ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к новым фармацевтическим композициям в форме самоэмульгирующейся композиции, которые обеспечивают высокую концентрацию и высокую пероральную биодоступность для липофильных, фармацевтически активных агентов.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Недавно было обнаружено, что некоторые соединения пиранона ингибируют ретровирусную протеазу и, следовательно, они применимы для лечения пациентов, инфицированных вирусом иммунодефицита человека (ВИЧ), который приводит к синдрому приобретенного иммунодефицита (СПИДу). В частности, было обнаружено, что соединение пиранона формулы I является особенно эффективным в качестве ингибитора ретровирусной протеазы.
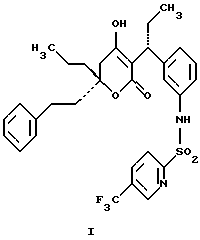
Однако подобно многим другим ингибиторам протеазы ВИЧ, эти соединения являются в типичном случае липофильными и, следовательно, слаборастворимыми в воде. Например, соединение формулы I имеет водорастворимость приблизительно 1 мкг/мл в буфере с рН 6,5 (близком к рН кишечника), что считается чрезвычайно низкой растворимостью в воде, и, как можно было бы ожидать, дает очень низкую пероральную биодоступность в форме свободной кислоты. Хорошо известно, что активное лекарственное вещество или терапевтический компонент, вводимый любым способом, должен обладать некоторой растворимостью в воде для системного всасывания и терапевтической ответной реакции. Слаборастворимые в воде соединения проявляют либо неполное, либо неустойчивое всасывание и, следовательно, производят минимальную ответную реакцию при желаемой дозе.
Были предприняты попытки идентификации солей соединений пиранона в твердой форме, которые могли бы улучшить растворимость в воде. Однако отвергающий эти попытки недостаток, который остается, заключается в том, что эти композиции в форме соли склонны к осаждению исходной свободной кислоты в желудочно-кишечном тракте и, следовательно, неспособны обеспечивать дозу в желаемой высокой концентрации для создания возможности удобного использования и в то же время удовлетворения требуемых критериев в отношении биодоступности.
Признавая эти проблемы, данное изобретение направлено на фармацевтические композиции в форме самоэмульгирующихся композиций, которые обеспечивают высокую концентрацию и высокую пероральную биодоступность для соединений пиранона. В частности, было обнаружено, что композиции данного изобретения позволяют приготовлять самоэмульгирующиеся композиции, содержащие пираноновый ингибитор ретровирусной протеазы в очень высокой концентрации до приблизительно 400 мг/г, для того чтобы сделать возможным пероральное введение, обеспечивая в то же самое время достижение улучшенной биодоступности, которая является по меньшей мере вдвое более высокой, чем биодоступность водной суспензии свободной кислоты.
Также было обнаружено, что композиции данного изобретения применимы к липофильным соединениям, как описано в этом изобретении.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В Международной публикации WO 95/30670 описаны соединения пиранона, применимые для лечения ретровирусных инфекций.
В Международной публикации WO 96/39142 описаны композиции, которые увеличивают биодоступность ингибиторов протеазы.
В Патентной заявке Великобритании GB 2222770 А описаны фармацевтические композиции, содержащие циклоспорин в преконцентрате микроэмульсии и в форме микроэмульсии.
В Патентной заявке Великобритании GB 2228198 А описаны фармацевтические композиции, содержащие циклоспорин в качестве активного ингредиента, триглицерид жирной кислоты, неполный эфир глицерина и жирной кислоты или полный или неполный сложный эфир пропиленгликоля или сорбита и поверхностно-активное вещество, имеющее HLB (гидрофильно-липофильный баланс) по меньшей мере 10.
В Патенте Великобритании GB 2257359 В описаны фармацевтические композиции, пригодные для перорального введения, содержащие циклоспорин, 1,2-пропиленгликоль, смешанный моно-, ди- и триглицерид и гидрофильное поверхностно-активное вещество.
В Патенте США 4230702 описана легко абсорбируемая в тонком кишечнике фармацевтическая композиция фармакологически активных агентов, которые per se являются слабо абсорбируемыми в тонком кишечнике.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Одной целью данного изобретения является обеспечение фармацевтической композиции, содержащей липофильный фармацевтически активный агент, которая обладает высокой пероральной биодоступностыо.
Другой целью данного изобретения является обеспечение фармацевтической композиции, содержащей высокую лекарственную нагрузку липофильного фармацевтически активного агента для удобного введения.
Другой целью данного изобретения является обеспечение фармацевтических композиций, которые проявляют адекватную физическую и химическую стабильность в самоэмульгирующейся композиции.
Еще одной целью данного изобретения является обеспечение жидкой композиции для мягких эластичных капсул.
Цели данного изобретения были достигнуты благодаря тому, что данное изобретение обеспечивает фармацевтические композиции в форме самоэмульгирующейся композиции, которые позволяют получить высокую нагрузку соединений пиранона (до приблизительно 400 мг/г) при одновременном достижении хорошей пероральной биодоступности.
Данное изобретение конкретно обеспечивает фармацевтическую композицию на основе применения определенной масляной фазы, содержащую:
(a) липофильный фармацевтически активный агент,
(b) смесь диглицерида и моноглицерида в соотношении от около 9:1 до около 6:4 по весу (диглицерид : моноглицерид), где диглицерид и моноглицерид являются эфирами моно- или диненасыщенных жирных кислот и глицерина, имеющими длину цепи шестнадцать-двадцать два атома углерода,
(с) один или несколько фармацевтически приемлемых растворителей и
(d) одно или несколько фармацевтически приемлемых поверхностно-активных веществ.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В соответствии с данным изобретением описаны фармацевтические композиции, содержащие соединение пиранона в качестве фармацевтически активного агента в носителе самоэмульгирующейся композиции.
Для цели данного изобретения термин "соединения пиранона" обозначает соединения формулы II
где R1 обозначает Н-; R2 обозначает С3-С5-алкил, фенил-(СН2)2-, het-SO2NH-(СН2)2-, циклопропил- (СН2)2-, F-фенил-(CH2)2-, het-SО2NН-фенил или F3С-(СН2)2-; или R1 и R2, взятые вместе, обозначают двойную связь; R3 обозначает R4-(СН2)n-CH(R5)-, Н3С-[O(СН2)2]2-СН2, С3-С5-алкил, фенил-(СН2)2-, het-SО2NН-(СН2)2-, (HOCH2)3C-NH-C(O)-NH-(CH2)3-, (НО2С)(H2N)СН-(СН2)2-С(O)-NH-(СН2)3-, пиперазин-1-ил-С(О)-NH-(СН2)3, НО3S(СН2)2-N(СН3)-С(О)-(СН2)6-С(О)-NH-(СН2)3-, циклопропил-(СН2)2-, F-фенил-(СН2)2-, het-SО2NН-фенил или F3С-(СН2)2-; n равно 0, 1 или 2; R4 обозначает фенил, het, циклопропил, Н3С-[O(СН2)2]2, het-SO2NH-, Br-, N3- или НО3S(СН2)2-N(СН3)-С(О)-(СН2)6-С(О)-NH-; R5 обозначает -СН2-СН3 или -СН2-циклопропил; R6 обозначает циклопропил, -СН3-СН2- или трет-бутил; R7 обозначает -NR8SO2-het, -NR8SO2-фенил, необязательно замещенный R9, -СН2-SО2-фенил, необязательно замещенный R9, или -СН2-SО2-het; R8 обозначает -Н или -СН3; R9 обозначает -CN, -F, -ОН или -NO2; где het обозначает 5-, 6- или 7-членное насыщенное или ненасыщенное кольцо, содержащее от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы; и в том числе любую бициклическую группу, в которой любое из вышеупомянутых гетероциклических колец конденсировано с бензольным кольцом или другим гетероциклом, необязательно замещенным -СН3, -CN, -ОН, -C(O)OC2H5, -СF3, -NH2 или -С(O)-NН2; или их фармацевтически приемлемую соль. Предпочтительным соединением формулы II является соединение формулы I.
Термин "соединения пиранона" относится также к соединениям формулы III и формулы IV

где R10 обозначает Н-, СН3О- или СН3О-[(СН2)2O]3-; R11 обозначает циклопропил или -СН2-СН(СН3)2; R12 обозначает -NR14SO2-фeнил, необязательно замещенный R15, -NR14SO2-het, СН2-SО2-фенил, необязательно замещенный R15, или -CH2-SO2-het; R13 обозначает -Н, -(СН2)2-СН3, -СН2-циклопропил или -СН2-фенил; R14 обозначает -Н или -СН3; R15 обозначает -CN, -F, -СН3, -СООН или -ОН; het обозначает 5, 6- или 7-членное насыщенное или ненасыщенное кольцо, содержащее от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы; и в том числе любую бициклическую группу, в которой любое из вышеупомянутых гетероциклических колец конденсировано с бензольным кольцом или другим гетероциклом, необязательно замещенным одним или двумя -СН3, -CN, С(O)ОС2Н5 или -ОН; или их фармацевтически приемлемой соли.
Эти соединения ингибируют ретровирусную протеазу и, следовательно, ингибируют репликацию вируса. Они применимы для лечения пациентов, инфицированных ретровирусом человека, таким как вирус иммунодефицита человека (штаммы ВИЧ-1 или ВИЧ-2) или вирусы Т-клеточного лейкоза человека (HTLV-I или HTLV-II), который приводит к синдрому приобретенного иммунодефицита (СПИДу) и/или родственным заболеваниям. Соединения формул I, II, III и IV описаны и заявлены и Международной заявке с номером PCT/US95/05219, включенной здесь в качестве ссылки, и могут быть получены в соответствии с процедурами, описанными в Международной публикации WO 95/30670. В частности, было обнаружено, что соединение пиранона формулы I является особенно эффективным в качестве ингибитора ретровирусной протеазы.
Термин "липофильные соединения", в применении здесь, относится к соединениям с LOG P≥2 (величина LOG P определяется как распределение в двухфазной системе, таким как коэффициент распределения между фазами октанола и воды; ее либо определяют экспериментально, либо рассчитывают при помощи коммерчески доступного программного обеспечения), низкой собственной растворимостью в воде (≤0,1 мг/мл) в области рН 1-8 и имеющим растворимость, в носителе самоэмульгирующейся композиции данного изобретения больше чем 1 мг/мл.
Типичные примеры липофильных соединений, которые пригодны для применения в данном изобретении, включают в себя, но не ограничиваются ими, соединения пиранона формул I, II, III или IV; циклоспорины, такие как природно встречающиеся циклоспорины А-Z, а также различные неприродные производные циклоспоринов или синтетические циклоспорины; липофильные стероиды, такие как медроксипрогестерон-ацетат, прогестерон или тестостерон, тиазолидиндионы, такие как троглитазон или пиоглитазон; сульфонилмочевины, такие как глибурид; азолы, такие как кетоконазол или итраконазол; камптотецины, такие как камптотецин, SN-38 или иринотекан-гидрохлорид (также называемый СРТ-11); таксаны, такие как паклитаксел, доцетаксел или PNU-1; простагландины, такие как PGE2α, PGE1 или PGE2; делавирдин-мезилат, витамин Е (α-токоферол), тирилазад-мезилат, гризеофульвин, фенитоин, ибупрофен, флурбипрофен, PNU-2, PNU-3 или PNU-4.
Термин "SN-38" относится к химическому соединению под названием (4S)-4,11-диэтил-4,9-дигидрокси-1Н-пирано[3', 4': 6,7] индолизино[1,2-b]хинолин-3,14(4Н,12Н)-дион.
Термин "PNU-1" относится к химическому соединению под названием [2aR-[2aα, 4аβ, 6β, 7β, 9(αR*, βS*), 11α, 12α, 12aα, 12bα]]-6,12b-бис(ацетилокси)-12-(бензоилокси)-2а, 4а, 5, 6, 7, 10, 11, 12, 12а, 12b-декагидро-11-гидрокси-4а, 8, 13, 13-тетраметил-5-оксо-7,11-метано-1Н-циклодека [3,4]бенз [1, 2-b] оксет-9-ил-β-[[[(1,1-диметилэтил) амино] карбонил] амино] -α-гидроксибензол-пропаноат, или (1S, 2S, 3R, 4R, 7R, 10R, 12R)-4,12-бис (ацетилокси)-15-[((2R, 3S)-3-{[(трет-бутиламино)карбояил]амино}-2-гидрокси-3-фенилпропаноил)окси] -1-гидрокси-10,14,17,17-тетраметил-11-оксо-6-оксатетрацикло[11, 3, 1,03,10, 04,7]гептадека-8,14-диен-2-илбензоат.
Термин "PNU-2" относится к химическому соединению под названием 1-[(2,4-ди-1-пирролидинил-9Н-пиримидо[4,5-b] индол-9-ил)ацетил]пирролидин, или 2-[2,4-ди(1-пирролидинил)-9Н-пиримидо[4,5-b] индол-9-ил] -1-(1-пирролидинил)-1-этанон.
Термин "PNU-3" относится к химическому соединению под названием (S)-1-[2-[4-[4-(аминокарбонил)фенил] -1-пиперазинил] этил]-3,4-дигидро-Н-метил-1Н-2-бензопиран-6-карбоксамид, или lH-2-бензопиран-6-карбоксамид, 1-[2-[4-[4-(аминокарбонил) фенил]-1-пиперазинил]этил]-3,4-дигидро-N-метил-, (S)- или (1S)-1-(2-[4-[4-(аминокарбонил)фенил] -1-пиперазинил)этил)-N-метил-3,4-дигидро-1H-изохромен-6-карбоксамид.
Термин "PNU-4" относится к химическому соединению под названием (-)-6-хлор-2-[(1-фуро[2,3-с] пиридин-5-илэтил)тио]-4-пиримидинамин, или 6-хлор-2-{ [(13)-1-фуро[2,3-с]пиридин-5-илэтил]сульфонил}-4-пиримидиниламин.
Все эти фармацевтически активные агенты известны в данной области и могут быть легко получены или приготовлены в соответствии с известными способами.
Например, природно встречающиеся циклоспорины могут быть получены в соответствии с процедурой, описанной в Тrаber et al. I, Helv. Chim. Acta. 60, 1247-1255 (1977); Traber et al. 2, Helv. Chim. Acta. 65 No. 162, 1655-1667 (1982); Kobel et al., Europ. J. Applied Microbiology and Biotechnology 14, 273-240 (1982); и von Wartburg et al., Progress in Allergy, No. 38, 28-45 (1986)].
Неприродные производные циклоспорина или синтетические циклоспорины могут быть получены в соответствии с процедурой, описанной в Патентах США с номерами 4108985, 4210581 и 4220641; Европейских публикациях патентов с номерами 0034567 и 0056782; Международной публикации патента с номером WO 86/02080; Wenger I, Transp. Proc. 15, Suppi. 1:2230 (1983); Wenger 2, Angew. Chem. Int. Ed. , 24, 77 (1985); и Wenger 3, Progress in the Chemistry of Organic Natural Products 50, 123 (1986).
Прогестерон и тестостерон являются общеизвестными и обсуждались в многочисленных публикациях.
Камптотецин может быть получен из стволовой древесины дерева Chinese tree согласно процедуре, описанной в М.Е. Wall et al., J. Am. Chem. Soc., vol. 88, p. 3888 (1966). Камптотецин может быть также получен в соответствии с процедурой, описанной в E.J. Corey, et al., ibid. 40. р. 2140 (1975), Stork, Schultz, J. Am. Chem. Soc., vol. 93, p. 4074 (1971); J.C. Bradley, G. Buchi, J. Org. Chem., vol. 41, p. 699 (1976); Т. Kametani et al., J. Chem. Soc. Perkin Trans. I, p. 1563 (1981).
Троглитазон может быть получен в соответствии с процедурой, описанной в Патенте США 4572912.
Пиоглитазон может быть получен в соответствии с процедурой, описанной в Патенте США 4687777.
Кетоконазол может быть получен в соответствии с процедурой, описанной в Патентах США 4144346 и 4223036.
Глибурид может быть получен в соответствии с процедурой, описанной в Патенте США 3454635.
Гризеофульвин может быть получен в соответствии с процедурами, описанными в Патенте США 3069328, Патенте США 3069329 и Groove et al., Chem. & Ind. (London), p. 219 (1951); и J. Chem. Soc. p. 3977 (1952).
Итраконазол может быть получен в соответствии с процедурой, описанной в Патенте США 4267179.
Паклитаксел может быть получен в соответствии с процедурой, описанной в R. A. Holton et al., J. Am. Chem. Soc., vol. 110, p. 6558 (1988); К.С. Nicolaou et al. . Nature, vol. 367, p. 630 (1994); D.G.I. Kingston et al. Studies in Organic Chemistry, vol. 26, "New Trends in Natural Products Chemistry 1986", Attaur-Rahman, P.W. Le Quesne, Eds. (Elsevier, Amsterdam, 1986), p. 219-235.
Медроксипрогестерон-ацетат может быть получен в соответствии с процедурой, описанной в Патенте США 3359287.
Тирилазад-мезилат может быть получен в соответствии с процедурой, описанной в Патенте США 5175281.
Делавирдин может быть получен в соответствии с процедурой, описанной в Международной патентной заявке РСТ 91/09849.
PNU-1 может быть получен в соответствии с процедурой, описанной в R.A. Johson et al., J. Med. Chem. vol. 40, р. 2810-2812 (1997).
PNU-2 может быть получен в соответствии с процедурой, описанной в Международной публикации с номером WO 93/20078.
PNU-3 может быть получен в соответствии с процедурой, описанной в Международной публикации с номером WO 97/02259.
PNU-4 может быть получен в соответствии с процедурой, описанной в Международной публикации с номером WO 96/135678.
Ибупрофен может быть получен в соответствии с процедурой, описанной в Патентах CDJA 3228831 и 3385886.
Флурбипрофен может быть получен в соответствии с процедурой, описанной в Патенте США 3755427.
Фенитоин может быть получен в соответствии с процедурой, описанной в Патенте США 2409754.
Иринотекан-гидрохлорид (СРТ-11) может быть получен в соответствии с процедурой, описанной в Патенте США 4604463.
PGE1 может быть получен в соответствии с процедурой, описанной в E.J. Corey et al., J. Am. Chem. Soc., 90, 3245-3247 (1968).
PGE2 может быть получен в соответствии с процедурой, описанной в Патенте США 3598858.
PGE2α может быть получен в соответствии с процедурой, описанной в Патенте США 3657327.
Термин "самоэмульгирующаяся композиция", в применении здесь, относится к концентрированной композиции, способной генерировать эмульсии или микроэмульсии при смешивании с достаточным количеством водной среды.
Эмульсии или микроэмульсии, получаемые по данному изобретению, являются общепринятыми растворами, содержащими гидрофильную фазу и липофилъную фазу. Микроэмульсии характеризуются также их термодинамической стабильностью, оптической прозрачностью и малым средним размером капелек, обычно менее около 0,15 микрон.
Термин "носитель самоэмульгирующейся композиции" относится к композиции, содержащей смесь диглицерида и моноглицерида в соотношении от около 9:1 до около 6:4 по весу (диглицерид : моноглицерид), где диглицерид и моноглицерид являются эфирами моно- или диненасыщенных жирных кислот и глицерина, имеющими длину цели шестнадцать-двадцать два атома углерода, один или несколько фармацевтически приемлемых растворителей и одно или несколько фармацевтически приемлемых поверхностно-активных веществ. Необязательно, носитель самоэмульгирующейся композиции может дополнительно содержать основной амин.
Диглицерид данного изобретения обозначает эфир жирной кислоты и глицерина, имеющий структурную формулу НОСН2-CH(O2CR)-CH2(O2CR) или (RCO2)СН2-СН(ОН)-CH2(O2CR), где R обозначает мононасыщенную или динасыщенную алкильную группу, имеющую пятнадцать-двадцать один атомов углерода. Предпочтительным диглицеридом является диолеин (R является мононенасыщенной алкильной группой с семнадцатью атомами углерода), дилинолеат (R является диненасыщенной алкильной группой с семнадцатью атомами углерода) или смесь диолеина и дилинолеата. Наиболее предпочтительным диглицеридом является диолеин.
Моноглицеридом данного изобретения является эфир жирной кислоты и глицерина, имеющий структурную формулу НОСН2-СН(ОН)-СН2(O2CR) или НОСН2-СН(О2СR)-СН2ОН, где R обозначает мононасыщенную или динасыщенную алкильную группу, имеющую пятнадцать-двадцать один атомов углерода. Предпочтительным моноглицеридом является моноолеин (R является мононенасыщенной алкильной группой с семнадцатью атомами углерода), монолинолеат (R является диненасыщенной алкильной группой с семнадцатью атомами углерода) или смесь моноолеина и монолинолеата. Наиболее предпочтительным моноглицеридом является моноолеин.
Смесь диглицерида и моноглицерида может быть приготовлена смешиванием индивидуальных диглицерида и моноглицерида в подходящем соотношении или частичным гидролизом триглицерида или реакцией переэтерификации триглицеридов, диглицеридов с глицерином.
Все глицериды данного изобретения являются известными и могут быть получены общепринятыми способами.
Количество активного ингредиента в этой композиции может широко варьироваться или корректироваться в зависимости от предполагаемого пути введения, активности конкретного используемого активного ингредиента, тяжести ретровирусной инфекции и требуемой концентрации. Однако, если желательно, липофильный фармацевтически активный агент может присутствовать в носителе самоэмульгирующейся композиции данного изобретения в количестве до около 400 мг/г с превосходной диспергируемостью и высокой пероральной биодоступностью in vivo, обычно достигающей 70-84% для крыс.
Композиции данного изобретения с высокой пероральной биодоступностью (84% для крыс) демонстрируют почти прозрачный или полупрозрачный раствор при разведении водой, что указывает на то, что образуется микроэмульсия.
Композиции данного изобретения с умеренно высокой биодоступностью (60-70% для крыс) обычно обнаруживают визуально мелкозернистую белую эмульсию без осаждения лекарственного средства при разведении водой, что указывает на то, что образуется эмульсия.
В одном аспекте данное изобретение специфически обеспечивает фармацевтическую композицию, основанную на применении определенной масляной фазы, содержащую:
(a) соединение пиранона формул I, II, III или IV в качестве фармацевтически активного агента,
(b) смесь диглицерида и моноглицерида в соотношении от около 9:1 до около 6: 4 по весу (диглицерид : моноглицерид), где диглицерид и моноглицерид являются эфирами моно- или диненасыщенных жирных кислот и глицерина, имеющими длину цепи шестнадцать-двадцать два атома углерода,
(c) один или несколько фармацевтически приемлемых растворителей и
(а) одно или несколько фармацевтически приемлемых поверхностно-активных веществ.
В другом аспекте, данное изобретение обеспечивает фармацевтическую композицию на основе применения конкретной масляной фазы, которая содержит:
(a) липофильный фармацевтически приемлемый активный агент, выбранный из группы, состоящей из циклоспоринов, медроксипрогестерон-ацетата, прогестерона, тестостерона, троглитазона, пиоглитазона, глибурида, кетоконазола, итраконазола, камптотецина, SN-38, иринотекан-гидрохлорида, паклитаксела, доцетаксела, PNU-1, PGE2α, PGE1, PGE2, делавирдинмезилата, витамина Е, тирилазад-мезилата, гризеофульвина, фенитоина, ибупрофена, флурбипрофена, PNU-2, PNU-3 и PNU-4,
(b) смесь диглицерида и моноглицерида в соотношении от около 9:1 до около 6: 4 по весу (диглицерид : моноглицерид), где диглицерид и моноглицерид являются эфирами моно- или диненасыщенных жирных кислот и глицерина, имеющими длину цепи шестнадцать-двадцать два атома углерода,
(с) один или несколько фармацевтически приемлемых растворителей и
(d) одно или несколько фармацевтически приемлемых поверхностно-активных веществ.
Кроме того, эти композиции могут дополнительно содержать фармацевтически приемлемый основной амин.
Термин "фармацевтически приемлемый", в применении здесь, обозначает те свойства, которые являются биологически совместимыми с проходящими лечение субъектами с фармакологической и токсикологической точки зрения.
Растворителями данного изобретения называют пропиленгликоль, полипропиленгликоль, полиэтиленгликоль (такой как ПЭГ300, 400, 600 и т.д.), глицерин, этанол, триацетин, диметилизосорбид, гликофурол, пропиленкарбонат, воду, диметилацетамид или их смесь.
Предпочтительным растворителем является пропиленгликоль или смесь, содержащая пропиленгликоль и 95% (об/об) этанол (далее этанол). В этой смеси пропиленгликоля и этанола пропиленгликоль находится в количестве от около 50 до около 95%.
Поверхностно-активными веществами данного изобретения называют неионогенные поверхностно-активные вещества, в том числе гидрогенизированное касторовое масло Полиоксил 40, продаваемое под товарным названием, среди прочих, Cremophor RH40; касторовое масло Полиоксил 35, продаваемое под товарным названием, среди прочих, Cremophor EL или Cremophor EL-Р; Полисорбаты; Solutol HS-15; Tagat TO; Пегликол 6-олеат; полиоксиэтиленстеараты; насыщенные полигликозилированные глицериды или полоксамеры; все они являются коммерчески доступными. Предпочтительным поверхностно-активным веществом является Cremophor RH40 или Creroophor EL.
Насыщенные полигликозилированные глицериды, в применении здесь, включают в себя Gelucire 44/14 или Gelucire 50/13.
Полиоксиэтиленстеараты, в применении здесь, включают в себя Полиоксил 6-стеарат, Полиоксил 8-стеарат, Полиоксил 12-стеарат и Полиоксил 20-стеарат.
Полиоксамеры, в применении здесь, включают в себя Полоксамер 124 и Полоксамер 188.
Полисорбаты, в применении здесь, включают в себя Полисорбат 20, Полисорбат 40, Полисорбат 60 и Полисорбат 80.
Термин "основной амин", в применении здесь, обозначает низшие алкиламины, такие как, например, этаноламин, диэтаноламин, триэтаноламин, диметиламиноэтанол, трис(гидроксиметил) аминометан или этилендиамин; четвертичные аммонии, такие как, например, холингидроксид; основные аминокислоты, такие как, например, аргинин, лизин или гуанидин. Предпочтительным низший алкиламином является диметиламиноэтанол или трис(гидроксиметил)аминометан.
Типичная композиция этого изобретения содержит:
(а) липофильный фармацевтически активный агент в количестве от около 1 до около 40% по весу общей композиции,
(b) смесь диглицерида и моноглицерида в соотношении от около 9:1 до около 6: 4 по весу (диглицерид : моноглицерид), где диглицерид и моноглицерид являются эфирами моно- или диненасыщенных жирных кислот и глицерина, имеющими длину цепи шестнадцать-двадцать два атома углерода в количестве от около 5 до около 40% по весу общей композиций,
(c) один или несколько фармацевтически приемлемых растворителей в количестве от около 10 до около 30% по весу общей композиции, и
(d) фармацевтически приемлемое поверхностно-активное вещество в количестве от около 10 до около 50% по весу общей композиции.
Необязательно, вышеупомянутая композиция может дополнительно содержать основной амин в количестве от около 0,1 до 10% по весу общей композиции.
Предпочтительными липофильными соединениями являются соединения пиранона формул I, II, III, IV или циклоспорин А.
Предпочтительная композиция этого изобретения содержит:
(a) липофильный фармацевтически активный агент в количестве от около 5% до около 30% по весу общей композиции,
(b) смесь диолеина и моноолеина в соотношении около 9:1 по весу (диолеин : моноолеин) в количестве от около 5 до около 35% по весу общей композиции,
(c) растворитель, содержащий пропиленгликоль или смесь пропиленгликоля и этанола в количестве от около 15 до около 25% по весу общей композиции, и
(d) поверхностно-активное вещество, содержащее Cremophor RH40 или Cremophor EL в количестве от около 30 до около 45% по весу обшей композиции.
Другая предпочтительная композиция этого изобретения содержит:
(a) липофильный фармацевтически активный агент в количестве от около 5 до около 30% по весу общей композиции,
(b) смесь диолеина и моноолеина в соотношении около 8:2 по весу (диолеин : моноолеин) в количестве от около 5 до около 35% по весу общей композиции,
(c) растворитель, содержащий пропиленгликоль или смесь пропиленгликоля и этанола в количестве от около 15 до около 25% по весу общей композиции, и
(d) поверхностно-активное вещество, содержащее Cremophor RH40 или Cremophor EL в количестве от около 30 до около 45% по весу общей композиции.
Необязательно, предпочтительные композиции содержат дополнительно основной амин в количестве от около 0,1 до около 7% по весу общей композиции.
В предпочтительных композициях данного изобретения, даже еще более предпочтительная композиция содержит соединение пиранона формулы I в количестве от около 20 до около 30% по весу общей композиции.
В предпочтительных композициях данного изобретения, даже еще более предпочтительная композиция содержит циклоспорин А в количестве от около 5 до около 15% по весу общей композиции.
В предпочтительных композициях данного изобретения смесь пропиленгликоля и этанола находится в соотношении около 1:1.
В предпочтительных композициях данного изобретения, даже еще более предпочтительная композиция содержит диметиламиноэтанол, трис(гидроксиметил)аминометан в количестве от около 0,1 до 7% по весу общей композиции.
В предпочтительных композициях данного изобретения, даже еще более предпочтительная композиция содержит смесь диолеина и моноолеина в соотношении около 8:2.
В частности, наиболее предпочтительная композиция данного изобретения содержит соединение пиранона формулы I.
Композиция данного изобретения может быть в форме жидкости для мягких эластичных капсул или твердых желатиновых капсул для перорального применения. Композиции могут быть также в форме жидкого раствора для перорального, парентерального, ректального или местного применения. Предпочтительной дозированной формой является форма жидкости для мягких эластичных капсул.
Если желательно, композиции данного изобретения могут дополнительно содержать общепринятые фармацевтические добавки, такие как агенты, сопутствующие поверхностно-активным веществам (например, лаурилсульфат натрия), красящие агенты, улучшающие вкус и запах агенты, ароматизаторы, консервирующие агенты, стабилизаторы, антиоксиданты и/или загущающие агенты.
Композиции данного изобретения могут быть приготовлены общепринятым образом, например растворением активного агента в растворителе, затем добавлением масляной фазы, поверхностно-активного вещества и необязательно основного амина. Затем полученный раствор готовят в виде желательной дозированной формы, такой как, например, мягкие эластичные капсулы или твердые желатиновые капсулы с использованием известной технологии приготовления.
Фармацевтические композиции данного изобретения будут более понятными в связи с нижеследующими примерами, которые предназначены для иллюстрации, а не для ограничения объема этого изобретения. Без дополнительной разработки, авторы изобретения считают, что специалист в этой области сможет, с использованием предшествующего описания и информации, обеспеченной ниже в примерах, применять на практике данное изобретение в его самом полном виде.
А. Общая процедура приготовления композиций данного изобретения
Лекарственное средство помещают в контейнер. Добавляют растворитель, содержащий пропиленгликоль, или смесь растворителей, выбранных из этанола (95%) и пропиленгликоля (1:1 по весу) и крышку затягивают. Контейнер помещают в водяную баню при приблизительно 60oС и встряхивают осторожно до растворения всего материала лекарственного средства. После охлаждения контейнера до комнатной температуры в контейнер добавляют подходящие количества смеси диглицерида (такого как диолеин) и моноглицерида (такого как моноолеин), поверхностно-активного вещества (такого как Cremophor RH40 или Cremophor EL) и необязательно основного амина (такого как этаноламин или диэтаноламин). Контейнер герметизируют и помещают в водяную баню при приблизительно 60oС и встряхивают осторожно до образования прозрачного раствора. Контейнер обычно оставляют в условиях окружающей среды до последующего применения.
В. Тест пероральной биодоступности.
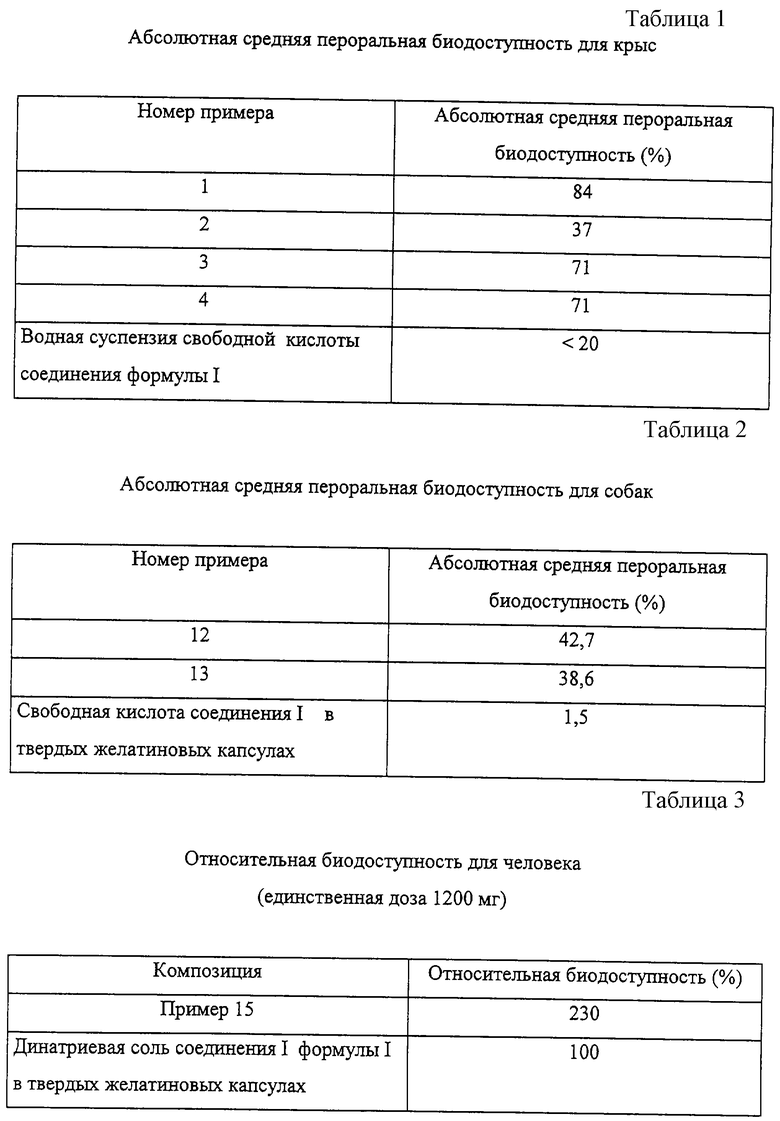
(i) Самцы крыс Sprague-Dawley были выбраны для исследования пероральной биодоступности in vivo. Каждую крысу подготавливали хирургической имплантацией постоянного катетера в верхнюю полую вену. Каждую крысу, весом в пределах 300-400 г, подвергали голоданию в течение ночи перед дозированием. Каждую композицию вводили перорально группе крыс (n=3) при дозе 20 мг/кг. Композиции с высокой концентрацией соединения формулы I (обычно 200-300 мг/мл) разводили в 100 раз водой и вводили непосредственно в желудок крысы с использованием перорального желудочного зонда. Последовательные пробы крови 0,25 мл получали из постоянного катетера при 0,25, 0,5, 1, 2, 4, 6, 8, 12 и 24 часах после дозирования. Эти пробы крови анализировали при помощи ВЖХ-анализа, специфического для тестируемых соединений. Концентрации лекарственного средства в крови тестируемых крыс наносили на график зависимости от времени после введения лекарственного средства внутривенным (i.v.) или пероральным путем и AUC (площадь под кривой зависимости концентрации в плазме от времени) интегрировали с применением правила трапеций для расчета абсолютной биодоступности, показанной в таблице 1.
Примеры 1 - 53 приведены в конце описания.

(ii) Самцов собак Beagle также выбирали для исследования пероральной биодоступности in vivo. Каждую собаку, весом в пределах 13,5-17,5 кг, подвергали голоданию перед дозированием. Каждую композицию вводили перорально группе собак (n=4) при дозе 20 мг/кг. Композицию высокой концентрации соединения формулы I (300 мг/г) инкапсулировали в желатиновые капсулы и вводили. Последовательные пробы крови 2 мл получали из яремной вены при 20, 40 минутах и 1, 2, 4, 6, 8, 12 и 24 часах после дозирования. Эти пробы крови анализировали при помощи ВЖХ-анализа, специфического для соединения формулы I. Концентрации соединения формулы I в крови тестируемых крыс наносили на график зависимости от времени и получали AUC для расчета абсолютной биодоступности. Результаты приведены в Таблице 2.
(iii) Десяти здоровым волонтерам вводили перорально восьмикратно по 150 мг (одна доза 1200 мг) динатриевой соли соединения формулы I, инкапсулированных в твердые желатиновые капсулы, в качестве фона. Недели спустя, той же группе перорально вводили четырежды по 300 мг (одна доза 1200 мг) соединения формулы I в композиции, представленной в Примере 15. Последовательные пробы крови волонтеров получали при 30 минутах и 1, 2, 4, 6, 8, 12 и 24 часах после дозирования. Эти пробы крови анализировали при помощи ВЖХ-анализа, специфического для соединения формулы I. Концентрации соединения формулы I в крови наносили на график зависимости от времени и получали AUC для расчета относительной биодоступности. Результаты приведены в Таблице 3.
Относительная биодоступность=АUСтест/АUСфон•100%
Данное изобретение обеспечивает желаемые результаты, как показано увеличенной абсолютной или относительной пероральной биодоступностью в Таблицах 1, 2 и 3. Кроме того абсолютная пероральная биодоступность циклоспорина А в композиции по Примеру 21 составила 23%, как определено в опытах на крысах (N=8).
Примеры 1-53 приведены в конце описания.


















Изобретение обеспечивает фармацевтическую композицию на основе применения определенной масляной фазы, которая содержит липофильный фармацевтически активный агент, смесь диглицерида и моноглицерида в отношении диглицерид : моноглицерид приблизительно 9:1 - 6:4, где диглицерид и моноглицерид являются эфирами моно- или диненасыщенных жирных кислот и глицерина, имеющими длину цепи шестнадцать - двадцать два атома углерода, один или несколько фармацевтически приемлемых растворителей и одно или несколько фармацевтически приемлемых поверхностно-активных веществ. Эта композиция находится в форме самоэмульгирующейся композиции, которая обеспечивает высокую концентрацию и высокую пероральную доступность для липофильных соединений пиранона. 2 с. и 29 з.п. ф-лы, 3 табл.
| Способ получения эмульсии жиров для внутривенных инъекций | 1980 |
|
SU1311604A3 |
| Способ получения лекарственных масляных эмульсий внутреннего применения | 1984 |
|
SU1346159A1 |
| US 5371109 A, 06.12.1994 | |||
| СПОСОБ ПОЛУЧЕНИЯ 1,3-ДИФТОРПРОПАНОЛА-2 | 0 |
|
SU267617A1 |

