В предлагаемом изобретении описаны производные эрголина, способ их получения, их применение в качестве лекарств и фармацевтических композиций, их содержащих.
Более конкретно, изобретение предлагает соединение формулы I,

в которой
или каждый R1 и R2 независимо означают Н; необязательно R10 и/или R11-замещенный фенил или -фенилС1-С4алкил; необязательно R10 и/или R11-замещенный гетероарил или -гетероарилС1-С4алкил; необязательно R10 и/или R11-замещенный гетероарил-N-оксид; необязательно R10-замещенный C1-С8алкил; необязательно R10-замещенный С2-С8алкенил; необязательно R10-замещенный С2-С8алкинил; необязательно R10-замещенный С3-С8циклоалкил или необязательно R10-замещенный С4-С8циклоалкенил;
или R1 и R2 образуют вместе с атомом азота, к которому они присоединены, необязательно R10-замещенное 3-8 членное кольцо, содержащее вдобавок к атому азота еще до 2-х гетероатомов, независимо выбранных из группы, состоящей из N, О или S;
где R10 представляет собой 1-4 заместителя, независимо выбранных из C1-С6алкила, C1-С6гидроксиалкила, C1-С6алкоксиалкила, C1-С6галоидалкила, С3-С6циклоалкила, С2-С6алкенила, С3-С6циклоалкенила, С2-С6алкинила, фенила, гетероарила, гетероарил-N-оксида, F, Cl, ВR, I, ОН, OR9, OCOR9, OCOOR9, OCONHR9, OCONR9R9, OSO2R9, COR9, СООН, COOR9, CONH2, CONHR9, CONR9R9, СF3, CHF2, CH2F, С1-C4алкилNH2, C1-C4алкилNHR9, C1-C4алкилNR9R9, CN, NO2, NH2, NHR9, NR9R9, NHCOR9, NR9COR9, NHCONHR9, NHCONH2, NR9CONHR9, NR9CONR9R9, NHCOOR9, NR9COOR9, NHSO2R9, N(SO2R9)2, NR9SO2R9, SR9, SOR9, SO2R9, SO2NH2, SO2NHR9, SO2NR9R9, или
R10 означает группу =O, присоединенную к фенильному или гетероарильному атому углерода, или может представлять собой одну или две группы =O, присоединенные к одному и тому же атому S в гетероариле, если они есть;
R11 представляет собой два соседних заместителя, образующих 4-7 членное сочлененное неароматическое кольцо, необязательно включающее до двух гетероатомов, независимо выбранных из N, О или S;
каждый R9 независимо означает C1-С6алкил; гидроксиС1-С6алкил; С3-С6циклоалкил; С2-С6алкенил; С2-С6алкинил; фенил; бензил; гетероарил; -СН2-гетероарил; или СF3; или два R9 вместе с атомом азота, к которому они присоединены, образуют необязательно R10-замещенное 4-8-членное кольцо, содержащее, в добавок к N атому, до двух гетероатомов, выбранных независимо из N, О и S;
R3 означает Н, OR1, CH3R1R2, (CH2)1-2NR1R2, CH2-CH2-OR1, CH2-CO-NR1R2 или CO-CH2R1R2;
R4 означает F, Cl, BR, I, OR1, NR1R2 или имеет одно из значений, приведенных для R1; и
R5 имеет одно из значений, приведенных для R1, в свободной форме или форме соли.
Любой алкил, алкенил или алкинил могут иметь линейное или разветвленное строение.
Под «гетероарилом» подразумевается ароматическая кольцевая система, включая моно-, ди- или трициклическую систему, которая содержит до 4-х гетероатомов, независимо выбранных из N, О или S, такая, например, как фурил, тиенил, пирролил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил, оксадиазолил, тиадиазолил, триазолил, тетразолил, пиридил, пиридазинил, пиримидинил, пиразинил, триазинил, тетразинил, индолил, бензотиофенил, бензофуранил, бензимидазолил, индазолил, бензотриазолил, бензотиазолил, бензоксазолил, хинолинил, изохинолинил, фталазинил, хиноксалинил, хиназолинил, циннолинил или нафтиридинил.
Предпочтительное сочлененное 4-7-членное неароматическое кольцо, представленное R11, означает сочлененное 5 или 6-членное неароматическое кольцо, необязательно содержащее 1 или 2 атома кислорода и включающее, например, -O-СН2-O- или -O-СН2-СН2-O- группы, присоединенные к двум соседним углеродным атомам.
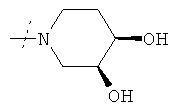
Соединения формулы I могут существовать в свободной форме или в форме соли, например аддитивных солей с, например, органическими или неорганическими кислотами, например хлористоводородной кислотой, уксусной кислотой, в тех случаях, когда R1, R2 и/или R3 включают необязательно замещенную аминогруппу или гетероциклический фрагмент, которые могут образовывать аддитивные соли. Когда соединения формулы I имеют один или более асимметрических центра в молекуле, например, когда пиперидиновое кольцо является замещенным, следует понимать, что изобретение включает все возможные оптические изомеры, также как рацематы, диастереоизомеры и их смеси.
В соединениях формулы I предпочтительны следующие значения или индивидуально или в любой комбинации:
1. каждый R1 и R2 независимо означают Н; необязательно R10-замещенный фенил; необязательно R10-замещенный гетероарил; необязательно R10-замещенный гетероарил-N-оксид; необязательно R10-замещенный C1-С6алкил;
необязательно R10-замещенный С2-С6алкенил; необязательно R10-замещенный С2-С6алкинил; необязательно R10-замещенный С3-С8циклоалкил или необязательно R10-замещенный С4-С6циклоалкенил;
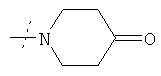
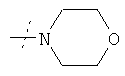
2. R1 и R2 образуют вместе с атомом азота, к которому они присоединены, необязательно R10-замещенное 3-6-членное кольцо, содержащее вдобавок к атому азота еще 1 гетероатом, выбранный независимо из группы, состоящей из N, О или S. Предпочтительно такое необязательно R10-замещенное 3-6-членное кольцо, которое содержит только один атом N, или два атома N, или один атом N и один атом О, более предпочтительно, когда это кольцо неароматическое. Примерами являются, например, кольцевые производные необязательно R10-замещенных азетидина, пирролина, пирролидина, пиперидина, пиперазина, кетопиперазина, тиазина, тиазиндиоксида, тетрагидропиридина, пиперидона, морфолина или азепина. Предпочтительно такие кольца замещены одной или двумя группами ОН, С1-С4алкил, С1-С4алкокси, СО-С1-С4алкил или карбамоильной.
3. R10 представляет собой 1-3 заместителя, независимо выбранных из группы, состоящей из С1-С3алкила, C1-С3гидроксиалкила, C1-С6алкоксиалкила, C1-С3галоидалкила, фенила, гетероарила, F, Cl, ОН;
4. R3 означает Н, OR1, -CH2-CH2-NR1R2, -CH2-CH2-OR1, -CH2-C(O)-NR1R2;
5. R4 означает F, Cl, Br; I, -OR1, -NR1R2 или имеет одно из значений, приведенных для R1;
6. R5 имеет одно из значений, приведенных для R1.
Предлагаемое изобретение также включает способ получения соединения формулы I, который включает
a) получение соединения формулы I, в которой каждый из R3 и R4 означает Н, реакцией соединения формулы II

в которой R1 и R2 означают то, что определено выше,
с образующим мочевину агентом, или
b) получение соединения формулы I, в которой каждый из R3 и R4 означает H, амидированием соединения формулы III
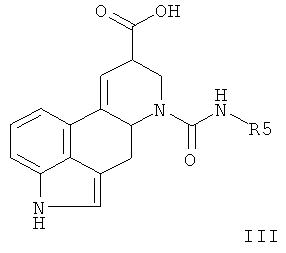
в которой R5 имеет значение, приведенное выше, или его функциональное производное, или
c) получение соединения формулы I, в которой каждый из R3 и R4 означает не Н, превращением соединения формулы I, в которой каждый из R3 и R4 означает Н,
и, если требуется, превращением конечного соединения формулы I, полученного в свободной форме, в форму требуемой соли и наоборот.
Образующий мочевину агент, используемый на стадии а) может быть, например, фосгеном, трифосгеном или трихлорметилформиатом, с последующим добавлением амина. Мочевину можно также получать по реакции соединения формулы II с изоцианатом.
Амидирование на стадии b) можно легко осуществить через активированное функциональное карбоксипроизводное, например ацилхлорид, смешанный ангидрид или симметричный ангидрид, с последующей реакцией с амином, или прямой реакцией, например, метилового эфира с амином при нагревании или микроволновом воздействии.
Соединения формулы II, применяемые в качестве исходных материалов, могут быть получены следующим образом:

R1 и R2 имеют значения, которые определены выше.
Соединения формулы III, используемые в качестве исходных материалов, могут быть получены следующим образом:
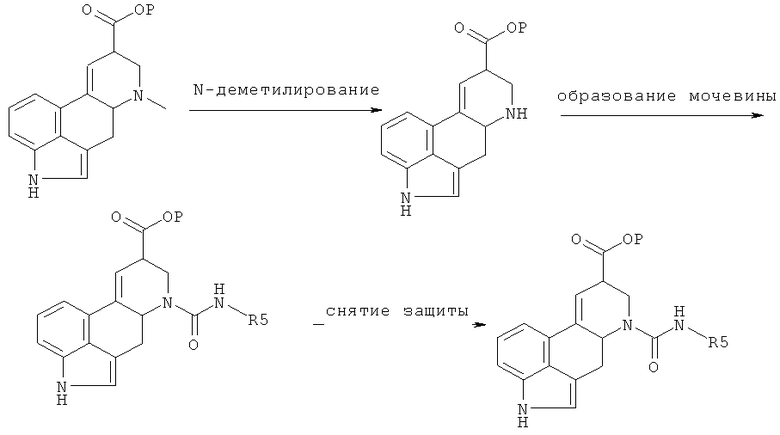
где R5 означает то, что определено выше, и Р означает защитную группу, например, метил, этил, трет-бутил, тритил, бензил, флуоренил, триметилсилилэтил или аллиловый эфир.
Приведенные выше реакции можно проводить в соответствии с методами, известными специалистам, или так, как это описано здесь далее. Удаление защитной группы Р может быть осуществлено кислотным или щелочным гидролизом, обработкой ионом фтора или гидрогенизацией.
В тех случаях, когда получение исходных веществ не описывается, эти соединения хорошо известны или могут быть получены аналогично способам, известным специалистам, или описанным здесь далее.
Следующие примеры иллюстрируют предлагаемое изобретение, но не ограничивают его.
Пример 1
9-Диэтиламид 7-фениламид (6аR,9R)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7,9-дикарбоновой кислоты
Смесь мезилата диэтиламида (6аR,9R)-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]хинолин-9-карбоновой кислоты (122 мг, 0,30 ммол) и изоцианатобензена (36 мг, 0,30 ммоль) в ацетоне (5 мл) перемешивают 3 час при 25°С. Растворитель удаляют, и остаток подвергают флэш-хроматографии (SiO2, циклогексан/трет-бутилметиловый эфир 1:0 до 2:3), получая озаглавленное соединение. МС/ЭР (масс-спектр/электрораспыление): 429 [М+Н]+
Соединения формулы
где R5 имеет значения, приведенные в Таблице 1, получают аналогичным образом.
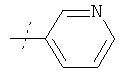


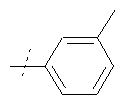
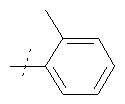
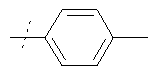

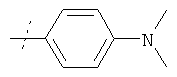


Пример 13
Фениламид (6аR,9R)-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индoлo[4,3-fg]xинoлин-7-кapбoнoвoй кислоты
Стадия 1: метиловый эфир (6аR,9R)-7-циано-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]хинолин-9-карбоновой кислоты
В 50 мл круглодонную колбу, содержащую раствор метилового эфира лизергиновой кислоты (1,0 г, 3,54 ммоль) в безводном дихлорметане (50 мл), медленно добавляют бромциан (2,02 г, 19,11 ммоль), и полученную реакционную смесь черного цвета перемешивают при комнатной температуре в течение 4 час, после чего ТСХ в 10% метанол/дихлорметан показала частичную конверсию исходного продукта в два продукта. Реакционную смесь перемешивают в течение двух дней и мониторируют с помощью ТСХ, не показывающей изменения. Затем реакционную смесь перемешивают при 50°С в течение 4 час, после чего экстрагируют винной кислотой и дихлорметаном. Водную фазу реэкстрагируют дихлорметаном (100 мл), и объединенные органические фазы промывают солевым раствором (200 мл), высушивают над MgSO4, фильтруют и концентрируют в вакууме, получая темное коричневое (цвет дегтя) масло. Очистку проводят колоночной нормально-фазной флэш-хроматографией (Biotage Flash 40, катридж 90 г), используя 40% этилацетат/гексан для отделения быстро проходящего продукта, выделяемого в виде бледножелтого твердого вещества. Продукт кристаллизуют из трет-бутилметилового эфира и медленно выпаривают. Полученный кристаллический материал фильтруют с отсасыванием, получая светложелтые кристаллы.
Стадия 2: метиловый эфир (6аR,9R)-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]хинолин-9-карбоновой кислоты
В круглодонную колбу на 100 мл, содержащую раствор метилового эфира (6аR,9R)-7-циано-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]хинолин-9-карбоновой кислоты (1,57 г, 5,35 ммоль) в уксусной кислоте (20 мл), добавляют воду (4 мл) и цинк (1,5 г). Реакционную смесь кипятят при 100°С 3 час, после чего анализ ТСХ в 20% метанол/ДХМ показал превращение исходного вещества в конечный продукт в виде смеси диастереомеров. Реакционную смесь фильтруют для удаления цинка, и бумажный фильтр тщательно промывают водой (200 мл) и этилацетатом (200 мл). Водную фазу подщелачивают добавлением твердого гидрокарбоната натрия. Затем фазы экстрагируют и дают разделиться. Водную фазу ре-экстрагируют этилацетатом (2×100 мл), объединенные органические фазы промывают насыщенным солевым раствором (200 мл), сушат (MgSO4), фильтруют с отсасыванием и концентрируют в вакууме, получая пену бежевого цвета. Очистку проводят нормально-фазной колоночной флэш-хроматографией (Biotage Flash 40, катридж 40 г), используя для разделения 10% метанол/дихлорметан. Продукт выделяют в виде пены бежевого цвета.
Стадия 3: метиловый эфир (6аR,9R)-7-фенилкарбамоил-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]хинолин-9-карбоновой кислоты
В круглодонную колбу на 100 мл, содержащую раствор метилового эфира (6аR,9R)-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]хинолин-9-карбоновой кислоты (0,8 г, 2,98 ммоль) в дихлорметане (20 мл), добавляют фенилизоцианат (0,45 мл, 4,5 ммоль, 1,5 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 16 час, после чего анализ ТСХ в 10% метанол/ДХМ показал превращение исходного соединения в продукт в виде смеси диастереомеров. Летучие вещества концентрируют в вакууме и очищают, используя нормально-фазную колоночную флэш-хроматографию (Biotage Flash 40, катридж 90 г), с применением 30% этилацетат/гексан, выделяя продукт в виде диастереомерной смеси 1:1.
Стадия 4: (6аR,9R)-7-фенилкарбамоил-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]xинoлин-9-кapбoнoвaя кислота
В круглодонную колбу на 100 мл, содержащую метиловый эфир (6aR,9R)-7-фенилкарбамоил-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]хинолин-9-карбоновой кислоты (0,8 г, 2,06 ммоль), добавляют метанол (12 мл), ТГФ (24 мл) и раствор гидрида лития (247 мг) в воде (12 мл). Реакционную смесь перемешивают 20 мин при комнатной температуре, после чего анализ ТСХ в 20% метанол/дихлорметан показал полное превращение исходного соединения. Цвет реакционной смеси изменился от светло-желтого до красно-фиолетового. Летучие вещества удаляют в вакууме (оставляя только воду), и водный раствор подкисляют добавлением 1 М НСl. Полученный осадок бежевого цвета фильтруют отсасыванием, и сгусток на фильтре промывают дистиллированной водой (50 мл). Затем сгусток с фильтра сушат в высоковакуумном шкафу при 50°С в течение 16 час, получая продукт.
Стадия 5: фениламид(6аR,9R)-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
В круглодонную колбу на 50 мл, содержащую суспензию (6aR,9R)-7-фенилкарбамоил-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]хинолин-9-карбоновой кислоты (0,2 г, 0,54 ммоль), добавляли PYBOP (0,307 г), дихлорметан (10 мл), пирролидин (0,054 мл, 0,64 ммоль, 1,2 экв.) и основание Хюнига (Hunigs, 0,187 мл, 1,07 ммоль, 2 экв.). Реакционную смесь перемешивают 3 час при комнатной температуре. Очистку проводят методом нормально-фазной колоночной флэш-хроматографии (Biotage Flash 40, катридж 40 г) с применением 50% этилацетат/гексан в качестве растворителя. Фениламид (6aR,9R)-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты выделяют в виде твердого вещества бежевого цвета. МС/ЭР: 427 (М+Н)+
Соединения формулы
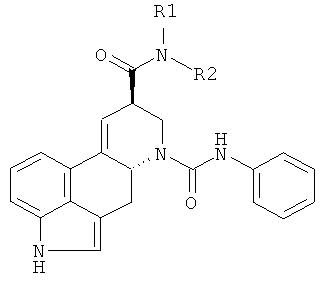
где R1 и R2 имеют значения, приведенные в Таблице 2, получают с использованием аналогичных способов и соответствующих аминов.




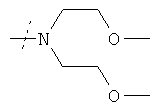
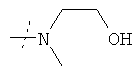
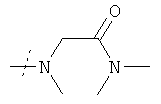
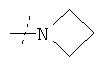

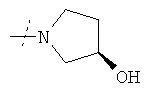
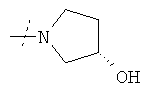
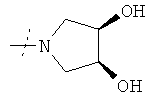
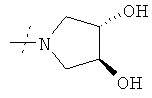

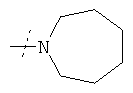






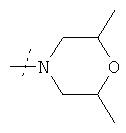
Пример 45
(2-Метоксифенил)амид (6аR,9R)-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
Стадия 1: ((6аR,9R)-7-Метил-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]хинолин-9-ил)пирролидин-1-илметанон
Лизергиновую кислоту (30 г, 111,80 ммоль) растворяют в 400 мл дихлорметана, охлаждают до 0°С - 5°С и добавляют триэтиламин (31,16 мл, 223,61 ммоль, 2 экв.) и пирролидин (18,66 мл, 223,61 ммоль, 2 экв.) и в течение 15 мин 1,5 экв. ангидрида пропанфосфоновой кислоты (50% в этилацетате). Реакционную смесь перемешивают 1 час при комнатной температуре, затем выливают ее на лед и экстрагируют дихлорметаном, органическую фазу высушивают над Na2SO4, упаривают, и остаток (28,6 г) очищают хроматографией на силикагеле, используя смесь дихлорметан: метанол 9:1 и получая ((6аR,9R)-7-метил-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]хинолин-9-ил)пирролидин-1-илметанон.
Стадия 2: (6аR,9R)-4,6,ба,7,8,9-гексагидроиндоло[4,3-fg]хинолин-9-илпирролидин-1-илметанон.
Продукт стадии 1 (6 г, 18,67 ммоль) растворяют в 180 мл дихлорметана при 0°С и добавляют 70% мета-хлорнадбензойную кислоту (5,522 г, 22,40 ммоль, 1,2 экв.). Через 10 мин образуется промежуточная N-окись, к которой добавляют FeSO4·7H2O (2,594 г, 9,33 ммоль, 0,5 экв.) в 12 мл метанола, прекращают охлаждение, и смесь перемешивают при комнатной температуре. Через 1 час 25 мин реакционную смесь экстрагируют 0,1 М раствором ЭДТА (доведенным предварительно до рН 9), высушивают над Na2SO4, упаривают и очищают хроматографией на силикагеле с элюентом дихлорметан: метанол: аммиак 93:6:1, получая (6аR,9R)-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]хинолин-9-ил)пирролидин-1-илметанон.
Стадия 3: (2-Метоксифенил)амид (6аR,9R)-9-(пирролидин-1-карбонил)-6,6a,8,9-тeтpaгидpo-4H-индoлo[4,3-fg]xинoлин-7-кapбoнoвoй кислоты
Продукт стадии 2 (5,087 г, 16,54 ммоль) растворяют в 80 мл тетрагидрофурана, добавляют 1-изоцианато-2-метоксибензен (1,79 мл, 16,5 ммоль) и перемешивают при комнатной температуре. Для того чтобы связать избыток изоцианата, добавляют 0,3 эквивалента 3-амино-1,2-пропандиола и перемешивают 2,5 час. Затем реакционную смесь промывают насыщенным раствором бикарбоната натрия и солевым раствором, сушат над Na2SO4 и частично упаривают. Кристаллизация приводит к получению (2-метоксифенил)амида(6аR,9R)-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты. МС/ЭР: 457 (М+Н)+
Соединения формулы

где R5 имеет значения, приведенные в таблице 3, получают по аналогичным методикам.







Пример 56
Фениламид 5-метил-9-(пиперидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индoлo[4,3-fg]xинoлин-7-кapбoнoвoй кислоты
Фениламид 5-метил-9-(пиперидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индoлo[4,3-fg]xинoлин-7-кapбoнoвoй кислоты получают по методикам, описанным в Примере 45, применяя 2-метиллизергиновую кислоту (вместо лизергиновой кислоты) и пиперидин (вместо пирролидина) на стадии 1 и фенилизоцианат на стадии 3. MS: 455 (M+H)+
Пример 57
(3-Фторфенил)амид 9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
Продукт стадии 2 примера 45 (62 г, 20 ммоль) растворяют в 3 мл дихлорметана и добавляют 0,14 мл (10 экв.) триэтиламина и 0,042 мл трихлорметилхлорформиата в 2 мл дихлорметана. Через 30 мин добавляют при комнатной температуре 0,14 мл триэтиламина (10 экв.) и 0,040 мл (2 экв.) 2-фторанилина. После перемешивания в течение 22 час при комнатной температуре реакционную смесь распределяют между 100 мл дихлорметана и насыщенным раствором бикарбоната натрия. Органический слой высушивают над Na2SO4 и выпаривают. Сырой продукт очищают хроматографией на силикагеле, элюируя смесью этилацетат/циклогексан 3:1 и получая (3-фторфенил)амид 9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты. МС/ЭР: 445 (М+Н)+
Соединения формулы
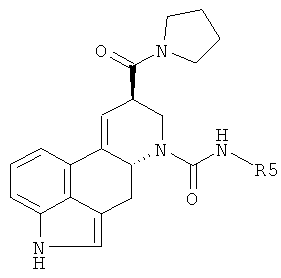
в которой R5 имеет значения, приведенные в таблице 4, получают по аналогичным методикам, используя подходящие амины в качестве реагентов.


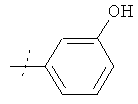

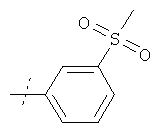
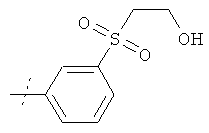


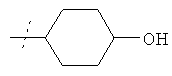
Пример 72
Фениламид (6аR,9R)-5-хлор-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
Соединение из примера 13 (64 мг, 0,15 ммоль) растворяют в 1 мл ДМФ, добавляют N-хлорсукцинимид (36 мг, 0,27 ммоль, 1,8 экв.) и перемешивают при комнатной температуре. Через 55 мин реакционную смесь очищают хроматографией на силикагеле, элюируя этилацетатом и получая фениламид (6аR,9R)-5-хлор-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты. МС: 461 (М+Н)+
Пример 73
Фениламид (6аR,9R)-5-иод-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
Фениламид (6аR,9R)-5-иод-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты получают по методике,
аналогичной описанной в примере 72, используя N-иодсукцинимид вместо N-хлорсукцинимида. МС: 553 (М+Н)+
Пример 74
Фениламид (6аR,9R)-5-бром-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
Стадия 1: (6аR,9R)-5-бром-7-метил-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]xинoлин-9-кapбoнoвaя кислота
Лизергиновую кислоту (8,05 г, 30 ммоль) суспендируют в диоксане и по каплям добавляют ТФУ (экзотермическая реакция). По каплям добавляют раствор брома (1,54 мл, 30 ммоль, 1,0 экв.) в хлороформе. Реакционную смесь охлаждают до 5°С, при этом кристаллизуется продукт. (6аR,9R)-5-Бром-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновую кислоту выделяют фильтрованием и перекристаллизовывают из диэтилового эфира. Rf=0,5, 10% МеОН: ДХМ, M+H+=346,348, t пл. >245 (с разложением).
Стадия 2: (6аR,9R)-5-бром-7-метил-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]xинoлин-9-ил)пиppoлидин-1-ил-метанон
В 50 мл круглодонную колбу, содержащую (6аR,9R)-5-бром-7-метил-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]хинолин-9-карбоновую кислоту (5,0 г, 13,38 ммоль) в ДМФ (20 мл), добавляют ГАТУ (HATU) (6,10 г, 16,06 ммоль, 1,2 экв.), и реакционную смесь перемешивают 1 час при комнатной температуре, затем добавляют пирролидин (2,24 мл, 26,78 ммоль, 2,0 экв), и реакционную смесь перемешивают следующие 2 час при комнатной температуре. ТСХ в 10% метанол/ДХМ показывает полную конверсию исходного материала (определяется по реагенту хлор/TBDM и УФ). В реакционную смесь добавляют 4 М НСl (150 мл) и воду (150 мл) и этилацетат (200 мл). После экстракции водную фазу ре-экстрагируют этилацетатом (2×200 мл), и комбинированные экстракты промывают насыщенным раствором бикарбоната (2×200 мл), водой (200 мл), насыщенным солевым раствором (200 мл), фильтруют (с отсасыванием), высушивают (MgSO4) и концентрируют в вакууме, получая темнокоричневое масло. Очистку проводят с помощью нормально-фазной колоночной флэш-хроматографии (Biotage Flash 40, катридж 90 г), используя градиент от 20% этилацетат/гексан к 100% этилацетату и затем к 5% метанол/этилацетат свыше 5 литров. Нужные фракции собирают, концентрируют в вакууме и оставляют в высоковакуумном шкафу на 3 час при 50°С, получая смесь (6аR,9R)-5-бром-7-метил-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]хинолин-9-карбоновой кислоты и диастереомер.
Стадия 3: ((R)-5-бром-7-метил-7-окси-4,6,6а,7,8,9-гексагидроиндоло[4,3-fg]xинoлин-9-ил)пиppoлидин-1-илмeтaнoн
1,1 г (2,75 ммоль) продукта стадии 2 растворяют в ТГФ (40 мл) с помощью ультразвукового оборудования, затем раствор охлаждают до -40°С с помощью смеси ацетон/сухой лед, по порциям добавляют м-хлорфенилбороновую кислоту (m-СРВА) (0,640 г, 3,75 ммоль, 1,35 экв.) в течение 30 мин, и полученной темнокоричневой реакционной смеси дают нагреться до 0°С при постоянном перемешивании (примерно 1,5 час). В реакционную смесь по каплям при 0°С добавляют раствор хлорида железа (II) (0,174 г, 1,37 ммоль, 0,5 экв.) в воде (10 мл). Через 1 час реакционной смеси дают медленно нагреться до комнатной температуры и затем перемешивают ее 2 час. Анализ ТСХ в 20% метанол/ДХМ показал полный расход промежуточной N-окиси. К реакционной смеси добавляют раствор бисульфита натрия (1 г) в воде (5 мл), и летучие вещества удаляют затем в вакууме, получая темную пену, которую очищают на Biotage Flash 40, используя смесь метанол/ДХМ от 2% до 8% с прибавлением 2% на литр растворителя и получая ((R)-5-бром-7-метил-7-окси-4,6,6а,7,8,9-гексагидроиндоло [4,3-fg] хинолин-9-ил)пирролидин-1-илметанон.
Стадия 4: Фениламид (6аR,9R)-5-бром-9-(пирролидин-1-карбонил)-б,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
Продукт стадии 3 (0,504 г, 1,32 ммоль) растворяют в безводном дихлорметане (20 мл), и раствор охлаждают до 0°С с помощью бани лед-соль. Затем добавляют основание Хюнига (0,940 мл, 0,21 ммоль) и фенилизоцианат (0,388 мл, 3,9 ммоль), и реакционной смеси дают нагреться до комнатной температуры. Перемешивание продолжают в течение 16 час, после чего ТСХ в 20% метанол/ДХМ показывает полное расходование исходного материала. Очистку проводят с помощью нормально-фазной колоночной флэш-хроматографии (Biotage Flash 40, катридж 40 г), используя градиент системы, от 20% этилацетат/гексан (1 литр) к 30% этилацетат/гексан (1 литр), затем к 50% этилацетат/гексан (1 литр) и, наконец, к 60% этилацетат/гексан (1 литр). Нужные фракции объединяют, концентрируют в вакууме и оставляют в высоковакуумном шкафу при 50°С на 3 час. β Фениламид (6аR,9R)-5-бром-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты выделяют в виде бесцветного твердого вещества. МС/ЭР: 505 (М+Н)+
Пример 75
Фениламид (6аR,9R)-5-фенил-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
В 5 мл сосуд для реакции в микроволновой печи, содержащий продукт примера 74 (0,05 г, 0,27 ммоль) в ДМЭ (1 мл), добавляют фенилбороновую кислоту (46 мг, 0,37 ммоль, 1,4 экв) и 2 М раствор карбоната натрия (1,50 мл). Реакционную смесь продувают азотом в течение 5 мин при перемешивании и добавляют катализатор (30 мг). Стенки сосуда промывают этанолом, продутым азотом (0,75 мл), и реакционную смесь продувают азотом следующие 5 мин. Сосуд закрывают крышкой и помещают в микроволновую печь при 100°С на 300 сек (фиксированное время). ТСХ в 10% метанол/ДХМ показывает полное расходование исходного материала (определяемое хлор/ТВДМ реагентом и УФ). Реакционную смесь распределяют между насыщенным раствором бикарбоната (20 мл) и дихлорметаном (20 мл). Органические слои сразу помещают в колонку и очищают нормально-фазной колоночной флэш-хроматографией (Biotage Flash 40, катридж 40 г), используя в качестве растворителя градиент смеси 30% этилацетат/гексан (1 литр) к 40% этилацетат/гексан (1 литр). Нужные фракции объединяют, концентрируют в вакууме и оставляют в высоковакуумном шкафу при 50°С на 3 час, выделяя продукт МС: 503 (М+Н)+
Соединения формулы
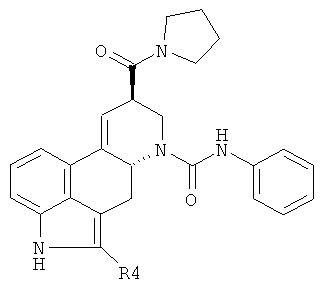
в которой R4 имеет значения, указанные в таблице 5, получают по аналогичным методикам.
Пример 78
(6аR,9R)-5-(3-Гидроксипроп-1-инил)-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло [4,3-fg] хинолин-7-карбоновая кислота
В 5 мл сосуд для проведения реакции в микроволновой печи, содержащий раствор соединения из примера 74 (50 мг, 0,1 ммоль), трифенилфосфин (3 мг, 0,001 ммоль, 0,1 экв.), иодид меди (1 мг, 0,005 ммоль, 0,05 экв.), триэтиламин (1 мл), пиридин (1 мл), добавляют пропаргиловый спирт (0,007 мл, 0,12 ммоль, 1,2 экв.), и реакционную смесь продувают азотом при перемешивании в течение 5 мин, затем добавляют катализатор - дихлорбис(трифенилфосфин)палладий (7 мг, 0,01 ммоль, 0,1 экв.), реакционную смесь продолжают продувать азотом еще 5 мин, после чего сосуд закрывают крышкой и помещают в микроволновую печь при 100°С на 300 сек (фиксированное время). Метод ЖХ-МС показывает некоторую степень превращения в продукт реакции. В реакционную смесь добавляют катализатор (7 мг, 0,01 ммоль, 0,1 экв.), пропаргиловый спирт (7 мл, 0,12 ммоль, 1,2 экв.) и иодид меди (I) (1 мг, 0,005 ммоль, 0,05 экв.), и помещают ее снова в микроволновую печь при 100°С на следующие 300 сек. Метод ЖХ-МС показывает 1:1 превращение исходного материала в продукт. Реакционную смесь помещают в микроволновую печь при 100°С на следующие 600 сек, после чего метод ЖХ-МС показывает превращение исходного материала в продукт. Очистку проводят с помощью нормально-фазной колоночной флэш-хроматографии (Biotage Flash 40, катридж 40 г), используя 50% этилацетат/гексан (2 литра). Нужные фракции собирают, концентрируют в вакууме и высушивают в высоковакуумном шкафу при 50°С в течение 3 час, выделяя полученный продукт. МС/ЭР 481 (М+H)+
Пример 79
Изопропиловый эфир [(6аR,9R)-7-фенилкарбамоил-9-(пирролидин-1-карбонил)-6а,7,8,9-тетрагидро-6Н-индоло[4,3-fg]хинолин-4-ил]уксусной кислоты
Продукт из примера 13 (5 г, 11,72 ммоль) растворяют в 100 мл дихлорметана и при комнатной температуре, добавляют 30 мл 40% водного раствора гидроксида натрия и 400 мг бензилтриэтиламмонийхлорида. Реакционную смесь охлаждают до 0-5°С, добавляют 6,23 мл (46,89 ммоль, 4 экв.) изопропилбромацетата и перемешивают 1 час. Реакционную смесь выливают на лед и экстрагируют CH2Cl2, органический слой промывают водой, сушат над Na2SO4 и упаривают. Остаток очищают хроматографией на силикагеле, элюируя трет-бутилметиловым эфиром и получая изопропиловый эфир [(6аR,9R)-7-фенилкарбамоил-9-(пирролидин-1-карбонил)-6а,7,8,9-тетрагидро-6Н-индоло[4,3-fg]хинолин-4-ил]уксусной кислоты. МС/ЭР: 527 (М+Н)+
Пример 80
[(6аR,9R)-7-Фенилкарбамоил-9-(пирролидин-1-карбонил)-6а,7,8,9-тетрагидро-6Н-индоло[4,3-fg]хинолин-4-ил]уксусная кислота
[(6аR,9R)-7-Фенилкарбамоил-9-(пирролидин-1-карбонил)-6а,7,8,9-тетрагидро-6Н-индоло[4,3-fg]хинолин-4-ил]уксусную кислоту получают как побочный продукт при синтезе, описанном в примере 79. МС/ЭР: 485 (М+Н)+
Соединения формулы
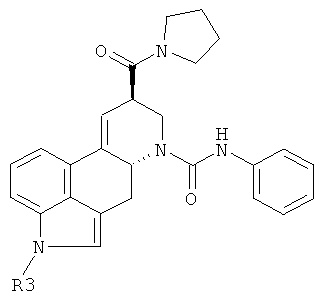
где R3 имеет значения, приведенные в таблице 6, получают способом, аналогичным описанному в примере 79.
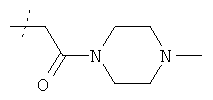
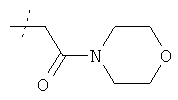
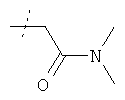

Соединения формулы

где R3 имеет значения, приведенные в таблице 7, получают способом, аналогичным описанному в примере 79, используя в качестве исходного соединения соединение из примера 1 вместо соединения из примера 13 и применяя подходящие алкилгалогениды.

Пример 87
Фениламид (6аR,9R)-4-(2-гидроксиэтил)-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
Соединение из примера 79 (3,99 г, 7,57 ммоль) растворяют в 100 мл тетрагидрофурана и при 0-5°С добавляют 989 мг (45,43 ммоль, 4 экв.) борогидрида лития. Реакционную смесь перемешивают 4,5 час при комнатной температуре, затем выливают в смесь лед/уксусная кислота (бурное выделение CO2) и экстрагируют дихлорметаном. Органический слой промывают водой, сушат над Na2SO4 и упаривают. Остаток очищают хроматографией на силикагеле, элюируя смесью дихлорметан: метанол 95:5, получая при элюировании желаемый β-изомер фениламида (6аR,9R)-4-(2-гидроксиэтил)-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло [4,3-fg] хинолин-7-карбоновой кислоты. МС/ЭР: 471 (М+Н)+
Пример 88
Фениламид (6аR,9R)-4-(2-морфолин-4-илэтил)-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
Стадия 1: 2-[(6аR,9R)-7-Фенилкарбамоил-9-(пирролидин-1-карбонил)-6а,7,8,9-тетрагидро-6Н-индоло[4,3-fg]хинолин-4-ил]этиловый эфир толуен-4-сульфокислоты
1,12 г (2,38 ммоль) соединения примера 87 растворяют в 40 мл дихлорметана, и при комнатной температуре добавляют 437 мг (3,58 ммоль, 1,5 экв.) диметиламинопиридина, после чего смесь охлаждают до 0-5°С. Затем добавляют 683 мг (3,589 ммоль, 1,5 экв.) 4-метилбензенсульфонилхлорида, и перемешивают реакционную смесь при комнатной температуре в течение 3,5 час, после чего выливают в лед и некоторое количество 6 н. серной кислоты и экстрагируют дихлорметаном. Органический слой сушат над Na2SO4 и упаривают. Остаток очищают хроматографией на силикагеле, получая 2-[(6аR,9R)-7-фенилкарбамоил-9-(пирролидин-1-карбонил)-6а,7,8,9-тетрагидро-6Н-индоло[4,3-fg]хинолин-4-ил]этиловый эфир толуен-4-сульфокислоты.
Стадия 2: Фениламид (6аR,9R)-4-(2-морфолин-4-илэтил)-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
80 мг (0,128 ммоль) продукта стадии 1 и 1 мл морфолина перемешивают при комнатной температуре в течение 16 час. Реакционную смесь очищают хроматографией на силикагеле, элюируя смесью ацетон/циклогексан 6:4, получая фениламид (6аR,9R)-4-(2-морфолин-4-илэтил)-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты. МС/ЭР: 540 (М+Н)+
Соединения формулы
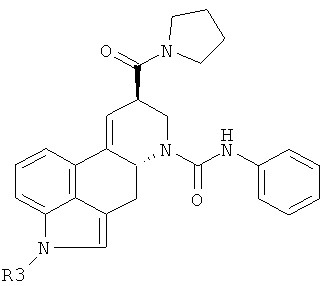
в которой R3 имеет значения, указанные в таблице 8, получают способом, подобным описанному в стадии 2 примера 88.
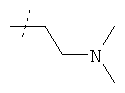
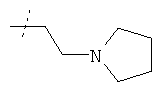
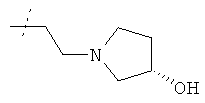
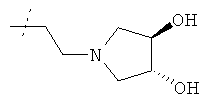


Пример 97
Фениламид (6аR,9R)-4-ацетил-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
Соединение примера 13 (80 мг, 0,18 ммоль) и диметиламинопиримидин (69 мг, 0,56 ммоль, 3 экв.) растворяют в 3 мл дихлорэтана и добавляют уксусный ангидрид (0,036 мл, 0,37 ммоль, 2 экв.). Реакционную смесь перемешивают 4 час при 65°С, затем распределяют между дихлорметаном и насыщенным водным раствором бикарбоната. Органический слой высушивают над Na2SO4 и упаривают. Сырой продукт очищают хроматографией на силикагеле, элюируя смесью дихлорметан/метанол 97:3 и получая фениламид (6аR,9R)-4-ацетил-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты. МС/ЭР: 469 (М+Н)+
Пример 98
Фениламид (6аR,9R)-4-гидрокси-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
Стадия 1: Фениламид (5аS,6аR,9R)-9-(пирролидин-1-карбонил)-5,5а,6,6а,8,9-гексагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
Соединение из примера 13 (351 мг, 0,82 ммоль) растворяют в 6 мл трифтоуксусной кислоты, добавляют триэтилсилан (0,407 мл, 2,47 ммоль, 3 экв.), и реакционную смесь перемешивают при комнатной температуре 1 час 10 мин. Затем реакционную смесь распределяют между этилацетатом и насыщенным водным раствором бикарбоната, органический слой промывают солевым раствором, высушивают над Na2SO4, упаривают и кристаллизуют из трет-бутилметилового эфира, получая фениламид (5аS,6аR,9R)-9-(пирролидин-1-карбонил)-5,5а,6,6а,8,9-гексагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты.
Стадия 2: Фениламид (6аR,9R)-4-гидрокси-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
Продукт стадии 1 (212 мг, 0,49 ммоль) растворяют в 12 мл метанола и добавляют дегидрат вольфрамата натрия (41 мг, 0,12 ммоль, 0,25 экв.) в нескольких каплях воды. Затем при 0°С добавляют 670 мл 30% Н2О2 (10 экв.) и перемешивают 50 мин при комнатной температуре. Реакционную смесь распределяют между дихлорметаном и насыщенным водным раствором бикарбоната, органический слой промывают солевым раствором, высушивают над Na2SO4, упаривают и очищают хроматографией на силикагеле, элюируя смесью трет-бутилметиловый эфир: циклогексан 9:1 и получая фениламид (6аR,9R)-4-гидрокси-9-(пирролидин-1-карбонил)-б,ба,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты. МС/ЭР: 441 (М+Н)+
Пример 99
Фениламид (6аR,9R)-4-метокси-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты
Продукт примера 98 (17 мг, 0,038 ммоль) растворяют в 2 мл смеси метанол: дихлорметан 1:1, и в реакционную смесь в течение 10 мин перегоняют свежеприготовленный раствор диазометана до образования устойчивой желтой окраски. Реакционную смесь упаривают и очищают хроматографией на силикагеле, используя смесь трет-бутилметиловый эфир: циклогексан 9:1 в качестве элюента. При упаривании продукт кристаллизуется, давая фениламид (6аR,9R)-4-метокси-9-(пирролидин-1-карбонил)-6,6а,8,9-тетрагидро-4Н-индоло[4,3-fg]хинолин-7-карбоновой кислоты. МС/ЭР: 458 (М+Н)+
Соединения формулы I в свободной форме или в форме фармацевтически приемлемой соли проявляют ценные фармакологические свойства, например, как антагонисты CXCR3, как это определено, например, в тестах in vitro, и в связи с этим показаны для лечения.
а). CXCR3 мембранный связывающий анализ
Анализ на связывание лиганда применяется для идентификации ингибиторов I-TAC, связанных с CXCR3 экспрессирующими мембранами. Клетки мембран получают из СНО клеток, трансфектированных человеческим CXCR3. Связывание меченого 125I CXCR3 лиганда, например I-TAC (CXCL11), с CXCR3 оценивают, применяя метод сцинцилляционного проксимального анализа (SPA) (Amersham Pharmacia Biotech). Буфер или серийные разбавленные растворы соединения инкубируют 2 час при комнатной температуре с меченым CXCR3 лигандом (например, I-TAC), CXCR3 экспрессирующими мембранами и покрытыми WGA (агглютинин проростков пшеницы) PVT шариками. Плашки затем центрифугируют и считывают показания на приборе Topcount (Packard). Данные представляют в виде концентрации соединения, требуемой для 50% ингибирования связывания 125I лиганда. В этом анализе соединения формулы I имеют значения IC50 в интервале 1 мкМ-1 нМ. Так, например, соединения из примеров 18, 23, 43, 59 и 79 имеют значения IС50, равные (нМ) 54, 61, 23, 43 и 145, соответственно.
b) CXCR3 функциональный анализ - активация Са2+
CXCR3 лигандо-индуцированную активацию Са2+ оценивают в CXCR3 трансфектированных L 1,2 клетках (рrе В клеточная линия мышей). Для этого клетки заполняют Са2+-чувствительным хромофором Fluo-4 (Molecular Probes). После промывания клетки преинкубируют с низкомолекулярными ингибиторами при комнатной температуре в течение 2 час. Временное повышение внутриклеточного Са2+ после добавления CXCR3 лиганда (например, I-TAC) замеряют на приборе (FLIPR), считывающем показания флуоресценции на плашке. Ингибирование CXCR3 лигандо индуцированной активности Са2+ в присутствии CXCR3 антагонистов представляют в виде значений IC50, т.е. концентрации соединения, понижающей максимальный Са2+ отклик на 50%. В этом анализе соединения формулы I имеют значения IС50 в интервале 1 мкМ-1 нМ. Так, например, соединения из примеров 18, 23, 43, 59 и 79 имеют значения IC50 18, 8, 16, 20 и 53 нМ, соответственно.
c) CXCR3 функциональный анализ - хемотаксис.
Направленную клеточную миграцию, индуцированную CXCR3 лигандами, например I-TAC, оценивают, применяя 96-луночные одноразовые камеры для хемотаксиса (Multiscreen MIC, Costar) с поликарбонатными мембранами, содержащими поры диаметром в 5 мкм. Хемокин (например, I-TAC) помещают на дно лунки камеры, и клетки (например, CXCR3 трансфектированные L 1,2 клетки) помещают на верхушку отделения камеры для хемотаксиса. Клеточной миграции через поры мембраны позволяют продолжаться 4 час при 37°С. Клетки, мигрирующие от верхушки отделения до дна отделения, определяют поточной цитометрией. Когда тестируют низкомолекулярные ингибиторы, соединения добавляют в оба отделения при одинаковых концентрациях. Серийные разбавления соединений тестируют для оценки их ингибиторного воздействия на CXCR3 зависимую клеточную миграцию. Концентрация низкомолекулярных CXCR3 ингибиторов, которая приводит к понижению количества мигрирующих клеток на 50%, определяется как IС50. В этом опыте соединения формулы I имеют значения IС50 в интервале 1 мкМ-1 нМ. Например, соединения примеров 18 и 43 имеют значения IС50 74 и 75 нМ, соответственно.
d) Эксперименты, проведенные на моделях мышей, показывают, что коррекция стенки сосуда после экспериментальной травмы (например, вызванной аллотрансплантацией) значительно понижена в отсутствие функционального CXCR3.
Соединения формулы I применимы, таким образом, в профилактике и/или лечении заболеваний или нарушений, опосредованных взаимодействиями между хемокинными рецепторами, например CXCR3, и их лигандами, например, при аутоиммунных заболеваниях, например ревматоидном артрите, системной красной волчанке, тиреоидите Хашимото, рассеянном склерозе, бульбоспинальном параличе, диабете I или II типа, и при нарушениях, связанных с васкулитом, пернициозной анемией, синдромом Шегрена, увеитом, псориазом, гнездной аллопецией и другими, аллергических заболеваниях, например аллергической астме, атопическом дерматите, аллергическом рините/конъюктивите, аллергическом контактном дерматите, воспалительных заболеваниях, необязательно с лежащими в основе аберрантными реакциями, например воспалительном заболевания кишечного тракта, болезни Крона или язвенном колите, инфекционно-аллергической астме, воспалительном поражении легких, воспалительном поражении печени, воспалительном гломерулярном поражении, атеросклерозе, остеоартрите, раздражающем контактном дерматите и далее экзематоидном дерматите, себорейном дерматите, кожных проявлениях иммунологически-опосредованных нарушений, воспалительном заболевании глаз, кератоконъюктивите, миокардите или гепатите, ишемическом/реперфузионном поражении, например инфаркте миокарда, инсульте, кишечной ишемии, почечной недостаточности или гемаррогическом шоке, травматическом шоке и других видах, раке, например, твердых опухолей или рака лимфатической системы, такого как лимфома Т-клеток или лейкемия Т-клеток, метастазах или ангиогенезе, инфекционных заболеваниях, например токсическом шоке (например, суперантиген индуцированном), септическом шоке, респираторном дистресс синдроме у взрослых, или при трансплантации, такой как острое или хроническое отторжение органа, ткани или клетки гомо- или гетеротрансплантатов или замедленной функции трансплантата. Под трансплантацией подразумеваются гомо- или гетеро-трансплантаты, например, клеток, тканей или твердых органов, например островков поджелудочной железы, стволовых клеток, тканей костного мозга, корнеальной ткани, нейрональной ткани, сердца, легкого, комбинации сердце-легкое, почек, печени, желудка, поджелудочной железы, трахеи или пищевода. Хроническое отторжение называют также заболеваниями сосудов трансплантата.
Для перечисленных выше применений требуемая доза будет, конечно, варьироваться в зависимости от типа введения, частного состояния, подвергаемого лечению, и желаемого эффекта. В общем случае удовлетворительные результаты достигаются при систематической дневной дозе, составляющей примерно 0,01-10 мг/кг веса тела. Показанная дневная доза для крупных млекопитающих, например людей, находится в интервале 0.5-1000 мг, обычно принимаемой, например, в разделенных дозах до четырех раз в день или в продленной форме. Подходящие формы стандартных доз для орального приема включают примерно 1-500 мг активного ингредиента.
Соединения формулы I могут быть введены обычным путем, в частности энтерально, например орально, например в форме таблеток или капсул, или парентерально, например, в форме инъектируемых растворов или суспензий, топикально, например, в форме примочек, гелей, мазей или кремов, или в назальной форме, или в форме суппозиториев. Фармацевтические композиции, включающие соединение формулы I в свободной форме или в форме фармацевтически приемлемой соли в комплексе с по крайней мере одним фармацевтически приемлемым носителем или разбавителем могут быть получены обычным методом смешивания с фармацевтически приемлемым носителем или разбавителем. Соединения формулы I могут вводиться в свободной форме или в форме фармацевтически приемлемой соли, как отмечалось выше. Такие соли могут быть получены обычным путем и проявляют такой же порядок активности, как и свободные формы соединений.
В соответствии с вышеупомянутым настоящее изобретение предлагает:
1.1 Способ профилактики или лечения нарушений или заболеваний, опосредованных взаимодействиями между хемокинными рецепторами и их лигандами, например, указанными выше, у субъекта, нуждающегося в таком лечении; этот способ включает введение указанному субъекту эффективного количества соединения формулы I или его фармацевтически приемлемой соли;
1.2 Способ профилактики или лечения воспалительных или аутоиммунных заболеваний, например, как указанные выше, у субъекта, нуждающегося в таком лечении, этот способ включает введение указанному субъекту эффективного количества соединения формулы I или его фармацевтически приемлемой соли;
2. Соединение формулы I или его фармацевтически приемлемая соль для применения в качестве лекарства, например, в любом из способов, указанных в п.п.1.1 или 1.2 выше;
3. Фармацевтическая композиция, например, для применения по любому из вышеуказанных п.п.1.1 или 1.2, содержащая соединение формулы I или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым разбавителем или носителем;
4. Соединение формулы I или его фармацевтически приемлемая соль для применения в получении фармацевтической композиции для применения по любому из вышеуказанных п.п.1.1 или 1.2.
Соединения формулы I могут быть введены как единственный активный ингредиент или в сочетании, например, как добавочное средство с другими лекарствами, например с иммуносупрессивными или иммуномодулирующими, или с другими противовоспалительными агентами, например, для лечения или профилактики острого или хронического отторжения алло- или ксенотрансплантата, или воспалительных или аутоиммунных нарушений, хемотерапевтическим агентом или противоинфекционным агентом, например антивирусным агентом, таким как, например, противоретровирусный агент, или антибиотик. Так, например, соединения формулы I могут применяться в комбинации с кальциневриновым ингибитором, например циклоспорином А или FK 506; mTOR ингибитором, например, рапамицином, 40-O-(2-гидроксиэтил)рапамицином, CCI779, АВТ578, или рапалогом (Rapalog), например, АР23573, АР23464, АР23675, АР23841, TAFA-93, биолимусом (biolimus) 7 или биолимусом 9; аскомицином (ascomycin), имеющим иммуносупрессивные свойства, например, АВТ-281, ASM981 и т.д.; кортикостероидами; циклофосфамидом; азатиоприном; метотриксатом; лефлунамидом; мизорибином; микофеноловой кислотой; микофенолятом мофетила; 15-дезоксиспергуалином (15-deoxyspergualine) или его иммуносупрессивным гомологом, аналогом или производным; агонистом или модулятором S1P рецептора, например, FTY720 или его аналогом; ингибитором РКС, например, описанным в WO 02/38561 или WO 03/82859, например соединением примера 56 или 70; моноклональными антителами к рецепторам лейкоцитов, например, МНС, CD2, CD3, CD4, CD7, CD8, CD11a/CD18, CD25, CD27, CD28, CD40. CD45, CD52, CD58, CD80, CD86, CD137, ICOS, CD150 (SLAM), OХ40, 4-1ВВ, или к их лигандам, например, CD 154, или их антагонистам; другими иммуномодулирующими соединениями, например рекомбинантными связующими молекулами, имеющими по крайней мере часть внеклеточного домена CTLA4 или его мутанта, например по крайней мере внеклеточную часть CTLA4 или его мутанта, присоединенные к нe-CTLA4 протеиновой последовательности, например, CTLA4Ig (например, обозначенный как АТСС68629) или его мутант, например, LEA29Y; ингибиторами адгезионных молекул, например LFA-1 антагонистами, ICAM-1 или -3 антагонистами, VCAM-4 антагонистами или VLA-4 антагонистами; или антихемокинными антителами, например анти-МСР-1 антителами, или антителами антихемокинных рецепторов или низкомолекулярными антагонистами хемокинного рецептора.
В тех случаях, когда соединения формулы I вводят в сочетании с другими иммуносупрессивными/иммуномодуляторными, противовоспалительными или химиотерапевтическими лекарствами, дозы добавляемого иммуносупрессора, иммуномодулятора, противовоспалительного или химиотерапевтического соединения будут, конечно, варьироваться в зависимости от типа применяемого со-лекарства, например в зависимости от того, является ли оно стероидом или кальциневриновым ингибитором, от специфичности применяемого лекарства, от состояния, подвергаемого лечению и т.д.
В соответствии с вышеизложенным настоящее изобретение предлагает следующий аспект:
5. Способ, как описано выше, включающий совместное введение, например совместно или поочередно, терапевтически эффективного количества соединения формулы I и по крайней мере второго лекарственного средства, например иммуносупрессивного, иммуномодулирующего, противовоспалительного, противоинфекционного или химиотерапевтического лекарства, например, как указано выше.
6. Фармацевтическая комбинация, например набор, содержащий а) первый агент, который означает антагонист CXCR3, например соединение формулы I, как здесь описано, в свободной форме или в форме фармацевтически приемлемой соли и b) по крайней мере один со-агент, например иммуносупрессивное, иммуномодулирующее, антивоспалительное, противоинфекционное или химиотерапевтическое лекарство. Набор может содержать инструкции по введению.
Термины «со-введение» или «комбинированное введение» или подобное, как здесь используется, означает введение выбранных терапевтических агентов одному пациенту и предполагает использование режимов лечения, при которых агенты необязательно вводят одним и тем же образом или в то же самое время.
Термин «фармацевтическая комбинация», как здесь используется, означает продукт, который получается от смешения или комбинирования более чем одного активного ингредиента и включает как фиксированную, так и нефиксированную комбинации активных ингредиентов. Термин «фиксированная комбинация» означает, что активные ингредиенты, например соединение формулы I и со-агент, вводятся пациенту одновременно в форме единой основы или дозы. Термин «нефиксированная комбинация» означает, что активные ингредиенты, например соединение формулы I и со-агент, вводятся пациенту как отдельные основы одновременно, конкурентно или последовательно без специфических ограничений времени, при этом такое введение обеспечивает терапевтически эффективные уровни 2 соединений в организме пациента. Это также относится к коктейлевой терапии, например введению 3 или более активных ингредиентов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ ИНФЕКЦИИ | 2021 |
|
RU2780103C1 |
| ПРОИЗВОДНЫЕ β-КАРБОЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1999 |
|
RU2210571C2 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ-ИНФЕКЦИИ | 2020 |
|
RU2780101C2 |
| PROTAC, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА ТАУ-БЕЛОК, И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2805523C2 |
| Способ получения производных изохинолина или их солей | 1979 |
|
SU1072808A3 |
| ЗАМЕЩЕННЫЕ N-ФЕНИЛПИРРОЛИДИНИЛМЕТИЛПИРРОЛИДИНАМИДЫ И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА Н3 ГИСТАМИНА | 2008 |
|
RU2477721C2 |
| Соединения, активные по отношению к бромодоменам | 2015 |
|
RU2743074C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ СРЕДСТВА ПРОТИВ ВИРУСОВ СЕМЕЙСТВА FLAVIVIRIDAE | 2004 |
|
RU2355687C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ КАТЕПСИНА S | 2001 |
|
RU2278863C2 |
| Способ получения производных пиридо (1,2-а) пиримидина или их фармацевтически приемлемых солей или их оптически активных изомеров | 1978 |
|
SU999973A3 |
Описаны производные эрголина формулы (I), в которой каждый R1 и R2 независимо друг от друга означают Н; необязательно R10 и/или R11-замещенный фенил или -фенилС1-С4алкил; необязательно R10 и/или R11-замещенный гетероарил или -гетероарилС1-С4алкил; необязательно R10 и/или R11-замещенный N-оксид гетероарила; необязательно R10-замещенный С1-С8алкил; необязательно R10-замещенный С2-С8алкенил; необязательно R10-замещенный С2-С8алкинил; необязательно R10-замещенный С3-С8циклоалкил, или необязательно R10-замещенный С4-С8 циклоалкенил; или r1 и r2 образуют вместе с атомом азота, к которому они присоединены, необязательно R10-замещенное 3-8-членное кольцо, содержащее в дополнение к атому азота до 2 гетероатомов, независимо выбранных из группы, состоящей из N, О и S; R3 означает Н; OR1; CH2R1R2; (CH2)1-2NR1R2; CH2-CH2-OR1; CH2-CO-NR1R2; или CO-CH2R1R2; R4 означает F; Cl; Br; I; OR1; NR1R2 или имеет одно из значений, приведенных для R1; и R5 имеет одно из значений, приведенных для R1, в свободной форме или в форме соли для профилактики или лечения нарушений или заболеваний, опосредованных взаимодействиями между хемокинными рецепторами и их лигандами. 3 н. и 11 з.п.ф-лы, 8 табл.

1. Соединение формулы I
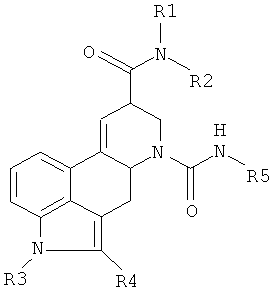
где каждый из R1 и R2 независимо друг от друга означают Н;
необязательно R10-замещенный С1-С8алкил; или С2-С8алкенил;
или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют необязательно R10-замещенное 3-6-членное кольцо, необязательно содержащее в дополнение к атому азота не более еще одного гетероатома, независимо выбранного из N, О и S, где указанное кольцо является насыщенным или содержит двойную связь;
или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют фрагмент формулы

 или
или 
R3 означает Н; ОН; OR9; СН2-фенил; (CH2)1-2NRR'; CH2-CO-NRR' где R и R' независимо представляют собой С1-С8алкил, или R и R' вместе с атомом азота, к которому они присоединены, образуют необязательно R10-замещенное 5-6-членное насыщенное или ненасыщенное кольцо, необязательно содержащее в дополнение к атому азота еще один гетероатом, независимо выбранный из N и О; СОСН3; C1-С6алкил; гидрокси-С1-С6алкил;
R4 означает F; Сl; Вr; I; Н; необязательно R10-замещенный фенил;
необязательно R10-замещенный гетероарил, где гетероарил представляет собой 6-членное ароматическое кольцо, содержащее гетероатом N; необязательно R10-замещенный С2-С8алкинил; C1-С6алкил;
R5 означает Н; необязательно R10-замещенный фенил или фенилС1-С4алкил; необязательно R10-замещенный гетероарил или гетероарилС1-С4алкил, где гетероарил означает фурил, тиенил, пирролил или пиридил; необязательно R10-замещенный С1-С8алкил; необязательно R10-замещенный С3-С8циклоалкил;
где R10 представляет собой 1-4 заместителя, независимо выбранных из группы, включающей C1-С6алкил; гидрокси-С1-С6алкил; C1-C6-(алкоксиалкил); галоген-С1-С6алкил; фенил; F; Сl; Вr; I; ОН; ОR9; СОR9; CONH2; CONHR9; CONHR9R9; СF3; CHF2; С1-С4алкил-NН2; С1-С4алкил-NHR9; C1-C4 алкил-NR9R9; CN; NR9R9; SO2R9; SO2NH2; и
каждый R9 независимо означает C1-С6алкил; гидрокси-С1-С6алкил;
или соединение, выбранное из:
(6аR,9R)-5-(3-гидроксипроп-1-инил)-9-(пирролидин-1-карбонил)-6,6а,8,9-тeтpaгидpo-4H-индoлo[4,3-fg]xинoлин-7-кapбoнoвoй кислоты, и
изопропилового эфира [(6аR,9R)-7-фенилкарбамоил-9-(пирролидин-1-карбонил)-6а,7,8,9-тетрагидро-6Н-индоло[4,3-fg]хинолин-4-ил]уксусной кислоты,
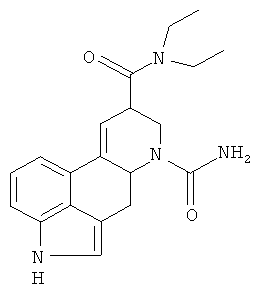
в свободной форме или в форме фармацевтически приемлемой соли, за исключением соединения формулы
2. Соединение по п.1 формулы

где остаток -NR1R2 имеет формулу
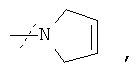
или его фармацевтически приемлемая соль.
3. Соединение по п.1 формулы
и где остаток -NR1R2 имеет формулу

или его фармацевтически приемлемая соль.
4. Соединение по п.1 формулы
где остаток -NR1R2 имеет формулу
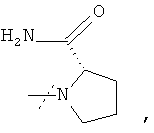
или его фармацевтически приемлемая соль.
5. Соединение по п.1 формулы
где остаток -NR1R2 имеет формулу

или его фармацевтически приемлемая соль.
6. Соединение по п.1 формулы
где остаток -NR1R2 имеет формулу
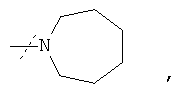
или его фармацевтически приемлемая соль.
7. Соединение по п.1 формулы

где остаток R5 имеет формулу

или его фармацевтически приемлемая соль.
8. Соединение по п.1 формулы
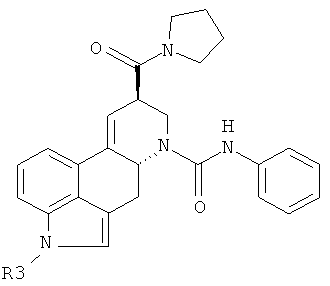
где остаток R3 имеет формулу
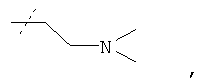
или его фармацевтически приемлемая соль.
9. Соединение по п.1 формулы
где остаток R3 имеет формулу
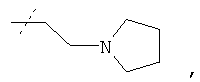
или его фармацевтически приемлемая соль.
10. Соединение по п.1 формулы
где остаток R3 имеет формулу

или его фармацевтически приемлемая соль.
11. Соединение по п.1 формулы
где остаток R3 имеет формулу

или его фармацевтически приемлемая соль.
12. Соединение по любому из пп.1-11 или его фармацевтически приемлемая соль для применения в качестве антагониста для лечения заболеваний, опосредованных хемокинным рецептором CXCR3.
13. Фармацевтическая композиция, для лечения заболеваний, опосредованных хемокинным рецептором CXCR3, включающая антагонист CXCR3 - соединение формулы I по любому из пп.1-11 или его фармацевтически приемлемую соль, вместе с одним или более фармацевтически приемлемыми разбавителями или носителями.
14. Применение соединения формулы I по любому из пп.1-11 или его фармацевтически приемлемой соли в качестве антагониста для приготовления лекарственного средства, предназначенного для лечения нарушений или заболеваний, опосредованных хемокинным рецептором CXCR3.
| NAKAHARA Y | |||
| et al | |||
| // Chemical and Pharm | |||
| Bull | |||
| Способ изготовления электрических сопротивлений посредством осаждения слоя проводника на поверхности изолятора | 1921 |
|
SU19A1 |
| WO 00/42071 A2, 20.07.2000. | |||

