Область техники
Настоящее изобретение относится к способу селективного получения фенилалканов, к фенилалкановым композициям на их основе и применению таких композиций.
Предшествующий уровень техники
Более тридцати лет назад многие моющие средства для домашней стирки получали на основе разветвленных алкилбензолсульфонатов (BABS). BABS производили из определенного типа алкилбензолов, называемых разветвленными алкилбензолами (ВАВ). Алкилбензолы (фенилалканы) относятся к общей категории соединений, содержащих алкильную группу, присоединенную к фенильной группе и отвечающих общей формуле (mi-алкилi)i-н-фенил-алкан. Алифатическая алкильная группа состоит из алифатической алкильной цепочки, на которую ссылаются, как на “алкан” в формуле (mi-алкилi)i-н-фенил-алкан. Среди различных цепочек алифатической алкильной группы, алифатическая алкильная цепочка представляет собой наиболее длинную прямую цепочку, содержащую атом углерода, соединенный с фенильной группой. Кроме этого, алифатическая алкильная группа может также содержать одно или более алкильных ответвлений, каждое из которых соединено с алифатической алкильной цепочкой и обозначено, как соответствующий (mi-алкил,), в формуле (m-iалкилi)i-н-фенил-алкан. Могут быть выбраны две или более цепочек одинаковой длины в качестве алифатической алкильной цепочки, причем такой подбор приводит к углеродной цепочке с максимальным количеством алкильных ответвлений. В приведенной выше формуле нижний символ i имеет значение в интервале от 1 до числа алкильных ответвлений и каждому значению i соответствует алкильное ответвление, присоединенное к углероду с номером mi алифатической алкильной цепочки. Фенильная группа присоединена к алифатической алкильной группе, конкретно к углеродному атому номер n алифатической алкильной цепочки. Алифатическая алкильная цепочка пронумерована от одного ее конца к другому, причем направление нумерации выбирается таким образом, чтобы положение фенильной группы имело наименьший возможный номер.
Стандартный способ, используемый в нефтехимической промышленности для получения ВАВ, заключается в олигомеризации легких олефинов, особенно пропилена, с получением разветвленных олефинов, содержащих 10-14 углеродных атомов с последующим алкилированием бензола разветвленными олефинами в присутствии такого катализатора, как HF. Хотя, полученный таким способом ВАВ содержит большое число алкил-фенил-алканов, отвечающих общей формуле (m-iалкилi)-н-фенил-алкан, с целью иллюстрации трех важнейших характеристик ВАВ достаточно рассмотреть лишь два примера ВАВ: m-алкил-m-алкил-n-фенилалканы, в которых m≠n, и m-алкил-m-фенилалканов, в которых m≥2.
Наиболее важной характеристикой ВАВ является тот факт, что для большинства ВАВ, к алифатической алкильной цепочке ВАВ обычно присоединено, по крайней мере, одно алкильное ответвление, как правило, три или более алкильных ответвлений. Вследствие этого ВАВ содержит относительно высокое количество первичных углеродных атомов в расчете на алифатическую алкильную группу, поскольку число первичных углеродных атомов, в расчете на алифатическую алкильную группу ВАВ, равно числу алкильных ответвлений плюс единица, в случае n=1, или плюс два, в случае n≥2, при условии, что сами алкильные ответвления являются неразветвленными. Если алкильное ответвление является разветвленным, то алифатическая алкильная группа в ВАВ имеет еще большее число первичных углеродных атомов. Таким образом, алифатическая алкильная группа в ВАВ обычно содержит три, четыре или более первичных углеродных атомов. Что касается алкильных ответвлений алифатической алкильной группы в ВАВ, то каждое алкильное ответвление обычно представляет собой метильное ответвление, хотя возможны этильное, пропильное и высшие алкильные ответвления.
Другой характеристикой ВАВ является тот факт, что фенильная группа ВАВ может быть присоединена к любому непервичному углеродному атому алифатической алкильной цепочки. Это типично для ВАВ, полученного стандартным нефтехимическим способом. За исключением 1-фенилалканов, образование которых, как известно, является нежелательным в связи с относительной неустойчивостью первичного карбениевого иона и пренебрежимо малым влиянием ответвлений в разветвленных парафинах, стадия олигомеризации приводит к образованию углерод-углеродной связи, неупорядоченно распределенной по длине алифатической алкильной цепочки, а в результате стадии алкилирования происходит, практически, случайное присоединение алкильной группы к атомам углерода в алифатический алкильной цепочке. Так, например, в случае фенилалкана с алифатической алкильной цепочкой, содержащей 10 углеродных атомов и полученного стандартным способом с использованием ВАВ, можно ожидать, что фенилалкановый продукт будет иметь практически случайное распределение 2-, 3-, 4- и 5-фенилалканов и, селективность процесса по такому фенилалкану, как 2-фенилалкан, должна составлять 25% при случайном распределении, однако на практике это значение составляет 10-40.
Третья характерная особенность ВАВ состоит в относительно высокой вероятности того, что один из углеродных атомов алифатической алкильной цепочки представляет собой четвертичный углеродный атом. Как проиллюстрировано первым примером ВАВ, четвертичным углеродным атомом может служить углеродный атом в алифатической алкильной группе, а не углеродный атом, связанный посредством углерод-углеродной связи с атомом углерода фенильной группы. Однако, как проиллюстрировано вторым примером ВАВ, четвертичный углеродный атом может также представлять собой углеродный атом, связанным посредством углерод-углеродной связи, с углеродом фенильной группы. В том случае, когда углеродный атом алкильной боковой цепочки связан не только с двумя различными углеродами алкильной боковой цепи и углеродным атомом алкильного ответвления, но и с углеродным атомом фенильной группы, полученный в результате алкил-фенил-алкан называют четвертичным алкил-фенил-алканом, или сокращено “кват” ("quat"). Таким образом, кваты включают алкил-фенил-алканы, отвечающие общей формуле m-алкил-m-фенил-алкан. Если четвертичным углеродным атомом является второй углеродный атом от конца алкильной боковой цепочки, то образовавшийся 2-алкил-2-фенил-алкан называют “концевым кватом”. Если четвертичным углеродным атомом является любой другой углеродный атом алкильной боковой цепочки, как этом имеет место во втором примере ВАВ, то полученный в результате алкил-фенил-алкан называют “внутренним кватом”. В известных способах получения ВАВ относительно большое количество ВАВ, обычно более 10 мол.%, составляют внутренние кваты.
Примерно тридцать лет назад стало ясно, что моющие средства, предназначенные для домашней стирки, полученные из ВАВ, постепенно загрязняют реки и озера. Исследования такой проблемы привели к заключению о том, что BABS медленно подвергаются биодеградации. Решение указанной проблемы привело к созданию моющих средств на основе алкилбензолсульфонатов (LABS), которые, как было установлено, быстрее подвергаются биодеградации, чем BABS. В настоящее время, моющие вещества на основе LABS применяются во всем мире. LABS производят из другого типа алкилбензолов, называемых линейными алкилбензолами (LAB). Стандартный процесс, используемый в нефтехимической промышленности для производства LAB, заключается в дегидрировании линейных парафинов в линейные олефины с последующим алкилированием бензола линейными олефинами в присутствии такого катализатора, как HF или твердого катализатора. LAB представляют собой фенилалканы, включающие линейную алифатическую алкильную группу и фенильную группу и отвечают общей формуле n-фенил-алкан. LAB не имеют алкильных ответвлений и, следовательно, линейная алифатическая алкильная группа обычно содержит два первичных углеродных атома (т.е. n≥2). Другой характерной особенностью LAB, получаемых стандартными способами, является тот факт, что фенильная группа в LAB обычно соединена с любым вторичным углеродным атомом линейной алифатической алкильной группы. В LAB, полученных с использованием в качестве катализатора HF, фенильная группа, с несколько большей вероятностью, присоединяется к вторичному углеродному атому, расположенному ближе к центру, а не к концу, как в случае линейных алифатических алкильных групп, тогда как в случае LAB полученных в процессе Detal™, примерно 25-35 мол.% н-фенилалканов представляют собой 2-фенилалканы.
В исследовании нескольких последних лет были идентифицированы некоторые модифицированные алкилбензолсульфонаты, на которые в тексте ссылаются, как на MABS, и которые отличаются по составу от алкилбензолсульфонатов, используемых в промышленности в настоящее время, включающих BABS и LABS, а также от всех алкилбензолсульфонатов, производимых на основе алкибензолов известными способами, включающими те, в которых осуществляют алкилирование ароматики с использованием таких катализаторов, как HF, хлористый алюминий, алюмосиликат, фторированный алюмосиликат, цеолиты и фторированные цеолиты. Кроме этого, MBAS отличаются от рассматриваемых алкилбензолсульфонатов улучшенными характеристиками, касающимися очистки при стирке, чистки сильно загрязненных поверхностей, и отличной эффективностью при стирке в жесткой и/или холодной воде, при этом их способность к биодеградации сравнима с LABS.
MABS могут быть получены путем сульфирования третьего типа алкилбензолов, называемых модифицированными алкилбензолами (МАВ), и при этом желаемые характеристики МАВ определяются желательной растворимостью, поверхностно-активными свойствами и способностью MABS к биодеградации. МАВ представляет собой фенилалкан, включающий легкоразветвленную алифатическую группу и фенильную группу, и отвечает общей формуле (m-iалкилi)i-n-фенил-алкан. Обычно МАВ имеет лишь одно алкильное ответвление и такое алкильное ответвление представляет собой метальную группу, предпочтительно, этильную или пропильную группы, в результате чего при наличии лишь одного алкильного ответвления и реализации условия n≠1, алифатическая алкильная группа в МАВ содержит три первичных углеродных атома. Однако алифатическая алкильная группа в МАВ может содержать два первичных углеродных атома, если имеется лишь одно алкильное ответвление и n=1, или четыре первичных углеродных атомов, если имеется два алкильных ответвления и n≠1. Таким образом, первый отличительный признак МАВ состоит в том, что алифатическая алкильная группа МАВ содержит промежуточное число первичных углеродных атомов по сравнению с ВАВ и LAB. Другой отличительный признак МАВ заключается в том, что рассматриваемый объект содержит большое количество 2-фенилалканов, т.е. 40-100% фенильных групп селективно присоединены к второму углеродному атому от конца алкильной боковой цепочки.
Наконец, еще одна особенность алкилата МАВ состоит в том, что МАВ содержит относительно низкое количество внутренних кватов. Некоторые внутренние кваты, как, например, 5-метил-5-фенилундекан, образуют MABS, демонстрирующие замедленную биодеградацию, тогда как такие концевые кваты, как 2-метил-2-фенилундеканы образуют MABS со способностью к биодеградации аналогичной способности LABS. Так, например, эксперименты по биодеградации показали, что при обработке в среде активированного ила в пористом вегетационном сосуде предельная степень биодеградации оказалась выше для 2-метил-2-ундецил [С14]бензолсульфоната натрия, чем для 5-метил-5-ундецил [С14]-бензолсульфоната натрия. См. статью Nielsen и др. "Bioderadation ofcoproducts of commercial linaer alkylbenzene sulfonate" в Environmental Science and Technology, т. 31, №12, 3397-3404 (1997). Относительно низкое количество, обычно менее 10 мол.% от МАВ, составляют внутренние кваты.
Поскольку MABS обладают преимуществами над другими алкилбензолсульфонатами проводятся исследования по разработке катализаторов и способов для селективного получения МАВ. Как предполагалось ранее, два из основных критериев процесса алкилирования в производства МАВ представляют собой селективность по 2-фенилалканам и селективность по четвертичным фенилалканам. Известные способы алкилирования, используемые в производстве LAB, с применением таких катализаторов, как хлористый алюминий или HF, не способны обеспечить получение МАВ с желаемой селективностью по 2-фенилалканам и внутренним кватам. В рассматриваемых известных способах, при реакции незначительно разветвленных олефинов (т.е. олефинов с практически такой же небольшой степенью разветвления, что и в алифатической алкильной группе МАВ) с бензолом, селективно образуются четвертичные фенилалканы. Один из механизмов, объясняющих такую селективность по четвертичным фенилалканам, предусматривает возможность превращения делинеаризованных в различной степени олефинов в промежуточные первичные, вторичные и третичные ионы карбения. Среди трех указанных ионов карбения ионы третичного карбения наиболее устойчивы и по этой причине их образование и последующая реакция с бензолом наиболее вероятны, в результате чего образуются четвертичные фенилалканы.
Один из предложенных способов получения МАВ включает три стадии. На первой стадии, парафиновое сырье подают в зону изомеризации с целью изомеризации парафинов и получения потока изомеризованного продукта, содержащего слаборазветвленные парафины (т.е. парафины с той же небольшой степенью разветвления, что и алифатическая алкильная группа МАВ). Далее, технологический поток изомеризата подают в зону дегидрирования, в которой слаборазветвленные парафины подвергают дегидрированию с образованием потока продукта дегидрирования, содержащего слаборазветвленные моноолефины (т.е. моноолефины с таким же слабым разветвлением, что и слаборазветвленные парафины и, следовательно, алифатическая аклильная группа МАВ). Наконец, поток продукт дегидрирования подают в зону алкилирования, в которой слаборазветвленные моноолефины в потоке продукта дегидрирования реагируют с бензолом с образованием МАВ.
Одна из проблем предложенного способа состоит в том, что в традиционных зонах реакции дегидрирования в олефины превращается лишь около 10 мас.% подаваемых парафинов, вследствие чего около 90 мас.% технологического потока из зоны дегидрирования составляют парафины как нормального, так и изостроения. Поскольку поток продукта из зоны дегидрирования подается в зону алкилирования, все рассматриваемые парафины также поступают в зону алкилирования. Хотя было бы желательно удалять парафины до подачи в зону алкилирования, трудность в разделении таких парафинов и олефинов с одинаковым числом атомов углерода препятствует такому решению. Обычно, в зоне алкилирования более 90 мас.% подаваемых моноолефинов превращаются в фенилалканы, тогда как входящие парафины оказываются практически инертными или нереакционоспособными. Таким образом, поток, покидающий зону алкилирования, содержит не только желаемый МАВ, но и указанные парафины. В соответствие со сказанным, проводятся исследования по созданию способов получения МАВ с эффективным выделением и использованием парафинов из потока, выходящего со стадии алкилирования.
Сущность изобретения
В соответствии с одним из аспектов, настоящее изобретение предусматривает способ получения фенилалканов, в особенности модифицированных алкилбензолов (МАВ), включающий стадии изомеризации парафина, дегидрирования парафинов и алкилирования фенильных соединений, в котором парафины, содержащиеся в потоке со стадии алкилирования, рециркулируют на стадию изомеризации и/или стадию дегидрирования. Подвергаемые рециркуляции парафины могут иметь линейную или нелинейную структуру, и они включают слаборазветвленные парафины. Поскольку рециркулируемые парафины могут быть превращены в слаборазветвленные олефины в настоящем изобретении осуществляется эффективное выделение парафинов из потока со стадии алкилирования и их использование для получения ценных фенилалкановых продуктов. Таким образом, в соответствии с рассматриваемым аспектом настоящего изобретения осуществляется увеличение выхода ценных продуктов в расчете на количество парафинового сырья, вводимого в процесс, и при этом исключается трудная стадия отделения парафинов от моноолефинов после осуществления стадии дегидрирования парафинов и до проведения стадии алкилирования.
Аспект настоящего изобретения, касающийся способа получения, имеет несколько целей. Главная цель настоящего изобретения состоит в получении фенилалканов, в особенности модифицированных алкилбензолов (МАВ), в результате изомеризации парафинов с последующим их дегидрированием в олефины, которыми осуществляют алкилирование ароматических соединений. Дополнительная цель настоящего изобретения заключается в повышении выхода фенилалканов в рассматриваемом процессе и, вследствие этого, в уменьшении количества парафинового сырья, требующегося для реализации процесса. Еще одна цель состоит в удалении непрореагировавших парафинов из фенилалканового продукта без необходимости в проведении трудного и/или дорогостоящего отделения парафинов от олефинов после стадии дегидрирования и перед стадией алкилирования.
В части, касающейся алкилирования детергентов, настоящее изобретение обеспечивает получение фенилалканов, удовлетворяющих непрерывно ужесточающимся требованиям к селективности по 2-фенилалканам и селективности по внутренним четвертичным фенилалканам с целью получения модифицированных алкилбензолов (МАВ). В свою очередь, МАВ могут быть подвергнуты сульфированию с получением модифицированных линейных алкилбензолсульфонатов (МАВS), обладающих улучшенной эффективностью очистки в твердой и/или холодной воде и сохраняющих способность к биодеградации, сравнимую со способностью линейных алкилбензолсульфонатов.
В соответствии с другим аспектом, настоящее изобретение предусматривает композиции на основе МАВ, получаемые способом настоящего изобретения. Предполагается, что МАВ, полученные способом настоящего изобретения, необязательно являются продуктами, которые могут быть получены известными способами, в которых не используют рециркуляцию парафинов. Не ограничиваясь конкретной теорией, считается, что в зоне дегидрирования степень конверсии разветвленных парафинов может превышать степень конверсии нормальных (линейных) парафинов, и/или, что степень конверсии тяжелых парафинов может быть более высокой, чем степень конверсии легких парафинов. В рассматриваемых случаях, концентрация линейных парафинов и/или более легких парафинов в рециркулируемом потоке парафинов может увеличиваться. Это, в свою очередь, может приводить к увеличению концентрации и предельного значения конверсии линейных и/или более легких парафинов в зоне дегидрирования до тех пор, пока скорость удаления из процесса линейных и/или более легких парафинов в результате дегидрирования и последующего алкилирования не сравняется со скоростью введения в зону дегидрирования рассматриваемых парафинов из зоны их изомеризации. Соответственно, при заданном значении степени конверсии олефинов в зоне алкилирования, алифатическая алкильная цепочка МАВ-продукта настоящего изобретения может стать менее разветвленной и/или более короткой, чем в известных способах. При сульфировании полученные в результате MABS могут проявлять аналогичную тенденцию к образованию менее разветвленной и/или более короткой алифатической алкильной цепочки, чем в известных способах. Таким образом, для заданной комбинации сырья способ настоящего изобретения может обеспечить получение особых МАВ-продуктов с алифатической алкильной цепочкой, имеющей специально подобранные степени разветвления, которые не обязательно аналогичны тем, что получают в известных способах. Согласно другому аспекту, настоящее изобретение предусматривает применение МАВ, получаемых способом изобретения, в качестве смазывающих агентов.
Раскрытие сущности изобретения
Парафиновое сырье, предпочтительно, включает молекулы неразветвленных (линейных) или нормальных парафинов, обычно содержащих 8-28, предпочтительно 8-15, и более предпочтительно 10-15 углеродных атомов. Два углеродных атома в молекуле неразветвленного парафина являются первичными углеродными атомами, а оставшиеся углеродные атомы представляют собой вторичные углеродные атомы.
Рассматриваемое парафиновое сырье может также содержать слаборазветвленный парафин представляющий собой парафин с общим числом углеродных атомов 8-28, три или четыре из которых являются первичными углеродными атомами, причем ни один из оставшихся углеродных атомов не является четвертичным углеродным атомом. Предпочтительно, когда слаборазветвленный парафин содержит 8-15, более предпочтительно, 10-15 углеродных атомов. Слаборазветвленный парафин обычно включает алифатический алкан, общей формулы (рi-алкилi)i-алкан и состоит из алифатической алкильной цепочки и одного или более алкильных ответвлений. Существует возможность подбора двух или более цепочек одинаковой длины в качестве алифатической алкильной цепочки, причем такой подбор обеспечивает цепочку с наивысшим числом алкильных ответвлений. Подстрочный символ i имеет значение от 1 до числа алкильных ответвлений и при каждом значении i соответствующее алкильное ответвление соединено с углеродом с номером pi алифатической алкильной цепочки. Алифатическая алкильная цепочка нумеруется от одного ее конца до другого, причем направление нумерации выбирают таким образом, чтобы углеродные атомы, имеющие алкильные ответвления, имели наименьшие, возможные номера.
Алкильное ответвление или ответвления обычно выбирают из метильных, этильных или пропильных групп, причем более короткие и нормальные разветвления являются предпочтительными. Предпочтительно, слаборазветвленный парафин имеет только одно алкильное ответвление, однако возможно наличие двух алкильных ответвлений. Слаборазветвленные парафины, имеющие два алкильных ответвления или четыре первичных атомов углерода, обычно составляют менее 40 мол.%, предпочтительно, менее 25 мол.% от общего количества слаборазветвленных парафинов. Слаборазветвленные парафины, имеющие одно алкильное ответвление или три первичных атома углерода составляют, предпочтительно, более 70 мол.% от общего количества слаборазветвленных парафинов.
Парафиновое сырье может также содержать более сильноразветвленные парафины, чем рассмотренные выше слаборазветвленные парафины. Предпочтительно минимизировать количество таких высокоразветвленных парафинов, вводимых в процесс. Молекулы парафина, содержащие, по крайней мере, один четвертичный атом углерода, обычно составляют менее 10 мол.%, предпочтительно, менее 5 мол.%, более предпочтительно менее 2 мол.% и наиболее предпочтительно менее 1 мол.% в расчете на количество парафинового сырья.
Обычно парафиновое сырье представляет собой смесь линейных и слаборазветвленных парафинов с различным числом углеродных атомов. Может использоваться любой подходящий способ получения указанного парафинового сырья. Предпочтительный способ представляет собой выделение неразветвленных (линейных) углеводородов или слаборазветвленных углеводородов из нефтяной фракции с пределами кипения керосина. UOP Molex™ процесс представляет собой апробированный, коммерчески состоятельный способ жидкофазного адсорбционного разделения парафинов нормального и изостроения и циклопарафинов с использованием технологии разделения UOP Sorbex. Другими подходящими процессами являются процесс UOP Kerosene Isosiv™ и парофазный адсорбционный процесс Еххоn, в котором в качестве десорбента используют аммиак. Потоки сырья для таких адсорбционных процессов могут быть получены экстракцией или подходящими способами олигомеризации. Для дополнительной информации, касающейся процессов UOP Molex™ и Kerosene Isosiv™ можно сослаться на книгу Handbook of Petroleum Refining Process. Robert A. Meyers, McGraw-Hill, New York, 1997.
Состав парафинового сырья может быть установлен методом газовой хроматографии в соответствие с работой Н. Schuiz с сотр., Chromatographia 1, 1968, 315.
Фенильное сырье включает способные к алкилированию фенильные соединения или какие-либо другие замещенные производные с более высокой молекулярной массой, чем бензол, включающие толуол, этилбензол, ксилол, фенол, нафталин, и т.п. Предпочтительным фенильным соединением является бензол.
В целях обсуждения, рассматриваемый процесс можно разделить на стадию изомеризации, стадию дегидрирования и стадию алкилирования. В стадии изомеризации, парафиновое сырье подают в зону скелетной изомеризации, где происходит уменьшение числа линейных молекул и повышается число первичных углеродных атомов в молекулах парафинового сырья. Число метальных ответвлений в алифатической алкильной цепи предпочтительно увеличивается на 2 или более предпочтительно на 1. Общее число углеродных атомов в молекуле парафина остается неизменным.
В стадии изомеризации поток материала, содержащего парафины, объединяют с рециркулирующим водородом. В результате такой операции образуется поток изомеризующего реагента, который нагревают и пропускают через слой подходящего катализатора изомеризации, находящегося при соответствующей температуре, давлении и т.п. Поток, выходящий из каталитического слоя, или поток, отходящий из реактора изомеризации, охлаждают, частично конденсируют и подают в сепаратор для системы пар - жидкость или для продукта. Сконденсированный материал, выходящий из сепаратора продукта, может вводиться в зону десорбционной сепарации, включающую отпарную колонну, в которой удаляются все компоненты, более летучие, чем наиболее легкий алифатический углеводород, который желательно подавать в секцию дегидрирования рассматриваемого процесса. С другой стороны, сконденсированный материал может подаваться на последующую стадию дегидрирования без предварительной отпарки, совместно с наиболее летучими алифатическими углеводородами, и поток дегидрированного продукта подвергают десорбции, с целью удаления всех компонентов, более летучих, чем наиболее легкий алифатический углеводород, который целесообразно подавать в секцию алкилирования. Парафинсодержащий поток, подаваемый из секции изомеризации в секцию дегидрирования, представляет собой поток изомеризованного продукта.
Скелетная изомеризация парафинового сырья может осуществляться любым известным способом или с использованием любого известного катализатора. Подходящие катализаторы включают металлы VIII группы (IUPAC 8-10) Периодической системы элементов и носитель. Подходящие металлы VIII группы включают платину и палладий, каждый из которых может использоваться в отдельности или совместно. В качестве носителя может использоваться материал, имеющий аморфную или кристаллическую структуру. Подходящие материалы-носители включают аморфный оксид алюминия, аморфные алюмосиликаты, ферриерит, ALPO-31, SAPO-11, SAPO-31, SAPO-37, SAPO-41, SM-3 и MgAPSO-31, каждый из которых может использоваться в отдельности или в комбинации с другими материалами. ALPO-31 раскрыт в патенте США US-A-4310440. SAPO-11, SAPO-31, SAPO-37 и SAPO-41 описаны в US-A-4440871. SM-3 раскрыт в US-A-4943424; US-A-5087347; US-A-5158665; и US-A-5208005. MgAPSO представляет собой частный случай MeAPSO, сокращенного обозначения металлалюмосиликатофосфатного молекулярного сита, в котором Me обозначает магний (Mg). Системы типа MeAPSO описаны в US-A-4793984, a MgAPSO описан в US-A-4758419. MgAPSO-31 является предпочтительным представителем систем типа MgAPSO, где цифра 31 обозначает MgAPSO со структурой 31. Катализатор изомеризации также может содержать модификатор, выбранный из группы, состоящей из лантана, церия, празеодима, неодима, самария, гадолиния, тербия и их смесей, в соответствие с US-A-5716897 и US-A-5851949. Предполагается, что другие подходящие материалы-носители включают ZSM-22, ZSM-23 и ZSM-35, использование которых в депарафинизации раскрыто в US-A-5246566 и в статье S.J. Miller, опубликованной в Microporous Materials 2 (1994) 439-449.
Скелетную изомеризацию парафинового сырья можно проводить в паровой фазе, жидкой фазе и комбинации указанных фаз. Углеводороды, предпочтительно, находятся в жидкой фазе. В системе может присутствовать избыток водорода относительного его количества, растворимого в жидких углеводородах. Парафиновое сырье в виде жидкости пропускают через неподвижный слой твердого катализатора в присутствии паров водорода. Обычно температура процесса изомеризации составляет 122-752°С. Давление процесса изомеризации обычно имеет значение в интервале от атмосферного до 13790 кПА (g), однако на практике, для минимизации капитальных и эксплуатационных расходов используют как можно более низкое давление. Молярное соотношение водород : углеводород обычно имеет значение выше 0,01:1, но, как правило, не более 10:1.
Поток изомеризованного продукта включает парафины с общим числом углеродных атомов, в расчете на молекулу парафина, 8-28, предпочтительно, 8-15, и более предпочтительно, 10-15. Обычно поток изомеризованного продукта содержит повышенные концентрации слаборазветвленных парафинов в расчете на общее содержание парафинов в потоке изомеризованного продукта по сравнению с концентрацией слаборазветвленных парафинов в парафиновом сырье, в расчете на общее содержание парафинов в сырье.
Слаборазветвленные парафины, содержащие два алкильных ответвления или четыре первичных углеродных атома, предпочтительно, составляют менее 40 мол.%, более предпочтительно, менее 30 мол.% от общего количества слаборазветвленных парафинов в потоке изомеризованного продукта. Слаборазветвленные парафины, содержащие одно алкильное ответвление или три первичных углеродных атома, предпочтительно, составляют более 70 мол.% от общего количества слаборазветвленных парафинов в потоке изомеризованного продукта. Слаборазветвленные парафины, содержащие 3 или 4 первичных углеродных атомов и не содержащие четвертичных углеродных атомов, предпочтительно, составляют более 25 мол.%, более предпочтительно, более 60 мол.% от количества потока изомеризованного продукта. Предпочтительными слаборазветвленными парафинами, содержащимися в потоке изомеризованного продукта, являются монометилалканы. При совместном присутствии в потоке изомеризованного продукта со слаборазветвленными парафинами содержание линейных парафинов может достигать 75 мол.%, но, как правило, оно составляет менее 40 мол.% от общего количества парафинов в потоке изомеризованного продукта. Количество парафиновых молекул, содержащих, по крайней мере, один четвертичный атом углерода, обычно составляет менее 10 мол.%, предпочтительно, менее 5 мол.%, более предпочтительно, менее 2 мол.%, и наиболее предпочтительно, менее 1 мол.% от количества потока изомеризованного продукта.
В секции дегидрирования, поток, содержащий парафины, объединяют с рециркулирующим водородом с образованием потока дегидрированного реагента, который нагревают и приводят в контакт с катализатором дегидрирования в виде неподвижного слоя, находящегося в соответствующих условиях дегидрирования. Поток, отходящий с неподвижного слоя катализатора, и поток, выходящий из реактора дегидрирования, охлаждают, частично конденсируют и подают в парожидкостной сепаратор. В парожидкостном сепараторе образуется паровая фаза, обогащенная водородом, и жидкая фаза, обогащенная углеводородом. Сконденсированную жидкую фазу, выведенную из сепаратора, подают в отпарную колонну, где удаляют все соединения, более летучие, чем наиболее легкие углеводороды, которые желательно подавать в секцию алкилирования. Затем олефинсодержащий поток подают из секции дегидрирования в секцию алкилирования и этот поток представляет собой продукт дегидрирования. Что касается дополнительной информации о LAB процессах в целом и процессах дегидрирования парафинов, в частности, можно упомянуть страницы 153-166 и 511-519 цитированной выше книги Meyers, на которую ссылаются в настоящем описании,
Катализатор дегидрирования может использоваться в виде движущегося слоя или псевдоожиженного слоя. Зона дегидрирования может включать одну или более реакционных зон, содержащих катализатор, снабженных теплообменниками, обеспечивающими поддержание требуемой температуры реакции на входе в каждую реакционную зону. Горячие потоки газа, обогащенного водородом, могут вводиться в пространства между реакционными зонами с целью подогрева потока, проходящего между реакционными зонами. Для дополнительной информации по этому вопросу, можно сослаться на US-A-5491275 и US-A-5689029. Каждая реакционная зона может работать в непрерывном или периодическом режимах. Каждая реакционная зона может содержать один или более каталитических слоев. Углеводороды могут контактировать с любым каталитическим слоем при их движении вверх, вниз или по радиусу, либо указанная операция может осуществляться в теплообменном реакторе. Для получения дополнительной информации, касающейся теплообменных реакторов, можно обратиться к содержанию патентов US-A-5405586 и US-A-5525311, на которые ссылаются в настоящем описании.
Катализаторы дегидрирования хорошо известны из предшествующего уровня техники, примерами которого могут служить US-A-3274387; US-A-3315007; US-A-3315008; US-A-3745112; US-A-4430517; US-A-4716143; US-A-4762960; US-A-4786625, и US-A-4827072. Однако предпочтительный катализатор представляет собой слоевую композицию, включающую внутреннюю сердцевину и внешний слой, связанный с внутренним ядром, причем внешний слой содержит тугоплавкий неорганический оксид, в котором однородно диспергирован, по крайней мере, один металл группы платины (группу VIII (IUPAC 8-10) и, по крайней мере, один металл-промотор, а в каталитической композиции диспергирован, по крайней мере, один модифицирующий металл. Предпочтительно, чтобы внешний слой был связан с внутренним ядром в такой степени, чтобы потери при истирании не превышали 10 мас.% в расчете на вес внешнего слоя. Для получения дополнительной информации по слоевым каталитическим композициям можно упомянуть заявку на патент США №09/185189 от 3 ноября 1998 г, на которую ссылаются в настоящем описании.
Условия проведения дегидрирования подбирают таким образом, чтобы уменьшить крекинг, изомеризацию и образование полиолефиновых побочных продуктов. Углеводород может находиться в жидкой фазе или в смешанной парожидкостной фазе, но предпочтительно, в паровой фазе. Условия дегидрирования включают температуру, обычно, в интервале 400-900°С, предпочтительно, 400-525°С, давление в интервале 1-1013 кПА (g) и LHSV в интервале 0,1-100 час-1. Давление поддерживают на как можно более низком значении, обычно оно имеет величину менее 345 кПА (g), в соответствии техническими ограничениями технологического оборудования, с целью максимально благоприятного смещения химического равновесия.
Поток продукта изомеризации может быть смешан с таким разбавителем как водород до, во время или после подачи в зону дегидрирования, при молярном соотношении водород : углеводород в интервале 0,1:1-40:1, предпочтительно, 1:1-10:1. Поток разбавляющего водорода, подаваемый в зону дегидрирования, обычно представляет собой рециркулирующий водород, выделенный из потока, отходящего из зоны дегидрирования.
Вода или материал, разлагающийся в условиях дегидрирования с образованием воды, например спирт, альдегид, эфир или кетон, могут непрерывно или периодически вводиться в зону дегидрирования в количестве, составляющем в расчете на водный эквивалент, 1-20000 вес.ч./млн от потока углеводородного сырья. Добавление воды в количестве 1-10000 вес.ч./млн обеспечивает наилучшие результаты в том случае, когда дегидрируемые парафины содержат 2-30 или более углеродных атомов.
Поток продукта дегидрирования, содержащий моноолефины, из процесса дегидрирования парафинов, обычно, представляет собой смесь непрореагировавших парафинов, линейных (неразветвленных) олефинов и разветвленных моноолефинов, включающих слаборазветвленные моноолефины. Обычно, 25-75 об.% олефинов в моноолефинсодержащем потоке из процесса дегидрирования парафина составляют линейные (неразветвленные) олефины.
Поток продукта дегидрирования может содержать высокоразветвленный моноолефин или линейный (неразветвленный) олефин, однако, предпочтительно, чтобы моноолефин представлял собой слаборазветвленный моноолефин. Слаборазветвленный моноолефин представляет собой моноолефин с общим числом углеродных атомов 8-28, три или четыре из которых являются первичными углеродными атомами, среди оставшихся углеродных атомов отсутствуют четвертичные углеродные атомы. Предпочтительно, слаборазветвленный моноолефин содержит 8-15, более предпочтительно, 10-15 углеродных атомов.
Слаборазветвленный моноолефин обычно включает алифатический алкен, отвечающий общей формуле (рiалкилi)i-q-алкен. Слаборазветвленный моноолефин имеет алифатическую алкенильную цепочку, представляющую собой самую длинную прямую цепочку, содержащую углерод-углеродную двойную связь слаборазветвленного моноолефина, и одно или более алкильных ответвлений, каждое из которых присоединено к алифатической алкенильной цепочке. Если существует возможность подбора двух или более цепочек одинаковой длины в качестве алифатической алкенильной цепочки, то такой подбор обеспечит цепочку с наибольшим числом алкильных ответвлений. Таким образом, подстрочный символ i имеет значение от 1 до числа алкильных ответвлений, причем для каждого значения i имеется соответствующее алкильное ответвление, присоединенное к углеродному атому с номером рi алифатической алкенильной цепочки. Двойная связь располагается между углеродом с номером q и углеродом с номером (q+1) алифатической алкенильной цепочки. Алифатическая алкенильная цепочка пронумерована в направлении от одного конца к другому, причем направление нумерации выбирают таким образом, чтобы углеродные атомы, несущие двойную связь, имели наименьшие возможные номера.
Слаборазветвленный моноолефин может представлять собой альфа моноолефин или винилиденовый моноолефин, но, предпочтительно, он представляет собой внутренний моноолефин. Используемый в тексте термин “внутренние олефины” охватывает дизамещенные внутренние олефины, имеющие химическую формулу R-CH=CH-R; тризамещенные внутренние олефины имеют химическую формулу R-C(R)=CH-R; a тетразамещенные олефины имеют химическую формулу R-C(R)=C(R)-R. Дизамещенные внутренние олефины включают бета внутренние олефины, отвечающие химической формуле R-СН=СН-(СН3). В каждой из приведенных в данном параграфе химических формул R представляет собой алкильную группу, которая может быть идентичной или отличаться от других алкильных групп, если они имеются в каждой из указанных формул.
В случае слаборазветвленных моноолефинов, отличных от винилиденовых олефинов, алкильное ответвление или ответвления слаборазветвленного моноолефина обычно выбирают из метальных, этильных и пропильных групп, причем укороченные и нормальные ответвления являются предпочтительными. В отличие от этого, в случае слаборазветвленных моноолефинов винилиденового типа алкильное ответвление, присоединенное к углеродному атому с номером 2 алифатической алкенильной цепочки, может быть выбрано не только из метальной, этильной и пропильной групп, но также из алкильных групп вплоть до тетрадецильных (С14) групп, хотя любые другие алкильные ответвления винилиденового олефина обычно выбирают из метальной, этильной и пропильной групп, причем укороченные и нормальные ответвления являются предпочтительными. Для всех слаборазветвленных моноолефинов предпочтительный моноолефин такого типа содержит только одно алкильное ответвление, хотя также возможно наличие двух алкильных ответвлений. Слаборазветвленные моноолефины, содержащие два алкильных ответвления или четыре первичных углеродных атома, обычно составляют менее 40 мол.%, и, предпочтительно, менее 30 мол.% от общего количества слаборазветвленных моноолефинов, причем оставшиеся слаборазветвленные моноолефины имеют одно алкильное ответвление. Слаборазветвленные моноолефины, имеющие одно алкильное ответвление или три первичных углеродных атома, предпочтительно, составляют более 70 мол.% от общего количества слаборазветвленных моноолефинов. Монометилалкены представляют собой предпочтительные слаборазветвленные моноолефины, содержащиеся в потоке продукта дегидрирования.
Винилиденовые моноолефины обычно представляют собой примесный компонент и обычно присутствуют в концентрации менее 0,5 мол.% и, как правило, менее 0,1 мол.% от количества олефинов в потоке продукта дегидрирования. Поэтому, все последующие ссылки на слаборазветвленные моноолефины и поток продукта дегидрирования предполагают отсутствие винилиденовых моноолефинов.
Состав смеси слаборазветвленных моноолефинов может быть установлен методом газовой хроматографии в соответствии с методикой ранее упомянутой статьи Schuiz и другие с использованием инжектора, снабженного гидрогенераторной вставной трубкой для гидрирования моноолефинов в парафины.
Помимо слаборазветвленных моноолефинов поток продукта дегидрирования может содержать другие ациклические соединения. Одним из преимуществ настоящего изобретения является тот факт, что поток продукта дегидрирования может непосредственно подаваться в секцию для проведения реакции алкилирования, несмотря на тот факт, что поток также содержит парафины с тем же числом углеродных атомов, что и слаборазветвленные моноолефины. Таким образом, настоящее изобретение делает необязательным отделение парафинов от моноолефинов перед подачей в секцию алкилирования. Другие ациклические соединения включают неразветвленные (линейные) олефины и моноолефины. Неразветвленные (линейные) олефины, которые могут загружаться в реактор, обычно имеют общее число углеродных атомов в расчете на молекулу парафина в интервале 8-28, предпочтительно, 8-15 и более предпочтительно 10-14 углеродных атомов. Два углеродных атома на молекулу неразветвленного олефина представляют собой первичные углеродные атомы, а оставшиеся углеродные атомы представляют собой вторичные углеродные атомы. Неразветвленный олефин может представлять собой альфа моноолефин, но, предпочтительно, представляет собой внутренний моноолефин. Содержание линейных олефинов может достигать 75 мол.%, но обычно оно составляет менее 40 мол.% от общего количества моноолефинов в потоке продукта дегидрирования.
Поток продукта дегидрирования может содержать, в среднем, около 3, или 2,25-4, или 3-4 первичных углеродных атомов в расчете на молекулу моноолефина в потоке продукта дегидрирования.
Линейные и/или нелинейные парафины в потоке продукта дегидрирования обычно имеют общее содержание углеродных атомов в расчете на молекулу парафина в интервале 8-28, предпочтительно 8-15 и более предпочтительно 10-14 углеродных атомов. Нелинейные парафины в потоке продукта дегидрирования могут содержать слаборазветвленные парафины и парафины, содержащие, по крайней мере, один четвертичный атом углерода. Ожидается, что рассматриваемые линейные и нелинейные парафины будут выполнять функции разбавителя на стадии алкилирования и не будут препятствовать проведению стадии алкилирования. Однако присутствие таких разбавителей в реакторе алкилирования обычно приводит к увеличению объемных скоростей технологических потоков.
Предпочтительно минимизировать в потоке продукта дегидрирования количество моноолефинов, которые более высоко разветвлены, чем слаборазветвленные моноолефины. Молекулы моноолефинов, содержащие, по крайней мере, один четвертичный углеродный атом, обычно составляют менее 10 мол.%, предпочтительно менее 5 мол.%, более предпочтительно менее 2 мол.% и наиболее предпочтительно менее 1 мол.% от потока продукта дегидрирования.
Слаборазветвленные моноолефины реагируют с фенильным соединением. В общем случае, слаборазветвленные моноолефины могут реагировать с другими фенильными соединениями помимо бензола, например с алкилированными, или как-либо иначе замещенными производными бензола, включающими толуол и этилбензол, однако бензол представляет собой предпочтительное фенильное соединение. Хотя стехиометрия реакции алкилирования требует расхода 1 моля фенильного соединения на моль моноолефинов, использование мольного соотношения 1:1 приводит к чрезмерной полимеризации олефина и полиалкилированию. С другой стороны, желательно использовать мольные соотношения фенильное соединение : моноолефин, как можно более близкие к 1:1 с тем, чтобы повысить утилизацию фенильного соединения и уменьшить количество рециркулирующего или непрореагировавшего фенильного соединения. Используемое молярное соотношение между фенильным соединением и общим количеством моноолефинов будет оказывать важное действие как на конверсию, так и на селективность реакции алкилирования. Общее молярное соотношение фенильное соединение : моноолефин обычно может иметь значение в интервале 2,5:1-50:1, как правило, 8:1-35:1.
Реакцию между фенильным производным и слаборазветвленным моноолефином проводят в условиях алкилирования в присутствии твердого катализатора алкилирования. Упомянутые условия алкилирования включают температура в интервале 80-200°С, обычно не выше 175°С. Поскольку алкилирование проводят, по крайней мере, в частично жидкой фазе, и, предпочтительно, в полностью жидкой фазе или при сверхкритических условиях, должны использоваться достаточные давления для удерживания реагентов в жидкой фазе. Требуемое давление обязательно зависит от природы олефина, фенильного производного и температуры, но обычно оно составляет 1379-6895 кПа (g), наиболее предпочтительно, 2069-3448 кПа (g).
Хотя условия алкилирования достаточны для алкилирования фенильного производного слаборазветвленными моноолефинами, предполагается, что в условиях алкилирования скелетная изомеризация слаборазветвленных моноолефинов реализуется лишь в незначительной степени. Под скелетной изомеризацией олефина в условиях алкилирования подразумевается изомеризация, происходящая в ходе алкилирования и изменяющая число углеродных атомов в алифатической алкенильной цепочке олефина, в алифатической алкильной цепочке фенилалканового продукта или в любом реакционном промежуточном продукте, который образуется, или является производным слаборазветвленного моноолефина до вывода фенилалканового продукта из условий алкилирования. Обычно, менее 15 мол.% и предпочтительно менее 10 мол.% олефина, алифатической алкильной цепочки и любого реакционного промежуточного продукта подвергаются скелетной изомеризации. Также считается, что любые другие олефины, содержащиеся в олефиновом сырье, подвергаются скелетной изомеризации лишь в минимальной степени. Таким образом, алкилирование, предпочтительно, происходит без существенной скелетной изомеризации слаборазветвленного моноолефина и степень разветвления слаборазветвленного моноолефина идентична степени слабого разветвления в алифатической алкильной цепочке молекулы фенилалканового продукта. Соответственно, число первичных атомов углерода в слаборазветвленном моноолефине, предпочтительно, равно числу первичных атомов углерода в молекуле фенилалкана. Однако количество первичных углеродных атомов в фенилалкановом продукте может несколько превышать или быть несколько меньшим числа первичных углеродных атомов в слаборазветвленном моноолефине.
Алкилирование фенильного соединения слаборазветвленными моноолефинами приводит к образованию (m-iалкилi)i-n-фенилалканов, алифатическая алкильная группа которых содержит два, три или четыре первичных углеродных атома в расчете на молекулу фенилалкана. Предпочтительно, алифатическая алкильная группа содержит три первичных углеродных атома на молекулу фенилалкана и более предпочтительно, когда один из трех первичных углеродных атомов представляет собой метальную группу на одном конце алифатической алкильной цепочки, второй первичный углеродный атом представляет собой метальную группу на другом конце цепочки, а третий первичный углеродный атом представляет собой изолированное метальное ответвление, соединенное с цепочкой. Обычно, 0-75 мол.% и предпочтительно 0-40 мол.% полученных (m-iалкилi)i-n-фенилалканов могут содержать 3 первичных углеродных атома в расчете на молекулу фенилалкана. Обычно, как можно большее количество полученных (m-iалкилi)i-n-фенилалканов, как правило, 25-100 мол.% могут содержать 3 первичных углеродных атома на молекулу фенилалкана. Обычно, 0-40 мол.% полученных (m-iалкилi)i-n-фенилалканов может содержать 4 первичных углеродных атома. Предпочтительными веществами являются монометилфенилалканы. Число первичных, вторичных и третичных углеродных атомов в расчете на молекулу полученного фенилалкана может быть определено по спектру многоимпульсного ядерного магнитного резонанаса (ЯМР) и неискаженным усилением в результате переноса поляризации (DEPT). Дополнительная информация по этому вопросу может быть получена из брошюры "High Resolution Multipulse NMR Spectrum Editing and DEPT", выпущенной Broker Instruments, Inc., Manning Park, Billerica, Massachusetts, USA.
Алкилирование фенильного производного слаборазветвленными олефинами обычно протекает с селективностью по 2-фенилалканам 40-100, и, предпочтительно, 60-100%, тогда как селективность по внутренним четвертичным фенилалканам обычно составляет менее 10%, предпочтительно, менее 5%. Четвертичные фенилалканы могут образовываться в результате алкилирования фенильного производного слаборазветвленными моноолефинами, содержащими, по крайней мере, один третичный углеродный атом. Полученный в результате фенилалкан может представлять собой внутренний углеводород или кват.
Алкилирование фенильного производного слаборазветвленными моноолефинами может осуществляться периодическим способом, или, предпочтительно, непрерывным способом. Катализатор алкилирования может использоваться в виде неподвижного слоя или псевдоожиженного слоя. Олефиновое сырье может подаваться в зону реакции снизу вверх, или сверху вниз, или даже горизонтально, как это имеет место в реакторе с радиальным слоем катализатора. Смесь бензола и олефинового сырья, содержащего слаборазветвленные моноолефины, вводят в реактор при общем молярном соотношении фенильное производное : моноолефин в интервале 2,5:1-50:1, хотя, как правило, используют интервал 8:1-35:1. Олефин может вводиться в несколько отдельных точек реакционной зоны и в каждой из таких зон молярное соотношение фенильное производное : моноолефин может быть выше, чем 50:1, хотя общее соотношение бензол : олефин остается в рамках указанного выше интервала. Общая сырьевая смесь, т.е. фенильное производное плюс олефиновое сырье, содержащее слаборазветвленные моноолефины, подается через плотной слой катализатора с часовой объемной скоростью по жидкому сырью (LHSV) в интервале 0,3-6 час-1, в зависимости от температуры алкилирования, продолжительности использования катализатора и т.п.
Более низкие значения LHSV в рамках указанного выше интервала являются предпочтительными. Температура в зоне реакции поддерживается в интервале 80-200°С, а давления обычно могут меняться в интервале 1379-6895 кПа (g) с целью обеспечения жидкой фазы или создания сверхкритических условий. После прохождения фенильного производного и олефинового сырья через реакционную зону эффлюент собирают и разделяют на непрореагировавшее фенильное производное, которое рециркулируют в точку подачи в зону реакции, парафин, который рециркулируют в установку дегидрирования и фенилалканы. Обычно, фенилалканы дополнительно разделяют на моноалкилбензолы, используемые в последующем сульфировании с образованием алкилбензолсульфонатов, и олигомеры плюс полиалкилбензолы. Поскольку, обычно реакция протекает до конверсии около 98% в расчете на моноолефин, с парафином рециркулируется лишь незначительное количество моноолефина.
В настоящем изобретении может использоваться любой подходящий катализатор алкилирования при условии, что он удовлетворяет требованиям, предъявляемым к конверсии, селективности и активности. Предпочтительные катализаторы алкилирования включают цеолиты с типом структуры, выбранным из BEA, MOR, MTW и NES. Такие цеолиты включают морденит, ZSM-4, ZSM-12, ZSM-20, оффретит, гмелинит, бета, NU-87 и готтардиит. Такие термины, касающиеся структуры цеолитов, как “тип цеолитной структуры” и “структура изотипической решетки” используются в тексте в соответствии с их определением и использованием W.M.Meier с сотр. в Atlas of Zeolite Structure Types, опубликованном Structure Commission of the International Zeolite Association издательством Elsevier, Boston, Massachusetts, USA, четвертое исправленное издание, 1996. Реакции алкилирования с использованием NU-87 и NU-85, представляющими собой проросшие формы цеолитов EU-1 и NU-87, описаны в патентах США 5041402 и 5446234, соответственно. Готтардиит, имеющий структуру изотипической решетки структурного типа NES цеолита, раскрыт в работах A. Alberti с сотр., в Eur.J. Mineral., 8, 69-75 (1996) и E.Galli с сотр., в Eur.J. Mineral., 8, 687-693 (1996). Наиболее предпочтительный катализатор алкилирования представляет собой морденит.
Цеолиты, подходящие для использования в качестве катализаторов алкилирования, обычно содержат, по крайней мере, 10% катионных центров, которые заняты ионами, отличными от ионов щелочных или щелочноземельных металлов. Такие другие ионы включают, но не ограничиваются ими, водород, аммоний, ионы редкоземельных элементов, цинка, меди и алюминий. Особенно предпочтительная группа элементов такого типа включает аммоний, водород, редкоземельные элементы или их комбинации. В соответствие с предпочтительным воплощением настоящего изобретения, указанные цеолиты переводят в преимущественно водородную форму, обычно, в результате замещения исходного иона щелочного металла или другого иона предшественником иона водорода, например ионами аммония, которые в результате обжига переводят в водородную форму. Указанный обмен удобно осуществлять путем взаимодействия цеолита с раствором соли аммония, например хлористого аммония, с использованием хорошо известной ионообменной технологии. Согласно некоторым техническим решениям используют такую степень замещения, чтобы получить цеолитный материал, в котором, по крайней мере, 50 процентов катионных центров заняты ионами водорода.
Цеолиты могут подвергаться различным химическим обработкам, включающим экстракцию алюминия (деалюминирование) и комбинацию с одним или более металлическими компонентами, например металлами IIIB (IUPAC 3), IVB (IUPAC 4), VIB (IUPAC 6), VIIB (IUPAC 7), VIII (IUPAC 8-10) и IIB (IUPAC 12) групп. В некоторых случаях предусматривается желательная термообработка цеолитов, включающая отпаривание или обжиг на воздухе, в среде водорода или такого инертного газа как азот или гелий. Подходящая обработка отпариванием включает контактирование цеолита с атмосферой, содержащей 5-100% пара при температуре 250-1000°С. Отпаривание можно осуществлять в течение 0,25-100 часов при давлении в интервале от субатмосферного до нескольких сот атмосфер.
В соответствие с настоящим изобретением, полезно вводить используемые цеолиты в другой материал, например материал-матрицу или связующее вещество, которые обладают устойчивостью к действию температуры и других условий, используемых при проведении процесса. Подходящие матричные материалы включают синтетические вещества, вещества естественного происхождения и такие неорганические материалы, как глина, оксид кремния, и/или оксиды металлов. Матричные материалы могут применяться в виде гелей, включающих смеси оксида кремния и оксидов металлов.
Гелевые смеси из оксида кремния и оксидов металлов могут иметь естественное происхождение или применяться в виде гелей или гелевых осадков. Глины природного происхождения, которые могут соединяться с цеолитами, используемыми в настоящем изобретении, включают глины из семейства монтмориллонита и каолина, причем указанное семейство включает суб-бентониты и каолины, традиционно известные как глины Dixie, McNamee-Georgia и Florida и др., основным минеральный компонентом которых может служить галлуизит, каолинит, диксит, накрит или анауксит. Указанные глины могут использоваться в качестве матричного материала в виде только что добытого материала или могут быть подвергнуты обжигу, кислотной обработке или химической модификации до их применения в качестве матричных материалов.
Помимо перечисленных материалов, цеолиты, используемые в настоящем изобретении, могут соединяться с такими пористыми матричными материалами, как оксид алюминия, смешанный оксид кремния и алюминия, смешанный оксид кремния и магния, смешанный оксид кремния и циркония, смешанный оксид кремния и тория, смешанный оксид кремния и бериллия, смешанный оксид кремния и титана и фосфат алюминия, а также такими тройными комбинациями, как смесь оксидов кремния, алюминия и тория, оксидов кремния, алюминия и циркония, оксидов кремния, алюминия и магния, оксидов кремния, магния и циркония. Указанный матричный материал может иметь форму совместного геля. Соотношения между цеолитом и матричным материалом могут изменяться в широких пределах, причем содержание цеолита обычно составляет 1-99 мас.%, как правило 5-80 мас.% и, предпочтительно, 30-80 мас.% от общей массы цеолита и матричного материала.
Цеолиты, используемые в катализаторах алкилирования, обычно имеют молярное соотношение решеточных оксида кремния и оксида алюминия в интервале 5:1-100:1. В том случае, когда цеолит катализатора алкилирования представляет собой морденит, его решеточное молярное соотношение оксид кремния : оксид алюминия составляет 12:1-90:1, предпочтительно, 12:1-25:1. Термин "решеточное молярное отношение оксид кремния: оксид алюминия" относится к молярному соотношению между оксидом кремния и оксидом алюминия, представляющему молярное отношение SiO2/Al2O3 в решетке цеолита.
В том случае, когда цеолиты готовят в присутствии органических катионов, они могут оказаться недостаточно каталитически активными для проведения реакции алкилирования. Не ограничиваясь конкретной теорией, можно предположить, что недостаточная каталитическая активность является результатом действия органических катионов из образующегося раствора, которые занимают внутрикристаллическое свободной пространство. Такие катализаторы можно активировать, например, путем нагревания в инертной атмосфере при 540°С в течение одного часа, проведения ионообменной реакции с солями аммония и обжига в атмосфере воздуха при 540°С. Наличие органических катионов в образующемся растворе может оказаться существенным фактором для образования конкретных цеолитов. Некоторые нейтральные цеолиты иногда могут превращаться в цеолиты желаемого типа с помощью различных методов активации и других методов обработки, например ионного обмена, отпаривания, удаления алюминия, и обжига. В том случае, когда цеолит синтезирован в форме на основе щелочного металла, его легко превратить в водородную форму, обычно путем промежуточного образования аммониевой формы в результате обмена на ион аммония и обжига аммониевой формы с образованием водородной формы. Хотя водородная форма цеолитов успешно катализирует реакцию, рассматриваемый цеолит может также частично находиться в форме, содержащей щелочной металл.
В зоне селективного алкилирования образуется соответствующий эффлюент, поступающий в разделительное устройство с целью регенерации продуктов реакции и рециркулируемых компонентов сырья. Поток, отходящий из зоны селективного алкилирования, поступает в колонну, орошаемую бензолом, в которой образуется выходящий сверху поток, содержащий бензол, и нижний поток, содержащий алкилат. Донный поток поступает в парафиновую колонну, в которой образуется верхний поток жидкости, содержащий непрореагировавшие парафины и донный поток, содержащий алкилат и высокомолекулярные побочные углеводороды, образующиеся в зоне селективного алкилирования. Рассматриваемый донный поток из парафиновой колонны может подаваться в колонну вторичной перегонки, в которой образуется выходящий сверху материальный поток алкилата, содержащий поверхностно-активный алкилат, и поток, выходящий со дна колонны вторичной перегонки, содержащий полимеризованные олефины и полиалкилированные бензолы (тяжелый алкилат). С другой стороны, если содержание тяжелого алкилата в донном потоке парафиновой колонны незначительно, потребность в колонне вторичной перегонки отпадает и донной поток из парафиновой колонны может выделяться, в виде целевого технологического потока поверхностно-активного алкилата.
В соответствии с настоящим изобретением, по крайней мере, часть головного жидкого потока из парафиновой колонны рециркулируют в зону изомеризации, зону дегидрирования или обе эти зоны. Предпочтительно, чтобы часть головного жидкого потока из парафиновой колонны, которую рециркулируют в зону изомеризации или зону дегидрирования, представляла собой аликвоту головного потока жидкости. Аликвотная часть головного потока жидкости представляет собой часть головного потока, которая имеет практически тот же состав, что головной поток жидкости. Головной поток из парафиновой колонны включает парафины с общим числом углеродных атомов, в расчете на молекулу парафина, обычно составляющим 8-28, предпочтительно, 8-15 и более предпочтительно, 10-15 углеродных атомов. Предпочтительно, по крайней мере, часть головного потока жидкости из парафиновой колонны рециркулируют только в зону дегидрирования. Обычно, 50-100 мас.% головного потока жидкости из парафиновой колонны рециркулируют в зону изомеризации и/или зону дегидрирования и, предпочтительно, весь головной поток жидкости из парафиновой колонны рециркулируют только в зону дегидрирования.
Независимо от того, осуществляют ли рециркуляцию в зоне изомеризации или зоне дегидрирования, головной поток из парафиновой колонны может содержать как неразветвленные (линейные) парафины, так и слаборазветвленные парафины, даже в том случае, когда на переработку поступают только неразветвленные парафины. Это происходит в связи с тем, что примерно 60-80 мас.% неразветвленных парафинов, вводимых в зону скелетной изомеризации, превращаются в слаборазветвленные парафины, в зоне дегидрирования 10-15 мас.% вводимых парафинов, обычно превращаются в олефины и фракция олефинов в потоке продуктов дегидрирования, представляющая собой слаборазветвленные олефины, примерно соответствует количеству парафинов в потоке изомеризованного продукта, представляющего собой слаборазветвленные парафины. Поскольку конверсия олефинов в зоне алкилирования обычно превышает 90 мас.% от подаваемого количества олефинов, как правило более 98 мас.%, и поскольку конверсия парафинов в зоне алкилирования практически равна нулю, эффлюент из зоны алкилирования будет содержать слаборазветвленные парафины.
Для иллюстрации сказанного на практике полезно рассмотреть начальную стадию процесса, когда в зону изомеризации подаются лишь линейные парафины и в указанной зоне происходит превращение х мас.% подаваемых на обработку неразветвленных парафинов в слаборазветвленные парафины. Слаборазветвленные парафины начинают образовываться в головном потоке парафиновой колонны. По мере рециркуляции слаборазветвленных парафинов в зону изомеризации состав смеси парафинов поданных в зону изомеризации, будет постепенно изменяться от смеси, содержащей только неразветвленные парафины, до смеси, содержащей неразветвленные и слаборазветвленные прарафины. Соответственно, далее, зона изомеризации может работать в условиях, когда конверсия нелинейных парафинов составляет величину менее x мас.%. С течением времени степень конверсии изомеризации может претерпевать дальнейшую корректировку до момента установления стационарного состояния, при котором скорость конверсии неразветвленных парафинов в слаборазветвленные парафины в зоне изомеризации, выраженная в молях на единицу времени, становится примерно равной низшему значению скорости, с которой МАВ фенилалканы выводятся из процесса.
Концентрация моноолефинов в головном потоке жидкости парафиновой колонны обычно составляет менее 0,3 мас.%. Моноолефины, содержащиеся в головном потоке жидкости, из парафиновой колонны могут рециркулироваться в зону изомеризации и/или зону дегидрирования. Концентрация парафинов в потоке жидкости из парафиновой колонны, содержащих, по крайней мере, один четвертичный атом углерода, предпочтительно, имеет минимальное значение.
Один из вариантов способа настоящего изобретения включает селективное гидрирование диолефинов, содержащихся в потоке продукта дегидрирования, поскольку диолефины могут образовываться в ходе каталитического дегидрирования парафинов. В результате селективного гидрирования диолефины превращают в моноолефины и образуют поток продукта селективного гидрирования олефинов, имеющий меньшую концентрацию диолефинов, чем поток продукта дегидрирования.
Другой вариант способа настоящего изобретения включает селективное удаление ароматических побочных продуктов, содержащихся в потоке продукта дегидрирования. Ароматические побочные продукты могут образовываться в ходе каталитического дегидрирования парафинов и такие побочные продукты могут оказывать ряд нежелательных воздействий на процесс. Подходящие зоны удаления ароматики включают зоны сорбционного разделения, содержащие такой сорбент как молекулярные сита и, в особенности, 13Х цеолит (натриевая форма цеолита X), а также зоны экстракции в системе жидкость - жидкость. Ароматические побочные продукты могут селективно удаляться из потока продукта дегидрирования, но также, или вместо этого, из потока продукта изомеризации и/или головного потока жидкости из парафиновой колонны, которые рециркулируют в зону изомеризации или зону дегидрирования. В том случае, когда рассматриваемый процесс включает зону селективного гидрирования диолефинов, ароматические побочные продукты могут селективно удаляться из потока продукта селективного гидрирования диолефинов. Хотя селективное удаления указанных ароматических побочных продуктов предпочтительно осуществлять непрерывно, такое селективное удаление может проводиться попеременно или периодически. Подробная информация, касающаяся селективного удаления ароматических побочных продуктов из процесса производства LAB, приведенная в патенте US-A-5276231, на которую ссылаются в настоящем описании, может рассматриваться как дополнительная информация. Предполагается, что специалист в данной области техники сумеет адаптировать данные патента US-A-5276231, касающиеся удаления ароматических побочных продуктов, для успешного удаления ароматических побочных продуктов из процесса производства МАВ.
Другим аспектом настоящего изобретения являются композиции на основе МАВ, получаемые с помощью описанного способа.
Согласно еще одному аспекту настоящего изобретения, предусматривается применение композиция на основе МАВ, получаемых описанным способом, в качестве смазок. Полагают, что рассматриваемые фенилалканы обладают такими свойствами, касающимися вязкости, зависимости вязкости от температуры и плотности, которые делают их привлекательными материалами для использования в качестве нефтяных смазок. Применение фенилалканов в качестве смазочных материалов описано, например, в статье E.R.Booser в Kirk-Othmer Encyclopedia of Chemical Technology, четвертое издание, т. 15. John Wiley and Sons, New York, США, 1995, стр. 463-517, на содержание которой можно сослаться как на материал, описывающий указанные смазки и их применение.
Приведенный чертеж демонстрирует предпочтительную обобщенную схему способа настоящего изобретения, включающую стадии изомеризации - дегидрирования - алкилирования.
Парафиновое сырье, включающее смесь нормальных парафинов C10-C13, загружается в систему по линии 12. Нормальные парафины в линии 12 смешивают с водородсодержащим потоком из линии 22 и смесь подают по линии 16. Смесь парафинов и водорода, подаваемую по линии 16, вначале нагревают в непрямом теплообменнике 18 и затем подают по линии 24 в печь 20. С другой стороны, вместо показанного на чертеже варианта, когда смешивание водородсодержащего потока в линии 22 с нормальными парафинами проводят сверху от теплообменника 18 и нагревателя 20, поток в линии 22 можно смешивать с нормальными парафинами в точке, расположенной между теплообменником 18 и нагревателем 20, или между нагревателем 20 и реактором 30. Полученную в результате смесь водорода и жидких парафинов подают по линии 26 в реактор изомеризации 30. В реакторе 30 парафины в результате присутствия катализатора изомеризации приводятся в такое состояние, которое обеспечивает превращение значительной части нормальных парафинов в слаборазветвленные парафины. В результате образуется отходящий поток из реактора изомеризации, отводимый по линии 28, который содержит смесь водорода, нормальных парафинов и слаборазветвленных парафинов. Эффлюент из реактора изомеризации вначале охлаждается в результате непрямого теплообмена в теплообменнике 18 и затем, после подачи по линии 32, подвергается дополнительному охлаждению в непрямом теплообменнике 34. Обеспечивается достаточное охлаждение для конденсации всех С10+ углеводородов с образованием потока жидкой фазы и отделения такого потока от оставшегося пара, обогащенного водородом. Затем поток эффлюента из реактора изомеризации подают по линии 36 в сосуд 38 для разделения системы пар - жидкость, в котором поток разделяют на поток паровой фазы, обогащенной водородом, выводимый по линии 40, и поток продукта изомеризации, отводимый по линии 50. Парофазный поток разделяют на очищающий поток, предназначенный для отвода легких углеводородов C1-C7 по линии 42, поток водорода, который рециркулируют по линии 44. Поток водорода из линии 44 объединяют с потоком водорода, подаваемым по линии 46. Комбинация потока водорода из линии 44 со свежим потоком в линии 46 обеспечивает создание рециркулирующего потока, подаваемого по линии 22.
Поток продукта изомеризации, отводимый снизу разделительного сосуда 38, содержит нормальные парафины, слаборазветвленные парафины и некоторое количество растворенного водорода. Затем поток продукта изомеризации, представляющий собой часть жидкой фазы эффлюента, из разделительного сосуда 38 подают по линии 50 и объединяют с рециркулирующими парафинами в линии 48. Объединенный поток парафинов подается по линии 54 и смешивается с рециркулирующим водородом из линии 82 с образованием смеси парафинов и водорода, которая подается по линии 56. Смесь парафинов и водорода, подаваемая по линии 56, вначале нагревают в непрямом теплообменнике 58 и затем подают по линии 62 в печь 60. Двухфазная смесь водорода и жидких парафинов, выходящая из печи 60, подается по линии 64 в реактор дегидрирования 70. В реакторе дегидрирования 70 парафины контактируют с катализатором дегидрирования в условиях, обеспечивающих конверсию значительного количества парафинов в соответствующие олефины. В результате этого образуется поток эффлюента из реактора дегидрирования, подаваемый по линии 66, который содержит смесь водорода, парафинов, моноолефинов, включающих слаборазветвленные моноолефины, диолефинов, углеводородов до С9 и ароматических углеводородов. Такой поток, отходящий из реактора дегидрирования, вначале охлаждают методом непрямого теплообмена в теплообменнике 58, подают по линии 68 и затем снова охлаждают в непрямом теплообменнике 72. Такое охлаждение оказывается достаточным для конденсации практически всех углеводородов С10+ в поток жидкой фазы и отделения потока жидкой фазы от оставшегося пара, обогащенного водородом. Эффлюент из реактора дегидрирования подается по линии 74 в сосуд для разделения системы пар - жидкость 80. В сепарационном сосуде 80 поток эффлюента из реактора дегидрирования разделяют на поток паровой фазы, обогащенный водородом, отводимый по линии 76, и поток продуктов дегидрирования, отводимый по линии 84. Поток паровой фазы разделяют на поток чистого водорода, отводимый по линии 78 и водородсодержащий поток, рециркулирующий по линии 82.
Поток продуктов дегидрирования, отводимый снизу сепарационного сосуда 80, содержит парафины, слаборазветвленные парафины, нормальные моноолефины, слаборазветвленные моноолефины, углеводороды до С9, диолефины, ароматические побочные продукты и некоторое количество растворенного водорода. Поток продуктов дегидрирования, представляющий собой эффлюент жидкой фазы из сепарационного сосуда 80, подают по линии 84 в реактор селективного гидрирования 86. В реакторе селективного гидрирования 86 поток продуктов дегидрирования приводится в контакт с катализатором селективного гидрирования в условиях, которые обеспечивают конверсию значительной части диоолефинов в соответствующие моноолефины. Такое превращение путем гидрирования может осуществляться с использованием водорода, растворенного в потоке продуктов дегидрирования и/или дополнительного свежего водород (не показано), подаваемого в реактор селективного гидрирования. В результате образуется поток эффлюента из реактора селективного гидрирования, подаваемый в линию 88, содержащий смесь водорода, нормальных парафинов, слаборазветвленных парафинов, нормальных моноолефинов, слаборазветвленных моноолефинов, углеводородов до С9 и ароматических побочных продуктов. Затем эффлюент из реактора селективного гидрирования подают по линии 88 в отпарную колонну 90. В такой колонне отпаривания углеводороды до С9, образовавшиеся в реакторе дегидрирования в качестве побочных продуктов и оставшийся растворенный водород, отделяются от С10+ углеводородов и концентрируются в головном потоке, выводимом из процесса по линии 94.
Остаток углеводородов, поданных в отпарную колонну 90, концентрируется в потоке эффлюента со стадии отпаривания, отводимом по линии 96. Затем поток эффлюента со стадии отпаривания подают в зону удаления ароматики 100. В этой зоне поток эффлюента со стадии отпаривания контактирует с адсорбентом в условиях, которые способствуют удалению ароматических побочных продуктов. Эффлюент из зоны удаления ароматики 100 подают в линии 98. Этот поток содержит смесь нормальных парафинов, слаборазветвленных парафинов, нормальных моноолефинов и слаборазветвленных моноолефинов и значительно более низкие концентрации ароматических побочных продуктов, чем поток эффлюента со стадии отпаривания. Полученную смесь объединяют с бензолом из линии 112 и по линии 102 подают в реактор алкилирования 104. В реакторе алкилирования, бензол и моноолефины приводятся в контакт с катализатором алкилирования в условиях, способствующих протеканию реакции алкилирования, с образованием фенилалканов.
Поток эффлюента из реактора алкилирования подают по линии 106 в колонну фракционирования бензола 110. Подаваемый поток содержит смесь бензола, нормальных парафинов, слаборазветвленных парафинов, фенилалканов, молекулы которых имеют одну фенильную часть и одну алифатическую алкильную часть, содержащую 1 или 2 первичных углеродных атома, и фенилалканов, молекулы которых содержат одну алифатическую алкильную часть и одну фенильную часть, причем алифатическая алкильная часть имеет 2, 3 или 4 первичных углеродных атомов и не содержит четвертичных углеродных атомов, за исключением четвертичного углеродного атома, связанного с фенильным фрагментом.
Другими словами, рассматриваемый поток содержит смесь бензола, нормальных парафинов, слаборазветвленных парафинов, LAB и МАВ. Такой поток разделяют в бензольной колонне фракционирования 110 на донный поток и головной поток, содержащий бензол и легкие газообразные продукты. Головной поток отводят по линии 107 и объединяют со свежим бензолом, подаваемым по линии 109. Объединенный поток подают по линии 108 в сепараторный барабан 120, из которого несконденсированные легкие газы, если они имеются, выводят по линии 114, а сконденсированная жидкость отводится по линии 116 для обеспечения флегмы для колонны 110 по линии 118 и бензола для рециркуляции по линии 112. По линии 122 остаточный поток эффлюента со стадии алкилирования из колоны 110 подается в парафиновую колонну 124, из которой донный поток, содержащий фенилалканы и тяжелые побочные продукты алкилирования, отводится по линии 126. Материал, подаваемый по линии 126, разделяют в колонне вторичной дистилляции 130 на донный поток 132, содержащий тяжелый алкилат, и головной поток продуктов алкилирования 128, содержащий фенилалкановые производные. Головной поток из парафиновой колонны 124 представляет собой рециркулирующий поток, который содержит смесь парафинов, которые рециркулируют в зону дегидрирования по линии 48. Хотя это и не показано на чертеже, некоторая часть головного потока их парафиновой колонны 124 может подаваться не в зону дегидрирования, а в зону изомеризации.
В качестве альтернативы схеме процесса, показанной на чертеже, головной поток из линии 48 может вводиться в зону дегидрирования в других точках, например по линии 62, линии 64 или через реактор 70. В том случае, когда местом ввода служит реактор дегидрирования 70, головной поток может вводиться в точке между входным отверстием линии 64 и выходным отверстием линии 66, вследствие чего головной поток может контактировать только с частью катализатора, находящегося в реакторе 70. Другой путь контактирования головного потока лишь с частью катализатора дегидрирования обеспечивается разделением реактора дегидрирования 70 на два или более промежуточных реактора, содержащих катализатор, последовательно соединенных друг с другом одной или более линиями, и вводом головного потока в линию между промежуточными реакторами. Предпочтительность наличия точки промежуточного ввода в реактор дегидрирования 70 будет определяться факторами, включающими содержание олефинов в головном потоке и требуемыми условиями реакции дегидрирования, включающими конверсию. Аналогичным образом, в техническом решении, предусматривающим ввод головного потока в зону изомеризации по линии 48, точка ввода может располагаться выше входа линии 26 в реактор изомеризации 30, в результате чего головной поток должен контактировать со всем количеством катализатора в реакторе изомеризации 30. Однако в зависимости от конверсии реакции изомеризации, степени разветвления головного потока в линии 48 и других факторов точка ввода может занимать промежуточное положение между входом линии 26 и выходом линии 28, вследствие чего будет обеспечиваться головной поток, контактирующий лишь с частью катализатора в реакторе изомеризации 30. Реактор изомеризации 30 может быть разделен на два или более мелких последовательных реактора, в результате чего головной поток может вводиться таким образом, что он будет проходить не через все реактора изомеризации. В результате анализа состава продукта изомеризации, продукта дегидрирования и потоков продуктов алкилирования выбор предпочтительной точки ввода рециркулирующего головного потока в процесс не составит труда для специалиста в данной области техники.
Сульфирование фенилалкановых производных в головном потоке продуктов алкилирования 128 может осуществляться описанным выше способом с получением фенилалкановых сульфокислот, которые могут быть подвергнуты нейтрализации в соответствие с описанными выше способами.
Примеры представлены для иллюстрации полезных свойств и преимуществ настоящего изобретения.
Примеры
Примеры 1 и 2 иллюстрируют применение предпочтительного катализатора изомеризации настоящего изобретения. В обоих примерах использовалась следующая методика. Образец катализатора изомеризации объемом в 20 см3 помещали в трубчатый реактор с внутренним диаметром 1,27 см. Катализатор изомеризации подвергали предварительному восстановлению путем контакта с 0,27 нм3/час водорода при давлении 69 кПа (g), при этом температуру катализатора поддерживали равной 110°С в течение 1 часа, повышали от 110 до 400°С в течение 3 часов и затем выдерживали при значении 400°С в течение 2 часов. После указанного предварительного восстановления катализатор изомеризации охлаждали до 150°С.
После этого катализатор тестировали на активность в реакции изомеризации с использованием в качестве сырья смеси нормальных парафинов С10-С14. Исходная реакционная смесь (“сырье”) пропускали над катализатором изомеризации с LHSV 5 час-1 при молярном отношении водорода к углеводородам 1,5:1 и при давлении 2447 кПа (g). Температуру катализатора регулировали таким образом, чтобы достичь желаемой конверсии линейных парафинов. Поток, отходящий из трубчатого реактора, подавали в сепаратор системы газ - жидкость и собирали жидкую фазу (“продукт”). Полученный продукт анализировали методом газовой хроматографии в соответствие с описанным выше.
В примерах 1 и 2 индивидуальные компоненты сырья и продукта, определенные методом газовой хроматографии, группировали по пяти классификациям: легкие продукты, содержащие 9 и менее углеродных атомов (С9-); линейный парафины, содержащие 10-14 углеродных атомов (“линейные”); монометилразветвленные парафины, содержащие 10-14 углеродных атомов в продукте (“моно”); диметилразветвленные парафины и этилразветвленные парафины, содержащие 10-14 углеродных атомов в продукте (“ди”); и тяжелые продукты, содержащие 15 или более углеродных атомов (C15+). Основываясь на таких пяти группах, рассчитывали следующие эксплуатационные характеристики:
i. Конверсия
Конверсия = 100×[1-(количество линейных в продукте) /(количество линейных в сырье)].
ii. Селективность по монометилпроизводным:
Селективность по монометилпроизводным: 100х[моно/(моно+ди)]
iii. Выход легких продуктов:
Выход легких продуктов: 100×[С9+(количество линейных в продукте)+ моно +ди +C15+)].
iv. Выход тяжелых продуктов:
Выход тяжелых продуктов = 100×[C15+/(C9-+(количество линейных в продукте) +
МОНО + ДИ + C15+)].
Пример 1
Катализатор примера 1 готовили совместной экструзией 0,39 мас.% Pt на носителе, включающем экструдат из 60 мас.% SAPO-11 и 40 мас.% оксида алюминия. В ходе изомеризации конверсия составила 73,4 мол.%, селективность по монометильным 55,5 мол.%, выход легких продуктов 7,9 мол.% и выход тяжелых продуктов 0,01 мол.%.
Пример 2
Катализатор для примера 2 готовили пропиткой смеси из 50 мас.% MgAPSO-31 и 50 мас.% оксида алюминия 0,26 мас.% Pt. В результате изомеризации конверсия составила 73,3 мол.%, селективность по монометильным 69,6 мол.%, выход легких продуктов составил 13,5 мол.%, а выход тяжелых продуктов менее 0,01 мол.%.
Данные, приведенные в примерах 1 и 2, демонстрируют хорошие значения конверсии и высокую селективность по монометилпарафинам, которые могут быть достигнуты с использованием катализаторов изомеризации, включающих SAPO-11 и MgAPSO-31.
ПРИМЕР 3
Пример 3 иллюстрирует предпочтительный катализатор дегидрирования, предназначенный для использования в настоящем изобретении, а также способ приготовления такого катализатора. Сферы из оксида алюминия готовили хорошо известным способом прикапывания в масле, описанным в патенте US-A-2620314, на который ссылаются в настоящем описании. Такой способ включает формирование гидрозоля алюминия путем растворения алюминия в хлористоводородной кислоте. Гексаметилентетрамин добавляли к золю для отвердевания последнего, с образованием сфер при диспергировании в виде капель на масляной бане при температуре 93°С. Капли оставляли в масляной бане до их осаждения и формирования сфер из гидрогеля. После извлечения полученных сфер из горячего масла их подвергали старению под давлением при 135°С и промывали разбавленным раствором гидроксида аммония, сушили при 110°С и обжигали при 650°С в течение 2 часов с образованием сфер из гамма-алюминия. Прокаленный оксид алюминия измельчали в мелкий порошок с размером частиц менее 200 микрон (0,2 мм).
После этого готовили суспензию путем смешивания 258 г алюминиевого золя (20 мас.% Аl2O3) и 6,5 г 50%-ного водного раствора хлорида олова и 464 г деионизированной воды и перемешивания до однородного распределения хлорида олова. К полученной смеси добавляли 272 г приготовленного выше порошка из оксида алюминия и суспензию перемешивали в течение 2 часов на шаровой мельнице, понижая максимальный размер частиц до значения менее 40 микрон (0,04 мм). Полученную суспензию (1000 г) в течение 17 минут распыляли с использованием устройств для грануляции и нанесения на 1 кг ядер из оксида альфа-алюминия, частицы которого имели средний диаметр около 1,05 мм, с образованием внешнего слоя толщиной около 74 микрон (0,074 мм). После завершения процесса оставалось 463 г суспензии, ядра которой не имели покрытия. Сферический носитель с нанесенным слоем сушили при 150°С в течение 2 часов и затем подвергали обжигу при 615°С в течение 4 часов с целью превращения псевдобемита во внешнем слое в гамма-оксид алюминия и превращения хлорида олова в оксид олова.
Прокаленный носитель с нанесенным слоем (1150 г) импрегнировали литием с использованием роторного импрегнатора, в результате контактирования подложки с водным раствором (при объемном отношении раствор : носитель 1:1), содержащим нитрат лития и 2 мас.% азотной кислоты в расчете на вес носителя. Пропитанный катализатор нагревали с использованием роторного импрегнатора до полного удаления раствора, сушили и снова прокаливали в течение 2 часов при 540°С.
Композит, содержащий литий и олово, пропитывали платиной в результате контактирования композита с водным раствором (объемное отношение раствор : носитель 1:1), содержащим хлорплатиновую кислоту и 1,2 мас.% хлористоводородной кислоты (в расчете на вес носителя). Пропитанный композит нагревали с применением роторного импрегнатора до полного удаления раствора, сушили, прокаливали в течение 2,5 часа при 540°С и восстанавливали в водороде при 500°С в течение 2 часов. Согласно данным элементного анализа полученный катализатор содержал 0,093 мас.% платины, 0,063 мас.% олова и 0,23 мас.% лития в расчете на общий вес катализатора. Распределение платины определяли микроаналитическом анализом с электронным зондом (ЕРМА), с использованием сканирующей электронной микроскопии, результаты которой показали, что платина однородно распределена только во внешнем слое.
Пример 4
Катализатор примера 3 тестировали на активность в отношении дегидрирования. В реактор с диаметром 1,27 см помещали 10 см3 катализатора и углеводородное сырье, содержащее 8,8 мас.% н-С10, 40,0 мас.% н-С11, 38,6 мас.% н-С12, 10,8 мас.% н-С13, 0,8 мас.% H-C14, и 1 об.% нелинейных углеводородов, пропускали над катализатором при давлении 138 кПа (g), молярном соотношении водород : углеводород 6:1 и LHSV 20 час-1.Впрыскивали воду с концентрацией 2000 ч/млн в расчете на массу углеводорода. В результате регулирования температуры реактора общую концентрацию нормальных олефинов в продукте (%TNO) поддерживали на значении 15 мас.%.
В проведенном испытании были получены следующие результаты. Селективность по TNO через 120 часов пропускания сырья, которую рассчитывали путем деления % TNO на общую конверсию, составила 94,6 мас.%. Концентрация нелинейных олефинов, которую рассчитывали, как 100% TNO составила 5,4 мас.%.
Полученные результаты показывают, что используемый в настоящем изобретении нанесенный катализатор обладает низкой скоростью дезактивации и высокой селективностью по нормальным олефинам. Поскольку углеводородное сырье в данном примере, в основном, состояло из нормальных парафинов, высокая селективность по TNO указывает на относительно низкую скелетную изомеризацию углеводородного сырья в ходе дегидрирования.
Пример 5
Пример 5 иллюстрирует катализатор алкилирования, предназначенный для использования в настоящем изобретении, который приготовлен обычным способом, используемым для приготовления катализаторов алкилирования. В качестве исходного материала использовали водородную форму морденита с соотношением SiO2/Аl2О3 около 18, на которую далее ссылаются, как на исходный морденит. 90 массовых частей исходного морденита смешивали с 10 весовыми частями порошка оксида алюминия. К полученной смеси добавляли подкисленный раствор для пептизации. Затем смесь экструдировали с помощью известных устройств. После завершения процесса экструзии полученный экструдат сушили и прокаливали. После завершения стадий сушки и прокаливания экструдат промывали водным раствором, содержащим 3 мас.% HCl в течение 2 часов при 66°С при объемном соотношении раствор : экструдат, равном 6:1. После промывки экструдат в течение 1 часа промывали водой при объемном соотношении раствор : экструдат, равном 5:1, и затем сушили.
Пример 6
Пример 6 иллюстрирует применение катализатора алкилирования, полученного в примере 5.
Использовали олефиновое сырье, включающее смесь монометил-олефинов С12 и имеющее состав, представленный в таблице 1.

Олефиновое сырье смешивали с бензолом с получением объединенного сырья, содержащего 93,3 мас.% бензола, 6,7 мас.% олефинового сырья, что соответствует молярному соотношению бензол : олефин около 30:1. В цилиндрический реактор с внутренним диаметром 22,2 мм загружали 75 см3 (53,0 г) экструдата, полученного в примере 5.
Объединенное сырье подавали в реактор и приводили в контакт с экструдатом при LHSV 2,0 час-1, общем давлении 3447 кПа (g) и температуре на входе в реактор 125°С. В этих условиях реактор футеровали в течение 24 часов и затем в течение 6 следующих часов собирали жидкий продукт.
Жидкий продукт селективной реакции анализировали методом 13С ядерного магнитного резонанса (ЯМР) с целью определения селективности по 2-фенилалканам и конечным четвертичным фенилалканам. Аналитический метод ЯМР обычно заключается в следующем. Образец фенилалкана в количестве 0,5 г разбавляли до 1,5 г безводным дейтерированным хлороформом. Аликвоту объемом в 0,3 мл разбавленной фенилалкановой смеси смешивали с 0,3 мл 0,1М раствора ацетилацетоната хрома (III) в дейтерированном хлороформе в 5 мм ампуле для ЯМР. В смесь добавляли небольшое количество тетраметилсилана (ТМС) в качестве эталона химического сдвига в 0,0 ч/млн. Спектр записывали на спектрометре Bruker ACP-300-FT-NMR, полученном от Bruker Instruments, Inc., Billerica, Massachusetts, США. Углеродный спектр записывали при напряженности поля 22727 Тесла или частоте 75,469 MГц в 5 мм зонде QNP при ширине отклонения 22727 Гц (301,1 ч./млн), записывая при этом около 65000 экспериментальных данных. Количественный углеродный спектр получали с использованием процесса стробируемого 1Н расщепления (расщепление с обратным затвором). Количественный 13С спектр записывали импульсами в 7,99 микросекунд (90°), при времени процесса 1,442 секунды, 5-секундной задержкой между импульсами, при мощности расщепления, с использованием смешанного импульса расщепления (CPD) порядка 18Н при ширине импульса 105 микросекунд (90°) и количестве сканов, по крайней мере, 2880. Используемое число сканов зависит от проведения отпарки бензола из жидкого продукта перед отбором упомянутого выше образца в количестве 0,5 г. Обработку данных проводили на Bruker PC с помощью программы WINMR-1D, версия 6, которую также поставляет компания Bruker Instruments, Inc. В ходе обработки данных использовали расширение данных на 1 Гц. Специфические пики интегрировали в области 152 ч./млн и 142 ч./млн. Идентификация химических сдвигов в пиках 13С ЯМР, относящихся к бензильному углероду изомерных фенилалканов, представлена в таблице 2. Используемый в тексте термин “бензильный углерод” обозначает атом углерода в фенильном кольце, связанный с алифатической алкильной группой.
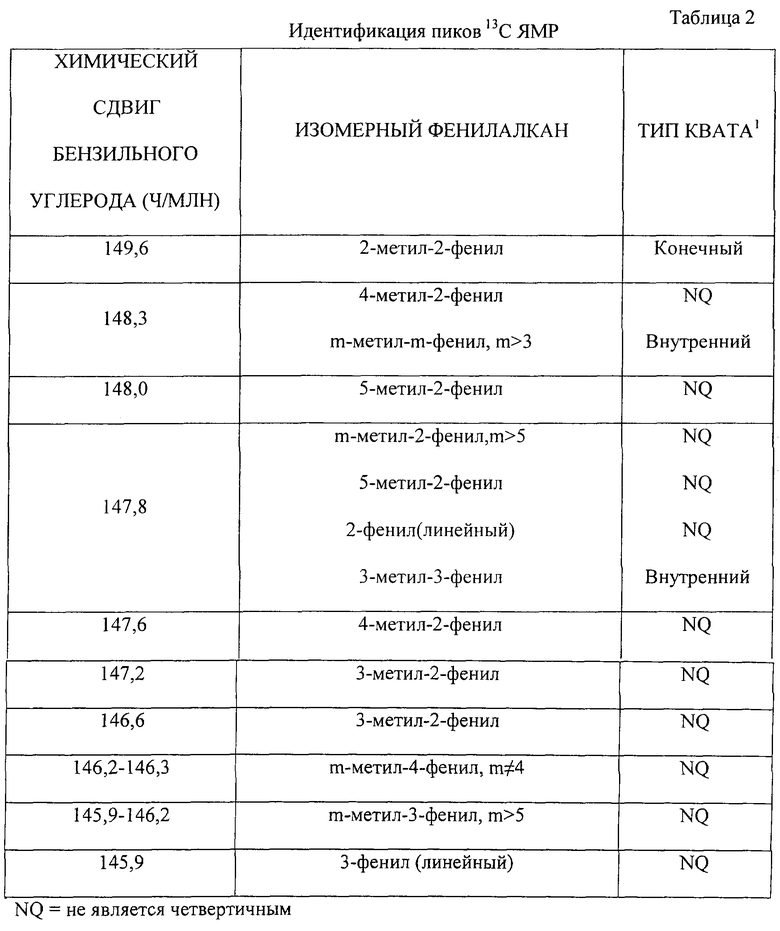
Пик при 148,3 ч./млн идентифицирован, как для 4-метил-2-фенилалканов, так и m-метил-m-фенилалканов (m>3). Однако при наличии m-метил-m-фенилалканов (m>3) в количестве более 1% эти вещества дают отчетливый пик, сдвинутый на 0,03 ч./млн относительно пика 4-метил-2-фенилалканов. Пик с 147,8 ч./млн, согласно данным таблицы 2, идентифицирован, как относящийся к 2-фенилалканам, с возможной интерференцией от 3-метил-3-фенилалканов.
Селективность по концевым фенилалканам рассчитывалась как частное от деления интеграла пика с 149,6 ч./млн на сумму интегралов всех пиков, перечисленных в таблице 2, помноженное на 100. Селективность по 2-фенилалканам может быть определена в том случае, если вклад от количества внутренних четвертичных фенилалканов в пики с 148,3 ч./млн и 147,8 ч./млн составляет менее 2%, в соответствии с обсуждаемыми ниже данными метода хромато-масс спектрометрии. В качестве первичного приближения такое условие удовлетворяется в том случае, когда сумма интегралов пиков, соответствующих 4-фенилалкану и 3-фенилалкану, при 146,2-146,3 ч./млн и 145,9-146,2 ч./млн (соответственно) мала относительно суммы интегралов всех пиков от 145,9 ч./млн до 149,6 ч./млн, а селективность по концевым четвертичным фенилалканам менее 10%. В рассматриваемом случае селективность по 2-фенилалканам рассчитывается, как частное от деления суммы интегралов пиков от 149,6 до 146,6 ч./млн на сумму интегралов всех пиков, перечисленных в таблице 2, помноженное на 100.
Жидкий продукт селективной реакции также анализировался методом хромато-масс-спектрометрии с целью определения селективности по внутренним четвертичным фенилалканам. Аналитический метод хромато-масс-спектрометрии состоит в следующем. Жидкий продукт анализировали на газовом хроматографе (GC) HP 5890 серии II, снабженном автоматическим устройством для нанесения образцов HP 7673 и масс-спектрометрическим (MS) детектором HP 5972. Для контроля характеристик процесса и полученных данных использовали HP Chemstation. HP 5890 серий II, HP 7673, HP 5972 и HP Chemstation, или любое другое аппаратное оборудование и программное обеспечение поставлены Hewlett Packard Company, Palo Alto, California, США. GC снабжен колонкой DB1HT (df=0,l мкм) размерами 30 м × 0,25 мм или ее эквивалентом, которая может быть поставлена J&W Scientific Incorporated, Folsom, California, США. В качестве газоносителя использовали гелий при постоянном давлении 103 кПа (g) и температуре 70°С. Температура испарителя поддерживалась равной 275°С. Температура подающей линии и MS источника поддерживались на значении 250°С. Использовалась следующая температурная программа: 70°С в течение 1 минуты, затем нагревание до 180°С со скоростью 1°С в минуту, затем нагревание до 275°С со скоростью 10°С в минуту и последующая выдержка при 275°С в течение 5 минут. MS настраивали с помощью HP Chemstation, программа которой регулировалась с помощью стандартной спектральной автонастройки. Начало сканирования MS составляло 50-550 Da с порогом =50. Концентрации внутренних четвертичных фенилалканов в жидком продукте определяли (т.е. селективный жидкий продукт подвергали количественному определению) с использованием метода добавления стандарта. Основная информация о методах добавления стандарта изложена в главе 7 книги, под названием Samples and Standards. B.W.Woodget с сотр., опубликованной ACOL, London by John Wily and Sons, New York, 1987.
Сначала готовили исходный раствор внутренних четвертичных фенилалканов и определяли его состав. Бензол алкилировали монометилалкеном с использованием такого неселективного катализатора как хлористый алюминий. Продукт неселективного алкилирования содержит смесь внутренних, четвертичных фенилалканов и рассматривается как базовый раствор внутренних четвертичных фенилалканов. С использованием метода GC идентифицировали наибольшие пики, соответствующие внутренним четвертичным фенилалканам в базовом растворе, и определяли их концентрации (т.е. проводили количественное определение компонентов базового раствора) с использованием пламенно-ионизационного детектора (FID). Концентрацию каждого внутреннего четвертичного фенилалкана рассчитывали путем деления площади пика рассматриваемого внутреннего фенилалкана на сумму площадей всех пиков.
Затем готовили реперный (spiking) раствор внутренних четвертичных фенилалканов. Аликвоту исходного раствора разбавляли дихлорметаном (хлористый метилен) с целью получения номинальной концентрации конкретного внутреннего, четвертичного фенилалкана (например, 3-метил-3-фенилдекана) 100 мас.ч./млн. Полученный в результате раствор обозначали как реперный раствор внутренних четвертичных фенилалканов. Концентрация любого другого внутреннего четвертичного фенилалкана в реперном растворе может быть выше или ниже 100 мас.ч./млн, в зависимости от концентрации такого внутреннего четвертичного фенилалкана в исходном растворе.
После этого готовили раствор образца. 0,05 г аликвоты селективного жидкого продукта помещали в мерную колбу емкостью 10 мл. Затем содержимое колбы разбавляли дихлорметаном, добавляя его до метки, соответствующей объему в 10 мл. Полученное в результате содержимое мерной колбы обозначали, как раствор образца.
Наконец, готовили результирующий раствор. 0,05 г аликвоты селективного жидкого продукта помещали в мерную колбу емкостью 10 мл. Затем содержимое колбы разбавляли до метки, соответствующей объему в 10 мл, реперным раствором. Полученное в результате содержимое колбы обозначали как результирующий раствор.
Раствор образца и результирующий раствор анализировали методом GC/MS с использованием указанных выше условий. В таблице 3 перечислены ионы, извлеченные из полной развертки MS спектра, причем данные по указанным ионам подвергали обработке и интегрированию с использованием программного обеспечения HP Chemstation. Указанные программные средства HP Chemstation использовали для определения площадей пиков, относящихся к индивидуальным ионам, соответствующим внутренним кватам, перечисленным в таблице 3.

Концентрацию каждого внутреннего четвертичного фенилалкана из перечисленных в таблице 3 рассчитывали по следующему уравнению:
С=S(A1/A2-A1),
в котором С - концентрация внутреннего четвертичного фенилалкана в растворе образца, мас.%;
S - концентрация внутреннего четвертичного фенилалкана в реперном растворе, мас.%;
A1 - площадь пика внутреннего четвертичного фенилалкана в растворе образца, единицы площади;
А2 - площадь пика внутреннего четвертичного фенилалкана в результирующем растворе, единицы площади.
Концентрации С и S имеют одинаковые размерности при условии, что площади A1 и А2 также имеют одинаковые размерности. После этого концентрацию каждого внутреннего четвертичного фенилалкана в селективном жидком продукте рассчитывали из концентрации такого внутреннего четвертичного фенилалкана в растворе образца, принимая во внимание эффект разбавления раствора образца дихлорметаном. Аналогичным образом рассчитывали концентрацию каждого из внутренних четвертичных фенилалканов, перечисленных в таблице 3. Общую концентрацию внутренних четвертичных фенилалканов в селективном жидком продукте, CIQPA, рассчитывали суммированием индивидуальных концентраций каждого из перечисленных в таблице 3 внутренних четвертичных фенилалканов.
Следует отметить, что селективный жидкий продукт может содержать внутренние четвертичные фенилалканы отличные от тех, что перечислены в таблице 3, например, m-метил-m-фенилалканы, где m>5, в зависимости от числа углеродных атомов в алифатических алкильных группах фенилалканов. Предполагается, что при использовании С12 олефинового сырья и условий, описанных в примере 6, концентрации таких других внутренних четвертичных фенилалканов относительно малы, по сравнению с содержанием внутренних четвертичных фенилалканов, перечисленных в таблице 3. В связи с этим в примере 6 общую концентрацию внутренних четвертичных фенилалканов в селективном жидком продукте, CIQPA, рассчитывали суммированием только тех индивидуальных концентраций каждого из внутренних четвертичных фенилалканов, что перечислены в таблице 3. Однако в том случае, когда олефиновое сырье содержит олефины с числом атомов углерода, например, до 28, общую концентрацию внутренних четвертичных фенилалканов в селективном жидком продукте, СIPQA, следует рассчитывать суммированием индивидуальных концентраций m-метил-m-фенилалканов, где m имеет значение в интервале 3-13. В общем случае, если олефиновое сырье содержит олефины с числом углеродных атомов x, то общую концентрацию внутренних четвертичных фенилалканов в селективном жидком продукте, СIPQA, рассчитывают суммированием индивидуальных концентраций m-метил-m-фенилалканов, где m имеет значение в интервале 3- х/2. Может быть идентифицирован, по крайней мере, один пик с отношением массы к заряду (m/z) выборочного иона, соответствующим каждому внутреннему четвертичному фенилалкану, на основании чего может быть определена концентрация каждого из внутренних четвертичных фенилалканов и последующим суммированием может быть получено значение СIPQA.
Селективность по внутренним четвертичным фенилалканам в селективном жидком продукте может быть рассчитана по следующей формуле:
Q=100(CIQPA/CMAB),
в которой Q - селективность по внутренним четвертичным фенилалканам;
СIPQA - концентрация внутренних четвертичных фенилалканов в селективном жидком продукта, мас.%;
СМАВ - концентрация модифицированных алкилбензолов в селективном жидком продукте, мас.%.
Концентрацию модифицированных алкилбензолов, СМАВ, в селективном жидком продукте определяли следующим методом. Во первых, методом газовой хроматографии определяли концентрацию примесей в селективном жидком продукте. В контексте, касающемся определения CМАВ, термин “примеси” обозначает компоненты селективного жидкого продукта, времена удерживания которых находятся вне специального интервала, используемого в газохроматографическом методе. Такие “примеси” обычно включают бензол, некоторые диалкилбензолы, олефины, парафины и т.д.
Для определения количества рассматриваемых примесей в селективном жидком продукте использовали следующий газохроматографический метод. Для определения примесей в селективном жидком продукте могут использоваться эквивалентное оборудование, эквивалентый способ приготовления образцов и эквивалентные параметры GС, отличающиеся от тех, что указаны ниже, при условии, что они обеспечивают результаты, эквивалентные указанным ниже.
Оборудование:
- Газовый хроматограф Hewlett Packard HP 5890 серия II, снабженный дозатором с делителем потока или без него и пламенно-ионизационным детектором (FID);
- J&W исследовательская капиллярная колонка DB-1HT, длиной 30 м, внутренним диаметром 0,25 мм, с толщиной пленки 0,1 ммкм;
- 11 мм перегородка Restek Corporation (США) Red lite Septa;
- 4 мм S-образный входной рукав из углешлака фирмы Restek;
- O-образная кольцо-прокладка для входной линии фирмы Hewlett Packard;
- хлористый метилен или его эквивалент чистоты HPLC от J.T.Baker Company (США);
- 2 мл ампулы для хроматографического автоматического устройства для нанесения образцов с загнутыми краями, или их эквиваленты.
Приготовление образца
- Образец в количестве 4-5 мг отвешивали в ампулу GC автоматического пробоотборника.
- В GC ампулу добавляли 1 мл хлористого метилена; ампулу закрывали гофрированной, футерованной тефлоном перегородкой (крышкой); и содержимое ампулы интенсивно перемешивали.
Параметры GC:
- Газ-носитель: водород
- Давление в головной части хроматографической колонки: 62 кПа
- Потоки: поток газа через колонку, 1 мл/мин; через разделительный вентиль, около 3 мл/мин; продувка прокладки, 1 мл/мин.
- Ввод пробы: автоматическое устройство для ввода пробы HP 7673, шприц на 10 мкл; объем вводимой пробы 1 мкл.
- Температура инжектора: 350°С
- Температура детектора: 400°С
- Термпрограммирование: начальная выдержка при 70°С в течение 1 минуты; нагревание со скоростью 1°С/мин; конечная выдержка в течение 10 минут при 180°С.
Используемый газохроматографический метод требует применения двух стандартов, которые тщательно перегоняют до чистоты выше 98 мол.%. Как правило, каждый стандарт представляет собой 2-фенилалкан. Один из 2-фенилалкановых стандартов, на который далее ссылаются, как на легкий стандарт, содержит в алифатической алкильной группе число углеродных атомов, которое, по крайней мере, на один углеродный атом меньше, чем олефин в олефиновом сырье, подаваемом в зону алкилирования, с минимальным числом углеродных атомов. Другой 2-фенилалкановый стандарт, на который ссылаются, как на тяжелый стандарт, содержит в алифатической алкильной группе, по крайней мере, на один углеродный атом больше, чем олефин в олефиновом сырье, подаваемом в зону алкилирования, с наибольшим числом углеродных атомов. Так например, если олефины в олефиновом сырье, подаваемом в зону алкилирования, содержат 10-14 углеродных атома, то подходящий легкий стандарт может представлять собой 2-фенилоктан, а подходящий тяжелый стандарт - 2-фенилпентадекан. Каждый из стандартов пропускают через хроматографическую колонку в указанных выше условиях для определения времени удерживания и два полученных значения времени удерживания определяли общий интервал времен удерживания компонентов смеси. Затем в таких же условиях проводят газохроматографический анализ аликвотного образца селективного жидкого продукта. Если более 90% площадей пиков GC попадают в указанный интервал времен удерживания, то считают, что количество примесей в селективном жидком продукте не превышает 10 мас.% от количества селективного жидкого продукта, и исключительно в целях упрощения расчета, селективность по внутренним четвертичным фенилалканам, СМАВ, принималась равной 100 мас.%.
Если же общее процентное количество площадей хроматографических пиков в рамках указанного интервала времен удерживания оказывается меньшим 90%, то считается, что количество примесей в селективном жидком продукте превышает 10 мас.% от массы селективного жидкого продукта. В этом случае, для определения CМАВ примеси удаляют из селективного жидкого продукта, для чего используют следующий метод дистилляции. Для удаления примесей из селективного жидкого продукта могут также использоваться эквивалентное оборудование, эквивалентные способы и эквивалентные условия дистилляции, отличающиеся от описанных ниже, но обеспечивающие получение эквивалентных результатов.
Для процесса дистилляции, предназначенного для удаления примесей из селективного жидкого продукта, используют 5-литровую, 3-горлую круглодонную колбу с магнитной мешалкой и небольшим количеством кипелок. На центральное горловое отверстие колбы помещают конденсатор Vigreux длиной 24,1 см. Сверху конденсатора Vigreux присоединяют охлаждаемый водой холодильник, снабженный контрольным термометром. К концу холодильника присоединяют вакуумный приемник. Один из отводов 5-литровой перегонной колбы закрывают стеклянной пробкой, а термометр помещают на другом отводе колбы. Колбу и конденсатор Vigreux оборачивают алюминиевой фольгой. В 5-литровую колбу загружают 2200-2300 г аликвотной части селективного жидкого продукта, содержащего около 10 мас.% примесей в соответствии с данными газохроматографического анализа. К вакуумному приемнику присоединяют линию от вакуумного насоса. Селективный жидкий продукт в 5-литровой колбе перемешивают и систему вакуумируют. После достижения максимального значения вакуума (остаточное давление, по крайней мере, 25,4 мм Hg или менее) селективный жидкий продукт нагревают с помощью электрического нагревательного кожуха.
После начала обогрева отбирают две фракции дистиллята. Одна фракция, которую далее обозначают, как фракция А, отбирается в температурном интервале от 25°С до точки кипения легкого стандарта. Другую фракцию, далее фракцию В, отбирают в температурном интервале от точки кипения легкого стандарта до точки кипения тяжелого стандарта. Легко кипящую фракцию А и низкокипящие остатки отбрасывают. Фракция В содержит модифицированные алкилбензолы и ее взвешивают. Описанный метод может при необходимости масштабироваться. Давления паров фенилалканов при различных температурах могут быть взяты из статьи в Industrial and Engineering Chemistry, т. 38, 194, начиная со стр. 320.
Затем аликвоту образца фракции В анализируют методом газовой хроматографии в указанных выше условиях. Если более 90% от общего числа хроматографических пиков во фракции В попадают в интервал времен удерживания, то количество примесей во фракции В полагают не превышающим 10 мас.% от селективного жидкого продукта, и исключительно в целях расчета селективности по внутренним четвертичным фенилалканам, СМАВ рассчитывают, как частное от деления веса фракции В на вес аликвотной части селективного жидкого продукта, загруженного в 5-литровую колбу в описанном выше методе дистилляции. Если же общий процент хроматографических пиков во фракции B в соответствующем интервале времен удерживания составляет менее 90%, то количество примесей во фракции В считают большим 10 мас.% от массы фракции В. В этом случае примеси, содержащиеся во фракции В, вновь удаляют с помощью описанного выше метода дитстилляции. Соответственно, низкокипящую фракцию (обозначаемую как фракцию С) и высококипящие кубовые остатки отбрасывают, фракцию (обозначаемую, как фракцию D), содержащую модифицированные алкилбензолы, выделяют и взвешивают, и аликвотный образец фракции В анализируют газохроматографическим методом. Если более 90% от общего числа хроматографических пиков во фракции D попадают в соответствующий интервал времен удерживания, то исключительно в целях расчета селективности по внутренним четвертичным фенилалканам, значение СМАВ рассчитывают, как частное от деления веса фракции D на вес аликвотной части селективного жидкого продукта, первоначально загруженного в 5-литровую колбу. В противном случае, дистилляцию фракции D и ее газохроматографический анализ повторяют.
Описанные выше методы дистилляции и газохроматографического анализа могут повторяться до получения фракции, содержащей модифицированные алкилбензолы и менее 10 мас.% примесей, при условии, что после каждой стадии дистилляции остается достаточное количество материала для последующего испытания указанными методами.
После определения значения СМАВ селективность по внутренним четвертичным фенилалканам, Q, может быть рассчитана по указанным выше формулам. Результаты проведенных анализов представлены в таблице 4:

При отсутствии размерной селективности, как это имеет место в случае использования таких катализаторов, как хлористый алюминий или HF, можно ожидать, что большая часть 2-метилундецена превращается в 2-метил-2-фенилундецен (т.е. концевой кват). Аналогичным образом, можно ожидать, что большая часть 6-метилундецена, 5-метилундецена, 4-метилундецена и 3-метилундецена превращается во внутренние кваты. Как предполагается, линейные олефины дают статистическое распределение 2-фенилдодекана, 3-фенилдодекана, 4-фенилдодекана, 5-фенилдодекана и 6-фенилдодекана.
Таким образом, если исключить из расчета легкие, тяжелые и другие алкилолефины, перечисленные в таблице 1, то селективность по 2-фенилалканам не превысит 17, а селективность по внутренним четвертичным фенилалканам достигнет 55. Данные, представленные в таблице 4, показывают, что селективность по 2-фенилалканам значительно выше, чем этого можно ожидать в отсутствие размерной селективности, а селективность по внутренним четвертичным алкилбензолам, полученным с использованием катализатора на основе морденита, значительно меньше того значения, которое можно было бы ожидать в отсутствие размерной селективности.
Применение: нефтехимия. Сущность: проводят изомеризацию парафинов, последующее дегидрирование изомеризованных парафинов и алкилирование фенилпроизводного слаборазветвленным олефином. Эффлюент из зоны алкилирования содержит парафины, которые рециркулируют на стадию изомеризации или стадию дегидрирования. Полученные фенилалканы могут применяться в качестве смазки. 2 с. и 7 з.п. ф-лы, 4 табл., 1 ил.
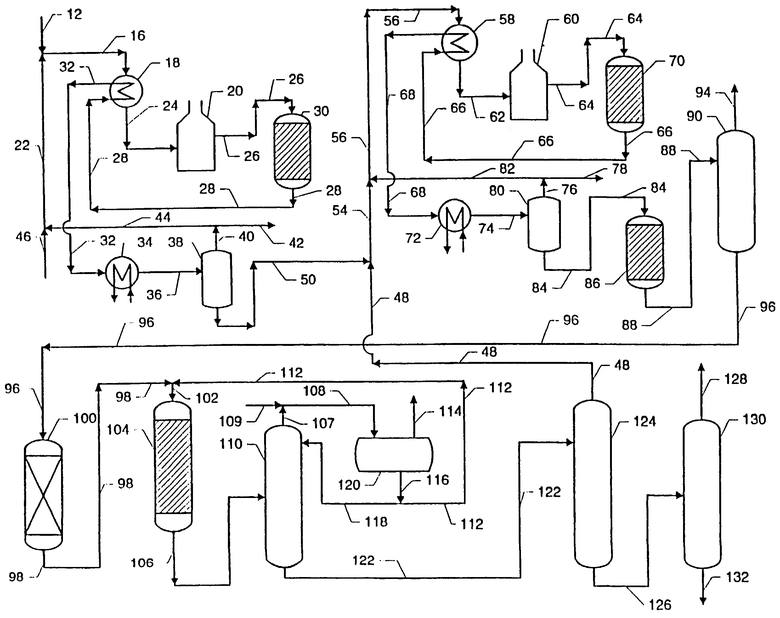
| US 5012021 A, 30.04.1991 | |||
| Гайковерт | 1972 |
|
SU491464A1 |
| Способ получения алкилароматических соединений | 1980 |
|
SU958404A1 |

