Настоящая заявка претендует на приоритет патентной заявки США Serial No. 60/428324, поданной 21 ноября 2002, которая полностью включена в данную работу.
Данное изобретение относится к производным 3-аминопиперидина, их промежуточным продуктам и способам получения.
Производные пирроло[2.3-d]пиримидина представляют собой ингибиторы протеинкиназ, таких как фермент Janus-киназа 3 (JAK3), и поэтому применяются в терапии как иммуносупрессанты для органов-трансплантатов, ксенотрансплантации, в терапии волчанки, рассеянного склероза, ревматоидного артрита, псориаза, диабета типа I и осложнений от диабета, рака, астмы, атопического дерматита, аутоиммунных расстройств щитовидной железы, язвенного колита, болезни Крона, болезни Альцгеймера, лейкемии и других показаний, где иммуносупрессия была бы желательной. Производные пирроло[2.3-d]пиримидина, фармацевтические композиции на их основе и способы их применения описаны в заявке Serial No. 09/732669, находящейся в процессе одновременного рассмотрения и поданной 8 декабря 2000 г., которая включена в данной работе в виде ссылок по всем позициям.
Как раскрыто в описании, данное изобретение в одном из аспектов относится к способам получения соединений формулы (Ia)
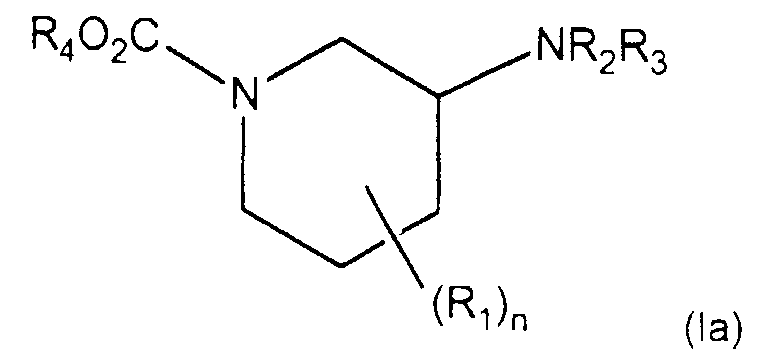
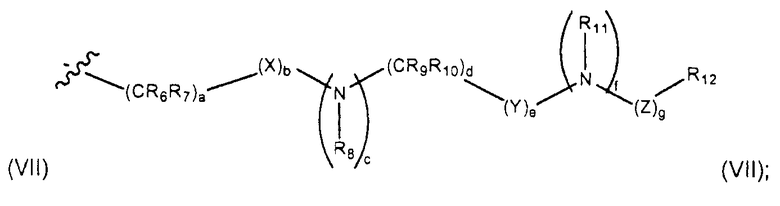
где R1 представляет собой карбокси, циано, дейтерий, (С1-С6)алкил, (С1-С6)алкокси, (С1-С6)ацил, (С1-С6)алкиламино, амино(С1-С6)алкил, (С1-С6)алкокси-СО-NH, (С1-С6)алкиламино-CO, (С2-С6)алкенил, (С2-С6)алкинил, (С1-С6)алкокси(С1-С6)алкил, (С1-С6)ацилокси(С1-С6)алкил, нитро, циано(С1-С6)алкил, нитро(С1-С6)алкил, трифторметил, трифторметил(С1-С6)алкил, (С1-С6)ациламино, (С1-С6)ациламино(С1-С6)алкил, (С1-С6)алкокси(С1-С6)ациламино, амино(С1-С6)ацил, амино(С1-С6)ацил(С1-С6)алкил, (С1-С6)алкиламино(С1-С6)ацил, ((С1-С6)алкил)2амино(С1-С6)ацил, R15R16N-CO-O-, R15R16N-CO-(С1-С6)алкил, (С1-С6)алкил-S(O)m, R15R16NS(O)m, R15R16NS(O)m(С1-С6)алкил, R15S(O)mR16N, R15S(O)mR16N(С1-С6)алкил или группу формулы (VII)
R2 представляет собой водород, (С1-С6)алкил, (С1-С6)алкилсульфонил, (С2-С6)алкенил или (С2-С6)алкинил, где алкил, алкенил и алкинил-группы необязательно замещены дейтерием, гидрокси, трифторметилом, (С1-С4)алкокси, (С1-С6)ацилокси, (С1-С6)алкиламино, ((С1-С6)алкил)2амино, циано, нитро, (С2-С6)алкенилом, (С2-С6)алкинилом или (С1-С6)ациламино; или R2 представляет собой (С3-С10)циклоалкил, где циклоалкильная группа необязательно замещена дейтерием, гидрокси, трифторметилом, (С1-С6)ацилокси, (С1-С6)ациламино, (С1-С6)алкиламино, ((С1-С6)алкил)2амино, циано, циано(С1-С6)алкилом, трифторметил(С1-С6)алкилои, нитро, нитро(С1-С6)алкилом или (С1-С6)ациламино;
R3 представляет собой водород, (С1-С6)алкил, (С3-С10)циклоалкил, (С2-С6)алкенил или (С2-С6)алкинил, где алкил, алкенил и алкинил-группы необязательно замещены дейтерием, гидрокси, галогеном, трифторметилом, (С1-С4)алкокси, (С1-С6)ацилокси, (С1-С6)алкиламино, (С1-С6)ациламино, ((С1-С6)алкил)2амино, (С2-С6)алкенилом, (С2-С6)алкинилом, циано, циано(С1-С6)алкилом, трифторметил(С1-С6)алкилом, нитро или нитро(С1-С6)алкилом;
R4 представляет собой (С1-С6)алкил, (С3-С10)циклоалкил, (С2-С6)алкенил или (С2-С6)алкинил, где алкил, алкенил и алкинил-группы необязательно замещены дейтерием, гидрокси, галогеном, амино, трифторметилом, (С1-С4)алкокси, (С1-С6)ацилокси, (С1-С6)алкиламино, (С1-С6)ациламино, ((С1-С6)алкил)2амино, (С2-С6)алкенилом, (С2-С6)алкинилом, циано, циано(С1-С6)алкилом, трифторметил(С1-С6)алкилом, нитро или нитро(С1-С6)алкилом;
R6, R7, R8, R9, R10 и R11 каждый независимо представляет собой водород или (С1-С6)алкил, необязательно замещенный дейтерием, гидрокси, трифторметилом, (С1-С4)ацилокси, (С1-С6)ациламино, (С1-С6)алкиламино, ((С1-С6)алкил)2амино, циано, циано(С1-С6)алкилом, трифторметил(С1-С6)алкилом, нитро, нитро(С1-С6)алкилом; R12 представляет собой карбокси, циано, амино, оксо, дейтерий, гидрокси, трифторметил, (С1-С6)алкил, трифторметил(С1-С6)алкил, (С1-С6)алкокси, (С1-С6)ацил, (С1-С6)алкиламино, ((С1-С6)алкил)2амино, амино(С1-С6)алкил, (С1-С6)алкокси-СО-NH, (С1-С6)алкиламино-СО-, (С2-С6)алкенил, (С2-С6)алкинил, гидрокси(С1-С6)алкил, (С1-С6)алкокси(С1-С6)алкил, (С1-С6)ацилокси(С1-С6)алкил, нитро, циано(С1-С6)алкил, нитро(С1-С6)алкил, (С1-С6)ациламино, (С1-С6)ациламино(С1-С6)алкил, (С1-С6)алкокси(С1-С6)ациламино, амино(С1-С6)ацил, амино(С1-С6)ацил(С1-С6)алкил, (С1-С6)алкиламино(С1-С6)ацил, ((С1-С6)алкил)2амино(С1-С6)ацил, R15R16N-CO-O, R15R16N-CO-(С1-С6)алкил, R15C(О)NH, R15OC(О)NH, R15NHC(О)NH, (С1-С6)алкил- S(O)m, (С1-С6)алкил-S(O)m-(С1-С6)алкил, R15R16NS(O)m, R15R16NS(O)m(С1-С6)алкил, R15S(O)mR16N или R15S(O)mR16N(С1-С6)алкил;
R15 и R16 каждый независимо представляет собой водород или (С1-С6)алкил;
Х представляет собой S(O)р, кислород, карбонил или -С(=N-циано)-;
Y представляет собой S(O)р или карбонил;
Z представляет собой S(O)р, карбонил, C(O)O- или C(O)NR-;
а принимает значение 0, 1, 2, 3 или 4;
b, c, e, f и g каждый независимо принимает значение 0 или 1;
d принимает значение 0, 1, 2 или 3;
m принимает значение 0, 1 или 2;
n принимает значение 1, 2, 3 или 4;
p принимает значение 0, 1 или 2;
где данный способ включает взаимодействие NHR2R3, N(CH3)R2H или N(CH2CH3)R2H с соединением формулы (IIa)
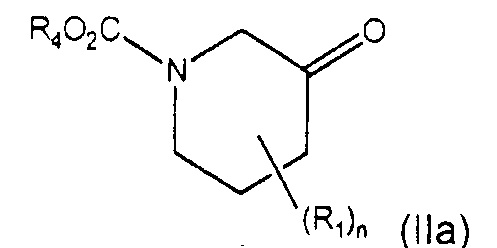
и восстановление соединения, полученного таким образом, с помощью восстановителя. В одном из вариантов осуществления восстановитель представляет собой борогидрид.
Кроме того, настоящее изобретение относится к получению соединения формулы (IIa) взаимодействием соединения формулы R4OH, воды или R4NH2 и соединения формулы (IIIa)
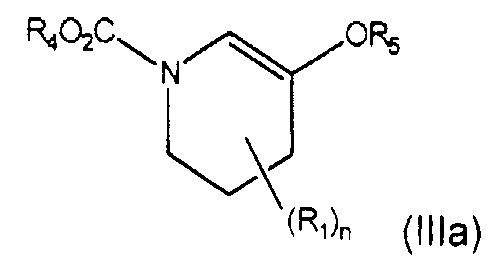
в которой R5 представляет собой CO(С1-С6)алкил.
Настоящее изобретение дополнительно относится к получению соединения формулы (IIIa) нагреванием соединения формулы (IVa)
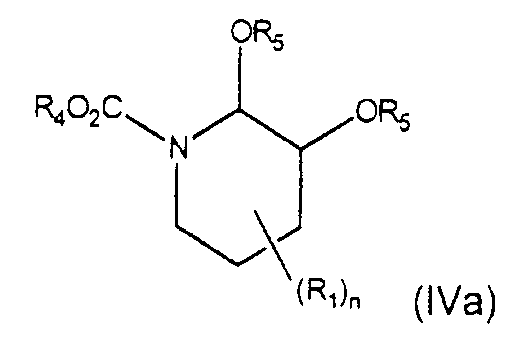
с соединением формулы (С1-С6)алкил-(С=О)-О-(С=О)-(С1-С6)алкил.
Дополнительно настоящее изобретение относится к получению соединения формулы (IVa) окислением соединения формулы (Va)
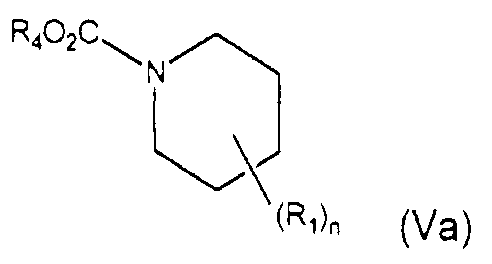
в соответствующих условиях для окисления. В одном из вариантов осуществления изобретения окисление проводят в условиях электрохимического окисления.
Настоящее изобретение также относится к получению соединения формулы (Va) взаимодействием соединения формулы WCO2R4 и соединения формулы (VIa)
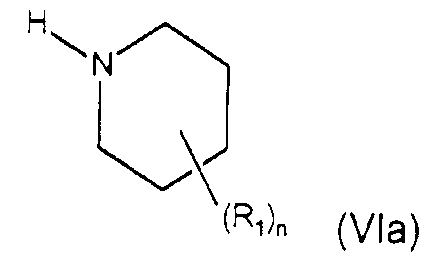
где W представляет собой галоген.
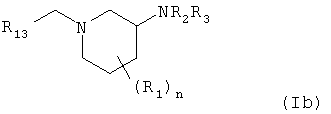
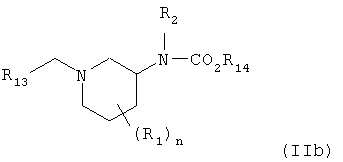
Второй аспект настоящего изобретения относится к способам получения соединения формулы (Ib)
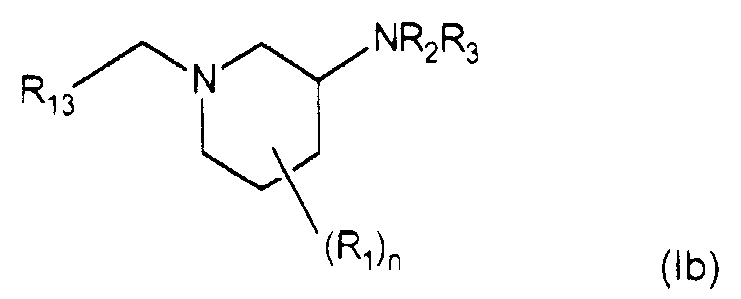
где R13 представляет собой (С2-С6)алкенил, (С2-С6)алкинил, (С6-С10)арил, (С1-С6)карбоалкокси, (С5-С9)гетероарил, (С6-С10)арил(С1-С6)алкил или (С5-С9)гетероарил(С1-С6)алкил, причем R13-группа необязательно замещена дейтерием, гидрокси, амино, трифторметилом, (С1-С6)алкилом, (С1-С4)алкокси, (С1-С6)алкиламино, ((С1-С6)алкил)2амино, (С2-С6)алкенилом, (С2-С6)алкинилом, трифторметил(С1-С6)алкилом, нитро или нитро(С1-С6)алкилом; и
где данный способ включает восстановление соединения формулы (IIb) восстановителем
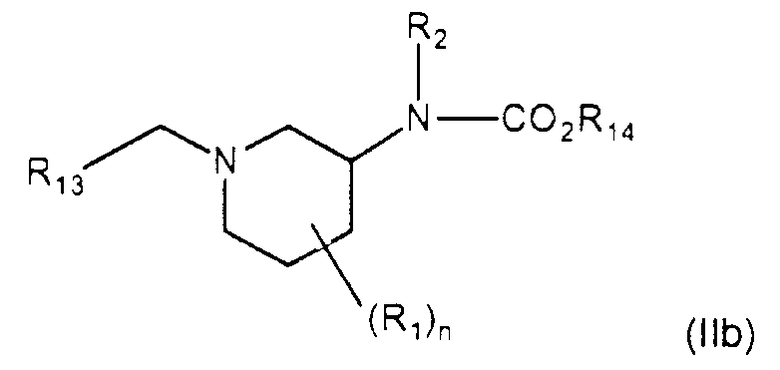
где R14 представляет собой (С1-С6)алкил, (С3-С10)циклоалкил, (С2-С6)алкенил или (С2-С6)алкинил, где алкил, алкенил и алкинил-группы необязательно замещены дейтерием, гидрокси, галогеном, амино, трифторметилом, (С1-С4)алкокси, (С1-С6)алкиламино, ((С1-С6)алкил)2амино, (С2-С6)алкенилом, (С2-С6)алкинилом, трифторметил(С1-С6)алкилом, нитро или нитро(С1-С6)алкилом.
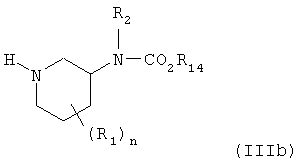
Настоящее изобретение также относится к получению соединения формулы (IIb) взаимодействием соединения формулы (IIIb)
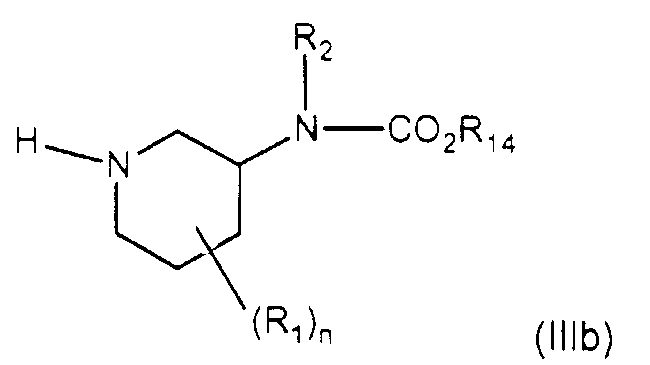
с альдегидом формулы R13-(C=O)-H и восстановлением соединения, полученного таким образом, восстановителем. В одном из вариантов осуществления восстановитель представляет собой литийалюминийгидрид.
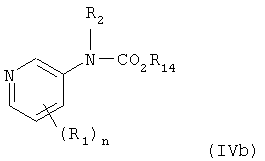
Кроме того, настоящее изобретение также относится к получению соединения формулы (IIIb) гидрированием соединения формулы (IVb)
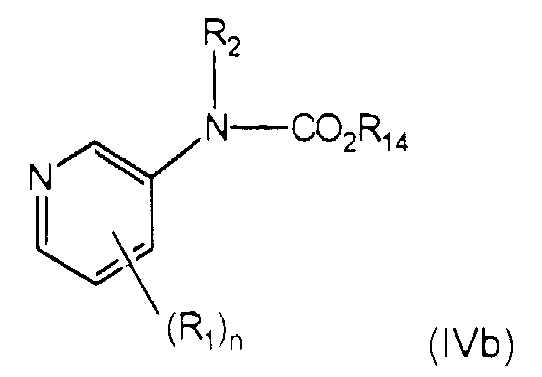
в присутствии катализатора. В одном из вариантов осуществления катализатор представляет собой Rh/Al2O3 или Rh/C.
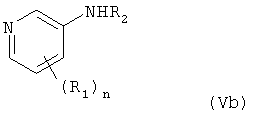
Настоящее изобретение также относится к получению соединения формулы (IVb) взаимодействием соединения формулы (Vb)
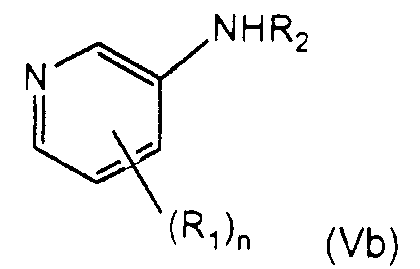
с (R14-O-(C=O))2O или R14-O-(C=O)-Х, где Х представляет собой галоген.
Кроме того, в дополнительных аспектах настоящее изобретение относится к соединениям, описанным здесь, включая соединения формул (Ia), (Ib) и (IIb).
В некоторых предпочтительных вариантах осуществления способов и соединений в соответствии с вышеупомянутыми аспектами настоящего изобретения R1 представляет собой (С1-С6)алкил и n принимает значение, равное единице; R2 и R3, каждый, представляет собой водород или (С1-С6)алкил; R4 представляет собой (С1-С6)алкил; и/или R13 представляет собой (С6-С10)арил.
В одном из вариантов осуществления соединение формулы (Ia) соответствует стереохимической формуле (Ia-1)
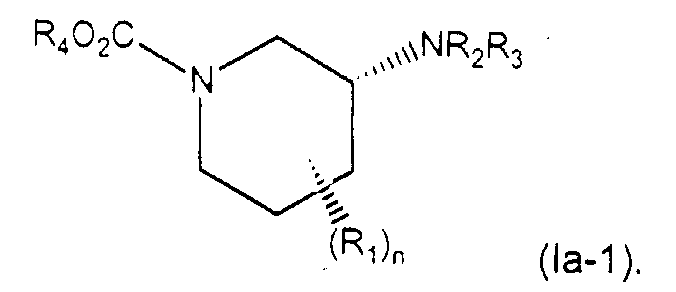
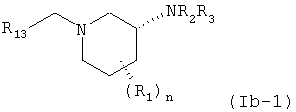
В другом варианте осуществления соединение формулы (Ib) соответствует стереохимической формуле (Ib-1)
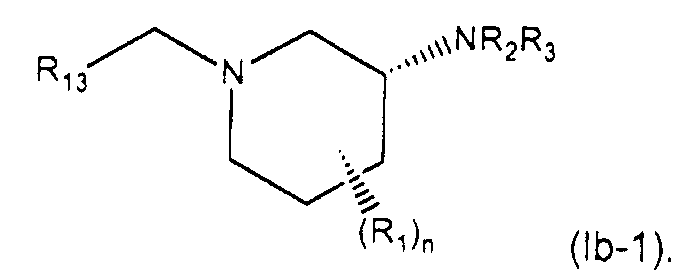
Приведенное выше описание и последующее подробное описание являются только иллюстративными и пояснительными и не ограничивают заявленное изобретение.
Настоящее изобретение может быть более легко понято отсылкой к следующему подробному описанию пояснительных вариантов осуществления изобретения и представленным примерам.
Данное изобретение не ограничено специфическими синтетическими способами получения, которые, конечно, могут различаться. Также терминология, используемая в данном изобретении, предназначена только для описания частных вариантов осуществления и не ограничивает объем изобретения.
В данном описании и в пунктах формулы изобретения используют термины, которые следует трактовать в соответствии со следующими значениями.
Если не оговорено особо, то алкильные и алкенильные группы, упоминаемые в описании, так же, как и алкильные фрагменты других групп, упомянутых в описании (например, алкокси), могут быть линейными или разветвленными, и они могут быть также циклическими (например, циклопропил, циклобутил, циклопентил, циклогексил или циклогептил) или быть линейными или разветвленными и содержать циклические фрагменты. Такие алкил- и алкокси-группы могут быть замещены одним, двумя или тремя атомами галогена и/или гидрокси, предпочтительно атомами фтора.
Если не оговорено особо, то галоген и галоид включают фтор, хлор, бром и йод.
Термин "(C3-C10)циклоалкил", используемый в данном изобретении, обозначает циклоалкильные группы, содержащие от нуля до двух степеней ненасыщенности, такие как циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, 1,3-циклогексадиен, циклогептил, циклогептенил, бицикло[3.2.1}октан, норборнанил и т.п.
Термин "(C2-C9)гетероциклоалкил", используемый в данном изобретении, обозначает пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидропиранил, пиранил, тиопиранил, азиридинил, оксиранил, метилендиоксил, хроменил, изоксазолидинил, 1,3-оксазолидин-3-ил, изотиазолидинил, 1,3-тиазолидин-3-ил, 1,2-пиразолидин-2-ил, 1,3-пиразолидин-1-ил, пиперидинил, тиоморфолинил, 1,2-тетрагидротиазин-2-ил, 1,3-тетрагидротиазин-3-ил, тетрагидротиадиазинил, морфолинил, 1,2-тетрагидродиазин-2-ил, 1,3-тетрагидродиазин-1-ил, тетрагидроазепинил, пиперазинил, хроманил и т.п. Специалисту в данной области будет ясно, что соединение указанных (C2-C9)гетероциклоалкильных колец осуществляется через углерод или sp3-гибридизованный гетероатом азота.
Термин "(C2-C9)гетероарил", используемый в данном изобретении, обозначает фурил, тиенил, тиазолил, пиразолил, изотиазолил, оксазолил, изоксазолил, пирролил, триазолил, тетразолил, имидазолил, 1,3,5-оксадиазолил, 1,2,4-оксадиазолил, 1,2,3-оксадиазолил, 1,3,5-тиадиазолил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, пиридил, пиримидил, пиразинил, пиридазинил, 1,2,4-триазинил, 1,2,3-триазинил, 1,3,5-триазинил, пиразоло[3.4-b]пиридинил, циннолинил, птеридинил, пуринил, 6,7-дигидро-5Н-[1]пириндинил, бензо[b]тиофенил, 5,6,7,8-тетрагидрохинолин-3-ил, бензоксазолил, бензотиазолил, бензизотиазолил, бензизоксазолил, бензимидазолил, тианафтенил, изотианафтенил, бензофуранил, изобензофуранил, изоиндолил, индолил, индолизинил, индазолил, изохинолил, хинолил, фталазинил, хиноксалинил, хиназолинил, бензоксазинил и т.п. Специалисту в данной области будет ясно, что соединение указанных (C2-C9)гетероарильных колец осуществляется через углерод или sp3-гибридизованный гетероатом азота.
Термин "арил", используемый в данном изобретении, обозначает фенил или нафтил.
Термином "фармацевтически приемлемый" обозначен продукт, который является подходящим биологически или по другим параметрам, т.е. продукт может быть введен индивидууму вместе с выбранным соединением, причем он не вызывает никаких нежелательных биологических эффектов или не взаимодействует с любыми другими компонентами фармацевтической композиции, в которой он содержится, разрушая их.
Термином "субъект" обозначен индивидуум. Предпочтительно субъект представляет собой млекопитающее, такое как примат, и более предпочтительно человек. Таким образом, "субъект" может включать одомашненных животных, скот и лабораторных животных.
Обычно "эффективное количество" или "эффективная доза" обозначает количество, необходимое для достижения желаемого результата или результатов (лечение или профилактика состояния). Специалист в данной области будет представлять, что эффективность и поэтому "эффективное количество" могут меняться для различных соединений, примененных в данном изобретении. Высококвалифицированный специалист в данной области может легко оценить эффективность данных соединений.
Если не оговорено особо, то числовые значения величин в данном описании и в формуле изобретения являются приблизительными. Изменения внутри величин могут быть связаны с калиброванием оборудования, ошибками оборудования, чистотой продуктов наряду с другими факторами. Кроме того, вариации могут быть возможны даже при получении одного и того же результата.
Следующие схемы реакций иллюстрируют получение соединений настоящего изобретения. Если не оговорено особо, то заместители в схемах реакций и в обсуждении, которые следуют, принимают значения, определенные выше.
Схема 1
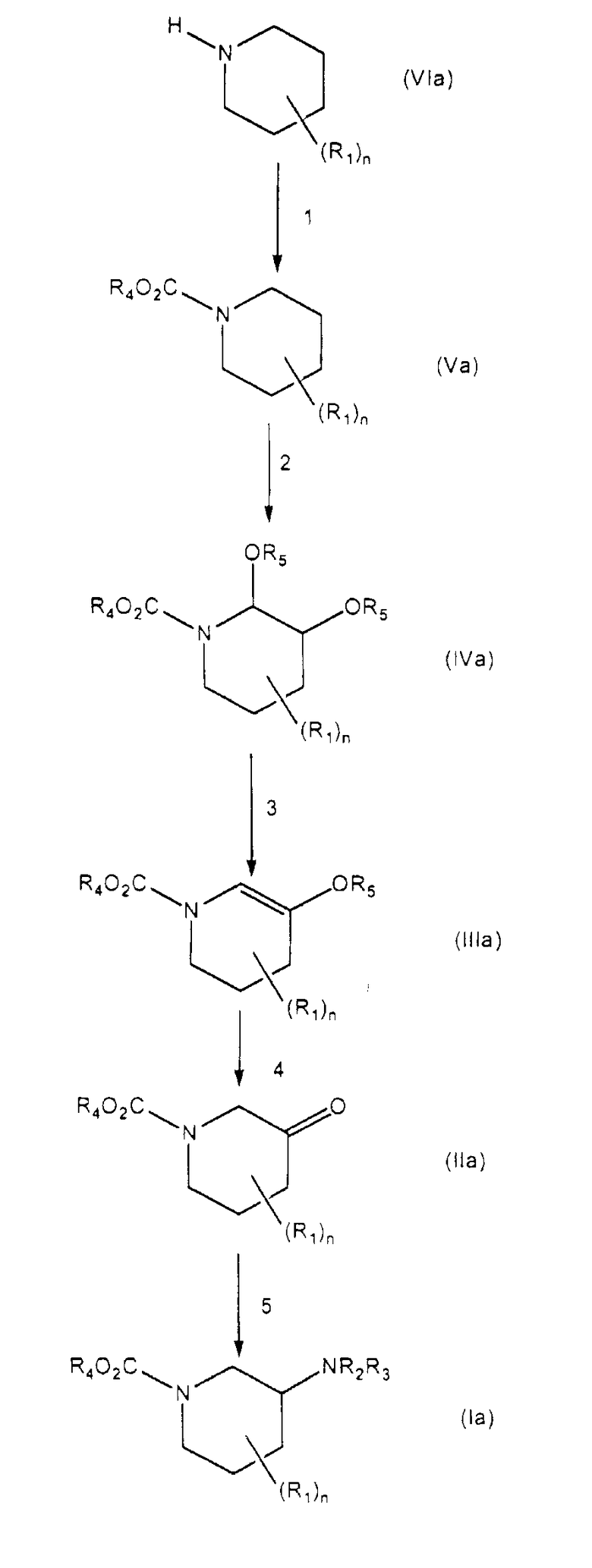
Схема 2
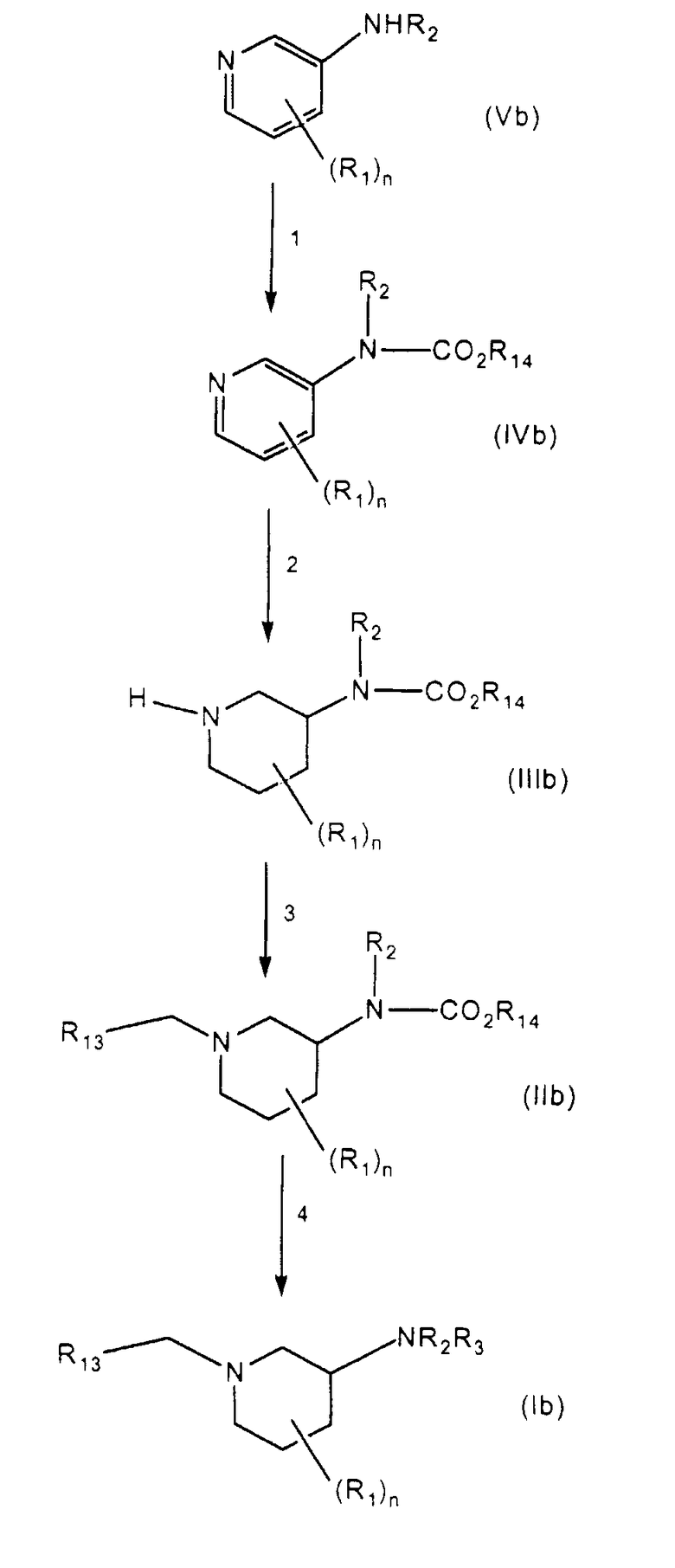
Схема 3
На стадии 1 схемы 1 соединение формулы (VIa) превращают в карбамат формулы (Va) взаимодействием с соединением формулы W-CO2R4 в системе растворителя. Система растворителя предпочтительно содержит амин, такой как триэтиламин, диизопропилэтиламин или другой третичный амин. Другие растворители могут быть также использованы, включая дихлорметан, тетрагидрофуран и метилтетрагидрофуран. Смесь затем охлаждают приблизительно до температуры от -80° до 25°C и медленно добавляют W-CO2R4 c контролем температуры. Смесь перемешивают в течение, по меньшей мере, одного часа, предпочтительно четырех и более часов.
На стадии 2 схемы 1 карбамат формулы (Va) окисляют для образования продукта окисления (IVa). Обычно реакция окисления приводит к смеси соединений, соответствующих формуле (IVa). Могут быть использованы любые подходящие условия окисления. Предпочтительные условия включают электрохимическое окисление, такое как проведение реакции окисления в растворе электролита в химическом источнике тока и проведение электролиза в источнике. В одном из вариантов осуществления электролитический раствор представляет собой смесь уксусной кислоты и ацетата калия. В другом варианте осуществления электролитический раствор включает уксусный ангидрид. Катод и анод могут быть сделаны из любого подходящего материала, включающего платину и ниобий. Затем смесь подвергают электролизу при соответствующем течении процесса до полного завершения реакции. Температура электролитического раствора может быть установлена при значениях ниже 60°C, предпочтительно ниже 40°С.
Продукт окисления (IVa) на стадии 3 схемы 1 нагревают с соединением формулы (С1-С6)алкил-(С=О)-О-(С=О)-(С1-С6)алкил, включающим уксусный ангидрид, для получения енаминоацетата формулы (IIIa). Температуру предпочтительно устанавливают примерно от 60 до примерно 160°C; более предпочтительно температуру поднимают до примерно >100°C, более предпочтительно до примерно >120°С. Смесь перемешивают в течение, по меньшей мере, двух часов, предпочтительно четырех и более часов.
На стадии 4 схемы 1 енаминоацетат (IIIa) превращают в кетопиперидин (IIa) взаимодействием с соединением формулы R4OH, водой или R4NH2. Предпочтительно данную реакцию проводят при температуре ниже 20°C, более предпочтительно ниже 5°С.
На стадии 5 схемы 1 кетопиперидин (IIa) превращают в аминопиперидин (Ia) взаимодействием с NHR2R3, N(CH3)R2R3 или N(CH2CH3)R2R3. Продукт реакции восстанавливают восстановителем для образования аминопиперидина (Ia). Реакции обычно проводят в растворителе, таком как метанол, при температуре окружающей среды в течение периода примерно от 12 часов до примерно 18 часов. Подходящие восстановители включают борогидриды, такие как цианоборогидрид натрия и борогидрид натрия.
На стадии 1 схемы 2 карбамат (IVb) получают взаимодействием соединения формулы (Vb) с соединением формулы (R14-O-(C=O))2O или R14-O-(C=O)-X, где Х представляет собой галоген. Предпочтительно температуру устанавливают ниже 0°С. Реакция в основном заканчивается в течение минут, обычно, по меньшей мере, в течение часа.
На стадии 2 схемы 2 карбамат (IVb) гидрируют для образования соединения формулы (IIIb). Гидрирование может также приводить к цис-транс-изомерам. Желательно применять некоторые катализаторы для обеспечения цис:транс селективности. Подходящие катализаторы гидрирования включают PtO2, Rh/C (несколько типов), RuO2, Rh/Al2O3, Ru/C (несколько типов), катализатор Линдлара (Lindlar's) и катализатор Вилькинсона (Wilkinson's). Подходящие растворители включают уксусную кислоту, пропанол, этанол, метанол/гидроксид аммония, ацетонитрил, тетрагидрофуран, циклогексан, гептаны, толуол, диметилформамид, воду. Обычно температуру реакции устанавливают выше комнатной температуры, предпочтительно выше 60°C, и давление увеличивают выше атмосферного давления с помощью газообразного водорода.
На стадии 3 схемы 2 соединение формулы (IIIb) подвергают взаимодействию с альдегидом формулы R13-(C=O)-H и восстанавливают восстановителем для образования соединения (IIb). Реакции обычно проводят в растворителе, таком как метанол, при температуре окружающей среды в течение периода примерно от 12 часов до примерно 18 часов. Подходящие восстановители включают борогидриды, такие как цианоборогидрид натрия и борогидрид натрия. В одном из вариантов осуществления восстановитель представляет собой триацетоксиборогидрид.
Наконец, на стадии 4 схемы 2 соединение формулы (IIb) восстанавливают до соединения формулы (Ib). Подходящие восстановители включают литийалюминийгидрид, витрид (красный-А)/(Vitride(Red-A)) и боран. Восстановление выполняют в растворителе, таком как тетрагидрофуран, диэтиловый эфир или метилтетрагидрофуран, предпочтительно при температурах примерно от -10 до примерно 100°C в течение примерно от 5 минут до примерно 48 часов.
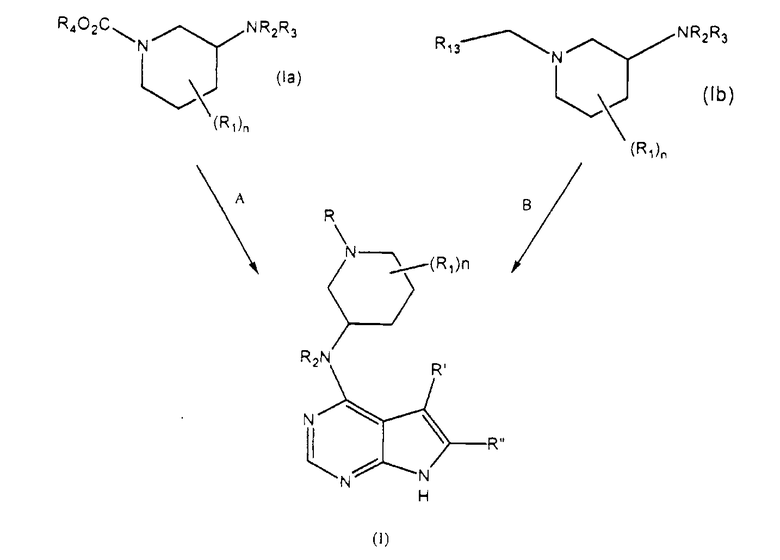
В реакциях A и B схемы 3 соединение формулы (Ia) или (Ib) соответствующим образом конденсируют с замещенным 4-хлор-пирроло[2.3-d]пиримидином формулы (А):
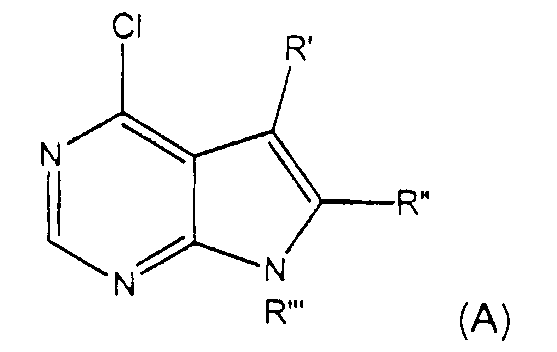
где R' и R′′ каждый независимо выбран из группы, включающей водород, дейтерий, амино, галоген, гидрокси, нитро, карбокси, (С2-С6)алкенил, (С2-С6)алкинил, трифторметил, трифторметокси, (С1-С6)алкил, (С1-С6)алкокси, (С3-С10)циклоалкил, где алкил, алкокси или циклоалкил-группы необязательно замещены одной-тремя группами, выбранными из галогена, гидрокси, карбокси, амино(С1-С6)алкилтио, (С1-С6)алкиламина, ((С1-С6)алкил)2амино, (С5-С9)гетероарила, (С2-С9)гетероциклоалкила, (С3-С9)циклоалкила или (С6-С10)арила; или R2 и R3 каждый независимо представляет собой (С3-С10)циклоалкил, (С3-С10)циклоалкокси, (С1-С6)алкиламино, ((С1-С6)алкил)2амино, (С6-С10)ариламино, (С1-С6)алкилтио, (С6-С10)арилтио, (С1-С6)алкилсульфинил, (С6-С10)арилсульфинил, (С1-С6)алкилсульфонил, (С6-С10)арилсульфонил, (С1-С6)ацил, (С1-С6)алкокси-CO-NH-, (С1-С6)алкиламино-СО-, (С5-С9)гетероарил, (С2-С9)гетероциклоалкил или (С6-С10)арил, где гетероарил, гетероциклоалкил и арил-группы необязательно содержат от одного до трех заместителей, таких как галоген, (С1-С6)алкил, (С1-С6)алкил-СО-NH-, (С1-С6)алкокси-CO-NH, (С1-С6)алкил-CO-NH-(С1-С6)алкил, (С1-С6)алкокси-CO-NH-(С1-С6)алкил, (С1-С6)алкокси-СО-NH-(С1-С6)алкокси, карбокси, карбокси(С1-С6)алкил, карбокси(С1-С6)алкокси, бензилоксикарбонил(С1-С6)алкокси, (С1-С6)алкоксикарбонил(С1-С6)алкокси, (С6-С10)арил, амино, амино(С1-С6)алкил, (С1-С6)алкоксикарбониламино, (С6-С10)арил(С1-С6)алкоксикарбониламино, (С1-С6)алкиламино, ((С1-С6)алкил)2амино, (С1-С6)алкиламино(С1-С6)алкил, ((С1-С6)алкил)2амино(С1-С6)алкил, гидрокси, (С1-С6)алкокси, (С1-С6)алкоксикарбонил, (С1-С6)алкоксикарбонил(С1-С6)алкил, (С1-С6)алкокси-CO-NH-, (С1-С6)алкил-CO-NH-, циано, (С5-С9)гетероциклоалкил, амино-СО-NH-, (С1-С6)алкиламино-СО-NH, ((С1-С6)алкил)2амино-СО-NH-, (С6-С10)ариламино-СО-NH-, (С5-С9)гетероариламино-СО-NH-, (С1-С6)алкиламино-СО-NH-(С1-С6)алкил, ((С1-С6)алкил)2амино-CO-NH-(С1-С6)алкил, (С6-С10)ариламино-СО-NH-(С1-С6)алкил, (С5-С9)гетероариламино-СО-NH-(С1-С6)алкил, (С1-С6)алкилсульфонил, (С1-С6)алкилсульфониламино, (С1-С6)алкилсульфониламино(С1-С6)алкил, (С6-С10)арилсульфонил, (С6-С10)арилсульфониламино, (С6-С10)арилсульфониламино(С1-С6)алкил, (С5-С9)гетероарил и (С2-С9)гетероциклоалкил; и
R′′′ представляет собой водород или защитную группу;
для получения 4-аминопирроло[2.3-d]пиримидинового производного формулы (I), в которой R представляет собой R4-O-(C=O)- или R13-(CH2)-.
Реакцию сочетания проводят в спиртовом растворителе, таком как трет-бутанол, метанол или этанол, или в других высококипящих органических растворителях, таких как диметилформамид, триэтиламин, 1,4-диоксан или 1,2-дихлорэтан, при температуре в интервале примерно от 60°С до примерно 120°С, предпочтительно примерно при 80°С. Обычно длительность протекания реакции составляет примерно от 2 часов до примерно 48 часов, предпочтительно примерно 16 часов.
Если R′′′ представляет собой защитную группу, то защитная группа может быть удалена на дополнительной стадии. Например, удаление защитной группы, которая представляет собой бензолсульфонил, проводят нагреванием продукта реакции конденсации А или B со щелочным основанием, таким как гидроксид натрия или гидроксид калия, в спиртовом растворителе, таком как метанол или этанол, или в смеси растворителей, такой как спирт/тетрагидрофуран или спирт/вода. Реакцию проводят при комнатной температуре в течение примерно от 15 минут до примерно 1 часа, предпочтительно 30 минут. Удаление защитной группы, которая представляет собой бензильную группу, проводят посредством обработки продукта реакции присоединения А или B натрием в аммиаке при температуре примерно -78°C в течение примерно от 15 минут до примерно 1 часа.
Если не оговорено особо, то давление для каждой из вышеуказанных реакций не является важным. Обычно реакции проводят при давлении примерно от одной до примерно трех атмосфер, предпочтительно при давлении окружающей среды (примерно одна атмосфера).
Соединения формул (Ia) и (Ib) способны к образованию широкого спектра различных солей с различными неорганическими и органическими кислотами.
Соединения настоящего изобретения, которые по природе являются основными, способны к образованию широкого спектра различных солей с различными неорганическими и органическими кислотами.
Хотя такие соли должны быть фармацевтически приемлемыми для введения животным, на практике часто желательно соединение настоящего изобретения первоначально выделять из реакционной смеси в виде фармацевтически неприемлемой соли и затем последнюю просто превращать в свободное основание обработкой щелочным реагентом и затем превращать последнее свободное основание в фармацевтически приемлемую кислотно-аддитивную соль. Кислотно-основные соли основных соединений данного изобретения легко приготавливают обработкой основного соединения по существу эквивалентным количеством выбранной минеральной или органической кислоты в водной среде или в подходящем органическом растворителе, таком как ацетон, метанол или этанол. После выпаривания растворителя легко получают желаемую соль в виде твердого вещества. Желаемая кислотная соль может быть также выделена из раствора свободного основания в органическом растворителе добавлением раствора соответствующей минеральной или органической кислоты.
Соединения настоящего изобретения, которые по природе являются кислотными, способны к образованию основных солей с различными фармакологически приемлемыми катионами. Примеры таких солей включают соли со щелочными и щелочно-земельными металлами и особенно соли кальция, натрия и калия. Все указанные соли приготавливают обычными методиками. Химические основания, которые применяют как реагенты для приготовления фармацевтически приемлемых основных солей данного изобретения, являются реагентами, которые образуют нетоксичные основные соли с кислотными соединениями настоящего изобретения. Такие нетоксичные основные соли включают соли таких фармакологически приемлемых катионов, как катионы натрия, калия, кальция и магния и др. Указанные соли могут быть легко приготовлены обработкой соответствующих кислотных соединений водными растворами, содержащими желаемые фармакологически приемлемые катионы, и последующим упариванием полученного раствора досуха, предпочтительно при пониженном давлении. Альтернативно, они могут быть также приготовлены смешиванием растворов кислотных соединений в низших алкановых спиртах и желаемых алкоксидов щелочных металлов вместе и затем упариванием полученного раствора досуха, как указано выше. В любом случае предпочтительно применяют стехиометрические количества реагентов для обеспечения полноты реакции и максимальных выходов целевого конечного продукта.
Соединения настоящего изобретения являются важными для получения соединения формулы I (в которой заместители принимают значения, определенные выше):
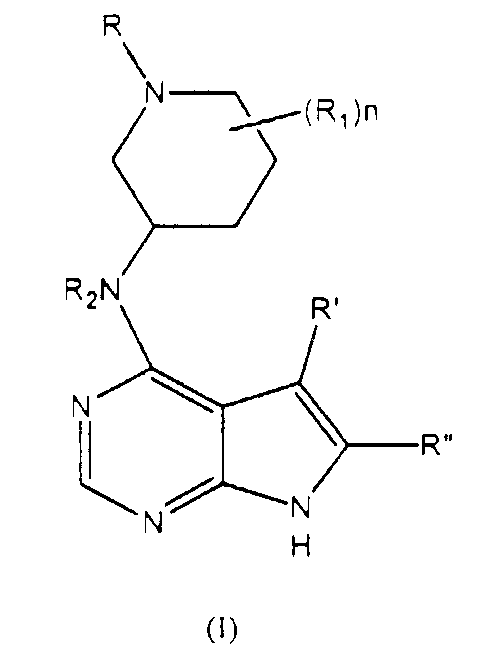
Соединения формулы I и его фармацевтически приемлемые соли (в дальнейшем также называемые вместе как "активные соединения") представляют собой ингибиторы протеинкиназ, таких как фермент Janus-киназа 3 (JAK3), и поэтому используются в терапии как иммуносупрессанты для лечения и профилактики отторжения органов-трансплантатов, ксенотрансплантации, волчанки, рассеянного склероза, ревматоидного артрита, псориаза, диабета типа I и осложнений от диабета, рака, астмы, атопического дерматита, аутоиммунных расстройств щитовидной железы, язвенного колита, болезни Крона, болезни Альцгеймера, лейкемии и других аутоиммунных заболеваний, острого и хронического отторжения органов-трансплантатов, отторжения трансплантата сердца, отторжения трансплантата легкого, отторжения трансплантата печени, отторжения трансплантата почки, отторжения трансплантата поджелудочной железы, отторжения трансплантата матки, отторжения трансплантата суставов, отторжения трансплантата ткани, отторжения трансплантата костного мозга, отторжения трансплантата конечностей, отторжения трансплантата роговицы, отторжения трансплантата кожи, отторжения трансплантата гепатоцитов, отторжения трансплантата печеночных клеток, отторжения трансплантата бета-клеток поджелудочной железы, отторжения трансплантата стволовых клеток, отторжения трансплантата невральных клеток, отторжения трансплантата кардиальных мышечных клеток, иммунозависимого бесплодия, подавления ВИЧ-репликации, гепатита B, гепатита С, интерстициального цистита, первичного билиарного цирроза печени, псориаза, псориатического артрита и ювенильного артрита у млекопитающих, включая человека.
Композиции настоящего изобретения могут быть приготовлены обычным способом с применением одного или нескольких фармацевтически приемлемых наполнителей. Таким образом, активные соединения данного изобретения могут быть приготовлены для орального, трансбуккального, интраназального, парентерального (например, внутривенного, внутримышечного или подкожного) или ректального введения или в форме, подходящей для введения ингаляцией или инсуффляцией. Активные соединения данного изобретения могут быть также приготовлены для пролонгированного высвобождения действующего вещества.
Для орального введения фармацевтические композиции можно использовать в форме, например, таблеток или капсул, приготовленных обычными способами с фармакологически приемлемыми носителями, такими как связывающие средства (например, предварительно желатинизированный кукурузный крахмал, поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнители (например, лактоза, микрокристаллическая целлюлоза или фосфат кальция); лубриканты (например, стеарат магния, тальк или кремнезем); диспергаторы (например, картофельный крахмал или натрийгликолят крахмала); или смачивающие средства (например, лаурилсульфат натрия). Таблетки могут быть с покрытием, достигаемым способами, хорошо известными в данной области. Жидкие препараты для орального введения можно использовать в форме, например, растворов, сиропов или суспензий, или они могут существовать как сухие продукты для сочетания с водой или другим подходящим наполнителем перед применением. Такие жидкие препараты могут быть приготовлены обычными способами вместе с фармацевтически приемлемыми добавками, такими как суспендирующие средства (например, сорбитоловый сироп, метилцеллюлоза или гидрогенизированные пищевые жиры); эмульгаторы (например, лецитин или аравийская камедь); неводные наполнители (например, миндальное масло, маслянистые сложные эфиры или этиловый спирт); консерванты (например, метил или пропил-п-гидроксибензоаты или сорбиновая кислота).
Для трансбуккального введения композиции можно использовать в форме таблеток или лепешек, приготовленных обычным способом.
Активные соединения данного изобретения могут быть приготовлены для парентерального введения инъекцией, включающего применение традиционных методик катетеризации или инфузии. Препараты для инъекции могут находиться в дозированной лекарственной форме, например, в ампулах или в емкостях с множеством доз с добавкой консерванта. Композиции могут быть приготовлены в таких формах, как суспензии, растворы или эмульсии в маслянистых или водных наполнителях, и могут содержать адъюванты, такие как суспендирующие, стабилизирующие и/или диспергирующие средства. Альтернативно активный ингредиент может быть в форме порошка для сочетания с подходящим наполнителем, например со стерильной антипирогенной водой, перед применением.
Активные соединения данного изобретения могут быть также приготовлены в виде композиций для ректального введения, таких как суппозитории или удерживающие клизмы, например, содержащие обычные основы для суппозиториев, такие как кокосовое масло или другие глицериды.
Для интраназального введения или введения ингаляцией активные соединения данного изобретения обычно поставляют в форме раствора или суспензии от насосного контейнера для разбрызгивания, которые накачиваются или выдавливаются пациентом, или поступают в виде аэрозоля из емкости под давлением или из распылителя с применением подходящего пропеллента, например дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае аэрозоля под давлением стандартная доза может быть определена с помощью впускного клапана для доставки отмеренного количества. Для применения в ингаляторе или инсуффляторе могут быть приготовлены капсулы или картриджи (сделанные, например, из желатина), содержащие порошкообразную смесь соединения данного изобретения и подходящую порошкообразную основу, такую как лактоза или крахмал.
Предполагаемая доза активных соединений данного изобретения для орального, парентерального или трансбуккального введения для среднестатистического взрослого человека при лечении состояний, относящихся к вышеуказанным (например, ревматоидный артрит), составляет от 0,1 до 1000 мг активного ингредиента на стандартную дозу, которая должна быть введена, например, от 1 до 4 раз в сутки.
Аэрозольные препараты для лечения состояний, относящихся к вышеуказанным (например, астма), для среднестатистического взрослого человека устанавливаются таким образом, что каждая отмеренная доза или "вдох" аэрозоля содержит от 20 мкг до 1000 мкг соединения данного изобретения. Общая суточная доза в случае аэрозоля будет находиться в интервале от 0,1 мг до 1000 мг. Введение может быть проведено несколько раз в сутки, например, 2, 3, 4 или 8 раз при поступлении 1, 2 или 3 доз каждый раз.
Активные соединения формулы (Ia-1) вводятся в фармацевтически приемлемой форме индивидуально или в комбинации с одним или несколькими дополнительными средствами, которые модулируют иммунную систему млекопитающих, или с противовоспалительными средствами, с агентами, которые могут включать, но без ограничения только ими, циклоспорин А (например, сандиммуне/Sandimmune® или неорал®), рапамицин, FK-506 (такролимус/tacrolimus), лефлуномид, деоксиспергуалин, микофенолят (например, целсепт®), азатиоприн (например, имуран®), даклизумаб (например, зенапакс®), ОКТ3 (например, ортоколон®), атгам/AtGam, аспирин, актаминофен, ибупрофен, напроксен, пироксам и противовоспалительные стероиды (например, преднизолон или дексаметазон), ЕРО, FK 778, Sdz-rad, стероиды, IVIG, ингибитор СОХ-2 (циклооксигеназы-2), NSAIDS (нестероидные противовоспалительные лекарственные средства), FTY720, базиликсимаб, донор-клетки, энеролимус/enerolimus, анти-CD28/CTLA-41g, ISTA-TX-247, ганцикловир, интерферон и альфа/ребиф, септра, анти-TNFS, ингибиторы Р38, антагонисты CCRI, антагонисты PDE4, липитор/статинс/lipitor/statins, ацикловир, рибовикин, ингибиторы протеазы/RTIs, инсулин, ритуксимаб, цетиризин или блокаторы Н1; причем такие средства могут быть введены как часть тех же самых лекарственных форм или в отдельных лекарственных формах, теми же или другими путями введения и теми же или различными схемами введения, соответствующими стандартной фармацевтической практике.
FK506 (такролимус) дают орально в дозе 0,10-0,15 мг/кг массы тела каждые 12 часов в течение первых послеоперационных 48 часов. Дозу контролируют по уровням такролимуса в сыворотке.
Циклоспорин А (сандиммуне оральный или внутривенный препарат, или неорал®, оральный раствор или капсулы) дают орально в дозе 5 мг/кг массы тела каждые 12 часов в течение послеоперационных 48 часов. Дозу контролируют по уровням циклоспорина в крови.
Активные средства могут быть приготовлены для пролонгированного высвобождения действующего вещества по способам, хорошо известным обычным специалистам в данной области. Примеры таких препаратов могут быть найдены в патентах США 3538214, 4060598, 4173626, 3119742 и 3492397.
Способность соединений формулы I или их фармацевтически приемлемых солей ингибировать Janus-киназу 3 и, соответственно, проявлять эффективность для лечения расстройств или состояний, связанных с участием Janus-киназы, показана в следующих оценочных опытах in vitro.
Количественное определение биологической активности
Определение ферментативной активности JAK3 (JH1:GST)
Для оценки активности JAK3-киназы используют протеин, экспрессируемый в SF9 клетках, инфицированных бакуловирусом (слияние протеина GST и каталитического домена человеческой JAK3), очищенный аффинной хроматографией на глутатион-сепахарозе. Субстрат для реакции представляет собой полиглутаминовую кислоту-тирозин (PGT (4:1), Sigma catalog #P0275), нанесенную на Nunc Maxi Sorp планшеты ночью при 37°C в концентрации 100 мкг/мл. Утром после нанесения планшеты промывают три раза и добавляют JAK3 в лунки, содержащие 100 мкл киназного буфера (50 мМ HEPES, pH 7,3, 125 мМ NaCl, 24 мМ MgCl2) + 0,2 мкМ АТР + 1 мМ Na ортованадат). Реакция протекает за 30 минут при комнатной температуре, и планшеты промывают более трех раз. Уровень фосфорилированного тирозина в данной лунке количественно определяют стандартным методом ELISA c использованием антифосфотирозиновых антител (ICN PY20, cat. #69-151-1).
Ингибирование разрастания IL-2(интерлейкин) зависимых Т-лимфоцитных бластных клеток человека
В данном опыте измеряется ингибирующий эффект соединений на разрастание IL-2 зависимых Т-лимфоцитных бластных клеток in vitro. Так как для сигнала от IL-2 рецептора требуется JAK3, то клеточно-активные ингибиторы JAK3 должны ингибировать разрастание IL-2 зависимых Т-лимфоцитных бластных клеток.
Клетки для данного анализа выделяли из свежей крови человека. После отделения моноядерных клеток с помощью Accuspin System-Histopaque-1077 (Sigma #A7054) первичные человеческие Т-лимфоциты выделяли негативным селективным отбором с помощью Lympho-Kwik T (One Lambda, Inc., Cat. # LK-50T). Т-лимфоциты культивировали в концентрации 1-2×106 клетка/мл в среде (RPMI+10% не активированная теплом сыворотка из икр плода (Hyclone Cat. #A-1111-L)+1% пенициллин/стрептомицин (Gibco)) и вызывали разрастание добавлением 10 мкг/мл РНА (Murex Diagnostics, Cat. #НА 16). После выдержки в течение 3 дней при 37°C в 5% СО2 клетки промывали 3 раза в среде, ресуспендировали до плотности 1-2×106 клетка/мл в среде плюс 100 единица/мл человеческого рекомбинантного IL-2 (R&D Systems, Cat. # 202-IL). Через 1 неделю клетки становятся IL-2 зависимыми и могут сохраняться до 3 недель подпиткой дважды в неделю равными объемами среда + 100 единица/мл IL-2.
Для оценки способности испытуемых соединений ингибировать разрастание IL-2 зависимых Т-лимфоцитов IL-2 зависимые клетки промывали 3 раза, ресуспендировали в среде и затем помещали (50000 клетка/лунка/0,1 мл) в плоскодонный 96-луночный микротитрованный планшет (Falcon # 353075). Из 10 мМ порции оцениваемого соединения в ДМСО готовили ряд двукратных разбавлений соединения, каждое из которых добавляли трижды в лунки, начиная с 10 мкМ. Через 1 час в каждую лунку добавляли 10 единица/мл IL-2. Планшеты затем инкубировали при 37°C с 5% СО2 в течение 72 часов. Планшеты затем подвергали вибрации с 3Н-тимидином (0,5 мкКи/лунка) (NEN Cat. # NET-027A) и инкубировали дополнительно 18 часов. Культуру затем убирали с планшета и определяли количество 3Н-тимидина, включенного в разросшиеся клетки с помощью сцинтилляционного счетчика Packard Top Count. Данные анализировали по зависимости % ингибирования разрастания от концентрации испытанного соединения. Величину IC50 определяли из полученной зависимости.
Эксперименты
Последующие примеры преследуют цель чисто пояснительную для данного изобретения и не предназначены для ограничения объема изобретения. Если не оговорено особо, то процент представляет собой мас.%, отнесенный к компоненту и общей массе композиции, температура дана в °С или является температурой окружающей среды, и давление равно или близко к атмосферному. Коммерческие реагенты применяли без дополнительной очистки. В данной работе использованы следующие сокращения:
Пример 1: Гидрохлоридная соль цис-(1-бензил-4-метилпиперидин-3-ил)метиламина
Метиловый эфир (4-метилпиридин-3-ил)карбаминовой кислоты
Синтез проводили добавлением 2 г (1 экв., 18,5 ммоль) 4-метилпиридин-3-иламина к раствору 6,55 г (3 экв., 55,5 ммоль) трет-бутоксида калия в 10 мл ТГФ (6,66 экв., 123 ммоль) при 0°С. После образования аниона в реакционную смесь добавляли 2,34 мл (1,5 экв., 27,7 ммоль) диметилкарбоната с такой скоростью, чтобы температура была ниже 0°С. Реакция завершалась в течение 30 минут. Красную взвесь гасили 50 мл воды (25 объемов) и продукт экстрагировали 50 мл этилацетата (25 объемов). Водный слой экстрагировали 50 мл этилацетата (25 объемов) и затем органические оранжевые слои концентрировали до получения оранжевого твердого вещества. Данные ЯМР показали, что трет-бутанол присутствует в продукте, поэтому твердые вещества обращали во взвесь с 10 мл толуола (5 объемов) и затем концентрировали досуха. Данную процедуру выполняли три раза и в результате получали очень чистое светло-оранжевое твердое вещество. Выход 89%. 1Н ЯМР: δ 8,90 (1Н, уш.с), 8,28 (1Н, д, J=4,8), 7,16-7,14 (1H, м), 6,54 (1Н, уш.с), 3,80 (3Н, с), 2,29 (3Н, с).
Метиловый эфир цис-(4-метилпиперидин-3-ил)карбаминовой кислоты
Гидрирование проводили при загрузке 5 г (1 экв., 30,1 ммоль) метилового эфира (4-метилпиридин-3-ил)карбаминовой кислоты, 50 мл этанола (10 объемов) и 2,5 г (0,5 экв.) родия на оксиде алюминия в автоклав для гидрирования. Гидрирование выполняли при 100 фунт/кв. дюйм водорода и 100°С в течение 24 часов и получали с количественным выходом только метиловый эфир цис-(4-метилпиперидин-3-ил)карбаминовой кислоты и его транс-изомер в соотношении 5:1. 1Н ЯМР: δ 5,6 (1Н, д, J=8,4 Гц), 3,66 (1Н, д, J=3,6 Гц), 3,65-3,57 (3Н, уш.с), 3,1 (2Н, д, J=9,6, второстепенный изомер)), 2,91-2,86 (2Н, м), 2,67-2,64 (1Н, м), 2,54-2,43 (1Н, м), 2,2-1,9 (1Н, м, второстепенный изомер), 1,78-1,65 (5Н, уш.с, второстепенный изомер), 1,65-1,59 (4Н, м, второстепенный изомер), 1,36-1,25 (2Н, м), 1,24-1,13 (2Н, м), 0,92 (3Н, д, J=6,8 Гц, второстепенный изомер), 0,83 (3Н, д, J=7,2).
Метиловый эфир цис-(1-бензил-4-метилпиперидин-3-ил)карбаминовой кислоты
Восстановительное аминирование метилового эфира цис-(4-метилпиперидин-3-ил)карбаминовой кислоты проводили добавлением 3,9 г (1 экв., 22,6 ммоль) указанного соединения к смеси 2,07 мл (0,9 экв., 20,4 ммоль) бензальдегида, 9,6 г (2 экв., 45,3 ммоль) триацетоксиборогидрида натрия и 39 мл метиленхлорида (10 объемов). Реакционную смесь перемешивали при 20°C и позволяли температуре экзотермически подняться до 30-35°С. Реакцию завершали с контролем GCMS в течение 30 минут. Реакцию гасили 78 мл насыщенного бикарбоната натрия (20 объемов), продукт экстрагировали 78 мл метиленхлорида (20 объемов) и концентрировали до чистого масла. Выход 70%. 1Н ЯМР: δ 7,2-7,3 (5Н, м, ароматические протоны), 5,50-5,48 (1Н, д, J=8,8 Гц), 3,77-3,75 (1Н, д, J=8,0 Гц)), 3,63 (3Н, с), 3,45-3,38 (2Н, м), 2,77-2,74 (2Н, д, J=11,2 Гц), 2,14-2,01 (1Н, м), 1,94-1,89 (1Н, м), 1,57-1,55 (1Н, уш.с), 1,37-1,20 (2Н, м), 0,876 (3Н, д, J=9,2 Гц).
(1-Бензил-4-метилпиперидин-3-ил)метиламин
Восстановление метилового эфира цис-(1-бензил-4-метилпиперидин-3-ил)карбаминовой кислоты проводили добавлением 2 г (1 экв., 7,62 ммоль) указанного субстрата к смеси 20 мл ТГФ (10 объемов) и 15,2 мл 1 М LAN (литийалюминийгидрид) (2 экв., 15,2 ммоль). Добавление выполняли с такой скоростью, чтобы температура достигла 30-40°C, и затем реакционной смеси позволяли охлаждаться до 20°С. Реакционную смесь обрабатывали гашением с помощью 40 мл сегнетовой соли (20 объемов) при температуре 35°С и затем экстрагировали продукт 2 раза 20 мл метиленхлорида (10 объемов). Фильтрат затем концентрировали до получения чистого и бесцветного масла. Выход 90%. Отмечено, что исходные продукты должны быть очищены для того, чтобы данная реакция проходила с высоким выходом. 1Н ЯМР: δ 7,2-7,3 (5Н, м, ароматические протоны), 3,54-3,51 (1Н, д, J=13,6 Гц), 3,40-3,37 (1Н, д, J=13,6 Гц), 2,70-2,62 (2Н, м), 2,39-2,36 (1Н, уш.с), 2,32 (3Н, с), 2,29-2,12 (1Н, уш.с), 2,10-2,00 (1Н, уш.с), 1,66 (1Н, уш.с), 1,47-1,43 (2Н, м), 1,32 (1Н, уш.с), 0,936-0,919 (3Н, д, J=6,8 Гц).
Гидрохлоридная соль цис-(1-бензил-4-метилпиперидин-3-ил)метиламина
Получение конечной соли проводили загрузкой 1,5 г (1 экв., 5,15 ммоль) цис-(1-бензил-4-метилпиперидин-3-ил)метиламина и 4,5 мл этанола (3 объема) в реакционный сосуд при 0°С. Затем в охлажденный до 0°C сосуд добавляли 0,93 мл 36% HCl (0,625 объема) так, чтобы температура была ниже 10°С. Затем из реакционной смеси концентрировали 3 мл (2 объема) этанола. К реакционной смеси добавляли 7,5 мл этилацетата (5 объемов), перемешивали 5 минут и затем удаляли в вакууме 6 мл (4 объема) этилацетата. К остатку снова добавляли 7,5 мл (5 объемов) этилацетата и снова проводили концентрирование смеси. Затем добавляли 4,5 мл (3 объема) ацетона и реакционную смесь медленно охлаждали до 0°C до образования белого твердого вещества. Выход 37,5%. 1Н ЯМР: δ 7,78-7,76 (2Н, д, J=8,0 Гц), 7,29-7,18 (5Н, м, ароматические протоны), 5,55 (1Н, с), 3,45-3,41 (1Н, д, J=13,2 Гц), 3,39-3,36 (1Н, д, J=13,2 Гц), 2,79 (1Н, уш.с), 2,63 (1Н, уш.с), 2,45 (3Н, с), 2,30 (2Н, с), 2,25-2,05 (1Н, м), 1,76 (1Н, уш.с), 1,40-1,39 (2Н, м), 0,875-0,845 (3Н, д, J=12 Гц).
Пример 2: Метиловый эфир 4-метил-3-метиламинопиперидин-1-карбоновой кислоты
Метиловый эфир 4-метилпиперидин-1-карбоновой кислоты
В трехгорлую круглодонную колбу добавляли 360 г 4-метилпиперидина, 470 мл триэтиламина и 390 мл метиленхлорида и смесь охлаждали на бане со льдом. К данной смеси медленно добавляли метилхлорформиат (260 мл) в метиленхлориде (215 мл) для установления температуры в реакционной среде в 20°C или ниже. Смесь перемешивали в течение ночи, затем добавляли 200 мл воды и разделяли слои. Органический слой промывали разбавленной HCl, насыщенным NaHCO3 и рассолом, затем органический слой сушили над сульфатом натрия и растворитель удаляли в вакууме. Продукт перегоняли при 90-93°C/˜10 мм и получали 338 г продукта.
Смесь метилового эфира 2,3-диацетокси-4-метилпиперидин-1-карбоновой кислоты и 3-ацетокси-2-гидрокси-4-метилпиперидин-1-карбоновой кислоты
В 500 мл стеклянный электролизер без диафрагмы устанавливали Pt анод с перфорацией в 60 см2 и перфорированный Pt-армированный Nb катод. Между электродами помещали полипропиленовый ситовый сепаратор. В электролизер добавляли 50 г метилового эфира 4-метилпиперидин-1-карбоновой кислоты, 40 г KOAc и 320 мл НОАс. Смесь подвергали электролизу при постоянном токе 6,0 А до прохождения через смесь 20 Ф/моль. Вольтаж в элекролизере изменялся от 13,5 до 20 В. Реакционную смесь погружали в холодную водяную баню для установления температуры реакционной смеси в интервале 35-40°С. Неочищенную смесь метилового эфира 2,3-диацетокси-4-метилпиперидин-1-карбоновой кислоты и 3-ацетокси-2-гидрокси-4-метилпиперидин-1-карбоновой кислоты обрабатывали на следующей стадии.
Метиловый эфир 5-ацетокси-4-метил-3,4-дигидро-2Н-пиридин-1-карбоновой кислоты
101,9 г (0,649 моль) полученной смеси метилового эфира 2,3-диацетокси-4-метилпиперидин-1-карбоновой кислоты и 3-ацетокси-2-гидрокси-4-метилпиперидин-1-карбоновой кислоты концентрировали при пониженном давлении до образования твердого вещества. Твердое вещество затем помещали в 2 л круглодонную колбу, снабженную впускным отверстием для азота, холодильником и термопарой. К данной смеси добавляли уксусный ангидрид (430 мл) и затем смесь кипятили с обратным холодильником 2 часа при 141°С. Раствор перемешивали ночь при комнатной температуре. Основную массу уксусного ангидрида удаляли при пониженном давлении и оставшееся количество удаляли добавлением Н2О (400 мл) и 5% раствора NaHCO3 до рН>7. Водную смесь экстрагировали этилацетатом (3×250 мл), органические слои объединяли и этилацетат удаляли при пониженном давлении. Фиксировали образование приблизительно 119,0 г метилового эфира 5-ацетокси-4-метил-3,4-дигидро-2Н-пиридин-1-карбоновой кислоты в виде густого коричневого масла. По данным ТСХ метиловый эфир 5-ацетокси-4-метил-3,4-дигидро-2Н-пиридин-1-карбоновой кислоты является главным компонентом превращения. Продукт сразу делили на две порции для проведения реакций гидролиза в различных условиях.
Метиловый эфир 4-метил-3-оксопиперидин-1-карбоновой кислоты (гидролиз с применением диметиламина)
В 1 л колбе растворяли 60 г неочищенного метилового эфира 5-ацетокси-4-метил-3,4-дигидро-2Н-пиридин-1-карбоновой кислоты в 100 мл МеОН и к полученному раствору добавляли 40 г (0,325 моль) 40% диметиламина в метаноле при 0°C в течение около 15 мин. Смесь оставляли перемешиваться в токе азота при комнатной температуре в течение ночи. По данным ТСХ продукт реакции - метиловый эфир 4-метил-3-оксопиперидин-1-карбоновой кислоты - получался в качестве основного компонента. Метанол удаляли при пониженном давлении и остаток экстрагировали дихлорметаном (3×100 мл). Дихлорметановый слой промывали водой (3×50 мл), органический слой отделяли и растворитель удаляли при пониженном давлении. Неочищенный остаток очищали колоночной хроматографией.
Продукт очищали на колонке с силикагелем (360 г). Элюирование смесью гексан/этилацетат 70:30 (5 л) приводило к неполярным полупродуктам. Непрерывное элюирование той же самой смесью растворителей (6 л), затем смесью гексан/этилацетат 50:50 (3 л) приводило к метиловому эфиру 4-метил-3-оксопиперидин-1-карбоновой кислоты в виде масла после удаления растворителей в вакууме. Выход: 11,9 г (0,0696 моль, 27%), ТСХ (SiO2, этилацетат/гексан 1:1) и ЯМР.
Метиловый эфир 4-метил-3-оксопиперидин-1-карбоновой кислоты (синтез с применением натрий карбонат/натрий бикарбонатного буфера)
В 1000 мл круглодонной колбе растворяли 50 г неочищенного метилового эфира 5-ацетокси-4-метил-3,4-дигидро-2Н-пиридин-1-карбоновой кислоты в 200 мл метанола. К 300 мл воды добавляли 30 г карбоната натрия и 30 г бикарбоната натрия (рН=10). Водный буфер добавляли к метанольному раствору. К смеси добавляли метанол до достижения легкости перемешивания и продолжали перемешивание при комнатной температуре в течение 18 часов. Смесь затем концентрировали для удаления метанола. К смеси добавляли воду для растворения всех солей. Затем смесь экстрагировали этилацетатом три раза. Органические экстракты объединяли, сушили над сульфатом магния, фильтровали и упаривали, получая неочищенный метиловый эфир 4-метил-3-оксопиперидин-1-карбоновой кислоты в виде масла, которое затем очищали, как описано ниже.
Полученное масло разделяли пополам и 26,9 г пропускали через колонку с силикагелем, содержащую 350 мл силикагеля. Растворитель элюировали градиентно, начиная с 5% этилацетата в гексане. Продукт элюировали 10% этилацетатом. Фракции с чистым продуктом, по данным ТСХ (SiO2, этилацетат/гексан 1:1), объединяли и концентрировали, получая 8,85 г (выход 32%). Дополнительные фракции с незначительными примесями, по данным ТСХ, в продукте объединяли и концентрировали. Также выделяли дополнительное количество менее чистого продукта (3,2 г, 10%).
Оставшуюся половину неочищенного продукта (26,9 г) перегоняли в высоком вакууме. Отбирали фракции, кипящие при 99-100°C /1 мм, и получали 12,1 г (выход 41%). Полученный продукт был сопоставим, по данным ТСХ и ЯМР, с образцом, очищенным хроматографией, но содержал незначительные неполярные примеси в соответствии с ТСХ (SiO2, этилацетат/гексан 1:1).
Метиловый эфир 4-метил-3-метиламинопиперидин-1-карбоновой кислоты (синтезированный с применением метиламин/натрийцианоборогидрида)
В 100 мл круглодонной колбе растворяли 4 г неочищенного метилового эфира 4-метил-3-оксопиперидин-1-карбоновой кислоты в 29 мл метанола. К перемешиваемой смеси добавляли 2 М метиламин (25 мл в метанольном растворе). После 1-часового перемешивания при комнатной температуре добавляли натрийцианоборогидрид (0,7 г) и перемешивали смесь 3 суток. Для гашения реакции добавляли при энергичном перемешивании воду (10 мл), затем медленно добавляли концентрированную HCl до тех пор, пока рН не становился сильнокислым (рН=˜1). Через 2 часа смесь концентрировали, водный раствор экстрагировали дважды метиленхлоридом. Органические экстракты объединяли, сушили над сульфатом магния, фильтровали и упаривали. Остаток пропускали через колонку с силикагелем с градиентом 50% этилацетата в гексане и в конце с 100% метанолом. Масса продукта составляет 600 мг. Из данных ЯМР (CDCl3) следовало, что целевое соединение - метиловый эфир 4-метил-3-метиламинопиперидин-1-карбоновой кислоты - получено в виде смеси диастереоизомеров с их соотношением, примерно равным 1:1, от разделения при 4-метил-группе. В ИК присутствует C=О полоса, характерная для карбамата.
Метиловый эфир 4-метил-3-метиламинопиперидин-1-карбоновой кислоты (синтезированный с применением метиламин/натрийборогидрида)
В 50 мл круглодонной колбе растворяли 2,0 г кетона - метилового эфира 4-метил-3-оксопиперидин-1-карбоновой кислоты - в 6 мл 2 М метиламина в метаноле. Раствор перемешивали 1 час, после чего концентрировали с помощью роторного испарителя. В отдельную 100 мл круглодонную колбу с 6 мл сухого тетрагидрофурана в атмосфере аргона добавляли 0,6 г борогидрида натрия и смесь охлаждали на бане со льдом. К суспензии медленно добавляли 3,6 мл ледяной уксусной кислоты при перемешивании и непрерывном охлаждении. Метиламин-кетоновый раствор, указанный выше, разбавляли абсолютным этанолом и медленно при непрерывном охлаждении на бане со льдом добавляли к раствору борогидрида натрия. После окончания добавления (около 1 ч) смеси позволяли нагреваться до комнатной температуры. Через 2 часа колбу оставляли на хранение при 4°C на ночной период. К перемешиваемому раствору добавляли 10 мл воды. Через 20 минут медленно добавляли концентрированную хлористо-водородную кислоту до достижения рН смеси около 2. Смесь затем концентрировали с помощью роторного испарителя. Водный раствор доводили до щелочного добавлением карбоната натрия и экстрагировали три раза этилацетатом. Органические слои объединяли, сушили над сульфатом магния, фильтровали и концентрировали с помощью роторного испарителя. Полученное масло очищали на колонке с силикагелем растворителем, содержащим 1% раствор гидроксида аммония и 3% метанол в метиленхлориде. Фракции с обнаруженным продуктом объединяли и растворитель удаляли с помощью роторного испарителя. Масса продукта составила 400 мг (выход 18,5%). Данные ТСХ сопоставимы с данными по метиловому эфиру 4-метил-3-метиламинопиперидин-1-карбоновой кислоты, приготовленному другим способом.
По тексту данной заявки привлечены различные публикации. Раскрытия данных публикаций во всей полноте, таким образом, включены в виде ссылок в настоящую заявку по всем позициям.
Будет очевидно специалистам в данной области, что могут быть сделаны различные модификации и вариации в настоящем изобретении без отклонения от объема или сущности изобретения. Другие варианты осуществления данного изобретения будут очевидными для специалистов в данной области из рассмотрения описания и практики изобретения, раскрываемых здесь. Это означает, что описания и примеры должны рассматриваться только как пояснительные к истинному объему и сущности данного изобретения, приведенным в следующей далее формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНАЦИИ, ВКЛЮЧАЮЩИЕ АГОНИСТ РЕЦЕПТОРА S1P И ИНГИБИТОР КИНАЗЫ JAK3 | 2005 |
|
RU2415678C2 |
| НОВЫЙ КЛАСС СЕЛЕКТИВНЫХ АГОНИСТОВ СОМАТОСТАТИНОВЫХ РЕЦЕПТОРОВ | 2014 |
|
RU2603962C2 |
| АНТАГОНИСТЫ ВИТРОНЕКТИНОВОГО РЕЦЕПТОРА, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 1997 |
|
RU2198892C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНА | 2011 |
|
RU2528386C2 |
| ПРОИЗВОДНЫЕ ИНДАЗОЛА КАК ИНГИБИТОРЫ ГОРМОНЧУВСТВИТЕЛЬНЫХ ЛИПАЗ | 2005 |
|
RU2370491C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНИЛПИПЕРАЗИНА, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ХЕМОКИНОВЫХ РЕЦЕПТОРОВ | 2006 |
|
RU2411241C2 |
| НОВЫЕ ИНГИБИТОРЫ РАССАСЫВАНИЯ КОСТЕЙ И АНТАГОНИСТЫ РЕЦЕПТОРА ВИТРОНЕКТИНА | 1997 |
|
RU2195460C2 |
| АМИДЫ ПИРИДИЛАЛКЕНОВЫХ И ПИРИДИЛАЛКИНОВЫХ КИСЛОТ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1997 |
|
RU2200734C2 |
| НОВЫЕ АНТРАНИЛАМИДОПИРИДИНМОЧЕВИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ РЕЦЕПТОРА VEGF | 2005 |
|
RU2415850C2 |
| ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ ИНГИБИТОР НАТРИЙЗАВИСИМОГО ПЕРЕНОСЧИКА ФОСФАТА | 2015 |
|
RU2740008C2 |
Изобретение относится к усовершенствованному способу получения производных 3-аминопиперидина - соединений формулы (Ib), которые используются для получения ингибиторов протеинкиназ
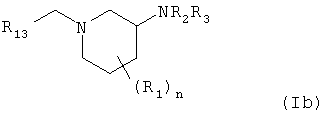
где R1 представляет собой (С1-С6)алкил; R2 представляет собой водород, (C1-С6)алкил; R3 представляет собой водород, (С1-С6)алкил; R13 представляет собой фенил; n принимает значение 1, 2, 3 или 4; который включает восстановление соединения формулы (IIb)
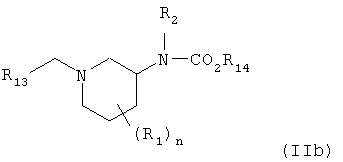
где R14 представляет собой (С1-С6)алкил, восстановителем. 6 з.п. ф-лы.
где R1 представляет собой (С1-С6)алкил;
R2 представляет собой водород, (С1-С6)алкил;
R3 представляет собой водород, (С1-С6)алкил;
R13 представляет собой фенил;
n принимает значение 1, 2, 3 или 4;
который включает восстановление соединения формулы (IIb),
где R14 представляет собой (С1-С6)алкил,
восстановителем.
с альдегидом формулы R13-(C=O)-H и восстановлением полученного соединения восстановителем.
в присутствии катализатора.
с (R14-O-(C=O))2O или R14-O-(C=O)-X, где X представляет собой галоген.
 ;
;
R1 представляет собой (С1-С6)алкил; n принимает значение, равное единице;
R2 и R3 каждый представляет собой водород или (С1-С6)алкил; R13 представляет собой фенил.
| US 5559128 А, 24.09.1996 | |||
| Способ электронного каротажа скважин и устройство для его осуществления | 1956 |
|
SU111864A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| Duhamel et al | |||
| Synthesis of Bicyclic Aza-enones via a Lewis Acid Catalysed Michael-type Addition with Silyl Enol Ethers bearing a Nitrogen Atom | |||
| Tetrahedron | |||
| Letters | |||
| Vol | |||
| Нивелир для отсчетов без перемещения наблюдателя при нивелировании из средины | 1921 |
|
SU34A1 |
| Пишущая машина для тюркско-арабского шрифта | 1922 |
|
SU24A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| JP 2002201192 А, 16.07.2002 | |||
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 3-АМИНОПИПЕРИДИНОВ | 1992 |
|
RU2105001C1 |

