиоактивно меченный паклитаксел в сочетании с пероральными дозами циклоспорина, вводимыми за 1 час до и сразу после введения дозы паклитаксела; причем на этом графике по оси ординат отложены мл.д. паклитаксела от 0 до 30.
Фиг.35 - график, иллюстрирующий уровни неизмененного радиоактивно меченного паклитаксела в образцах цельной крови, взятых через 1-12 часов после введения дозы у крыс Группы А, определенной выше для Фиг.32, и у крыс Группы D, определенной выше для Фиг.34, причем на этом графике по оси ординат отложены мл.д. паклитаксела от 0,000 до 5,000.
Фиг.36-41 схематически иллюстрируют способ экстракции и распределение радиоактивности, зарегистрированной в композите (гомогенате) от различных органов крыс Групп А и С, соответственно, определенных выше для Фиг.32.
Фиг.42 - график, иллюстрирующий уровни паклитаксела, зарегистрированные в образцах плазмы, взятых через определенные промежутки времени у группы из десяти крыс на третий и на четвертый дни эксперимента, где этим крысам два раза в день вводили пероральную дозу (5 мг/кг) циклоспорина, а затем, через 1 час, вводили комбинацию той же самой пероральной дозы циклоспорина + пероральную дозу (3 мг/кг) паклитаксела.
Подробное описание изобретения
В общих чертах настоящее изобретение относится к повышению пероральной абсорбции и биологической доступности фармакологически активных агентов после их перорального введения, особенно агентов, которые плохо абсорбируются или вообще не абсорбируются из желудочно-кишечного тракта или кишечника. В своем предпочтительном варианте настоящее изобретение относится (а) к способу повышения пероральной биологической доступности противоопухолевых агентов, в частности паклитаксела (известного в настоящее время под товарным знаком TAXOL® Bristol-Myers Squibb Oncology Division) и его производных; других таксанов; полусинтетического аналога паклитаксела, доцетаксела (N-дебензоил-N-трет-бутоксикарбонил-10-деацетилпаклитаксел), производимого под товарным знаком TAXOTERE®, Rhone-Poulenc Rorer S.A.; и этопозида; (b) к лекарственным формам и наборам для перорального введения противоопухолевых агентов и других лекарственных средств, которые до настоящего времени вводились лишь парентерально; и (с) к способам лечения пациентов, страдающих раком, с использованием указанных пероральных форм или их комбинаций.
Термины "пероральная биологическая доступность" и "биологическая доступность при пероральном введении", используемые в настоящем описании, относятся к системной доступности (т.е., уровни в крови/плазме) данного количества лекарственного средства, перорально вводимого пациенту.
Паклитаксел представляет собой натуральный дитерпеновый продукт, выделенный из тихоокеанского (коротколистного) тиса (Taxus brevifolia). Он является членом таксанового семейства терпенов. Впервые паклитаксел был выделен в 1971 г. Wani и др. (J. Am. Chem. Soc., 93: 2325, 1971), которые охарактеризовали его структуру химическими методами и методами рентгеновской кристаллографии. Один из механизмов активности паклитаксела заключается в его способности к связыванию тубулина и, тем самым, к ингибированию роста раковых клеток. Schiff et al., Proc. Natl. Acad. Sci. USA, 77: 1561-1565 (1980); Schiff et al., Nature, 277: 665-667 (1979); Kumar, J. Biol. Chem., 256: 10435-10441 (1981).
Паклитаксел был разрешен в США для клинического использования при лечении не поддающегося лечению рака яичника (Markman et al., Yale Journal of Biology amd Medicine, 64: 583, 1991; McGuire et al., Ann. Intern. Mod., 111; 273, 1989). Паклитаксел является эффективным для химиотерапии некоторых видов рака, включая рак молочной железы (Holmes et al., J. Nat. Cancer Inst., 83; 1797, 1991), и был также разрешен для лечения рака молочной железы. Он является потенциальным кандидатом для получения препарата, который может быть использован для лечения опухолей кожи (Einzig et al., Proc. Am. Soc. Clim Oncol., 20, 46), а также карциномы головы и шеи (Forastire et al., Sem. Oncol., 20: 56, 1990). Это соединение может быть также использовано для лечения поликистоза почек (Woo et al., Nature, 368; 750, 1994), рака легких и малярии.
Паклитаксел обладает лишь очень небольшой растворимостью в воде, что создает большие проблемы при разработке подходящих препаратов для инъекций и инфузий, используемых в противораковой химиотерапии. Некоторые препараты паклитаксела для внутривенных инфузий были разработаны с использованием CREMOPHOR EL™ (полиэтоксилированное касторовое масло) в качестве носителя лекарственного средства, поскольку паклитаксел почти не растворяется в воде. Так, например, паклитаксел, использованный в клинических испытаниях под защитой NCl был изготовлен в виде препарата в 50% CREMOPHOR EL™ и в 50% дегидратированного спирта. Однако сам CREMOPHOR EL™, при его введении внутривенно, является токсичным и вызывает вазодилатацию, одышку, летаргию, гипотензию и смерть у собак. Полагают, что CREMOPHOR EL™ является ответственным за реакции аллергического типа, наблюдаемые при введении паклитаксела.
В целях улучшения растворимости паклитаксела и разработки более безопасных клинических препаратов были проведены исследования, направленные на синтез аналогов паклитаксела, где 2- и/или 7-положения дериватизированы группами, которые должны усиливать растворимость. Эти исследования привели к получению пролекарственных соединений, которые обладают большей растворимостью в воде, чем исходные соединения, и которые обнаруживают цитотоксические свойства после активации. Одной из важных групп таких пролекарственных соединений являются 2'-ониевые соли паклитаксела и доцетаксела, в частности соли мезилата 2'-метилпиридиния (2'-MPM).
Паклитаксел очень плохо абсорбируется при пероральном введении (менее, чем 1%) (см. Eiseman et al., Second NCI Workshop on Taxol and Taxus (сент. 1992); Suffness et al., в Taxol Science and Applications (CRC Press 1995). Eiseman и др. показали, что биологическая доступность паклитаксела после перорального введения составляет 0%, a Suffness и др. сообщают, что пероральное введение паклитаксела, по всей вероятности, неэффективно, поскольку при дозе 160 мг/кг/день он не обнаруживал заметной противоопухолевой активности. Кроме того, не был разработан эффективный способ перорального введения паклитаксела (т.е., способ, позволяющий повысить биологическую доступность паклитаксела при пероральном введении) или других таксанов или аналогов паклитаксела, таких как доцетаксел, которые обладают противоопухолевой активностью. Исходя из этого до настоящего времени паклитаксел не использовался для перорального введения человеку и поэтому не использовался должным образом для лечения заболеваний, восприимчивых к паклитакселу.
Доксетаксел изготавливается промышленностью в виде парентерального препарата под товарным знаком TAXOTERE® и применяется для лечения рака молочной железы. До настоящего времени в научной литературе не встречалось каких-либо ссылок или указаний на пероральную абсорбцию доксетаксела у животных или человека.
Этопозид представляет собой полусинтетическое производное подофиллотоксина и используется для лечения некоторых опухолевых заболеваний, в частности рака половых органов (например, рака яичек (тестикулярный рак)) и мелкоклеточного рака легких (Loehrer. Sem. One., 19, №6, supp. 14, rr. 48-52, 1992). Пероральный препарат этопозида изготавливается промышленностью (в виде капсул VEPESID®, Bristol-Myers Squibb Oncology), но этот препарат не обладает хорошей пероральной абсорбируемостью (среднее значение пероральной биологической доступности для капсул этопозида составляет приблизительно 50%).
Циклоспорины представляют группу неполярных циклических олигопептидов (некоторые из которых обладают иммунодепрессивной активностью), продуцируемых микроорганизмами рода Topycladium, включая, например, Topycladium inflatum Gams (именуемый ранее Trichoderma polysporum). Topycladium terricola и другие низшие грибы. Был идентифицирован главный представитель этой группы соединений, циклоспорин А (циклоспорин или CsA) вместе с несколькими его менее крупными метаболитами, например, циклоспоринами В-Z, некоторые из которых обнаруживают значительно меньшую иммунодепрессивную активность, чем циклоспорин А. Был также получен ряд синтетических и полусинтетических аналогов. В общих чертах, см. Jegorov и др., Phytochemistry, 38: 403-407 (1995). Настоящее изобретение охватывает натуральные, полусинтетические и синтетические аналоги циклоспоринов.
Циклоспорины представляют собой нейтральные, липофильные, циклические ундекапептиды с молекулярными массами, составляющими около 1200. Обычно они используются внутривенно или перорально в качестве иммунодепрессантов, главным образом при трансплантации органов и некоторых других состояниях. Циклоспорины, особенно циклоспорин А (циклоспорин), являются известными ингибиторами Р- гликопротеина, действующего как откачивающий насос, а также некоторых расщепляющих ферментов Р450, однако до настоящего времени не было разработано эффективной схемы для клинического применения этого свойства циклоспоринов, такой, чтобы она была осуществима и целесообразна с клинической и с коммерческой точки зрения, или чтобы она давала возможность проводить контрольные испытания.
С механистической точки зрения перорально введенный циклоспорин способен ингибировать Р-гликопротеиновый насос в верхнем отделе тонкой кишки, являющимся участком, в котором происходит наибольшая абсорбция лекарственного средства. При внутривенном введении лекарственное средство, подвергающееся высокой степени метаболизма, такое как циклоспорин, очевидно не способно оставаться интактным в той области кишечника, в которой обычно абсорбируются лекарственные средства. После парентерального введения циклоспорин экстрагируется печенью и поступает в желчные протоки и кишечник, дистально расположенные по отношению к указанной области оптимальной абсорбции. Одно из неожиданных открытий настоящего изобретения заключается в том, что иммуносупрессия, наблюдаемая при использовании некоторых циклоспоринов, не обязательно является необходимым условием для повышения пероральной биологической доступности терапевтических агентов. Так, например, циклоспорин F повышает пероральную биологическую доступность паклитаксела, даже не обладая, как сообщается в литературе, иммунодепрессивной активностью. См. Stewart et al., Transplantation Proceedings, 20: (Supp. 3) 989-992 (1988); Granelli-Piperno et al. Transplantation 46: 53-60S (1988).
Кетоконазол представляет собой широко используемое фунгицидное имидазоловое производное, которое также, до некоторой степени, используется для лечения карциномы предстательной железы. Было показано, что кетоконазол, в соответствии с одним из своих свойств, способен подавлять в высокорезистентных клетках карциномы человека KB (Siegsmund et al., J.Urology, 151; 485-491, 1994) и, кроме того, он может также ингибировать действие ферментов, катализирующих метаболизм лекарственных соединений, например таких ферментов, как цитохром Р450.
Было обнаружено, что многие фармацевтические агенты, имеющие плохие профили пероральной абсорбции, могут быть с успехом введены перорально и при этом обнаруживать достаточную системную абсорбцию и терапевтическую активность при условии, что пероральное введение будет сопровождаться пероральным введением определенной дозы циклоспорина или других агентов, о которых известно, что они ингибируют лекарственную полирезистентность, лекарственно-транспортную активность Р-гликопротеинового внутриклеточного насоса, а также некоторых усиливающих агентов, способность которых ингибировать Р- гликопротеиновый транспорт пока еще не установлена. Еще одно неожиданное открытие настоящего изобретения заключается в том, что при тех же самых условиях пероральное введение приводит к более благоприятному фармакокинетическому профилю, к лучшему проникновению в ткани и к более высокому объему распределения целевого терапевтического агента.
В исследованиях с использованием животных было обнаружено, что некоторые агенты, подавляющие лекарственную полирезистентность, такие как циклоспорин и кетоконазол, при их пероральном введении непосредственно после и/или до введения лекарственных средств, таких как паклитаксел и этопозид, повышают абсорбцию этих лекарственных средств из кишечника до неожиданно высокого уровня, что позволяет достигать соответствующих терапевтических условий. Однако при этом остается не совсем ясным, можно ли объяснить полученные результаты подавлением Р-гликопротеинового насоса.
Другим возможным объяснением наблюдаемой повышенной биологической доступности паклитаксела и этопозида является тот факт, что между циклоспорином и паклитакселом может иметь место взаимодействие на уровне ферментов, катализирующих метаболизм лекарственных соединений. Известно, что оба эти агента подвергаются высокой степени метаболизма под действием системы цитохрома Р-450 (например, Р450 3А), которая концентрируется в печени, а также в тонком кишечнике. Предполагается, что циклоспорин, который был введен первым, может ингибировать эти ферменты так, чтобы паклитаксел, который является неполярным и липофильным соединением, мог абсорбироваться. При отсутствии такого локального ингибирования паклитоксел подвергается метаболизму с образованием более полярных метаболитов, которые не должны проходить через клетки слизистой оболочки. Невозможность продемонстрировать фармакокинетическое взаимодействие между циклоспорином и паклитакселом в случае введения циклоспорина за 3 часа до внутривенного введения паклитаксела, дает основание предположить, что местом вышеуказанного взаимодействия является полость кишечника. Однако даже это теоретическое обоснование не может объяснить неожиданное открытие авторов настоящего изобретения, заключающееся в том, что некоторые ингибиторы Р-гликопротеина (например, циклоспорины и кетоконазол) способствуют повышению пероральной биологической доступности специфических целевых лекарственных средств до достаточно высокого уровня, тогда как другие агенты, которые, как известно, также являются ингибиторами активного Р-гликопротеина, обнаруживают незначительную активность в качестве усилителей пероральной абсорбции для тех же самых целевых лекарственных средств.
В соответствии с теорией ингибирование кишечного метаболизма целевого агента не должно приводить или должно приводить к незначительному увеличению уровня целевого агента в общем кровотоке при его внутривенном введении. Кроме того, поскольку главным эффектом агента, повышающего пероральную абсорбцию, может быть локальный эффект в полости кишечника, то для достижения нужного эффекта должны быть эффективны субтерапевтические дозы. Это соображение является очень важным, если принять во внимание, что в случае использования в качестве усиливающего агента циклоспорина, обладающего сильной иммунодепрессивной активностью, введение высоких доз этого агента может создать проблемы, связанные с его токсичностью. Поэтому обнаружение авторами настоящего изобретения того факта, что циклоспорины, не обладающие иммунодепрессивной активностью, такие как циклоспорин F, могут также действовать как пероральные усилители абсорбции, имеет очень большое клиническое значение.
Важно отметить, что хотя нами была высказана гипотеза относительно механизмов действия, лежащих в основе настоящего изобретения, однако, в действительности, нам не известны механизмы, объясняющие обсуждаемые выше неожиданно обнаруженные факты, но это не мешает практическому использованию настоящего изобретения любым специалистом.
Способ настоящего изобретения, направленный на повышение пероральной биологической доступности целевого терапевтического агента, обладающего низкой пероральной усвояемостью в обычных условиях (т.е. его средняя биологическая доступность составляет 50% или менее), предусматривает пероральное введение млекопитающему (человеку или животному) агента, повышающего пероральную абсорбцию или биологическую доступность целевого агента одновременно с введением этого целевого агента, либо до его введения, либо одновременно с введением и до перорального введения этого целевого агента в целях повышения количества и продолжительности абсорбции интактного целевого агента в кровоток.
Такими перорально вводимыми усиливающими абсорбцию агентами, которые могут быть использованы в целях настоящего изобретения, но не ограничиваются ими, являются следующие соединения:
- циклоспорины, включая циклоспорины А-Z, а особенно циклоспорин А (циклоспорин), циклоспорин F, циклоспорин D, дигидроциклоспорин А, дигидроциклоспорин С, ацетилциклоспорин A, PSC-833, SDZ-NIM 8112 (SDZ-NIM 811 - (Me-11e-4)циклоспорин, противовирусный циклоспорин, не являющийся иммунодепрессантом) (оба производятся фармацевтической корпорацией Sandoz Pharmaceutical Corp), и родственные олигопептиды, продуцируемые микроорганизмами рода Topycladium. Структуры циклоспоринов А описаны ниже в Таблице 1;
- фунгициды - кетоконазол;
- лекарственные средства, действующие на сердечно-сосудистую систему - MS-209 (от BASF), амиодарон, нифедипин, резерпин, хинидин, никардипин, этакриновая кислота, пропафенон, резерпин, амилорид;
- натуральные продукты против мигрени - эргоалкалоиды;
- антибиотики - цефоперазон, тетрациклин, хлорохин, фосфомицин;
- антипаразитарные средства - ивермектин;
- ингибиторы полилекарственной резистентности - VX-710 и VX-853 (Vertex Pharmaceuticals Incorporated);
- ингибиторы тирозинкиназы - генистеин и родственные изофлавоноиды, кверцетин;
- ингибиторы протеинкиназы С - калфостин;
- индукторы апоптоза - керамиды;
- агенты, обладающие активностью против рецепторов эндорфина - морфин, другие представители соединений класса морфинов, другие опиаты и антагонисты опиатов, включая (но не ограничиваясь ими) налогсон, налтрексон и налмефен.
К классу перорально вводимых целевых терапевтических агентов, пероральная абсорбция которых возрастает при введении усиливающих агентов, относятся, но не ограничиваются ими, следующие соединения:
- паклитаксел, другие таксаны, доцетаксел, их производные и пролекарства, в частности их 2 - МРМ-соли и другие 2'-метилпиридиниевые соли;
- другие химиотерапевтические агенты, которые имеют низкую или в высокой степени нестабильную пероральную биологическую доступность, включая этопозид, камптотецин, СРТ-11 (Pharmacia и Upjohn), топетекан (SmithKline Beecham), доксорубин, винкристин, даунорубицин, митоксантрон и колхицин, все указанные агенты, очевидно, подвержены воздействию Р-гликопротеинового насоса;
- другие лекарственные средства, которые, как было показано, подвержены действию Р-гликопротеина, но которые могут перорально абсорбироваться в присутствии ингибитора Р- гликопротеина в кишечнике, например такие, как ганцикловир, фоскамет, камптотецин и производные камптотецина.

В соответствии с настоящим изобретением интервал доз усиливающего агента, вводимого совместно с целевым агентом, составляет от около 0,1 до около 15 мг/кг веса пациента. Под понятием "совместное введение" усиливающего агента подразумевается, в основном, одновременное введение с целевым агентом (либо менее чем за полчаса до введения, либо менее чем через полчаса после введения целевого агента, либо вместе с введением целевого агента), введение примерно за 0,5-24 часа до введения целевого агента либо сочетание того и другого режима введения, т.е. одна или несколько доз одного и того же или различных усиливающих агентов могут быть введены, по крайней мере, за 0,5 часа до введения целевого агента и одна доза может быть введена одновременно с введением целевого агента (либо вместе с целевым агентом, либо непосредственно после его введения). Кроме того, понятие "совместное введение" подразумевает введение более одной дозы целевого агента через 24 часа после введения дозы усиливающего агента, другими словами, усиливающий агент (агенты) необязательно должен быть снова введен до введения или одновременно с введением целевого агента, он может также вводиться периодически в процессе курса лечения.
Интервал доз для перорального введения целевого агента может варьироваться для каждого конкретно используемого лекарственного средства в зависимости от его терапевтического индекса, а также в зависимости от режима, требуемого для лечения данного заболевания, от состояния здоровья пациента и т.п. Способ настоящего изобретения позволяет вводить паклитаксел перорально в дозах, составляющих от около 20 мг/м2 до около 1000 мг/м2 (в расчете на площадь поверхности тела пациента) или примерна от 2 до 30 мг/кг (в расчете на вес тела пациента) в виде одноразовой дозы или дробных доз (2-3) в день и поддерживать уровни паклитаксела в плазме человека в пределах от 50 до 500 нг/мл в течение продолжительного периода времени (например, 8-12 часов) после введения каждой пероральной дозы. Эти уровни являются, по крайней мере, сравнимыми с уровнями, которые достигаются при 96-часовой внутривенной инфузии таксола (которая вызывает у пациента значительные неудобства, дискомфорт, потерю ориентации во времени, возможные инфекции и т.п.). Кроме того, указанные уровни паклитаксела в плазме являются более чем достаточными для достижения нужных фармакологических эффектов целевого лекарственного агента, например ингибирования деградации тубулина (которое происходит при уровнях около 0,1 мкМ или около 85 нг/мл) и ингибирования изопренилирования белка (которое происходит при уровнях около 0,03 мкМ или около 25 нг/мл), которые непосредственно связаны с их противоопухолевыми эффектами благодаря ингибированию онкогенных функций и других сигнал-трандуцирующих белков, играющих решающую роль в регуляции клеточного роста.
В некоторых случаях предпочтительно вводить пациенту более высокую, ударную, начальную дозу целевого агента для достижения пиковых уровней этого агента в крови с последующим введением более низких поддерживающих доз.
Два или более различных усиливающих агента и/или два или более различных целевых агента могут быть введены вместе, поочередно или периодически при осуществлении всех различных аспектов способа настоящего изобретения.
Настоящее изобретение также относится к способу лечения млекопитающих, страдающих раком, опухолями, саркомой Капоши, злокачественными заболеваниями, неконтролируемой пролиферацией тканей или клеток, вторичной по отношению к повреждению ткани и любыми другими патологическими состояниями, восприимчивыми к воздействию паклитаксела, таксанов, доцетаксела, этопозида, их пролекарств и производных, паклитаксела 2'-МРМ, и доцетаксела 2' -МРМ путем перорального введения лекарственных форм, содержащих один или несколько из указанных агентов. Среди всех типов карцином особенно эффективному лечению с использованием перорального паклитаксела, доцетаксела, других таксанов и их пролекарств и производных поддаются гепатоцеллюлярная карцинома и метастазы в печени, а также рак желудочно-кишечного тракта, поджелудочной железы и легких. Примерами незлокачественных заболеваний, которые могут быть подвергнуты эффективному лечению путем перорального введения вышеуказанных активных агентов в соответствии с настоящим изобретением, являются неконтролируемая, вторичная по отношению к повреждению тканей пролиферация тканей или клеток; поликистоз почек; малярия, включая паразитарную хлорохин- и пириметамин-резистентную малярию (Pouvelle et al., J. Clin. Invest., 44: 413-417, 1994).
В соответствии с настоящим изобретением противоопухолевые агенты, которые ранее могли быть введены только парентерально, могут теперь вводиться перорально с биологической доступностью, достаточной для обеспечения фармакологически активных концентраций лекарственных средств в крови, являющихся особенно эффективными для лечения пациентов, страдающих первичными опухолями и метастазами. Благодаря предварительному и/или одновременному введению MDR-ингибиторов или других усилителей биологической усвояемости указанные активные ингредиенты могут проникать в кишечную стенку и быстро попадать в портальный кровоток, обеспечивая, тем самым, более высокую начальную локальную концентрацию химиотерапевтических агентов в печени (значительно более высокую локальную концентрацию, чем это обычно достигается при терапии путем внутривенного вливания), чем в общем кровотоке или в большинстве других органов за 7 дней. Кроме того, следует отметить, что более высокие уровни паклитаксела в печени после перорального введения могут не отражаться в повышении его уровней в плазме из-за эффекта его первичного высокого прохождения в печени. Благодаря селективно продуцируемым высоким концентрациям противоопухолевых агентов в крови способ настоящего изобретения является особенно подходящим для лечения рака печени (например, гепатоцеллюлярной карциномы и метастазов в печени), рака желудочно-кишечного тракта (например, рака толстой кишки и прямой кишки) и рака легких.
Аналогичным образом после перорального введения в соответствии с настоящим изобретением, в желудочно-кишечном тракте, поджелудочной железе и легких были обнаружены (после анализа тканевого распределения) более высокие уровни (по сравнению с уровнями в общем кровотоке и в большинстве других органов) паклитаксела через 24 часа после введения. Благодаря этому факту пероральное введение паклитаксела может быть очень эффективным при лечении рака желудочно-кишечного тракта, поджелудочной железы и легких.
Особенно неожиданными и заслуживающими внимания являются Фиг.21-24. В некоторых случаях настоящее изобретение относится к способу достижения сравнимых, а иногда и более высоких локальных концентраций паклитаксела в тканях при пероральном введении, чем при внутривенном введении. Это соответствует более высокому объему распределения терапевтического агента. Кроме того, было показано, что пероральное введение усиливающего агента до и непосредственно после введения целевого агента (в случае использования циклоспорина и паклитаксела, см. Фиг.20) приводит к продуцированию даже более высокой, чем при внутривенном введении, концентрации целевого агента в моче. Поэтому такое совместное пероральное введение усиливающего агента и выбранного целевого агента должно быть эффективно в случае лечения пациентов, страдающих опухолями или метастазами мочеполовых путей.
Согласно настоящему изобретению помимо более высокой, чем достигаемая ранее, локальной концентрации активных ингредиентов в печени, распределение в плазме и тканях активных целевых агентов, вводимых перорально вместе с усиливающими агентами в соответствии с настоящим изобретением, неожиданно оказалось таким же, как и распределение, наблюдаемое при внутривенном введении. Серия экспериментов, проведенных с использованием экспериментальных животных, показала, что стабильные уровни паклитаксела в плазме достигаются на третий день после перорального введения вместе с CsA. Эти стабильные уровни целевого агента сравнимы с уровнями, достигаемыми при 96-часовой инфузии паклитаксела. У пациентов с дефицитом таксана, страдающих раком молочной железы с метастазами и подвергнутых лечению путем непрерывного 96-часового вливания каждые три недели, наблюдалась 27%-ная скорость реакции (Seidiaan et al., J.Clin.Oncol., 14; 1877, 1996). Предполагается, что аналогичные результаты могут быть достигнуты с использованием способов настоящего изобретения, которые не вызывают побочных эффектов, обычно наблюдаемых при длительной внутривенной инфузии, например таких, как ощущение дискомфорта и беспокойства, а также риск инфекционного заражения.
Кроме того, что очень важно, при пероральном введении, осуществляемом в соответствии с настоящим изобретением, концентрация паклитаксела и других вышеперечисленных противоопухолевых агентов в крови на фазе элиминации приблизительно аналогична соответствующей концентрации, наблюдаемой при внутривенном введении, и эти высокие терапевтические эффективные уровни могут поддерживаться в течение 8-12 часов после каждого введения. Увеличение экскреции лекарственного средства из мочи после перорального введения в присутствии CsA не только подтверждает повышенную пероральную абсорбцию паклитаксела, но также обеспечивает доставку большего количества лекарственного средства в мочеполовые пути для лечения ракового заболевания.
Пероральные лекарственные формы целевых агентов, биологическая доступность которых возрастает при совместном введении усиливающих агентов, могут быть изготовлены в виде стандартных лекарственных препаратов, таких как таблетки, капсулы, гранулы, гелевые микрокапсулы, драже, жидкости (например, растворы, суспензии или эликсиры), пастилки, и любые другие пероральные лекарственные формы, обычно применяемые в фармацевтической практике. Жидкие препараты могут содержать, например, паклитаксел или другой таксан в наполнителе, включающем CREMOPHOR EL или другое полиэтоксилированное касторовое масло, спирт и/или полиоксиэтилированный сорбитанмоноолеат (например, TWEEN® 80, ICI Americas,Inc.). Каждая лекарственная форма включает эффективное количество целевого агента (например, эффективное противоопухолевое или антибластомное количество противоопухолевого или антибластомного агента) и фармацевтически инертные ингредиенты, например стандартные эксипиенты, носители, наполнители, связующие вещества, дезинтегрирующие агенты, растворители, солюбилизирующие агенты, подслащивающие агенты, красители и любые другие неактивные ингредиенты, которые обычно используются при изготовлении фармацевтических лекарственных форм для перорального введения. Многие из таких лекарственных форм и пероральные носители, перечисленные выше в списке неактивных ингредиентов, описаны в Remington's Pharmaceutical 17th edition (1985). Каждая лекарственная форма также содержит фармакологически эффективное количество, например эффективное антибластомное или противоопухолевое количество одного из целевых лекарственных средств.
Точное количество каждого из целевых лекарственных средств в пероральных лекарственных формах варьируется в зависимости от возраста, веса, заболевания и состояния здоровья пациента. Так, например, лекарственные препараты паклитаксела могут содержать количества паклитаксела, достаточные для получения суточной дозы примерно 20-1000 мг/м2 (в расчете на поверхность тела пациента) или примерно 2-30 мг/кг (в расчете на вес тела пациента) в виде разовой дозы или дробных (2-3) доз. Пероральные лекарственные препараты этопозида могут содержать количества этопозида, достаточные для получения суточной дозы примерно 20-200 мг/м2 (в расчете на среднюю поверхность тела пациента) в виде разовой суточной дозы или дробных (2-3) доз.
Как указывалось выше, некоторые целевые агенты, несмотря на их относительно низкую или неадекватную пероральную биологическую доступность, являются коммерчески доступными в виде пероральных лекарственных форм. Так, например, имеются в продаже капсулы VEPESID®, содержащие 50 мг (каждая) этопозида.
При выборе схемы лечения для каждого конкретного пациента, которому требуется вводить пероральные лекарственные формы настоящего изобретения, содержащие целевое лекарственное средство, необходимо учитывать повышенную биологическую доступность этого лекарственного средства, обусловленную одновременным и/или предварительным введением усиливающих агентов. Так например, хотя рекомендованная производителем доза в капсулах VEPESID®, предназначенных для лечения мелкоклеточного рака легких, в два раза превышает внутривенную дозу (округленная до 50 мг), однако повышенная биологическая доступность этопозида, обеспечиваемая предварительным и/или одновременным введением усиливающих агентов, таких как циклоспорин, позволяет значительно снизить дозу перорального этопозида и достичь при этом тех же самых эффективных уровней лекарственного средства в крови с большей продолжительностью и стабильностью действия и без повышения (а иногда даже со снижением) токсических побочных эффектов. При пероральном введении можно избежать высоких пиковых уровней лекарственного средства в крови, так как именно эти пиковые уровни ответственны за некоторые токсические побочные эффекты. На основании полученных нами экспериментальных данных (см. Фиг.18 и 19 и Таблицу 6), свидетельствующих о том, что пероральная абсорбция этопозида в присутствии циклоспорина является почти полной (около 96%), пероральная суточная доза этопозида для лечения рака яичек должна составлять около 50-100 мг/м2, а для лечения мелкоклеточного рака легких эта доза должна составлять около 35-50 мг/м2 в расчете на поверхность тела пациента.
Схема введения лекарственных препаратов при лечении заболеваний способами настоящего изобретения, например, при лечении паклитакселвосприимчивых заболеваний путем перорального введения препаратов паклитаксела совместно с введением усиливающих агентов может быть соответствующим образом скорректирована с учетом физиологических особенностей пациента и серьезности его заболевания. Предпочтительная схема применения перорального препарата паклитаксела предусматривает (а) ежедневное введение пациенту, нуждающемуся в таком введении, 1-3 равных разделенных доз, обеспечивающих суточную дозу примерно 20-1000 мг/м (в расчете на поверхность тела пациента), причем такое ежедневное введение продолжается в течение 1-4 дней, следующих друг за другом каждые 2-3 недели, либо (b) введение в течение одного дня каждую неделю. Первая схема применения аналогична использованию 96-часовой инфузии паклитаксела каждые 2-3 недели, которая до некоторой степени рассматривается как предпочтительная схема внутривенного введения. Предпочтительная схема перорального введения этопозида с совместным введением усиливающих агентов предусматривает ежедневное введение пациенту, нуждающемуся в таком введении, 1-3 равные разделенные дозы, обеспечивающие суточную дозу примерно 50-100 мг/м2 (в расчете на поверхность тела пациента) для лечения пациентов, страдающих раком яичек; суточную дозу примерно 35-50 мг/м2 для лечения пациентов, страдающих мелкоклеточным раком легких, причем в каждом случае такое ежедневное введение продолжается в течение 5-21 дней с интервалом примерно 2-3 недели между каждым курсом лечения.
В соответствии с настоящим изобретением пероральное введение сильнодействующих химиотерапевтических агентов может, во многих случаях, в действительности снижать побочные токсические эффекты, обычно наблюдаемые при использовании внутривенной терапии. В отличие от внутривенной инфузии, при которой быстро и внезапно продуцируются высокие уровни активного агента в крови, при абсорбции активного агента через стенку кишечника (стимулированной усиливающими агентами) происходит постепенное нарастание концентраций активного агента в крови до стабильного стационарного уровня, который поддерживается при этом значении или значении, близком к идеальному интервалу, в течение длительного периода времени.
В соответствии с другим своим аспектом настоящее изобретение относится к комбинированным пероральным лекарственным формам, которые содержат фиксированные количества, по крайней мере, одного усиливающего агента и, по крайней мере, одного целевого агента. Так, например, указанные лекарственные формы могут быть изготовлены в виде таблеток, капсул, микрокапсул, гелевых капсул, пилюль, жидкостей, пастилок и любых других стандартных пероральных лекарственных форм, содержащих в качестве активных ингредиентов эффективное количество агента, повышающего пероральную биологическую доступность противоопухолевого или антибластомного агента, вместе с этим противоопухолевым агентом, а также подходящие неактивные ингредиенты. Один из таких комбинированных продуктов содержит от около 0,1 до около 15 мг/кг одного или нескольких циклоспоринов A, D, С, F и G, дигидро CsA, дигидро CsC и ацетил CsA, вместе с около 20-1000 мг/м2 (в расчете на среднюю поверхность тела пациента) паклитаксела, доцетаксела, других таксанов либо производных паклитаксела или доцетаксела, таких как паклитаксел 2'-MPM или доцетаксел 2' -MPM. Другая подходящая лекарственная форма содержит от около 0,1 до около 15 мг/кг циклоспорина или циклоспорина D или F в сочетании с около 20-200 мг/м2 этопозида.
Совместное введение усиливающих агентов вместе с целевыми лекарственными средствами не только стимулирует пероральную биологическую доступность указанных агентов, но также позволяют использовать их при лечении опухолей, локализованных в областях, в высокой степени защищенных MDP, например, в яичках и головном мозге. Таким образом, в другом своем аспекте настоящее изобретение относится к способу доставки противоопухолевых средств к месту локализации опухоли, защищенному MDP, где указанный способ предусматривает совместное введение усиливающих агентов и противоопухолевых агентов, что позволяет проводить лечение опухолей головного мозга, таких как многообразные формы глиобластомы.
В еще одном аспекте настоящее изобретение относится к способу доставки активного метаболита, паклитаксела к очагу заболевания в терапевтическом количестве, необходимом для лечения заболеваний, восприимчивых к паклитакселу. Были идентифицированы главные in vivo-метаболиты паклитаксела, а в частности, нижеследующие гидроксилированные метаболиты паклитаксела А, В и С:
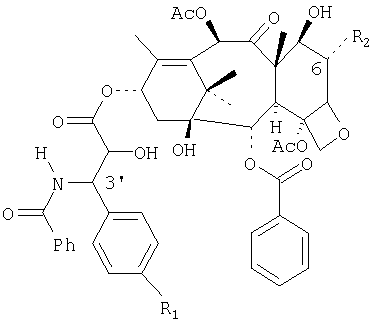
A: R1=H, R2=OH; В: R1=OH, R2=OH; С: R1=OH, R2=OH (Паклитаксел: R1=H, R2=H).
В некоторых in vitro-тестах было обнаружено, что метаболит В, указанный выше (именуемый в литературе также метаболитом М4), имеет более высокий терапевтический индекс (отношение уровня токсической концентрации к уровню эффективной концентрации), чем паклитаксел, для некоторых опухолевых клеточных линий человека. Настоящее изобретение дает возможность осуществлять доставку повышенных количеств метаболита В и других активных метаболитов паклитаксела к местам локализации опухолей благодаря тому, что после перорального введения весь введенный паклитаксел проникает в печень и подвергается там метаболизму с помощью печеночных микросом, в результате чего в общий кровоток поступает большее количество каждого метаболита, чем это достигается при внутривенном введении.
В еще одном аспекте настоящее изобретение относится к наборам для лечения млекопитающих, страдающих нарушениями, восприимчивыми к воздействию любых фармакологически активных целевых агентов, пероральная абсорбция и биологическая доступность которых возрастает под влиянием усиливающих агентов. Эти наборы включают одну или несколько пероральных лекарственных форм, по крайней мере, одного усиливающего агента, и одну или несколько пероральных лекарственных форм, по крайней мере, одного целевого агента либо одну или несколько лекарственных форм, содержащих оба указанных агента.
Так, например, набор настоящего изобретения может содержать одну или несколько таких лекарственных форм, как таблетки, капсулы, микрокапсулы, гелевые капсулы, или жидкие препараты, содержащие циклоспорин или кетоконазол; и одну или несколько таких лекарственных форм, как таблетки, капсулы, микрокапсулы, гелевые капсулы или жидкие препараты, содержащие паклитаксел или этопозид в дозах, интервалы которых описаны выше. Указанные наборы могут применяться в стационарах, клиниках, врачебных кабинетах или на дому у пациентов для облегчения совместного введения усиливающих и целевых агентов. Эти наборы должны также содержать вкладыш-инструкцию с указанием доз для совместного введения усиливающих и целевых агентов.
Указанные наборы могут также включать комбинации различных усиливающих агентов и/или комбинации целевых агентов. Так, например, набор может включать соответственно пероральные лекарственные формы, содержащие циклоспорин и кетоконазол в качестве усиливающих агентов, и лишь один паклитаксел в качестве целевого агента или комбинацию паклитаксела с другим противоопухолевым средством. При этом, вторым целевым агентом должно быть (подобно паклитакселу) лекарственное средство, которое также обладает плохой пероральной биологической доступностью, но которое, при совместном введении с усиливающими агентами, может достигать терапевтически эффективных уровней в крови после его перорального введения. Целевой агент может присутствовать вместе с усиливающим агентом в одной и той же лекарственной форме либо он может присутствовать в виде отдельной лекарственной формы.
Нижеследующие примеры иллюстрируют различные аспекты настоящего изобретения и демонстрируют неожиданное и значительное увеличение пероральной абсорбции целевых агентов. Эти примеры никоим образом не ограничивают настоящего изобретения и не претендуют на описание каких-то конкретных усиливающих или целевых агентов, интервалов доз, процедур тестов или других параметров, которые исключительно должны использоваться на практике настоящего изобретения.
ПРИМЕР 1
Восемнадцать (18) здоровых крыс Spraque Dawley, весом 225-275 граммов каждая, в возрасте примерно 6-8 недель произвольно были разделены на три группы по 6 животных в каждой. Первой группе из шести крыс вводили внутривенно паклитаксел в дозе 9 мг/кг. Вторая группа получала разовую пероральную дозу паклитаксела, равную 9 мг/кг. А третья группа получала разовую пероральную дозу циклоспорина, равную 5 мг/кг, а через один час эта группа получала пероральную дозу циклоспорина в 5 мг/кг и паклитаксела в 9 мг/кг.
Пробы крови брали из хвостовой вены каждой крысы через 0,5; 1; 2; 3; 4 и 6 часов после введения дозы паклитаксела. У внутривенно обработанных крыс первой группы брали дополнительную пробу крови через 8 часов после введения дозы паклитаксела. Каждую пробу крови центрифугировали, и отделяли сыворотку. Затем для каждого временного интервала получали одну репрезентативную пробу путем объединения шести проб на группу. Все пробы были проанализированы на неизмененный паклитаксел с помощью ЖХ/МС с нижним пределом количественного определения 50 пг/мл.
Результаты исследований графически проиллюстрированы на Фиг.1 и 2. На Фиг.1 представлены сравнительные графики для всех трех групп крыс, а на Фиг.2 представлены графики лишь для второй и третьей групп, которым перорально вводили паклитаксел. Из этих графиков можно видеть, что в отсутствие циклоспорина биологическая доступность паклитаксела в сыворотке составляет менее 1%, однако при введении крысам (третья группа) циклоспорина за один час до введения комбинированной дозы циклоспорина/паклитаксела биологическая доступность паклитаксела возрастает до 6-7%.
В нижеприведенной Таблице 2 представлены данные определения значений площади под кривой (AUC), вычисленных для трех групп крыс. Эти данные показывают, что значения AUC0-6 час для третьей группы, получившей циклоспорин и паклитаксел, почти в 8 раз превышает значения AUC для второй группы крыс, получившей лишь один пероральный паклитаксел.
ПРИМЕР 2
Сорок (40) здоровых крыс Spraque Dawley, имеющих такие же данные, что и крысы, использованные в эксперименте, описанном в Примере 1, были произвольно разделены на четыре группы по десять крыс в каждой, обозначенные A, F, G и Н. В нижеприведенной Таблице 3 проиллюстрирована обработка каждой испытуемой группы крыс и указаны временные интервалы для каждого введения дозы.
1
5
Пробы крови брали из хвостовой вены каждой крысы через 0,25; 0,5; 1; 2; 3; 4; 5; 6; 8; 12 и 24 часа после введения паклитаксела. После соответствующей обработки проб и получения объединенной пробы для каждой группы плазму от каждой пробы анализировали на неизмененный паклитаксел.
Результаты этого анализа графически проиллюстрированы на Фиг.3 и 4. На Фиг.3 показаны для сравнения две кривые зависимости концентрации от времени для Группы А, которой вводили предварительную дозу циклоспорина, а через один час - комбинированную дозу паклитаксела-циклоспорина, и для Группы F, которой вводили предварительную дозу циклоспорина, а затем через 1 час только пероральную дозу паклитаксела. На Фиг.4 показано сравнение между результатами, полученными для Групп G и Н, обеим из которых вводили внутривенно паклитаксел, а Группа G, кроме того, за три часа до введения паклитаксела получала пероральную дозу циклоспорина. Как показано на Фиг.4, эти две группы обнаруживали, в основном, идентичные уровни паклитаксела в плазме в одни и те же временные интервалы.
В Таблице 4 представлены значения AUC для всех четырех групп крыс, участвовавших в данном эксперименте. Как видно из Таблицы, значения AUC для Групп G и Н, в основном, идентичны, а значения AUC для Группы А на 25-30% превышают значения для Группы F, что свидетельствует об эффективности предварительного введения циклоспорина, а затем совместного введения циклоспорина с паклитакселом.
ПРИМЕР 3
Восемнадцать (18) здоровых крыс Spraque Dawley, имеющих такие же физические данные, что и крысы, использованные в эксперименте, описанном в Примере 1, были произвольно разделены на три группы по шесть крыс в каждой, Группы А, В и С. Группе А внутривенно (IV) вводили радиоактивно меченный паклитаксел; Группе В перорально вводили 3Н-меченный паклитаксел; а Группе С вводили перорально дозу циклоспорина, а затем, через один час, вводили комбинированную пероральную дозу циклоспорина и пероральную дозу радиоактивно меченного паклитаксела.
Пробы крови брали из хвостовой вены каждой крысы через интервалы времени, указанные в Примере 2. Эти пробы сохраняли в виде цельной крови. Кроме того, через 2-24 часа после введения дозы паклитаксела, у каждой крысы брали пробы мочи. Пробы крови и мочи анализировали на радиоактивность.
На Фиг.5 представлены сравнительные уровни паклитаксела в пробах цельной крови, взятых из Групп А, В и С. Сравнение уровней паклитаксела для отдельных членов Группы В и С проиллюстрировано на Фиг.6 и 1, соответственно.
В этом эксперименте пероральная абсорбция радиоактивности (выраженная в эквивалентах паклитаксела) в цельной крови в отсутствие циклоспорина (Группа В) составляла около 10%, а при введении циклоспорина (Группа С) она составляла около 40%. Абсорбцию радиоактивности определяли путем измерения AUC после внутривенного и перорального введения радиоактивно меченного паклитаксела. Формально биологическую доступность паклитаксела не измеряли в этом эксперименте, поскольку для этого необходимо провести анализ на неизмененное лекарственное средство для каждого периода времени. Хотя для одного временного параметра был проведен стандартный ВЭЖХ-анализ радиоактивности, экстрагированной из плазмы, который показал, что, по крайней мере, 32% радиоактивности, присутствующей в плазме, приходится на неизмененный паклитаксел. Профиль радиоактивности ВЭЖХ-экстракта плазмы для животных Группы С, обнаруживающий, в основном, один пик (которым является паклитаксел), показан на Фиг.28. AUC, Сmax, Тmax и другие данные, полученные в этом эксперименте, представлены в Таблице 5.
Для крыс, обработанных, как описано в Примере 3, определяли AUC для полной радиоактивности. Исходя из отношения AUCp.o./AUCi.v. для временных интервалов до бесконечности пероральная абсорбция в присутствии циклоспорина возрастает до 54,7% по сравнению с 16,4%-ной абсорбцией в отсутствие циклоспорина (Таблица 5А). С использованием аналогичных анализов для неизмененного паклитаксила в крови было установлено, что биологическая доступность паклитаксела составляет 25,7% в присутствии циклоспорина и 1,7% в отсутствие циклоспорина (Таблица 5В). Выведение лекарственного средства из организма оказалось неожиданно аналогичным для всех трех групп. Объем распределения паклитаксела в группе, которой вводили циклоспорин и перорально паклитаксел, примерно на 50% превышал соответствующий объем в группе, которой вводили паклитаксел внутривенно.
В экспериментах, описанных в нижеследующих Примерах 4-5, были использованы крысы Spraque Dawley с теми же самыми физическими характеристиками, что и крысы в эксперименте Примера 1, и эти крысы были разделены на три группы по три крысы (самца) в каждой. За 12-14 часов до введения доз всех крыс держали на голодной диете. По окончании периода голодания крысам, которым должны быть введены усиливающие агенты, сначала вводили эти агенты, а затем, через 1 час, им вводили радиоактивно меченный (3Н)-паклитаксел (9 мг/кг) и одновременно дозы усиливающего агента. Крысам, которым не должны быть введены усиливающие агенты, после голодания вводили радиоактивно меченный паклитаксел.
У каждого животного брали пробу крови через 0,5; 1; 2; 3; 4; 5; 6; 7; 8; 12 и 24 часа после введения дозы паклитаксела. Мочу брали через 4-24 часа после введения дозы. Затем для каждой крысы определяли полную радиоактивность в крови и моче и вычисляли средние значения для каждой группы.
ПРИМЕР 4
Трем группам крыс вводили соответственно 10 мг/кг верапамила перорально, 5 мг/кг прогестерона перорально, и 10 мг/кг дипирамидона перорально в качестве усиливающих агентов, причем эти усиливающие агенты вводили сначала отдельно, а затем, через 1 час, вместе с пероральной дозой паклитаксела. Сравнение кривых зависимости концентрации в цельной крови от времени (эквиваленты концентрации от времени), построенных для всех трех групп, показано на Фиг.8. Представленные данные иллюстрируют почти похожие (в грубом приближении) результаты, которые были получены с использованием верапамила и дипиридамона в качестве усиливающих агентов, тогда как данные биологической доступности, полученные с использованием прогестерона, показали заметно более низкие значения.
На Фиг.9 представлены для сравнения кривые зависимости концентрации паклитаксела от времени, построенные для группы крыс, которым вводили верапамил (10 мг/кг) в качестве усиливающего агента; для группы животных предыдущего эксперимента, которым был перорально введен один паклитаксел (9 мг/кг), и для другой группы животных, которым был перорально введен циклоспорин (5 мг/кг) сначала за 1 час до, а затем сразу после введения пероральной дозы паклитаксела (9 мг/кг). Группа, получившая циклоспорин, обнаружила значительно более высокие уровни паклитоксела в крови, чем другие группы, на протяжении почти всего 24-часового периода.
На Фиг.10 и 11 проиллюстрировано параллельное сравнение с Фиг.9, за исключением того, что вместо значений для группы, которой вводили верапамил (Фиг.9), сравнивали значения для группы, которой вводили прогестерон (Фиг.10), и для группы, которой вводили дипиридамол (Фиг.11).
ПРИМЕР 5
Трем группам крыс вводили соответственно 100 мг/кг верапамила перорально, 5 мг/кг мегестеролацетата перорально и 50 мг/кг кетоконазола перорально в качестве усиливающих агентов, причем эти усиливающие агенты вводили отдельно, а затем, через 1 час, их вводили вместе с пероральной дозой радиоактивно меченного паклитаксела. Графическое сравнение кривых временной зависимости концентрации паклитаксела в цельной крови (измеренной в виде зависимости эквивалентов концентрации от времени), построенных для всех трех групп, показано на Фиг.12. Представленные данные иллюстрируют почти сходные (в грубом приближении) результаты, полученные с использованием верапамила и мегестеролацетата в качестве усиливающих агентов, тогда как данные биологической доступности, полученные с использованием кетоконазола, показали значительно более высокие значения за первые 12 часов.
На Фиг.13 проиллюстрировано графическое сравнение кривой зависимости концентрации радиоактивности от времени, полученной для группы крыс, которым вводили верапамил (100 мг/кг) в качестве усиливающего агента, с кривыми зависимости, полученными в предыдущем эксперименте для группы крыс, которым перорально вводили один паклитаксел (9 мг/кг), и для группы крыс, которым перорально вводили циклоспорин (5 мг/кг) за 1 час до, а затем сразу после введения пероральной дозы радиоактивно меченного паклитаксела (9 мг/кг).
На Фиг.14 и 15 проиллюстрировано параллельное графическое сравнение с Фиг.13, но для групп, которым вводили мегестеролацетат (Фиг.14) и кетоконазол (Фиг.15) вместо группы, которой вводили верапамил (Фиг.13).
На Фиг.16 проиллюстрировано графическое сравнение зависимости концентрации радиоактивности от времени, определенных для группы крыс, которым вводили 10 мг/кг верапамила (как описано в Примере 4), и для группы крыс, которым вводили 100 мг/кг верапамила (как описано в Примере 5).
На Фиг.17 проиллюстрировано графическое сравнение зависимости концентрации радиоактивности от времени, определенных для группы крыс, которым вводили 5 мг/кг прогестерона, как описано в Примере 4, и для группы крыс, которым вводили 5 мг/кг мегестеролацетата, как описано в Примере 5.
На Фиг.16 и 17 также показаны те же самые профили, изображенные на Фиг.13-15, для исследуемых групп, которым вводили лишь один радиоактивно меченный паклитаксел, и перорально радиоактивно меченный паклитаксел сразу после введения и через 1 час после введения 5 мг/кг циклоспорина.
Были проведены исследования данных дозовой зависимости для циклоспорина. Увеличение дозы до 10 мг/кг и 20 мг/кг для введения циклоспорина за 1 час и одновременно с введением паклитаксела приводило к пероральной абсорбции радиоактивности примерно до 45%. Этот результат отличается от результата, полученного для кетоконазола, где дозы до 50 мг/кг, которые были введены за 1 час до и одновременно с паклитакселом, не приводили к дальнейшему увеличению пероральной абсорбции радиоактивности (см. Фиг.17А и 17В).
Средние фармакокинетические параметры для исследуемых групп животных, обсуждаемые в Примерах 4 и 5, представлены в Таблице 6 (Эксперимент Примера 4 обозначен в Таблице 6 как протокол NP951202, а эксперимент Примера 5 обозначен как протокол NP960101.).
Данные, полученные в экспериментах Примеров 4 и 5 и представленные в Таблице 6 и на Фиг.8-17В, ясно свидетельствуют об эффективности циклоспорина в качестве агента, усиливающего пероральную биологическую доступность активного агента, и о его преимуществе по сравнению с верапамилом, прогестероном или мегестеролацетатом как при низких, так и при высоких дозах, особенно в первые 12 часов после введения дозы паклитаксела. Эти данные также указывают на то, что кетоконазол, хотя и не является таким же эффективным, как циклоспорин, однако также обладает заметной активностью, стимулирующей пероральную абсорбцию паклитаксела.
ПРИМЕР 6
Три группы крыс, по три крысы-самца в каждой, держали на голодной диете за 16-18 часов до введения доз. По окончании периода голодания одной группе крыс вводили пероральную дозу (5 мг/кг) циклоспорина. Затем, через 1 час, этой группе вводили перорально 5 мг/кг циклоспорина и 1 мг/кг радиоактивно меченного 3Н-этопозида. Двум другим группам, после голодания вводили только 1 мг/кг 3Н-этопозида внутривенно, 1 мг/кг 3Н-этопозида перорально соответственно. Процедуры взятия проб крови и мочи и определение полной радиоактивности осуществляли так же, как описано в Примерах 4 и 5, за исключением того, что у крыс, которым вводили этопозид внутривенно, пробы крови брали еще через 0,033 и 0,25 часа. Результаты эксперимента представлены в Таблице 7.
На Фиг.18 и 19 графически проиллюстрированы зависимости средней концентрации этопозида в цельной крови от времени, определенные для трех исследуемых групп. На Фиг.18 по оси ординат отложены эквиваленты концентраций этопозида от 0 до 0,2 (м.д.), а на Фиг.19 по оси ординат отложены эквиваленты этопозида от 0 до 0,2 (мл.д.) для более наглядной иллюстрации различий между значениями, полученными для рассматриваемых трех групп.
Данные, представленные в Таблице 7 и на Фиг.18 и 19, показали, что циклоспорин является эффективным агентом, повышающим пероральную биологическую доступность этопозида, особенно в первые 12 часов после введения доз.
1/РО(Р) 5/РО(с)
ПРИМЕР 7
В другой серии экспериментов три группы крыс (по три крысы-самца в каждой) держали на голодной диете в течение 16-18 часов до введения доз. По истечении времени голодания одной группе крыс вводили пероральную дозу кетоконазола (2 мг/кг). Затем, через 1 час, этой группе перорально вводили 2 мг/кг кетоконазола и 1 мг/кг радиоактивно меченного 3Н-этопозида. Две другие группы были обработаны точно так же, за исключением того, что после голодания, этим группам перорально вводили 10 и 50 мг/кг кетоконазола соответственно до и сразу после введения 3Н-этопозида. Процедуры взятия проб крови и мочи и определения полной радиоактивности осуществляли так же, как описано в Примерах 4 и 5. Полученные данные представлены в Таблице 7А. Таким образом, в отличие от действия циклоспорина, который почти вдвое повышает пероральную абсорбцию радиоактивного паклитаксела, кетоконазол, вводимый в широком диапазоне доз, не обнаруживал способности к повышению пероральной абсорбции этопозида по сравнению с данными, полученными в случае введения лишь одного этопозида.
ПРИМЕР 7А
Были проведены исследования баланса экскреции паклитаксела у крыс. Три группы крыс, по 4-5 крыс-самцов в каждой, держали на голодной диете в течение 12-14 часов до введения доз. По истечении времени голодания одной группе крыс вводили пероральную дозу (5 мг/кг) циклоспорина. Затем, через 1 час, этой группе перорально вводили 5 мг/кг циклоспорина и 9 мг/кг радиоактивно меченного паклитаксела. Двум другим группам вводили после голодания лишь 9 мг/кг радиоактивно меченного паклитаксела внутривенно и 9 мг/кг радиоактивно меченного паклитаксела перорально.
Мочу и экскременты брали у каждого животного при следующих интервалах: через 0-2, 2-4, 4-8, 8-12, 12-24, 24-36, 36-48, 48-72, 72-96, 96-120, 120-144 и 144-168 часов после введения дозы. Образцы ткани брали через 168 часов после введения дозы. Определение полной радиоактивности осуществляли так же, как описано в Примерах 4 и 5.
На Фиг. 20 проиллюстрировано графическое сравнение среднего кумулятивного процента от дозы паклитаксела, обнаруженного в экскрементах и моче испытуемых животных в течение 168-часового периода времени. Группа крыс, которой вводили циклоспорин до и вместе с пероральным паклитакселом, обнаруживала значительно более низкий процент дозы в экскрементах, чем две другие группы, и значительно более высокий процент дозы в моче, чем две другие группы, что свидетельствует о том, что у животных, обработанных циклоспорином, значительно большее количество перорального паклитаксела диффундировало через стенку кишечника и попадало в общий кровоток. Кроме того, тот факт, что процент дозы в моче был значительно выше у крыс, обработанных пероральным циклоспорином и паклитакселом, чем у крыс, которым внутривенно вводили паклитаксел, свидетельствует о том, что совместное пероральное введение способствует прохождению более высокой концентрации радиоактивности через мочеполовые пути.
На Фиг.21-2.4 представлены гистограммы, иллюстрирующие средние величины (мл.д.) паклитаксела, полученные для ряда тканей, взятых от крыс трех групп, где Группа А представляет животных, которым внутривенно вводили паклитаксел. Группа В представляет животных, которым перорально вводили паклитаксел, а Группа С представляет животных, которым вводили циклоспорин. Полученные графики показали, что уровни паклитаксела, зарегистрированные в различных тканях, взятых у мышей Группы С, в грубом приближении сравнимы с уровнями паклитаксела, наблюдаемыми у крыс Группы А, которым внутривенно вводили паклитаксел, за исключением печени, где уровень паклитаксела у обработанной циклоспорином группы в два раза превышал уровень паклитаксела у группы, которой вводили паклитаксел внутривенно. Уровни паклитаксела, обнаруженные в тканях у крыс Группы В (которым вводили перорально лишь один паклитаксел), были очень низкими, а в большинстве случаев значительно меньше половины от уровней, наблюдаемых в любой другой группе.
Данные, полученные в этом эксперименте, представлены в Таблицах 8 и 9.
ПРИМЕР 8
Было также проведено другое исследование распределения паклитаксела в тканях крыс. Две группы крыс (по 10 крыс-самцов в каждой) держали на голодной диете в течение 12-14 часов до введения доз. При истечении времени голодания одной группе крыс вводили пероральную дозу (5 мг/кг) циклоспорина. Затем, через 1 час, этой группе перорально вводили 5 мг/кг циклоспорина и 9 мг/кг радиоактивно меченного паклитаксела. Другой группе после голодания вводили внутривенно лишь 9 мг/кг радиоактивно меченного паклитаксела.
Образцы тканей брали через 24 часа после введения дозы. Определение полной радиоактивности определяли так же, как описано в Примерах 4 и 5.
В Таблице 9А представлены величины (мл.д.) радиоактивности, выделяемой паклитакселом и зарегистрированной в различных тканях, взятых у крыс из двух исследуемых групп. Одна группа включала животных, которым внутривенно вводили паклитаксел, а вторая группа включала животных, которым вводили паклитаксел вместе с циклоспорином, вводимым за 1 час до и сразу после введения паклитаксела. Уровни паклитаксела, обнаруженные в различных тканях, взятых у обработанных циклоспорином крыс, были, в грубом приближении, сравнимы с уровнем паклитаксела, наблюдаемыми у крыс, которым паклитаксел был введен внутривенно, за исключением селезенки, поджелудочной железы и желудочно-кишечного тракта, где уровни паклитаксела у обработанной циклоспорином группы примерно в два раза превышали уровни паклитаксела для группы, которой вводили паклитаксел внутривенно.
В Таблице 9В проиллюстрировано сравнение концентраций неизмененного паклитаксела в различных органах для группы, которой внутривенно вводили лишь один паклитаксел, и для группы, которой вводили перорально паклитаксел в сочетании с циклоспорином. В легких и желудочно-кишечном тракте после перорального введения обнаруживались более высокие концентрации неизмененного паклитаксела, чем после внутривенного введения.
ПРИМЕР 9
Повторяли процедуру, описанную в Примерах 4 и 5, за исключением того, что трем группам крыс (по три крысы-самца в каждой) перорально вводили соответственно дозы (5 мг/кг) циклоспорина D, циклоспорина G и циклоспорина А; причем эти дозы вводили отдельно, а затем сразу через 1 час после введения пероральной дозы (9 мг/кг) радиоактивно меченного паклитаксела. На Фиг. 25 проиллюстрировано графическое сравнение зависимостей концентрации радиоактивности в цельной крови от времени, определенных для всех трех групп. Хотя все три циклоспорина показали значительную активность в стимулировании пероральной абсорбции паклитаксела, однако наибольшую активность, способствующую повышению биологической доступности паклитаксела, обнаружил циклоспорин D, который из всех трех испытуемых циклоспоринов обладает наименьшей иммунодепрессантной активностью (Jeffery, Clin. Biochem., 24: 15-21 (1991)).
ПРИМЕР 10
Был проведен ряд экспериментов, в которых повторяли процедуры, описанные в Примерах 4 и 5, и в которых группам, состоящим из трех крыс-самцов каждая, перорально вводили 5-10 мг/кг разных циклоспоринов отдельно, а затем, через 1 час после введения пероральной дозы (8 мг/кг) радиоактивно меченного паклитаксела. В Таблице 10 проиллюстрировано сравнение AUC и % абсорбции, полученных в этих экспериментах, каждый из которых идентифицирован номером протокола, начинающегося с префикса "NP".
ПРИМЕР 11
Повторяли процедуру, описанную в Примерах 4 и 5, за исключением того, что трем группам крыс, состоящим из трех крыс-самцов каждая, перорально вводили соответственно 5 мг/кг циклоспорина А, 50 мг/кг кетоконазола и 5 мг/кг циклоспорина А+50 мг/кг кетоконазола; причем все дозы вводили отдельно, а затем через 1 час после введения пероральной дозы (9 мг/кг) радиоактивно меченного паклитаксела. Графическое сравнение полученных результатов проиллюстрировано на Фиг.26. Группа, получившая комбинацию кетоконазола и циклоспорина А, неожиданно обнаружила в течение почти 24-часового периода времени значительно более высокие уровни радиоактивности, чем группы, получившие только один из указанных усиливающих агентов.
ПРИМЕР 12
Повторяли процедуру, описанную в Примерах 4 и 5, за исключением того, что трем группам крыс (по три крысы-самца в каждой) перорально вводили соответственно 100 мг/кг каптоприла отдельно и через два часа после введения пероральной дозы (9 мг/кг) радиоактивно меченного паклитаксела; 5 мг/кг циклоспорина отдельно и через один час после введения пероральной дозы (9 мг/кг) радиоактивно меченного паклитаксела; и 9 мг/кг радиоактивно меченного паклитаксела. Графическое сравнение полученных результатов проиллюстрировано на Фиг.27.
Вышеописанные эксперименты выявили ранее неизвестные и неожиданные факты, которые имеют большое значение для клинической терапии многих заболеваний, в частности некоторых видов рака:
1. Некоторые ингибиторы MDR (Р-гликопротеина), а также другие агенты, не являющиеся известными как ингибиторы MDR, могут быть введены перорально в целях эффективного повышения пероральной биологической доступности активных лекарственных агентов, которые до сих пор вводились только парентерально, так как после перорального введения они не могли достигать терапевтических уровней в крови.
2. Совместное введение усиливающих агентов настоящего изобретения вместе с целевыми лекарственными средствами, имеющими плохую пероральную биологическую доступность, позволяет достигать стабильно поддерживаемых уровней целевых лекарственных средств в крови, сравнимых с уровнями, достигаемыми при терапии путем внутривенной инфузии, но без резкого первоначального их возрастания, а поэтому с меньшим риском возникновения токсических побочных эффектов.
3. Совместное пероральное введение усиливающих агентов и целевых лекарственных агентов, по сравнению с внутривенным введением, способствует повышению соответствующей концентрации целевого агента в печени, легких и желудочно-кишечном тракте, что делает этот новый метод введения особенно эффективным для лечения опухолей и метастазов в печени.
4. Пероральное введение усиливающего агента до введения совместных пероральных доз усиливающего агента и целевого агента способствует повышению биологической доступности целевого лекарственного средства до значительно более высокой степени, чем просто совместное введение усиливающего и целевого агентов без предварительного введения усиливающего агента. Такое введение позволяет достичь терапевтических уровней целевого лекарственного агента в плазме.
5. Циклоспорины, особенно циклоспорины A, D и F, являются более эффективными агентами для повышения биологической доступности противоопухолевых агентов, чем такие ингибиторы MDR, как верапамил и прогестерон. Кетоконазол обладает заметной активностью, направленной на повышение пероральной биологической доступности лекарственного средства, но меньшей, чем циклоспорины.
В общих чертах можно сказать, что различные аспекты настоящего изобретения впервые позволяют практически осуществить пероральное введение лекарственных форм широко используемых фармацевтических агентов, а особенно противораковых лекарственных средств, таких как паклитаксел, родственные таксаны и этопозид, которые ранее могли эффективно вводиться лишь путем внутривенного вливания. Использование таких пероральных лекарственных форм в клинической терапии рака обеспечивает пациенту комфорт, удобство и безопасность, а также способствует экономии средств, затрачиваемых на лечение пациентами, больницами, правительством и частными медицинскими страховыми компаниями.
Кроме того, данное описание настоящего изобретения представляет соответствующую информацию относительно выбора целевых и усиливающих агентов, а также времени, схемы и дозы их введения. Эти сведения, способы и композиции настоящего изобретения позволяют врачам-клиницистам поддерживать стабильные терапевтические уровни лекарственных средств, которые требуют применения узкого интервала концентраций во избежание излишних и часто вредных пиков и спадов в уровнях концентраций в крови. Кроме того, повышенный объем распределения паклитаксела в присутствии циклоспорина позволяет предположить, что должно существовать большое количество лекарственных средств с противоопухолевой активностью.
Помимо полилекарственной резистентности, обусловленной Р-гликопротеином, кодируемым геном MDP1, существует и другой ген, который, как было недавно установлено, несет фенотип полилекарственной резистентности в некоторых лабораторных системах: ген, кодирующий белок, ассоциированный с полилекарственной резистентностью, MRP (см., например, Zaman et al., Proc.NatI. Acad. Sci. USA, 91: 8822-8826,: 1994).
Об этом новом гене и его белковом продукте, мембрано-ассоциированном 190 кДа-гликопротеине, мало что известно. Хотя оба гена MRP и MDR1 кодируют мембранные гликопротеины, которые могут действовать как переносчики многих лекарственных агентов, однако в их функциях имеются и различия, которые, вероятно, обусловлены субстратами и прогностическим значением этих двух генов. Так, например, экспрессия гена MRP, но не гена MDR1, является хорошим показателем плохого клинического прогноза для пациентов с нейробластомой. Предполагаемая функция MRP-ассоциированных белков заключается в том, что они служат откачивающим насосом для конъюгатов глутатиона S. Таким образом, молекулы, которые подвергаются конъюгированию с глутатионом, должны быть восприимчивы к воздействию MRP-ассоциированной системы.
Пероральная биологическая доступность фармакологически активных агентов (или воздействие этих агентов на опухоль), к которым вырабатывается резистентность под действием MRP- ассоциированных белков, может быть увеличена путем совместного перорального введения ингибиторов MRP. Предпочтительный вариант осуществления такого способа повышения биологической доступности лекарственного средства предусматривает пероральное введение одного или нескольких ингибиторов MRP до перорального совместного введения одного или нескольких ингибиторов MRP и одного или нескольких целевых агентов, к которым вырабатывается MRP-опосредованная резистентность.
Примерами целевых агентов такого типа являются (но не ограничиваются ими) винкаалкалоиды (например, винкристин), антрациклины, эпидофиллотоксины (например, этопозид) и различные таксаны. Примерами ингибиторов MRP, которые могут повышать пероральную биологическую доступность целевых агентов, являются (но не ограничиваются ими) циклоспорины, кетоконазол и экспериментальные лекарственные средства VX-710 и vx-853 (Vertex Pharmaceuticals, Inc., Cambridge, MA). Структуры VX-710 и VX-853, а также другие родственные соединения, которые раскрываются в патенте США №5192773.
Другой способ повышения пероральной биологической доступности лекарственных агентов, к которым вырабатывается MRP-опосредованная резистентность, предусматривает совместное введение с этими агентами глутатиона или соединений, которые образуют конъюгированные с глутатионом продукты, препятствующие функционированию MRP-системы и повышающие абсорбцию целевых агентов из кишечника, или повышающие системное воздействие целевых агентов, подвергаемых MRP-опосредованному транспорту.
Еще одной системой, способной нести полилекарственную резистентность, является так называемый белок, ассоциированный с резистентностью в легких (LRP), поскольку он впервые был идентифицирован в полирезистентной клеточной линии рака легких. Этот белок является главным структурным белком так называемого сводчатого аппарата и представляет собой широко распространенную цитоплазматическую рибонуклеопротеиновую частицу, которая была передана человеку от миксомицетов. Ингибирование этой системы может также положительно повлиять на биологическую доступность некоторых агентов. Наибольшая экспрессия LRP была обнаружена в эпителиальных клетках с секреторными и экскреторными функциями, а также в клетках, подвергаемых постоянному воздействию ксенобиотиков, таких как клеточные линии бронхов и тонкого кишечника (Scheffer et al., Nature Medicine, 1: 578-582, 1955). Поэтому указанная система может также служить мишенью в способе повышения биологической доступности лекарственных средств.
Таким образом, как было указано выше, способы, композиции и наборы, используемые в целях настоящего изобретения, могут быть соответствующим образом адаптированы согласно условиям практического использования.
При этом следует отметить, что вышеописанные варианты настоящего изобретения имеют лишь иллюстративный, а не ограничительный характер, а поэтому в него могут быть внесены различные изменения, не выходящие за рамки существа и объема изобретения.
Все, что является новым и, желательно, защищенным патентом, заявлено в нижеследующей формуле изобретения.





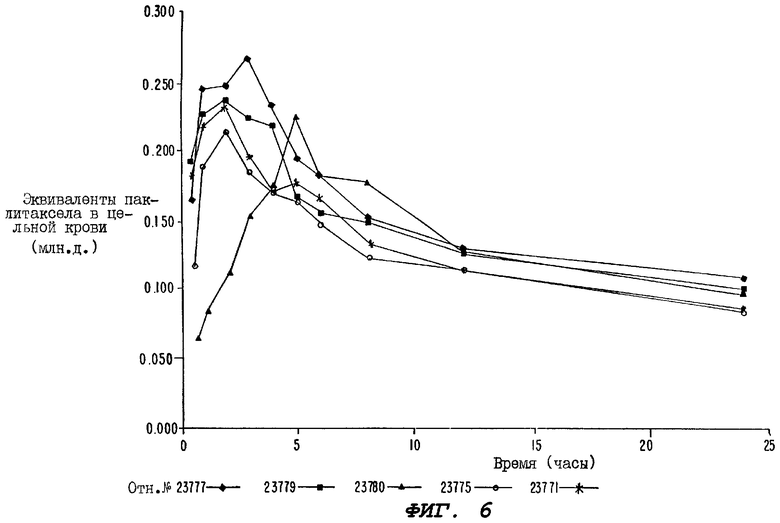


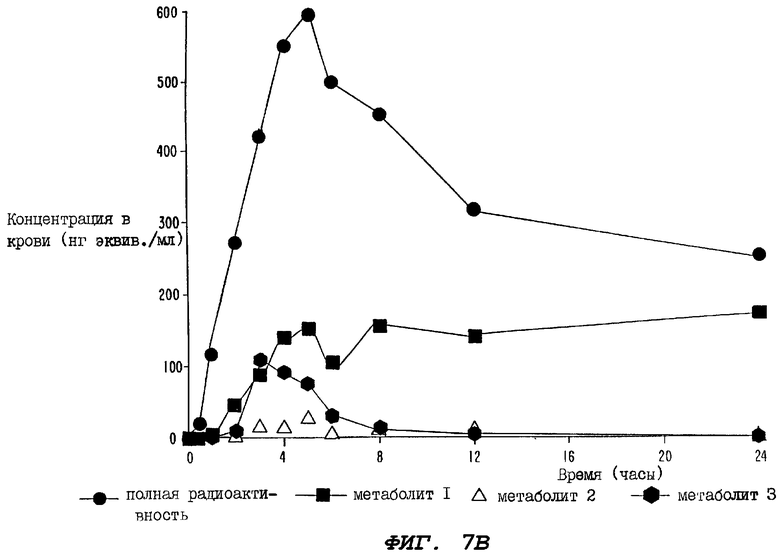
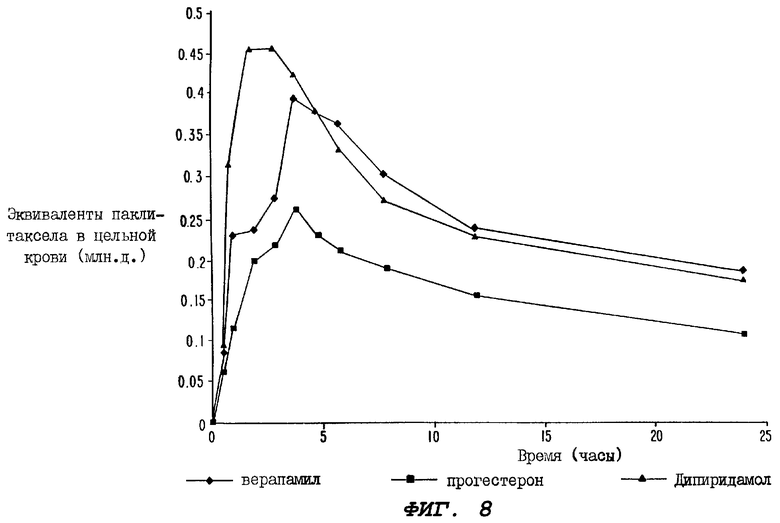
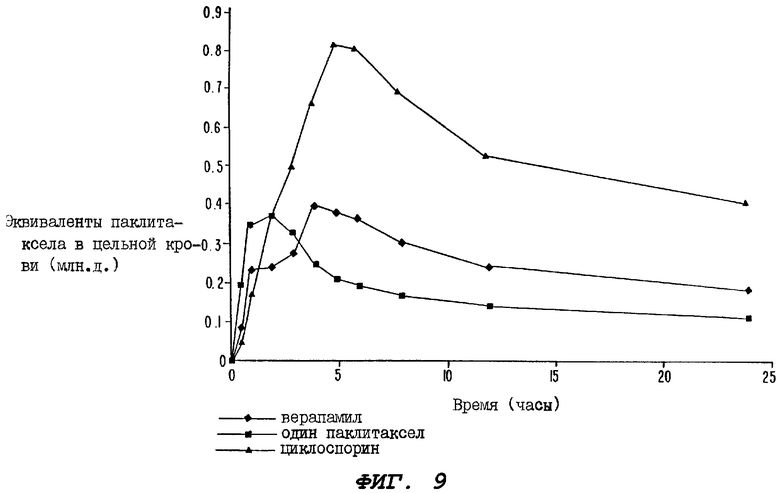
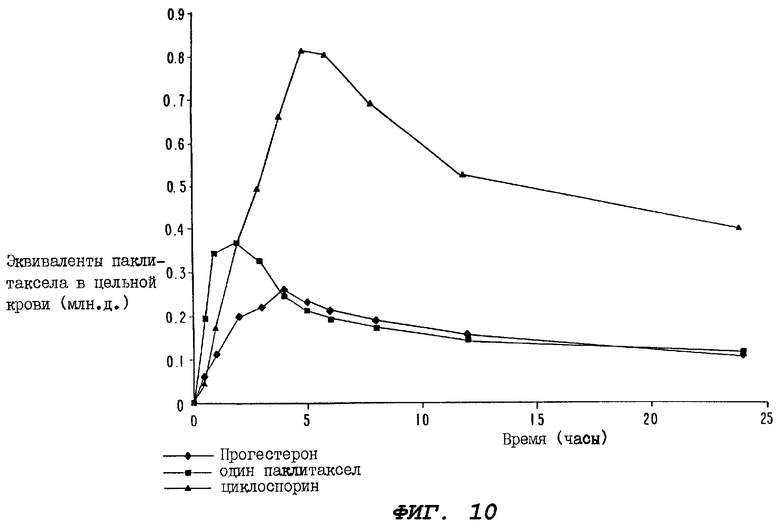



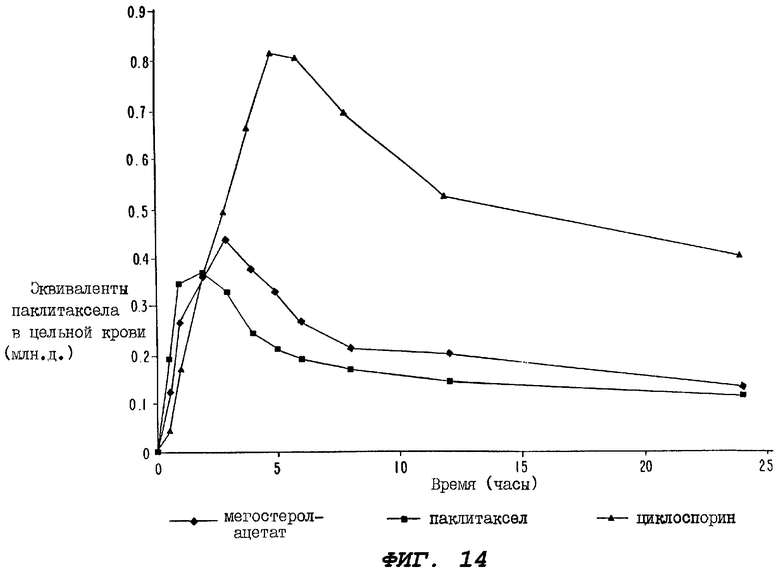
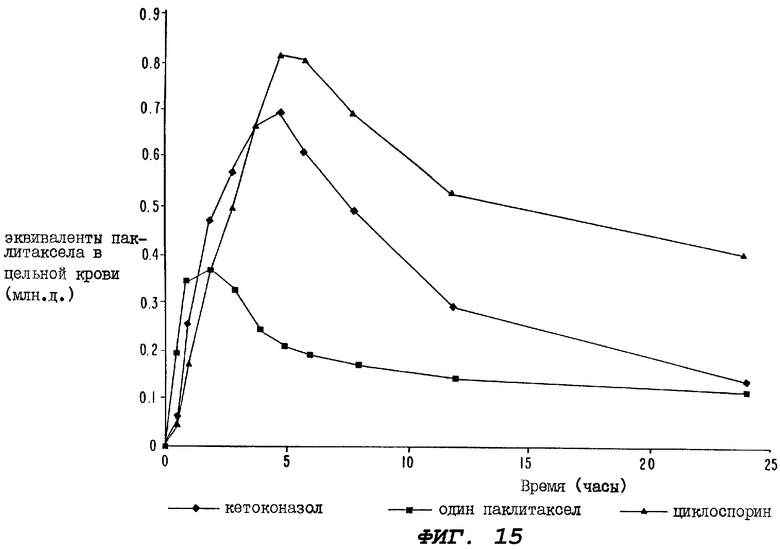


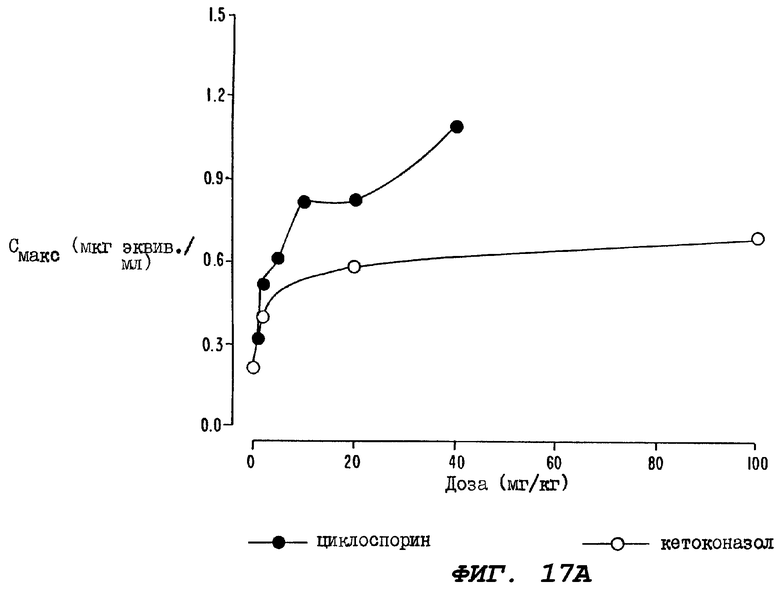

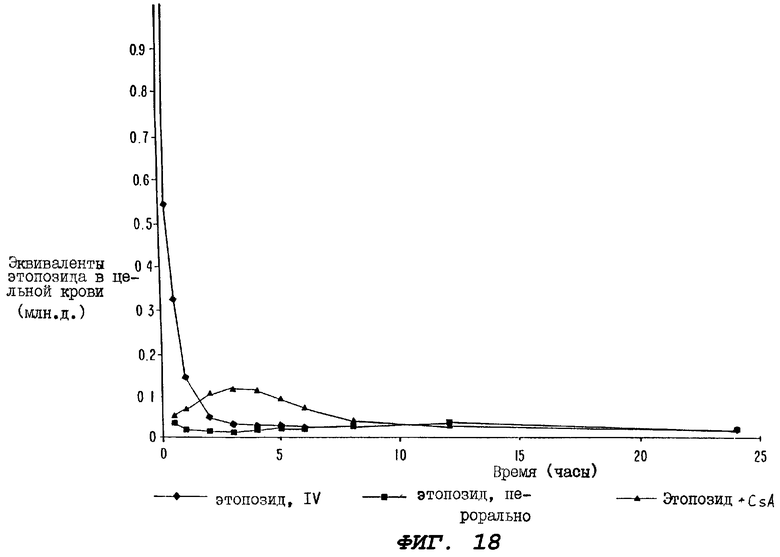

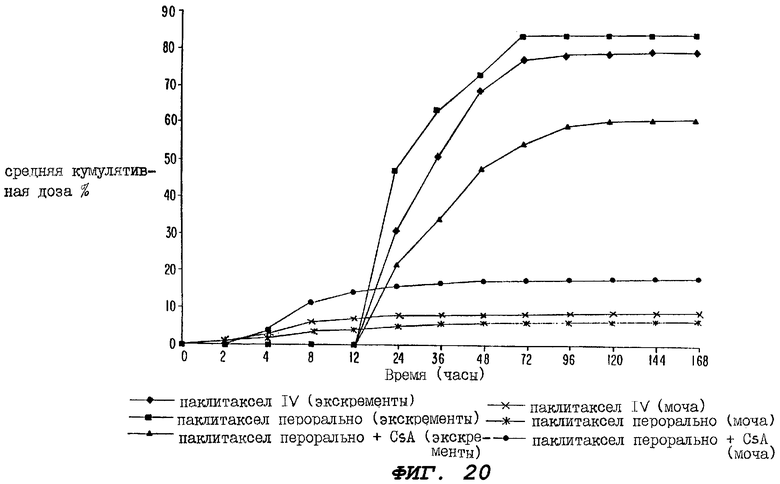
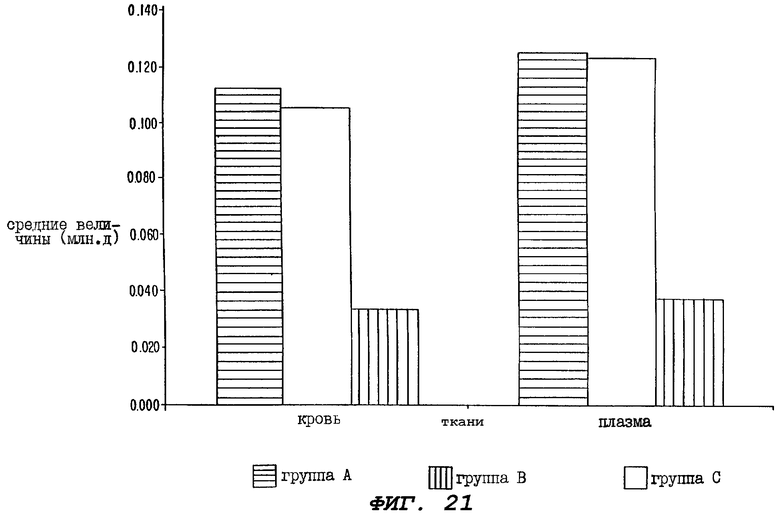

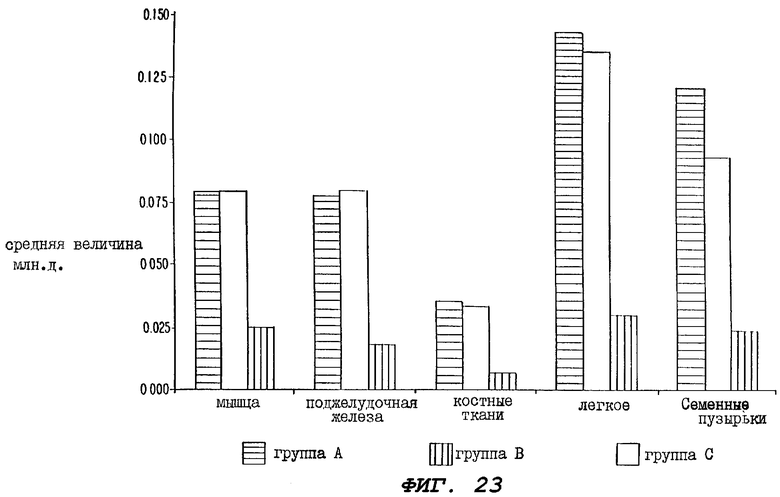



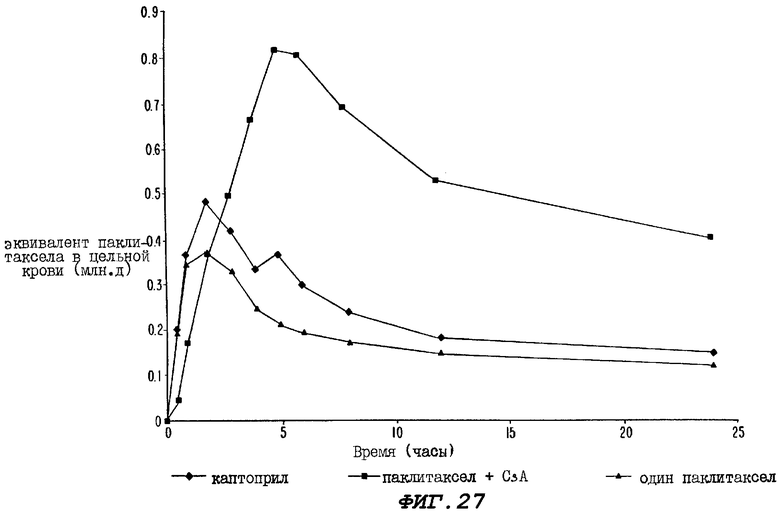
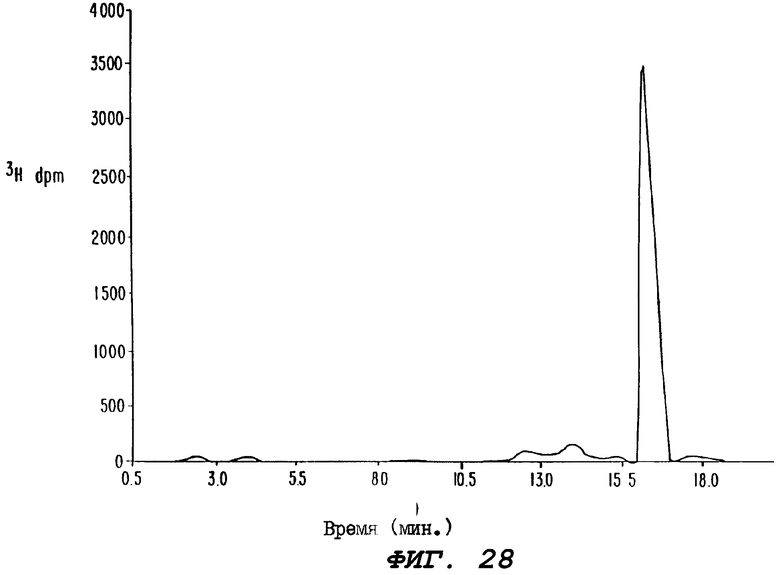
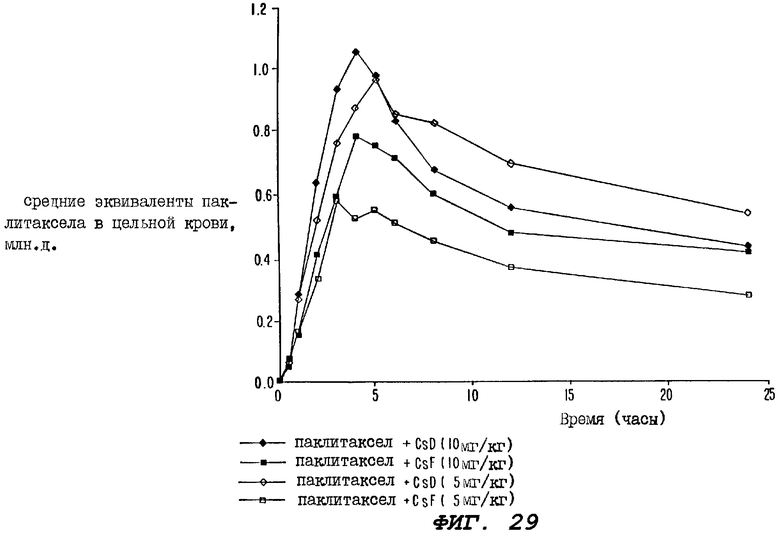
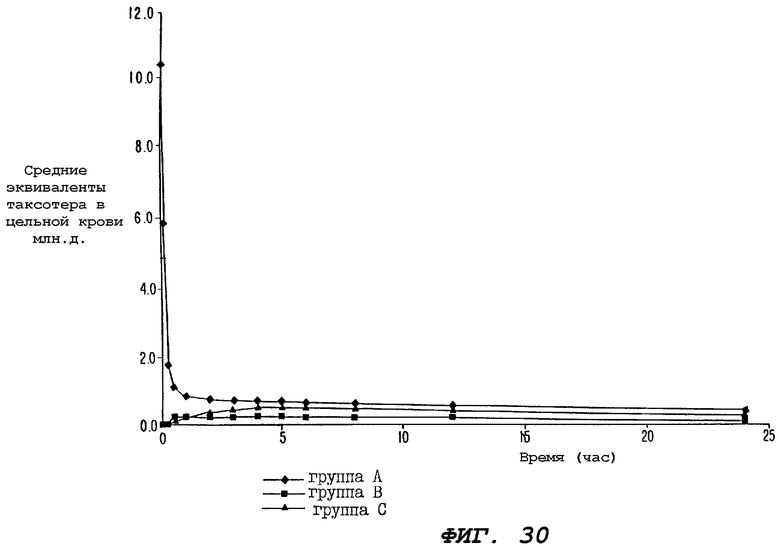


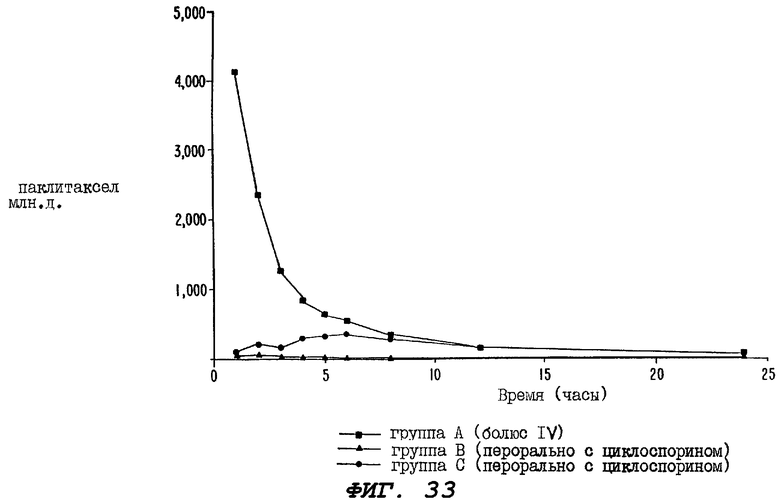

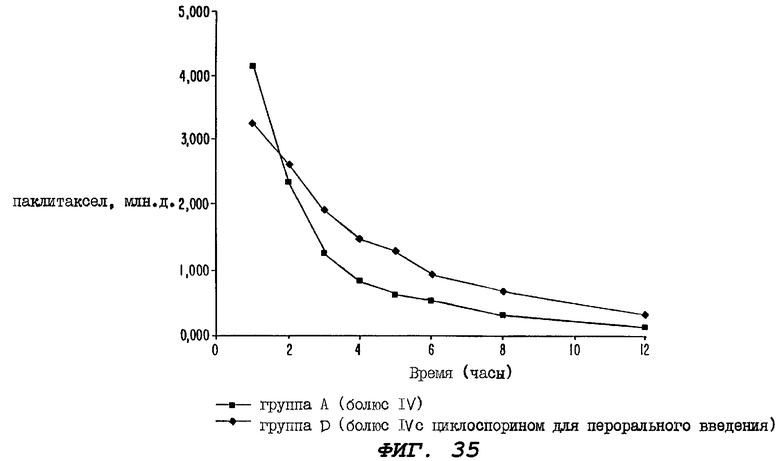
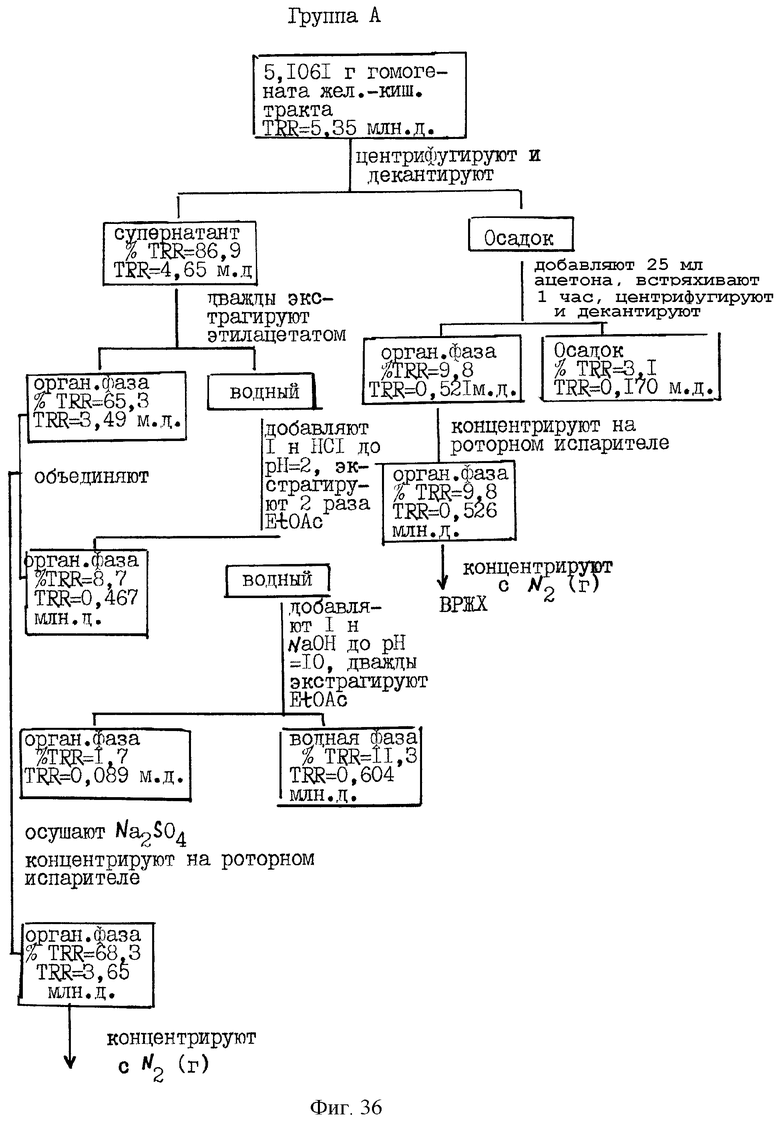


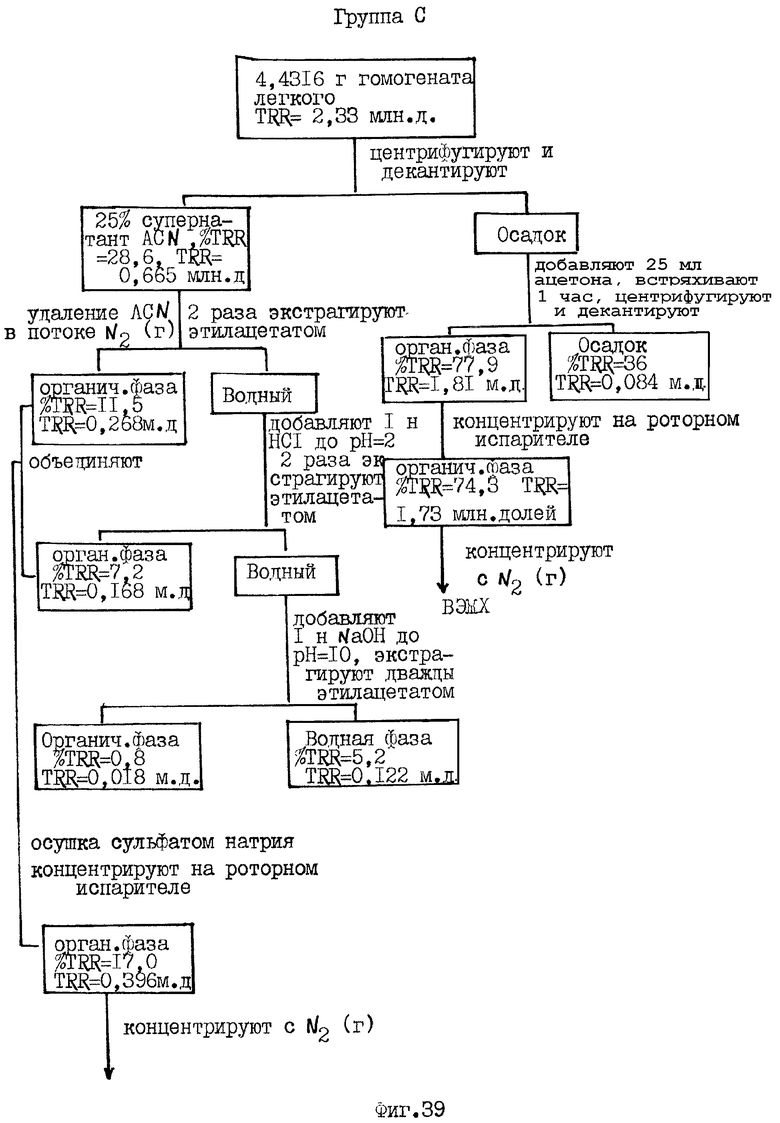
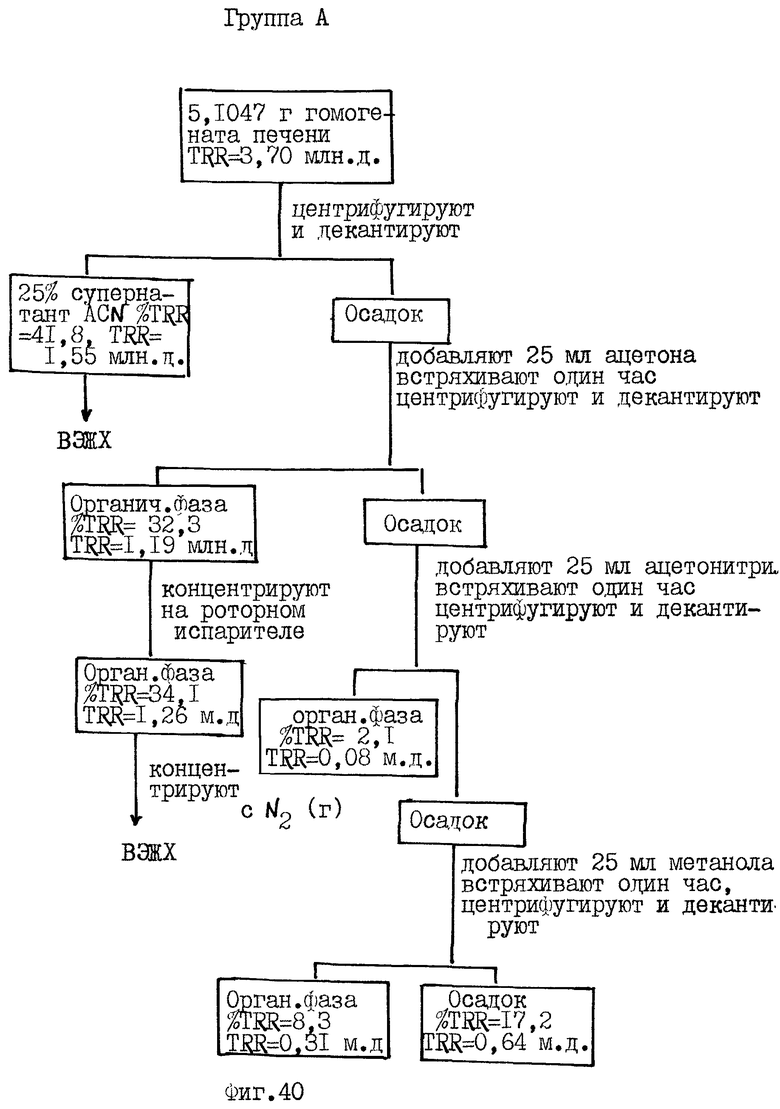

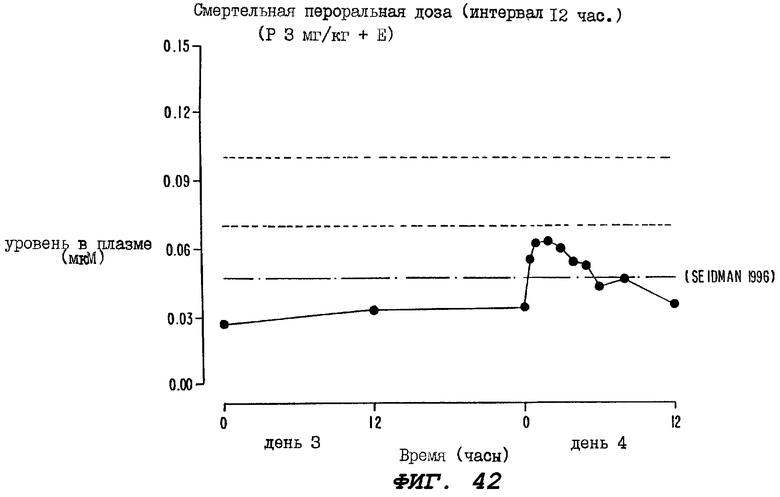
Изобретение относится к области медицины, фармакологии, онкологии, и касается способов и лекарственных форм для противоопухолевого лечения и повышения пероральной биологической доступности таксанов. Для этого таксан вводят в сочетании с агентом, усиливающим биологическую доступность таксана при пероральном введении субъекту, причем таксан достигает уровней терапевтической активности у субъекта. В качестве агента, усиливающего пероральную биодоступность таксана, используют кетоконазол - ингибитор цитохрома Р450. Изобретение обеспечивает повышение биодоступности таксана при его пероральном введении субъекту в субтерапевтических дозах, что приводит к снижению токсичности лечения. 4 н. и 29 з.п. ф-лы, 42 ил., 10 табл.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Противоопухолевая химиотерапия | |||
| Справочник./Под ред | |||
| проф | |||
| Н.И.Переводчиковой | |||
| - М.: Медицина, 1986, с.6-9. | |||

