Настоящее изобретение относится к пиримидиновым производным или к их фармацевтически приемлемым солям или in vivo гидролизуемым сложным эфирам, которые обладают активностью, направленной на ингибирование клеточного цикла, и в соответствии с этим являются эффективными ингибиторами пролиферации клеток (таких как раковые клетки), и поэтому они могут быть использованы в способах лечения человека или животного. Настоящее изобретение также относится к способам получения указанных пиримидиновых производных, к фармацевтическим композициям, содержащим эти производные, и к их применению в изготовлении лекарственных средств для ингибирования пролиферации клеток у теплокровного животного, такого как человек.
Семейство внутриклеточных белков, называемых циклинами, играет центральную роль в клеточном цикле. Синтез и деградация циклинов жестко регулируется, так что уровень их экспрессии варьируется на протяжении клеточного цикла. Циклины связываются с циклин-зависимыми сериновыми/треониновыми киназами (CDK), и это связывание играет важную роль в стимуляции активности CDK (таких как CDK1, CDK2, CDK4 и/или CDK6) в клетке. Хотя точно неизвестно, каким образом все эти факторы сочетаются для регуляции CDK-активности, однако, баланс между двумя этими факторами определяет способность данной клетки продолжать свой жизненный цикл.
Недавнее объединение исследований, направленных на онкогены и гены супрессии опухолей, привело к идентификации регуляции начала клеточного цикла как ключевой контрольной точки митогенеза в опухолях. Кроме того, очевидно, что CDK являются более поздними участниками ряда путей передачи сигнала онкогенами. Нарушение CDK-активности путем активации циклинов и/или делеции эндогенных ингибиторов является, очевидно, важным связующим звеном между митогенными путями передачи сигнала и пролиферацией опухолевых клеток.
В связи с этим было установлено, что ингибитор киназ, участвующих в клеточном цикле, в частности ингибиторы CDK2, CDK4 и/или CDK6 (которые действуют в S-фазе, G1-S-фазе и G1-S-фазе соответственно), должен действовать как селективный ингибитор пролиферации клеток, таких как раковые клетки млекопитающих.
Настоящее изобретение основано на неожиданном обнаружении того факта, что некоторые пиримидиновые соединения селективно ингибируют действие киназ CDK2, CDK4 и CDK6 на клеточный цикл и поэтому обладают способностью ингибировать пролиферацию клеток. Предполагается, что это свойство имеет важное значение для лечения патологических состояний, ассоциированных с нарушением клеточных циклов и пролиферации клеток, таких как рак (твердые опухоли и лейкоз), фибропролиферативные расстройства и расстройства, ассоциированные с нарушением дифференцировки клеток, псориаз, ревматоидный артрит, саркома Капоши, гемангиома, острая и хроническая нефропатия, атерома, атеросклероз, артериальный рестеноз, аутоиммунные заболевания, острые и хронические воспалительные заболевания, болезни костей и глазные болезни с пролиферацией сосудов сетчатки.
В соответствии с этим настоящее изобретение относится к соединению формулы (I)
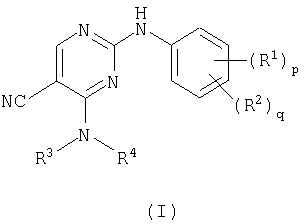
где R1 представляет галоген, нитро, циано, гидрокси, амино, карбокси, карбамоил, меркапто, С1-6алкил, С2-6алкенил или С2-6алкинил;
р=0-4, где R1 могут быть одинаковыми или различными;
R2 представляет сульфамоил или группу В-Е-;
q=0-2, где R2 могут быть одинаковыми или различными и где р+q=1-5;
R3 представляет водород, С1-6алкил, С2-6алкенил, С2-6алкинил или С3-8циклоалкил, где R3 может быть необязательно замещен у атома углерода одним или несколькими М;
R4 представляет С1-6алкил, С2-6алкенил, С2-6алкинил, С3-8циклоалкил или гетероциклил, где R4 может быть необязательно замещен одним или несколькими М и где, если указанный гетероциклил содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из Z,
или R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют гетероциклическое кольцо, необязательно замещенное у атома углерода одним или несколькими М, где, если указанное гетероциклическое кольцо содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из Q;
В выбран из С1-6алкила, С2-6алкенила, С2-6алкинила, С3-8циклоалкила, С3-8циклоалкилС1-6алкила, фенила, гетероциклической группы, фенилС1-6алкила или (гетероциклическая группа)С1-6алкила, где В может быть необязательно замещен у атома углерода одним или несколькими D и где, если указанная гетероциклическая группа содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из G;
Е представляет -С(О)-, -N(Rа)С(О)-, -С(О)N(Rа)-, -S(O)r-, -SO2N(Ra)- или -N(Ra)SO2-, где Rа представляет водород или С1-6алкил, необязательно замещенный одним или несколькими D, и r равно 1-2;
D независимо выбран из галогена, нитро, циано, гидрокси, трифторметила, трифторметокси, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-6алкила, С2-6алкенила, С2-6алкинила, С1-6алкокси, С1-6алканоила, С1-6алканоилокси, N-(С1-6алкил)амино, N,N-(С1-6алкил)2амино, С1-6алканоиламино, N-(С1-6алкил)карбамоила, N,N-(С1-6алкил)2карбамоила, С1-6алкилS(O)a, где а равно 0-2, С1-6алкоксикарбонила, N-(С1-6алкил)сульфамоила и N,N-(С1-6алкил)2сульфамоила, где D может быть необязательно замещен у атома углерода одним или несколькими V;
М независимо выбран из галогена, нитро, циано, гидрокси, трифторметокси, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-6алкила, С2-6алкенила, С2-6алкинила, С1-6алкокси, С1-6алканоила, С1-6алканоилокси, N-(С1-6алкил)амино, N,N-(С1-6алкил)2амино, С1-6алканоиламино, N-(С1-6алкил)карбамоила, N,N-(С1-6алкил)2карбамоила, С16алкилS(O)a, где а равно 0-2, С1-6алкоксикарбонила, N-(С1-6алкил)сульфамоила, N,N-(С1-6алкил)2сульфамоила, С3-8циклоалкила, фенила или гетероциклической группы, где М может быть необязательно замещен у атома углерода одним или несколькими Р и где, если указанная гетероциклическая группа содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из Т;
Р, Х и V независимо выбраны из галогена, нитро, циано, гидрокси, трифторметокси, трифторметила, амино, карбокси, карбамоила, меркапто, сульфамоила, метила, этила, метокси, этокси, ацетила, ацетокси, метиламино, этиламино, диметиламино, диэтиламино, N-метил-N-этиламино, ацетиламино, N-метилкарбамоила, N-этилкарбамоила, N,N-диметилкарбамоила, N,N-диэтилкарбамоила, N-метил-N-этилкарбамоила, метилтио, этилтио, метилсульфинила, этилсульфинила, мезила, этилсульфонила, метоксикарбонила, этоксикарбонила, N-метилсульфамоила, N-этилсульфамоила, N,N-диметилсульфамоила, N,N-диэтилсульфамоила или N-метил-N-этилсульфамоила и
G, Q, T и Z независимо выбраны из С1-4алкила, С1-4алканоила, С1-4алкилсульфонила, С1-4алкоксикарбонила, карбамоила, N-(С1-6алкил)карбамоила, N,N-(С1-4алкил)карбамоила, бензила, бензилоксикарбонила, бензоила и фенилсульфонила, где G, Q и Т могут быть необязательно замещены у атома углерода одним или несколькими Х;
или к его фармацевтически приемлемой соли или к in vivo гидролизуемому сложному эфиру.
В описании данной заявки термин "алкил" означает прямые и разветвленные алкильные группы, но он может также означать и отдельные алкильные группы, такие как "пропил", которые являются специфичными только для прямой цепи. Так, например, термин "С1-6алкил" включает С1-4алкил, С1-3алкил, пропил, изопропил и трет-бутил. Однако данный термин означает также отдельные алкильные группы, такие как "пропил", которые являются специфичными только для прямой цепи, и отдельные разветвленные алкильные группы, такие как "изопропил", которые являются специфичными только для разветвленной цепи. Аналогичное определение относится также и к другим радикалам, например, термин "фенилС1-6алкил" включает фенилС1-4алкил, бензил, 1-фенилэтил и 2-фенилэтил. Термин "галоген" означает фтор, хлор, бром и йод.
Если указано, что необязательные заместители выбраны из "одной или нескольких" групп, то подразумевается, что это понятие включает все заместители, выбранные из одной конкретной группы, или все заместители, выбранные из двух или нескольких конкретных групп.
Термин "гетероциклическая группа" означает насыщенное, частично насыщенное или ненасыщенное моно- или бициклическое кольцо, содержащее 4-12 атомов, из которых, по крайней мере, один атом выбран из азота, серы или кислорода, и которые, если не оговорено особо, могут быть связаны с углеродом или с азотом, где группа -СН2- может быть необязательно заменена -С(О)-, а атом азота в кольце может необязательно нести С1-6алкильную группу и образовывать четвертичное соединение, либо атом азота и/или атом серы в кольце могут быть необязательно окислены с образованием N-оксида и/или S-оксидов. Подходящими примерами таких "гетероциклических групп" являются морфолино, пиперидил, пиридил, пиранил, пирролил, изотиазолил, индолил, хинолил, тиенил, 1,3-бензодиоксолил, тиадиазолил, пиперазинил, тиазолидинил, пирролидинил, тиоморфолино, пирролинил, гомопиперазинил, 3,5-диоксапиперидинил, тетрагидропиранил, имидазолил, пиримидил, пиразинил, пиридазинил, изоксазолил, N-метилпирролил, 4-пиридон, 1-изохинолон, 2-пирролидон, 4-тиазолидон, пиридин-N-оксид и хинолин-N-оксид. "Гетероциклическая группа" предпочтительно представляет насыщенное, частично насыщенное или ненасыщенное моно- или бициклическое кольцо, содержащее 5 или 6 атомов, из которых, по крайней мере, один выбран из азота, серы или кислорода, и, если не оговорено особо, может быть связан с углеродом или азотом, группа -СН2- может быть необязательно заменена -С(О)-, а атом серы в кольце может быть необязательно окислен с образованием S-оксидов. В другом аспекте настоящего изобретения термин "гетероциклическая группа" предпочтительно означает насыщенное, частично насыщенное или ненасыщенное моноциклическое кольцо, содержащее 5 или 6 атомов, из которых, по крайней мере, один выбран из азота, серы или кислорода, и, если не оговорено особо, может быть связан с углеродом или азотом, группа -СН2- может быть необязательно заменена -С(О)-, а атом серы в кольце может быть необязательно окислен с образованием S-оксидов. Более предпочтительной "гетероциклической группой" является тетрагидрофурил, пиридил, пирролидинонил, морфолино, имидазолил, пиперидинил или пирролидинил. Особенно предпочтительной "гетероциклической группой" является тетрагидрофурил или морфолино. В другом аспекте настоящего изобретения конкретной "гетероциклической группой" является морфолино, тетрагидрофурил, пиперидинил, пиридил, имидазолил, пиперазинил, пирролидинил, триазолил, диоксанил и диоксоланил.
Если R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют гетероциклическое кольцо, то указанное "гетероциклическое кольцо" представляет собой насыщенное, частично насыщенное или полностью ненасыщенное моно- или бициклическое кольцо, содержащее 4-12 атомов, из которых, по крайней мере, один атом представляет азот, с которым связаны R3 и R4, а другими атомами являются либо все атомы углерода, либо атомы углерода и 1-3 гетероатома, выбранных из азота, серы или кислорода, где группа -СН2- может быть необязательно заменена -С(О)-, а атом азота в кольце может необязательно нести С1-6алкильную группу и образовывать четвертичное соединение, либо атом азота и/или атом серы в кольце могут быть необязательно окислены с образованием N- и/или S-оксида. При этом следует отметить, что если R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют "гетероциклическое кольцо", то этот атом азота не является кватернизированным, т.е. образуется нейтральное соединение. Подходящими примерами такой "гетероциклической группы" являются азетидинил, морфолино, пиперидил, пиперазинил, пирролидинил, тиоморфолино, пирролинил, гомопиперазинил, пирролил, пиразолил, пиразолинил, имидазолил, имидазолинил, имидазолидинил, пиразолидинил и триазолил. Предпочтительной "гетероциклической группой" является морфолино. В другом аспекте настоящего изобретения предпочтительной "гетероциклической группой" является морфолино, пиперидино, пирролидин-1-ил или пиперазин-1-ил.
Термин "гетероциклил" означает насыщенное моно- или бициклическое кольцо, содержащее 4-12 атомов, из которых, по крайней мере, один атом выбран из азота, серы или кислорода и которые, если не оговорено особо, могут быть связаны с углеродом или с азотом, где группа -СН2- может быть необязательно заменена -С(О)-, а атом серы в кольце может быть необязательно окислен с образованием S-оксида(ов). Примерами "гетероциклила" являются морфолинил, пиперидил, пиперазинил, тиазолидинил, пирролидинил, тиоморфолинил, 1,1-диоксотиоморфолинил, гомопиперазинил, тетрагидропиранил, 2-пирролидон и 4-тиазолидон. В одном из аспектов настоящего изобретения термин "гетероциклил" представляет насыщенное моноциклическое кольцо, содержащее 5 или 6 атомов, из которых, по крайней мере, один атом выбран из азота, серы или кислорода, и, если не оговорено особо, может быть связан с углеродом или азотом, группа -СН2- может быть необязательно заменена -С(О)-, а атом серы в кольце может быть необязательно окислен с образованием S-оксида(ов). В другом аспекте настоящего изобретения термин "гетероциклил" означает тетрагидрофурил.
Примером "С1-6алканоилокси" является ацетокси. Примеры "С1-6алкоксикарбонила" включают С1-4алкоксикарбонил, метоксикарбонил, этоксикарбонил, н- и трет-бутоксикарбонил. Примеры "С1-6алкокси" включают метокси, этокси и пропокси. Примеры "С1-6алканоиламино" включают формамидо, ацетамидо и пропиониламино. Примеры "С1-6алкилS(O)a, где а равно 0-2" включают С1-6алкилсульфонил, метилтио, этилтио, метилсульфинил, этилсульфинил, мезил и этилсульфонил. Примеры "С1-6алкилS(O)r, где r равно 1-2" включают метилсульфинил, этилсульфинил, мезил и этилсульфонил. Примеры "С1-6алканоила" включают С1-4алканоил, пропионил и ацетил. Примеры "N-С1-6алкиламино" включают метиламино и этиламино. Примеры "N,N-(С1-6алкил)2амино" включают ди-N-метиламино, ди-(N-этил)амино и N-этил-N-метиламино. Примерами "С2-6алкенила" являются винил, аллил и 1-пропенил. Примерами "С2-6алкинила" являются этинил, 1-пропинил и 2-пропинил. Примерами "N-(С1-6алкил)сульфамоила" являются N-(метил)сульфамоил и N-(этил)сульфамоил. Примерами "N-(С1-6алкил)2сульфамоила" являются N,N-(диметил)сульфамоил и N-(метил)-N-(этил)сульфамоил. Примерами "N-(С1-6алкил)карбамоила" являются N-(С1-4алкил)карбамоил, метиламинокарбонил и этиламинокарбонил. Примерами "N,N-(С1-6алкил)2карбамоила" являются N,N-(С1-4алкил)2карбамоил, диметиламинокарбонил и метилэтиламинокарбонил. Примерами "С3-8циклоалкила" являются циклопропил, циклобутил, циклопропил и циклогексил. Примеры "(гетероциклическая группа)С1-6алкила" включают пиридилметил, 3-морфолинопропил и 2-пиримид-2-илэтил. Примерами "С3-8циклоалкилС1-6алкила" являются циклопропилэтил, циклобутилметил, 2-циклопропилпропил и циклогексилэтил.
Подходящей фармацевтически приемлемой солью соединения настоящего изобретения является, например, кислотно-аддитивная соль соединения настоящего изобретения, которое является достаточно основным, например кислотно-аддитивная соль, образованная, например, неорганической или органической кислотой, например хлористоводородной, бромистоводородной, серной, фосфорной, трифторуксусной, лимонной или малеиновой кислотой. Кроме того, подходящей фармацевтически приемлемой солью соединения настоящего изобретения, которое является достаточно кислотным, является соль щелочного металла, например соль натрия или калия, соль щелочноземельного металла, например соль кальция или магния, аммониевая соль или соль, образованная органическим основанием, которое дает физиологически приемлемый катион, например соль, образованная метиламином, диметиламином, триметиламином, пиперидином, морфолином или трис-(2-гидроксиэтил)амином.
Соединения формулы (I) могут быть введены в форме пролекарства, которое разлагается в организме человека или животного с образованием соединения формулы (I). Примеры пролекарств включают in vivo гидролизуемые сложные эфиры соединения формулы (I).
In vivo гидролизуемым сложным эфиром соединения формулы (I), содержащим карбокси- или гидроксигруппу, является, например, фармацевтически приемлемый сложный эфир, который гидролизуется в организме человека или животного с образованием исходной кислоты или спирта. Подходящие фармацевтически приемлемые сложные эфиры для карбокси включают С1-6алкоксиметиловые эфиры, например метоксиметил, С1-6алканоилоксиметиловые эфиры, например пивалоилоксиметил, фталидиловые сложные эфиры, С3-8циклоалкоксикарбонилоксиС1-6алкиловые эфиры, например 1-циклогексилкарбонилоксиэтил; 1,3-диоксолен-2-онилметиловые эфиры, например 5-метил-1,3-диоксолен-2-онилметил; и С1-6алкоксикарбонилоксиэтиловые эфиры, например 1-метоксикарбонилоксиэтил, и такие эфиры могут быть образованы у любой карбоксигруппы соединений настоящего изобретения.
In vivo гидролизуемые сложные эфиры соединения формулы (I), содержащего гидроксигруппу, включают сложные эфиры неорганических кислот, такие как эфиры фосфорной кислоты и α-ацилоксиалкиловые эфиры, и родственные соединения, которые в результате in vivo-гидролиза сложного эфира разлагаются с образованием исходной гидроксигруппы. Примеры α-ацилоксиалкиловых эфиров включают ацетоксиметокси и 2,2-диметилпропионилоксиметокси. Подходящие группы, образующие in vivo гидролизуемый сложный эфир у гидроксигруппы, включают алканоил, бензоил, фенилацетил и замещенный бензоил и фенилацетил, алкоксикарбонил (с образованием эфиров алкилкарбоновой кислоты), диалкилкарбамоил и N-(диалкиламиноэтил)-N-алкилкарбамоил (с образованием карбаматов), диалкиламиноацетил и карбоксиацетил. Примеры заместителей у бензоила включают морфолино и пиперазин-1-ил, связанные с атомом азота в кольце посредством метиленовой группы в положении 3 или 4 данного бензоильного кольца.
Некоторые соединения формулы (I) могут иметь хиральные центры и/или геометрические изомерные центры (Е- и Z-изомеры), и при этом следует отметить, что настоящее изобретение относится ко всем указанным оптическим изомерам, диастереоизомерам и геометрическим изомерам, которые обладают CDK-ингибирующей активностью.
Настоящее изобретение относится к любым и ко всем таутомерным формам соединений формулы (I), которые обладают CDK-ингибирующей активностью.
Следует также отметить, что некоторые соединения формулы (I) могут присутствовать в сольватированной, а также в несольватированной формах, таких как, например, гидратированные формы. Следует отметить, что настоящее изобретение относится ко всем указанным сольватированным формам, которые обладают CDK-ингибирующей активностью.
В соответствии с другим аспектом настоящее изобретение относится к соединению формулы (I)
где R1 представляет галоген, нитро, циано, гидрокси, амино, карбокси, карбамоил, меркапто, С1-6алкил, С2-6алкенил или С2-6алкинил;
р=0-4, где R1 могут быть одинаковыми или различными;
R2 представляет сульфамоил или группу В-Е-;
q=0-2, где R2 могут быть одинаковыми или различными и где р+q=1-5;
R3 представляет водород, С1-6алкил, С2-6алкенил, С2-6алкинил или С3-8циклоалкил, где указанные С1-6алкил, С2-6алкенил, С2-6алкинил или С3-8циклоалкил могут быть необязательно замещены одним или несколькими М;
R4 представляет С1-6алкил, С2-6алкенил, С2-6алкинил или С3-8циклоалкил, где указанные С1-6алкил, С2-6алкенил, С2-6алкинил или С3-8циклоалкил могут быть необязательно замещены одним или несколькими М,
или R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют гетероциклическое кольцо, необязательно замещенное у атома углерода одним или несколькими М, где, если указанное гетероциклическое кольцо содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из Q;
В выбран из С1-6алкила, С2-6алкенила, С2-6алкинила, С3-8циклоалкила, С3-8циклоалкилС1-6алкила, фенила, гетероциклической группы, фенилС1-6алкила или (гетероциклическая группа)С1-6алкила, где указанные С1-6алкил, С2-6алкенил, С2-6алкинил, С3-8циклоалкил, С3-8циклоалкилС1-6алкил, фенил, гетероциклическая группа, фенилС1-6алкил или (гетероциклическая группа)С1-6алкил могут быть необязательно замещены у атома углерода одним или несколькими D и где, если указанная гетероциклическая группа содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из G;
Е представляет -С(О)-, -N(Rа)С(О)-, -С(О)N(Rа)-, -S(O)r-, -SO2N(Ra)- или -N(Ra)SO2-, где Rа представляет водород или С1-6алкил, необязательно замещенный одним или несколькими D, и r равно 1-2;
D независимо выбран из галогена, нитро, циано, гидрокси, трифторметила, трифторметокси, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-6алкила, С2-6алкенила, С2-6алкинила, С1-6алкокси, С1-6алканоила, С1-6алканоилокси, N-(С1-6алкил)амино, N,N-(С1-6алкил)2амино, С1-6алканоиламино, N-(С1-6алкил)карбамоила, N,N-(С1-6алкил)2карбамоила, С1-6алкилS(O)a, где а равно 0-2, С1-6алкоксикарбонила, N-(С1-6алкил)сульфамоила и N,N-(С1-6алкил)2сульфамоила, где D может быть необязательно замещен у атома углерода группой, выбранной из V;
М независимо выбран из галогена, нитро, циано, гидрокси, трифторметокси, амино, карбокси, карбамоила, меркапто, сульфамоила, С1-6алкила, С2-6алкенила, С2-6алкинила, С1-6алкокси, С1-6алканоила, С1-6алканоилокси, N-(С1-6алкил)амино, N,N-(С1-6алкил)2амино, С1-6алканоиламино, N-(С1-6алкил)карбамоила, N,N-(С1-6алкил)2карбамоила, С1-6алкилS(O)a, где а равно 0-2, С1-6алкоксикарбонила, N-(С1-6алкил)сульфамоила, N,N-(С1-6алкил)2сульфамоила, С3-8циклоалкила, фенила или гетероциклической группы, где М может быть необязательно замещен у атома углерода группой, выбранной из Р, и где в случае, если указанная гетероциклическая группа содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из Т, и
Р и V независимо выбраны из галогена, нитро, циано, гидрокси, трифторметокси, трифторметила, амино, карбокси, карбамоила, меркапто, сульфамоила, метила, этила, метокси, этокси, ацетила, ацетокси, метиламино, этиламино, диметиламино, диэтиламино, N-метил-N-этиламино, ацетиламино, N-метилкарбамоила, N-этилкарбамоила, N,N-диметилкарбамоила, N,N-диэтилкарбамоила, N-метил-N-этилкарбамоила, метилтио, этилтио, метилсульфинила, этилсульфинила, мезила, этилсульфонила, метоксикарбонила, этоксикарбонила, N-метилсульфамоила, N-этилсульфамоила, N,N-диметилсульфамоила, N,N-диэтилсульфамоила или N-метил-N-этилсульфамоила;
G, Q и T независимо выбраны из С1-4алкила, С1-4алканоила, С1-4алкилсульфонила, С1-4алкоксикарбонила, карбамоила, N-(С1-4алкил)карбамоила, N,N-(С1-4алкил)карбамоила, бензила, бензилоксикарбонила, бензоила и фенилсульфонила,
и/или
к его фармацевтически приемлемой соли или к in vivo гидролизуемому сложному эфиру.
Предпочтительные значения для R1, R2, R3, R4, p и q определены ниже. Указанные значения могут быть использованы, где это необходимо, в любых определениях, в формуле изобретения или в вариантах осуществления изобретения, определенных выше или ниже.
R1 предпочтительно представляет галоген или С1-2алкил.
Более предпочтительно R1 представляет фтор, хлор или метил.
В частности, R1 представляет фтор или хлор.
Предпочтительно p равно 0-2, где R1 могут быть одинаковыми или различными.
Более предпочтительно p равно 0 или 1.
В одном из аспектов настоящего изобретения p предпочтительно равно 0.
В другом аспекте настоящего изобретения p предпочтительно равно 1.
В еще одном аспекте настоящего изобретения p предпочтительно равно 2, где R1 могут быть одинаковыми или различными.
Предпочтительно, если p равно 1, а R1 находится в мета-положении по отношению к аминогруппе анилина формулы (I).
R2 предпочтительно представляет сульфамоил или группу В-Е-, где
В выбран из С1-6алкила, С2-6алкенила, С3-8циклоалкила, С3-8циклоалкилС1-6алкила, фенилС1-6алкила или (гетероциклическая группа)С1-6алкила, где указанные С1-6алкил, С2-6алкенил, С3-8циклоалкил, С3-8циклоалкилС1-6алкил, фенилС1-6алкил или (гетероциклическая группа)С1-6алкил могут быть необязательно замещены у атома углерода одним или несколькими D, и где, если указанная гетероциклическая группа содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из G;
Е представляет -N(Ra)SO2-, где Rа представляет водород;
D независимо выбран из галогена, гидрокси, С1-6алкокси или N-(С1-6алкил)амино, N,N-(С1-6алкил)2амино и
G представляет С1-4алкил.
Более предпочтительно R2 представляет сульфамоил или группу В-Е-, где
В выбран из С1-6алкила или (гетероциклическая группа)С1-6алкила, где указанные С1-6алкил или (гетероциклическая группа)С1-6алкил могут быть необязательно замещены у атома углерода одним или несколькими D;
Е представляет -N(Ra)SO2-, где Rа представляет водород;
D независимо выбран из гидрокси или N-(С1-6алкил)амино.
В частности, R2 выбран из сульфамоила, N-(3-гидрокси-2,2-диметилпропил)сульфамоила,
N-(3-(N-изопропиламино)пропил)сульфамоила или N-(тетрагидрофур-2-илметил)сульфамоила.
В другом аспекте настоящего изобретения R2 предпочтительно представляет сульфамоил или группу В-Е;
В выбран из С1-6алкила, С2-6алкенила, С2-6алкинила или (гетероциклическая группа)С1-6алкила, где В может быть необязательно замещен у атома углерода одним или несколькими D и где, если указанная гетероциклическая группа содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из G;
Е представляет -S(О)r- или -N(Ra)SO2-, где Rа представляет водород или С1-6алкил и r равно 2;
D независимо выбран из галогена, циано, гидрокси, амино, С1-6алкила, С1-6алкокси, N-(C1-6алкил)амино, N,N-(С1-6алкил)2амино, С1-6алканоиламина и С1-6алкилS(O)a, где а равно 0-2 и где D может быть необязательно замещен у атома углерода группой, выбранной из V;
V выбран из гидрокси и диметиламино и
G выбран из С1-4алкила.
В другом аспекте настоящего изобретения, более предпочтительно, R2 представляет сульфамоил или группу В-Е-;
В выбран из метила, этила, пропила, бутила, 2,2-диметилпропила, пентила, аллила, 2-пропинила, пирролидин-2-илметила, пирид-3-илметила, 1,4-диоксан-2-илметила, пирид-2-илметила, 2-морфолиноэтила, 2-1,3,4-триазол-2-илэтила, 2-пиперидиноэтила, 2-пирид-2-илэтила, 2-пирид-4-илэтила, 2-пирролидин-1-илэтила, 2-имидазол-4-илэтила, 3-имидазол-1-илпропила, 3-морфолинопропила, 3-пиперидинопропила или тетрагидрофур-2-илметила, где В может быть необязательно замещен у атома углерода одним или несколькими D и где, если указанная гетероциклическая группа содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из G;
Е представляет -S(О)r- или -N(Ra)SO2-, где Rа представляет водород или С1-6алкил и r равно 2;
D независимо выбран из фтора, циано, гидрокси, амино, метила, метокси, этокси, изопропиламино, диметиламино, диэтиламино, ацетамидо, этилтио и мезила, где D может быть необязательно замещен у атома углерода группой, выбранной из V;
V выбран из гидрокси и диметиламино и
G выбран из этила.
В другом аспекте настоящего изобретения R2, в частности, представляет сульфамоил, мезил, этилсульфонил,
2-этоксиэтилфульфонил, пропилсульфонил,
3-изопропиламинопропилсульфонил, 4-изопропиламинобутилсульфонил,
N-(тетрагидрофур-2-илметил)сульфамоил,
N-(пирид-3-илметил)сульфамоил, N-(пирид-2-илметил)сульфамоил,
N-(1,4-диоксан-2-илметил)сульфамоил, N-(метил)сульфамоил,
N-(2-метоксиэтил)сульфамоил, N-(2-этилтиоэтил)сульфамоил,
N-(2-морфолиноэтил)сульфамоил, N-(2-пиперидиноэтил)сульфамоил,
N-(2-пирид-2-илэтил)сульфамоил,
N-(2-пирролидин-1-илэтил)сульфамоил,
N-(2-имидазол-4-илэтил)сульфамоил,
N-(2-изопропиламиноэтил)сульфамоил, N-(2-мезилэтил)сульфамоил,
N-[2-(2-гидроксиэтокси)этил]сульфамоил,
N-[2-(1-этилпирролидин-2-ил)этил]сульфамоил,
N-(2-пирид-2-илэтил)сульфамоил, N-(2-диэтиламиноэтил)сульфамоил,
N-(2-пирид-4-илэтил)сульфамоил, N-(2-ацетамидоэтил)сульфамоил,
N-(2-диметиламиноэтил)сульфамоил,
N-2-[(5-метил-1,3,4-триазол-2-ил)этил]сульфамоил,
N-(2-гидроксиэтил)сульфамоил, N-(2-цианоэтил)сульфамоил,
N-(2-диэтиламиноэтил)-N-(метил)сульфамоил,
N-(2-метоксиэтил)-N-(метил)сульфамоил,
N-(2,2,2-трифторэтил)сульфамоил,
N-(3-гидрокси-2,2-диметилпропил)сульфамоил,
N-(3-изопропиламинопропил)сульфамоил,
N-(3-метоксипропил)сульфамоил,
N-(3-имидазолил-1-илпропил)сульфамоил,
N-(2-гидрокси-3-аминопропил)сульфамоил,
N-(3-гидроксипропил)сульфамоил, N-(3-этоксипропил)сульфамоил,
N-[3-(2-диметиламиноэтил)пропил]сульфамоил,
N-(3-морфолинопропил)сульфамоил, N-(2-гидроксипропил)сульфамоил,
N-(2-гидрокси-3-пиперидинопропил)сульфамоил,
N-(3-пиперидинопропил)-N-(метил)сульфамоил,
N-(2-гидроксибутил)сульфамоил, N-(пентил)сульфамоил,
N-(5-гидроксипентил)сульфамоил, N-(аллил)сульфамоил или
N-(2-пропинил)сульфамоил.
Предпочтительно Е представляет -NHSO2-.
В другом аспекте настоящего изобретения Е предпочтительно представляет -NHSO2-, -N(Ме)SO2- или -SO2-.
Предпочтительно q равно 0 или 1.
В одном из аспектов настоящего изобретения q предпочтительно равно 0.
В другом аспекте настоящего изобретения q предпочтительно равно 1.
В еще одном аспекте настоящего изобретения q предпочтительно равно 2; где R2 могут быть одинаковыми или различными.
Предпочтительно р+q=1 или 2.
Более предпочтительно р+q=1.
Предпочтительно, если q равно 1, то R2 находится в мета- или пара-положении по отношению к аминогруппе в анилине формулы (I).
Более предпочтительно, если q равно 1, то R2 находится в пара-положении по отношению к аминогруппе в анилине формулы (I).
Предпочтительно R3 представляет водород, С1-6алкил, С2-6алкенил или С2-6алкинил, где указанные С1-6алкил, С2-6алкенил или С2-6алкинил могут быть необязательно замещены одним или несколькими М, и
R4 представляет С1-6алкил, С2-6алкенил или С2-6алкинил, где указанные С1-6алкил, С2-6алкенил или С2-6алкинил могут быть необязательно замещены одним или несколькими М;
или R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют гетероциклическое кольцо, необязательно замещенное у атома углерода одним или несколькими М, где, если указанное гетероциклическое кольцо содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из Q.
Более предпочтительно R3 представляет водород или С1-6алкил, где указанный С1-6алкил может быть необязательно замещен одним или несколькими М, и
R4 представляет С1-6алкил или С2-6алкенил, где указанные С1-6алкил или С2-6алкенил могут быть необязательно замещены одним или несколькими М,
или R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют гетероциклическое кольцо, где
М независимо выбран из галогена, циано, С1-6алкокси, С1-6алкоксикарбонила или гетероциклической группы.
В частности, R3 представляет водород или С1-4алкил и
R4 представляет С1-4алкил или С2-4алкенил, где указанные С1-4алкил или С2-4алкенил могут быть необязательно замещены одним или несколькими М,
или R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют морфолино, где
М независимо выбран из фтора, циано, метокси, этокси, этоксикарбонила или морфолино.
Более предпочтительно R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют морфолино, изобутиламино, этиламино, 2-фторэтиламино, 3-этоксипропиламино, бутиламино, (N-метил)аллиламино, (N-метил)этоксикарбонилметиламино, (N-метил)-2-цианоэтиламино, N,N-диэтиламино, (N-метил)-2-метоксиэтиламино, 2,2,2-трифторэтиламино, N,N-ди-(2-цианоэтил)амино или 3-морфолинопропиламино.
В другом аспекте настоящего изобретения R3 предпочтительно представляет водород или С1-6алкил, где R3 может быть необязательно замещен одним или несколькими М, и
R4 представляет С1-6алкил, С2-6алкенил, С2-6алкинил или С3-8циклоалкил, где R4 может быть необязательно замещен одним или несколькими М,
или R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют гетероциклическое кольцо, необязательно замещенное у атома углерода одним или несколькими М, где, если указанное гетероциклическое кольцо содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из Q;
М независимо выбран из галогена, циано, гидрокси, С1-6алкила, С1-6алкокси, N,N-(С1-6алкил)2амино, С1-6алкоксикарбонила, С3-8циклоалкила или гетероциклической группы, где М может быть необязательно замещен у атома углерода группой, выбранной из Р;
Р и Х независимо выбраны из гидрокси и метокси и
Q выбран из С1-4алкила, С1-4алкилсульфонила или С1-4алкоксикарбонила, где G может быть необязательно замещен у атома углерода одним или несколькими Х.
В другом аспекте настоящего изобретения более предпочтительно R3 представляет водород, метил или этил, где R3 может быть необязательно замещен одним или несколькими М, и
R4 представляет метил, этил, бутил, изобутил, пропил, аллил, 2-пропинил, циклопропил или циклогексил, где R4 может быть необязательно замещен одним или несколькими М,
или R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют морфолино, пиперидино, пирролидин-1-ил или пиперазин-1-ил, необязательно замещенные у атома углерода одним или несколькими М, где указанный пиперазин-1-ил может быть необязательно замещен у атома азота группой, выбранной из Q;
М независимо выбран из фтора, циано, гидрокси, метила, метокси, этокси, диметиламино, метоксикарбонила, этоксикарбонила, бутоксикарбонила, циклопропила, тетрагидрофурила, пиридила, имидазолила, диоксоланила или морфолино, где М может быть необязательно замещен у атома углерода группой, выбранной из Р;
Р и Х независимо выбраны из гидрокси и метокси и
Q выбран из метила, этила, изопропила, этилсульфонила или этоксикарбонила, где G может быть необязательно замещен у атома углерода одним или несколькими Х.
В частности, R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют изобутиламино, этиламино,
2-фторэтиламино, 3-этоксипропиламино, бутиламино,
2,2,2-трифторэтиламино, 3-морфолинопропиламино,
циклопропиламино, циклопропилметиламино, циклогексиламино,
тетрагидрофур-2-иламино, 2-диметиламиноэтиламино,
цианометиламино, пирид-3-илметиламино,
бутоксикарбонилметиламино, 2-(метоксикарбонил)этиламино,
2-гидроксиэтиламино, метиламино, 2-пропиниламино,
2-метоксиэтиламино, 2-имидазол-4-илэтиламино,
2-(2-гидроксиэтокси)этиламино, 2,3-дигидроксипропиламино,
2,2-диметилдиоксолан-4-илметиламино, пропиламино,
N-метил-N-аллиламино, N-метил-N-этоксикарбонилметиламино,
N-метил-N-(2-цианоэтил)амино, диэтиламино,
N-метил-N-(2-метоксиэтил)амино, бис-(2-цианоэтил)амино,
N-этил-N-циклогексиламино, N-метил-N-(2,2,2-трифторэтил)амино,
N-метил-N-(2-пропинил)амино, морфолино, 2,6-диметилморфолино,
3,5-диметилпиперидино, пиперидино,
4-(2-метоксиэтил)пиперазин-1-ил, 4-метилпиперазин-1-ил,
4-изопропилпиперазин-1-ил, 4-этилсульфонилпиперазин-1-ил,
4-этоксикарбонилпиперазин-1-ил, 4-(2-гидроксиэтил)пиперазин-1-ил
и 3-гидроксипирролидин-1-ил.
Поэтому в одном аспекте настоящее изобретение относится к соединению формулы (I), описанному выше, где
p равно 0;
R2 представляет сульфамоил или группу В-Е, где
В выбран из С1-6алкила или (гетероциклическая группа)С1-6алкила, где указанные С1-6алкил или (гетероциклическая группа)С1-6алкил необязательно замещены у атома углерода одним или несколькими D;
Е представляет -N(Ra)SO2-, где Rа представляет водород;
D независимо выбран из гидрокси или N-(С1-6алкил)амино;
q равно 1.
R3 представляет водород или С1-6алкил, где указанный С1-6алкил может быть необязательно замещен одним или несколькими М, и
R4 представляет С1-6алкил или С2-6алкенил, где указанные С1-6алкил или С2-6алкенил могут быть необязательно замещены одним или несколькими М,
или R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют гетероциклическое кольцо, где
М независимо выбран из галогена, циано, С1-6алкокси, С1-6алкоксикарбонила или гетероциклической группы,
или к его фармацевтически приемлемой соли или in vivo гидролизуемому сложному эфиру.
Поэтому в другом аспекте настоящее изобретение относится к соединению формулы (I), описанному выше, где
p равно 0;
R2 представляет сульфамоил, N-(3-гидрокси-2,2-диметилпропил)сульфамоил,
N-(3-(N-изопропиламино)пропил)сульфамоил или N-(тетрагидрофур-2-илметил)сульфамоил;
q равно 1;
R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют морфолино, изобутиламино, этиламино, 2-фторэтиламино, 3-этоксипропиламино, бутиламино, (N-метил)аллиламино, (N-метил)этоксикарбонилметиламино, (N-метил)-2-цианоэтиламино, N,N-диэтиламино, (N-метил)-2-метоксиэтиламино, 2,2,2-трифторэтиламино, N,N-ди-(2-цианоэтил)амино или 3-морфолинопропиламино,
или к его фармацевтически приемлемой соли или in vivo гидролизуемому сложному эфиру.
Поэтому в следующем аспекте настоящее изобретение относится к соединению формулы (I) (описанному выше), где
p равно 0.
В другом аспекте настоящего изобретения R2 предпочтительно представляет сульфамоил или группу В-Е;
В выбран из С1-6алкила, С2-6алкенила, С2-6алкинила или (гетероциклическая группа)С1-6алкила, где В может быть необязательно замещен у атома углерода одним или несколькими D и где, если указанная гетероциклическая группа содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из G;
Е представляет -S(O)r- или -N(Ra)SO2-, где Rа представляет водород или С1-6алкил и r равно 2;
D независимо выбран из галогена, циано, гидрокси, амино, С1-6алкила, С1-6алкокси, N-(С1-6алкил)амино, N,N-(С1-6алкил)2амино, С1-6алканоиламино и С1-6алкилS(O)a, где а равно 0-2 и где D может быть необязательно замещен у атома углерода группой, выбранной из V;
V выбран из гидрокси и диметиламино;
G выбран из С1-4алкила;
q равно 1;
R3 представляет водород или С1-6алкил, где R3 может быть необязательно замещен одним или несколькими М, и
R4 представляет С1-6алкил, С2-6алкенил, С2-6алкинил или С3-8циклоалкил, где R4 может быть необязательно замещен одним или несколькими М,
или R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют гетероциклическое кольцо, необязательно замещенное у атома углерода одним или несколькими М, где, если указанное гетероциклическое кольцо содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из Q;
М независимо выбран из галогена, циано, гидрокси, С1-6алкила, С1-6алкокси, N,N-(С1-6алкил)2амино, С1-6алкоксикарбонила, С3-8циклоалкила или гетероциклической группы, где М может быть необязательно замещен у атома углерода группой, выбранной из Р;
Р и Х независимо выбраны из гидрокси и метокси;
Q выбран из С1-4алкила, С1-4алкилсульфонила или С1-4алкоксикарбонила, где G может быть необязательно замещен у атома углерода одним или несколькими Х;
или к его фармацевтически приемлемой соли или к in vivo гидролизуемому сложному эфиру.
Поэтому в следующем дополнительном аспекте настоящее изобретение относится к соединению формулы (I) (описанному выше), где
p равно 0;
R2 представляет сульфамоил или группу В-Е;
В выбран из метила, этила, пропила, бутила, 2,2-диметилпропила, пентила, аллила, 2-пропинила, пирролидин-2-илметила, пирид-3-илметила, 1,4-диоксан-2-илметила, пирид-2-илметила, 2-морфолиноэтила, 2-1,3,4-триазол-2-илэтила, 2-пиперидиноэтила, 2-пирид-2-илэтила, 2-пирид-4-илэтила, 2-пирролидин-1-илэтила, 2-имидазол-4-илэтила, 2-имидазол-1-илпропила, 3-морфолинопропила, 3-пиперидинопропила или тетрагидрофур-2-илметила, где В может быть необязательно замещен у атома углерода одним или несколькими D и где, если указанная гетероциклическая группа содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из G;
Е представляет -S(O)r- или -N(Ra)SO2-, где Rа представляет водород или С1-6алкил и r равно 2;
D независимо выбран из фтора, циано, гидрокси, амино, метила, метокси, этокси, изопропиламино, диметиламино, диэтиламино, ацетамидо, этилтио и мезила, где D может быть необязательно замещен у атома углерода группой, выбранной из V;
V выбран из гидрокси и диметиламино;
G выбран из этила;
q равно 1;
R3 представляет водород, метил или этил, где R3 может быть необязательно замещен одним или несколькими М, и
R4 представляет метил, этил, бутил, изобутил, пропил, аллил, 2-пропинил, циклопропил или циклогексил, где R4 может быть необязательно замещен одним или несколькими М,
или R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют морфолино, пиперидино, пирролидин-1-ил или пиперазин-1-ил, необязательно замещенные у атома углерода одним или несколькими М, где указанный пиперазин-1-ил может быть необязательно замещен у атома азота группой, выбранной из Q;
М независимо выбран из фтора, циано, гидрокси, метила, метокси, этокси, диметиламино, метоксикарбонила, этоксикарбонила, бутоксикарбонила, циклопропила, тетрагидрофурила, пиридила, имидазолила, диоксоланила или морфолино, где М может быть необязательно замещен у атома углерода группой, выбранной из Р;
Р и Х независимо выбраны из гидрокси и метокси и
Q выбран из метила, этила, изопропила, этилсульфонила или этоксикарбонила, где G может быть необязательно замещен у атома углерода одним или несколькими Х;
или к его фармацевтически приемлемой соли или к in vivo гидролизуемому сложному эфиру.
Поэтому в другом дополнительном аспекте настоящее изобретение относится к соединению формулы (I) (указанному выше), где
p равно 0;
R2 представляет сульфамоил, мезил, этилсульфонил,
2-этоксиэтилсульфонил, пропилсульфонил,
3-изопропиламинопропилсульфонил, 4-изопропиламинобутилсульфонил,
N-(тетрагидрофур-2-илметил)сульфамоил,
N-(пирид-3-илметил)сульфамоил, N-(пирид-2-илметил)сульфамоил,
N-(1,4-диоксан-2-илметил)сульфамоил, N-(метил)сульфамоил,
N-(2-метоксиэтил)сульфамоил, N-(2-этилтиоэтил)сульфамоил,
N-(2-морфолиноэтил)сульфамоил, N-(2-пиперидиноэтил)сульфамоил,
N-(2-пирид-2-илэтил)сульфамоил,
N-(2-пирролидин-1-илэтил)сульфамоил,
N-(2-имидазол-4-илэтил)сульфамоил,
N-(2-изопропиламиноэтил)сульфамоил, N-(2-мезилэтил)сульфамоил,
N-[2-(2-гидроксиэтокси)этил]сульфамоил,
N-[2-(1-этилпирролидин-2-ил)этил]сульфамоил,
N-(2-пирид-2-илэтил)сульфамоил, N-(2-диэтиламиноэтил)сульфамоил,
N-(2-пирид-4-илэтил)сульфамоил, N-(2-ацетамидоэтил)сульфамоил,
N-(2-диметиламиноэтил)сульфамоил,
N-2-[(5-метил-1,3,4-триазол-2-ил)этил]сульфамоил,
N-(2-гидроксиэтил)сульфамоил, N-(2-цианоэтил)сульфамоил,
N-(2-диэтиламиноэтил)-N-(метил)сульфамоил,
N-(2-метоксиэтил)-N-(метил)сульфамоил,
N-(2,2,2-трифторэтил)сульфамоил,
N-(3-гидрокси-2,2-диметилпропил)сульфамоил,
N-(3-изопропиламинопропил)сульфамоил,
N-(3-метоксипропил)сульфамоил,
N-(3-имидазол-1-илпропил)сульфамоил,
N-(2-гидрокси-3-аминопропил)сульфамоил,
N-(3-гидроксипропил)сульфамоил, N-(3-этоксипропил)сульфамоил,
N-[3-(2-диметиламиноэтил)пропил]сульфамоил,
N-(3-морфолинопропил)сульфамоил, N-(2-гидроксипропил)сульфамоил,
N-(2-гидрокси-3-пиперидинопропил)сульфамоил,
N-(3-пиперидинопропил)-N-(метил)сульфамоил,
N-(2-гидроксибутил)сульфамоил, N-(пентил)сульфамоил,
N-(5-гидроксипентил)сульфамоил, N-(аллил)сульфамоил или
N-(2-пропинил)сульфамоил;
q равно 1, и R2 находится в пара-положении по отношению к аминогруппе в анилине формулы (I), и
R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют изобутиламино, этиламино, 2-фторэтиламино, 3-этоксипропиламино, бутиламино, 2,2,2-трифторэтиламино, 3-морфолинопропиламино, циклопропиламино, циклопропилметиламино, циклогексиламино, тетрагидрофур-2-илметил, 2-диметиламиноэтиламино, цианометиламино, пирид-3-илметиламино, бутоксикарбонилметиламино, 2-(метоксикарбонил)этиламино, 2-гидроксиэтиламино, метиламино, 2-пропиниламино, 2-метоксиэтиламино, 2-имидазол-4-илэтиламино, 2-(2-гидроксиэтокси)этиламино, 2,3-дигидроксипропиламино, 2,2-диметилдиоксолан-4-илметиламино, пропиламино, N-метил-N-аллиламино, N-метил-N-этоксикарбонилметиламино, N-метил-N-(2-цианоэтил)амино, диэтиламино, N-метил-N-(2-метоксиэтил)амино, бис-(2-цианоэтил)амино, N-этил-N-циклогексиламино, N-метил-N-(2,2,2-трифторэтил)амино, N-метил-N-(2-пропинил)амино, морфолино, 2,6-диметилморфолино, 3,5-диметилпиперидино, пиперидино, 4-(2-метоксиэтил)пиперазин-1-ил, 4-метилпиперазин-1-ил, 4-изопропилпиперазин-1-ил, 4-этилсульфонилпиперазин-1-ил, 4-этоксикарбонилпиперазин-1-ил, 4-(2-гидроксиэтил)пиперазин-1-ил и 3-гидроксипирролидин-1-ил,
или к его фармацевтически приемлемой соли или in vivo гидролизуемому сложному эфиру.
В другом аспекте настоящего изобретения соединениями настоящего изобретения являются любые соединения примеров 1-17 или их фармацевтически приемлемая соль или in vivo гидролизуемый сложный эфир.
В другом аспекте настоящего изобретения соединениями настоящего изобретения являются любые соединения примеров или их фармацевтически приемлемая соль или их in vivo гидролизуемый сложный эфир.
В дополнительном аспекте настоящего изобретения предпочтительными соединениями являются соединения примеров 24, 38, 58, 59, 60, 63, 67, 73, 95 или 126 или их фармацевтически приемлемая соль или их in vivo гидролизуемый сложный эфир.
В предпочтительных аспектах настоящее изобретение относится к соединению формулы (I) или к его фармацевтически приемлемой соли.
В другом аспекте настоящее изобретение относится к способу получения соединения формулы (I) или его фармацевтически приемлемой соли или in vivo гидролизуемого сложного эфира, причем указанный способ (где R1, R2, R3, R4, p и q, если не оговорено особо, определены в формуле (I)) предусматривает
а) взаимодействие пиримидина формулы (II)
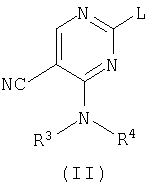
где L представляет заменяемую группу,
с анилином формулы (III)
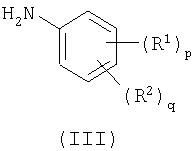
b) взаимодействие пиримидина формулы (IV)
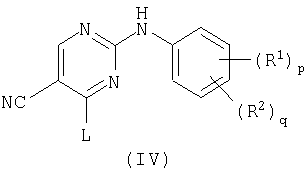
где L представляет заменяемую группу,
с амином формулы (V)
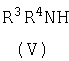
или
с) взаимодействие соединения формулы (VI)
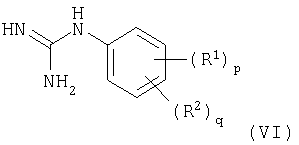
с соединением формулы (VII)
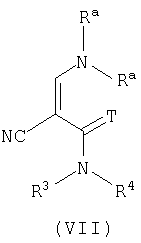
где Т представляет О или S, Rа могут быть одинаковыми или различными и выбраны из С1-6алкила;
d) для соединения формулы (I), где R2 представляет сульфамоил или группу В-Е и Е представляет -NHSO2-, взаимодействие пиримидина формулы (VIII)
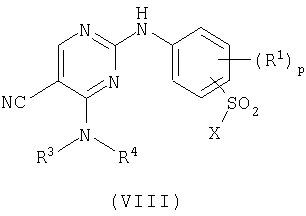
где Х представляет заменяемую группу,
с амином формулы (IX)
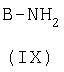
е) превращение соединения формулы (Х)
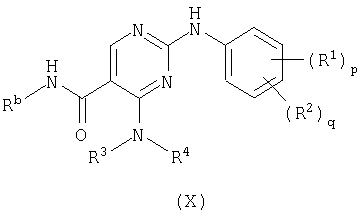
где Rb представляет водород или трет-бутил,
в соединение формулы (I)
и затем, если необходимо:
(i) превращение соединения формулы (I) в другое соединение формулы (I);
(ii) удаление любых защитных групп;
(iii) получение фармацевтически приемлемой соли или in vivo гидролизуемого сложного эфира.
L представляет заменяемую группу и обычно L представляет, например, галоген, (необязательно замещенный)арилокси или сульфонилоксигруппу, например хлор, бром, фенокси, метансульфонилокси или толуол-4-сульфонилоксигруппу.
Х представляет заменяемую группу, и подходящим значением Х является, например, галоген, например фтор, хлор или бром. Предпочтительно Х представляет фтор.
Предпочтительно Т представляет S.
Конкретные условия для указанных выше реакций описаны ниже.
а) и b) Пиримидины формулы (II) и анилины формулы (III) и пиримидины формулы (IV) и амины формулы (V) могут быть вместе подвергнуты взаимодействию:
(i) в присутствии подходящего растворителя, например кетона, такого как ацетон, или спирта, такого как этанол или бутанол, или ароматического углеводорода, такого как толуол или N-метилпирролидин, необязательно в присутствии подходящей кислоты, например неорганической кислоты, такой как хлористоводородная кислота или серная кислота, или органической кислоты, такой как уксусная кислота или муравьиная кислота (или подходящая кислота Льюиса), и при температуре в пределах от 0°С до температуры дефлегмации, предпочтительно при температуре дефлегмации, или
(ii) в стандартных условиях Бухвальда (например, см., J. Am. Chem. Soc., 118, 7215; J. Am. Chem. Soc., 119, 8451; J. Org. Chem., 62, 1568 и 6066), например, в присутствии ацетата палладия, в подходящем растворителе, например ароматическом растворителе, таком как толуол, бензол или ксилол, с подходящим основанием, например, неорганическим основанием, таким как карбонат цезия, или органическим основанием, таким как трет-бутоксид калия, в присутствии подходящего лиганда, такого как 2,2'-бис(дифенилфосфино)-1,1'-бинафтил, и при температуре в пределах от 25 до 80°С.
Пиримидины формулы (II) и (IV) и анилины формулы (III) и амины формулы (V) являются коммерчески доступными соединениями, либо они описаны в литературе, либо они могут быть получены стандартными способами, известными в данной области.
с) Соединения формулы (VI) и соединения формулы (VII) вместе подвергают взаимодействию в подходящем растворителе, таком как N-метилпирролидинон или бутанол, при температуре в пределах 100-200°С, а предпочтительно в пределах 150-170°С. Данную реакцию предпочтительно проводят в присутствии подходящего основания, такого как, например, метоксид натрия или карбонат калия.
Соединения формул (VI) и (VII) являются коммерчески доступными соединениями, либо они описаны в литературе, либо они могут быть получены стандартными способами, известными в данной области.
d) Соединения формулы (VIII) и соединения формулы (IX) могут быть вместе подвергнуты взаимодействию в присутствии основания, например неорганического основания, такого как карбонат цезия, в присутствии инертного растворителя, такого как толуол или тетрагидрофуран, или в присутствии органического основания, такого как избыток соединения (IX), и при температуре в пределах 25-80°С.
Соединения формулы (VIII), где Х представляет фтор, могут быть получены в соответствии со следующей схемой:
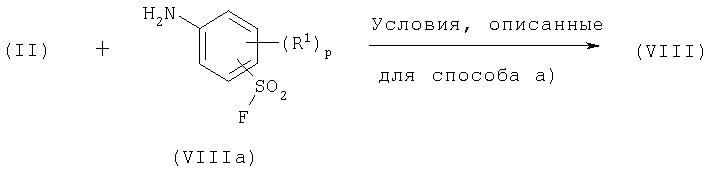
Соединения формулы (VIIIa) и (IX) являются коммерчески доступными соединениями, либо они описаны в литературе, либо они могут быть получены стандартными способами, известными в данной области.
е) Соединения формулы (Х) могут быть вместе превращены в соединение формулы (I) в стандартных условиях, например, в присутствии трифторуксусного ангидрида (где Rb представляет водород) или тионилхлорида при температуре в пределах 25-100°С.
Соединения формулы (Х) могут быть получены в соответствии со схемой 1 или схемой 2:
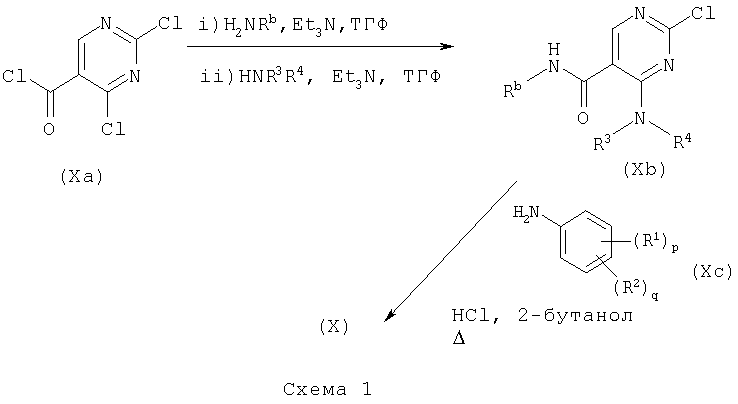
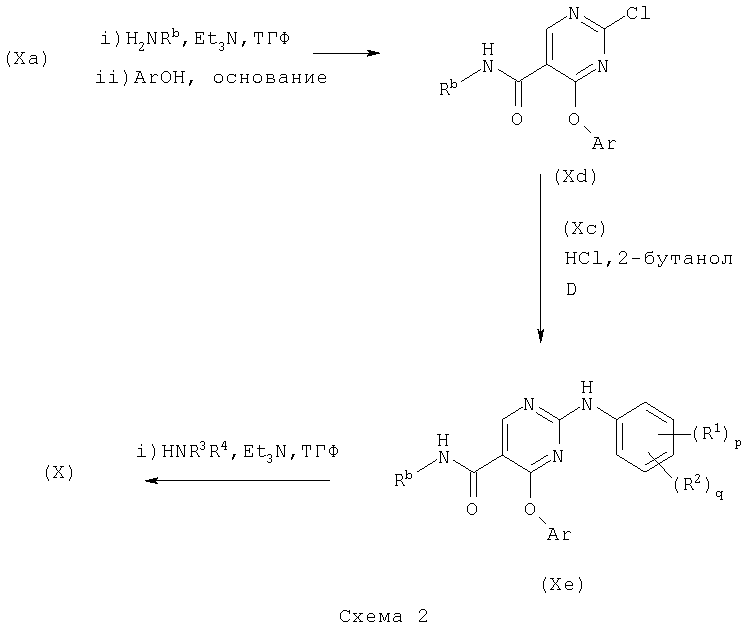
где Ar представляет необязательно замещенный арил, например фенил.
Соединения формул (Ха) и (Хс) являются коммерчески доступными соединениями, либо они описаны в литературе, либо они могут быть получены стандартными способами, известными в данной области.
При этом следует отметить, что в соединениях настоящего изобретения некоторые из различных заместителей в кольце могут быть введены стандартными реакциями ароматического замещения, либо они могут образованы стандартными модификациями функциональных групп, либо до, либо непосредственно после проведения указанных выше реакций, и такие реакции входят в один из аспектов настоящего изобретения, относящихся к способу. Указанными реакциями и модификациями являются, например, введение заместителя по реакции ароматического замещения, восстановление заместителей, алкилирование заместителей и окисление заместителей. Реагенты и реакционные условия для каждой реакции хорошо известны в данной области химии. Конкретные примеры реакций ароматического замещения включают введение нитрогруппы с использованием концентрированной азотной кислоты, введение ацильной группы с использованием, например, ацилгалогенида и кислоты Льюиса (такой как трихлорид алюминия) в условиях Фриделя-Крафтса; введение алкильной группы с использованием алкилгалогенида и кислоты Льюиса (такой как трихлорид алюминия) в условиях Фриделя-Крафтса; введение галогена. Конкретные примеры модификаций включают восстановление нитрогруппы в аминогруппу, например, каталитическим гидрированием в присутствии никелевого катализатора или обработкой железом в присутствии хлористоводородной кислоты при нагревании; окисление алкилтио до алкилсульфинила или алкилсульфонила.
Следует также отметить, что в некоторых указанных здесь реакциях может оказаться необходимой/желательной защита каких-либо реакционноспособных групп в данных соединениях. Примеры, где необходима или желательна защита и подходящие методы защиты известны специалистам в данной области. Стандартные защитные группы могут быть использованы в соответствии со стандартной практикой (для иллюстрации см., T.W.Green, Protective Groups in Organic Synthesis, John Wiley & Sons, 1991). Таким образом, если реагентами являются такие группы, как амино, карбокси или гидрокси, то в некоторых указанных здесь реакциях может оказаться желательной защита данных групп.
Подходящей защитной группой для амино или алкиламиногруппы является, например, ацильная группа, например, алканоильная группа, такая как ацетил, алкоксикарбонильная группа, например метоксикарбонильная, этоксикарбонильная или трет-бутоксикарбонильная группа, арилметоксикарбонильная группа, например бензилоксикарбонил, или ароильная группа, например бензоил. В случае использования указанных выше защитных групп условия снятия защиты могут варьироваться в зависимости от данной защитной группы. Так, например, ацильная группа, такая как алканоильная, или алкоксикарбонильная группа, или ароильная группа могут быть удалены, например, гидролизом с использованием подходящего основания, такого как гидроксид щелочного металла, например гидроксид лития или натрия. Альтернативно ацильная группа, такая как трет-бутоксикарбонильная группа, может быть удалена, например, обработкой подходящей кислотой, такой как хлористоводородная, серная или фосфорная кислота, или трифторуксусная кислота, а арилметоксикарбонильная группа, такая как бензилоксикарбонильная группа, может быть удалена, например, гидрированием в присутствии катализатора, такого как палладий на угле, или обработкой кислотой Льюиса, например, трис(трифторацетатом) бора. Подходящей альтернативной защитной группой для первичной аминогруппы является, например, фталоильная группа, которая может быть удалена обработкой алкиламином, например диметиламинопропиламином или гидразином.
Подходящей защитной группой для гидроксигруппы является, например, ацильная группа, например алканоильная группа, такая как ацетил, ароильная группа, например бензоил, или арилметильная группа, например бензил. В случае использования указанных выше защитных групп условия снятия защиты могут варьироваться в зависимости от используемой защитной группы. Так, например, ацильная группа, такая как алканоильная группа, или ароильная группа могут быть удалены, например, гидролизом с использованием подходящего основания, такого как гидроксид щелочного металла, например, гидроксид лития или натрия. Альтернативно арилметильная группа, такая как бензильная группа, может быть удалена, например, гидрированием в присутствии катализатора, такого как палладий на угле.
Подходящей защитной группой для карбоксигруппы является, например, этерифицирующая группа, например метильная или этильная группа, которая может быть удалена, например, гидролизом с использованием основания, такого как гидроксид натрия; или, например, трет-бутильная группа, которая может быть удалена, например, обработкой кислотой, например органической кислотой, такой как трифторуксусная кислота, или, например, бензильная группа, которая может быть удалена, например, гидрированием в присутствии катализатора, такого как палладий на угле.
Защитные группы могут быть удалены на любой из обычно используемых стадий синтеза стандартными методами, хорошо известными в данной области химии.
Как указано выше, соединения настоящего изобретения обладают активностью, ингибирующей пролиферацию клеток, такой как противораковая активность, которая, очевидно, обусловлена CDK-ингибирующей активностью данного соединения. Эти свойства могут быть оценены, например, с использованием описанной ниже методики.
Анализ
Были использованы следующие сокращения:
HEPES - N-[2-гидроксиэтил]пиперазин-N'-[2-этансульфоновая кислота]
DTT - дитиотреитол
PMSF - фенилметилсульфонилфторид.
Указанные соединения были протестированы в in vitro киназном анализе в 96-луночном планшете с использованием набора для сцинтилляционного проксимального анализа (SPA, полученного от Amersham) для измерения включения [γ-33-Р]-аденозинтрифосфата в тестируемый субстрат (GST-белок ретинобластомы; GST-Rb). В каждую лунку помещали тестируемое соединение (разведенное в ДМСО и в воде до нужных концентраций), а в контрольные лунки помещали либо росковитин в качестве контрольного ингибитора, или ДМСО в качестве позитивного контроля.
В каждую лунку добавляли приблизительно 0,2 мкл частично очищенного фермента CDK2/циклина Е (количество зависит от ферментативной активности), разведенного в 25 мкл буфера для инкубирования, а затем добавляли 20 мкл смеси GST-Rb/АТР/АТР33 (содержащей 0,5 мкг GST-Rb и 0,2 мкМ АТР и 0,14 мкКи [γ-33-Р]-аденозинтрифосфата в буфере для инкубирования) и полученную смесь слегка встряхивали, а затем инкубировали при комнатной температуре в течение 60 минут.
Затем в каждую лунку добавляли 150 мкл раствора для прекращения реакции, содержащего (0,8 мг/лунка PVT-сфер с белком А, SPA (Amersham)), 20 пМ/лунка кроличьего иммуноглобулина IgG против глутатион-трансферазы (полученного от Molecular Probes), 61 мМ EDTA и 50 мМ HEPES, рН 7,5, содержащего 0,05% азида натрия.
Планшеты герметично закрывали материалом для заклеивания планшетов Topseal-S, затем оставляли на два часа и центрифугировали при 2500 об/мин, 1124хg, в течение 5 минут. Планшеты считывали на счетчике Topcount, каждую лунку в течение 30 секунд.
Буфер для инкубирования, используемый для разведения фермента и субстратных смесей, содержал 50 мМ HEPES, рН 7,5, 10 мМ MnCl2, 1 мМ DTT, 100 мкМ ванадата натрия, 100 мкМ NaF, 10 мМ глицерофосфата натрия, BSA (конечная концентрация 1 мг/мл).
Тестируемый субстрат
В данном анализе использовали только часть белка ретинобластомы (Science, 1987, Mar 13; 235 (4794):1394-1399; Lee W.H., Bookstein R. Hong F., Young L.J., Shew J.Y., Lee E.Y.), присоединенного к GST-метке. Затем осуществляли ПЦР гена ретинобластомы, кодирующего аминокислоты 379-928 (полученного из плазмиды ретинобластомы АТСС pLRbRNL), и эту последовательность клонировали в гибридный вектор pGEX 2T (Smith D.B. & Johnson, K.S. Gene 67, 31 (1988), содержащий промотор tac для вызываемой экспрессии, внутренний ген lac Iq для использования в любом хозяине E.coli и кодирующую область для расщепления тромбина (полученный от Pharmacia Biotech), который был использован для амплификации аминокислот 792-928. Эту последовательность снова клонировали в pGEX 2Т.
Полученную таким образом последовательность 792-928 ретинобластомы экспрессировали в E.coli (клетки BL21 (DE3) pLysS) с использованием стандартной техники вызываемой экспрессии и очищали, как описано ниже.
Пасту E.coli ресуспендировали в 10 мл/г буфера NETN (50 мМ Tris, рН 7,5, 120 мМ NaCl, 1 мМ EDTA, 0,5% об/об NP-40, 1 мМ PMSF, 1 мкг/мл лейпептина, 1 мкг/мл апротинина и 1 мкг/мл пепстатина) и обрабатывали ультразвуком в течение 2×45 секунд на 100 мл гомогената. После центрифугирования супернатант загружали на колонку с 10 мл глутатион-сефарозы (Pharmacia Biotech, Herts, UK) и промывали буфером NETN. После промывки киназным буфером (50 мМ HEPES, рН 7,5, 10 мМ MgCl2, 1 мМ DTT, 1 мМ PMSF, 1 мкг/мл лейпептина, 1 мкг/мл апротинина и 1 мкг/мл пепстатина) белок элюировали 50 мМ восстановленного глутатиона в киназном буфере. Фракции, содержащие GST-Rb(792-927) объединяли и в течение ночи диализовали против киназного буфера. Конечный продукт анализировали с помощью электрофореза в ПААГ (полиакриламидном геле) с додецилсульфатом натрия (ДСН) с использованием 8-16% Tris-глициновых гелей (Novex, San Diego, USA).
CDK2 и циклин Е
Открытые рамки считывания CDK2 и циклина Е выделяли с помощью ПЦР с обратной транскриптазой с использованием клеток HeLa и активированной Т-клеточной мРНК в качестве матрицы, а затем клонировали в экспрессирующий вектор, происходящий от насекомого рVL1393 (полученный от Invitrogen 1995, каталожный номер V1392-20). Затем CDK2 и циклин Е подвергали двойной экспрессии [с использованием стандартной техники коинфицирования вирусом Baculogold] в системе клеток насекомого SF21 (коммерчески доступные клетки Spodoptera Frugiperda, полученные из ткани яичника совки травяной).
Пример продуцирования циклина Е/CDK2
В следующем ниже примере подробно описано продуцирование циклина Е/CDK2 в клетках SF21 (в Т100+10% FBS (TCS)+0,2% Плюроника) с двойной множественностью заражения (m.o.i.=3) для каждого вируса циклина Е и СDK2.
Клетки SF21, культивированные в роллер-флаконах до плотности 2,33×106 клеток/мл, использовали для инокуляции 10×500 мл клеточной культуры в роллер-флаконах с плотностью клеток 0,2×106 клеток/мл. Эти роллер-флаконы инкубировали на роллерном стенде при 28°С.
Через 3 дня (72 часа) клетки подсчитывали, и их среднее число для 2 флаконов составляло 1,86×106 клеток/мл (99% жизнеспособных клеток). Затем культуры инфицировали двумя вирусами при m.o.i.=3 для каждого вируса.
Перед добавлением в культуры эти вирусы смешивали вместе и культуры снова помещали на роллерный стенд при 28°С.
Через 2 дня (48 ч) после инфицирования собирали 5 литров культуры. Общее число клеток при каждом сборе составляло 1,58×106 клеток/мл (99% жизнеспособных клеток). Данные клетки центрифугировали при 2500 об/мин, 30 мин, 4°С на центрифуге Heraeus Omnifuge 2.0 RS в 250-миллилитровых партиях. Супернатант отбрасывали.
Частичная совместная очистка CDK2 и циклина Е
Клетки SF21 ресуспендировали в буфере для лизиса (50 мМ Tris, рН 8,2, 10 мМ MgCl2, 1 мМ DTT, 10 мМ глицерофосфата, 0,1 мМ ортованадата натрия, 0,1 мМ NaF, 1 мМ PMSF, 1 мкг/мл лейпептина и 1 мкг/мл апротинина) и гомогенизировали в течение 2 минут в 10-миллилитровом гомогенизаторе Даунса. После центрифугирования супернатант загружали на анионообменную колонку Poros HQ/M 1.4/100 (PE Biosystem, Hertford, UK). CDK2 и циклин Е совместно элюировали, начиная с градиента 0-1 М NaCl (пропуская в буфере для лизиса без ингибиторов протеазы) в 20 объемах колонки. Совместное элюирование контролировали с помощью Вестерн-блот-анализа с использованием антител против CDK2 и против циклина Е (Santa Cruz Biotechnology, California, US).
Аналогичным образом могут быть разработаны анализы для оценки ингибирования CDK4 и CDK6. CDK2 (EMBL рег. №Х62071) может быть использована вместе с циклином А или циклином Е (см. EMBL рег. №М73812), и более подробные описания данных анализов приводятся в публикации Международной заявки РСТ № WO 99/212845, в соответствующих разделах "Биохимическая и биологическая оценка", которые приводятся в настоящем описании в качестве ссылки.
Хотя фармакологические свойства соединений формулы (I) варьируются с изменением структуры, однако, в основном, активность соединений формулы (I) может быть продемонстрирована при концентрациях IC50 или при дозах в пределах от 250 мкМ до 1 нМ.
При тестировании в указанном выше in vitro-анализе CDK2-ингибирующая активность соединения примера 1, которую измеряли как IC50, составляла IC50=0,148 мкМ.
In vivo-активность соединений настоящего изобретения может быть определена стандартными методами, например путем измерения ингибирования клеточного роста и оценки цитотоксичности.
Ингибирование клеточного роста может быть измерено путем окрашивания клеток сульфородамином В (SRB), флуоресцентным красителем, который окрашивает белки и поэтому может быть использован для оценки количества белка (т.е. клеток) в лунке (см., Boyd M.R. (1989) Status of the NCI preclinical antitumour drug discovery screen. Prin. Prac Oncol. 10:1-12). Так, например, подробное описание измерения ингибирования клеточного роста приводится ниже.
Клетки высевали в 96-луночные планшеты в соответствующую среду в объеме 100 мкл, которая представляла собой модифицированную по способу Дульбекко среду Игла для MCF-7, SK-UT-1B и SK-UT-1. Клетки оставляли на ночь для связывания, а затем добавляли ингибирующие соединения в различных концентрациях при максимальной концентрации 1% ДМСО (об/об). Перед добавлением дозы соединения контрольный планшет оценивали на число клеток. Клетки инкубировали при 37°С (5% CO2) в течение трех дней.
По истечении трех дней в планшеты добавляли ТСА до конечной концентрации 16% (об/об). Затем планшеты инкубировали при 4°С в течение 1 часа, супернатант удаляли и планшеты промывали водопроводной водой. После сушки в течение 30 минут при 37°С добавляли 100 мкл красителя SRB (0,4% SRB в 1% уксусной кислоте). Избыток SRB удаляли и планшеты промывали 1% уксусной кислотой. Связанный с белком SRB солюбилизировали в 10 мМ Tris, рН 7,5, и встряхивали в течение 30 минут при комнатной температуре. OD (оптическую плотность) считывали при 540 нм и по графику зависимости 1/2 log концентрации ингибитора от оптической плотности определяли концентрацию, которая давала 50%-ное ингибирование клеточного роста. Концентрацию соединения, которая снижала оптическую плотность до величины, ниже величины, полученной при высевании клеток в начале эксперимента, определяли как степень токсичности.
Обычно значения IC50 для соединений настоящего изобретения при их тестировании в анализе с использованием SRB составляли в пределах от 1 мМ до 1нМ.
В соответствии с другим аспектом настоящее изобретение относится к фармацевтической композиции, содержащей пиримидиновое соединение формулы (I) или его фармацевтически приемлемую соль или in vivo гидролизуемый сложный эфир, определенные выше, в сочетании с фармацевтически приемлемым разбавителем или носителем.
Указанная композиция может быть изготовлена в форме, подходящей для перорального введения, например в форме таблетки или капсулы; для парентеральной инъекции (включая внутривенные, подкожные, внутримышечные, внутрисосудистые инъекции или вливания) в форме стерильного раствора, суспензии или эмульсии; для местного применения в форме мази или крема; для ректального введения в форме суппозитория.
Вообще говоря, указанные выше композиции могут быть изготовлены стандартным способом с использованием стандартных носителей.
Соединения формулы (I), в основном, вводят теплокровному животному в виде разовой лекарственной формы в дозе, составляющей 5-5000 мг на квадратный метр поверхности тела животного, т.е. приблизительно 0,1-100 мг/кг, и такая концентрация обычно обеспечивает терапевтически эффективную дозу. Разовая лекарственная форма, такая как таблетка или капсула, обычно содержит, например, 1-250 мг активного ингредиента. Предпочтительная суточная доза составляет 1-50 мг/кг. Однако при необходимости указанная суточная доза может варьироваться в зависимости от хозяина, подвергаемого лечению, от конкретного способа введения и от тяжести заболевания, подвергаемого лечению. В соответствии с этим оптимальная доза может быть определена специалистом, который лечит данного конкретного пациента.
В соответствии со следующим аспектом настоящее изобретение относится к соединению формулы (I) или к его фармацевтически приемлемой соли или к in vivo гидролизуемому сложному эфиру, определенным выше, которые могут быть использованы в способе терапевтического лечения человека или животного.
Было обнаружено, что соединения настоящего изобретения или их фармацевтически приемлемая соль или in vivo гидролизуемый сложный эфир являются эффективными ингибиторами клеточного цикла (ингибиторами пролиферации клеток), свойства которых, по всей вероятности, обусловлены их CDK-ингибирующими свойствами. В соответствии с этим предполагается, что соединения настоящего изобретения могут быть использованы для лечения заболеваний или патологических состояний, опосредованных отчасти или только ферментами CDK, т.е. указанные соединения могут быть использованы для продуцирования CDK-ингибирующего эффекта у теплокровного животного, нуждающегося в таком лечении. Таким образом, соединения настоящего изобретения позволяют осуществлять способ лечения, направленный на пролиферацию злокачественных клеток и характеризующийся ингибированием ферментов CDK, т.е. указанные соединения могут быть использованы для продуцирования антипролиферативного эффекта, опосредованного только или частично ингибированием CDK. Предполагается, что указанные соединения настоящего изобретения обладают широким спектром противораковых свойств, поскольку CDK участвуют во многих процессах, вызывающих наиболее распространенные раковые заболевания человека, такие как лейкоз и рак молочной железы, легких, толстой кишки, прямой кишки, желудка, предстательной железы, мочевого пузыря, поджелудочной железы и яичника. Поэтому ожидается, что соединение настоящего изобретения будет обладать противораковой активностью в отношении перечисленных раковых заболеваний. Кроме того, ожидается, что соединения настоящего изобретения будут обладать активностью против ряда лейкозов, лимфоидных злокачественных опухолей и твердых опухолей, таких как карциномы и саркомы в тканях, таких как печень, почки, предстательная железа и поджелудочная железа. В частности, предполагается, что такие соединения настоящего изобретения преимущественно замедляют рост первичных опухолей и рецидивов твердых опухолей, например, толстой кишки, молочной железы, предстательной железы, легких и кожи. Более конкретно, предполагается, что такие соединения настоящего изобретения или их фармацевтически приемлемые соли или in vivo гидролизуемый сложный эфир ингибируют рост первичных опухолей и рецидивов твердых опухолей, который ассоциируется с действием CDK, а в частности тех опухолей, рост и размножение которых в значительной степени зависят от CDK, включая, например, некоторые опухоли толстой кишки, молочной железы, предстательной железы, легких, вульвы и кожи.
Кроме того, ожидается, что соединения настоящего изобретения будут обладать активностью против других заболеваний широкого ряда, ассоциированных с пролиферацией клеток, включая, лейкозы, фибропролиферативные расстройства и расстройства, ассоциированные с нарушением дифференцировки клеток, псориаз, ревматоидный артрит, саркому Капоши, гемангиому, острую и хроническую нефропатию, атерому, атеросклероз, артериальный рестеноз, аутоиммунные заболевания, острые и хронические воспалительные заболевания, болезни костей и глазные болезни с пролиферацией сосудов сетчатки.
Таким образом, в соответствии с данным аспектом настоящее изобретение относится к определенному здесь соединению формулы (I) или к его фармацевтически приемлемой соли или in vivo гидролизуемому сложному эфиру, используемым в целях изготовления лекарственного средства, и к применению соединения формулы (I) или его фармацевтически приемлемой соли или in vivo гидролизуемого сложного эфира, определенных выше, при изготовлении лекарственного средства в целях продуцирования эффекта ингибирования клеточного цикла (ингибирования пролиферации клеток) у теплокровного животного, такого как человек. В частности, указанный ингибирующий эффект продуцируется путем предотвращения начала или дальнейшего развития клетки, т.е. ее прохождения через S-фазу, и путем ингибирования CDK2, CDK4 и/или CDK6, а особенно CDK2.
В соответствии со следующим аспектом настоящее изобретение относится к соединению формулы (I) или к его фармацевтически приемлемой соли или in vivo гидролизуемому сложному эфиру, определенным выше и используемым для лечения раковых заболеваний (твердых опухолей и лейкозов), фибропролиферативных расстройств и расстройств, ассоциированных с нарушением дифференцировки клеток, псориаза, ревматоидного артрита, саркомы Капоши, гемангиомы, острой и хронической нефропатии, атеромы, атеросклероза, артериального рестеноза, аутоиммунных заболеваний, острых и хронических воспалительных заболеваний, болезней костей и глазных болезней с пролиферацией сосудов сетчатки, а особенно для лечения рака.
В соответствии со следующим аспектом настоящее изобретение относится к способу продуцирования эффекта ингибирования клеточного цикла (ингибирования пролиферации клеток) у теплокровного животного, такого как человек, нуждающегося в таком лечении, где указанный способ предусматривает введение данному животному эффективного количества соединения, описанного непосредственно выше. В частности, указанный ингибирующий эффект заключается в предотвращении начала или дальнейшего развития клетки, т.е. ее прохождения через S-фазу, и обусловлен ингибированием CDK2, CDK4 и/или CDK6, а особенно CDK2.
В соответствии с другим аспектом настоящее изобретение относится к способу продуцирования эффекта ингибирования клеточного цикла (ингибирования пролиферации клеток) у теплокровного животного, такого как человек, нуждающегося в таком лечении, где указанный способ предусматривает введение данному животному эффективного количества описанного выше соединения формулы (I) или его фармацевтически приемлемой соли или in vivo гидролизуемого сложного эфира. В частности, ингибирующий эффект заключается в предотвращении начала или дальнейшего развития клетки, т.е. ее прохождения через S-фазу, и обусловлен ингибированием CDK2, CDK4 и/или CDK6, а особенно CDK2.
В соответствии с дополнительным аспектом настоящее изобретение относится к способу лечения раковых заболеваний (твердых опухолей и лейкозов), фибропролиферативных расстройств и расстройств, ассоциированных с нарушением дифференцировки клеток, псориаза, ревматоидного артрита, саркомы Капоши, гемангиомы, острой и хронической нефропатии, атеромы, атеросклероза, артериального рестеноза, аутоиммунных заболеваний, острых и хронических воспалительных заболеваний, болезней костей и глазных болезней с пролиферацией сосудов сетчатки, у теплокровного животного, такого как человек, нуждающегося в таком лечении, где указанный способ предусматривает введение данному животному эффективного количества описанного выше соединения формулы (I) или его фармацевтически приемлемой соли или in vivo гидролизуемого сложного эфира, описанных выше.
В частности, настоящее изобретение относится к способу лечения раковых заболеваний у теплокровного животного, такого как человек, нуждающегося в таком лечении, где указанный способ предусматривает введение данному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или in vivo гидролизуемого сложного эфира, описанных выше.
В еще одном аспекте настоящее изобретение относится к фармацевтической композиции, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль или in vivo гидролизуемый сложный эфир, определенные выше, в сочетании с фармацевтически приемлемым разбавителем или носителем и которая может быть использована для продуцирования эффекта ингибирования клеточного цикла (пролиферации клеток) у теплокровного животного, такого как человек.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль или in vivo гидролизуемый сложный эфир, определенные выше, в сочетании с фармацевтически приемлемым разбавителем или носителем и которая может быть использована для лечения раковых заболеваний (твердых опухолей и лейкозов), фибропролиферативных расстройств и расстройств, ассоциированных с нарушением дифференцировки клеток, псориаза, ревматоидного артрита, саркомы Капоши, гемангиомы, острой и хронической нефропатии, атеромы, атеросклероза, артериального рестеноза, аутоиммунных заболеваний, острых и хронических воспалительных заболеваний, болезней костей и глазных болезней с пролиферацией сосудов сетчатки, у теплокровного животного, такого как человек.
В еще одном аспекте настоящее изобретение относится к фармацевтической композиции, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль или in vivo гидролизуемый сложный эфир, определенные выше, в сочетании с фармацевтически приемлемым разбавителем или носителем и которая может быть использована для лечения рака у теплокровного животного, такого как человек.
Предупреждение начала синтеза ДНК путем ингибирования главной S-фазы, в которой инициируется активность такого фермента, как CDK2, может быть также использовано для защиты нормальных клеток организма от токсического воздействия цикл-специфических фармацевтических агентов. Ингибирование CDK2 или 4 предупреждает прохождение нормальными клетками последующих стадий клеточного цикла, что должно ограничивать токсическое воздействие цикл-специфических фармацевтических агентов, которые действуют в S-фазе, G2-фазе или в фазе митоза. Такая защита может обеспечивать предупреждение выпадения волос, обычно ассоциированное с действием указанных агентов.
Поэтому в другом аспекте настоящее изобретение относится к определенному здесь соединению формулы (I) или к его фармацевтически приемлемой соли или in vivo гидролизуемому сложному эфиру, используемым в качестве агента для защиты клеток.
Поэтому в следующем аспекте настоящее изобретение относится к определенному здесь соединению формулы (I) или к его фармацевтически приемлемой соли или in vivo гидролизуемому сложному эфиру, используемым для предупреждения выпадения волос при лечении злокачественных заболеваний фармацевтическими агентами.
Примеры фармацевтических агентов для лечения злокачественных заболеваний, которые, как известно, вызывают выпадение волос, включают алкилирующие агенты, такие как ифосфамид и циклофосфамид; антиметаболиты, такие как метотрексат, 5-фторурацил, гемцитабин и цитарабин; винкаалкалоиды и их аналоги, такие как винкристин, винбластин, виндезин, винорелбин; таксаны, такие как паклитаксел и доцетаксел; ингибиторы топоизомеразы I, такие как иринтотекан и топотекан; цитотоксические антибиотики, такие как доксорубицин, даунорубицин, митоксантрон, актиномицин-D и митомицин; другие агенты, такие как этопозид и третиноин.
В другом аспекте настоящего изобретения соединение формулы (I) или его фармацевтически приемлемая соль или in vivo гидролизуемый сложный эфир могут быть введены в сочетании с одним или несколькими указанными выше фармацевтическими агентами. В этом случае соединение формулы (I) может быть введено системными или несистемными способами. В частности, соединение формулы (I) может быть введено несистемными способами, например путем местного введения.
Поэтому в другом варианте осуществления настоящее изобретение относится к способу предупреждения выпадения волос при лечении одного или нескольких злокачественных заболеваний фармацевтическими агентами у теплокровного животного, такого как человек, где указанный способ предусматривает введение данному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или in vivo гидролизуемого сложного эфира.
В другом варианте осуществления настоящее изобретение относится к способу предупреждения выпадения волос при лечении одного или нескольких злокачественных заболеваний фармацевтическими агентами у теплокровного животного, такого как человек, где указанный способ предусматривает введение данному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или in vivo гидролизуемого сложного эфира при одновременном, поочередном или раздельном введении эффективного количества указанного фармацевтического агента.
В соответствии с другим аспектом настоящее изобретение относится к фармацевтической композиции для предупреждения выпадения волос при лечении злокачественных заболеваний фармацевтическими агентами, где указанная композиция содержит соединение формулы (I) или его фармацевтически приемлемую соль или in vivo гидролизуемый сложный эфир и указанный фармацевтический агент в сочетании с фармацевтически приемлемым разбавителем или носителем.
В соответствии с другим аспектом настоящее изобретение относится к набору, содержащему соединение формулы (I) или его фармацевтически приемлемую соль или in vivo гидролизуемый сложный эфир и фармацевтический агент для лечения злокачественных заболеваний, который, как известно, вызывает выпадение волос.
В соответствии с другим аспектом настоящее изобретение относится к набору, содержащему:
а) соединение формулы (I) или его фармацевтически приемлемую соль или in vivo гидролизуемый сложный эфир, в первой разовой лекарственной форме;
b) фармацевтический агент для лечения злокачественных заболеваний, которые, как известно, вызывают выпадение волос, во второй разовой лекарственной форме, и
с) емкость для первой и второй лекарственных форм.
В соответствии с другим вариантом настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли или in vivo гидролизуемого сложного эфира в изготовлении лекарственного средства для предупреждения выпадения волос при лечении злокачественных заболеваний фармацевтическими агентами.
В соответствии с другим аспектом настоящее изобретение относится к комбинированному лечению для предупреждения выпадения волос, которое предусматривает введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или in vivo гидролизуемого сложного эфира, необязательно в сочетании с фармацевтически приемлемым разбавителем или носителем, при одновременном, поочередном или раздельном введении теплокровному животному, такому как человек, эффективного количества фармацевтического агента для лечения злокачественных заболеваний.
Как указывалось выше, размер дозы, необходимой для терапевтического или профилактического лечения конкретного заболевания, ассоциированного с пролиферацией клеток, должен варьироваться в зависимости от хозяина, подвергаемого лечению, от конкретного способа введения и от тяжести заболевания, подвергаемого лечению. Предусматривается, что разовая доза должна находится в пределах, например, 1-100 мг/кг, предпочтительно 1-50 мг/кг.
CDK-ингибирующая активность, определенная выше, может быть реализована путем отдельной терапии, либо она может быть реализована путем введения, помимо соединения настоящего изобретения, одного или нескольких других веществ, и/или путем осуществления дополнительной терапии. Такое комбинированное лечение может быть осуществлено путем одновременного, поочередного или раздельного введения отдельных компонентов, используемых в данной терапии. В области клинической онкологии обычно практикуется использование комбинации различных форм для лечения конкретного ракового пациента. В клинической онкологии другим(и) компонентом(ами) такой комбинированной терапии могут быть хирургические операции, лучевая терапия и химиотерапия. В указанной химиотерапии могут быть использованы терапевтические агенты трех главных категорий:
(i) другие ингибирующие клеточный цикл агенты, которые действуют в соответствии с теми же самыми механизмами или механизмами, отличающимися от тех, по которым действуют соединения, определенные выше;
(ii) цитостатические агенты, такие как антиэстрогены (например, тамоксифен, торемифен, ралоксифен, дролоксифен, иодоксифен), прогестогены (например, ацетат мегестрола), ингибиторы ароматазы (например, анастрозол, летразол, воразол, эксеместан), антипрогестогены, антиандрогены (например, флутамид, нилутамид, бикалутамид, ацетат ципротерона), агонисты и антагонисты LHRH (например, ацетат гозерелина, лупролид), ингибиторы тестостерон-5α-дигидроредуктазы (например, финастерид), антиинвазивные агенты (например, ингибиторы металлопротеиназы, такие как маримастат, и ингибиторы функции рецептора урокиназы, являющейся активатором плазминогена) и ингибиторы функции факторов роста (такими факторами роста являются, например, ингибиторы тромбоцитарного фактора роста и гепатоцитарного фактора роста, включая антитела против факторов роста, антитела против рецепторов факторов роста, ингибиторы тирозинкиназы и ингибиторы серин/треонинкиназ);
(iii) антипролиферативные/противоопухолевые лекарственные средства и их комбинации, используемые в клинической онкологии, такие как антиметаболиты (например, антифолаты, такие как метотрексат, фторопиримидины, такие как аналоги 5-фторурацила, пурина и аденозина, арабинозид цитозина); противоопухолевые антибиотики (например, антрациклины, такие как доксорубицин, дауномицин, эпирубицин и идарубицин, митомицин-С, дактиномицин, митрамицин); производные платины (например, цисплатин, карбоплатин); алкилирующие агенты (например, азотистый иприт, мелфалан, хлорамбуцил, бусулфан, циклофосфамид, ифосфамид, нитрозомочевины, тиотепа); антимитотические агенты (например, винкаалкалоиды, такие как винкристин, и таксоиды, такие как таксол, таксотер); ингибиторы топоизомеразы (например, эпиподофиллотоксины, такие как этопозид и тенипозид, амсакрин, топотекан). В соответствии с данным аспектом настоящее изобретение относится к фармацевтическому продукту, который содержит соединение формулы (I), определенное выше, и другое противоопухолевое вещество, определенное выше, и который используется для комбинированного лечения рака.
Помимо применения в терапии соединения формулы (I) и их фармацевтически приемлемые соли могут быть также использованы в качестве фармакологических средств для разработки и стандартизации in vitro и in vivo тест-систем, применяемых для оценки действия ингибиторов активности клеточного цикла у лабораторных животных, таких как кошки, собаки, кролики, обезьяны, крысы и мыши, и являющихся частью исследований для поиска новых терапевтических агентов.
В указанных выше других фармацевтических композициях, процессах, методах, применениях и вариантах изготовления лекарственных средств могут быть также использованы альтернативные и предпочтительные варианты описанных здесь соединений настоящего изобретения.
Примеры
Настоящее изобретение проиллюстрировано на следующих ниже, но не ограничивающих примерах, в которых, если не оговорено особо:
(i) температуры даны в градусах Цельсия (°С); процедуры осуществляют при комнатной температуре или при температуре окружающей среды, то есть при температуре в пределах 18-25°С;
(ii) органические растворы сушат над безводным сульфатом магния; выпаривание растворителя осуществляют на роторном испарителе при пониженном давлении (600-4000 паскаль; 4,5-30 мм рт.ст.) при температуре бани до 60°С;
(iii) термин "хроматография" означает флэш-хроматографию на силикагеле; тонкослойную хроматографию (ТСХ) осуществляют на пластинах с силикагелем;
(iv) в основном, за ходом реакции наблюдают с помощью ТСХ, а время реакции приводится лишь для иллюстрации;
(v) конечные продукты имеют удовлетворительные спектры протонного ядерного магнитного резонанса (ЯМР) и/или данные масс-спектров;
(vi) выходы приведены лишь для иллюстрации и представляют собой необязательно выходы, которые могут быть получены трудоемкими способами выделения, причем, если требовались большие количества продукта, то стадии получения повторяли;
(vii) данные ЯМР, если они указаны, даны в форме дельта-величин для главных характерных протонов, выражены в миллионных долях (м.д.) по отношению к тетраметилсилану (ТМС), используемому в качестве внутреннего стандарта, и определены при 300 МГц с использованием пердейтеродиметилсульфоксида (ДМСО-d6), используемого в качестве растворителя, если не оговорено особо;
(viii) химические символы имеют свое обычное значение, при этом используются единицы и символы в системе СИ;
(ix) отношения растворителей выражены в величинах объем:объем (об/об) и
(х) масс-спектры были получены химической ионизацией (ХИ) с энергией электронов 70 электрон-вольт путем прямого облучения пробы, где указанную ионизацию осуществляли злектронным ударом (EI), бомбардировкой быстрыми атомами (FAB) или электронапылением (ESP), при этом приводятся величины для m/z; и, в основном, регистрировались только ионы, которые имели массу исходного иона, и если не оговорено особо, то образовывался масс-ион (МН)+;
(xi) если не оговорено особо, то соединения, содержащие асимметрически замещенный атом углерода и/или серы, не разделяли;
(xii) в том случае, если описываемый синтез был аналогичен синтезу, описанному в предыдущем примере, то используемые количества имели миллимолярное отношение, эквивалентное отношениям, используемым в предыдущем примере;
(xvi) были использованы следующие сокращения:
Пример 1
5-Циано-4-морфолино-2-{4-[N-(3-изопропиламинопропил)сульфамоил]анилино}пиримидин
2-Хлор-5-циано-4-морфолинопиримидин (метод 55; 425 мг, 1,90 ммоль), 4-[N-(3-изопропиламинопропил)сульфамоил]анилин (метод 1; 514 мг, 1,90 ммоль) и 1 М эфирный раствор хлористого водорода (189 мкл, 3,79 ммоль) в 2-бутаноле (2 мл) нагревали при 95°С в течение 15 часов. Смесь оставляли для охлаждения, добавляли двуокись кремния и летучие вещества выпаривали. Остаток очищали хроматографией, элюируя смесью ДХМ/метанольный раствор аммиака (100:0) с увеличением полярности до (92:8), и полученный продукт перекристаллизовывали из метанола, получая указанное в заголовке соединение (164 мг, 19%). ЯМР: 0,89 (д, 6H), 1,44 (м, 2H), 2,40 (т, 2H), 2,56 (м, 1H), 2,76 (т, 2H), 3,68-3,74 (м, 4H), 3,83-3,90 (м, 4H), 7,69 (д, 2H), 7,83 (д, 2H), 8,49 (с, 1H); m/z: 460,
Примеры 2-11
В соответствии с методикой примера 1 с использованием 4-[N-(3-изопропиламинопропил)сульфамоил]анилина (метод 1) и соответствующего исходного 4-замещенного пиримидинового продукта, получали следующие соединения.
Пример 12
5-Циано-4-(3-морфолинопропиламино)-2-{4-[N-(тетрагидрофур-2-илметил)сульфамоил]анилино}пиримидин
2-Хлор-5-циано-4-(3-морфолинопропиламино)пиримидин (метод 66; 525 мг, 1,87 ммоль), 4-[N-(тетрагидрофур-2-илметил)сульфамоил]анилин (метод 2; 430 мг, 1,68 ммоль) и 1 М эфирный раствор хлористого водорода (1,87 мл, 1,87 ммоль) в 2-бутаноле (3 мл) нагревали при 95°С в течение 3 часов. Смесь оставляли для охлаждения, полученный осадок отфильтровывали и промывали этилацетатом. Неочищенное твердое вещество очищали хроматографией, элюируя смесью ДХМ/метанольный раствор аммиака (100:0) с увеличением полярности до (97:3). Полученный продукт перекристаллизовывали из метанола, получая указанное в заголовке соединение (111 мг). ЯМР: 1,50 (м, 1H), 1,67-1,88 (м, 5H), 2,28-2,40 (м, 6H), 2,73 (т, 2H), 3,48 (м, 2H), 3,51-3,60 (м, 5H), 3,67 (м, 1H), 3,77 (м, 1H), 7,50 (т, 1H), 7,69 (д, 2H), 7,91 (д, 2H), 7,93 (с, 1H), 8,38 (с, 1H), 10,13 (шир.с, 1H); m/z: 502.
Примеры 13-19
В соответствии с методикой примера 12 с использованием 4-[N-(тетрагидрофур-2-илметил)сульфамоил]анилина (метод 2) и соответствующего исходного 4-замещенного пиримидинового продукта, получали следующие соединения.
Примеры 20-28
В соответствии с методикой примера 12 с использованием 4-[N-(2-метоксиэтил)сульфамоил]анилина (метод 5) и соответствующего исходного 4-замещенного пиримидинового продукта, получали следующие соединения.
Примеры 29-34
В соответствии с методикой примера 12 с использованием 4-[N-(3-метоксипропил)сульфамоил]анилина (метод 4) и соответствующего исходного 4-замещенного пиримидинового продукта получали следующие соединения.
Примеры 35-39
В соответствии с методикой примера 12 с использованием 4-н-бутиламино-2-хлор-5-цианопиримидина (метод 60) и соответствующего исходного анилинового соединения получали следующие соединения.
Примеры 39-40
В соответствии с методикой примера 12 с использованием 2-хлор-5-циано-4-(2,2,2-трифторэтиламино)пиримидина (метод 95) и соответствующего исходного анилинового соединения получали следующие соединения.
Примеры 41-42
В соответствии с методикой примера 12 с использованием 2-хлор-5-циано-4-этиламинопиримидина (метод 56) и соответствующего исходного анилинового соединения получали следующие соединения.
Пример 43
5-Циано-4-бутиламино-2-{4-[N-(тетрагидрофур-2-илметил)сульфамоил]анилино}пиримидин
4-Бутиламино-5-N-трет-бутилкарбамоил-2-{4-[N-(тетрагидрофур-2-илметил)сульфамоил]анилино}пиримидин (метод 67; 200 мг, 0,40 ммоль) в тионилхлориде (1 мл) нагревали при 90°С в течение 12 часов. Смесь оставляли для охлаждения, а затем выпаривали на двуокиси кремния. Остаток очищали хроматографией, элюируя смесью ДХМ/метанол (100:0) с увеличением полярности до (97:3). Продукт перекристаллизовывали из метанола, получая указанное в заголовке соединение (33 мг). ЯМР: 0,90 (т, 3H), 1,34 (м, 2H), 1,41-1,63 (м, 3H), 1,66-1,88 (м, 3H), 2,73 (т, 2H), 3,40 (м, 2H), 3,53 (м, 1H), 3,64 (м, 1H), 3,77 (м, 1H), 7,51 (т, 1H), 7,67 (д, 2H), 7,88 (шир.с, 1H), 7,91 (д, 2H), 8,37 (с, 1H), 10,13 (шир.с, 1H); m/z: 431.
Примеры 44-54
В соответствии с методикой примера 43 с использованием исходного 4-замещенного амидопиримидинового соединения получали следующие соединения.
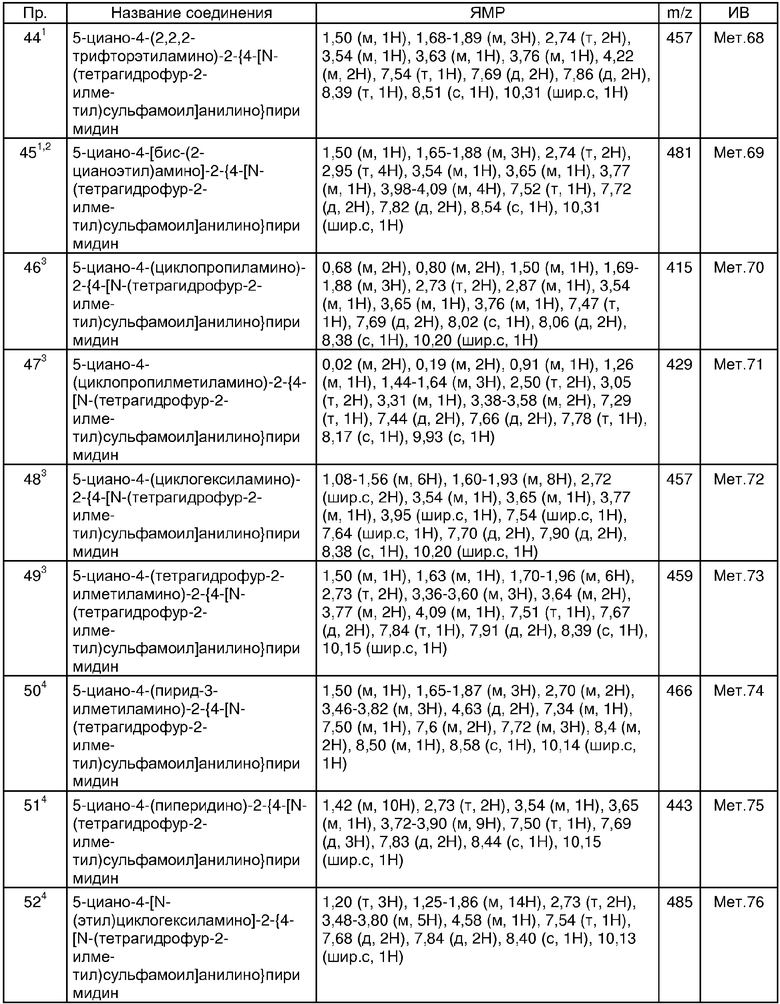
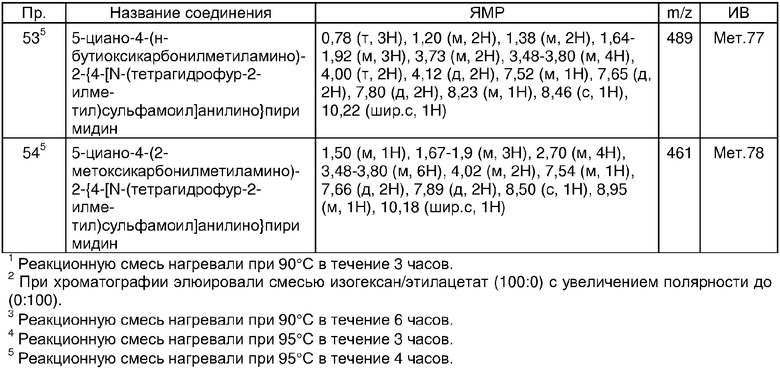
Примеры 55-56
В соответствии с методикой примера 1 2-хлор-5-циано-4-морфолинопиримидин (метод 55) обрабатывали соответствующим анилином, получая следующие соединения.
доступ.
Пример 57
4-[4-(2-Гидроксиэтил)пиперазин-1-ил]-5-циано-2-{4-[N-(3-метоксипропил)сульфамоил]анилино}пиримидин
4-[4-(2-Ацетоксиэтилпиперазин-1-ил)]-5-циано-2-{4-[N-(3-метоксипропил)сульфамоил]анилино}пиримидин (метод 119; 290 мг) растворяли в метаноле (3 мл), добавляли 35% аммиак (1,5 мл) и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Летучие вещества выпаривали и остаток очищали хроматографией на силикагеле, элюируя смесью ДХМ/метанол (98:2) с увеличением полярности до (95:5). Полученное твердое вещество растирали со смесью этилацетат/диэтиловый эфир и отфильтровывали, получая указанное в заголовке соединение (19 мг). ЯМР: 1,55 (м, 2H), 2,45 (2H), 2,55 (шир.т, 4H), 2,75 (кв, 2H), 3,14 (с, 3H), 3,26 (2H), 3,50 (кв, 2H), 3,88 (шир.т, 4H), 4,42 (т, 1H), 7,35 (т, 1H), 7,68 (д, 2H), 7,84 (д, 2H), 8,48 (с, 1H), 10,21 (с, 1H); m/z: 476.
Пример 58
5-Циано-4-циклопропиламино-2-{4-[N-(2-метоксиэтил)сульфамоил]анилино}пиримидин
2-Хлор-5-циано-4-циклопропиламинопиримидин (метод 96; 295 мг, 1,28 ммоль) растворяли в горячем 2-бутаноле (3 мл). Затем добавляли горячий раствор 4-[N-(2-метоксиэтил)сульфамоил]анилина (метод 5; 207 мг, 0,90 ммоль) в 2-бутаноле (3 мл) и реакционную смесь перемешивали и нагревали при 90°С в течение 1 часа. Затем реакционную смесь охлаждали и летучие вещества выпаривали с получением твердого вещества. Твердый остаток растирали с горячим этилацетатом, охлаждали и полученное твердое вещество отфильтровывали, получая указанное в заголовке соединение (270 мг, 77%). ЯМР: 0,68 (м, 2H), 0,80 (м, 2H), 2,88 (м, 3H), 3,14 (с, 3H), 3,28 (м, 2H), 7,48 (шир.с, 1H), 7,70 (д, 2H), 8,05 (д, 2H), 8,12 (шир.с, 1H) 8,41 (с, 1H), 10,31 (с, 1H); m/z: 389.
Примеры 59-72
В соответствии с методикой примера 58 с использованием исходного 2-хлор-5-циано-4-циклопропиламинопиримидина (метод 96) и соответствующего анилина получали следующие соединения.
Пример 73
5-Циано-4-этиламино-2-{4-[N-(3-метоксипропил)сульфамоил]анилино}пиримидин
5-Циано-4-этиламино-2-[(4-фторсульфонил)анилино]пиримидин (метод 111; 50 мг, 0,16 ммоль), 3-метоксипропиламин (28 мг, 0,31 ммоль) и полимер, несущий 4-(диметиламино)пиридин (поставляемый Argonout Technologies Inc; 1,45 ммоль/л) (218 мг, 0,31 ммоль) в NMP (1 мл) нагревали при 100°С в течение 18 часов. Летучие вещества удаляли выпариванием и остаток растирали с эфиром. Полученное твердое вещество очищали хроматографией c обращенной фазой (Waters xterra, колонка 19×50 мм) (0,1% муравьиной кислоты в градиенте 0-95% ацетонитрила в воде), получая указанное в заголовке соединение (16 мг, 26%). ЯМР: 1,18 (т, 3H), 1,57 (м, 2H), 2,75 (м, 2H), 3,13 (с, 3H), 3,25 (м, 2H), 3,43 (м, 2H), 7,35 (м, 1H), 7,65 (д, 2H), 7,82 (м, 1H), 7,94 (д, 2H), 8,39 (с, 1H), 10,13 (с, 1H); m/z: 391.
Примеры 74-100
В соответствии с методикой примера 73 5-циано-4-этиламино-2-[(4-фторсульфонил)анилино]пиримидин (метод 111) обрабатывали соответствующим амином с получением следующих соединений.
Пример 101
5-Циано-4-н-бутиламино-2-{4-[N-(этилтиоэтил)сульфамоил]анилино}пиримидин
5-Циано-4-бутиламино-2-[(4-фторсульфонил)анилино]пиримидин (метод 112; 200 мг, 0,57 ммоль), 2-(этилтио)этиламин (480 мг, 4,56 ммоль), триэтиламин (58 мг, 0,57 ммоль) и 4-(диметиламино)пиридин (7 мг, 0,06 ммоль) в 1-бутаноле (6 мл) нагревали при 95°С в течение 24 часов. Затем смесь оставляли для охлаждения и растворитель удаляли выпариванием. Полученный остаток перекристаллизовывали из этанола с получением указанного в заголовке соединения (130 мг). ЯМР: 0,90 (т, 3H), 1,10 (т, 3H), 1,35 (м, 2H), 1,59 (м, 2H), 2,40-2,60 (м, 4H), 2,90 (т, 2H), 3,40 (кв, 2H), 7,70 (д, 2H), 7,80-8,88 (м, 3H), 8,40 (с, 1H); m/z: 434.
Примеры 102-108
В соответствии с методикой примера 101 с использованием исходного 5-циано-4-бутиламино-2-(4-фторсульфонил)анилино]пиримидина (метод 112) и соответствующих аминов, получали следующие соединения.
Пример 109
5-Циано-4-(2,2,2-трифторэтиламино)-2-{4-[N-(2-имидазо-4-илэтил)сульфамоил]анилино}пиримидин
5-Циано-4-(2,2,2-трифторэтиламино)-2-[(4-фторсульфонил)анилино]пиримидин (метод 113; 200 мг, 0,53 ммоль), гистамин (466 мг, 4,24 ммоль), триэтиламин (54 мг, 0,53 ммоль) и 4-(диметиламино)пиридин (7 мг, 0,05 ммоль) в 2-бутаноле (6 мл) нагревали при 95°С в течение 48 часов. Затем смесь оставляли для охлаждения и растворитель удаляли выпариванием. Остаток перекристаллизовывали из этанола с получением указанного в заголовке соединения (80 мг). ЯМР: 2,70 (м, 2H), 3,10 (т, 2H), 4,30 (м, 2H), 6,70 (с, 1H), 7,40 (с, 1H), 7,70 (д, 2H), 7,90 (д, 2H), 8,50 (с, 1H); m/z: 466.
Примеры 110-114
В соответствии с методикой примера 109 с использованием исходного 5-циано-4-(2,2,2-трифторэтиламино)-2-[(4-фторсульфонил)анилино]пиримидина (метод 113) и соответствующих аминов получали следующие соединения.
Пример 115
5-Циано-4-(2,3-дигидроксипропиламино)-2-{4-[N-(3-метоксипропил)сульфамоил]анилино}пиримидин
Суспензию 2-хлор-4-(4-хлорфенокси)-5-цианопиримидина (метод 115; 179 мг, 0,68 ммоль) и 4-[N-(3-метоксипропил)сульфамоил]анилина (метод 4; 149 мг, 0,61 ммоль) в 2-бутаноле (6 мл) нагревали и перемешивали при 50°С в течение 5 часов. Затем добавляли 3-амино-1,2-пропандиол (309 мг, 3,4 ммоль) и температуру повышали до 90°С, после чего реакционную смесь перемешивали при этой температуре еще 18 часов. Растворитель удаляли выпариванием и оставшееся твердое вещество перекристаллизовывали из этанола с получением указанного в заголовке соединения (104 мг, 39%). ЯМР: 1,55 (м, 2H), 2,75 (м, 2H), 3,15 (с, 3H), 3,25 (м, 2H), 3,40 (м, 3H), 3,60 (м, 1H), 3,75 (м, 1H), 4,70 (т, 1H), 5,85 (м, 1H), 7,35 (т, 1H), 7,55 (шир.с, 1H), 7,70 (д, 2H), 7,95 (д, 2H), 8,40 (с, 1H), 10,20 (с, 1H); m/z: 436.
Примеры 116-129
В соответствии с методикой примера 115 с использованием исходного 2-хлор-4-(4-хлорфенокси)-5-цианопиримидина (метод 115), соответствующего анилиносульфонамида/сульфона (см. столбец ИВ) и соответствующих аминов получали следующие соединения.
пиримидин
пиримидин
Пример 130
5-Циано-4-этиламино-2-{4-[N-(2-гидрокси-3-пиперидинопропил)сульфамоил]анилино}пиримидин
4-[N-(2-Гидрокси-3-пиперидинопропил)сульфамоил]анилин (метод 118; 200 мг, 0,64 ммоль) растворяли в метаноле (4 мл). Затем добавляли 1 М раствор хлористого водорода в эфире (1,28 мл, 1,28 ммоль) и смесь слегка нагревали для получения раствора. Летучие вещества частично выпаривали до получения объема приблизительно 1,5 мл. После этого добавляли 2-бутанол (5 мл) и мутный раствор нагревали до 50°С. Затем порциями добавляли 2-хлор-5-циано-4-этиламинопиримидин (метод 56; 233 мг, 1,28 ммоль) и реакционную смесь нагревали при 95°С в течение 20 часов. Горячую надосадочную жидкость декантировали от твердого остатка и раствор оставляли для охлаждения. Полученный осадок отфильтровывали, промывали диэтиловым эфиром и суспендировали в воде (приблизительно 35 мл). Доводили рН суспензии до рН>10 2 М водным раствором гидроксида натрия и экстрагировали этилацетатом (3×15 мл). Экстракты сушили, летучие вещества выпаривали и остаток очищали хроматографией на двуокиси кремния, элюируя смесью ДХМ/метанольный раствор аммиака (100:0) с увеличением полярности до (85:15). Полученный продукт растирали с диэтиловым эфиром и отфильтровывали, получая указанное в заголовке соединение (30 мг). ЯМР: 1,15 (т, 3H), 1,35 (м, 2H), 1,43 (м, 4H), 2,10-2,35 (м, 6H), 2,68 (м, 1H), 2,83 (м, 1H), 3,48 (квин, 2H), 3,56 (м, 1H), 4,59 (с, 1H) 7,36 (шир.т, 1H), 7,70 (д, 2H), 7,88 (т, 1H), 7,95 (д, 2H), 8,40 (с, 1H) 10,16 (с, 1H); m/z: 460.
Примеры 131-132
В соответствии с методикой примера 130 с использованием 4-[N-(2-гидрокси-3-пиперидинопропил)сульфамоил]анилина (метод 118) и соответствующего пиримидинового исходного соединения получали следующие соединения.
Получение исходных соединений
Исходные соединения для указанных выше примеров являются либо коммерчески доступными, либо они могут быть легко получены стандартными методами из известных соединений. Так, например, некоторые из исходных соединений, используемых в описанных выше реакциях, могут быть получены в соответствии со следующими иллюстративными, но не ограничивающими реакциями.
Метод 1
4-[N-(3-Изопропиламинопропил)сульфамоил]анилин
Сульфанилилфторид (6,5 г, 37,1 ммоль), N-изопропил-1,3-пропандиамин (5,71 мл, 40,8 ммоль) и триэтиламин (5,69 мл, 40,8 ммоль) в N-бутаноле (15 мл) кипятили с обратным холодильником в течение 10 часов. Смесь оставляли для охлаждения, добавляли двуокись кремния и летучие вещества выпаривали. Остаток очищали хроматографией, элюируя смесью ДХМ/метанольный раствор аммиака (100:0) с увеличением полярности до (90:10), и получали указанное в заголовке соединение в виде прозрачного масла, которое кристаллизовалось при стоянии (8,81 г, 88%). ЯМР: 0,89 (д, 6H), 1,43 (м, 2H), 2,41 (т, 2H), 2,58 (м, 1H), 2,68 (т, 2H), 3,16 (с, 2H), 5,85 (с, 2H), 6,58 (д, 2H), 7,38 (д, 2H); m/z: 272.
Методы 2-9
Следующие соединения получали в соответствии с указаниями метода 1.
Метод 10
N-(2-Метоксиэтил)-N-метил-4-нитробензолсульфонамид
Перемешиваемый раствор триэтиламина (2,4 мл, 17,23 ммоль) и N-(2-метоксиэтил)метиламина (1,5 г, 16,85 ммоль) в ДХМ (20 мл) охлаждали на водяной бане со льдом. Затем по каплям добавляли раствор 4-нитробензолсульфонилхлорида (3,2 г, 14,45 ммоль) в ДХМ (20 мл). Реакционную смесь перемешивали в течение одного часа, охлаждающую баню удаляли и смесь перемешивали еще 3 часа. Реакционную смесь промывали 1 М хлористоводородной кислотой (40 мл), водой (30 мл), а затем насыщенным раствором соли. Летучие вещества выпаривали с получением указанного в заголовке соединения (3,8 г). m/z: 245.
Методы 11-14
В соответствии с указаниями метода 10 с использованием исходного 4-нитробензолсульфонилхлорида и соответствующего коммерчески доступного амина получали следующие соединения.
Метод 15
N-Метил-4-нитро-N-(3-пиперидинопропил)бензолсульфонамид
4-Нитро-N-(3-пиперидинопропил)бензолсульфонамид (метод 14; 1 г, 3,06 ммоль) перемешивали в атмосфере азота при комнатной температуре. Затем порциями добавляли гидрид натрия (60% дисперсия в минеральном масле) (122 мг, 3,05 ммоль). Реакционную смесь перемешивали в течение 10 минут, а затем по каплям добавляли иодметан (210 мкл, 3,38 ммоль). Через 2 часа добавляли дополнительное количество гидрида натрия (<30 мг) и реакционную смесь перемешивали в течение 15 минут, а затем оставляли на ночь. Летучие вещества удаляли выпариванием, добавляли воду (20 мл) и раствор экстрагировали этилацетатом (2×30 мл). Органические слои объединяли, промывали водой и летучие вещества выпаривали с получением указанного в заголовке соединения (830 мг). ЯМР: 1,34 (м, 2H), 1,45 (м, 4H), 1,60 (квинтет, 2H), 2,19 (м, 2H), 2,24 (м, 4H), 2,72 (с, 3H), 3,02 (т, 2H), 8,02 (д, 2H), 8,40 (д, 2H); m/z: 342.
Метод 16
4-[N-(2-Метоксиэтил)-N-(метил)сульфамоил]анилин
N-(2-Метоксиэтил)-N-метил-4-нитробензолсульфонамид (метод 10; 3,8 г) восстанавливали гидрированием в этаноле (100 мл) в присутствии 10% палладия на угле (400 мг) под давлением 3 бар. Катализатор удаляли фильтрованием и летучие вещества выпаривали с получением указанного в заголовке соединения (3,2 г). m/z: 245.
Методы 17-20
Следующие соединения получали в соответствии с указаниями метода 16.
Метод 21
1-Нитро-4-[4-(фталимидо)бутилсульфид]фенил
4-Нитротиофенол (1,9 г, 12,26 ммоль) перемешивали в ДМФ в атмосфере азота и охлаждали (водяная баня со льдом). Затем порциями добавляли гидрид натрия (60% дисперсия в минеральном масле; 400 мг, 10 ммоль) и смесь перемешивали в течение 10 минут. Затем добавляли N-(4-бромбутил)фталимид (2,8 г, 9,93 ммоль) в ДМФ (10 мл) и реакционную смесь нагревали при 90°С в течение 1,5 часов, охлаждали до комнатной температуры и оставляли на ночь при комнатной температуре. Летучие вещества удаляли выпариванием, добавляли воду (20 мл) и раствор экстрагировали этилацетатом (50+25 мл). Органические слои объединяли, промывали водой (20 мл) и насыщенным раствором соли, сушили и летучие вещества выпаривали. Полученную смолу дважды растирали с изогексаном. Растворитель декантировали с получением указанного в заголовке соединения в виде твердого вещества (3,8 г). ЯМР: 1,64 (м, 2H), 1,74 (м, 2H), 3,12 (т, 2H), 3,60 (т, 2H), 7,45 (д, 2H), 7,81 (м, 4H), 8,06 (д, 2H).
Метод 22
Следующее соединение получали в соответствии с указаниями метода 21.
Метод 23
1-Нитро-4-[4-(фталимидо)бутилсульфонил]фенил
Раствор оксида хрома (VI) (3,5 г, 35,0 ммоль) в воде (3 мл) и в ледяной уксусной кислоте (12,5 мл) по каплям в течение 15-20 минут добавляли к раствору 1-нитро-4-[4-(фталимидо)бутилсульфид]фенила (метод 21; 3,5 г, 9,83 ммоль) в ледяной уксусной кислоте (17,5 мл), нагретому при 90-100°С. Затем смесь нагревали при 100°С в течение 3,5 часов. Реакционную смесь охлаждали и выливали на измельченный лед (250 г). Твердое вещество отфильтровывали и промывали водой. Затем твердое вещество сушили тройной азеотропной перегонкой с метанолом, получая указанное в заголовке соединение (3,4 г). m/z: 389.
Метод 24
1-Нитро-4-[3-(фталимидо)пропилсульфонил]фенил
1-Нитро-4-[3-(фталимидо)пропилсульфид]фенил (метод 22; 600 мг) растворяли в ДХМ (20 мл) и в метаноле (1 мл). Затем порциями в течение 10 минут добавляли 3-хлорпербензойную кислоту (70%; 1,4 г). Через 75 минут добавляли ДХМ (10 мл), насыщенный раствор бикарбоната натрия (10 мл) и воду (10 мл) и раствор перемешивали в течение 20 минут, а затем снова добавляли насыщенный раствор натриевой соли. Органический слой отделяли и промывали водой и насыщенным раствором соли, сушили и выпаривали досуха, получая указанное в заголовке соединение (795 мг). ЯМР: 1,90 (м, 2H), 3,55 (м, 2H), 3,64 (т, 2H), 7,81 (с, 4H), 8,17 (д, 2H), 8,41 (д, 2H).
Метод 25
1-Нитро-4-(4-аминобутилсульфонил)фенил
1-Нитро-4-[4-(фталимидо)бутилсульфонил]фенил (метод 23; 3 г, 7,73 ммоль) нагревали при 90°С в ацетонитриле (30 мл) и метаноле (10 мл). Затем добавляли гидразингидрат (0,76 мл, 15,7 ммоль) и реакционную смесь нагревали в течение 1,5 часов, охлаждали и оставляли на ночь при комнатной температуре. Полученное твердое вещество удаляли фильтрованием и промывали метанолом. Объединенные фильтраты выпаривали, получая указанное в заголовке соединение (2,3 г). m/z: 259.
Метод 26
4-(3-Аминопропилсульфонил)-1-нитрофенил
Указанное в заголовке соединение получали из соединения метода 24, используя указания метода 25.
Метод 27
1-Нитро-4-[4-(изопропиламино)бутилсульфонил]фенил
1-Нитро-4-(4-аминобутилсульфонил)фенил (метод 25; 2 г, 7,75 ммоль) перемешивали в метаноле (20 мл) и добавляли ацетон (2,3 мл). Затем порциями в течение 5 минут добавляли цианоборогидрид натрия (730 мг, 11,62 ммоль) и реакционную смесь перемешивали в течение 2,5 часов. После этого добавляли воду (15 мл), органические растворители удаляли выпариванием, снова добавляли воду и раствор экстрагировали этилацетатом (130 мл). Органические слои промывали водой (25 мл) и насыщенным раствором соли. Летучие вещества выпаривали и остаток очищали хроматографией на нейтральной окиси алюминия (активность II), элюируя ДХМ с увеличением полярности до метанол:ДХМ (3:97), в результате чего получали указанное в заголовке соединение (1,26 г). m/z: 301.
Метод 28
Следующее соединение получали в соответствии с указаниями метода 27.
Метод 29
4-[4-(Изопропиламино)бутилсульфонил]анилин
1-Нитро-4-[4-(изопропиламино)бутилсульфонил]фенил (метод 27; 1,2 г, 4 ммоль) растворяли в этаноле (20 мл) и в 1 М хлористоводородной кислоте (6 мл) и гидрировали под давлением в 1 атмосферу и в присутствии 10% палладия на угле (400 мг) в течение 4 часов. Катализатор удаляли фильтрованием и летучие вещества выпаривали с получением указанного в заголовке соединения в виде пены. m/z: 271.
Метод 30
Следующее соединение получали в соответствии с указаниями метода 29.
Метод 31
2,4-Дихлор-5-хлорформилпиримидин
5-Карбокси-2,4-дигидроксипиримидин (19,0 г, 0,12 моль), пентахлорид фосфора (83,5 г, 0,40 моль) и фосфорилхлорид (28,3 мл, 0,30 моль) нагревали при 114°С в течение 5 часов. Полученный раствор охлаждали в течение ночи и летучие вещества удаляли выпариванием. Полученный остаток очищали вакуумной перегонкой с получением указанного в заголовке соединения в виде прозрачного масла (17,85 г, 70%). m/z: 211.
Метод 32
5-N-трет-Бутилкарбамоил-2-хлор-4-морфолинопиримидин
Раствор 2,4-дихлор-5-хлорформилпиримидина (метод 31; 500 мг, 2,36 ммоль) в безводном ТГФ (5 мл) охлаждали до -15°С. Затем медленно добавляли трет-бутиламин (250 мкл, 2,36 ммоль) и триэтиламин (330 мкл, 2,36 ммоль) в безводном ТГФ так, чтобы температура поддерживалась ниже -10°С. Полученную смесь перемешивали при -10°С в течение 2 часов, оставляли для нагревания до комнатной температуры и перемешивали еще 30 минут. Затем добавляли морфолин (208 мкл, 2,36 ммоль) и триэтиламин (330 мкл, 2,36 ммоль) в безводном ТГФ (1 мл) и полученную смесь перемешивали при комнатной температуре в течение 12 часов. Осадок удаляли фильтрованием и фильтрат выпаривали с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (570 мг, 78%). ЯМР: 1,30 (с, 9H), 3,52-3,58 (м, 4H), 3,60-3,67 (м, 4H), 8,00 (с, 1H), 8,21 (шир.с, 1H); m/z: 299.
Методы 33-54
В соответствии с указаниями метода 32 и с использованием в качестве исходных соединений 2,4-дихлор-5-хлорформилпиримидина (метод 31), трет-бутиламина и аминов получали следующие промежуточные соединения.
Метод 55
2-Хлор-5-циано-4-морфолинопиримидин
5-N-трет-Бутилкарбамоил-2-хлор-4-морфолинопиримидин (метод 32; 560 мг, 1,88 ммоль) в тионилхлориде (4 мл) нагревали при 90°С в течение 2 часов. Смесь охлаждали до комнатной температуры, летучие вещества выпаривали и осадок подвергали азеотропной перегонке с толуолом, получая указанное в заголовке соединение в виде коричневого твердого вещества (425 мг). ЯМР: 3,66-3,74 (м, 4Н), 3,85-3,93 (м, 4Н), 8,63 (с, 1Н); m/z: 225, 227.
Метод 56
2-Хлор-5-циано-4-этиламинопиримидин
5-N-трет-Бутилкарбамоил-2-хлор-4-этиламинопиримидин (метод 34; 255 мг, 1,00 ммоль) и тионилхлорид (240 мкл, 3,29 ммоль) в толуоле (2 мл) нагревали при 90°С в течение 15 часов. Смесь охлаждали до комнатной температуры, добавляли двуокись кремния и летучие вещества выпаривали. Остаток очищали хроматографией при элюировании смесью ДХМ/метанол (100:0) с увеличением полярности до (97:3), получая указанное в заголовке соединение (86 мг). ЯМР: 1,12 (т, 3Н), 3,39 (м, 2Н), 8,52 (с, 1Н), 8,55 (шир.с, 1Н).
Методы 57-66
Следующие соединения получали в соответствии с указаниями метода 56 с использованием соответствующего исходного пиримидинового соединения.
Метод 67
4-н-Бутиламино-5-N-трет-бутилкарбамоил-2-{4-[N-(тетрагидрофур-2-илметил)сульфамоил]анилино}пиримидин
5-н-Бутиламино-5-N-трет-бутилкарбамоил-2-хлорпиримидин (метод 37; 282 мг, 1,0 ммоль), 4-[N-(тетрагидрофур-2-илметил)сульфамоил]анилин (метод 2; 241 мг, 0,95 ммоль) и 1 М эфирный раствор хлористого водорода (993 мкл, 1,0 ммоль) в 2-бутаноле (2 мл) нагревали при 90°С в течение 3 часов. Смесь охлаждали до комнатной температуры, добавляли двуокись кремния и летучие вещества выпаривали. Остаток очищали хроматографией при элюировании смесью ДХМ/метанол (100:0) с увеличением полярности до (95:5), получая указанное в заголовке соединение в виде белого твердого вещества (372 мг). m/z: 505.
Методы 68-78
Все следующие соединения получали в соответствии с указаниями метода 67 с использованием 4-[N-(тетрагидрофур-2-илметил)сульфамоил]анилина (метод 2) и соответствующего пиримидина.
Метод 79
5-N-трет-Бутилкарбамоил-2,4-дихлорпиримидин
Раствор 2,4-дихлор-5-хлорформилпиримидина (метод 31; 9,8 г, 46,3 ммоль) в безводном ТГФ (50 мл) охлаждали до -15°С. Затем медленно добавляли трет-бутиламин (5,2 мл, 49,3 ммоль) и триэтиламин (6,9 мл, 49,5 ммоль) в безводном ТГФ (20 мл) так, чтобы температура поддерживалась ниже -10°С. Полученную смесь перемешивали при -10°С в течение 2 часов, оставляли для нагревания до комнатной температуры и перемешивали еще 30 минут. Осадок удаляли фильтрованием и фильтрат выпаривали с получением вязкого масла, а затем выпаривали в высоком вакууме, получая указанное в заголовке соединение в виде твердого вещества (10,48 г, 90%). ЯМР: 1,49 (с, 9Н), 6,19 (шир.с, 1Н), 8,86 (с, 1Н); m/z: 248.
Метод 80
5-N-трет-Бутилкарбамоил-2-хлор-4-[4-(2-гидроксиэтил)пиперазин-1-ил]пиримидин
Раствор триэтиламина (210 мкл, 1,5 ммоль) и 1-(2-гидроксиэтил)пиперазина (195 мг, 1,5 ммоль) в безводном ТГФ (3 мл) добавляли к раствору 5-N-трет-бутилкарбамоил-2,4-дихлорпиримидина (метод 79; 372 мг, 1,5 ммоль) в безводном ТГФ (2 мл). Смесь перемешивали при комнатной температуре в течение 5 часов. Полученный осадок удаляли фильтрованием, фильтровальный слой промывали сухим эфиром (5 мл) и фильтрат выпаривали с получением указанного в заголовке соединения. m/z: 342 (1×Cl).
Методы 81-94
Следующие производные получали из 5-N-трет-бутилкарбамоил-2,4-дихлорпиримидина (метод 79) и соответствующего амина, используя указания метода 80.
Метод 95
2-Хлор-5-циано-4-(2,2,2-трифторэтиламино)пиримидин
Раствор 5-N-трет-бутилкарбамоил-2-хлор-4-(2,2,2-трифторэтиламино)пиримидина (метод 43; 8,5 г, 0,027 моль) в тионилхлориде (100 мл) кипятили с обратным холодильником в течение 4 часов. Тионилхлорид удаляли выпариванием и полученную смолу растирали со смесью изогексан/этилацетат (95:5), отфильтровывали и сушили с получением указанного в заголовке соединения. ЯМР: 4,20 (м, 2Н), 8,70 (с, 1Н), 9,10 (шир.т, 1Н); m/z: 236.
Метод 96
2-Хлор-5-циано-4-циклопропиламинопиримидин
5-N-трет-Бутилкарбамоил-2-хлор-4-циклопропиламинопиримидин (метод 46; 2,40 г, 8,92 ммоль) нагревали при 80-90°С с тионилхлоридом (10 мл) в течение 4-6 часов. Тионилхлорид удаляли выпариванием и остаток растирали со смесью эфир/этилацетат с получением указанного в заголовке соединения (2,00 г), которое использовали без дополнительной очистки.
Методы 97-110
Следующие соединения получали, используя указания метода 96, из соответствующего исходного пиримидинового соединения. Конечные продукты не были охарактеризованы, а были использованы непосредственно в последующих реакциях.
Метод 111
5-Циано-4-этиламино-2-[4-(фторсульфонил)анилино]пиримидин
2-Хлор-5-циано-4-этиламинопиримидин (метод 56; 6,35 г, 34,88 ммоль) и сульфанилилфторид (6,11 г, 34,88 ммоль) в 2-бутаноле (120 мл) нагревали при 95°С в течение 4 часов, а затем перемешивали при комнатной температуре в течение 48 часов. Летучие вещества выпаривали и остаток растирали с эфиром, получая указанное в заголовке соединение (10,46 г, 93%). ЯМР: 1,20 (т, 3Н), 3,45 (м, 2Н), 8,00 (д, 2Н), 8,13 (д, 2Н), 8,41 (с, 1Н), 10,52 (с, 1Н); m/z: 321.
Методы 112-113
Следующие соединения получали, используя указания метода 111, из соответствующего исходного пиримидинового соединения.
Метод 114
5-N-трет-Бутилкарбамоил-2-хлор-4-(4-хлорфенокси)пиримидин
Гидрид натрия (105 мг, 2,63 ммоль) добавляли к раствору 4-хлорфенола (338 мг, 2,63 ммоль) в безводном ТГФ (10 мл). После прекращения выделения пузырьков газа раствор медленно добавляли к раствору 5-N-трет-бутилкарбамоил-2,4-дихлорпиримидина (метод 79; 680 мг, 2,74 ммоль) в безводном ТГФ (15 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли выпариванием и полученное твердое вещество суспендировали в диэтиловом эфире. Нерастворившиеся вещества удаляли фильтрованием и фильтрат промывали 2 М раствором гидроксида натрия, раствором лимонной кислоты, водой и насыщенным раствором соли, а затем сушили. Летучие вещества удаляли выпариванием с получением указанного в заголовке соединения (880 мг, 99%). m/z: 339.
Метод 115
2-Хлор-4-(4-хлорфенокси)-5-цианопиримидин
Раствор 5-N-трет-бутилкарбамоил-2-хлор-4-(4-хлорфенокси)пиримидина (метод 114; 200 мг, 0,59 ммоль) в тионилхлориде (5 мл, 69 ммоль) кипятили с обратным холодильником (95°С) в течение 18 часов. Тионилхлорид удаляли выпариванием, получая указанное в заголовке соединение (210 мг). ЯМР: 7,35 (д, 2H), 7,55 (д, 2H), 9,20 (с, 1H).
Метод 116
4-[N-(2,3-Эпоксипропил)сульфамоил]нитробензол
п-Нитробензолсульфонамид (6,06 г, 30 ммоль) порциями добавляли к раствору гидроксида натрия (1,32 г, 33 ммоль) в воде (60 мл) при комнатной температуре. Затем по каплям быстро добавляли эпибромгидрин (4,5 г, 33 ммоль) и раствор перемешивали при комнатной температуре в течение 24 часов. Смесь подкисляли 2 М хлористоводородной кислотой до рН 1 и экстрагировали ДХМ (2×30 мл). Объединенные органические экстракты сушили сульфатом натрия, фильтровали и летучие вещества удаляли выпариванием. Полученное желтое масло очищали хроматографией на двуокиси кремния, элюируя смесью ДХМ/метанол (100:0) с увеличением полярности до (99:1), в результате чего получали указанное в заголовке соединение в виде светло-желтого твердого вещества (2,5 г, 32%). ЯМР (CDCl3): 2,65 (м, 1H), 2,80 (т, 1H), 3,10 (м, 2H), 3,50 (м, 1H), 5,08 (шир.с, 1H), 8,08 (д, 2H), 8,38 (д, 2H); m/z: 257.
Метод 117
4-[N-(2-Гидрокси-3-пиперидинопропил)сульфамоил]нитробензол
4-[N-(2,3-Эпоксипропил)сульфамоил]нитробензол (метод 116, 1 г, 3,88 ммоль) и пиперидин (0,34 г, 4 ммоль) в 1-пропаноле (75 мл) кипятили с обратным холодильником в течение 20 часов. Смесь охлаждали и растворитель выпаривали. Полученное желтое масло растирали с диэтиловым эфиром, отфильтровывали и сушили в вакууме, получая указанное в заголовке соединение (1,2 г, 90%). ЯМР: 1,26-1,50 (м, 6H), 2,10-2,28 (м, 6H), 2,70 (дд, 1H), 2,90 (дд, 1H), 3,55 (м, 1H), 8,04 (д, 2H), 8,40 (д, 2H); m/z: 344.
Метод 118
4-[N-(2-Гидрокси-3-пиперидинопропил)сульфамоил]анилин
К суспензии 4-[N-(2-гидрокси-3-пиперидинопропил)сульфамоил]нитробензола (метод 117; 1,2 г, 3,5 ммоль) в этаноле (100 мл) порциями добавляли формиат аммония (1,1 г, 17,46 ммоль), а затем суспензию катализатора 10% палладия на угле (0,7 г) в этаноле. Смесь кипятили с обратным холодильником в атмосфере азота в течение 2 часов. Смесь охлаждали и катализатор удаляли фильтрованием через диатомовую землю. Слой на фильтре промывали этанолом и объединенные фильтраты выпаривали. Остаток растирали с диэтиловым эфиром, отфильтровывали и сушили, получая указанное в заголовке соединение (0,93 г, 85%) в виде светло-зеленого твердого вещества. ЯМР: 1,3-1,5 (м, 6H), 2,10-2,32 (м, 6H), 2,60 (м, 1H), 2,75 (м, 1H), 3,52 (м, 1H), 4,50 (шир.с, 1H), 5,85 (с, 2H), 6,59 (д, 2H), 6,98 (шир.с, 1H), 7,39 (д, 2H); m/z: 314.
Метод 119
4-[4-(2-Ацетоксиэтилпиперазин-1-ил)]-5-циано-2-{4-[N-(3-метоксипропил)сульфамоил]анилино}пиримидин
Тионилхлорид (1,5 мл) добавляли к 4-[4-(2-ацетоксиэтилпиперазин-1-ил)]-5-N-трет-бутилкарбамоил-2-{4-[N-(3-метоксипропил)сульфамоил]анилино}пиримидину (метод 120; 570 мг) и реакционную смесь нагревали при 80°С в течение 3 часов. Смесь охлаждали, летучие вещества выпаривали, остаток растирали с этилацетатом и отфильтровывали, получая указанное в заголовке соединение (290 мг). m/z: 518.
Метод 120
4-[4-(2-Ацетоксиэтилпиперазин-1-ил)]-5-N-трет-бутилкарбамоил-2-{4-[N-(3-метоксипропил)сульфамоил]анилино}пиримидин
5-N-трет-Бутилкарбамоил-4-[4-(2-гидроксиэтилпиперазин-1-ил)]-2-{4-[N-(3-метоксипропил)сульфамоил]анилино}пиримидин (метод 121; 1 г) растворяли в пиридине (5 мл) при комнатной температуре, добавляли 4-диметиламинопиридин (˜5 мг), а затем по каплям добавляли уксусный ангидрид (0,29 мл, 3 ммоль). Реакционную смесь перемешивали в течение 18 часов. Летучие вещества выпаривали, добавляли воду и реакционную смесь экстрагировали EtOAc (2×20 мл), промывали насыщенным раствором соли, сушили и выпаривали с получением смолы (400 мг). Остаток растворяли в ДХМ (10 мл), добавляли триэтиламин (300 мкл, 4,1 ммоль), а затем по каплям добавляли ацетилхлорид (100 мкл, 1,15 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, затем летучие вещества выпаривали. Остаток суспендировали в этилацетате, нерастворившиеся вещества удаляли фильтрованием и фильтрат выпаривали, получая указанное в заголовке соединение в виде масла (570 мг), которое использовали без дополнительной очистки.
Метод 121
5-N-трет-Бутилкарбамоил-4-[4-(2-гидроксиэтилпиперазин-1-ил)]-2-{4-[N-(3-метоксипропил)сульфамоил]анилино}пиримидин
5-N-трет-Бутилкарбамоил-2-хлор-4-[4-(2-гидроксиэтил)пиперазин-1-ил]пиримидин (метод 80; 1,5 ммоль) и 4-[N-(3-метоксипропил)сульфамоил]анилин (метод 4; 330 мг, 1,35 ммоль) в 2-бутаноле (5 мл) нагревали при 95°С в течение 5 минут. Затем добавляли 1 М эфирный раствор хлористого водорода (1,5 мл, 1,5 ммоль) и нагревание продолжали при 95°С в течение 2 часов. Летучие вещества выпаривали, остаток растирали с этилацетатом и отфильтровывали, получая указанное в заголовке соединение (1 г). m/z 550.
Пример 133
Ниже приведены иллюстративные примеры типичных фармацевтических лекарственных форм, содержащих соединение формулы (I) или его фармацевтически приемлемую соль или in vivo гидролизуемый сложный эфир (далее называемые соединением Х), для лечения или профилактики заболеваний у человека.
(5% мас./об. пасты)
Примечания
Указанные выше композиции могут быть получены стандартными методами, хорошо известными в данной области фармацевтики. Таблетки (а)-(с) могут быть покрыты энтеросолюбильным покрытием стандартными способами, например покрытием из фталата ацетатцеллюлозы.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИМИДАЗОЛО-5-ИЛ-2-АНИЛИНОПИРИМИДИНЫ КАК АГЕНТЫ ДЛЯ ИНГИБИРОВАНИЯ ПРОЛИФЕРАЦИИ КЛЕТОК, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПРОДУЦИРОВАНИЯ | 2001 |
|
RU2284327C2 |
| ИМИДАЗО[1,2-А]ПИРИДИНОВЫЕ И ПИРАЗОЛ[2,3-А]ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2000 |
|
RU2248976C2 |
| ПРОИЗВОДНОЕ ИНДОЛИЗИНА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНСКИХ ЦЕЛЯХ | 2010 |
|
RU2529868C2 |
| ТРИАЗИНОВОЕ ПРОИЗВОДНОЕ И ВКЛЮЧАЮЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНАЛЬГЕТИЧЕСКОЙ АКТИВНОСТЬЮ | 2011 |
|
RU2565073C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ N-ФЕНИЛ-2-ГИДРОКСИ-2-МЕТИЛ-3,3,3-ТРИФТОРПРОПАНАМИДА, ПОВЫШАЮЩИЕ АКТИВНОСТЬ ПИРУВАТДЕГИДРОГЕНАЗЫ | 2000 |
|
RU2255085C2 |
| Имидазопиридиновые соединения в качестве ингибиторов PAD | 2018 |
|
RU2782743C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ И СПОСОБ ЛЕЧЕНИЯ МЛЕКОПИТАЮЩИХ С ИХ ИСПОЛЬЗОВАНИЕМ | 2002 |
|
RU2310655C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PRMT5 | 2018 |
|
RU2797822C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА, ОБЛАДАЮЩИЕ СВОЙСТВАМИ АНТАГОНИСТА СRTH2 (ВАРИАНТЫ) | 2004 |
|
RU2361865C2 |
| Гетероциклические соединения в качестве ингибиторов PAD | 2018 |
|
RU2764243C2 |
Изобретение относится к новым производным пиримидина общей формулы (I) или его фармацевтически приемлемым солям или in vivo гидролизуемым сложным эфирам, обладающим свойствами селективного ингибитора циклинзависимых киназ, таких как CDK-2, и ингибирующим пролиферацию клеток. Соединения могут быть использованы для приготовления лекарственных средств для лечения раковых заболеваний. В соединениях формулы (I)
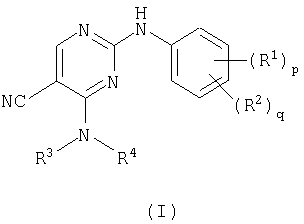
R1 представляет галоген; р=0 или 1; R2 представляет сульфамоил или группу В-Е-; q=0 или 1, где p+q=1; R3 представляет водород, С1-6алкил, где R3 может быть необязательно замещен у атома углерода одним или несколькими M; R4 представляет С1-6алкил, С2-6алкенил, С2-6алкинил, С3-8циклоалкил, где R4 может быть необязательно замещен одним или несколькими М, или R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют гетероциклическое кольцо, необязательно замещенное у атома углерода одним или несколькими М, где, если указанное гетероциклическое кольцо содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из Q; B выбран из С1-6алкила, С2-6алкенила, С2-6алкинила, С2-6циклоалкила, С3-8циклоалкилС1-6алкила, фенилС1-6алкила или (гетероциклическая группа)С1-6алкила, где В может быть необязательно замещен у атома углерода одним или несколькими D и где, если указанная гетероциклическая группа содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из G; E представляет -S(O)r- или -N(Ra)SO2-, где Ra представляет водород или C1-6алкил и r равно 2; D независимо выбран из галогена, нитро, циано, гидрокси, амино, C1-6алкила, C1-6алкокси, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, С1-6алканоиламино, С1-6алкилS(O)а, где a равно 0-2, где D может быть необязательно замещен у атома углерода одним или несколькими V; М независимо выбран из галогена, нитро, циано, гидрокси, C1-6алкила, C1-6алкокси, N,N-(C1-6алкил)2амино, C1-6алкоксикарбонила, C3-8циклоалкила или гетероциклической группы, где М может быть необязательно замещен у атома углерода одним или несколькими Р; Р, Х и V независимо выбраны из гидрокси, метила, метокси, диметиламино; G и Q независимо выбраны из C1-4алкила, C1-4алкилсульфонила, C1-4алкоксикарбонила, где Q может быть необязательно замещен у атома углерода одним или несколькими X. Изобретение также относится к способам получения соединений, фармкомпозициям на их основе и способу ингибирования пролиферации клеток. 9 н. и 6 з.п. ф-лы, 27 табл.
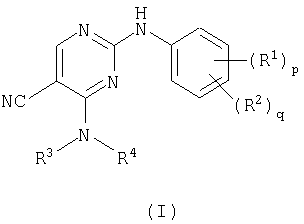
где R1 представляет галоген;
р=0 или 1;
R2 представляет сульфамоил или группу В-Е-;
q=0 или 1, где p+q=1;
R3 представляет водород, C1-6алкил, где R3 может быть необязательно замещен у атома углерода одним или несколькими М;
R4 представляет C1-6алкил, C2-6алкенил, C2-6алкинил или C3-8циклоалкил, где R4 может быть необязательно замещен одним или несколькими М;
или R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют гетероциклическое кольцо, необязательно замещенное у атома углерода одним или несколькими М, где, если указанное гетероциклическое кольцо содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из Q;
В выбран из C1-6алкила, C2-6алкенила, C2-6алкинила, C3-8циклоалкила, C3-8циклоалкилC1-6алкила, фенилC1-6алкила или (гетероциклическая группа)С1-6алкила, где В может быть необязательно замещен у атома углерода одним или несколькими D, и где, если указанная гетероциклическая группа содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из G;
Е представляет -S(O)r- или -N(Ra)SO2-, где Ra представляет водород или C1-6алкил, и r равно 2;
D независимо выбран из галогена, нитро, гидрокси, амино, C1-6алкила, C1-6алкокси, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, C1-6алканоиламино, C1-6алкилS(O)а, где а равно 0-2, где D может быть необязательно замещен у атома углерода одним или несколькими V;
М независимо выбран из галогена, нитро, циано, гидрокси, C1-6алкила, C1-6алкокси, N,N-(C1-6алкил)2амино, C1-6алкоксикарбонила, C3-8циклоалкила или гетероциклической группы, где М может быть необязательно замещен у атома углерода одним или несколькими Р;
Р, Х и V независимо выбраны из гидрокси, метила, метокси, диметиламино; и
G и Q независимо выбраны из C1-4алкила, C1-4алкилсульфонила, C1-4алкоксикарбонила, где Q может быть необязательно замещен у атома углерода одним или несколькими X;
где «гетероциклическая группа» означает насыщенное или частично насыщенное или ненасыщенное 5-6-членное кольцо, содержащее от 1 до 3 гетероатомов, выбранных из азота и/или кислорода, и которое может быть конденсировано с бензольным кольцом, и которые, если неоговорено особо, могут быть связаны с углеродом или азотом, где группа -СН2- может быть необязательно заменена -С(O)-, а атом азота в кольце может необязательно нести C1-6-алкильную группу,
если R3 и R4 вместе с атомом азота, с которым они связаны образуют гетероциклическое кольцо, то указанное «гетероциклическое кольцо» представляет собой насыщенное или частично насыщенное 5-6-членное кольцо, содержащее 1 атом азота и 1-2 дополнительных гетероатома, выбранных из азота и кислорода, при этом указанное кольцо может быть конденсировано с бензольным кольцом, и в которых группа -CH2- может быть необязательно заменена -C(O)-, а атом азота в кольце может необязательно нести C1-6-алкильную группу,
или его фармацевтически приемлемая соль или in vivo гидролизуемый сложный эфир.
Е представляет -S(O)r или -N(Ra)SO2-, где Ra представляет водород или C1-6алкил и r равно 2;
D независимо выбран из галогена, циано, гидрокси, амино, C1-6алкила, C1-6алкокси, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, С1-6алканоиламино и С1-6алкилS(O)а, где а равно 0-2, и где D может быть необязательно замещен у атома углерода группой, выбранной из V;
V выбран из гидрокси и диметиламино; и
G выбран из С1-4алкила;
или его фармацевтически приемлемая соль или in vivo гидролизуемый сложный эфир.
R4 представляет С1-6алкил, С2-6алкенил, С2-6алкинил или С2-6циклоалкил, где R4 может быть необязательно замещен одним или несколькими М;
или R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют гетероциклическое кольцо, необязательно замещенное у атома углерода одним или несколькими М, где, если указанное гетероциклическое кольцо содержит группу -NH-, то атом азота может быть необязательно замещен группой, выбранной из Q;
М независимо выбран из галогена, циано, гидрокси, С1-6алкила, С1-6алкокси, N,N-(С1-6алкил)2амино, C1-6алкоксикарбонила, С3-8циклоалкила или гетероциклической группы, где М может быть необязательно замещен у атома углерода группой, выбранной из P;
Р и Х независимо выбраны из гидрокси и метокси; и
Q выбран из C1-4алкила, C1-4алкилсульфонила или C1-4алкоксикарбонила, где Q может быть необязательно замещен у атома углерода одним или несколькими X;
или его фармацевтически приемлемая соль или in vivo гидролизуемый сложный эфир.
p равно 0;
R2 представляет сульфамоил, мезил, этилсульфонил,
2-этоксиэтилсульфонил, пропилсульфонил,
3-изопропиламинопропилсульфонил, 4-изопропиламинобутилсульфонил,
N-(тетрагидрофур-2-илметил)сульфамоил,
N-(пирид-3-илметил)сульфамоил, N-(пирид-2-илметил)сульфамоил,
N-(1,4-диоксан-2-илметил)сульфамоил, N-(метил)сульфамоил,
N-(2-метоксиэтил)сульфамоил, N-(2-этилтиоэтил)сульфамоил,
N-(2-морфолиноэтил)сульфамоил, N-(2-пиперидиноэтил)сульфамоил,
N-(2-пирид-2-илэтил)сульфамоил,
N-(2-пирролидин-1-илэтил)сульфамоил,
N-(2-имидазол-4-илэтил)сульфамоил,
N-(2-изопропиламиноэтил)сульфамоил, N-(2-мезилэтил)сульфамоил,
N-[2-(2-гидроксиэтокси)этил]сульфамоил,
N-[2-(1-этилпирролидин-2-ил)этил]сульфамоил,
N-(2-пирид-2-илэтил)сульфамоил, N-(2-диэтиламиноэтил)сульфамоил,
N-(2-пирид-4-илэтил)сульфамоил, N-(2-ацетамидоэтил)сульфамоил,
N-(2-диметиламиноэтил)сульфамоил,
N-2-[(5-метил-1,3,4-триазол-2-ил)этил]сульфамоил,
N-(2-гидроксиэтил)сульфамоил, N-(2-цианоэтил)сульфамоил,
N-(2-диэтиламиноэтил)-N-(метил)сульфамоил,
N-(2-метоксиэтил)-N-(метил)сульфамоил,
N-(2,2,2-трифторэтил)сульфамоил,
N-(3-гидрокси-2,2-диметилпропил)сульфамоил,
N-(3-изопропиламинопропил)сульфамоил,
N-(3-метоксипропил)сульфамоил,
N-(3-имидазол-1-илпропил)сульфамоил,
N-(2-гидрокси-3-аминопропил)сульфамоил,
N-(3-гидроксипропил)сульфамоил, N-(3-этоксипропил)сульфамоил,
N-(3-морфолинопропил)сульфамоил,
N-(2-гидроксипропил)сульфамоил,
N-(2-гидрокси-3-пиперидинопропил)сульфамоил,
N-(3-пиперидинопропил)-N-(метил)сульфамоил,
N-(2-гидроксибутил)сульфамоил, N-(пентил)сульфамоил,
N-(5-гидроксипентил)сульфамоил, N-(аллил)сульфамоил или
N-(2-пропинил)сульфамоил;
q равно 1, a R2 находится в пара-положении по отношению к аминогруппе в анилине формулы (I); и
R3 и R4, взятые вместе с атомом азота, с которым они связаны, образуют изобутиламино, этиламино, 2-фторэтиламино,
3-этоксипропиламино, бутиламино, 2,2,2-трифторэтиламино,
3-морфолинопропиламино, циклопропиламино, циклопропилметиламино,
циклогексиламино,тетрагидрофур-2-илметиламино,
2-диметиламиноэтиламино, цианометиламино, пирид-3-илметиламино,
бутоксикарбонилметиламино, 2-(метоксикарбонил)этиламино,
2-гидроксиэтиламино, метиламино, 2-пропиниламино,
2-метоксиэтиламино, 2-имидазол-4-илэтиламино,
2-(2-гидроксиэтокси)этиламино, 2,3-дигидроксипропиламино,
2,2-диметилдиоксолан-4-илметиламино, пропиламино,
N-метил-N-аллиламино, N-метил-N-этоксикарбонилметиламино,
N-метил-N-(2-цианоэтил)амино, диэтиламино,
N-метил-N-(2-метоксиэтил)амино, бис-(2-цианоэтил)амино,
N-этил-N-циклогексиламино, N-метил-N-(2,2,2-трифторэтил)амино,
N-метил-N-(2-пропинил)амино, морфолино, 2,6-диметилморфолино,
3,5-диметилпиперидино, пиперидино,
4-(2-метоксиэтил)пиперазин-1-ил, 4-метилпиперазин-1-ил,
4-изопропилпиперазин-1-ил, 4-этилсульфонилпиперазин-1-ил,
4-этоксикарбонилпиперазин-1-ил, 4-(2-гидроксиэтил)пиперазин-1-ил
и 3-гидроксипирролидин-1-ил;
или его фармацевтически приемлемая соль или in vivo гидролизуемый сложный эфир.
5-циано-4-н-бутиламино-2-(4-мезиланилино)пиримидина;
5-циано-4-этиламино-2-{4-[N-(2-метоксиэтил)сульфамоил]анилино}пиримидина;
5-циано-4-этиламино-2-{4-[N-(3-метоксипропил)сульфамоил]анилино}пиримидина;
5-циано-4-циклопропиламино-2-{4-[N-(2-метоксиэтил)сульфамоил]анилино} пиримидина;
5-циано-4-циклопропиламино-2-{4-[N-(3-метоксипропил)сульфамоил]анилино}пиримидина;
5-циано-4-циклопропиламино-2-[4-(этилсульфонил)анилино]пиримидина;
5-циано-4-циклопропиламино-2-(4-мезиланилино)пиримидина;
5-циано-4-этиламино-2-[4-(N-метилсульфамоил)анилино]пиримидина;
5-циано-4-циклопропиламино-2-{4-[N-(2-метоксиэтил)-N-(метил)сульфамоил]анилино}пиримидина и
5-циано-4-(этиламино)-2-(4-этилсульфониланилино)пиримидина;
или их фармацевтически приемлемой соли или in vivo гидролизуемого сложного эфира.
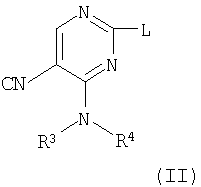
где L представляет заменяемую группу с анилином формулы (III)
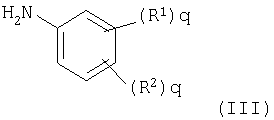
и затем, если необходимо,
i) превращение соединения формулы (I) в другое соединение формулы (I);
ii) удаление любых защитных групп;
iii) получение фармацевтически приемлемой соли или in vivo гидролизуемого сложного эфира.
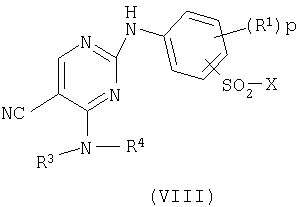
где Х представляет заменяемую группу, с амином формулы (IX)

и затем, если необходимо,
i) превращение соединения формулы (I) в другое соединение формулы (I);
ii) удаление любых защитных групп;
iii) получение фармацевтически приемлемой соли или in vivo гидролизуемого сложного эфира.
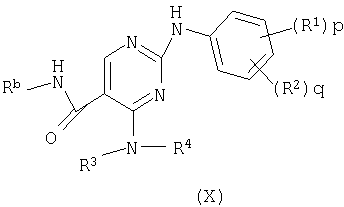
Rb представляет водород или трет-бутил, в соединение формулы (I);
и затем, если необходимо,
i) превращение полученного соединения формулы (I) в другое соединение формулы (I);
ii) удаление любых защитных групп;
iii) получение фармацевтически приемлемой соли или in vivo гидролизуемого сложного эфира.
| WO 00/39101 A1, 06.07.2000 | |||
| Способ получения 5-замещенных 2,4-диаминопиримидинов или их кислотно-аддитивных солей | 1987 |
|
SU1535379A3 |
| DIANE H.BOSCHELLI et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Journal of medicinal chemistry, v.41, no.22, p.4365-4377. | |||

