Настоящее изобретение относится к новым производным дибензо[b,f]азепина и их получению. Соединения по настоящему изобретению используются в качестве промежуточных материалов при получении фармацевтических препаратов.
Более подробно изобретение относится к соединениям формулы I.
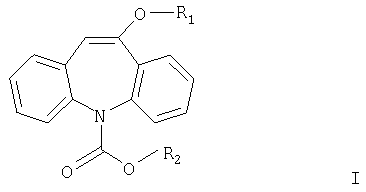
где R1 означает С1-С4алкил и R2 означает С1-С4алкил или фенил.
Соединения формулы I используются в качестве исходных материалов при получении фармацевтического препарата окскарбазепина (Trileptal®) формулы IV (см. ниже), который используется в качестве противосудорожного средства, например, при лечении эпилепсии.
Окскарбазепин может быть получен из соединений формулы I, например, по следующей схеме реакций:
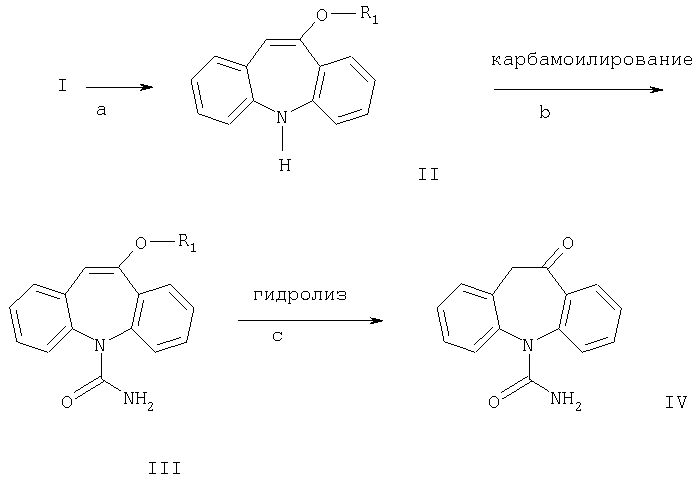
Реакции a, b и с можно проводить с использованием известных методов, например, как описано в примере, стадии е-з.
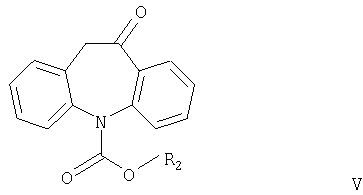
Другим аспектом настоящего изобретения является способ получения соединений формулы I, в котором соединение формулы V
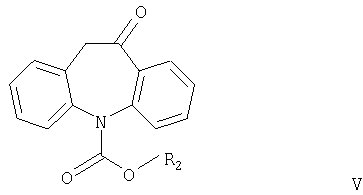
где R2 имеет значения, указанные выше, взаимодействует с соединением формулы R1OH или (R1O)3CH, причем R1 имеет значения, указанные выше.
Реакцию можно проводить по известному методу, например, как описано в примере, стадии г, г' г".
Соединения формулы V никогда не были описаны в литературе и также являются частью настоящего изобретения, наряду со способом их получения.
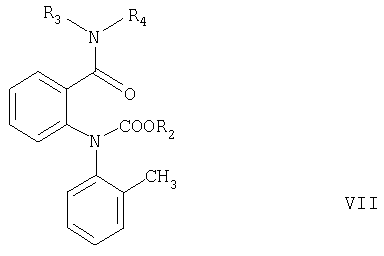
Согласно настоящему изобретению соединения формулы V могут быть получены замыканием цикла в соединениях формулы VI или VII 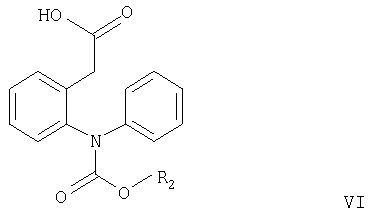
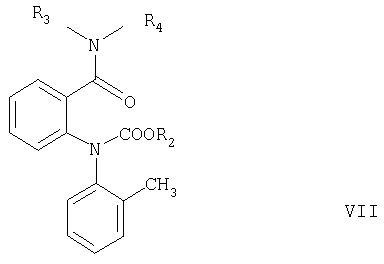
где R2 имеет значения, указанные выше, а R3 и R4 независимо означают С1-С4алкил.
Замыкание цикла в соединении формулы VI обычно проводят в кислотных условиях, например, как описано в примере, стадия в2. Если полученное соединение формулы V используют для получения соединения формулы I, то его предпочтительно не выделяют, а вводят в реакцию in situ с образованием соединения формулы I, например, как описано в примере, стадия д.
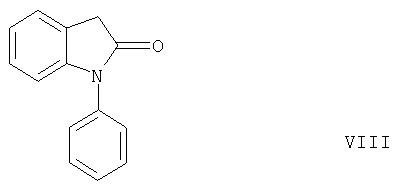
Неожиданно было обнаружено, что такая циклизация приводит к образованию соединений формулы V, а не к образованию 5-членного лактама формулы VIII,
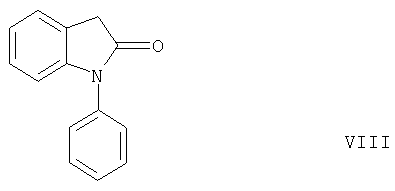
с расщеплением группы -COOR2, как следовало ожидать согласно статье J.W.Schulenberg и др., J. Amer. Chem. Soc. 82, 2035 (1960) благодаря электроноакцепторным свойствам группы -COOR2.
Замыкание цикла в соединении формулы VII обычно проводят в сильно щелочных условиях, например, как описано в примере, стадия в1.
Соединения формулы VI, а также соединения формулы VII, где R2 не является трет-бутильной группой, если R3 и R4 оба означают изопропил, также являются новыми соединениями и частью настоящего изобретения, наряду со способом их получения.
Согласно настоящему изобретению соединения формулы VI могут быть получены взаимодействием соединения формулы VIII в сильно основных условиях с соединением формулы X-COOR2, где R2 имеет значения, указанные выше, а Х означает хлор или метокси. Реакцию проводят обычным способом, например, как описано в примере, стадия а2.
Согласно настоящему изобретению соединения формулы VII могут быть получены взаимодействием соединения формулы IX
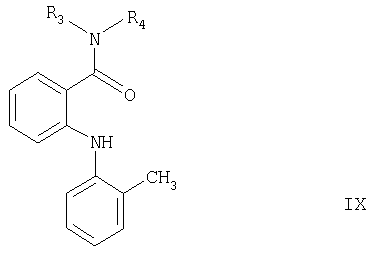
где R3 и R4 имеют значения, указанные выше, с соединением формулы Cl-COOR2, причем R2 имеет значения, указанные выше, но не является трет-бутильной группой, если R3 и R4 оба означают изопропил. Реакцию можно проводить обычным способом, как описано в примере, стадия 6.
Соединения формулы IX также являются новыми и частью настоящего изобретения. Они могут быть получены взаимодействием соединения формулы Х
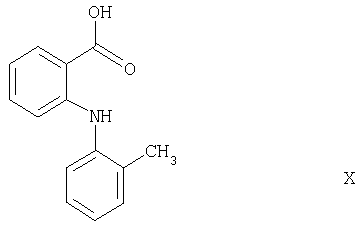
с соединением формулы R3-NH-R4, причем R3 и R4 имеют значения, указанные выше, обычным способом, как описано в примере, стадия а1.
Исходные соединения формул VIII и Х являются известными соединениями.
Другим аспектом настоящего изобретения является способ получения соединений формулы III карбамоилированием соединения формулы II. Карбамоилирование соединения формулы II, где R означает метил, описано в WO 96/21649. Согласно данному описанию используют цианаты металлов в присутствии неорганических кислот или относительно сильных карбоновых кислот и растворитель.
Неожиданно было обнаружено, что данное карбамоилирование также может быть достигнуто в мягких условиях с использованием уксусной кислоты. Присутствие сильной кислоты и дополнительного растворителя не требуется. Ввиду относительно низкой стабильности соединений формулы II отсутствие сильной кислоты является значительным преимуществом. В связи с этим существенно увеличивается выход.
Согласно настоящему изобретению разработан способ получения соединения формулы III карбамоилированием соединения формулы II в присутствии цианата металла, причем реакцию проводят с использованием уксусной кислоты в присутствии большого избытка цианата металла и в отсутствие дополнительного растворителя.
Цианатом металла предпочтительно является цианат натрия или калия. "Большой избыток" цианата металла означает по меньшей мере 0,2 экв., предпочтительно от 0,2 до 0,5 экв. Такой избыток является ключевым условием для проведения реакции с повышенным выходом по сравнению с известным способом карбамоилирования. Реакцию можно проводить, например, как описано в примере, стадия ж.
Изобретение иллюстрируется следующим примером.
Пример
а1) N,N-Диметил-2-орто-толиламинобензамид
2-орто-Толиламинобензойную кислоту (101 г, 0,444 моля) суспендировали в толуоле (800 мл) и нагревали до 58°С. Затем в течение 20 мин добавляли раствор тионилхлорида (57,6 г, 0,484 моля, 1,1 экв.) в толуоле (100 мл). Смесь медленно нагревали до 82°С (в течение 1 ч) и концентрировали в вакууме. К полученному остатку добавляли толуол (800 мл) и раствор концентрировали в вакууме. Неочищенный хлорангидрид растворяли в толуоле (500 мл) и раствор охлаждали до 3°С. Затем в течение 45 мин добавляли раствор диметиламина (61,3 мл 40%-ного водного раствора, 1,1 экв.), гидроксида натрия (77 г 30%-ного водного раствора, 1,3 экв.) и воды (240 мл). Полученную суспензию перемешивали при 3°С в течение 30 мин, а затем нагревали до 30°С. Фазы разделяли и водную фазу экстрагировали толуолом (100 мл). Объединенные органические фазы дважды промывали водой (200 мл), упаривали досуха и дегазировали в вакууме в течение 1 ч (30 мбар, 60°С). Полученный в виде масла продукт затвердевает при хранении (104,7 г, выход 93,5%). Затем полученное соединение, указанное в названии, может быть перекристаллизовано из циклогексана.
б) Метиловый эфир (2-диметилкарбамоилфенил)-орто-толилкарбаминовой кислоты
N,N-Диметил-2-орто-толиламинобензамид (104,7 г, 0,412 моля, 1 экв.) растворяли в толуоле (800 мл), охлаждали до -8°С и медленно (в течение 1 ч), поддерживая температуру ниже 0°С, добавляли раствор н-бутиллития в гексане (257 мл, 1,6 М раствор, 1 экв). Полученную таким образом суспензию оранжевого цвета перемешивали при -8°С в течение 30 мин, а затем в течение 30 мин добавляли метилхлорформиат (42,8 г, 1,1 экв.). Суспензию перемешивали при 5°С в течение 1 ч, а затем реакцию останавливали добавлением насыщенного водного раствора бикарбоната натрия (500 мл). Фазы разделяли и водную фазу экстрагировали толуолом (200 мл). Объединенные органические фазы дважды промывали водой (200 мл), упаривали досуха и дегазировали в вакууме в течение 1 ч (30 мбар, 60°С). Неочищенное соединение (133,8 г) растворяли в этилацетате (240 мл) при 50°С и добавляли гексан (990 мл). Раствор охлаждали до 25°С (30 мл), при этом начинался процесс кристаллизации. Суспензию перемешивали при 25°С в течение 1 ч, охлаждали до 3°С и перемешивали при указанной температуре еще в течение 4 ч. После фильтрования твердое вещество промывали холодным гексаном и сушили в вакууме в течение 16 ч (50°С, 50 мбар). При этом получали указанное в названии соединение в виде твердого вещества светло-желтого цвета (98,0 г, суммарный выход в расчете на 2-орто-толиламинобензойную кислоту составляет 70,5%).
в1) Метиловый эфир 10-оксо-10,11-дигидродибензо[b,f]азепин-5-карбоновой кислоты
Диизопропиламин (11,66 г, 0,115 моля, 1,2 экв.) растворяли в ТГФ (150 мл), раствор охлаждали до -10°С и медленно (в течение 40 мин), поддерживая температуру ниже 0°С, добавляли раствор н-бутиллития в гексане (72 мл, 1,6 М раствор, 1,2 экв.). Затем к полученному раствору в течение 45 мин добавляли раствор метилового эфира (2-диметилкарбамоилфенил)-орто-толилкарбаминовой кислоты (30,2 г, 0,096 моля, 1 экв.) в ТГФ (80 мл). Реакционную смесь перемешивали при -5°С в течение 1 ч, после чего реакцию останавливали добавлением воды (30 мл). Смесь концентрировали в вакууме и к масляному остатку добавляли воду (220 мл) и этилацетат (220 мл). Фазы быстро перемешивали перед разделением. Органическую фазу промывали водным раствором серной кислоты (300 мл 1 М раствора) и дважды водой (300 мл). Органическую фазу упаривали досуха, при этом получали 23,0 г (выход 89,5%) указанного в названии соединения в виде масла оранжевого цвета, которое затвердевало при хранении.
а2) [2-(Метоксикарбонилфениламино)фенил]уксусная кислота
Смесь 1-фенил-1,3-дигидроиндол-2-она (80 г, 382 ммоля), гидроксида натрия (16,06 г, 402 ммоля) и тетрагидрофурана (113 мл) кипятили с обратным холодильником (при 67°С) в течение 5 ч. Раствор разбавляли другой порцией тетрагидрофурана (169 мл) и охлаждали до -10°С. Затем к смеси при указанной температуре добавляли 20%-ный раствор бутиллития в циклогексане (122,3 г, 382 ммоля), диметилкарбонат (51,7 г, 573 ммоля) и раствор перемешивали при -10°С в течение 2 ч. Затем добавляли концентрированную хлористоводородную кислоту (38 мл) и воду (125 мл) и органические растворители выпаривали при пониженном давлении. После добавления к суспензии толуола (345 мл) водную фазу подкисляли хлористоводородной кислотой (34 мл) до рН 1,5. После разделения фаз при 75°С органическую фазу промывали другой порцией воды (120 мл) и концентрировали при пониженном давлении, остаток кристаллизовали при 0°С, при этом получали 81,2 г (75%) очищенного соединения, указанного в названии.
г) Метиловый эфир 10-метоксидибензо[b,f]азепин-5-карбоновой кислоты
Неочищенный метиловый эфир 10-оксо-10,11-дигидродибензо[b,f]азепин-5-карбоновой кислоты (22,3 г, 0,083 моля, 1 экв.) растворяли в метаноле (112 мл) при 50°С. Затем добавляли каталитическое количество пара-толуолсульфоновой кислоты (0,445 мг) и триметилортоформиат (11,5 мл, 1,25 экв.). Реакцию проводили в течение 5 ч, затем отгоняли метанол при непрерывном добавлении свежих порций метанола. После перегонки 100 мл метанола смесь охлаждали до 25°С в течение 1 ч. Затем суспензию охлаждали до 3°С в течение 20 мин, перемешивали при указанной температуре в течение 1 ч и фильтровали. Твердо вещество промывали холодным метанолом и высушивали в вакууме в течение 15 ч (50°С, 50 мбар). При этом получали указанное в названии очищенное соединение в виде порошка светло-желтого цвета (18,02 г, выход 80,8%).
в2) Метиловый эфир 10-оксо-10,11-дигидродибензо[b,f]азепин-5-карбоновой кислоты
Смесь [2-(метоксикарбонилфениламино)фенил]уксусной кислоты (16 г, 55,5 ммоля) и полифосфорной кислоты (29 г, 167 ммолей в расчете на P2O5) нагревали до 100°С в течение 4 ч. Затем к реакционной смеси при температуре 85-100 С при перемешивании по каплям добавляли воду (41 мл) и охлаждали. Затем при 65°С добавляли толуол (41 мл) и смесь перемешивали в течение 30 мин. Две фазы разделяли и промывали. Органические фазы концентрировали, продукт кристаллизовали при 0°С, при этом получали 12 г (80%) указанного в названии очищенного соединения.
г') Метиловый эфир 10-метоксидибензо[b,f]азепин-5-карбоновой кислоты
Суспензию метилового эфира 10-оксо-10,11-дигидродибензо[b,f]азепин-5-карбоновой кислоты (15 г, 56 ммолей) в метаноле (75 мл) нагревали до 60°С и добавляли каталитическое количество пара-толуолсульфоновой кислоты (0,213 г, 1,1 ммоля). После добавления триметил-орто-формиата (6,25 г, 58,9 ммоля) полученный раствор перемешивали при 60-70°С в течение 4 ч. В ходе реакции продукт выпадал в осадок в виде кристаллов белого цвета. Смесь охлаждали до комнатной температуры, кристаллы отделяли фильтрованием и высушивали, при этом получали 15,5 г (98%) очищенного соединения, указанного в названии.
г") Метиловый эфир 10-этоксидибензо[b,f]азепин-5-карбоновой кислоты
Суспензию метилового эфира 10-оксо-10,11-дигидродибензо[b,f]азепин-5-карбоновой кислоты (15 г, 56 ммолей) в этаноле (75 мл) нагревали до 60°С и добавляли каталитическое количество пара-толуолсульфоновой кислоты (0,213 г, 1,1 ммоля). После добавления триэтилортоформиата (8,73 г, 58,9 ммоля) полученный раствор перемешивали при 60-70°С в течение 4 ч. В ходе реакции продукт выпадал в осадок в виде кристаллов белого цвета. Смесь охлаждали до комнатной температуры, кристаллы отделяли фильтрованием и высушивали, при этом получали 16,0 г (97%) очищенного соединения, указанного в названии.
д) Метиловый эфир 10-метоксидибензо[b,f]азепин-5-карбоновой кислоты
Смесь [2-(метоксикарбонилфениламино)фенил]уксусной кислоты (16 г, 55,5 ммоля) и полифосфорной кислоты (29 г, 167 ммолей в расчете на P2O5) нагревали до 100°С в течение 4 ч. К реакционной смеси при 65°С по каплям при перемешивании добавляли метанол (50 мл). Полученную суспензию охлаждали до комнатной температуры, кристаллы отделяли фильтрованием, промывали метанолом (40 мл) и высушивали, при этом получали 12,2 г (80%) указанного в названии очищенного соединения в виде кристаллов белого цвета.
е) 10-Метокси-5Н-дибензо[b,f]азепин
Смесь метилового эфира 10-метоксидибензо[b,f]азепин-5-карбоновой кислоты (19 г, 67,5 ммоля), поли(этиленгликоля) 200 (20 мл) и 50%-ного раствора гидроксида натрия (13 мл, 246 ммолей) нагревали до 100°С в течение 4 ч. Затем добавляли воду (30 мл), суспензию охлаждали до 20°С и фильтровали. Осадок на фильтре промывали водой и сушили при 60°С/30 мбар, при этом получали 14,7 г (98%) очищенного соединения, указанного в названии.
ж) Амид 10-метоксидибензо[b,f]азепин-5-карбоновой кислоты
Уксусную кислоту (150 мл) по каплям добавляли к перемешиваемой смеси 10-метокси-5Н-дибензо[b,f]азепина (25,0 г, 112 ммолей) и NaOCN (9,25 г, 142 ммоля) в атмосфере азота при комнатной температуре. После перемешивания в течение 7 ч полученную суспензию указанного в названии соединения желтого цвета (по данным ЖХВР площадь пика соединения составляла >95%) использовали для синтеза амида 10-оксо-10,11-дигидродибензо[b,f]азепин-5-карбоновой кислоты. Соединение, указанное в заголовке, выделяли добавлением 1н. NaOH до рН ≥8 и последующей экстракцией толуолом. Объединенные экстракты сушили и концентрировали в вакууме, при этом получали указанное в заголовке соединение в виде твердого вещества светло-желтого цвета (выход ≥75%).
з) Амид 10-оксо-10,11-дигидродибензо[b,f]азепин-5-карбоновой кислоты
К смеси в уксусной кислоте, полученной на стадии ж), добавляли воду (12,5 мл, 694 ммоля) и 100%-ную H2SO4 (˜7,5 мл, 140 ммолей) до достижения рН≤1.
После перемешивания в течение 17 ч добавляли воду (275 мл). Осадок соединения, указанного в названии, отделяли фильтровыванием и высушивали в вакууме (суммарный выход в расчете на исходный 10-метокси-5Н-дибензо[b,f]азепин составлял ≥78%).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ЭРГОЛИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛИГАНДОВ ХЕМОКИННОГО РЕЦЕПТОРА | 2006 |
|
RU2416613C2 |
| ПРОИЗВОДНЫЕ 8-ХИНОЛИНКСАНТИНА И 8-ИЗОХИНОЛИНКСАНТИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОЭСТЕРАЗЫ PDE 5 | 2001 |
|
RU2269529C2 |
| СПОСОБ СИНТЕЗА ПЕПТИДОВ, СОДЕРЖАЩИХ ПОДСТРУКТУРУ 4-ГИДРОКСИПРОЛИНА | 2003 |
|
RU2392265C2 |
| ДИПЕПТИДЫ, СОДЕРЖАЩИЕ НА N-КОНЦЕВОМ АМИНОКИСЛОТНОМ ОСТАТКЕ 2-ТИОАЦИЛЬНУЮ ГРУППУ, В КАЧЕСТВЕ ИНГИБИТОРОВ ВАЗОПЕПТИДАЗЫ | 2002 |
|
RU2298559C2 |
| АМИНОЗАМЕЩЕННЫЕ АНАЛОГИ ДИАРИЛ [a,d] ЦИКЛОГЕПТЕНА В КАЧЕСТВЕ МУСКАРИНОВЫХ АГОНИСТОВ И СПОСОБЫ ЛЕЧЕНИЯ ПСИХОНЕВРОЛОГИЧЕСКИХ РАССТРОЙСТВ | 2004 |
|
RU2394030C2 |
| ПРОИЗВОДНЫЕ ТЕТРАЛОНА В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ АГЕНТОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2288220C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА Ха | 2004 |
|
RU2346944C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАК ИНГИБИТОРЫ КАТЕПСИНА S | 2005 |
|
RU2424239C2 |
| СПИРОЦИКЛИЧЕСКИЕ НИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕАЗЫ | 2012 |
|
RU2621695C2 |
| СПИРОЦИКЛИЧЕСКИЕ НИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕАЗЫ | 2007 |
|
RU2478620C2 |
Изобретение относится к способу получения соединения формулы III
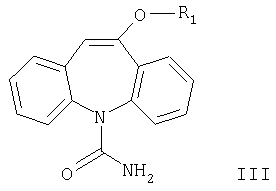
где R1 означает С1-С4алкил, карбамоилированием соединения формулы II
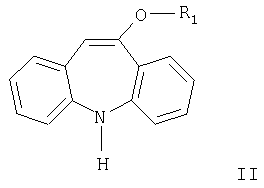
где R1 имеет значения, указанные выше, цианатом металла, причем реакцию проводят с использованием уксусной кислоты в присутствии большого избытка цианата металла и в отсутствие дополнительного растворителя. 11 н.п. ф-лы.
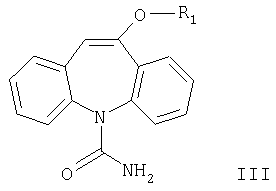
где R1 означает С1-С4алкил, карбамоилированием соединения формулы II
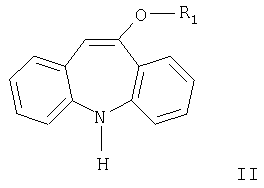
где R1 имеет значения, указанные выше, цианатом металла, причем реакцию проводят с использованием уксусной кислоты в присутствии большого избытка цианата металла и в отсутствии дополнительного растворителя.
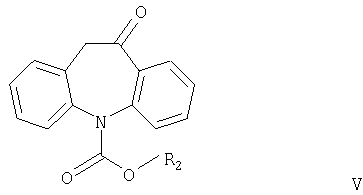
где R2 означает С1-С4алкил или фенил, включающий замыкание цикла в кислотных условиях в соединении формулы VI
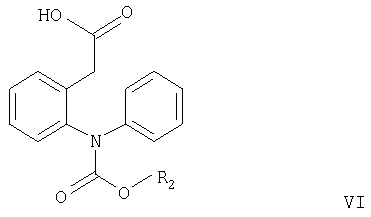
где R2 имеет значения, указанные выше.
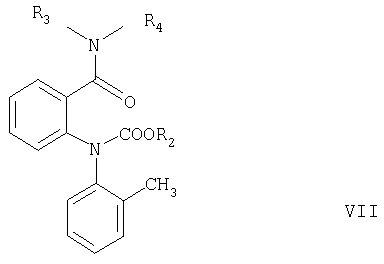
где R2 означает С1-С4алкил или фенил, а R3 и R4 независимо означают С1-С4алкил.
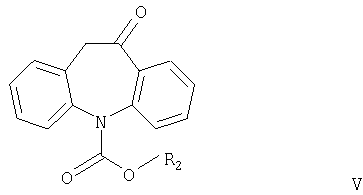
где R2 означает С1-С4алкил или фенил.
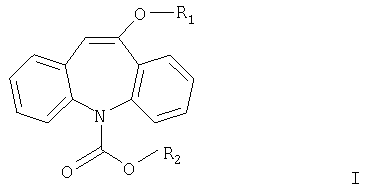
где R1 означает С1-С4алкил, a R2 означает С1-С4алкил или фенил.
где R2 означает С1-С4алкил или фенил.
в сильно щелочных условиях с соединением формулы Х-COOR2, где R2 означает С1-С4алкил или фенил, а Х означает хлор или метокси.
где R2 означает С1-С4алкил или фенил, а R3 и R4 независимо означают С1-С4алкил, при условии, что R2 не означает трет-бутил, если R3 и R4 оба означают изопропил.
где R1 означает С1-С4алкил, включающий следующие стадии:
замыкание цикла в сильно щелочных условиях в соединении формулы VII
где R2 означает С1-С4алкил или фенил, а R3 и R4 независимо означают С1-С4алкил, с получением соединения формулы V
где R2 означает С1-С4алкил или фенил,
взаимодействие соединения формулы V с соединением формулы R1OH или (R1O)3СН, где R1 означает С1-С4алкил, с получением соединения формулы I
где R1 означает С1-С4алкил, а R2 означает С1-С4алкил или фенил,
восстановление соединения формулы I с получением соединения формулы II
где R1 имеет значения, указанные выше,
карбамоилирование соединения формулы II цианатом металла, причем реакцию проводят с использованием уксусной кислоты в присутствии большого избытка цианата металла и в отсутствии дополнительного растворителя, с получением соединения формулы III.
где R1 означает С1-С4алкил, включающий следующие стадии:
взаимодействие соединения формулы VIII
в сильно щелочных условиях с соединением формулы X-COOR2, где R2 означает С1-С4алкил или фенил, а Х означает хлор или метоксигруппу, с получением соединения формулы VI
где R2 означает С1-С4алкил или фенил,
замыкание цикла в кислотных условиях в соединении формулы VI с получением соединения формулы V
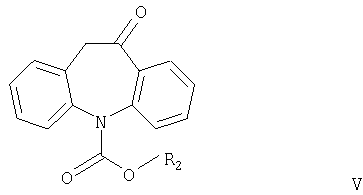
где R2 означает С1-С4алкил или фенил,
взаимодействие соединения формулы V с соединением формулы R1OH или (R1O)3СН, где R1 означает С1-С4алкил, с получением соединения формулы I
где R1 означает С1-С4алкил, а R2 означает С1-С4алкил или фенил,
восстановление соединения формулы I с получением соединения формулы II
где R1 имеет значения, указанные выше,
карбамоилирование соединения формулы II цианатом металла, причем реакцию проводят с использованием уксусной кислоты в присутствии большого избытка цианата металла и в отсутствии дополнительного растворителя, с получением соединения формулы III.
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| ШАРОВОЙ КРАН | 1990 |
|
RU2011087C1 |
| RU 95122817 А, 27.09.1997. | |||

