Предпосылки создания изобретения
Изобретение относится к способам и композициям для перорального введения пациентам фармацевтических агентов, которые слабо абсорбируются из желудочно-кишечного тракта, и к способам лечения больных путем перорального введения подобных агентов. Основной аспект настоящего изобретения относится к способам и композициям для перорального введения пациентам паклитакселя и родственных таксанов.
Многие полезные фармакологически активные соединения не могут эффективно вводиться перорально пациентам из-за их слабой или неустойчивой системной абсорбции из желудочно-кишечного тракта. Таким образом все эти фармакологические агенты обычно применяются внутривенным путем, требуя врачебного вмешательства или участия другого профессионального медицинского работника, создавая значительные неудобства и возможную местную травму и даже требуя введения в условиях клиники с применением хирургического вмешательства в случае некоторых инфузий типа IV.
Один из важных классов цитотоксических агентов, которые в норме при пероральном введении человеку не являются биодоступными, представляет собой таксаны, которые включают паклитаксель, его производные и аналоги. Паклитаксель (обычно обозначаемый Bristol-Myers Squibb Oncology Division как TAXOL®) представляет собой природный дитерпеновый продукт, выделенный из Тихоокеанского тисового дерева (Taxus brevifolia). Он является представителем таксанового семейства терпенов. Впервые он был выделен в 1971 году Wani et al. (J. Am. Chem. Soc., 93:2325, 1971), который охарактеризовал его структуру с помощью химических методов и методом рентгеновской кристаллографии. Один из механизмов его активности относится к способности паклитакселя связывать тубулин, подавляя таким образом рост опухолевых клеток. Schiff et al., Proc. Natl. Acad. Sci. USA, 77:1561-1565 (1980); Schiff et al. Nature, 277:667 (1979); Kumar, J. Biol. Chem., 256:10435-10441 (1981).
Паклитаксель разрешен в США для клинического использования в лечении резистентного рака яичника (Markman et al., Yale Journal of Biology and Medicine, 64:583, 1991; McGuire et al., Ann. Intern. Med., 111:273, 1989). Он эффективен для проведения химиотерапии в случае нескольких видов неоплазм, включая молочную железу (Holmes et al., J. Nat., Cancer Inst., 83:1797, 1991), и разрешен также для лечения рака молочной железы. Он является возможным кандидатом для лечения неопластических образований на коже (Einzig et al. , Proc. Am. Soc. Clin. Оncol., 20:46), рака легкого и карциномы головы и шеи (Forastire et al., Sem. Oncol., 20:56, 1990). Это соединение также демонстрирует возможность лечения поликистозного заболевания почек (Woo et al. , Nature, 368:750, 1994) и малярии.
Растворимость паклитакселя в воде очень невелика, и это создает значительные проблемы в создании готовых препаративных форм, подходящих для противораковой химиотерапии. Некоторые готовые препаративные формы паклитакселя для инфузий типа IV из-за нерастворимости паклитакселя в воде приготовлены с использованием CREMOPHOR ELTM (полиэтоксилированного касторового масла) в качестве носителя. Например, паклитаксель, использовавшийся в клинических анализах на фоне NC1, представлял собой готовую препаративную форму из 50% CREMOPHOR ELTM и 50% дегидратированного спирта. Однако сам CREMOPHOR ELTM при внутривенном введении является токсичным и у собак приводит к расширению кровеносных сосудов, затрудненному дыханию, летаргии, гипотензии и смерти. Также его считают по меньшей мере отчасти ответственным за реакции аллергического типа, наблюдающиеся во время введения паклитакселя, хотя существуют некоторые основания считать, что паклитаксель сам может вызывать острые реакции даже в отсутствие CREMOPHOR.
В попытках повысить растворимость паклитакселя и создать более безопасную готовую препаративную форму были проведены исследования с целью синтеза аналогов паклитакселя, которые по 2' и/или 7-положениям замещены группами, повышающими растворимость в воде. В результате были получены пролекарственные соединения, которые растворимы в воде более, чем родительское соединение, и которые при активации проявляют цитотоксические свойства. Одна важная группа таких пролекарств включает 2'-ониевые соли паклитакселя и доцетакселя, в частности соли 2'-метилпиридинмезилата (2'-MPM).
При пероральном введении паклитаксель абсорбируется очень слабо (менее 1%), смотри Eiseman et al., Second NCI Workshop on Taxol and Taxus (Sept. 1992); Suffness et a1., in Taxol Science and Applications (GRC Press, 1995). Eiseman et al. указывают, что при пероральном введении паклитаксель обладает биодоступностью в 0%, и Suffness et al. сообщают, что пероральное дозирование паклитакселя не кажется им возможным, так как при пероральном применении до 160 мг/кг в день не обнаружено признаков противораковой активности. Более того, не разработано эффективного способа для того, чтобы сделать эффективным пероральное введение паклитакселя (то есть способа повышения пероральной биодоступности паклитакселя) или других пероральных таксанов или аналогов паклитакселя, таких как доцетаксель, который проявляет противоопухолевую активность. По этой причине до настоящего времени паклитаксель не применялся перорально в лечении больных людей и, конечно, не входил в курс лечения заболеваний, на которые можно воздействовать с помощью паклитакселя.
Доцетаксель (N-дебензоил-N-трет-бутоксикарбонил-10-деацетилпаклитаксель) является коммерчески доступным в виде препарата TAXOTERE® (Rhone-Poulenc-Rorer S.A.) в парентеральной форме для лечения рака молочной железы. К настоящему времени в научной литературе нет ссылок на абсорбцию доцетакселя при пероральном введении животным или человеку.
Предполагают, что в некоторых случаях слабая или отсутствующая биодоступность лекарственного средства, такого как паклитаксель, после перорального применения является результатом активности полилекарственного переносчика, связанного с мембраной Р-гликопротеина, который действует как энергетически зависимый переносчик или поточный насос для снижения внутриклеточной аккумуляции лекарственного средства путем вытеснения ксенобиотических веществ из клетки. Этот Р-гликопротеин определен в нормальных тканях секреторного эндотелия, таких как билиарная выстилка, реснитчатая кайма проксимальных канальцев почки и поверхности внутреннего просвета кишечника, сосудистые эндотелиальные клетки, покрывающие гематоэнцефалический барьер, плаценту и яички.
Считают, что поточный насос Р-гликопротеина предупреждает проникновение некоторых фармацевтических соединений в клетки слизистой оболочки тонкого кишечника и, таким образом, абсорбцию в систему циркуляции. Показано, что ряд известных нецитотоксичных фармакологических агентов подавляет Р-гликопротеин, включая помимо прочих циклоспорин А (также известный как циклоспорин), верапамил, тамоксифен, хинидин и фенотиазины. Многие из этих исследований имели целью достигнуть большей аккумуляции цитотоксических лекарственных средств в опухолевых клетках при их внутривенном применении. Действительно, проведены клинические испытания для изучения влияния циклоспорина на фармакокинетику и токсичность паклитакселя (Fisher et al., Рroc. Am. Soc. Clin. Oncol. , 13: 143, 1994); доксорубицина (Bartlett er al., J. Clin. Onс., 12: 835-842, 1994) и этопозида (Lum et al., J. Clin. Onс., 10:1635-1642, 1992), все из которых являются противораковыми агентами, известными как вещества для полилекарственной устойчивости (MDR). Эти испытания показали, что пациенты, получившие внутривенно циклоспорин до или вместе с противораковыми лекарственными средствами, имеют более высокий уровень в крови этих лекарственных средств, предположительно благодаря снижению клиренса тела, и показали ожидаемую токсичность при значительно более низких дозах. Эти открытия скорее всего показывают, что одновременное применение циклоспорина подавляет MDR-действие Р-гликопротеина, вызывая большую внутриклеточную аккумуляцию терапевтического агента. Обсуждение общих вопросов фармакологического использования для клинического применения ингибиторов Р-гликопротеина, смотри у Lum et al., Drug Resist. Clin. Onс. Hemat., 9:319-336 (1995); Schinkel et al., Eur. J. Cancer., 31A: 1295-1298 (1995).
В обсужденных выше исследованиях, относящихся к использованию циклоспорина для повышения уровня в крови фармацевтических агентов, активные противоопухолевые агенты и циклоспорин вводились внутривенно. В этих публикациях не сделано никаких предположений относительно того, что циклоспорин может вводиться перорально для существенного повышения биодоступности применяемых перорально противораковых лекарственных средств и других фармацевтических агентов, которые сами по себе слабо абсорбируются из кишечника, без получения высоко токсичных побочных эффектов. Действительно, в 1995 году в работах, приведенных в ссылках выше, Lum et al. показали, что совместное введение IV типа ингибиторов MDR и химиотерапевтических агентов повышает уровень токсичности и усиливает у больных серьезные побочные эффекты. Schinkel et а1. кратко касаются того факта, что MDR1 и Р-гликопротеин имеются в изобилии в клетках слизистой оболочки кишечника и что это может влиять на биодоступность при пероральном введении лекарственных средств, являющихся субстратом Р-гликопротеина, но не предполагают и не подразумевают, что пероральное введение MDR-супрессирующих агентов сможет улучшить биодоступность агентов, недоступных при пероральном применении. Кроме того, как Lum et al., Schinkel et al. предупреждают, ингибиторы Р-гликопротеина могут значительно повысить токсичность у получающих химиотерапию больных, и, таким образом, должны использоваться с осторожностью.
В более ранних публикациях Schinkel et al. показывал, что абсорбция при пероральном введении вирмектина была повышена у гомозиготных мышей из-за расщепления MDR1 гена по сравнению с нормальными мышами, демонстрируя, что Р-гликопротеин играет основную роль в снижении биодоступности этого агента (Cell, 77: 491-502, 1994). Кроме того, это исследование также показало, что проникновение винбластина в различные ткани было повышено у мутантных мышей.
Ни одно из этих опубликованных исследований не предлагало какого-либо режима проведения лечения человека с помощью перорального введения слабо биодоступных лекарственных средств, таких как паклитаксель, например, указывая ожидаемую величину доз и время применения лекарственных средств-мишеней и агентов, усиливающих биодоступность, которые наилучшим образом подходят для усиления абсорбции при пероральном введении каждого целевого лекарственного средства или класса лекарственных средств.
Имеющиеся в настоящем уровне техники способы повышения кишечной абсорбции лекарственных средств, применявшихся до сих пор только парентерально, обычно сфокусированы на использовании усилителей проникающей способности и растворимости в качестве вспомогательных агентов, или на совместном назначении путем перфузии в просвет тонкого кишечника или внутривенном введении ингибиторов Р-гликопротеина, например, Leu et al., Cancer Chemother. Pharmacol., 35:432-436, 1995 (перфузия или инфузия типа IV хинидина подавляет выход этопозида в просвет желудочно-кишечного тракта из крови). Но эти способы имеют различные недостатки. Агенты, повышающие растворимость и проницаемость, часто или непригодны для употребления, или неэффективны при пероральном применении в требуемых дозах и могут влиять на фармакологическую активность лекарства-мишени. Парентеральное введение ингибиторов Р-гликопротеина в терапевтических (или околотерапевтических) дозах в организм человека может вызвать серьезные клинические последствия. В случае хинидина, например, введение типа IV может вызвать аритмии, расширение периферических сосудов, желудочно-кишечные расстройства и им подобные. Наиболее важно, что они не показывают, каким образом применять человеку какие-либо противораковые агенты пероральным путем.
В опубликованной РСТ заявке WO 95/20980 (опубликованной 10 августа 1995 г. ) Benet et al. раскрывает изложенный способ повышения биодоступности гидрофобных фармацевтических соединений при их пероральном введении. Этот способ включает в себя пероральное введение больным подобных соединений одновременно со средством, повышающим биодоступность и содержащим ингибитор фермента цитохрома Р450 3А или ингибитор опосредованного Р-гликопротеином транспорта через мембрану. Benet et al., однако, не предлагает фактически никаких средств для определения того, какой усиливающий биодоступность агент будет улучшать способность конкретного фармацевтического соединения-мишени, а также не указывает ни конкретную величину доз, ни график или режим приема усиливающих агентов или агентов-мишеней. В действительности, хотя заявка Benet et al. перечисляет множество возможных усилителей (Р450 3А ингибиторы) и лекарств-мишеней (Р450 3А субстратов), только одна комбинация усиливающего агента и агента-мишени, кетоконазола в качестве усилителя и циклоспорина А в качестве лекарства-мишени, явно подтверждена экспериментальными данными.
При описании общей характеристики соединений, которые могут быть использованы в качестве агентов, усиливающих биодоступность путем снижения транспортной активности Р-гликопротеина, Benet et al. указывает, что они представляют из себя гидрофобные соединения, которые обычно, но не обязательно, содержат два копланарных ароматических кольца, положительно заряженную азотсодержащую группу или карбонильную группу - класс веществ, который включает огромное количество соединений, большинствo из которых не обеспечивает желаемой активности по повышению абсорбции в случае конкретного целевого агента. Более того, классы лечебных агентов, раскрытых Benet et аl., включают большое количество фармакологических агентов, перечисленных в Physician's Desk Reference. Эти включенные сюда критерии не имеют ценности для практикующих врачей в поиске безопасных, практичных и эффективных способов перорального введения конкретных фармацевтических агентов.
Еще одним недостатком раскрытия Benet et аl. является стандарт для определения улучшения биодоступности лекарственного средства, обладающего слабой абсорбцией при пероральном введении. Benet et al. указывает, что любой агент, ингибирующий Р-гликопротеин, и который при наличии его в кишечнике в данной концентрации снижает транспорт через мембрану Родамина 123 с помощью Р-гликопротеина в мембранных везикулах реснитчатой каймы или Р-гликопротеинсодержащих клеток на 10% или более, можно считать агентом, повышающим биодоступность, и он может быть использован в практическом применении изобретения. Но только 10%-ное повышение абсорбции из кишечника агента, который в другом случае вообще не абсорбируется, недостаточно для того, чтобы сделать агент терапевтически полезным для любых целей. Действительно, по рекомендациям Федерального Ведомства по лекарствам и пищевым продуктам (FDA) две фармацевтические готовые препаративные формы, содержащие один и тот же активный ингредиент, но различающиеся по уровню своей биодоступности на -20%/+25%, все еще считаются биоэквивалентными, так как для большинства лекарственных средств различие -20%/+25% в концентрации активного ингредиента в крови не является клинически значимым. Approved Drug Products with Therapeutic Equivalence Evaluations (Dept. of HH3, 14th ed., 1994). Когда FDA указывает, что две готовые препаративные формы биоэквивалентны, врачи и фармацевты считают их свободно заменяемыми одна другой.
В целом, Benet et at. не дает рекомендаций, которым могут следовать специалисты в области медицины и фармакологии для определения подходящего сочетания лечебного средства и агента, усиливающего его биодоступность, или создания определенного режима лечения и графиков приема, которые бы сделали агенты-мишени терапевтически эффективными при пероральном применении пациентом. Benet et a1. также не предлагают указаний, каким-либо образом показывающих, как паклитаксель или другие таксаны могут применяться человеком перорально с терапевтической эффективностью и приемлемой токсичностью.
Таким образом, существует потребность в безопасном, но эффективном способе усиления системной доступности при пероральном введении пациенту лекарственного средства, которое обычно назначается только парентерально из-за того, что оно не абсорбируется в достаточных количествах или последовательности при введении его пероральным путем, который еще не предложен в предшествующем уровне техники.
К удивлению, в настоящее время обнаружено и экспериментально доказано, что антинеопластические агенты класса таксанов, в частности паклитаксель, могут применяться человеком перорально, причем достигается значительный и терапевтический уровень в крови при отсутствии нежелательной токсичности или вредных побочных эффектов, что наблюдается даже без проведения премедикации для предупреждения вредных реакций.
Настоящее изобретение в своем главном аспекте относится к пероральному введению одного таксана или их комбинации пациенту, страдающему от болезненных состояний, на которые можно воздействовать с помощью этих агентов. Предпочтительное воплощение изобретения представляет собой способ усиления для человека пероральной биодоступности таксанов, которые слабо абсорбируются или совсем не абсорбируются из желудочно-кишечного тракта или кишечника, путем предварительного и/или одновременного введения человеку пероральным путем одного агента или сочетания агентов ("усиливающих агентов"), эффективных в подавлении клеточной полилекарственной устойчивости. При их предварительном применении агент или агенты, усиливающие биодоступность, должны быть назначены в достаточном количестве и в достаточно короткий период времени до применения таксана, биодоступность которого должна быть повышена ("средство-мишень" или "агент-мишень") таким образом, чтобы соответствующий уровень усиливающего агента появился в месте всасывания во время применения агента-мишени с целью эффективного подавления активности полилекарственных транспортных веществ, метаболических ферментов и/или других факторов, которые предотвращают или ингибируют кишечную всасываемость агента-мишени. Второй аспект или воплощение изобретения относится к способу лечения больного, страдающего от заболевания, на которое можно воздействовать с помощью таксана, путем перорального введения таксанов, которые ранее были доступны только при парентеральном введении. Еще один аспект или воплощение представляет собой способ предотвращения или снижения гиперчувствительности и аллергических реакций у больного, получающего таксановую терапию.
Фиг.1 представляет собой график, отражающий уровни циркуляции паклитакселя в образцах, полученных: (а) нижняя кривая - в период от 6 до 8 часов от одной группы крыс, получивших перорально только паклитаксель, (b) верхняя кривая - в течение периода в 24 часа от второй группы крыс, получивших перорально циклоспорин А за один час до совместного перорального применения циклоспорина А и перорального паклитакселя.
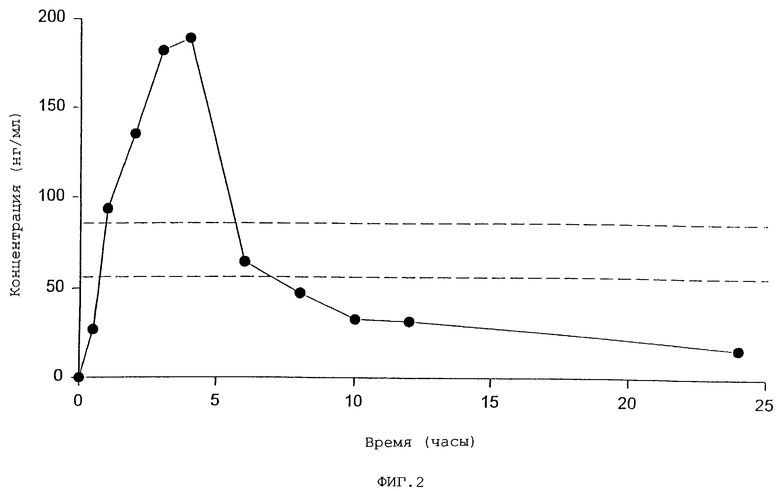
Фиг. 2 представляет график, отражающий уровни паклитакселя в образцах плазмы пациентов, получивших паклитаксель перорально после приема двух доз перорального циклоспорина А, первый прием за один час до приема паклитакселя и второй прием непосредственно перед паклитакселем.
Фиг.3 представляет собой график, отражающий уровни паклитакселя в образцах плазмы второго пациента, получившего перорально паклитаксель в том же режиме, как описано для фиг.2.
Фиг.4 представляет собой график, показывающий сравнение следующих кривых уровня паклитакселя в плазме: определенного через 24 часа у крыс (фиг.1) и у людей (фиг. 2 и 3), получивших паклитаксель после перорального приема двух доз циклоспорина А.
Настоящее изобретение в своем главном аспекте относится к пероральному применению антинеопластических агентов класса таксанов, в частности паклитакселя, их производных, аналогов и пролекарств, и полусинтетического аналога паклитакселя доцетакселя - (N-дебензоил-N-трет-бутоксикарбонил-10-деацетил-паклитаксель) пациентами, страдающими от болезненных состояний, на которые можно воздействовать с помощью таксанов. Предпочтительные воплощения или аспекты изобретения включают (а) способы перорального введения таксанов, которые ранее применялись только парентерально, с биодоступностью, достаточной для достижения терапевтического уровня в крови; (b) способы лечения пациентов, страдающих от болезненных состояний, на которые можно воздействовать с помощью таксанов, путем перорального применения таксанов и (с) способы профилактики или снижения гиперчувствительности и аллергических реакций у больных, получающих таксановую терапию.
Термин "биодоступность" при использовании здесь обозначает системную доступность (то есть уровни в крови/плазме) данного количества лекарственного средства, полученного больным.
Обнаружено, что таксаны, обладающие слабым профилем пероральной абсорбции, могут назначаться больным перорально с достижением соответствующей системной абсорбции и пероральной биодоступности для получения терапевтического уровня в плазме. Действительно, мы назначали таксан паклитаксель человеку, страдающему раковым заболеванием, перорально и убедились, что терапевтический уровень паклитакселя в крови достигается у этих больных в течение ожидаемого периода времени.
Мы наблюдали в исследованиях на животных, что определенные агенты, такие как циклоспорин А, при введении перорально сразу после и/или до лекарственного средства, такого как паклитаксель, повышают абсорбцию последнего лекарственного средства из кишечника в неожиданной и удивительной степени, в результате чего достигается его терапевтический уровень. Однако не совсем очевидно, что эти наблюдаемые результаты происходят благодаря подавлению Р-гликопротеинового насоса.
Следует подчеркнуть, что настоящее изобретение не ограничивается использованием какого-либо определенного перорального агента, повышающего биодоступность, одновременно с пероральным таксаном для того, чтобы последний стал биодоступным для пациента. Изобретение в более широком смысле состоит в пероральном применении таксанов пациентами и не ограничивается какими-либо конкретными усилителями, количеством дозы или режимом или использованием определенного биологического механизма или фармацевтической методики для того, чтобы сделать таксаны доступными при пероральном введении человеку.
Предпочтительное воплощение способа изобретения для перорального применения человеком паклитакселя, его производных, аналогов и пролекарств и других таксанов включает пероральное введение больным человеком агентов, повышающих пероральную абсорбцию или биодоступность, одновременно с или предварительно, или как одновременно, так и предварительно до перорального применения для повышения количества абсорбции интактного агента-мишени в кровоток.
Предпочтительное воплощение включает пероральное применение следующих усиливающих агентов, которые могут быть использованы на практике, но не ограничивается ими:
циклоспорины, включая циклоспорины от А до Z, но в частности циклоспорин А (циклоспорин), циклоспорин F, циклоспорин D, дигидроциклоспорин А, дигидроциклоспорин С, ацетилциклоспорин А, РSС-833, SDZ-NIM 8111 (оба от Sandoz Pharmaceutical Corp.). 1 SDZ-NIM 811 представляет собой (Me-Ilе-4)-циклоспорин, противовирусный неиммуносупрессивный циклоспорин. Структура циклоспоринов A-Z описана в таблице .
Класс перорально применяемых целевых терапевтических агентов, пероральная абсорбция которых повышается с помощью усиливающих агентов, включает, не ограничиваясь ими, следующие:
паклитаксель, другие таксаны, доцетаксель и производные и пролекарства всех вышеперечисленных, в частности 2'-МРМ соли и другие соли 2'-метилпиридиния.
Циклоспорины представляют собой неполярные циклические олигопептиды (некоторые из которых обладают иммуносупрессивной активностью), продуцируемые родом Topycladium, включая, например, Topycladium inflatum Cams (ранее обозначавшийся как Trichoderma polysporum), Topycladium terricola и другие fungi imperfecti (несовершенные грибы). Основной компонент, циклоспорин А, (циклоспорин или СsА) определен наряду с несколькими другими меньшими метаболитами, например циклоспорины от В до Z, некоторые из которых проявляют значительно меньшую иммуносупрессивную активность, чем циклоспорин А. Также получен ряд синтетических и полусинтетических аналогов. Смотри в целом Jegorov et al., Phytochemistry, 38: 403-407 (1995). Настоящее изобретение охватывает природные, полусинтетические и синтетические аналоги циклоспоринов.
Циклоспорины представляют из себя нейтральные, липофильные, циклические ундекапептиды с молекулярным весом около 1200. Они применяются перорально или внутривенно как иммуносупрессанты преимущественно при трансплантации органов и при некоторых других состояниях. Циклоспорины, в частности циклоспорин (циклоспорин А), являются известными ингибиторами Р-гликопротеинового поточного насоса и других транспортных насосов, а также определенных Р-450 расщепляющих ферментов, но к настоящему времени не разработано никаких эффективных режимов для применения этого свойства клинически с точки зрения клинической и коммерческой пригодности или одобрения регулирующими органами.
Одно из удивительных открытий изобретения состоит в том, что иммуносупрессия, наблюдаемая при применении некоторых циклоспоринов, не является обязательно связанной с улучшением пероральной биодоступности терапевтических агентов. Так циклоспорин F усиливает пероральную биодоступность паклитакселя, даже несмотря та то, что в соответствии с данными литературы, он не проявляет иммуносупрессивной активности. Stewart et al., Transplantation Proceedings, 20: (Supp.3) 989-992 (1988); Granelli-Piperno et al., Transplantation, 46: 53S-60S (1988).
Другое возможное объяснение наблюдаемой повышенной биодоступности паклитакселя состоит в том, что может существовать взаимодействие на уровне ферментов лекарственного метаболизма для циклоспорина и паклитакселя. Известно, что оба агента сильно метаболизируются цитохромной Р-450 системой (например Р-450 3А), которая сконцентрирована в печени, а также в тонкой кишке. Возможно, что циклоспорин, который применяется первым, может подавлять эти ферменты таким образом, что паклитаксель, являющийся неполярным и липофильным, может абсорбироваться. В отсутствие этого местного ингибирования паклитаксель метаболизировался бы в более полярный метаболит, который не проникал бы через клетки слизистой оболочки.
Когда целевой агент применяется внутривенно, теоретическое ингибирование кишечного метаболизма целевого агента может иметь небольшой эффект или его отсутствие в повышении системного уровня в крови. Более того, так как первичный эффект усиливающего агента по пероральной абсорбции может быть местным эффектом в просвете кишки, дозы, являющиеся субтерапевтическими (например, с точки зрения иммуносупрессии) должны быть эффективными в достижении желаемого эффекта. Это является важным соображением в случае таких усиливающих агентов, как циклоспорин, который обладает сильной иммуносупрессивной активностью, и могут существовать проблемы с токсичностью при применении в высоких дозах. Огромное клиническое значение имеет наше наблюдение, что не являющийся иммуносупрессивным циклоспорин, такой как циклоспорин F, может все же функционировать как пероральный усилитель.
Важно отметить, что в то время как мы выдвигаем гипотезу о механизме действия, лежащем в основе нашего изобретения, мы не знаем точно механизма(ов), ответственного(ых) за это удивительное, обсужденное здесь открытие; это не мешает квалифицированному специалисту применять описанное изобретение на практике. Возможно, что ни один из предполагаемых механизмов, ни некоторые из них, ни все они, не играют роли в экспериментально или клинически подтвержденном усилении оральной биодоступности таксанов, в частности паклитакселя.
Уровень доз усиливающего агента для совместного
применения с агентом-мишенью в соответствии с изобретением составляет от около 0,1 до около 20 мг/кг веса тела больного. Под "совместным введением" усиливающего агента понимается введение в значительной степени одновременно с агентом-мишенью (или менее чем за 0,5 часа до, или менее чем через 0,5 часа после, или вместе), от около 0,5 до около 72 часов до применения целевого агента, или двух, то есть с одной или большим количеством доз того же самого или разных усиливающих агентов, данных по крайней мере за 0,5 часа до, и одной дозы, данной в значительной степени одновременно с (или вместе, или немедленно до или после) целевого агента. Кроме того, в "совместное введение" входит введение более чем одной дозы целевого агента в течение 72 часов после приема дозы усиливающего агента, другими словами, усиливающий агент(ы) не нуждается в повторном приеме до или с каждым следующим приемом целевого агента, но может применяться периодически в течение курса лечения.
Диапазон доз для перорального введения таксана, как целевого агента, варьируется в зависимости от соединения, основываясь на его терапевтическом индексе, требованиях болезненного состояния, которое лечится, состояния больного и так далее. Способ изобретения делает возможным применять паклитаксель и другие таксаны перорально в дозе от около 20 мг/м2 до около 1000 мг/м2 (основываясь на площади поверхности тела больного) или около 20-30 мг/кг (основываясь на весе тела больного) как за один раз, так и разделяя дневную дозу на 2-3 приема, поддерживая уровень паклитакселя в плазме человека на уровне 50-500 нг/мл в течение длительного периода времени (например, 8-12 часов), после каждого перорального приема дозы. Эти уровни являются по меньшей мере сопоставимыми с теми, которые достигались с помощью 96-часовой IV инфузионной таксоловой терапии (которая вызывает у пациента большое неудобство, дискомфорт, потерю времени, возможность инфицирования и т.д.). Более того, такой плазменный уровень паклитакселя более чем достаточен для обеспечения желаемой фармакологической активности целевого лекарства, например, для ингибирования тубулинового расщепления (которое происходит при уровне около 0,1 мкм или около 85 нг/мл) и для ингибирования изопренилирования белка (которое происходит при уровне около 0,03 мкм или около 25 нг/мл), которое прямо связано с их противоопухолевыми эффектами путем ингибирования онкогенных факторов и других передающих сигнал белков, которые играют центральную роль в регулировании клеточного роста.
Предпочтительные дозированные количества для перорального введения паклитакселя и других таксанов, применяемых в соответствии с изобретением, составляют около 50-200 мг/м2 или 2-6 мг/кг.
В некоторых случаях может быть удобно назначать больному более высокие начальные дозы целевого агента для достижения пика уровня в крови с последующим применением более низких продерживающих доз. Два или большее количество различных усиливающих агентов и/или два или большее количество различных целевых агентов могут применяться вместе, альтернативно или чередуясь во всех различных аспектах способа данного изобретения.
Настоящее изобретение также касается способов лечения больных людей, страдающих раком, опухолями, саркомой Капоши, злокачественной, бесконтрольной тканевой или клеточной пролиферацией, вторичной по отношению к повреждению ткани, и любыми другими заболеваниями, на которые можно воздействовать с помощью паклитакселя, таксанов, доцетакселя и/или пролекарства и производных перечисленных выше агентов, таких как паклитаксель 2'-МРН и доцетаксель 2'-МРМ с применением пероральных дозированных форм, содержащих один или большее количество этих агентов. Среди видов карцином, которые можно лечить особенно эффективно с помощью перорально вводимого паклитакселя, доцетакселя, других таксанов и их пролекарств и производных, можно назвать гепатоцеллюлярную карциному, метастазы в печени, раковые заболевания желудочно-кишечного тракта, поджелудочной железы, простаты и легкого и саркому Капоши. Примерами нераковых заболеваний, которые можно эффективно лечить этими активными агентами, применяемыми перорально в соответствии с настоящим изобретением, являются вторичная бесконтрольная тканевая или клеточная пролиферация после повреждений ткани, поликистозное заболевание почек, воспалительные заболевания (например, артрит) и малярия, включая хлорохин- и пириметаминустойчивых малярийных паразитов (Pouvelle et a1., J. Clin. Invest, 44: 413-417, 1994).
Противоопухолевые агенты, которые до настоящего времени назначаются больному человеку только парентерально, могут теперь вводиться человеку в соответствии с настоящим изобретением пероральным путем с соответствующей биодоступностью для обеспечения фармакологически активной концентрации в крови, что будет особенно эффективно в лечении больных с первичными опухолями и метастазами. Активные ингредиенты будут проходить через кишечную стенку в результате предварительного и/или одновременного применения циклоспориновых усилителей и будут быстро попадать в портальную циркуляцию, обеспечивая более высокий уровень начальной концентрации химиотерапевтических агентов в печени (выше местной концентрации, которой обычно достигают при применении IV инфузионной терапии), чем в общей системной циркуляции или в большинстве других органов в первый и седьмой дни.
Более того, следует отметить, что более высокие уровни паклитакселя в печени после перорального применения могут вызвать повышение уровня в плазме вследствие высокого первого проходящего эффекта печени. Способ изобретения, состоящий в выборочном получении высокой концентрации в крови противоопухолевых агентов, является особенно полезным в лечении рака печени (например, гепатоцеллюлярной карциномы и метастазов в печени), раковых заболеваний желудочно-кишечного тракта (например, ободочной кишки, прямой кишки), раковых заболеваний легкого.
Плазменный уровень активных целевых агентов, вводимых перорально с соответствующими усиливающими агентами, как предлагается в настоящем изобретении, является замечательным и удивительно подобным тому, который наблюдался при введении по типу IV. Ряд исследований на экспериментальных животных показал, что неизменное состояние плазменных уровней паклитакселя достигается путем перорального применения одновременно с CsA к третьему дню по графику. Уровни агента-мишени, достигшие устойчивого состояния, сравнимы с таковыми, полученными у больных при 96-часовой инфузии паклитакселя по IV типу. 27% скорость ответа обнаружена у больных с недостатком таксана с метастатическим раком грудной железы, который лечили длительным 96-часовым введением каждые три недели (Seidman et al., J. Clin. Oncol., 14: 1877, 1996). Полагают, что подобный результат можно получить при использовании способов лечения настоящего изобретения, не вызывая дискомфорта, неудобства и риска, сопутствующих пролонгированной инфузии по типу IV.
Данные, отраженные на фиг. 1-4, являются особенно заслуживающими внимания. Как обсуждено более подробно в приведенных ниже примерах, данные, отраженные на фиг. 1, были получены при изучении применения паклитакселя на крысах, но данные, отраженные на фиг. 2 и 3, отражают действительные уровни концентрации паклитакселя по прошествии времени в плазме двух пациентов, которые получили паклитаксель перорально в соответствии с настоящим изобретением, то есть путем одновременного перорального введения циклоспоринового усиливающего агента. Данные, полученные на человеке, замечательны не только потому, что они впервые отражают, насколько обнаружено в литературе, что паклитаксель назначался перорально человеку, нуждающемуся в лечении паклитакселем, но также потому, что терапевтические уровни концентрации в плазме достигались и поддерживались в течение примерно 24 часов; конечно, уровни лекарственного средства, наблюдаемые в плазме пациентов, были сравнимы с уровнями, полученными при введении по типу IV, и применяемые способы не вызвали серьезных местных или системных побочных эффектов.
Кроме данных исследования на животных (крысах), приведенных ниже в примерах и отраженных на фиг. 1 и 4, мы проводили обширную серию исследований на крысах, когда паклитаксель и другие таксаны применялись перорально вместе с циклоспорином А и другими усиливающими биодоступность циклоспоринами С, D, F и G, и результаты этих исследований были сообщены и проиллюстрированы в одновременно рассматриваемой родительской заявке, серийный номер 08/733142. Кроме того, эффект от перорального введения таксанов, в особенности паклитакселя, животным одновременно с пероральным применением циклоспоринов был сопоставим в родительской заявке с применением того же самого агента-мишени самого по себе, при введении по типу IV или пероральным путем, и применением вместе с целевым лекарством других возможных, но менее эффективных агентов, усиливающих биодоступность. Раскрытие и экспериментальные примеры заявки серийного номера 08/733142 включены здесь в виде ссылки.
В настоящее время продемонстрировано, что у крыс фармакокинетический профиль паклитакселя, введенного одновременно с пероральным циклоспорином А, вполне сопоставим с профилем пациентов, получающих препараты таким же образом. Действительно, фиг. 4 отражает наложение одинаковых графиков кривых плазменной концентрации паклитакселя спустя 24 часа после перорального совместного приема двух доз усилителя (циклоспорина А) с интервалом 1 час, принятых отдельно от перорального паклитакселя, принятого после второй дозы усилителя, причем названные данные получены в результате 24-часового исследования на крысах, отраженного на фиг. 1, и исследования на больных людях, отраженного на фиг. 2 и 3. Можно наблюдать, что три кривые на графике фиг. 4 (одна для крыс и две для человека) имеют очень похожую конфигурацию, показывая, что результаты, полученные на людях, подтверждаются результатами исследования на животных.
Рассматриваемая заявка не уменьшает и не умаляет важности и уместности данных, полученных на крысах. Крысы представляют собой общепринятую модель для изучения фармакокинетики и профиля абсорбции химиотерапевтических агентов. Однако из-за известных межвидовых различий никакой клинический или медицинский работник не сможет назначить человеку паклитаксель или другие таксаны перорально с уверенностью, основанной только на данных животных без каких-либо клинических экспериментов на человеке. Мы имеем, в противоположность общепринятой технике, точный и действительно подтвержденный способ, по которому таксаны могут безопасно и эффективно применяться человеком перорально. С врачебной точки зрения данное изобретение представляет собой значительное улучшение по отношению к предшествующему уровню техники и показывает, что фармакологические свойства таксана, такого как паклитаксель, могут быть использованы в клинической практике без непременного использования внутривенного катетера и времени, затраченного в больнице или химиотерапевтической клинике, без сопутствующих затрат, неудобств и риска инфицирования больного и даже без проведения премедикации для исключения гиперчувствительности или аллергических реакций и без возможных неблагоприятных эффектов от самой премедикации.
Пероральные дозированные формы целевых агентов, чья биодоступность повышается путем совместного введения с усиливающими агентами, могут представлять собой формы общепринятых таблеток, капсул (мягкогелевые или твердогелевые), капельки, гелевые капельки, пилюли, жидкости (например, растворы, суспензии и эликсиры), порошки, пастилки, размельченные частицы или системы осмотической доставки и другие пероральные дозированные формы, известные в фармацевтической технике. Жидкие препаративные формы могут включать, например, паклитаксель или другой таксан в носителе, включающем CREMOPHOR EL или другое полиэтоксилированное касторовое масло, спирт и/или моноолеат полиоксиэтилированного сорбитана (например, TWEEN® 80, ICI Americans, Inc.) с или без ароматизатора. Каждая дозированная форма, включает эффективное количество таксанового целевого агента и фармацевтически инертные ингредиенты, то есть общепринятые эксципенты, носители, наполнители, связывающие вещества, дезинтегрирующие агенты, растворители, растворяющие агенты, подсластители, красящие агенты и любые другие неактивные ингредиенты, которые обычно включаются в фармацевтические дозированные формы для орального применения. Многие из таких дозированных форм и оральных наполнителей немедленно после перечисления неактивных ингредиентов были внесены в Remington's Pharmaceutical Sciences, 17th edition (1985).
Точное количество каждого лекарства-мишени в пероральных дозированных формах варьируется в зависимости от возраста, веса, заболевания и состояния больного. Например, дозированные формы паклитакселя или других таксанов могут содержать соответствующее количество целевого агента для обеспечения дневной дозы около 20-1000 мг/м2 (беря за основу площадь поверхности тела больного) или около 2-30 мг/кг (беря за основу вес тела больного), в виде одной дневной дозы или поделенной на 2-3 приема. Предпочтительные количества доз составляют около 50-200 мг/м2 или около 2-6 мг/кг.
Для способа лечения в соответствии с настоящим изобретением, например для лечения заболеваний, на которые можно воздействовать с помощью паклитакселя, дозированными формами перорального паклитакселя, вводимыми одновременно с усиливающими агентами, график доз можно так же составить с учетом количества для пациента с определенными характеристиками и состоянием заболевания. Предпочтительный график приема перорального паклитакселя представляет собой (а) прием дневной дозы нуждающимся в ней больным, разделенной на 1-3 равных дозы, составляющей около 20-1000 мг/м2 (беря за основу площадь поверхности тела), предпочтительно около 50-200 мг/м2, причем подобный прием продолжается 1-4 последовательных дня каждые 2-3 недели, или (b) прием в течение примерно одного дня каждую неделю. Первый график сравним с использованием 96-часовой инфузии паклитакселя каждые 2-3 недели, что считается предпочтительным IV режимом лечения.
Пероральное введение таксанов в соответствии с настоящим изобретением может действительно во многих случаях снизить токсические побочные эффекты по сравнению с применяемой в настоящее время терапией типа IV. Абсорбция активного агента через кишечную стенку (с помощью усиливающего агента) обеспечивает более постепенное появление уровня в крови и стабильное, постоянное его поддерживание на идеальном уровне или близко к идеальному уровню в течение длительного периода времени, что является более предпочтительным, чем получение внезапного и быстрого высокого уровня концентрации в крови, как обычно происходит в случае инфузии по типу IV.
Согласно другому аспекту изобретения предлагается сочетание пероральной дозированной формы, которая содержит постоянное количество по крайней мере одного усиливающего агента и по крайней мере одного целевого агента. Например, такая дозированная форма может представлять собой таблетки, капсулы, капельки, гелевые капельки, пилюли, жидкости, пастилки и любые другие общепринятые пероральные дозированные формы, содержащие в качестве активных ингредиентов эффективное количество перорального усилителя биодоступности противоопухолевого или антинеопластического агента, а также подходящие неактивные ингредиенты. Одно такое сочетание включает от около 0,1 до около 30 мг/кг одного или более циклоспоринов A,D, С, F и G, дигидро CsA, дигидро CsC и ацетил CsA вместе с около 20-1000 мг/м2 (беря за основу среднюю площадь поверхности тела больного) и предпочтительно около 50-200 мг/м2 паклитакселя, доцетакселя, других таксанов или производных паклитакселя или доцетакселя, таких как паклитаксель 2'-МРМ или доцетаксель 2'-МРМ.
Совместное введение усиливающих агентов с целевым лекарством способствует не только оральной биодоступности этих агентов, но также делает возможным их использование в лечении опухолей на участках, защищенных MDR, то есть яичек и мозга. Другой аспект настоящего изобретения, таким образом, представляет собой способ доставки противоопухолевых лекарственных средств к участкам опухолей, защищенных MDR, путем перорального совместного применения усиливающих агентов и противоопухолевых агентов, делая их способными лечить опухоли мозга, такие как мультиформная глиобластома.
Дополнительные преимущества настоящего изобретения касаются области безопасности. Из-за своих физико-химических свойств паклитаксель должен быть растворим в смеси Cremophor/этанол, при этом наполнитель может быть способен на некоторые реакции аллергического типа, испытываемые больными в ответ на терапию паклитакселем. Используются и другие растворяющие агенты, но ни один из них не является таким подходящим, как Cremophor/этанол. Паклитаксель должен вводиться больному медленно с участием медицинского персонала при постоянной бдительности по поводу серьезных реакций гиперчувствительности.
Для стандартного внутривенного режима премедикации в общем требуются Н-1 и Н-2 блокаторы и стероиды. Однако, даже когда растворитель Сremophor/этанол не применяется, внутривенные таксаны все же могут вызвать серьезные реакции, следующие за внутривенным введением. Таким образом, применение доцетакселя связано с анасаркой и другими реакциями. Лечение, позволяющее устранить или снизить необходимость премедикации в таких ситуациях, клинически очень ценно.
Настоящее изобретение в одном из своих воплощений предлагает способ предупреждения или снижения гиперчувствительности или аллергических реакций у пациентов, получающих лечение таксанами. Способ состоит в пероральном применении таксанов больными. Пероральное применение путем раскрываемого сейчас способа значительно менее напоминает внутривенную терапию с точки зрения получения подобных вредных реакций. Действительно, мы давали паклитаксель больным людям (смотри примеры 2 и 3) без премедикации (то есть Н-1 или Н-2 блокаторами и стероидами), и при достижении терапевтического уровня в системе циркуляции реакций гиперчувствительности не наблюдалось.
Кроме того, применение паклитакселя связано с различными видами токсичности и побочными эффектами. Два наиболее значимых вида токсичности представляют собой нейтропению и невропатию. Различные клинические данные показали, что желательно сохранять концентрацию в системе циркуляции в определенном "окне" с целью создания максимальной противоопухолевой активности и получения минимальных побочных эффектов. Считают, что для многих видов опухоли низкая, но длительная экспозиция опухолевых клеток в теле приводит к лучшим клиническим результатам. Таким образом, можно было бы ожидать, что уровни около 0,03 мкмолей ингибируют изопренилирование белка раковых клеток, и уровни около 0,1 мкмоля блокируют разъединение микроканальцев. Есть клинические данные, показывающие, что постоянное внутривенное введение в течение нескольких дней для создания "окна" от около 0,05 до 0,1 мкмоль в системе циркуляции может свести к минимуму токсичность и вызвать регрессию опухоли, даже если это такой пациент, на опухоль которого не воздействует 3-часовой режим введения. При применяющемся в настоящее время режиме 3-часовой инфузии паклитакселя достигается такой пик концентрации в плазме, который значительно превышает эти уровни.
Настоящее изобретение также делает возможным давать паклитаксель сравнительно редко в течение дня (например, примерно дважды в день) и в соответствии с таким графиком, который невозможен или практически неосуществим при внутривенном введении. Использование усилителя (например, циклоспорина А) способствует пероральной абсорбции первой дозы паклитакселя, и если вторая доза паклитакселя должна быть дана позже в этот же день, не будет необходимости в дополнительном приеме циклоспорина А. Таким образом, паклитаксель можно давать периодически в виде одной дозы по постоянному графику (еженедельно, два раза в неделю и т.д.) или постоянно в течение нескольких последовательных дней (например, четырех) каждые 2-4 недели с целью поддержать уровень в границах безопасного и эффективного "окна". Следующие примеры иллюстрируют различные аспекты изобретения и демонстрируют неожиданное, очень существенное повышение абсорбции, достигаемое при пероральном применении паклитакселя. Эти примеры, однако, не имеют целью каким-либо образом ограничить изобретение или рекомендовать конкретные агенты-усилители или целевые агенты, уровень доз, аналитические методики или другие параметры, которые должны быть использованы исключительно для примера практического применения изобретения.
ПРИМЕР 1
Шесть (6) здоровых крыс Sprague Dawley, все весом от 225 до 215 грамм и в возрасте приблизительно от шести до восьми недель получали одну пероральную дозу паклитакселя в размере 9 мг/кг. Образцы крови собирали из хвостовой вены каждой крысы через 0,5, 1, 2, 3, 4 и 6 часов после приема паклитакселя. Индивидуальные образцы центрифугировали и отделяли сыворотку. Для каждого интервала времени шесть образцов смешивали, получая один характерный образец. Все образцы анализировали на неизменный паклитаксель с помощью жидкостной хроматографии/масс-спектрометрии с низким разрешением количества в 50 пг/мл.
Эти результаты исследования графически иллюстрируются на нижней кривой фиг. 1, которая показывает, что биодоступность примененного перорально паклитакселя в сыворотке составляет менее 1%.
ПРИМЕР 2
Десять (10) здоровых крыс Sprague Dawley с такими же характеристиками, как в исследовании, описанном в примере 1, получали 5 мг/кг перорального циклоспорина А с последующим получением спустя 1 час еще одной дозы а 5 мг/кг циклоспорина А и 9 мг/кг перорального паклитакселя.
Образцы крови собирали из хвостовой вены каждой крысы через 0,25, 0,5, 1, 2, 3, 4, 5, 6, 8, 12 и 24 часа после приема паклитакселя. После соответствующей обработки образцов и создания одного общего образца для группы плазму из каждого образца исследовали на неизмененный паклитаксель.
Результаты этого исследования графически проиллюстрированы на верхней кривой фиг. 1. Можно наблюдать, что уровни паклитакселя в плазме в этой группе животных в течение первых шести часов в несколько раз выше, чем у крыс примера 1, которые получали только паклитаксель, что уровни, соответствующие или выше терапевтических уровней "мишени", поддерживались в течение восьми (8) часов после приема и что значительный плазменный уровень поддерживался в течение 24 часов.
ПРИМЕР 3
71-летний мужчина, страдающий раком простаты в течение 3 лет, согласился получать перорально паклитаксель и усилитель в виде циклоспорина А. Площадь поверхности его тела составляла 2,04 квадратных метра, его вес составлял приблизительно 84 килограмма. После голодания в течение ночи он получил 2 пероральные дозы циклоспорина A (Sandimmune 5 мг/кг) по отдельности с промежутком в один час. Немедленно после второй дозы больной выпил дозу паклитакселя на основе раствора Cremophor/спирт, содержащего 180 мг растворенной в 120 мл 5% декстрозы в воде, то есть 2,0 мг/кг веса тела или около 90 мг/м2 площади тела. Стандартная премедикация, которая обычно проводится при кратковременной инфузии таксанов, не проводилась. После выпивания раствора больной отмечал, что вкус был неприятный. Он отмечал слегка жидкий стул в течение нескольких часов. Он также сообщал о некотором покраснении через несколько часов после приема, которое может быть связано с временным прекращением приема противогипертензивных препаратов. Дальнейшее клиническое течение без особенностей.
Образцы плазмы, полученные с частыми интервалами после приема паклитакселя исследовались с помощью жидкостной хроматографии/масс-спектрометрии. Результаты уровня плазмы спустя некоторое время показаны на фиг. 2. Пик был достигнут примерно через 4 часа после приема и уровень около 0,07 мкмолей был достигнут в период от одного до пяти часов. Уровни, сопоставимые с таковыми у больных раком молочной железы, получающими 96-часовую внутривенную инфузию паклитакселя (0,05 микромолей) присутствуют в течение 10-12 часов (Seidman et al., J. Clin. Oncol., 14:1877, 1996).
ПРИМЕР 4
75-летний пожилой мужчина, страдающий раком простаты в течение нескольких лет, получил пероральную дозу паклитакселя и циклоспорина А. Площадь поверхности его тела составляла 1,82 квадратных метра,и вес тела составлял приблизительно 72 кг. После голодания в течение ночи он получил циклоспорин А в таком же режиме (Sandimmune 5 мг/кг) и перорально паклитаксель (180 мг), как больной в примере 1, что эквивалентно 2,5 мг/кг или 100 мг/м2 паклитакселя у этого больного. И на этот раз стандартная премедикация, которая применяется при кратковременной инфузии таксанов, не проводилась. После выпивания раствора больной отметил, что вкус его неприятный. Он отмечал несколько жидкий стул в течение нескольких часов. После приема также наблюдалось умеренное падение кровяного давления, которое могло быть связано со вторичной вазовагальной реакцией вследствие его голодания и забора крови. Для предосторожности больной получал около 100 мл физраствора внутривенно. После завтрака он почувствовал себя значительно лучше, и дальнейшее клиническое течение без особенностей.
Образцы плазмы были получены через небольшие интервалы после приема паклитакселя и исследовались с помощью жидкостной хроматографии/масс-спектрометрии. Результаты плазменного уровня показаны на фиг. 3. Пик составлял почти 0,3 мкмоля и появлялся на 4 часу после приема. Уровни выше 0,07 мкмоля достигались в интервале времени от одного до десяти часов. Уровни, сопоставимые с таковыми у больных раком молочной железы, получающих 96-часовую внутривенную инфузию паклитакселя, присутствуют в течение примерно 12-15 часов.
Как отмечалось прежде, фиг. 4 представляет соединение уровней концентрации паклитакселя, определенных по истечении некоторого времени у крыс (верхняя кривая на фиг. 1) и у человека (кривые на фиг. 2 и 3), получавших паклитаксель перорально после двух доз перорального циклоспорина с интервалом в один час в соответствии с настоящим изобретением. Можно наблюдать, что уровни концентрации, достигнутые у человека, не только прежде всего подтверждают эффективность настоящего изобретения в создании биодоступности паклитакселя при пероральном применении, но они превышают уровни концентрации, достигнутые на модели крыс. Эти результаты неожиданны и удивительны, и пока мы не продемонстрировали клиническую эффективность данного способа на человеке, нельзя было их предсказать, основываясь на каких-либо данных предшествующего уровня техники, касающихся циклоспорина или других возможных усиливающих агентов или касающихся паклитакселя, его производных, аналогов, пролекарств или других таксанов.
Таким образом показано, что предлагаемый способ осуществляет различные цели изобретения и хорошо приспосабливается к условиям практического использования.
Понятно, что все обсужденные здесь аспекты могут рассматриваться в качестве иллюстрации, а не в смысле ограничения, так как на основе вышеприведенного изобретения возможны различные воплощения, а также могут быть внесены различные изменения в приведенные выше воплощения.
Заявленное в качестве нового и желательного для защиты в письмах - патенте приведено в следующей формуле изобретения.



Изобретение относится к медицине и может быть использовано при пероральном введении пациентам фармацевтических агентов, которые слабо абсорбируются из желудочно-кишечного тракта. Способ представляет собой предотвращение или снижение гиперчувствительности или аллергических реакций у пациента, получающего таксановую терапию, при лечении заболевания, поддающегося лечению таксаном, включающий пероральное совместное введение таксана и агента, усиливающего биодоступность при пероральном введении, без предварительного введения лекарственного средства для предотвращения гиперчувствительности или аллергических реакций, причем таксан достигает у пациента терапевтически активных уровней. Предложенный способ позволяет повысить биодоступность препарата. 52 з.п.ф-лы, 4 ил., 1 табл.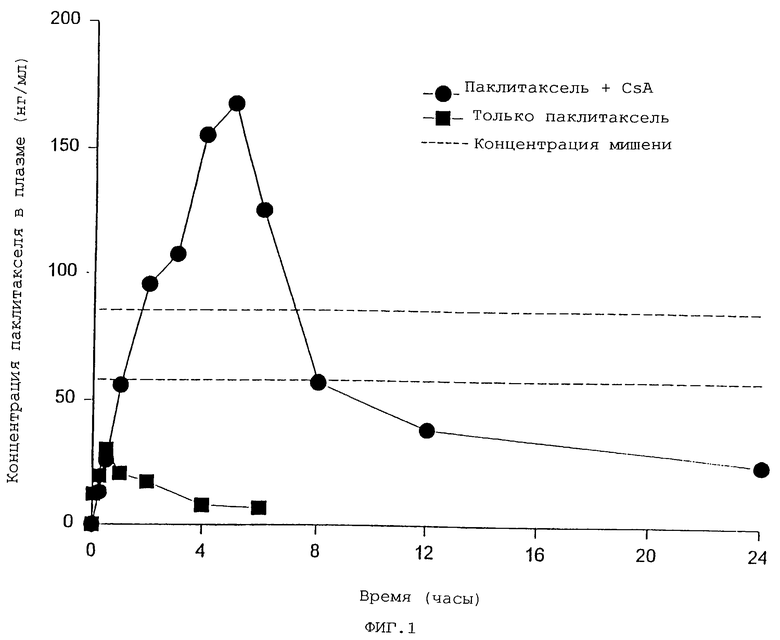
| RU 95105981 A1, 27.10.1996 | |||
| Lum BL, Gosland MP | |||
| MDR expression in normal tissues | |||
| Pharmacologic implications for the clinical use of P-glycoprotein inhibitors, Hematol Oncol Clin North Am 1995 Apr; 9(2):319-336 | |||
| Nicolaou КС, Riemer C, Kerr MA, Rideout D, Wrasidio W | |||
| Design, synthesis and biological activity of protaxols, Nature 1993 Jul 29; 364(6436):464-466. |

