Предшествующий уровень техники
Область изобретения
Настоящее изобретение относится к новому, по существу энантиомерно чистому диарилметилпиперазиновому соединению, применяемому в качестве рецепторсвязывающего вещества, например в качестве опиоидного соединения мю- и/или дельта-рецептора, опосредующего аналгезию; в качестве терапевтического агента для совместного введения с различными другими биоактивными композициями, включая анестетики, барбитураты, анальгетики и так далее, для уменьшения, лечения, обращения или предупреждения опосредованного лекарством угнетения дыхания, которое может быть прямо или косвенно вызвано применением таких различных биоактивных композиций; в качестве конъюгата в агонистическом/антагонистическом спаривании для подтверждения/анализа рецепторной и нейротрансмиттерной функции; и в качестве терапевтического агента, применяемого в борьбе с лекарственным привыканием, хроническим алкоголизмом, сердечными расстройствами, передозировкой лекарств, психическим заболеванием, кашлем, отеком легкого, диареей, респираторными и желудочно-кишечными расстройствами.
Описание уровня техники
При изучении биохимии опиоидов было идентифицировано многообразие эндогенных опиоидных соединений и не эндогенных опиоидных соединений. В этих исследованиях значительные усилия были сфокусированы на понимании механизма действия опиоидного лекарства, особенно того, как он связан с опиатными рецепторами клеток и дифференцированных тканей.
Опиоидные лекарства обычно классифицируют по их селективности связывания в отношении рецепторов клеток и дифференцированных тканей, с которыми конкретные лекарственные вещества связываются в качестве лиганда. Эти рецепторы включают в себя мю (μ), дельта (δ), сигма (σ) и каппа (κ) рецепторы.
Хорошо известные наркотические опиаты, такие как морфин и его аналоги, являются селективными для опиатного мю-рецептора. Мю-рецепторы опосредуют аналгезию, угнетение дыхания и подавление транзита пищи по желудочно-кишечному тракту; каппа-рецепторы опосредуют аналгезию и седацию; а сигма-рецепторы опосредуют различные биологические активности.
Существование опиоидного дельта-рецептора является относительно недавним открытием, которое было сделано после того, как были выделены и охарактеризованы эндогенные энкефалиновые пептиды, представляющие собой лиганды для дельта-рецептора. Исследования последнего десятилетия дали значительную информацию о дельта-рецепторе. Дельта-рецепторы опосредуют аналгезию, но, по-видимому, не подавляют транзит пищи по желудочно-кишечному тракту таким образом, как это характерно для мю-рецепторов.
Опиоидные агенты часто характеризуют как либо агонисты, либо антагонисты. Агонисты и антагонисты представляют собой агенты, которые распознают и связываются с рецепторами, влияя на (либо инициируя, либо блокируя) биохимические/физиологические последствия, процесс, известный как трансдукция. Агонисты ингибируют или подавляют выходы нейротрансмиттеров в ткани, содержащие рецепторы, например, подавляя болевые ответы, либо затрагивая другие явления, связанные с выходом. Антагонисты также связываются с рецепторами, но не подавляют выходы нейротрансмиттеров. Таким образом, антагонисты связываются с сайтами рецепторов и блокируют связывание агонистических веществ, которые селективны для того же самого рецептора.
Опиоидные диарилметилпиперазины, обладающие как мю, так и дельта-рецепторной активностью, описаны в патенте США № 5658908 (Chang et al.). Однако синтез этих соединений, имеющих по меньшей мере один асимметричный атом углерода, в лаборатории, неизменно приводит к рацемической смеси, не проявляющей никакой оптической активности. Напротив, соединения, встречающиеся в природе, которые обладают асимметричным атомом углерода, почти неизменно оптически активны.
При описании оптически активного соединения префиксы D и L, либо R и S используют для обозначения абсолютной конфигурации молекулы относительно его хирального центра(ов). Префиксы (+) и (-) либо d и l используют для обозначения направления вращения плоскополяризованного света соединением, причем (-) или l обозначает, что соединение является левовращающим. Соединение с префиксом (+) или d является правовращающим. Многие органические соединения существуют в оптически активных формах, то есть они обладают способностью вращать плоскость плоскополяризованного света.
Для данной химической структуры различные оптически активные соединения называют стереоизомерами и они идентичны, за исключением того, что они являются зеркальными изображениями один другого. Конкретный стереоизомер также может быть назван энантиомером, и смесь таких изомеров называют энантиомерной или рацемической смесью.
Стереохимическая чистота является важной в области фармацевтических препаратов, где многие из наиболее принятых лекарств проявляют хиральность. Иллюстративным примером является I-форма пропранолола, про которую известно, что она является в 100 раз более сильнодействующей, чем d-энантиомер. Более того, оптическая чистота является важной, поскольку некоторые изомеры могут действительно быть даже скорее вредными, чем просто инертными. Например, предполагается, что d-энантиомер талидомида является безопасным и эффективным успокаивающим средством, при назначении для контроля утренней тошноты во время беременности, и что соответствующий I-энантиомер является сильнодействующим тератогеном.
В то время как в вышеупомянутом патенте Chang et al. признано, что диарилметилпиперазины могут иметь оптически активные формы и конкретные энантиомерные формы могут быть синтезированы, никакого примера заявляемой в настоящем изобретении оптически активной формы не было дано. Хотя раньше в целом было сделано заключение, что описанные рацемические смеси диарилметилпиперазинов и содержащиеся энантиомеры проявляли сходную активность, авторами настоящего изобретения было открыто, что существуют значительные неожиданные преимущества при использовании энантиомерно чистого диарилметилпиперазина по настоящему изобретению.
Сущность изобретения
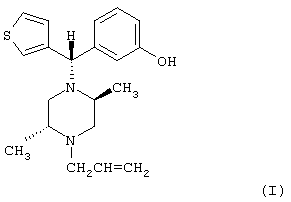
Настоящее изобретение относится к соединению, имеющему структуру формулы (I):
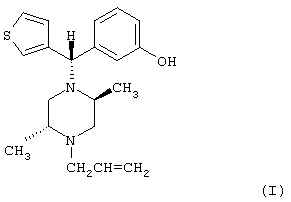
имеющему название по ИЮПАК (Международный союз теоретической и прикладной химии) (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол, или его фармацевтически приемлемым эфирам или солям.
Примеры фармацевтически приемлемых эфиров соединения формулы (I) включают в себя эфиры карбоновой кислоты гидроксильной группы в соединении формулы (I), в которых некарбонильная группировка карбоново-кислотной части эфирной группировки выбрана из алкила с прямой или разветвленной цепью (например, н-пропила, трет-бутила, н-бутила), алкоксиалкила (например, метоксиметила), арилалкила (например, бензила), арилоксиалкила (например, феноксиметила) и арила (например, фенила); алкил-, арил-, или арилалкилсульфонила (например, метансульфонила); эфиров аминокислоты (например, L-валильного или L-изолейцильного); эфиров дикарбоновой кислоты (например, гемисукцината); карбонатных эфиров (например, этоксикарбонила); эфиров карбаминовой кислоты (например, диметиламинокарбонила, (2-аминоэтил)аминокарбонила); и неорганических эфиров (например, моно-, ди- или трифосфата).
Примеры фармацевтически приемлемых солей соединения формулы (I) включают в себя соли, полученные от соответствующего основания, как например щелочного металла (например, натрия, калия), щелочно-земельного металла (например, кальция, магния), аммония и NX4 + (где Х представляет собой C1-С4алкил). Фармацевтически приемлемые соли аминогруппы включают в себя соли органических карбоновых кислот, таких как уксусная, лимонная, молочная, винная, малеиновая, лактобионовая, фумаровая и янтарная кислота; органических сульфоновых кислот, таких как метансульфоновая, этансульфоновая, изэтионовая, бензолсульфоновая и пара-толуолсульфоновая кислота; и неорганических кислот, таких как соляная, бромистоводородная, серная, фосфорная и сульфаминовая кислота. Фармацевтически приемлемые соли соединения, имеющего гидроксильную группу, состоят из аниона указанного соединения в комбинации с подходящим катионом, таким как Na+, NH4 + или NX4 + (где Х представляет собой, например, C1-С4алкильную группу).
Для терапевтического применения соли соединения формулы (I) должны быть фармацевтически приемлемыми, то есть они должны представлять собой соли, полученные от фармацевтически приемлемой кислоты или основания. Однако соли кислот или оснований, которые не являются фармацевтически приемлемыми, могут также найти применение, например, в получении или очистке фармацевтически приемлемого соединения. Все соли, независимо от того получены они или нет от фармацевтически приемлемой кислоты или основания, входят в объем настоящего изобретения.
Соединение формулы (I) применимо в качестве экзогенного соединения, склонного к комбинации с рецептором, и может быть использовано для связывания с опиоидным рецептором. Более того, соединение может представлять собой конъюгат в агонистической/антагонистической паре, что может быть использовано для трансдукционного анализа нейротрансмиттерной функции в соответствующих клеточных системах или системах дифференцированной ткани. В дополнение к рецепторному анализу, дифференциальному связыванию и специфичным применениям для клеточного, гистологического и телесного мониторинга и целей оценки соединение вышеуказанной формулы (I) проявляет свойства специфической биоактивности, делающие его полезным в качестве лечебного агента для различных физиологических и патологических состояний.
Молекула формулы (I) опосредует аналгезию с пониженным угнетением дыхания, и, кроме того, она полезна для лечения диареи, психического заболевания, апноэ, когнитивных расстройств, сердечных расстройств, кашля, отека легких, желудочно-кишечных расстройств, повреждения спинного мозга и лекарственного привыкания.
Дополнительно, настоящее изобретение относится в одном аспекте к фармацевтической композиции, содержащей (i) терапевтический агент, представляющий собой агонист мю-рецептора, опосредующий дыхательный, мышечный или тошнотворный побочный эффект и (ii) эффективное для снижения лечения или предупреждения побочных эффектов количество соединения формулы:
(-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола, или его эфиров или солей.
В дополнение, до степени, в которой дегенерация или дисфункция опиоидных рецепторов присутствует или вовлечена в болезненное состояние, в которое вовлечены тканевые или дискретные клеточные локусы, меченные изотопом варианты опиоидного соединения по настоящему изобретению могут найти использование в диагностических применениях и применениях визуализации, например диагностических методиках, в которые вовлечены позитронные эмиссионные томограммы (PET) головного мозга.
Еще один аспект настоящего изобретения включает в себя способ опосредования аналгезии, при котором вводят эффективное количество соединения, представляющего собой агонист опиоидного рецептора формулы
(-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола, или его эфиров или солей.
Соединение формулы (I) можно вводить в фармацевтически приемлемом носителе.
Еще один аспект настоящего изобретения относится к способу лечения пациента, нуждающегося в нем, с помощью терапевтического агента, представляющего собой агонист опиоидного мю-рецептора, с ослаблением угнетения дыхания, присущего его введению, при котором пациенту вводят указанный терапевтический агент, представляющий собой агонист опиоидного мю-рецептора, эффективное количество агониста опиоидного рецептора для ослабления угнетения дыхания, причем соединение, представляющее собой агонист опиоидного рецептора, имеет формулу:
(-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол, или его эфиры или соли.
Еще один аспект настоящего изобретения относится к фармацевтической композиции, содержащей:
(а) эффективное количество биоактивного агента для лечения состояния, выбранного из группы, состоящей из лекарственного привыкания, хронического алкоголизма, передозировки лекарств, психического заболевания, кашля, отека легкого, желудочно-кишечных расстройств, артрита, псориаза, астмы, воспалительного заболевания кишечника, расстройств дыхательной функции, функционального заболевания кишечника, синдрома раздраженной кишки, диареи, функционального растяжения, функциональной боли, неязвенной диспепсии, отторжения трансплантированного органа, отторжения кожного трансплантата, сердечных расстройств, психических расстройств, эмоциональных расстройств, когнитивных расстройств; рвоты; угнетения дыхания; угрей и поражений кожи; и
(б) эффективное количество соединения, представляющего собой формулу:
(-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола, или его фармацевтически приемлемой соли или эфиров.
Краткое описание чертежей
На Фиг.1 показано влияние рацемической смеси DPI-1197W92 на аналгезию и угнетение дыхания у тестируемых животных.
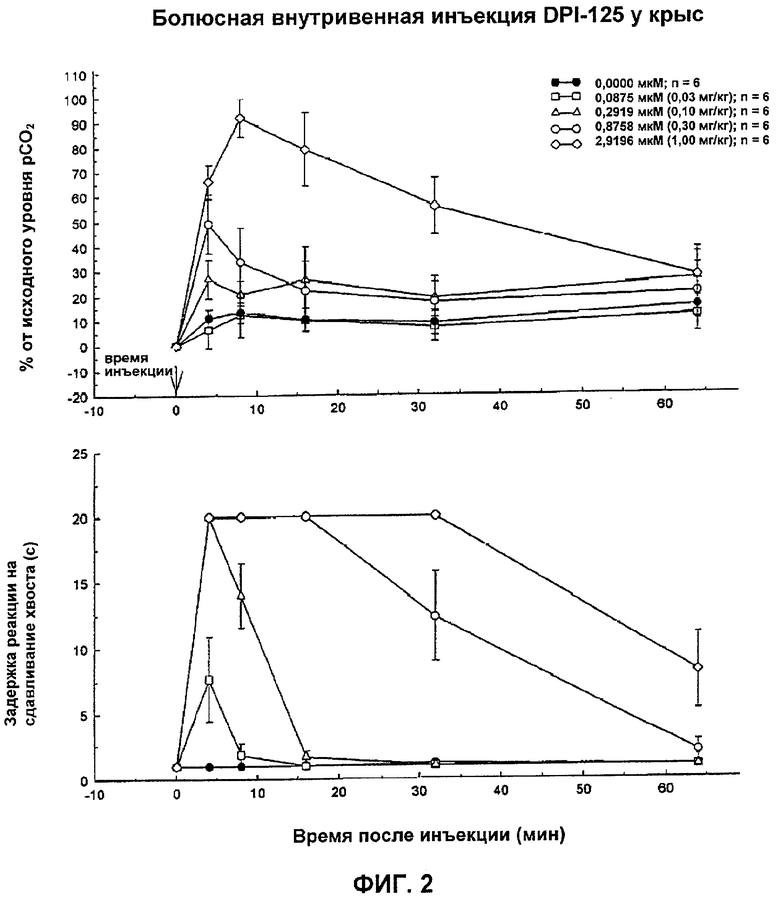
На Фиг.2 показаны высокоэффективные результаты влияния (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола, соединения по настоящему изобретению, на аналгезию и угнетение дыхания у тестируемых животных.
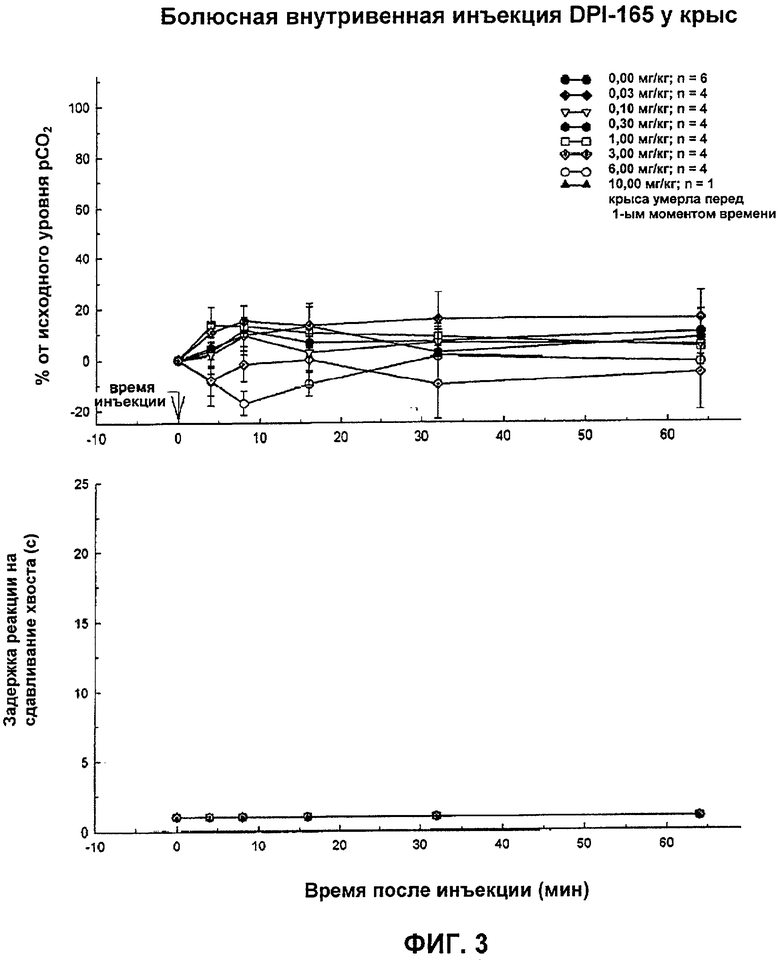
На Фиг.3 показано влияние DPI-165 на аналгезию и угнетение дыхания у тестируемых животных.
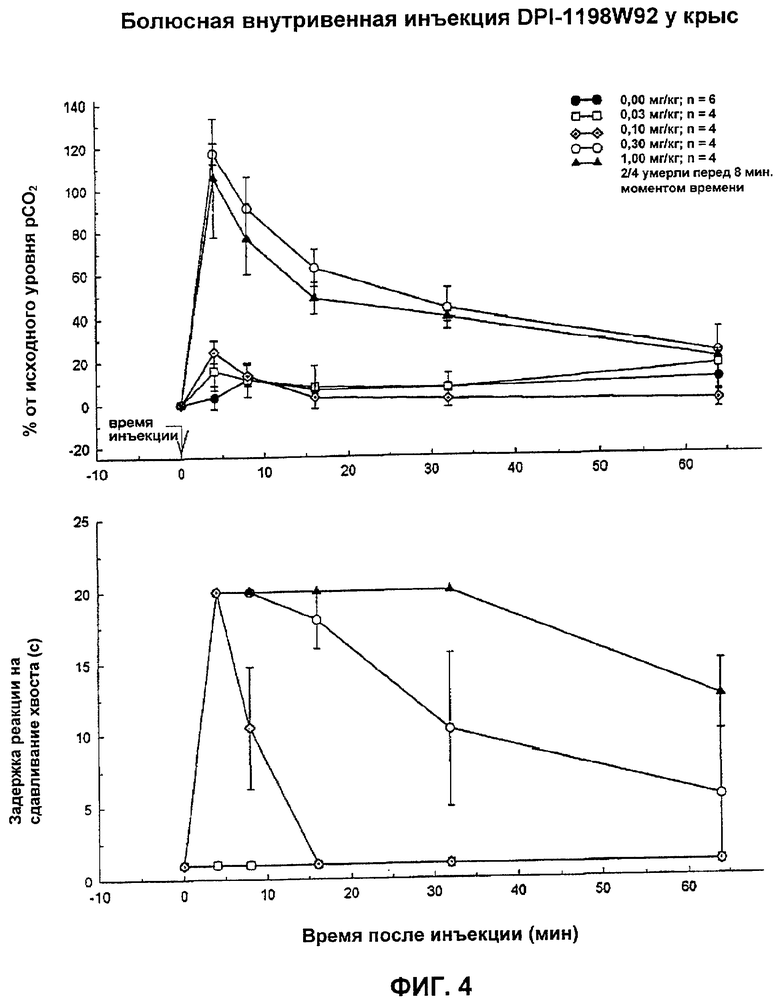
На Фиг.4 показано влияние рацемической смеси DPI-1198W92 на аналгезию и угнетение дыхания у тестируемых животных.
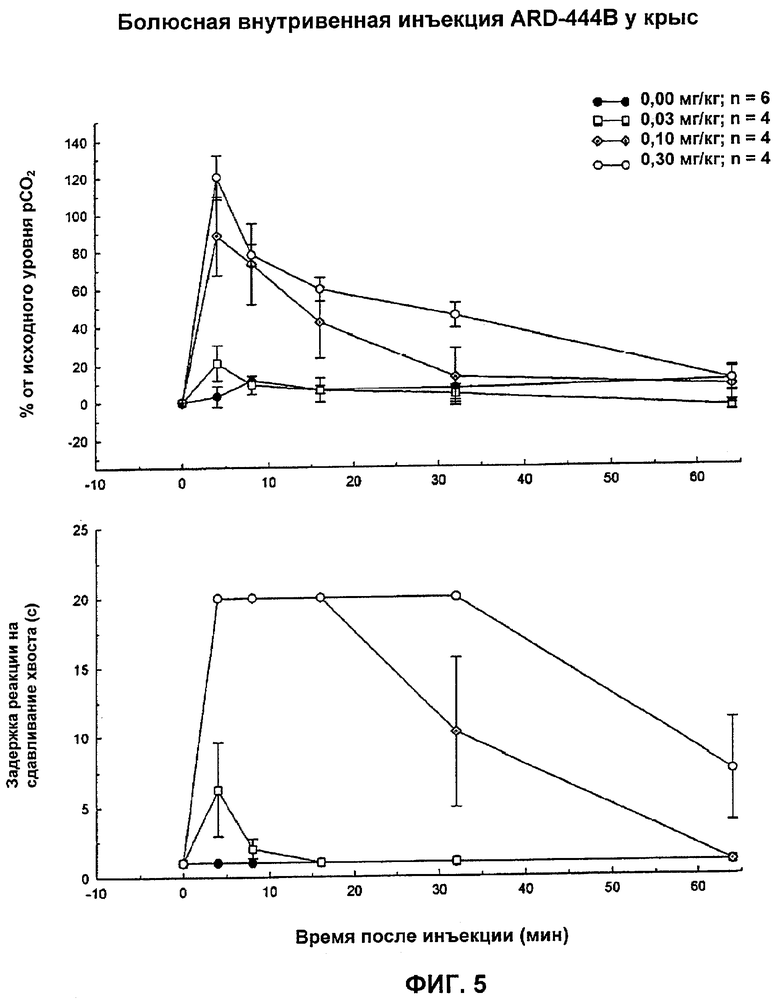
На Фиг.5 показано влияние ARD-444 на аналгезию и угнетение дыхания у тестируемых животных.
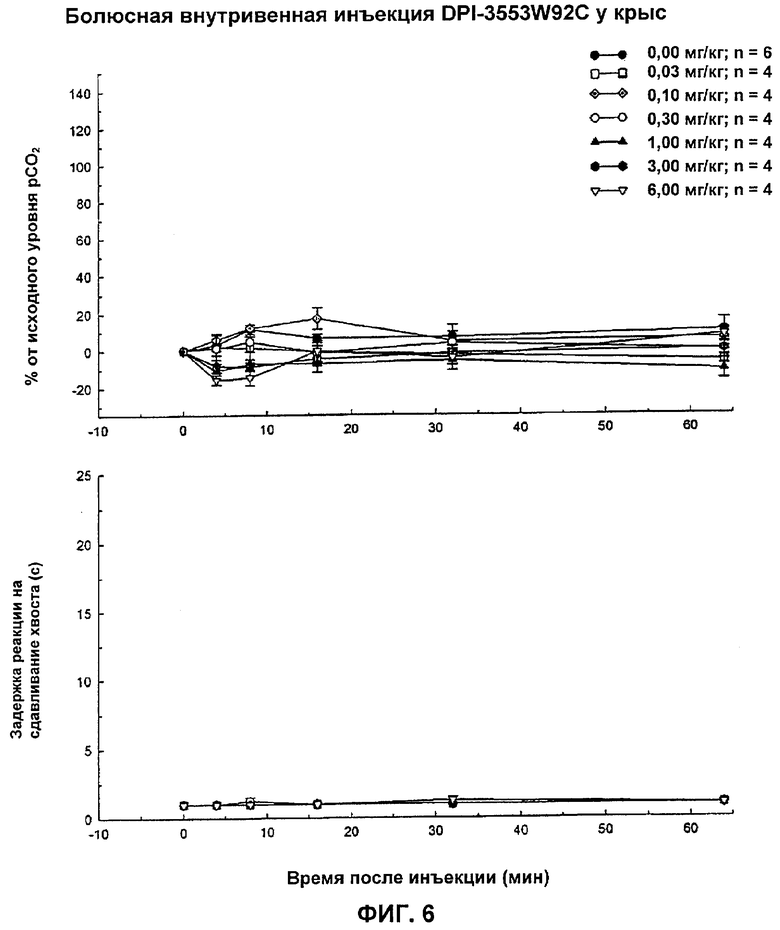
На Фиг.6 показано влияние DPI-3553W92 на аналгезию и угнетение дыхания у тестируемых животных.
Детальное описание изобретения
Настоящее изобретение относится к по существу энантиомерно чистому соединению формулы:
(-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенолу, или его эфирам или солям.
Соединение формулы (I), иногда здесь и далее называемое DPI-125, действует как дельта-опиоидный агонист в дельта-рецепторном подтипе выносящих сосудах мыши, а также как агонист при дельта-рецепторе в головном мозге мыши, подтипе дельта-рецептора, эмпирически отличимом от дельта-рецептора в выносящих сосудах мыши. Это соединение обладает активностью при мю-опиоидном рецепторе и проявляет некоторое сродство к каппа-рецептору.
В in vitro тестах на агонистическую/антагонистическую активность, таких как тесты сродства рецепторного связывания и тесты подавления электрически стимулированных мышечных сокращений, соединение формулы (I) проявляет эффективность на протяжении диапазона от наномолярной до микромолярной концентраций.
Соединение формулы (I) и его эфиры и соли обладают фармацевтической активностью, включая, среди прочего, аналгетическую активность, и полезны при лечении животных, например млекопитающих, таких как люди, для состояний, при которых аналгезия желательна.
Способ лечения боли у животного, нуждающегося в таком лечении, включает в себя введение животному эффективного индуцирующего аналгезию количества соединения формулы (I) или его эфира или соли.
В дополнение, соединение формулы (I) и его эфиры или соли обладают свойственной терапевтической применимостью для лечения состояний, включая предупреждение или лечение воспалительных заболеваний, таких как артрит, псориаз, астма или воспалительное заболевание кишечника, расстройств дыхательной функции, желудочно-кишечных расстройств, таких как функциональное заболевание кишечника, функциональных ЖК (желудочно-кишечные) расстройств, таких как синдром раздраженной кишки, функциональная диарея, функциональное растяжение, функциональная боль, неязвенная диспепсия или другие сопутствующие расстройства моторики или секреции, в качестве анальгетиков для лечения боли, включая не соматическую боль, в качестве иммуносупрессантов для предупреждения отторжения в трансплантате органа и кожном трансплантате, сердечных расстройств, лекарственного привыкания и хронического алкоголизма, передозировки лекарств/алкоголя, психических, эмоциональных и когнитивных расстройств; кашля; отека легкого; рвоты; угнетения дыхания; и желудочно-кишечных расстройств.
Соответственно, в настоящем изобретении рассматривается способ лечения животного субъекта, имеющего такое состояние(я) и нуждающегося в таком лечении, при котором такому животному вводят эффективное количество соединения по настоящему изобретению, которое терапевтически эффективно для указанного состояния.
Субъекты, которых следует лечить способами по настоящему изобретению, включают в себя как субъекты, представляющие собой человека, так и субъекты, представляющие собой животное, не являющееся человеком (например, птица, собака, кошка, корова, лошадь), и представляют собой, предпочтительно млекопитающие субъекты и наиболее предпочтительно человеческие субъекты.
В зависимости от конкретного состояния, которое следует лечить, животным субъектам можно вводить соединение формулы (I) в любых подходящих терапевтически эффективных и безопасных дозировках, которые можно легко определить в пределах квалификации данной области техники, и без лишних экспериментов.
В целом, хотя эффективная дозировка соединения по изобретению для терапевтического применения может широко варьировать при широком применении изобретения, в зависимости от конкретного применения, состояния или вовлеченного болезненного состояния, как легко определить в пределах квалификации данной области, подходящие терапевтические дозы соединения формулы (I) или его эфиров или солей для каждой из соответствующих композиций, описанных здесь, и для достижения терапевтической пользы при лечении каждого из состояний, описанных здесь, типично должны быть в диапазоне от 1 микрограмма (мкг) до 100 миллиграмм (мг) на килограмм массы тела реципиента в сутки, предпочтительно в диапазоне от 5 мкг до 75 мг на килограмм массы тела в сутки, и наиболее предпочтительно в диапазоне от 10 мкг до 50 мг на килограмм массы тела в сутки.
Желаемая доза предпочтительно представлена в виде двух, трех, четырех, пяти, шести или более субдоз, которые вводят с соответствующими интервалами на протяжении дня. Эти субдозы можно вводить в стандартных лекарственных формах, например, содержащих от 10 мкг до 1000 мг, предпочтительно от 50 мкг до 500 мг, более предпочтительно от 50 мкг до 250 мг и наиболее предпочтительно от 50 мкг до 10 мг активного ингредиента на стандартную лекарственную форму. Альтернативно, если состояние реципиента этого требует, дозы можно вводить в виде непрерывной инфузии.
Способ введения и формы дозировки будут, естественно, влиять на терапевтическое количество соединения, которое желаемо и эффективно для данного лечебного применения.
Например, перорально вводимые дозировки типично соответствуют по меньшей мере двукратным, например 2-10-кратным, уровням дозировки, используемым при парентеральных способах введения для того же самого активного ингредиента. При пероральном введении для индукции аналгезии уровни дозировки для соединения(й) по изобретению могут быть порядка 5-200 мг/70 кг массы тела/сутки.
Уровни дозировки для интратекального введения обычно составляют порядка примерно 10% от уровней, характерных для уровней дозировки для парентерального введения. В таблеточных лекарственных формах типичные уровни дозы активного агента, подходящие для индукции аналгезии, составляют порядка 10-100 мг на таблетку.
Соединение формулы (I) можно вводить само по себе, а также в форме фармацевтически приемлемых простых эфиров, сложных эфиров, солей и других его физиологически функциональных производных.
В настоящем изобретении также рассматриваются фармацевтические препараты как для ветеринарного, так и для человеческого медицинского применения, которые содержат в качестве активного агента молекулу формулы (I).
В таких фармацевтических препаратах активный агент предпочтительно используют вместе с одним или более чем одним фармацевтически приемлемым носителем(ями) для него и возможно любыми другими терапевтическими ингредиентами. Носитель(и) должны быть фармацевтически приемлемыми в смысле совместимости с другими ингредиентами препарата и не быть чрезмерно вредными для его реципиента. Активный агент предложен в количестве, эффективном для достижения желаемого фармакологического эффекта, как описано выше, и в количестве, подходящем для достижения желаемой суточной дозы.
Препараты включают в себя таковые, подходящие для парентерального, а также не парентерального введения, и конкретные модальности введения включают в себя пероральное, ректальное, местное, назальное, офтальмическое, подкожное, внутримышечное, внутривенное, чрескожное, спинальное, интратекальное, внутрисуставное, внутриартериальное, субарахноидальное, сублингвальное введение в слизистую оболочку полости рта, бронхиальное, лимфатическое и внутриматочное введение. Препараты, подходящие для парентерального и перорального введения, предпочтительны.
Когда активный агент формулы (I) используют в препарате, представляющем собой жидкий раствор, препарат преимущественно можно вводить парентерально. Когда активный агент используют в препарате жидкой суспензии, препарат можно преимущественно вводить перорально, ректально или бронхиально.
Когда активный агент используют непосредственно в форме порошкообразного твердого вещества, активный агент можно преимущественно вводить перорально или сублингвально. Альтернативно, его можно вводить бронхиально, посредством распыления порошка в газе-носителе, с образованием газообразной дисперсии порошка, которая вдыхается пациентом из дыхательного контура, содержащего подходящее распылительное устройство.
В некоторых применениях может быть преимущественным использовать активное соединение формулы (I) в «векторизованной» форме, как, например, путем инкапсуляции активного агента в липосому или другую инкапсулирующую среду, либо путем фиксации активного соединения, например путем ковалентного связывания, хелатирования или ассоциативной координации, на подходящей биомолекуле, такой как те, которые выбраны из белков, липопротеинов, гликопротеинов и полисахаридов.
Препараты, содержащие активное соединение формулы (I), могут удобным образом быть представлены в стандартных лекарственных формах и могут быть приготовлены с помощью любого из способов, хорошо известных в области фармации. Такие способы обычно включают в себя стадию приведения активного соединения в контакт с носителем, который составляют один или более чем один вспомогательный ингредиент. Обычно препараты готовят путем однородного и тесного приведения активного соединения в контакт с жидким носителем, тонко измельченным твердым носителем, либо обоими, а затем, если необходимо, формование продукта в формы дозировки желаемого препарата.
Препараты по настоящему изобретению, подходящие для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы, облатки, таблетки или лепешки, причем каждая содержит предопределенное количество активного ингредиента в виде порошка или гранул; либо суспензии в водной жидкости или неводной жидкости, такой как сироп, эликсир, эмульсия или доза жидкого лекарства.
Таблетка может быть приготовлена путем прессования или формования, возможно с одним или более чем одним вспомогательными ингредиентами. Прессованные таблетки могут быть приготовлены путем прессования в подходящей машине, причем активное соединение находится в свободно текучей форме, такой как порошок или гранулы, которые возможно смешивают со связующим веществом, дезинтегрантом, смазывающим веществом, инертным разбавителем, поверхностно-активным агентом или освобождающим агентом. Формованные таблетки, состоящие из смеси порошкообразного активного соединения с подходящим носителем, могут быть приготовлены путем формования в подходящей машине.
Сироп может быть приготовлен путем добавления активного соединения к концентрированному водному раствору сахара, например сахарозы, к которому может быть также добавлен любой вспомогательный ингредиент(ы). Такой вспомогательный ингредиент(ы) может включать в себя ароматизаторы, подходящие консерванты, агенты для замедления кристаллизации сахара и агенты для увеличения растворимости любого другого ингредиента, такого как многоатомный спирт, например глицерин или сорбит.
Препараты, подходящие для парентерального введения, представляют собой стерильный водный препарат активного соединения, который предпочтительно изотоничен крови реципиента (например, физиологический солевой раствор). Такие препараты могут включать в себя суспендирующие агенты и загущающие агенты, липосомы или другие системы микрочастиц, которые разработаны, чтобы направлять соединение к компонентам крови или одному или более чем одному органу. Препараты могут быть представлены в форме единичной дозы или множественной дозы.
Препараты назального аэрозоля представляют собой очищенные водные растворы активного соединения формулы (I) с консервантами и изотоническими агентами. Такие препараты предпочтительно подводят до рН и изотонического состояния, совместимого со слизистыми оболочками носа.
Препараты для ректального введения могут быть представлены в виде суппозитория с подходящим носителем, таким как масло какао, гидрогенизированные жиры или гидрогенизированные жирные карбоновые кислоты.
Офтальмические препараты готовят способом, сходным с таковым для назального аэрозоля, за исключением того, что рН и изотонические факторы предпочтительно подводят таким образом, чтобы соответствовать таковым глаза.
Местные препараты содержат активное соединение формулы (I), растворенное или суспендированное в одной или более чем одной среде, такой как минеральное масло, вазелин, многоатомные спирты или другие основы, используемые для местных фармацевтических препаратов.
Чрескожные препараты могут быть приготовлены путем включения активного агента формулы (I) в тиксотропный или желатиновый носитель, такой как целлюлозная среда, например метилцеллюлоза или гидроксиэтилцеллюлоза, причем полученный препарат затем упаковывают в чрескожное устройство, адаптированное, чтобы гарантировать кожный контакт с кожей носителя устройства.
В дополнение к вышеуказанным ингредиентам, препараты по настоящему изобретению могут дополнительно включать в себя один или более чем один вспомогательный ингредиент(ы), выбранный из разбавителей, буферов, ароматизаторов, связующих веществ, дезинтегрантов, поверхностно-активных агентов, загустителей, смазывающих веществ, консервантов (включая антиоксиданты) и тому подобного.
В настоящем изобретении также рассматривается способ получения (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола или его сложного эфира или соли, чтобы синтезировать по существу энантиомерно чистый агонист опиоидного рецептора, который по существу свободен от его стереоизомера.
Такое соединение желательно готовят в по существу чистой энантиомерной форме с энантиочистотой по меньшей мере 98% ЕЕ и наиболее предпочтительно по меньшей мере 99% ЕЕ. Величины энантиомерного избытка (enantiomeric excess) обеспечивают количественную меру избытка процентного количества основного изомера над процентным количеством неосновного (второстепенного) изомера, который присутствует с ним, и могут быть легко определены с помощью подходящих методов, хорошо известных и принятых в данной области техники, как, например, хиральная жидкостная хроматография высокого давления (ВЭЖХ), хиральная газовая хроматография (ГХ), ядерный магнитный резонанс (ЯМР), с использованием хиральных реагентов сдвига, и тому подобное.
Энантиомерно чистое соединение по настоящему изобретению можно вводить совместно с биоактивным агентом, который опосредует угнетение дыхания, таким как агонист мю-рецептора, например различные анальгетики и анестетики и барбитураты. Подавляющее большинство используемых в настоящее время высокоэффективных анальгетиков, включая морфин, алфантанил, морфин-6-глюкоронид, оксиморфон, гидроморфон, оксикодон, гидрокодон, фентанил, меперидин, суфентанил и кодеин, представляют собой соединения, связывающие мю-рецептор. Как хорошо известно, эти соединения, хотя и являются высокоэффективными при опосредовании аналгезии, обладают сопутствующими побочными эффектами, включая подавление дыхания. Применение (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола или его сложного эфира или соли по настоящему изобретению может предупредить, уменьшить, ослабить или даже устранить или обратить состояния, при которых аналгезия индуцирует угнетение дыхания, как, например, угнетение дыхания, представляющее собой побочные эффекты, обычно сопровождающие применение соединений, связывающих мю-рецептор.
Таким образом, в настоящем изобретении рассматривается совместное введение (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола с лекарственными агентами, опосредующими угнетение дыхания, при котором соединение по настоящему изобретению вводят в количестве, эффективном для борьбы, например значительного ослабления и предпочтительно по существу устранения, с угнетением дыхания, присущим применению агента, опосредующего угнетение дыхания.
Таким образом, соединения по настоящему изобретению обладают широкой применимостью в хирургических и клинических медицинских применениях для борьбы с нежелательным эпизодом угнетения дыхания, представляющего собой побочный эффект, для применения таких широко используемых лекарств, как морфин и фентанил.
Дополнительно, в настоящем изобретении предложены фармацевтические композиции, содержащие комбинацию эффективного количества опиатного анальгетика и эффективного количества (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола или его сложного эфира или соли, в композиции для борьбы с эффектом угнетения дыхания агентом, опосредующим угнетение дыхания. Применение заявленного соединения для борьбы с угнетением дыхания и в комбинированном «коктейле» фармацевтических композиций более полно обсуждается ниже.
В такой комбинации опиатный агент (или другое соединение, опосредующее угнетение дыхания) и соединение формулы (I) для борьбы с угнетением дыхания, или его сложный эфир или соль, дозировка опиатного агента для индукции аналгезии и дозировка соединения формулы (I) или его сложного эфира или соли для уменьшения, лечения или предупреждения угнетения дыхания могут быть определены независимо. Независимый контроль дозировок для этих двух функций обеспечивает большую гибкость при лечении конкретных пациентов. Этот независимый контроль является одним из преимуществ комбинированных фармацевтических композиций по настоящему изобретению.
Комбинированные фармацевтические композиции по настоящему изобретению, таким образом, содержат комбинацию (1) эффективного количества терапевтического агента, обладающего (побочным) эффектом угнетения дыхания, например опиатного анальгетика, и (2) эффективного количества (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола или его сложного эфира или соли для уменьшения, лечения или предупреждения угнетения дыхания.
В дополнение к способам опосредования аналгезии и лечения, уменьшения или предупреждения угнетения дыхания, в настоящем изобретении также предложены способы скрининга и характеристики соединений, подавляющих угнетение дыхания, при которых проводят анализы обращения активности соединений-кандидатов, подавляющих угнетение дыхания, которые в рецепторной ткани трансдукционно опосредуют эффект подавления угнетения дыхания в ответ на композицию, угнетающую дыхание.
Анализы обращения активности проводят сравнительным образом, в отсутствии и в присутствии антиподавляющего соединения формулы (I) или его сложного эфира или соли, для определения, обращена ли заметным образом подавляющая (угнетение дыхания) активность соединения-кандидата в рецепторной системе путем присутствия антиподавляющего соединения формулы (1) или его сложного эфира или соли. Если это так, то этот анализ показывает соединение-кандидата, подавляющее угнетение дыхания, как обладающего потенциальной биоэффективностью для подавления эффектов угнетения дыхания, свойственных применению других терапевтических агентов.
Настоящее изобретение дополнительно определено путем ссылки на следующие примеры, описывающие в деталях получение соединения и композиций, содержащих его. Специалистам в данной области должно быть очевидно, что многие модификации могут быть осуществлены на практике без отступления от цели и смысла изобретения.
ПРИМЕР 1
Ниже приведена схема синтеза для получения (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола.
Раствор 3-бромфенола (400 г, 2,31 моль), трет-бутилхлордиметилсилана (391 г, 2,54 моль) и имидазола (346 г, 5,08 моль) в 5000 мл дихлорметана перемешивали в течение ночи при комнатной температуре. Реакционный раствор выливали в 2000 мл воды и слои разделяли. Органический слой промывали 1 N водным раствором гидроксида натрия (3×1500 мл) и водой (2×1500 мл) перед пропусканием через подложку из силикагеля (400 g, диоксид кремния 60, 230-400 меш). Силикагель промывали дихлорметаном (2×500 мл), фильтраты объединяли и растворитель удаляли при пониженном давлении с получением 669 г (98,4%) 3-(бромфенокси)-трет-бутилдиметилсилана в виде прозрачной бледно-желтой жидкости. ЯМР (300 МГц, CDCl3): δ 0.2 (s, 6H); 1.0 (s, 9Н); 6.75 (m, 1H); 7.0 (br s, 1H);7.1 (m, 2H).
3-трет-Бутилдиметилсилилоксифенилмагния бромид образовывался путем медленного добавления смеси 3-бромфенокси-трет-бутилдиметилсилана (118 г, 400 ммоль) и дибромэтана (15 г, 80 ммоль) в 400 мл свободного от ингибитора безводного тетрагидрофурана к раствору магниевой стружки (15,5 г, 640 ммоль) в 800 мл свободного от ингибитора безводного тетрагидрофурана при кипячении с обратным холодильником. После перемешивания в течение одного часа при кипячении с обратным холодильником светло-коричневую прозрачную смесь охлаждали до комнатной температуры.
Дважды перегнанный тиофен-3-карбоксальдегид (2,46 г, 22 ммоль), бензотриазол (2,62 г, 22 ммоль), (2R,5S)-1-аллил-2,5-транс-диметилпиперазин (3,39 г, 22 ммоль, Chirotech Technology, Ltd., Cambridge, England) и пара-толуолсульфоновой кислоты моногидрат (209 мг, 1,1 ммоль) растворяли в 125 мл толуола и нагревали до мягкой дефлегмации. Водотолуоловую азеотропную смесь собирали в ловушку Дина-Старка в течение курса 2,5 часов. Оставшийся растворитель удаляли под вакуумом. Остаток растворяли в 25 мл безводного свободного от ингибитора тетрагидрофурана и к этому добавляли раствор 3-трет-бутилдиметилсилилоксифенилмагния бромида в тетрагидрофуране (125 мл, 0,32 М) в атмосфере азота при 20-25°С.
Реакционную смесь перемешивали при 40°С в течение 2 часов и затем гасили реакцию добавлением 25 мл насыщенного раствора NH4Cl. Добавляли безводный сульфат магния (˜5 г) и Целит (˜10 г). Смесь перемешивали и фильтровали и растворитель удаляли при пониженном давлении. Остаток растворяли в этилацетате (200 мл) и промывали 1N раствором NaOH (3×100 мл), водой (1×100 мл) и солевым раствором (1×100 мл). Раствор затем концентрировали при пониженном давлении.
Темный остаток растворяли в 50 мл безводного тетрагидрофурана и добавляли тетрабутиламмония фторида дигидрат (8,63 г, 33 ммоль). После перемешивания в течение 2 часов реакционную смесь концентрировали и остаток растворяли в 100 мл этилацетата. Смесь экстрагировали разбавленным раствором NaHCO3 (3×75 мл) и водой (1×75 мл). Органический слой разбавляли 100 мл метил-трет-бутилового эфира и экстрагировали 1% раствором лимонной кислоты (3×100 мл). Объединенные водные экстракты фильтровали под вакуумом через 0,45 микронный мембранный фильтр и фильтрат подводили до рН 8,5, используя 50% раствор NaOH, перед тем как его экстрагировали дихлорметаном (2×100 мл). Раствор сушили азеотропно, при концентрировании при пониженном давлении. Полученное стекловидное твердое вещество желто-коричневого цвета (3,6 г, 10,5 ммоль, 47,8%) кристаллизовали из 43 мл смеси 2-пропанола и воды в соотношении 45:55 и перекристаллизовывали из 20 мл смеси 2-пропанола и воды в соотношении 1:1 с получением пушистых белых игольчатых кристаллов (2,1 г, 6,13 ммоль, 28% основываясь на хиральном пиперазине), [α]20=-8,33° (абс. этанол, с=1,0).
1H ЯМР (500 МГц, d6-DMSO): δ 9.32 (s, 1H), 7.44 (dd, J=3,2; 4,9 Гц, 1Н), 7.15 (s, 1H), 7.13 (t, J=8,25 Гц, 1H), 6.98 (d, J=4,9 Гц, 1H), 6.66-6.70 (m, 3H), 5.73-5.81 (m, 1H), 5.15 (d, J=17,1 Гц; 1 Н), 5.09 (d, J=10,5 Гц, 1 Н), 5.02 (s, 1 Н), 3.20 (br d, J=10,2 Гц, 1 Н), 2.78 (dd, J=7,3; 7,5 Гц, 1 Н), 2.68 (dd, J=2,6; 11,3 Гц, 1 Н), 2.59 (dd, J=1; 9,3 Гц, 1Н), 2.44 (brs, 2H), 2.02 (t, J=8,6 Гц, 1H), 1.81 (t, J=8,1 Гц, 1H), 1.09 (d, J=6 Гц, 3H), 0.91 (d, J=6 Гц, 3Н).
Рассчитано для С20Н26N2OS: С, 70,14; Н, 7,65; N, 8,18; S, 9,36%.
Найдено: С, 70,19; Н, 7,58; N, 8,12; S, 9,33%.
Настоящее изобретение включает в себя синтезированное выше соединение и его применение, где соединение формулы (I) обладает неожиданной эффективностью при сравнении с рацемической смесью, включающей его или его энантиомер. На первый взгляд можно предположить, что все энантиомеры и/или рацемические смеси должны были бы обладать сходными in vivo или in vitro профилями, однако это не всегда так, как показано в следующих примерах 2-6.
ПРИМЕР 2
Две стереоизомерно родственные рацемические смеси и входящие в них энантиомеры оценивали на in vitro сродство к опиоидному рецептору в оболочках головного мозга крыс (μ и δ опиоид) и мозжечка морских свинок (κ опиоидный рецептор). Оболочки для связывания радиолиганда готовили либо из целого головного мозга крысы, либо из мозжечка морской свинки, поставляемых Pel-Freeze Biological Inc. (Rogers, AR.). Ткани гомогенизировали в 50 мМ TRIS (Трис[гидрооксиметил]аминометан) буфере (рН 7,4), содержащем 50 мкг/мл ингибитора трипсина из соевых бобов, 1 мМ EDTA (этилендиаминтетрауксусная кислота), и 100 мкМ PMSF (фенилметилсульфонила фторид). Гомогенизированные ткани головного мозга центрифугировали при 500×g в течение 30 минут (4°С) для удаления больших обломков. Супернатант политронно озвучивали в течение 10 секунд (Р.Е. установки 2, 4°С). Затем добавляли раствор сахарозы до конечной концентрации 0,35 M с использованием 10 mM TRIS-сахарозного буфера (pH 7,4) и оболочки головного мозга затем центрифугировали при 40000×g в течение 30 минут (4°С). Капли осадков оболочек затем промывали дважды в 10 мМ TRIS буфере (рН 7,4), содержащем 50 мкг/мл ингибитора трипсина из соевых бобов, 1 мМ EDTA и 100 мкМ PMSF.
Анализы связывания радиолиганда осуществляли в 10 мМ TRIS буфере (рН 7,4), содержащем 50 мкг/мл ингибитора трипсина из соевых бобов, 1 мМ EDTA, 5 мМ MgCl2 и 100 мкМ PMSF. Меченный тритием DAMGO (μ), Deltorphin II (δ) или U69593 (κ), закупленные от New England Nuclear, использовали в качестве лигандов в конкурентных экспериментах (2-3×10-10 М конечные концентрации) с неспецифическим связыванием, определяемым с помощью 0,5×10-6 М Налоксона (Naloxone) (закуплен от SIGMA Chemical Co.). Все анализы связывания осуществляли при комнатной температуре в течение 90 минут, а затем оканчивали быстрым фильтрованием на GF/C стекловолоконных фильтрах (Whatman, Hillsboro, OR) 50 мМ TRIS буфером (4°С, рН 7,4), используя полуавтоматический харвестер клеток Brandel (Model M48, Brandel, Gaithersburg, MD). Фильтры промывали дважды 50 мМ TRIS буфером (4°С, рН 7,4), и эти фильтры помещали в жидкий сцинтилляционный коктейль и связанную радиоактивность подсчитывали на сцинтилляционном счетчике Beckman LS 6500. Эффективность соединений при ингибировании связывания радиомеченых DAMGO (μ), Deltorphin II (δ) или U69593 (κ) определяли из полных кривых концентрация-эффект. С помощью компьютерной программы Prism (GraphPad Software Inc., San Diego, CA) величины ИК50 определяли с использованием одноместного нелинейного регрессионного анализа данных связывания радиолиганда. Величины ИК50 затем переводили в величины Ki, используя уравнение Ченга-Прусофа (Cheng Y and Prusoff W H (1973) Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 percent inhibition (150) of a enzymatic reaction. Biochem Pharm 22:3099-3108.)
Тестировали следующие соединения:
Соединение 1
(DPI-1197W92) рацемическая смесь (±)3-((R*)-((2R*,5S*)-4-алил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола, которая включает в себя энантиомеры (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол и (+)3-((R)-((2R,5S)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол (RRS и SSR).
Соединение 2
(DPI-125) энантиомер по настоящему изобретению (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол (SSR).
Соединение 3
(DPI-165) энантиомер, включенный в Соединение 1, (+)3-((R)-((2R,5S)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол (RRS).
Соединение 4
(DPI-1198W92) рацемическая смесь (±)3-((R*)-((2S*,5R*)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола, которая включает в себя энантиомеры (+)3-((R)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол и (-)3-((S)-((2R,5S)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол (RSR и SRS).
Соединение 5
(ARD-444) энантиомер, включенный в соединение 4: (+)-3-((R)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол (RSR).
Соединение 6
(DPI-3553W92) энантиомер, включенный в соединение 4: (-)3-((S)-((2R,5S)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол (SRS).
Результаты анализов связывания радиолигандов приведены ниже в таблице I:
(RRS и SSR)
Результаты: очевидно, что каждое соединение проявляет четкое и различное сродство связывания к различным типам протестированных рецепторов. Сильное и повышенное сродство соединения DPI-125 к обоим мю- и дельта-рецепторам показано с помощью очень низкой концентрации, необходимой для подавления связывания меченых соединений. Кi DPI-125 составляет приблизительно 1/1000 от Ki его энантиомера DPI-165.
ПРИМЕР 3
Соединение формулы (I) и соединения 1, 3, 4, 5 и 6, как определено выше, оценивали на in vitro активность опиоидного рецептора в различных рецепторных системах, включая выносящие сосуды мыши (Mouse Vas Deferens ЭД50), и подвздошную кишку морской свинки (Guinea Pig lleum ЭД50). Процедуры анализа, используемые для таких определений рецепторной активности, приведены ниже.
In vitro биоанализы: выносящие сосуды мыши ((Mouse vasa deferentia (MVD)), CD-1 штамм, Harlan, Raleigh, NC) выделяли из мыши и суспендировали между платиновыми электродами с 0,5 г напряжения в кюветах для органов, содержащих модифицированный свободный от Mg++ буфер Кребса следующего состава (миллимолярный): NaCl 117,5; KCl 4,75; CaCl2 2,6; КН2PO4 1,20; NaHCO3 24,5; и глюкоза 11. Буфер насыщали 95% O2/5% CO2 и хранили при 37°С. Ткани стимулировали при сверхмаксимальном напряжении серией импульсов 10 Гц в течение 400 мс (миллисекунд), интервал между сериями 10 секунд, и 1,0 мс продолжительностью импульса при максимальном напряжении. Активность дельта рецептора определяли путем добавления соответствующих концентраций тестируемого соединения в кюветы для органов и давая возможность максимального ответа перед добавлением следующей более высокой концентрации. Активность мю-рецептора определяли сходным образом, но в присутствии 3 мкМ TIPP (высокоселективный дельта антагонист; Р.W.Schiller, Т.M.-D.Nguyen, G.Weltrowska, В.С.Wilkes, В.J.Marsden, С.Lemieux, and N.N.Chung, Proc. Natl. Acad. Sci. 89, 11871 (1992)) и 15 нМ нор-BNI (селективный каппа-антагонист; Р.S.Portoghese, A.W.Lipkowski, and A.E.Takemori, Life Sci. 40, 1287 (1987)).
Интактные подвздошные кишки (примерно 3 см длиной) выделяли из морской свинки и суспендировали с 1 г напряжения в кюветах, как описано для выносящих сосудов. Подвздошные кишки стимулировали электрическими прямоугольными импульсами 0,1 Гц с продолжительностью импульса 1 мс при сверх максимальном напряжении.
Процентное ингибирование электрически индуцированных мышечных сокращений определяли для соединений с варьирующими кумулятивными концентрациями. Величины ЭД50 экстраполировали с кривых, показывающих дозовую концентрацию, нанесенную на график против ответа (J.A.H.Lord, A.A.Waterfield, J.Hughes, H.W.Kosterlitz, Nature 267, 495, (1977)). Результаты приведены в таблице II, как показано ниже:
Рацемическая смесь (RRS и SSR)
Рацемическая смесь (RSR и SRS)
ПРИМЕР 4
Аналгезию анализировали у крыс с использованием теста сдавливания хвоста с одновременным мониторингом газов капиллярной крови (pCO2). Во время периода этого тестирования также получали величины угнетения дыхания. Самцов крыс (Wistar Hannover 200-300 г) анестезировали 2% изофлураном (J.A.Webster, Inc., Sterling, Massachusetts). Бедренную артерию канюлировали с помощью РЕ50 трубки для отбора образцов крови. Яремную наружную вену также канюлировали с помощью силиконовой (Silastic) трубки для инъекции лекарства. После операции анестезирующие газы удаляли и крысе давали возможность покоя в пластмассовом ящике для отдыха (рестрейнере) в течение 60 минут для установления базовых величин газов в крови.
Соединения 1-6 вводили внутривенно. Ноцицептивную реакцию и величины дыхания получали в течение 1-2 часового периода. Каннюляцию бедренной артерии использовали для забора артериальной крови в шприц, предварительно смоченный гепарином. Образцы затем анализировали с помощью газового анализатора крови (Ph/Gas Analyzer Synthesis 25 Model, Instrumentation Laboratory) для оценки эффектов угнетения дыхания. Объем крови, отбираемой каждый раз, составлял 0,15 см3. Шприцы вставляли немедленно и газы крови анализировали в течение 5 минут. Кровь, подвергавшуюся действию воздуха на кончике шприца, вытесняли. Кровь перемешивали путем спокойного переворачивания и аликвоты по 0,10 см3 инъецировали в газовый анализатор крови.
Газовый анализатор поддерживался в хорошем состоянии и функционировал. Калибровки (низкая, нормальная и высокая) делали в начале каждого дня тестирования. Линии для образцов, ко-оксиметр и электроды для газов крови регулярно чистили в конце каждого дня тестирования. Калибровку гематокрита (высокую и низкую) осуществляли по плану на недельной основе, а трубку, клапан для образца и сдавливания заменяли на ежемесячной основе.
Гемостатический зажим помещали на хвост (один дюйм от кончика хвоста) в течение короткого периода времени до тех пор, пока не наступала реакция избегания (то есть резкое движение хвоста либо выражение голосом). Время ожидания (задержку) реакции избегания регистрировали с помощью секундомера. Предельное время 20 с использовали для предупреждения ненужного повреждения ткани. Крыс наблюдали на ноцицептивные реакции выражения голосом или болезненных движений тела. Общее время, затраченное, чтобы вызвать болевую реакцию, регистрировали как задержку сдавливания хвоста, в секундах. Мониторинг газов крови осуществляли приблизительно в те же самые моменты времени, что и тест сдавливания хвоста.
Величины ЭД50 для эффективности аналгезии и угнетения дыхания определяли, чтобы вычислить индекс безопасности или терапевтический индекс, который определяют как ЭД50 угнетения дыхания, деленная на ЭД50 аналгезии. Анальгетическую эффективность (половину максимальной эффективной дозы, ЭД50) определяли с помощью дозы, при которой половина животных не показывала никакой ноцицептивной реакции на давление гемостатического зажима в течение 20 секунд. Как показано в таблице III, индекс безопасности (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола неожиданно много больше, чем любой из рацемических смесей и любого из других протестированных энантиомеров.
угнетения дыхания, мг/кг
Как можно видеть благодаря вышеуказанным результатам, индекс безопасности для (-)3-((S)-((2S,5R)-4-аллил-2,5-метил-1-пиперазинил)(3-тиенил)метил)фенола по меньшей мере в три (3) раза больше, чем любой из рацемических смесей, и по меньшей мере в шесть раз больше, чем любого из других протестированных энантиомерных соединений. Таким образом, применение (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола обеспечивает благотворные эффекты аналгезии с существенно сниженным риском угнетения дыхания.
ПРИМЕР 5
Результаты теста сдавливания хвоста и уровней CO2, определенных из отобранных образцов крови, приведены на графиках 1-6. Следующая Таблица IV иллюстрирует результаты в простом формате, чтобы показать неожиданную и превосходную эффективность заявленного в настоящем изобретении энантиомерного соединения (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола.
DPI-1197W92
Рацемическая смесь (RRS и SSR)
DPI-125
Заявленный энантиомер (SSR)
DPI-165
Энантиомер (RRS)
DPI-1198W92
Рацемическая смесь (RSR и SRS)
ARD-444
Энантиомер (RSR)
DPI-3553W92
Энантиомер (SRS)
Как показано на Фигурах 1-6, энантиомерно чистое заявленное соединение (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол (DPI-125) безопасно поддерживает аналгезию в течение продолжительного времени без летального угнетения дыхания. Двумя другими соединениями, которые поддерживают аналгезию в течение более 32 минут, были рацемическая смесь DPI-1198W92 (Соединение 4 (RSR и SRS)) и один из ее компонентов ARD-444 (Соединение 5, RSR), но некоторые из тестируемых субъектов умерли во время режима тестирования вследствие полного угнетения дыхания (дыхательная недостаточность). DPI-165 (Соединение 3 (RRS)) и DPI-3553W92 (Соединение 6 (SRS)) не давали никакого измеримого анальгетического эффекта и никакого эффекта на pCO2 уровни в крови и, следовательно, никакого угнетения дыхания. DPI-1197W92 (Соединение 1 (RRS и SSR)) давало только ограниченную аналгезию в течение короткого периода времени и в течение 32 минут на протяжении периода тестирования никакого анальгетического эффекта не сохранялось. Эти результаты четко показывают неожиданную эффективность заявленного соединения по отношению к другим тестируемым соединениям. (-)3-((S)-((2S,5R)-4-Аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол обеспечивает продолжительную аналгезию сверх любого другого тестируемого соединения с существенно более низким угнетением дыхания и смертностью при анальгетическом эффекте одинаковой эффективности.
ПРИМЕР 6
Эксперименты осуществляли для определения эффектов заявленного соединения (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола на угнетение дыхания и аналгезию, индуцированную внутривенной инфузией алфентанила или фентанила, оба из которых являются сильнодействующими мю-агонистами. Эффекты угнетения дыхания измеряли с помощью анализа газов крови крысы на pCO2 уровни. Образцы крови крыс отбирали и анализировали на содержание CO2 после непрерывной внутривенной инфузии алфентанила (6 мг/мин) и внутривенной болюсной инъекции различных доз заявленного соединения (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола.
За уровнями CO2 в крови наблюдали в качестве показателя угнетения дыхания как результат введения алфентанила и (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола (DPI-125). Аналгезию также оценивали с помощью способа сдавливания хвоста в те же самые моменты времени, когда отбирали кровь для определения блокады угнетения дыхания с помощью (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола (DPI-125), но не аналгезию, индуцированную алфентанилом.
В целом, заявленное соединение (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол клинически полезно при операционном, послеоперационном применении и применении при хронической боли для ослабления угнетения дыхания и поддержания анальгетических эффектов анальгетиков мю опиоидных рецепторов.
ПРИМЕР 7
Далее описана процедура, которая обеспечивает наблюдения за рвотными эффектами (-)-3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола (DPI-125). Трем взрослым кобелям гончей независимо вводили (-)-3-((S)-((2S, 5R,)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол (DPI-125) в увеличивающихся дозировках. Дозы выбирали, чтобы получить уровни в крови, согласующиеся с аналгезией от сильной до чрезмерной, и которые могли бы обеспечить очевидные фармакологические эффекты, включая летаргию и седацию. Соединение растворяли в стерильном 5% растворе декстрозы, который забуферивали с помощью буфера уксусная кислота/ацетат натрия, и вводили внутривенно в медленной болюсной инъекции в течение 1-2 минут. Сначала трем собакам вводили дозировки тестируемого объема забуференной 5% декстрозы, чтобы убедиться в отсутствии реакции на растворитель. Во-вторых, (-)3-((S)-((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол (DPI-125) вводили в однократных инъекциях в увеличивающихся дозировках 0,1, 0,3 и 0,5 мг/кг с промежуточным семидневным периодом перед дачей следующей более высокой дозы. Никаких позывов на рвоту или рвоты не наблюдалось ни у одной из собак при любых дозах.
Тошнота и рвота являются обычными и ожидаемыми вредными последствиями традиционных мю-опиатов, таких как морфин и фентанил, а также для смешанных дельта/мю-опиоидных анальгетиков как у собак, так и у людей. Собаки рассматриваются в качестве вида, особенно чувствительного к прорвотным эффектам опиатов. Из результатов вышеуказанного теста очевидно, что фармацевтическая композиция по настоящему изобретению, содержащая (-)-3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол (DPI-125) превосходна по безопасности и неожиданно устраненным негативным побочным эффектах тошноты и рвоты, которые обычно имеют место при введении опиоидного анальгетика.
Обычно мю-агонисты дают существенный благотворный эффект анальгезии и много вредных побочных эффектов, таких как угнетение дыхания, тошнота, привыкание и зависимость. Возможность применения соединения (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола для блокирования нежелательных побочных эффектов мю-агонистов позволяет лечащим врачам увеличивать введение анальгетиков благодаря уменьшенному беспокойству относительно угнетения дыхания. Пациенты испытывают меньшую боль после операции и нуждаются в меньшем послеоперационном уходе со стороны персонала больницы. Общий образ жизни пациентов, принимающих мю-опиоиды, может быть значительно улучшен с помощью параллельного применения соединения (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола.
Лечение сердечных расстройств
Для проиллюстрирации того, что (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил(3-тиенил)метил)фенол обладает кардиозащитным действием при использовании в программе предварительного лечения, самцов крыс обезболивали уретаном (1,2 г/кг интраперитонеально). Когда достигался хирургический уровень, осуществляли трахеотомию (трубка-240), и животным вводили катетер (трубка-50) в яремную вену (для внутривенного введения соединения и красителя) и в сонную артерию (для измерения кровяного давления). Животное помещали в аппарат для исскуственной вентиляции лугких (Harvard, модель 683), присоединенный к источнику O2. Дыхание осуществлялось в темпе 38-42 вдоха в минуту. Катетер сонной артерии был присоединен к датчику давления РТ300 через 3-ходовой шприцевой клапан для измерения артериального кровяного давления и частоты сердечных сокращений.
Торакотомию осуществляли слева в 5-м межреберном пространстве, с последующей перикардотомией и регулировкой левого ушка предсердия, чтобы выявить положение левой коронарной артерии. Через внутривенный катетер вводили энантиомер (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил(3-тиенил)метил)фенол (DPI-125) или 5%-ную глюкозу (контроль носителя). Лигатуру пропускали ниже левого ушка предсердия к правой части левого желудочка. Концы швов продевали через полиэтиленовую трубку с фланцами на одном конце (так что конец с фланцем был расположен близко к стенке желудочка) с образованием петли. Через пять минут после введения лекарственного средства коронарную артерию пережимали, туго натягивая нить и зажимая петлю на эпикардиальной поверхности кровоостанавливающим зажимом. Окклюзию коронарной артерии контролировали по цианозу эпикарда и уменьшению кровяного давления. Окклюзия продолжалась в течение 30 минут. Реперфузию инициировали, растягивая петлю, и подтверждали визуально по гиперемическому отклику эпикарда. Период реперфузии составлял 60-90 минут. В конце реперфузионного периода коронарную артерию снова пережимали, используя петлю, и через внутривенный катетер вводили краситель патентованный синий (0,4 мл 10%-ного мас./об. раствора в физиологическом растворе). Сердце извлекали немедленно после того, как краситель распространялся с кровотоком. Предсердие и левый желудочек быстро отделяли, и оставшийся левый желудочек рассекали на 4-5 срезов. Области, определяемые как нормальные (окрашенные синим), отделяли от областей риска (AAR, не окрашенные синим), и ткани помещали в отдельные 20 мл сосуды, содержащие 100 мМ KH2PO4 и 0,187% 2,3,5-трифенилтетразолия хлорида (ТТС), и инкубировали при 37°С в течение 5-10 минут. ТТС использовали как индикатор для разделения жизнеспособной и нежизнеспособной ткани (Klein H.H., et al., Virchows Arch. (1981) 393:287-297). Эта методика позволяла визуализировать нормальную, неишемизированную область, область риска (AAR) и величину инфаркта. Ткани затем помещали в отдельные сосуды, содержащие 10%-ный забуференный раствор формальдегида, на ночь для фиксации. Зоны инфаркта отсекали от неинфарктных зон. Неинфарктные и инфарктные ткани взвешивали, и величину инфаркта (IS) рассчитывали как процент (%) от площади риска.
Энантиомерное соединение по настоящему изобретению (30 мгк/кг и 50 мкг/кг) сравнивали с фентанилом в концентрации 5 мкг/кг, которая является приблизительно двойным анальгетическим эквивалентом дозе 50 мкг/кг DPI-125 (две анальгетические эффективные дозы ED50) и максимальной дозой фентанила, которую можно вводить безопасно. Анальгетическая ЭД50 для смешанного мю-дельта анальгетика DPI-125 у крыс составляет 50 мкг/кг. Концентрации лекарственного средства в носителе регулировали так, чтобы все испытываемые инъекции выполнялись с объемом 1,0 мл/кг.
Результаты представлены ниже в Таблице I. Как видно, энантиомерное диарилметилпиперазиновое соединение по настоящему изобретению (DPI-125) обеспечивает статистически значимый кардиозащитный эффект в дозе 50 мг/кг. Фентанил не обладал никаким эффектом по сравнению с контролем. Таким образом, величины инфаркта значительно уменьшалась относительно контрольной группы. Более конкретно, предварительная обработка 50 мкг/кг DPI-125 вызывала 35%-ное уменьшение величины инфаркта по сравнению с величиной инфаркта, наблюдаемой после предварительной обработки фентанилом или 5%-ной глюкозой (контроль носителя).
Таким образом, результаты показывают, что энантиомерное диарилметилпиперазиновое соединение по настоящему изобретению обеспечивает кардиозащиту, и что сердечная мышца при ишемии повреждается меньше, когда животных предварительно обрабатывают (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил(3-тиенил)метил)фенолом (DPI-125), заявленным в настоящем изобретении.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ОСЛАБЛЕНИЯ РЕСПИРАТОРНОЙ ДЕПРЕССИИ И СОПУТСТВУЮЩИХ ПОБОЧНЫХ ЭФФЕКТОВ МЮ-ОПИОИДНЫХ СОЕДИНЕНИЙ | 1997 |
|
RU2201231C2 |
| СПОСОБ ВОССТАНОВЛЕНИЯ НАРУШЕННЫХ ФУНКЦИЙ ОПИОИДНЫХ РЕЦЕПТОРОВ | 1996 |
|
RU2307833C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА ИЛИ ПИПЕРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2194702C2 |
| 4-ЗАМЕЩЕННЫЕ-2-ФЕНОКСИФЕНИЛАМИНОВЫЕ МОДУЛЯТОРЫ ДЕЛЬТА-ОПИОИДНЫХ РЕЦЕПТОРОВ | 2010 |
|
RU2553453C2 |
| ПИРАЗИНЫ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ МОДУЛЯТОРОВ ДЕЛЬТА-ОПИОИДНЫХ РЕЦЕПТОРОВ | 2010 |
|
RU2543484C2 |
| ПРИМЕНЕНИЕ СИГМА-ЛИГАНДОВ ПРИ ИНДУЦИРОВАННОЙ ОПИОИДАМИ ГИПЕРАЛГЕЗИИ | 2011 |
|
RU2589899C2 |
| 3,4,4-ТРЕХЗАМЕЩЕННЫЕ ПИПЕРИДИНИЛ-N-АЛКИЛКАРБОКСИЛАТЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2145958C1 |
| НОВЫЕ СОЕДИНЕНИЯ | 2000 |
|
RU2258703C2 |
| КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ АГОНИСТОМ ТОЛЛ-ПОДОБНОГО РЕЦЕПТОРА (TLR7) И ИНГИБИТОРОМ СБОРКИ КАПСИДА ВИРУСА ГЕПАТИТА В | 2016 |
|
RU2718917C2 |
| ПРОИЗВОДНЫЕ 3-КАРБОКСИПРОПИЛ-АМИНОТЕТРАЛИНА И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АНТАГОНИСТОВ MU-ОПИОИДНОГО РЕЦЕПТОРА | 2008 |
|
RU2482107C2 |
Изобретение относится к области медицины и фармакологии и касается нового энантиомерно чистого (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола, содержащей его фармацевтической композиции, обладающей агонистической активностью в отношении мю- и/или дельта-опиоидных рецепторов, способов лечения боли, кашля, функциональной диареи, функциональной боли, сердечных расстройств, рвоты, способа осуществления рецептор-опосредованной анальгезии, способа уменьшения, лечения и предупреждения опосредованного лекарством угнетения дыхания, способа скрининга соединений, подавляющих опиоидное угнетение дыхания, фармацевтической композиции, содержащей биоактивный агент для лечения кашля, диареи, функциональной боли, сердечных расстройств, рвоты, угнетения дыхания и (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенол, и способа осуществления реакции, опосредованной опиодными рецепторами. Изобретение обеспечивает повышенную эффективность лечения. 12 н. и 5 з.п. ф-лы, 6 ил., 4 табл.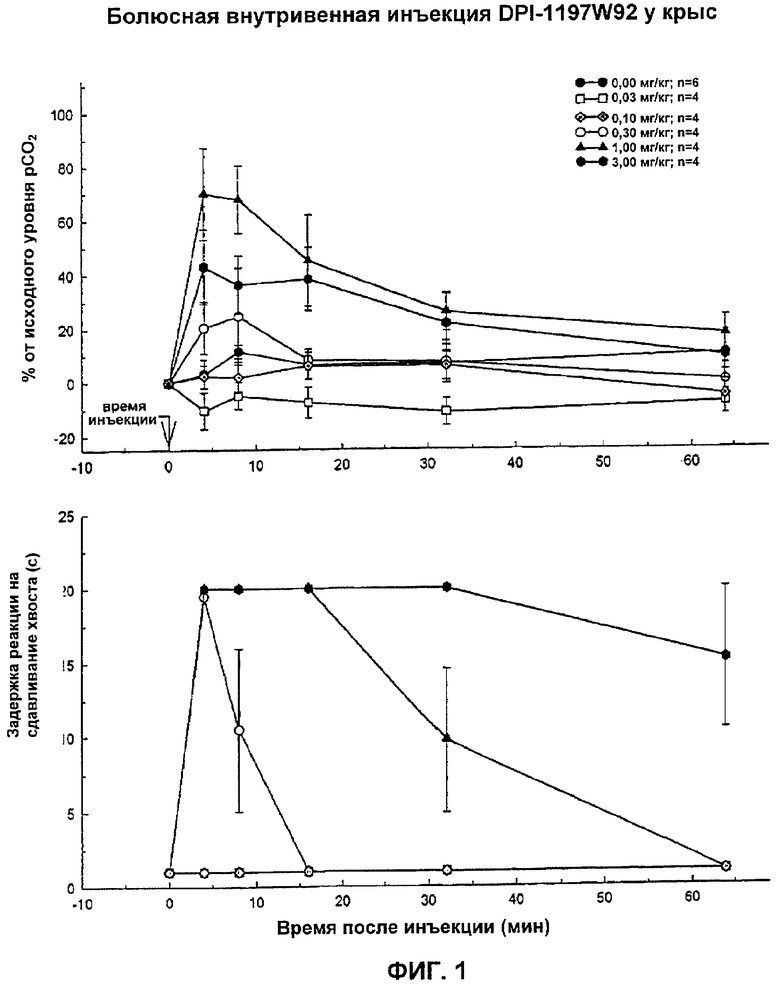
или его фармацевтически приемлемый эфир или соль.
(1) эффективное количество агента, связывающегося с мю-опиоидными рецепторами, который воздействует на мю-опиоидные рецепторы и обладает побочным эффектом угнетения дыхания, и
(2) эффективное количество (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола формулы
или его фармацевтически приемлемого сложного эфира или соли, причем это соединение действует как ослабляющее побочный эффект угнетения дыхания агентом, связывающимся с мю-опиоидными рецепторами, без устранения терапевтической эффективности этого агента, связывающегося с мю-опиоидными рецепторами.
или его фармацевтически приемлемого сложного эфира или соли, для определения, обращена ли существенно активность соединения-кандидата в тканевом сайте из-за присутствия антиподавляющего соединения формулы (I), таким образом показывая соединение-кандидата, подавляющего угнетение дыхания, как обладающего потенциальной биоэффективностью для подавления эффектов угнетения дыхания, свойственных применению других терапевтических агентов.
(а) эффективное количество биоактивного агента для лечения состояния, выбранного из группы, состоящей из кашля, диареи, функциональной боли, сердечных расстройств, рвоты, угнетения дыхания, и
(б) эффективное количество (-)3-((S)-((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)(3-тиенил)метил)фенола формулы
или его фармацевтически приемлемого сложного эфира или соли.
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| US 5658908, А, 19.04.1997 | |||
| US 5854249, А, 29.12.1998 | |||
| RU 2133744, C1, 27.07.1999. | |||

