Настоящее изобретение в общем относится к каталитическим реакциям алкилирования и, в частности, к катализаторам цеолитового типа.
Алкилароматические соединения являются важным семейством веществ, которые используют в качестве сырьевых материалов во многих областях промышленности, таких как области пластификаторов, полимерных материалов, инсектицидов, для предотвращения агломерации удобрений в сельском хозяйстве, в производстве текстильных материалов и волокон, в производстве кожи и мехов, гербицидов, в промышленных способах очистки, в фотографической промышленности, в производстве клеев и противопожарных продуктов, таких как увлажняющие агенты, в электрохимических способах удаления грязи и жира с поверхности вещества и в биологически разлагаемых моющих средствах (Surfactants in Consumers Product, Theory, Technology and Application, edited by J. Falbe, Springer Verlag, 1987).
Обычный способ, используемый в нефтехимической промышленности для производства алкилароматических соединений, в особенности для применения в моющих средствах, состоит из дегидрогенизации линейных парафинов с получением линейных моноолефинов и затем осуществления алкилирования бензола указанными моноолефинами. Линейный алкилбензолсульфонат (ЛАС) получают путем сульфонирования линейного алкилбензола (ЛАБ) и последующей нейтрализации соответствующих сульфоновых кислот (Н-ЛАС). Линейные олефины, использованные в этом способе, имеют от девяти до шестнадцати атомов углерода. Стадию алкилирования проводят в жидкой фазе в присутствии катализаторов типа Фриделя-Крафта, например фтористоводородной кислоты. Способ в присутствии HF хорошо известен и используется в промышленности, давая высокий выход (>99 мас.%) ЛАБ с относительно низкой селективностью для 2-фенил изомера, меньшей чем 20%. Объединенный способ получения ЛАБ описан в Handbook of Petroleum Refining Process, published by Robert A. Meyers, 1986, p.1-23, включенной здесь в качестве ссылки. В патенте US 5276231 описывают промежуточные стадии способа получения ЛАБ, такие как селективную гидрогенизацию диолефиновых побочных продуктов, образующихся при дегидрогенизации парафинов, и удаление нелинейных побочных продуктов из потока стадии дегидрогенизации. Использование HF имеет некоторые недостатки, так как оно требует бережного обращения и специального оборудования в силу ее коррозионной активности, что приводит к увеличению текущих и эксплуатационных расходов; вследствие этого была предпринята попытка разработки альтернативных катализаторов на основе твердых веществ кислотного характера.
В предшествующем уровне техники описано множество твердых кислотных катализаторов для получения фенилалканов, таких как синтетические фожазиты (цеолиты X и Y), цеолиты L, ZSM-5, ZSM-18, ZSM-20, морденит и оффретит. Современные разработки, в которых используют несколько видов цеолитов, аморфные алюмосиликаты и другие виды материалов в качестве катализаторов алкилирования, описаны для получения ЛАБ с содержанием 2-фенил изомера в диапазоне от 20 до 80 мас.% (J.L.Berna Tejero, A.Moreno Danvila, патент US 5157158, 1992 и J.L.G. De Almedia, M.Dufaux, Y.Ben Taarit, C.Naccache, Journal of the American Oil Chemist′s Society, Vol.71, no.7, 675-694, 1 1994). Твердые цеолиты, упомянутые здесь в качестве катализаторов, определены в классификации Atlas of Zeolite Framework Types, W.M.Meier, D.H.Olson and С.H.Baerlocher, 5th revised edition, 2001, Elsevier, на которую ссылаются в настоящем изобретении.
Один из кристаллических алюмосиликатов, который можно использовать в качестве катализатора в этом виде реакций, является цеолитом семейства FAU, обозначенным как цеолит Y, состав которого является следующим:
Na56[Al56Si136O384]:250H2O
Цеолит Y имеет пористую трехмерную структуру с порами, расположенными перпендикулярно друг другу в плоскостях x, y и z. Диаметр пор велик, примерно 7,4 Å, при этом отверстие определено кольцом из 12-ти атомов кислорода и ведет в большую полость диаметром 12,5 Å. Он структурно включает трехмерный каркас из тетраэдров SiO2 и AlO4, соединенных друг с другом посредством общих атомов кислорода. Когда четырехвалентный ион кремния изоморфно замещен трехвалентным атомом алюминия в кремниевом каркасе, включающем кремниевые тетраэдры, получающийся отрицательный заряд должен быть стабилизирован соседним положительным ионом, таким как протон. Последний может быть получен путем диссоциации воды, образующей гидроксильную группу на атоме алюминия. Получающаяся структура является кислотой Брэнстеда (С.N.Satterfield, Heterogeneous Catalysts in Practice, McGraw-Hill Inc., 1980, page 152). При нагревании последней вода экстрагируется из состава и активные центры кислоты Брэнстеда преобразуются в активные центры кислоты Льюиса. Дегидратированные цеолиты могут иметь как активные центры кислоты Брэнстеда, так и активные центры кислоты Льюиса, в зависимости от ионного обмена и условий термической обработки. В результате их микропористой структуры цеолиты могут производить намного большее число доступных активных кислотных центров на площади поверхности твердого вещества в отличие от стандартного катализатора на основе аморфного алюмосиликата, используемого при алкилировании.
На практике, когда ароматическое соединение алкилируют, используя твердые кислотные катализаторы, образуются диалкилированные, так же как и моноалкилированные, продукты, в особенности, когда реакция протекает в условиях высокой степени конверсии и ароматическое соединение является бензолом. Помимо кислотности, цеолит Y обладает другим важным свойством, относящимся к каталитической активности. Из-за их кристаллической структуры цеолитовые каналы или поры являются микроскопически малыми, следовательно, их малые размеры и их молекулярная конфигурация обладают экстраординарным влиянием на селективность реакции, поскольку они могут предотвращать образование более объемных соединений, таких как некоторые виды тяжелых алкилатов, также известных как диалкилбензолы. Этот эффект называют селективностью 0, обусловленной размерами пор.
Хорошо известно, что присутствие редкоземельных элементов может сильно влиять на силу и плотность кислотных активных центров в цеолитовом материале (L.В.Zinner, К.Zinner, М.Ishige and A.S.Araujo, Catalytic activity of lanthanide-doped Y Zeolite on the alkylation of benzene with 1-dodecene model reaction, Journal of Alloys and Compounds, 193, 65-67, 1993). Алюмосиликат после ионного обмена с редкоземельными элементами является активированным кристаллическим катализатором, в котором атомная структура модифицирована, поскольку он обладает хемосорбированными или ионносвязанными катионами редкоземельных металлов. Редкоземельные катионы можно получить исходя из соли одного металла или, предпочтительно, из смеси солей различных металлов, таких как легкие редкоземельные хлориды или хлориды дидима (La, Ce, Pr, Nd, CAS: 11098-090-1). В большинстве применений редкоземельные смеси являются предпочтительными, так как их можно легко получить промышленно, и они намного более рентабельны, чем очищенные редкоземельные металлы. Р.В.Venua et al. опубликовали в Journal of Catalysts, 4, 81-98, 1966 возможное применение высококислотных твердых катализаторов, таких как фожазиты типа цеолита X и Y с ионным обменом с редкоземельными металлами, обозначаемые REY и REX, для алкилирования бензола с олефинами. В бразильском патенте P.I. 9204326-7 показано применение катализатора на основе цеолитового фожазита, в частности цеолита Y, активированного Ca и лантанидами, в частности La, Ce, Nd или Gd. В патенте заявлен способ в твердо-жидкостном реакторе периодического действия, действующем в условиях полного перемешивания, с суперкислотным катализатором, действующим при температурах, настолько низких, как 80°C.
Другим катализатором алкилирования, который часто используют в этих реакциях, является цеолит со структурой типа MOR, морденит, который характеризуется молярным отношением SiO2/Al2O3, равным 10. Состав его типичной элементарной ячейки представляет собой:
Na8[(AlO2)8(SiO2)40]×24H2O
Кристаллическая структура показывает систему каналов из 12-членных колец со свободным диаметром 6,7×7 Å, и эти основные каналы соединены между собой посредством малых каналов, ограниченных 8-членными кольцами с отверстием 2,9×5,7 Å. Эти небольшие каналы препятствуют прохождению большинства органических молекул, в результате пористый каркас морденита в основном выглядит как однонаправленная пористая структура. Способ деалюминирования морденита путем гидрообработки, за которой следует обработка кислотой, может модифицировать его структуру и каталитические характеристики, увеличивая адсорбционную емкость и скорость диффузии молекул, как описано в ссылке, озаглавленной "Alquilation du benzene par le 1-dodecene sur catalyseurs zeolithiques", J.L.Almeida, PhD, тезисы, представленные в Universite Claude Bernard-Lyon 1, 1994. В следующих патентах US 2003/0166481-A1, US 6521804-B1, US 6133492 и US 6562776-B1, включенных здесь в качестве ссылок, описывается способ алкилирования, в котором катализатор морденитового типа можно смешивать с различными катализаторами, которые не приводят к получению высокого содержания 2-фенил изомера, следовательно, количества каждого катализатора необходимо подбирать для получения требуемых уровней 2-фенил изомера в продукте.
Способ получения алкилароматических соединений или алкилатов посредством алкилирования бензола олефинами с длинной цепью характеризуется, в показателях селективности, содержанием тяжелых алкилатов, содержанием 2-фенил изомера и также содержанием разветвленных алкилатов. Эти химические вещества, определяющие эффективность способа и качество продукта, в основном зависят от типа катализатора, качества исходного материала и рабочих условий способа.
Группа тяжелых алкилатов включает совокупность химических веществ с молекулярной массой, превышающей массу моноалкилированного продукта. Они обычно образованы диакилбензолами, дифенилалканами, олигомеризованными олефинами и алкилатами этих олигомеризованных олефинов, образуемых в течение стадии алкилирования. Эти продукты в основном получают путем алкилирования моноалкилбензола, предварительно образованного при помощи олефина. Дифенилалканы образуют путем алкилирования бензола диолефином, который был дегидроциклизирован. Образование этого типа продуктов нежелательно, так как они уменьшают общий выход способа и могут ухудшить качество получаемого алкилата, таким образом снижая экономическую целесообразность. Дополнительно существуют другие соединения, которые необходимо рассмотреть, например некоторые алкилполиароматические продукты, образующиеся путем алкилирования моноолефинами полиароматических соединений, полученных на стадии дегидрогенизации. Эти продукты можно извлечь совместно с наиболее тяжелыми ЛАБ соединениями, так как диапазон температуры кипения их фракции перекрывается с диапазоном температуры кипения фракции указанных ЛАБ. Эти продукты существенно снижают качество сульфонированного конечного продукта Н-ЛАС, так как цветность после сульфонирования очень сильно возрастает.
2-фенил изомеры являются алкилированными молекулами, имеющими ароматическое кольцо, соединенное с алкильной цепью в позиции 2 цепи. Содержание 2-фенил изомера определяют как массовое процентное содержание 2-фенил изомера в смеси ЛАБ или ЛАС и вычисляют по следующей формуле:
2-фенил изомер [%]=(масса 2-фенил изомера)×100/(общая масса ЛАБ или ЛАС)
Смеси ЛАБ с содержанием внешних изомеров (2+3 фенил), превышающим 60 мас.% характеризуются тем, что обеспечивают ЛАС с улучшенной поверхностной активностью после сульфонирования и нейтрализации. Эти ЛАС, однако, имеют важный недостаток, обусловленный их низкой растворимостью в холодной воде, низкой стабильностью при условиях высокой вязкости. Смеси ЛАС, включающие более 60 мас.% внешних изомеров (2+3 фенил), стремятся к образованию сильно нерастворимых гелей с высокой вязкостью, делая сложным обращение с ними и их обработку. По этой причине желательно включать гидротроп с целью улучшения растворимости конечного поверхностно-активного вещества, когда содержание 2-фенил изомера выше 60 мас.% Хотя существует множество патентов, связанных с применением гидротропов, один из них рассматривают как наиболее рекомендуемый для этого способа. PCT/ES 2005000169 относится к способу получения подходящего гидротропа исходя из предварительно дегидрогенизированных парафинов, в частности исходя из их побочных продуктов, извлекаемых в течение стадии очистки моноолефинов.
Наконец, разветвленные алкилаты можно определить как процентное содержание всех моноалкилатов, в которых алкильная цепь, связанная с ароматическим кольцом, не является линейной или нормальной алкильной группой, однако является разветвленной. Эти ненормальные алкильные группы обладают радикалами, такими как метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил и различные варианты гексила, гептила, октила, связанные в любом месте с алкильной цепью за исключением концов цепи. Разветвленные алкилаты образуются путем алкилирования из таких разветвленных парафинов, которые присутствуют в свежих исходных парафинах, или путем процессов перегруппировки алкилов, возникающих в течение стадий дегидрогенизации и алкилирования. Разветвленные алкилаты включают алкилаты, имеющие один из атомов углерода алифатической алкильной группы в положении четвертичного углерода. Атом четвертичного углерода в алкильной цепи определяют как углерод, присоединенный к четырем другим атомам углерода, и один из этих атомов может быть связан с атомом углерода фенильной группы, образуя четвертичный алкилфенилалкан. Если четвертичный углерод является вторым атомом углерода в алкильной цепи, то четвертичный углерод, присутствующий в полученном 2-алкил-2-фенилалкане, можно назвать «конечным четвертичным углеродом». Известно, что эти вещества подобно простым разветвленным алкилатам имеют скорость биоразложения, аналогичную той, которую имеет алкилбензолсульфонат. Однако, когда четвертичный углерод является любым другим атомом углерода алкильной цепи, например 5-метил-5-фенилалкан, его называют «внутренним четвертичным углеродом», и соответствующий алкилбензолсульфонат имеет намного более низкую скорость биоразложения. В статьях, озаглавленных "Iso-branching of LAS biodegradation study of two model compounds", L.Cavalli, G.Cassani, M.Lazzarin, C.Maraschin, G.Nuzzi, J.L.Berna, J.Bravo, J.Ferrer, A.Masno, Toxicology & Environmental Chemistry, Vol.54, page 167-186, 1966 и "Biodegradation of co-products of commercial LAS", A.M.Nielsen, L.N.Britton, L.Cavalli, J.L.Berna, The Cler 20 Review, Vol.1.2, N.1, p.14-27, 1996, представлено научное доказательство биоразлагаемого поведения этих алкилбензольных производных.
Реакцию алкилирования можно окончательно оценить с помощью следующих показателей: степень конверсии, селективность по моноалкилбензолу, распределение изомеров и скорость дезактивации.
1) степень конверсии алкилирования или, более конкретно, относительная степень конверсии: В реакции алкилирования, рассматриваемой в этом изобретении, бензол всегда используют со стехиометрическим избытком по отношению к олефинам. Относительную степень конверсии можно определить как долю лимитирующих реагентов, в этом случае олефина, которые расходуются для получения всех продуктов, таким образом:
Степень конверсии=[(NA0-Na)/NA0]×100,
где NA0 является числом молей олефина на входе в реактор и NA является числом молей того же реагента на выходе из реактора.
2) Селективность по моноалкилбензолу определяют как
Se/маб=[Wмаб/(Wлег+Wмаб+Wтяж)]×100,
где Wмаб является массой полученного рассматриваемого моноалкилбензола, Wлег является массой побочных продуктов, более легких, чем самый легкий рассматриваемый моноалкилбензол, и Wтяж является общей массой всех веществ, чьи молекулярные массы выше, чем молекулярные массы полученных моноалкилатов, и как было указано, они обычно образуются из диалкилбензолов, дифенилалканов, олигомеризованных олефинов и алкилатов этих олигомеризованных олефинов. Массы или массовые процентные содержания каждого семейства продуктов можно использовать для вычисления селективности.
3) Распределение изомеров. Среди полученных моноалкилбензолов распределение изомеров можно определить как массовое процентное содержание каждого типа полученного изомера, такого как 2-фенил, 3-фенил…6-фенил изомеры, а также разветвленных алкилатов.
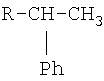 линейный 2-фенил изомер (R = линейный)
линейный 2-фенил изомер (R = линейный)
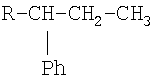 линейный 3-фенил изомер (R = линейный)
линейный 3-фенил изомер (R = линейный)
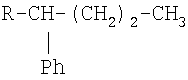 линейный 6-фенил изомер (R = линейный)
линейный 6-фенил изомер (R = линейный)
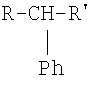 разветвленный n-фенил изомер (разветвленный R или R′, n=2-6)
разветвленный n-фенил изомер (разветвленный R или R′, n=2-6)
Распределение изомеров играет важную роль для общей экономической обоснованности способа производства, для качества конечного сульфонированного продукта и также для его скорости биоразложения.
4) Скорость дезактивации катализатора: эту переменную определяют как отношение активности катализатора при рассматриваемом времени и активности катализатора в нулевой момент времени:
Скорость дезактивации катализатора=активность катализатора (t)/активность катализатора (t=0)
Активность катализатора рассматривают в показателях степени конверсии:
Скорость дезактивации катализатора=степень конверсии (t)/степень конверсии (t=0).
Дезактивация катализатора обусловлена либо отравлением активных центров (необратимая дезактивация), либо процессами загрязнения (образование, обусловленное столкновением на активных центрах или порах катализатора, обычно обратимое посредством регулируемого сжигания или пиролиза).
Поэтому существует потребность в способе получения хорошо растворимого линейного алкилбензолсульфоната с максимизированными моющими свойствами, чрезвычайно низкой цветностью после сульфонирования, оптимальным поведением в окружающей среде и максимально эффективными условиями использования сырьевых материалов.
Настоящее изобретение относится к очень эффективному способу получения сильно растворимого линейного алкилбензольного сульфоната с регулируемым содержанием 2-фенил изомера и чрезвычайно низкой цветностью после сульфонирования, в котором используют каталитическую систему, основанную на высокостабильных твердых катализаторах, с высокой селективностью по линейным моноалкилированным соединениям, такому который решает проблемы, описанные в настоящем уровне техники.
Поэтому первый аспект изобретения относится к способу получения высокорастворимого линейного алкилбензолсульфоната с содержанием 2-фенил изомера от 18 до 70 мас.% путем каталитического алкилирования ароматического соединения алкилирующим агентом, включающему следующие стадии:
1) каталитическую дегидрогенизацию линейного парафинового сырья, при этом получают линейные моноолефины, непреобразованные парафины и определенное количество побочных продуктов, таких как диолефины и нелинейные соединения;
2) селективную гидрогенизацию диолефинов, полученных в качестве побочного продукта на стадии (1), в моноолефины, при этом получают сырьевой алкилирующий агент;
3) очистку сырьевого алкилирующего агента путем отделения нелинейных продуктов, содержащихся в выходящем потоке стадии (2), при этом получают очищенный алкилирующий агент, образованный моноолефинами и парафинами;
4) обработку нелинейных продуктов, извлеченных на стадии (3), с образованием гидротропного предшественника;
5) алкилирование ароматического углеводорода моноолефинами, присутствующими в очищенном алкилирующем агенте, путем сочетания двух способов алкилирования на основе:
а) способа алкилирования с катализатором, образующим сырьевое линейное алкилароматическое соединение с максимальным содержанием 2-фенил изомера, составляющим 20 мас.%;
6) способа алкилирования с катализатором, образующим сырьевое линейное алкилароматическое соединение с минимальным содержанием 2-фенил изомера, составляющим 20 мас.%;
б) разделение на фракции выходящего потока стадии (5) для отделения непрореагировавших ароматических соединений, парафинов и наиболее тяжелых побочных продуктов рассматриваемых алкилароматических соединений;
7) очистку фракции рассматриваемых алкилароматических соединений, поступающих со стадии (6);
8) сульфонирование очищенных линейных алкилароматических соединений, полученных на стадии (7);
9) нейтрализацию линейной алкилсульфоновой кислоты, полученной на стадии (8).
Этот способ отличается тем, что катализатор, образующий максимально 20% 2-фенил изомера, включает цеолит типа FAU, от 0,01 до 0,15 мас.% по меньшей мере одного металла, выбираемого из группы, состоящей из Li, Na, K, Mg или Ca, и от 0,1 до 8,0 мас.% по меньшей мере одного редкоземельного металла, выбираемого из группы, состоящей из La, Ce, Pr, Nd, Pm, Sm или Eu; и тем, что катализатор, образующий минимально 20% 2-фенил изомера, включает цеолит типа MOR, от 0,01 до 0,20 мас.% по меньшей мере одного металла, выбираемого из группы, состоящей из Li, Na, К, Mg или Ca, максимально 0,01% Na, и от 0 до 0,5 мас.% по меньшей мере одного металла, выбираемого из группы, состоящей из Ti, Zr, Hf.
В настоящем изобретении цеолиты типа FAU или EMT-FAU относятся к группе цеолитов с изотипическими структурами, соответствующими структуре типа FAU, таким как цеолит Y, цеолит Na-X, кремнистый цеолит Na-Y, цеолит Linde X, цеолит Linde Y, цеолит ZSM-3 и цеолит ZSM-20, предпочтительно цеолит Linde Y или цеолит Y.
В настоящем изобретении цеолиты типа MOR относятся к группе цеолитов с изотипическими структурами, соответствующими структурному типу MOR, таким как морденит, цеолит Na-D и цеолит Ca-Q, более предпочтительно мордениту.
В конкретном воплощении изобретения катализатор, образующий максимально 20 мас.% 2-фенил изомера, включает:
а) цеолит с изотипической структурой типа FAU, предпочтительно цеолит Y;
б) от 0,01 до 0,15 мас.% по меньшей мере одного металла, выбираемого из группы, состоящей из Li, Na, K, Mg или Ca, предпочтительно примерно 0,08 мас.% Na;
в) от 0,1 до 8 мас.% по меньшей мере одного редкоземельного металла, выбираемого из группы, состоящей из La, Ce, Pr, Nd, Pm, Sm или Eu, предпочтительно от 3 до 4 мас.% La, от 0,5 до 1,5 мас.% Ce, от 0,1 до 1 мас.% Pr и от 1 до 2 мас.% Nd.
В конкретном воплощении изобретения катализатор, образующий максимально 20 мас.% 2-фенил изомера по своей структуре и текстуре характеризуется:
а) порошковой рентгенограммой, отличающейся тем, что наиболее интенсивный пик появляется при угле 2θ, соответствующем 6,2°, а остальные основные пики появляются при углах дифракции 2θ, соответствующих 23,6°; 27°; 31,3°; 15,6°; 20,3°; 18,6° и 30°, указанных в порядке уменьшения интенсивности соответствующих пиков;
б) общим молярным отношением кремний/алюминий от 1:1 до 3:1, предпочтительно 1,6:1,0;
в) суммарной удельной поверхностью (по БЭТ от 400 до 700 м2/г, предпочтительно 650 м2/г;
г) суммарной удельной поверхностью микропор от 350 до 600 м2/г, предпочтительно 525 м2/г;
д) удельным объемом микропор от 0,1 до 0,3 мл/г, предпочтительно 0,21 мл/г;
е) распределением макропор, в котором диаметр макропор находится в диапазоне от 20 до 2000 ангстрем, предпочтительно от 20 до 80 ангстрем.
В конкретном воплощении изобретения катализатор, образующий минимально 20 мас.% 2-фенил изомера, включает:
а) цеолит с изотипической структурой MOR-типа,
б) от 0,01 до 0,2 мас.% по меньшей мере одного металла, выбираемого из группы, состоящей из Li, Na, K, Mg или Ca, предпочтительно, примерно 0,07 мас.% Mg,
в) максимально 0,01% Na,
г) от 0 до 0,5 мас.% по меньшей мере одного металла, выбираемого из группы, состоящей из Ti, Zr, Hf, предпочтительно 0,25 мас.% Ti.
В частном воплощении изобретения катализатор, образующий минимально 20 мас.% 2-фенил изомера по своей структуре и текстуре характеризуется:
а) порошковой рентгенограммой, отличающейся тем, что наиболее интенсивный пик появляется при угле 2θ, соответствующем 25,6°, а остальные основные пики появляются при углах дифракции 2θ, соответствующих 22,3°; 26,3°; 19,6°; 13,5° и 27,7°, указанных в порядке уменьшения интенсивности соответствующих пиков;
б) общим молярным отношением кремний/алюминий от 1,5:1,0 до 3,5:1,0, предпочтительно 2,8:1,0;
в) суммарной удельной поверхностью (по БЭТ) от 150 до 600 м2/г, предпочтительно 490 м2/г;
г) суммарной удельной поверхностью микропор от 300 до 450 м2/г, предпочтительно 380 м2/г;
д) удельным объемом микропор от 0,01 до 0,2 мл/г, предпочтительно 0,15 мл/г;
е) распределением макропор, в котором диаметр макропор находится в диапазоне от 20 до 800 ангстрем, предпочтительно от 20 до 300 ангстрем.
В конкретном воплощении настоящего изобретения парафины стадии (1) включают алканы с прямой цепью, содержащие от 8 до 30 атомов углерода, предпочтительно от 10 до 16 атомов углерода, более предпочтительно от 10 до 14 атомов углерода. Эти парафины можно дегидронезировать и очистить посредством любого способа, описанного в известном уровне техники.
В конкретном воплощении настоящего изобретения ароматический углеводород представляет собой ароматический углеводород, выбираемый из группы: толуол, ксилол, бензол или их смеси, предпочтительно, он является бензолом.
В конкретном воплощении настоящего изобретения ароматическое соединение и олефин смешивают перед реакцией алкилирования стадии (5) в молярном соотношении бензол: олефин, составляющем от 5:1 до 70:1, предпочтительно от 10:1 до 30:1, более предпочтительно от 10:1 до 15:1. В конкретном воплощении эта смесь дополнительно включает от 0 до 0,3 мас.% нелинейных соединений. В конкретном воплощении эта смесь дополнительно включает от 0 до 0,1 мас.% воды.
В конкретном воплощении настоящего изобретения реакцию алкилирования стадии (5) проводят в реакторе с размещением катализатора, выбираемого из группы, состоящей из неподвижного каталитического слоя с одним катализатором, по меньшей мере одного неподвижного каталитического слоя с двумя полностью перемешанными различными твердыми катализаторами, по меньшей мере двух обособленных неподвижных каталитических слоев, каждый из которых содержит одинаковый катализатор, по меньшей мере двух обособленных неподвижных каталитических слоев, каждый из которых содержит различные катализаторы, и реактора с псевдоожиженным слоем с одним или более различных катализаторов.
В конкретном воплощении реакции алкилирования проводят одновременно в конфигурации реактора, включающей по меньшей мере одну конфигурацию, выбираемую из группы: независимый реактор, по меньшей мере два реактора, соединенные параллельно, по меньшей мере два реактора, соединенные последовательно, и их сочетания.
В конкретном воплощении очистку на стадии (3) осуществляют посредством технологий очистки, известных специалистам в области техники, например таких, как гидрогенизация, разделение на фракции и селективная адсорбция. В случае селективной адсорбции адсорбирующий слой формируют по меньшей мере из одного материала, выбираемого из группы, состоящей из цеолитов, диоксида кремния, силикагеля, макропористого силиката магния, активированного оксида алюминия, алюмосиликатов, глин, молекулярных сит, ацетата целлюлозы, геля макропористого полистирола, активированного угля и органоселективных полимерных мембран.
В конкретном воплощении очистку на стадии (4) осуществляют посредством технологий очистки, известных специалистам в области техники, например таких, как гидрогенизация, разделение на фракции и селективная адсорбция. В случае селективной адсорбции адсорбирующий слой формируют по меньшей мере из одного материала, выбираемого из группы, состоящей из цеолитов, диоксида кремния, силикагеля, макропористого силиката магния, активированного оксида алюминия, алюмосиликатов, глин, молекулярных сит, ацетата целлюлозы, геля макропористого полистирола, активированного угля и органоселективных полимерных мембран.
В конкретном воплощении очистку на стадии (6) осуществляют посредством технологий очистки, известных специалистам в области техники, например таких, как гидрогенизация, разделение на фракции и селективная адсорбция. В предпочтительном воплощении очистку на стадии осуществляют посредством селективной адсорбции при помощи селективного адсорбента типа глины, характеризуемого:
а) общим молярным соотношением диоксид кремния: оксид алюминия от 3:1 до 6:1, предпочтительно от 3,5:1,0 до 5,5:1,0, более предпочтительно 4,2:1;
б) содержанием от 1 до 4 мас.% Fe, предпочтительно 2,1 мас.%;
в) от 0,5 до 2 мас.% K, предпочтительно 1,2 мас.%;
г) от 0,2 до 2 мас.% Mg, предпочтительно 0,7 мас.%;
д) от 0,1 до 1,0 мас.% TiO2, предпочтительно 0,3 мас.%;
е) суммарной удельной поверхностью, выраженной как поверхность по БЭТ, составляющей от 150 до 500 м2/г, предпочтительно от 200 до 400 м2/г, более предпочтительно 260 м2/г;
ж) совокупным объем пор от 0,1 до 2,0 мл/г, предпочтительно 0,4 мл/г;
з) распределением макропор, в котором диаметр макропор находится в диапазоне от 20 до 800 ангстрем, предпочтительно от 20 до 200 ангстрем, более предпочтительно от 20 до 100 ангстрем, при среднем диаметре по показателю объема пор с центром на 34 Ангстремах.
В конкретном воплощении гидротропный предшественник стадии (4) добавляют к алкилароматическому потоку перед очисткой на стадии (7), очисткой, сульфонированием и/или нейтрализацией, таким образом проводя совместную очистку и сульфонирование.
В конкретном воплощении гидротропный предшественник стадии (4) очищают, сульфонируют и/или нейтрализуют и впоследствии добавляют к алкилароматическому сульфонату, полученному на стадии (8).
В конкретном воплощении нейтрализацию на стадии (9) выполняют посредством щелочного вещества, включающего один или более катионов, выбираемых из группы: Na, K, NH4+, Mg, Ca, Ba, или посредством замещенных аммонийных щелочей.
В конкретном воплощении температура реакции составляет от 20 до 400°C.
В конкретном воплощении объемная скорость составляет от 1 час-1 до 15 час-1.
Другой аспект настоящего изобретения относится к способу получения выскорастворимого линейного алкилбензолсульфоната с содержанием 2-фенил изомера, составляющим по меньшей мере 18 мас.%, посредством каталитического алкилирования ароматического соединения алкилирующим агентом, как описано ранее, в котором стадии (1), (2), (3) и (4) являются необязательными.
В более конкретном воплощении олефины представляют собой альфа-олефины и включают от 18 до 28 атомов углерода, предпочтительно от 20 до 24 атомов углерода, и могут дополнительно включать максимально 10 мас.%, разветвленных олефинов.
В более конкретном воплощении настоящего изобретения ароматическое соединение и альфа-олефины смешивают перед реакцией алкилирования стадии (5) в молярном соотношении ароматическое соединение: олефин, составляющем от 5:1 до 70:1.
В более конкретном воплощении эта смесь дополнительно включает от 0 до 0,01 мас.% воды.
Другой аспект настоящего изобретения относится к подходящим чистящим композициям для средств мытья посуды, очистителей для твердых покрытий, жидких моющих средств, порошковых моющих средств, чистящих средств в форме паст, гелей, моющих брусков, включающим:
а) от 1 до 99 мас.% растворимого линейного алкилбензолсульфоната, полученного посредством способа согласно любому из описанных выше;
б) от 99 до 1 мас.% других ингредиентов моющих средств, выбранных из группы, состоящей из производных жирных спиртов, сопутствующих поверхностно-активных веществ, структурирующих агентов, растворителей, добавок и их смесей, таких как производные жирных спиртов, жирные кислоты, алкилсульфаты, этаноламин, аминоксид, щелочные карбонаты, этанол, изопропанол, вода, хвойное масло, хлорид натрия, силикат натрия, полимеры, алкоксилаты спиртов, пербораты, цеолиты, щелочные сульфаты, энзимы, гидротропы, красящие вещества, душистые вещества, консерванты, блескообразователи, вещества, способствующие смешиванию (incorporators), полиакрилаты, эфирные масла, щелочи, эфирсульфонаты, водорастворимые разветвленные алкилбензолсульфонаты, алкилфенолалкоксилаты, амины жирных кислот, альфа-олефинсульфонаты, парафинсульфонаты, бетаины, хелатообразующие агенты, этоксилаты таллового амина, полиэфираминэтоксилаты, блоксополимеры этиленоксида и пропиленоксида, малопенящиеся спиртовые поверхностно-активные вещества на основе этиленоксида/пропиленоксида, метилэфирсульфонаты, алкилполисахариды, н-метилглюкамиды, сульфонированный алкилдифенилоксид и полиэтиленгликоль.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
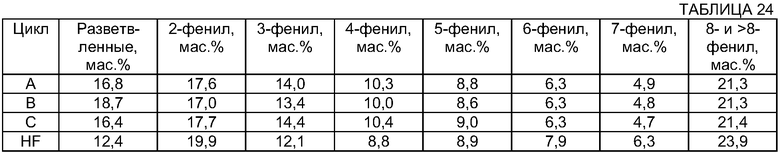
На Фиг.1 показана схема реакции по настоящему изобретению в форме технологической схемы.
На Фиг.2 показана схема реакции по настоящему изобретению в форме технологической схемы, в которой опущены стадии каталитической дегидрогенизации парафина, гидрогенизации диолефина, очистки алкилирующего агента и очистки и добавления гидротропного предшественника.
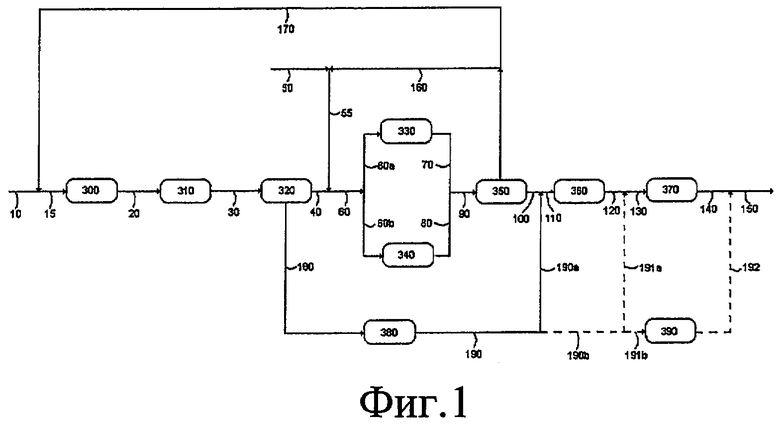
На Фиг.3 одновременно показаны две хроматограммы полученных продуктов, полученные посредством газовой хроматографии с детектором ионизации пламени (ГХ-ДИП), после разделения непрореагировавшего бензола и парафинов, когда выполняют алкилирование бензола очищенным алкилирующим агентом, используя два различных катализатора по отдельности: катализатор, используемый обычным образом в аналогичном способе алкилирования (катализатор В), и один из катализаторов, применение которых заявлено в этом патенте (катализатор A). Хроматограмма конкретно относится к времени удерживания тяжелого алкилата.
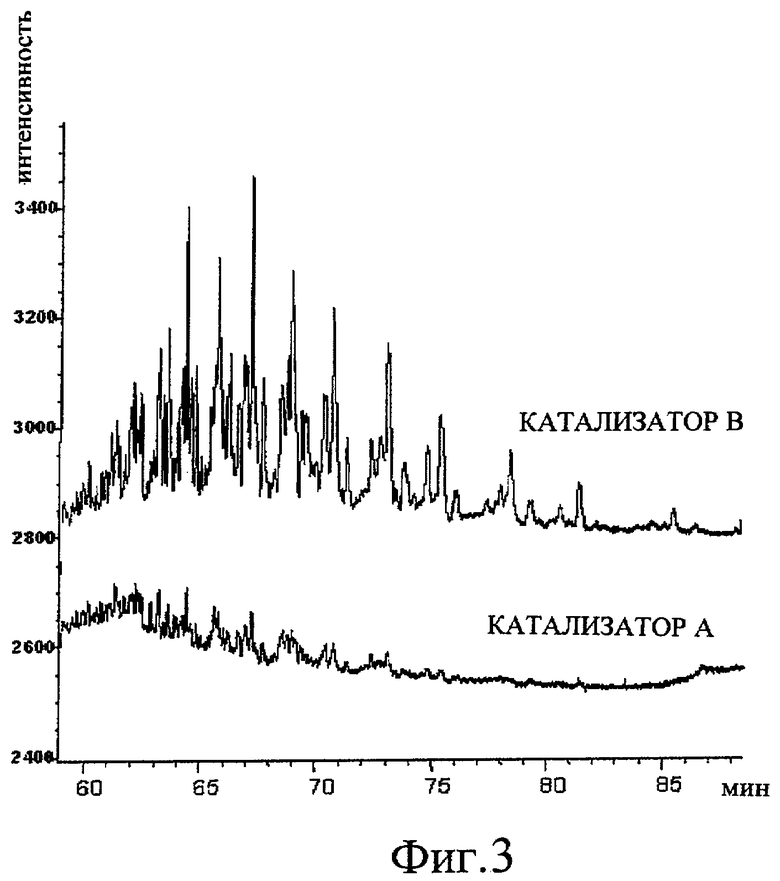
На Фиг.4 одновременно показаны две хроматограммы полученных продуктов, полученные посредством ГХ-ДИП, после разделения непрореагировавшего бензола и парафинов, когда выполняют алкилирование бензола очищенным алкилирующим агентом, используя один из катализаторов, заявленных в этом патенте.
На Фиг.1 показана неограничивающая схема реализации этого изобретения.
Поток 15 представляет собой парафиновое сырье, подаваемое в блок дегидрогенизации, и включает свежую парафиновую смесь, поток 10, вместе с непреобразованными парафинами, отделяемыми в блоке очистки сырьевого алкилбензола, блоке 350, и направляемыми рециклом в потоке 170. В блоке 300 дегидрогенизации частично преобразуют подаваемое парафиновое сырье в смесь моноолефинов, парафинов и различных побочных продуктов, таких как диолефины и нелинейные соединения. В блок очистки олефинов, блок 310, подают выходящий поток из блока дегидрогенизации через поток 20, увеличивая чистый выход моноолефинов путем конверсии некоторых побочных продуктов, образуемых в блоке дегидрогенизации, главным образом диолефинов, в моноолефины. Получающийся поток, поток 30, обрабатывают в блоке 320, содержащем селективный адсорбент для устранения нелинейных соединений, получаемых в процессе дегидрогенизации. Свежий бензол закачивают в процесс через поток 50 и смешивают с повторно используемым бензолом, который не прореагировал (поток 160), поступающим из блока очистки сырьевого алкилбензола, блока 350. Смесь этих двух потоков образует поток подаваемого бензола (поток 55), который смешивают с выходящим потоком (поток 40) из блока 320 селективной адсорбции с образованием потока 60, состоящего из моноолефинов, бензола и парафинов и подаваемого в блок алкилирования.
Поток 60 разделяют на два одинаковых потока, 60a и 60b, которые подают в два различных реактора алкилирования, блоки 330 и 340 соответственно; в реакторе 330 алкилирования используют катализатор, образующий сырьевой линейный алкилбензол (поток 70) с максимальным содержанием 2-фенил изомера по меньшей мере 20 мас.%, в то время как в реакторе 340 алкилирования используют катализатор, образующий сырьевой линейный алкилбензол (поток 80) с содержанием 2-фенил изомера по меньшей мере 20 мас.%. Потоки 70 и 80 смешивают в различных пропорциях для получения потока, потока 90, содержащего линейный алкилбензол с изменяемым содержанием 2-фенил изомера, непрореагировавший бензол, парафины и побочные продукты. Поток 90 подают в блок очистки сырьевого алкилбензола, блок 350, в котором непрореагировавший бензол, парафины и побочные продукты, более тяжелые, чем линейный алкилбензол, перегоняют с целью получения относительно чистого линейного алкилбензола (поток 100). После этого поток 100 подают на конечную стадию очистки алкилбензола, в блок 360, который содержит селективный адсорбент для устранения ароматических соединений, которые еще присутствуют в небольших количествах в относительно чистом алкилбензоле в силу того факта, что диапазон температуры кипения перекрывается с диапазоном температуры кипения представляющего интерес алкилбензола.
Нелинейные соединения, извлеченные в блоке 320, закачивают в блок 380 специальной обработки через поток 180. Этот блок 380 включает такие стадии, как разделение на фракции, гидрогенизация и/или селективная адсорбция, для преобразования нелинейных примесей в гидротропном предшественнике (поток 190). В зависимости от рабочих условий способа данного изобретения поток 190 можно провести через потоки 190a и 190b или его нельзя использовать, если содержание 2-фенил изомера линейного алкилбензола составляет менее 60 мас.%. Относительно чистый поток линейного алкилбензола, поток 100, и поток 190a гидротропного предшественника можно таким образом смешать, образуя поток 110; их очищают совместно в блоке 360 и затем направляют в потоках 120 и 130 в блок 370, где их сульфонируют/нейтрализуют совместно, получая конечный поток, поток 150. Потоки 190a можно также опустить, так что относительно чистый линейный алкилбензол, поступающий из блока 350, поток 100-110, отдельно очищают в блоке 360, и после его очистки (поток 120), затем смешивают его с гидротропным предшественником, поставляемым потоками 190b и 191a, образуя поток 130, таким образом сульфонируя и нейтрализуя совместно в блоке 370, таким образом образуя конечный поток, поток 150. Потоки 190a и 191a можно также опустить, так что только один относительно чистый алкилбензол потока 100 очищают (блок 360), сульфонируют и нейтрализуют (блок 370), отдельно получая поток алкилбензолсульфоната. Этот поток 140 затем смешивают с потоком 192, относящимся к гидротропу, который отдельно сульфонируют и нейтрализуют в блоке 390 через потоки 190, 190b и 191b, таким образом образуя конечный поток 150. Поток 150 включает высокорастворимый линейный алкилбензолсульфонат с регулируемым содержанием 2-фенил изомера и низкой цветностью после сульфонирования. Добавку гидротропного вещества (в любом из этих вариантов) выполняют, когда содержание 2-фенил изомера превышает 60 мас.%.
На Фиг.2 показана другая неограничивающая схема реализации этого изобретения, когда стадии 1), 2), 3) и 4) являются необязательными.
Поток 10 представляет собой подачу свежего альфа-олефина в способ. Поток 20 представляет собой подачу свежего ароматического соединения, такого как бензол, в способ. Поток 20 смешивают с потоком 60, который представляет собой рециркулируемый избыток ароматического соединения, после его выделения из способа алкилирования в ректификационной колонне, блоке 120. Поток 30, который представляет собой подачу ароматического соединения, образуют путем смешивания потоков 20 и 60. Этот поток 30 смешивают с потоком 10 в блоке 100 смешивания с целью получения смеси ароматического соединения и олефинов при требуемом молярном соотношении ароматическое соединение/олефин, потока 40. Этот поток подают в реактор алкилирования, блок 110, где катализатор размещают в реакторе с неподвижным слоем, так что алкилирование ароматического соединения осуществляют с помощью олефинов. Выходящий поток из реактора алкилирования, поток 50, состоит из непрореагировавшего ароматического соединения и алкилароматического соединения, получаемого в течение алкилирования. Поток 50 разделяют в ректификационной колонне, блоке 120, с целью отделения непрореагировавшего ароматического соединения, в ее верхней части, поток 60, и алкилароматического соединения в ее нижней части, поток 70. Поток 60 возвращают обратно в способ, как упоминалось ранее, в то время как поток 70 сульфонируют в реакторе пленочного типа (falling-film reactor) газообразным SO3, блоке 130. Сульфонированный продукт, поток 80, состоит из высокочистой линейной алкилароматической сульфокислоты с определенным содержанием 2-фенил изомера. Этот продукт можно затем нейтрализовать в блоке 140 основными солями кальция, бария, натрия, магния и аммиака в присутствии сильно ионизируемого соединения, такого как фенол, с целью получения высокочистого алкилароматического сульфоната, как нейтрального, так и предпочтительно сверхщелочного, в зависимости от количества основания, используемого в нейтрализации.
Данное изобретение описано дополнительно, только с целью иллюстрации, посредством следующих примеров, которые не следует рассматривать как ограничивающие область настоящего изобретения.
Примеры
Пример 1
В этом примере показано преимущество использования цеолита Y, допированного 7 мас.% редкоземельных металлов, таких как La, Ce, Pr и Nd (названного катализатором A) при алкилировании бензола смесью олефинов/парафинов по сравнению с использованием промышленного аморфного фторированного диоксида кремния-оксида алюминия (названного катализатором B), широко используемого в этой реакции в промышленном масштабе. Оба катализатора измельчали и просеивали с получением эквивалентного распределения размеров частиц. Бензол сушили с помощью молекулярного сита, чтобы минимизировать добавление воды, и его затем смешивали со смесью моноолефинов и парафинов, которые поступали из дегидрогенизации парафинов и селективной гидрогенизации диолефинов. Испытания на опытной установке для алкилирования были выполнены в изотермическом реакторе с неподвижным слоем с 24-часовыми реакционными циклами, за которыми следовали циклы регенерации катализатора посредством промывки бензолом в течение такого же периода времени. Стандартный цикл включает 24-часовой реакционный цикл при 140°C, ЧОСЖ (часовая объемная скорость жидкости)=11 час-1 и молярном отношении бензол:олефин 30:1, за которым следует цикл промывки бензолом в течение такого же периода времени. В Табл.1 представлены масса катализатора, условия промывки и т.д.
Композиция исходного сырьевого материала, относящегося к смеси олефинов и парафинов, суммирована в Табл.2.
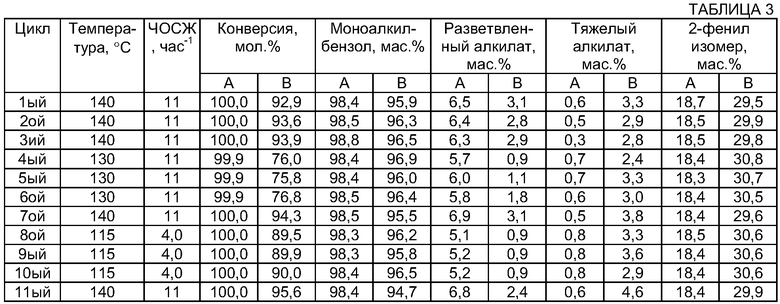
Эту смесь парафинов и олефинов смешивали с сухим бензолом до достижения требуемого молярного отношения бензол:олефин. Оценку катализатора выполняли в пяти различных последовательностях реакционных циклов. После осуществления каждого реакционного цикла выходящий продукт алкилирования разгоняли за три стадии, используя три ректификационные колонны (первая работала при атмосферном давлении, в то время как две другие работали под вакуумом). Первая колонна работала при атмосферном давлении и в ней отделяли непрореагировавший бензол в ее верхней части, в то время как соединения, присутствующие в ее нижней части, подавали во вторую колонну. Во второй колонне отделяли парафины в ее верхней части, в то время как соединения, присутствующие в ее нижней части, подавали в третью колонну. В третьей колонне отделяли алкилбензол в ее верхней части, а тяжелый алкилат - в ее нижней части. Анализы (выполненные посредством ГХ-ДИП) относятся к соединениям, подаваемым в третью колонну. В первой последовательности реакционных циклов были независимо испытаны оба катализатора, используя одинаковые рабочие условия. Для каждого катализатора последовательность включала семь циклов, которые выполняли при ЧОСЖ 11 час-1, при температуре 130 и 140°C, за которыми следовали три цикла при 155°C и ЧОСЖ 4 час-1. Далее был выполнен стандартный цикл при 140°C и ЧОСЖ 11 час-1 с целью проверки деактивации катализатора. Во всех этих циклах молярное отношение бензола к олефину поддерживали при значении 30. Все эти условия и результаты представлены в Табл.3.
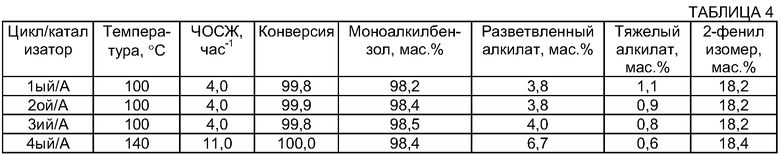
Во второй последовательности реакционных циклов катализатор A испытывали для того, чтобы проанализировать его активность при более низких температурах. Эта последовательность включала три реакционных цикла, которые выполняли при ЧОСЖ 4 час-1, однако теперь при 100°C, за которыми следовал стандартный цикл (ЧОСЖ=11 час-1; Т=140°C). Во всех этих циклах молярное отношение бензола к олефину поддерживали при значении 30. Все условия и результаты представлены в Табл.4.
В третьей последовательности реакционных циклов оба катализатора испытывали независимо для того, чтобы проанализировать их скорость дезактивации. Чтобы сильнее воздействовать на стабильность катализатора после стандартного цикла (ЧОСЖ=11 час-1; Т=140°C), нормальный период реакции в 24 часа расширяли до 60 часов без стандартной промывки бензолом. Во всех этих циклах молярное отношение бензола к олефину поддерживали при значении 30. Каталитическую активность анализировали в показателях степени конверсии. Все результаты и условия представлены в Табл.5.
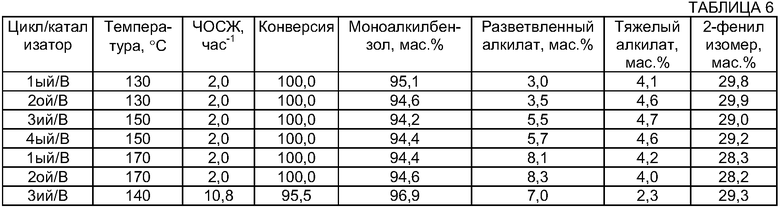
Четвертая последовательность реакционных циклов, в которой испытывали катализатор B, включала шесть реакционных циклов, которые выполняли при ЧОСЖ 2 час-1 (с целью достижения 100% конверсии), при различных температурах, за этим следовал стандартный цикл (ЧОСЖ=11 час-1; Т=140°C). Во всех этих испытаниях молярное отношение бензола к олефину поддерживали при значении 30. Все эти условия и результаты представлены в Табл.6.
Пятая последовательность реакционных циклов, в которой испытывали только катализатор A, включала шесть реакционных циклов, их выполняли с различными ЧОСЖ, температурами и молярным отношением бензола к олефину с целью анализа влияния последней переменной на конверсию и селективность реакции. Все результаты и условия представлены в Табл.7.

Как можно видеть в Табл.3, активность катализатора A намного выше активности катализатора B, так как катализатор A достигает 100% степени конверсии почти во всех циклах. Даже когда температура составляет 100°C (ЧОСЖ=4 час-1, Табл.4), с катализатором A достигают приблизительно 100% степени конверсии, в то время как с катализатором B не достигают 100% степени конверсии до очень низких пространственных скоростей и температур выше 130°C (Табл.6). Для того чтобы подвергнуть активность катализатора нагрузке, нормальный период реакции 24 часа расширяли до 60 часов в третьей последовательности (Табл.5) и, в случае катализатора A даже после 50-го часа реакции наблюдаемая конверсия была выше 99%, в то время как с катализатором B дезактивацию наблюдали после 24 часов. Это подразумевает, что катализатор A намного более устойчив к дезактивации по сравнению с катализатором B, поэтому этот катализатор позволяет работать в более длинных реакционных циклах, уменьшая число промывок бензолом в течение срока службы катализатора и, в заключение, уменьшая эксплуатационные расходы.
Другое важное преимущество катализатора A связано с получением тяжелого алкилата. Как можно видеть в Табл.3, катализатор A производит значительно меньшее количество тяжелых субпродуктов, чем катализатор B (по меньшей мере, на 80% меньше), достигая менее 1% тяжелого алкилата во всех этих испытаниях, когда молярное отношение бензола к олефину составляет 30:1.
На Фиг.3 показаны результаты ГХ-ДИП продуктов, полученных при испытаниях с молярным отношением бензола к олефину, равным 30 (после разделения бензола и парафина), относящейся к продуктам, полученным с катализатором A и катализатором B. В частности, они соответствуют временам удерживания тяжелого алкилата.
Как можно видеть на Фиг.3, катализатор A образует намного менее интенсивные пики в хроматографической области, соответствующей соединениям алкилбензольного типа (тяжелые алкилаты), чем аморфный катализатор B. Эти соединения имеют от 26 до 32 атомов углерода и они образуются путем реакции моноалкилбензола с олефином, образуя диалкилбензольное соединение (такое, как дидецилбензол, дидодецилбензол, децилдодецилбензол и т.д.). Преимущество использования катализатора A состоит в том, что его внутренняя микропористая структура включает ограничения размера и формы, минимизирующие образование этих побочных продуктов, так как их размер больше, чем диаметр пор катализатора.
С целью анализа влияния молярного отношения бензола к олефину на распределение продукта были выполнены эксперименты, результаты которых представлены в Табл.7. Как видно из Табл.7, когда это отношение уменьшается, количество тяжелых побочных продуктов возрастает, однако даже с испытанным минимальным молярным отношением в этой последовательности (молярное отношение бензола к олефину, равное 10, Табл.7), когда Табл.7 сравнивают с Табл.3, количество тяжелых побочных продуктов, образованных посредством катализатора A (примерно 3%), меньше или равно количеству, получаемому посредством катализатора B при более высоком молярном отношении (молярное отношение бензола к олефину, равное 30). Таким образом, катализатор A позволяет работать при более низком молярном отношении бензола к олефину, чем катализатор B, что предполагает более низкие расходы на последующей стадии отделения непрореагировавшего бензола.
Наконец, как можно видеть в Табл.3, катализатор A производит относительно большое количество разветвленного алкилата по сравнению с катализатором B. Однако катализатор A является более активным, чем катализатор B, таким образом не нужно эксплуатировать реакционную систему при настолько высоких температурах, как 130-140°C (как можно наблюдать во всех результатах, по мере того, как температура возрастает, содержание разветвленных углеводородов увеличивается) для достижение 100% степени конверсии. Это означает, что когда реакционная система работает при промышленно разумной объемной скорости (ЧОСЖ=4 час-1) и поддержании температуры при 100°C (Табл.4), достигают 100% степени конверсии и содержание разветвленных углеводородов в алкилате уменьшается до 4 мас.% приемлемого значения и аналогичного достигаемому, когда катализатор B работает при 100% степени конверсии (Табл.6).
Пример 2
Этот пример показывает преимущества очистки смеси олефинов/парафинов, поступающей со стадий дегидрогенизации и селективной гидрогенизации, перед тем как они реагируют с бензолом на стадии алкилирования. Эту реакцию катализировали цеолитом Y, допированным 7 мас.% редкоземельных металлов, таких как La, Ce, Pr и Nd (названным катализатором A). В этом примере катализатор A размещали на неподвижном слое с целью ускорения алкилирования бензола очищенными и неочищенными смесями олефин/парафин.
Стадию очистки выполняют, используя адсорбирующий слой, в котором размещено определенное количество некоторого молекулярного сита. Выбранное молекулярное сито является цеолитом 13X, широко используемым в способах селективного удаления ароматических компонентов из смеси олефинов и парафинов. Смесь олефинов и парафинов пропускают через слой для достижения селективной адсорбции нелинейных соединений, поступающих из предыдущей стадии дегидрогенизации (или присутствующих в свежей подаче парафина и/или повторно используемого парафина). После насыщения слой промывают парафинами с короткой цепью для десорбции олефинов и парафинов, которые могут оставаться в порах, и затем их транспортируют вместе с бензолом с целью извлечения адсорбированных ароматических соединений, которые можно использовать как гидротропные предшественники. Рабочие условия для стадии очистки для опытной установки представлены в Табл.8.
Смесь олефинов и парафинов, поступающая из стадий дегидрогенизации и гидрогенизации, содержала приблизительно 2 мас.% ароматических соединений. Ее состав после стадии очистки представлен в Табл.9.
После очистки смесь олефинов и парафинов смешивали с сухим бензолом для получения выбранного молярного отношения. Затем эту смесь использовали в качестве подаваемого сырья для стадии алкилирования. Рабочие условия для стадии алкилирования представлены в Табл.10. Другие параметры, связанные с неподвижным слоем, являются теми же самыми, как и параметры, представленные в Примере 1 (катализатор A), так как алкилирование очищенными и неочищенными смесями выполняют в том же реакторе и при тех же условиях, определенных в этом Примере. Процесс разгонки выполняли для разделения бензола, парафинов, алкилбензолов и тяжелого алкилата, как определено в Примере 1. Газохроматографические анализы относятся к потоку, подаваемому в третью колонну. Легкие продукты анализировали посредством ГХ-ДИП на верхнем потоке первой колонны.
Другое испытание выполняли с целью анализа влияния стадии очистки на распределение продуктов, сохраняя все рабочие параметры при тех же значениях, как и раньше, однако используя неочищенную смесь. Распределение продуктов, относящееся к алкилированию с очищенными и неочищенными смесями, представлено в Табл.11.
Как можно видеть в Табл.11, количество тяжелого алкилата, образованного при использовании очищенных смесей, уменьшается на 86 мас.% по сравнению с неочищенными смесями. На Фиг.4 показаны хроматограммы, соответствующие ГХ-ДИП полученных продуктов, после разделения бензола и парафинов, когда используют очищенные и неочищенные смеси.
Верхняя линия соответствует использованию неочищенной смеси, в то время как нижняя линия соответствует использованию очищенной смеси. При сравнении обеих хроматограмм становится ясно, что линии интенсивности пиков, соответствующих тяжелому алкилату, получаемому при использовании очищенных смесей, намного ниже, чем при использовании неочищенных смесей. Эффект селективной адсорбции в основном влияет на конечное содержание «наилегчайших» тяжелых алкилатов, расположенных на временах удерживания менее 62 минут, как показано на Фиг.4. Эти продукты в основном являются соединениями из 20-22 атомов углерода, такими как алкилфлуорены, алкилантрацены, алкилфенантрены, инданы, индены и т.д. Они в основном образуют путем алкилирования полиароматических соединений, образуемых на стадии дегидрогенизации (таких, как антрацен, фенантрен, флуорены и т.д.) C10-C14 олефинами. Очистка олефинов и парафинов адсорбирующим слоем включает устранение этих продуктов, которые непосредственно образуются на стадии дегидрогенизации или содержатся в свежем или повторно используемом сырье, а также устранение тех полиароматических соединений, которые могут взаимодействовать с C10-C14 олефинами на стадии алкилирования.
Как можно видеть в Табл.11, использование очищенных смесей также включает уменьшение конечного количества легких побочных продуктов. Количество образующихся легких побочных продуктов снижается на 44 мас.% при использовании очищенных смесей.
Пример 3
Этот пример показывает стабильность цеолита Y, допированного редкоземельными металлами, когда его используют в качестве катализатора при алкилировании бензола олефинами. Длинную последовательность циклов, в которых использовали цеолит Y, допированный редкоземельными металлами, выполняли с целью анализа скорости их дезактивации. Было выполнено тридцать стандартных циклов, как прежде было определено в Примере 1, при ЧОСЖ=8 час-1 и Т=140°C. После каждого 24-часового реакционного цикла выполняли промывочный цикл в течение 24 часов. Результаты представлены в Табл.12 и они усреднены по рассматриваемым циклам.
Как можно видеть в Табл.12, в течение этой последовательности циклов реакции/регенерации средняя степень конверсии всегда выше 99,5% и не было зафиксировано никакой дезактивации в третьем десятке циклов по отношению к начальным циклам. Это означает, что цеолит Y, допированный редкоземельными металлами, является очень стабильным катализатором, поддерживающим высокую степень конверсии с течением времени и не показывающим каких-либо признаков дезактивации.
Пример 4
Этот пример показывает преимущества использования двух параллельных изотермических реакторов с неподвижным слоем, один из которых загружен ранее используемым цеолитом Y, допированным 7 мас.% редкоземельными металлами, такими как La, Ce, Pr и Nd (называемый катализатором А) и другой загружен промышленным кристаллическим морденитом, называемым катализатором С, при регулировании распределения предварительно очищенного подаваемого сырья между двумя реакторами и смешивании получаемых выходящих потоков с целью получения регулируемого содержания 2-фенил изомера в результирующем выходящем потоке.
Определенное количество катализатора А помещали в один из реакторов с неподвижным слоем (называемым слоем А), в то время как другой реактор с неподвижным слоем (называемый слоем С) загружали определенным количеством промышленного кристаллического морденита (катализатор С). Состав подаваемого сырья был одинаковым для обоих реакторов. Поток подаваемого сырья получали путем смешивания очищенной смеси олефинов и парафинов (композиция смеси определена в Примере 2) с подходящим количеством сухого бензола с получением требуемого молярного отношения бензола к олефину. Указанный начальный поток всегда поддерживали при постоянном расходе. После этого поток разделяли на два подпотока посредством трехходового клапана, каждый поток подавали в реактор (после стадии предварительного нагревания) с целью дозирования переменного потока в каждый реактор посредством регулирования клапана (однако сохраняя постоянным общий расход). Выходящие потоки из каждого реактора смешивали, таким образом получая конечный выходящий поток, который анализировали (после разделения бензола и парафинов посредством разгонки, как было показано в Примере 1) с помощью ГХ-ДИП. В этом Примере как состав подаваемого сырья, так и давление реакции поддерживали постоянными для обоих реакторов, в то время как температура реакции была различной в каждом реакторе и расход подаваемого сырья в каждом реакторе изменяли с целью модифицирования конечного содержания 2-фенил изомера. Рабочие условия представлены в Табл.13.
Необходимо принять во внимание, что часовая объемная скорость жидкости (ЧОСЖ), относящаяся к каждому реактору, изменяется, когда изменяют процентное содержание исходного подаваемого сырья, дозируемого в каждый реактор, от 2,7 час-1 (когда 25% исходного подаваемого сырья проходит через такой реактор) до 10,8 час-1 (когда 100% исходного подаваемого сырья проходит через один реактор). Распределение продукта в конечном выходящем потоке при изменении расходов подаваемого сырья представлено в Табл.14.
Как можно видеть в Табл.14, изменение потока, подаваемого в каждый реактор, подразумевает изменение конечного содержания 2-фенил изомера от 18 мас.% (полученного, когда все исходное подаваемое сырье проходит через слой катализатора А) до 70 мас.% (полученного, когда все исходное подаваемое сырье проходит через слой морденита). Во всех случаях количество тяжелого алкилата, регистрируемого в конечном выходящем потоке, было меньше или равно 1,6% масс. и количество разветвленного алкилата всегда было меньше 6,5% масс. Оба катализатора показывают очевидную стабильность по отношению к изменениям ЧОСЖ (в показателях степени конверсии и селективности) в диапазоне ЧОСЖ, который был проверен.
Пример 5
Этот пример показывает преимущества очистки сырьевого алкилбензола перед стадией сульфонирования с целью сведения к минимуму цветности после сульфонирования конечной сульфоновой кислоты.
Очищенную смесь олефинов и парафинов, смешанную с сухим бензолом, использовали в качестве сырья, как описано в Примерах 2 и 4. Было выбрано молярное отношение бензола к олефину, равное 20:1. Алкилирование выполняли посредством двух параллельных изотермических реакторов с неподвижным слоем, как в Примере 4. Катализаторы и рабочие условия были в точности такими же, как в Примере 4. Для каждого реактора была выбрана дозировка подаваемого сырья в количестве 50% исходного потока. Сырьевой алкилбензол был получен путем фракционирования выходящего потока, поступающего со стадии алклирования. Однако разгонка слегка отличалась от разгонки, использованной в предыдущих Примерах. В этом примере были использованы четыре ректификационные колонны. Первая колонна работала при атмосферном давлении и в ней отделяли непрореагировавший бензол в ее верхней части, в то время как соединения, которые присутствовали на дне колонны, подавали во вторую колонну. Во второй колонне, которая работала в условиях вакуума, отделяли парафины в ее верхней части, в то время как соединения, которые присутствовали в нижней части колонны, подавали в третью колонну. В третьей колонне, которая работала в условиях вакуума, отделяли моноалкилбензол в ее верхней части, а тяжелый алкилат в ее нижней части. Этот сырьевой моноалкилбензол, полученный в верхней части третьей колонны и содержащий определенное количество тяжелого алкилата, подавали в адсорбционный слой. После выполнения очистки выходящий поток из адсорбционного слоя подавали в четвертую ректификационную колонну. В этой колонне отделяли легкие побочные продукты, полученные в течение стадии очистки (в основном бензол, полученный посредством реакций переалкилирования, на уровнях значений частей на миллион) в ее верхней части, в то время как соединения в ее нижней части представляли собой чистые моноалкилбензолы. Эти высокочистые моноалкилбензолы затем сульфонировали, используя разбавленную SO3 в сухом воздухе в качестве сульфонирующего агента, в многотрубном пленочном реакторе и их затем выдерживали для созревания и подвергали гидролизу для завершения реакции.
Как было упомянуто перед этим, стадию очистки выполняли посредством неподвижного слоя, в котором было помещено определенное количество промышленной кислой глины. В этом примере используемая глина характеризовалась массовым отношением диоксида кремния к оксиду алюминия, равным 4,9, она была частично нейтрализована 1,4 мас.% K2O и 1,2 мас.% MgO, а также содержала 2,9 мас.% Fe2O3 и 0,5 мас.% TiO2. Эту глину предварительно активировали посредством пропускания потока горячего инертного газа с целью устранения адсорбированной воды. Условия активации и эксплуатации представлены в Табл.15.
Средние условия сульфонирования представлены в Табл.16.
Конечную цветность после сульфонирования анализировали, используя колориметр Клетта-Саммерсона. С целью устранения влияния стадии очистки на конечную цветность после сульфонирования алкилбензол также сульфонировали при тех же условиях без стадии очистки. Цветность после сульфонирования сырьевого сульфонированного алкилбензола и очищенного алкилбензола представлена в Табл.17.
Как видно из Табл.17, очистка сырьевого алкилбензола посредством слоя селективного адсорбента подразумевает уменьшение конечной цветности после сульфонирования примерно на 80%. Когда цветность после сульфонирования уменьшают, возрастает качество нейтрализованного продукта, особенно когда его используют в жидких моющих составах, так как он может препятствовать визуальному эффекту красителей, содержащихся в моющем составе.
Пример 6
Этот пример показывает преимущества добавления гидротропного предшественника, полученного при обработке нелинейных соединений, извлеченных на стадии очистки смеси после алкилирования, к алкилбензолу перед стадией его очистки селективной адсорбцией. Оба потока смешивали, затем очищали кислой глиной и перегоняли для отделения легких побочных продуктов и сульфонировали, как описано в Примере 4, и наконец, нейтрализовали водным гидроксидом натрия в стехиометрическом количестве.
Были получены три образца алкилбензола с различным содержанием 2-фенил изомера на стадии алкилирования (как определено в Примере 2) с целью оценки гидротропного эффекта на содержание 2-фенил изомера. Образцы обозначены S1, S2, S3, и содержание 2-фенил изомера в них представлено в Табл.18.
Гидротропный эффект гидротропного предшественника оценивали в показателях температуры помутнения при охлаждении (ТПО) конечного продукта, разбавленного водой, до получения обычных промышленных концентраций. С целью оценки влияния гидротропного предшественника были изготовлены образцы, содержащие 90 мас.% алкилбензола и 10 мас.% этого гидротропного предшественника, и затем они были разбавлены в воде. Другой хорошо известный гидротроп, ксилолсульфонат натрия, добавляли к образцам без гидротропного предшественника (90% чистого натриевого сульфоната алкилбензола (sodium sulfonate alkylbenzene) с переменным содержанием 2-фенил изомера и 10% ксилолсульфоната натрия) и затем его разбавляли водой до тех же концентраций, как и в предыдущих образцах. Результаты представлены в Табл.19.
Как можно видеть в Табл.19, рассматриваемый в этом патенте гидротроп уменьшает ТПО конечного продукта во всех образцах, и во всех случаях его влияние намного более сильное, чем гидротропное влияние КСН, что особенно интересно, когда содержание 2-фенил изомера является более высоким.
Пример 7
Этот пример показывает поведение цеолита Y, допированного 7 мас.% редкоземельных металлов, таких как La, Ce, Pr, Nd, при алкилировании бензола линейными альфа-олефинами с длинной цепью по сравнению с результатами, полученными, когда используют HF - катализатор, широко используемый в промышленном масштабе для аналогичного способа.
Бензол высушивали молекулярным ситом для сведения к минимуму добавки воды и затем смешивали со смесью линейных альфа-олефинов с длинной цепью. В случае твердого катализатора эксперименты по алкилированию на пилотной установке выполняли в изотермическом реакторе с неподвижным слоем с 24-часовыми циклами реакции, за которыми следовали циклы промывания бензолом в течение того же периода времени. Стандартные циклы включают 24-часовой реакционный цикл с молярным отношением бензола к олефину, равным 50:1, за которым следует цикл промывания бензолом в течение того же периода времени. Рабочие условия представлены в Табл.20.
Смесь альфа-олефинов, использованная в качестве сырьевой композиции, представлена ниже в Табл.21.
Были выполнены двенадцать циклов реакции алкилирования, используя цеолит Y. Первые три цикла были выполнены при ЧОСЖ, равной 8 час-1, и Т=150°C (цикл А). Затем три цикла были выполнены при ЧОСЖ, равной 6 час-1, и Т=150°С (цикл В). Последние четыре цикла были выполнены при ЧОСЖ, равной 4 час-1, два из них при Т=150°С и последние два при более низкой температуре, Т=140°С (цикл С). Все эти циклы выполняли с целью анализа влияния температуры и объемной скорости на выход катализатора. Продукты анализировали посредством газовой хроматографии (ГХ) и детектора ионизации пламени (ДИП).
Алкилирование с HF выполняли в охлаждаемом реакторе периодического действия с непрерывным перемешиванием с учетом того, что алкилирование является экзотермической реакцией и необходимо отводить тепло из реактора с целью сохранения постоянной требуемой температуры реакции, что является характерным в настоящем уровне техники. Определенное количество ранее рассмотренной смеси линейных альфа-олефинов (состав показан в Табл.20) смешивали с сухим бензолом до тех пор, пока выбранное молярное отношение бензола к олефину не вводили в реактор. Эту смесь бензола и олефинов предварительно нагревали до выбранной температуры реакции. Затем определенный объем жидкого HF вводили в реактор до тех пор, пока не достигали выбранного отношения HF к олефину. Время реакции доводили до 10 минут с целью получения ЧОСЖ=6 час-1, которая типична для настоящего уровня техники в области алкилирования с помощью HF. Рабочие условия реакции алкилирования с помощью HF представлены в Табл.22.
Результаты, приведенные в Табл.23, относятся к среднему распределению продукта для этих циклов (названных циклами А, В и С) при использовании твердого катализатора с различными условиями ЧОСЖ и температуры. Результаты, соответствующие технологии HF, расположены в нижней части Табл.23 для сравнения выходов с HF и твердым катализатором.
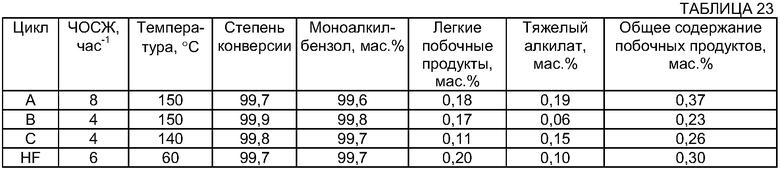
Как видно в Табл.23, активность цеолита Y, допированного редкоземельными металлами, всегда выше 99,7% при рассмотрении в показателях степени конверсии олефина. Это означает, что цеолит Y после ионного обмена с редкоземельными металлами является высокоактивным. Когда анализировали полученные побочные продукты, оба катализатора (модифицированный цеолит Y и HF) обладали высокой селективностью в отношении сведения к минимуму легких побочных продуктов и тяжелого алкилата. Общее количество побочных продуктов, полученных посредством цеолита Y, ниже 0,4 мас.% в трех оцениваемых циклах, а в циклах В и С даже ниже, чем при использовании HF, следовательно, это ясно показывает, что цеолит Y, допированный редкоземельными металлами, является высокоселективным при технологии с использованием реактора с неподвижным слоем.
Распределение изомеров, соответствующее предыдущим экспериментам, представлено в Табл.24. Как и в Табл.23, строка результатов в нижней части соответствует результатам, относящимся к реакции с HF при рабочих условиях, которые были определены прежде в Табл.22.
В Табл.24 распределение изомеров относительно аналогично при сравнении продуктов, полученных с применением цеолита Y, и продуктов, полученных с применением HF. Основное отличие связано с содержанием разветвленного алкилата. Как можно видеть, количество разветвленного алкилата, получаемого, когда используют цеолит Y, допированный редкоземельными металлами, составляет примерно на 4 мас.% больше, чем количество, полученное, когда используют HF в качестве катализатора. Это небольшое отличие не является важным при условии, что разветвленный алкилат не влияет на свойства детергента. По отношению к содержанию 2-фенил изомера можно видеть, что с помощью HF технологии получают немного большее количество 2-фенил изомера, чем в случае модифицированного цеолита Y.
Хотя существует небольшое отличие между распределением продукта, связанного с каждой технологией, было ясно показано, что цеолит Y, допированный редкоземельными металлами, способен катализировать алкилирование бензола линейными C20-24 олефинами с высокой активностью в показателях степени конверсии олефина и с высокой селективностью по моноалкилбензолам.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ МОНОАЛКИЛИРОВАННЫХ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ С ВЫСОКОЙ ЛИНЕЙНОСТЬЮ И РЕГУЛИРУЕМОЙ ИЗОМЕРИЕЙ | 2007 |
|
RU2460715C2 |
| СПОСОБ ОЧИСТКИ АЛКИЛАРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2006 |
|
RU2415834C2 |
| СПОСОБ РЕГУЛИРОВКИ СОДЕРЖАНИЯ 2-ФЕНИЛЬНОГО ИЗОМЕРА ЛИНЕЙНОГО АЛКИЛБЕНЗОЛА И КАТАЛИЗАТОР, ИСПОЛЬЗУЕМЫЙ В ЭТОМ СПОСОБЕ | 2011 |
|
RU2538974C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛБЕНЗОЛОВ НА ТВЕРДОКИСЛОТНОМ КАТАЛИЗАТОРЕ | 2007 |
|
RU2447051C2 |
| СПОСОБ ИЗОМЕРИЗАЦИИ УГЛЕРОДНОГО СКЕЛЕТА ОЛЕФИНОВ (ВАРИАНТЫ) | 1998 |
|
RU2186756C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛАРИЛЬНЫХ СОЕДИНЕНИЙ И ИХ СУЛЬФОНАТОВ | 2004 |
|
RU2344114C2 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ ПОЛУЧЕНИЯ АЛКИЛБЕНЗОЛОВ, ПРИМЕНЯЕМЫХ В ПРОИЗВОДСТВЕ МОЮЩИХ СРЕДСТВ, С ПОМОЩЬЮ ТРАНСАЛКИЛИРОВАНИЯ | 2008 |
|
RU2453522C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛАЛКАНОВ И СМАЗКА НА ИХ ОСНОВЕ | 2000 |
|
RU2237647C2 |
| АЛКИЛИРОВАНИЕ ДЛЯ ПОЛУЧЕНИЯ МОЮЩИХ СРЕДСТВ С ИСПОЛЬЗОВАНИЕМ КАТАЛИЗАТОРА, ПОДВЕРГНУТОГО ОБМЕНУ С РЕДКОЗЕМЕЛЬНЫМ ЭЛЕМЕНТОМ | 2010 |
|
RU2510639C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛАРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 1973 |
|
SU398026A1 |

Изобретение относится к способу получения высокорастворимого алкилароматического сульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% посредством каталитического алкилирования ароматического соединения очищенным алкилирующим агентом, включающему следующие стадии: 1) каталитическое дегидрирование линейного парафинового сырья; 2) селективное гидрирование диолефинов, полученных в качестве побочного продукта на стадии (1), до моноолефинов; 3) очистку сырьевого алкилирующего агента, полученного на стадии (2), отделение нелинейных продуктов, содержащихся в выходящем потоке стадии (2); 4) обработку нелинейных продуктов, извлеченных на стадии (3) для образования гидротропного предшественника; 5) алкилирование ароматического углеводорода моноолефинами, присутствующими в очищенном алкилирующем агенте, посредством сочетания следующих способов алкилирования: а) способ алкилирования с катализатором, образующим сырьевое линейное алкилароматическое соединение с максимальным содержанием 2-фенил изомера, составляющим 20 мас.%; б) способ алкилирования с катализатором, образующим сырьевое линейное алкилароматическое соединение с минимальным содержанием 2-фенил изомера, составляющим 20 мас.%; 6) разделение на фракции выходящего потока стадии (5) с целью разделения непрореагировавших ароматических соединений, парафинов и наиболее тяжелых побочных продуктов линейных алкилароматических соединений; 7) очистку фракции линейных алкилароматических соединений, поступающих со стадии (6); 8) сульфонирование очищенных линейных алкилароматических соединений, полученных на стадии (7); 9) нейтрализацию линейной алкилсульфоновой кислоты, полученной на стадии (8), отличающемуся тем, что катализатор, образующий максимально 20 мас.% 2-фенил изомера, включает цеолит типа FAU, от 0,01 до 0,15 мас.% по меньшей мере одного металла, выбранного из группы, состоящей из Li, Na, K, Mg или Са, и от 0,1 до 8 мас.% по меньшей мере одного редкоземельного металла, выбранного из группы, состоящей из La, Се, Pr, Nd, Pm, Sm или Eu; а катализатор, образующий минимально 20 мас.% 2-фенил изомера, включает цеолит типа MOR, от 0,01 до 0,2 мас.% по меньшей мере одного металла, выбранного из группы, состоящей из Li, Na, K, Mg или Са, при максимальном содержании Na, равном 0,01%, и от 0 до 0,5 мас.% по меньшей мере одного металла, выбранного из группы, состоящей из Ti, Zr, Hf. Технический результат - получение алкилароматического сульфоната с чрезвычайно низкой цветностью после сульфонирования с максимизированными моющими свойствами. 23 з.п. ф-лы, 24 табл., 4 ил.
1. Способ получения высокорастворимого алкилароматического сульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% посредством каталитического алкилирования ароматического соединения очищенным алкилирующим агентом, включающий следующие стадии:
1) каталитическое дегидрирование линейного парафинового сырья;
2) селективное гидрирование диолефинов, полученных в качестве побочного продукта на стадии (1), до моноолефинов;
3) очистку сырьевого алкилирующего агента, полученного на стадии (2), отделение нелинейных продуктов, содержащихся в выходящем потоке стадии (2);
4) обработку нелинейных продуктов, извлеченных на стадии (3) для образования гидротропного предшественника;
5) алкилирование ароматического углеводорода моноолефинами, присутствующими в очищенном алкилирующем агенте, посредством сочетания следующих способов алкилирования:
а) способ алкилирования с катализатором, образующим сырьевое линейное алкилароматическое соединение с максимальным содержанием 2-фенил изомера, составляющим 20 мас.%;
б) способ алкилирования с катализатором, образующим сырьевое линейное алкилароматическое соединение с минимальным содержанием 2-фенил изомера, составляющим 20 мас.%;
6) разделение на фракции выходящего потока стадии (5) с целью разделения непрореагировавших ароматических соединений, парафинов и наиболее тяжелых побочных продуктов линейных алкилароматических соединений;
7) очистку фракции линейных алкилароматических соединений, поступающих со стадии (6);
8) сульфонирование очищенных линейных алкилароматических соединений, полученных на стадии (7);
9) нейтрализацию линейной алкилсульфоновой кислоты, полученной на стадии (8),
отличающийся тем, что катализатор, образующий максимально 20 мас.% 2-фенил изомера, включает цеолит типа FAU, от 0,01 до 0,15 мас.% по меньшей мере одного металла, выбранного из группы, состоящей из Li, Na, K, Mg или Са, и от 0,1 до 8 мас.% по меньшей мере одного редкоземельного металла, выбранного из группы, состоящей из La, Се, Pr, Nd, Pm, Sm или Eu; а катализатор, образующий минимально 20 мас.% 2-фенил изомера, включает цеолит типа MOR, от 0,01 до 0,2 мас.% по меньшей мере одного металла, выбранного из группы, состоящей из Li, Na, K, Mg или Са, при максимальном содержании Na, равном 0,01%, и от 0 до 0,5 мас.% по меньшей мере одного металла, выбранного из группы, состоящей из Ti, Zr, Hf.
2. Способ получения высокорастворимого алкилароматического сульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором катализатор, образующий максимально 20 мас.% 2-фенил изомера, включает:
а) цеолит изотопической структуры типа FAU;
б) от 0,01 до 0,15 мас.% по меньшей мере одного металла, выбранного из группы, состоящей из Li, Na, K, Mg или Са;
в) от 0,1 до 8 мас.% по меньшей мере одного редкоземельного металла, выбранного из группы, состоящей из La, Се, Pr, Nd, Pm, Sm или Eu.
3. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по любому из пп.1 и 2, в котором катализатор, образующий максимально 20 мас.% 2-фенил изомера, характеризуется:
а) порошковой рентгенограммой, отличающейся тем, что наиболее интенсивный пик появляется при угле 2θ, соответствующем 6,2°, а остальные основные пики появляются при углах дифракции 2θ, соответствующих 23,6; 27; 31,3; 15,6; 20,3; 18,6 и 30°, указанных в порядке уменьшения интенсивности соответствующих пиков;
б) общим молярным отношением кремний/алюминий от 1:1 до 3:1;
в) общей удельной поверхностью (БЭТ) от 400 до 700 м2/г;
г) удельной поверхностью микропор от 350 до 600 м2/г;
д) удельным объемом микропор от 0,1 до 0,3 мл/г;
е) распределением макропор, в котором диаметр макропор находится в интервале от 20 до 2000 Å.
4. Способ получения высокорастворимого алкилароматического сульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором катализатор, образующий минимально 20 мас.% 2-фенил изомера, включает:
а) цеолит изотипической структуры типа MOR;
б) от 0,01 до 0,20 мас.% по меньшей мере одного металла, выбранного из группы, состоящей из Li, Na, K, Mg или Са;
в) максимально 0,01% Na;
г) от 0 до 0,5 мас.% по меньшей мере одного металла, выбранного из группы, состоящей из Ti, Zr, Hf.
5. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором катализатор, образующий минимально 20 мас.% 2-фенил изомера, характеризуется:
а) порошковой рентгенограммой, отличающейся тем, что наиболее интенсивный пик появляется при угле 2θ, соответствующем 25,6°, а остальные основные пики появляются при углах дифракции 2θ, соответствующих 22,3; 26,3; 19,6; 13,5 и 27,7°, указанных в порядке уменьшения интенсивности соответствующих пиков;
б) общим молярным отношением кремний/алюминий от 1,5:1 до 3,5:1;
в) общей удельной поверхностью (БЭТ) от 150 до 600 м2/г;
г) удельной поверхностью микропор от 300 до 450 м2/г;
д) удельным объемом микропор от 0,1 до 0,2 мл/г;
е) распределением макропор, в котором диаметр макропор находится в интервале от 20 до 800 Å.
6. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором используемые парафины включают от 8 до 30 атомов углерода.
7. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором ароматический углеводород выбирают из группы, состоящей из толуола, ксилола, бензола или их смесей.
8. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором ароматический углеводород и очищенный алкилирующий агент смешивают перед реакцией алкилирования стадии (5) при молярном соотношении ароматический углеводород:олефин от 5:1 до 70:1.
9. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.8, в котором смесь ароматического углеводорода и очищенного алкилирующего агента содержит максимально 0,3 мас.% нелинейных соединений, отличных от ароматического углеводорода.
10. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.8, в котором смесь ароматического углеводорода и очищенного алкилирующего агента содержит от 0 до 0,1 мас.% воды.
11. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором реакции алкилирования стадии (5) осуществляют одновременно.
12. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором катализатор, используемый в реакции алкилирования размещают в реакторе в компоновке, выбранной из группы, состоящей из таких компоновок, как: неподвижный слой с одним катализатором, неподвижный слой с двумя полностью перемешанными различными катализаторами, по меньшей мере два различных неподвижных слоя с одинаковым катализатором в каждом из них, по меньшей мере два различных неподвижных слоя с различным катализатором в каждом из них, суспензионный реактор с одним или более различными катализаторами.
13. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором алкилирование выполняют в конфигурации реактора, включающей по меньшей мере одну из конфигураций реактора, выбранных из группы: независимый реактор, по меньшей мере два реактора, соединенные параллельно, по меньшей мере два реактора, соединенные последовательно, и их сочетания.
14. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором очистку выполняют посредством селективной адсорбции, и/или гидрирования, и/или разделения на фракции.
15. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.14, в котором селективную адсорбцию выполняют посредством селективного адсорбента типа глины.
16. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.15, в котором глина характеризуется:
а) общим молярным соотношением диоксид кремния-оксид алюминия от 3:1 до 6:1;
б) содержанием от 1 до 4 мас.% Fe,
в) от 0,5 до 2,0 мас.% K, предпочтительно примерно 1,2 мас.%,
г) от 0,2 до 2,0 мас.% Mg, предпочтительно примерно 0,7 мас.%,
д) от 0,1 до 1,0 мас.% TiO2, предпочтительно примерно 0,3% масс.;
е) удельной поверхностью, выраженной как удельная поверхность по БЭТ, от 150 до 500 м2/г;
ж) совокупным объемом пор от 0,1 до 2 мл/г;
з) распределением макропор, в котором диаметр макропор составляет от 20 до 800Å.
17. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором гидротропный предшественник, получаемый на стадии (4), добавляют к потоку алкилароматических соединений, когда содержание 2-фенил изомера в алкилароматических соединениях больше и равно 60 мас.% добавляя его:
а) непосредственно перед стадией (6);
б) сразу после стадии (6).
18. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором гидротропный предшественник, получаемый на стадии (4), сульфонируют и нейтрализуют отдельно, и впоследствии добавляют к конечному алкилароматическому соединению, когда содержание 2-фенил изомера в нем больше и равно 60 мас.%.
19. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором нейтрализацию на стадии (9) выполняют посредством щелочного соединения, включающего один или более катионов, выбранных из группы Na, K, NH4+, Mg, Са, Ва или замещенных аммонийных щелочей.
20. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором стадии (1), (2), (3) и (4) являются необязательными.
21. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.20, в котором олефины стадии (5) представляют собой линейные альфа-олефины.
22. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.20, в котором олефины стадии (5) содержат от 9 до 30 атомов углерода.
23. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.1, в котором температура реакции составляет от 20 до 400°С.
24. Способ получения высокорастворимого линейного алкилбензолсульфоната с регулируемым содержанием 2-фенил изомера от 18 до 70 мас.% по п.20, в котором объемная скорость составляет от 1 до 15 ч-1.
| US 20030166481 A1, 04.09.2003 | |||
| В.THOMAS et al, J | |||
| Mater | |||
| Sci., vol.41, no.5, 09.03.2006, p.1611-1616 | |||
| S.SIVASANKER et al., Journal of Catalysis, vol.138, no.1, 1992, p.386-390 | |||
| Пустотелое долото | 1934 |
|
SU40551A1 |
| A.R.A.S.DESHMUNKH et al, Catalysis Letters, vol.64, no.2-4, 2000, p.247-250 | |||
| RU 2005123322 A, 27.01.2006 | |||
| RU 99115479 A, 27.04.2001 | |||
| RU 2001131562 A, 20.07.2003. | |||

