Область техники
Изобретение относится к способу селективного получения композиций фенилалканов, к композициям и к использованию данных композиций.
Предшествующий уровень техники
Более чем тридцать лет назад из разветвленных алкилбензолсульфонатов (BABS) получили множество бытовых моющих средств для стирки. BABS изготавливают из определенного типа алкилбензолов, называемого разветвленными алкилбензолами (ВАВ). Алкилбензолы (фенилалканы) входят в общую категорию соединений, имеющих алифатическую алкильную группу, соединенную с фенильной группой, и описываемых общей формулой (mi-алкилi)i-n-фенилалкан. Алифатическая алкильная группа также может состоять из одного, либо нескольких образуемых алкильными группами разветвлений, обозначаемых соответствующим «(mi-алкиломi)i».
Стандартный способ, используемый в нефтехимической промышленности для получения ВАВ, заключается в олигомеризации легких олефинов, в особенности пропилена, с получением разветвленных олефинов, содержащих от 10 до 14 атомов углерода, а после этого алкилировании бензола под действием разветвленных олефинов в присутствии катализатора, такого как HF. Несмотря на то что продукт ВАВ содержит большое количество алкилфенилалканов, описываемых общей формулой (mi-алкилi)i-n-фенилалкан, двумя примерами ВАВ являются m-алкил-m-алкил-n-фенилалканы, где m≠n, и m-алкил-m-фенилалканы, где m≥2.
Наиболее примечательной общей характеристикой ВАВ является то, что у большой доли ВАВ к алифатической алкильной цепи ВАВ в общем случае присоединено, по меньшей мере, одно разветвление в виде алкильной группы, а более часто три или более разветвлений в виде алкильных групп. Если какое-либо разветвление в виде алкильной группы самое по себе будет разветвлено, то тогда алифатическая алкильная группа в ВАВ будет иметь еще больше первичных атомов углерода. Таким образом, алифатическая алкильная группа в ВАВ обычно будет иметь три, четыре или более первичных атомов углерода. Каждое разветвление в виде алкильной группы обычно представляет собой разветвление в виде метильной группы несмотря на то, что возможны разветвления и в виде этильной, пропильной, либо высшей алкильной групп.
Еще одной типичной характеристикой ВАВ является присоединение фенильной группы к любому непервичному атому углерода алифатической алкильной цепи. Исключая 1-фенилалканы, образование которых, как известно, неблагоприятно вследствие относительной нестойкости первичного иона карбения, и пренебрегая относительно малозначительным влиянием разветвлений в разветвленных парафинах, можно сказать, что стадия олигомеризации приводит к получению двойной связи углерод-углерод, которая случайным образом распределена по длине алифатической алкильной цепи, а стадия алкилирования приводит к почти что случайному распределению мест присоединения фенильной группы к атому углерода вдоль алифатической алкильной цепи. Таким образом, например, ВАВ, который содержит алифатическую алкильную цепь, имеющую 10 атомов углерода, как ожидается, будет представлять собой смесь с приблизительно случайным распределением 2-, 3-, 4- и 5-фенилалканов и селективность по получению 2-фенилалкана будет равна 25, если распределение будет полностью случайным, но обычно она заключена в диапазоне от 10 до 40.
У ВАВ в общем случае имеется один из четвертичных атомов углерода, представляющий собой один из атомов углерода алифатической алкильной группы. Четвертичный атом углерода через связь углерод-углерод может быть связан с атомом углерода в фенильной группе. И в данном случае молекулу называют «четвертичным алкилфенилалканом», или же далее в настоящем документе просто «четвертичным соединением», и она включает алкилфенилалканы, описываемые общей формулой m-алкил-m-фенилалкан. «Концевые четвертичные соединения» образуют 2-алкил-2-фенилалканы, у которых четвертичным является второй атом углерода от конца алкильной боковой цепи. «Четвертичные соединения», имеющие четвертичный атом углерода в других положениях, представляют собой в результате алкилфенилалкан, называемый «внутренним четвертичным соединением». Известные способы получения ВАВ приводят к образованию относительно высокой доли «внутренних четвертичных соединений», обычно превышающей 10% (моль.).
Бытовые моющие средства для стирки, полученные из BABS, постепенно загрязняли реки и озера. Было установлено, что биоразложение BABS протекает медленно. Остроту проблемы уменьшило использование линейных алкилбензолсульфонатов (LABS), биоразложение которых протекает быстрее, чем биоразложение BABS. LABS получают из линейных алкилбензолов (LAB). В нефтехимической промышленности LAB получают в результате дегидрирования линейных парафинов с получением линейных олефинов, которые после этого алкилируют под действием бензола в присутствии HF, либо твердого катализатора. LAB содержат линейную алифатическую алкильную группу и фенильную группу, и они описываются общей формулой n-фенилалкан. LAB не имеют разветвлений в виде алкильных групп и обычно содержат два первичных атома углерода (то есть n≥2). В стандартном способе получения LAB происходит присоединение фенильной группы к любому вторичному атому углерода в линейной алифатической алкильной группе. Алкилирование с использованием катализатора HF отличается несколько большей вероятностью присоединения фенильной группы ко вторичному атому углерода, расположенному недалеко от середины линейной алифатической алкильной группы. В LAB, полученном по способу Detal™, 2-фенилалканы составляют приблизительно 25-35% (моль.) от n-фенилалканов.
В недавнем исследовании идентифицировали модифицированные алкилбензолсульфонаты "MABS", которые отличаются от всех алкилбензолсульфонатов, коммерчески используемых в настоящее время, и от всех алкилбензолсульфонатов, получаемых по прежним способам получения алкилбензолов, в том числе и тех, в которых в качестве катализатора использовали HF, хлорид алюминия, диоксид кремния-оксид алюминия, фторированный диоксид кремния-оксид алюминия, цеолиты и фторированные цеолиты. MABS по сравнению с LABS отличаются улучшенными эксплуатационными характеристиками для очищения при стирке, эксплуатационными характеристиками для очищения твердых поверхностей и превосходной эффективностью в жесткой и/или холодной воде при том, что в то же время их биоразложимость сопоставима с биоразложимостью LABS.
MABS можно получать в результате сульфирования третьего типа алкилбензолов, называемого модифицированными алкилбензолами (МАВ), и желательные характеристики МАВ определяются желательными свойствами растворимости, способности выступать в роли поверхностно-активного вещества и биоразложимости MABS. МАВ включает большое количество фенилалканов, некоторые из которых могут относиться к LAB и ВАВ, но попадающие одновременно в разные группы фенилалканы не являются желательными фенилалканами для МАВ. Фенилалканы у МАВ представляют собой фенилалканы, содержащие легкоразветвленную алифатическую алкильную группу и фенильную группу, и они описываются общей формулой (mi-алкилi)i-n-фенилалкан. Фенилалканы у МАВ обычно имеют только одно разветвление в виде алкильной группы, и, когда n≠1, МАВ включает три первичных атома углерода. Предпочтительный фенилалкан у МАВ представляет собой монометилфенилалкан. Однако МАВ может иметь и два первичных атома углерода, если будет иметься только одно разветвление в виде алкильной группы, а n=1, или же четыре первичных атома углерода, если будет иметься два разветвления в виде алкильных групп, a n≠1. Таким образом, первой характеристикой МАВ является среднее количество первичных атомов углерода в алифатических алкильных группах фенилалканов, промежуточное между количеством в ВАВ и в LAB. МАВ характеризует также и высокая доля 2-фенилалканов, а именно, то, что от 40 до 100% фенильных групп селективно присоединены ко второму атому углерода алкильной боковой цепи.
В качестве заключительной характеристики алкилат у МАВ отличается относительно низкой долей «внутренних четвертичных соединений», обычно меньшей 10% (моль.). Некоторые «внутренние четвертичные соединения», такие, как 5-метил-5-фенилундекан, приводят к получению MABS с более медленным протеканием биоразложения. MABS с «концевыми четвертичными соединениями», такими, как 2-метил-2-фенилундекан, демонстрируют биоразложение, подобное тому, что и у LABS. Смотрите статью, озаглавленную "Biodegradation of Coproducts of Commercial Linear Alkylbenzene Sulfonate", by A.M.Nielsen et al., in Environmental Science and Technology, vol.31, №12, 3397-3404 (1997).
Международные публикации в рамках договора о патентной кооперации №№ WO 99/05082, WO 99/05084, 99/05241 и WO 99/05243, где все четыре были опубликованы 4 февраля 1999 года, описывают способы алкилирования уникально легкоразветвленных, либо делинеаризованных алкилбензолов. Международная публикация в рамках договора о патентной кооперации № WO 99/07656, опубликованная 18 февраля 1999 года, описывает способы получения таких алкилбензолов с использованием адсорбционного разделения.
Вследствие наличия у MABS преимуществ в сравнении с другими алкилбензолсульфонатами проводятся поиски катализаторов и способов, которые приводили бы к получению МАВ при обеспечении селективности по получению 2-фенилалканов и селективности по предотвращению получения внутренних четвертичных фенилалканов.
Краткое изложение изобретения
В одном аспекте данное изобретение представляет собой способ получения фенилалканов, в особенности, модифицированных алкилбензолов (МАВ), в результате адсорбционного разделения, дегидрирования и алкилирования. Способ отличается составом пары адсорбента и десорбента, используемой в способе. Используемым адсорбентом является силикалит, а десорбент содержит линейный C5-C8 линейный парафин, C5-C8 циклопарафин и/или предпочтительно разветвленный парафин, такой, как изооктан.
В варианте реализации способа поток парафинового исходного сырья, содержащий первый ациклический парафин, отличающийся количеством атомов углерода в диапазоне C8-C28 и имеющий 2, либо 3 первичных атома углерода, с первой концентрацией и второй ациклический парафин, перепускают в зону адсорбции. Зону адсорбции образует слой адсорбента, содержащего силикалит, находящийся в условиях, способствующих прохождению адсорбции при проведении селективной адсорбции, по меньшей мере, части ациклического парафина, имеющего 2, либо 3 первичных атома углерода. Со слоем адсорбента вводят в контакт поток десорбента, включающий, по меньшей мере, одного представителя, выбираемого из C5-C8 циклопарафина, С5-С8 нормального парафина и C5-C8 разветвленного парафина. Из зоны адсорбции извлекают экстракт адсорбции, отличающийся второй концентрацией первого ациклического углеводорода, которая превышает первую концентрацию. По меньшей мере, часть экстракта адсорбции перепускают в зону дегидрирования, которая функционирует в условиях прохождения дегидрирования, достаточных для обеспечения дегидрирования первого ациклического парафина. Из зоны дегидрирования извлекают поток продуктов дегидрирования, содержащий C8-C28 ациклический моноолефин, имеющий 2, либо 3 первичных атома углерода. Ароматическое исходное сырье, содержащее фенильное соединение и, по меньшей мере, часть потока продуктов дегидрирования, перепускают в зону алкилирования, которая функционирует в условиях прохождения алкилирования, достаточных для алкилирования фенильного соединения под действием ациклического моноолефина в присутствии катализатора алкилирования. Зона алкилирования делает возможным извлечение фенилалкана, включающего молекулу, содержащую одно фенильное звено и одно C8-C28 алифатическое алкильное звено, имеющее 2, либо 3 первичных атома углерода и не имеющее четвертичных атомов углерода за исключением случаев «четвертичных соединений». Алкилирование характеризуется селективностью по получению 2-фенилалканов в диапазоне от 40 до 100 и селективностью по получению внутренних четвертичных фенилалканов, меньшей 10. В предпочтительном варианте реализации алкилирование характеризуется селективностью по получению «не четвертичных соединений», меньшей 10, а более предпочтительно меньшей 1.
В предпочтительном варианте реализации способа данное изобретение приводит к получению композиции МАВ, содержащей фенилалканы, имеющие одну фенильную группу и одну алифатическую алкильную группу. Фенилалканы, кроме этого, характеризуются средней массой алифатических алкильных групп в фенилалканах в диапазоне между массой С10 алифатической алкильной группы и C13 алифатической алкильной группы; содержанием фенилалканов, имеющих фенильную группу, присоединенную во 2-м и/или 3-м положении алифатической алкильной группы, превышающим 55% (масс.) от количества фенилалканов; и средним уровнем разветвления алифатических алкильных групп в фенилалканах в диапазоне от 0,25 до 1,3 разветвления в виде алкильной группы на одну молекулу фенилалкана, если сумма содержаний 2-фенилалканов и 3-фенилалканов будет больше 55% (масс.) и меньше, либо равна 85% (масс.) от количества фенилалканов, или же средним уровнем разветвления алифатических алкильных групп в фенилалканах в диапазоне от 0,4 до 1,3 разветвления в виде алкильной группы на одну молекулу фенилалкана, если сумма концентраций 2-фенилалканов и 3-фенилалканов будет больше 85% (масс.) от количества фенилалканов. В дополнение к этому, алифатические алкильные группы у фенилалканов включают, в первую очередь, линейные алифатические алкильные группы и моноразветвленные алифатические алкильные группы, и где разветвления в виде алкильных групп на алифатической алкильной цепи у алифатических алкильных групп включают, в первую очередь, небольшие заместители, такие, как разветвления в виде метильных групп, разветвления в виде этильных групп, либо разветвления в виде пропильных групп, и где разветвления в виде алкильных групп присоединены в любом положении на алифатической алкильной цепи у алифатических алкильных групп при том условии, что фенилалканы, имеющие, по меньшей мере, один четвертичный атом углерода в алифатической алкильной группе, составляют меньше 20% от количества фенилалканов.
Данное изобретение при его использовании для алкилирования с получением моющего средства приводит к получению моющих средств, которые удовлетворяют становящимся все более жесткими требованиям по селективности по получению 2-фенилалканов и селективности по получению внутренних четвертичных фенилалканов при получении МАВ, которые можно сульфировать с получением MABS с улучшенной эффективностью по очищению в жесткой и/или холодной воде и биоразложимостью, сопоставимой с биоразложимостью LAS.
В другом аспекте способ данного изобретения приводит к получению конкретных композиций продуктов МАВ и MABS со специально заданным разветвлением на атомах углерода, которое отличается от того, что получали по способам предшествующего уровня техники. В еще одном аспекте данные полученные МАВ и MABS можно использовать в качестве смазочного материала, либо присадки к смазочному материалу, соответственно.
Краткое описание чертежей
Фигура 1 демонстрирует вариант реализации изобретения.
Фигура 2 демонстрирует профиль концентраций для разделения в импульсном испытании.
Подробное описание изобретения
В способе изобретения используют исходную смесь, содержащую парафин и исходное сырье, содержащее фенильное соединение. Исходная смесь включает ациклические парафины, содержащие от 8 до 28 атомов углерода. Ациклический парафин предпочтительно представляет собой «легкоразветвленный парафин», который в соответствии с тем, как это используется в настоящем документе, обозначает парафин, имеющий три, либо четыре первичных атома углерода и не имеющий четвертичных атомов углерода. Обычно легкоразветвленный парафин характеризуется совокупным количеством атомов углерода в диапазоне от 9 до 16, предпочтительно от 10 до 14 атомов углерода, а в высшей степени предпочтительно от 10 до 13 атомов углерода. Легкоразветвленный парафин в общем случае включает алифатический алкан, описываемый общей формулой (рi-алкилi)i-алкан.
Легкоразветвленные парафины в общем случае составляют более 30% (моль.), а предпочтительно более 70% (моль.) от количества исходной смеси. Алкильные группы разветвлений в общем случае включают метильные, этильные и пропильные группы, при этом более короткие разветвления предпочтительны. Предпочтительно легкоразветвленный парафин имеет только одно разветвление в виде алкильной группы и составляет предпочтительно более 85% (моль.) от совокупного количества легкоразветвленных парафинов. Легкоразветвленные парафины, имеющие либо два разветвления в виде алкильных групп, либо четыре первичных атома углерода, в общем случае составляют менее 30% (моль.), а предпочтительно менее 15% (моль.) от совокупного количества легкоразветвленных парафинов.
Исходная смесь также может содержать и одну, либо несколько молекул неразветвленных (линейных), или нормальных парафинов со средним количеством атомов углерода, приходящимся на одну молекулу парафина, в общем случае в диапазоне от 8 до 28 атомов углерода, а в высшей степени предпочтительно от 10 до 13 атомов углерода. Концентрация неразветвленных парафинов в исходной смеси зачастую превышает 0,3% (моль.).
В дополнение к легкоразветвленным и неразветвленным парафинам в исходной смеси могут присутствовать и другие, более высокоразветвленные ациклические соединения. Однако после дегидрирования такие высокоразветвленные парафины имеют тенденцию образовывать высокоразветвленные моноолефины, которые после алкилирования имеют тенденцию образовывать ВАВ. Например, молекулы парафинов, содержащие, по меньшей мере, один четвертичный атом углерода, имеют тенденцию после дегидрирования с последующим алкилированием образовывать фенилалканы, которые в алифатической алкильной части будут иметь четвертичный атом углерода, и которые не будут являться «четвертичными соединениями». Поэтому количество данных высокоразветвленных парафинов, отправляемых на переработку по данному способу, предпочтительно сводят к минимуму. Молекулы парафинов, содержащие, по меньшей мере, один четвертичный атом углерода, в общем случае составляют менее 10% (моль.), предпочтительно менее 5% (моль.), более предпочтительно менее 2% (моль.), а наиболее предпочтительно менее 1% (моль.) от количества исходной смеси.
Получение исходной смеси не представляет собой существенный элемент данного изобретения, и возможно использование любого подходящего способа получения исходной смеси. Поскольку диапазон количества атомов углерода у исходной смеси, желательной для получения МАВ, обычно заключен в пределы от 9 до 16, то данный диапазон соответствует парафинам, кипящим в диапазоне температур кипения керосина, поэтому керосиновые фракции образуют подходящие прекурсоры для исходной смеси. Способы получения керосинов по своей внутренней сущности не являются точными, и они приводят к получению смеси соединений. Исходные смеси данного способа могут содержать определенные количества парафинов, имеющих несколько разветвлений, и парафинов, имеющих несколько атомов углерода в разветвлениях, циклопарафинов, разветвленных циклопарафинов, либо других соединений с температурами кипения, относительно близкими к температуре изомера желательного соединения. Керосиновые фракции содержат очень большое количество различных углеводородов, и поэтому исходная смесь в настоящем способе может содержать 200, или более различных соединений, в том числе и ощутимые количества ароматических углеводородов. Для фракций, извлеченных из сырой нефти в результате фракционирования, обычно будет необходимо провести гидрирование для удаления серы и/или азота перед подачей на переработку по способу настоящего изобретения.
Однако представляется, что в сравнении с олигомеризацией, либо другими формами синтеза подходящую смесь с меньшей стоимостью позволит получить разделение, и поэтому оно будет предпочтительным источником исходной смеси. Предпочтительный способ получения исходной смеси заключается в отделении неразветвленных (линейных) углеводородов, либо легкоразветвленных углеводородов от фракции нефти с диапазоном температур кипения керосина. Известно несколько способов, которые приводят к такому разделению. Один способ - способ UOP Molex™ - представляет собой признанный, проверенный в промышленности способ жидкофазного адсорбционного отделения нормальных парафинов от изопарафинов, циклопарафинов и ароматики при использовании технологии разделения UOP Sorbex. Другим подходящим, признанным и проверенным способом является способ UOP Kerosene Isosiv™, в котором используют парофазную адсорбцию для отделения нормальных парафинов от парафинов, не являющихся нормальными, при использовании молекулярных сит в емкости адсорбера. Смотрите главы 10.3, 10.6 и 10.7 в книге, озаглавленной Handbook of Petroleum Refining Process, Second Edition, edited by Robert A.Meyers, published by McGraw-Hill, New York, 1997.
Поток рафината в способе адсорбционного разделения, таком, как способ UOP Molex™, в котором селективно извлекают неразветвленные (линейные) парафины из потока экстракта, представляет собой в особенности предпочтительную исходную смесь для способа настоящего изобретения. Поток рафината после переработки по такому способу не будет содержать загрязняющих примесей, таких, как серо-, либо азотсодержащие соединения, и он также будет отличаться подходящей низкой концентрацией неразветвленных парафинов и олефинов. Использование такого потока рафината в качестве исходной смеси делает возможной интеграцию способа настоящего изобретения в существующую установку получения LAB при последовательном проведении двух стадий адсорбционного разделения. Поток извлеченных при разделении нормальных парафинов и исходную смесь после этого можно подвергать переработке различными способами. Например, каждого представителя, выбираемого из потока неразветвленных парафинов и исходной смеси, можно подвергать переработке независимо с использованием дегидрирования и алкилирования ароматики с получением двух отдельных продуктов. В альтернативном варианте поток неразветвленных парафинов и исходную смесь можно использовать для получения желательной смеси парафинов. То есть, поток, подаваемый в зону дегидрирования способа настоящего изобретения, может содержать продукт зоны разделения способа настоящего изобретения плюс от 10 до 50% (об.) неразветвленных парафинов.
Состав смеси линейных, легкоразветвленных и разветвленных парафинов, а также олефинов можно определить с помощью аналитических методов, которые хорошо известны специалисту в области газовой хроматографии. Статья, написанная авторами Н.Schuiz, et al. и опубликованная, начиная со страницы 315, в журнале Chromatographia 1, 1968, описывает аппарат, представляющий собой газовый хроматограф с программируемой температурой, и метод, который пригоден для идентификации компонентов сложных смесей парафинов, либо олефинов. Специалист в соответствующей области может разделить и идентифицировать компоненты в смеси парафинов при использовании по существу аппарата и метода, описанных в статье у Schulz et al.
Содержащее ароматику исходное сырье, подаваемое на переработку по способу настоящего изобретения, включает фенильное соединение, которым является бензол тогда, когда способ представляет собой алкилирование с получением моющего средства. В более общем случае фенильное соединение из ароматического исходного сырья может быть алкилированными, либо другим образом замещенными производными, или же иметь более высокую молекулярную массу в сравнении с бензолом, включая толуол, этилбензол, ксилол, фенол, нафталин и тому подобное.
В секции адсорбционного разделения из исходной смеси извлекают ациклические, легкоразветвленные парафины. Данное разделение можно проводить в периодическом, либо непрерывном режимах, в том числе и при использовании двух, или более слоев адсорбента при циклической операции. В данном режиме для разделения используют один, либо несколько слоев, тогда как другой слой регенерируют.Значительные эксплуатационные и экономические преимущества достигаются при проведении разделения в непрерывном режиме. Технология с симулированным подвижным слоем (SMB) представляет собой предпочтительный способ проведения процесса в непрерывном режиме и достижения однородности продуктов. При разделении из исходной смеси предпочтительно извлекают монометилпарафины.
Установки адсорбционного разделения по способу SMB, предназначенные для симулирования перемещения адсорбента по отношению к потоку исходного сырья, хорошо известны. Данное симулирование проводят с использованием признанной коммерческой технологии, где адсорбент удерживают фиксированным на одном месте в виде нескольких подслоев, находящихся в одной, либо нескольких цилиндрических камерах для адсорбента. Позиции, в которых потоки, участвующие в способе, поступают в камеры и покидают их, медленно смещаются от подслоя к подслою по длине камер для адсорбента, так что потоки по мере продвижения по ходу рабочего цикла поступают в различные подслои, либо различные подслои покидают.Обычно имеется, по меньшей мере, четыре потока (исходное сырье, десорбент, экстракт и рафинат), используемых в данной методике, и местоположения, в которых потоки исходного сырья и десорбента поступают в камеру, а потоки экстракта и рафината покидают камеру, одновременно смещаются в одном и том же направлении с установленными интервалами. Для каждого подслоя обычно используют только одну линию, и каждая линия слоя транспортирует один из четырех технологических потоков в некоторой точке цикла. Циклическое продвижение входящих и выходящих потоков при данном симулировании можно осуществить при использовании системы распределительных труб, либо при помощи клапанов с вращающимся диском. Данное симулирование обычно включает использование насоса с переменной производительностью, который подает жидкость, покидающую один конец емкости (емкостей) с адсорбентом, на другой конец в одном непрерывном цикле.
Способы с симулированным подвижным слоем обычно включают, по меньшей мере, три, либо четыре отдельные стадии, которые реализуют последовательно в раздельных зонах в массе адсорбента. Каждую из данных зон обычно формирует несколько подслоев, при этом количество слоев, приходящихся на одну зону, находится в диапазоне от 2, либо 3 вплоть до величины в пределах от 8 до 10. Наиболее широко распространенные на практике коммерческие технологические установки обычно включают 24 слоя. Все слои содержатся в одной, либо нескольких вертикальных емкостях, совокупно называемых в настоящем документе камерой для адсорбента. Общая методика, используемая при проведении адсорбционного разделения по способу с симулированным подвижным слоем, хорошо описывается на странице 70 в работе the September 1970 edition of Chemical Engineering Progress (vol.66, №9), как это продемонстрировано в работах US-A-3,040,777 и US-A-3,422,848.
В ходе стадии адсорбции данного способа исходную смесь, содержащую смесь соединений, водят в контакт с адсорбентом в условиях прохождения адсорбции и с использованием адсорбента проводят селективную адсорбцию и удерживание одного соединения, либо нескольких соединений, или же класса соединений, в то время как другие соединения исходной смеси остаются относительно не адсорбированными. Обычно адсорбируют желательное соединение. Исходная смесь может содержать большое количество соединений, в том числе изомеров желательного соединения. Например, смешанный поток ксилольного исходного сырья может содержать этилбензол и/или С9 ароматику, и при использовании подходящей пары адсорбент/десорбент, эксплуатируемой в подходящих условиях, его можно подвергнуть переработке для извлечения конкретного изомера. Для различных разделений используют различные комбинации сита/десорбент. Например, для извлечения п-ксилола из ксилольных смесей предпочтительными адсорбентами являются Х цеолиты, говоря конкретно, Х цеолиты с ионами бария, либо бария и калия, введенными в ходе ионного обмена в центры цеолитов, способные к ионному обмену.
После этого на следующей стадии способа неадсорбированные компоненты (рафинат) исходной смеси удаляют из свободного пространства между частицами адсорбента и с поверхности адсорбента в виде потока рафината. Затем адсорбированное соединение извлекают из адсорбента в результате введения его на стадии десорбции в контакт с потоком, содержащим вещество десорбента, в условиях прохождения десорбции. Десорбент вытесняет желательное соединение с получением потока экстракта, который обычно перепускают в зону фракционирования для извлечения желательного соединения из десорбента потока экстракта. В некоторых случаях желательный продукт способа может находиться в потоке рафината, а не в потоке экстракта.
Для целей данного описания различные термины, используемые в настоящем документе, получают следующие определения. «Исходной смесью» является смесь, содержащая один, либо несколько компонентов экстракта и один, либо несколько компонентов рафината, разделяемых при помощи секции адсорбции способа настоящего изобретения. Термин «поток исходного сырья» указывает на поток исходной смеси, подаваемый для введения в контакт с адсорбентом. «Компонент экстракта» представляет собой соединение, либо класс соединений, которые адсорбент адсорбирует более селективно. «Компонент рафината» представляет собой соединение, либо класс соединений, которые адсорбируются менее селективно. Термин «вещество десорбента» обозначает вещество, способное десорбировать компонент экстракта. Термин «поток рафината» обозначает поток, удаляемый из слоя адсорбента после адсорбции соединений экстракта, который может варьироваться от по существу 100% вещества десорбента до по существу 100% компонентов рафината. Термин «поток экстракта» обозначает поток, десорбируемый под действием вещества десорбента и удаляемый из слоя адсорбента и варьирующийся в диапазоне от по существу 100% вещества десорбента до по существу 100% компонентов экстракта.
Устройства для разделения, обычно колонны для фракционной перегонки, позволяют извлекать все количество, либо части потока экстракта и потока рафината. Поток, содержащий нежелательное соединение, можно отправить на изомеризацию для повторного использования. Поток экстракта может быть обогащен желательным соединением, либо только может содержать его в повышенной концентрации. Использование в отношении технологического потока термина «обогащенный» предполагает указание на концентрацию указанного соединения, либо класса соединений, превышающую 50% (моль.).
На современном уровне техники стало обычной практикой группирование различных слоев в адсорбционных камерах в несколько зон. В зоне I - зоне адсорбции - вводят поток исходного сырья в контакт с адсорбентом. В зоне II - зоне очистки - нежелательные изомеры удаляют в виде рафината. В хоне III - зоне десорбции - десорбент высвобождает желательные изомеры из адсорбента для извлечения из потока экстракта. Зона IV содержит определенное количество адсорбента, расположенного между зоной I и зоной III, который разделяет зоны I и III и частично удаляет десорбент из адсорбента. Поток жидкости через зону IV предотвращает загрязнение зоны III жидкостью зоны I вследствие сонаправленности потока и симулированного перемещения адсорбента от зоны III к зоне I. Более детальное разъяснение способов с симулированным подвижным слоем приводится в разделе Adsorption, Liquid Separation в работе Kirk-Othmer Encyclopedia of Chemical Technology.
Цели данного изобретения достигаются при использовании новой пары адсорбент-десорбент, включающей адсорбент на основе силикалита и десорбент, содержащий разветвленный парафин; линейный парафин и/или циклопарафин; линейный парафин и разветвленный парафин; либо линейный парафин, циклопарафин и разветвленный парафин. Предпочтительным десорбентом является C5-C8 разветвленный парафин. Предпочтительным разветвленным парафином для десорбента является изооктан.
Предпочтительный адсорбент включает силикалит. Силикалит хорошо описывается в статье "Silicalite, A New Hydrophobic Crystalline Silica Molecular Sieve," Nature, vol.271, Feb.9, 1978, которая включается в настоящий документ для справки в связи с описанием в ней силикалита и приведением его характеристик. Силикалит представляет собой гидрофобные кристаллические молекулярные сита на основе диоксида кремния, имеющие структуру, относящуюся к типу MFI, в виде пересекающихся изогнутых ортогональных каналов, сформированных с двумя поперечно пересекающимися геометриями, круговой 6 Å и эллиптической 5,1-5,7 Å для основной оси. Это придает силикалиту как молекулярным ситам с селекцией по размерам высокую селективность. Вследствие отсутствия в его структуре, образованной из диоксида кремния, алюминия силикалиту не свойственны ионообменные свойства. Таким образом, силикалит не является цеолитом.
Для практической реализации настоящего изобретения не требуется проведения значительных изменений рабочих условий, состава адсорбента, либо десорбента в камерах для адсорбента, либо в ходе проведения различных стадий способа. То есть, адсорбент предпочтительно остается при одних и тех же температуре и давлении в ходе всего технологического процесса.
Активный компонент адсорбента обычно используют в виде небольших агломератов, обладающих высокими механической прочностью и стойкостью к истиранию. Агломераты содержат активный адсорбирующий материал, диспергированный в аморфной неорганической матрице, называемой связующим и отличающейся наличием в ней каналов и пустот, которые делают возможным доступ текучей среды к адсорбирующему материалу. Способы формования из кристаллических порошков таких агломератов включают добавление неорганического связующего, в общем случае глины, содержащей диоксид кремния и оксид алюминия, к высокочистому порошку адсорбента во влажной смеси. Подходящим связующим является диоксид кремния. Частицы адсорбента могут иметь форму экструдатов, таблеток, макросфер, либо гранул с желательным диапазоном частиц, предпочтительно от 16 до 60 меш (стандартный меш США) (от 1,9 мм до 250 микрон). В общем случае в качестве связующих используют глины, относящиеся к типу каолина, водопроницаемые органические полимеры, либо диоксид кремния.
Специалисты в соответствующей области должны понимать, что на эксплуатационные характеристики конкретного адсорбента зачастую значительное влияние оказывают несколько переменных, не связанных с его составом, таких, как рабочие условия, состав потока исходного сырья и содержание воды в адсорбенте. Одной такой переменной является содержание воды в адсорбенте, которое в настоящем документе выражают в единицах, принятых в признанном испытании на определение потери массы при прокаливании (LOI). В испытании LOI содержание летучих веществ в адсорбенте на основе цеолита определяют по разнице масс, получаемых до и после высушивания образца адсорбента при 500°С при продувке инертным газом, таким, как азот, в течение периода времени, достаточного для достижения постоянной массы. В способе настоящего изобретения предпочитается, чтобы содержание воды в адсорбенте при определении по методу LOI при 900°С составляло в результате величину менее, чем 7,0% (масс.), а предпочтительно в диапазоне от 0 до 4,0% (масс.).
Силикалит, либо другой микропористый активный компонент адсорбента обычно будет иметься в виде небольших кристаллов, присутствующих в частицах адсорбента в количествах в диапазоне от 75 до 98% (масс.) частицы в расчете на состав, не включающий летучих веществ. Не включающие летучих веществ составы в общем случае определяют после того, как адсорбент будет прокален при 900°С для того, чтобы отогнать из него все летучие вещества. Остающийся адсорбент в общем случае будет представлять собой неорганическую матрицу связующего.
В настоящем изобретении перепускают исходную смесь, содержащую один, либо несколько монометилразветвленных углеводородов и, по меньшей мере, один не являющийся нормальным углеводород с подобным количеством атомов углерода, но с другой структурой, через один, либо несколько слоев адсорбента, которые селективно адсорбируют монометилразветвленный углеводород и позволяют другим компонентам пройти через зону адсорбции. В некоторый момент времени движение потока исходного сырья через слой адсорбента прекращают и зону адсорбции промывают для удаления не адсорбированных веществ, окружающих адсорбент.После этого перепускание десорбента через слой адсорбента приводит к извлечению желательного изомера.
Селективность (β) пары адсорбент/десорбент определяют как соотношение количеств двух компонентов в адсорбированной фазе, поделенное на соотношение количеств тех же самых двух компонентов в не адсорбированной фазе в равновесных условиях. Относительную селективность получают по уравнению:
Селективность = (массовый процент С/массовый процент DА)/(массовый процент С/массовый процент DU),
где С и D представляют собой количества двух компонентов потока исходного сырья, выраженные через массовый процент, а подстрочные индексы А и U представляют собой адсорбированную и не адсорбированную фазы, соответственно. Равновесные условия определяют тогда, когда поток исходного сырья, проходящий через слой адсорбента, не изменяет своего состава, другими словами, когда в совокупности переноса вещества между не адсорбированной и адсорбированной фазами не происходит. Относительную селективность можно выразить не только для одного соединения потока исходного сырья в сопоставлении с другим, но ее также можно выразить и через соотношение между любым компонентом исходной смеси и веществом десорбента.
Важной характеристикой адсорбента является скорость обмена десорбента на компонент экстракта из исходной смеси, либо, другими словами, относительная скорость десорбции компонента экстракта. Более значительные скорости обмена приводят к уменьшению количества вещества десорбента, необходимого для удаления компонента экстракта, и, следовательно, эксплуатационных затрат в способе. Скорости обмена зачастую зависят от температуры. В идеальном случае вещества десорбента должны характеризоваться селективностью по отношению к компонентам экстракта, равной, либо несколько меньшей 1, для того, чтобы десорбировать компоненты экстракта как класс с разумными расходами вещества десорбента, и так, чтобы компоненты экстракта могли бы вытеснять вещество десорбента на последующих стадиях адсорбции.
В способах непрерывного жидкофазного адсорбционного разделения вещество десорбента необходимо рационально выбрать удовлетворяющим многим критериям. Если говорить о селективности, то адсорбент должен характеризоваться большей селективностью для всех компонентов экстракта по отношению к компоненту рафината в сравнении с селективностью для вещества десорбента по отношению к компоненту рафината. Вещества десорбента также должны быть совместимыми с конкретным адсорбентом и конкретной исходной смесью и иметь разумную стоимость.
Условия проведения адсорбции в общем случае включают диапазон температур от 20°С до 250°С, при этом более предпочтителен диапазон от 40°С до 150°С. В высшей степени предпочтительны температуры от 80°С до 140°С. Условия проведения адсорбции также предпочтительно включают давление, достаточное для выдерживания текучих сред технологического процесса в жидком состоянии; где данное давление может находиться в диапазоне от атмосферного до 4137 кПа (600 фунт/дюйм2) избыточного давления. Условия проведения десорбции в общем случае включают те же самые температуры и давления, что и использованные для создания условий проведения адсорбции.
Предпочтительный десорбент содержит смесь нормального парафина, циклопарафина (нафтена) и/или разветвленного парафина. Предпочтительными циклопарафинами являются циклопентан, циклогексан и метилциклогексан. Предпочтительными нормальными парафинами являются н-пентан и н-гексан. Нормальные парафины являются сильными десорбентами, а н-гексан фактически является самым сильным десорбентом из данных соединений. Для регулирования силы потока десорбента зачастую желательной является смесь нормальных парафинов и циклопарафинов, либо нормальных парафинов и изооктана. Данные смеси могут содержать от 10 до 90% (об.) циклопарафина, либо изооктана, при этом остальным веществом является нормальный парафин.
Поток экстракта содержит парафины с совокупным количеством атомов углерода, приходящимся на одну молекулу парафина, в общем случае в диапазоне от 8 до 28, предпочтительно от 8 до 15, а более предпочтительно от 10 до 15 атомов углерода. Поток экстракта содержит более высокую концентрацию легкоразветвленных парафинов в расчете на совокупное количество парафинов в потоке экстракта в сравнении с концентрацией легкоразветвленных парафинов в исходной смеси в расчете на совокупное количество парафинов в исходной смеси. Легкоразветвленные парафины, имеющие либо два разветвления в виде алкильных групп, либо четыре первичных атома углерода, в общем случае составляют менее 60% (моль.), предпочтительно менее 30% (моль.), а более предпочтительно менее 15% (моль.) от совокупного количества легкоразветвленных парафинов в части потока экстракта, которую подают в зону дегидрирования данного способа. Легкоразветвленные парафины, имеющие одно разветвление либо в виде алкильной группы, либо, что более желательно, в виде метильной группы, предпочтительно составляют более 85% (моль.) от совокупного количества легкоразветвленных парафинов в части потока экстракта, запускаемого в зону дегидрирования. В случае присутствия в потоке экстракта вместе с легкоразветвленными парафинами и линейных парафинов их содержание не должно быть большим 75% (моль.) от совокупного количества парафинов в той части потока экстракта, которую пропускают в зону дегидрирования. Молекулы парафинов, содержащие, по меньшей мере, один четвертичный атом углерода, в общем случае составляют менее 10% (моль.), предпочтительно менее 5% (моль.), более предпочтительно менее 2% (моль.), а наиболее предпочтительно менее 1% (моль.) от той части потока экстракта, которую подают в зону дегидрирования.
Конфигурацию секции дегидрирования можно создать по существу такой, как изображенная на чертеже. Говоря кратко, поток, содержащий парафины, объединяют с рециркулирующим водородом и формируют поток реагентов для дегидрирования, который нагревают и вводят в контакт с катализатором дегидрирования в неподвижном слое, выдерживаемом в условиях прохождения дегидрирования. Поток продуктов, идущий от неподвижного слоя катализатора, который в настоящем документе называют потоком продуктов из реактора дегидрирования, охлаждают, частично конденсируют и пропускают в парожидкостной сепаратор. Парожидкостной сепаратор позволяет получить обогащенную водородом фазу пара и обогащенную углеводородом фазу жидкости. Конденсированную жидкую фазу, извлеченную из сепаратора, подают в колонну для отгонки легких фракций, в которой удаляют все соединения, которые являются более летучими по сравнению с самым легким углеводородом, который было бы желательно подать в секцию алкилирования. Результирующий проток, содержащий олефин, который подают из секции дегидрирования в секцию алкилирования данного способа, в настоящем документе называют потоком продуктов дегидрирования.
Данное изобретение не ограничивается какой-либо одной конкретной схемой потоков для секции дегидрирования, которая может включать катализатор дегидрирования в подвижном, либо неподвижном слое, содержащие катализатор зоны реакции с теплообменниками между ними и введение горячих газовых потоков, обогащенных водородом. Углеводороды можно вводить в контакт с любым слоем катализатора при подаче потока снизу вверх, сверху вниз, либо в радиальном направлении.
Катализаторы дегидрирования хорошо известны на предшествующем уровне техники, и их примеры приводятся в работах US-A-3,274,287; US-A-3,315,007; US-A-3,315,008; US-A-3,745,112; US-A-4,430,517; US-A-4,716,143; US-A-4,762,960; US-A-4,786,625; и US-A-4,827,072. Представляется, что выбор конкретного катализатора дегидрирования для успеха данного изобретения критическим моментом не является. Предпочтительный катализатор представляет собой слоистую композицию с внутренним ядром, связанным с наружным слоем, включающим тугоплавкий неорганический оксид, содержащий, по меньшей мере, один однородно диспергированный металл платиновой группы (группа VIII (IUPAC 8-10)) и, по меньшей мере, один металл промотора, и где в композиции катализатора диспергируют, по меньшей мере, один металл модификатора. Предпочтительно наружный слой связан с внутренним ядром в такой степени, что потери при истирании будут меньше 10% (масс.) в расчете на массу наружного слоя.
Условия дегидрирования выбирают такими, чтобы свести к минимуму прохождение крекинга и образование полиолефиновых побочных продуктов. Обычные условия проведения дегидрирования в результате не будут приводить к какой-либо ощутимой изомеризации углеводородов в реакторе дегидрирования. Углеводород может вступать в контакт с катализатором, находясь в жидкой фазе, смешанной парожидкостной фазе, либо предпочтительно в паровой фазе. Условия проведения дегидрирования включают температуру, в общем случае находящуюся в диапазоне от 400°С (752°F) до 900°С (1652°F), а предпочтительно от 400°С (752°F) до 525°С (977°F), давление, в общем случае находящееся в диапазоне от 1 кПа избыточного давления (0,15 фунт/дюйм2 избыточного давления) до 1013 кПа избыточного давления (147 фунт/дюйм2 избыточного давления), и величину LHSV, находящуюся в диапазоне от 0,1 до 100 час-1. В соответствии с тем, как это используется в настоящем документе, аббревиатура «LHSV» обозначает часовую объемную скорость жидкости, которую определяют как объемный расход жидкости за один час, поделенный на объем катализатора. В общем случае для нормальных парафинов чем меньше будет молекулярная масса, тем выше будет температура, необходимая для сопоставимой степени превращения. В зоне дегидрирования выдерживают давление, настолько низкое, насколько это будет практически возможно, обычно меньшее 345 кПа избыточного давления (50 фунт/дюйм2 избыточного давления), соответствующее ограничениям, существующим для оборудования, для того, чтобы довести до максимума преимущества химического равновесия.
К потоку экстракта можно примешать вещество разбавителя до, во время, либо после подачи в зону дегидрирования. Веществом разбавителя может быть водород, водяной пар, метан, этан, диоксид углерода, азот, аргон и тому подобное либо их смесь. Водород представляет собой предпочтительный разбавитель. Обычно, когда в качестве разбавителя используют водород, то его используют в количествах, достаточных для поддержания мольного соотношения водорода и углеводорода в диапазоне от 0,1:1 до 40:1.
В зону дегидрирования можно непрерывно, либо периодически добавлять воду, либо вещество, разложимое с образованием воды в условиях прохождения дегидрирования, такое, как спирт, альдегид, простой эфир, либо кетон, в количестве, рассчитанном по эквивалентной воде в диапазоне от 1 до 20000 ч./млн (масс.) для потока экстракта. Диапазон добавления воды от 1 до 10000 ч./млн (масс.) приводит к получению наилучших результатов при дегидрировании парафинов, имеющих от 2 до 30, или более атомов углерода.
Поток продуктов дегидрирования обычно представляет собой смесь непрореагировавших парафинов, линейных (неразветвленных) олефинов и разветвленных моноолефинов с включением легкоразветвленных моноолефинов. Обычно от 0 до 75% (моль.), а предпочтительно от 0 до 50% (моль.) олефинов в моноолефинсодержащем потоке, покидающем технологический процесс дегидрирования парафинов, представляют собой линейные (неразветвленные) олефины. Продукт дегидрирования также может содержать моноолефины, характеризующиеся совокупным количеством атомов углерода в диапазоне от 8 до 28, при этом предпочтительно менее 10% (моль.), а предпочтительно менее 1% (моль.) моноолефинов содержат четвертичные атомы углерода.
Поток продуктов дегидрирования может содержать высокоразветвленный моноолефин, либо линейный (неразветвленных) олефин, но предпочтительно легкоразветвленный моноолефин. Термин «легкоразветвленный моноолефин» в соответствии с тем, как это используется в настоящем документе, обозначает моноолефин с совокупным количеством атомов углерода в диапазоне от 8 до 28, из которых три, либо четыре атома углерода являются первичными атомами углерода, и ни один из остальных атомов углерода не является четвертичным атомом углерода. Предпочтительно легкоразветвленный моноолефин характеризуется совокупным количеством атомов углерода в диапазоне от 8 до 15, а более предпочтительно от 10 до 15 атомов углерода.
Легкоразветвленный моноолефин в общем случае включает алифатический алкен, описываемый общей формулой (рi-алкилi)i-q-алкен. Легкоразветвленный моноолефин может быть альфа-моноолефином, либо винилиденовым моноолефином, но обычно им является внутренний моноолефин. В соответствии с тем, как это используется в настоящем документе, термин «альфа-олефины» обозначает олефины, описываемые химической формулой R-CH=CH2. Термин «внутренние олефины» в соответствии с тем, как это используется в настоящем документе, включает бизамещенные внутренние олефины, описываемые химической формулой R-CH=CH-R; тризамещенные внутренние олефины, описываемые химической формулой R-C(R)=CH-R; и тетразамещенные олефины, описываемые химической формулой R-C(R)=C(R)-R. Бизамещенные внутренние олефины включают бета-внутренние олефины, описываемые химической формулой R-СН=СН-СН3. В соответствии с тем, как это используется в настоящем документе, термин «винилиденовые олефины» обозначает олефины, описываемые химической формулой R-C(R)=CH2. Подходящие легкоразветвленные моноолефины включают октены, нонены, децены, ундецены, додецены, тридецены, тетрадецены, пентадецены, гексадецены, гептадецены, октадецены, нонадецены, эйкозены, генэйкозены, докозены, трикозены, тетракозены, пентакозены, гексакозены, гептакозены и октакозены.
В случае легкоразветвленных моноолефинов разветвление, либо разветвления в виде алкильных групп у легкоразветвленного моноолефина в общем случае выбирают из метильной, этильной и пропильной групп, при этом предпочтительны более короткие и нормальные разветвления. У всех вариантов легкоразветвленных моноолефинов, перепускаемых в секцию алкилирования, легкоразветвленный моноолефин предпочтительно имеет только одно разветвление в виде алкильной группы, но также возможны и два разветвления в виде алкильных групп. Легкоразветвленные моноолефины, имеющие либо два разветвления в виде алкильных групп, либо четыре первичных атома углерода, в общем случае составляют менее 30% (моль.), а предпочтительно менее 15% (моль.) от совокупного количества легкоразветвленных моноолефинов, перепускаемых в секцию алкилирования, при этом остальные легкоразветвленные моноолефины, перепускаемые в секцию алкилирования, имеют одно разветвление в виде алкильной группы. Моноолефины, имеющие либо два разветвления в виде алкильных групп, либо четыре первичных атома углерода и четвертичный атом углерода, в общем случае составляют менее 10% (моль.), а предпочтительно менее 1% (моль.) от совокупного количества легкоразветвленных моноолефинов, перепускаемых в секцию алкилирования. Легкоразветвленные моноолефины, имеющие либо одно разветвление в виде алкильной группы, либо три первичных атома углерода, предпочтительно составляют более 85% (моль.) от совокупного количества легкоразветвленных моноолефинов, перепускаемых в секцию алкилирования. Легкоразветвленные моноолефины, имеющие только одно разветвление в виде алкильной группы, и у которых единственное разветвление в виде алкильной группы является метильной группой, в настоящем документе называются монометилалкенами, и они представляют собой предпочтительный компонент потока продуктов дегидрирования.
Несмотря на то что в потоке продуктов дегидрирования могут присутствовать и винилиденовые моноолефины, обычно они являются второстепенным компонентом и имеют концентрацию, обычно меньшую 0,5% (моль.), а более часто меньшую 0,1% (моль.) от количества олефинов в потоке продуктов дегидрирования.
Структуры скелетов моноолефинов в смеси, содержащей легкоразветвленные моноолефины, можно определить, используя аналитические методы, которые хорошо известны специалисту в области газовой хроматографии, и которые не требуется описывать подробно в настоящем документе. Специалист в соответствующей области может модифицировать аппарат и метод из ранее упомянутой статьи авторов Schulz et al., введя в оснащение инжектор с трубой вставки в гидрогенизатор для того, чтобы производить гидрирование легкоразветвленных моноолефинов с получением легкоразветвленных парафинов в инжекторе. После этого легкоразветвленные парафины отделяют и идентифицируют по существу при использовании аппарата и метода, описанных в статье авторов Schulz et al. Однако данные аппарат и метод не позволяют определить местоположение двойной связи углерод-углерод в любом из моноолефинов в смеси.
В дополнение к легкоразветвленному моноолефину посредством потока продуктов дегидрирования в секцию алкилирования можно запускать и другие ациклические соединения. Одно из преимуществ данного изобретения заключается в том, что поток, содержащий легкоразветвленные моноолефины, можно перепускать непосредственно в секцию проведения реакции алкилирования несмотря на то, что данный поток также содержит и ациклические парафины, имеющие то же самое количество атомов углерода, что и легкоразветвленные моноолефины. Таким образом, данное изобретение устраняет необходимость отделения парафинов от моноолефинов перед их перепусканием в секцию алкилирования. Другие ациклические соединения включают неразветвленные (линейные) олефины и моноолефины. Неразветвленные (линейные) олефины, которые можно загружать на переработку по способу данного изобретения, характеризуются совокупным количеством атомов углерода, приходящимся на одну молекулу парафина, в общем случае в диапазоне от 8 до 28, предпочтительно от 8 до 15, а более предпочтительно от 10 до 13 атомов углерода. Неразветвленным олефином может быть альфа-моноолефин, но предпочтительно им является внутренний моноолефин. В случае присутствия в потоке продуктов дегидрирования вместе с легкоразветвленными моноолефинами и линейных олефинов их содержание не должно быть большим 75% (моль.) от совокупного количества моноолефинов в потоке продуктов дегидрирования, но в общем случае оно меньше 60% (моль.) от совокупного количества моноолефинов в потоке продуктов дегидрирования.
Вследствие возможного присутствия в потоке продуктов дегидрирования в дополнение к легкоразветвленным моноолефинам и линейных моноолефинов суммарный поток продуктов дегидрирования может в среднем иметь менее 3, либо от 3 до 3,4 первичных атомов углерода, приходящихся на одну молекулу моноолефина в потоке продуктов дегидрирования. В зависимости от относительных соотношений количеств линейных и легкоразветвленных моноолефинов поток продуктов дегидрирования, либо сумма всех моноолефинов, которые перепускают в зону алкилирования, может иметь от 2,25 до 3,4 первичных атомов углерода, приходящихся на одну молекулу моноолефина.
Линейные и/или нелинейные парафины, которые подают в секцию алкилирования посредством потока продуктов дегидрирования, характеризуются совокупным количеством атомов углерода, приходящимся на одну молекулу парафина, в общем случае находящимся в диапазоне от 8 до 28, предпочтительно от 8 до 15, а более предпочтительно от 10 до 13 атомов углерода. Нелинейные парафины в потоке продуктов дегидрирования могут включать легкоразветвленные парафины и также могут включать парафины, имеющие, по меньшей мере, один четвертичный атом углерода. Такие линейные и нелинейные парафины, как представляется, выступают в роли разбавителя на стадии алкилирования и по существу не оказывают влияния на стадию алкилирования. Молекулы моноолефинов, содержащие, по меньшей мере, один четвертичный атом углерода, в общем случае составляют менее 10% (моль.), предпочтительно менее 5% (моль.), более предпочтительно менее 2% (моль.), а наиболее предпочтительно менее 1% (моль.) от потока продуктов дегидрирования, либо от суммы всех моноолефинов, которые подают в зону алкилирования.
В секции алкилирования моноолефины из потока продуктов дегидрирования вступают в реакцию с фенильным соединением. В общем случае моноолефины могут вступать в реакцию с бензолом, либо с замещенными производными бензола, в том числе с толуолом и этилбензолом. В случае алкилирования с получением моющего средства предпочтительным фенильным соединением является бензол. Реакцию фенильного соединения и моноолефинов обычно проводят в присутствии твердого катализатора алкилирования.
Как представляется, в секции алкилирования происходит только минимальная изомеризация скелета олефинов. Минимальная изомеризация скелета моноолефинов обозначает то, что в общем случае менее 25 моль.%, а предпочтительно менее 10 моль.% олефина, алифатической алкильной цепи и любого промежуточного продукта в реакции претерпевают изомеризацию скелета. Таким образом, степень легкого разветвления у легкоразветвленного моноолефина идентична степени легкого разветвления алифатической алкильной цепи в молекуле продукта фенилалкана, а количество первичных атомов углерода также по существу остается тем же самым. Наконец, несмотря на то, что образование продукта 1-фенилалкана в условиях прохождения алкилирования незначительно, количество первичных атомов углерода в продукте фенилалкане будет несколько меньшим в сравнении с количеством первичных атомов углерода в легкоразветвленном моноолефине.
Алкилирование фенильного соединения под действием легкоразветвленных моноолефинов приводит к получению (mi-алкилi)i-n-фенилалканов, где алифатическая алкильная группа содержит два, три, либо четыре первичных атома углерода, приходящихся на одну молекулу фенилалкана. Предпочтительно алифатическая алкильная группа содержит три первичных атома углерода, приходящихся на одну молекулу фенилалкана, а более предпочтительно один из трех первичных атомов углерода присутствует в метильной группе на одном конце алифатической алкильной цепи, второй первичный атом углерода присутствует в метильной группе на другом конце цепи, а третий первичный атом углерода присутствует в единственном разветвлении в виде метильной группы, присоединенном к цепи. В общем случае от 0% (моль.) до 75% (моль.), а предпочтительно от 0% (моль.) до 50% (моль.) полученных (mi-алкилi)i-n-фенилалканов могут иметь 2 первичных атома углерода, приходящихся на одну молекулу фенилалкана. Обычно от 25 моль.% до 100 моль.% полученных (mi-алкилi)i-n-фенилалканов могут иметь 3 первичных атома углерода, приходящихся на одну молекулу фенилалкана. В общем случае от 0 моль.% до 40 моль.% полученных (mi-алкилi)i-n-фенилалканов могут иметь 4 первичных атома углерода. Таким образом, предпочтительны (m-метил)-n-фенилалканы, имеющие только одно разветвление в виде метильной группы, и они в настоящем документе называются монометилфенилалканами. Предполагается, что количество первичных, вторичных и третичных атомов углерода, приходящихся на одну молекулу продукта фенилалкана, можно определить при помощи редактирования и улучшения отношения сигнал-шум без искажений под действием переноса поляризации (DEPT), проведенных для спектра многоимпульсного ядерного магнитного резонанса (ЯМР) высокого разрешения, что описывается в брошюре, озаглавленной "High Resolution Multipulse NMR Spectrum Editing and DEPT", которая распространяется с выходными данными Bruker Instruments, Inc., Manning Park, Billerica, Massachusetts, USA, и которая включается в настоящий документ для справки.
Алкилирование фенильного соединения под действием моноолефинов характеризуется селективностью по получению 2-фенилалканов, в общем случае находящейся в диапазоне от 40 до 100, а предпочтительно от 60 до 100, и селективностью по получению внутреннего четвертичного фенилалкана, в общем случае меньшей 10, а предпочтительно меньшей 5.
Алкилирование фенильного соединения под действием моноолефинов характеризуется селективностью по получению фенилалканов, содержащих четвертичные атомы углерода в «не четвертичных соединениях», меньшей 10, а предпочтительно меньшей 1. Подходящую аппроксимацию для оценки селективности по получению таких четвертичных фенилалканов можно провести при использовании следующей формулы:
Т=100(СQO/СO),
где Т = селективность по получению четвертичных атомов углерода в «не являющихся четвертичными соединениях»,
CQO = число молей моноолефинов, имеющих четвертичный атом углерода, поступающих в зону селективного алкилирования,
СO = число молей моноолефинов, поступающих в зону селективного алкилирования.
Значения CQO и СO можно определить при использовании мольных расходов моноолефинов, поступающих в зону селективного алкилирования, и ранее упомянутых модифицированного аппарата и метода авторов Schulz et al. Селективность Т можно оценить при использовании данной формулы, если каждый моноолефин, поступающий в зону селективного алкилирования, будет характеризоваться одинаковой вероятностью алкилирования фенильного соединения вне зависимости от того, будет ли моноолефин иметь четвертичный атом углерода. В качестве первого приближения данное условие удовлетворяется в случае, когда более 40% (масс.) моноолефинов, поступающих в зону селективного алкилирования, являются легкоразветвленными моноолефинами, либо нормальными моноолефинами.
Алкилирование фенильного соединения под действием моноолефинов можно провести в варианте периодического способа, но предпочтителен непрерывный способ. Катализатор алкилирования можно использовать в виде уплотненного слоя, либо псевдоожиженного слоя в режиме восходящего потока, нисходящего потока, либо горизонтального потока. Бензол и поток продуктов дегидрирования, содержащий легкоразветвленные моноолефины, который поступает в зону алкилирования, обычно характеризуются совокупным мольным соотношением фенильное соединение: моноолефин в диапазоне от 2,5:1 до 50:1, а более часто от 8:1 до 35:1. Части потока продуктов дегидрирования можно подавать в несколько раздельных точек в пределах зоны реакции алкилирования, и в каждой зоне мольное соотношение фенильное соединение: моноолефин будет превышать 50:1. Однако полное соотношение количеств бензол:олефин, используемое в описанном выше варианте данного изобретения, будет оставаться в пределах указанного диапазона. Совокупное объединенное исходное сырье перепускают через уплотненный слой при часовой объемной скорости жидкости (LHSV) в диапазоне от 0,3 до 6 час-1. Условия проведения алкилирования включают температуру в диапазоне от 80°С (176°F) до 225°С (437°F). Предпочтительно алкилирование, по меньшей мере, частично протекает в жидкой фазе, а предпочтительно либо в полностью жидкой фазе, либо в сверхкритических условиях. Требуемое давление обязательным образом зависит от олефина, фенильного соединения и температуры, но обычно оно находится в диапазоне 1379-6895 кПа избыточного давления (200-1000 фунт/дюйм2 избыточного давления), а наиболее часто 2069-3448 кПа избыточного давления (300-500 фунт/дюйм2 избыточного давления). После прохождения реакции алкилирования не вступившего в реакцию олефина обычно остается немного, и реакция обычно идет со степенью превращения, по меньшей мере, равной 98% в расчете на моноолефин.
Может быть использован любой катализатор алкилирования, который удовлетворяет требованиям к степени превращения, селективности и активности. Предпочтительные катализаторы алкилирования включают цеолиты со структурами цеолитов ВЕА, MOR, MTW, либо NES. Такие цеолиты включают морденит, ZSM-4, ZSM-12, ZSM-20, оффретит, гмелинит, бета, NU-87 и готтардиит. Данные типы структур цеолитов, термин «тип структуры цеолита» и термин «изотипическая структура каркаса» используются в настоящем документе так, как они были определены и использовались в работе Atlas of Zeolite Structure Types, by W.M.Meier, et al., опубликованной от имени Комиссии по структуре международной ассоциации по цеолитам с выходными данными Elsevier, Boston, Massachusetts, USA, Fourth Revised Edition, 1996. Алкилирование с использованием NU-87 и NU-85, которые представляют собой продукты срастания цеолитов EU-1 и NU-87, описывается в работах US-A-5,041,402 и US-A-5,446,234 соответственно. Готтардиит, который имеет изотопическую структуру каркаса для типа структуры цеолита NES, описывается в статьях A.Alberti et al., Eur. J. Mineral., 8, 69-75 (1996) и Е.Galli et al., Eur. J. Mineral., 8, 687-693 (1996). Наиболее предпочтительно катализатор алкилирования включает морденит.
У цеолитов, пригодных для использования в катализаторе алкилирования в настоящем изобретении, в общем случае, по меньшей мере, 10 процентов их катионных центров занято ионами, отличными от ионов щелочных, либо щелочноземельных металлов. Такие другие ионы включают ионы алюминия, цинка, меди, алюминия, а предпочтительно аммония, водорода, редких земель, либо их комбинации. В предпочтительном варианте реализации цеолиты превращают в преимущественно водородную форму в общем случае в результате замещения первоначально присутствующих в них ионов на прекурсоры водородных ионов, например, ионы аммония, что после прокаливания приводит к получению водородной формы. Данный обмен удобно проводить в результате введения цеолита в контакт с раствором аммониевой соли, например, хлорида аммония, при использовании хорошо известных методик ионного обмена. В определенных вариантах реализации замещение приводит к получению материала цеолита, в котором, по меньшей мере, 50 процентов катионных центров будут заняты ионами водорода. Несмотря на то, что водородная форма цеолита роль катализатора в реакции выполняет успешно, цеолит частично также может присутствовать и в форме, содержащей щелочной металл.
Цеолиты можно подвергать различным типам химической переработки, включая удаление алюминия (деалюминирование) и комбинирование с одним, либо несколькими компонентами, содержащими металл, такой, как металлы группы IIIB (IUPAC 3), IVB (IUPAC 4), VIB (IUPAC 6), VIIB (IUPAC 7), VIII (IUPAC 8-10) и IIB (IUPAC 12). Цеолиты в некоторых случаях можно подвергать тепловой обработке, в том числе обработке паром, либо прокаливанию на воздухе, в среде водорода, либо инертного газа, например, азота, или гелия. Подходящая обработка водяным паром включает введение цеолита в контакт с атмосферой, содержащей от 5 до 100% пара, при температуре в диапазоне от 250°С (482°F) до 1000°С (1832°F) в течение промежутка времени в диапазоне от 0,25 до 100 часов при давлении в диапазоне от субатмосферного до нескольких сотен атмосфер.
Цеолит могут включать материал матрицы, либо связующее, которые обладают стойкостью к воздействию температуры и других условий, используемых в способе. Подходящие материалы матрицы включают синтетические вещества, вещества, встречающиеся в природе, и неорганические материалы, такие, как глина, диоксид кремния и/или оксиды металлов. Встречающиеся в природе глины, подходящие для образования композиции с цеолитом, включают те, что относятся к семействам монтмориллонита и каолина. В дополнение к упомянутым выше материалам, с цеолитом, используемым в данном изобретении, композицию может составлять материал пористой матрицы, такой, как оксид алюминия, диоксид кремния-оксид алюминия, диоксид кремния-оксид магния, диоксид кремния-оксид циркония, диоксид кремния-оксид тория, диоксид кремния-оксид бериллия, диоксид кремния-оксид титана и фосфат алюминия, а также тройные комбинации, такие, как диоксид кремния-оксид алюминия-оксид тория, диоксид кремния-оксид алюминия-оксид циркония, диоксид кремния-оксид алюминия-оксид магния и диоксид кремния-оксид магния-оксид циркония. Относительные доли компонентов материала матрицы могут варьироваться в широких пределах, при этом массовое содержание цеолита находится в диапазоне от 1% до 99%, обычно от 5% до 80 масс.%, а предпочтительно от 30% до 80% в расчете на совокупную массу цеолита и материала матрицы.
Цеолиты в катализаторе алкилирования в общем случае характеризуются мольным соотношением диоксид кремния:оксид алюминия для каркаса в диапазоне от 5:1 до 100:1. Морденит, пригодный для использования при алкилировании, характеризуется мольным соотношением диоксид кремния:оксид алюминия для каркаса в общем случае в диапазоне от 12:1 до 90:1, а предпочтительно от 12:1 до 25:1. Термин «мольное соотношение диоксид кремния: оксид алюминия для каркаса» обозначает мольное соотношение SiO2 и Al2О3 в каркасе цеолита.
Цеолиты, полученные в присутствии органических катионов, могут оказаться недостаточно каталитически активными для алкилирования. Как полагают, недостаточная каталитическая активность является результатом присутствия органических катионов из образующего материал раствора, которые занимают свободный объем внутри кристалла. Активировать такие катализаторы могут нагревание в инертной атмосфере при 540°С (1004°F) в течение одного часа, проведение ионного обмена под действием аммониевых солей и прокаливание при 540°С (1004°F) на воздухе. Температуры прокаливания, превышающие 540°С (1004°F), могут привести к разложению аммиака, присутствующего в катализаторе.
В зоне реакции алкилирования получают продукт реакции алкилирования, который поступает на установку разделения для извлечения продуктов и отправляемых на повторное использование соединений исходного сырья. Продукт реакции алкилирования подают в колонну для отгонки бензола для получения потока дистиллята, содержащего бензол, предназначенного для отправки на повторное использование в зоне реакции алкилирования, и потока кубового остатка, содержащего продукт фенилалкан. Данный поток кубового остатка подают в колонну для отгонки парафинов для получения потока дистиллята, содержащего не прореагировавшие парафины, и потока кубового остатка, содержащего продукт фенилалканы и любые более высокомолекулярные углеводородные побочные продукты, образованные в зоне реакции алкилирования. Поток кубового остатка из колонны для отгонки парафинов можно подавать в колонну для вторичной перегонки для получения потока дистиллята в виде продукта фенилалкана, содержащего МАВ, и потока кубового остатка колонны для вторичной перегонки, содержащего заполимеризованные олефины и полиалкилированные бензолы (тяжелый алкилат). В альтернативном варианте достаточно низкий уровень содержания тяжелого алкилата в потоке кубового остатка из колонны для отгонки парафинов делает ненужной колонну для вторичной перегонки, и поток кубового остатка из колонны для отгонки парафинов можно извлекать в виде чистого потока МАВ, который впоследствии можно подвергать сульфированию с получением MABS.
Возможны несколько вариантов способа настоящего изобретения. Селективное гидрирование может приводить к насыщению диолефинов, присутствующих в потоке продуктов дегидрирования, с получением желательных моноолефинов. В способе можно селективно удалять вредные ароматические побочные продукты, содержащиеся в потоке продуктов дегидрирования, которые могут образоваться в ходе каталитического дегидрирования парафинов, и которые могут вызвать деактивирование катализатора в секции алкилирования, уменьшение селективности по получению желательных фенилалканов и накопление неприемлемых концентраций. В зоне селективного удаления можно извлекать ароматические побочные продукты из потока экстракта, потока продуктов дегидрирования, потока жидкости дистиллята из колонны для отгонки парафинов, потока из зоны дегидрирования, либо, где это будет уместно, потока продуктов селективного гидрирования диолефинов.
Данный способ может привести к получению предпочтительной композиции МАВ в результате адсорбционного разделения парафинов со средней массой в диапазоне от массы С10 парафина до массы C13 парафина с получением парафинов экстракта, характеризующихся средним уровнем разветвления в диапазоне от 0,25 до 1,3, либо от 0,4 до 1,3 разветвления в виде алкильной группы, приходящегося на одну молекулу парафина. Данные парафины экстракта, в первую очередь, образованы линейными парафинами и моноразветвленными парафинами, а разветвления в виде алкильных групп на алифатической алкильной цепи парафинов экстракта, в первую очередь, образованы небольшими заместителями, такими, как разветвления в виде метильных групп, разветвления в виде этильных групп, либо разветвления в виде пропильных групп. Парафины экстракта подвергают дегидрированию с получением соответствующих моноолефинов, которые алкилируют фенильное соединение с получением фенилалканов. Получающиеся в результате фенилалканы имеют такие характеристики, что фенилалканы, у которых фенильная группа присоединена ко 2-му и/или 3-му положению алифатической алкильной группы, составляют более 55% (масс.) от количества фенилалканов, а фенилалканы, имеющие, по меньшей мере, один четвертичный атом углерода в алифатической алкильной группе, составляют менее 20% от количества фенилалканов.
Сульфирование фенилалканов, полученных по способам данного изобретения, можно провести в результате введения соединений фенилалканов в контакт с любой из хорошо известных систем сульфирования, в том числе с теми, что описываются в работе Detergent Manufacture Including Zeolite Builders and Other New Materials, by Marshall Sittig, Noyes Data Corporation, Park Ridge, New Jersey, 1979 и в работе Volume 56 of "Surfactant Science" series, Marcel Dekker, Inc., New York, NY, 1996. Сульфирование соединений фенилалканов приводит к получению продукта сульфирования, содержащего фенилалкансульфоновые кислоты. После сульфирования нейтрализация продукта сульфирования под действием любой подходящей щелочи, такой, как щелочи натрия, калия, аммония, магния, кальция и замещенного аммония и их смеси, приводит к получению продукта нейтрализации, содержащего фенилалкансульфонаты.
В еще одном аспекте настоящего изобретения данное изобретение представляет собой использование композиций МАВ, полученных по способам, описанным в настоящем документе, в качестве смазочных материалов. Представляется, что данные фенилалканы характеризуются вязкостными свойствами, температурной зависимостью вязкости и плотностью, которые делают их выгодными для использования в качестве нефтяных смазочных материалов. Использование фенилалканов в качестве смазочных материалов описывается, например, в статье Е.R.Booser in Kirk-Othmer Encyclopedia of Chemical Technology. Fourth Edition, Volume 15, John Wiley and Sons, New York, New York, USA, 1995, pp.463-517, ссылка на которую делается в том, что касается описания таких смазочных материалов и их использования.
В еще одном аспекте данное изобретение представляет собой использование композиций MABS, полученных по способам, описанным в настоящем документе, в качестве присадок к смазочным материалам. Представляется, что фенилалкансульфонаты в виде либо нормальных солей, либо основных солей фенилалкансульфоновых кислот, полученных в соответствии с тем, что описывается в настоящем документе, обладают способностью уменьшать, либо предотвращать образование отложений в двигателях, функционирующих при высоких температурах. Термин «нормальная соль» кислоты обозначает соль, которая содержит стехиометрическое количество металла, необходимое для нейтрализации присутствующих кислотных группы, либо групп, а термин «основная соль» обозначает соль, которая содержит больше металла, чем это требуется для реакции нейтрализации. Избыточный металл в виде основных солей, как представляется, способен вызывать нейтрализацию продуктов сгорания, образующихся вследствие окисления масла, и продуктов сгорания топлива при «пропуске газа через поршневые кольца». Фенилалкансульфонаты и их использование в качестве присадок к смазочным материалам, в частности в качестве присадок, предотвращающих образование осадка, описывается, например, в упомянутой выше статье автора Booser; в работе Lubricant Additives, by С.V.Smalheer and R.К.Smith, The Lezius-Hiles Co., Cleveland, Ohio, USA, 1967, pp.2-3; и в статье авторов R.W.Watson and Т.F.McDonnell, Jr., озаглавленной "Additives - The Right Stuff for Automotive Engine Oils", in Fuels and Lubricants Technology: An Overview SP-603, Society of Automotive Engineers, Warrendale, Pennsylvania, USA, October 1984, pp.17-28.
Чертеж демонстрирует предпочтительную компоновку для интегрированной схемы разделения-дегидрирования-алкилирования данного изобретения. Следующее далее описание чертежа не предполагает исключения других компоновок для схемы технологического процесса данного изобретения, и оно не подразумевает ограничения данного изобретения, предложенного в формуле изобретения.
Если обратиться теперь к чертежу, то можно сказать, что по линии 12 производят подачу исходной смеси, содержащей смесь С10-С13, в том числе легкоразветвленные парафины, более высокоразветвленные парафины и нормальные (неразветвленные) парафины, в зону адсорбционного разделения 20, где в качестве десорбента используют нормальный парафин и/или циклопарафин и/или изооктан. По линии 20 подают поток рафината, содержащий более высокоразветвленные парафины и циклопарафин, либо изооктан, из зоны адсорбционного разделения 20 в колонну рафината 30. Условия в колонне рафината 30 позволяют получить поток дистиллята 32, содержащий вещество десорбента, и поток кубового остатка, содержащий более высокоразветвленные парафины, подаваемый в линию 28. По линии 34 пропускают поток экстракта, содержащий легкоразветвленные парафины, нормальные парафины и вещества десорбента, из зоны адсорбционного разделения 20 в колонну экстракта 40. В колонне экстракта 40 извлекают вещества десорбента с подачей в линию 46. Объединенные потоки дистиллятов в линиях 32 и 46 отправляют десорбент на повторное использование через линию 44. Колонна экстракта 40 также позволяет получить в линии 52 поток кубового остатка, содержащий легкоразветвленные парафины и нормальные парафины.
В линии 52 производят смешивание потока кубового остатка из колонны экстракта 40 с отправленным на повторное использование водородом из линии 82 с получением смеси парафинов и водорода, которую пропускают через линию 56. В непрямом теплообменнике 58 нагревают содержимое линии 56, которое пропускают через линию 62 в огневой подогреватель 60. По линии 64 подогретый поток транспортируют до реактора дегидрирования 70. В реакторе дегидрирования 70 парафины вводят в контакт с катализатором дегидрирования в условиях, которые обеспечивают достижение значительной степени превращения парафинов в соответствующие олефины. В линиях 66 и 68 поток продуктов реактора дегидрирования, содержащий смесь водорода, парафинов, моноолефинов, в том числе легкоразветвленных моноолефинов, диолефинов, С9-минус углеводородов и ароматических углеводородов, охлаждают в результате последовательного перепускания через теплообменники 58 и 72. Данное охлаждение приводит к конденсации по существу всех С4-плюс углеводородов с получением потока жидкой фазы и к отделению потока жидкой фазы от остающегося пара, обогащенного водородом. По линии 74 перепускают продукт реактора дегидрирования в емкость парожидкостного разделения 80, в которой продукт разделяют на поток паровой фазы, обогащенной водородом, удаляемый через линию 76, и поток продуктов дегидрирования, удаляемый через линию 84. По линии 76 производят подачу водорода, отправленного на повторное использование, в линию 82, а в линии 78 извлекают поток продукта в виде чистого водорода.
По линии 84 подают среду, отбираемую в нижней части емкости разделения 80, содержащую нормальные парафины, легкоразветвленные парафины, нормальные моноолефины, легкоразветвленные моноолефины, С9-минус углеводороды, диолефины, ароматические побочные продукты и некоторое количество растворенного водорода, в реактор селективного гидрирования 86. В реакторе селективного гидрирования 86 поток продуктов дегидрирования водят в контакт с катализатором селективного гидрирования в условиях, в которых происходит превращение значительного количества диолефинов в соответствующие моноолефины, и для прохождения реакции в нем используют водород, растворенный в потоке продуктов дегидрирования, и/или дополнительное восполняющее количество водорода (не показано). По линии 88 транспортируют поток продуктов реактора селективного гидрирования, содержащий смесь водорода, нормальных парафинов, легкоразветвленных парафинов, нормальных моноолефинов, легкоразветвленных моноолефинов, С9-минус углеводородов и ароматических углеводородных побочных продуктов, в колонну для отгонки легких фракций 90. В колонне для отгонки легких фракций 90 в линию 94 извлекают совокупный поток дистиллята, который содержит С9-минус углеводороды, полученные в реакторе дегидрирования в качестве побочных продуктов, и любые оставшиеся количества растворенного водорода, их отделяют и концентрируют в виде совокупного потока дистиллята, удаляемого из технологического процесса через линию 94.
По линии 96 подают оставшиеся С10-плюс углеводороды из колонны отгонки легких фракций 90 в зону удаления ароматики 100, где данный продукт вводят в контакт с адсорбентом для удаления ароматических побочных продуктов. По линии 98 из зоны удаления ароматики 100 транспортируют продукт, содержащий смесь нормальных парафинов, легкоразветвленных парафинов, нормальных моноолефинов и легкоразветвленных моноолефинов, и где данный продукт характеризуется значительно уменьшенной концентрацией ароматических побочных продуктов в сравнении с потоком продуктов после отгонки легких фракций. После добавления бензола через линию 112 по линии 102 пропускают смесь в реактор алкилирования 104, в котором смесь вводят в контакт с катализатором алкилирования в условиях, способствующих протеканию алкилирования с получением фенилалканов.
По линии 106 транспортируют поток продуктов реактора алкилирования в колонну фракционной перегонки с отгонкой бензола 110. Данный поток содержит смесь бензола, нормальных парафинов, легкоразветвленных парафинов и МАВ. МАВ характеризуются как фенилалканы, которые в дополнение к фенильной части имеют одну алифатическую алкильную часть, имеющую либо 1, либо 2 первичных атома углерода, или же 2, 3 либо 4 первичных атома углерода и не имеющую четвертичных атомов углерода за исключением «четвертичных соединений». В колонне 110 разделяют продукт на поток кубового остатка и поток дистиллята, содержащий бензол и возможно легкие газы. В линии 107 объединяют поток дистиллята линии 107 с восполняющим количеством бензола из линии 109 с подачей в линию 108, по которой объединенный поток подают в барабан-сепаратор 120. По линии 114 из барабана 120 удаляют несконденсированные легкие газы, в то время как по линии 116 производят подачу сконденсированной жидкости в качестве флегмы в колонну 110 через линию 118 и подачу бензола для отправки на повторное использование по линии 112. По линии 122 транспортируют оставшийся поток продуктов алкилирования из колонны 110 в колонну для отгонки парафинов 124, из которой по линии 48 отбирают поток дистиллята, содержащий смесь парафинов и в общем случае менее 0,3 масс.% моноолефинов. По линии 126 производят транспортирование потока кубового остатка из колонны для отгонки парафинов, содержащего фенилалканы и побочные продукты в виде тяжелых алкилатов, в колонну для вторичной перегонки 130, в которой разделяют поток кубового остатка из колонны для отгонки парафинов на поток кубового остатка 132, содержащий тяжелый алкилат, и поток дистиллята в виде продукта алкилата 128, содержащий соединения фенилалканы. Сульфирование соединений фенилалканов в потоке дистиллята в виде продукта алкилата 128 приведет к получению фенилалкансульфоновых кислот, которые можно нейтрализовать.
ПРИМЕР 1
В методике «импульсного испытания» проводят испытание адсорбентов при использовании конкретной исходной смеси и вещества десорбента с измерением адсорбционной способности, селективности, разрешения и скорости обмена для адсорбента. Базовый аппарат для импульсного испытания состоит из цилиндрической камеры для адсорбента с объемом, приблизительно равным 70 куб.см, имеющей впускное и выпускное отверстия на противоположных концах камеры. Камера находится в устройстве для регулирования температуры, а оборудование для регулирования давления выдерживает в камере постоянное предварительно заданное давление. В результате присоединения оборудования для проведения количественных и качественных аналитических определений, такого, как рефрактометры, поляриметры и хроматографы, к линии выпускного отверстия камеры станут возможными количественное детектирование и/или качественное определение одного, либо нескольких компонентов в исходящем потоке, покидающем камеру для адсорбента. Импульсное испытание начинается при пропускании через камеру для адсорбента вещества десорбента для того, чтобы заполнить адсорбент до равновесия с конкретным веществом десорбента, после этого производят импульсный ввод пробы исходной смеси, иногда разбавленной десорбентом, в течение периода времени продолжительностью в одну, либо несколько минут, а затем возобновляют элюирование компонентов исходной смеси потоком десорбента так, как при проведении хроматографической операции в системе жидкой и твердой сред. Исходящий поток можно анализировать непрерывно, и/или можно собирать фракции исходящего потока и анализировать их впоследствии по отдельности, что позволит вычертить линии огибающих для пиков соответствующих компонентов в виде зависимости концентрации компонента от количества исходящего потока.
Из информации, полученной с помощью импульсного испытания, обычно можно оценить эксплуатационные характеристики системы адсорбент/десорбент, выраженные через объем удерживания компонента экстракта, либо рафината, селективность для одного компонента по отношению к другому, продолжительность стадии, разрешение между компонентами и скорость десорбции компонента экстракта под действием десорбента. Расстояние между центром огибающей для пика компонента экстракта, либо рафината и огибающей для пика компонента свидетеля, либо какой-либо другой известной точкой отсчета будет определять объем удерживания для экстракта, либо рафината, выраженный через объем десорбента в кубических сантиметрах, прокачанный в течение временного интервала, соответствующего расстоянию между огибающими для пиков.
В таблице 1 перечисляются переменные и результаты для маломасштабных «импульсных испытаний», проведенных для оценки различных десорбентов и условий в отношении нескольких исходных смесей. Вещества с маркировкой Рафинат А и В представляют собой потоки рафинатов из коммерческих установок адсорбционного разделения, в которых извлекают из фракции С10-С14 углеводородов нормальные парафины. В столбце Десобент в таблице 1 указывается объемный процент каждого компонента десорбента в соответствии с указаниями в сносках.
С6 указывает на циклогексан.
N6 указывает на н-гексан.
Во всех испытаниях использовали адсорбент, содержащий 80% силикалита и 20% связующего на основе диоксида кремния, и хроматографическую колонку с объемом адсорбента 70 мл. Скорость течения через колонку составляла 1,21 куб.см/мин.
Принимая во внимание очень большое количество различных соединений в импульсе исходной смеси и ограничения, свойственные простой методике импульсных испытаний, эффективность разделения определяли в результате сбора фракций исходящего потока каждые две минуты и анализа каждой фракции. Первоначальные фракции имели высокие концентрации десорбента, а за ними следовали фракции с высокими концентрациями более высокоразветвленных углеводородов, не являющихся нормальными. Желательные ациклические углеводороды, содержащие только 3 первичных атома углерода (то есть монометилуглеводороды), имели тенденцию концентрироваться во фракциях, собираемых в конце импульса. В таблице 2 приводятся концентрации (в массовых процентах) ациклических парафинов, содержащих только 3 первичных атома углерода (то есть монометилпарафинов), присутствующих в нескольких различных фракциях в эксперименте №9937-06.
В эксперименте №9937-06 объединяли жидкость, собранную в качестве фракций №№ от 19 до 100. Анализ показал, что объединена жидкость характеризуется содержаниями различных структурных классов соединений, выраженными через массовые проценты без учета в расчете наличия десорбента, как это демонстрирует таблица 3.
В эксперименте №9937-17 объединяли жидкость, собранную в качестве фракций №№ от 23 до 50. Анализ показал, что жидкость характеризуется выраженными через массовые проценты содержаниями различных структурных классов соединений, как это демонстрирует таблица 4:
Анализ образца, образованного в результате объединения жидкости из фракций от 23 до 48 из эксперимента 9953-6, выявил 67% ациклических парафинов, имеющих только 3 первичных атома углерода (то есть монометилразветвленных соединений) и 9,3% ациклических парафинов, имеющих только 2 первичных атома углерода (то есть нормальных парафинов).
При сопоставлении этих данных с эксплуатационными характеристиками, обычно желательными при проведении разделений в коммерческих масштабах, необходимо осознавать, что лучшая селективность будет получена в результате проведения оптимизации в отношении состава адсорбента, состава десорбента и рабочих условий. Кроме этого, эксплуатационные характеристики способа будет улучшать использование технологии с симулированным подвижным слоем (SMB), либо еще лучше технологии разделения в периодическом режиме.
ПРИМЕР 2
В данном примере для представительной смеси чистых С10 компонентов проводили операцию импульсного испытания, в которой использовали предварительный импульс C8 изопарафина. В испытании использовали исходную смесь, содержащую равные объемы 3,3,5-триметилгептана, 2,6-диметилоктана, 2-метилнонана, нормального декана и 1,3,5-триметилбензола, в колонке для импульсных испытаний с объемом 70 куб.см и при температуре, выдерживаемой равной 120°С (248°F). Скорость течения через колонку составляла 1,1 куб.см/мин. В испытании использовали адсорбент на основе силикалита и смесь 70/30% (об.) нормального гептана и изооктана в качестве десорбента. В испытании в испытательный контур вводили 40 мл изооктана в виде предварительного импульса непосредственно перед вводом пробы исходной смеси.
Фигура 2 графически представляет результаты в виде графика зависимости относительных концентраций компонентов от времени, измеряемом через объем собранного исходящего потока. Фигура 2 демонстрирует подходящее разделение между монометилпарафином и нормальным парафином, с одной стороны, и ди- и триметилпарафинами, с другой стороны. Несмотря на то, что использование предварительного импульса, как представляется, улучшает выделение полосы монометилпарафина в исходящем потоке, подходящая отдельная полоса монометилпарафина получается в результате присутствия в десорбенте изооктана, даже и в отсутствие предварительного импульса.
ПРИМЕР 3
Использовали олефиновый поток.
Состав олефинового потока
2«Линейные олефины» включают С12 линейные олефины.
3«Другие алкилолефины» включают диметил-, триметил- и другие C12 олефины.
4«Тяжелые» включают димеры и тримеры C12 олефинов.
Смесь олефинового потока, содержащего композицию монометиловых C12 олефинов и характеризующегося составом, показанным в таблице 5, с бензолом образовывала объединенный поток, состоящий из 93,3 масс.% бензола и 6,7 масс.% олефинового потока, что соответствовало мольному соотношению бензола и олефина 30:1. В цилиндрический реактор с внутренним диаметром 22,2 мм (0,875 дюйма) загружали 75 куб. см (53,0 г) катализатора, полученного в результате экструдирования морденита-оксида алюминия при использовании водородной формы морденита, у которой SiO2/Al2О3 равно 18.
Объединенный поток перепускали в реактор и вводили в контакт с экструдатом при LHSV 2,0 час-1, полном давлении 500 фунт/дюйм2 избыточного давления (3447 кПа избыточного давления) и температуре на входе в реактор 125°С (257°F). При данных условиях реактор функционировал в течение периода времени продолжительностью 24 часа, а после этого в течение последующего периода времени продолжительностью 6 часов собирали селективный жидкий продукт.
Селективный жидкий продукт анализировали по методу 13С ядерного магнитного резонанса (ЯМР) для того, чтобы определить селективность по получению 2-фенилалканов и концевых четвертичных фенилалканов. Аналитический метод ЯМР обычно заключается в разбавлении образца 0,5 г смеси фенилалканов до массы 1,5 г при использовании безводного дейтерированного хлороформа, смешивании в ампуле для ЯМР диаметром 5 мм аликвоты разбавленной смеси фенилалканов объемом 0,3 миллилитра с 0,3 миллилитра 0,1 М ацетилацетоната хрома (III) в дейтерированном хлороформе, добавлении к смеси небольшого количества тетраметилсилана (TMS) в качестве точки отсчета химического сдвига 0,0 м.д. и проведении измерения спектра углерода с использованием спектрометра Bruker ACP-300 FT-NMR, приобретаемого у компании Bruker Instruments, Inc., Billerica, Massachusetts, USA, при напряженности поля 7,05 тесла, либо при 75,469 МГц для зонда QNP (зонд с четырьмя ядрами) 5 мм при ширине развертки 22727 Гц (301,1 м.д.) для сбора 65000 точек данных. Количественный спектр углерода получали при использовании во время сбора данных стробированной 1H развязки (инвертированная стробированная развязка). Количественный 13С спектр регистрировали с импульсами 7,99 микросекунд (90°), временем сбора данных 1,442 секунды, задержкой между импульсами 5 секунд, мощностью блока развязки, при использовании композитной импульсной развязки (CPD), равной 18Н, при ширине импульса 105 микросекунд (90°) и, по меньшей мере, 2880 сканированиях. Количество сканирований зависело от того, был ли отогнан бензол из жидкого продукта перед отбором упомянутого выше образца 0,5 г. Обработку данных проводили с использованием программного обеспечения The Bruker PC software WINNMR-1D, Version 6.0. В ходе обработки данных для них использовали уширение линии в 1 Гц. Конкретные пики интегрировали в области между 152 м.д. и 142 м.д. Таблица 6 демонстрирует идентификацию пиков 13С ЯМР для химических сдвигов у бензильного атома углерода изомеров фенилалканов. Термин «бензильный атом углерода» обозначает атом углерода в кольце фенильной группы, связанный с алифатической алкильной группой.
Идентификация пиков 13С ЯМР
Пик в области 148,3 м.д. идентифицировали как принадлежащий как 4-метил-2-фенилалканам, так и m-метил-m-фенилалканам (m>3). Однако присутствие m-метил-m-фенилалканов (m>3) с содержанием более 1% обнаруживает их по отчетливому пику, смещенному на 0,03 м.д. в сторону сильного поля от пика для 4-метил-2-фенилалканов. Пик в области 147,8 м.д. идентифицировали как принадлежащий 2-фенилалканам, как показано в таблице 6, при возможных помехах от 3-метил-3-фенилалканов.
Селективность по получению концевых четвертичных фенилалканов вычисляли в результате деления интеграла пика в области 149,6 м.д. на сумму интегралов всех пиков, перечисленных в таблице 6, и умножения на 100. Селективность по получению 2-фенилалкана можно оценить, если количество внутренних четвертичных фенилалканов, вносящих свой вклад в пики в области 148,3 м.д. и в области 147,8 м.д., будет меньше 2%, как это определили с использованием описанного далее метода газовой хроматографии/массе-спектрометрии. В качестве первого приближения данное условие выполняется, когда сумма интегралов пиков 4-фенилалкана и 3-фенилалкана в области 146,2-146,3 м.д. и в области 145,9-146,2 м.д. (соответственно) будет мала в сравнении с суммой интегралов всех пиков в диапазоне от 145,9 м.д. до 149,6 м.д., а селективность по получению концевых четвертичных фенилалканов будет меньше 10. В таком случае селективность по получению 2-фенилалкана вычисляли в результате деления суммы интегралов пиков в диапазоне от 149,6 до 146,6 м.д. на сумму интегралов всех пиков, перечисленных в таблице 6, и умножения на 100.
Анализ селективного жидкого продукта по методу газовой хроматографии/масс-спектрометрии также позволил определить селективность по получению внутренних четвертичных фенилалканов. Аналитический метод газовой хроматографии/масс-спектрометрии обычно состоит в проведении анализа жидкого продукта при помощи газового хроматографа (ГХ) HP 5890 Series II, оборудованного автоматическим пробоотборником HP 7673 и детектором масс-спектрометра (МС) HP 5972, используемого вместе с HP Chemstation для управления сбором и анализом данных, где оба устройства приобретаются у компании Hewlett Packard Company, Palo Alto, California, USA. ГХ имеет колонку DB1HT (df=0,1 мкм) 30 метров × 0,25 мм, либо эквивалент, приобретаемые у компании J&W Scientific Incorporated, 91 Blue Ravine Road, Folsom, California, USA. В режиме постоянного давления подавали газообразный носитель гелий при 15 фунт/дюйм2 избыточного давления (103 кПа избыточного давления) и 70°С (158°F), тогда как температуру инжектора выдерживали равной 275°С (527°F). Температуры линии передачи и источника МС выдерживали равными 250°С (482°F). Использовали следующую программу для температуры печки: 70°С (158°F) в течение 1 минуты, после этого до 180°С (356°F) при 1°С в минуту (1,8°F в минуту), после этого до 275°С (527*F) при 10°С в минуту (18°F в минуту), после этого выдерживание при 275°С (527°F) в течение 5 минут. Программное обеспечение у HP Chemstation производило настройку для МС при помощи установки в программном обеспечении на стандартную автоматическую настройку для спектров. Детектор МС проводил сканирование в диапазоне 50-550 Да с пороговой величиной = 50.
Концентрации внутренних четвертичных фенилалканов в селективном жидком продукте определяли (то есть, получали количественные характеристики селективного жидкого продукта) при использовании способа добавления стандарта. Глава 7 в книге, озаглавленной Samples and Standards, by В.W.Woodget et al., опубликованной от имени компании ACOL, London с выходными данными John Wiley and Sons, New York, 1987, приводит обоснование данного способа.
Сначала формировали маточный раствор внутренних четвертичных фенилалканов и получали его количественные характеристики в результате алкилирования бензола под действием монометилалкена при использовании неселективного катализатора, такого, как хлорид алюминия. Маточный раствор, содержащий данный неселективный жидкий продукт данного алкилирования, содержал смесь внутренних четвертичных фенилалканов. По стандартной методологии ГХ идентифицировали самые большие пики, соответствующие маточному раствору, и определяли концентрации внутренних четвертичных фенилалканов в маточном растворе (то есть, получали количественные характеристики маточного раствора) при использовании пламенно-ионизационного детектора (FID). Времена удерживания для пиков внутренних четвертичных фенилалканов уменьшались по мере того, как увеличивался индекс m в формуле m-метил-m-фенилалкан, и по мере того, как уменьшалось количество атомов углерода в алифатической алкильной группе внутреннего четвертичного фенилалкана. Концентрацию каждого внутреннего четвертичного фенилалкана рассчитывали в результате деления площади пика данного внутреннего четвертичного фенилалкана на сумму площадей всех пиков.
После этого получали раствор маркера, содержащий внутренние четвертичные фенилалканы, проводя разбавление составляющей аликвоту части маточного раствора дихлорметаном (метиленхлоридом) до получения номинальной концентрации, равной 100 м.д. (масс.) представляющего интерес одного конкретного внутреннего четвертичного фенилалкана (например, 3-метил-3-фенилдекана). Концентрация любого другого конкретного внутреннего четвертичного фенилалкана в растворе маркера может быть большей, либо меньшей 100 масс.(м.д.) в зависимости от концентрации данного внутреннего четвертичного фенилалкана в маточном растворе.
В-третьих, получали раствор образца в результате добавления 0,05 г составляющей аликвоту части селективного жидкого продукта в мерную колбу объемом 10 миллилитров и после этого разбавления ее содержимого дихлорметаном вплоть до метки 10 миллилитров.
В-четвертых, получали результирующий раствор в результате добавления 0,05 г составляющей аликвоту части селективного жидкого продукта в мерную колбу объемом 10 миллилитров и разбавления ее содержимого раствором маркера вплоть до метки 10 миллилитров.
Как раствор образца, так и результирующий раствор анализировали при помощи ГХ/МС при описанных выше условиях. Таблица 7 приводит данные, полученные из полного сканирования по методу МС, графически обработанные и проинтегрированные с использованием программного обеспечения для HP Chemstation. Программное обеспечение для HP Chemstation определяло площади пиков индивидуальных выделенных ионов, которые соответствовали «внутренним четвертичным соединениям», перечисленным в таблице 7.
Соотношение массы и заряда иона для пиков выделенных ионов
Концентрацию каждого внутреннего четвертичного фенилалкана в таблице 7 рассчитывали с использованием следующей формулы:
С=S(A1/(A2-A1)),
где С = концентрация внутреннего четвертичного фенилалкана в растворе образца, масс.%,
S = концентрация внутреннего четвертичного фенилалкана в растворе маркера, масс.%,
А1 = площадь пика внутреннего четвертичного фенилалкана в растворе образца, единицы площади,
A2 = площадь пика внутреннего четвертичного фенилалкана в результирующем растворе, единицы площади.
При использовании согласованных единиц из концентрации каждого внутреннего четвертичного фенилалкана в растворе образца, принимая во внимание влияние разбавления дихлорметаном, присутствующим в растворе образца, рассчитывали концентрацию данного внутреннего четвертичного фенилалкана в селективном жидком продукте. Таким образом рассчитывали концентрацию в селективном жидком продукте каждого из внутренних четвертичных фенилалканов, приведенных в таблице 7. Полную концентрацию внутренних четвертичных фенилалканов в селективном жидком продукте CIQPA рассчитывали в результате суммирования индивидуальных концентраций каждого из внутренних четвертичных фенилалканов из таблицы 7.
Селективный жидкий продукт может содержать внутренние четвертичные фенилалканы, отличные от тех, что перечислены в таблице 7, такие, как m-метил-m-фенилалканы, где m>5 в зависимости от количества атомов углерода в алифатических алкильных группах фенилалканов. Представляется, что в случае С12 олефинового потока и условий данного примера концентрации таких других внутренних четвертичных фенилалканов будут относительно низкими в сравнении с концентрациями внутренних четвертичных фенилалканов, перечисленных в таблице 7. Для данного примера совокупную концентрацию внутренних четвертичных фенилалканов в селективном жидком продукте CIQPA рассчитывали в результате суммирования только индивидуальных концентраций каждого из внутренних четвертичных фенилалканов, приведенных в таблице 7. Однако, если олефиновый поток включал бы олефины, содержащие, скажем, вплоть до 28 атомов углерода, то тогда совокупную концентрацию внутренних четвертичных фенилалканов в селективном жидком продукте CIQPA рассчитывали бы в результате суммирования индивидуальных концентраций m-метил-m-фенилалканов, где m находится в диапазоне от 3 до 13. В еще более общем случае, если олефиновый поток будет содержать олефины, имеющие x атомов углерода, то тогда совокупную концентрацию внутренних четвертичных фенилалканов в селективном жидком продукте CIQPA будут рассчитывать в результате суммирования индивидуальных концентраций m-метил-m-фенилалканов, где m находится в диапазоне от 3 до x/2. Без проведения излишних экспериментов можно идентифицировать, по меньшей мере, один пик с соотношением массы и заряда (m/z) выделенного иона, соответствующего каждому внутреннему четвертичному фенилалкану, так что для получения CIQPA можно определить, а после этого суммировать концентрации всех внутренних четвертичных фенилалканов.
Селективность по получению внутренних четвертичных фенилалканов в селективном жидком продукте рассчитывали при использовании следующей формулы:
Q=100(CIQPA/CMAB),
где Q = селективность по получению внутренних четвертичных фенилалканов,
СIQPA = концентрация внутренних четвертичных фенилалканов в селективном жидком продукте, % (масс.),
CМАВ = концентрация модифицированных алкилбензолов в селективном жидком продукте, % (масс.).
Концентрацию модифицированных алкилбензолов CМАВ в селективном жидком продукте определяли при использовании метода газовой хроматографии для того, чтобы установить концентрацию примесей в селективном жидком продукте. При определении СМАВ «примеси» обозначают компоненты селективного жидкого продукта, которые выпадают из конкретного диапазона времен удерживания, используемого в методе газовой хроматографии. «Примеси» в общем случае включают бензол, некоторые диалкилбензолы, олефины, парафины и тому подобное.
По методу газовой хроматографии определяли количество примесей в селективном жидком продукте. Для определения количества примесей в селективном жидком продукте также можно использовать и эквивалентное оборудование, эквивалентное получение образцов и эквивалентные параметры ГХ, которые будут другими, но которые приведут к получению результатов, эквивалентных тем, что описаны ниже.
Оборудование:
- Газовый хроматограф Hewlett Packard HP 5890 Series II, оборудованный щелевым/бесщелевым инжектором и пламенно-ионизационным детектором (FID).
- Капиллярная колонка J&W Scientific DB-1HT, длиной 30 метров, с внутренним диаметром 0,25 мм, с толщиной пленки 0,1 микрометр, каталожный номер 1221131.
- Мембрана 11 мм Restek Red lite, каталожный номер 22306. (Приобретается у компании Restek Corporation, 110 Benner Circle, Bellefonte, Pennsylvania, USA).
- Гибкая муфта на входе 4 мм Restek с материалом карбофрит, каталожный номер 20799-209.5.
- Уплотнительное кольцо для вкладыша на входе Hewlett Packard, каталожный номер 5180-4182.
- Метиленхлорид, марки для ВЭЖХ, J.Т.Baker, каталожный номер 9315-33, либо эквивалент. (Приобретается у компании J.Т.Baker Co., 222 Red School Lane, Phillipsburg, New Jersey, USA).
- Ампулы автоматического пробоотборника для газового хроматографа объемом 2 мл с пережатым верхом, либо эквивалент.
Приготовление образца:
- Отвешивают 4-5 мг образца в ампулу автоматического пробоотборника ГХ объемом 2 мл.
- Добавляют в ампулу ГХ 1 мл метиленхлорида; герметизируют при помощи заглушек (колпачков) 11 мм с тефлоновым покрытием для ампул с пережатием, HP инвентарный номер 5181-1210 (приобретается у компании Hewlett Packard Company), с использованием устройства для пережимания, HP инвентарный номер 8710-0979 (приобретается у компании Hewlett Packard Company); и хорошо перемешивают.
- После этого образец готов для ввода в ГХ.
Параметры ГХ:
- Газообразный носитель: водород.
- Давление нагнетания в колонке: 62,1 кПа (9 фунт/дюйм2).
- Потоки: течение в колонке, 1 мл/мин; щелевое отверстие, 3 мл/мин; промывка мембраны, 1 мл/мин.
- Ввод пробы: автоматический пробоотборник HP 7673, шприц объемом 10 микролитров, ввод 1 микролитра пробы.
- Температура вводимой пробы: 350°С (662°F).
- Температура детектора: 400°С (752°F).
- Программа для температуры печки: первоначальное выдерживание при 70°С (158°F) в течение 1 минуты; скорость нагревания 1°С в минуту (1,8°F в минуту); заключительное выдерживание при 180°С (356°F) в течение 10 минут.
В данном методе газовой хроматографии необходимо иметь два стандарта, свежеперегнанных до степени чистоты, превышающей 98% (моль.), и в общем случае содержащих 2-фенилалкан. «Легкий стандарт» 2-фенилалкана в своей алифатической алкильной группе имеет, по меньшей мере, на один атом углерода меньше в сравнении с количеством атомов углерода в олефине из олефинового потока, запускаемого в зону алкилирования, который имеет наименьшее количество атомов углерода. «Тяжелый стандарт» стандарта 2-фенилалкана в своей алифатической алкильной группе имеет, по меньшей мере, на один атом углерода больше в сравнении с количеством атомов углерода в олефине в олефиновом потоке, запускаемом в зону алкилирования, который имеет наибольшее количество атомов углерода. Например, если олефины в олефиновом потоке, запускаемом в зону алкилирования, будут иметь от 10 до 14 атомов углерода, то тогда подходящие стандарты будут включать 2-фенилоктан в качестве легкого стандарта и 2-фенилпентадекан в качестве тяжелого стандарта.
Методы газовой хроматографии, использующие условия, описанные выше, позволяют определить время удерживания для каждого стандарта, а времена удерживания для двух стандартов, в свою очередь, определяют диапазон времен удерживания. После этого по методу газовой хроматографии с использованием упомянутых выше условий анализировали образец аликвоты селективного жидкого продукта. Если более 90% от совокупной площади под кривой ГХ будет попадать в пределы диапазона времени удерживания, то тогда предполагается, что содержание примесей в селективном жидком продукте не превышает 10 масс.% от количества селективного жидкого продукта, и исключительно для целей вычисления селективность по получению внутренних четвертичных фенилалканов СМАВ принимали равной 100 масс.%.
Если процент совокупной площади под кривой ГХ, попадающей в пределы диапазона времени удерживания, не будет превышать 90%, то тогда предполагается, что содержание примесей в селективном жидком продукте превышает 10 масс.% от количества селективного жидкого продукта. В данном случае для того, чтобы определить СМАВ, примеси отгоняли из селективного жидкого продукта в результате добавления от 2200 до 2300 г селективного жидкого продукта в 5-литровую, 3-горлую круглодонную колбу с соединительными элементами 24/40, содержащую магнитную мешалку и несколько керамических кипятильников. В среднем горле колбы размещали конденсатор Vigreux длиной 9-1/2 дюйма (24,1 см) с соединительным элементом 24/40, а к верхней части устройства Vigreux присоединяли охлаждаемый водой конденсатор, оснащенный калиброванным термометром. К концу конденсатора присоединяли вакуумную приемную колбу. В одном боковом горле колбы помещали пробку, а из другого бокового горла выходил калиброванный термометр. Колбу и устройство Vigreux окружала обертка из алюминиевой фольги. В приемную колбу подавали вакуум из вакуумной линии тогда, когда в колбе перемешивали селективный жидкий продукт, и после достижения максимального вакуума (по меньшей мере 25,4 мм ртутного столба по вакуумметру, либо меньше) при помощи электрической нагревательной рубашки нагревали селективный жидкий продукт.
После того, как начинали нагревание, сначала собирали «фракцию А» в диапазоне от 25°С (77°F) до температуры кипения легкого стандарта при давлении в верхней части устройства Vigreux, проводя измерения при помощи калиброванного термометра в верхней части устройства Vigreux. После этого собирали «фракцию В» в диапазоне от температуры кипения легкого стандарта до температуры кипения тяжелого стандарта с измерением обеих величин при помощи калиброванного термометра в верхней части устройства Vigreux при давлении в верхней части устройства Vigreux. Низкокипящую фракцию А и высококипящий кубовый остаток отбрасывали. Фракция В содержала представляющие интерес модифицированные алкилбензолы, и ее взвешивали. Подходящие температуры для сбора фракций А и В можно определить из статьи, написанной авторами Samuel В.Lippincott and Margaret M.Lyman, опубликованной в журнале Industrial and Engineering Chemistry, vol.38, 1946, начиная со страницы 320.
После этого образец аликвоты фракции В анализировали по методу газовой хроматографии с использованием упомянутых выше условий. Если более 90% от совокупной площади под кривой ГХ для фракции В будет попадать в пределы диапазона времени удерживания, то тогда предполагается, что содержание примесей во фракции В не превышает 10 масс.% от количества селективного жидкого продукта, и исключительно для целей вычисления селективности по получению внутренних четвертичных фенилалканов СМАВ будут вычислять в результате деления массы собранной фракции В, на массу составляющей аликвоту части селективного жидкого продукта, загруженной в 5-литровую колбу в упомянутом выше способе перегонки. Если процент от совокупной площади под кривой ГХ для фракции В, попадающий в пределы диапазона времени удерживания, будет меньше 90%, то тогда предполагается, что содержание примесей во фракции В превышает 10 масс.% от количества фракции В, и опять проводили удаление примесей с использованием описанного выше способа перегонки с отдалением низкокипящей фракции С и кубового остатка от фракции D, содержащей представляющие интерес модифицированные алкилбензолы. Фракцию D извлекали, взвешивали и анализировали по методу газовой хроматографии для определения, удовлетворяет ли она тому же самому критерию 90% для попадания совокупной площади под кривой ГХ для фракции в пределы диапазона времени удерживания. Если соответствие критерию достигалось, то тогда по ранее описанной методике рассчитывали СМАВ. Если соответствия критерию не было, то тогда для фракции D повторяли способы перегонки и газовой хроматографии.
Описанные выше способы перегонки и газовой хроматографии можно повторять до тех пор, пока не будет собрана фракция, содержащая представляющие интерес модифицированные алкилбензолы и имеющая менее 10 масс.% примесей, при том условии, что после каждой перегонки будет оставаться количество вещества, достаточное для проведения последующих испытаний по данным методам. После этого, как только СМАВ будет определена, с использованием упомянутой выше формулы рассчитывают селективность по получению внутренних четвертичных фенилалканов Q.
Результаты данных анализов продемонстрированы в таблице 8:
Анализ жидкого продукта
В отсутствие селективности, зависящей от формы реагентов, предполагается, что наиболее значительная часть 2-метилундецена будет образовывать 2-метил-2-фенилундекан (то есть, «концевое четвертичное соединение»). Подобным же образом, наиболее значительные части 6-метилундецена, 5-метилундецена, 4-метилундецена и 3-метилундецена, как предполагается, будут образовывать «внутренние четвертичные соединения». Как ожидается, линейные олефины будут приводить к получению статистического распределения 2-фенилдодекана, 3-фенилдодекана, 4-фенилдодекана, 5-фенилдодекана и 6-фенилдодекана. Таким образом, если из вычислений исключить «легкие», «тяжелые» и «другие алкилолефины», перечисленные в таблице 5, то тогда селективность по получению 2-фенилалкана не будет превышать 17, а селективность по получению внутренних четвертичных фенилалканов будет приближаться к 55. Таблица 8 демонстрирует, что селективность по получению 2-фенилалкана значительно превышает величину, ожидаемую в отсутствие селективности, зависящей от формы реагентов, и что селективность по получению внутренних четвертичных алкилбензолов, полученная при использовании морденитового катализатора, намного меньше селективности по получению внутренних четвертичных алкилбензолов, которая предполагалась в отсутствие селективности, зависящей от формы реагентов.
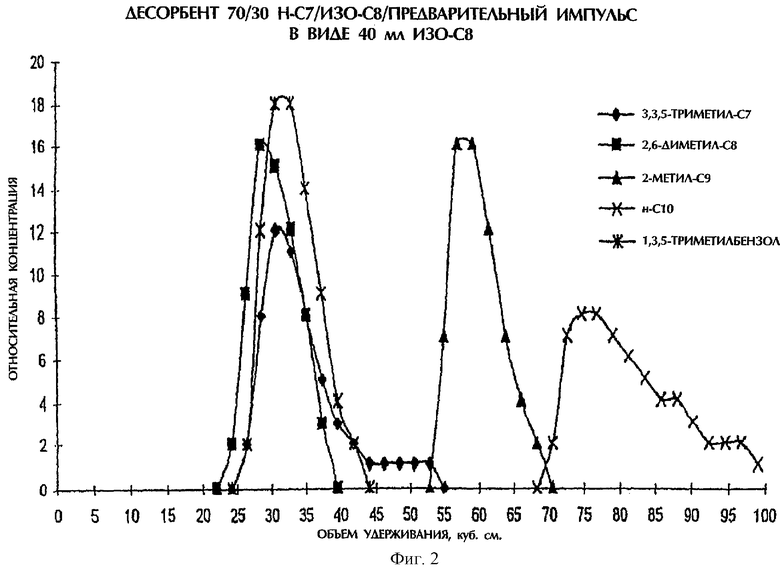
Изобретение относится к процессу каталитического алкилирования высшими моноолефинами бензола или его производных (толуола и этилбензола) с получением фенилалканов. Способ включает следующие стадии: a) подачу потока исходного сырья, содержащего первый ациклический парафин С8-С28 и имеющий 2, либо 3 первичных атома углерода, с первой концентрацией и, по меньшей мере, второй ациклический парафин, в зону адсорбции, для проведения селективной адсорбции на силикалите, введения в контакт с потоком десорбента, и извлечения из зоны адсорбции экстракта адсорбции со второй концентрацией первого ациклического парафина, которая превышает первую концентрацию; b) подачу части экстракта адсорбции в зону дегидрирования и извлечения из зоны дегидрирования потока продуктов, содержащего ациклический моноолефин, C8-C28, имеющий 2, либо 3 первичных атома углерода; c) подачу исходного сырья, содержащего фенильное соединение и часть потока продуктов дегидрирования, содержащих ациклический моноолефин, в зону алкилирования, в присутствии катализатора алкилирования, с получением фенилалкана, содержащего одну С8-С28 алифатическую алкильную группу; d) извлечение фенилалкана из зоны алкилирования. Композиция модифицированного алкилбензола включает алкилбензол, где звено модифицированного алкилбензола получают изложенным выше способом, и предназначена для использования в качестве смазочного материала, либо присадки к смазочному материалу, или в качестве моющего средства, либо компонента моющего средства. Технический результат - усовершенствование технологии алкилирования фенильного соединения легкоразветвленными моноолефинами. 2 н. и 7 з.п. ф-лы, 2 ил., 8 табл.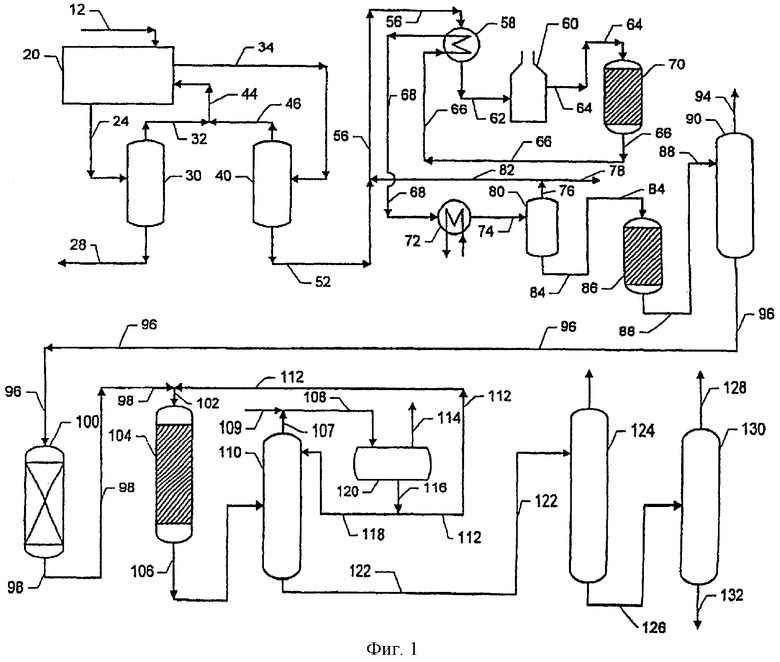
a) подачу потока исходного сырья, содержащего первый ациклический парафин, отличающийся диапазоном атомов углерода C8-C28 и имеющий 2 либо 3 первичных атома углерода с первой концентрацией, и, по меньшей мере, второй ациклический парафин, в зону адсорбции, включающую слой адсорбента, содержащего силикалит в условиях, способствующих прохождению адсорбции, для проведения селективной адсорбции, по меньшей мере, части первого ациклического парафина, введения в контакт со слоем адсорбента потока десорбента, содержащего, по меньшей мере, один компонент, выбираемый из группы, состоящей из C5-C8 циклопарафина, C5-C8 нормального парафина и С5-С8 разветвленного парафина, и извлечения из зоны адсорбции экстракта адсорбции со второй концентрацией первого ациклического парафина, которая превышает первую концентрацию;
b) подачу, по меньшей мере, части экстракта адсорбции в зону дегидрирования для дегидрирования первого ациклического парафина и извлечения из зоны дегидрирования потока продуктов дегидрирования, содержащего ациклический моноолефин, имеющий диапазон атомов углерода C8-C28 и имеющий 2 либо 3 первичных атома углерода;
c) подачу исходного сырья, содержащего фенильное соединение, и подачу, по меньшей мере, части потока продуктов дегидрирования, содержащих ациклический моноолефин, в зону алкилирования в условиях прохождения алкилирования, достаточных для алкилирования фенильного соединения под действием ациклического моноолефина в присутствии катализатора алкилирования, с получением фенилалкана, содержащего молекулу, включающую одну фенильную часть и одну C8-C28 алифатическую алкильную часть, где алифатическая алкильная часть содержит 2 либо 3 первичных атома углерода и не содержит четвертичных атомов углерода, которые не связаны через связь углерод-углерод с атомом углерода фенильной части; причем алкилирование характеризуется селективностью по получению 2-фенилалканов в диапазоне от 40 до 100 и селективностью алкилирования по получению внутренних четвертичных фенилалканов меньше 10; и
d) извлечение фенилалканов из зоны алкилирования.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Способ получения алкилароматических соединений | 1980 |
|
SU958404A1 |

