Область изобретения
Настоящее изобретение относится к некоторым новым производным (2S)-3-(4-{2-[амино]-2-оксоэтокси}фенил)-2-этоксипропионовой кислоты, способам получения таких соединений, их применению в лечении клинических состояний, в том числе липидных расстройств (дислипидемий), ассоциированных или не ассоциированных с инсулинорезистентностью и другими проявлениями метаболического синдрома, способам их терапевтического применения и фармацевтическим композициям, содержащим их.
Предшествующий уровень техники
Метаболический синдром, включая сахарный диабет 2 типа, относится к совокупности проявлений, в том числе инсулинорезистентности с сопутствующими гиперинсулинемией, возможно, сахарным диабетом 2 типа, артериальной гипертензией, центральным (висцеральным) ожирением, дислипидемией, обнаруживаемой по ненормальным уровням липопротеинов, типично характеризуемым повышенными концентрациями ЛОНП (липопротеинов очень низкой плотности), мелкими плотными частицами ЛНП (липопротеинов низкой плотности) и пониженными концентрациями ЛВП (липопротеинов высокой плотности), и пониженным фибринолизом.
Недавние эпидемиологические исследования подтвердили, что люди с инсулинорезистентностью подвержены значительно повышенному риску заболеваемости сердечно-сосудистой системы и смертности, особенно страдая от инфаркта миокарда и удара. Состояния, связанные с атеросклерозом при сахарном диабете 2 типа, являются причиной до 80% всех смертей.
В клинической медицине знают о необходимости увеличения чувствительности к инсулину у пациентов с метаболическим синдромом и, таким образом, устранения дислипидемии, которая, как считают, приводит к ускоренному прогрессированию атеросклероза. Однако в настоящее время не существует универсальной общепринятой постановки диагноза с однозначными фармакотерапевтическими показаниями.
Известен S-энантиомер соединения формулы С, приведенной ниже,
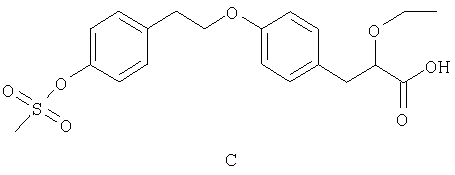
2-этокси-3-[4-(2-{4-метансульфонилоксифенил}этокси)фенил]пропионовой кислоты (публикация согласно РСТ WO 99/62872). Сообщается, что это соединение является модулятором активируемых пролифератором пероксисом рецепторов (PPAR, обзор рецепторов PPAR: T.M.Willson et al, J. Med. Chem. 2000, vol 43, 527) и обладает объединенной агонистической активностью по отношению к PPARα/PPARγ (Structure, 2001, том 9, 699, P.Cronet et al). Это соединение эффективно при лечении состояний, ассоциированных с инсулинорезистентностью.
В настоящее время неожиданно обнаружен ряд соединений, которые являются мощными модуляторами PPARα.
Описание изобретения
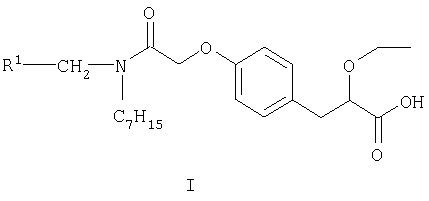
Согласно настоящему изобретению предложен S-энантиомер соединения формулы I
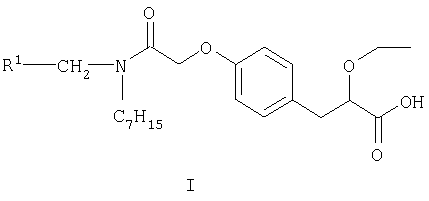
где R1 представляет собой 2,4-дифторфенил или циклогексил, а также его фармацевтически приемлемые соли, сольваты, кристаллические формы и пролекарства.
Термин "пролекарство", используемый в этом описании, включает в себя производные группы карбоновой кислоты, которые в млекопитающем, в частности человеке, превращаются в группу карбоновой кислоты, или ее соль или конъюгат. Не будучи связанным с теорией, следует понимать: считается, что ассоциируемая с пролекарствами активность главным образом является результатом активности соединения формулы I, в которое превращаются пролекарства. Пролекарства могут быть получены в соответствии со стандартной методологией в рамках возможностей специалистов данной области техники. Из предшествующего уровня техники известны различные карбокси-пролекарства. Примеры таких производных пролекарств описаны:
a) "Design of Prodrugs", edited by H.Bundgaard (Elsevier, 1985) и "Methods in Enzymology". 42: 309-396, edited by K.Widder, et al. (Academic Press, 1985);
6) "A Textbook of Drug Design and Development", edited by Krogsgaard-Larsen and H.Bundgaard, Chapter 5 "Design and Application of Prodrugs", by H.Bundgaard, p.113-191 (1991);
в) H.Bundgaard, Advanced Drug Delivery Reviews, 8: 1-38 (1992);
г) H.Bundgaard, et al., Journal of Pharmaceutical Sciences, 77: 285 (1988);
д) N.Kakeya, et al., Chem. Pharm. Bull., 32: 692 (1984).
Упомянутые выше документы ((а)-(д)) включены в этот документ посредством ссылки.
Сложные эфиры, расщепляемые in vivo, являются только одним из типов пролекарств исходной молекулы. Гидролизуемый (или расщепляемый) in vivo сложный эфир соединения формулы (I), которое содержит карбоксигруппу, представляет собой, например, фармацевтически приемлемый сложный эфир, который гидролизуется в организме человека или животного с образованием исходной кислоты. Фармацевтически приемлемые сложные эфиры, подходящие для карбоксигруппы, включают в себя С1-6алкоксиметиловые эфиры, например метоксиметиловый; С1-6алканоилоксиметиловые эфиры, например пивалоилоксиметиловый; фталидиловые эфиры; С3-8циклоалкоксикарбонилоксиС1-6алкиловые эфиры, например 1-циклогексилкарбонилоксиэтиловый; 1,3-диоксолен-2-онилметиловые эфиры, например 5-метил-1,3-диоксолен-2-онилметиловый; и C1-6алкоксикарбонилоксиэтиловые эфиры, например 1-метоксикарбонилоксиэтиловый; и могут быть образованы у любой карбоксигруппы соединений по этому изобретению.
Соединения формулы I обладают активностью лекарственных средств. В частности, соединения формулы I являются очень мощными агонистами PPARα. Кроме того, соединения формулы I также являются агонистами PPARγ. Термин агонисты, используемый в этом документе, включает в себя частичные агонисты.
Конкретными соединениями по изобретению являются:
(2S)-3-(4-{2-[(циклогексилметил)(гептил)амино]-2-оксоэтокси}фенил)-2-этоксипропионовая кислота, и
(2S)-3-(4-{2-[(2,4-дифторбензил)(гептил)амино]-2-оксоэтокси}фенил)-2-этоксипропионовая кислота,
и их фармацевтически приемлемые соли, сольваты и кристаллические формы.
В настоящем описании выражение "фармацевтически приемлемые соли" предназначено для определения основных солей, таких как соли щелочных металлов, соли щелочно-земельных металлов, аммониевые соли, соли с основными аминокислотами и соли с органическими аминами, но не ограничивается ими.
Также следует понимать, что некоторые соединения по настоящему изобретению могут существовать в сольватированной форме, например гидратированной, а также в несольватированных формах. Следует понимать, что настоящее изобретение охватывает все такие сольватированные формы. Некоторые соединения по настоящему изобретению могут существовать в виде таутомеров. Следует понимать, что настоящее изобретение охватывает все такие таутомеры.
Способы получения
Соединения по изобретению могут быть получены так, как изложено ниже. Однако изобретение не ограничивается этими способами, соединения также могут быть получены, как это описано в предшествующем уровне техники для структурно-родственных соединений. Реакции можно проводить в соответствии со стандартными методиками или как описано в экспериментальной части.
Соединения формулы I могут быть получены путем взаимодействия S-энантиомера соединения формулы II
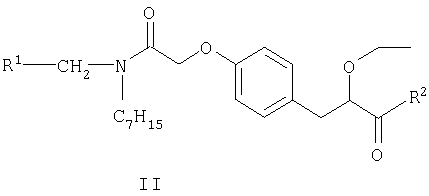
в котором R1 является таким, как определено выше, и R2 представляет собой защитную группу для карбоксильной гидроксигруппы, как описано в книге "Protective Groups in Organic Synthesis", 2nd Edition (1991), Greene and Wuts, с реагентом для удаления защитной группы. Защитной группой также может быть смола, такая как смола Ванга (Wang resin) или 2-хлортритилхлоридная смола. Защитные группы могут быть удалены в соответствии с методиками, хорошо известными специалистам в данной области техники. Одной из таких защитных групп R2 является группа, представляющая собой С1-6алкоксигруппу или арилалкоксигруппу, например бензил, так, что COR2 представляет собой сложный эфир. Такие сложные эфиры можно подвергать взаимодействию с реагентом для удаления защитной группы, например, гидролизующим реагентом, например гидроксидом лития в смеси тетрагидрофурана (ТГФ) и воды, при температуре в пределах от 0 до 100°С, с образованием соединений формулы I.
Соединения формулы II могут быть получены путем взаимодействия S-энантиомера соединения формулы III
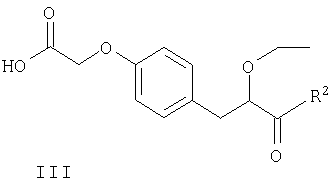
в котором R2 является таким, как определено выше, с соединением формулы IV
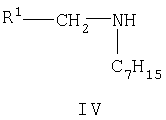
в котором R1 является таким, как определено выше, в инертном растворителе, например дихлорметане, в присутствии агента сочетания, например карбодиимида (например, 1-(3-диметиламинопропил)-3-этилкарбодиимида), и, возможно, в присутствии катализатора, например основного катализатора (например, 4-диметиламинопиридина), при температуре в пределах от -25 до 150°С.
Соединения формулы III и IV могут быть получены способами, описанными в примерах, или аналогичными способами, известными специалистам в данной области техники.
Соединения формулы II и III являются полезными промежуточными соединениями при получении соединений формулы I, и полагают, что они являются новыми. Соединения формулы II и III заявлены в этом документе в качестве еще одного аспекта изобретения. S-энантиомеры соединений формулы II и III являются предпочтительными.
Соединения по изобретению могут быть выделены из их реакционных смесей с использованием традиционных методик.
Специалисты в данной области техники должны понимать, что для получения соединений по изобретению альтернативным и в некоторых случаях более удобным способом, отдельные технологические операции, упомянутые выше, можно выполнять в другом порядке, и/или отдельные реакции можно проводить на другой стадии общего пути (то есть химические превращения можно осуществлять с промежуточными соединениями, отличными от тех, которые приведены выше для конкретной реакции).
Выражение "инертный растворитель" относится к растворителю, который не взаимодействует с исходными веществами, реагентами, промежуточными соединениями или продуктами в такой степени, чтобы оказывать неблагоприятное влияние на выход целевого продукта.
Фармацевтические препараты
Соединения по изобретению обычно будут вводить пероральным, парентеральным, внутривенным, внутримышечным, подкожным или другими способами введения при помощи инъекции, трансбуккальным, ректальным, вагинальным, трансдермальным и/или назальным путем и/или посредством ингаляции в форме фармацевтических препаратов, включающих в себя активный ингредиент или в виде свободной кислоты, или в виде фармацевтически приемлемой соли, полученной присоединением органического или неорганического основания, в фармацевтически приемлемой лекарственной форме. В зависимости от расстройства, пациента, подлежащего лечению, и пути введения композиции можно вводить в различных дозах.
Подходящие суточные дозы соединений по изобретению при терапевтическом лечении человека находятся в пределах 0,0001-100 мг/кг массы тела, предпочтительно 0,001-10 мг/кг массы тела.
Пероральные препараты являются предпочтительными, в особенности таблетки или капсулы, которые можно приготавливать способами, известными специалистам в данной области техники, обеспечивая дозы активного соединения в пределах от 0,5 мг до 500 мг, например 1 мг, 3 мг, 5 мг, 10 мг, 25 мг, 50 мг, 100 мг и 250 мг.
Таким образом, согласно еще одному аспекту изобретения предложен фармацевтический препарат, включающий в себя любое из соединений по изобретению или их фармацевтически приемлемых производных в смеси с фармацевтически приемлемыми адъювантами, разбавителями и/или носителями.
Пример получения фармацевтической композиции и изготовления фармацевтического препарата в форме таблетки.
ПРИМЕР: Изготовление таблеток
Активный ингредиент 1 смешивают с ингредиентами 2, 3, 4 и 5 в течение приблизительно 10 минут. Затем добавляют стеарат магния, полученную смесь перемешивают в течение приблизительно 2 минут и прессуют в форме таблетки, которую затем можно покрыть пленочной оболочкой.
Фармакологические свойства
Настоящие соединения формулы (I) являются полезными для профилактики и/или лечения клинических состояний, ассоциированных с врожденной или индуцированной пониженной чувствительностью к инсулину (инсулинорезистентностью) и с сопутствующими метаболическими расстройствами (также известными как метаболический синдром). Эти клинические состояния будут включать в себя общее ожирение, абдоминальное ожирение, артериальную гипертензию, гиперинсулинемию, гипергликемию, диабет 2 типа и дислипидемию, обычно появляющиеся вместе с инсулинорезистентностью, но не ограничиваться ими. Эта дислипидемия, также известная как атерогенный липопротеиновый профиль, характеризуется умеренно повышенными уровнями неэтерифицированных жирных кислот, повышенными уровнями частиц, обогащенных триглицеридами липопротеинов очень низкой плотности (ЛОНП), высокими уровнями Аро В, низкими уровнями липопротеинов высокой плотности (ЛВП), связанными с низкими уровнями частиц Аро AI и высокими уровнями Аро В в присутствии мелких плотных частиц липопротеинов низкой плотности (ЛНП), фенотип В.
Полагают, что соединения по настоящему изобретению являются полезными при лечении пациентов с комбинированной или смешанной гиперлипидемиями или разными степенями гипертриглицеридемий и постпрандиальной дислипидемии с другими проявлениями метаболического синдрома или без них.
Полагают, что лечение настоящими соединениями благодаря их антидислипидемическим и противовоспалительным свойствам снижает заболеваемость сердечно-сосудистой системы и смертность, связанную с атеросклерозом. Сердечно-сосудистые болезненные состояния включают в себя макроангиопатии различных внутренних органов, вызывающие инфаркт миокарда, застойную сердечную недостаточность, церебрально-васкулярное заболевание и периферическую артериальную недостаточность нижних конечностей. Также полагают, что соединения формулы I благодаря своему эффекту изменения чувствительности к инсулину предотвращают или замедляют развитие диабета 2 типа из метаболического синдрома и "диабета беременности". Таким образом, ожидается, что будет замедляться развитие продолжительных осложнений, ассоциированных с хронической гипергликемией при сахарном диабете, таких как микроангиопатии, вызывающие заболевание почек, повреждение сетчатки и заболевание периферических сосудов нижних конечностей. Более того, кроме лечения сердечно-сосудистой системы соединения могут быть полезными при лечении различных состояний, ассоциированных или не ассоциированных с инсулинорезистентностью, таких как синдром поликистоза яичника, ожирение, рак и воспалительные состояния, включая нейродегенеративные расстройства, такие как умеренное ухудшение когнитивных способностей, болезнь Альцгеймера, болезнь Паркинсона и рассеянный склероз.
Полагают, что соединения по настоящему изобретению могут быть полезными для контролирования уровней глюкозы у пациентов, страдающих от диабета 2 типа.
В настоящем изобретении предложен способ лечения или профилактики дислипидемий, синдрома инсулинорезистентности и/или метаболических расстройств (таких, как определено выше), при котором нуждающемуся в этом млекопитающему (в частности, человеку) вводят соединение формулы I.
В настоящем изобретении предложен способ лечения или профилактики диабета 2 типа, при котором нуждающемуся в этом млекопитающему (в частности, человеку) вводят эффективное количество соединения формулы I.
В следующем аспекте настоящего изобретения предложено применение соединения формулы I в качестве лекарственного средства.
В еще одном аспекте настоящего изобретения предложено применение соединения формулы I в производстве лекарственного средства для лечения инсулинорезистентности и/или метаболических расстройств.
Комбинированная терапия
Соединения по изобретению можно комбинировать с другими терапевтическими агентами, которые являются полезными в лечении расстройств, ассоциированных с развитием и прогрессированием атеросклероза, таких как гипертензия, гиперлипидемии, дислипидемии, диабет и ожирение. Соединения по изобретению можно комбинировать с еще одним терапевтическим агентом, который уменьшает соотношение ЛНП:ЛВП, или агентом, который приводит к снижению циркулирующих уровней ЛНП-холестерина. Для пациентов с сахарным диабетом соединения по изобретению также можно комбинировать с терапевтическими агентами, используемыми для лечения осложнений, связанных с микроангиопатиями.
Соединения по изобретению можно использовать совместно с другими терапиями для лечения метаболического синдрома или диабета 2 типа и осложнений, ассоциированных с ним, которые включают в себя бигуанидные лекарственные средства, например метформин, фенформин и буформин, инсулин (синтетические аналоги инсулина, амилин) и пероральные антигипергликемические препараты (которые подразделяются на прандиальные регуляторы глюкозы и ингибиторы альфа-глюкозидазы). Примером ингибитора альфа-глюкозидазы является акарбоза, или воглибоза, или миглитол. Примером прандиального регулятора глюкозы является репаглинид или натеглинид.
В другом аспекте изобретения соединение формулы I или его фармацевтически приемлемые соль, сольват, сольват такой соли или пролекарство можно вводить вместе с другим PPAR-модулирующим агентом. PPAR-модулирующие агенты включают в себя агонист PPARα и/или PPARγ или его фармацевтически приемлемые соли, сольваты, сольваты таких солей или пролекарства, но не ограничиваются ими. Подходящие агонисты PPARα и/или PPARγ, их фармацевтически приемлемые соли, сольваты, сольваты таких солей или пролекарства хорошо известны в данной области технике. Они включают в себя соединения, описанные в WO 01/12187, WO 01/12612, WO 99/62870, WO 99/62872, WO 99/62871, WO 98/57941, WO 01/40170, J. Med. Chem, 1996, 39, 665, Expert Opinion on Therapeutic Patents, 10 (5), 623-634 (в частности, соединения, описанные в патентных заявках, перечисленных на странице 634) и J. Med. Chem., 2000, 43, 527, которые все включены в этот документ посредством ссылки. В частности, агонист PPARα и/или PPARγ относится к NN 622/Ragaglitazar, BMS 298585, WY-14643, клофибрату, фенофибрату, безафибрату, гемфиброзилу и ципрофибрату, GW 9578, циглитазону, троглитазону, пиоглитазону, росиглитазону, эглитазону, проглитазону, BRL-49634, KRP-297, JTT-501, SB 213068, GW 1929, GW 7845, GW 0207, L-796449, L-165041 и GW 2433. В частности, агонист PPARα и/или PPARγ относится к (5)-2-этокси-3-[4-(2-{4-метансульфонилоксифенил}этокси)-фенил]пропионовой кислоте и ее фармацевтически приемлемым солям.
Кроме того, комбинацию по изобретению можно использовать вместе с сульфонилмочевиной, например глимепиридом, глибенкламидом (глибуридом), гликлазидом, глипизидом, гликвидоном, хлорпропамидом, толбутамидом, ацетогексамидом, гликопирамидом, карбутамидом, глибонуридом, глизоксепидом, глибутиазолом, глибузолом, глигексамидом, глимидином, глипинамидом, фенбутамидом, толциламидом и толазамидом. Предпочтительно сульфонилмочевиной является глимепирид или глибенкламид (глибурид). Более предпочтительно сульфонилмочевиной является глимепирид. Таким образом, настоящее изобретение включает в себя введение соединения по настоящему изобретению вместе с одним, двумя или более чем двумя существующими терапевтическими агентами, описанными в этом абзаце. Дозы других существующих терапевтических агентов для лечения диабета 2 типа и связанных с ним осложнений известны специалистам в данной области техники и утверждены к применению регулятивными органами, например FDA (Управление по контролю за продуктами и лекарствами (США)), и их можно найти в "Orange Book", опубликованной FDA. Альтернативно, можно применять меньшие дозы как результат преимущества комбинации.
Настоящее изобретение также включает в себя соединение по настоящему изобретению в сочетании с агентом, понижающим уровень холестерина. Агенты, понижающие уровень холестерина, упоминаемые в этой заявке, включают в себя ингибиторы HMG-CoA-редуктазы (3-гидрокси-3-метилглутарил-кофермент А-редуктазы), но не ограничиваются ими. Подходящим ингибитором HMG-CoA-редуктазы является статин, выбранный из группы, состоящий из аторвастатина, бервастатина, церивастатина, далвастатина, флувастатина, итавастатина, ловастатина, мевастатина, никостатина, нивастатина, правастатина и симвастатина, или его фармацевтически приемлемая соль, особенно натриевая или кальциевая, или его сольват, или сольват такой соли. Конкретным статином является аторвастатин или его фармацевтически приемлемые соль, сольват, сольват такой соли или пролекарство. Более конкретным статином является кальциевая соль аторвастатина. Однако особенно предпочтительным статином является соединение с химическим названием (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил(метилсульфонил)-амино]-пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновая кислота, [также известная как (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[N-метил-N-(метилсульфонил)-амино]пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновая кислота] или его фармацевтически приемлемые соль, или сольват, или сольват такой соли. Соединение (Е)-7-[4-(4-фторфенил)-6-изопропил-2-[метил-(метилсульфонил)-амино]-пиримидин-5-ил](3R,5S)-3,5-дигидроксигепт-6-еновая кислота и его кальциевая и натриевая соли описаны в европейской патентной заявке ЕР-А-0521471 и "Bioorganic and Medicinal Chemistry", (1997), 5 (2), 437-444. Этот последний статин в настоящее время известен под международным непатентованным наименованием розувастатин.
В настоящей заявке термин "агент, понижающий уровень холестерина" также включает в себя химические модификации ингибиторов HMG-CoA-редуктазы, такие как сложные эфиры, пролекарства и метаболиты, активные или неактивные.
Настоящее изобретение также включает в себя соединение по настоящему изобретению в сочетании с ингибитором системы транспорта желчных кислот в подвздошной кишке (IBAT-ингибитором).
Подходящие соединения, обладающие IBAT-ингибиторующей активностью, описаны; смотри, например, соединения, раскрытые в WO 93/16055, WO 94/18183, WO 94/18184, WO 96/05188, WO 96/08484, WO 96/16051, WO 97/33882, WO 98/07449, WO 98/03818, WO 98/38182, WO 99/32478, WO 99/35135, WO 98/40375, WO 99/35153, WO 99/64409, WO 99/64410, WO 00/01687, WO 00/47568, WO 00/61568, WO 00/62810, WO 01/68906, DE 19825804, WO 00/38725, WO 00/38726, WO 00/38727, WO 00/38728, WO 00/38729, WO 01/68906, WO 01/66533, WO 02/32428, WO 02/50051, ЕР 864582, ЕР 489423, ЕР 549967, ЕР 573848, ЕР 624593, ЕР 624594, ЕР 624595 и ЕР 624596; содержание этих патентных заявок включено в этот документ посредством ссылки.
Конкретными классами IBAT-ингибиторов, подходящих для применения по настоящему изобретению, являются бензотиепины, и соединения, описанные в формуле изобретения, в частности в п.1, WO 00/01687, WO 96/08484 и WO 97/33882 включены в этот документ посредством ссылки. Другими подходящими классами IBAT-ингибиторов являются 1,2-бензотиазепины, 1,4-бензотиазепины и 1,5-бензотиазепины. Кроме того, подходящим классом IBAT-ингибиторов являются 1,2,5-бензотиадиазепины.
Одним конкретным подходящим соединением, обладающим IBAT-ингибирующей активностью, является (3R,5R)-3-бутил-3-этил-1,1-диоксидо-5-фенил-2,3,4,5-тетрагидро-1,4-бензотиазепин-8-ил-β-D-глюкопиранозидуроновая кислота (ЕР 864582). Другие подходящие IBAT-ингибиторы включают в себя один из:
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-1'-фенил-1'-[N'-(карбоксиметил)карбамоил]метил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N'-(карбоксиметил)карбамоил]-4-гидроксибензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-1'-фенил-1'-[N'-(2-сульфоэтил)карбамоил]метил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-1'-фенил-1'-[N'-(2-сульфоэтил)карбамоил]метил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N'-(2-сульфоэтил)карбамоил]-4-гидроксибензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-α-[N'-сульфоэтил)карбамоил]-4-гидроксибензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-α-[N'-(2-карбоксиэтил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N'-(2-карбоксиэтил)карбамоил]-4-гидроксибензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-α-[N'-(5-карбоксипентил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N'-(2-карбоксиэтил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{α-[N'-(2-сульфоэтил)карбамоил]-2-фторбензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{(R)-α-[N'-(R)-(2-гидрокси-1-карбоксиэтил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N'-(R)-(2-гидрокси-1-карбоксиэтил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-{N-{(R)-α-(N'-{(R)-1-[N"-(R)-(2-гидрокси-1-карбоксиэтил)карбамоил]-2-гидроксиэтил}карбамоил)бензил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{α-[N'-(карбоксиметил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-(N-{α-[N'-((этокси)(метил)фосфорил-метил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3-бутил-3-этил-5-фенил-7-метилтио-8-{N-[(R)-α-(N'-{2-[(гидрокси)(метил)фосфорил]этил}карбамоил)бензил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N'-(2-метилтио-1-карбоксиэтил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-{N-[(R)-α-(N'-{2-[(метил)(этил)фосфорил]этил}карбамоил)-4-гидроксибензил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-{N-[(R)-α-(N'-{2-[(метил)(гидрокси)фосфорил]этил}карбамоил)-4-гидроксибензил]карбамоилметокси}-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[(R)-N'-(2-метилсульфинил-1-карбоксиэтил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метокси-8-[N-{(R)-α-[N'-(2-сульфоэтил)карбамоил]-4-гидроксибензил}карбамоилметокси]-2,3,4,5-тетрагидро-1,5-бензотиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((R)-1-карбокси-2-метилтио-этил)карбамоил]-4-гидроксибензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбокси-2-(R)-гидроксипропил)карбамоил]-4-гидроксибензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбокси-2-метилпропил)карбамоил]-4-гидроксибензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбоксибутил)карбамоил]-4-гидроксибензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбоксипропил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбоксиэтил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбокси-2-(R)-гидроксипропил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-(2-сульфоэтил)карбамоил]-4-гидроксибензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбоксиэтил)карбамоил]-4-гидроксибензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((R)-1-карбокси-2-метилтиоэтил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-{(S)-1-[N-((S)-2-гидрокси-1-карбоксиэтил)карбамоил]пропил}карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбокси-2-метилпропил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-((S)-1-карбоксипропил)карбамоил]-4-гидроксибензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-[N-((R/S)-α-{N-[1-(R)-2-(S)-1-гидрокси-1-(3,4-дигидроксифенил)проп-2-ил]карбамоил}-4-гидроксибензил)карбамоилметокси]-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина,
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-(2-(S)-3-(R)-4-(R)-5-(R)-2,3,4,5,6-пентагидроксигексил)карбамоил]-4-гидроксибензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина и
1,1-диоксо-3,3-дибутил-5-фенил-7-метилтио-8-(N-{(R)-α-[N-(2-(S)-3-(R)-4-(R)-5-(R)-2,3,4,5,6-пентагидроксигексил)карбамоил]бензил}карбамоилметокси)-2,3,4,5-тетрагидро-1,2,5-бензотиадиазепина
или их фармацевтически приемлемых солей, сольватов, сольватов таких солей или пролекарств.
Согласно другому аспекту настоящего изобретения предложено комбинированное лечение, при котором теплокровному животному, такому как человек, нуждающемуся в таком терапевтическом лечении, вводят эффективное количество соединения формулы I или его фармацевтически приемлемых соли, сольвата, сольвата такой соли или пролекарства, возможно, вместе с фармацевтически приемлемым разбавителем или носителем, с одновременным, последовательным или раздельным введением одного или более чем одного из следующих агентов, выбранных из:
СЕТР-ингибитора (ингибитора белка переноса холестериловых эфиров), например, таких, которые упомянуты и описаны в WO 00/38725, стр.7, строка 22, - стр.10, строка 17, которые включены в этот документ посредством ссылки;
антагониста абсорбции холестерина, например азетидинонов, таких как SCH 58235 и таких, которые описаны в US 5767115, которые включены в этот документ посредством ссылки;
МТР-ингибитора (ингибитора микросомального переносящего белка), например, таких, которые описаны в Science, 282, 751-54, 1998, которые включены в этот документ посредством ссылки;
производного никотиновой кислоты, включая продукты медленного высвобождения и комбинированные продукты, например никотиновую кислоту (ниацин), аципимокс и ницеритрол;
фитостерольного соединения, например станолов;
пробукола;
соединения от ожирения, например орлистата (ЕР 129748) и сибутрамина (GB 2184122 и US 4929629);
антигипертензивного соединения, например ингибитора ангиотензин-конвертирующего фермента (АСЕ), антагониста рецепторов ангиотензина II, адреноблокатора, альфа-адреноблокатора, бета-адреноблокатора, смешанного альфа/бета-адреноблокатора, адренергического стимулятора, блокатора кальциевых каналов, АТ-1-блокатора, салуретика, диуретика или сосудорасширяющего средства;
СВ1-антагониста или обратного агониста, например, как описано в WO 01/70700 и ЕР 65635;
антагониста меланинконцентрирующего гормона (МСН);
PDK-ингибитора; или
модуляторов ядерных рецепторов, например LXR, FXR, RXR и RORальфа;
или их фармацевтически приемлемых соли, сольвата, сольвата такой соли или пролекарства, возможно, вместе с фармацевтически приемлемым разбавителем или носителем.
Конкретные АСЕ-ингибиторы или их фармацевтически приемлемые соли, сольваты, сольваты таких солей или пролекарства, в том числе активные метаболиты, которые можно использовать в сочетании с соединением формулы I, включают в себя следующие соединения: алацеприл, алатриоприл, кальций алтиоприл, анковенин, беназеприл, гидрохлорид беназеприла, беназеприлат, бензоилкаптоприл, каптоприл, каптоприл-цистеин, каптоприл-глутатион, церанаприл, цераноприл, церонаприл, цилазаприл, цилазаприлат, делаприл, делаприл-дикислота, эналаприл, эналаприлат, энаприл, эпикаптоприл, фороксимитин, фосфеноприл, фозеноприл, натрий фозеноприл, фозиноприл, натрий фозиноприл, фозиноприлат, фозиноприловая кислота, глюкоприл, геморфин-4, идраприл, имидаприл, индолаприл, индолаприлат, либензаприл, лизиноприл, лициумин А, лициумин В, миксанприл, моэксиприл, моэксиприлат, мовелтиприл, мурацеин А, мурацеин В, мурацеин С, пентоприл, периндоприл, периндоприлат, пивалоприл, пивоприл, квинаприл, гидрохлорид квинаприла, квинаприлат, рамиприл, рамиприлат, спираприл, гидрохлорид спираприла, спираприлат, спироприл, гидрохлорид спироприла, темокаприл, гидрохлорид темокаприла, тепротид, трандолаприл, трандолаприлат, утибаприл, забициприл, забициприлат, зофеноприл и зофеноприлат, но не ограничены ими. Предпочтительными АСЕ-ингибиторами для применения в настоящем изобретении являются рамиприл, рамиприлат, лизиноприл, эналаприл и эналаприлат. Более предпочтительными АСЕ-ингибиторами для применений в настоящем изобретении являются рамиприл и рамиприлат.
Предпочтительные антагонисты ангиотензина II, их фармацевтически приемлемые соли, сольваты, сольваты таких солей или пролекарства для применения в сочетании с соединением формулы I включают в себя соединения: кандесартан, кандесартан цилексетил, лозартан, валсартан, ирбесартан, тазосартан, телмисартан и эпросартан, но не ограничены ими. Особенно предпочтительными антагонистами ангиотензина II или их фармацевтически приемлемыми производными для применения в настоящем изобретении являются кандесартан и кандесартан цилексетил.
Таким образом, согласно еще одному аспекту изобретения предложен способ лечения диабета 2 типа и связанных с ним осложнений у теплокровного животного, такого как человек, нуждающегося в таком лечении, при котором указанному животному вводят эффективное количество соединения формулы I или его фармацевтически приемлемых соли, сольвата, сольвата такой соли или пролекарства с одновременным, последовательным или раздельным введением эффективного количества одного из дополнительных соединений, описанных в разделе "комбинированная терапия", или его фармацевтически приемлемых соли, сольвата, сольвата такой соли или пролекарства.
Таким образом, согласно другому аспекту изобретения предложен способ лечения гиперлипидемических состояний у теплокровного животного, такого как человек, нуждающегося в таком лечении, при котором указанному животному вводят эффективное количество соединения формулы I или его фармацевтически приемлемых соли, сольвата, сольвата такой соли или пролекарства с одновременным, последовательным или раздельным введением эффективного количества одного из дополнительных соединений, описанных в разделе "комбинированная терапия", или его фармацевтически приемлемых соли, сольвата, сольвата такой соли или пролекарства.
Согласно еще одному аспекту изобретения предложена фармацевтическая композиция, которая включает в себя соединение формулы I или его фармацевтически приемлемые соль, сольват, сольват такой соли или пролекарство и одно из дополнительных соединений, описанных в разделе "комбинированная терапия", или его фармацевтически приемлемые соль, сольват, сольват такой соли или пролекарство вместе с фармацевтически приемлемым разбавителем или носителем.
Согласно еще одному аспекту настоящего изобретения предложен набор, включающий в себя соединение формулы I или его фармацевтически приемлемые соль, сольват, сольват такой соли или пролекарство и одно из дополнительных соединений, описанных в разделе "комбинированная терапия", или его фармацевтически приемлемые соль, сольват, сольват такой соли или пролекарство.
Согласно другому аспекту настоящего изобретения предложен набор, включающий в себя:
а) соединение формулы I или его фармацевтически приемлемые соль, сольват, сольват такой соли или пролекарство в первой стандартной лекарственной форме,
б) одно из дополнительных соединений, описанных в разделе "комбинированная терапия", или его фармацевтически приемлемые соль, сольват, сольват такой соли или пролекарство во второй стандартной лекарственной форме,
в) контейнерное средство для хранения указанных первой и второй лекарственных форм.
Согласно другому аспекту настоящего изобретения предложен набор, включающий в себя:
а) соединение формулы I или его фармацевтически приемлемые соль, сольват, сольват такой соли или пролекарство, вместе с фармацевтически приемлемым разбавителем или носителем в первой стандартной лекарственной форме,
б) одно из дополнительных соединений, описанных в разделе "комбинированная терапия", или его фармацевтически приемлемые соль, сольват, сольват такой соли или пролекарство, во второй стандартной лекарственной форме,
в) контейнерное средство для хранения указанных первой и второй лекарственных форм.
Согласно еще одному аспекту изобретения предложено применение соединения формулы I или его фармацевтически приемлемых соли, сольвата, сольвата такой соли или пролекарства и одного из дополнительных соединений, описанных в разделе "комбинированная терапия", или его фармацевтически приемлемых соли, сольвата, сольвата такой соли или пролекарства в производстве лекарственного средства для применения в лечении метаболического синдрома или диабета 2 типа и связанных с ним осложнений у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложено применение соединения формулы I или его фармацевтически приемлемых соли, сольвата, сольвата такой соли или пролекарства и одного из дополнительных соединений, описанных в разделе "комбинированная терапия", или его фармацевтически приемлемых соли, сольвата, сольвата такой соли или пролекарства в производстве лекарственного средства для применения в лечении гиперлипидемических состояний у теплокровного животного, такого как человек.
Согласно другому аспекту настоящего изобретения предложено комбинированное лечение, при котором теплокровному животному, такому как человек, нуждающемуся в таком терапевтическом лечении, вводят эффективное количество соединения формулы I или его фармацевтически приемлемых соли, сольвата, сольвата такой соли или пролекарства, возможно, вместе с фармацевтически приемлемым разбавителем или носителем с одновременным, последовательным или раздельным введением эффективного количества одного из дополнительных соединений, описанных в разделе "комбинированная терапия", или его фармацевтически приемлемых соли, сольвата, сольвата такой соли или пролекарства, возможно, вместе с фармацевтически приемлемым разбавителем или носителем.
Примеры
1H-ЯМР- и 13С-ЯМР-измерения выполняли на спектрометрах Varian Mercury 300 или Varian UNITY plus 400, 500 или 600 с частотой 300, 400, 500 и 600 МГц (для 1Н) соответственно и 75, 100, 125 и 150 МГц (для 13С) соответственно. Измерения осуществляли по дельта-шкале (δ).
Если не указано иначе, химические сдвиги выражены в миллионных долях (ppm) с использованием растворителя в качестве внутреннего стандарта.
Сокращения
ДМСО - диметилсульфоксид
ТГФ - тетрагидрофуран
ДМАП - диметиламинопиридин
t - триплет
s - синглет
d - дублет
q - квартет
m - мультиплет
bs - широкий синглет
Пример 1
(2S)-3-(4-{2-Циклогексилметил)(гептил)амино]-2-оксоэтокси}фенил)-2-этоксипропионовая кислота
(1) Этиловый эфир (2S)-3-{4-[2-(бензилокси)-2-оксоэтокси]фенил}-2-этоксипропионовой кислоты
К раствору этилового эфира (2S)-2-этокси-3-(4-гидроксифенил)пропионовой кислоты (23,8 г, 100 ммоль, полученного, как описано в WO 99/62872) в ацетонитриле (200 мл) добавляли безводный карбонат калия (31,9 г, 231 ммоль), а затем бензилбромацетат (17,4 мл, 11 ммоль) и реакционную смесь нагревали с обратным холодильником в течение ночи. Реакционную смесь оставляли охлаждаться до комнатной температуры, нерастворимые соли отфильтровывали и раствор концентрировали под вакуумом. Остаток переносили в этилацетат (300 мл) и органическую фазу промывали водным раствором NaHCO3 (3×100 мл) и рассолом (100 мл), сушили над безводным MgSO4 и концентрировали под вакуумом. Очистка на силикагеле метиленхлоридом в качестве элюента и сбор чистых фракций привели в результате к 22,4 г (58%) желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 1.16 (t, 3Н), 1.22 (t, 3Н), 2.93-2.97 (m, 2H), 3.35 (m, 1H), 3.60 (m, 1H), 3.97 (m, 1H), 4.16 (q, 2H), 4.64 (s, 2H), 5.23 (s, 2H), 6.82 (d, 2H), 7.15 (d, 2H), 7.32-7.39 (m, 5H).
13С ЯМР (100 МГц, CDCl3): δ 14.3, 15.2, 38.6, 60.9, 65.6, 66.3, 67.0, 80.4, 114.6, 128.5, 128.6, 128.7, 130.6, 135.3, 156.7, 169.0, 172.6.
(2) {4-[(2S)-2,3-Диэтокси-3-оксопропил]фенокси}уксусная кислота
К раствору этилового эфира (2S)-3-{4-[2-(бензилокси)-2-оксоэтокси]фенил}-2-этоксипропионовой кислоты (22,33 г, 57,8 ммоль) в свежеперегнанном ТГФ (290 мл) добавляли Pd/C (10%, 3,1 г) и реакционную смесь подвергали гидрированию при атмосферном давлении и комнатной температуре в течение ночи. Смесь фильтровали через подушку из Целита и фильтрат концентрировали под вакуумом, получая 16,6 г (97%) светло-желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 1.15 (t, 3Н), 1.21 (t, 3H), 2.93-2.98 (m, 2H), 3.35 (m, 1H), 3.60 (m, 1H), 3.97 (m, 1H), 4.16 (q, 2H), 4.65 (s, 2H), 6.84 (d, 2H), 7.17 (d, 2H), 8.48 (bs, 1H).
13C ЯМР (100 МГц, CDCl3): δ 14.3, 15.1, 38.5, 61.0, 65.1, 66.4, 80.3, 114.6, 130.7, 130.9, 156.4, 172.7, 173.7.
(3) N-(Циклогексилметил)гептанамид
К раствору аминометилциклогексана (0,34 г, 3,0 ммоль) в метиленхлориде (30 мл) добавляли гептановую кислоту (0,39 г, 3 ммоль) и ДМАП (0,37 г, 3,0 ммоль), а затем гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (0,57 г, 3,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь разбавляли метиленхлоридом (100 мл) и органическую фазу промывали 5%-ным раствором HCl (3×75 мл), водным раствором NaHCO3 (75 мл) и рассолом (75 мл) и сушили над Na2SO4. Концентрирование под вакуумом привело в результате к 0,62 г (92%) масла, которое затем закристаллизовалось.
1H ЯМР (400 МГц, CDCl3): δ 0.84-0.98 (m, 5H), 1.08-1.36 (m, 8H), 1.44 (m, 1H), 1.56-1.78 (m, 8H), 2.16 (t, 2H), 3.09 (t, 2H), 5.45 (bs, 1H).
13C ЯМР (100 МГц, CDCl3): δ 14.1, 22.7, 26.0, 26.6, 29.1, 31.0, 31.7, 37.1, 38.1,45.8, 173.2.
(4) Гидрохлорид N-(циклогексилметил)-N-гептиламина
N-(Циклогексилметил)гептанамид (0,58 г, 2,6 ммоль) сушили азеотропной перегонкой с толуолом (один раз), переносили в свежеперегнанный ТГФ (23 мл) и охлаждали на ледяной бане в атмосфере аргона. Добавляли боран (3,2 мл 2 М раствора метилсульфидного комплекса в диэтиловом эфире) и ледяную баню удаляли через 15 минут. Реакционную смесь нагревали с обратным холодильником в течение четырех часов, а затем оставляли охлаждаться до комнатной температуры. Осторожно добавляли 1,2 мл 10%-ного раствора HCl и смесь перемешивали в течение ночи. Концентрирование под вакуумом с последующим добавлением ледяного ТГФ (приблизительно 15 мл) привело в результате к образованию белого осадка. Добавляли воду (приблизительно 3 мл), затем толуол (приблизительно 10 мл) и смесь концентрировали под вакуумом. К остатку добавляли ледяной ТГФ (приблизительно 15 мл) и полученный осадок отфильтровывали и сушили лоб вакуумом с образованием 2,96 г сырого продукта в виде белой соли. Это вещество использовали на следующей стадии без какой-либо дополнительной очистки.
1H ЯМР (400 МГц, CD3OD): δ 0.87-0.98 (m, 5H), 0.97-1.11 (m, 2H), 1.15-1.45 (m, 11H), 1.65-1.86 (m, 8H), 2.84 (d, 2H), 2.93-3.01 (m, 2H).
13C ЯМР (100 МГц, CD3OD): δ 14.3, 23.6, 26.6, 27.0, 27.1, 27.6, 29.9, 31.5, 32.7, 36.4, 55.0.
(5) Этиловый эфир (2S)-3-(4-{2-[(циклогексилметил)(гептил)амино]-2-оксоэтокси}фенил)-2-этоксипропионовой кислоты
К раствору {4-[(2S)-2,3-диэтокси-3-оксопропил]фенокси}уксусной кислоты (0,108 г, 0,36 ммоль) в метиленхлориде (3,6 ммоль) добавляли гидрохлорид N-(циклогексилметил)-N-гептиламина (0,090 г, 0,36 ммоль) и ДМАП (0,098 г, 0,80 ммоль), а затем гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (0,070 г, 0,36 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь разбавляли метиленхлоридом (25 мл) и органическую фазу промывали 5%-ным раствором HCl (3×25 мл), водным раствором NaHCO3 (25 мл) и рассолом (25 мл), сушили над Na2SO4 и концентрировали под вакуумом. Очистка на предварительно наполненной силикагелем колонке (Isolute® SPE Column, 5 г Si/25 мл) с использованием в качестве элюента метанола (градиент 0-1%) в метиленхлориде привела в результате к 0,103 г (58%) бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 0.83-0.97 (m, 5H), 1.11-1.33 (m, 17H), 1.45-1.80 (m, 8H), 2.88-3.00 (m, 2H), 3.14 и 3.19 (2d, 2H, ротамеры), 3.24-3.39 (m, 3H), 3.58 (m, 1H), 3.95 (m, 1H), 4.15 (q, 2H), 4.64 и 4.66 (2s, 2H, ротамеры), 6.84 и 6.84 (2d, 2H, ротамеры), 7.14 (d, 2H).
13С ЯМР (100 МГц, CDCl3): δ 14.2, 14.3, 15.2, 22.7, 26.0, 26.0, 26.5, 26.5, 27.0, 27.0, 27.2, 28.9, 29.1, 31.0, 31.2, 31.9, 36.1, 37.3, 38.6, 46.4, 48.0, 51.7, 53.3, 60.9, 66.3, 67.5, 67.7, 80,4, 114.6, 114.7, 130.2, 130.5, 157.1, 157.1, 167.8; 167.9, 172.6. (Число пиков больше числа атомов углерода вследствие ротамеров).
(6) (2S)-3-(4-{2-Г[Циклогексилметил)(гептил)амино]-2-оксоэтокси}фенил)-2-этоксипропионовая кислота
К раствору этилового эфира (2S)-3-(4-{2-[(циклогексилметил)(гептил)амино]-2-оксоэтокси}фенил)-2-этоксипропионовой кислоты (0,031 г, 0,057 ммоль) в ТГФ (2,0 мл) добавляли воду (2,0 мл) и гидроксид лития (0,006 г, 0,26 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь подкисляли 2 М HCl и экстрагировали этилацетатом (4×25 мл). Объединенную органическую фазу промывали рассолом (25 мл), сушили над Na2SO4 и концентрировали под вакуумом, получая 0,027 г (93%) бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 0.82-0.99 (m, 5H), 1.10-1.35 (m, 14H), 1.46-1.82 (m, 8H), 2.94 (m, 1H), 3.05 (m, 1H), 3.15 и 3.21 (2d, 2H, ротамеры), 3.25-3.46 (m, 3Н), 3.61 (m, 1H), 4.02 (m, 1H), 4.66 и 4.68 (2s, 2H, ротамеры), 6.85 (2d, 2H), 7.16 (d, 2H), 7.77 (bs, 1H).
13С ЯМР (100 МГц, CDCl3): δ 14.2, 15.1, 22.7, 26.0, 26.0, 26.4, 26.5, 27.0, 27.0, 27.2, 28.9, 29.1, 31.0, 31.2, 31.9, 36.1, 37.2, 38.0, 46.6, 48.0, 51.8, 53.4, 66.8, 67.3, 67.5, 79.9, 114.7, 114.8, 129.9, 130.6, 157.1, 157.2, 168.2, 168.3, 175.2. (Число пиков больше числа атомов углерода вследствие ротамеров).
Пример 2
(2S)-3-(4-{2-[(2,4-Дифторбензил)(гептил)амино]-2-оксоэтокси}фенил)-2-этоксипропионовая кислота
(1) N-(2,4-Дифторбензил)гептанамид
К раствору 2,4-дифторбензиламина (0,43 г, 3,0 ммоль) в метиленхлориде (30 мл) добавляли гептановую кислоту (0,39 г, 3,0 ммоль) и ДМАП (0,37 г, 3,0 ммоль), а затем гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (0,58 г, 3,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь разбавляли метиленхлоридом (100 мл) и органическую фазу промывали 5%-ным раствором HCl (3×75 мл), водным раствором NaHCO3 (75 мл) и рассолом (75 мл) и сушили над Na2SO4. Концентрирование под вакуумом привело в результате к 0,63 г (82%) желтого масла.
1H ЯМР (400 МГц, CDCl3): δ 0.83-0.91 (m, 3H), 1.22-1.35 (m, 6H), 1.56-1.68 (m, 2H), 2.19 (t, 2H), 4.43 (d, 2H), 5.80 (bs, 1H), 6.75-6.88 (m, 2H), 7.33 (m, 1H).
13С ЯМР (100 МГц, CDCl3): δ 14.1, 22.6, 25.7, 29.0, 31.6, 36.8, 37.1, 104.0 (t), 111.5 (dd), 131.5 (dd), 173.2. (о непротонированных атомах углерода не сообщается).
(2) Гидрохлорид N-(2,4-дифторбензил)-N-гептиламина
N-(2,4-Дифторбензил)гептанамид (0,55 г, 2,2 ммоль) сушили азеотропной перегонкой с толуолом (один раз), переносили в свежеперегнанный ТГФ (19 мл) и охлаждали на ледяной бане в атмосфере аргона. Добавляли боран (2,7 мл 2 М раствора диметилсульфидного комплекса в диэтиловом эфире) и ледяную баню удаляли через 15 минут. Реакционную смесь нагревали с обратным холодильником в течение четырех часов, а затем оставляли охлаждаться до комнатной температуры. Осторожно добавляли 1,0 мл 10%-ного раствора HCl и смесь перемешивали в течение ночи. Концентрирование под вакуумом с последующим добавлением ледяного ТГФ (приблизительно 15 мл) привело к образованию осадка, который отфильтровывали и сушили под вакуумом, получая 0,81 г сырого продукта в виде соли белого цвета с желтоватым оттенком. Это вещество использовали на следующей стадии без какой-либо дополнительной очистки.
1H ЯМР (400 МГц, CD3OD): δ 0.88-0.95 (m, 3H), 1.27-1.45 (m, 8H), 1.66-1.79 (m, 2H), 3.03-3.10 (m, 2H), 4.27 (s, 2H), 7.06-7.17 (m, 2H), 7.62 (m, 1H).
13C ЯМР (100 МГц, CD3OD): δ 14.3, 23.6, 27.1, 27.5, 29.8, 32.7, 45.0 (d), 48.9, 105.4 (t), 113.3 (dd), 134.8 (dd).
(3) Этиловый эфир (2S)-3-(4-{2-[(2,4-дифторбензил)(гептил)амино]-2-оксоэтокси}фенил)-2-этоксипропионовой кислоты
К раствору {4-[(2S)-2,3-диэтокси-3-оксопропил]фенокси}уксусной кислоты (0,104 г, 0,35 ммоль) в метиленхлориде (3,5 мл) добавляли гидрохлорид N-{2,4-дифторбензил)-N-гептиламина (0,098 г, 0,35 ммоль) и ДМАП (0,094 г, 0,77 ммоль), а затем гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (0,067 г, 0,35 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь разбавляли метиленхлоридом (50 мл) и органическую фазу промывали 5%-ным раствором HCl (3×25 мл), водным раствором NaHCO3 (25 мл) и рассолом (25 мл), сушили над Na2SO4 и концентрировали под вакуумом. Очистка на предварительно наполненной силикагелем колонке (Isolute® SPE Column, 5 г Si/25 мл) с использованием в качестве элюента метанола (градиент 0-1%) в метиленхлориде привела в результате к 0,066 г (36%) бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 0.81-0.90 (m, 3Н), 1.15 (t, 3H), 1.17-1.31 (m, 11H), 1.43-1.65 (m, 2H), 2.89-3.00 (m, 2H), 3.24-3.39 (m, 3Н), 3.59 (m, 1H), 3.96 (m, 1H), 4.15 (q, 2H), 4.60 (s, 2H), 4.69 и 4.70 (2s, 2H, ротамеры), 6.73-6.88 (m, 4H), 7.08-7.22 и 7.22-7,31 (2m, 3Н, ротамеры).
13С ЯМР (100 МГц, CDCl3): δ 14.1, 14.3, 15.1, 22.6, 26.9, 27.1, 28.7, 29.0, 31.8, 38.5, 41.5, 44.3, 46.1, 47.2, 60.9, 66.3, 67.5, 68.1, 80.3, 103.6 (t), 104.2 (t), 111.6 (dd), 114.4, 114.6, 119.8 (dd), 120.3 (dd), 129.6 (dd), 130.4, 130.6, 131.7 (dd), 156.7, 156.9, 168.2, 168.3, 172.5. (Число пиков больше числа атомов углерода вследствие ротамеров. О фторированных атомах углерода не сообщается).
(4) (2S)-3-(4-{2-[((2,4-Дифторбензил)(гептил)амино]-2-оксоэтокси}фенил)-2-этоксипропионовая кислота
К раствору этилового эфира (2S)-3-(4-{2-[(2,4-дифторбензил)(гептил)амино]-2-оксоэтокси}фенил)-2-этоксипропионовой кислоты (0,047 г, 0,090 ммоль) в ТГФ (2,0 мл) добавляли воду (2,0 мл) и гидроксид лития (0,010 мг, 0,42 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали под вакуумом, подкисляли 2 M HCl и экстрагировали этилацетатом (4×25 мл). Объединенную органическую фазу промывали рассолом (25 мл), сушили над Na2SO4 и концентрировали под вакуумом, получая 0,044 г (89%) бесцветного масла.
1H ЯМР (400 МГц, CDCl3): δ 0.83-0.93 (m, 3Н), 1.17 (t, 3Н), 1.20-1.35 (m, 8H), 1.45-1.67 (m, 2H), 2.90-3.14 (m, 2H), 3.26-3.35 (m, 2H), 3.42 (m, 1H), 3.63 (m, 1H), 4.04 (m, 1H), 4.63 (s, 2H), 4.74 (s, 2H), 6.75-6.90 (m, 4H), 7.11-7.22 и 7.25-7.35 (2m, 3Н, ротамеры), 9.13 (bs, 1H).
13С ЯМР (100 МГц, CDCl3): δ 14.1, 15.1, 22.6, 26.9, 27.1, 28.6, 29.0, 31.8, 38.0, 41.6, 44.3, 46.2, 47.3, 66.8, 67.3, 68.0, 79.8, 103.7 (t), 104.3 (t), 104.3, 111.7 (dd), 114.6, 114.7, 119.7 (dd), 120.1 (dd), 129.7 (m), 130.1, 130.7, 131.8 (dd), 156.8, 157.0, 168.6, 168.7, 175.6. (Число пиков больше числа атомов углерода вследствие ротамеров. О фторированных атомах углерода не сообщается).
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
Препараты
Соединения растворяли в ДМСО с получением 16 мМ исходных растворов. Перед исследованиями исходные растворы дополнительно разводили в ДМСО и культуральных средах.
ОСНОВНЫЕ ХИМИЧЕСКИЕ РЕАКТИВЫ И РЕАГЕНТЫ
Люциферазный реагент для анализа приобретали у Packard, США. Рестрикционные ферменты были предоставлены Boehringer, a Vent полимераза - New England Biolabs.
КЛЕТОЧНЫЕ ЛИНИИ И УСЛОВИЯ КУЛЬТИВИРОВАНИЯ КЛЕТОК
U2-OS (остеогенная саркома человека) предоставлена Американской коллекцией типовых культур (АТСС), США. Клетки размножали и вновь замораживали по партиям из шестого пассажа. Клетки культивировали на модифицированной по способу Дульбекко среде Игла (DMEM) с 25 мМ глюкозой, 2 мМ глутамином или 4 мМ L-аланил-L-глутамином, 10% фетальной телячьей сывороткой при 5% CO2. Использовали физиологический раствор с фосфатным буфером (PBS) без добавления кальция или магния. Все реагенты для культивирования клеток были предоставлены Gibco (США), а 96-луночные планшеты для культивирования клеток приобретали у Wallach.
ПЛАЗМИДНЫЕ КОНСТРУКЦИИ ДЛЯ ГЕТЕРОЛОГИЧНОЙ ЭКСПРЕССИИ
Стандартные методики работы с рекомбинантными ДНК выполняли, как описано Ausubel (7). Люциферазный репортерный вектор, pGL5UAS (клон состоит из пяти копий ДНК-связывающей последовательности GAL4, 5'-CGACGGAGTACTGTCCTCCGAGCT-3', клонированной по Sacl/Xhol-сайтам pGLS-промотора (Promega). Sacl/Xhol-фрагмент, содержащий UAS-сайты, конструировали с использованием ренатурированных перекрывающихся олигонуклеотидов.
Используемые экспрессирующие векторы основаны на pSG5 (Stratagene). Все векторы содержат EcoRI/Nhel-фрагмент, кодирующий ДНК-связывающий домен GAL4 (кодирующий последовательность с 1-й по 145-ю аминокислоту, согласно последовательности Р04386 из базы данных), с последующим лигированием с сохранением рамки считывания с фрагментом, кодирующим последовательность ядерной локализации Т-антигена вируса полиомы. Последовательность ядерной локализации конструировали, используя ренатурированные перекрывающиеся олигонуклеотиды, приводящие к образованию Nhel/Kpnl липких концов (5'-CTAGCGCTCCTAGAAGAAACGCAAGGTTGGTAC-3'). Лиганд-связывающие домены человеческих и мышиных PRAPα и человеческих и мышиных PRAPγ амплифицировали методом полимеразной цепной реакции (ПЦР) в виде Kpnl/BamHI-фрагментов и клонировали в рамку считывания ДНК-связывающего домена GAL4 и последовательности ядерной локализации. Последовательности всех используемых плазмидных конструкций подтверждали секвенированием. Для временных трансфекций использовали следующие экспрессирующие векторы:
ВРЕМЕННЫЕ ТРАНСФЕКЦИИ
Замороженные исходные клетки из шестого пассажа размораживали и размножали до восьмого пассажа перед трансфекциями. Конфлюэнтные клетки обрабатывали трипсином, промывали и осаждали центрифугированием при 270×g в течение 2 минут. Дебрис ресуспендировали в холодном PBS до концентрации клеток приблизительно 18×106 клеток/мл. После добавления ДНК клеточную суспензию инкубировали на льду в течение приблизительно 5 минут перед электропорацией при 230 В, 960 мкФ в Gene Pulser™ (Bio-Rad) в партиях по 0,5 мл. В целом, 50 мкг ДНК добавляли в каждую партию с 0,5 мл клеток, включая 2,5 мкг экспрессирующего вектора, 25 мкг репортерного вектора и 22,5 мкг неспецифичной ДНК (pBluescript, Stratagene).
После электропорации клетки разбавляли до концентрации 320000 клеток/мл в DMEM без фенолового красного и в 96-луночные планшеты высевали примерно 25000 клеток на лунку. Для того чтобы дать возможность клеткам адаптироваться, засеянные планшеты перед добавлением анализируемых соединений инкубировали при 37°С в течение 3-4 часов. В анализах PPARα, для того чтобы избежать фоновой активации компонентами жирных кислот фетальной телячьей сыворотки (FCS), в среду для культивирования клеток добавляли FCS, очищенную смолой-древесным углем. FCS, очищенную смолой-древесным углем, получали следующим образом: 10 г древесного угля и 25 г анионообменной смолы аналитической степени чистоты размером 200-400 меш (Bio-Rad) добавляли к 500 мл FCS, инактивированной теплом, и раствор выдерживали на магнитной мешалке при комнатной температуре в течение ночи. На следующий день FCS центрифугировали и методику очищения сыворотки повторяли в течение 4-6 часов. После второй обработки FCS центрифугировали и стерилизовали через фильтр, чтобы удалить остатки древесного угля и смолы.
МЕТОДИКА АНАЛИЗА
Исходные растворы соединений в DMSO разводили в подходящих концентрационных пределах в мастер-планшетах (master plate). Из мастер-планшетов соединения разводили в культуральных средах, получая растворы анализируемых соединений с конечными дозами.
После доведения количества среды для культивирования клеток до 75 мкл в каждой лунке добавляли 50 мкл раствора анализируемого соединения. Временно трансфицированные клетки подвергали воздействию соединений приблизительно за 24 часа перед выполнением люциферазного анализа. Для люциферазных анализов к каждой лунке вручную добавляли 100 мкл реагента для анализа и планшеты оставляли приблизительно на 20 минут для того, чтобы произошел лизис клеток. После лизиса люциферазную активность измеряли счетчиком 1420 Multiwell, Victor, Wallach.
Эталонные соединения
TZD пиоглитазон использовали в качестве эталонного соединения для активации как человеческого, так и мышиного PPARγ. 5,8,11,14-эйкозатетраеновую кислоту (ETYA) использовали в качестве эталонного соединения для человеческого PPARα.
Расчеты и анализ
Для расчета значений ЕС50 строили кривую зависимости концентрация-эффект. Используемые значения выводили из среднего значения двух или трех независимых измерений (после вычитания среднего фонового значения) и выражали в процентах от максимальной активации, полученной при использовании эталонного соединения. Значения наносили на график по отношению к логарифму концентрации анализируемого соединения. Значения ЕС50 оценивали посредством линейного интеркалирования между экспериментальными точками и расчета концентрации, требуемой для достижения 50% от максимальной активации, полученной при использовании эталонного соединения.
Соединения формулы I имеют значения EC50 менее 0,5 мкмоль/л для PPARα, а предпочтительные соединения имеют значения ЕС50 менее 0,05 мкмоль/л для PPARα. Соединения формулы I представляют собой селективную группу соединений, которые являются более мощными по отношению к PPARα, чем к PPARγ. Полагают, что это соотношение является важным для фармакологической активности соединений и их терапевтического профиля.
Кроме того, соединения по настоящему изобретению проявляют улучшенные свойства DMPK (метаболизма лекарственных веществ и фармакокинетики), например, они проявляют улучшенную метаболическую стабильность in vitro, а также показывают подходящие кривые зависимости доза-ответ in vivo. Эти соединения также обладают многообещающим токсикологическим профилем.
Данные, полученные в in vivo тестах на мышах линии ob/ob, показывают, что агонисты PPARα эффективно понижают уровни глюкозы, инсулина, триглицеридов в организме, а также предотвращают увеличение массы тела. Следовательно, можно сделать вывод о том, что агонисты PPARα можно использовать для лечения гипергликемии, резистентности к инсулину и, следовательно, диабета, а также дислипидемии у млекопитающих.
Изобретение относится к S-энантиомерам соединения формулы I
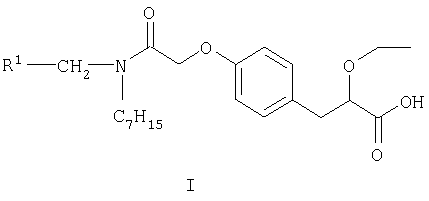
где R1 представляет собой 2,4-дифторфенил или циклогексил и его фармацевтически приемлемые соли и сольваты. S-энантиомеры соединения формулы I выбраны из (2S)-3-(4-{2-[(циклогексилметил)(гептил)амино]-2-оксоэтокси}фенил)-2-этоксипропионовой кислоты и (2S)-3-(4-{2-[(2,4-дифторбензил)(гептил)амино]-2-оксоэтокси}фенил)-2-этокси-пропионовой кислоты и их фармацевтически приемлемых солей и сольватов. Изобретение относится к фармацевтическому препарату, обладающему агонистической активностью в отношении PPARα (активируемых пролифератором цероксисом альфа-рецепторов человека), включающему в себя соединение формулы I в смеси с фармацевтически приемлемыми адъювантами, разбавителями и/или носителями. S-энантиомер соединения формулы I применяют в производстве лекарственного средства для лечения липидных расстройств (дислипидемии), ассоциированных или не ассоциированных с инсулинорезистентностью. Также данное соединение используют в лечении или профилактики липидных расстройств (дислипидемии), ассоциированных или не ассоциированных с инсулинорезистентностью, и диабета 2 типа. Технический результат - производные замещенной фенилпропионовой кислоты как агонисты активируемого пролифератором пероксисом альфа-рецептора человека (PPARα). 11 н.п. ф-лы.
где R1 представляет собой 2,4-дифторфенил или циклогексил,
и его фармацевтически приемлемые соли и сольваты.
(2S)-3-(4-{2-[(циклогексилметил)(гептил)амино]-2-оксоэтокси}фенил)-2-этоксипропионовой кислоты и
(2S)-3-(4-{2-[(2,4-дифторбензил)(гептил)амино]-2-оксоэтокси}фенил)-2-этокси-пропионовой кислоты и их фармацевтически приемлемых солей и сольватов.
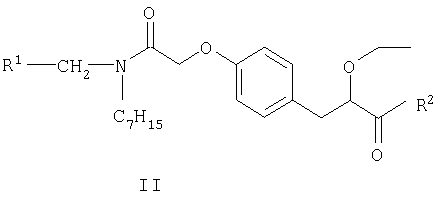
в котором R1 представляет собой 2,4-дифторфенил или циклогексил и R2 представляет собой защитную группу для карбоксильной гидроксигруппы,
подвергают взаимодействию с реагентом для удаления защитной группы.
в котором R1 представляет собой 2,4-дифторфенил или циклогексил и R2 представляет собой защитную группу для карбоксильной гидроксигруппы.
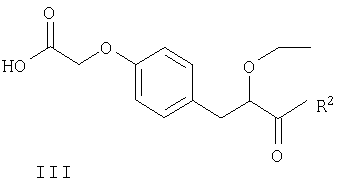
в котором R2 представляет собой защитную группу для карбоксильной гидроксигруппы.
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| WO 00/59889 A1, 12.10.2000 | |||
| WO 00/75103 A1, 14.12.2000 | |||
| WO 00/61582 A1, 19.10.2000 | |||
| ПРОИЗВОДНЫЕ АМИДОКАРБОНОВОЙ КИСЛОТЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ СНИЖЕНИЯ ГЛЮКОЗЫ В КРОВИ | 1998 |
|
RU2176999C2 |

