Настоящее изобретение относится к пиримидиновым производным или к их фармацевтически приемлемым солям или к их сложным эфирам, гидролизующимся in vivo, которые обладают ингибиторной активностью по отношению к клеточному циклу, и соответственно являются пригодными для использования благодаря их активности против пролиферации клеток (например, против рака) и по этой причине являются пригодными для использования в способах лечения организма человека или животного. Настоящее изобретение также относится к способам получения указанных пиримидиновых производных, к фармацевтическим композициям, содержащим их, и к их использованию при производстве лекарственных средств, используемых для продуцирования воздействия против пролиферации клеток у теплокровного животного, такого как человек.
Семейство внутриклеточных белков, называемых циклинами, играет центральную роль в клеточном цикле. Синтез и деградация циклинов плотно контролируется, так что их уровень экспрессирования флуктуирует в течение клеточного цикла. Циклины связываются с циклин-зависимыми серин/фреонин киназами (CDK), и это связывание является самым важным для активности CDK (таких как CDK1, CDK2, CDK4 и/или CDK6) в клетке. Хотя конкретные детали относительно того, как каждый из этих факторов объединяется для регуляции активности CDK, являются малопонятными, равновесие между ними двумя диктует, будет ли клетка развиваться в пределах клеточного цикла или нет.
Последние исследования конвергенции онкогена и гена суппрессора опухоли идентифицируют вхождение в клеточный цикл в качестве ключевой точки для контроля митогенеза опухолей. Более того, CDK, видимо, находятся в цикле после некоторого количества сигнальных путей онкогена. Разрегуляция активности CDK путем положительной регуляции циклинов и/или стирания эндогенных ингибиторов, видимо, является важной осью между митогенными сигнальными путями и пролиферацией клеток опухоли.
Соответственно было обнаружено, что ингибитор клеточных циклокиназ, в особенности ингибиторы CDK2, CDK4 и/или CDK6 (которые работают в S-фазе, G1-S и G1-S фазе соответственно), должен представлять собой ценность в качестве ингибитора пролиферации клеток, такой как рост раковых клеток у млекопитающих.
Настоящее изобретение основывается на обнаружении того факта, что определенные пиримидиновые соединения неожиданно ингибируют воздействия клеточных циклокиназ, демонстрируя селективность по отношению к CDK2, CDK4 и CDK6, и, таким образом, обладают свойствами, направленными против пролиферации клеток. Такие свойства, как ожидается, представляют ценность при лечении болезненных состояний, связываемых с отклонениями в клеточных циклах и в пролиферации клеток, таких как раковые заболевания (солидные опухоли и различные виды лейкемии), фибропролиферативные и дифференциационные расстройства, псориаз, ревматоидный артрит, саркома Капоши, хемангиома,острые и хронические невропатии, атерома, атеросклероз, артериальный рестеноз, аутоиммунные заболевания, острое и хроническое воспаление, заболевания костей и заболевания глаз с пролиферацией ретинальных сосудов.
Соответственно настоящее изобретение предусматривает соединение формулы (I):
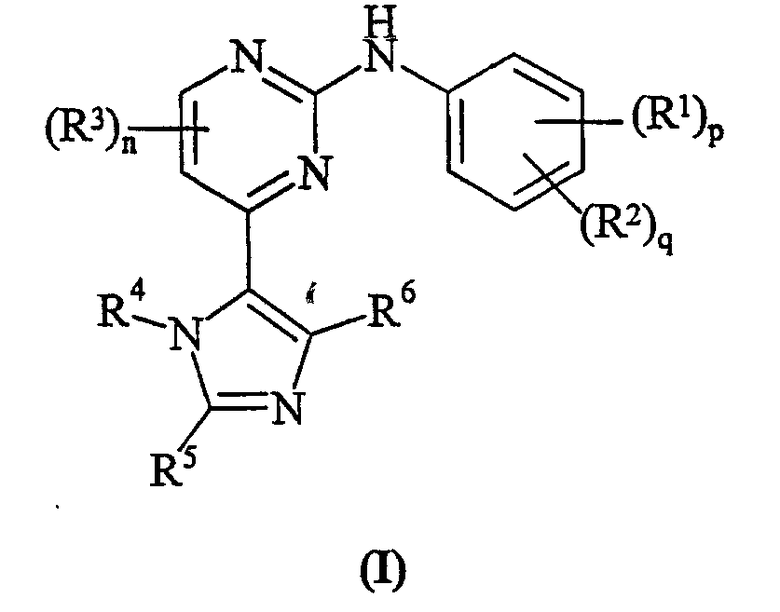
где:
R1 представляет собой галоген, нитро, циано, гидрокси, амино, карбокси, карбамоил, меркапто, C1-6алкил,C1-6алкокси, C2-6алкенил или C2-6алкинил;
p равно 0-4; где значения R1 могут быть одинаковыми или различными;
R2 представляет собой сульфамоил или группу Ra-Rb-;
q равно 0-2; где значения R2 могут быть одинаковыми или различными; и где p + q = 0-5;
R3 представляет собой галоген, нитро, циано, гидрокси, трифторметил, трифторметокси, амино, карбокси, карбамоил, меркапто, сульфамоил, C1-3алкил, C2-3алкенил, C2-3алкинил, C1-3алкокси, C1-3алканоил, N-(C1-3алкил)амино, N,N-(C1-3алкил)2амино, C1-3алканоиламино, N-(C1-3алкил)карбамоил, N,N-(C1-3алкил)2карбамоил, C1-3алкилS(O)а, где а равно 0-2, N-(C1-3алкил)сульфамоил или N,N-(C1-3алкил)2сульфамоил; где R3 может быть необязательно замещенным на атоме углерода одним или несколькими Rc;
n равно 0-2, где значения R3 могут быть одинаковыми или различными;
R4 представляет собой водород, C1-6алкил, C2-6алкенил, C2-6алкинил, C3-8циклоалкил, фенил или гетероциклическую группу, связанную с углеродом; где R4 может быть необязательно замещенным на атоме углерода одним или несколькими Rd; и где, если указанная гетероциклическая группа содержит остаток -NH-, его азот может быть необязательно замещенным группой, выбранной из Rn;
R5 и R6 являются независимо выбранными из водорода, галогена, нитро, циано, гидрокси, трифторметокси, амино, карбокси, карбамоила, меркапто, сульфамоила, C1-6алкила, C2-6алкенила, C2-6алкинила, C1-6алкокси, C1-6алканоила, C1-6алканоилокси, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, C1-6алканоиламино, N-(C1-6алкил)карбамоила, N,N-(C1-6алкил)2карбамоила, C1-6алкилS(O)а, где а равно 0-2, C1-6алкоксикарбонила, N-(C1-6алкил)сульфамоила, N,N-(C1-6алкил)2сульфамоила, C1-6алкилсульфониламино, C3-8циклоалкила или 4-7 членной насыщенной гетероциклической группы; где R5 и R6 независимо друг от друга могут быть необязательно замещенными на атоме углерода одним или несколькими Re; и где, если указанная 4-7 членная насыщенная гетероциклическая группа содержит остаток -NH-, его азот может быть необязательно замещенным группой, выбранной из Rf;
Ra является выбранным из C1-6алкила, C2-6алкенила, C2-6алкинила, C3-8циклоалкила, C3-8циклоалкилС1-6алкила, фенила, гетероциклической группы, фенилС1-6алкила или (гетероциклическая группа)C1-6алкила; где Ra может быть необязательно замещенным на атоме углерода одним или несколькими Rg; и где, если указанная гетероциклическая группа содержит остаток -NH-, его азот может быть необязательно замещенным группой, выбранной из Rh;
Rb представляет собой -C(O)-, -N(Rm)C(O)-, -C(O)N(Rm)-, -S(O)r-, -OC(O)N(Rm)SO2-, -SO2N(Rm)- или -N(Rm)SO2-; где Rm представляет собой водород или C1-6алкил, необязательно замещенный одним или несколькими Ri, и r равно 1-2;
Rd, Rg и Ri являются независимо выбранными из галогена, нитро, циано, гидрокси, амино, карбокси, карбамоила, меркапто, сульфамоила, C1-6алкила, C2-6алкенила, C2-6алкинила, C1-6алкокси, C1-6алкоксиС1-6алкокси, C1-6алкоксиС1-6алкоксиС1-6алкокси, C1-6алканоила, C1-6алканоилокси, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, C1-6алканоиламино, N-(C1-6алкил)карбамоила, N,N-(C1-6алкил)2карбамоила, C1-6алкилS(O)а, где а равно 0-2, C1-6алкоксикарбонила, N-(C1-6алкил)сульфамоила, N,N-(C1-6алкил)2сульфамоила, C1-6алкилсульфониламино, C3-8циклоалкила, фенила, гетероциклической группы, фенилС1-6алкил-R°-, (гетероциклическая группа)C1-6алкил-R°-, фенил-R°- или (гетероциклическая группа)-R°-; где Rd, Rg и Ri независимо друг от друга могут быть необязательно замещенными на атоме углерода одним или несколькими Rj; и где, если указанная гетероциклическая группа содержит остаток -NH-, его азот может быть необязательно замещенным группой, выбранной из Rk;
Rо представляет собой -O-, -N(Rp)-, -C(O)-, -N(Rp)C(O)-, -C(O)N(Rp)-, -S(O)S-, -SO2N(Rp)- или -N(RP)SO2-; где Rp представляет собой водород или C1-6алкил, и s равно 0-2;
Rf, Rh, Rk и Rn являются независимо выбранными из C1-4алкила, C1-4алканоила, C1-4алкилсульфонила, C1-4алкоксикарбонила, карбамоила, N-(C1-4алкил)карбамоила, N,N-(C1-4алкил)карбамоила, бензила, бензилоксикарбонила, бензоила и фенилсульфонила; где Rf, Rh, Rk и Rn, независимо друг от друга, могут быть необязательно замещенными на атоме углерода одним или несколькими Rl; и
Rc, Re, Rl и Rj являются независимо выбранными из галогена, нитро, циано, гидрокси, трифторметокси, трифторметила, амино, карбокси, карбамоила, меркапто, сульфамоила, метила, этила, метокси, этокси, ацетила, ацетокси, метиламино, этиламино, диметиламино, диэтиламино, N-метил-N-этиламино, ацетиламино, N-метилкарбамоила, N-этилкарбамоила, N,N-диметилкарбамоила, N,N-диэтилкарбамоила, N-метил-N-этилкарбамоила, метилтио, этилтио, метилсульфинила, этилсульфинила, мезила, этилсульфонила, метоксикарбонила, этоксикарбонила, N-метилсульфамоила, N-этилсульфамоила, N,N-диметилсульфамоила, N,N-диэтилсульфамоила или N-метил-N-этилсульфамоила; или их фармацевтически приемлемую соль или их сложный эфир, гидролизующийся in vivo.
В другом аспекте настоящего изобретения предусматривается соединение формулы (I):
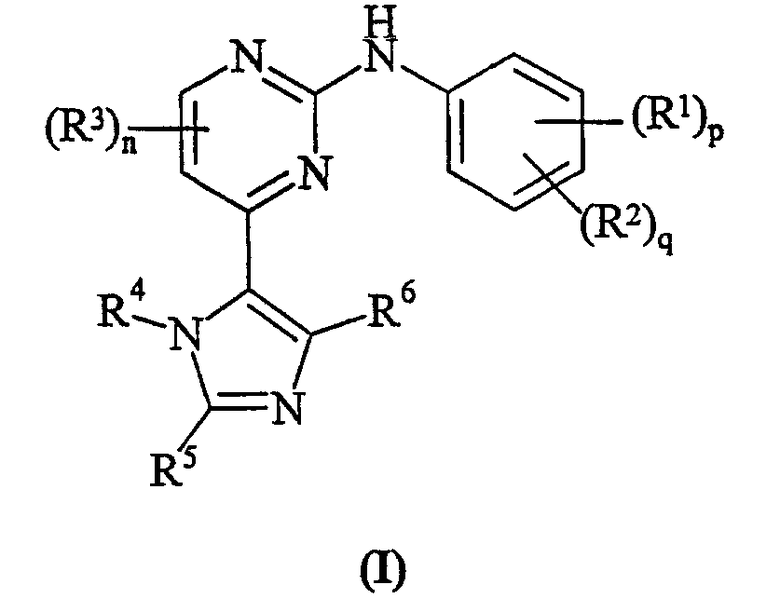
где:
R1 представляет собой галоген, нитро, циано, гидрокси, амино, карбокси, карбамоил, меркапто, C1-6алкил, C1-6алкокси, C2-6алкенил или C2-6алкинил;
p равно 0-4; где значения R1 могут быть одинаковыми или различными;
R2 представляет собой сульфамоил или группу Ra-Rb-;
q равно 0-2; где значения R2 могут быть одинаковыми или различными; и где p + q = 0-5;
R3 представляет собой галоген, нитро, циано, гидрокси, трифторметил, трифторметокси, амино, карбокси, карбамоил, меркапто, сульфамоил, C1-3алкил, C2-3алкенил, C2-3алкинил, C1-3алкокси, C1-3алканоил, N-(C1-3алкил)амино, N,N-(C1-3алкил)2амино, C1-3алканоиламино, N-(C1-3алкил)карбамоил, N,N-(C1-3алкил)2карбамоил, C1-3алкилS(O)а, где а равно 0-2, N-(C1-3алкил)сульфамоил или N,N-(C1-3алкил)2сульфамоил; где R3 может быть необязательно замещенным на атоме углерода одним или несколькими Rc;
n равно 0-2, где значения R3 могут быть одинаковыми или различными;
R4 представляет собой водород, C1-6алкил, C2-6алкенил, C2-6алкинил, C3-8циклоалкил, фенил или гетероциклическую группу, связанную с углеродом; где R4 может быть необязательно замещенным на атоме углерода одним или несколькими Rd; и где, если указанная гетероциклическая группа содержит остаток -NH-, его азот может быть необязательно замещенным группой, выбранной из Rn;
R5 и R6 являются независимо выбранными из водорода, галогена, нитро, циано, гидрокси, трифторметокси, амино, карбокси, карбамоила, меркапто, сульфамоила, C1-6алкила, C2-6алкенила, C2-6алкинила, C1-6алкокси, C1-6алканоила, C1-6алканоилокси, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, C1-6алканоиламино, N-(C1-6алкил)карбамоила, N,N-(C1-6алкил)2карбамоила, C1-6алкилS(O)а, где а равно 0-2, C1-6алкоксикарбонила, N-(C1-6алкил)сульфамоила, N,N-(C1-6алкил)2сульфамоила, C1-6алкилсульфониламино, C3-8циклоалкила или 4-7-членной насыщенной гетероциклической группы; где R5 и R6 независимо друг от друга могут быть необязательно замещенными на атоме углерода одним или несколькими Re; и где, если указанная 4-7-членная насыщенная гетероциклическая группа содержит остаток -NH-, его азот может быть необязательно замещенным группой, выбранной из Rf;
Ra является выбранным из C1-6алкила, C2-6алкенила, C2-6алкинила, C3-8циклоалкила, C3-8циклоалкилС1-6алкила, фенила, гетероциклической группы, фенилС1-6алкила или (гетероциклическая группа)C1-6алкила; где Ra может быть необязательно замещенным на атоме углерода одним или несколькими Rg; и где, если указанная гетероциклическая группа содержит остаток -NH-, его азот может быть необязательно замещенным группой, выбранной из Rh;
Rb представляет собой -C(O)-, -N(Rm)C(O)-, -C(O)N(Rm)-, -S(O)r-, -SO2N(Rm)- или -N(Rm)SO2-;
где Rm представляет собой водород или C1-6алкил, необязательно замещенный одним или несколькими Ri, и r равно 1-2;
Rd, Rg и Ri являются независимо выбранными из галогена, нитро, циано, гидрокси, амино, карбокси, карбамоила, меркапто, сульфамоила, C1-6алкила, C2-6алкенила, C2-6алкинила, C1-6алкокси, C1-6алканоила, C1-6алканоилокси, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, C1-6алканоиламино, N-(C1-6алкил)карбамоила, N,N-(C1-6алкил)2карбамоила, C1-6алкилS(O)а, где а равно 0-2, C1-6алкоксикарбонила, N-(C1-6алкил)сульфамоила, N,N-(C1-6алкил)2сульфамоила, C1-6алкилсульфониламино, C3-8циклоалкила, фенила, гетероциклической группы, фенил-R°- или (гетероциклическая группа)-R°-; где Rd, Rg и Ri независимо друг от друга могут быть необязательно замещенными на атоме углерода одним или несколькими Rj; и где, если указанная гетероциклическая группа содержит остаток -NH-, его азот может быть необязательно замещенным группой, выбранной из Rk;
Rо представляет собой -O-, -N(Rp)-, -C(O)-, -N(Rp)C(O)-, -C(O)N(Rp)-, -S(O)s-, -SO2N(Rp)- или -N(Rp)SO2-; где Rp представляет собой водород или C1-6алкил, и s равно 0-2;
Rf, Rh, Rk и Rn являются независимо выбранными из C1-4алкила, C1-4алканоила, C1-4алкилсульфонила, C1-4алкоксикарбонила, карбамоила, N-(C1-4алкил)карбамоила, N,N-(C1-4алкил)карбамоила, бензила, бензилоксикарбонила, бензоила и фенилсульфонила; где Rf, Rh и Rk независимо друг от друга могут быть необязательно замещенными на атоме углерода одним или несколькими Rl; и
Rc, Re, Ri и Rj являются независимо выбранными из галогена, нитро, циано, гидрокси, трифторметокси, трифторметила, амино, карбокси, карбамоила, меркапто, сульфамоила, метила, этила, метокси, этокси, ацетила, ацетокси, метиламино, этиламино, диметиламино, диэтиламино, N-метил-N-этиламино, ацетиламино, N-метилкарбамоила, N-этилкарбамоила, N,N-диметилкарбамоила, N,N-диэтилкарбамоила, N-метил-N-этилкарбамоила, метилтио, этилтио, метилсульфинила, этилсульфинила, мезила, этилсульфонила, метоксикарбонила, этоксикарбонила, N-метилсульфамоила, N-этилсульфамоила, N,N-диметилсульфамоила, N,N-диэтилсульфамоила или N-метил-N-этилсульфамоила;
или их фармацевтически приемлемую соль, или их сложный эфир, гидролизующийся in vivo.
В настоящем описании термин "алкил" включает в себя алкильные группы как с прямой, так и с разветвленной цепью, но ссылки на индивидуальные алкильные группы, такие как "пропил", указывают только на вариант с прямой цепью. Например, "C1-6алкил" включает в себя C1-4алкил, C1-3алкил, пропил, изопропил и трет-бутил. Однако ссылки на индивидуальные алкильные группы, такие как "пропил", указывают только на вариант с прямой цепью, а ссылки на индивидуальные алкильные группы с разветвленной цепью, такие как "изопропил", указывают только на вариант с разветвленной цепью. Подобное же условие применяется к другим радикалам, например "фенилС1-6алкил" включает в себя фенилС1-4алкил, бензил, 1-фенилэтил и 2-фенилэтил. Термин "галоген" относится к фтору, хлору, брому и йоду.
Там, где необязательные заместители выбираются из "одной или нескольких" групп, необходимо понять, что это определение включает в себя все заместители, выбираемые только из одной из указанных групп или заместители, выбираемые из двух или из нескольких указанных групп.
"Гетероциклическая группа" представляет собой насыщенное, частично насыщенное или ненасыщенное, моно- или бициклическое кольцо, содержащее 4-12 атомов, из которых, по меньшей мере, один атом выбирается из азота, серы или кислорода, который, если не указано иного, может быть связан с атомами углерода или азота, где группа -CH2- может необязательно быть заменена -C(O)-, кольцевой атом азота может необязательно нести на себе C1-6алкильную группу и образовывать четвертичное соединение, или кольцевой атом азота и/или серы может необязательно быть окислен, с образованием N-оксидов и/или S-оксидов. Примеры и соответствующие значения термина "гетероциклическая группа" представляют собой морфолино, пиперидил, пиридил, пиранил, пирролил, изотиазолил, индолил, хинолил, тиенил, 1,3-бензодиоксолил, тиадиазолил, пиперазинил, тиазолидинил, пирролидинил, тиоморфолино, пирролинил, гомопиперазинил, 3,5-диоксапиперидинил, тетрагидропиранил, имидазолил, пиримидил, пиразинил, пиридазинил, изоксазолил, N-метилпирролил, 4-пиридон, 1-изохинолон, 2-пирролидон, 4-тиазолидон, пиридин-N-оксид и хинолин-N-оксид. Предпочтительно "гетероциклическая группа" представляет собой насыщенное, частично насыщенное или ненасыщенное, моно- или бициклическое кольцо, содержащее 5 или 6 атомов, из которых, по меньшей мере, один атом выбирается из азота, серы или кислорода, он может, если не указано иного, быть связанным с атомами углерода или азота, группа -CH2- может необязательно быть заменена -C(O)-, и кольцевой атом серы может быть необязательно окислен с образованием S-оксидов. Более предпочтительно "гетероциклическая группа" представляет собой тетрагидрофурил, пиридил, пирролидинонил, морфолино, имидазолил, пиперидинил или пирролидинил. В особенности, "гетероциклическая группа" представляет собой тетрагидрофурил или морфолино. В другом аспекте настоящего изобретения, в особенности, "гетероциклическая группа" представляет собой тетрагидрофуран-2-ил, 2-оксопирролидин-1-ил, фуран-2-ил, оксазолил, морфолино, пиперидинил, тиазолил, пиразинил, изоксазолил, тетрагидропиран, пиридил, изоксазолил, изотиазолил, 1,2,5-тиадиазолил, фталимидо.
"4-7-Членная насыщенная гетероциклическая группа" представляет собой насыщенное моноциклическое кольцо, содержащее 4-7 атомов, из которых, по меньшей мере, один атом выбирается из азота, серы или кислорода, который может, если не указано иного, быть связан с атомами углерода или азота, где группа -CH2- может необязательно быть заменена -C(O)-, и атом серы может быть необязательно окислен с образованием S-оксидов. Примеры и соответствующие значения термина "гетероциклическая группа" представляют собой морфолино, пиперидил, 1,4-диоксанил, 1,3-диоксоланил, 1,2-оксатиоланил, имидазолидинил, пиразолидинил, пиперазинил, тиазолидинил, пирролидинил, тиоморфолино, гомопиперазинил и тетрагидропиранил.
Пример "C1-6алканоилокси" представляет собой ацетокси. Примеры "C1-6алкоксикарбонила" включают в себя C1-4алкоксикарбонил, метоксикарбонил, этоксикарбонил, н- и трет-бутоксикарбонил. Примеры "C1-6алкокси" включают в себя C1-4алкокси, C1-3алкокси, метокси, этокси и пропокси. Примеры "C1-6алканоиламино" включают в себя формамидо, ацетамидо и пропиониламино. Примеры "C1-6алкилS(O)а, где а равно 0-2" включают в себя C1-6алкилсульфонил, метилтио, этилтио, метилсульфинил, этилсульфинил, мезил и этилсульфонил. Примеры "C1-6алкилS(O)r, где r равно 1-2" включают в себя метилсульфинил, этилсульфинил, мезил и этилсульфонил. Примеры "C1-6алканоила" включают в себя C1-6алканоил, пропионил и ацетил. Примеры "N-C1-6алкиламино" включают в себя метиламино и этиламино. Примеры "N,N-(C1-6алкил)2амино" включают ди-N-метиламино, ди-(N-этил)амино и N-этил-N-метиламино. Примеры "C2-6алкенила" представляют собой винил, аллил и 1-пропенил. Примеры "C2-6алкинила" представляют собой этинил, 1-пропинил и 2-пропинил. Примеры "N-(C1-6алкил)сульфамоила" представляют собой N-(метил)сульфамоил и N-(этил)сульфамоил. Примеры "N-(C1-6алкил)2сульфамоила" представляют собой N,N-(диметил)сульфамоил и N-(метил)-N-(этил)сульфамоил. Примеры "N-(C1-6алкил)карбамоила" представляют собой N-(C1-4алкил)карбамоил, метиламинокарбонил и этиламинокарбонил. Примеры "N,N-(C1-6алкил)2карбамоила" представляют собой N,N(C1-4алкил)2карбамоил, диметиламинокарбонил и метилэтиламинокарбонил. Примеры "C3-8циклоалкила" представляют собой циклопропил, циклобутил, циклопропил и циклогексил. Примеры "(гетероциклическая группа)C1-6алкила" включают в себя пиридилметил, 3-морфолинопропил и 2-пиримид-2-илэтил. Примеры "C3-8циклоалкилС1-6алкила" представляют собой циклопропилэтил, циклобутилметил, 2-циклопропилпропил и циклогексилэтил.
Пригодная для использования фармацевтически приемлемая соль соединения по настоящему изобретению представляет собой, например, кислотно-аддитивную соль соединения по настоящему изобретению, которое является достаточно основным, например, кислотно-аддитивную соль, например, неорганической или органической кислоты, например, хлористоводородной, бромистоводородной, серной, фосфорной, трифторуксусной, лимонной или малеиновой кислоты. Кроме того, пригодная для использования фармацевтически приемлемая соль соединения по настоящему изобретению, которое является достаточно кислотным, представляет собой соль щелочного металла, например, соль натрия или калия, соль щелочноземельного металла, например, соль кальция или магния, соль аммония или соль органического основания, которое дает физиологически приемлемый катион, например, соль метиламина, диметиламина, триметиламина, пиперидина, морфолина или трис-(2-гидроксиэтил)амина.
Сложный эфир, гидролизующийся in vivo, соединения формулы (I), содержащего карбокси или гидроксигруппу, представляет собой, например, фармацевтически приемлемый сложный эфир, который гидролизуется в организме человека или животного с получением исходной кислоты или спирта. Пригодные для использования фармацевтически приемлемые сложные эфиры для карбокси включают в себя C1-6алкоксиметиловые сложные эфиры, например, метоксиметиловый, C1-6алканоилоксиметиловые сложные эфиры, например, пивалоилоксиметиловый, фталидиловые сложные эфиры, C3-8циклоалкоксикарбонилоксиС1-6алкиловые сложные эфиры, например, 1-циклогексилкарбонилоксиэтиловый; 1,3-диоксолен-2-онилметиловые сложные эфиры, например, 5-метил-1,3-диоксолен-2-онилметиловый; и C1-6алкоксикарбонилоксиэтиловые сложные эфиры, например, 1-метоксикарбонилоксиэтиловый, и могут быть образованы на любой карбоксигруппе в соединениях по настоящему изобретению.
Сложный эфир, гидролизующийся in vivo, соединения формулы (I), содержащего гидроксигруппу, включает в себя неорганические сложные эфиры, такие как фосфатные сложные эфиры, и α-ацилоксиалкиловые простые эфиры, и родственные соединения, которые в результате гидролиза сложного эфира in vivo распадаются с получением исходной гидроксигруппы. Примеры α-ацилоксиалкиловых простых эфиров включают в себя ацетоксиметокси и 2,2-диметилпропионилоксиметокси. Выбор групп, образующих сложные эфиры, гидролизующиеся in vivo, для гидрокси, включает в себя алканоил, бензоил, фенилацетил, и замещенный бензоил и фенилацетил, алкоксикарбонил (с получением алкилкарбонатных сложных эфиров), диалкилкарбамоил и N-(диалкиламиноэтил)-N-алкилкарбамоил (с получением карбаматов), диалкиламиноацетил и карбоксиацетил. Примеры заместителей на бензоиле включают в себя морфолино и пиперазино, связанные с кольцевым атомом азота через метиленовую группу в 3- или 4-положении бензоильного кольца.
Некоторые соединения формулы (I) могут иметь хиральные центры и/или геометрические изомерные центры (E-и Z-изомеры), и необходимо понять, что настоящее изобретение охватывает все такие оптические изомеры, диастереоизомеры и геометрические изомеры, которые обладают ингибиторной активностью по отношению к CDK.
Настоящее изобретение относится к любой таутомерной форме соединений формулы (I), которые обладают ингибиторной активностью по отношению к CDK, и ко всем им вместе. В частности, специалист в данной области заметит, что там, где R4 представляет собой водород, имидазольное кольцо, изображенное на формуле (I), может таутомеризоваться.
Необходимо также понять, что определенные соединения формулы (I), могут существовать как в сольватированной, так и в несольватированной формах, например, в таких формах, как гидратированные формы. Необходимо понять, что настоящее изобретение охватывает все сольватированные формы, которые обладают ингибиторной активностью по отношению к CDK.
Предпочтительные значения R1, R2, R3, R4, R5, R6, n, p и q являются следующими. Такие значения могут использоваться там, где это необходимо, вместе с любым из определений, пунктов формулы изобретения или воплощений, определенных выше или ниже.
Предпочтительно R1 представляет собой галоген, амино, C1-6алкил или C1-6алкокси.
Более предпочтительно R1 представляет собой галоген, C1-4алкил или C1-4алкокси.
Конкретно R1 представляет собой хлор, C1-3алкил или C1-3алкокси.
Более конкретно R1 представляет собой хлор.
В другом аспекте настоящего изобретения, R1 предпочтительно представляет собой галоген, амино, C1-6алкил или C1-6алкокси.
В другом аспекте настоящего изобретения, R1 более предпочтительно представляет собой хлор, амино, метил или метокси.
Предпочтительно p равно 0-2; где значения R1 могут быть одинаковыми или различными.
Более предпочтительно p равно 0 или 1.
В одном из аспектов настоящего изобретения предпочтительно p равно 0.
В другом аспекте настоящего изобретения предпочтительно p равно 1.
Предпочтительно когда p равно 1, R1 представляет собой группу, находящуюся в мета- или пара-положении по отношению к -NH-группе анилина формулы (I).
Более предпочтительно когда p равно 1, R1 находится в мета-положении по отношению к -NH-группе анилина формулы (I).
Предпочтительно R2 представляет собой сульфамоил или группу Ra-Rb-; где
Ra является выбранным из C1-6алкила, C3-8циклоалкила, C3-8циклоалкилС1-6алкила, фенила, гетероциклической группы, фенилС1-6алкила или (гетероциклическая группа)C1-6алкила; где Ra может быть необязательно замещенным на атоме углерода одним или несколькими Rg;
Rb представляет собой -N(Rm)C(O)-, -C(O)N(Rm)-, -SO2N(Rm)-или -N(Rm)SO2-; где Rm представляет собой водород;
Rg является выбранным из галогена, гидрокси, амино, карбамоила, C1-6алкила или C1-6алкокси; и
Rj является выбранным из галогена или гидрокси.
Более предпочтительно R2 представляет собой сульфамоил или группу Ra-Rb-; где
Ra является выбранным из C1-6алкила, C3-8циклоалкилС1-6алкила, фенилС1-6алкила или (гетероциклическая группа)C1-6алкила; где Ra может быть необязательно замещенным на атоме углерода одним или несколькими Rg;
Rb представляет собой -N(Rm)SO2-; где Rm представляет собой водород;
Rg является выбранным из галогена, гидрокси, карбамоила или C1-6алкокси; и
Rj является выбранным из гидрокси.
Конкретно R2 представляет собой сульфамоил, N-(тетрагидрофуран-2-илметил)сульфамоил, N-[3-(2-оксопирролидин-1-ил)пропил]сульфамоил, N-(3-метоксипропил)сульфамоил, N-(4-фторбензил)сульфамоил, N-(циклопропилметил)сульфамоил, N-пропилсульфамоил, N-(2,3-дигидроксипропил)сульфамоил, N-[2-(2-гидроксиэтокси)этил]сульфамоил, N-(фуран-2-илметил)сульфамоил, N-(2-гидроксиэтил)сульфамоил или N-(карбамоилметил)сульфамоил.
В другом аспекте настоящего изобретения R2 предпочтительно представляет собой сульфамоил или группу Ra-Rb-; где
Ra является выбраннымиз C1-6алкила, C2-6алкенила, C2-6алкинила, C3-8циклоалкила, фенила или гетероциклической группы; где Ra может быть необязательно замещенным на атоме углерода одним или несколькими Rg;
Rb представляет собой -N(Rm)C(O)-, -C(O)N(Rm)-, -S(O)r, -OC(O)N(Rm)SO2-, -SO2N(Rm)- или -N(Rm)SO2-; где Rm представляет собой водород или C1-6алкил, и r равно 2;
Rg является выбранным из галогена, гидрокси, амино, циано, карбамоила, C1-6алкила, C1-6алкокси, C1-6алкоксиС1-6алкокси, C1-6алкоксиС1-6алкоксиС1-6алкокси, N,N-(C1-6алкил)2амино, C1-6алкилS(O)а, где а равно 2, C3-8циклоалкила, фенила, гетероциклической группы, фенилС1-6алкил-Rо- или (гетероциклическая группа)-Rо-; где Rg может быть необязательно замещенным на атоме углерода одним или несколькими Rj;
Rо представляет собой -O-; и
Rj является выбранным из галогена, гидрокси, метила или метокси.
В другом аспекте настоящего изобретения R2 более предпочтительно представляет собой сульфамоил или группу Ra-Rb-; где
Ra является выбранным из метила, этила, пропила, трет-бутила, пентила, 1,1-диметилпропила, 2,2-диметилпропила, аллила, 2-пропинила, циклопропила, циклобутила, фенила или оксазолила; где Ra может быть необязательно замещенным на атоме углерода одним или несколькими Rg;
Rb представляет собой -N(Rm)C(O)-, -C(O)N(Rm)-, -S(O)2-, -OC(O)N(Rm)SO2-, -SO2N(Rm)- или -N(Rm)SO2-; где Rm представляет собой водород или метил;
Rg является выбранным из фтора, гидрокси, амино, циано, карбамоила, метила, метокси, этокси, изопропокси, этоксиэтокси, этоксиэтоксиэтокси, N,N-диметиламино, мезила, циклопропила, фенила, тетрагидрофуранила, 2-оксопирролидинила, 1,3-диоксоланила, морфолино, пиперидинила, фурана, тиазолила, пиразинила, изоксазолила, тетрагидропирана, пиридила, бензилокси, изоксазолилокси, изотиазолилокси, 1,2,5-тиадиазолилокси, где Rg может быть необязательно замещенным на атоме углерода одним или несколькими Rj; и
Rj является выбранным из фтора, гидрокси, метила или метокси.
В другом аспекте настоящего изобретения конкретно R2 представляет собой сульфамоил, N-(трет-бутоксикарбонил)сульфамоил, N-(тетрагидрофур-2-илметил)сульфамоил, N-(циклопропилметил)сульфамоил, N-(фур-2-илметил)сульфамоил, N-(цианометил)сульфамоил, N-(2,2-диметил-1,3-диоксолан-4-илметил)сульфамоил, N-(карбамоилметил)сульфамоил, N-метилсульфамоил, N-(4-фторбензил)сульфамоил, N-(пиридин-2-илметил)сульфамоил, N-(пиридин-3-илметил)сульфамоил, N-(4-метилтиазол-2-ил)сульфамоил, N-(3-метилизоксазол-5-илметил)сульфамоил, N-(тетрагидропиран-2-илметил)сульфамоил, N-(2-метилпиразин-5-ил)сульфамоил, N-[2-(2-гидроксиэтокси)этил]сульфамоил, N-(2-гидроксиэтил)сульфамоил, N-(2,2,2-трифторэтил)сульфамоил, N-(2-метоксиэтил)сульфамоил, N-(2-мезилэтил)сульфамоил, N-(2-бензилоксиэтил)сульфамоил, N-(2,2-диметоксиэтил)сульфамоил, N-[2-(N,N-диметиламино)этил]сульфамоил, N-(2-пиперидин-1-илэтил)сульфамоил, N-[2-(метоксиметокси)этил]сульфамоил, N-этилсульфамоил, N-[2-(2-метоксиэтокси)этил]сульфамоил, N-{2-[2-(2-метоксиэтокси)этокси]этил}сульфамоил, N-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}этил)сульфамоил, N-(2-пиридин-2-илэтил)сульфамоил, N-(2-пиридин-4-илэтил)сульфамоил, N-(2-изоксазол-3-илоксиэтил)сульфамоил, N-(2-изотиазол-3-илоксиэтил)сульфамоил, N-(2-1,2,5-тиадиазол-3-илоксиэтил)сульфамоил, N-метил-N-(2-метоксиэтил)сульфамоил, N-[3-(2-оксопирролидин-1ил)пропил]сульфамоил, N-(3-метоксипропил)сульфамоил, N-пропилсульфамоил, N-(2,3-дигидроксипропил)сульфамоил, N-(3-морфолинопропил)сульфамоил, N-[3-(N,N-диметиламино)пропил]сульфамоил, N-(3,3,3-трифторпропил)сульфамоил, N-(2,2-диметил-3-гидроксипропил)сульфамоил, N-(3-гидроксипропил)сульфамоил, N-(3-этоксипропил)сульфамоил, N-(2-гидроксипропил)сульфамоил, N-(3-изопропоксипропил)сульфамоил, N-(3-изопропокси-2-гидроксипропил)сульфамоил, N-(3-изоксазол-3-илоксипропил)сульфамоил, N-(3-изотиазол-3-илоксипропил)сульфамоил, N-(3-1,2,5-тиадиазол-3-илоксипропил)сульфамоил, N-(1,1-диметилпропил)сульфамоил, N-метил-N-(3-морфолинопропил)сульфамоил, N-бутилсульфамоил, N-трет-бутилсульфамоил, N-(2-гидроксибутил)сульфамоил, N-метил-N-трет-бутилсульфамоил, N-пентилсульфамоил, N-(5-гидроксипентил)сульфамоил, N-(4,5-диметилоксазол-2-ил)сульфамоил, N-(циклопропил)сульфамоил, N-(циклобутил)сульфамоил, N-(3-трифторметилфенил)сульфамоил, N-аллилсульфамоил, N-(2-пропинил)сульфамоил, N-метилкарбамоил, ацетамидо, мезиламино или мезил.
В другом аспекте настоящего изобретения более конкретно R2 представляет собой N-(циклопропилметил)сульфамоил, N-(2,2,2-трифторэтил)сульфамоил, N-(2-метоксиэтил)сульфамоил, N-(3-метоксипропил)сульфамоил, N-(циклопропил)сульфамоил или N-(циклобутил)сульфамоил.
Предпочтительно q равно 0 или 1.
В одном из аспектов настоящего изобретения предпочтительно q равно 0.
В другом аспекте настоящего изобретения предпочтительно q равно 1.
Предпочтительно, когда q равно 1, R2 находится в мета- или пара-положении по отношению к -NH-группе анилина формулы (I).
Более предпочтительно когда q равно 1, R2 находится в пара-положении по отношению к -NH-группе анилина формулы (I).
Предпочтительно p + q = 0-3.
Более предпочтительно p + q равно 0-2.
Конкретно p + q равно 0 или 1.
В одном из аспектов настоящего изобретения предпочтительно p + q равно 0.
В другом аспекте настоящего изобретения предпочтительно p + q равно 1.
Предпочтительно R3 представляет собой галоген.
Более предпочтительно R3 представляет собой бром.
В другом аспекте настоящего изобретения предпочтительно R3 представляет собой бром или хлор.
Предпочтительно n равно 0 или 1.
В одном из аспектов настоящего изобретения более предпочтительно n равно 0.
В другом аспекте настоящего изобретения более предпочтительно n равно 1.
Предпочтительно, когда n равно 1, R3 находится в 5-положении пиримидинового кольца.
Предпочтительно R4 представляет собой водород, C1-6алкил, C2-6алкенил, C2-6алкинил; где R4 может быть необязательно замещенным на атоме углерода одним или несколькими Rd; где Rd является таким, как определено здесь ниже.
Более предпочтительно R4 представляет собой водород или C1-6алкил; где R4 может быть необязательно замещенным на атоме углерода одним или несколькими Rd;
Rd является выбранным из амино, C1-6алкокси, C1-6алканоиламино, C1-6алкилсульфониламино, фенила, гетероциклической группы, или (гетероциклическая группа)-R°-; где Rd может быть необязательно замещенным на атоме углерода одним или несколькими Rj;
R° представляет собой -C(O)N(Rp)-; где Rp представляет собой водород; и
Rj представляет собой галоген.
Конкретно R4 представляет собой водород или C1-6алкил; где R4 может быть необязательно замещенным на атоме углерода одним или несколькими Rd;
Rd является выбранным из амино, C1-6алкокси, фенила или гетероциклической группы.
Более конкретно R4 представляет собой водород, метил, этил, бензил, 2-фталимидоэтил, 2-аминоэтил или 2-метоксиэтил.
Конкретно предпочтительный R4 представляет собой метил или этил.
В другом аспекте настоящего изобретения предпочтительно R4 представляет собой водород, C1-6алкил или C2-6алкенил; где R4 может быть необязательно замещенным на атоме углерода одним или несколькими Rd; где
Rd является выбранным из галогена, амино, C1-6алкокси, C1-6алканоиламино, C1-6алкилсульфониламино, фенила или гетероциклической группы.
В другом аспекте настоящего изобретения более предпочтительно R4 представляет собой водород, метил, этил, изопропил или 3-бутенил; где R4 может быть необязательно замещенным на атоме углерода одним или несколькими Rd; где
Rd является выбранным из фтора, амино, метокси, ацетамидо, мезиламино, фенила или фталимидо.
В другом аспекте настоящего изобретения конкретно R4 представляет собой водород, метил, этил, изопропил, 3-бутенил, бензил, 2-фталимидоэтил, 2-аминоэтил, 2-метоксиэтил, 2-ацетамидоэтил, 2-мезиламиноэтил или 2,2,2-трифторэтил.
В другом аспекте настоящего изобретения более конкретно R4 представляет собой метил, этил или изопропил.
Предпочтительно R5 и R6 являются независимо выбранными из водорода или C1-6алкила.
Более предпочтительно R5 и R6 являются независимо выбранными из водорода или метила.
Конкретно R5 является выбранным из водорода или метила, и R6 представляет собой водород.
В другом аспекте настоящего изобретения предпочтительно R5 и R6 являются независимо выбранными из водорода или C1-6алкила; где R5 и R6 независимо друг от друга могут быть необязательно замещенными на атоме углерода одним или несколькими Re; где
Re является выбранным из галогена или метокси.
В другом аспекте настоящего изобретения более предпочтительно R5 и R6 являются независимо выбранными из водорода, метила, этила или изопропила; где R5 и R6 независимо друг от друга могут быть необязательно замещенными на атоме углерода одним или несколькими Re; где
Re является выбранным из фтора или метокси.
В другом аспекте настоящего изобретения более предпочтительно R5 и R6 являются независимо выбранными из водорода, метила, этила, изопропила, трифторметила или метоксиметила.
В другом аспекте настоящего изобретения более предпочтительно R5 представляет собой метил или изопропил, и R6 представляет собой водород.
По этой причине в другом аспекте настоящего изобретения, предусматривается соединение формулы (I) (как показано выше) где:
R1 представляет собой хлор;
p равно 0 или 1;
R2 представляет собой сульфамоил или группу Ra-Rb-;
Ra является выбранным из C1-6алкила, C3-8циклоалкилС1-6алкила, фенилС1-6алкила или (гетероциклическая группа)C1-6алкила; где Ra может быть необязательно замещенным на атоме углерода одним или несколькими Rg;
Rb представляет собой -N(Rm)SO2; где Rm представляет собой водород;
Rg является выбранным из галогена, гидрокси, карбамоила или C1-6алкокси;
Rj является выбранным из гидрокси;
q равно 0 или 1;
p + q равно 0 или 1;
n равно 0;
R4 представляет собой водород или C1-6алкил; где R4 может быть необязательно замещенным на атоме углерода одним или несколькими Rd;
Rd является выбранным из амино, C1-6алкокси, фенила или гетероциклической группы; и
R5 и R6 являются независимо выбранными из водорода или C1-6алкила; или их фармацевтически приемлемая соль или их сложный эфир, гидролизующийся in vivo.
По этой причине в дальнейшем аспекте настоящего изобретения предусматривается соединение формулы (I) (как изображено выше), где:
R1 представляет собой хлор;
p равно 0 или 1; и, когда p равно 1, R1 находится в мета-положении по отношению к -NH-группе анилина формулы (I);
R2 представляет собой сульфамоил, N-(тетрагидрофуран-2-илметил)сульфамоил, N-[3-(2-оксопирролидин-1-ил)пропил]сульфамоил, N-(3-метоксипропил)сульфамоил, N-(4-фторбензил)сульфамоил, N-(циклопропилметил)сульфамоил, N-пропилсульфамоил, N-(2,3-дигидроксипропил)сульфамоил, N-[2-(2-гидроксиэтокси)этил]сульфамоил, N-(фуран-2-илметил)сульфамоил, N-(2-гидроксиэтил)сульфамоил или N-(карбамоилметил)сульфамоил;
q равно 0 или 1; и, когда q равно 1, R2 находится в пара-положении по отношению к -NH-группе анилина формулы (I);
p + q равно 1;
n равно 0;
R4 представляет собой метил или этил; и
R5 является выбранным из водорода или метила, и R6 представляет собой водород; или его фармацевтически приемлемая соль, или его сложный эфир, гидролизующийся in vivo.
По этой причине в дальнейшем дополнительном аспекте настоящего изобретения предусматривается соединение формулы (I) (как изображено выше), где:
R1 представляет собой галоген, амино, C1-6алкил или C1-6алкокси;
p равно 0-2; где значения R1 могут быть одинаковыми или различными;
R2 представляет собой сульфамоил или группу Ra-Rb-; где
Ra является выбранным из C1-6алкила, C2-6алкенила, C2-6алкинила, C3-8циклоалкила, фенила или гетероциклической группы; где Ra может быть необязательно замещенным на атоме углерода одним или несколькими Rg;
Rb представляет собой -N(Rm)C(O)-, -C(O)N(Rm)-, -S(O)r-, -OC(O)N(Rm)SO2-, -SO2N(Rm)- или -N(Rm)SO2-; где Rm представляет собой водород или C1-6алкил, и r равно 2;
Rg является выбранным из галогена, гидрокси, амино, циано, карбамоила, C1-6алкила, C1-6алкокси, C1-6алкоксиС1-6алкокси, C1-6алкоксиС1-6алкоксиС1-6алкокси, N,N-(C1-6алкил)2амино, C1-6алкилS(O)а, где а равно 2, C3-8циклоалкила, фенила, гетероциклической группы, фенилС1-6алкил- Rо - или (гетероциклическая группа)-R°-; где Rg может быть необязательно замещенным на атоме углерода одним или несколькими Rj;
Rо представляет собой -O-;
Rj является выбранным из галогена, гидрокси, метила или метокси;
q равно 0 или 1;
R3 представляет собой галоген;
n равно 0 или 1;
R4 представляет собой водород, C1-6алкил или C2-6алкенил; где R4 может быть необязательно замещенным на атоме углерода одним или несколькими Rd; где
Rd является выбранным из галогена, амино, C1-6алкокси, C1-6алканоиламино, C1-6алкилсульфониламино, фенила или гетероциклической группы; и
R5 и R6 являются независимо выбранными из водорода или C1-6алкила; где R5 и R6 независимо друг от друга могут быть необязательно замещенными на атоме углерода одним или несколькими Re; где
Re является выбранным из галогена или метокси,
или его фармацевтически приемлемая соль, или его сложный эфир, гидролизующийся in vivo.
По этой причине, в другом дальнейшем дополнительном аспекте настоящего изобретения, предусматривается соединение формулы (I) (как изображено выше), где:
R1 представляет собой хлор, амино, метил или метокси,
p равно 0-2; где значения R1 могут быть одинаковыми или различными;
R2 представляет собой сульфамоил, N-(тетрагидрофур-2-илметил)сульфамоил, N-(циклопропилметил)сульфамоил, N-(фур-2-илметил)сульфамоил, N-(2,2-диметил-1,3-диоксолан-4-илметил)сульфамоил, N-(цианометил)сульфамоил, N-(карбамоилметил)сульфамоил, N-метилсульфамоил, N-(4-фторбензил)сульфамоил, N-(пиридин-2-илметил)сульфамоил, N-(пиридин-3-илметил)сульфамоил, N-(4-метилтиазол-2-ил)сульфамоил, N-(3-метилизоксазол-5-илметил)сульфамоил, N-тетрагидропиран-2-илметил)сульфамоил, N-(2-метилпиразин-5-ил)сульфамоил, N-[2-(2-гидроксиэтокси)этил]сульфамоил, N-(2-гидроксиэтил)сульфамоил, N-(2,2,2-трифторэтил)сульфамоил, N-(2-метоксиэтил)сульфамоил, N-(2-мезилэтил)сульфамоил, N-(2-бензилоксиэтил)сульфамоил, N-(2,2-диметоксиэтил)сульфамоил, N-[2-(N,N-диметиламино)этил]сульфамоил, N-(2-пиперидин-1-илэтил)сульфамоил, N-[2-(метоксиметокси)этил]сульфамоил, N-этилсульфамоил, N-[2-(2-метоксиэтокси)этил]сульфамоил, N-{2-[2-(2-метоксиэтокси)этокси]этил}сульфамоил, N-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}этил)сульфамоил, N-(2-пиридин-2-илэтил)сульфамоил, N-(2-пиридин-4-илэтил)сульфамоил, N-(2-изоксазол-3-илоксиэтил)сульфамоил, N-(2-изотиазол-3-илоксиэтил)сульфамоил, N-(2-1,2-5-тиадиазол-3-илоксиэтил)сульфамоил, N-метил-N-(2-метоксиэтил)сульфамоил, N-[3-(2-оксопирролидин-1-ил)пропил]сульфамоил, N-(3-метоксипропил)сульфамоил, N-пропилсульфамоил, N-(2,3-дигидроксипропил)сульфамоил, N-(3-морфолинопропил)сульфамоил, N-[3-(N,N-диметиламино)пропил]сульфамоил, N-(3,3,3-трифторпропил)сульфамоил, N-(2,2-диметил-3-гидроксипропил)сульфамоил, N-(3-гидроксипропил)сульфамоил, N-(3-этоксипропил)сульфамоил, N-(2-гидроксипропил)сульфамоил, N-(3-изопропоксипропил)сульфамоил, N-(3-изопропокси-2-гидроксипропил)сульфамоил, N-(3-изоксазол-3-илоксипропил)сульфамоил, N-(3-изотиазол-3-илоксипропил)сульфамоил, N-(3-1,2-5-тиадиазол-3-илоксипропил)сульфамоил, N-(1,1-диметилпропил)сульфамоил, N-метил-N-(3-морфолинопропил)сульфамоил, N-бутилсульфамоил, N-трет-бутилсульфамоил, N-(2-гидроксибутил)сульфамоил, N-метил-N-трет-бутилсульфамоил, N-пентилсульфамоил, N-(5-гидроксипентил)сульфамоил, N-(4,5-диметилоксазол-2-ил)сульфамоил, N-(циклопропил)сульфамоил, N-(циклобутил)сульфамоил, N-(3-трифторметилфенил)сульфамоил, N-аллилсульфамоил, N-(2-пропинил)сульфамоил, N-метилкарбамоил, ацетамидо, мезиламино или мезил;
q равно 0 или 1;
R3 представляет собой бром или хлор;
n равно 0 или l;
R4 представляет собой водород, метил, этил, изопропил, 3-бутенил, бензил, 2-фталимидоэтил, 2-аминоэтил, 2-метоксиэтил, 2-ацетамидоэтил, 2-мезиламиноэтил или 2,2,2-трифторэтил;
R5 и R6 являются независимо выбранными из водорода, метила, этила, изопропила, трифторметила или метоксиметила;
или его фармацевтически приемлемая соль, или его сложный эфир, гидролизующийся in vivo.
В другом аспекте настоящего изобретения предпочтительные соединения по настоящему изобретению представляют собой любое соединение из представленных примеров или его фармацевтически приемлемую соль, или его сложный эфир, гидролизующийся in vivo.
Еще в одном аспекте настоящего изобретения, предпочтительные соединения настоящего изобретения представляют собой соединения, полученные в примерах 25, 37, 42, 43, 53, 67, 121, 122, 123 и 136.
Предпочтительные аспекты настоящего изобретения представляют собой такие аспекты, которые относятся к соединению формулы (I) или к его фармацевтически приемлемой соли.
Другой аспект настоящего изобретения предусматривает способ получения соединения формулы (I) или его фармацевтически приемлемой соли или его сложного эфира, гидролизующегося in vivo, этот способ (где R1, R2, R3, R4, R5, R6, n, p и q, если не указано иного, являются такими, как определено в формуле (I)), состоит из:
Способа а) взаимодействия пиримидина формулы (II):
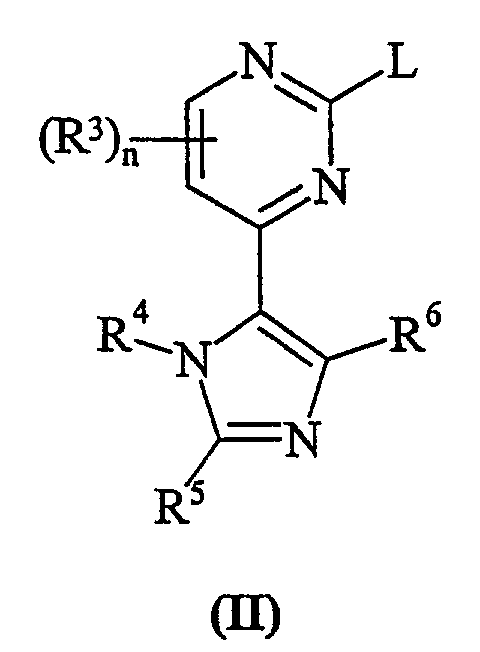
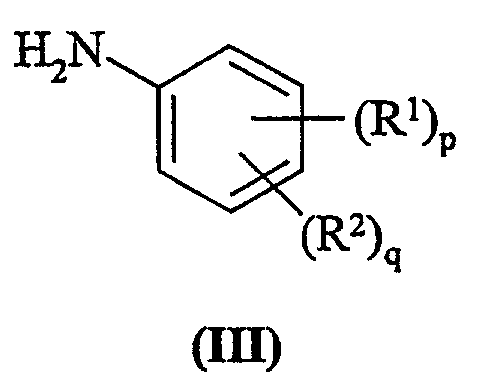
где L представляет собой заменяемую группу; с анилином формулы (III):
или
Способа b) взаимодействия соединения формулы (IV):
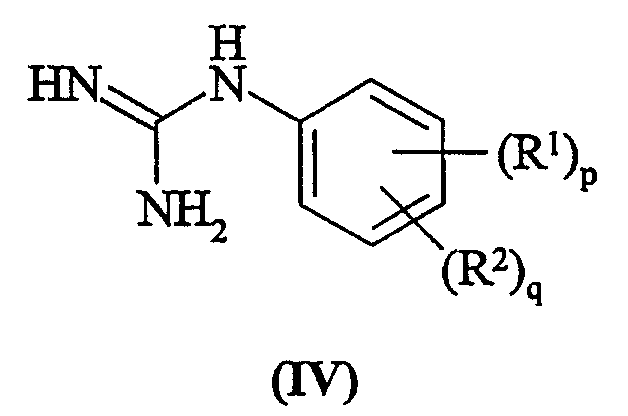
с соединением формулы (V):
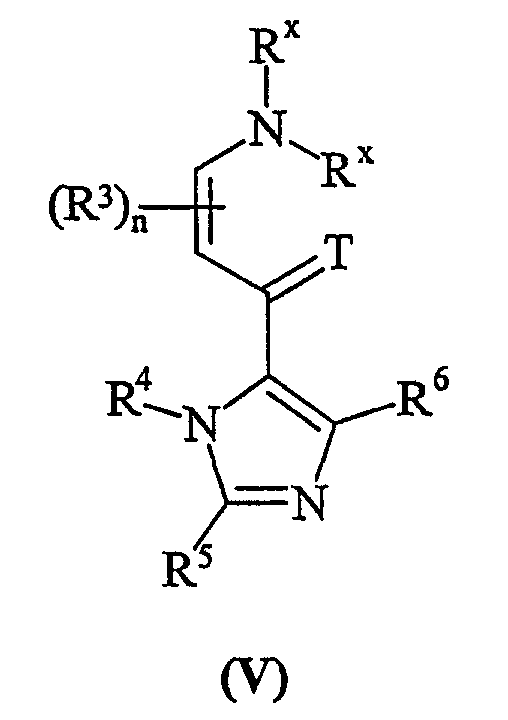
где T представляет собой O или S; Rx могут быть одинаковыми или различными, и являются выбранными из С1-6алкила;
Способа c) для соединений формулы (I), где R2 представляет собой сульфамоил или группу Ra-Rb-, и Rb представляет собой -NHSO2-; взаимодействия пиримидина формулы (VI):
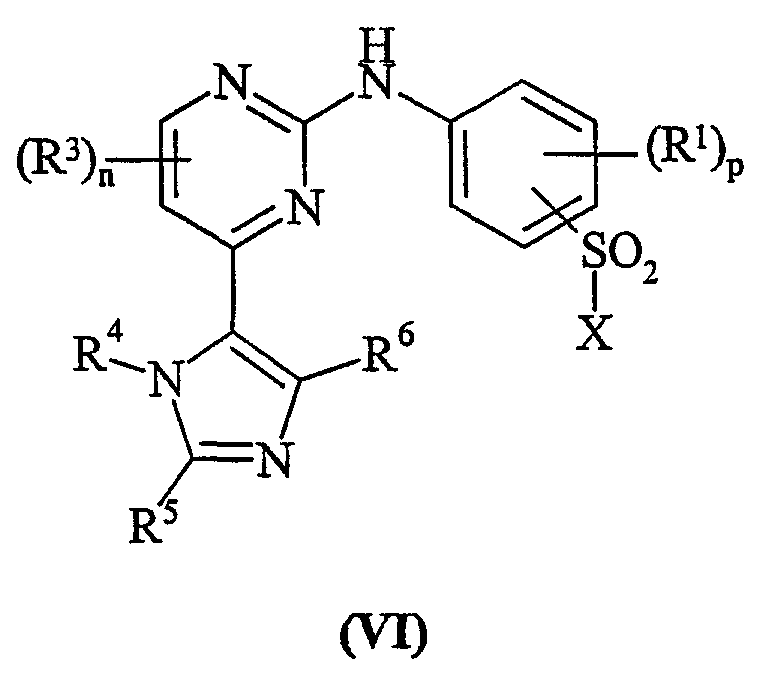
где X представляет собой заменяемую группу; с амином формулы (VII):
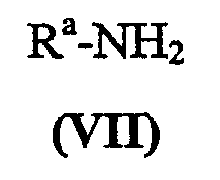
Способа d) для соединений формулы (I); взаимодействия пиримидина формулы (VIII)
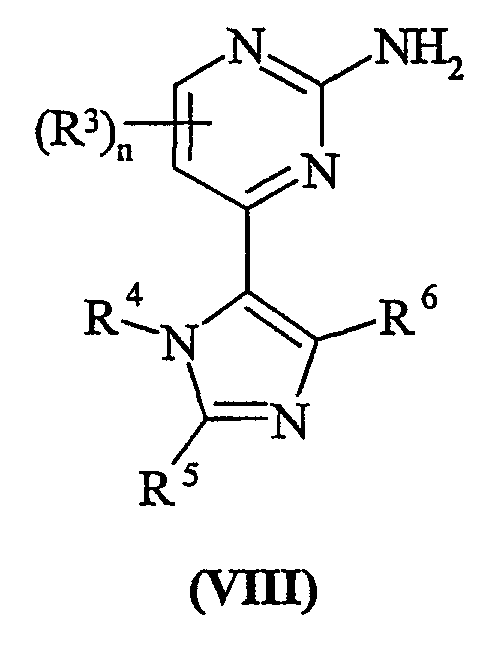
с соединением формулы (IX):
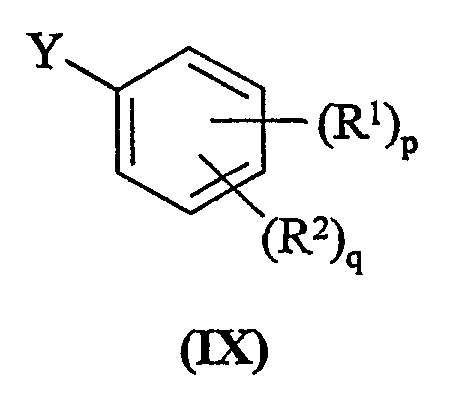
где Y представляет собой заменяемую группу;
и после этого, если необходимо:
i) преобразования соединения формулы (I) в другое соединение формулы (I);
ii) удаления любых защитных групп;
iii) образования фармацевтически приемлемой соли или сложного эфира, гидролизующегося in vivo.
L представляет собой заменяемую группу, пригодные для использования значения для L представляют собой, например, галогеновую или сульфонилоксигруппу, например хлор, бром, метансульфонилокси или толуол-4-сульфонилоксигруппу.
X представляет собой заменяемую группу, пригодные для использования значения для X представляют собой, например, группу фтора или хлора. Предпочтительно X представляет собой фтор.
Y представляет собой заменяемую группу, пригодные для использования значения для Y представляют собой, например, галогеновую или сульфонилокси группу, например, бром, йод или трифторметансульфонилоксигруппу. Предпочтительно Y представляет собой йод.
Конкретные условия реакции для указанных выше реакций являются следующими.
Способ а) Пиримидины формулы (II) и анилины формулы (III) могут взаимодействовать вместе:
i) в присутствии соответствующего растворителя, например, кетона, такого как ацетон, или спирта, такого как этанол или бутанол, или ароматического углеводорода, такого как толуол или N-метилпирролидин, необязательно в присутствии соответствующей кислоты, например, неорганической кислоты, такой как хлористоводородная кислота или серная кислота, или органической кислоты, такой как уксусная кислота или муравьиная кислота (или соответствующей кислоты Льюиса) и при температуре в пределах от 0°C до температуры кипения, предпочтительно при температуре кипения; или
ii) при стандартных условиях Бухвальда (например, смотри J.Am.Chem.Soc., 118, 7215; J.Am.Chem.Soc., 119, 8451; J.Org.Chem., 62, 1568 and 6066), например, в присутствии ацетата палладия, в соответствующем растворителе, например, в ароматическом растворителе, таком как толуол, бензол или ксилол, с соответствующим основанием, например, с неорганическим основанием, таким как карбонат цезия, или с органическим основанием, таким как трет-бутоксидкалий, в присутствии соответствующего лиганда, такого как 2,2'-бис(дифенилфосфино)-1,1'-бинафтил, и при температуре в интервале от 25 до 80°C.
Пиримидины формулы (II), где L представляет собой хлор, могут быть приготовлены в соответствии со схемой 1:
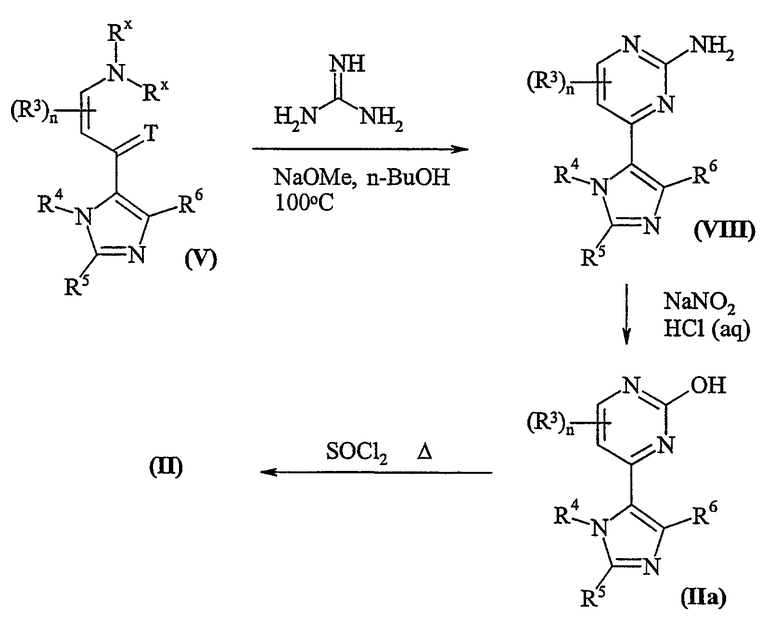
Схема 1
Анилины формулы (III) являются коммерчески доступными соединениями, или они известны из литературы, или они приготавливаются с помощью стандартных способов, известных в данной области.
Способ b) Соединения формулы (IV) и соединения формулы (V) взаимодействуют вместе в соответствующем растворителе, таком как N-метилпирролидинон или бутанол, при температуре в интервале 100-200°C, предпочтительно в интервале 150-170°C. Взаимодействие предпочтительно проводится в присутствии соответствующего основания, такого, например, как гидрид натрия, метоксид натрия или карбонат калия.
Соединения формулы (V) могут быть приготовлены в соответствии со схемой 2:
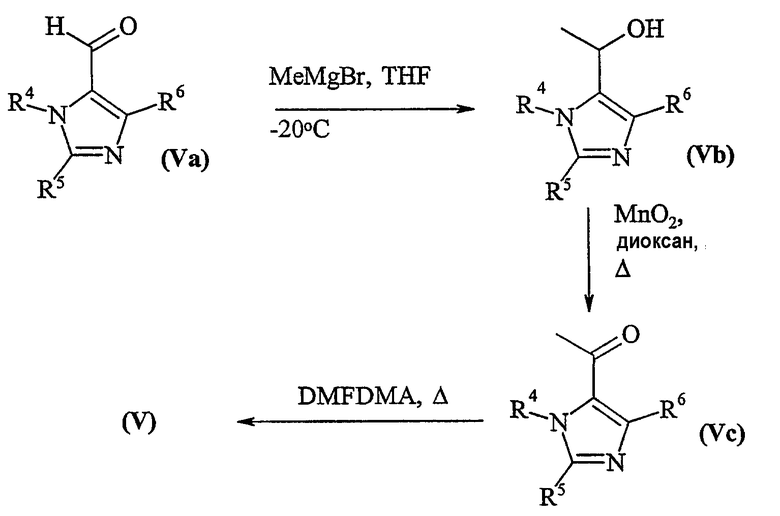
Схема 2
Соединения формулы (IV) и (Va) являются коммерчески доступными соединениями, или они известны из литературы, или они приготавливаются с помощью стандартных способов, известных в данной области.
Способ c) Соединения формулы (VI) и амины формулы (VII) могут взаимодействовать вместе в присутствии инертного растворителя, такого как N-метилпирролидинон или пиридин, в присутствии основания, например, неорганического основания, такого как карбонат цезия, или в присутствии органического основания, такого как избыток (VII), и при температуре в интервале от 25 до 80°C.
Соединения формулы (VI) (где X представляет собой хлор) могут быть приготовлены в соответствии со схемой 3:
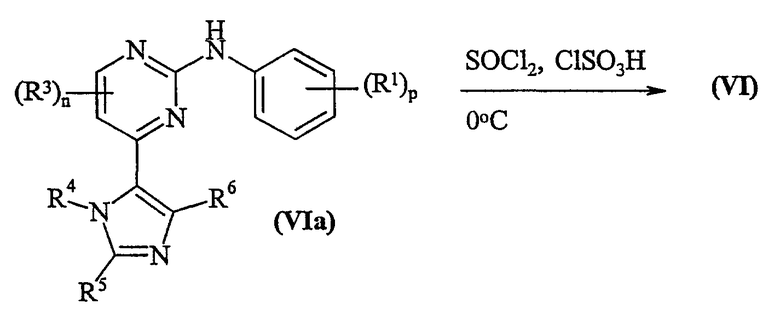
Схема 3
Соединения формулы (VIa) могут быть приготовлены в соответствии со способом а, способом b или способом d, где q равно 0.
Способ d) Соединения формулы (VIII) и амины формулы (IX) могут взаимодействовать вместе при стандартных условиях Бухвальда, как описано в способе а.
Синтез соединений формулы (VIII) описан в схеме 1.
Соединения формулы (IX) являются коммерчески доступными соединениями, или они известны из литературы, или они приготавливаются с помощью стандартных способов, известных в данной области.
Амины формулы (VI) являются коммерчески доступными соединениями, или они известны из литературы, или они приготавливаются с помощью стандартных способов, известных в данной области.
Некоторые из различных кольцевых заместителей в соединениях по настоящему изобретению могут быть введены с помощью стандартных реакций ароматического замещения или генерироваться с помощью обычных модификаций функциональных групп, либо до, либо непосредственно после процессов, рассмотренных выше, и, как таковые, включаются в аспект способа настоящего изобретения. Такие взаимодействия и модификации включают в себя, например, введение заместителя посредством реакции ароматического замещения, восстановления заместителей, алкилирования заместителей и окисления заместителей. Реагенты и реакционные условия для таких процедур являются хорошо известными в области химии. Конкретные примеры реакций ароматического замещения включают в себя введение нитрогруппы с использованием концентрированной азотной кислоты, введение ацильной группы с использованием, например, ацилгалогенида и кислоты Льюиса (такой как трихлорид алюминия) в условиях Фриделя-Крафтса; введение алкильной группы с использованием алкилгалогенида и кислоты Льюиса (такой как трихлорид алюминия) в условиях Фриделя-Крафтса; и введение группы галогена. Конкретные примеры модификаций включают в себя восстановление нитрогруппы до аминогруппы, например, путем каталитического гидрирования с помощью никелевого катализатора, или обработку с помощью железа в присутствии хлористоводородной кислоты при нагревании; окисление алкилтио до алкилсульфинила или алкилсульфонила.
Можно также заметить, что в некоторых из реакций, рассмотренных здесь, может быть необходимым/желательным защищать какие-либо чувствительные группы в соединениях. Случаи, где защита является необходимой или желательной, и соответствующие способы для защиты известны специалистам в данной области. Могут быть использованы обычные защитные группы в соответствии со стандартной практикой (иллюстрации смотри в T.W. Green, Protective Groups в Organic Synthesis, John Wiley and Sons, 1991). Таким образом, если реагенты включают в себя такие группы, как амино, карбокси или гидрокси, может оказаться желательным защищать группу в некоторых из реакций, рассматриваемых здесь.
Пригодная для использования защитная группа для амино или алкиламиногруппы представляет собой, например, ацильную группу, например, алканоильную группу, такую как ацетильная, алкоксикарбонильную группу, например, метоксикарбонильную, этоксикарбонильную или трет-бутоксикарбонильную группу, арилметоксикарбонильную группу, например, бензилоксикарбонильную или ароильную группу, например, бензоильную. Условия снятия защиты указанных выше защитных групп при необходимости различаются в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или алкоксикарбонильная группа, или ароильная группа, может быть удалена, например, путем гидролиза с помощью соответствующего основания, такого как гидроксид щелочного металла, например, гидроксид лития или натрия. Альтернативно, ацильная группа, такая как трет-бутоксикарбонильная группа, может быть удалена, например, путем обработки с помощью соответствующей кислоты, такой как хлористоводородная, серная или фосфорная кислота, или трифторуксусная кислота, и арилметоксикарбонильная группа, такая как бензилоксикарбонильная группа, может быть удалена, например, путем гидрирования над катализатором, таким как палладий-на-угле, или путем обработки с помощью кислоты Льюиса, например, бортрис(трифторацетата). Соответствующая альтернативная защитная группа для первичной аминогруппы представляет собой, например, фталоильную группу, которая может быть удалена путем обработки с помощью алкиламина, например, диметиламинопропиламина, или с помощью гидразина.
Соответствующая защитная группа для гидроксигруппы представляет собой, например, ацильную группу, например, алканоильную группу, такую как ацетильная, ароильную группу, например, бензоильную, или арилметильную группу, например, бензильную. Условия для снятия защиты с указанных выше защитных групп будут изменяться в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или ароильная группа, может быть удалена, например, путем гидролиза с помощью соответствующего основания, такого как гидроксид щелочного металла, например, гидроксид лития или натрия. Альтернативно, арилметильная группа, такая как бензильная группа, может быть удалена, например, путем гидрирования над катализатором, таким как палладий-на-угле.
Соответствующая защитная группа для карбоксигруппы представляет собой, например, этерифицирующую группу, например, метильную или этильную группу, которая может быть удалена, например, путем гидролиза с помощью основания, такого как гидроксид натрия, или, например, трет-бутильную группу, которая может быть удалена, например, путем обработки с помощью кислоты, например, органической кислоты, такой как трифторуксусная кислота, или, например, бензильную группу, которая может быть удалена, например, путем гидрирования над катализатором, таким как палладий-на-угле.
Защитные группы могут быть удалены на любой удобной стадии синтеза с использованием обычных методик, хорошо известных в данной области.
Как утверждалось выше, соединения, определяемые в настоящем изобретении, обладают активностью против пролиферации клеток, такой как противораковая активность, которая, как предполагается, возникает в связи с ингибиторной активностью соединения по отношению к CDK. Эти свойства могут быть оценены, например, с использованием процедуры, приведенной ниже:
Анализы
Используются следующие сокращения:
HEPES представляет собой N-[2-гидроксиэтил]пиперазин-N'-[2-этансульфоновую кислоту]
DTT представляет собой дитиофреитол
PMSF представляет собой фенилметилсульфонилфторид
Соединения исследуются с помощью анализа киназы in vitro в 96 луночном формате с использованием Scintillation Proximity Assay (SPA - получено от Amersham) для измерения включения [γ-33-P]-аденозинтрифосфата в исследуемый субстрат (белок ретинобластомы - GST; GST-Rb). В каждую лунку помещается соединение, подлежащее исследованию (разбавленное в ДМСО и в воде, для корректировки концентраций), а в контрольные лунки либо росковитин, в качестве ингибиторного контроля, либо ДМСО, в качестве положительного контроля.
В каждую лунку добавляют примерно 0,2 мкл частично очищенного фермента CDK2/циклин E (количество зависит от активности фермента), разбавленного в 25 мкл инкубационного буфера, а затем 20 мкл смеси GST-Rb/ATP/ATP33 (содержит 0,5 мкг GST-Rb и 0,2 мкМ ATP, и 0,14 мккюри [γ-33-P]-аденозинтрифосфата в инкубационном буфере), и полученную в результате смесь осторожно встряхивают, а затем инкубируют при комнатной температуре в течение 60 минут.
Затем в каждую лунку добавляют по 150 мкл стоп-раствора, содержащего (0,8 мг/лунка шариков Protein A-PVT SPA (Amersham)), 20 пМ/лунка антиглютатион трансферазы, IgG кролика (получен от Molecular Probes), 61 мМ EDTA и 50 мМ HEPES, pH 7,5, содержащего 0,05% азида натрия.
Планшеты герметизируют с помощью уплотнений для планшетов Topseal-S, оставляют на два часа, а затем центрифугируют при 2500 об/мин, 1124хg, в течение 5 минут. Производят отсчеты планшетов на Topcount, по 30 секунд на каждую лунку.
Инкубационный буфер, используемый для разбавления смесей фермента и субстрата, содержит 50 мМ HEPES, pH 7,5, 10 мМ MnCl2, 1 мМ DTT, 100 мкМ ванадата натрия, 100 мкМ NaF, 10 мМ глицерофосфата натрия, BSA (конечная концентрация 1 мг/мл).
Исследуемый субстрат
В этом анализе используется только часть белка ретинобластомы (Science 1987 Marl3;235(4794):1394-1399; Lee W.H., Bookstein R., Hong F., Young L.J., Shew J.Y., Lee E.Y.), слитого с меткой GST. Осуществляют PCR гена ретинобластомы, кодирующего аминокислоты 379-928, (получены от плазмида ретинобластомы ATCC pLRbRNL), и последовательность клонируется в вектор слияния pGEx 2T (Smith D.B. and Johnson, K.S. Gene 67, 31 (1988): который содержит промотор tac для индуцируемой экспрессии, внутренний ген lac Iq для использования в какой-либо клетке-хозяине E.Coli, и кодирующий регион для расщепления тромбина (получен от Pharmacia Biotech), который используется для амплификации аминокислот 792-928. Эта последовательность опять клонируется в pGEx 2T.
Последовательность 792-928 ретинобластомы, полученная таким образом, экспрессируется в E.Coli (клетки BL21 (DE3) pLysS), используя методики индуцируемой экспрессии, и очищают следующим образом.
Пасту E.coli повторно суспендируют в 10 мл/г буфера NETN (50 мМ Tris, pH 7,5, 120 мМ NaCl, 1 мМ EDTA, 0,5% об/об NP-40, 1 мМ PMSF, 1 мкг/мл лейпептина, 1 мкг/мл апротинина и 1 мкг/мл пепстатина) и обрабатывают ультразвуком в течение 2 x 45 секунд на 100 мл гомогената. После центрифугирования супернатант загружают в 10 мл глютатиона в колонку Sepharose (Pharmacia Biotech, Herts, UK) и промывают буфером NETN. После промывки киназным буфером (50 мМ HEPES, pH 7,5, 10 мМ MgC12, 1 мМ DTT, 1 мМ PMSF, 1 мкг/мл лейпептина, 1 мкг/мл апротинина и 1 мкг/мл пепстатина), белок элюируют с помощью 50 мМ восстановленного глютатиона в киназном буфере. Фракции, содержащие GST-Rb (792-927), собирают и подвергают диализу в течение ночи с киназным буфером снаружи. Конечный продукт анализируют с помощью гель-электрофореза (полиакриламидный гель), с использованием додецилсульфата натрия (SDS), используя гели на основе 8-16% Трис-глицина (Novex, San Diego, USA).
CDK2 и циклин E
Открытые рамки считывания CDK2 и циклина E выделяют с помощью PCR с обратной траскриптазой, используя мРНК клеток HeLa и активированных T лимфоцитов в качестве шаблонов, и клонируют в вектор экспрессии насекомых pVL1393 (получают от Invitrogen 1995, номер по катологу: V1392-20). Затем CDK2 и циклин E дуально экспрессируют [используя стандартную технику совместного инфицирования вирусом Baculogold] в клеточную систему насекомых SF21 (клетки Spodoptera Frugiperda, полученные из ткани яичников Fall Army Worm - коммерчески доступны).
Пример продуцирования циклина E/CDK2
Следующий далее пример приводит детали продуцирования циклина E/CDK2 в клетках SF21 (в TC100 + 10% FBS(TCS) + 0,2% Pluronic), получивших дуальное инфицирование MOI 3 каждым вирусом, циклином E и CDK2.
Клетки SF21, выращенные в культуре во вращающемся флаконе до количества 2,33 x 106 клеток/мл, используются для инокуляции 10 x 500 мл вращающихся колб при 0,2 x 106 клеток/мл. Вращающиеся флаконы инкубируют на вращающейся стойке при 28°C.
Через 3 дня (72 час), производят подсчет клеток, и среднее значение для 2 флаконов, как обнаружено, составляет 1,86 x 106 клеток/мл (99% жизнеспособных). Затем культуры инфицируются дуальными вирусами при MOI 3 для каждого вируса.
Вирусы смешиваются вместе перед добавлением к культурам, и культуры возвращают на вращающуюся стойку при 28°C.
Через 2 дня (48 час) после инфицирования собирают 5 литров культуры. Общее количество клеток при сборе составляет 1,58 x 106 клеток/мл (99% жизнеспособность). Клетки центрифугируют при 2500 об/мин, 30 мин, 4C, в Heraeus Omnifuge 2,0 RS, порциями по 250 мл. Супернатант сливают.
Частичная совместная очистка CDK2 и циклина E
Клетки SF21 повторно суспендируют в лизирующем буфере (50 мМ Tris, pH 8,2, 10 мМ MgCl2, 1 мМ DTT, 10 мМ глицерофосфата, 0,1 мМ ортованадата натрия, 0,1 мМ NaF, 1 мМ PMSF, 1 мкг/мл лейпептина и 1 мкг/мл апротинина) и гомогенизируют в течение 2 минут в 10 мл гомогенизаторе Dounce. После центрифугирования, супернатант загружают в анионообменную колонку Poros HQ/M 1,4/100 (PE Biosystems, Hertford, UK). CDK2 и циклин E совместно элюируются в начале прохождения 0-1M градиента NaCl (осуществляют в лизирующем буфере минус ингибиторы протеазы), в 20 объемах колонки. Совместное элюирование отслеживается с помощью вестерн-блоттинга с использованием антител как против CDK2, так и против циклина E (Santa Cruz Biotechnology, California, US).
По аналогии могут быть построены анализы, сконструированные для оценки ингибирования CDK4 и CDK6. CDK2 (№ доступа EMBL X62071) может быть использован вместе с циклином A или циклином E (смотри EMBL, номер доступа M73812), и дальнейшие детали таких анализов содержатся в Международной публикации PCT № WO99/21845, соответствующие разделы Биохимическая и Биологическая Оценка из которой включаются сюда в качестве ссылки.
Хотя фармакологические свойства соединений формулы (I) изменяются вместе со структурными изменениями, в целом, активность, которой обладают соединения формулы (I), может быть продемонстрирована при концентрациях или дозах IC50 в пределах от 250 мкМ до 1 нМ.
При исследовании в указанном выше анализе in vitro ингибиторная активность CDK2 из примера 14 измерена как IC50 = 0,146 мкМ.
Активность in vivo соединений по настоящему изобретению может быть оценена с помощью стандартных методик, например, путем измерения ингибирования роста клеток и оценки цитотоксичности.
Ингибирование роста клеток может быть измерено путем окрашивания клеток с помощью сульфородамина B (SRB), флуоресцентного красителя, который окрашивает белки, и, как следствие, дает оценку количества белка (то есть клеток) в лунке (смотри Boyd, M.R.(1989) Status of the NCI preclinical antitumour drug discovery screen. Prin. Prac Oncol 10:1-12). Таким образом, при измерении ингибирования роста клеток предусматриваются следующие детали:
Клетки в соответствующей среде в объеме 100 мкл помещаются в 96 луночные планшеты; среды представляют собой модифицированные Дюльбеко среды Игла для MCF-7, SK-UT-1B и SK-UT-1. Клеткам дают возможность прикрепиться в течение ночи, затем добавляют ингибиторные соединения при различных концентрациях, при максимальной концентрации 1% ДМСО (объем/объем). Контрольный планшет анализируется для получения значения для количества клеток перед дозировкой. Клетки инкубируются при 37°C, (5% CO2) в течение трех дней.
По прохождении трех дней, в планшеты добавляют TCA до конечной концентрации 16% (объем/объем). Затем планшеты инкубируют при 4°C в течение 1 часа, супернатант удаляют, и планшеты промывают в водопроводной воде. После сушки добавляют 100 мкл красителя SRB (0,4% SRB в 1% уксусной кислоте), в течение 30 минут, при 37°C. Избыток SRB удаляют и планшеты промывают в 1% уксусной кислоте. SRB, связанный с белком, солюбилизируют в 10 мМ Tris, pH 7,5, и встряхивают в течение 30 минут при комнатной температуре. Значения OD регистрируют при 540 нм, и концентрация ингибитора, вызывающая 50% ингибирование роста, определяется по полулогарифмическому графику концентрации ингибитора как функции коэффициента поглощения. Концентрация соединения, которая понижает оптическую плотность до значения, более низкого, чем то, которое получается, когда клетки помещаются в планшет в начале эксперимента, дает значение для токсичности.
Типичные значения IC50 для соединений по настоящему изобретению, когда они исследуются в анализе с использованием SRB, находятся в пределах от 1 мМ до 1 нМ.
В соответствии с дальнейшим аспектом настоящего изобретения предусматривается фармацевтическая композиция, которая содержит пиримидиновое производное формулы (I), или его фармацевтически приемлемую соль, или его сложный эфир, гидролизующийся in vivo, как определено выше, в сочетании с фармацевтически приемлемым разбавителем или носителем.
Композиция может находиться в форме, пригодной для использования при пероральном введении, например, в виде таблетки или капсулы, для парентеральной инъекции (включая внутривенную, подкожную, внутримышечную, внутрисосудистую инъекцию или вливание), в виде стерильного раствора, суспензии или эмульсии, для местного введения, в виде мази или крема, или для ректального введения, в виде суппозитория.
Как правило, указанные выше композиции могут быть приготовлены обычным способом, с использованием обычных разбавителей.
Соединение формулы (I), как правило, может быть введено теплокровному животному, при единичной дозе, находящейся в пределах 5-5000 мг на квадратный метр поверхности тела животного, то есть примерно 0,1-100 мг/кг, и это, как правило, обеспечивает терапевтически эффективную дозу. Единичная дозированная форма, такая как таблетка или капсула, должна, как правило, содержать, например, 1-250 мг активного ингредиента. Предпочтительно используется дневная доза в пределах 1-50 мг/кг. Однако ежедневная доза при необходимости будет меняться в зависимости от подвергающегося лечению субъекта, конкретного способа введения и тяжести заболевания, которое подвергается лечению. В соответствии с этим оптимальная доза может быть определена лечащим врачом, который лечит конкретного пациента.
В соответствии с дальнейшим аспектом настоящего изобретения создается соединение формулы (I) или его фармацевтически приемлемая соль, или его сложный эфир, гидролизующийся in vivo, как определено выше, для использования в способе лечения организма человека или животного с помощью терапии.
Авторы обнаружили, что соединения, определяемые в настоящем изобретении, или их фармацевтически приемлемая соль или их сложный эфир, гидролизующийся in vivo, являются эффективными ингибиторами клеточного цикла (агентами против пролиферации клеток), как предполагается, это свойство возникает из-за их свойств относительно ингибирования CDK. Соответственно соединения по настоящему изобретению, как ожидается, являются пригодными для использования при лечении заболеваний или медицинских состояний, опосредуемых полностью или частично, ферментами CDK, то есть соединения могут использоваться для оказания ингибиторного воздействия на CDK у теплокровного животного, нуждающегося в таком лечении. Таким образом, соединения по настоящему изобретению предусматривают способ лечения пролиферации злокачественных клеток, характеризуемый ингибированием ферментов CDK, то есть эти соединения могут быть использованы для оказания противопролиферативного воздействия, опосредуемого полностью или частично ингибированием CDK. Такое соединение по настоящему изобретению, как ожидается, обладает широким набором противораковых свойств, поскольку CDK участвуют в развитии многих распространенных раковых заболеваниях у людей, таких как лейкемия и рак груди, легких, толстой кишки, прямой кишки, желудка, простаты, мочевого пузыря, поджелудочной железы и яичников. Таким образом, ожидается, что соединение по настоящему изобретению будет обладать противораковой активностью по отношению к этим раковым заболеваниям. В дополнение к этому, ожидается, что соединение по настоящему изобретению будет обладать активностью против ряда лейкемий, злокачественных заболеваний лимфатической системы и солидных опухолей, таких как карциномы и саркомы, в таких тканях, как печень, почки, простата и поджелудочная железа. Конкретно такие соединения по настоящему изобретению, как ожидается, преимущественно замедляют рост первичных и вторичных солидных опухолей, например, толстой кишки, груди, простаты, легких и кожи. Более конкретно такие соединения по настоящему изобретению или их фармацевтически приемлемая соль, или их сложный эфир, гидролизующийся in vivo, как ожидается, ингибируют рост тех первичных и вторичных солидных опухолей, которые ассоциируются с CDK, особенно тех опухолей, которые существенно зависят от CDK при их росте и распространении, включая, например, определенные опухоли толстой кишки, груди, простаты, легких, женских наружных половых органов и кожи.
В дополнение к этому ожидается, что соединение по настоящему изобретению будет обладать активностью против других заболеваний, связанных с пролиферацией клеток, в широком диапазоне других болезненных состояний, включая различные виды лейкемии, фибропролиферативные и дифференциационные расстройства, псориаз, ревматоидный артрит, саркому Капоши, хемангиома, острые и хронические невропатии, атерому, атеросклероз, артериальный рестеноз, аутоиммунные заболевания, острые и хронические воспаления, заболевание костей и заболевания глаз с пролиферацией ретинальных сосудов.
Таким образом, в соответствии с этим настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли или его сложному эфиру, гидролизующемуся in vivo, как определено выше, для использования в качестве лекарственного средства; и использованию соединения формулы (I) или его фармацевтически приемлемой соли, или его сложного эфира, гидролизующегося in vivo, как определено выше, для получения лекарственного средства для продуцирования ингибиторного воздействия на клеточный цикл (против пролиферации клеток) у теплокровного животного, такого как человек. В частности, ингибиторное воздействие производится путем предотвращения входа или развития в S фазе путем ингибирования CDK2, CDK4 и/или CDK6, в особенности, CDK2.
В соответствии с дальнейшей особенностью настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, или его сложному эфиру, гидролизующемуся in vivo, как определено здесь выше, для получения лекарственного средства, предназначенного для лечения раковых заболеваний (солидных опухолей и различных видов лейкемий), фибропролиферативных и дифференциационных расстройств, псориаза, ревматоидного артрита, саркомы Капоши, хемангиомы, острых и хронических невропатий, атеромы, атеросклероза, артериального рестеноза, аутоиммунных заболеваний, острых и хронических воспалений, заболевания костей и заболевания глаз с пролиферацией ретинальных сосудов, в частности, при лечении раковых заболеваний.
Кроме того, настоящее изобретение относится к способу продуцирования ингибиторного воздействия на клеточный цикл (против пролиферации клеток) у теплокровного животного, такого как человек, нуждающегося в таком лечении, который включает в себя введение указанному животному эффективного количества соединения, как определено непосредственно выше. В частности, ингибиторное воздействие осуществляется путем предотвращения входа или развития в S фазе путем ингибирования CDK2, CDK4 и/или CDK6, в особенности, CDK2.
Настоящее изобретение также относится к способу продуцирования ингибиторного воздействия на клеточный цикл (против пролиферации клеток) у теплокровного животного, такого как человек, нуждающегося в таком лечении, который включает в себя введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, или его сложного эфира, гидролизующегося in vivo, как определено здесь выше. В частности, ингибиторное воздействие осуществляется путем предотвращения входа или развития в S фазе путем ингибирования CDK2, CDK4 и/или CDK6, в особенности, CDK2.
Более того, настоящее изобретение относится к способу лечения раковых заболеваний (солидных опухолей и различных видов лейкемий), фибропролиферативных и дифференциационных расстройств, псориаза, ревматоидного артрита, саркомы Капоши, хемангиомы, острых и хронических невропатий, атеромы, атеросклероза, артериального рестеноза, аутоимунных заболеваний, острого и хронического воспаления, заболевания костей и заболевания глаз с пролиферацией ретинальных сосудов, у теплокровных животных, таких как человек, нуждающихся в таком лечении, который включает в себя введение указанным животным эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, или его сложного эфира, гидролизующегося in vivo, как определено здесь выше.
В частности, предусматривается способ лечения ракового заболевания у теплокровного животного, такого как человек, нуждающегося в таком лечении, который включает в себя введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, или его сложного эфира, гидролизующегося in vivo, как определено здесь выше.
В дальнейшем аспекте настоящего изобретения предусматривается фармацевтическая композиция, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль, или его сложный эфир, гидролизующийся in vivo, как определено здесь выше, в сочетании с фармацевтически приемлемым разбавителем или носителем, пригодная для продуцирования ингибиторного воздействия на клеточный цикл (против пролиферации клеток) у теплокровного животного, такого как человек.
В дальнейшем аспекте настоящего изобретения предусматривается фармацевтическая композиция, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль, или его сложный эфир, гидролизующийся in vivo, как определено здесь выше, в сочетании с фармацевтически приемлемым разбавителем или носителем, пригодная при лечении раковых заболеваний (солидных опухолей и различных видов лейкемий), фибропролиферативных и дифференциационных расстройств, псориаза, ревматоидного артрита, саркомы Капоши, хемангиомы, острых и хронических невропатий, атеромы, атеросклероза, артериального рестеноза, аутоимунных заболеваний, острых и хронических воспалений, заболевания костей и заболевания глаз с пролиферацией ретинальных сосудов, у теплокровного животного, такого как человек.
Далее настоящее изобретение относится к фармацевтической композиции, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль, или его сложный эфир, гидролизующийся in vivo, как определено здесь выше, в сочетании с фармацевтически приемлемым разбавителем или носителем, пригодной при лечении ракового заболевания у теплокровного животные, такого как человек.
Предотвращение синтеза ДНК клетками путем ингибирования основных инициирующих активностей при вхождении в S фазу, таких как инициация CDK2, может быть полезным также при защите нормальных клеток организма от токсичности циклоспецифичных фармацевтических агентов. Ингибирование CDK2 или 4 будет предотвращать развитие клеточного цикла у нормальных клеток, что могло бы ограничивать токсичность циклоспецифичных фармацевтических агентов, которые действуют в S фазе, в G2 фазе или при митозе. Такая защита может приводить к предотвращению потери волос, обычно связанной с применением этих агентов.
По этой причине в дальнейшем аспекте настоящего изобретения предусматривается соединение формулы (I), как определено выше, или его фармацевтически приемлемая соль, или его сложный эфир, гидролизующийся in vivo, для использования в качестве агента для защиты клеток.
По этой причине в дальнейшем аспекте настоящего изобретения предусматривается соединение формулы (I), как определено здесь выше или его фармацевтически приемлемая соль, или его сложный эфир, гидролизующийся in vivo, для использования при предотвращении потери волос, происходящей при лечении злокачественных состояний с помощью фармацевтических агентов.
Примеры фармацевтических агентов для лечения злокачественных состояний, которые, как известно, вызывают потерю волос, включают в себя алкилирующие агенты, такие как ифосфамид и циклофосфамид; антиметаболиты, такие как метотрексат, 5-фтороурацил, гемцитабин и цитарабин; vinca алкалолиды и аналоги, такие как винкристин, винбалстин, виндезин, винорелбин; таксаны, такие как паклитаксель и доцетаксель; ингибиторы топоизомеразы I, такие как иринтотекан и топотекан; цитоксичные антибиотики, такие как доксорубицин, даунорубицин, митоксантрон, актиномицин-D и митомицин; и другие, такие как этопозид и третиноин.
В другом аспекте настоящего изобретения соединение формулы (I) или его фармацевтически приемлемая соль, или его сложный эфир, гидролизующийся in vivo, могут вводиться в сочетании с одним или несколькими из указанных выше фармацевтических агентов. В этом случае соединение формулы (I) может вводиться с помощью системных или несистемных средств. В частности, соединение формулы (I) может вводиться с помощью несистемных средств, например, местного введения.
Поэтому настоящее изобретение касается способа предотвращения потери волос во время лечения одного или более злокачественных состояний у теплокровного животного, такого как человек, с помощью фармацевтических агентов, включающего введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или in vivo гидролизуемого эфира.
Кроме того, изобретение касается способа предотвращения потери волос во время лечения одного или более злокачественных состояний у теплокровного животного, такого как человек, фармацевтическими агентами, путем введения указанному животному эффективного количества соединения формулы (I), или его фармацевтически приемлемой соли или его сложного эфира, гидролизующегося in vivo, с эффективным количеством указанного фармацевтического агента, при одновременном, последовательном или раздельном введении.
В соответствии с дальнейшим аспектом настоящего изобретения предусматривается фармацевтическая композиция, пригодная для предотвращении потери волос, возникающей в связи с лечением злокачественных состояний с помощью фармацевтических агентов, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль, или его сложный эфир, гидролизующийся in vivo, и указанный фармацевтический агент, в сочетании с фармацевтически приемлемым разбавителем или носителем.
В соответствии с дальнейшим аспектом настоящего изобретения предусматривается набор, содержащий соединение формулы (I) или его фармацевтически приемлемую соль, или его сложный эфир, гидролизующийся in vivo, и фармацевтический агент для лечения злокачественных состояний, о котором известно, что он вызывает потерю волос.
В соответствии с дальнейшим аспектом настоящего изобретения предусматривается набор, содержащий:
a) соединение формулы (I) или его фармацевтически приемлемую соль, или его сложный эфир, гидролизующийся in vivo, в первой стандартной дозированной форме;
b) фармацевтический агент для лечения злокачественных состояний, о котором известно, что он вызывает потерю волос; во второй стандартной дозированной форме; и
c) контейнер для размещения указанных первой и второй дозированных форм.
В соответствии с другой особенностью настоящего изобретения, предусматривается применение соединения формулы (I) или его фармацевтически приемлемой соли или его сложного эфира, гидролизующегося in vivo, при производстве лекарственного средства для предотвращения потери волос во время лечения злокачественных состояний с помощью фармацевтических агентов.
В соответствии с дальнейшим аспектом настоящего изобретения предусматривается комбинированное лечение для предотвращения потери волос, включающее в себя введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или его сложного эфира, гидролизующегося in vivo, необязательно вместе с фармацевтически приемлемым разбавителем или носителем с одновременным, последовательным или раздельным введением эффективного количества фармацевтического агента для лечения злокачественных состояний теплокровному животному, такому как человек.
Как указано выше, размер дозы, необходимой для терапевтического или профилактического лечения конкретного заболевания, связанного с пролиферацией клеток, с необходимостью будет изменяться в зависимости от объекта лечения, способа введения и тяжести заболевания, подвергающегося лечению. Предполагается стандартная доза, находящаяся в диапазоне, например, 1-100 мг/кг, предпочтительно 1-50 мг/кг.
Ингибиторная активность по отношению к CDK, определенная выше, может применяться в качестве единственного вида терапии или может включать в себя, в дополнение к соединению по настоящему изобретению одно или несколько других веществ и/или видов лечения. Такое комбинированное лечение может быть достигнуто путем одновременного, последовательного или раздельного введения индивидуальных компонентов лечения. В области медицинской онкологии нормальной практикой является использование сочетания различных форм лечения для лечения каждого пациента с раковым заболеванием. В медицинской онкологии другим компонентом (компонентами) такого комбинированного лечения в дополнение к лечению с ингибированием клеточного цикла, определенному выше, может быть хирургия, радиационная терапия или химиотерапия. Такая химиотерапия может включать три главные категории терапевтического агента:
(i) другие агенты, ингибирующие клеточный цикл, которые работают по тем же самым механизмам, что и описанные выше, или по другим;
(ii) цитостатические агенты, такие как антиэстрогены (например, тамоксифен, торемифен, ралоксифен, дролоксифен, йодоксифен), прогестогены (например, мегестрол ацетат), ингибиторы ароматазы (например, анастрозол, летразол, воразол эксеместан), антипрогестогены, антиандрогены (например, флутамид, нилутамид, бикалютамид, ципротерон ацетат), агонисты и антагонисты LHRH (например, гозерелин ацетат, люпролид), ингибиторы тестостерон 5α-дигидроредуктазы (например, финастерид), антиинвазивные агенты (например, ингибиторы металлопротеиназы, подобные маримастату, и ингибиторы активатора функции рецептора плазминогена урокиназы) и ингибиторы функции фактора роста (такие факторы роста включают в себя, например, фактор роста, полученный из тромбоцитов, и фактор роста гепатоцитов, такие ингибиторы включают в себя антитела факторов роста, антитела рецепторов фактора роста, ингибиторы тирозин киназы и ингибиторы серин/фреонин киназы); и
(iii) лекарственные средства против пролиферации новообразований и их сочетания, которые используются в медицинской онкологии, такие как антиметаболиты (например, антифолиаты, подобные метотрексату, фторпиримидины, подобные 5-фторурацилу, аналоги пурина и аденозина, цитозин арабинозид); противоопухолевые антибиотики (например, антрациклины, подобные доксорубицину, дауномицину, эпирубицину и идарубицину, митомицин-C, дактиномицин, митрамицин); соединения платины (например, цисплатин, карбоплатин); алкилирующие агенты (например, азотистый иприт, мельфалан, хлорамбуцил, бусульфан, циклофосфамид, ифосфамид, нитрозомочевины, тиотепа); антимитотические агенты (например, винка алкалоиды, подобные винкристину, и таксоиды, подобные таксолу, таксотере); ингибиторы топоизомеразы (например, эпиподофиллотоксины, подобные этопозиду и тенипозиду, амсакрин, топотекан). В соответствии с этим аспектом настоящего изобретения предусматривается фармацевтический продукт, содержащий соединение формулы (I), как определено выше, и дополнительное противоопухолевое вещество, как определено выше, для комбинированного лечения ракового заболевания.
В дополнение к их использованию в терапевтической медицине соединения формулы (I) и их фармацевтически приемлемые соли являются также полезными в качестве фармакологических инструментов при разработке и стандартизации систем исследования in vitro и in vivo для оценки воздействия ингибиторов, на их активность относительно клеточного цикла, на лабораторных животных, таких как коты, собаки, кролики, обезьяны, крысы и мыши, в качестве части исследования новых терапевтических агентов.
Для указанной выше другой фармацевтической композиции, способа, процесса, использования и особенностей производства лекарственных средств, также заявляются альтернативные и предпочтительные воплощения соединений по настоящему изобретению, описанных здесь.
ПРИМЕРЫ
Теперь изобретение будет иллюстрироваться с помощью следующих далее неограничивающих примеров, в которых, если не утверждается иного:
(i) температуры приведены в градусах Цельсия (°C); операции осуществляют при комнатной температуре или температуре окружающей среды, то есть при температуре в пределах 18-25°C;
(ii) органические растворы сушат над безводным сульфатом магния; выпаривание растворителя осуществляют с использованием роторного испарителя при пониженном давлении (600-4000 Паскалей; 4,5-30 мм рт.ст.), при температуре бани вплоть до 60°C;
(iii) хроматография означает флэш-хроматографию на силикагеле; тонкослойную хроматографию (ТСХ) осуществляют на пластинах, покрытых силикагелем;
(iv) как правило, после осуществления реакций, производят ТСХ, и времена реакций приведены только для иллюстрации;
(v) конечные продукты имеют удовлетворительные спектры протонного ядерного магнитного резонанса (ЯМР) и/или масс-спектры;
(vi) выходы приведены только для иллюстрации, и они не обязательно являются теми, которые могут быть получены при тщательной разработке способа; если требуется больше материала, приготовления повторяют;
(vii) когда они приведены, данные ЯМР приводятся в форме значений дельта для главных диагностических протонов, они приводятся в миллионных долях (м.д.) по отношению к тетраметилсилану (TMS), в качестве внутреннего стандарта, определяются при 300 МГц с использованием пердейтерий диметил сульфоксида (ДМСО-d6) в качестве растворителя, если не указано иного;
(viii) химические символы имеют их обычные значения; используются единицы и символы СИ;
(ix) отношения растворителей приведены в объем/объем (об./об.); и
(x) масс-спектры получают при энергии электронов 70 электронвольт, в режиме химической ионизации (CI), используя прямую экспозицию образца; там, где указано, ионизация осуществляется с помощью электронного удара (EI), бомбардировкой быстрыми атомами (FAB) или электрораспылением (ESP); приводятся значения для m/z; как правило, сообщается только об ионах, которые показывают исходную массу; и если не утверждается иного, ион сравнения представляет собой (MH)+;
(xi) если не утверждается иного, соединения, содержащие асимметрично замещенный атом углерода и/или серы, не разрешаются;
(xii) там, где синтез описывается, как аналогичный тому, который описан в предыдущем примере, используемые количества являются эквивалентами миллимолярных отношений по отношению к тем, которые используются в предыдущем примере;
(xvi) используются следующие сокращения:
ТГФ тетрагидрофуран;
ДМФ N,N-диметилформамид;
DMFDMA диметилформамид диметилацеталь;
EtOAc этилацетат;
MeOH метанол;
EtOH этанол;
DCM дихлорметан; и
ДМСО диметилсульфоксид.
xvii) там, где упоминается колонка Isolute SCX-2, это означает "ионнообменный" экстракционный картридж для адсорбции основных соединений, то есть полипропиленовую трубку, содержащую сильный катионобменный сорбент на основе бензолсульфоновой кислоты, используемый в соответствии с инструкциями производителя, получен от International Sorbent Technologies Limited, Dyffryn Business Park, Hengeod, Mid Glamorgan, UK, CF82 7RJ;
xviii) там, где упоминается аминовая колонка Isolute, это означает "ионнообменный" экстракционный картридж для адсорбции кислотных соединений, то есть полипропиленовую трубку, содержащую аминосилан, ковалентно связанный с частицами из окиси кремния, используемую в соответствии с инструкциями производителя, получен от International Sorbent Technologies Limited, Dyffryn Business Park, Hengeod, Mid Glamorgan, UK, CF82 7RJ;
xix) там, где упоминается колонка Chemelut, это означает экстракционный картридж для удаления воды, то есть полипропиленовую трубку, содержащую диатомовую землю, используемую в соответствии с инструкциями производителя, получен от Varian, Harbor City, California, USA.
Пример 1
2-(3-Хлоранилино)-4-(2-метилимидазол-5-ил)пиримидин
Гидрид натрия (45 мг 60% суспензии в минеральном масле, 1,12 ммоль) добавляют к перемешиваемой суспензии 5-(3-диметиламинопроп-2-ен-1-оил)-2-метилимидазола (100 мг, 0,56 ммоль) и 3-хлорфенилгуанидина (95 мг, 0,56 ммоль) в сухом 1-бутаноле (4,0 мл) в атмосфере азота. Смесь перемешивают при температуре окружающей среды в течение 15 минут, затем нагревают при 126°C в течение 26 час. Реакционной смеси дают возможность охладиться и летучие продукты удаляют путем выпаривания. Остаток суспендируют в воде (20 мл), и добавляют уксусную кислоту (67 мкл), и раствор экстрагируют DCM (3 x 20 мл). Экстракты объединяют, сушат (Na2SO4), и растворитель удаляют путем выпаривания. Остаток очищают с помощью колоночной хроматографии, элюируя DCM/MeOH (100:0, с увеличением полярности до 92:8), с получением указанного в заголовке соединения, 33 мг (21%), в виде твердого продукта. ЯМР: 2,35 (с, 3H), 6,95 (д, 1H), 7,23 (д, 1H), 7,30 (т, 1H), 7,67 (с, 1H), 7,72 (с, 1H), 8,05 (с, 1H), 8,43 (д, 1H), 9,62 (с, 1H), 12,15 (с, 1H); m/z: 286.
Пример 2
2-(3-(Хлоранилино)-4-(1,2-диметилимидазол-5-ил)пиримидин
5-(3-Диметиламинопроп-2-ен-1-оил)-1,2-диметилимидазол (способ 1; 111 мг, 0,58 ммоль) и 3-хлорфенилгуанидин (97 мг, 0,58 ммоль) обрабатывают, как описано в примере 1, с получением указанного в заголовке соединения, 51 мг (29%), в виде твердого продукта. ЯМР: 2,40 (с, 3H), 3,97 (с, 3H), 6,98 (д, 1H), 7,15 (д, 1H), 7,30 (т, 1H), 7,58 (д, 1H), 7,67 (с, 1H), 7,97 (с, 1H), 8,40 (д, 1H), 9,68 (с, 1H); m/z: 300.
Пример 3
2-Анилино-(2-метилимидазол-5-ил)пиримидин
Гидрид натрия (167 мг 60% суспензии в минеральном масле, 4,18 ммоль) добавляют к перемешиваемой суспензии 5-(3-диметиламинопроп-2-ен-1-оил)-2-метилимидазола (250 мг, 1,39 ммоль) и фенилгуанидин бикарбоната (275 мг, 1,39 ммоль), суспендируют в сухом 1-бутаноле (10 мл), в атмосфере азота и смесь перемешивают и нагревают в атмосфере азота, при 126°C, в течение 18 час. Реакционной смеси дают возможность охладиться, а затем добавляют фенилгуанидин бикарбонат (275 мг, 1,39 ммоль) и гидрид натрия (111 мг 60% суспензия в минеральном масле, 2,78 ммоль) и смесь перемешивают и нагревают при 126°C в течение дополнительных 20 час. Затем реакционную смесь извлекают, как описано в примере 1, с получением указанного в заголовке соединения 159 мг, (46%) в виде твердого продукта. ЯМР: 2,33 (с, 3H), 6,92 (т, 1H), 7,18 (д, 1H), 7,27 (т, 2H), 7,67 (с, 1H), 7,80 (д, 2H), 8,36 (д, 1H), 9,37 (с, 1H), 12,12 (с, 1H); m/z: 252.
Пример 4
4-(2-Метилимидазол-5-ил)-2-(4-сульфамоиланилино)пиримидин
Тионилхлорид (2,0 мл) добавляют к 2-анилино-4-(2-метилимидазол-5-ил)пиримидину (пример 3; 98 мг, 0,39 ммоль), охлажденному при 0°C, в атмосфере азота. Добавляют хлорсульфоновую кислоту (104 мкл, 1,56 ммоль), и смесь перемешивают при 0°C в течение 30 минут. Избыток тионилхлорида удаляют путем выпаривания и остаток обрабатывают смесью ТГФ (4,0 мл) и концентрированного водного раствора аммония (1,0 мл). Смесь перемешивают в течение 15 минут и летучие продукты удаляют путем выпаривания. Остаток растирают с водой и осажденный твердый продукт собирают путем фильтрования, промывают дистиллированной водой и сушат в вакууме с получением указанного в заголовке соединения, 62 мг, (48%). ЯМР: 2,33 (с, 3H), 7,10 (с, 2H), 7,24 (д, 1H), 7,72 (m, 3H), 7,95 (д, 2H), 8,43 (д, 1H), 9,83 (с, 1H); m/z: 331.
Пример 5
2-Анилино-4-(1,2-диметилимидазол-5-ил)пиримидин
5-(3-Диметиламинопроп-2-еноил)-1,2-диметилимидазол (способ 1; 314 мг, 1,62 ммоль) и фенилгуанидин бикарбонат (321 мг, 1,62 ммоль) обрабатывают, как описано в примере 1, с получением указанного в заголовке соединения, 113 мг, (26%), в виде твердого продукта. ЯМР: 2,37 (с, 3H), 3,93 (с, 3H), 6,95 (т, 1H), 7,08 (д, 1H), 7,28 (т, 2H), 7,59 (с, 1H), 7,69 (д, 2H), 8,35 (д, 1H), 9,43 (с, 1H); m/z: 266.
Пример 6
4-(1,2-Диметилимидазол-5-ил)-2-(4-сульфамоиланилино)пиримидин
Тионилхлорид (2,0 мл) добавляют к 2-анилино-4-(1,2-диметилимидазол-5-ил)пиримидину (пример 5; 94 мг, 0,36 ммоль), охлажденному при 0°C, в атмосфере азота. Добавляют хлорсульфоновую кислоту (94 мкл, 1,56 ммоль) и смесь перемешивают при 0°C в течение 30 минут, затем дают возможность нагреться и перемешивают в течение двух часов при температуре окружающей среды, а затем нагревают при 90°C в течение одного часа. Избыток тионилхлорида удаляют путем выпаривания и остаток азеотропно отгоняют вместе с толуолом. Полученный сырой сульфонилхлорид обрабатывают смесью ТГФ (4,0 мл), воды (2,0 мл) и концентрированного водного раствора аммония (1,0 мл). Смесь перемешивают в течение 15 минут и летучие продукты удаляют путем выпаривания. Остаток растирают с водой (5 мл) и осажденный твердый продукт собирают путем фильтрования, промывают дистиллированной водой и сушат в вакууме. Сырой продукт затем суспендируют и перемешивают в DCM (10 мл), содержащем несколько капель MeOH. Твердый продукт собирают путем фильтрования, промывают DCM и сушат в вакууме с получением указанного в заголовке соединения, 67 мг (54%). ЯМР: 2,38 (с, 3H), 3,96 (с, 3H), 7,13 (с, 2H), 7,20 (д, 1H), 7,63 (с, 1H), 7,73 (д, 2H), 7,88 (д, 2H), 8,43 (д, 1H), 9,88 (с, 1H); m/z: 345.
Пример 7
4-(1-Бензил-2-метилимидазол-5-ил)-2-(3-хлоранилино)пиримидин
Метоксид натрия (36,8 мг, 0,68 ммоль) добавляют к перемешиваемой суспензии 1-бензил-5-(3-диметиламинопроп-2-ен-1-оил)-2-метилимидазола (способ 5; 153 мг, 0,57 ммоль) и 3-хлорфенилгуанидина (106 мг, 0,62 ммоль) в сухом 1-бутаноле (1,0 мл) в атмосфере азота. Реакционную смесь нагревают с обратным холодильником в течение 4 часов, затем дают ей возможность охладиться. Летучие продукты удаляют путем выпаривания и остаток распределяют между EtOAc и насыщенным водным раствором бикарбоната натрия. Органическую фазу отделяют, сушат и растворитель удаляют путем выпаривания. Остаток очищают с помощью колоночной хроматографии, элюируя DCM и 7M метанольным раствором аммония (97:3), с получением указанного в заголовке соединения, 73 мг (34%). ЯМР: 2,35 (с, 3H), 5,78 (с, 2H), 6,84-7,00 (м, 5H), 7,07 (т, 1H), 7,15-7,30 (м, 4H), 7,56-7,65 (м, 2H), 8,29 (д, 1H); m/z 374.
Пример 8
2-(3-Хлоранилино)-4-[1-(2-метоксиэтил)имидазол-5-ил]пиримидин гидрохлорид
Трифторметилсульфоновый ангидрид (0,16 мл, 0,93 ммоль) добавляют к раствору 2-метоксиэтанола (73,7 мл, 0,88 ммоль) и диизопропилэтиламина (0,20 мл, 1,17 ммоль) в DCM (1 мл), при -20°C, и раствор перемешивают в течение 30 минут. Затем эту смесь добавляют к раствору 2-(3-хлоранилино)-4-(1-трифенилметилимидазол-4-ил)пиримидина (способ 2; 300 мг, 0,58 ммоль) в DCM (5 мл) при -20°C, и реакционной смеси дают возможность нагреться и перемешивают в течение 2 часов при температуре окружающей среды. Смесь распределяют между EtOAc и насыщенным водным раствором бикарбоната натрия. Органическую фазу отделяют, сушат и летучие продукты удаляют путем выпаривания. Остаток очищают с помощью колоночной хроматографии, элюируя DCM и 7M метанольным раствором аммония (99,5:0,5, с увеличением полярности до 96:4). Очищенный продукт растворяют в простом эфире и обрабатывают эфирным раствором хлористого водорода. Преципитат собирают путем фильтрования, промывают простым эфиром и сушат с получением указанного в заголовке соединения, 132 мг (69%). ЯМР: 3,17 (с, 3H), 3,63 (т, 2H), 4,96 (т, 2H), 5,86 (ушир.с, 1H), 7,04 (д, 1H), 7,28-7,44 (m, 2H), 7,60 (д, 1H), 7,88 (с, 1H), 8,56 (с, 1H), 8,64 (д, 1H), 9,28 (с, 1H), 10,0 (с, 1H); m/z: 330.
Пример 9
2-(3-Хлоранилино)-4-(имидазол-5-ил)пиримидин
Смесь 2-(3-хлоранилино)-4-(1-трифенилметилимидазол-4-ил)пиримидина (способ 2; 256 мг, 0,5 ммоль) в MeOH (3 мл) и 2M хлористоводородной кислоты (1 мл) перемешивают в течение 15 минут. Летучие продукты удаляют путем выпаривания, и остаток распределяют между EtOAc и насыщенным водным раствором бикарбоната натрия. Органический слой отделяют, сушат и растворитель удаляют путем выпаривания. Остаток очищают с помощью колоночной хроматографии, элюируя DCM и 7M метанольным раствором аммония (99,5:0,5, с увеличением полярности до 93:7), с получением указанного в заголовке соединения, 102 мг (75%), в виде твердого продукта. ЯМР: 6,95 (дд, 1H), 7,25-7,33 (м, 2H), 7,73 (дд, 1H), 7,81 (д, 2H), 8,06 (с, 1H), 8,46 (д, 1H), 9,68 (с, 1H), 12,48 (ушир.с, 1H); m/z: 270.
Пример 10
2-(3-Хлоранилино)-4-[1-(2-фталимидоэтил)имидазол-5-ил]пиримидин
2-Фталимидоэтилтрифлат (660 мг, 2,04 ммоль) добавляют к раствору 2-(3-хлоранилино)-4-(1-трифенилметилимидазол-4-ил)пиримидина (способ 2; 1,00 г, 1,95 ммоль) в DCM (5 мл), и реакционную смесь перемешивают в течение 4 часов. Растворитель удаляют путем выпаривания и к остатку добавляют MeOH (6 мл) и 2M хлористоводородную кислоту (1,5 мл). Смесь перемешивают в течение 5 минут, летучие продукты удаляют путем выпаривания и остаток распределяют между EtOAc и насыщенным водным раствором бикарбоната натрия. Полученный преципитат собирают путем фильтрования, промывают водой и EtOAc и сушат с получением указанного в заголовке соединения, 350 мг (40%), в виде твердого продукта. ЯМР: 3,81-3,96 (м, 2H), 4,77-4,92 (м, 2H), 6,98 (д, 1H), 7,06 (д, 1H), 7,31 (т, 1H), 7,37 (д, 1H), 7,63-7,80 (м, 6H), 7,92 (с, 1H), 8,27 (д, 1H), 9,50 (с, 1H); m/z: 443.
Пример 11-12
Следующие далее соединения приготавливают с помощью способа, аналогичного тому, что описывается в примере 10, с использованием соответствующих исходных материалов1, но при извлечении органический слой отделяют, сушат, растворитель удаляют путем выпаривания и остаток очищают с помощью колоночной хроматографии, элюируя DCM и 7M метанольным раствором аммония (99,5:0,5, с увеличением полярности до 93:7).
Пример 13
4-[1-(2-Аминоэтил)имидазол-5-ил]-2-(3-хлоранилино)пиримидин
Гидразин гидрат (54 мл, 1,73 ммоль) добавляют к суспензии 2-(3-хлоранилино)-4-[1-(2-фталимидоэтил)имидазол-5-ил]пиримидина (пример 10; 163 мг, 0,37 ммоль) в EtOH (5 мл) и смесь нагревают с обратным холодильником в течение 2 часов. Смеси дают возможность охладиться, летучие продукты удаляют путем выпаривания и остаток очищают с помощью колоночной хроматографии, элюируя DCM и 7M метанольным раствором аммония (90:10) с получением указанного в заголовке соединения, 69 мг (59%), в виде твердого продукта. ЯМР: 1,41 (ушир.с, 2H), 2,99 (т, 2H), 4,55 (т, 2H), 7,00-7,09 (м, 2H), 7,22-7,35 (м, 3H), 7,65-7,70 (м, 2H), 7,73-7,78 (м, 1H), 8,39 (д, 1H); m/z: 315.
Пример 14
2-Анилино-4-(1-метилимидазол-5-ил)пиримидин
Метоксид натрия (2,63 г, 48,7 ммоль) добавляют к раствору 5-(3-диметиламинопроп-2-ен-1-оил)-1-метилимидазола (способ 4; 2,91 г, 16,2 ммоль) и фенилгуанидин бикарбоната (3,52 г, 17,9 ммоль) в 2-пропаноле (14 мл) и реакционную смесь нагревают с обратным холодильником в течение 3 часов. Реакционной смеси дают возможность охладиться и распределяют между EtOAc и насыщенным водным раствором бикарбоната натрия. Органическую фазу отделяют, сушат и растворитель удаляют путем выпаривания. Остаток очищают с помощью колоночной хроматографии, элюируя DCM и 7M метанольным раствором аммония (97:3) с получением указанного в заголовке соединения, 2,57 г (64%), в виде твердого продукта; m/z: 252.
Пример 15
4-(1-Метилимидазол-5-ил)-2-(4-сульфамоиланилино)пиримидин
Хлорсульфоновую кислоту (0,48 мл, 7,16 ммоль) добавляют к суспензии 2-анилино-4-(1-метилимидазол-5-ил)пиримидина (пример 14; 449 мг, 1,79 ммоль) в тионилхлориде (9 мл), охлажденной при 0°C. Смеси дают возможность нагреться до температуры окружающей среды, затем нагревают с обратным холодильником в течение 30 минут. Летучие продукты удаляют путем выпаривания и остаток сушат ввысоком вакууме. К остатку добавляют 7M метанольный раствор аммония (30 мл) и смесь перемешивают в течение 10 минут. Летучие продукты удаляют путем выпаривания с получением указанного в заголовке соединения, 360 мг (61%) в виде твердого продукта. ЯМР: 4,04 (с, 3H), 7,15 (с, 2H), 7,27 (д, 1H), 7,73 (д, 2H), 7,84-7,91 (м, 3H), 8,06 (с, 1H), 8,50 (д, 1H), 9,92 (с, 1H); m/z: 331.
Пример 16
2-{4-[N-(3-Метоксипропил)сульфамоил]анилино}-4-(1-метилимидазол-5-ил)пиримидин
Хлорсульфоновую кислоту (0,22 мл, 3,18 ммоль) добавляют к суспензии 2-анилино-4-(1-метилимидазол-5-ил)пиримидина (пример 14; 200 мг, 0,80 ммоль) в тионилхлориде (4 мл), охлажденной при 0°C. Смеси дают возможность нагреться до температуры окружающей среды, перемешивают в течение 15 минут, затем нагревают с обратным холодильником в течение 20 минут. Летучие продукты удаляют путем выпаривания и твердый остаток сушат в высоком вакууме. Остаток суспендируют в пиридине (3 мл), охлаждают до -20°C, и добавляют диизопропилэтиламин (0,56 мл, 3,98 ммоль), а затем 3-метоксипропиламин (0,16 мл, 1,60 ммоль). Реакционной смеси дают возможность нагреться до температуры окружающей среды и перемешивают в течение 30 минут. Добавляют EtOAc (15 мл) и смесь промывают насыщенным водным раствором бикарбоната натрия (15 мл), а затем насыщенным раствором соли (15 мл). Растворитель удаляют путем выпаривания и остаток очищают с помощью колоночной хроматографии, элюируя DCM и 2М метанольным раствором аммония (100:0, с увеличением полярности до 85:15) с получением указанного в заголовке соединения, 89 мг (28%), в виде твердого продукта. ЯМР: 1,75 (м, 2Н), 2,76 (кв, 2Н), 3,14 (с, 3Н), 3,22-3,30 (м, 2Н), 4,01 (с, 3Н), 7,25 (д, 1Н), 7,34 (т, 1Н), 7,70 (д, 2Н), 7,77 (с, 1Н), 7,83 (с, 1Н), 7,91 (д, 2Н), 8,47 (д, 1Н), 9,92 (с, 1Н); m/z: 403.
Примеры 17-25
Следующие далее соединения приготавливают способом, аналогичным тому, что описывается в примере 16, с использованием соответствующих промежуточных соединений.
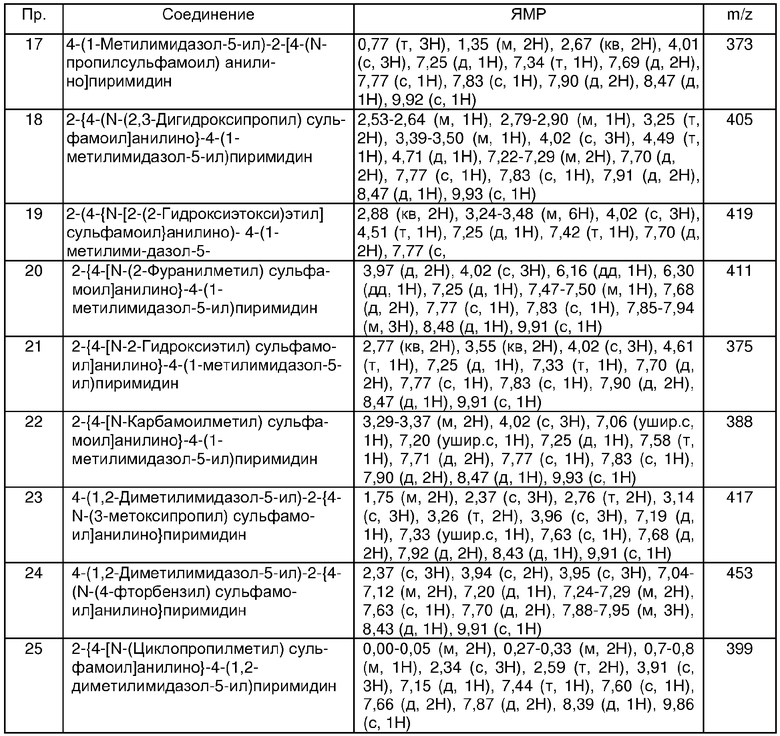
Пример 26
4-(1,2-Диметилимидазол-5-ил)-2-(4-{N-[3-(пирролидин-2-он-1-ил)пропил]сульфамоил}анилино)пиримидин
Эфирный раствор хлористого водорода (1 мл 1M раствора, 1,0 ммоль) добавляют к раствору 4-{N-[3-(пирролидин-2-он-1-ил)пропил]сульфамоил}анилина (способ 13, 300 мг, 1,0 ммоль) в MeOH (минимальный объем). Летучие продукты удаляют путем выпаривания и к остатку добавляют цианамид (50 мг, 1,2 ммоль), а затем диметилацетамид (0,5 мл). Смесь нагревают до 100°C в течение 30 минут. Добавляют 5-(3-диметиламинопроп-2-еноил)-1,2-диметилимидазол (способ 1; 180 мг, 0,93 ммоль) и метоксид натрия (110 мг, 2,0 ммоль) и смесь нагревают с обратным холодильником в течение одного часа. Смеси дают возможность охладиться и распределяют между EtOAc и водным раствором бикарбоната натрия. Органический слой отделяют, промывают насыщенным раствором соли, сушат (Na2SO4) и летучие продукты удаляют путем выпаривания. Остаток очищают с помощью колоночной хроматографии, элюируя DCM и 7M метанольным раствором аммония (96:4), с получением указанного в заголовке соединения, 220 мг (50%). ЯМР: 1,48-1,58 (м, 2H), 1,79-1,89 (м, 2H), 2,14 (т, 2H), 2,37 (с, 3H), 2,68 (кв, 2H), 3,10 (т, 2H), 3,21 (т, 2H), 3,95 (с, 3H), 7,19 (д, 1H), 7,34 (т, 1H), 7,63 (с, 1H) 7,69 (д, 2H), 7,92 (д, 2H) 8,43 (д, 1H), 9,92 (с, 1H); m/z: 470.
Пример 27
Следующее далее соединение приготавливают способом, аналогичным тому, что описывается в примере 26, с использованием соответствующих промежуточных соединений.
пиримидин
Пример 28
2-Анилино-4-(1-этил-2-метилимидазол-5-ил)пиримидин
5-(3-Диметиламинопроп-2-ен-1-оил)-1-этил-2-метилимидазол (способ 16; 2,10 г, 10,1 ммоль), фенилгуанидин бикарбонат (2,2 г, 11,1 ммоль) и метоксид натрия (1,2 г, 22,2 ммоль) суспендируют в безводном DMA (15 мл), и смесь нагревают при 110°C в течение 18 часов. Реакционной смеси дают возможность охладиться до температуры окружающей среды, и выливают в воду (50 мл). Раствор экстрагируют EtOAc (2 x 50 мл). Объединенные экстракты промывают водой (2 x 50 мл), а затем насыщенным раствором соли (2 x 50 мл), сушат и летучие продукты удаляют путем выпаривания. Остаток растирают с простым эфиром, собирают путем фильтрования и сушат на воздухе, с получением указанного в заголовке соединения (1,48 г, 53%) в виде красновато-коричневого твердого продукта. ЯМР: 1,17 (т, 3H), 2,38 (с, 3H), 4,52 (кв, 2H), 6,93 (т, 1H), 7,08 (д, 1H), 7,27 (т, 2H), 7,60 (с, 1H), 7,62 (д, 2H), 8,35 (д, 1H), 9,35 (с, 1H); m/z 280.
Примеры 29-33
Следующие далее соединения синтезируют аналогично способу примера 28.
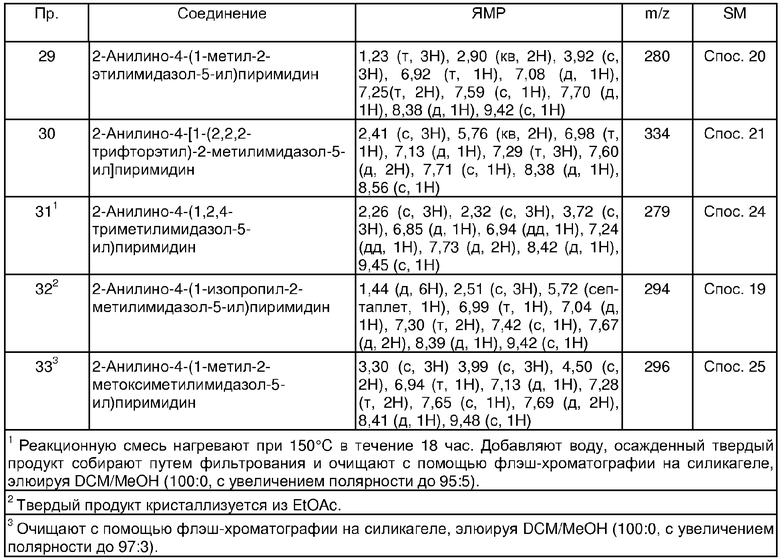
Пример 34
4-(1,2-Диметилимидазол-5-ил)-2-(4-мезиламиноанилино)пиримидин
Метансульфонилхлорид (0,055 мл, 0,71 ммоль) добавляют к раствору 4-(1,2-диметилимидазол-5-ил)-2-(4-аминоанилино)пиримидина (пример 165; 0,18 г, 0,64 ммоль) и пиридина (0,052 мл, 0,64 ммоль) в DCM (2,0 мл), охлажденному при 4°C. Смеси дают возможность нагреться до температуры окружающей среды. Смесь распределяют между насыщенным водным раствором бикарбоната натрия и EtOAc. Органический слой отделяют, летучие продукты выпаривают и остаток очищают с помощью колоночной хроматографии на силикагеле, элюируя DCM/7M метанольным раствором аммония (96:4), с получением указанного в заголовке соединения (0,15 г, 65%) в виде твердого продукта. ЯМР: 2,36 (с, 3H), 2,90 (с, 3H), 3,91 (с, 3H), 7,06 (д, 1H), 7,14 (д, 2H), 7,57 (с, 1H), 7,64 (д, 2H), 8,33 (д, 1H), 9,37 (ушир.с, 1H), 9,42 (с, 1H); m/z 359.
Пример 35
4-(1,2-Диметилимидазол-5-ил)-2-{4-[N-(2-метоксиэтил)сульфамоил]анилино}пиримидин
Натрий трет-бутоксид (1,04 г, 10,8 ммоль) добавляют к дегазированному раствору 2-амино-4-(1,2-диметилимидазол-5-ил)пиримидина (способ 26; 567 мг, 3 ммоль), N-(2-метоксиэтил)-4-йодбензолсульфонамида (способ 40; 1,54 г, 4,5 ммоль), трис(дибензилиденацетон)дипалладия(0) (72 мг, 0,15 ммоль) и 2,2'-бис(дифенилфосфино)-1,1'-бинафтила (102 мг, 0,15 ммоль) в диоксане (36 мл), и смесь нагревают при 80°C в течение ночи. Реакционную смесь охлаждают до комнатной температуры, и добавляют MeOH (5 мл), и смесь выливают в колонку Isolute SCX-2, элюируя сначала MeOH (10 x 30 мл), а затем продукт элюируют 2% метанольным раствором аммония (10 x 30 мл). Растворитель удаляют путем выпаривания и остаток растворяют в EtOAc (100 мл), промывают водой (3 x 100 мл), а затем насыщенным раствором соли (100 мл), сушат и растворитель удаляют путем выпаривания с получением указанного в заголовке соединения (1,0l г, 84%) в виде пены. ЯМР: 2,40 (с, 3H), 3,07 (кв, 2H), 3,20 (с, 3H), 3,38 (т, 2H), 3,86 (с, 3H), 5,00 (т, 1H), 6,95 (д, 1H), 7,47 (с, 2H), 7,71 (м, 4H), 8,36 (д, 1H); m/z 403.
Примеры 36-72
Следующие далее соединения синтезируют аналогично способу примера 35.
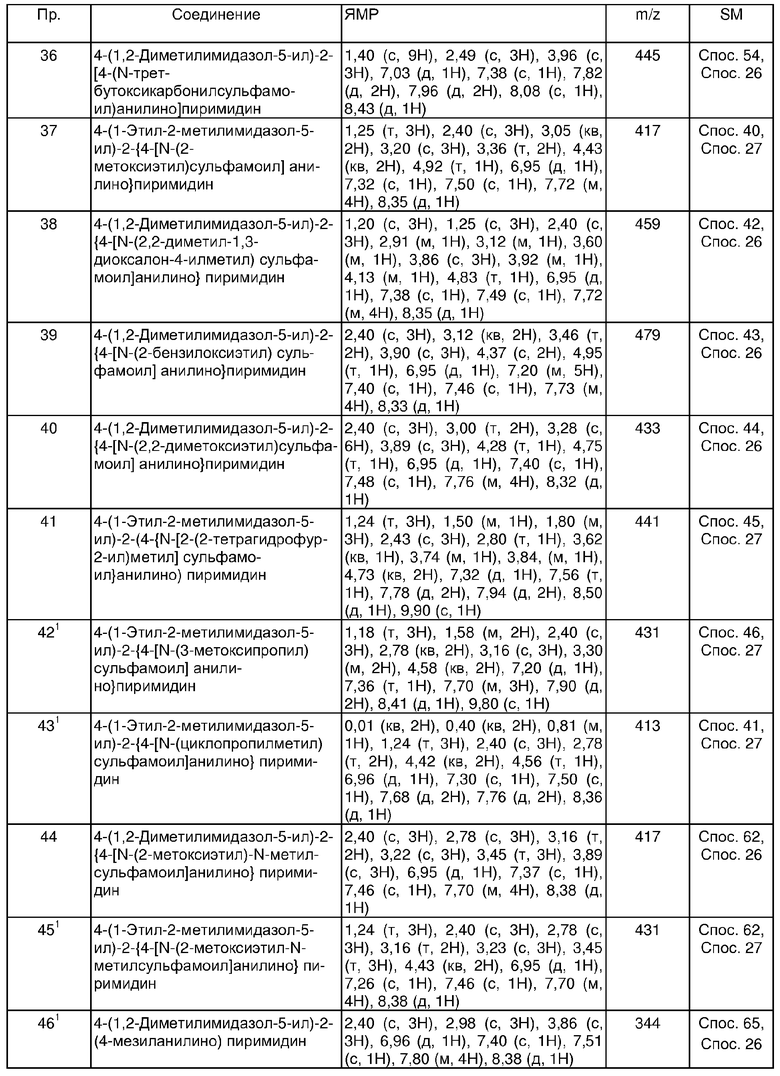
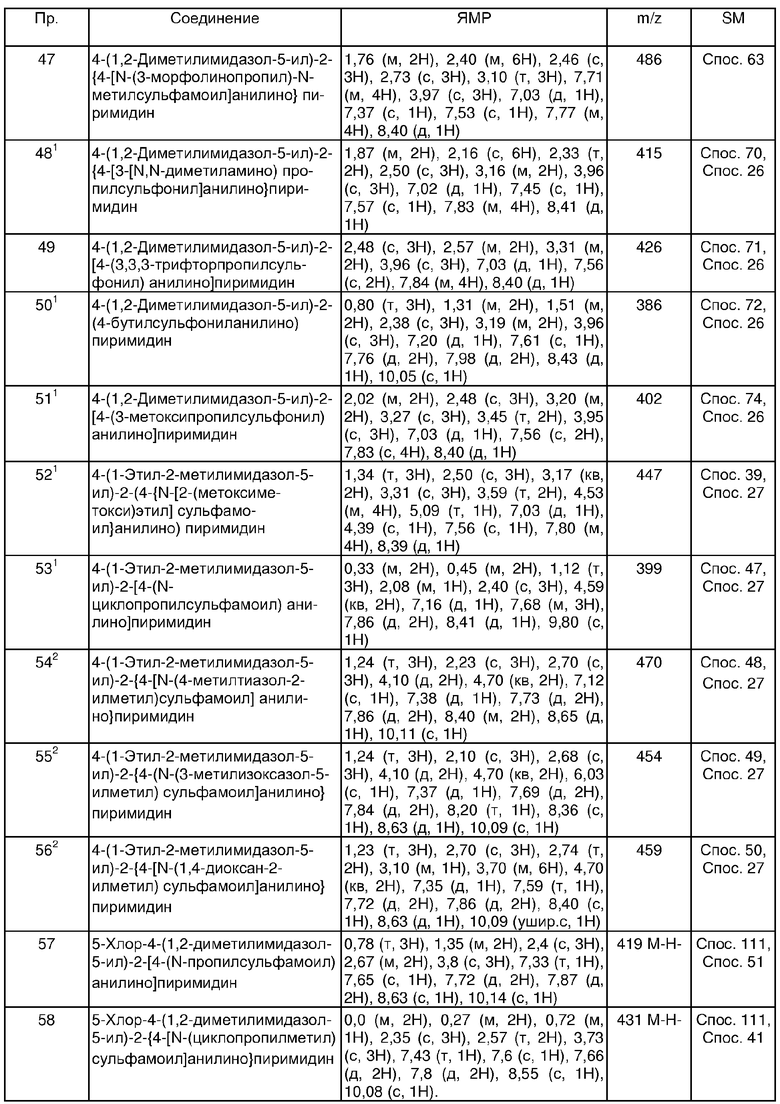
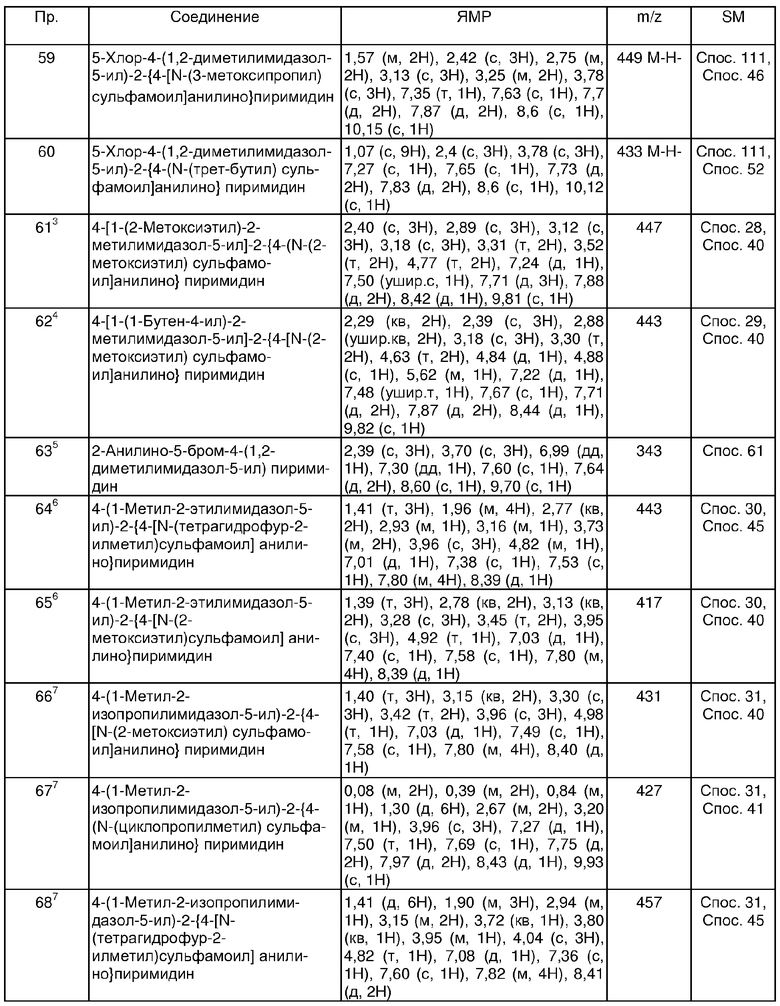
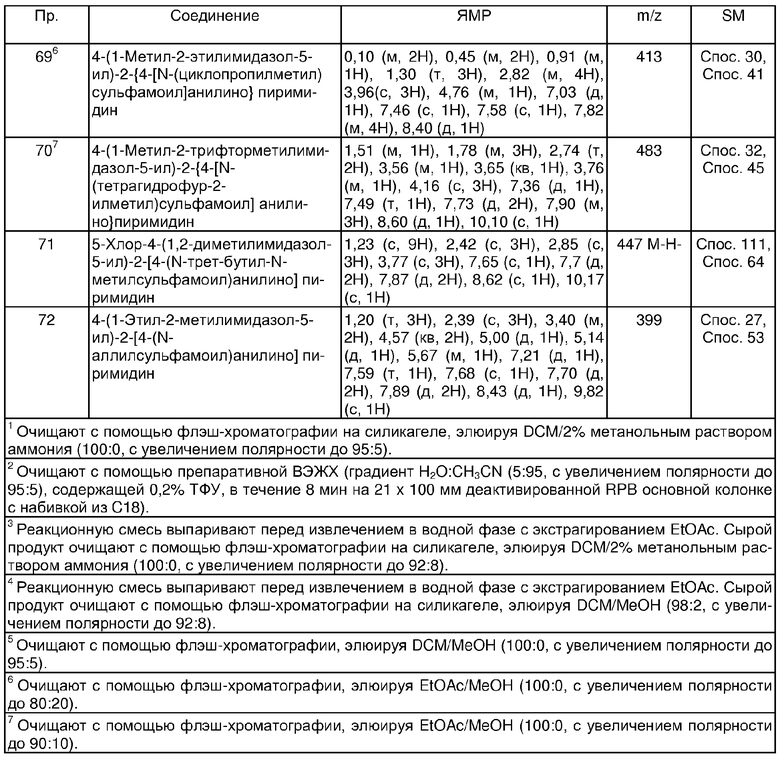
Пример 73
4-(1,2-Диметилимидазол-5-ил)-2-(4-{N-[2-(2-метоксиэтокси)этил]сульфамоил}анилино)пиримидина гидрохлорид
1M Эфирный раствор хлористого водорода (4 мл) добавляют к раствору 4-(1,2-диметилимидазол-5-ил)-2-(4-(N-трет-бутоксикарбонил)-N-[2-(2-метоксиэтокси)этил]сульфамоил)анилино)пиримидина (способ 55; 77 мг, 0,14 ммоль) в безводном диоксане (2 мл), и смесь перемешивают при температуре окружающей среды в течение 5 дней. Летучие продукты удаляют путем выпаривания и остаток растирают c простым эфиром, собирают путем фильтрования, промывают простым эфиром (2 x 10 мл) и сушат с получением указанного в заголовке соединения (65 мг (96%)) в виде желтого твердого продукта. ЯМР: 2,70 (с, 3H), 2,86 (м, 2H), 3,18 (с, 3H), 3,36 (м, 4H), 3,42 (м, 2H), 4,08 (с, 3H), 7,38 (д, 1H), 7,58 (с, 1H), 7,74 (д, 2H), 7,93 (д, 2H), 8,40 (с, 1H), 8,69 (д, 1H), 10,25 (с, 1H); m/z 447.
Примеры 74-75
Следующие далее соединения синтезируют аналогично способу примера 73.
Пример 76
4-(1,2-Диметилимидазол-5-ил)-2-{4-[N-(2-мезилэтил)сульфамоил]анилино}пиримидин
4-Диметиламинопиридин (3 мг, 0,025 ммоль) и 2-(метилсульфонил)этанамин (200 мкл, 2 ммоль) добавляют к раствору 4-(1,2-диметилимидазол-5-ил)-2-(4-(фторсульфонил)анилино)пиримидина (способ 59; 87 мг, 0,25 ммоль) в NMP (1 мл) и смесь нагревают при 100°С в течение 18 час. Смеси дают возможность охладиться до температуры окружающей среды и растворитель удаляют путем выпаривания. Остаток очищают с помощью препаративной ЖХ-МС (постоянный поток 5% об/об (35% NH3 в МеОН) с градиентом H2O: CH3CN) (5:95, с увеличением полярности до 95:5) в течение 7,5 мин), с получением указанного в заголовке соединения (91 мг, 81%) в виде твердого продукта. ЯМР: 2,38 (с, 3Н), 2,97 (с, 3Н), 3,11 (м, 2Н), 3,21 (м, 2Н), 3,95 (с, 3Н), 7,20 (д, 1Н), 7,61 (с, 1Н), 7,75 (м, 3Н), 7,95 (д, 2Н), 8,43 (д, 1Н), 9,95 (с, 1Н); m/z 451.
Примеры 77-79
Следующие далее соединения синтезируют аналогично способу примера 76.
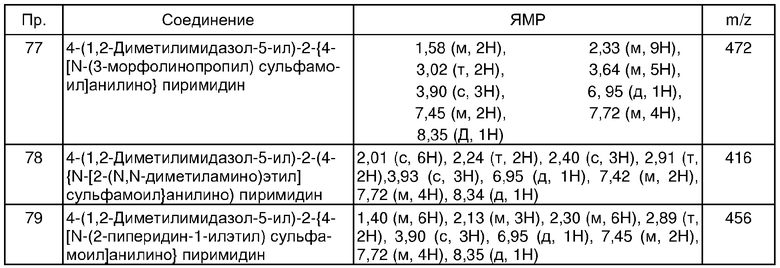
Пример 80
4[1-(2-Метоксиэтил)-2-метилимидазол-5-ил]-{4-[N-(тетрагидрофур-2-илметил)сульфамоил]анилино}пиримидин
Смесь 4-[1-(2-метоксиэтил)-2-метилимидазол-5-ил]-2-N-(4-фторсульфониланилино)пиримидина (способ 60; 200 мг, 0,51 ммоль) и диметиламинопиридина, нанесенного на полистирол (800 мг: 1,6 ммоль/г смолы) в 1-метил-2-пирролидоне (4 мл) перемешивают в течение 10 минут при температуре окружающей среды. Добавляют тетрагидрофурфуриламин (258 мг, 2,55 ммоль) и реакционную смесь нагревают при 90°C в течение 40 часов, затем, при 100°C в течение 48 часов. Летучие продукты удаляют путем выпаривания, и остаток очищают с помощью колоночной хроматографии на силикагеле, элюируя DCM/MeOH (99:1, с увеличением полярности до 96:4), для получения очищенногопродукта (120 мг), растирают с простым эфиром, собирают путем фильтрования и сушат при 80°C в вакууме с получением указанного в заголовке соединения (55 мг, 23%). ЯМР: 1,52 (м, 1H), 1,70-1,88 (м, 3H), 2,39 (с, 3H), 2,75 (м, 2H), 3,10 (с, 3H), 3,49 (т, 2H), 3,55 (м, 1H), 3,67 (м, 1H), 3,78 (м, 1H), 4,74 (т, 2H), 7,23 (д, 1H), 7,49 (т, 1H), 7,70 (д, 3H), 7,85 (д, 2H), 8,42 (д, 1H), 9,79 (с, 1H); m/z 473.
Примеры 81-82
Следующие далее соединения синтезируют аналогично примеру 80.
Пример 83
4-(1-Этил-2-метилимидазол-5-ил)-2-(4-(N-(гидроксиэтил)сульфамоил)анилино)пиримидин
Хлорсульфоновую кислоту (150 мкл, 2,16 ммоль) добавляют по каплям к раствору 2-анилино-4-(1-этил-2-метилимидазол-5-ил)пиримидина (пример 28; 150 мг, 0,54 ммоль) в тионилхлориде (3 мл), охлажденном при 0°C и смесь перемешивают при 0°C в течение 10 минут, затем нагревают при 90°C в течение 90 минут. Летучие продукты удаляют путем выпаривания и остаток сушат в высоком вакууме (<2 мм рт.ст) в течение 1 часа. Полученный твердый продукт помещают в атмосферу азота и добавляют раствор этаноламина (494 мг, 8,1 ммоль) в MeOH (3 мл). Смесь перемешивают в течение 30 минут и летучие продукты выпаривают в вакууме. Добавляют воду (20 мл) и осажденный твердый продукт собирают путем фильтрования, промывают водой (2 x 10 мл) и простым эфиром (2 x 10 мл) и сушат в вакууме при 60°C с получением указанного в заголовке соединения (177 мг, 81%) в виде бежевого твердого продукта. ЯМР: 1,22 (т, 3H), 2,41 (с, 3H), 2,80 (с, 2H), 3,38 (кв, 2H), 4,63 (м, 3H), 7,20 (д, 1H), 7,36 (с, 1H), 7,77 (с, 1H), 7,82 (д, 2H), 7,91 (д, 2H), 8,34 (д, 1H), 9,85 (с, 1H); m/z 403.
Примеры 84-125
Следующие далее соединения синтезируют аналогично способу примера 83.
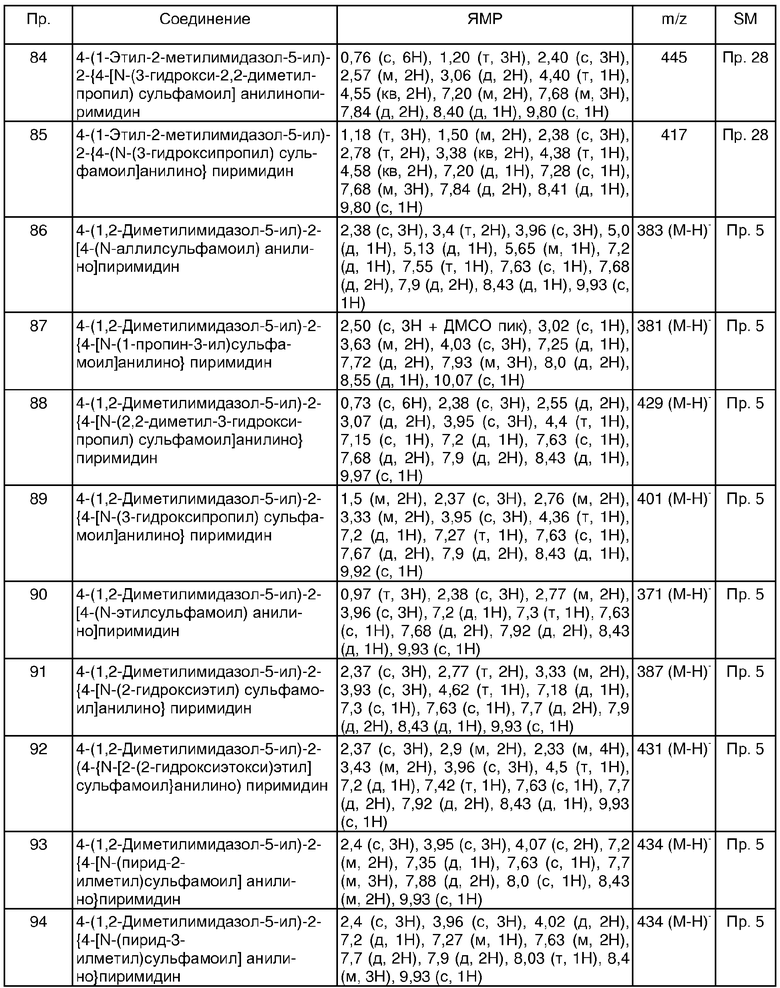
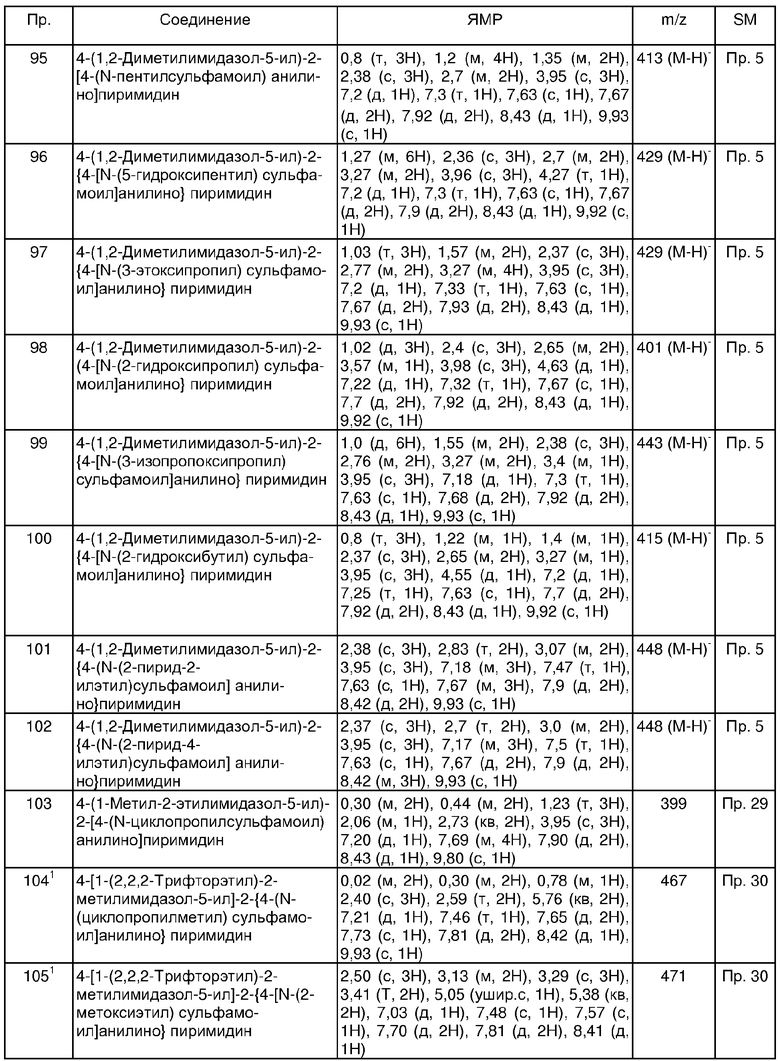
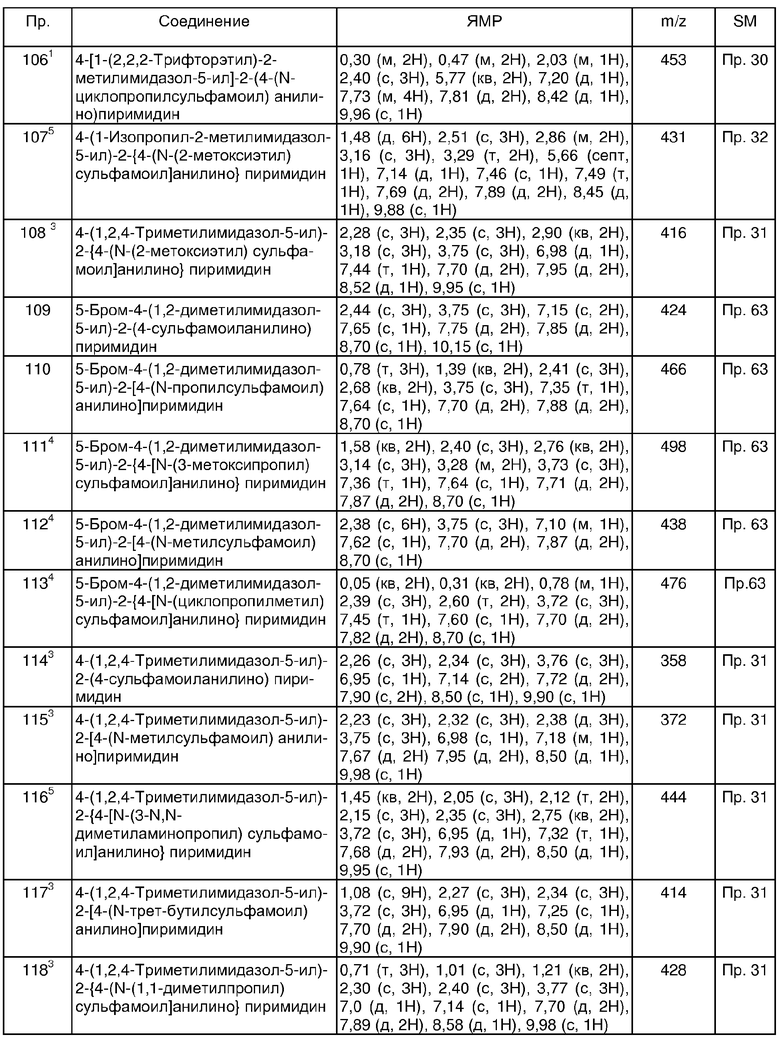
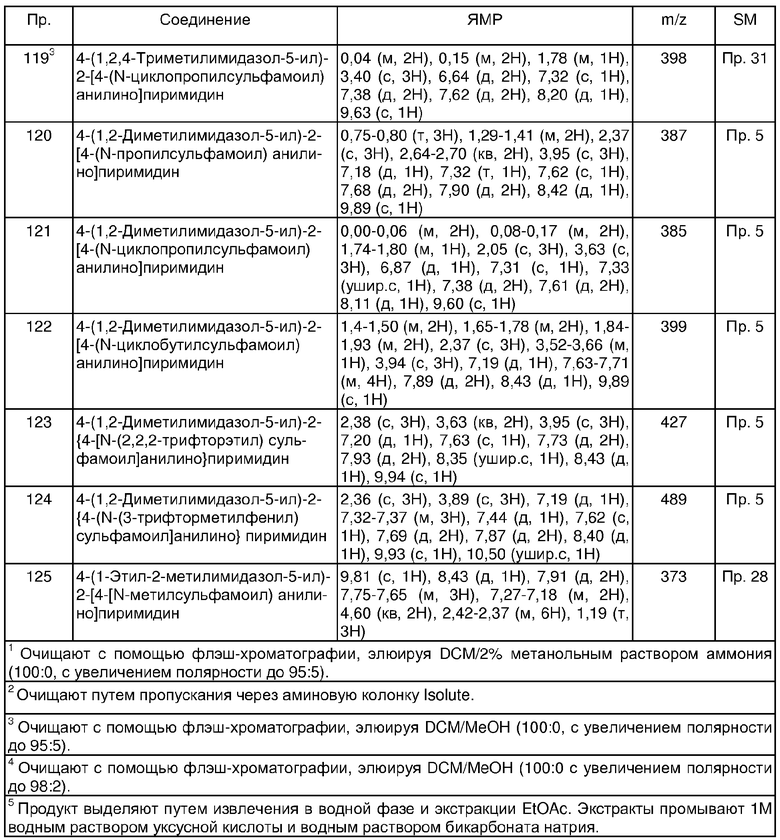
Пример 126
4-(1-Этил-2-метилимидазол-5-ил)-2-(4-{N-[2-(2-гидроксиэтокси)этил]сульфамоил}анилино)пиримидин
Хлорсульфоновую кислоту (150 мкл, 2,16 ммоль) добавляют по каплям к раствору 2-анилино-4-(1-этил-2-метилимидазол-5-ил)пиримидина (пример 28; 150 мг, 0,54 ммоль) в тионилхлориде (3 мл), охлажденному до 0°C, и смесь перемешивают в течение 10 минут при 0°C, затем нагревают при 90°C в течение 90 минут. Летучие продукты удаляют путем выпаривания, и полученный твердый продукт помещают в высокий вакуум (<2 мм рт.ст.) в течение 1 час. Полученный твердый продукт помещают в атмосферу азота, и осторожно добавляют раствор 2-(2-аминоэтил)этанола (114 мг, 1,08 ммоль) и диэтилметиламина в MeOH (3 мл). Раствор перемешивают в течение 30 минут и летучие продукты выпаривают. Добавляют воду (20 мл) и осажденный твердый продукт собирают путем фильтрования и промывают водой (2 x 10 мл). Остаток растворяют в MeOH (5 мл) и загружают в аминовую колонку Isolute, элюируют MeOH (30 мл), и фракции, содержащие продукт, выпаривают с получением указанного в заголовке соединения (190 мг, 79%) в виде бежевого твердого продукта. ЯМР: 1,18 (т, 3H), 2,39 (с, 3H), 2,89 (т, 2H), 3,15 (м, 7H), 4,38 (кв, 2H), 7,21 (д, 1H), 7,71 (м, 3H), 7,89 (д, 2H), 8,41 (д, 1H), 9,82 (с, 1H); m/z 447.
Примеры 127-144
Следующие далее соединения синтезируют аналогично способу примера 126.
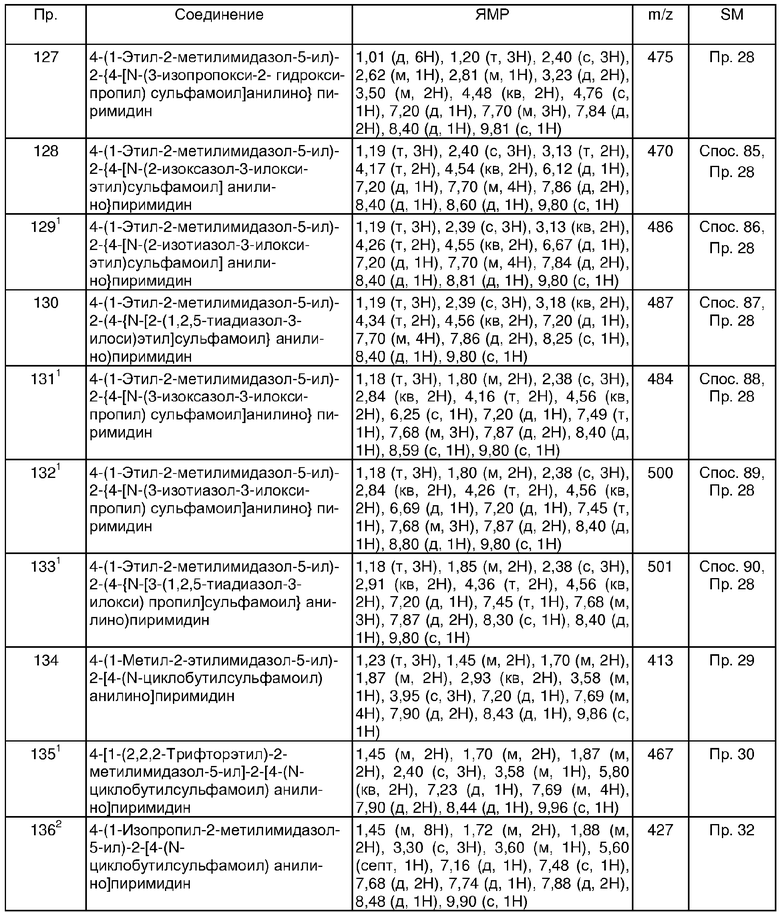
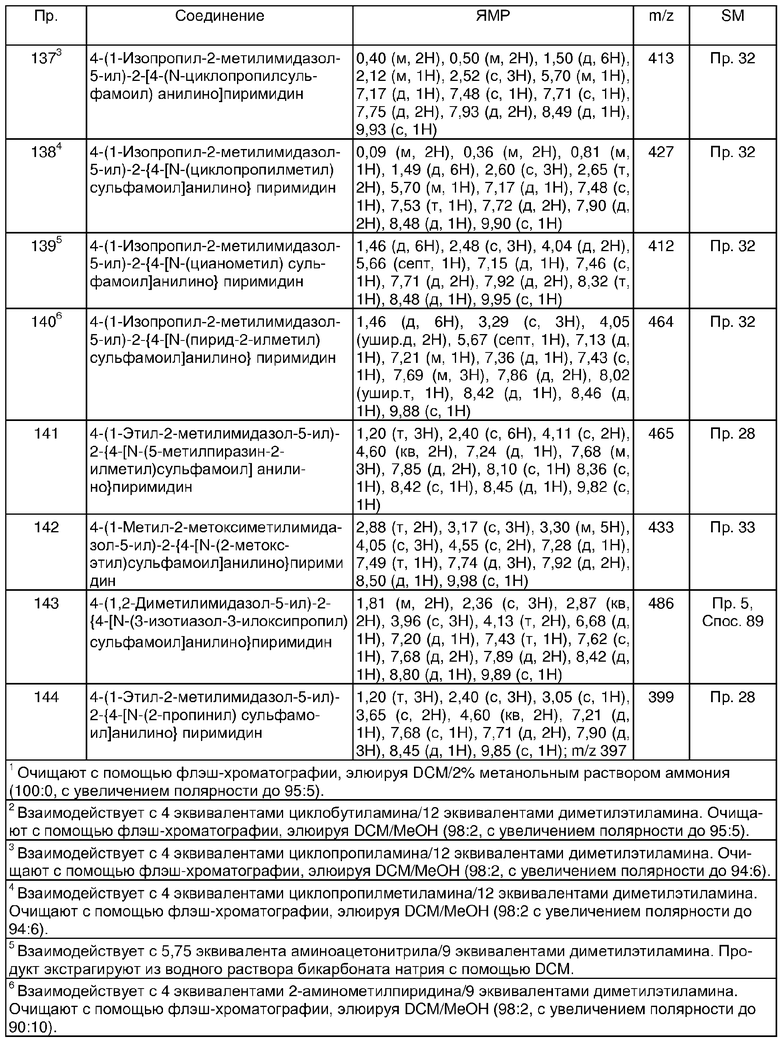
Пример 145
5-Бром-4-(1,2-диметилимидазол-5-ил)-2-{4-[N-(2-метоксиэтил)сульфамоил]анилино}пиримидин
Бром (8 мкл, 0,14 ммоль) добавляют к раствору 4-(1,2-диметилимидазол-5-ил)-2-{4-[N-(2-метоксиэтил)сульфамоил]анилино}пиримидина (пример 35; 52 мг, 0,13 ммоль) в ледяной уксусной кислоте (2 мл), нагретого до 60°C. Смесь нагревают при 60°C в течение 4 часов, затем растворитель удаляют путем выпаривания. Остаток растворяют в DCM (20 мл), промывают насыщенным водным раствором бикарбоната натрия (20 мл), сушат (колонка Chemelut1005) и очищают с помощью флэш-хроматографии, элюируя DCM/2% метанольным раствором аммония (100:0, с увеличением полярности до 97:3) с получением указанного в заголовке соединения (37 мг, 60%) в виде белой пены. ЯМР: 2,40 (с, 3H), 3,06 (кв, 2H), 3,20 (с, 3H), 3,36 (т, 2H), 3,68 (с, 3H), 5,00 (т, 1H), 7,56 (с, 1H), 7,67 (д, 2H), 7,73 (д, 2H), 7,80 (с, 1H), 8,53 (с, 1H); m/z 483.
Примеры 146-148
Следующие далее соединения синтезируют аналогично способу примера 145.
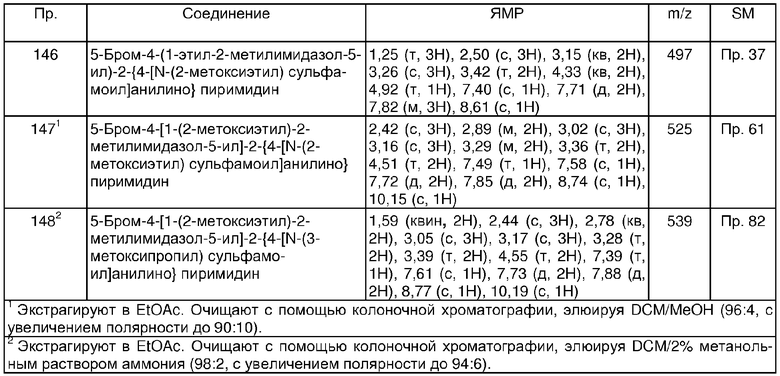
Пример 149
5-Хлор-4-(1-этил-2-метилимидазол-5-ил)-2-{4-[N-(2-метоксиэтил)сульфамоил]анилино}пиримидин
N-Хлорсукцинимид (80 мг, 0,6 ммоль) добавляют к раствору 4-(1-этил-2-метилимидазол-5-ил)-2-{4-[N-(2-метоксиэтил) сульфамоил]анилино}пиримидина (пример 37; 208 мг, 0,5 ммоль) в ледяной уксусной кислоте (5 мл) и смесь нагревают при 60°C в течение 3 часов. Растворитель выпаривают и остаток растворяют в DCM (30 мл), промывают насыщенным водным раствором бикарбоната натрия (20 мл), водный слой экстрагируют DCM (20 мл). Экстракты в DCM объединяют, сушат (колонка Chemelut 1005) и растворитель выпаривают. Остаток очищают с помощью флэш-хроматографии на силикагеле, элюируя DCM/2% метанольным раствором аммония (100:0, с увеличением полярности до 97:3) с получением указанного в заголовке соединения (110 мг, 44%) в виде белой пены. ЯМР: 1,24 (т, 3H), 2,45 (с, 3H), 3,09 (кв, 2H), 3,28 (с, 3H), 3,40 (т, 2H), 4,32 (т, 2H), 4,92 (т, 1H), 7,40 (с, 1H), 7,72 (д, 2H), 7,83 (д, 2H), 7,88 (с, 1H), 8,49 (с, 1H); m/z 451.
Примеры 150-153
Следующие далее соединения синтезируют аналогично способу примера 149.
Пример 154
4-(1,2-Диметилимидазол-5-ил)-2-{4-[N-(2,3-дигидроксипропил)сульфамоил]анилино}пиримидин
Воду (0,5 мл), а затем ТФУ (2,5 мл), добавляют к раствору 4-(1,2-диметилимидазол-5-ил)-2-{4-[N-(2,2-диметил-1,3-диоксалон-4-илметил)сульфамоил]анилино}пиримидина (пример 38,119 мг, 0,26 ммоль) в DCM (2 мл) и смесь перемешивают при температуре окружающей среды в течение 1 часа. Растворитель выпаривают и к остатку добавляют 1M эфирного раствора хлористого водорода (5 мл) и эфира (20 мл). Полученный преципитат собирают путем фильтрования и сушат в вакууме. Твердый продукт суспендируют в MeOH (2 мл), добавляют 1M водный раствор гидроксида лития (2 мл) и смесь перемешивают в течение 1 часа при температуре окружающей среды. Реакционную смесь выливают в колонку Isolute SCX-2, промывают MeOH (10 x 15 мл) и продукт элюируют 2% метанольным раствором аммония (5 x 15 мл). Растворитель удаляют путем выпаривания с получением указанного в заголовке соединения (66 мг, 61%) в виде белого твердого продукта. ЯМР: 2,38 (с, 3H), 2,60 (м, 1H), 2,83 (м, 1H), 3,25 (м, 2H), 3,43 (м, 1H), 3,95 (с, 3H), 4,48 (т, 1H), 4,70 (д, 1H), 7,20 (м, 2H), 7,62 (с, 1H), 7,69 (д, 2H), 7,90 (д, 2H), 8,41 (д, 1H), 9,90 (с, 1H); m/z 419.
Пример 155
5-Хлор-4-(1,2-диметилимидазол-5-ил)-2-(4-сульфамоиланилино)пиримидин
Смесь 5-хлор-4-(1,2-диметилимидазол-5-ил)-2-{4-[N-(трет-бутил)сульфамоил]анилино}пиримидина (пример 60; 116 мг, 0,267 ммоль), трифторуксусной кислоты (2,7 мл), воды (0,3 мл) и анизола (145 мкл, 1,34 ммоль), перемешивают при температуре окружающей среды в течение 72 часов. Затем смесь концентрируют путем выпаривания и остаток обрабатывают водой и эфиром. Осажденный твердый продукт собирают путем фильтрования, промывают водой и простым эфиром и сушат с получением указанного в заголовке соединения (87 мг, 86%) в виде белого твердого продукта. ЯМР: 2,4 (с, 3H), 3,78 (с, 3H), 7,15 (с, 2H), 7,65 (с, 1H), 7,73 (д, 2H), 7,83 (д, 2H), 8,6 (с, 1H), 10,11 (с, 1H); m/z 378 (M-H)-.
Пример 156
Следующие далее соединения синтезируют аналогично способу примера 155.
Пример 157
5-Бром-4-(1-метилимидазол-5-ил)-2-(4-сульфамоиланилино)пиримидин
Бром (75,5 мг, 0,47 ммоль) добавляют к раствору 4-(1-метилимидазол-5-ил)-2-(4-сульфамоиланилино)пиримидина (пример 15; 0,14 г, 0,42 ммоль) и ацетата натрия (41,7 мг, 0,51 ммоль) в уксусной кислоте (4 мл), и смесь перемешивают в течение 1 часа. Летучие продукты выпаривают, и остаток распределяют между EtOAc и насыщенным водным раствором бикарбоната калия. Органическую фазу отделяют и сушат. Остаток предварительно абсорбируют на силикагеле и очищают с помощью колоночной хроматографии на силикагеле, элюируя DCM/2% метанольным раствором аммония (9:1) с получением указанного в заголовке соединения (91 мг, 52%). ЯМР: 10,14 (с, 1H), 8,75 (с, 1H), 7,90-7,69 (м, 4H), 7,17 (с, 2H), 3,84 (с, 3H); m/z 409.
Пример 158
2-(3-Хлоранилино)-4-[1-(2-ацетамидоэтил)имидазол-5-ил]пиримидин
Уксусный ангидрид (0,58 мкл, 1,0 ммоль) добавляют к раствору 2-(3-хлоранилино)-4-[1-(2-аминоэтил)имидазол-5-ил]пиримидина (пример 13; 0,30 г, 0,63 ммоль) в пиридине (2 мл) при 0°C. Смеси дают возможность нагреться до температуры окружающей среды и перемешивают в течение 2 часов. Добавляют 7M метанольного раствора аммония (0,5 мл) и смесь разбавляют EtOAc (10 мл). Преципитат удаляют путем фильтрования и фильтрат предварительно абсорбируют на силикагеле, и очищают с помощью колоночной хроматографии на силикагеле, элюируя DCM/2% метанольным раствором аммония (11:1) с получением указанного в заголовке соединения (88 мг, 39%) в виде белого твердого продукта. ЯМР: 9,68 (с, 1H), 8,43 (д, 1H), 8,03-7,96 (м, 2H), 7,81 (с, 1H), 7,79 (с, 1H), 7,60 (дд, 1H), 7,33 (т, 1H), 7,12 (д, 1H), 6,98 (дд, 1H), 4,56-4,46 (м, 2H), 3,44-3,37 (м, 2H), 1,80 (с, 3H); m/z 357.
Пример 159
Следующее далее соединение синтезируют аналогично способу примера 158 с использованием соответствующего сульфонилхлорида вместо уксусного ангидрида.
Пример 160
4-(1,2-Диметилимидазол-5-ил)-2-[4-[N-метилсульфамоил)анилино]пиримидин
N-Метил-4-аминобензолсульфонамид (способ 110; 250 мг, 1,3 ммоль) растворяют в MeOH, (3 мл) и добавляют 1M HCl в эфире (1,3 мл, 1,3 ммоль). Добавляют цианамид (68 мг, 1,6 ммоль) вместе с DMA (0,5 мл). Смесь нагревают до 100°C в течение 30 мин. К ней добавляют 5-(3-диметиламинопроп-2-ен-1-оил)-1,2-диметилимидазол (способ 15; 230 мг, 1,2 ммоль) и метоксид натрия (150 мг, 2,6 ммоль) и нагревают до 180°C в течение 1 часа. Реакционную смесь выливают в насыщенный раствор бикарбоната натрия, и полученный твердый продукт собирают. Твердый продукт растирают с горячим ДМФ и фильтруют. Фильтрат выпаривают в вакууме и очищают с помощью флэш-хроматографии на силикагеле, элюируя DCM/2% метанольным раствором аммония (100:0, с увеличением полярности до 85:15) с получением белого твердого продукта, который дигерируют вместе с ацетонитрилом с получением указанного в заголовке соединения в виде твердого продукта (84 мг, 20%). ЯМР: 2,38 (д, 6H), 3,95 (с, 3H), 7,19 (д, 2H), 7,63 (с, 1H), 7,68 (д, 2H), 7,93 (д, 2H), 8,43 (д, 1H), 9,91 (с, 1H); m/z 359.
Примеры 161-164
Следующие далее соединения синтезируют аналогично способу примера 160.
Пример 165
4-(1,2-Диметилимидазол-5-ил)-2-(4-аминоанилино)пиримидин
Гидроксид натрия (1,2 г, 3,0 ммоль) добавляют к раствору 4-(1,2-диметилимидазол-5-ил)-2-(4-ацетамидоанилино)пиримидина (пример 164; 1,25 г, 3,88 ммоль) в изопропаноле (12 мл) и воде (0,5 мл) и смесь нагревают с обратным холодильником в течение 90 минут. Смеси дают возможность охладиться и ее распределяют между насыщенным водным раствором бикарбоната натрия и EtOAc. Органический слой отделяют и летучие продукты выпаривают. Остаток очищают с помощью колоночной хроматографии на силикагеле, элюируя DCM/7M метанольным раствором аммония (96:4) с получением указанного в заголовке соединения (0,75 г, 69%) в виде коричневого твердого продукта. ЯМР: 2,33 (с, 3H), 3,85 (с, 3H), 4,75 (ушир.с, 2H), 6,51 (д, 2H), 6,92 (д, 1H), 7,22 (д, 2H), 7,51 (с, 1H), 8,22 (д, 1H), 8,90 (с, 1H); m/z 28.
Приготовление исходных материалов:
Исходные материалы для примеров выше либо являются коммерчески доступными, либо легко приготавливаются с помощью стандартных способов из известных материалов. Например, следующие далее реакции представляют собой иллюстрацию, но не ограничение, некоторых из исходных материалов, используемых в указанных выше реакциях.
Способ 1
5-(3-Диметиламинопроп-2-еноил)-1,2-диметилимидазол
5-(3-Диметиламинопроп-2-еноил)-2-метилимидазол (350 мг, 1,95 ммоль) суспендируют в DMFDMA (14 мл) и смесь перемешивают и нагревают при 100°C в течение 56 час. Избыток DMFDMA удаляют путем выпаривания и остаток очищают с помощью хроматографии, элюируя DCM/MeOH (94:6) с получением указанного в заголовке соединения 111 мг, (29%) в виде твердого продукта. ЯМР (CDCl3): 2,40 (с, 3H), 3,00 (с, 6H), 3,88 (с, 3H), 5,50 (д, 1H), 7,47 (с, 1H), 7,65 (д, 1H); m/z: 194.
Способ 2
2-(3-Хлоранилино)-4-(1-трифенилметилимидазол-4-ил)пиримидин
4-(3-Диметиламинопроп-2-ен-1-оил)-1-трифенилметилимидазол (способ 3) обрабатывают 3-хлорфенилгуанидином при условиях аналогичных тем, которые описываются в примере 7, с получением указанного в заголовке соединения; m/z: 514.
Способ 3
4-(3-Диметиламинопроп-2-ен-1-оил)-1-трифенилметилимидазол
Суспензию 4-ацетил-1-трифенилметилимидазола (способ 6; 11,9 г, 33,9 ммоль) в DMFDMA (30 мл) нагревают с обратным холодильником в течение 24 часов. Раствору дают возможность охладиться и преципитат собирают путем фильтрования с получением указанного в заголовке соединения 10,7 г, (78%). M/z: 408.
Способы 4-5
Следующие далее соединения приготавливают с помощью процедуры способа 3.
Способ 6
4-Ацетил-1-трифенилметилимидазол
Раствор 4-(1-гидроксиэтил)-1-трифенилметилимидазола (способ 10; 30,5 г, 86 ммоль) в диоксане (500 мл) нагревают до 100°C. Добавляют диоксид марганца (63,6 г, 0,73 моль) по частям, так что поддерживается умеренная дефлегмация. Смеси дают возможность немного охладиться и неорганические твердые продукты удаляют путем фильтрования. Летучие продукты удаляют из фильтрата путем выпаривания с получением указанного в заголовке соединения 30,3 г, (99%) в виде твердого продукта. ЯМР: 2,55 (с, 3H), 7,04-7,40 (м, 15H), 7,43 (с, 1H), 7,57 (с, 1H).
Способы 7-8
Следующие далее соединения приготавливают с помощью процедуры способа 6.
Способ 9
5-(1-Гидроксиэтил)-1-метилимидазол
Метилмагнийбромид (100 мл, 3M раствор в диэтиловом эфире, 0,30 моль) добавляют по каплям к раствору 5-формил-1-метилимидазола (14,5 г, 0,13 моль) в ТГФ (750 мл), охлаждают до -20°C таким образом, что температура реакции поддерживается ниже 3°C. Смеси дают возможность нагреться до температуры окружающей среды и осторожно добавляют воду (150 мл). Водную смесь непрерывно экстрагируют EtOAc. Экстракт в EtOAc сушат, и летучие продукты удаляют путем выпаривания с получением указанного в заголовке соединения 14,4 г, (88%) в виде твердого продукта. ЯМР: 1,41 (д, 3H), 4,65-4,77 (м, 1H), 4,96-5,11 (м, 1H), 6,72 (с, 1H), 7,47 (с, 1H).
Способы 10-11
Следующие далее соединения приготавливают с помощью процедуры способа 9.
Способ 12
1-Бензил-5-формил-2-метилимидазол
Бензилбромид (21,4 мл, 0,18 моль) осторожно добавляют к смеси 4-формил-2-метилимидазола (18,1 г, 0,16 моль) и карбоната калия (45,0 г, 0,33 моль) в ДМФ (100 мл) при 0°C и реакционной смеси дают возможность нагреться до температуры окружающей среды. Затем смесь распределяют между EtOAc и насыщенным водным раствором бикарбоната натрия, органическую фазу отделяют и сушат. Летучие продукты удаляют путем выпаривания с получением указанного в заголовке соединения в виде сырой смесь региоизомеров 32,0 г, (99%). M/z: 201.
Способ 13
4-{N-[3-(Пирролидин-2-он-1-ил)пропил]сульфамоил}анилин
Сульфанилфторид (6,5 г, 37,1 ммоль), 3-(пирролидин-2-он-1-ил)пропиламин (5,79 г, 40,8 ммоль) и триэтиламин (5,69 мл, 40,8 ммоль) в н-бутаноле (15 мл) нагревают с обратным холодильником в течение 10 часов. Смеси дают возможность охладиться, добавляют окись кремния и летучие продукты выпаривают. Остаток очищают с помощью хроматографии, элюируя DCM/MeOH (100:0), с увеличением полярности до (90:10), с получением указанного в заголовке соединения. М/z: 297.
Способ 14
Следующее далее соединение приготавливают с использованием процедуры способа 13.
Способ 15
5-(3-Диметиламинопроп-2-ен-1-оил)-1,2-диметилимидазол
2-Метил-4-ацетилимидазол (129 г, 1,04 моль) растворяют в смеси ДМФ (900 мл) и ДМФ.DMA (1,5 л) и смесь нагревают с обратным холодильником в атмосфере азота в течение 18 часов. Реакционной смеси дают возможность охладиться до температуры окружающей среды, продукт кристаллизуется. Твердый продукт собирают путем фильтрования, промывают ДМФ, DMA, а затем простым эфиром и сушат в вакууме при 40°C с получением указанного в заголовке соединения (115 г, 57%) в виде бледно-коричневогокристаллического твердого продукта. ЯМР: 2,13 (с, 3H), 2,95 (с, 6H), 3,78 (с, 3H), 5,56 (д, 1H), 7,50 (д, 1H), 7,53 (с, 1H); m/z 94.
Способы 16-25
Следующие далее соединения синтезируют способом, аналогичным способу 15.
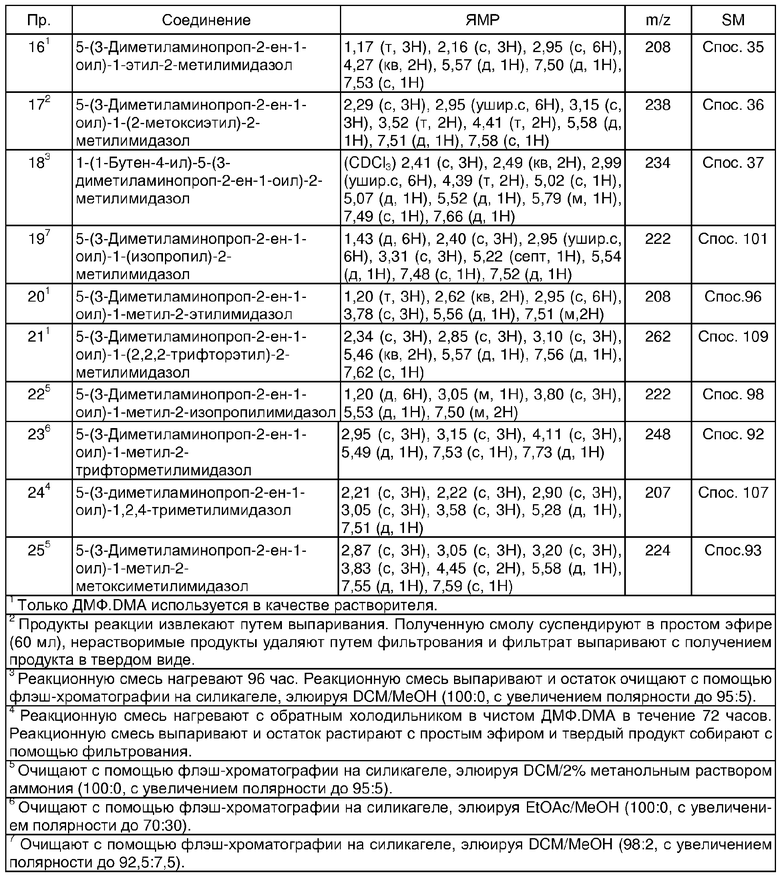
Способ 26
2-Амино-4-(1,2-диметилимидазол-5-ил)пиримидин
5-(3-Диметиламинопроп-2-ен-1-оил)-1,2-диметилимидазол (способ 15; 2,8 г, 14,5 ммоль) и гуанидингидрохлорид (3,5 г, 36,3 ммоль) суспендируют в 1-бутаноле (30 мл). Метоксид натрия (3,1 г, 58 ммоль) добавляют за один раз и смесь нагревают с обратным холодильником, в атмосфере азота, в течение 18 часов. Реакционной смеси дают возможность охладиться до температуры окружающей среды, предварительно абсорбируют на силикагеле и очищают с помощью колоночной хроматографии на силикагеле, элюируя DCM/2% метанольным раствором аммония (100:0, с увеличением полярности до 95:5) с получением указанного в заголовке соединения (2,3 г, 84%). ЯМР 2,16 (с, 3H), 3,93 (с, 3H), 6,52 (с, 2H), 6,80 (д, 1H), 7,47 (с, 1H), 8,17 (д, 1H); m/z 190.
Способы 27-32
Следующие далее соединения синтезируют способом аналогичным способу 26.
Способ 33
1-(Трифенилметил)-2-метил-4-(2-гидроксиэтил)имидазол
Трифенилметилхлорид (24,5 г, 88 ммоль) в ДМФ (100 мл) добавляют по каплям в течение 1 часа к раствору 2-метил-4-(2-гидроксиэтил)имидазола (10 г, 80 ммоль) и триэтиламина в ДМФ (100 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 18 час, а затем летучие продукты удаляют путем выпаривания. Полученный твердый продукт перетирают с водой (3 x 500 мл) и эфиром (200 мл), собирают путем фильтрования и сушат в вакууме при 60°C с получением указанного в заголовке соединения (23,7 г, 80%) в виде бледно-желтого твердого продукта. ЯМР 1,43 (д, 3H), 1,62 (с, 3H), 2,53 (с, 1H), 4,80 (кв, 1H), 6,59 (с, 1H), 7,13 (м, 6H), 7,37 (м, 9H); m/z 369.
Способ 34
1-(Трифенилметил)-2-метил-4-ацетилимидазол
1-(Трифенилметил)-2-метил-4-(2-гидроксиэтил)имидазол (способ 33; 23,7 г, 64 ммоль) суспендируют в хлороформе (180 мл) в атмосфере азота. Активированный оксид марганца(IV) (27,8 г, 320 моль) добавляют за один раз, и смесь нагревают с обратным холодильником в течение 3 часов. Реакционной смеси дают возможность охладиться, затем фильтруют через пад из диатомовой земли и пад тщательно промывают хлороформом. Фильтрат выпаривают с получением указанного в заголовке соединения (23,4 г, 100%) в виде бледно-желтого порошка. ЯМР 1,71 (с, 3H), 2,53 (с, 3H), 7,13 (м, 6H), 7,37 (м, 9H), 7,52 (с, 1H); m/z 367.
Способ 35
1-Этил-2-метил-5-ацетилимидазол
Этилтрифлат (11 мл, 83,2 ммоль) добавляют по каплям в течение 15 минут к раствору 1-(трифенилметил)-2-метил-4-ацетилимидазола (способ 34; 23,4 г, 64 ммоль) в DCM (300 мл) и смесь перемешивают в течение 5 часов при температуре окружающей среды. Раствор разбавляют DCM (100 мл) и экстрагируют 1M водным раствором лимонной кислоты (5 x 75 мл). Водные экстракты объединяют, твердый продукт подщелачивают бикарбонатом натрия и экстрагируют DCM (5 x 75 мл). Органические экстракты объединяют, сушат и выпаривают с получением указанного в заголовке соединения (8,59 г, 88%) в виде бледно-желтого масла. ЯМР 1,32 (т, 3H), 2,41 (с, 6H), 4,29 (кв, 2H), 7,68 (с, 1H); m/z 153.
Способ 36
1-(2-Метоксиэтил)-2-метил-5-ацетилимидазол
Раствор 2-метоксиэтилтрифлата (приготавливают в количестве 6 ммоль из 2-метоксиэтанола и трифторметансульфонового ангидрида с помощью способа, опубликованного в Synthesis 1982 85) в DCM (20 мл) добавляют по каплям к раствору 1-(трифенилметил)-2-метил-4-ацетилимидазола (способ 34; 1,5 г, 4 ммоль) в DCM (5 мл), и смесь перемешивают в течение 40 часов при температуре окружающей среды. Летучие продукты удаляют путем выпаривания с получением твердого продукта (2,4 г), который очищают с помощью флэш-хроматографии на силикагеле, элюируя DCM/MeOH (100:0, с увеличением полярности до 95:5), с получением указанного в заголовке соединения (660 мг, 88%) в виде твердого продукта. ЯМР (CDC13) 1,31 (с, 3H), 1,49 (с, 3H), 2,02 (с, 3H), 2,43 (м, 2H), 3,31 (м, 2H), 6,87 (с, 1H); m/z 183.
Способ 37
1-(1-Бутен-4-ил)-2-метил-5-ацетилимидазол
Указанное в заголовке соединение синтезируют способом, аналогичным способу 36, с использованием трифлата, полученного из циклопропанметанола. Указанное в заголовке соединение получают в виде масла после флэш-хроматографии на силикагеле, элюируя DCM/MeOH (100:0, с увеличением полярности до 96:4). ЯМР (CDC13) 2,43 (м, 8H), 4,32 (т, 2H), 5,02 (м, 1H), 5,08 (с, 1H), 5,74 (м, 1H), 7,69 (с, 1H); m/z 179.
Способ 38
{N-[2-(Метоксиметокси)этил)]карбамоилоксиметил}фенил
Хлорметилметиловый простой эфир (5 мл, 65 ммоль) осторожно добавляют к раствору [N-(2-гидроксиэтил)карбамоилоксиметил] фенила (6,45 г, 33 ммоль) и диизопропилэтиламина (12 мл, 70 ммоль) в DCM (50 мл) и реакционную смесь перемешивают при температуре окружающей среды в течение 4 часов. Летучие продукты удаляют путем выпаривания и остаток растворяют в EtOAc (100 мл), промывают 1M водным раствором лимонной кислоты (2 x 50 мл), насыщенным водным раствором бикарбоната натрия (50 мл), а затем насыщенным раствором соли (50 мл), сушат и выпаривают с получением указанного в заголовке соединения (7,64 г, 97%) в виде бесцветного масла. ЯМР 3,34 (с, 3H), 3,42 (кв, 2H), 3,61 (т, 2H), 4,60 (с, 3H), 5,14 (м, 3H), 7,34 (м, 5H); m/z 262 (M+Na)+.
Способ 39
N-[2-(Метоксиметокси)этил1-4-йодбензолсульфонамид
Суспензию {N-[2-(метоксиметокси)этил)]карбамоилоксиметил}фенила (способ 38, 2,4 г, 10 ммоль) и 10% палладия на угле (300 мг) в ТГФ (20 мл) перемешивают в атмосфере водорода при температуре окружающей среды в течение 18 часов. Катализатор удаляют путем фильтрования и фильтрат помещают в атмосферу азота. Добавляют триэтиламин (1 мл, 7,5 ммоль) и 4-йодфенилсульфонилхлорид (1,82 г, 6 ммоль) и смесь перемешивают при температуре окружающей среды в течение 2 часов. Реакционную смесь выливают в смесь EtOAc (30 мл) и 1M водного раствора лимонной кислоты (30 мл). Фазы разделяют и водную фазу промывают EtOAc (30 мл). Органические экстракты объединяют, промывают 1M водным раствором лимонной кислоты (2 x 30 мл), насыщенным раствором соли (30 мл), сушат и летучие продукты удаляют путем выпаривания с получением указанного в заголовке соединения (2,18 г, 98%) в виде воскообразного твердого продукта. ЯМР 3,15 (кв, 2H), 3,31 (с, 3H), 3,59 (т, 2H), 4,53 (с, 2H), 4,96 (т, 1H), 7,58 (д, 2H), 7,90 (д, 2H); m/z 370 (M-H)-.
Способ 40
N-(2-Метоксиэтил)-4-йодбензолсульфонамид
Раствор 4-йодфенилсульфонилхлорида (3,64 г, 12 ммоль) в DCM (30 мл) добавляют по каплям к раствору 2-метоксиэтиламина (1,3 мл, 15 ммоль) и триэтиламина (2 мл, 15 ммоль) в DCM (60 мл), охлаждают на ледяной бане до 0°C. Затем смеси дают возможность нагреться до температуры окружающей среды, и перемешивают в течение 1 часа. Растворитель удаляют путем выпаривания и полученное в результате масло растворяют EtOAc (100 мл) и промывают 1 н. водным раствором лимонной кислоты (2 x 100 мл), насыщенным раствором соли (100 мл) и сушат. Летучие продукты удаляют путем выпаривания с получением указанного в заголовке соединения (4,1 г, 100%) в виде прозрачного масла. ЯМР: 3,12 (2H, кв), 3,28 (3H, с), 3,44 (2H, т), 4,90 (1H, т), 7,57 (2H, д), 7,81 (2H, д); m/z: 342.
Способы 41-53
Следующие далее соединения синтезируют способом, аналогичным способу 40.
(M-H)-
Способ 54
N-трет-Бутоксикарбонил-4-йодбензолсульфонамид
Раствор ди-трет-бутил дикарбоната (10 г, 46 ммоль) в DCM (80 мл) добавляют по каплям в течение 15 мин к перемешиваемому раствору 4-йодбензолсульфонамида (11,3 г, 40 ммоль), 4-диметиламинопиридина (488 мг, 4 ммоль) и триэтиламина (6,2 мл, 44 ммоль) в DCM (50 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 2 часов, а затем растворитель удаляют путем выпаривания. Остаток растворяют в EtOAc (240 мл), промывают 1M водным раствором лимонной кислоты (2 x 160 мл), насыщенным раствором соли (160 мл), сушат и удаляют растворитель путем выпаривания с получением оранжевого твердого продукта. Сырой продукт перекристаллизовывают из EtOAc/изогексана, собирают путем фильтрования, дважды промывают изогексаном и сушат с получением указанного в заголовке соединения (10,25 г, 67%) в виде беловатых кристаллов. ЯМР 1,40 (с, 9H), 7,71 (д, 2H), 7,90 (д, 2H); m/z 382 (M-H)-.
Способ 55
4-(1,2-Диметилимидазол-5-ил)-2-(4-{N-(трет-бутоксикарбонил)-N-[2-(2-метоксиэтокси)этил]сульфамоил}анилино)пиримидин
2-(2-Метоксиэтокси)этанол (50 мкл, 0,4 ммоль), а затем диизопропила азодикарбоксилат (0,1 мл, 0,4 ммоль), добавляют к перемешиваемому раствору 4-(1,2-диметилимидазол-5-ил)-2-{4-[N-(третбутоксикарбонил)сульфамоил]анилино}пиримидина (пример 36; 90 мг, 0,2 ммоль) и трифенилфосфина (105 мг, 0,4 ммоль) в безводном ТГФ (4 мл), в атмосфере азота, при 0°C. Реакционной смеси дают возможность нагреться до температуры окружающей среды и перемешивают в течение 1 часа. Смесь выливают непосредственно в колонку Isolute SCX-2, элюируют сначала MeOH (8 x 15 мл), а затем продукт элюируют 2% метанольным раствором аммония (6 x 15 мл). Растворитель выпаривают и остаток растворяют в EtOAc (25 мл), промывают насыщенным водным раствором бикарбоната натрия (2 x 25 мл), сушат и выпаривают растворитель с получением указанного в заголовке соединения (77 мг, 71%) в виде желтого масла. ЯМР: 1,38 (с, 9H), 2,49 (с, 3H), 3,38 (с, 3H), 3,56 (м, 2H), 3,68 (м, 2H), 3,76 (т, 2H), 3,96 (с, 3H), 4,06 (т, 2H), 7,03 (д, 1H), 7,49 (с, 1H), 7,58 (с, 1H), 7,78 (д, 2H), 7,93 (д, 2H), 8,40 (д, 1H); m/z 547.
Способы 56-57
Следующие далее соединения синтезируют способом, аналогичным способу 55.
Способ 58
4-Йодбензолсульфонилфторид
18-Краун-6 (0,5 г) и фторид калия (11,6 г, 200 ммоль) добавляют к раствору йодбензолсульфонилхлорида (30,3 г, 100 ммоль) в ацетонитриле (100 мл) и суспензию перемешивают в течение 18 часов при температуре окружающей среды. Нерастворимые продукты удаляют путем фильтрования и растворитель удаляют из фильтрата путем выпаривания. Остаток растворяют в EtOAc (300 мл), промывают водой (2 x 150 мл), насыщенным раствором соли (100 мл), сушат и выпаривают растворитель с получением указанного в заголовке соединения (27,54 г, 96%) в виде белого твердого продукта. ЯМР 7,70 (д, 2H), 8,01 (д, 2H); m/z 286.
Способ 59
4-(1,2-Диметилимидазол-5-ил)-2-[4-(фторсульфонил)анилино]пиримидин
Карбонат цезия (2,3 г, 7,2 ммоль) добавляют к дегазированному раствору 2-амино-4-(1,2-диметилимидазол-5-ил)пиримидина (способ 26; 756 мг, 4 ммоль), 4-йодсульфонилфторида (способ 58; 1,50 г, 5,2 ммоль), трис(дибензилиденацетон)дипалладия(0) (92 мг, 0,18 ммоль) и 2,2'-бис(дифенилфосфино)-1,1'-бинафтила (124 мг, 0,18 ммоль) в диоксане (36 мл) в атмосфере азота. Смесь нагревают при 80°C в течение 18 час, а затем дают ей возможность охладиться до температуры окружающей среды. Смесь выливают в воду (50 мл) и экстрагируют DCM (2 x 50 мл). Органические экстракты объединяют, промывают насыщенным раствором соли (50 мл), сушат и выпаривают растворитель. Остаток предварительно абсорбируют на силикагеле и очищают с помощью колоночной хроматографии на силикагеле, элюируя DCM/2% метанольным раствором аммония (100:0, с увеличением полярности до 97:3) с получением указанного в заголовке соединения (984 мг, 71%) в виде бледно-желтого твердого продукта. ЯМР 2,38 (с, 3H), 3,96 (с, 3H), 7,28 (д, 1H), 7,65 (с, 1H), 8,00 (д, 2H), 8,13 (с, 2H), 8,47 (д, 1H), 10,32 (с, 1H); m/z 348.
Способ 60
4-[1-(2-Метоксиэтил)-2-метилимидазол-5-ил]-2-N-(4-фторсульфониланилино)пиримидин
Указанное в заголовке соединение синтезируют из соединения способа 28, способом, аналогичным способу 59, за исключением того, что реакционную смесь выпаривают перед извлечением водной фазой и экстрагированием EtOAc. Сырой продукт очищают с помощью колоночной хроматографии на силикагеле, элюируя DCM/MeOH (98:2, с увеличением полярности до 96:4). ЯМР: (CDC13) 2,52 (с, 3H), 3,27 (с, 3H), 3,61 (т, 2H), 4,68 (т, 2H), 7,11 (д, 1H), 7,52 (с, 1H), 7,61 (с, 1H), 7,89 (д, 2H), 7,96 (д, 2H), 8,41 (д, 1H); m/z 392.
Способ 61
2-Амино-5-бром-4-(1,2-диметилимидазол-5-ил)пиримидин
Указанные в заголовке соединения синтезируют из соединения способа 26, способом, аналогичным способу примера 145, за исключением того, что реакционную смесь нагревают при 60°C в течение 1,5 час, разбавляют водой и подщелачивают 2M водным раствором гидроксида натрия. Полученный твердый продукт собирают путем фильтрования и сушат в вакууме при 60°C. ЯМР: 2,38 (с, 3H), 3,72 (с, 3H), 6,84 (с, 2H), 7,55 (с, 1H), 8,38 (с, 1H); m/z 269.
Способ 62
N-(2-Метоксиэтил)-N-метил-4-йодбензолсульфонамид
Гидрид натрия (144 мг, 3,6 ммоль) добавляют по частям к раствору N-(2-метоксиэтил)-4-йодбензолсульфонамида (способ 40, 1 г, 3 ммоль) в ТГФ (10 мл) и смесь перемешивают при температуре окружающей среды в течение 15 минут. Добавляют йодметан (230 мкл, 3,6 ммоль) и реакционную смесь перемешивают в течение 18 часов. Осторожно добавляют воду (30 мл) и смесь экстрагируют эфиром (40 мл). Объединенные органические слои промывают насыщенным раствором соли (50 мл), сушат и выпаривают летучие продукты. Остаток очищают с помощью флэш-хроматографии на силикагеле, элюируя изогексан/EtOAc (100:0, с увеличением полярности до 10:1) с получением указанного в заголовке соединения (730 мг, 69%) в виде прозрачного масла. ЯМР: 2,78 (с, 3H), 3,16 (т, 2H), 3,22 (с, 3H), 3,45 (т, 3H), 7,42 (д, 2H), 7,80 (д, 2H); m/z 356.
Способы 63-64
Следующие далее соединения синтезируют способом, аналогичным способу 62.
Способ 65
4-Мезилбромбензол
К раствору 4-бромтиоанизола (22,3 г, 11 ммоль) в DCM (250 мл) частями по 10 г добавляют м-хлорпероксибензойную кислоту (40 г, 23 ммоль). Преципитат удаляют путем фильтрования и промывают DCM. Фильтрат выпаривают в вакууме и полученный твердый продукт перекристаллизовывают из EtOH (около 180 мл) с получением указанного в заголовке соединения в виде бесцветных кристаллов 11,7 г (45%). Т.пл. 103-106°C.
Способ 66
N-(3-Морфолинопропил)-4-йодбензолсульфонамид
4-Йодфенилсульфонилхлорид (3,03 г, 10 ммоль) в DCM (30 мл) по каплям добавляют в течение 15 минут к раствору 4-(3-аминопропил)морфолина (1,75 мл, 12 ммоль) и триэтиламина (1,7 мл, 12 ммоль) в DCM (50 мл), охлажденному на ледяной бане. Смеси дают возможность нагреться до температуры окружающей среды и перемешивают в течение 15 минут. Добавляют воду (50 мл) и фазы разделяют. Органический слой промывают водой (50 мл) и насыщенным раствором соли (50 мл), сушат (колонка Chemelut 1010) и выпаривают с получением указанного в заголовке соединения (4,10 г, 100%) в виде бежевого твердого продукта. ЯМР: 1,70 (м, 2H), 2,43 (м, 6H), 3,14 (т, 2H), 3,71 (м, 4H), 7,08 (с, 1H), 7,58 (д, 2H), 7,85 (д, 2H); m/z 411.
Способ 67
1-[3-[N,N-Диметиламино)пропилтио]-4-бромбензол
3-(Диметиламино)пропилхлорида гидрохлорид (3,48 г, 22 ммоль) по частям добавляют к суспензии 4-бромтиофенола (3,78 г, 20 ммоль) и карбоната калия (5,52 г, 40 ммоль) в ДМФ (40 мл) и реакционную смесь нагревают до 60°C в течение 15 минут. Смеси дают возможность охладиться до температуры окружающей среды, выливают в воду (100 мл) и экстрагируют EtOAc (2 x 100 мл). Экстракты объединяют, промывают насыщенным раствором соли (3 x 100 мл), сушат (колонка Chemelut 1010) и выпаривают с получением указанного в заголовке соединения (5,25 г, 96%) в виде бледно-желтого масла. ЯМР: 1,76 (м, 2H), 2,20 (с, 6H), 2,35 (т, 2H), 2,93 (т, 2H), 7,18 (д, 2H), 7,38 (д, 2H); m/z 276.
Способ 68
1-(3,3,3-Трифторпропилтио)-4-бромбензол
3-Бром-1,1,1-трифторпропан (640 мкл, 6 ммоль) добавляют к смеси 4-бромтиофенола (945 мг, 5 моль) и карбоната калия (760 мг, 5,5 ммоль) в ДМФ (5 мл) и реакционную смесь нагревают при 40°C в течение 1 часа. Смеси дают возможность охладиться до температуры окружающей среды, выливают в воду (50 мл) и экстрагируют EtOAc (2 x 30 мл). Экстракты объединяют, промывают насыщенным раствором соли (3 x 30 мл), сушат (колонка Chemelut 1010) и выпаривают с получением указанного в заголовке соединения (1,36 г, 95%) в виде бледно-желтого масла. ЯМР: 2,56 (м, 2H), 3,13 (т, 2H), 7,31 (д, 2H), 7,51 (д, 2H); m/z 285 (M+).
Способ 69
1-(1-Бутилтио)-4-бромбензол
Указанные в заголовке соединения синтезируют способом, аналогичным способу 68. ЯМР 0,85 (т, 3H), 1,38 (м, 2H), 1,51 (м, 2H), 2,96 (т, 2H), 7,23 (д, 2H), 7,46 (д, 2H); m/z 244 (M+).
Способ 70
1-[3-(N,N-Диметиламино)пропилсульфонил]-4-бромбензол
Оксон (14 г, 23 ммоль) добавляют к раствору 1-[3-(N,N-диметиламино)пропилтио]-4-бромбензола (способ 67; 5,24 г, 19,1 ммоль) в MeOH (150 мл) и воде (30 мл) и смесь перемешивают при температуре окружающей среды в течение 90 минут. Реакционную смесь выливают в колонку Isolute SCX-2, промывают MeOH (6 x 40 мл) и элюируют продукт 2% метанольным растворомаммония (10 x 40 мл). Растворитель выпаривают и остаток очищают с помощью флэш-хроматографии на силикагеле, элюируя DCM/2% метанольным раствором аммония (100:0, с увеличением полярности до 94:6), с получением указанного в заголовке соединения (4,68 г, 80%) в виде бледно-желтого масла. ЯМР: 1,62 (м, 2H), 2,03 (с, 6H), 2,19 (т, 2H), 3,32 (м, 2H), 7,81 (м, 4H); m/z 306.
Способ 71
1-(3,3,3-Трифторпропилсульфонил)-4-бромбензол
Оксон (3,7 г, 6 ммоль) добавляют к раствору 1-(3,3,3-трифторпропилтио)-4-бромбензола (способ 68, 1,36, 4,75 ммоль) в MeOH (25 мл) и воде (5 мл) и смесь перемешивают при температуре окружающей среды в течение 18 часов. MeOH выпаривают, добавляют воду (20 мл) и смесь экстрагируют DCM. Экстракты сушат (колонка Chemelut CE1005), и растворитель удаляют путем выпаривания с получением указанного в заголовке соединения (1,43 г, 95%) в виде белого твердого продукта. ЯМР: 2,62 (м, 2H), 3,67 (м, 2H), 7,86 (с, 4H); m/z 316 (M+).
Способ 72
1-(1-Бутилсульфонил)-4-бромбензол
Указанное в заголовке соединение синтезируют из соединения по способу 69 способом, аналогичным способу 71. ЯМР: 0,80 (т, 3H), 1,31 (м, 2H), 1,47 (м, 2H), 3,29 (т, 2H), 7,78 (д, 2H), 7,86 (д, 2H); m/z 276 (M+).
Способ 73
3-Метокси-1-пропанолметансульфонат
Метансульфонилхлорид (1,75 мл, 22 ммоль) добавляют к раствору 3-метокси-1-пропанола (1,81 г, 20 ммоль) и триэтиламина (3,35 мл, 24 ммоль) в DCM (40 мл), охлажденного на ледяной бане, и смесь перемешивают при температуре окружающей среды в течение 18 часов. Добавляют DCM (25 мл) и воду (50 мл), и фазы разделяют и водный слой экстрагируют DCM (25 мл). Экстракты объединяют, промывают водой (50 мл) и насыщенным раствором соли (50 мл), сушат (колонка Chemelut CE1010) и выпаривают с получением указанного в заголовке соединения 3,25 г (97%) в виде бледно-желтого масла. ЯМР: 2,00 (м, 2H), 3,01 (с, 3H), 3,35 (с, 3H), 3,49 (т, 2H), 4,38 (т, 2H).
Способ 74
1-(3-Метоксипропилсульфонил)-4-бромбензол
Карбонат калия (2,8 г, 20 ммоль) добавляют к раствору 3-метоксипропан-1-ил метансульфоната (способ 73; 3,25 г, 19,3 ммоль) и 4-бромтиофенола (3,48 г, 18,4 ммоль) в ДМФ (30 мл) и смесь нагревают при 40°C в течение 4 часов. Смеси дают возможность охладиться до температуры окружающей среды, выливают в воду (100 мл) и экстрагируют EtOAc (2 x 50 мл). Экстракты объединяют, промывают насыщенным водным раствором бикарбоната натрия (50 мл) и насыщенным раствором соли (2 x 50 мл), сушат (колонка Chemelut CE1010) и удаляют летучие продукты путем выпаривания. Остаток растворяют в MeOH (150 мл) и по частям добавляют воду (30 мл) и оксон (13,4 г, 21,6 ммоль). Смесь перемешивают при температуре окружающей среды в течение 18 часов. MeOH выпаривают, добавляют воду (50 мл) и раствор экстрагируют DCM (3 x 50 мл). Экстракты объединяют, промывают насыщенным раствором соли (50 мл), сушат (колонка Chemelut CE1010) и выпаривают. Остаток очищают с помощью флэш-хроматографии на силикагеле, элюируя изогексан:EtOAc (100:0, с увеличением полярности до 90:10) с получением указанного в заголовке соединения (3,32 г, 62%) в виде бесцветного масла. ЯМР: 1,95 (м, 2H), 3,19 (м, 2H), 3,26 (с, 3H), 3,41 (т, 2H), 7,70 (д, 2H), 7,78 (д, 2H).
Способ 75
3-Гидроксиизоксазол
Гидроксиламина гидрохлорид (35 г, 0,5 моль) добавляют к раствору гидроксида натрия (58 г, 1,45 моль) в воде (580 мл). Добавляют MeOH (600 мл), а затем по частям добавляют этилпропиолят (38 мл, 0,37 моль) и полученный раствор перемешивают при температуре окружающей среды в течение 6 дней. Смесь подкисляют до pH 2 концентрированной хлористоводородной кислотой, а затем насыщенным хлоридом натрия. Раствор экстрагируют DCM (8 x 500 мл), экстракты объединяют, сушат и выпаривают растворитель. Твердый остаток промывают горячим изогексаном (3 x 300 мл) конечной суспензии дают возможность охладиться и полученный твердый продукт собирают путем фильтрования, сушат в вакууме с получением указанного в заголовке соединения (11,16 г, 35%) в виде кристаллизовавшегося белого твердого продукта. ЯМР: 6,04 (с, 1H), 8,43 (с, 1H), 11,16 (с, 1H); m/z 85 (M+).
Способ 76
3-Оксо-2,3-дигидроизотиазол
Глицинамид. HCl (1 моль) суспендируют в ДМФ (500 мл) и по каплям добавляют SO2Cl2 (300 мл) в течение 1,5 часа с охлаждением, поддерживая температуру реакции в пределах между 5 и 10°C. Реакционную смесь перемешивают при 10-15°C в течение 6 часов, в этот момент осторожно добавляют воду (500 мл). Твердый продукт удаляют путем фильтрования и фильтрат экстрагируют простым эфиром (2 л). Эфирный раствор промывают насыщенным раствором соли (200 мл) и выпаривают в вакууме с получением бледно-желтого твердого продукта (132 г) - A. Водный слой экстрагируют DCM (2 x 600 мл). Порции DCM объединяют и промывают эфиром и водой. Органический слой промывают насыщенным раствором соли и выпаривают в вакууме с получением кремового твердого продукта (18 г) - B. A и B объединяют, растворяют в эфире, сушат и добавляют активированный древесный уголь. Раствор фильтруют и фильтрат выпаривают в вакууме с получением бледно-желтого твердого продукта (104,3 г). Этот твердый продукт растирают с изогексаном с получением указанного в заголовке соединения (91,3 г, 90%). Т.пл.: 102-105°C.
Способ 77
Этинилкарбамоил
К жидкому аммонию (300 мл) в течение 2 часов добавляют метилпропиолат (52,4 г, 0,62 моль), поддерживая температуру при -70°C. Аммонию дают возможность испариться и реакционную смесь выпаривают в вакууме с получением указанного в заголовке соединения (43 г), которое используют без дополнительной очистки. Т.пл.: 54-55°C.
Способ 78
3-Оксо-2,3-дигидро-1,2,5-тиадиазол
К перемешиваемому раствору этинилкарбамоила (способ 77; 43 г, 0,62 моль) в воде (310 мл), охлажденному на ледяной бане, за один раз добавляют тиосульфат аммония (92,35 г, 0,62 моль). Реакционной смеси дают возможность нагреться до комнатной температуры в течение 5 часов. К реакционной смеси быстро, в течение 10 минут, добавляют раствор йода (79,2 г, 0,31 моль) в MeOH (1 л) с получением темного раствора. Добавляют аммоний тиосульфат, до тех пор, пока не получат желтый раствор. Растворитель выпаривают примерно до 400 мл и смесь экстрагируют простым эфиром (3 x 300 мл). Эфирный раствор промывают насыщенным раствором соли (100 мл), пропускают через бумагу для разделения фаз и выпаривают в вакууме с получением указанного в заголовке соединения в виде бледно-оранжевого твердого продукта (32,8 г, 52%). Т.пл.: 70-71°C.
Способ 79
3-[2-(трет-Бутоксикарбониламино)этокси]изоксазол
Диизопропилазодикарбоксилат (1,1 мл, 5,5 ммоль) добавляют по каплям к раствору 2-(трет-бутоксикарбониламино)этанола (850 мкл, 5,5 ммоль), 3-гидроксиизоксазола (способ 75; 425 мг, 5 ммоль) и трифенилфосфина (1,44 г, 5,5 ммоль) в ТГФ (20 мл) и смесь перемешивают при температуре окружающей среды в течение 18 часов. Растворитель выпаривают и остаток очищают с помощью флэш-хроматографии на силикагеле, элюируя изогексан:EtOAc (100:0, с увеличением полярности до 4:1) с получением указанного в заголовке соединения (506 мг, 44%) в виде белого твердого продукта. ЯМР: 1,43 (с, 9H), 3,56 (м, 2H), 4,32 (м, 2H), 4,90 (с, 1H), 5,98 (с, 1H), 8,16 (с, 1H); m/z 229.
Способы 80-84
Следующие далее соединения синтезируют способом, аналогичным способу 79, с использованием соответствующего амина и гетероцикла в качестве исходных материалов.
Способ 85
3-(2-Аминоэтокси)изоксазола гидрохлорид
4M Хлористый водород в диоксане (10 мл) добавляют к раствору 3-[2-(трет-бутоксикарбониламино)этокси]изоксазола (способ 79; 500 мг, 2,2 ммоль) в диоксане (10 мл), и смесь перемешивают при температуре окружающей среды в течение 3 дней. Полученный твердый продукт собирают путем фильтрования, промывают эфиром и сушат с получением указанного в заголовке соединения (298 мг, 83%) в виде белого твердого продукта. ЯМР 3,20 (м, 2H), 4,39 (т, 2H), 6,13 (с, 1H), 8,30 (с, 3H), 8,69 (с, 1H); m/z 129.
Способы 86-90
Следующие далее соединения синтезируют способом, аналогичным способу 85.
Способы 91-94
Следующие далее соединения синтезируют с помощью процедуры, как описано в JOC 1987, 2714-2716.
Способы 95-109
Следующие далее соединения приготавливают с использованием процедур, аналогичных тем, что описываются в JOC 1987, 2714-2726.
Способ 110
N-Метил-4-аминобензолсульфонамид
4-Аминобензолсульфонилфторид (200 мг, 1,14 ммоль) растворяют в растворе метиламина в EtOH (3 мл, избыток) и нагревают до 80°C в течение 45 минут, затем охлаждают до комнатной температуры и оставляют перемешиваться в течение ночи. Растворитель выпаривают в вакууме и азеотропно отгоняют вместе с эфиром с получением указанного в заголовке соединения в виде твердого продукта (160 мг, 75%). ЯМР: 2,12 (с, 3H), 5,85 (с, 2H), 6,59 (д, 2H), 7,37 (д, 2H); m/z 187.
Способ 111
2-Амино-4-(1,2-диметилимидазол-5-ил)-5-хлорпиримидин
2-Амино-4-(1,2-диметилимидазол-5-ил)пиримидин (способ 26; 378 мг, 2 ммоль) и N-хлорсукцинимид (267 мг, 2 ммоль) растворяют в ледяной уксусной кислоте (7 мл) в атмосфере азота. Реакционную смесь нагревают при 65°C в течение 18 часов, когда добавляют дополнительный N-хлорсукцинимид (89 мг, 0,66 ммоль), и реакционную смесь нагревают при 65°C в течение дополнительных 2 часов. Летучие продукты удаляют путем выпаривания и остаток растворяют в воде (10 мл). Значение pH раствора доводят до 11-12 путем добавления 40% водного раствора гидроксида натрия. Осажденный твердый продукт собирают путем фильтрования и осторожно промывают водой, сушат в вакууме при 60°C с получением указанного в заголовке соединения (344 мг, 77%) в виде желтого твердого продукта. ЯМР 2,35 (с, 3H), 3,75 (с, 3H), 4,83 (с, 2H), 7,53 (с, 1H), 8,27 (с, 1H); m/z 224.
Пример 166
Следующее далее иллюстрирует наиболее типичные фармацевтические дозированные формы, содержащие соединение формулы (I) или его фармацевтически приемлемую соль, или его сложный эфир, гидролизующийся in vivo (здесь и далее, соединение X), для терапевтического или профилактического использования у людей:
(5% мас./об. пасты)
(5% мас./об. пасты)
(5% мас./об. пасты
буфера)
Замечания
Указанные выше препараты могут быть получены с помощью обычных процедур, хорошо известных в области фармацевтики. Таблетки (а)-(c) могут быть снабжены покрытием для энтерального всасывания с помощью обычных средств, например, путем создания покрытия из ацетатфталата целлюлозы.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРИМИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ (ВАРИАНТЫ) И СПОСОБ ИНГИБИРОВАНИЯ | 2001 |
|
RU2299201C2 |
| ИМИДАЗО[1,2-А]ПИРИДИНОВЫЕ И ПИРАЗОЛ[2,3-А]ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2000 |
|
RU2248976C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНЫХ ТИРОЗИНКИНАЗ | 2009 |
|
RU2518089C2 |
| 1,5-БЕНЗОТИАЗЕПИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТИГИПЕРЛИПИДЕМИЧЕСКИХ СРЕДСТВ | 2001 |
|
RU2302414C2 |
| ИНГИБИТОРЫ FGFR4 | 2014 |
|
RU2715708C2 |
| СОЛИ ТИАЗОЛИДИНДИОНА СО СНИЖЕННЫМ СРОДСТВОМ К PPAR ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ ОБМЕНА ВЕЩЕСТВ | 2010 |
|
RU2564661C2 |
| Бициклические гетероциклические соединения и их применения в терапии | 2012 |
|
RU2662827C2 |
| ПРОИЗВОДНЫЕ ГИДРОКСИБЕНЗАМИДА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ Hsp90 | 2006 |
|
RU2458919C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА И ХИНАЗОЛИНА СО СРОДСТВОМ К 5HT-РЕЦЕПТОРАМ | 2004 |
|
RU2402533C2 |
| 2-ПИРИДИЛЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ALK5 И/ИЛИ ALK4 | 2012 |
|
RU2612958C2 |
Изобретение относится к соединениям формулы (I) или их фармацевтически приемлемым солям или сложным эфирам, гидролизующимся in vivo, обладающим ингибирующей клеточный цикл активностью, селективной в отношении к CDK-2 CDK-4 и CDK-6. Соединения могут быть использованы для лечения рака. В формуле (I):
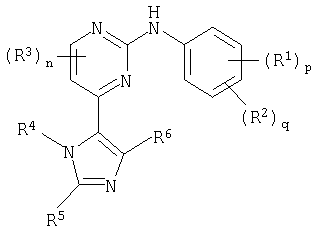
R1 представляет собой галоген, амино, C1-6алкил, C1-6алкокси; р равно 0-4; где значения R1 могут быть одинаковыми или различными; R2 представляет собой сульфамоил или группу Ra-Rb; q равно 0-2; где значения R2 могут быть одинаковыми или различными; и где р+q=0-5; R3 представляет собой галоген или циано; n равно 0-2, где значения R3 могут быть одинаковыми или различными; R4 представляет собой водород, C1-6алкил, С2-6алкенил, С2-6алкинил, С3-8циклоалкил, фенил или гетероциклическую группу, связанную с углеродом; где R4 может быть необязательно замещенным на атоме углерода одним или несколькими Rd; R5 и R6 являются независимо выбранными из водорода, галогена, C1-6алкила, С2-6алкенила или С3-8циклоалкила; где R5 и R6 независимо друг от друга могут быть необязательно замещенными на атоме углерода одним или несколькими Re; Ra является выбранным из C1-6алкила, С2-6алкенила, С2-6алкинила, С3-8циклоалкила, С3-8циклоалкилС1-6алкила, фенила, гетероциклической группы, фенилС1-6алкила или (гетероциклическая группа)С1-6алкила; где R3 может быть необязательно замещенным на атоме углерода одним или несколькими Rg; и где, если указанная гетероциклическая группа содержит остаток -NH-, его азот может быть необязательно замещенным группой, выбранной из Rh; Rb представляет собой -N(Rm)C(O)-, -C(O)N(Rm)-, -S(O)r-, -OC(O)N(Rm)SO2-, -SO2N(Rm)- или -N(Rm)SO2-; где Rm представляет собой водород или C1-6алкил, и r равно 1-2. Изобретение также относится к способам получения соединений, фармацевтической композиции, способу ингибирования и применению соединений. 12 н. и 12 з.п. ф-лы.
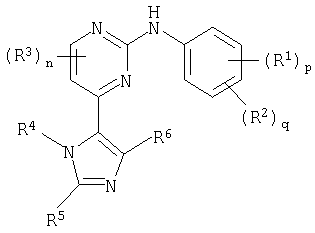
где R1 представляет собой галоген, амино, C1-6алкил, C1-6алкокси;
р равно 0-4, где значения R1 могут быть одинаковыми или различными;
R2 представляет собой сульфамоил или группу Ra-Rb-;
q равно 0-2, где значения R2 могут быть одинаковыми или различными и где р+q=0-5;
R3 представляет собой галоген или циано;
n равно 0-2, где значения R3 могут быть одинаковыми или различными;
R4 представляет собой водород, C1-6алкил, С1-6алкенил, С3-8циклоалкил или гетероциклическую группу, связанную с углеродом, где R4 может быть необязательно замещенным на атоме углерода одним или несколькими Rd;
R5 и R6 являются независимо выбранными из водорода, галогена, C1-6алкила, С2-6алкенила или С3-8циклоалкила, где R5 и R6 независимо друг от друга могут быть необязательно замещенными на атоме углерода одним или несколькими Re;
Ra является выбранным из C1-6алкила, С2-6элкенила, С2-6алкинила, С3-8циклоалкила, С3-8циклоалкилС1-6алкила, фенила, гетероциклической группы, фенилС1-6алкила или (гетероциклическая группа)С1-6алкила, где Rа может быть необязательно замещенным на атоме углерода одним или несколькими Rg и где, если указанная гетероциклическая группа содержит остаток -NH-, его азот может быть необязательно замещенным группой, выбранной из Rh;
Rb представляет собой -N(Rm)С(О)-, -C(O)N(Rm)-, -S(O)r-, -OC(O)N(Rm)SO2-, -SO2N(Rm)- или -N(Rm)SO2-, где Rm представляет собой водород или C1-6алкил и r равно 1-2;
Rd и Rg являются независимо выбранными из галогена, циано, гидрокси, амино, карбокси, карбамоила, C1-6алкила, C1-6алкокси, С1-6алкоксиС1-6алкокси, C1-6алкоксиС1-6алкоксиС1-6алкокси, N,N-(С1-6алкил)2амино, С1-6алканоиламино, С1-6алкил-S(O)а, где а равно 0-2, C1-6алкоксикарбонила, C1-6алкилсульфониламино, С3-8циклоалкила, фенила, гетероциклической группы, фенилС1-6алкил-Rо-, (гетероциклическая группа)С1-6алкил-Rо-, фенил-Ro- или (гетероциклическая группа)-Rо-,
где Rd и Rg независимо друг от друга могут быть необязательно замещенными на атоме углерода одним или несколькими Rj;
Rо представляет собой -О-;
Rh выбран из C1-6алкила или бензила и
Re и Rj являются независимо выбранными из галогена, гидрокси, метокси, этокси, этиламино, диметиламино, метилтио, этилтио, метилсульфинила, этилсульфинила и этилсульфонила;
причем гетероциклическая группа представляет насыщенное или ненасыщенное, моно- или бициклическое кольцо, содержащее 4-12 атомов, из которых, по крайней мере, один атом выбран из азота, серы или кислорода и который может быть связан с атомами углерода или азота, где группа -CH2- может необязательно быть заменена -С(O)-группой;
или его фармацевтически приемлемая соль, или его сложный эфир, гидролизующийся in vivo.
С3-8циклоалкила, фенила или гетероциклической группы, где Ra может быть необязательно замещенным на атоме углерода одним или несколькими Rg;
Rb представляет собой -N(Rm)C(O)-, -C(O)N(Rm)-, -S(O)r-, -OC(O)N(Rm)SO2-, -SO2N(Rm)- или -N(Rm)SO2-, где Rm представляет собой водород или C1-6алкил и r равно 2;
Rg является выбранным из галогена, гидрокси, амино, циано, карбамоила, C1-6алкила, C1-6алкокси, С1-6алкоксиС1-6алкокси, С1-6алкоксиС1-6алкоксиС1-6алкокси, N,N-(C1-6алкил)2амино, С1-6алкил-S(O)а, где а равно 2, С3-8циклоалкила, фенила, гетероциклической группы, фенилС1-6алкил-Ro- или (гетероциклическая группа)-Ro-, где Rg может быть необязательно замещенным на атоме углерода одним или несколькими Rj;
Ro представляет собой -О- и
Rj является выбранным из галогена, гидрокси, метила или метокси;
причем, гетероциклическая группа представляет насыщенное или ненасыщенное, моно- или бициклическое кольцо, содержащее 4-12 атомов, из которых, по крайней мере, один атом выбран из азота, серы или кислорода и который может быть связан с атомами углерода или азота, где группа -СН2- может необязательно быть заменена -С(O)-группой;
или его фармацевтически приемлемая соль, или его сложный эфир, гидролизующийся in vivo.
или его фармацевтически приемлемая соль, или его сложный эфир, гидролизующийся in vivo.
или его фармацевтически приемлемая соль, или его сложный эфир, гидролизующийся in vivo.
R1 представляет собой хлор, амино, метил или метокси;
р равно 0-2, где значения R1 могут быть одинаковыми или различными;
R2 представляет собой сульфамоил, N-(тетрагидрофур-2-илметил)сульфамоил, N-(циклопропилметил)сульфамоил, N-(фур-2-илметил)сульфамоил, N-(2,2-диметил-1,3-диоксолан-4-илметил)сульфамоил, N-(цианометил)сульфамоил, N-(карбамоилметил)сульфамоил, N-метилсульфамоил, N-(4-фторбензил)сульфамоил, N-(пиридин-2-илметил)сульфамоил, N-(пиридин-3-илметил)сульфамоил, N-(4-метилтиазол-2-ил)сульфамоил, N-(3-метилизооксазол-5-илметил)сульфамоил, N-(тетрагидропиран-2-илметил)сульфамоил, N-(2-метилпиразин-5-ил)сульфамоил, N-[2-(2-гидроксиэтокси)этил]сульфамоил, N-(2-гидроксиэтил)сульфамоил, N-(2,2,2-трифторэтил)сульфамоил, N-(2-метоксиэтил)сульфамоил, N-(2-мезилэтил)сульфамоил, N-(2-бензилоксиэтил)сульфамоил, N-(2,2-диметоксиэтил)сульфамоил, N-[2-(N,N-диметиламино)этил]сульфамоил, N-(2-пиперидин-1-илэтил)сульфамоил, N-[2-(метоксиметокси)этил]сульфамоил, N-этилсульфамоил, N-[2-(2-метоксиэтокси)этил]сульфамоил, N-{2-[2-(2-метоксиэтокси)этокси]этил}сульфамоил, N-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}этил)сульфамоил, N-(2-пиридин-2-илэтил)сульфамоил, N-(2-пиридин-4-илэтил)сульфамоил, N-(2-изоксазол-3-илоксиэтил)сульфамоил, N-(2-изотиазол-3-илоксиэтил)сульфамоил, N-(2-1,2,5-тиадиазол-3-илоксиэтил)сульфамоил, N-метил-N-(2-метоксиэтил)сульфамоил, N-[3-(2-оксопирролидин-1-ил)пропил]сульфамоил, N-(3-метоксипропил)сульфамоил, N-пропилсульфамоил, N-(2,3-дигидроксипропил)сульфамоил, N-(3-морфолинопропил)сульфамоил, N-[3-(N,N-диметиламино)пропил]сульфамоил, N-(3,3,3-трифторпропил)сульфамоил, N-(2,2-диметил-3-гидроксипропил)сульфамоил, N-(3-гидроксипропил)сульфамоил, N-(3-этоксипропил)сульфамоил, N-(2-гидроксипропил)сульфамоил, N-(3-изопропоксипропил)сульфамоил, N-(3-изопропокси-2-гидроксипропил)сульфамоил, N-(3-изоксазол-3-илоксипропил)сульфамоил, N-(3-изотиазол-3-илоксипропил)сульфамоил, N-(3-1,2,5-тиадиазол-3-илоксипропил)сульфамоил, N-(1,1-диметилпропил)сульфамоил, N-метил-N-(3-морфолинопропил)сульфамоил, N-бутилсульфамоил, N-трет-бутилсульфамоил, N-(2-гидроксибутил)сульфамоил, N-метил-N-трет-бутилсульфамоил, N-пентилсульфамоил, N-(5-гидроксипентил)сульфамоил, N-(4,5-диметилоксазол-2-ил)сульфамоил, N-(циклопропил)сульфамоил, N-(циклобутил)сульфамоил, N-(3-трифторметилфенил)сульфамоил, N-аллилсульфамоил, N-(2-пропинил)сульфамоил, N-метилкарбамоил, ацетамидо, мезиламино или мезил;
q равно 0 или 1;
R3 представляет собой бром или хлор;
n равно 0 или 1;
R4 представляет собой водород, метил, этил, изопропил, 3-бутенил, бензил, 2-фталимидоэтил, 2-аминоэтил, 2-метоксиэтил, 2-ацетамидоэтил, 2-мезиламиноэтил или 2,2,2-трифторэтил;
R5 и R6 являются независимо выбранными из водорода, метила, этила, изопропила, трифторметила или метоксиметила,
или его фармацевтически приемлемая соль, или его сложный эфир, гидролизующийся in vivo.
2-{4-[N-(циклопропилметил)сульфамоил]анилино}-4-(1,2-диметилимидазол-5-ил)пиримидина;
4-(1-этил-2-метилимидазол-5-ил)-2-{4-[N-(2-метоксиэтил)сульфамоил]анилино}пиримидина;
4-(1-этил-2-метилимидазол-5-ил)-2-{4-[N-(3-метоксипропил)сульфамоил]анилино}пиримидина;
4-(1-этил-2-метилимидазол-5-ил)-2-{4-[N-(циклопропилметил)сульфамоил]анилино}пиримидина;
4-(1-этил-2-метилимидазол-5-ил)-2-[4-[N-циклопропилсульфамоил)анилино]пиримидина;
4-(1-метил-2-изопропилимидазол-5-ил)-2-{4-(N-(циклопропилметил)сульфамоил]анилино}пиримидина;
4-(1,2-диметилимидазол-5-ил)-2-[4-(N-циклопропилсульфамоил)анилино]пиримидина;
4-(1,2-диметилимидазол-5-ил)-2-[4-(N-циклобутилсульфамоил)анилино]пиримидина;
4-(1,2-диметилимидазол-5-ил)-2-{4-[N-2,2,2-трифторэтил)сульфамоил]анилино}пиримидина;
и
4-(1-изопропил-2-метилимидазол-5-ил)-2-[4-(N-циклобутилсульфамоил)анилино]пиримидина,
или его фармацевтически приемлемая соль, или его сложный эфир, гидролизующийся in vivo.
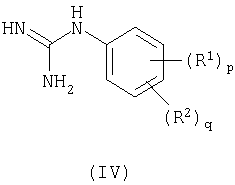
с соединением формулы (V)
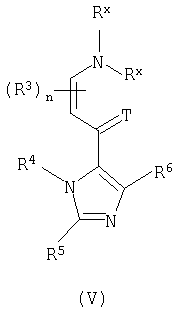
где Т является О или S;
Rx может быть одинаковым или различным и выбран из C1-6алкила и,
если необходимо, последующее превращение соединения формулы (I) в другое соединение формулы (I);
удаление любой защитной группы;
образование фармацевтически приемлемой соли или in vivo гидролизуемого эфира.
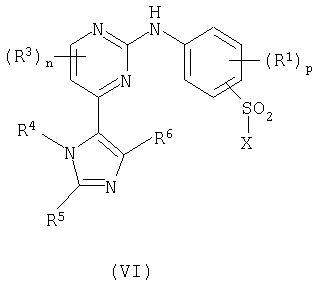
где Х является замещаемой группой,
с амином формулы (VII)

если необходимо, последующее превращение соединения формулы (I) в другое соединение формулы (I);
удаление любой защитной группы;
образование фармацевтически приемлемой соли или in vivo гидролизуемого эфира.
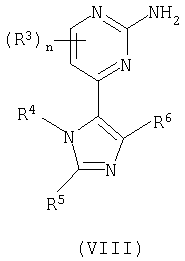
с соединением формулы (IX)
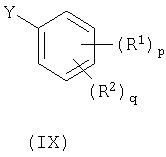
где Y является замещаемой группой,
и, если необходимо, последующее превращение соединения формулы (I) в другое соединение формулы (I);
удаление любой защитной группы;
образование фармацевтически приемлемой соли или in vivo гидролизуемого эфира.
| US 5859041 A, 12.01.1999 | |||
| ТРИ-ЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ, ОБЛАДАЮЩИЕ ТЕРАПЕВТИЧЕСКИМИ СВОЙСТВАМИ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2140918C1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| WO 00/026209 A1, 11.05.2000 | |||
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |

