Настоящее изобретение относится к соединениям, которые повышают активность пируватдегидрогеназы (ПДГ), способам их получения, 25 фармацевтическим композициям, которые их содержат в качестве активного компонента, способам лечения болезненных состояний, связанных со сниженной активностью ПДГ, к их применению в качестве лекарственных средств и к их применению для приготовления лекарственных средств для применения для повышения активности ПДГ в теплокровных животных, таких как люди. В 30 частности, настоящее изобретение относится к соединениям, полезным для лечения сахарного диабета, заболевания периферических сосудов и ишемии миокарда у теплокровных животных, таких как люди, более предпочтительно, к применению этих соединений для приготовления лекарственных средств для применения для лечения сахарного диабета у теплокровных животных, таких как люди.
В тканях аденозинтрифосфат (АТФ) обеспечивает энергию для синтеза сложных молекул и в мышцах для их сокращения. АТФ образуется при расщеплении высокоэнергетических субстратов, таких как глюкоза или свободные жирные кислоты с длинной цепью. В окислительных тканях, таких как мышцы, большинство АТФ образуется из ацетил СоА, при его вхождении в цикл лимонной кислоты, соответственно, запасы ацетил СоА имеют решающее значение для образования АТФ в окислительных тканях. Ацетил СоА образуется как при β-окислении жирных кислот, так и в результате метаболизма глюкозы по гликолитическому пути метаболизма. Ключевым регуляторным ферментом для контролирования уровня образования ацетил СоА из глюкозы является ПДГ, которая катализирует окисление пирувата до ацетил СоА и углекислого газа, что сопровождается восстановлением никотинамидадениндинуклеотида (НАД) до НАДН.
При болезненных состояниях, таких как оба типа сахарного диабета, а именно инсулин-независимом (Тип 2) и инсулин-зависимом (Тип 1) сахарном диабете, увеличивается окисление липидов, что сопровождается уменьшением усвоения глюкозы, что приводит к гипергликемии. Уменьшение усвоения глюкозы при диабете 1 и 2 типов связано с уменьшением активности. Кроме того, еще одним свидетельством уменьшения активности ПДГ может быть то, что повышение концентрации пирувата приводит к повышенной доступности лактата в качестве субстрата для глюконеогенеза в печени. Также можно обоснованно ожидать, что повышение активности ПДГ может приводить к повышению уровня окисления глюкозы и следовательно суммарного усвоения глюкозы, уменьшая при этом выработку глюкозы печенью. Другим фактором, который вносит свой вклад в развитие сахарного диабета, является снижение выработки инсулина, что, как было показано, связано с уменьшенной активностью ПДГ в β-клетках поджелудочной железы (на генетической модели сахарного диабета у грызунов Zhou и др. (1996) Diabetes 45: 580-586).
При окислении глюкозы образуется больше АТФ на моль кислорода, чем при окислении жирных кислот. В условиях, когда потребность в энергии может превышать запасы энергии, таких как ишемия миокарда, перемежающаяся хромота, ишемия головного мозга и реперфузия (Zaidan и др., 1998; J. Neurochem. 70: 233-241), сдвиг равновесия утилизации субстрата в сторону метаболизма глюкозы путем повышения активности ПДГ может приводить, как ожидается, к улучшению возможностей для поддержания уровней АТФ и следовательно, функционирования.
Средство, которое способно повышать активность ПДГ, также может быть полезным, как предполагают, при лечении состояний, при которых имеет место избыток циркулирующей молочной кислоты, таких как определенные виды сепсиса.
Для дихлоруксусной кислоты (ДХК), которая при остром введении повышает у животных активность ПДГ (Vary и др., 1988; Circ. Shock, 24: 3-18), показано, что она обладает предсказанными действиями относительно уменьшения гликемии (Stacpoole и др., 1978; N. Engl. J. Med. 298: 526-530) и для лечения ишемии миокарда (Bersin и Stacpoole 1997; American Heart Journal, 134: 841-855) и молочной ацидемии (Stacpoole и др., 1983; N. Engl. J. Med. 309: 390-396).
ПДГ представляет собой внутримитохондриальный многоферментный комплекс, который состоит из множества копий нескольких субъединиц, включающих три активных фермента E1, E2 и Е3, необходимых для осуществления превращения пирувата в ацетил СоА (Patel и Roche 1990; FASEB J., 4: 3224-3233). E1 катализирует необратимое выделение СО2 из пирувата; E2 образует ацетил СоА и Е3 восстанавливает НАД до НАДН. Два дополнительных активных фермента связаны с комплексом: специфическая киназа, которая способна фосфорилировать Е1 по трем остаткам серина, и слабо связанная специфическая фосфатаза, которая обладает противоположным фосфорилированию действием. Фосфорилирование только одного из трех остатков вызывает инактивацию Е1. Доля ПДГ в ее активном (дефосфорилированном) состоянии определяется равновесием между активностями киназы и фосфатазы. Активность киназы может регулироваться in vivo при помощи относительных концентраций метаболических субстратов, таких как НАД/НАДН, СоА/ацетилСоА и аденозиндифосфата (АДФ)/АТФ, а также доступностью самого пирувата.
Соединение, которое повышает активность ПДГ, потенциально может являться эффективным при лечении болезненных состояний, связанных с нарушением усвоения глюкозы, таких как сахарный диабет, ожирение, (Curto и др., 1997; Int. J. Obes. 21: 1137-1142), и молочная ацидемия. Кроме того, предполагается, что такое соединение также может быть эффективным при заболеваниях, при которых ограничены запасы высокоэнергетических субстратов в тканях, таких как заболевания периферических сосудов (включая перемещающуюся хромоту), сердечную недостаточность и определенные миопатии сердца, мышечную слабость, гиперлипидемии и атеросклероз (Stacpoole и др., 1978; N. Engl. J. Med. 298: 52-530). Соединение, которое активирует ПДГ, также может быть полезным для лечения болезни Альцгеймера (AD) (J Neural Transm (1998) 105, 855-870).
В европейских патентных публикациях №№617010 и 524781 описаны соединения, способны вызывать расслабление гладких мышц мочевого пузыря и которые могут применятся для лечения позывов к мочеиспусканию. В международных заявках WO 9944618, WO 9947508, WO 9962506, WO 9962873, WO 01/17942, WO 01/17955 и WO 01/17956 описаны соединения, которые повышают активность ПДГ. Соединения согласно настоящему изобретению конкретно не описаны в любой из вышеупомянутых заявок, и нами неожиданно было обнаружено, что эти соединения обладают полезными свойствами в отношении одной или нескольких их фармацевтических активностей (особенно в качестве соединений, которые повышают активность пируватдегидрогеназы) и/или имеют фармакокинетические, действующие, метаболические и токсикологические профили, которые делают их особенно пригодными для введения в условиях in vivo теплокровному животному, такому как человек.
Таким образом, настоящее изобретение относится к соединению формулы (I):
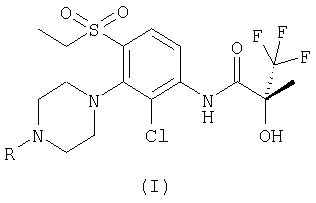
где R представляет собой метил или мезил;
или его фармацевтически приемлемой соли или сложному эфиру, который способен к гидролизу в условиях in vivo.
В одном варианте осуществления изобретения R представляет собой метил.
В другом варианте осуществления изобретения R представляет собой мезил.
Другие варианты осуществления изобретения относятся к соединению или его фармацевтически приемлемой соли.
Также подразумевается, что соединение формулы (I) и его фармацевтически приемлемые соли и сложные эфиры, которые способны к гидролизу в условиях in vivo, могут существовать в виде сольватов, а также в несольватированных формах, таких как, например, гидратированные формы. Также подразумевается, что под объем изобретения подпадают все такие сольватированные формы, которые повышают активность ПДГ.
Соединение формулы (I) и его физиологически приемлемые соли и сложные эфиры, которые способны к гидролизу в условиях in vivo, могут быть получены при помощи любых известных способов, которые пригодны для получения химически сходных соединений. Такие способы включают, например, описанные в европейских заявках на патенты, опубликованных под номерами 0524781, 0617010, 0625516, и в GB 2278054 и в международных заявках WO 9323358, WO 9738124, WO 9944618, WO 9947508, WO 9962506, WO 9962873, WO 01/17942, WO 01/17955 и WO 01/17956.
Другим объектом настоящего изобретения является способ получения соединений формулы (I) или его фармацевтически приемлемой соли или сложного эфира, который способен к гидролизу в условиях in vivo, при этом способ (где изменяемые группы имеют значения, указанные для формулы (I), если специально не указано иначе) предусматривает:
(а) снятие защиты с защищенного соединения формулы (II):
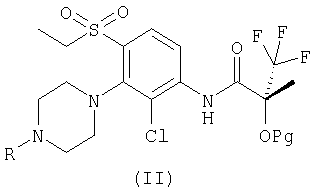
где Pg представляет собой спиртовую защитную группу;
(б) окисление соединения формулы (III):
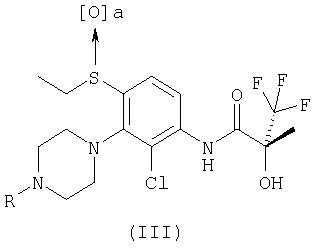
где а представляет собой 0 или 1;
(в) взаимодействие соединения формулы (IV):
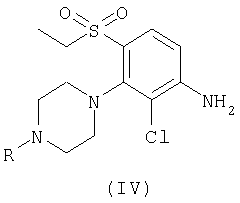
с кислотой формулы (V):
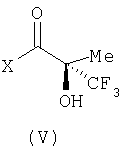
где Х представляет собой ОН;
(г) взаимодействие анилина формулы (IV) с активированным производным кислоты формулы (V);
(д) реагирование соединения формулы (VI):
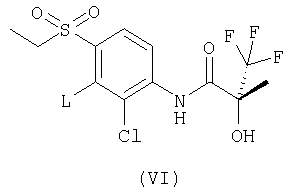
где L представляет собой вытесняемую группу; с 4-мезилпиперазином или 4-метилпиперазином;
(е) для соединений формулы (I), где R представляет собой метил; метилирование соединения формулы (VII):
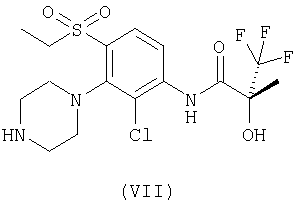
(ж) для соединений формулы (I), где R представляет собой мезил; мезилирование соединения формулы (VII);
(з) хлорирование соединения формулы (VIII):
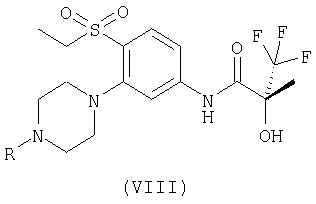
(и) преобразование функциональной группы на хлор для соединения формулы (IX):
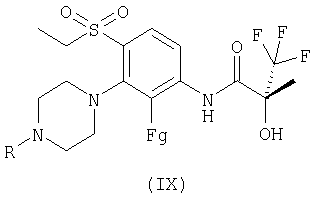
где Fg представляет собой функциональную группу;
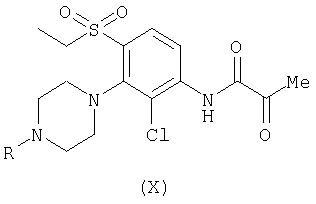
(к) присоединение металлоорганического реагента к соединению формулы (X):
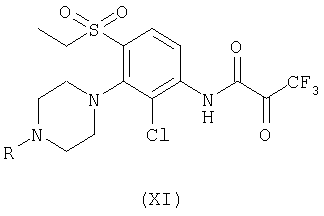
(л) присоединение металлоорганического реагента к соединению формулы (XI):
(м) присоединение соединения формулы (V), где Х представляет собой NH2, к соединению формулы (XII):
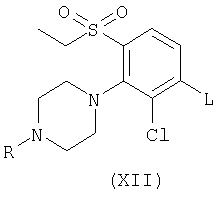
где L представляет собой вытесняемую группу;
(н) перегруппировку Смайсла соединения формулы (XIII):
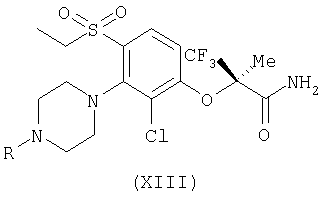
или
(о) разделение смеси (R) и (S) энантиомеров соединений формулы (I) для получения (R)-энантиомера;
и затем, при необходимости, получение фармацевтически приемлемой соли или сложного эфира, способного к гидролизу в условиях in vivo.
Подходящими значениями для Pg является бензильная группа, силильная группа (например, триалкилсилильная группа или алкилдифенилсилильная группа) или ацетилзащитная группа.
Если формула (V) представляет собой активированное производное кислоты, подходящими значениями для Х являются галоген (например, хлор или бром), ангидриды, арилокси (например, 4-нитрофенокси или пентафторфенокси) или имидазол-1-ил.
L представляет собой вытесняемую группу. Подходящими значениями для L являются фтор, хлор, бром, нитро, метансульфонат и трифторметансульфонат.
Fg представляет собой функциональную группу. Подходящей функциональной группой является аминогруппа, которая может быть взаимопревращена при помощи диазотирования и реакции соли диазония с хлоридом в присутствии медного катализатора.
Конкретными условиями для вышеуказанных реакций являются следующие:
Способ (а)
Примерами подходящих реагентов для снятия защиты со спирта формулы (II) являются:
1) если Pg представляет собой бензил:
(i) водород в присутствии палладиевого/углеродного катализатора, то есть гидрогенолиз; или
(ii) бромид водорода или йодид водорода;
2) если Pg представляет собой силильную защитную группу:
(i) фторид тетрабутиламмония; или
(ii) фтористоводородная или соляная кислота;
3) если Pg представляет собой ацетил:
i) слабая водная щелочь, например, гидроокись лития; или
ii) аммиак или амин, такой как диметиламин.
Реакцию можно проводить в подходящем растворителе, таком как этанол, метанол, ацетонитрил или диметилсульфоксид и подходящая температура для осуществления реакции находится в интервале от -40 до 100°С.
Соединения формулы (II) могут быть получены согласно следующей схеме:
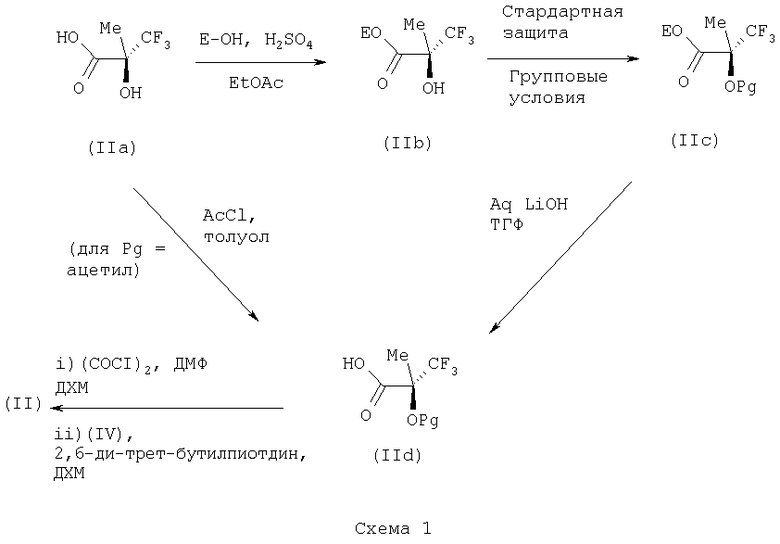
Е представляет собой карбоксизащитную группу. Подходящими значениями для Е являются C1-С6алкил, такой как метил и этил.
Соединение формулы (IIa) является коммерчески доступным соединением.
Способ (б)
Подходящими окислителями являются перманганат калия, OXONE™, перйодат натрия, надкислоты (такие как, например, 3-хлорпероксибензойная кислота или перуксусная кислота), перекись водорода, ТРАР (перрутенат тетрапропиламмония) или кислород. Реакцию можно проводить в подходящем растворителе, таком как диэтиловый эфир, ДХМ, метанол, этанол, вода, уксусная кислота, или смесях двух или более этих растворителей. Реакцию обычно осуществляют при температуре в интервале от -40 до 100°С.
Соединения формулы (III) могут быть получены согласно следующей схеме:
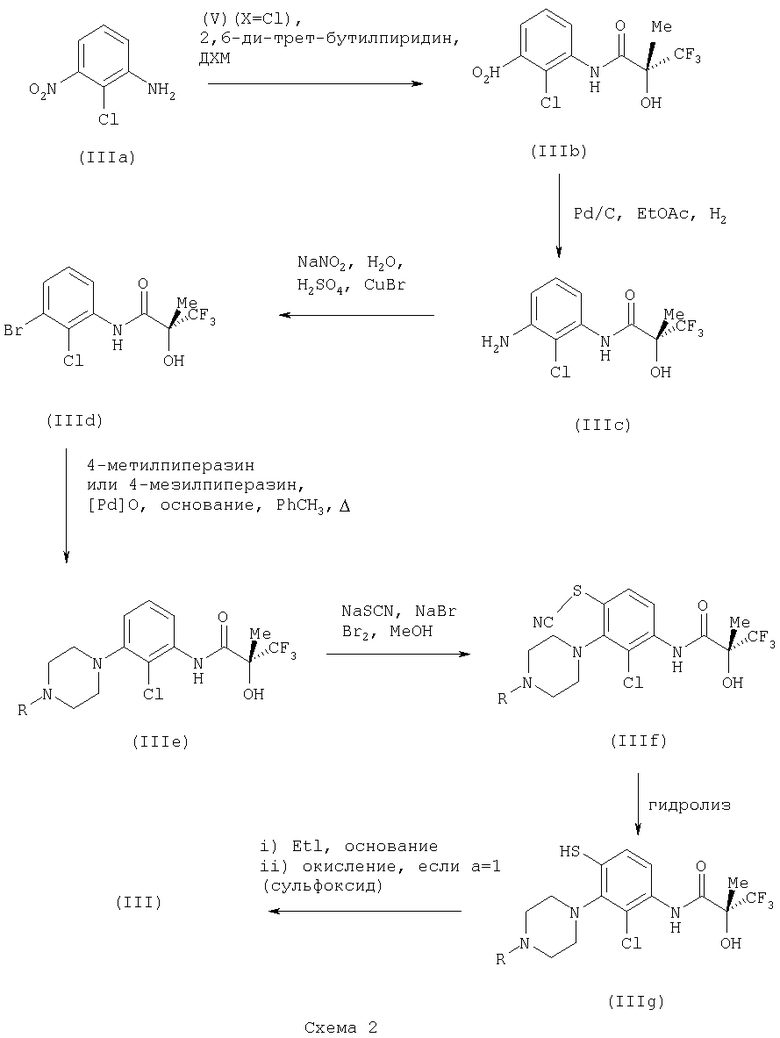
Соединение формулы (IIIa) является коммерчески доступным соединением.
Способ (в)
Реакцию можно осуществлять в присутствии подходящего связующего вещества. Стандартные амидные связующие реагенты, известные в данной области, могут применяться в качестве подходящих связующих реагентов, например, такие условия, как описанные выше для взаимодействий (IIIa) и (V) или (IV) и (IId), или, например дициклогексил-карбодиимид, необязательно в присутствии катализатора, такого как диметиламинопиридин или 4-пирролидинопиридин, необязательно в присутствии основания, например триэтиламина, пиридина, или 2,6-ди-алкил-пиридинов (таких как 2,6-лутидин или 2,6-ди-трет-бутилпиридин) или 2,6-дифенилпиридина. Подходящими растворителями являются диметилацетамид, ДХМ, бензол, ТГФ и ДМФА. Сочетание обычно может осуществляться при температуре в интервале от -40 до 40°С.
Соединения формулы (IV) могут быть получены согласно следующей схеме:
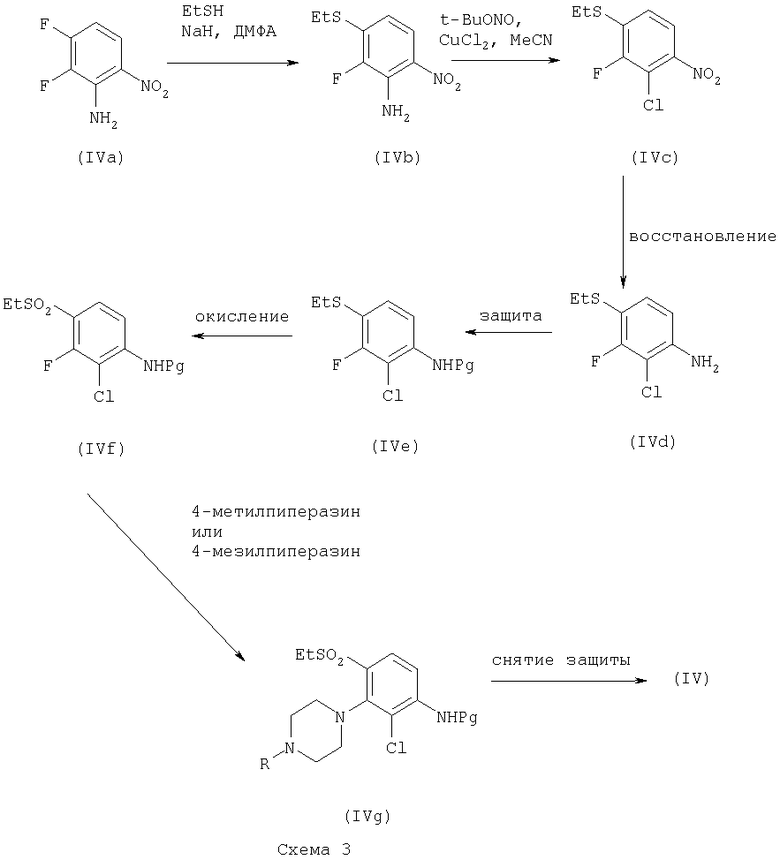
Pg представляет собой аминозащитную группу, такую как описанную далее.
Соединения формулы (IVa) и (V) являются коммерчески доступными соединениями.
Например, разделение кислоты формулы (V) может осуществляться при помощи любого из известных способов получения оптически активных форм (например, путем перекристаллизации хиральной соли {например, заявка WO 9738124}, путем ферментативного разделения или хроматографического разделения при помощи хиральной неподвижной фазы). Например, (R)-(+) разделенная кислота может быть получена при помощи способа согласно схеме 2, описанной в опубликованной международной заявке на патент WO 9738124 для получения (S)-(-) кислоты, то есть при помощи способа традиционного повторного разделения, описанного в европейской патентной заявке №ЕР 0524781, также для получения (S)-(-) кислоты, за исключением того, что (1S,2R)-норэфедрин применяется вместо (S)-(-)-1-фенилэтиламина. Хиральная кислота также может быть получена при помощи способа ферментативного разделения, как описано в Tetrahedron Asymmetry, 1999, 10, 679.
Способ (г)
Это взаимодействие может осуществляться необязательно в присутствии основания, например, триэтиламина, пиридина, 2,6-ди-алкил-пиридинов (таких как 2,6-лутидин или 2,6-ди-трет-бутилпиридин) или 2,6-дифенилпиридина. Подходящими растворителями являются диметилацетамид, ДХМ, бензол, ТГФ и ДМФА. Сочетание обычно может осуществляться при температуре в интервале от -40 до 40°С.
Способ (д)
Это реакцию можно осуществлять путем взаимодействия 1-мезилпиперазина (US 6140351) или 1-метилпиперазина (1-20 молярных эквивалентов, предпочтительно 2-10 эквивалентов) с (VI) в растворителе, таком как N-метил-2-пирролидон или диметилацетамид, или неразбавленных, при нагревании до температуры от 40 до 160°С.
Соединения формулы (VI), в которых L представляет собой фтор, могут быть получены согласно следующей схеме.
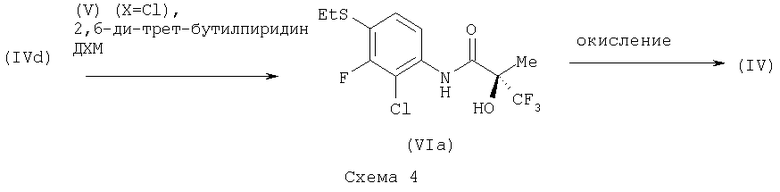
Способ (е)
Соединение (VII) может быть метилировано при помощи формальдегида и восстановителя, такого как борогидрид натрия или триацетоксиборогидрид натрия в подходящем растворителе, таком как 1,2-дихлорэтан, ДХМ или ТГФ, при температуре в диапазоне от 0°С до температуры флегмы, предпочтительно при комнатной или приблизительно при комнатной температуре. Альтернативно соединение (VII) может быть метилировано при помощи метилирующего средства, такого как метилйодид или диметилсульфат в растворителе, таком как ацетон или ДМФА в присутствии основания, такого как бикарбонат натрия, карбонат натрия или гидроокись натрия, необязательно при защите гидроксильной группы. Получение соединения (VII) описано далее в способе 1.
Способ (ж)
Соединение (VII) может быть мезилировано при помощи подходящего средства, такого как хлорид метансульфонила, в присутствии основания, такого как триэтиламин, в подходящем растворителе, таком как ДХМ, ТГФ, пиридин или этилацетан, при температуре в диапазоне от -40°С до температуры флегмы, предпочтительно при комнатной или приблизительно при комнатной температуре.
Способ (з)
Хлорирование может осуществляться, например, при помощи N-хлорсукцинимида в растворителе, таком как ДХМ, ацетонитрил, изопропанол или ДМФА при температуре в диапазоне от 0°С до температуры флегмы, или при помощи хлора в присутствии катализатора, такого как трихлорид железа, в подходящем растворителе, таком как уксусная кислота, ДМФА или ацетонитрил, при температуре в диапазоне от -20°С до 40°С, предпочтительно при комнатной или ниже комнатной температуре, с последующим разделением требуемого продукта от нежелательных изомерных примесей, если таковые образуются.
Соединения формулы (VIII) могут быть получены согласно следующей схеме:
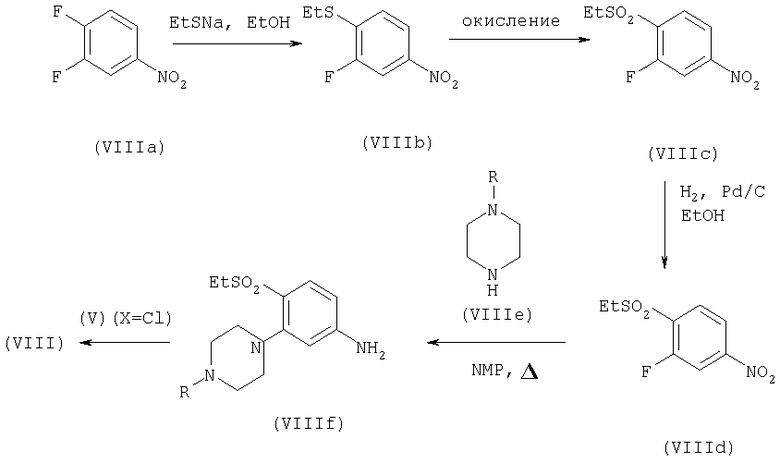
Соединение (VIIIa) является коммерчески доступным.
Способ (и)
Для этих взаимопревращений функциональных групп используются реагенты и условия реакций, которые хорошо известны в данной области химии.
Например, взаимопревращение функциональной группы (IX), где Fg представляет собой NH2, в соединение формулы (I) может осуществляться при помощи диазотирования, например с трет-бутилнитритом и т.д. в присутствии катализатора, такого как хлорид меди, в растворителе, таком как ацетонитрил, при температуре в диапазоне от 0°С до температуры флегмы, предпочтительно при комнатной или приблизительно при комнатной температуре. Альтернативно превращение может осуществляться путем диазотирования с нитритной солью в присутствии кислоты, такой как HCl или серная кислота, в растворителе, таком как вода, уксусная кислота или смеси двух растворителей, при температуре от -20 до 40°С, с последующей реакцией таким образом образованной соли диазония с хлоридом одновалентной меди в том же растворителе при температуре в интервале от 0°С до температуры флегмы.
Соединения формулы (IX) могут быть получены согласно следующей схеме:
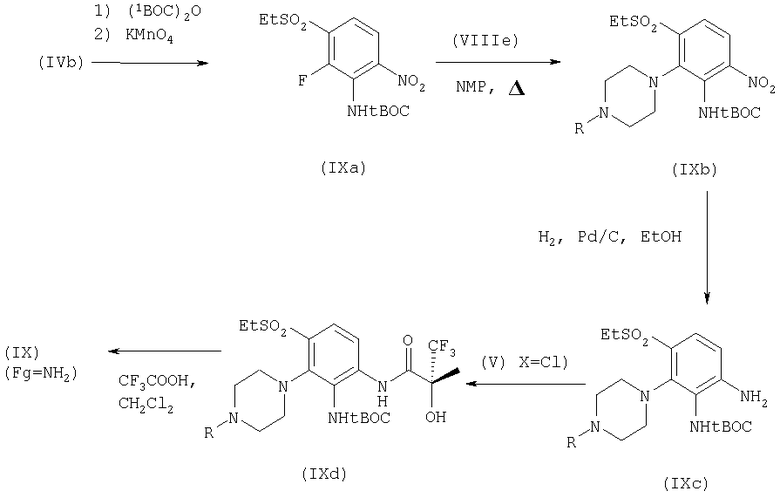
Способ (к)
Реакцию можно осуществлять путем добавления подходящего реагента, такого как CF3SiMe3 (реагент Рупперта (Ruppert's reagent). Tetrahedron, 2000, 56(39), 7613), которая может происходить асимметрично при использовании подходящего катализатора, такого как хиральный цинхоний-фторидный катализатор, (Tetrahedron Lett., 1994, 35, 3137), и последующего подкисления водной рабочей среды для вызывания гидролиза третичного простого силильного спиртового эфира, образованного в реакции. Эту реакцию можно осуществлять в подходящем растворителе, таком как толуол, при температуре в диапазоне от -100°С до комнатной температуры до температуры флегмы, предпочтительно от -78°С до комнатной температуры.
Соединения формулы (X) могут быть получены при помощи способа (в), то есть путем взаимодействия соединения (IV) с пировиноградной кислотой вместо кислоты формулы (V).
Способ (л)
Реакцию можно осуществлять путем прибавления подходящего металлоорганического реагента, такого как CH3MgBr или СН3CeCl2 в растворителе, таком как простой эфир или ТГФ при температуре от -120 до 40°С в присутствии хирального катализатора, такого как TADDOL (тетраарилдиметилдиоксоландиметанол, например, где арил представляет собой фенил или 2-нафтил; Angew. Chem. Int. Ed. Engl., 1992, 31, 84-6).
Соединения формулы (XI) могут быть получены при помощи способа (в), то есть путем взаимодействия соединения (IV) с трифторпировиноградной кислотой (Tetrahedron Lett., 1989, 30(39), 5243) вместо кислоты формулы (V).
Способ (м)
Реакцию можно осуществлять при помощи взаимодействия соединения формулы (XII) с дианионом, полученным путем обработки соединения формулы (V), где Х представляет собой NH2, с двумя молярными эквивалентами основания, такого как гидрид натрия, в подходящем растворителе, таком как ТГФ или NMP, при температуре в интервале 20-160°С.
Соединение формулы (XII), где L представляет собой Cl, может быть получено, например, путем диазотирования соединения формулы (IV) согласно методу, описанному в способе (и).
Способ (н)
Перегруппировку Смайсла можно осуществлять путем обработки соединения формулы (XIV) основанием, таким как гидрид натрия, в растворителе, таком как ДМФА.
Соединение формулы (XIV) может быть получено из соединения формулы (XII), где L представляет собой Cl, при реагировании с соединением формулы (V), где Х представляет собой NH2, с одним молярным эквивалентом основания, такого как гидрид натрия, в растворителе, таком как ТГФ или NMP, при температуре в интервале 20-160°С.
Способ (о)
Необходимая оптически активная форма соединения формулы (I) может быть получена путем разделения смеси соединения формулы (I) и его соответствующего (S) энантиомера при использовании обычных методик, хорошо известных специалисту в данной области техники, например, кристаллизацией, ферментативным разделением или хроматографическим разделением энантиомеров.
Необходимые исходные вещества для вышеописанных методик, если они не являются коммерчески доступными, могут быть получены при помощи методик, выбранных из стандартных технологий органической химии, технологий, аналогичных синтезу известных структурно сходных соединений, или технологий, аналогичных вышеописанным методикам, или методикам, описанным в примерах.
Следует отметить, что многие исходные вещества для вышеуказанных способов синтеза являются коммерчески доступными и/или хорошо описанными в научной литературе, или могут быть получены из коммерчески доступных соединений при помощи модификаций способов, описанных в научной литературе. В качестве основного руководства для условий осуществления реакции и реагентов авторы ссылаются на Advanced Organic Chemistry, 4-ое издание, автор Jerry March, опубликованную John Wiley & Sons 1992.
Также следует принять во внимание, что для некоторых реакций, описанных в настоящем изобретении, может являться необходимым/желательным защищать любые чувствительные группы соединений. Случаи, когда такая защита необходима или желательная, известны специалисту в данной области техники, так же как и походящие способы для такой защиты. Обычные защитные группы могут применятся в соответствии с общепринятой практикой (например, см. T.W.Greene, Protective Groups in Organic Synthesis, John Wiley и Sons, 1991).
Примерами подходящей защитной группы для гидроксильной группы являются, например, ацильная группа, например алканоильная группа, такая как ацетил, ароильная группа, например бензоил, силильная группа, такая как триметилсилил или арилметильная группа, например бензил. Условия снятия защиты для вышеупомянутых защитных групп главным образом будут зависеть от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или ароильная группа, могут быть отщеплены, например, при помощи гидролиза с подходящим основанием, таким как гидроокись щелочного металла, например гидроокись лития или натрия. Альтернативно, силильная группа, такая как триметилсилил, может быть отщеплена, например, при помощи фторида или аквокислоты; или арилметилльная группа, такая как бензильная группа, может быть отщеплена, например, путем гидрирования в присутствии катализатора, такого как палладий на угле.
Подходящими защитными группами для аминогруппы являются, например, ацильная группа, например алканоильная группа, такая как ацетил, алкоксикарбонильная группа, например, метоксикарбонильная, этоксикарбонильная или трет-бутоксикарбонильная группа, арилметоксикарбонильная группа, например бензилоксикарбонил, или ароильная группа, например бензоил. Условия снятия защиты для вышеупомянутых защитных групп главным образом будут зависеть от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или алкоксикарбонильная группа или ароильная группа могут быть отщеплены, например, путем гидролиза с подходящим основанием, таким как гидроокись щелочного металла, например гидроокись лития или натрия. Альтернативно, ацильная группа, такая как трет-бутоксикарбонильная группа может быть отщеплена, например, путем обработки подходящей кислотой, такой как соляная, серная или фосфорная кислота или трифторуксусная кислота и арилметоксикарбонильная группа, такая как бензилоксикарбонильная группа, может быть отщеплена, например, путем гидрирования в присутствии катализатора, такого как палладий на угле или путем обработки кислотой Льюиса, например, трис(трифторацетатом) бора. Подходящей альтернативной защитной группой для первичной аминогруппы является, например, фталоильная группа, которая может быть отщеплена путем обработки с алкиламином, например, диметиламинопропиламином или 2-гидроксиэтиламином, или с гидразином.
Защитные группы могут быть отщеплены на любой подходящей стадии синтеза при помощи обычных методик, хорошо известных в области химии, или они могут быть отщеплены на поздней стадии реакции или подготовки.
Соединение формулы (I) может образовывать стабильные кислотные или основные соли, и в таких случаях введение соединения в виде соли может являться целесообразным, и фармацевтически приемлемые соли могут быть получены при помощи обычных способов, таких как описаны далее. Примерами фармацевтически приемлемых солей являются органические кислото-аддитивные соли, образованные с кислотами, которые образуют физиологически приемлемый анион, например, тозилат, метансульфонат и α-глицеролфосфат. Подходящими образованными неорганическими солями также являться сульфат, нитрат и хлорид.
Фармацевтически приемлемые соли могут быть получены при помощи стандартных методик, известных в данной области, например путем реагирования соединения формулы (I) (и в некоторых случаях сложного эфира) с подходящей кислотой, которая дает физиологически приемлемый анион. Также представляется возможным получать соли с соответствующим щелочным металлом (например, натрием, калием или литием) или щелочноземельным металлом (например, кальцием) путем обработки соединения формулы (I) (и в некоторых случаях сложного эфира) одним эквивалентом гидроокиси щелочного металла или алкоксида или половиной эквивалента гидроокиси щелочноземельного металла или алкоксида (например, этоксида или метоксида) в водной среде с последующими обычными методиками очистки.
Сложный эфир соединения формулы (I), который способен к гидролизу в условиях in vivo, представляет собой, например, фармацевтически приемлемый сложный эфир, который гидролизируется в организме человека или животного с образованием исходной кислоты или спирта.
Подходящие сложные эфиры соединения формулы (I), которые способны к гидролизу в условиях in vivo, образованные с гидроксильной группой, включают неорганические сложные эфиры, такие как фосфатные сложные эфиры и α-ацилоксиалкильные эфиры. Примерами α-ацилоксиалкильных эфиров являются ацетоксиметокси и 2,2-диметилпропионилокси-метокси. Другими группами, которые образуют сложные эфиры, способны к гидролизу в условиях in vivo, для гидроксигруппы включают алканоил, бензоил, фенилацетил и замещенные бензоил и фенилацетил, алкоксикарбонил (для получения алкилкарбонатного сложного эфира), диалкилкарбамоил и N-(диалкиламиноэтил-N-алкилкарбамоил (для получения карбаматов), диалкиламиноацетил и карбоксиацетил. Примерами заместителей для бензоила являются морфолиногруппа и пиперазиногруппа, связанные с атомом азота кольца при помощи метиленовой группы в 3-м или 4-м положении бензоильного кольца.
Объектом настоящего изобретения является выявление соединений, которые повышают активность ПДГ. Такие свойства могут быть оценены, например, при помощи одного или более способов тестирования, известных из литературы, например, таких, которые изложены в заявке WO 9962506; а именно исследование (а) - повышение активности ПДГ в условиях in vitro, исследование (b) - повышение активности ПДГ в условиях in vitro на выделенных первичных клетках и исследование (с) повышение активности ПДГ в условиях in vivo и все эти исследования включены в настоящее изобретение путем ссылки. Альтернативно, эти свойства могут быть оценены при помощи следующего эксперимента.
Повышение активности ПДГ в условиях in vitro
При помощи этого анализа определялась способность тестируемого соединения повышать активность ПДГ. кДНК, которая кодирует киназу ПДГ, может быть получена при помощи полимеразной цепной реакции (ПЦР) и затем клонирована. Ее можно экспрессировать в подходящей экспрессионной системе и получать полипептид, обладающий ПДГ-киназной активностью. Например, ПДГкиназа2 человека (rPDHK2), полученная при экспрессии рекомбинатного белка в Escherichia coli (E. Coli), обнаруживает ПДГ-киназную активность.
rPDHK2 человека (регистр, номер L42451.1 в банке генов) клонировали и экспрессировали при помощи способа, описанного в Baker и др. (2000) J. Biol. Chem. 275,15773-15781. В экспрессируемый белок вводили сайт расщепления протеазой, как описано в этой ссылке. Другие известные киназы ПДГ для применения в исследованиях могут быть клонированы и экспрессированы подобным образом. Для экспрессии активной rPDHK2, клетки штамма E. coli BL21 (DE3) трансформировали вектором рЕТ28А, содержащим кДНК rPDHK2. Этот вектор включает метку на 6-His в белок на его N-конце. Клетки E. coli выращивали в ферментере до значения оптической плотности, равного 12 (550 нм) при температуре 37°С, уменьшая температуру до 22°С до тех пор, пока оптическая плотность не достигала 15, и экспрессию белка индуцировали путем добавления 0,5 мМ изопропилтио-β-галактозидазы. Клетки выращивали в течении 3 часов при температуре 22°С и собирали путем центрифугирования. Массу ресуспендированных клеток лизировали путем гомогенизации при высоком давлении и нерастворимое вещество удаляли путем центрифугирования при 26000×g в течение 30 минут. Меченный по 6-His белок удаляли из супернатанта при помощи кобальтовой хелатирующей полимерной матрицы (TALON: Clontech), которую промывали в 20 мМ N-[2-гидроксиэтил]пиперазин-N'-[2-этансульфоновой кислоты (HEPES), 500 мМ NaCl, 1% (об./об.) этиленгликоля, 0,1% (мас./об.) Pluronics F-68 рН 8,0, до постепенного ступенчатого элюирования связанного белка при помощи подобного буфера при добавлении 100 мМ имидазола рН 8,0. Элюированные фракции, содержащие меченный по 6-His белок, объединяли, добавляли этилендиаминтетрауксусную кислоту (EDTA) и дитиотретоил (DTT) до конечной концентрации 1 мМ и метку отщепляли путем прибавления прецизионной протеазы (Amersham Pharmacia Biotech). Эту протеазу удаляли при помощи глутатион-сефарозы (Amersham Pharmacia Biotech). Немеченный белок диализировали в буфере для хранения, состоящему из 20 мМ HEPES-Na, 150 мМ хлорида натрия, 0,5 мМ EDTA, 1% (мас./об.) Pluronics F68, 1% (об./об.) этиленгликоля рН 8,0 и хранили а аликвотах при температуре -80°С.
Каждую новую партию исходного фермента ПДГК титровали в исследовании для определения концентрации, которая обеспечивает приблизительно 75%-ное ингибирование ПДГ в условиях анализа. Исходный фермент (обычно в концентрации 20 мкг/мл) оставляли для связывания в течение 24 часов при температуре 4°С с ПДГ (ПДГ из сердца свиньи, Sigma Р7032) (0,05 Ед/мл) в буфере, содержащем 50 мМ 3-[N-морфолино]пропансульфоновой кислоты (MOPS), 20 мМ ортофосфата дикалия, 60 мМ хлорида калия, 2 мМ хлорида магния, 0,4 мМ этилендиаминтетрауксусной кислоты (EDTA), 0,2% Pluronic F68, 1 мМ дитиотретоила (DTT), рН 7,3.
Для исследования активности новых соединений, соединения растворяли в 5% ДМСО и 5 мкл переносили в отдельные лунки 384-луночних плашек для исследования. Контрольные лунки содержали 5 мкл 5% ДМСО вместо соединения. Для определения максимального значения реакции ПДГ вторая серия контрольных лунок включала 5 мкл известного ингибитора в конечной концентрации в киназной реакции 10 мкМ.
Добавляли 40 мкл раствора предварительно связанного фермента и реакцию фосфорилирования инициировали путем прибавления 5 мкл 10 мкММ АТФ в вышеуказанном буфере. Через 45 минут при комнатной температуре определяли остаточную активность ПДГ путем прибавления субстратов (2,5 мМ кофермента А, 2,5 мМ пирофосфата тиамина (кокарбоксилазы), 2,5 мМ пирувата натрия, 6 мМ НАД) в объеме 40 мкл и плашки инкубировали на протяжении 90 минут при температуре окружающей среды. Получение восстановленного НАД (НАДН) определяли путем измерения оптической плотности при 340 нм при помощи спектрофотометра для анализа плашек. Значения ЕС50 для тестируемого соединения определяли обычным путем на основании полученных результатов для 12 разных концентраций соединения.
В соответствии с другим вариантом осуществления, настоящее изобретение обеспечивает фармацевтическую композицию, которая содержит соединение формулы (I), как описано здесь выше, или его фармацевтически приемлемую соль или сложный эфир, который способен к гидролизу в условиях in vivo, в сочетании с фармацевтически приемлемым наполнителем или носителем.
Композиция может находиться в форме, подходящей для перорального введения, например в виде таблетки или капсулы, для парантерального введения (включая внутривенное, подкожное, внутримышечное, внутрисосудистое введение или инфузию) например, в виде стерильного раствора, суспензии или эмульсии, для местного введения, например в виде мази или пасты, или для ректального введения, например, в виде суппозитория. В целом, вышеприведенные композиции могут быть приготовлены обычным способом при использовании обычных наполнителей.
Композиции согласно настоящему изобретению предпочтительно представлены в виде стандартной дозированной формы. Обычно соединение вводится теплокровному животному в стандартной дозе в диапазоне 5-5000 мг на м2 поверхности тела животного, то есть приблизительно 0,1-100 мг/кг.
Стандартная доза предположительно находится в диапазоне, например, 1-100 мг/кг, предпочтительно 1-50 мг/кг и обычно она является терапевтически эффективной дозой. Лекарственная форма, которая содержит стандартную дозу, такая как таблетка или капсула, обычно включает, например, 1-250 мг активного компонента.
В соответствии с другим вариантом осуществления, настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли или сложному эфиру, который способен к гидролизу в условиях in vivo, как определено выше, для применения в способе терапевтического лечения человека или животного.
Нами было обнаружено, что соединения по настоящему изобретению повышают активность ПДГ и, следовательно, представляют интерес, поскольку снижают уровень глюкозы в крови.
Другим вариантом осуществления настоящего изобретения является соединение формулы (I) и его фармацевтически приемлемые соли или сложные эфиры, способные к гидролизу в условиях in vivo, для применения в качестве лекарственного средства.
Таким образом, изобретение относится к соединению формулы (I), или его фармацевтически приемлемой соли или сложному эфиру, который способен к гидролизу в условиях in vivo, для применения в качестве лекарственного средства для повышения активности ПДГ у теплокровного животного, такого как человек.
Более конкретно, изобретение относится к соединению формулы (I), или его фармацевтически приемлемой соли или сложному эфиру, который способен к гидролизу в условиях in vivo, для применения в качестве лекарственного средства для лечения сахарного диабета у теплокровного животного, такого как человек.
Более предпочтительно, изобретение относится к соединению формулы (I), или его фармацевтически приемлемой соли или сложному эфиру, который способен к гидролизу в условиях in vivo, для применения в качестве лекарственного средства для лечения сахарного диабета, заболевания периферических сосудов и ишемии миокарда у теплокровного животного, такого как человек.
Таким образом, другим вариантом осуществления изобретения является применение соединения формулы (I), или его фармацевтически приемлемой соли или сложного эфира, который способен к гидролизу в условиях in vivo, для приготовления лекарственного средства для применения для повышения активности ПДГ у теплокровного животного, такого как человек.
Следовательно, другим вариантом осуществления изобретения является применение соединения формулы (I), или его фармацевтически приемлемой соли или сложного эфира, который способен к гидролизу в условиях in vivo, для приготовления лекарственного средства для применения для лечения сахарного диабета у теплокровного животного, такого как человек.
Таким образом, другим вариантом осуществления изобретения является применение соединения формулы (I), или его фармацевтически приемлемой соли или сложного эфира, который способен к гидролизу в условиях in vivo, для приготовления лекарственного средства для применения для лечения сахарного диабета, заболеваний периферических сосудов и ишемии миокарда у теплокровного животного, такого как человек.
В соответствии с другим вариантом осуществления, изобретение обеспечивает фармацевтическую композицию, которая содержит соединение формулы (I), как описано здесь выше, или его фармацевтически приемлемую соль или сложный эфир, который способен к гидролизу в условиях in vivo, в сочетании с фармацевтически приемлемым наполнителем или носителем для применения для повышения активности ПДГ у теплокровного животного, такого как человек.
В соответствии с другим вариантом осуществления, изобретение обеспечивает фармацевтическую композицию, которая содержит соединение формулы (I), как описано здесь выше, или его фармацевтически приемлемую соль или сложный эфир, который способен к гидролизу в условиях in vivo, в сочетании с фармацевтически приемлемым наполнителем или носителем для применения для лечения сахарного диабета у теплокровного животного, такого как человек.
В соответствии с другим вариантом осуществления, изобретение обеспечивает фармацевтическую композицию, которая содержит соединение формулы (I), как описано здесь выше, или его фармацевтически приемлемую соль или сложный эфир, который способен к гидролизу в условиях in vivo, в сочетании с фармацевтически приемлемым наполнителем или носителем для применения для лечения сахарного диабета, заболеваний периферических сосудов и ишемии миокарда у теплокровного животного, такого как человек.
В соответствии с другим вариантом осуществления, изобретение обеспечивает способ повышения активности ПДГ у теплокровного животного, такого как человек, которое нуждается в таком лечении, который предусматривает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или сложного эфира, который способен к гидролизу в условиях in vivo, как описано здесь выше.
В соответствии с другим вариантом осуществления, изобретение обеспечивает способ лечения сахарного диабета у теплокровного животного, такого как человек, которое нуждается в таком лечении, который предусматривает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или сложного эфира, который способен к гидролизу в условиях in vivo, как описано здесь выше.
В соответствии с другим вариантом осуществления, изобретение обеспечивает способ лечения сахарного диабета, заболеваний периферических сосудов и ишемии миокарда у теплокровного животного, такого как человек, которое нуждается в таком лечении, который предусматривает введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или сложного эфира, который способен к гидролизу в условиях in vivo, как описано здесь выше.
Как указывалось выше, величина дозы, необходимой для терапевтического или профилактического лечения конкретного болезного состояния обычно изменяется в зависимости от организма, который подвергается лечению, пути введения и тяжести заболевания, которое поддается лечению. Предпочтительно применяемая дневная доза находится в диапазоне 1-50 мг/кг. Однако дневная доза обычно изменяется в зависимости от организма, который подвергается лечению, особенности пути введения и тяжести заболевания, которое поддается лечению. Следовательно, оптимальная доза может быть определена лечащим врачом индивидуально для каждого пациента.
Как указывалось выше, соединения, раскрытые в настоящем изобретении, представляют интерес в связи с их способностью повышать активность ПДГ. Следовательно, соединения согласно изобретению могут быть полезными для лечения ряда болезненных состояний, включая сахарный диабет, заболевания периферических сосудов (включая перемежающуюся хромоту), сердечную недостаточность и определенные миопатии сердца, ишемию миокарда, ишемию головного мозга и реперфузию, мышечную слабость, гиперлипидемии, болезнь Альцгеймера и/или атеросклероз. Альтернативно, такие соединения согласно изобретению могут быть полезными при ряде болезненных состояний, включая заболевания периферических сосудов (включая перемежающуюся хромоту), сердечную недостаточность и определенные миопатии сердца, ишемию миокарда, ишемию головного мозга и реперфузию, мышечную слабость, гиперлипидемии, болезнь Альцгеймера и/или атеросклероз, особенно заболевания периферических сосудов и ишемию миокарда.
Дополнительно к их применению в терапевтической медицине, соединения формулы (I) и их фармацевтически приемлемые соли и сложные эфиры, которые способны к гидролизу в условиях in vivo, также полезны в качестве фармацевтических средств для развития и стандартизации тестируемых систем в условиях in vitro и in vivo для оценки действий веществ, которые повышают активность ПДГ у лабораторных животных, таких как коты, собаки, кролики, обезьяны, крысы и мыши, для поиска новых терапевтических средств.
Далее изобретение иллюстрируется следующими примерами, которые не ограничивают его объем, в которых, если специально не указано иначе:
(i) температура приведена в градусах Цельсия (°С); действия осуществляются при комнатной температуре или при температуре окружающей среды, то есть при температуре в диапазоне 18-25°С и в атмосфере инертного газа, такого как аргон;
(ii) органические растворы высушивают над безводным сульфатом магния, испарение растворителя осуществляют на роторном испарителе при пониженном давлении (600-4000 Па; 4,5-30 мм рт.ст.) при температуре бани до 60°С;
(iii) хроматография представляет собой флеш-хроматографию на силикагеле; где под картриджем Biotage подразумевают картридж, содержащий KP-SIL™ диоксид кремния, 60Å, с размером частичек 32-63 мкМ, поставляемый Biotage, отделом Dyax Corp., 1500 Avon Street Extended, Charlottesville, VA 22902, США;
(iv) в целом, ход реакций отслеживают при помощи ТСХ и время реакции приведено только с целью иллюстрации;
(v) выходы представлены только с целью иллюстрации и необязательно, что их можно получить при тщательном осуществлении способа; приготовление может повторяться, если необходимы дополнительные вещества;
(vi) данные ЯМР, если они приведены, представлены в виде дельта-значений основных определяющих протонов, представленных в виде част. на млн (ppm) относительно тетраметилсилана (ТМС) в качестве внутреннего эталона, определенных при 300 МГц (если не указано иначе) с применением обработанного дейтерием диметилсульфоксида (ДМСО-δ6) в качестве растворителя; и пики мультиплетности представлены следующим образом: s, синглет; d, дублет; dd, дублет дублетов; t, триплет; tt, тройной триплет; q, квартет; tq, тройной квартет; m, мультиплет; br, широкий;
(vii) химические символы имеют обычные значения; применяются единицы и символы CI;
(viii) соотношение растворителей представлено в виде объемных значений (об./об.);
(ix) масс-спектрометрический анализ (MS) осуществляли при энергии электронов 70 эВ методом химической ионизации (CI), используя зонд прямого облучения; где указанную ионизацию осуществляли путем ионизации электронным ударом (El), бомбардировки быстрыми атомами (FAB) или электрораспылительной ионизации (ESP); представлены значения для массы/заряда (m/z); обычно приведены только для ионов, которые указаны в исходной массе и, если не указано иначе, значения представлены в кавычках в виде (М-Н)-;
(х) использованы следующие сокращения:
(xi) если (R) или (S) стереохимия приведена в кавычках в начале наименования, то указанная стереохимия относится к -NH-С(O)-С*(Ме)(CF3)(ОН) центру, как изображено в формуле (I).
Пример 1
(R)-N-[2-Хлор-4-этилсульфонил-3-(4-метилпиперазин-1-ил)фенил]-2-гидрокси-2-метил-3,3,3-трифторпропанамид
Формальдегид (0,77 г) и триацетоксиборогидрид натрия (1,00 г) добавляли к перемешиваемому раствору (R)-N-(2-хлор-4-этилсульфонил-3-пиперазин-1-илфенил)-2-гидрокси-2-метил-3,3,3-трифторпропанамида (0.467 г; Способ 1) в 1,2-дихлорэтане (9 мл). Реакционную смесь перемешивали при температуре окружающей среды на протяжении 16 часов, затем добавляли 1М раствор NaOH (20 мл) и продукт экстрагировали с ДХМ (3×30 мл). Объединенные органические экстракты высушивали и летучее вещество удаляли путем испарения. Остаток перекристаллизовывали из EtOAc/изогексана, получая указанное в заглавии соединение (0,315 г) в виде твердого вещества. ЯМР: 1,11 (3Н, t), 1,60 (3Н, s), 2,10-2,18 (2Н, m), 2,21 (3Н, s), 2,70-2,82 (4Н, m), 3,53 (2Н, q), 3,55-3,62 (2Н, m), 7,91 (1Н, d), 8,07 (1H, brs), 8,23 (1H, d), 9,94 (1H, brs); m/z: 456.
Пример 2
(R)-N-[2-Хлор-4-этилсульфонил-3-(4-метилпиперазин-1-ил)фенил]-2-гидрокси-2-метил-3,3,3-трифторпропанамид (альтернативное получение)
1-Метилпиперазин (0,102 г) добавляли к перемешиваемому раствору (R)-N-(4-этилсульфонил-3-фтор-2-хлорфенил)-2-гидрокси-2-метил-3,3,3-трифторпропанамида (пример 15 заявки WO 01/17956; 0,096 г) в NMP (1 мл). Реакционную смесь нагревали при 130°С в течение 24 часов. Реакционную смесь охлаждали и затем добавляли насыщенный раствор хлорида аммония (100 мл). Продукт экстрагировали с диэтиловым эфиром (3×100 мл). Органические экстракты высушивали и летучее вещество удаляли путем испарения. Остаток очищали путем хроматографии на картридже Biotage (8 г диоксида кремния), элюируя с 5% метанолом/ДХМ, получая указанное в заглавии соединение (0,086 г) в виде твердого вещества. ЯМР: 1,11 (3Н, t), 1,60 (3Н, s), 2,10-2,18 (2Н, m), 2,21 (3Н, s), 2,70-2,82 (4Н, m), 3,53 (2Н, q), 3,55-3,62 (2Н, m), 7,91 (1H, d), 8,07 (1H, brs), 8,23 (1H, d), 9,94 (1H, brs); m/z: 456.
Пример 3
(R)-N-[2-Хлор-4-этилсульфонил-3-(4-мезилпиперазин-1-ил)фенил]-2-гидрокси-2-метил-3,3,3-трифторпропанамид
Триэтиламин (0,091 г) и метансульфонилхлорид (0,124 г) добавляли к перемешиваемой суспензии (R)-N-(2-хлор-4-этилсульфонил-3-пиперазин-1-илфенил)-2-гидрокси-2-метил-3,3,3-трифторпропанамида (0,401 г; Способ 1) в ДХМ (10 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 2 часов и затем добавляли насыщенный раствор хлорида аммония (20 мл) и продукт экстрагировали с ДХМ (3×30 мл). Органические экстракты высушивали и летучее вещество удаляли путем испарения. Остаток очищали путем хроматографии на картридже Biotage (8 г диоксида кремния), элюируя с 50-70% EtOAc/изогексана, получая указанное в заглавии соединение (0,215 г) в виде твердого вещества. ЯМР (CDCl3): 1,26 (3Н, t), 1,78 (3Н, s), 2,86 (3Н, s), 3,01-3,18 (4Н, m), 3,39 (2Н, q), 3,68 (1H, s), 3,75-3,87 (4Н, m), 8,01 (1H, d), 8,57 (1H, d), 9,62 (1H, brs); m/z: 520.
Пример 4
(R)-N-[2-Хлор-4-этилсульфонил-3-(4-мезилпиперазин-1-ил)фенил]-2-гидрокси-2-метил-3,3,3-трифторпропанамид (альтернативное получение)
1-Метансульфонилпиперазина (0,370 г) добавляли к перемешиваемому раствору (R)-N-(4-этилсульфонил-3-фтор-2-хлорфенил)-2-гидрокси-2-метил-3,3,3-трифторпропанамида (пример 15 заявки WO 01/17956; 0,213 г) в NMP (2 мл). Реакционную смесь нагревали при 150°С в течение 48 часов, охлаждали и затем добавляли насыщенный раствор хлорида аммония (100 мл). Продукт экстрагировали с диэтиловым эфиром (3×100 мл). Органические экстракты высушивали и летучее вещество удаляли путем испарения. Остаток очищали путем хроматографии на картридже Biotage (8 г диоксида кремния), элюируя с 50-70% EtOAc/изогексана. Продукт перекристаллизовывали из EtOAc/изогексана, получая указанное в заглавии соединение (0,167 г) в виде твердого вещества. ЯМР (CDCl3): 1,26 (3Н, t), 1,78 (3Н, s), 2,86 (3Н, s), 3,01-3,18 (4Н, m), 3,39 (2Н, q), 3.68 (1H, s), 3,75-3,87 (4H, m), 8,01 (1Н, d), 8,57 (1H, d), 9,62 (1H, brs); m/z: 520.
Исходное вещество
Способ 1
(R)-N-(2-Хлор-4-этилсульфонил-3-пиперазин-1-илфенил)-2-гидрокси-2-метил-3,3,3-трифторпропанамид
Трет-бутиловый эфир 1-пиперазинкарбоксилата (6,12 г) добавляли к перемешиваемому раствору (R)-N-(4-этилсульфонил-3-фтор-2-хлорфенил)-2-гидрокси-2-метил-3,3,3-трифторпропанамида (пример 15 заявки WO 01/17956; 4,14 г) в NMP (15 мл). Реакционную смесь нагревали при 150°С в течение 24 часов, охлаждали и затем добавляли насыщенный раствор хлорида аммония (300 мл). Продукт экстрагировали с диэтиловым эфиром (3×300 мл). Органические экстракты высушивали и летучее вещество удаляли путем испарения. Остаток очищали путем хроматографии на картридже Biotage (90 г диоксида кремния), элюируя с 70% EtOAc/изогексана. Продукт растворяли в трифторуксусной кислоте (12 мл), затем перемешивали при температуре окружающей среды в течении 30 минут. Реакционную смесь растворяли в EtOAc (200 мл), затем промывали 1 М раствором NaOH (300 мл). Органические экстракты высушивали и летучее вещество удаляли путем испарения, получая указанное в заглавии соединение (3,52 г) в виде твердого вещества. ЯМР: 1,12 (3Н, t), 1,60 (3Н, s), 2,74-2,86 (6Н, m), 3,48-3,59 (4Н, m), 7,89 (1H, d), 8,22 (1H, d); m/z: 442.
Описываются производные замещенного N-фенил 2-гидрокси-2-метил-3,3,3-трифторпропанамида формулы (I):
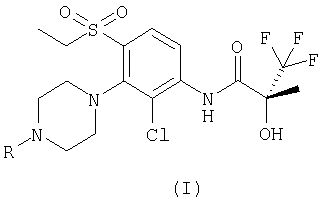
где R представляет собой метил или мезил или его соль. Также описаны способы их получения, фармацевтические композиции, их содержащие, и их применение для повышения активности ПДГ у теплокровного животного. Представленные соединения полезны для лечения сахарного диабета у теплокровных животных. 8 н. и 2 з.п. ф-лы.
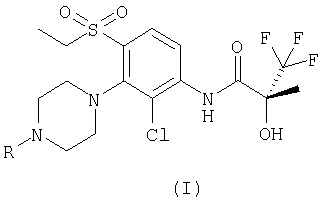
где R представляет собой метил или мезил;
или его фармацевтически приемлемая соль.
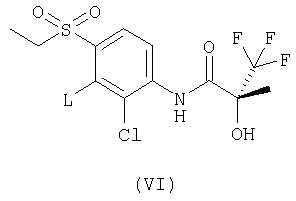
где L представляет собой вытесняемую группу, с 4-мезилпиперазином или 4-метилпиперазином, последующее разделение, при необходимости, смеси (R) и (S) энантиомеров соединений формулы (I) с получением (R)-энантиомера соединения формулы (I), и затем, при необходимости, получение фармацевтически приемлемой соли.
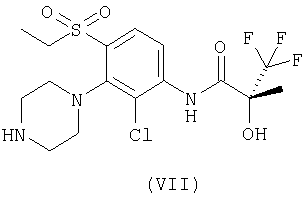
с получением соответствующих соединений формулы (I), в которых R представляет собой метил или мезил, соответственно, последующее разделение, при необходимости, смеси (R) и (S) энантиомеров соединений формулы (I) с получением (R)-энантиомера соединения формулы (I), и затем, при необходимости, получение фармацевтически приемлемой соли.
| WO 9947508 A1, 23.09.1999 | |||
| Цифровой автоматический компенсатор | 1958 |
|
SU117956A2 |
| АМИДЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СВОЙСТВАМИ ОТКРЫВАТЕЛЯ КАНАЛОВ ДЛЯ КЛЕТОЧНОГО КАЛИЯ | 1992 |
|
RU2074173C1 |

