УРОВЕНЬ ТЕХНИКИ
Область изобретения
Изобретение относится к соединениям 3-карбоксипропил-аминотетралина, которые могут быть использованы в качестве антагонистов mu-опиоидного рецептора. Изобретение также относится к фармацевтическим композициям, включающим такие соединения, к способам применения таких соединений для лечения или облегчения медицинских состояний, опосредуемых активностью mu-опиоидного рецептора, и к способам и промежуточным соединениям, которые могут быть использованы для получения таких соединений.
Уровень техники
Теперь стало в целом понятно, что эндогенные опиоиды играют сложную роль в физиологии желудочно-кишечного тракта. Рецепторы опиоидов экспрессируются в организме повсеместно, как в центральной нервной системе, так и в периферических областях, включая желудочно-кишечный тракт (ЖКТ).
Соединения, которые функционируют как агонисты опиоидных рецепторов, примером-прототипом которых является морфин, являются основами анальгетической терапии для лечения боли, имеющей интенсивность от умеренной до тяжелой. К сожалению, использование аналгезирующих опиоидных средств часто сочетается с неблагоприятными эффектами в отношении ЖКТ, в целом называемыми индуцированной опиоидами дисфункцией кишечника (OBD). OBD включает такие симптомы, как запор, сниженное опорожнение желудка, боль и дискомфорт в животе, вздутие, тошнота и гастроэзофагеальный рефлюкс. Как центральные, так и периферические опиоидные рецепторы вероятно участвуют в замедлении гастроинтестинального транзита после применения опиоида. Однако ряд данных свидетельствует, что периферические опиоидные рецепторы в ЖКТ прежде всего ответственны за неблагоприятные эффекты опиоидов на функционирование ЖКТ.
Так как побочные эффекты опиоидов преимущественно опосредуются периферическими рецепторами, тогда как аналгезия является центральной по происхождению, периферический селективный антагонист может потенциально блокировать нежелательные гастроинтестинальные побочные эффекты, не препятствуя полезным центральным эффектам аналгезии или ослабляя синдром отмены на уровне центральной нервной системы.
Из трех главных подтипов опиоидного рецептора, обозначаемых как mu, delta и kappa, большинство клинически используемых опиоидных анальгетиков оказывают анальгетическое действие и изменяют моторику ЖКТ предположительно через активацию mu-опиоидного рецептора. Соответственно, периферические селективные антагонисты mu-опиоида, как ожидают, будут пригодны для лечения вызываемой опиоидами дисфункции кишечника. Предпочтительные средства будут демонстрировать значительное связывание на mu-опиоидных рецепторах in vitro и будут активны in vivo в гастроинтестинальных моделях животных.
Послеоперационная непроходимость кишечника (POI) представляет собой нарушение, связанное со сниженной моторикой ЖКТ, которое встречается после брюшной или другой хирургии. Симптомы POI подобны таковым OBD. Кроме того, так как пациенты хирургии в течение и после хирургической операции часто получают лечение опиоидными анальгетиками, продолжительность POI может сопровождаться снижением моторики ЖКТ, связанной с использованием опиоидов. Поэтому также ожидается, что антагонисты mu-опиоидов, пригодные для лечения OBD, будут также пригодны для лечения POI.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Изобретение относится к новым соединениям, которые обладают активностью антагонистов mu-опиоидного рецептора, и к промежуточным соединениям для их получения.
Соответственно, изобретение относится к соединению формулы (I):
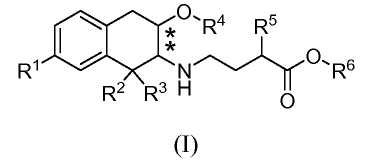
в которой
R1 обозначает -ORa или -C(O)NRbRc;
R2, R3 и R4 обозначают, каждый независимо, C1-3алкил;
R5 выбран из C1-6алкила, фенила, циклогексила, -(CH2)1-3-циклогексила и
-(CH2)1-3-фенила;
Ra, Rb и Rc обозначают, каждый независимо, водород или C1-3алкил; и
R6 обозначает водород или C1-3алкил; и
в которой заместители в хиральных центрах, отмеченных звездочками, находятся в конфигурации транс;
или к его фармацевтически приемлемой соли.
Изобретение также относится к фармацевтической композиции, включающей соединение по изобретению и фармацевтически приемлемый носитель.
Изобретение также относится к способу лечения заболевания или состояния, связанного с активностью mu-опиоидного рецептора, например нарушения, связанного со сниженной моторикой желудочно-кишечного тракта, такого как вызванная опиоидом дисфункция кишечника и послеоперационная непроходимость кишечника, включающему введение млекопитающему терапевтически эффективного количества соединения или фармацевтической композиции по изобретению.
Соединения по изобретению могут также использоваться в качестве инструментальных средств для исследования, то есть для изучения биологических систем или проб, или для изучения активности других химических соединений. Соответственно, в другом из аспектов способа изобретение относится к способу применения соединения формулы (I), или его фармацевтически приемлемой соли, в качестве инструмента для исследования в целях изучения биологической системы или пробы, или для обнаружения новых соединений, имеющих активность mu-опиоидного рецептора, включающему введение биологической системы или пробы в контакт с соединением по изобретению и определения эффектов, вызванных соединением на биологической системе или пробе.
В отдельных и различных аспектах изобретение также относится к способам синтеза и к промежуточным соединениям, описанным здесь, которые могут быть использованы для получения соединений по изобретению.
Изобретение также относится к соединению по изобретению, как описано здесь, для применения в медицинской терапии, а также к применению соединения по изобретению в получении состава или лекарственного средства для лечения заболевания или состояния, связанного с активностью mu-опиоидного рецептора, например нарушения, связанного со сниженной моторикой желудочно-кишечного тракта, у млекопитающего.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение относится к 3-карбоксипропил-аминотетралиновым антагонистам mu-опиоидного рецептора формулы (I), к их фармацевтически приемлемым солям и к промежуточным соединениям для их получения. Следующие заместители и значения представляют собой репрезентативные примеры различных аспектов этого изобретения. Эти репрезентативные значения дополнительно определяют такие аспекты и не предназначены для исключения других значений или для ограничения объема изобретения.
В частном аспекте R1 обозначает -ORa или -C(O)NRbRc.
В другом частном аспекте R1 обозначает -OH или -C(O)NH2.
В еще одном частном аспекте R1 обозначает -C(O)NH2.
В частном аспекте R2, R3 и R4 обозначают, каждый независимо, C1-3алкил.
В другом частном аспекте R2 и R3 обозначают, каждый независимо, метил или этил.
В других аспектах R2 и R3 обозначают этил; или R2 и R3 обозначают метил.
В частном аспекте R4 обозначает метил.
В частном аспекте R5 выбран из C1-6алкила, фенила, циклогексила, -(CH2)1-3-циклогексила и -(CH2)1-3-фенила.
В другом частном аспекте R5 выбран из C3-5алкила, циклогексила, -(CH2)1-3-циклогексила и -(CH2)1-3-фенила. Репрезентативные группы представителя R5 в рамках этого аспекта включают, но не ограничены ими, н-пентил, н-бутил, 2,2-диметилпропил, 2-метилпропил, 1-метилэтил, циклогексил, циклогексилметил, 4-фенилбутил и фенилметил.
В еще одном частном аспекте R5 обозначает циклогексилметил.
В частном аспекте R6 обозначает водород или C1-3алкил.
В другом аспекте R6 обозначает водород, то есть соединения представляют собой карбоновые кислоты.
Было показано, что карбоновые кислоты по изобретения являются мощными антагонистами mu-опиоидного рецептора.
В других аспектах R6 обозначает C1-3алкил, или R6 обозначает метил, то есть соединения представляют собой сложные эфиры.
Как описано ниже, сложные эфиры по изобретению являются полезными промежуточными соединениями для получения карбоновых кислот по изобретению. Кроме того, было показано, что сложноэфирные соединения, в которых R1 обозначает -C(O)NH2, R2 и R3 обозначают этил, R4 обозначает метил, R5 обозначает 2-метилпропил или циклогексилметил, и R6 обозначает метил, являются мощными антагонистами mu-опиоидного рецептора.
Изобретение также относится к соединениям из Примеров 1-16.
Используемый здесь принцип присвоения химических названий проиллюстрирован для соединения Примера 1
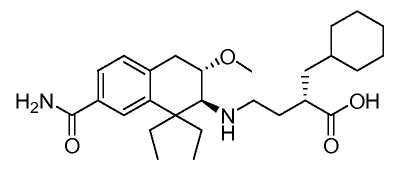
которое представляет собой (S)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидронафталин-2-иламино)-2-(циклогексилметил)масляную кислоту согласно соглашениям IUPAC, используемым в программном обеспечении AutoNom (MDL Information Systems, GmbH, Франкфурт, Германия). Для удобства, бициклическая 1,2,3,4-тетрагидронафталин-2-иламиногруппа альтернативно указана здесь под обычным названием "аминотетралин".
Все соединения по изобретению находятся в конфигурации транс относительно двух хиральных центров, обозначенных звездочками в формуле (I):
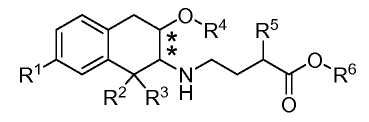
В дополнение к стереохимии аминотетралиновой группы, соединения по изобретению могут содержать хиральный центр в атоме углерода, к которому присоединен заместитель R5. Соединения могут быть чистым диастереомером, например (2S),(3S) диастереомер соединения Примера 1, изображенного выше, или смесями (2S),(3S) диастереомера и (2R),(3R) диастереомера. Такие диастереомерные смеси обозначены здесь приставкой транс. Соответственно, изобретение включает чистые диастереомеры, смеси диастереомеров, рацемические смеси и стереоизомерно обогащенные смеси изомеров, если не указано иное. Когда стереохимия соединения определена, специалисту будет понято, что незначительные количества других стереоизомеров могут присутствовать в композициях по изобретению, если не указано иное, при условии, что пригодность композиции в каком-либо качестве в целом не исключается в силу присутствия таких других изомеров.
В другом аспекте изобретение относится к соединению формулы (Ia):
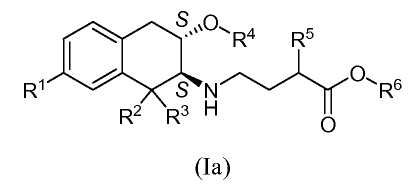
в котором стереохимия в хиральных центрах представляет собой (2S),(3S) и в котором R1, R2, R3, R4, R5 и R6 принимают любое из значений, описанных выше.
В частном аспекте изобретение относится к соединению формулы (Ia), в которой:
R1 обозначает -C(O)NH2;
R2 и R3 обозначают этил;
R4 обозначает метил;
R5 выбран из C3-5алкила, циклогексила, -(CH2)1-3-циклогексила и -(CH2)1-3-фенила; и
R6 обозначает водород или метил;
или к его фармацевтически приемлемой соли.
При введении млекопитающему, медицинские соединения обычно метаболизируются в организме до форм, которые могут быть выделены. Как описано ниже в разделе примеров, метаболическое превращение соединений по изобретению исследовали, инкубируя соединение по изобретению с сохраненными замораживанием человеческими гепатоцитами и сравнивая полученные метаболиты с соединениями известной структуры. Полученные результаты подтверждают заключение, что основной метаболит гидроксила соединения из Примера 1 замещен гидроксилом в положении 4 циклогексильного кольца.
Поэтому в еще одном аспекте изобретение относится к соединению формулы (Ib), в которой R обозначает гидроксил:
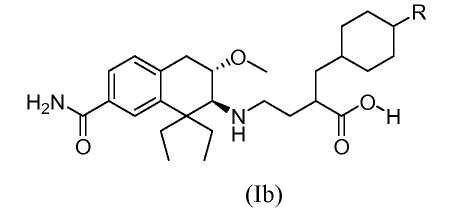
причем соединение формулы (Ib), в которой R обозначает гидроксил, получен in vivo путем введения человеку соединения формулы (Ib), в которой R является водородом.
Определения
При описании соединений, композиций и способов по изобретению следующие термины имеют следующие значения, если не указано иное.
Термин "алкил" означает одновалентную насыщенную углеводородную группу, которая может быть прямой или разветвленной или представлять собой комбинации этого. Если не указано иное, такие алкильные группы обычно содержат от 1 до 10 атомов углерода. Репрезентативные алкильные группы включают, например, метил, этил, н-пропил (n-Pr), изопропил (i-Pr), н-бутил (n-Bu), втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, 2,2-диметилпропил, 2-метилбутил, 3-метилбутил, 2-этилбутил, 2,2-диметилпентил, 2-пропилпентил и т.п.
Термин "соединение" означает соединение, которое было искусственно получено или получено любым другим способом, таким как метаболизм in vivo.
Термин “терапевтически эффективное количество” означает количество, достаточное для лечения эффекта при введении пациенту.
Термин "лечение" в рамках изобретения означает лечение заболевания, нарушения или медицинского состояния у пациента, такого как млекопитающее (особенно человек), которое включает один или более из следующих эффектов:
(a) предотвращение возникновения заболевания, нарушения или медицинского состояния, то есть профилактическое лечение пациента;
(b) облегчение заболевания, нарушения или медицинского состояния, то есть устранение или регрессию заболевания, нарушения или медицинского состояния у пациента, включая противодействие эффектам других терапевтических средств;
(c) подавление заболевания, нарушения или медицинского состояния, то есть замедление или остановку развития заболевания, нарушения или медицинского состояния у пациента; или
(d) облегчение симптомов заболевания, нарушения или медицинского состояния у пациента.
Термин “фармацевтически приемлемая соль” означает соль, полученную из кислоты или основания, которая является приемлемой для введения пациенту, такому как млекопитающее. Такие соли могут быть получены из фармацевтически приемлемых неорганических или органических кислот и из фармацевтически приемлемых оснований. Как правило, фармацевтически приемлемые соли соединений согласно настоящему изобретению получают из кислот.
Соли, полученные из фармацевтически приемлемых кислот, включают, но не ограничены ими, уксусную, адипиновую, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, гликолевую, бромистоводородную, хлористоводородную, молочную, малеиновую, яблочную, миндальную, метансульфоновую, муциновую, азотную, щавелевую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую, ксинафоевую (1-гидрокси-2-нафтойную кислоту), нафталин-1,5-дисульфоновую кислоту и т.п.
Термин "защитная группа аминогруппы" означает защитную группу, подходящую для предотвращения нежелательных реакций на азоте аминогруппы. Репрезентативные защитные группы аминогруппы включают, но не ограничены ими, формил; ацильные группы, например алканоильные группы, такие как ацетил и три-фторацетил; алкоксикарбонильные группы, такие как трет-бутоксикарбонил (Вос); арилметоксикарбонильные группы, такие как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметильные группы, такие как бензил (Bn), тритил (Tr), и 1,1-ди-(4'-метоксифенил)метил; силильные группы, такие как триметилсилил (TMS) и трет-бутилдиметилсилил (TBDMS); и т.п.
Термин "защитная группа гидрокси-группы" означает защитную группу, подходящую для предотвращения нежелательных реакций в гидроксильной группе. Репрезентативные защитные группы гидрокси-группы включают, но не ограничены ими, алкильные группы, такие как метил, этил и трет-бутил; ацильные группы, например алканоильные группы, такие как ацетил; арилметильные группы, такие как бензил (Bn), п-метоксибензил (PMB), 9-флуоренилметил (Fm) и дифенилметил (бензгидрил, DPM); силильные группы, такие как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS); и т.п.
Общие процедуры синтеза
Соединения по изобретению могут быть получены из легко доступных исходных материалов с использованием следующих общих способов и процедур. Хотя частный аспект настоящего изобретения проиллюстрирован на схемах ниже, специалисту понятно, что все аспекты настоящего изобретения могут быть осуществлены с использованием способов, описанных здесь, или при использовании других способов, реагентов и исходных материалов, известных специалисту. Следует также понимать, что там, где указан типичный или предпочтительный режим процесса (то есть температуры, время реакции, молярные отношения реагентов, растворители, давление и т.д.), другой режим процесса может также использоваться, если не указано иное. Оптимальные условия реакции могут варьировать в зависимости от конкретных используемых реагентов или растворителя, но такие условия могут быть определены специалистом в соответствии с обычными процедурами оптимизации.
Дополнительно, как будет очевидным специалисту, обычные защитные группы могут быть необходимыми для того, чтобы воспрепятствовать некоторым функциональным группам участвовать в нежелательных реакциях. Выбор подходящей защитной группы для конкретной функциональной группы, а также подходящих условий для защиты и снятия защиты является известным в данной области техники. Например, многочисленные защитные группы и их введение и удаление описаны в T. W. Greene and G. M. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999, и в цитируемых там ссылках.
В типичном способе синтеза сложные эфиры по изобретению формулы (I), в которой R6 обозначает C1-3алкил, получали как показано в Схеме A. (Заместители и переменные, показанные на следующих схемах, имеют определения, приведенные выше, если не указано иное).
Схема А
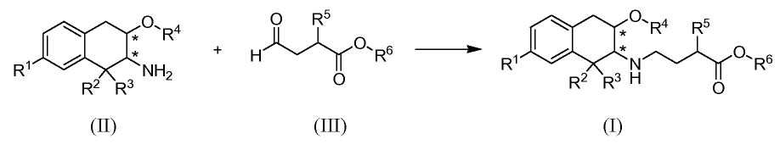
На Схеме A промежуточное соединение (II) восстановительно N-алкилировано реакцией с альдегидом (III) с получением продукта (I). Реакцию обычно проводят, вводя промежуточное соединение (II) в контакт с от приблизительно 1 до приблизительно 2 эквивалентов альдегида формулы (III) в подходящем инертном разбавителе, таком как дихлорметан, метанол или 2-метилтетрагидрофуран, в присутствии от приблизительно 1 до приблизительно 5 эквивалентов восстановителя. Реакцию обычно проводят при температуре в диапазоне от приблизительно 0°C до температуры окружающей среды в течение от приблизительно получаса до приблизительно 3 часов или до в основном полного завершения реакции. Типичные восстановители включают триацетоксиборгидрид натрия, борогидрид натрия и цианоборгидрид натрия.
Альдегид (III) может быть получен in situ из соответствующего бисульфитного аддукта (III'):
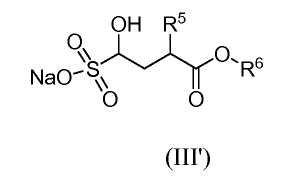
реакцией с основанием, таким как гидроксид натрия, непосредственно перед реакцией с аминотетралином (II).
Карбоновые кислоты по изобретению формулы (I), в которой R6 является водородом, получают из вышеуказанных сложных эфиров, вводя соответствующий сложный эфир в контакт с избытком основания, например, от приблизительно 4 до приблизительно 6 эквивалентов основания, такого как гидроксид натрия в метаноле. Реакцию проводят при температуре от приблизительно 25 до приблизительно 50°C в течение времени от приблизительно 2 до приблизительно 24 часов или до в основном полного завершения реакции.
Альтернативно, карбоновая кислота по изобретению может быть получена процессом, в котором в R6 используют защитную группу гидроксигруппы и который включает конечную стадию удаления защитной группы, как описано ниже в Примере 17.
Пример процедуры получения аминотетралинового промежуточного соединения (II), в котором переменная R1 обозначает -C(O)NH2, проиллюстрирован на Схеме B
Схема B

где P1 обозначает защитную группу гидроксигруппы, P2 обозначает защитную группу аминогруппы, и -OTf обозначает трифторметан сульфонат (обычно трифлат). Примечание "Rac" показывает, что соединение представляет собой рацемическую смесь специфической изображенной структуры и структуры, имеющей противоположную стереохимию в хиральных центрах.
Небольшие алкильные группы могут быть использованы в качестве защитных групп P1. При использовании алкила для P1 азиридиновое промежуточное соединение 1 может быть введено в реакцию с HBr с получением промежуточного соединения 2, которое предпочтительно выделяют в твердой форме в форме соли HBr. Обычно промежуточное соединение 1 вводят в контакт с избытком, например от приблизительно 12 до приблизительно 18 эквивалентов, HBr. Эффективность реакции может быть улучшена включением катализатора фазового переноса. Реакцию обычно проводят при температуре от приблизительно 90 до приблизительно 110°C в течение от приблизительно 10 до приблизительно 20 часов или до в основном полного завершения реакции. Используя Вос, например, в качестве защитной группы P2 , промежуточное соединение 3 получают затем, обрабатывая 2 основанием, преобразуя азиридиновое кольцо in situ, добавляя от приблизительно 1 до приблизительно 1,3 эквивалента ди-трет-бутилдикарбоната (обычно (Вос)2O) в обычных условиях реакции, получая промежуточное соединение 3.
Альтернативно, группу P1 азиридинового промежуточного соединения 1 удаляют в две стадии реакцией с HBr или BBr3 и последующей обработкой основанием, чтобы получить промежуточное соединение 2a:
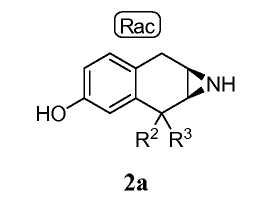
которое затем защищают на азоте азиридина, например реакцией с (Boc)2O, получая промежуточное соединение 3.
Затем амино-защищенный азиридин 3 вводят в контакт с большим избытком спирта R4OH в присутствии умеренного кислотного катализатора, такого как пиридий тозилат, получая промежуточное соединение 4.
Промежуточное аминотетралиновое соединение формулы (II), в которой R1 обозначает -ОН, может быть получено удалением защитной группы от промежуточного соединения 4. Например, когда защитная группа P2 является Вос, промежуточное фенольное соединение формулы (II) получают, обрабатывая соединения 4 кислотой. Точно так же промежуточное аминотетралиновое соединение формулы (II), в которой R1 обозначает -ORa, где Ra обозначает C1-3алкил, может быть получено аналогично исходя из промежуточного соединения формулы (I), в которой P1 является желаемой небольшой алкильной группой, и исключением начальной стадии удаления защитной группы.
Остальные стадии на Схеме B относятся к превращению гидроксизамещенного аминотетралина 4 в карбоксамидзамещенное промежуточное соединение 7 и к конечной стадии удаления защитной группы. Гидроксил промежуточного соединения 4 сначала превращают в трифлат, вводя 4 в контакт в инертном разбавителе с от приблизительно 1 до приблизительно 2 эквивалентов трифторметан сульфонилхлорида в присутствии от приблизительно 1 до приблизительно 3 эквивалентов основания, такого как триэтиламин, получая промежуточное соединение 5. Реакция 5 с цианидом цинка в присутствии катализатора на основе переходного металла приводит к промежуточному соединению 6. Эту реакцию обычно проводят при температуре от приблизительно 80 до 120°C в инертной атмосфере в течение от приблизительно получаса до приблизительно 2 часов или до в основном полного завершения реакции.
Затем нитрил промежуточного соединения 6 гидролизуют до карбоксамида промежуточного соединения 7. Как описано ниже в примерах, в одном способе синтеза нитрил 6 вводят в контакт с от приблизительно 5 до приблизительно 8 эквивалентами моногидрата пербората натрия в инертном разбавителе, таком как метанол. Реакцию проводят при температуре от приблизительно 50 до приблизительно 60°C в течение от приблизительно 12 до приблизительно 24 часов или до в основном полного завершения реакции. Альтернативные способы гидролиза нитрила до амида включают использование платинового катализатора, в частности гидридо(диметилфосфоновая кислота-kP)[водород-бис(диметилфосфинито-kP)]платины (II), и обработку пероксидом водорода, как описано в примерах ниже. Наконец, от промежуточного соединения 7 удаляют защитную группу обычной обработкой кислотой, получая аминотетралин формулы (II).
Промежуточное соединение формулы (II), в которой R1 обозначает -C(O)NRbRc, где Rb и Rc обозначают алкил, может быть получено из промежуточного соединения 6 превращением нитрила в карбоновую кислоту путем гидролиза в присутствии основания с последующим амидным сочетанием с амином формулы HNRbRc.
Индивидуальные энантиомеры формулы (II) могут быть разделены с использованием хирального вспомогательного соединения. Схема C иллюстрирует использование хирального вспомогательного соединения, представляющего собой 4-нитро-фенил (R)-1-фенил-этиловый эфир (8):
Схема С
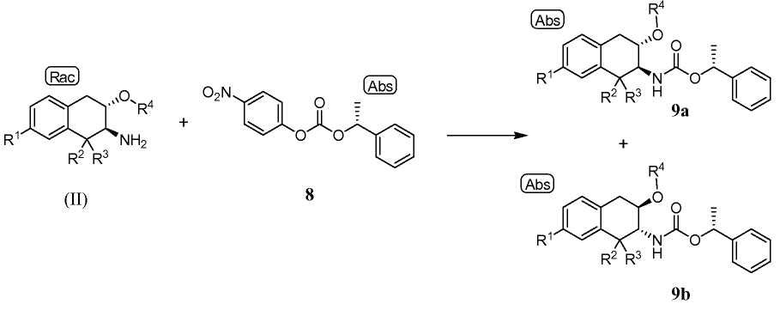
для получения пары нерацемических диастереомеров 9a и 9b, которые могут быть разделены. Примечание "Abs" обозначает определенное показанное хиральное соединение. Рацемический аминотетралин (II) вводят в контакт с от приблизительно 0,8 до приблизительно 1,2 эквивалентов хирального вспомогательного соединения 8 в инертном разбавителе в присутствии от приблизительно 2 до приблизительно 4 эквивалентов основания, такого как триэтиламин, с получением диастереомерной смеси промежуточных соединений 9a и 9b. Реакцию обычно проводят при температуре от приблизительно 80 до приблизительно 95°C в течение от приблизительно 4 до приблизительно 20 часов или до в основном полного завершения реакции. Диастереомеры 9a и 9b могут быть разделены высокоэффективной жидкостной хроматографией (ВЭЖХ) и собраны отдельно или кристаллизацией, в которой диастереомер 9a селективно кристаллизуется, оставляя преимущественно диастереомер 9b в растворе. Наконец, карбаматная группа может быть удалена от выделенных диастереомеров 9a и 9b обработкой кислотой с получением индивидуальных энантиомеров аминотетралина (II). Хиральные вспомогательные соединения 8 могут быть получены реакцией (R)-1-фенилэтанола с п-нитрофенил хлороформиатом, как описано ниже в примерах.
Промежуточное азиридиновое соединение 1, используемое в Схеме B, может быть получено реакцией замещенного 3,4-дигидро-1Н-нафталин-2-он:

с галогеналкилом, чтобы добавить алкильные заместители R2 и R3 в положение 2, обработкой солью гидроксиламина для превращения карбокси в оксим и последующей обработкой литий-алюминийгидридом или другим восстановителем для превращения оксима в азиридин 1, как описано, например, в US 6 844 368 и в Примере получения 14, ниже.
Альдегид (III), используемый в Схеме A, предпочтительно получают из соответствующей карбоновой кислоты 10, как показано на Схеме D:
Схема D
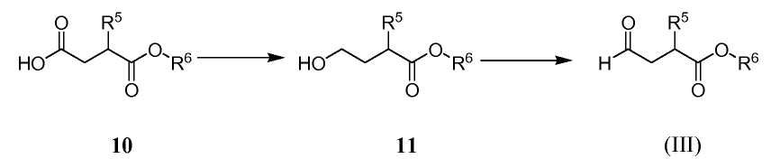
где R6 обозначает C1-3алкил. Восстановление гидрида бора карбоновой кислоты 10 приводит к спирту 11. Реакцию обычно проводят, вводя кислоту 10 в контакт с приблизительно 2 эквивалентами комплекса боран-тетрагидрофуран в тетрагидрофуране при температуре от приблизительно -5 до приблизительно 0°C. Спирт 11 затем окисляют до альдегида (III). Реагенты, которые могут быть использованы для окисления, включают диметилсульфоксид, активированный комплексом пиридин-триоксид серы и кипохлоритом натрия с 2,2,6,6-тетраметилпиперидин-1-оксиловым (TEMPO) катализатором. Если желательно, спирт 11 может быть преобразован в бисульфитный аддукт (III') без выделения альдегида (III) путем добавления бисульфита натрия после стадии окисления.
Дальнейшие детали относительно конкретных условий реакции и других процедур для получения репрезентативных соединений по изобретению или промежуточных соединений для их получения описаны ниже в примерах.
Соответственно, в одном аспекте способа изобретение относится к способу получения соединения формулы (I), или его соли, включающему (a) введение соединения формулы (II) в реакцию с соединением формулы (III), в которой R6 обозначает C1-3алкил, и, (b), когда R6 обозначает водород, вводя продукт стадии (a) в контакт с избытком основания, получая соединение формулы (I) или его соль.
В других аспектах изобретение относится к новому промежуточному соединению формулы 2 или его гидробромиду и к способу получения гидробромида соединения 2 в твердой форме, включающему введение соединения формулы 1 в реакцию с HBr и выделение продукта в твердой форме.
Фармацевтические композиции
Соединения 3-карбоксипропил-аминотетралина по изобретению обычно вводят пациенту в форме фармацевтической композиции или состава. Такие фармацевтические композиции могут вводиться пациенту любым приемлемым путем введения, включая, но не ограничиваясь ими, пероральный, ректальный, влагалищный, носовой, путем ингаляции, топический (включая чрескожный) и парентеральный способы введения.
Соответственно, в одном из аспектов композиций изобретение относится к фармацевтической композиции, включающей фармацевтически приемлемый носитель или эксципиент и терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли. В случае необходимости, такие фармацевтические композиции могут содержать, если желательно, другие терапевтические средства и/или средства для получения состава. Применительно к композициям, "соединение по изобретению" может также быть указанно как "активный агент". В рамках изобретения термин "соединение по изобретению" включает соединения формулы (I), а также варианты, воплощенные в формуле (Ia). "Соединение по изобретению" включает, кроме того, фармацевтически приемлемые соли и сольваты соединения, если не указано иное.
Фармацевтические композиции по изобретению обычно содержат терапевтически эффективное количество соединения согласно настоящему изобретению или его фармацевтически приемлемой соли. Специалисту понятно, однако, что фармацевтическая композиция может содержать больше, чем терапевтически эффективное количество, то есть композиции внавес, или меньше, чем терапевтически эффективное количество, то есть индивидуальные унифицированные дозы, предназначенные для многократного введения для достижения терапевтически эффективного количества.
Как правило, такие фармацевтические композиции содержат от приблизительно 0,1 до приблизительно 95 вес.% активного агента; предпочтительно, от приблизительно 5 до приблизительно 70 вес.%; и более предпочтительно от приблизительно 10 до приблизительно 60 вес.% активного агента.
Любой обычный носитель или эксципиент может использоваться в фармацевтических композициях по изобретению. Выбор конкретного носителя или эксципиента, или комбинаций носителей или эксципиентов, будет зависеть от способа введения, используемого для лечения конкретного пациента, или от типа медицинского состояния или болезненного состояния. В этом отношении, получение подходящей фармацевтической композиции для конкретного способа введения находится в рамках квалификации специалиста. Дополнительно, носители или эксципиенты, используемые в фармацевтических композициях по изобретению, являются коммерчески доступными. В качестве дальнейшей иллюстрации, обычные методики получения составов описаны в Remington: The Science and Practice of Pharmacy, 20th Edition, Lippincott Williams & White, Baltimore, Maryland (2000); и H.C. Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 7th Edition, Lippincott Williams & White, Baltimore, Maryland (1999).
Репрезентативные примеры материалов, которые могут служить фармацевтически приемлемыми носителями, включают, но не ограничены ими, следующее: сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза, такие как микрокристаллическая целлюлоза и ее производные, такие как натриевая соль карбоксиметилцеллюлозы, этилцеллюлоза и ацетилцеллюлоза; порошковый трагакант; солод; желатин; тальк; эксципиенты, такие как в форме масла какао и воски для суппозиториев; масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; многоатомные спирты, такие как глицерин, сорбит, маннит и полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар-агар; буферные средства, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический солевой раствор; раствор Рингера; этиловый спирт; фосфатные буферные растворы; и другие нетоксичные совместимые вещества, используемые в фармацевтических композициях.
Фармацевтические композиции обычно получают, тщательно и тесно смешивая или перемешивая активный агент с фармацевтически приемлемым носителем и одним или более дополнительными ингредиентами. Полученная однородно смешанная смесь может затем быть составлена или заполнена в таблетки, капсулы, пилюли и т.п. с использованием обычных процедур и оборудования.
Фармацевтические композиции по изобретению предпочтительно упаковывают в форме лекарственных форм. Термин "стандартная лекарственная форма" относится к физически дискретной единице, подходящей для введения пациенту, то есть каждая единица содержит предварительно определенное количество активного агента, вычисленное таким образом, чтобы оно производило желаемый терапевтический эффект, индивидуально или в комбинации с одной или более дополнительными единицами. Например, такие стандартные лекарственные формы могут быть капсулами, таблетками, пилюлями и т.п., или упаковками форм, подходящих для парентерального введения.
В одном варианте осуществления фармацевтические композиции по изобретению являются подходящими для перорального введения. Подходящие фармацевтические композиции для перорального введения могут быть в форме капсул, таблеток, пилюль, таблеток для рассасывания, облаток, драже, порошков, гранул; или в форме раствора или суспензии в водной или неводной жидкости; или в форме жидкой эмульсии масло-в-воде или вода-в-масле; или в форме эликсира или сиропа; и т.п.; при этом каждая такая форма содержит предварительно определенное количество соединения согласно настоящему изобретению в качестве активного ингредиента.
Когда фармацевтические композиции по изобретению предназначены для перорального введения в твердой лекарственной форме (то есть в форме капсул, таблеток, пилюль и т.п.), они обычно включают активный агент и один или более фармацевтически приемлемых носителей, таких как цитрат натрия или дикальций фосфат. В случае необходимости или альтернативно, такие твердые лекарственные формы могут также включать: наполнители или экстендеры, такие как крахмалы, микрокристаллическая целлюлоза, лактоза, сахароза, глюкоза, маннит и/или кремневая кислота; связующие, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинил пирролидон, сахароза и/или гуммиарабик; увлажнители, такие как глицерин; дезинтеграторы, такие как агар-агар, карбонат кальция, картофельный крахмал или крахмал тапиоки, альгиновую кислоту, некоторые силикаты и/или карбонат натрия; ингибиторы растворения, такие как парафин; ускорители абсорбции, такие как четвертичные аммониевые основания; смачивающие вещества, такие как цетиловый спирт и/или моностеарат глицерина; абсорбирующие вещества, такие как каолин и/или бентонитовая глина; лубриканты, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия, и/или их смеси; красители; и буферные средства.
Агенты, способствующие высвобождению, смачивающие вещества, средства для создания покрытий, подсластители, вкусовые агенты и отдушки, консерванты и антиоксиданты могут также присутствовать в фармацевтических композициях по изобретению. Примеры фармацевтически приемлемых антиоксидантов включают: водорастворимые антиоксиданты, такие как аскорбиновая кислота, цистеин гидрохлорид, бисульфат натрия, метабисульфат натрия, сульфит натрия и т.п.; маслорастворимые антиоксиданты, такие как аскорбил пальмитат, бутилированный гидроксианизол, бутилированный гидрокситолуол, лецитин, пропилгаллат, альфа-токоферол и т.п.; и агенты для образования хелатных соединений с металлами, такие как лимонная кислота, этилендиамин тетрауксусная кислота, сорбит, винная кислота, фосфорная кислота и т.п. Средства для создания покрытий на таблетках, капсулах, пилюлей и т.п. включают используемые для энтеросолюбильных покрытий, такие как фталат ацетилцеллюлозы, фталат поливинилацетата, гидроксипропилфталат метилцеллюлозы, сополимеры метакриловой кислоты и эфиров метакриловой кислоты, тримеллитат ацетилцеллюлозы, карбоксиметил этилцеллюлозу, сукцинат ацетата гидроксипропил метилцеллюлозы и т.п.
Фармацевтические композиции по изобретению могут также быть составлены для получения медленного или контролируемого высвобождения активного агента с использованием, например, гидроксипропилметилцеллюлозы в варьирующих соотношениях; или других полимерных матриц, липосом и/или микросфер. Кроме того, фармацевтические композиции по изобретению могут в случае необходимости содержать рентгеноконтрастные агенты и могут быть составлены так, чтобы они высвобождали активный ингредиент только, или избирательно, в определенной части желудочно-кишечного тракта, в случае необходимости, отсроченным образом. Примеры покрывающих композиций, которые могут использоваться, включают полимерные вещества и воски. Активный агент может также быть в микроинкапсулированной форме, если необходимо, с одним или более вышеописанных эксципиентов.
Подходящие жидкие лекарственные формы для перорального введения включают, в качестве иллюстрации, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Жидкие лекарственные формы обычно включают активный агент и инертный разбавитель, такой как, например, вода или другие растворители, средства для растворения и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (например, семян хлопчатника, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры жирной кислоты и сорбитана, и их смеси. Суспензии, в дополнение к активному ингредиенту, могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант и их смеси.
Соединения по изобретению могут также вводиться парентерально (например, внутривенной, подкожной, внутримышечной или интраперитонеальной инъекцией). Для парентерального введения активный агент обычно смешивают с подходящим носителем для парентерального введения, включая, например, стерильный водный раствор, солевой раствор, низкомолекулярные спирты, такие как пропиленгликоль, полиэтиленгликоль, растительные масла, желатин, эфиры жирной кислоты, такие как этилолеат и т.п. Парентеральные составы могут также содержать один или более антиоксидантов, ожижающих агентов, стабилизаторов, консервантов, смачивающих веществ, эмульгаторов, буферных агентов или диспергирующих агентов. Эти составы могут быть сделаны стерильными при помощи стерильной среды для инъекций, стерилизующего агента, фильтрации, облучения или нагревания.
Альтернативно, фармацевтические композиции по изобретению составляют для введения ингаляцией. Подходящие фармацевтические композиции для введения ингаляцией обычно находятся в форме аэрозоля или порошка. Такие композиции обычно вводят, используя известные устройства для доставки, такие как ингалятор с отмериваемым дозированием, сухой порошковый ингалятор, небулайзер или подобное устройство для доставки.
При введении ингаляцией с использованием герметичного контейнера фармацевтические композиции по изобретению обычно включают активный ингредиент и подходящий пропеллент, такой как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, диоксид углерода или другой подходящий газ. Дополнительно, фармацевтическая композиция может быть в форме капсулы или картриджа (например, из желатина), включающей(его) соединение по изобретению и порошок, подходящий для использования в порошковом ингаляторе. Подходящие порошковые основы включают, например, лактозу или крахмал.
Соединения по изобретению могут также вводиться чрескожно с использованием известных чрескожных систем доставки и эксципиентов. Например, активный агент может быть смешан с усилителями проникновения, такими как пропиленгликоль, монолаурат полиэтиленгликоля, азациклоалкан-2-онами и т.п., и включен в пластырь или подобную систему доставки. Дополнительные эксципиенты, включая желирующие средства, эмульгаторы и буферы, могут использоваться в таких чрескожных композициях, если желательно.
Если желательно, соединения по изобретению могут вводиться в комбинации с одним или более другими терапевтическими средствами. В этом варианте осуществления соединение по изобретению либо физически смешивают с другим терапевтическим средством, получая композицию, содержащую оба средства; либо каждое средство присутствует в отдельных и различных композициях, которые вводят пациенту одновременно или последовательно в любом порядке.
Например, соединение формулы I может быть скомбинировано со вторым терапевтическим средством с использованием обычных процедур и оборудования с получением композиции, включающей соединение формулы I и второе терапевтическое средство. Дополнительно, терапевтические средства могут быть скомбинированы с фармацевтически приемлемым носителем, образуя фармацевтическую композицию, включающую соединение формулы I, второе терапевтическое средство и фармацевтически приемлемый носитель. В этом варианте осуществления компоненты композиции обычно смешивают или перемешивают, чтобы создать физическую смесь. Эту физическую смесь затем вводят в терапевтически эффективном количестве, используя любой из путей, описанных здесь.
Альтернативно, терапевтические средства могут оставаться разделенными перед введением пациенту. В этом варианте осуществления средства физически не смешивают вместе перед введением, но вводят одновременно или в отдельные моменты времени в виде отдельных композиций. При отдельном введении, средства вводят в достаточно близкие моменты времени, получая желаемый терапевтический эффект. Такие композиции могут быть упакованы отдельно или могут быть упакованы вместе в виде набора. Два терапевтических средства в наборе могут вводиться тем же самым путем введения или разными путями введения.
В частности, соединения по изобретению могут быть скомбинированы опиоидными терапевтическими анальгетиками. Как описано выше, использование опиоидных анальгетиков часто связано с нежелательными побочными эффектами, такими как, например, запор, сниженное опорожнение желудка, боль в животе, вздутие, тошнота и гастроэзофагеальный рефлюкс. Эти неблагоприятные эффекты могут быть достаточно тяжелыми, что ограничивает дозу опиоидного аналгезирующего средства, которое может быть доставлено пациенту, до субоптимального уровня. Совместное введение соединения по изобретению с опиоидом, вероятно, уменьшит или предотвратит побочные эффекты, таким образом, увеличивая полезность аналгезирующего средства для облегчения боли.
Аналгезирующие опиоидные средства, которые могут использоваться в комбинации с соединениями согласно настоящему изобретению, включают, но не ограничены ими, морфин, гидроморфон, оксиморфон, петидин, кодеин, дигидрокодеин, оксиконтин, оксикодон, гидрокодон, суфентанил, фентанил, ремифентанил, бупренорфин, буторфанол, трамадол, метадон, героин, пропоксифен, меперидин, леворфенол, пентазоцин и комбинации аналгезирующих опиоидных средств с ибупрофеном или параоксиацетанилидом. Соединения по изобретению могут использоваться в дозах в пределах от приблизительно 0,05 до приблизительно 100 мг в сутки для среднего пациента массой 70 кг, при введении в комбинации с аналгезирующим опиоидным средством в его терапевтической дозе, например при введении в комбинации с оксикодоном в дозе от приблизительно 5 мг до приблизительно 160 мг в сутки.
Кроме того, прокинетические средства, действующие через механизмы, отличные от антагонизма mu-опиоидного рецептора, могут использоваться в комбинации с соединениями по изобретению. Например, агонисты 5-HT4 рецептора, такие как тегасерод, рензаприд, мосаприд, прукалоприд, 1-изопропил-1Н-индазол-3-карбоновая кислота {(1S,3R,5R)-8-[2-(4-ацетилпиперазин-1-ил)этил]-8-азабицикло[3.2.1]окт-3-ил}амид, {(1S,3R,5R)-8-[(R)-2-гидрокси-3-(метансульфонил-метил-амино)пропил]-8-азабицикло[3.2.1]окт-3-ил}амид 1-изопропил-2-оксо-1,2-дигидрохинолин-3-карбоновой кислоты или метиловый эфир 4-(4-{[(2-изопропил-1Н-бензоимидазол-4-карбонил)амино]метил}-пиперидин-1-илметил)пиперидин-1-карбоновой кислоты, могут использоваться в качестве второго терапевтического средства.
Дополнительные полезные прокинетические средства включают, но не ограничены ими, агонисты 5-HT3 рецептора (например, пумосетраг), антагонисты 5-HT1A рецептора (например, AGI 001), альфа-2-дельта лиганды (например, PD-217014), открыватели хлоридных каналов (например, любипростон), антагонисты допамина (например, итоприд, метаклопрамид, домперидон), агонисты ГАМК-B (например, бакловен, AGI 006), агонисты каппа-опиоида (например, асимадолин), мускариновые антагонисты M1 и M2 (например, акотиамид), агонисты мотилина (например, митемцинал), активаторы гуанилатциклазы (например, MD-1100) и агонисты грелина (например, Tzp 101, RC 1139).
Многочисленные дополнительные примеры таких терапевтических средств известны в данной области техники, и любые такие известные терапевтические средства могут быть использованы в комбинации с соединениями по изобретению. Вторичное средство (средства), когда оно включено, присутствует в терапевтически эффективном количестве, то есть в любом количестве, которое производит терапевтически благоприятное воздействие при совместном использовании с соединением по изобретению. Подходящие дозы для других терапевтических средств, вводимых в комбинации с соединением по изобретению, находятся обычно в диапазоне от приблизительно 0,05 мкг/сутки до приблизительно 100 мг/сутки.
Соответственно, фармацевтические композиции по изобретению в случае необходимости включают второе терапевтическое средство, как описано выше.
Следующие примеры иллюстрируют репрезентативные фармацевтические композиции согласно настоящему изобретению.
Пример состава A: Твердые желатиновые капсулы для перорального введения
Соединение по изобретению (50 г), высушенную распылением лактозу (200 г) и стеарат магния (10 г) тщательно перемешивали. Полученную композицию загружали в твердую желатиновую капсулу (260 мг композиции на капсулу).
Пример состава B: Твердые желатиновые капсулы для перорального введения
Соединение по изобретению (20 мг), крахмал (89 мг), микрокристаллическую целлюлозу (89 мг) и стеарат магния (2 мг) тщательно перемешивали и затем пропускали через сито №45 меш U.S. Полученную композицию загружали в твердую желатиновую капсулу (200 мг композиции на капсулу).
Пример состава C: Желатиновые капсулы для перорального введения
Соединение по изобретению (10 мг), полиоксиэтиленсорбитан моноолеат (50 мг) и порошок крахмала (250 мг) тщательно перемешивали и затем загружали в желатиновую капсулу (310 мг композиции на капсулу).
Пример состава D: Таблетки для перорального введения
Соединение по изобретению (5 мг), крахмал (50 мг) и микрокристаллическую целлюлозу (35 мг) пропускали через сито №45 меш U.S. и тщательно перемешивали. С полученными порошками смешивали раствор поливинилпирролидона (10 вес.% в воде, 4 мг), и эту смесь затем пропускали через сито №14 меш U.S. Полученные гранулы высушивали при 50-60°C и пропускали через сито №18 меш U.S. Затем к гранулам добавляли карбоксиметилкрахмал натрия (4,5 мг), стеарат магния (0,5 мг) и тальк (1 мг), которые предварительно пропускали через сито №60 меш U.S. После смешивания смесь прессовали на машине для таблетирования, получая таблетку массой 100 мг.
Пример состава E: Таблетки для перорального введения
Соединение по изобретению (25 мг), микрокристаллическую целлюлозу (400 мг), дымящий диоксид кремния (10 мг) и стеариновую кислоту (5 мг) тщательно перемешивали и затем прессовали, получая таблетки (440 мг композиции на таблетку).
Пример состава F: Таблетки с одной насечкой для перорального введения
Соединение по изобретению (15 мг), кукурузный крахмал (50 мг), кроскармеллозу натрия (25 мг), лактозу (120 мг) и стеарат магния (5 мг) тщательно перемешивали и затем прессовали, получая таблетку с одной насечкой (215 мг композиций на таблетку).
Пример состава G: Суспензия для перорального введения
Следующие ингредиенты тщательно перемешивали, получая суспензию для перорального введения, содержащую 100 мг активного ингредиента на 10 мл суспензии:
Пример состава Н: Сухая порошковая композиция
Измельченное соединение по изобретению (1 мг) смешивали с лактозой (25 мг) и затем загружали в желатиновый картридж для ингаляции. Содержимое картриджа вводили, используя порошковый ингалятор.
Пример состава J: Инъецируемый состав
Соединение по изобретению (0,1 г) смешивали с буферным раствором 0,1М цитрата натрия (15 мл). Полученный раствор доводили до рН 6 с использованием 1н. водного раствора соляной кислоты или 1н. водного раствора гидроксида натрия. Затем добавляли стерильный нормальный солевой раствор в цитратном буфере, получая полный объем 20 мл.
Пример состава K: Таблетки с одной насечкой для перорального введения
Соединение по изобретению (10 мг), оксикодон гидрохлорид (10 мг), кукурузный крахмал (50 мг), кроскармеллозу натрия (25 мг), лактозу (120 мг) и стеарат магния (5 мг) тщательно перемешивали и затем прессовали, получая таблетку с одной насечкой (220 мг композиций на таблетку).
Пример состава L: Инъецируемый состав
Соединение по изобретению (0,1 г) и оксикодон гидрохлорид (0,1 г) смешивали с буферным раствором 0,1М цитрата натрия (15 мл). Полученный раствор доводили до рН 6 с использованием 1н. водного раствора соляной кислоты или 1н. водного раствора гидроксида натрия. Затем добавляли стерильный нормальный солевой раствор в цитратном буфере, получая полный объем 20 мл.
Следует понимать, что любая форма соединений по изобретению (то есть свободное основание, фармацевтическая соль или сольват), которая является подходящей для конкретного способа введения, может использоваться в фармацевтических композициях, описанных выше.
Полезность
Соединения 3-карбоксипропил-аминотетралина по изобретению являются антагонистами рецептора mu-опиоида, и поэтому ожидается, что они могут быть использованы для лечения медицинских состояний, опосредуемых рецепторами mu-опиоида или связанных с активностью mu-опиоидного рецептора, то есть медицинских состояний, которые облегчаются в результате лечения антагонистом mu-опиоидного рецептора. В частности, ожидается, что соединения по изобретению могут быть использованы для лечения неблагоприятных эффектов, связанных с использованием аналгезирующих опиоидных средств, то есть таких симптомов, как запор, сниженное опорожнение желудка, боль в животе, вздутие, тошнота и гастроэзофагеальный рефлюкс, которые все вместе называются индуцируемой опиоидами дисфункцией кишечника. Также ожидается, что антагонисты mu-опиоидного рецептора по изобретению могут быть использованы для лечения послеоперационной непроходимости кишечника, нарушения, связанного со сниженной моторикой желудочно-кишечного тракта, которая имеет место после брюшной или другой хирургии. Кроме того, предполагается, что соединения-антагонисты mu-опиоидного рецептора могут использоваться для прекращения индуцируемой опиоидами тошноты и рвоты. Далее, антагонисты mu-опиоидного рецептора, демонстрирующие определенное центральное проникновение, могут быть использованы в лечении зависимости от, или склонности к, наркотических препаратов, алкоголя или азартных игр, или в профилактике, лечении и/или уменьшении ожирения.
Поскольку соединения по изобретению усиливают моторику желудочно-кишечного (гастроинтестинального) тракта на моделях животных, ожидается, что эти соединения могут быть использованы для лечения нарушений ЖКТ, вызванных сниженной моторикой у млекопитающих, включая человека. Такие нарушения моторики ЖКТ включают, в качестве иллюстрации, хронический запор, синдром раздраженного кишечника с преобладающей картиной запора (C-IBS), диабетический и идиопатический гастропарез и функциональную диспепсию.
В одном аспекте поэтому изобретение относится к способу усиления моторики желудочно-кишечного тракта у млекопитающего, включающему введение млекопитающему терапевтически эффективного количества фармацевтической композиции, включающей фармацевтически приемлемый носитель и соединение по изобретению.
Когда соединения по изобретению используют для лечения нарушения, связанного со сниженной моторикой ЖКТ или других состояний, опосредуемых рецепторами mu-опиоида, их обычно вводят перорально в виде единственной суточной дозы или нескольких доз в сутки, хотя могут использоваться другие формы введения. Например, особенно когда соединения по изобретению используют для лечения послеоперационной непроходимости кишечника, они могут вводиться парентерально. Количество активного средства, вводимого на дозу, или общее количество, вводимое в сутки, будет обычно определено врачом в свете релевантных обстоятельств, включая состояние, подлежащее лечению, выбранный путь введения, конкретное вводимое соединение и его относительную активность, возраст, массу тела и реакцию индивидуального пациента, серьезность симптомов у пациента и т.п.
Подходящие дозы для лечения нарушений, связанных со сниженной моторикой ЖКТ, или других нарушений, опосредуемых рецепторами mu-опиоида, составляют от приблизительно 0,0007 до приблизительно 20 мг/кг/сутки активного агента, включая от приблизительно 0,0007 до приблизительно 1,4 мг/кг/сутки. Для среднего человека массой 70 кг это составляет от приблизительно 0,05 до приблизительно 100 мг в сутки активного агента.
В одном аспекте изобретения соединения по изобретению используют для лечения индуцируемой опиоидами дисфункции кишечника. Когда соединения по изобретению используют для лечения индуцируемой опиоидами дисфункции кишечника, их обычно вводят перорально в виде единственной суточной дозы или нескольких доз в сутки. Предпочтительно, доза для лечения индуцируемой опиоидами дисфункции кишечника составляет от приблизительно 0,05 до приблизительно 100 мг в сутки.
В другом аспекте изобретения соединения по изобретению используют для лечения послеоперационной непроходимости кишечника. Когда соединения по изобретению используют для лечения послеоперационной непроходимости кишечника, их обычно вводят перорально или внутривенно в виде единственной суточной дозы или нескольких доз в сутки. Предпочтительно, доза для лечения послеоперационной непроходимости кишечника составляет от приблизительно 0,05 до приблизительно 100 мг в сутки.
Изобретение также относится к способу лечения млекопитающего, имеющего заболевание или состояние, связанное с активностью mu-опиоидного рецептора, включающему введение млекопитающему терапевтически эффективного количества соединения по изобретению или фармацевтической композиции, включающей соединение по изобретению.
Как описано выше, соединения по изобретению являются антагонистами mu-опиоидного рецептора. Изобретение поэтому также относится к способу противодействия рецептору mu-опиоида у млекопитающего, включающему введение млекопитающему соединения по изобретению.
Антагонисты mu-опиоидного рецептора по изобретению могут вводиться в комбинации с другим терапевтическим средством или средствами, в частности в комбинации с аналгезирующими опиоидными средствами или с прокинетическими средствами, действующими через не-mu-опиоидные механизмы. Соответственно, в другом аспекте способы и композиции по изобретению далее включают терапевтически эффективное количество аналгезирующего опиоидного средства или другого прокинетического средства. Способы по изобретению включают, например, способ уменьшения или предотвращения побочного эффекта, связанного с использованием опиоидного средства, у млекопитающего, включающий введение млекопитающему опиоидного средства и соединения по изобретению.
Кроме того, соединения по изобретению также могут быть использованы в качестве инструментальных средств для исследования или изучения биологических систем или проб, имеющих mu-опиоидные рецепторы, или для обнаружения новых соединений, имеющих активность mu-опиоидного рецептора. Любая подходящая биологическая система или проба, имеющая mu-опиоидные рецепторы, может использоваться в таких исследованиях, которые могут проводиться как in vitro, так и in vivo. Примеры биологических систем или проб, подходящих для таких исследований, включают, но не ограничены ими, клетки, клеточные экстракты, плазматические мембраны, образцы ткани, млекопитающих (таких как мыши, крысы, морские свинки, кролики, собаки, свиньи и т.д.) и т.п. Эффекты контактирования биологической системы или пробы, включающей рецептор mu-опиоида, с соединением по изобретению определяют, используя обычные процедуры и оборудование, такие как тест связывания меченого лиганда и функциональный тест, описанный здесь, или другие функциональные тесты, известные в данной области техники. Такие функциональные тесты включают, но не ограничены ими, опосредованные лигандом изменения во внутриклеточном циклическом аденозинмонофосфате (цАМФ), опосредованные лигандом изменения в активности фермента аденилилциклазы, опосредованные лигандом изменения включения аналогов гуанозинтрифосфата (ГТФ), таких как [35S]ГТФγS (гуанозин 5'-O-(γ-тио)трифосфат) или ГТФ-Eu, в изолированные мембраны через катализируемый рецептором обмен аналогов ГТФ на аналоги ГДФ, и опосредованные лигандом изменения в свободных внутриклеточных ионах кальция. Подходящая концентрация соединения по изобретению для таких исследований обычно составляет от приблизительно 1 нМ до приблизительно 500 нМ.
При использовании соединений по изобретению в качестве инструментальных средств исследования для обнаружения новых соединений, имеющих активность mu-опиоидного рецептора, данные по связыванию или функциональные данные тестируемого соединения или группы тестируемых соединений сравнивают со связыванием с mu-опиоидным рецептором или функциональными данными для соединения по изобретению, чтобы идентифицировать тестируемые соединения, которые имеют высокую связывающую или функциональную активность. Этот аспект изобретения включает, как отдельные варианты осуществления, как получение данных для сравнения (с использованием подходящих тестов), так и анализ данных тестирования для идентификации представляющих интерес тестируемых соединений.
Среди других свойств было обнаружено, что соединения по изобретению показывают мощное связывание на mu-опиоидных рецепторах и небольшой агонизм или отсутствие агонизма с mu-рецепторными функциональными тестами. Поэтому соединения по изобретению являются мощными антагонистами mu-опиоидного рецептора. Далее, соединения по изобретению продемонстрировали преимущественно периферическую активность по сравнению с активностью в центральной нервной системе на моделях животных. Поэтому можно ожидать, что эти соединения обращают индуцированное опиоидами снижение моторики ЖКТ, не препятствуя полезным центральным эффектами аналгезии. Эти свойства, а также полезность соединений по изобретению, могут быть показаны с использованием различных тестов in vitro и in vivo, известных специалисту. Репрезентативные тесты описаны более подробно в следующих примерах.
ПРИМЕРЫ
Следующие примеры синтеза и биологические примеры предлагаются для иллюстрации изобретения и не должны никоим образом рассматриваться как ограничение объема изобретения. В примерах, приведенных ниже, следующие аббревиатуры имеют следующие значения, если не указано иное. Аббревиатуры, не определенные ниже, имеют их общепринятые значения.
CAN = ацетонитрил
AcOH = уксусная кислота
Вос = трет-бутоксикарбонил
(Вос)2O = ди-трет-бутил дикарбонат
DCM = дихлорметан
DIPEA=N,N-диизопропилэтиламин
DMF=N,N-диметилформамид
ДМСО = диметилсульфоксид
EtOAc = этилацетат
EtOH = этанол
HATU = N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)уроний гексафторфосфат
MeOH = метанол
MeTHF = 2-метил-тетрагидрофуран
MTBE = метил-трет-бутиловый эфир
RT = температура окружающей среды
TFA = трифторуксусная кислота
THF = тетрагидрофуран
Реактивы и растворители были приобретены у коммерческих поставщиков (Aldrich, Fluka, Sigma и т.д.) и использовались без дальнейшей очистки. Реакции проводили в атмосфере азота, если не указано иное. Прогресс реакционных смесей проверяли тонкослойной хроматографией (TLC), аналитической высокоэффективной жидкостной хроматографией (анал. ВЭЖХ) и масс-спектрометрией. Реакционные смеси обрабатывали, как описано конкретно в каждой реакции; обычно их очищали экстракцией и другими способами очистки, такими как зависимое от температуры или растворителя осаждение. Кроме того, реакционные смеси обычно очищали препаративной ВЭЖХ, обычно используя наполнители для колонок Microsorb C18 и Microsorb BDS и обычные элюенты. Исследование продуктов реакции обычно осуществляли масс-спектрометрией и 1H-ЯМР. Для измерения способом ЯМР, образцы растворяли в дейтеризованном растворителе (CD3OD, CDCl3 или ДМСО-d6), и спектры 1H-ЯМР получали с помощью прибора Varian Gemini 2000 (400 МГц) в стандартных условиях наблюдения. Масс-спектрометрическую идентификацию соединений осуществляли способом ионизации с электрораспылением (ESMS) с использованием оборудования Applied Biosystems (Foster City, CA) модель API 150 EX или Agilent (Palo Alto, CA) модель 1200 LC/MSD.
Пример получения 1: трет-бутиловый эфир 7,7-диэтил-5-гидрокси-1a,2,7,7a-тетрагидро-1-аза-циклопропа[b]нафталин-1-карбоновой кислоты
a. 7-амино-6-бром-8,8-диэтил-5,6,7,8-тетрагидронафталин-2-ол гидробромид
В колбу добавляли 7,7-диэтил-5-метокси-1a,2,7,7a-тетрагидро-1Н-1-аза-циклопропа[b]нафталин (268 г, 1,16 моль) и бромид водорода (1,97 л, 17,38 моль), затем тетра-N-бутиламмоний бромид (38 г, 0,12 моль). Реакционную смесь нагревали при 100°C в течение ночи при перемешивании, охлаждали до температуры окружающей среды и затем вливали в перемешиваемый этилацетат (2,5 л). Продукт выделяли фильтрацией, остаток после фильтрации промывали этилацетатом (2 ×200 мл) и высушивали, получая сырой продукт (370 г) в форме твердого вещества пурпурного цвета. Сырой продукт суспендировали в этаноле (1,50 л), затем нагревали при 80°C в течение 30 мин. Полученную суспензию охлаждали до температуры окружающей среды в течение 1 часа и фильтровали. Колбу и остаток после фильтрации промывали этанолом (2 ×100 мл) и затем этилацетатом (100 мл) и высушивали в течение ночи, получая целевое соединение в форме твердого вещества (275 г, чистота ~96%).
b. трет-бутиловый эфир 7,7-диэтил-5-гидрокси-1a,2,7,7a-тетрагидро-1-аза-циклопропа[b]нафталин-1-карбоновой кислоты
К суспензии 7-амино-6-бром-8,8-диэтил-5,6,7,8-тетрагидронафталин-2-ол гидробромида (20,0 г, 52,8 ммоль) и этилацетата (200 мл) добавляли 1М гидроксида натрия в воде (106 мл). Реакционную смесь перемешивали при 25°C в течение 2 часов, добавляли ди-трет-бутилдикарбонат (15 г, 68 ммоль) в этилацетате (5 мл), и реакционную смесь перемешивали при температуре окружающей среды в течение 2 часов. После удаления двух третей этилацетата (135 мл) добавляли гептан (135 мл), и полученную суспензию перемешивали при температуре окружающей среды в течение 30 минут и затем при 5°C в течение ночи. Суспензию фильтровали, и остаток после фильтрации промывали водой (100 мл), промывали гептаном (50 мл) и высушивали под вакуумом, получая целевое соединение (14,3 г).
Пример получения 2: трет-бутиловый эфир транс-(7-циано-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты
a. трет-бутиловый эфир транс-(1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты
К суспензии трет-бутилового эфира 7,7-диэтил-5-гидрокси-1a,2,7,7a-тетрагидро-1-аза-циклопропа[b]нафталин-1-карбоновой кислоты (170,0 г, 535,6 ммоль) и метанола (1700 мл) добавляли пиридиний п-толуолсульфонат (13,4 г, 53,6 ммоль), и реакционную смесь перемешивали при 40°C в течение 4 часов. Объем уменьшали упариванием на роторном испарителе до ~300 мл, получая густую белую суспензию. Продукт выделяли фильтрацией; остаток после фильтрации промывали холодным метанолом (50 мл) и высушивали в воздухе в течение 3 часов, получая целевое соединение (150 г). Фильтрат уменьшали до ~50 мл и перемешивали при 0°C в течение 2 часов, фильтровали и высушивали, получая дополнительный продукту (25 г).
b. 7-трет-бутоксикарбониламино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-иловый эфир транс-трифтор-метансульфоновой кислоты
Смесь трет-бутилового эфира транс-(1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты (195,0 г, 0,558 моль), триэтиламина (160 мл, 1,1 моль) и этилацетата (2000 мл) перемешивали при температуре окружающей среды в течение 15 минут и охлаждали до 0°C, после чего медленно добавляли трифтор-метансульфонил хлорид (150 г, 0,89 моль), поддерживая внутреннюю температуру ниже 4°C. Полученную суспензию перемешивали при 0°C в течение 1 часа. Добавляли дополнительный триэтиламин (16 мл), затем, медленно, дополнительный трифторметансульфонил хлорид (15,0 г), поддерживая температуру ниже 5°C. Реакционную смесь перемешивали при температуре окружающей среды в течение дополнительного часа. Добавляли разбавленный солевой раствор (1,0 л), и реакционную смесь перемешивали в течение 10 минут при температуре окружающей среды. Слои разделяли; органический слой промывали разбавленным NaHCO3 (1,0 л) и затем концентрировали до ~350 мл упариванием на роторном испарителе при 28°C и перемешивали при температуре окружающей среды в течение 30 мин. Добавляли гептан (700 мл), и полученную суспензию перемешивали при температуре окружающей среды в течение 30 минут, охлаждали до 4°C и перемешивали в течение 1 часа. Твердые частицы отфильтровывали, промывали гептаном и затем высушивали под вакуумом, получая целевое соединение (193,0 г, чистота >97%). Фильтрат концентрировали, суспендировали в смеси изопропил ацетата и гептана (1:3, 60 мл) в течение 30 минут, фильтровали и высушивали, получая дополнительный продукт (45,0 г, чистота >97%).
c. трет-бутиловый эфир транс-(7-циано-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты
7-трет-бутоксикарбониламино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-иловый эфир трифтор-метансульфоновой кислоты (236,6 г, 0,49 моль) растворяли в N,N-диметилформамиде (851 мл, 10,99 моль) и воде (23,8 мл, 1,32 моль) при температуре окружающей среды. Раствор продували азотом в течение 5 минут, и затем подключали к вакууму на 5 мин. Очистку азотом и обработку вакуумом повторяли дважды. К реакционной смеси при перемешивании добавляли цианид цинка (34,2 г, 0,29 моль), трис(дибензилиденацетон)дипалладий (0) (4,4 г, 4,8 ммоль) и 1,1'-бис(дифенилфосфино)ферроцен (5,4 г, 9,7 ммоль). Реакционную смесь продували азотом в течение 5 минут, нагревали в атмосфере азота при 110°C в течение 1 часа, охлаждали до температуры окружающей среды и затем фильтровали через целит. Фильтрованную реакционную смесь добавляли медленно в воду (3 л), охлаждали до 0°C при перемешивании, перемешивали в течение 30 минут при 0°C и затем фильтровали. Остаток после фильтрации промывали водой (500 мл) и высушивали в воздухе в течение 2 часов, суспендировали в этаноле (1 л) при перемешивании в течение 1 часа и затем фильтровали, получая целевое соединение (165,0 г, чистота >96%). Фильтрат высушивали (21,6 г) и растворяли в этаноле (110 мл) при перемешивании в течение 1 часа, и полученную суспензию фильтровали и высушивали под вакуумом, получая дополнительный продукт (10,2 г, чистота >98%).
Пример получения 3: трет-бутиловый эфир транс-(7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты
Суспензию продукта Примера получения 2 (160,0 г, 446,3 ммоль) и метанола (3,3 л) нагревали при 55°C в течение 15 минут, добавляли моногидрат пербората натрия (280 г, 2800 ммоль) и воду (330 мл), и реакционную смесь нагревали при 55°C в течение ночи. Добавляли дополнительный моногидрат пербората натрия (90 г), и реакционную смесь нагревали при 55°C в течение ночи, затем охлаждали до температуры окружающей среды, и неорганические твердые частицы отфильтровывали. Фильтрат переносили в колбу на 5 л, и большую часть растворителя удаляли упариванием на роторном испарителе. К полученной суспензии добавляли воду (1,1 л) и этилацетат (450 мл), и реакционную смесь перемешивали при температуре окружающей среды в течение 20 мин. Реакционную смесь фильтровали, и остаток после фильтрации промывали водой (200 мл) и затем этилацетатом (200 мл) и высушивали, получая целевое соединение (123 г, чистота ~95%). Фильтрат концентрировали досуха и высушивали под вакуумом, получая дополнительный продукт (18 г, чистота 65%).
Пример получения 4: трет-бутиловый эфир транс-(7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты
К смеси трет-бутилового эфира транс-(7-циано-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты (33,0 г, 92 ммоль), этанола (45 мл), DMF (25 мл) и воды (7,5 мл) добавляли гидридо(диметилфосфоновая кислота-kP)[водород-бис(диметилфосфинито-kP)]платину (II) (0,25 г, 0,58 ммоль) и реакционную смесь нагревали при 80°C в течение 24 часов. Реакционную смесь охлаждали до температуры окружающей среды и концентрировали досуха под вакуумом, получая целевое соединение (36,3 г), которое использовали без дальнейшей очистки. (m/z): [M+H]+ рассчит. для C21H32N2O4 377,24; найдено 377,8. 1H ЯМР (d6-ДМСО, 400 мГц) δ (ч/млн): 7,92 (с, 1H), 7,64 (м, 2H), 7,26 (с, 1H), 7,14 (д, J=7,9 Гц, 1H), 6,64 (д, J=9,4 Гц) 3,81 (т, J=10,0 Гц), 3,58 (м, 1H), 3,30 (с, 3H), 2,58 (дд, J=16,9 Гц, 9,4 Гц, 1H), 1,82 (м, 1H), 1,56-1,45 (м, 4H), 1,41 (с, 9H), 0,58 (м, 6H).
Пример получения 5: трет-бутиловый эфир транс-(7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты
К раствору трет-бутилового эфира транс-(7-циано-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты (8,5 г, 24 ммоль) в ДМСО (105 мл) добавляли K2CO3 (4,98 г, 36 ммоль), и смесь перемешивали до растворения всех твердых частиц. К раствору добавляли 30% пероксид водорода (12,2 мл, 120 ммоль) частями по 0,5 мл за 45 минут со скоростью, позволяющей поддерживать температуру 30-35°C. Реакционную смесь разбавляли водой (200 мл) и изопропил ацетатом (500 мл), и добавляли метабисульфит натрия (10 г) для восстановления избытка пероксида. Слои разделяли, и водный слой экстрагировали изопропил ацетатом (3 ×150 мл) и смесью 10% MeOH/изопропил ацетат (2 ×100 мл). Объединенные органические слои промывали водой (3 ×150 мл) и насыщенным NaCl (100 мл), высушивали Na2SO4 и концентрировали, получая целевое соединение (9,4 г). (m/z): [M+H]+ рассчит. для C21H32N2O4 377,24; найдено 377,6.
Пример получения 6: амид транс-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты
Ацетил хлорид (278,8 мл, 3920 ммоль) добавляли по каплям к этанолу (382 мл, 6530 ммоль) при -5°C за 2 часа, поддерживая внутреннюю температуру ниже 20°C. Полученный раствор добавляли частями за 15 минут, поддерживая внутреннюю температуру ниже 30°C, к суспензии трет-бутилового эфира транс-(7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты (123,0 г, 327 ммоль) и этанола (500 мл), которая была охлаждена до 10°C. Реакционную смесь перемешивали при температуре окружающей среды в течение 2 часов и концентрировали до ~200 мл упариванием на роторном испарителе. Добавляли этилацетат (200 мл), и полученную суспензию перемешивали при 0°C в течение 30 минут, фильтровали и высушивали, получая гидрохлорид целевого соединения (102 г, чистота >98%) в форме твердого вещества белого цвета.
Пример получения 7: 4-нитрофенил-(R)-1-фенилэтиловый эфир угольной кислоты
Смесь (R)-1-фенилэтанола (60,6 г, 0,496 моль), пиридина (42,5 мл, 0,526 моль) и 2-метилтетрагидрофурана (600 мл) охлаждали до 0°C, и п-нитрофенил хлорформиат (100 г, 0,496 моль) добавляли за 15 минут, поддерживая внутреннюю температуру ниже 5°C. Реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение 2 часов. К реакционной смеси добавляли 1,0М HCl в воде (300 мл). Слои разделяли. Органический слой промывали 1н. HCl (300 мл) и солевым раствором (300 мл), фильтровали, концентрировали досуха упариванием на роторном испарителе и высушивали под вакуумом, получая целевое соединение (140 г) в форме прозрачного желтого масла.
Пример получения 8: амид (6S,7S)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты
a. (R)-1-фенилэтиловый эфир ((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты
Смесь 4-нитрофенил-(R)-1-фенилэтилового эфира угольной кислоты (102 г, 357 ммоль), N,N-диметилформамида (200 мл) и триэтиламина (32,7 мл, 235 ммоль) перемешивали при температуре окружающей среды в течение ночи. К реакционной смеси добавляли гидрохлорид амида транс-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты (100 г, 320 ммоль), N,N-диметилформамид (320 мл) и триэтиламин (98,0 мл, 703 ммоль). Реакционную смесь нагревали при 85°C в течение 5 часов и затем перемешивали при температуре окружающей среды в течение ночи. Приблизительно 90% DMF удаляли перегонкой при 70°C, и полученное густое масло охлаждали до температуры окружающей среды и затем разделяли между этилацетатом (1,5 л) и разбавленным солевым раствором (500 мл). Органический слой промывали 1M NaOH (3 ×500 мл) и высушивали Na2SO4. Большую часть растворителя удаляли упариванием на роторном испарителе, добавляли 3 объема этилацетата, и полученную суспензию перемешивали при температуре окружающей среды в течение 30 минут, фильтровали и высушивали, получая целевое соединение (48 г, химическая и оптическая чистота >99%).
Фильтрат промывали 1M NaOH (200 мл) и затем разбавленным солевым раствором (2×200 мл). Большую часть растворителя удаляли упариванием на роторном испарителе, получая густое масло, к которому добавляли этилацетат (100 мл). Добавляли щепотку гранул целевого соединения, и реакционную смесь охлаждали при 0°C после перемешивания в течение ~30 мин. Полученную тонкодисперсную суспензию перемешивали в течение 5 минут и фильтровали; колбу и остаток после фильтрации промывали этилацетатом (2 ×15 мл), получая дополнительное целевое соединение (4,1 г, 97% химическая и >99%-ная оптическая чистота, 38% объединенный выход).
b. амид (6S,7S)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты
Ацетил хлорид (193 мл, 2710 ммоль) добавляли по каплям к этанолу (260 мл, 4500 ммоль) при -5°C за 40 минут, поддерживая внутреннюю температуру ниже 30°C. Полученный раствор добавляли за 5 минут при 10°C к смеси (R)-1-фенилэтилового эфира ((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты (49,0 г, 115 ммоль) и этанола (200 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение ночи и концентрировали до ~100 мл упариванием на роторном испарителе. Добавляли этилацетат (100 мл), и полученную суспензию перемешивали при 0°C в течение 30 минут и фильтровали. Остаток после фильтрации промывали этилацетатом и высушивали, получая гидрохлорид целевого соединения (30 г, чистота >99%). Объем фильтрата уменьшали почти досуха. Добавляли изопропиловый спирт (20 мл), и полученную густую суспензию перемешивали в течение 30 минут и фильтровали. Остаток после фильтрации промывали этилацетатом (2×20 мл) и высушивали под вакуумом в течение ночи, получая дополнительный продукт (5,5 г, чистота >97%). 1H ЯМР (ДМСО-d6): δ ч/млн 0,49 (т, 3H), 0,63 (т, 3H), 1,62 (кв, 2H), 1,89 (м, 1H), 2,09 (м, 1H), 2,60 (дд, 1H), 3,22 (м, 1H), 3,41 (с, 3H), 3,50 (дд, 1H), 3,82 (кв, 1H), 7,19 (д, 1H), 7,31 (ушир., 1H), 7,70 (д, 1H), 7,71 (с, 1H), 7,98 (ушир., 1H), 8,15 (ушир., 3H).
Пример получения 9: транс-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-ол
К раствору трет-бутилового эфира транс-(1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты (6,0 г, 17,2 ммоль) в дихлорметане (60 мл) добавляли раствор 4,0н. HCl в диоксане (21,5 мл, 86 ммоль) в течение приблизительно 2 мин. После перемешивания при температуре окружающей среды в течение ночи, реакционную смесь концентрировали при пониженном давлении и высушивали под вакуумом, получая гидрохлорид целевого соединения (5,5 г). (m/z): [M+H]+ рассчит. для C15H23NO2 250,36; найдено 250,2. 1H ЯМР (d6-ДМСО, 400 мГц) δ (ч/млн) 9,26 (с, 1H), 8,09 (ушир.с, 3H), 6,92 (д, J=8,0 Гц, 1H), 6,61 (м, 2H), 3,77 (м, 1H), 3,41 (с, 3H), 3,30 (дд, J=15,8 Гц, 5,9 Гц, 1H), 3,17 (м, 1H), 2,43 (дд, J=15,5 Гц, 9,6 Гц, 1H), 1,85 (м, 2H), 1,66-1,50 (м, 2H), 0,66 (т, J=7,4 Гц, 3H), 0,54 (т, J=7,1 Гц, 3H).
Пример получения 10: (6S,7S)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-ол и (6R,7R)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-ол
a. (R)-1-фенилэтиловый эфир ((2R,3R)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты (RR) и (R)-1-фенилэтиловый эфир ((2S,3S)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты (SS)
Смесь транс-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-ол гидрохлорида (1,00 г, 3,5 ммоль), 4-нитрофенил-(R)-1-фенилэтилового эфира угольной кислоты (800 мг, 2,8 ммоль), триэтиламина (707 мг, 7,0 ммоль) и DMF (3,5 мл) нагревали при 90°C. Через 4 часа добавляли дополнительную часть 4-нитрофенил-(R)-1-фенилэтилового эфира угольной кислоты (200 мг, 0,7 ммоль), и нагревание продолжали еще 3 часа. Реакционную смесь охлаждали и оставляли на ночь при температуре окружающей среды. DMF удаляли при пониженном давлении, и остаток растворяли в этилацетате (25 мл). Органические фракции промывали 10%-ным карбонатом натрия и насыщенным хлоридом натрия, высушивали с помощью Na2SO4 и концентрировали досуха. Остаток растворяли в метаноле (6 мл), и добавляли 1,0 н. раствор гидроксида натрия в метаноле (3,0 мл, 3,0 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут, после чего добавляли 50%-ный водный раствор уксусной кислоты (2 мл). Реакционную смесь концентрировали приблизительно до 4 мл и добавляли 50%-ный водный раствор ацетонитрила (15 мл).
Сырые диастереомеры разделяли препаративной ВЭЖХ и собирали отдельно. Сырой продукт растворяли в смеси 1:1 ацетонитрил/вода и выделяли в следующих условиях: колонка: Microsorb C18 100A 8 мкм; объемная скорость потока: 50 мл/мин; Растворитель A: >99% воды, 0,05% TFA; Растворитель B: >99% ацетонитрила, 0,05% TFA; Градиент (время(мин)/% B): 0/15, 4/15, 8/40, 60/55. Чистые фракции каждого объединяли, и ацетонитрил удаляли при пониженном давлении. Продукт экстрагировали в дихлорметан (3 ×30 мл), органические экстракты высушивали с помощью Na2SO4 и концентрировали, получая целевое соединение.
RR: 435 мг (выход 39%) (m/z): [M+H]+ рассчит. для C24H31NO4 398,52; найдено 398,2. 1H ЯМР (d6-ДМСО, 400 мГц) δ (ч/млн) 9,01 (с, 1H), 7,37-7,26 (м, 5H), 7,05 (д, J=9,8 Гц, 1H), 6,86 (д, 8,2, 1H), 6,52 (дд, J=8,0, 2,4 Гц, 1H), 6,48 (д, J=2,3 Гц, 1H), 5,70 (кварт., J=6,7 Гц, 1H), 3,77 (т, J=10,3 Гц, 1H), 3,55 (м, 1H), 3,32 (с, 3H), 3,17 (дд, J=15,9, 6,0 Гц, 1H), 2,43 (м, 1H), 1,57-1,52 (м, 2H), 1,56 (д, J=6,7 Гц, 3H), 1,44-1,33 (м, 2H), 0,60 (т, J=7,4 Гц, 3H), 0,51 (т, J=7,0 Гц, 3H).
SS: 363 мг (выход 32%) (m/z): [M+H]+ рассчит. для C24H31NO4 398,52; найдено 398,2. 1H ЯМР (d6-ДМСО, 400 мГц) δ (ч/млн) 9,02 (с, 1H), 7,39-7,24 (м, 5H), 7,03 (д, J=9,7 Гц, 1H), 6,85 (д, 8,3, 1H), 6,53 (дд, J=8,1, 2,6 Гц, 1H), 6,48 (д, J=2,2 Гц, 1H), 5,69 (кварт., J=6,7 Гц, 1H), 3,75 (т, J=10,6 Гц, 1H), 3,52 (м, 1H), 3,27 (с, 3H), 3,14 (дд, J=15,9, 5,9 Гц, 1H), 2,37 (дд, J=15,7, 9,5, 1H), 1,65-1,41 (м, 4H), 1,46 (д, J=6,6 Гц, 3H), 0,64-0,60 (м, 6H).
b. (6S,7S)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-ол
(R)-1-фенилэтиловый эфир ((2S,3S)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты (635 мг, 1,60 ммоль) обрабатывали 4,0н. HCl в диоксане (6,0 мл, 24 ммоль) и перемешивали при температуре окружающей среды. Через 3 дня растворитель удаляли при пониженном давлении, и остаточное твердое вещество растирали с 50%-ным дихлорметаном в гептане (4 мл). Твердое вещество собирали на воронке Buchner и высушивали под вакуумом, получая гидрохлорид целевого соединения (462 мг). (m/z): [M+H]+ рассчит. для C15H23NO2 250,36; найдено 250,2. 1H ЯМР (d6-ДМСО, 400 мГц) δ (ч/млн) 9,23 (с, 1H), 8,02 (ушир.с, 3H), 6,92 (д, J=8,2 Гц, 1H), 6,61 (м, 2H), 3,77 (м, 1H), 3,41 (с, 3H), 3,30 (м, 1H), 3,17 (м, 1H), 2,44 (дд, J=15,9 Гц, 9,8 Гц, 1H), 1,85 (м, 2H), 1,62-1,52 (м, 2H), 0,66 (т, J=7,2 Гц, 3H), 0,55 (т, J=7,0 Гц, 3H).
c. (6R,7R)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-ол
В соответствии с процедурой предыдущей стадии, используя (R)-1-фенилэтиловый эфир ((2R,3R)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты, получали гидрохлорид целевого соединения. (m/z): [M+H]+ рассчит. для C15H23NO2 250,36; найдено 250,4. 1H ЯМР (d6-ДМСО, 400 мГц) δ (ч/млн) 9,23 (с, 1H), 8,02 (ушир.с, 3H), 6,92 (д, J=8,2 Гц, 1H), 6,61 (м, 2H), 3,77 (м, 1H), 3,41 (с, 3H), 3,30 (м, 1H), 3,17 (м, 1H), 2,44 (дд, J=15,7 Гц, 10,2 Гц, 1H), 1,84 (м, 2H), 1,62-1,52 (м, 2H), 0,66 (т, J=7,4 Гц, 3H), 0,55 (т, J=7,0 Гц, 3H).
Пример получения 11: (R)-1-фенилэтиловый эфир ((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты (SS) и (R)-1-фенилэтиловый эфир ((2R,3R)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты (RR)
Смесь 4-нитрофенил-(R)-1-фенилэтилового эфира угольной кислоты (7,35 г, 25,6 моль), гидрохлорида амида транс-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты (4,0 г, 13 ммоль) и триэтиламина (5,3 мл, 38 моль) в DMF (13 мл) нагревали при 85°C. Через 2,5 часа реакционную смесь охлаждали и перемешивали при температуре окружающей среды в течение ночи. Растворитель удаляли под вакуумом, и остаток очищали хроматографией на силикагеле с элюированием EtOAc в DCM (градиент от 10% до 50%), получая смесь, содержащую целевое соединение (6,96 г). Смесь диастереомеров разделяли препаративной ВЭЖХ в условиях, описанных в Примере получения 10(a) за исключением использования следующего градиента (время(мин)/% B): 0/5, 4/5, 8/37, 60/42. Чистые фракции каждого изомера объединяли и лиофилизировали, получая целевое соединение.
SS: 1,4 г (26%) (m/z): [M+H]+ рассчит. для C25H32N2O4 425,24; найдено 425,6.
RR: 1,5 г (28%) (m/z): [M+H]+ рассчит. для C25H32N2O4 425,24; найдено 425,4.
Дифрактометрия рентгеновских лучей монокристалла диастереомера SS
SS (3 мг) растворяли в ацетонитриле (100 мл) в открытой ампуле для ВЭЖХ, которую частично погружали в ампулу на 20 мл, содержащую смесь 1:9 ацетонитрил:вода (4 мл). Ампулу на 20 мл закрывали и держали при температуре окружающей среды, получая крупные двоякопреломляющие игольчатые кристаллы SS.
Данные структуры дифракции рентгеновских лучей в кристалле получали для монокристалла с размерами 0,44 ×0,13 ×0,10 мм, используя облучение Mo Kα (λ=0,71073 Å) на дифрактометре Nonius KappaCCD, оборудованном графитовым кристаллом и монохроматором падающего луча света, и анализировали на LINUX PC, используя программное обеспечение SHELX97. Получали следующие параметры решетки: элементарная ячейка является гексагональной с размерами=17,451 Å, b=17,451 Å, c=19,822 Å, α=90,00°, β=90,00°, γ=120,00°, объем ячейки (V)=5228 Å3, пространственная группа P 3121. Молекула содержит три хиральных центра. От известной конфигурации R углерода, несущего фенильную группу:

другие два центра были определены как имеющие конфигурации S.
Остальные кристаллы были проанализированы порошковой дифракцией рентгеновских лучей. Пики порошковой дифракции рентгеновских лучей, предсказанные на основании кристаллографических данных производного монокристалла, согласовывались с наблюдаемыми пиками порошковой дифракции рентгеновских лучей.
Пример получения 12: амид (6R,7R)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты
(R)-1-фенилэтиловый эфир ((2R,3R)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты (1,7 г, 4,0 ммоль) обрабатывали 4,0н. HCl в диоксане (20 мл, 80 ммоль) и перемешивали при температуре окружающей среды. Через 24 часа растворитель удаляли при пониженном давлении, и остаточное твердое вещество растирали с 50%-ным дихлорметаном в гексане (15 мл). Твердое вещество собирали на воронке Бюхнера, промывали 50%-ным дихлорметаном в гексане (10 мл) и высушивали под вакуумом, получая целевое соединение в форме гидрохлорида (1,2 г). (m/z): [M+H]+ рассчит. для C16H24N2O2 277,19; найдено 277,4. 1H NMR (d6-ДМСО, 400 мГц) δ (ч/млн) 8,19 (ушир, с, 3H), 7,98 (с, 1H), 7,70 (м, 2H), 7,32 (с, 1H) 7,19 (д, J=7,8 Гц, 1H), 3,83 (м, 1H), 3,47 (м, 1H), 3,42 (с, 3H), 3,23 (м, 1H), 2,63 (дд, J=16,8 Гц, 9,7 Гц, 1H) 2,06 (м, 1H), 1,88 (м, 1H) 1,64 (кварт., J=7,7 Гц, 2H), 0,62 (т, J=7,5 Гц, 3H), 0,50 (т, J=7,0 Гц, 3H).
Пример получения 13: (S)-4-циклогексил-1-гидрокси-3-метоксикарбонил-бутан-1-сульфонат натрия
a: метиловый эфир (S)-2-циклогексилметил-4-гидрокси-масляной кислоты
Смесь 1-метилового эфира (S)-2-циклогексилметил-янтарной кислоты (60,0 г, 263 ммоль) и тетрагидрофурана (600 мл) перемешивали при температуре окружающей среды и затем охлаждали до -5°C в течение 30 мин. К реакционной смеси добавляли по каплям за 45 минут 1,0М гидрида бора в тетрагидрофуране (520 мл), поддерживая внутреннюю температуру ниже 0°C. К реакционной смеси добавляли по каплям MeOH (100 мл), чтобы остановить реакцию. Реакционную смесь концентрировали приблизительно до 100 мл упариванием на роторном испарителе. Добавляли (трифторметил)бензол (200 мл) и объем уменьшали до 25 мл упариванием на роторном испарителе. К полученному густому маслу добавляли (трифторметил)бензол (100 мл), и объем уменьшали до ~25 мл, получая сырой целевой продукт (56,3 г).
b. (S)-4-циклогексил-1-гидрокси-3-метоксикарбонил-бутан-1-сульфонат натрия
Смесь метилового эфира (S)-2-циклогексилметил-4-гидрокси-масляной кислоты (44,8 г, 209 ммоль) и DCM (310 мл) охлаждали до 5°C при перемешивании. К реакционной смеси добавляли раствор бромида калия (2,5 г, 21 ммоль) и бикарбонат натрия (2,4 г, 29 ммоль) в дистиллированной воде (130 мл), и затем 2,2,6,6-тетраметилпиперидин-1-оксил (ТЕМРО) (0,33 г, 2,1 ммоль), с последующим добавлением гипохлорита натрия (140 мл, 210 ммоль) со скоростью 130 мл/ч, поддерживая внутреннюю температуру в диапазоне от 6 до 8°C. Реакционную смесь перемешивали в течение 15 минут и добавляли DCM (200 мл). Слои разделяли, и органический слой промывали насыщенным солевым раствором (200 мл), и высушивали Na2SO4.
К органическому слою добавляли EtOAc (40 мл), с последующим добавлением бисульфита натрия (21,8 г, 209 ммоль). Реакционный раствор концентрировали, чтобы удалить половину DCM (~175 мл) упариванием на роторном испарителе. К реакционному раствору добавляли воду (2 мл) и перемешивали при температуре окружающей среды в течение ночи. Полученную суспензию фильтровали; остаток после фильтрации высушивали под вакуумом в течение ночи, получая целевое соединение (61,9 г). 1H ЯМР (ДМСО-d6): δ (ч/млн) 0,78 (м, 2H), 0,95-1,20 (м, 4H), 1,33 (м, 1H), 1,40-1,95 (м, 5H), 2,45-2,65 (м, 1H), 3,21 (м, 2H), 3,45 (с, 3H), 3,6-3,8 (м, 1H), 5,18 (д, 1H).
Пример получения 14: амид транс-7-амино-8,8-диметил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты
a. 7-метокси-1,1-диметил-3,4-дигидро-1Н-нафталин-2-он
Суспензию трет-бутоксида натрия (21,1 г, 220 ммоль) в THF (100 мл) охлаждали до 0°C. Раствор 7-метокси-3,4-дигидро-1Н-нафталин-2-она (17,6 г, 100 ммоль) и метилйодида (30,1 г, 220 ммоль) в THF (100 мл) добавляли по каплям за 40 минут, и реакционную смесь нагревали до температуры окружающей среды в течение 10 мин. Добавляли воду (200 мл) и EtOAc (600 мл). Слои разделяли, органический слой промывали водой (5 ×100 мл) и насыщенным NaCl (100 мл), фильтровали и высушивали Na2SO4, получая целевое соединение (20 г).
b. 7-метокси-1,1-диметил-3,4-дигидро-1Н-нафталин-2-он оксим
К раствору 7-метокси-1,1-диметил-3,4-дигидро-1Н-нафталин-2-она (25,4 г, 98 ммоль) в метаноле (175 мл) добавляли раствор гидрохлорида гидроксиламина (20,5 г, 295 ммоль) и ацетата натрия (24,2 г, 295 ммоль) в воде (175 мл), и реакционную смесь нагревали при 70°C в течение 3 часов, и охлаждали на льду в течение 30 мин. Твердое вещество собирали на воронке Бюхнера, перемешивали метанолом (125 мл) при 50°C в течение 30 минут и затем перемешивали при температуре окружающей среды в течение ночи. Реакционную смесь охлаждали до 0°C; твердое вещество собирали на воронке Бюхнера, промывали холодным метанолом (20 мл) и высушивали под вакуумом, получая целевое соединение (14,7 г).
c. (1aS,7aR)-4-метокси-2,2-диметил-1a,2,7,7a-тетрагидро-1Н-1-аза-циклопропа[b]-нафталин
К раствору 7-метокси-1,1-диметил-3,4-дигидро-1Н-нафталин-2-он оксима (15,3 г, 70 ммоль) в THF (240 мл) добавляли диэтиламин (18 мл). Реакционную смесь охлаждали до 0°C, и раствор на 2,0 м. литий-алюминийгидрида в THF (100 мл, 200 ммоль) добавляли медленно за 20 минут, чтобы контролировать скорость выделения водорода. Реакционную смесь нагревали до 70°C в течение 1 часа, охлаждали до 0°C и добавляли Na2SO4 10H2O (20 г), солевой раствор (60 мл) и EtOAc (300 мл). Твердое вещество промывали EtOAc (4×100 мл); объединенные органические слои промывали водой (4×100 мл) и солевым раствором (100 мл), высушивали Na2SO4 и концентрировали, получая сырой целевой продукт (14,3 г). Сырой продукт растворяли в EtOAc (500 мл), экстрагировали 0,1н. HCl (100 мл), затем 0,3н. HCl (225 мл). К водному слою добавляли карбонат натрия (8 г, 75 ммоль), и водный слой экстрагировали EtOAc (4×200 мл). Органические слои объединяли, высушивали с помощью Na2SO4 и концентрировали, получая целевое соединение в форме масла (10,1 г), которое при отстаивании кристаллизовалось в форме твердого вещества желто-коричневого цвета. (m/z): [M+H]+ рассчит. для C13H17NO 204,14; найдено 204,2.
d. трет-бутиловый эфир транс-(7-гидрокси-3-метокси-1,1-диметил-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты
Используя процедуру, подобную используемой в Примерах получения 1(b) и 2(a), получали целевое соединение. 1H ЯМР (d6-ДМСО, 400 мГц) δ (ч/млн): 9,04 (с, 1H), 6,83 (д, J=8,2 Гц, 1H), 6,69 (д, J=9,4 Гц, 1H), 6,65 (д, J=2,5 Гц, 1H), 6,52 (дд, J=8,2, 2,5 Гц, 1H), 3,50 (м, 1H), 3,45 (м, 1H), 3,30 (с, 3H), 3,15 (м, 1H), 2,55 (м, 1H), 1,34 (с, 9H), 1,16 (с, 3H), 1,00 (с, 3H).
e. трет-бутиловый эфир транс-(7-карбамоил-3-метокси-1,1-диметил-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты.
Используя процедуру, подобную используемой в Примерах получения 2(b), 2(c) и 5, получали целевое соединение. (m/z): [M+H]+ рассчит. для C19H28N2O4 349,21; найдено 349,1.
f. амид транс-(7-амино-6-метокси-8,8-диметил-5,6,7,8-тетрагидронафталин-2-карбоновой кислоты
К суспензии трет-бутилового эфира транс-(7-карбамоил-3-метокси-1,1-диметил-1,2,3,4-тетрагидро-нафталин-2-ил)-карбаминовой кислоты (9,22 г, 26.4 ммоль) в DCM (100 мл) медленно добавляли 4н. HCl в диоксане (25 мл, 100 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 15 часов, концентрировали досуха, растирали с DCM (25 мл) в течение 30 минут, фильтровали, промывали DCM (3×15 мл) и высушивали под вакуумом. Добавляли этанол (100 мл), и реакционную смесь концентрировали под вакуумом, получая соль HCl целевого соединения (7,17 г) в форме белого порошка. (m/z): [M+H]+ рассчит. для C14H20N2O2 249,16; найдено 249,1. 1H ЯМР (d6-ДМСО, 400 мГц) δ (ч/млн): 8,18 (с, 3H), 8,00 (с, 1H), 7,92 (д, J=1,6 Гц, 1H), 7,66 (дд, J=8,0 Гц, 1,8 Гц, 1H), 7,32 (с, 1H), 7,17 (д, J=8,0 Гц, 1H), 3,70 (м, 1H), 3,44 (с, 3H), 3,43 (м, 1H), 3,22 (м, 1H), 2,67 (дд, J=16,4 Гц, 10,2 Гц), 1,50 (с, 3H), 1,24 (с, 3H).
Пример получения 15: метиловый эфир (S)-2-циклогексилметил-4-оксо-масляной кислоты.
a. метиловый эфир (S)-2-циклогексилметил-4-гидрокси-масляной кислоты
Смесь 1-метилового эфира (S)-2-циклогексилметил-янтарной кислоты (484 мг, 2,12 ммоль) и тетрагидрофурана (10 мл) перемешивали при температуре окружающей среды и затем охлаждали до 0°C. К реакционной смеси по каплям за 5 мин добавляли 1,0М гидрида бора в тетрагидрофуране (4,2 мл). Через 2 часа добавляли по каплям MeOH, чтобы остановить реакцию. Реакционную смесь перемешивали в течение 30 минут при температуре окружающей среды и затем концентрировали досуха. Сырой остаток суспендировали в MeOH, концентрировали досуха и очищали на SiO2 (40 г), используя 5-10% MeOH/DCM в качестве элюента, получая целевое соединение (0,32 г) в форме прозрачного масла: 1H ЯМР (CDCl3, 400 мГц) δ (ч/млн): 3,69-3,65 (м, 5H), 2,65 (м, 1H), 1,85-1,59 (м, 8H), 1,32-1,12 (м, 5H), 0,87 (м, 2H).
b. метиловый эфир (S)-2-циклогексилметил-4-оксо-масляной кислоты
Смесь продукта предыдущей стадии (0,32 г, 1,49 ммоль), N,N-диизопропилэтиламина (0,65 мл, 3,7 ммоль), диметилсульфоксида (0,26 мл, 3,7 ммоль) и дихлорметана (20 мл, 0,3 моль) охлаждали до 0°C и промывали азотом. Комплекс триоксид серы-пиридин (0,59 г, 3,7 ммоль) добавляли в токе азота, и реакционную смесь перемешивали в течение 1,5 часов. К реакционной смеси добавляли 0,1н. HCl. Органический слой промывали 0,1н. HCl (2×) и солевым раствором (2×), высушивали Na2SO4, фильтровали и концентрировали досуха, получая целевое соединение (0,305 г) в форме прозрачного масла, которое использовали без дальнейшей очистки. 1H ЯМР (CDCl3, 400 мГц) δ (ч/млн): 9,75 (с, 1H), 3,69 (с, 3H), 3,02-2,96 (м, 1H), 2,84 (дд, J=18,0, 9,0 Гц, 1H), 2,55 (дд, J=18, 4,7 Гц, 1H), 1,79-1,12 (м, 11H), 0,93-0,84 (м, 2H).
Пример получения 16: Альдегидные реагенты
Согласно способу Примера получения 14 с использованием подходящего сложного метилового эфира вместо 1-метилового эфира (S)-2-циклогексилметил-янтарной кислоты, получали следующие альдегиды:
метиловый эфир (R)-2-циклогексилметил-4-оксо-масляной кислоты: 1H ЯМР (CDCl3, 400 мГц) δ (ч/млн): 9,76 (с, 1H), 3,69 (с, 3H), 3,01-2,96 (м, 1H), 2,84 (дд, J=18,0, 9,0 Гц, 1H), 2,55 (дд, J=18, 4,7 Гц, 1H), 1,79-1,14 (м, 11H), 0,92-0,96 (м, 2H);
метиловый эфир (S)-2-циклогексил-4-оксо-масляной кислоты: 1H ЯМР (CDCl3, 400 мГц) δ (ч/млн): 9,78 (с, 1H), 3,69 (с, 3H), 2,91 (дд, J=18, 10 Гц, 1H), 2,80-2,75 (м, 1H), 2,56 (дд, J=18, 3,5 Гц, 1H), 1,84-0,98 (м, 11H);
метиловый эфир (S)-2-пентил-4-оксо-масляной кислоты: 1H ЯМР (CDCl3, 400 мГц) δ (ч/млн): 9,76 (с, 1H), 3,70 (с, 3H), 2,92-2,84 (м, 2H), 2,80-2,75 (м, 1H), 2,58-2,54 (м, 1H), 1,67-1,63 (м, 1H), 1,57-1,48 (м, 2H), 1,28 (ушир.с, 6H), 0,89-0,86 (м, 3H);
метиловый эфир (S)-2-фенилпропил-4-оксо-масляной кислоты: 1H ЯМР (CDCl3, 400 мГц): подтверждено присутствие альдегида с пиком 9,75 (с, 1H);
метиловый эфир (S)-2-изобутил-4-оксо-масляной кислоты: 1H ЯМР (CDCl3, 400 мГц) δ (ч/млн): 9,76 (с, 1H), 3,70 (с, 3H), 3,1-2,94 (м, 1H), 2,88-2,81 (м, 1H), 2,62-2,52 (м, 1H), 1,66-1,26 (м, 3H), 0,97-0,88 (м, 6H);
метиловый эфир (R)-2-изобутил-4-оксо-масляной кислоты: 1H ЯМР (CDCl3, 400 мГц) δ (ч/млн): 9,76 (с, 1H), 3,70 (с, 3H), 2,96-2,93 (м, 1H), 2,84 (дд, J=18, 9,0 Гц, 1H), 2,55 (дд, J=18, 4,5 Гц, 1H), 1,64-1,25 (м, 3H), 0,94-0,86 (м, 6H);
метиловый эфир (S)-2-изопропил-4-оксо-масляной кислоты. 1H ЯМР (CDCl3, 400 мГц): подтверждено присутствие альдегида с пиком 9,79 (с, 1H);
метиловый эфир (S)-4,4-диметил-2-(2-оксо-этил)-пентановой кислоты: 1H ЯМР (CDCl3, 400 мГц) δ (ч/млн): 9,73 (с, 1H), 3,69 (с, 3H), 2,80 (дд, J=17,8, 8,0 Гц, 1H), 2,57 (дд, J=18, 5,8 Гц, 1H), 1,79 (дд, J=14, 8,41 Гц, 1H), 1,50-1,40 (м, 1H), 1,25 (дд, J=14, 3,7 Гц, 1H), 0,91 (с, 9H);
метиловый эфир (R)-4,4-диметил-2-(2-оксо-этил)-пентановой кислоты: 1H ЯМР (CDCl3, 400 мГц) δ (ч/млн): 9,73 (с, 1H), 3,69 (с, 3H), 2,80 (дд, J=17,8, 8,0 Гц, 1H), 2,57 (дд, J=17,8, 5,67 Гц, 1H), 1,79 (дд, J=14,1, 8,41 Гц, 1H), 1,50-1,41 (м, 1H), 1,25 (дд, J=14,1, 3,7 Гц, 1H), 0,91 (с, 9H);
метиловый эфир (S)-бензил-4-оксо-масляной кислоты: 1H ЯМР (CDCl3, 400 мГц) δ (ч/млн): подтверждено присутствие альдегида с пиком 9,70 (с, 1H);
метиловый эфир (R)-2-бутил-4-оксо-масляной кислоты: 1H ЯМР (CDCl3, 400 мГц) δ (ч/млн): 9,76 (с, 1H), 3,69 (с, 3H), 2,91-2,87 (м, 1H), 2,57-2,54 (м, 1H), 1,70-1,25 (м, 6H), 0,92-0,87 (м, 3H).
Пример получения 17: (S)-3-бензилоксикарбонил-4-циклогексил-1-гидрокси-бутан-1-сульфонат натрия
a. метиловый эфир (S)-2-циклогексилметил-4,4-диметокси-масляной кислоты
К суспензии (S)-4-циклогексил-1-гидрокси-3-метоксикарбонил-бутан-1-сульфоната натрия (400,0 г, 1,26 моль) и метанола (2 л) добавляли 4,0М HCl в 1,4-диоксане (400 мл), и реакционную смесь перемешивали в течение 15 мин. Добавляли триметоксиметан (340 мл, 3,11 моль), и реакционную смесь нагревали при 50°C в течение ночи и затем охлаждали до температуры окружающей среды. Белые твердые частицы отфильтровывали и отбрасывали. Большую часть растворителя удаляли из фильтрата упариванием на роторном испарителе. Добавляли этилацетат (800 мл), что усиливало осаждение. Белый осадок удаляли фильтрацией. Растворитель удаляли из фильтрата упариванием на роторном испарителе и затем под высоким вакуумом при температуре окружающей среды в течение ночи, получая целевое соединение (211 г) в форме густого масла. 1H ЯМР (400 МГц, ДМСО-d6): δ (ч/млн) 4,25 (т, 1H), 3,57 (с, 3H), 3,18 (с, 6H), 2,43 (м, 1H), 1,55-1,81 (м, 2H), 1,50-1,72 (м, 5H), 1,20-1,48 (м, 2H), 1,05-1,21 (м, 4H), 0,71-0,92 (м, 2H).
b. (S)-2-циклогексилметил-4,4-диметокси-бутират калия
Гидроксид калия (289,6 г, 2322 ммоль) добавляли к раствору продукта предыдущей стадии (200,0 г, 0,77 моль) в метаноле (700 мл) в виде одной части, и реакционную смесь перемешивали при температуре окружающей среды в течение 20 часов. Медленно добавляли хлорид водород (130 мл, 1,5 моль) до достижения реакционной смесью рН~8 (изменение цвета с зеленоватого на оранжевый), что приводило к осаждению тонкодисперсных твердых частиц. Твердые частицы удаляли фильтрацией. Растворитель удаляли из фильтрата. К сырому продукту добавляли ацетонитрил (1 л), и полученную суспензию перемешивали при температуре окружающей среды в течение ночи. Густую суспензию фильтровали, остаток после фильтрации промывали ацетонитрилом (50 мл) и высушивали, получая первый выход целевого соединения (133 г) в форме твердого вещества грязно-белого цвета. Растворитель удаляли из фильтрата, который затем высушивали под вакуумом, получая приблизительно 100 г пастообразного твердого вещества. Добавляли MTBE (500 мл), и твердые частицы перемешивали при температуре окружающей среды в течение ночи, получая густую суспензию, которую фильтровали и высушивали под высоким вакуумом, получая второй выход целевого соединения (82 г). 1H ЯМР (400 МГц, ДМСО-d6): δ (ч/млн) 4,28 (дд, 1H), 3,12 (с, 3H), 3,15 (с, 3H), 1,95 (м, 1H), 1,75 (м, 1H), 1,51-1,65 (м, 6H), 1,22-1,39 (м, 2H), 1,05-1,20 (м, 4H), 0,85-0,93 (м, 1H), 0,65-0,81 (м, 2H).
c: бензиловый эфир (S)-2-циклогексилметил-4,4-диметокси-масляной кислоты
Бензилбромид (50,54 мл, 424,9 ммоль) добавляли к суспензии продукта предыдущей стадии (150,0 г, 531,1 ммоль) в ацетонитриле (2,0 л) в виде одной части, и гетерогенную реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Добавляли дополнительный бензилбромид (5,05 мл, 42,49 ммоль), и реакционную смесь перемешивали при температуре окружающей среды в течение 18 часов. Твердые частицы удаляли фильтрацией. Фильтрат высушивали упариванием на роторном испарителе и затем под высоким вакуумом в течение ночи, получая целевое соединение (162 г). 1H ЯМР (400 МГц, ДМСО-d6): δ 7,22-7,40 (м, 5H), 5,0-5,15 (кв, 2H), 4,23 (т, 1H), 3,15 (с, 3H), 3,17 (с, 3H), 2,52 (м, 1H), 1,78 (м, 1H), 1,69 (м, 1H), 1,45-1,61 (м, 6H), 1,20-1,43 (м, 2H), 1,0-1,15 (м, 4H), 0,70-0,83 (м, 2H).
d. (S)-3-бензилоксикарбонил-4-циклогексил-1-гидрокси-бутан-1-сульфонат натрия
К смеси продукта предыдущей стадии (160,0 г, 478,4 ммоль) и ацетонитрила (1,0 л) добавляли 1,0М HCl в воде (1,2 л) и реакционную смесь нагревали при 35-40°C в течение 2 часов. Добавляли этилацетат (1,2 л), фазы разделяли, и органический слой промывали солевым раствором (1 л). К влажному органическому слою добавляли бисульфит натрия (74,7 г, 718 ммоль), и реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Большую часть растворителя удаляли упариванием на роторном испарителе и добавляли ацетонитрил (1 л), и полученную суспензию перемешивали при температуре окружающей среды в течение ночи. Полученную густую белую суспензию фильтровали, остаток после фильтрации промывали ацетонитрилом (2 ×100 мл) и высушивали под вакуумом, получая целевое соединение (200 г, чистота >98%) в форме твердого вещества белого цвета. 1H ЯМР (400 МГц, ДМСО-d6): δ 7,23-7,41 (м, 5H), 5,30 (д, 1H), 4,98-5,18 (кв., 2H), 3,75-3,88 (м, 1H), 3,60-3,79 (м, 1H), 2,05 (м, 0,5H), 1,45-1,82 (м, 2,5H), 1,45-1,60 (м, 5H), 1,20-1,42 (м, 2H), 1,0-1,17 (м, 4H), 0,69-0,82 (м, 2H).
Пример 1: (S)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляная кислота
a. метиловый эфир (S)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляной кислоты
К суспензии (S)-4-циклогексил-1-гидрокси-3-метоксикарбонил-бутан-1-сульфоната натрия (25,8 г, 81,5 ммоль) и 2-метил-тетрагидро-фурана (300 мл) добавляли 1,0М NaOH в воде (76,1 мл), и реакционную смесь перемешивали в течение 20 минут при температуре окружающей среды. К реакционной смеси добавляли гидрохлорид амида (6S,7S)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты (17,0 г, 54,3 ммоль); реакционную смесь перемешивали в течение 40 минут при температуре окружающей среды, добавляли в 4 частях триацетоксиборгидрид натрия (46,1 г, 217 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение ночи после первых двух частей. Добавляли воду (200 мл) и MeTHF (100 мл); фазы разделяли, и органический слой промывали 1М NaOH (2 ×200 мл), разбавленным солевым раствором (200 мл), высушивали с помощью Na2SO4 и растворитель удаляли, получая сырое промежуточное соединение (22 г) в форме стекловидного твердого вещества желтого цвета.
Сырой продукт очищали хроматографией с обратной фазой, используя 4-дюймовую колонку Microsorb 100-10 BDS. Сырой продукт растворяли в смеси растворителей 1:1 ацетонитрил:1М водный раствор HCl (150 мл) и элюировали мобильной фазой вода (0,1% HCl)/ацетонитрил (градиент 10-40%). Чистые фракции (>98%) объединяли, большую часть ацетонитрила удаляли упариванием на роторном испарителе, подщелачивали до рН ~12 с помощью твердого Na2CO3, и очищенный продукт экстрагировали MeTHF (3×1 л). Объединенные органические слои высушивали с помощью Na2SO4 и растворитель удаляли, получая целевое соединение (16,5 г).
b. (S)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляная кислота
К раствору метилового эфира (S)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляной кислоты (12,0 г, 25,4 ммоль) в метаноле добавляли 5,0М NaOH (25 мл), и реакционную смесь нагревали при 30°C в течение 8 часов и затем при 25°C в течение ночи. Большую часть метанольного растворителя удаляли упариванием на роторном испарителе при 25°C, добавляли воду (100 мл) и изопропил ацетат (100 мл), и полученную смесь перемешивали в течение 15 мин. Два нижних слоя из трех слоев экстрагировали изопропилацетатом (100 мл). Нижние слои охлаждали до -5°C и добавляли MeTHF (200 мл) и затем добавляли частями концентрированную HCl (~15 мл) до рН~2. Фазы разделяли, водный слой промывали MeTHF (100 мл), и объединенные органические слои высушивали с помощью Na2SO4. Большую часть органического растворителя удаляли упариванием на роторном испарителе, добавляли этилацетат (200 мл), и объем уменьшали до 50 мл. Добавляли этилацетат (200 мл), и полученную суспензию перемешивали/растирали при температуре окружающей среды в течение 3 часов. Продукт фильтровали в атмосфере азота и высушивали под вакуумом в течение 48 часов, получая гидрохлорид целевого соединения (11 г, чистота 98,2%) в форме твердого вещества белого цвета. 1H ЯМР (ДМСО-д6): δ (ч/млн) 0,54 (т, 3H), 0,63 (т, 3H), 0,82 (м, 2H), 1,05-1,3 (м, 6H), 1,45 (м, 1H), 1,55-2,0 (м, 10H), 2,40 (м, 1H), 2,67 (дд, 1H), 3,06 (м, 1H), 3,22 (м, 1H), 3,30 (дд, 1H), 3,41 (с, 3H), 3,45 (дд, 1H), 4,05 (м, 1H), 7,19 (д, 1H), 7,50 (ушир., 1H), 7,69 (д, 1H), 7,70 (с, 1H), 7,95 (ушир., 2H), 9,26 (ушир., 1H).
Пример 2: метиловый эфир (S)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляной кислоты
Метиловый эфир (S)-2-циклогексилметил-4-оксо-масляной кислоты (822 мг, 3,38 ммоль), гидрохлорид амида (6S,7S)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидронафталин-2-карбоновой кислоты (1,01 г, 3,23 ммоль) и триэтиламин (326 мг, 3,23 ммоль) растворяли в дихлорметане (15 мл) и метаноле (10 мл). Добавляли триацетоксиборгидрид натрия (1,03 г, 4,85 ммоль). В течение 3 часов добавляли в двух частях дополнительный триацетоксиборгидрид натрия (900 мг, 4,2 моль) и метиловый эфир (S)-2-циклогексилметил-4-оксо-масляной кислоты (550 мг, 2,6 ммоль), и реакционную смесь перемешивали в течение 1 часа после последнего добавления. Добавляли насыщенный бикарбонат натрия (60 мл), и реакционную смесь экстрагировали дихлорметаном (4×50 мл). Органический слой высушивали Na2SO4 и концентрировали. Полученное масло растворяли в метаноле (50 мл) и концентрировали. Сырой продукт очищали препаративной ВЭЖХ. Фракции, содержащие чистый продукт, объединяли, и ацетонитрил удаляли при пониженном давлении. Добавляли карбонат натрия (1,3 г, 12,3 ммоль), и продукт экстрагировали в дихлорметан (3×200 мл). К водному слою добавляли дополнительную часть сульфата натрия (15 г, 140 ммоль) и экстрагировали дихлорметаном (3×200 мл). Органические экстракты объединяли, высушивали с помощью Na2SO4 и концентрировали, получая целевое соединение в форме свободного основания (1,16 г, 76%-ный выход).
(m/z): [M+H]+ рассчит. для C28H44N2O4, 473,67; найдено 473,4. 1H ЯМР (CDCl3, 400 мГц) δ (ч/млн): 7,66 (д, J=1,8 Гц, 1H), 7,50 (дд, J=8,0 Гц, 1,9 Гц, 1H), 7,13 (д, J=8,0 Гц, 1H), 3,65 (с, 3H), 3,58 (м, 1H), 3,47 (с, 3H), 3,31 (дд, J=16,7, 6,1 Гц, 1H), 2,97 (м, 1H), 2,74-2,64 (м, 3H), 2,57 (м, 1H), 1,90-1,53 (м, 14H), 1,33-1,11 (м, 6H), 0,89-0,83 (м, 3H), 0,69 (т, J=7,6 Гц, 3H), 0,59 (т, J=7,3 Гц, 3H).
Пример 3: (S)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляная кислота
Смесь продукта Примера 2 (685 мг, 1,43 ммоль), 10н. NaOH (0,87 мл, 8,7 ммоль), метанола (4,5 мл) и воды (0,45 мл) нагревали при 55°C в течение 2 часов. Смесь охлаждали до температуры окружающей среды, разбавляли 50%-ным водным раствором уксусной кислоты и очищали препаративной ВЭЖХ. Чистые фракции объединяли с чистыми фракциями от другой загрузки (масштаб 0,79 ммоль) и лиофилизировали, получая целевое соединение (1,02 г, 80%-ный выход) в форме соли TFA. (m/z): [M+H]+ рассчит. для C27H42N2O4, 459,32; найдено 459,8. 1H ЯМР (d6-ДМСО, 400 мГц) δ (ч/млн): 8,92 (ушир.с, 1H), 7,97 (с, 1H), 7,70 (м, 3H), 7,35 (с, 1H), 7,22 (д, J=8,4 Гц, 1H), 4,01 (м, 1H), 3,48 (дд, J=16,4, 5,7 Гц, 1H), 3,42 (с, 3H), 3,36 (м, 1H), 3,27 (м, 1H), 3,10 (м, 1H), 2,70 (дд, J=16,8, 10,2 Гц, 1H), 2,43 (м, 1H), 2,15 (м, 1H), 1,90 (м, 2H), 1,69-1,59 (м, 8H), 1,49 (м, 1H), 1,28-1,09 (м, 5H) 0,86, (м, 2H), 0,66 (т, J=7,4 Гц, 3H), 0,58 (т, J=7,0 Гц, 3H).
Пример 4: метиловый эфир (S)-4-((2R,3R)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляной кислоты
Согласно процедуре Примера 2 с использованием гидрохлорида амида (6R,7R)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидронафталин-2-карбоновой кислоты получали целевое соединение. (m/z): [M+H]+ рассчит. для C28H44N2O4, 473,34; найдено 473,4.
Пример 5: метиловый эфир (R)-4-((2R,3R)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляной кислоты
Гидрохлорид амида (6R,7R)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты (0,16 г, 0,51 ммоль) растворяли в дихлорметане (4,4 мл) и метаноле (2 мл) при температуре окружающей среды. Добавляли метиловый эфир (R)-2-циклогексилметил-4-оксо-масляной кислоты (0,22 г, 1,0 ммоль), затем триэтиламин (0,071 мл, 0,51 ммоль) и триацетоксиборгидрид натрия (0,16 г, 0,77 ммоль). В течение 2 часов добавляли дополнительный триацетоксиборгидрид натрия (0,16 г). Добавляли насыщенный раствор бикарбоната натрия, и реакционную смесь экстрагировали DCM. Органический экстракт промывали солевым раствором (2×), высушивали над сульфатом натрия, фильтровали и концентрировали досуха. Сырой продукт растворяли в смеси 1:1 AcOH/H2O и очищали препаративной ВЭЖХ, получая целевое соединение в форме соли TFA (161 мг, выход 53,6%). (m/z): [M+H]+ рассчит. для C28H44N2O4, 473,33; найдено 473,4. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 8,92 (ушир.с, 1H), 7,95 (с, 1H), 7,70 (с, 2H), 7,68 (с, 1H), 7,33 (с, 1H), 7,20 (д, J=8,4 Гц, 1H), 4,02-3,96 (м,1H), 3,61 (с, 1H), 3,46 (дд, J=16,8, 5,87 Гц, 1H), 3,40 (с, 3H), 3,35-3,31 (м, 1H), 3,20-3,08 (м, 2H), 2,68 (дд, J=16,4, 9,78 Гц, 1H), 2,51-2,58 (м, 1H), 2,16-2,11 (м, 1H), 1,96-1,86 (м, 2H), 1,77-1,59 (м, 9H), 1,50-1,44 (м, 1H), 1,16-1,10 (с, 4H), 0,88-0,83 (м, 2H), 0,64 (т, J=7,4 Гц, 3H), 0,56 (т, J=7,4 Гц, 3H).
Пример 6: метиловый эфир (R)-4-((2S,3SR)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляной кислоты
Согласно процедуре Примера 5 с использованием гидрохлорида амида (6S,7S)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты получали целевое соединение. (m/z): [M+H]+ рассчит. для C28H44N2O4, 473,33; найдено 473,4. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 8,90 (ушир.c, 1H), 7,96 (с, 1H), 7,74 (ушир.с, 1H), 7,70 (с, 1H), 7,68 (с, 1H), 7,32 (с, 1H), 7,20 (д, J=8,4 Гц, 1H), 4,04-4,97 (м, 1H), 3,60 (с, 1H), 3,47 (дд, J=16,8, 5,67 Гц, 1H), 3,40 (с, 3H), 3,33-3,22 (м, 2H), 3,03 (м, 1H), 2,69-2,57 (м, 2H), 2,17-2,11 (м, 1H), 1,99-1,96 (м, 1H), 1,86-1,59 (м, 9H), 1,50-1,43 (м, 1H), 1,32-1,25 (м, 1H), 1,17-1,06 (с, 4H), 0,89-0,80 (м, 2H), 0,64 (т, J=7,4 Гц, 3H), 0,56 (т, J=7,4 Гц, 3H).
Пример 7: метиловый эфир (S)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-4-метил-пентановой кислоты (7-a) и (S)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-4-метил-пентановая кислота (7-b)
Гидрохлорид амида (6S,7S)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты (0,19 г; 0,62 ммоль) растворяли в (3 мл) и метаноле (1,85 мл) при температуре окружающей среды. Добавляли метиловый эфир (S)-2-изобутил-4-оксо-масляной кислоты (0,212 г, 1,23 ммоль), затем триэтиламин (0,086 мл, 0,62 ммоль) и затем триацетоксиборгидрид натрия (0,20 г, 0,92 ммоль). В течение 3 часов добавляли дополнительное количество метилового эфира (S)-2-изобутил-4-оксо-масляной кислоты (0,070 г) и триацетоксиборгидрида натрия (0,15 г). Добавляли насыщенный раствор бикарбоната натрия, и реакционную смесь экстрагировали DCM. Органический экстракт высушивали с использованием сульфата натрия, фильтровали и концентрировали досуха. Сырой остаток растворяли в метаноле (3 мл) и 5н. NaOH (0,15 мл). Реакционную смесь нагревали при 50°C в течение 17 часов, затем охлаждали до температуры окружающей среды, разбавляли смесью 1:1 AcOH/H2O (3 мл) и очищали препаративной ВЭЖХ, получая соль TFA целевых соединений.
7-A: (35,3 мг, 10,2% за 2 стадии). (m/z): [M+H]+ рассчит. для C25H40N2O4, 433,30; найдено 433,4. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 8,94 (ушир.с, 1H), 7,95 (с, 1H), 7,69 (с, 2H), 7,68 (с, 1H), 7,29 (с, 1H), 7,16 (д, J=8,6 Гц, 1H), 4,01 (м, 1H), 3,61 (с, 3H), 3,46 (дд, J=16,6, 5,67 Гц, 1H), 3,40 (с, 3H), 3,35-3,30 (м, 1H), 3,12-3,09 (м, 2H), 2,67 (дд, J=16,4, 9,78 Гц, 1H), 2,5 (м, 1H), 2,16-2,10 (м, 1H), 1,92-1,87 (м, 2H), 1,78-1,59 (м, 3H), 1,52-1,45 (м, 2H), 1,29-1,22 (м, 1H), 0,85 (т, J=6,7, 6H), 0,63 (т, J=7,4 Гц, 3H), 0,56 (т, J=7,4 Гц, 3H).
7-B: (30 мг, 8,8% за 2 стадии). (m/z): [M+H]+ рассчит. для C24H38N2O4, 419,28; найдено 419,6. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 12,42 (ушир.с, 1H), 8,89 (ушир.с, 1H), 7,95 (с, 1H), 7,70 (с, 2H), 7,68 (с, 1H), 7,33 (с, 1H), 7,19 (д, J=8,6 Гц, 1H), 4,0 (м,1H), 3,49-3,47 (м, 1H), 3,40 (с, 3H), 3,36-3,30 (м, 1H), 3,24 (ушир.с, 1H), 3,1 (ушир.с, 1H), 2,65 (дд, J=16,4, 9,78 Гц, 1H), 2,42-2,41 (м, 1H), 2,16-2,10 (м, 1H), 1,94-1,81 (м, 2H), 1,80-1,42 (м, 6H), 1,27-1,21 (м, 1H), 0,93-0,80 (м, 8H), 0,64 (т, J=7,4 Гц, 3H), 0,56 (т, J=7,4 Гц, 3H).
Пример 8:
Согласно процедуре Примера 7 с использованием подходящего сложного метилового эфира вместо метилового эфира (S)-2-изобутил-4-оксо-масляной кислоты получали соли TFA следующих соединений:
8-A: метиловый эфир (R)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-4-метил-пентановой кислоты: (m/z): [M+H]+ рассчит. для C25H40N2O4, 433,30; найдено 433,6. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 8,84 (ушир.s, 1H), 7,93 (с, 1H), 7,68 (с, 2H), 7,66 (с, 1H), 7,31 (с, 1H), 7,18 (д, J=8,6 Гц, 1H), 3,98 (м, 1H), 3,59 (с, 3H), 3,46 (дд, J=16,6, 5,28 Гц, 1H), 3,38 (с, 3H), 3,32-3,23 (м, 2H), 3,02 (ушир.с, 1H), 2,63 (дд, J=16,2, 9,4 Гц, 1H), 2,55 (м, 1H), 2,16-2,10 (м, 1H), 1,97-1,95 (м, 1H), 1,83-1,59 (м, 3H), 1,49-1,43 (м, 2H), 1,26-1,23 (м, 1H), 0,84 (т, J=6,1 Гц, 6H), 0,63 (т, J=7,4 Гц, 3H), 0,54 (т, J=7,4 Гц, 3H).
8-B: (R)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-4-метил-пентановая кислота: (m/z): [M+H]+ рассчит. для C24H38N2O4, 419,28; найдено 419,4. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 12,41 (ушир.с, 1H), 8,88 (ушир.с, 1H), 7,93 (с, 1H), 7,68 (с, 2H), 7,66 (с, 1H), 7,31 (с, 1H), 7,18 (д, J=8,4 Гц, 1H), 4,01-3,94 (м,1H), 3,46 (дд, J=16,6, 16,6 Гц, 1H), 3,38 (с, 3H), 3,32-3,26 (м, 1H), 3,04 (ушир.с, 1H), 2,65 (дд, J=16,6, 9,79 Гц, 1H), 2,42-2,37 (м, 1H), 2,15-2,02 (м, 1H), 1,95-1,92 (м, 1H), 1,81-1,42 (м, 6H), 1,24-1,17 (м, 1H), 0,85 (т, J=6,2 Гц, 6H), 0,63 (т, J=7,4 Гц, 3H), 0,54 (т, J=7,4 Гц, 3H).
8-C: метиловый эфир (R)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-3-метилмасляной кислоты: (m/z): [M+H]+ рассчит. для C24H38N2O4, 419,28; найдено 419,4.
8-D: (R)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-3-метилмасляная кислота: (m/z): [M+H]+ рассчит. для C23H36N2O4, 405,27; найдено 405,4. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 112,39 (ушир.с, 1H), 8,92 (ушир.с, 1H), 7,94 (с, 1H), 7,68 (с, 2H), 7,67 (с, 1H), 7,32 (с, 1H), 7,19 (д, J=8,4 Гц, 1H), 4,01-3,96 (м,1H), 3,46 (дд, J=16,6, 5,68 Гц, 1H), 3,39 (с, 3H), 3,34-3,31 (м, 1H), 3,25 (ушир.с, 1H), 3,04 (ушир.с, 1H), 2,64 (дд, J=16,4, 9,59 Гц, 1H), 2,23-2,09 (м, 2H), 1,98-1,59 (м, 6H), 0,89-0,86 (м, 4H), 0,63 (т, J=7,4 Гц, 3H), 0,55 (т, J=7,4 Гц, 3H).
8-E: метиловый эфир (R)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-4,4-диметил-пентановой кислоты: (m/z): [M+H]+ рассчит. для C26H42N2O4, 447,31; найдено 447,6.
8-F: (R)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-4,4-диметил-пентановая кислота: (m/z): [M+H]+ рассчит. для C25H40N2O4, 433,30; найдено 433,2. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 8,90 (ушир.с, 1H), 7,94 (с, 1H), 7,69 (с, 2H), 7,67 (с, 1H), 7,32 (с, 1H), 7,19 (д, J=8,4 Гц, 1H), 4,01-3,94 (м,1H), 3,46 (дд, J=17,0, 5,87 Гц, 1H), 3,39 (с, 3H), 3,33-3,27 (м, 1H), 3,25 (ушир.с, 1H), 3,03 (ушир.с, 1H), 2,64 (дд, J=16,4, 9,78 Гц, 1H), 2,31-2,35 (м, H), 2,16-2,10 (м, 1H), 1,97-1,94 (м, 1H), 1,80-1,57 (м, 5H), 1,18 (дд, J=13,8, 2,93 Гц, 1H), 0,85 (м, 9H), 0,65 (т, J=7,4 Гц, 3H), 0,54 (т, J=7,4 Гц, 3H).
Пример 9: (S)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-гептановая кислота
Гидрохлорид амида (6S,7S)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты (0,20 г; 0,64 ммоль) растворяли в дихлорметане (3 мл) и метаноле (1,92 мл) при температуре окружающей среды. Добавляли метиловый эфир (S)-2-пентил-4-оксо-масляной кислоты (0,24 г, 1,3 ммоль), затем триэтиламин (0,089 мл, 0,64 ммоль) и триацетоксиборгидрид натрия (0,20 г, 0,96 ммоль). В течение 90 минут добавляли дополнительный триацетоксиборгидрид натрия (0,070 г, 0,33 ммоль). Добавляли насыщенный раствор бикарбоната натрия, и реакционную смесь экстрагировали DCM. Органический слой высушивали сульфатом натрия, фильтровали и концентрировали досуха (0,40 г). Сырой остаток растворяли в MeOH (3 мл) и добавляли 5н. NaOH (0,40 мл). Реакционную смесь нагревали при 50°C в течение 5 часов, затем охлаждали при температуре окружающей среды, перемешивая в течение ночи. Сырую реакционную смесь разбавляли смесью 1:1 AcOH/H2O (3 мл) и очищали препаративной ВЭЖХ, получая целевое соединение в форме соли TFA (32 мг, 8,8% за 2 стадии). (m/z): [M+H]+ рассчит. для C25H40N2O4, 433,30; найдено 433,4. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 7,91 (с, 1H), 7,66 (с, 2H), 7,64 (с, 1H), 7,29 (с, 1H), 7,16 (д, J=8,6 Гц, 1H), 3,97-3,91 (м,1H), 3,43 (дд, J=16,4, 5,48 Гц, 1H), 3,36 (с, 3H), 3,31-3,17 (м, 2H), 3,02 (м, 1H), 2,63 (дд, J=16,4, 9,78 Гц, 1H), 2,34-2,26 (м, 1H), 2,12-2,00 (м, 1H), 1,88-1,82 (м, 2H), 1,74-1,51 (м, 3H), 1,49-1,36 (м, 2H), 1,20 (с, 6H), 0,80 (т, J=6,7, 4H), 0,60 (т, J=7,4 Гц, 3H), 0,52 (т, J=7,4 Гц, 3H).
Пример 10
Согласно процедуре Примера 9 с использованием подходящего сложного метилового эфира вместо метилового эфира (S)-2-пентил-4-оксо-масляной кислоты получали соли TFA следующих соединений:
10-A: (S)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-5-фенил-пентановая кислота: (m/z): [M+H]+ рассчит. для C29H40N2O4, 481,30; найдено 481,4. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 12,39 (ушир.с, 1H), 8,87 (ушир.с, 1H), 7,92 (с, 1H), 7,65 (м, 3H), 7,29 (с, 1H), 7,22-7,05 (м, 7H), 3,98-3,91 (м,1H), 3,43 (дд, J=16,6, 5,67 Гц, 1H), 3,35 (с, 3H), 3,31-3,17 (м, 2H), 3,04 (ушир.с, 1H), 2,63 (дд, J=16,6, 9,59 Гц, 1H), 2,52 (т, J=6,6 Гц, 2H), 2,34 (м, 1H), 2,12-2,06 (м, 1H), 1,88-1,82 (м, 2H), 1,74-1,41 (м, 7H), 1,49-1,36 (м, 2H), 1,20 (с, 6H), 0,80 (т, J=6,7, 3H), 0,60 (т, J=7,4 Гц, 3H), 0,514 (т, J=7,4 Гц, 3H).
10-B: (S)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-4,4-диметил-пентановая кислота: (m/z): [M+H]+ рассчит. для C25H40N2O4, 433,30; найдено 433,6. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 12,39 (ушир.с, 1H), 8,87 (ушир.с, 1H), 7,93 (с, 1H), 7,66 (с, 2H), 7,64 (с, 1H), 7,31 (с, 1H), 7,18 (д, J=8,2 Гц, 1H), 3,96 (м,1H), 3,43 (дд, J=16,8, 5,87 Гц, 1H), 3,38 (с, 3H), 3,29 (ушир.с, 1H), 3,18-3,10 (м, 1H), 2,66 (дд, J=16,4, 9,97 Гц, 1H), 2,32 (м, 1H), 2,14-2,08 (м, 1H), 1,87 (м, 2H), 1,76-1,58 (м, 4H), 1,18 (м, 1H), 0,84 (с, 9H), 0,62 (т, J=7,4 Гц, 3H), 0,54 (т, J=7,4 Гц, 3H).
10-C: (S)-2-бензил-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляная кислота: (m/z): [M+H]+ рассчит. для C27H36N2O4, 453,27; найдено 453,1. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 8,86 (ушир.с, 1H), 7,93 (с, 1H), 7,68 (с, 1H), 7,67 (с, 2H), 7,31-7,16 (м, 5H), 7,17 (д, J=8,2 Гц, 1H), 4,0-3,93 (м,1H), 3,46 (дд, J=16,8, 5,87 Гц, 1H), 3,38 (с, 3H), 3,35-3,26 (м, 2H), 3,04 (ушир.с, 1H), 2,91-2,86 (м, 1H), 2,76-2,61 (м, 2H), 2,17-2,07 (м, 1H), 1,89-1,83 (м, 2H), 1,72-1,55 (м, 3H), 0,61 (т, J=7,4 Гц, 3H), 0,51 (т, J=7,4 Гц, 3H).
10-D: (R)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-гексановая кислота: (m/z): [M+H]+ рассчит. для C27H36N2O4, 419,28; найдено 419,3. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 7,97 (с, 1H), 7,72 (с, 2H), 7,70 (с, 1H), 7,34 (с, 1H), 7,22 (д, J=8,1 Гц, 1H), 4,04-3,98 (м,1H), 3,48 (дд, J=16,8, 5,68 Гц, 1H), 3,42 (с, 3H), 3,37-3,25 (м, 2H), 3,09-3,05 (м, 1H), 2,68 (дд, J=16,6, 9,78 Гц, 1H), 2,44-2,40 (м, 1H), 2,29-2,13 (м, 1H), 2,01-1,98 (м, 1H), 1,86-1,62 (м, 4H), 1,55-1,46 (м, 2H), 1,34-1,22 (м, 6H), 0,87 (т, J=6,8, 4H), 0,66 (т, J=7,4 Гц, 3H), 0,58 (т, J=7,4 Гц, 3H).
10-E: (S)-4-((2R,3R)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляная кислота: (m/z): [M+H]+ рассчит. для C27H42N2O4, 459,31; найдено 459,4. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 12,42 (ушир.с 1H), 8,84 (ушир.с 1H), 7,95 (с, 1H), 7,69-7,65 (м, 3H), 7,33 (с, 1H), 7,20 (д, J=8,6 Гц, 1H), 4,04-3,96 (м,1H), 3,47 (дд, J=16,8, 5,86 Гц, 1H), 3,40 (с, 3H), 3,35-3,22 (м, 2H), 3,06 (ушир.с 1H), 2,66 (дд, J=16,4, 9,39 Гц, 1H), 2,32 (м, 1H), 2,17-2,12 (м, 1H), 1,96-1,90 (м, 1H), 1,81-1,60 (м, 9H), 1,50-1,44 (м, 1H), 1,29-1,07 (м, 5H), 0,91-0,81 (м, 2H), 0,64 (т, J=7,4 Гц, 3H), 0,55 (т, J=7,4 Гц, 3H).
10-F: (R)-4-((2R,3R)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляная кислота: m/z): [M+H]+ рассчит. для C27H42N2O4, 459,31; найдено 459,8.
10-G: (R)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляная кислота: (m/z): [M+H]+ рассчит. для C27H42N2O4, 459,31; найдено 459,4. 1H ЯМР (ДМСО-d6, 400 мГц) δ (ч/млн): 12,4 (ушир.с 1H), 8,82 (ушир.с 1H), 7,91 (с, 1H), 7,66-7,61 (м, 3H), 7,29 (с, 1H), 7,16 (д, J=8,4 Гц, 1H), 4,06-3,93 (м,1H), 3,43 (дд, J=17,2, 6,06 Гц, 1H), 3,36 (с, 3H), 3,31-3,25 (м, 2H), 3,0 (ушир.с 1H), 2,65-2,58 (м, 1H), 2,13-2,07 (м, 1H), 1,89 (м, 1H), 1,71-1,57 (м, 9H), 1,45-1,41 (м, 1H), 1,28-1,03 (м, 5H), 0,86-0,78 (м, 2H), 0,61 (т, J=7,4 Гц, 3H), 0,52 (т, J=7,4 Гц, 3H).
Пример 11: транс-(S)-4-(7-карбамоил-3-метокси-1,1-диэтил-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексил-масляная кислота
Согласно процедуре Примера 9 с использованием метилового эфира (S)-2-циклогексил-4-оксо-масляной кислоты и рацемического соединения гидрохлорида амида транс-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты получали соль TFA целевого соединения. (m/z): [M+H]+ рассчит. для C26H40N2O4, 445,30; найдено 445,4.
Пример 12: (S)-2-циклогексилметил-4-((2S,3S)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляная кислота
a. метиловый эфир (S)-2-циклогексилметил-4-((2S,3S)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляной кислоты
К суспензии (S)-4-циклогексил-1-гидрокси-3-метоксикарбонил-бутан-1-сульфоната натрия (158 мг, 0,5 ммоль) в 2-MeTHF (2 мл) добавляли 2н. NaOH (0,22 мл, 0,44 ммоль). Реакционную смесь перемешивали в течение 20 минут, в течение которых все твердые частицы растворялись. Добавляли (6S,7S)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-ол гидрохлорид (100 мг, 0,35 ммоль), и смесь перемешивали при температуре окружающей среды в течение 30 мин. Триацетоксиборгидрид натрия (425 мг, 2,0 ммоль) добавляли в 4 частях за 3 часа. Спустя тридцать минут после последнего добавления добавляли EtOAc (15 мл), и смесь промывали 5%-ным водным раствором карбоната натрия (2 ×5 мл) и насыщенным раствором хлорида натрия (5 мл). Органический слой высушивали с Na2SO4 и концентрировали, получая целевое соединение (159 мг). (m/z): [M+H]+ рассчит. для C27H43NO4, 446,33; найдено 446,6.
b. (S)-2-циклогексилметил-4-((2S,3S)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляная кислота
Раствор продукта предыдущей стадии (159 мг, 0,36 ммоль), метанола (1,5 мл), воды (0,20 мл) и 10н. NaOH (0,21 мл, 2,1 ммоль) нагревали при 50°C. Через 4 часа реакционную смесь охлаждали до температуры окружающей среды, разбавляли 50%-ным водным раствором уксусной кислоты и очищали препаративной ВЭЖХ, получая целевое соединение в форме соли TFA (110 мг, 58% за 2 стадии). (m/z): [M+H]+ рассчит. для C26H41NO4, 432,31; найдено 432,8. 1H ЯМР (CD3OD, 400 мГц) δ (ч/млн): 6,97 (д, J=8,2 Гц, 1H), 6,65 (дд, J=8,4, 2,5 Гц, 1H), 6,62 (д, J=2,4 Гц, 1H), 4,01 (м, 1H), 3,48 (с, 3H), 3,47-3,37 (м, 3H), 3,22 (м, 1H), 2,58 (дд J=15,9, 10,2 Гц, 2H), 2,10 (м, 1H), 1,97 (м, 2H), 1,82-1,60 (м, 10H), 1,37-1,18 (м, 6H), 0,92 (м, 2H), 0,80 (т, J=7,4 Гц, 3H), 0,71 (т, J=7,2 Гц, 3H).
Пример 13: (S)-2-циклогексилметил-4-((2R,3R)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляная кислота
Согласно процедуре Примера 12 с использованием (6R,7R)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-ол гидрохлорида на стадии (a) получали целевое соединение. (m/z): [M+H]+ рассчит. для C26H41NO4, 432,31; найдено 432,8. 1H ЯМР (CD3OD, 400 мГц) δ (ч/млн): 6,97 (д, J=8,2 Гц, 1H), 6,65 (дд, J=8,3, 2,5 Гц,1H), 6,62 (д, J=2,6 Гц, 1H), 4,01 (м, 1H), 3,48 (с, 3H), 3,47-3,37 (м, 3H), 3,20 (м, 1H), 2,58 (дд J=16,0, 10,0 Гц, 2H), 2,13-1,95 (м, 2H), 1,95-1,60 (м, 10H), 1,38-1,19 (м, 6H), 0,92 (м, 2H), 0,80 (т, J=7,4 Гц, 3H), 0,71 (т, J=7,3 Гц, 3H).
Пример 14: (R)-2-циклогексилметил-4-((2S,3S)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляная кислота
a. метиловый эфир (R)-2-циклогексилметил-4-((2S,3S)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляной кислоты
(6S,7S)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-ол гидрохлорид (100 мг, 0,35 ммоль), метиловый эфир (R)-2-циклогексилметил-4-оксо-масляной кислоты (90 мг, 0,42 ммоль) и триэтиламин (35 мг, 0,035 ммоль) растворяли в дихлорметане (2,0 мл) и перемешивали при температуре окружающей среды в течение 30 мин. Добавляли триацетоксиборгидрид натрия (1,03 г, 4,85 ммоль), и реакцию проверяли ВЭЖХ. Через 3 часа добавляли дополнительные количества метилового эфира (R)-2-циклогексилметил-4-оксо-масляной кислоты (20 мг, 0,1 ммоль) и триацетоксиборгидрида натрия (30 мг, 0,14 ммоль). Спустя тридцать минут после последнего добавления добавляли EtOAc (15 мл), и смесь промывали 5%-ным водным раствором карбоната натрия (2×5 мл) и насыщенным раствором хлорида натрия (5 мл). Органический слой высушивали Na2SO4 и концентрировали, получая целевое соединение (190 мг). (m/z): [M+H]+ рассчит. для C27H43NO4, 446,33; найдено 446,6.
b. (R)-2-циклогексилметил-4-((2S,3S)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляная кислота
Согласно процедуре Примера 12 стадии (b) выделяли соль TFA целевого соединения. (m/z): [M+H]+ рассчит. для C26H41NO4, 432,31; найдено 432,8. 1H ЯМР (CD3OD, 400 мГц) δ (ч/млн): 6,97 (д, J=8,5 Гц, 1H), 6,65 (дд, J=8,3, 2,3 Гц, 1H), 6,62 (д, J=2,3 Гц, 1H), 4,01 (м, 1H), 3,48 (с, 3H), 3,47-3,37 (м, 3H), 3,22 (м, 1H), 2,58 (дд, J=16,0, 10,0 Гц, 2H), 2,15-1,95 (м, 2H), 1,95-1,60 (м, 10H), 1,38-1,18 (м, 6H), 0,92 (м, 2H), 0,80 (т, J=7,6 Гц, 3H), 0,71 (т, J=7,5 Гц, 3H).
Пример 15: (R)-2-циклогексилметил-4-((2R,3R)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляная кислота
Согласно процедуре Примера 14 с использованием (6R,7R)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-ол гидрохлорида на стадии (a) получали целевое соединение. (m/z): [M+H]+ рассчит. для C26H41NO4, 432,31; найдено 432,8. 1H ЯМР (CD3OD, 400 мГц) δ (ч/млн): 6,97 (д, J=8,2 Гц, 1H), 6,65 (дд, J=8,5, 2,5 Гц, 1H), 6,62 (д, J=2,3 Гц, 1H), 4,01 (м, 1H), 3,48 (с, 3H), 3,47-3,37 (м, 3H), 3,22 (м, 1H), 2,58 (дд J=15,7, 10,2 Гц, 2H), 2,10 (м, 1H), 1,98 (м, 2H), 1,79-1,60 (м, 10H), 1,37-1,18 (м, 6H), 0,92 (м, 2H), 0,80 (т, J=7,4 Гц, 3H), 0,71 (т, J=7,2 Гц, 3H).
Пример 16
Согласно процедуре Примера 9 с использованием подходящего сложного метилового эфира вместо метилового эфира (S)-2-пентил-4-оксо-масляной кислоты и рацемического соединения гидрохлорида амида транс-7-амино-8,8-диметил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты получали соли TFA следующих соединений:
16-A: транс-(S)-4-(7-карбамоил-1,1-диметил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляная кислота: (m/z): [M+H]+ рассчит. для C25H38N2O4, 431,28; найдено 431,2.
16-B: транс-(S)-4-(7-карбамоил-3-метокси-1,1-диметил-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексил-масляная кислота: (m/z): [M+H]+ рассчит. для C24H36N2O4, 417,27; найдено 417,4.
Пример 17: (S)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляная кислота
a. гидрохлорид бензилового эфира (S)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляной кислоты
К суспензии (S)-3-бензилоксикарбонил-4-циклогексил-1-гидрокси-бутан-1-сульфоната натрия (160 г, 400 ммоль) добавляли продукт Примера получения 17 в MeTHF (2,0 л) и воде (600 мл), 1,0М NaOH в воде (400 мл), и реакционную смесь перемешивали при температуре окружающей среды в течение 90 мин. Фазы разделяли, и раствор концентрировали до объема ~300 мл.
Полученный концентрированный раствор добавляли к суспензии гидрохлорида амида (6S,7S)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты (100,0 г, 319,7 ммоль) в DMF (1 л). Полученную суспензию перемешивали при температуре окружающей среды в течение 2 часов, реакционную смесь охлаждали до 0°C с последующим добавлением частями триацетоксиборгидрида натрия (169 г, 799 ммоль) за 15 мин. Реакционную смесь перемешивали при температуре окружающей среды в течение ночи, охлаждали до 10°C и затем добавляли 1,0М NaOH в воде (3 л) и этилацетате (5 л). Реакционную смесь перемешивали в течение 10 минут, фазы разделяли, и органический слой промывали разбавленным солевым раствором (1:1, 2 л). К органическому слою добавляли 1,0М HCl в воде (520 мл, 520 ммоль) и большую часть этилацетата удаляли упариванием на роторном испарителе. Добавляли воду (500 мл) и этанол (1 л), и объем медленно уменьшали упариванием на роторном испарителе до ~1 л. Полученную текучую суспензию грязно-белого цвета перемешивали при температуре окружающей среды в течение ночи. Продукт выделяли фильтрацией, колбу и остаток после фильтрации промывали водой (2 ×200 мл) и затем высушивали, получая целевое соединение (175 г) в форме твердого вещества белого цвета (чистота ~99%, 90%-ный выход в рассчете на аминотетралиновый реагент). 1H ЯМР (400 МГц, ДМСО-d6): δ (ч/млн) 9,33 (ушир, 1H), 8,09 (ушир, 1H), 7,98 (с, 1H), 7,70 (с, 1H), 7,68 (д, 1H), 7,28-7,36 (м, 2H), 7,19 (д, 1H), 5,10 (кв, 2H), 4,04 (м, 1H), 3,45 (дд, 1H), 3,38 (с, 3H), 3,25 (м, 2H), 3,05 (м, 1H), 2,62 (м, 2H), 1,95-2,15 (м, 2H), 1,61-1,82 (м, 3H), 1,50-1,61 (м, 4H), 1,42-1,50 (м, 1H), 1,24-1,32 (м, 1H), 0,98-1,18 (м, 4H), 0,71-0,89 (м, 2H), 0,63 (т, 3H), 0,52 (т, 3H)
b. (S)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляная кислота
Продукт предыдущей стадии (175,0 г, 299 ммоль) разделяли между этилацетатом (2,5 л), водой (1 л) и 1,0М NaOH в воде (300 мл, 299 ммоль). Фазы разделяли, органический слой промывали разбавленным солевым раствором (1:1, 250 мл) и высушивали сульфатом натрия. Растворитель удаляли упариванием на роторном испарителе, и полученный продукт высушивали в течение ночи под высоким вакуумом, получая промежуточное соединение в форме свободного основания бензилового эфира (S)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляной кислоты (~160 г) в форме липкого твердого вещества.
Промежуточное соединение в форме свободного основания растворяли в смеси ацетонитрила (1,6 л) и воды (300 мл). К половине раствора (1 л) добавляли 10%-ный палладий (10 г, 9 ммоль) на (влажном) углероде. Реакционную смесь продували азотом и затем водородом в течение 2 минут, и затем подвергали действию 10-15 фунт/кв.дюйм H2 в течение 3 часов при температуре окружающей среды. Реакционную смесь фильтровали через целит, и колбу и остаток после фильтрации промывали ацетонитрилом (50 мл). Желтоватый фильтрат перемешивали с модифицированным тиолом диоксидом кремния (10 г) при температуре окружающей среды в течение 2 часов и затем фильтровали через целит. Большую часть растворителя удаляли упариванием на роторном испарителе при 25°C. Добавляли ацетонитрил (500 мл), и большую часть растворителя удаляли упариванием на роторном испарителе. Добавляли дополнительный ацетонитрил (500 мл), что приводило к быстрому осаждению липких твердых частиц. Реакционную смесь энергично перемешивали при температуре окружающей среды в течение ночи, что приводило к текучей суспензии грязно-белого цвета. Продукт выделяли фильтрацией; остаток после фильтрации промывали ацетонитрилом (2 ×50 мл) и затем высушивали под вакуумом, получая целевое соединение в форме кристаллического твердого вещества (56 г, чистота 98,8%). Содержание воды 0,49% (вес./вес.). 1H ЯМР (400 МГц, ДМСО-d6): δ (ч/млн) 7,89 (ушир, 1H), 7,65 (с, 1H), 7,60 (д, 1H), 7,22 (ушир, 1H), 7,11 (д, 1H), 3,55 (м, 1H), 3,38 (с, 3H), 3,25 (дд, 1H), 2,95 (м, 1H), 2,59 (д, 1H), 2,49 (м, 2H), 1,81 (м, 2H), 1,49-1,63 (м, 5H), 1,41-1,50 (м, 2H), 1,05-1,25 (м, 4H), 0,72-0,90 (м, 2H), 0,45 (т, 3H), 0,57 (т, 3H).
Пример 18: 4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-(транс-4-гидрокси-циклогексилметил)-масляная кислота (A) и 4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-(цис-4-гидрокси-циклогексилметил)-масляная кислота (B)
a. 1-метиловый эфир 2-[4-(трет-бутил-диметил-силанилокси)-циклогексилметилен]-янтарной кислоты
Раствор диметилового эфира янтарной кислоты (730 мг, 5,0 ммоль) и 4-(трет-бутил-диметил-силанилокси)-циклогексанкарбальдегида (1,00 г, 4,1 ммоль) добавляли за 25 минут к 1,0M раствору трет-бутоксида калия в трет-бутаноле (4,4 мл, 4,4 ммоль). Реакционную смесь нагревали при 50°C в течение 50 минут, охлаждали до температуры окружающей среды и концентрировали под вакуумом. Остаток растворяли в воде (25 мл) и промывали EtOAc (2×10 мл). Водный слой подкисляли 6 н. HCl (2,0 мл, 12 ммоль) и экстрагировали EtOAc (2×20 мл), высушивали (Na2SO4) и концентрировали. Сырой продукт очищали флэш-хроматографией (25% EtOAc/DCM), получая целевое соединение (560 мг) в форме смеси олефиновых изомеров (~1:1) и смеси цис и транс изомеров по циклогексильному кольцу (~1:1). 1H ЯМР (CDCl3, 400 мГц) δ (ч/млн) олефиновые пики в 6,88 (д, J=10,2 Гц) 6,77 (д, J=10,0 Гц) пики CHOTBS в 3,96 ч/млн (ушир.с) (цис-изомер, экваториальный водород, 3,55 (м) (транс-изомер, аксиальный водород).
b. 1-метиловый эфир 2-[4-(трет-бутил-диметил-силанилокси)-циклогексилметил]-янтарной кислоты
К раствору продукта предыдущей стадии (560 мг, 1,6 ммоль) в EtOAc (15 мл) добавляли 10%-ный Pd/C (50% воды, сухой вес 165 мг). Реакционную смесь взбалтывали под 50 фунт/кв.дюйм водорода в течение 16 часов. Реакционную смесь фильтровали через целит, промывали EtOAc (5×5 мл), MeOH (3×5 мл) и DCM (3×5 мл). Объединенные фильтраты концентрировали досуха при пониженном давлении. Сырой продукт очищали флэш-хроматографией (25% EtOAc/DCM), получая целевое соединение (245 мг) как ~1:1 смесь цис и транс изомеров по циклогексильному кольцу. 1H ЯМР (CDCl3, 400 мГц) δ (ч/млн) пики CHOTBS в 3,92 ч/млн (ушир.с) (цис-изомер, экваториальный водород), 3,50 (м) (транс-изомер, аксиальный водород)
c. метиловый эфир 2-[4-(трет-бутил-диметил-силанилокси)-циклогексилметил]-4-гидроксимасляной кислоты
Раствор продукта предыдущей стадии (245 мг, 0,683 ммоль) в THF (2,0 мл) охлаждали на льду, и раствор гидрида бора в 1,0М THF (1,4 мл) добавляли за 5 мин. Реакционную смесь перемешивали при 0°C в течение 1,5 часов и затем гасили добавлением по каплям MeOH (10 мл). Смесь концентрировали при пониженном давлении. Добавляли дополнительный MeOH (10 мл), и смесь концентрировали при пониженном давлении, получая сырой целевой продукт (228 мг), который использовали непосредственно на следующей стадии.
d. метиловый эфир 2-[4-(трет-бутил-диметил-силанилокси)-циклогексилметил]-4-оксо-масляной кислоты
Продукт предыдущей стадии (228 мг, 0,66 ммоль) растворяли в DCM (7,0 мл). Добавляли диметилсульфоксид (218 мг, 2,8 ммоль) и DIPEA (361 мг, 2,8 ммоль), и смесь охлаждали до -10°C. Комплекс триоксид серы-пиридин (223 мг, 1,4 ммоль) добавляли в форме твердого вещества, и реакционную смесь перемешивали при -10°C в течение 1,5 часов. Добавляли DCM (20 мл), затем 0,5 н. HCl (10 мл). Слои разделяли, и водный слой экстрагировали DCM (2×10 мл). Объединенные органические слои промывали водой (3×10 мл) и насыщенным раствором NaCl (10 мл), затем высушивали (Na2SO4) и концентрировали, получая целевое соединение (220 мг). 1H ЯМР (CDCl3, 400 мГц) показала пики альдегида в 9,81 и 9,75 ч/млн.
e. метиловый эфир 2-[4-(трет-бутил-диметил-силанилокси)-циклогексилметил]-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляной кислоты
Раствор амида (6S,7S)-7-амино-8,8-диэтил-6-метокси-5,6,7,8-тетрагидро-нафталин-2-карбоновой кислоты (100 мг, 0,32 ммоль), продукта предыдущей стадии (140 мг, 0,41), триэтиламина (33 мг, 0,33 ммоль) в DCM (2,0 мл) и MeOH (0,5 мл) перемешивали при температуре окружающей среды в течение 35 мин. Добавляли триацетоксиборгидрид натрия (135 мг, 0,64 ммоль), и реакцию проверяли ВЭЖХ. Дополнительные части триацетоксиборгидрида натрия добавляли в моменты времени 1 час (50 мг) и 1,5 часа (100 мг), и дополнительную часть альдегида добавляли в момент времени 1,75 часа (80 мг). Спустя пятнадцать минут после последнего добавления добавляли DCM (20 мл) и насыщенный раствор NaHCO3 (10 мл). Слои разделяли, и водный слой экстрагировали DCM (2×10 мл). Объединенные органические экстракты высушивали (Na2SO4) и концентрировали, получая сырой целевой продукт (283 мг).
f. метиловый эфир 4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-(транс-4-гидрокси-циклогексилметил)-масляной кислоты (f1) и метиловый эфир 4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-(цис-4-гидрокси-циклогексилметил)-масляной кислоты (f2)
Часть сырого продукта предыдущей стадии (28 мг, 0,32 ммоль) растворяли в 50%-ном водном растворе AcOH (0,5 мл). Через 16 часов продукты разделяли препаративной ВЭЖХ, получая целевое соединение.
f1 (элюирующий первым) (3,3 мг) (m/z): [M+H]+ рассчит. для C28H44N2O5 489,33; найдено 489,6. 1H ЯМР (CD3OD, 400 мГц) δ (ч/млн) 7,71 (д, J=1,6 Гц, 1H), 7,65 (д, J=8,0, 1H), 7,20 (д, J=8,0 Гц, 1H), 4,02 (м, 1H), 3,63 (с, 3H), 3,51 (дд, J=16,5, 5,5 Гц, 1H), 3,44 (с, 3H), 3,44-3,30 (м, 3H), 3,14 (м, 1H), 2,70 (м, 1H), 2,54 (м, 1H), 2,24 (м, 1H), 2,00-1,50 (м, 9H), 1,30 (м, 2H), 1,12 (м, 3H), 0,91 (м, 2H), 0,71 (т, J=7,5 Гц, 3H), 0,62 (т, J=7,2 Гц).
f2 (элюирующий вторым) (7,3 мг) (m/z): [M+H]+ рассчит. для C28H44N2O5 489,33; найдено 489,6. 1H ЯМР (CD3OD, 400 мГц) δ (ч/млн) 7,71 (д, J=1,6 Гц, 1H), 7,65 (дд, J=8,0, 1,4 Гц, 1H), 7,20 (д, J=8,2 Гц, 1H), 4,02 (м, 1H), 3,79 (ушир.с, 1H) 3,62 (с, 3H), 3,51 (дд, J=16,7, 5,7 Гц, 1H), 3,44 (с, 3H), 3,44-3,30 (м, 2H), 3,15 (м, 1H), 2,68 (м, 1H), 2,57 (м, 1H), 2,24 (м, 1H), 2,05-1,80 (м, 2H), 1,70-1,55 (м, 6H), 1,50-1,10 (м, 8H), 0,71 (т, J=7,4 Гц, 3H), 0,62 (т, J=7,3 Гц, 3H).
g. 4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-(транс-4-гидрокси-циклогексилметил)-масляная кислота (A)
К раствору метилового эфира 4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-(транс-4-гидрокси-циклогексилметил)-масляной кислоты (f1) (27,4 мг, 0,045 ммоль) в MeOH (0,50 мл) добавляли воду (32 мкл) и 10 н. NaOH (32 мкл, 0,32 ммоль). Смесь нагревали при 50°C. Через 15 часов реакционную смесь охлаждали до температуры окружающей среды, растворяли в 50%-ном водном растворе AcOH (6 мл) и очищали препаративной ВЭЖХ, получая целевое соединение (13,8 мг) в форме лиофилизованного порошка. (m/z): [M+H]+ рассчит. для C27H42N2O5 475,32; найдено 475,2. 1H ЯМР (CD3OD, 400 мГц) δ (ч/млн) 7,78 (д, J=1,6 Гц, 1H), 7,72 (дд, J=8,0 Гц, 1,7 Гц, 1H), 7,28 (д, J=8,2 Гц, 1H), 4,09 (м, 1H), 3,57 (дд, J=16,7, 5,9 Гц, 1H), 3,52 (с, 3H), 3,52-3,42 (м, 3H), 3,23 (м, 1H), 2,78 (м, 1H), 2,58 (м, 1H), 2,31 (м, 1H), 2,05-1,60 (м, 10H), 1,40-1,15 (м, 4H), 1,00 (м, 2H), 0,79 (т, J=7,5 Гц, 3H), 0,70 (дт, J=7,3, 1,3 Гц, 3H).
h. 4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-(цис-4-гидрокси-циклогексилметил)-масляная кислота (B)
К раствору метилового эфира 4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-(цис-4-гидрокси-циклогексилметил)-масляной кислоты (f2) (56 мг, 0,11 ммоль) в MeOH (0,50 мл) добавляли воду (66 мкл) и 10н. NaOH (66 мкл, 0,66 ммоль). Смесь нагревали при 50°C. Через 15 часов реакционную смесь охлаждали до температуры окружающей среды, растворяли в 50%-ном водном растворе AcOH (6 мл) и очищали препаративной ВЭЖХ, получая целевое соединение (54 мг) в форме лиофилизованного порошка. (m/z): [M+H]+ рассчит. для C27H42N2O5 475,32; найдено 475,2. 1H ЯМР (CD3OD, 400 мГц) δ (ч/млн) 7,78 (д, J=1,8 Гц, 1H), 7,72 (дд, J=8,0 Гц, 1,7 Гц, 1H), 7,28 (д, J=8,0 Гц, 1H), 4,09 (м, 1H), 3,88 (ушир.с, 1H), 3,58 (дд, J=16,6, 5,6 Гц, 1H), 3,52 (с, 3H), 3,52-3,46 (м, 2H) 3,30-3,20 (м, 1H), 2,77 (м, 1H), 2,59 (м, 1H), 2,31 (м, 1H), 2,05-1,92 (м, 2H), 1,79-1,65 (м, 6H), 1,60-1,35 (м, 8H), 0,79 (т, J=7,5 Гц, 3H), 0,70 (дт, J=7,2, 1,6 Гц).
Пример 19: Исследование метаболитов
Образец (S)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляной кислоты, соединения Примера 1, инкубировали в течение 4 часов при 37°C с сохраненными замораживанием человеческими гепатоцитами в ДМСО в концентрации 1,2 миллиона клеток/мл. Аликвоты полученного раствора смешивали с одним объемом смеси 97% ацетонитрила/3% TFA и замораживали при -20°C. После размораживания образцы центрифугировали в течение 10 минут при 20800×g и 4°C. Супернатанты собирали, разбавляли 3 объемами воды и анализировали ВЭЖХ вместе с масс-спектрометрией (LC/MS) в условиях, описанных ниже. Для идентификации метаболита аликвоты после разбавления объединяли отдельно с соединением A и с соединением B Примера 18 и анализировали LC/MS. Полученные ионные экстракционные хроматограммы при 475,3±0,5 a.m.u. коррелируют с интерпретацией, что основной гидроксильный метаболит соединения Примера 1 является соединением транс-4-гидрокси A, хотя соединение цис-4-гидрокси B не может быть исключено.
Agilent модель 1100 HPLC с колонкой Luna C18(2) 100A 5 мкм; скорость потока: 0,25 мл/мин; Растворитель A: 95% воды, 5% ацетонитрила, 0,05% TFA; Растворитель B: >95% ацетонитрила, 5% воды, 0,05% TFA; Градиент (время(мин)/% B): 0/7, 5/7, 60/25, 61/100, 63,5/100, 64/7, 70/7. Тройной четырехполюсный масс-спектрометр Applied Biosystems модель API3000.
Тест 1: Тест связывания меченого лиганда с опиоидными рецепторами Mu человека, Delta человека и Kappa морской свинки
a. Мембранный препарат
Клетки CHO-K1 (Яичника Китайского хомячка), стабильно трансфицированные кДНК человеческого mu-опиоидного рецептора или каппа-рецептора морской свинки, выращивали в среде, состоящей из среды Хэма-F12, дополненной 10%-ным FBS, 100 единиц/мл пенициллина - 100 мг/мл стрептомицина и 800 мг/мл Geneticin во влажном инкубаторе с 5% CO2 при 37°C. Уровни экспрессии рецептора (Bmax ~2,0 и ~0,414 пмоль/мг белка соответственно) определяли, используя [3H]-дипренорфин (удельная активность ~ 50-55 Ci/ммоль) в мембранном тесте связывания меченого лиганда.
Клетки выращивали до слияния 80-95% (<25 пассажей субкультуры). Для пассажей линии клеток мономолекулярный слой клеток инкубировали в течение 5 минут при температуре окружающей среды и собирали механическим перемешиванием в 10 мл PBS, дополненной 5 мМ EDTA. После повторного суспендирования клетки переносили в 40 мл свежей среды для выращивания и центрифугировали в течение 5 минут при 1000 об/мин и повторно суспендировали в свежей среде для выращивания в подходящем соотношении.
Для мембранного препарата клетки собирали нежным механическим перемешиванием с 5 мМ EDTA в PBS, затем центрифугировали (2500 g в течение 5 минут). Осадки повторно суспендировали в Тестовом буфере (50 мМ 4-(2-гидроксиэтил)пиперазин-1-этансульфоновой кислоты N-(2-гидроксиэтил)пиперазин-N'-(2-этансульфоновая кислота) (HEPES)), рН 7,4, и гомогенизировали политронным гомогенизатором на льду. Полученные гомогенаты центрифугировали (1200 g в течение 5 минут), осадки отбрасывали и супернатант центрифугировали (40000 g в течение 20 минут). Осадки промывали один раз путем ресуспендирования в Тестовом буфере, после чего дополнительно центрифугировали (40000 g в течение 20 минут). Конечные осадки повторно суспендировали в Тестовом буфере (эквивалент 1 T-225 колба/1 мл тестового буфера). Концентрацию белка определяли, используя набор Bio-Rad Bradford Protein Assay, и мембраны сохраняли в виде замороженных аликвот при -80°C до использования.
Мембраны с человеческим дельта-опиоидным рецептором (hDOP) получали от Perkin Elmer. Kd и Bmax для этих мембран, определенные путем анализа в тесте насыщения связывания [3H]-Natrindole-меченого лиганда, составляли 0,14 нМ (pKd=9,85) и 2,2 пмоль/мг белка соответственно. Концентрацию белка определяли, используя набор Bio-Rad Bradford Protein Assay. Мембраны сохраняли в виде замороженных аликвот при -80°C до использования.
b. Тесты связывания меченого лиганда
Тесты связывания меченого лиганда осуществляли в полипропиленовом тестовом планшете с 96 лунками глубиной 1,1 мл Axygen в полном объеме теста 200 мл, содержащем соответствующее количество мембранного белка (~3, ~2 и ~20 мг для mu, делта и каппа соответственно) в Тестовом буфере, дополненном 0,025% бычьего сывороточного альбумина (BSA). Исследования насыщения связывания для определения значений Кд меченого лиганда осуществляли, используя [3H]-дипренорфин в 8-12 различных концентрациях в пределах от 0,001 нМ до 5 нМ. Тесты смещения для определения значений pKi соединений осуществляли с [3H]-дипренорфином в концентрации 0,5, 1,2 и 0,7 нМ для mu, делта и каппа соответственно и одиннадцатью концентрациями соединения в пределах от 10 пМ до 100 мМ.
Данные связывания анализировали анализом нелинейной регрессии с Пакетом программ GraphPad Prism Software package (GraphPad Software, Inc., San Diego, CA) с использованием модели с 3 параметрами для конкуренции с одним участком. Минимум кривой фиксировали для значения неспецифического связывания, определяемого в присутствии 10 мкМ налоксона. Значения Ки для тестируемых соединений вычисляли, в Prism, от наилучшего приближения значения IC50 и значения Kd меченого лиганда, используя уравнение Cheng-Prusoff (Ki=IC50/(1+([L]/Kd)), где [L]=концентрация [3H]-дипренорфина. Результаты выражали в виде отрицательного десятичного логарифма значений Ki, pKi.
Тестируемые соединения, имеющие более высокое значение pKi в этих тестах, имеют более высокое связывающее сродство с mu, дельта или каппа-опиоидным рецептором. В этих тестах проверяли целевые соединения, описанные в Примерах 1-16. Все соединения имели значение pKi от приблизительно 8,7 до приблизительно 10,9 в человеческом mu-опиоидном рецепторе. Например, соединения Примеров 1, 9, 10-G и 12 имели значения pKi 9,4, 9,2, 9,6 и 9,7 соответственно. Соединения по изобретению также показали значения pKi от приблизительно 7,5 до приблизительно 10,3 в человеческих дельта-рецепторах и каппа-опиоидных рецепторах морской свинки.
Тест 2: Агонист-опосредованная активация mu-опиоидного рецептора в мембранах, полученных из клеток CHO-K1, экспрессирующих человеческий mu-опиоидный рецептор
В этом тесте потенциал и значения свойственной активности тестируемых соединений определяли, измеряя количество связанного [35S]ГТФγS после активации рецептора в мембранах, полученных от клеток CHO-K1, экспрессирующих человеческий mu-опиоидный рецептор.
a. Пример получения мембраны mu-опиоидного рецептора:
Мембраны человеческого mu-опиоидный рецептор (hMOP) либо получали как описано выше, либо приобретали у Perkin Elmer. pKd и Bmax для приобретенных мембран, определенные исследованиями в тесте насыщения связывания [3H]-дипренорфин-меченого лиганда, составляли 10,06 и 2,4 пмоль/мг белка соответственно. Концентрацию белка определяли, используя набор Bio-Rad Bradford Protein Assay. Мембраны сохраняли в виде замороженных аликвот при -80°C до использования.
b. Тест нуклеотидного обмена человеческого mu [35S]ГТФγS
Мембраны получали, как описано выше, и до начала теста аликвоты разбавляли в концентрации 200 мг/мл в Тестовом буфере (50 мМ HEPES, рН 7,4 при 25°C), затем гомогенизировали в течение 10 секунд, используя гомогенизатор Polytron. Тестируемые соединения получали в виде сток-растворов на 10 мМ в ДМСО, разбавляли до 400 мкМ в Тестовом буфере, содержащем 0,1% BSA, и затем осуществляли последовательные (1:5) разбавления, получая десять концентраций соединения в пределах от 40 пМ до 80 мМ. ГДФ и [35S]ГТФγS разбавляли до 40 мкМ и 0,4 нМ соответственно, в Тестовом буфере. Тест проводили в полном объеме 200 мкл, содержащем 10 мг мембранного белка, при этом количество тестируемого соединения составляло от 10 пМ до 20 мкМ, 10 мкМ ГДФ и 0,1 нМ [35S]ГТФγS, разбавленного в 10 мМ MgCl2, 25 мМ NaCl и 0,0125% BSA (конечные тестируемые концентрации). В каждый планшет включали кривую концентрация-ответ DAMGO (Tyr-D-Ala-Gly-(метил)Phe-Gly-ол) (в пределах от 12,8 пМ до 1 мМ).
Тарелки для теста готовили непосредственно перед проведением теста после добавления 50 мкл раствора NaCl/MgCl2/ГДФ, 50 мл тестируемого соединения и 50 мл [35S]ГТФγS. Тест инициировали добавлением 50 мл мембранного белка и инкубировали в течение 30 минут при температуре окружающей среды. Реакцию завершали фильтрацией на фильтровальных планшетах GF/B с 96 лунками, предварительно блокированных 0,3% полиэтиленимина, используя устройство Packard Filtermate, и промывали ледяным Тестовым буфером (3×200 мл). Тарелки высушивали в течение ночи до определения индексов связания путем жидкостной сцинтилляции на приборе Packard Topcount. Носитель: ДМСО, чтобы не превысить 1%-ную конечную тестируемую концентрацию.
Количество связанного [35S]ГТФγS пропорционально степени активации mu-опиоидных рецепторов тестируемым соединением. Свойственную активность (IA), выраженную в процентах, определяли как отношение количества связанного [35S]ГТФγS, наблюдаемого для активации тестируемым соединением, к количеству, наблюдаемому для активации DAMGO, который, как предполагают, является полным агонистом (IA=100). Все соединения карбоновой кислоты формулы (I) были проверены в этом тесте и продемонстрировали свойственные активности менее чем приблизительно 22. Например, соединения Примеров 1, 9, 10-G и 12 имели значения IA -8, -2, 7 и -5 соответственно. Кроме того, сложные эфиры Примеров 2, 4, 5, 6 и 7-A показали значения IA -5, 6, 17, 19 и 8 соответственно. Таким образом, соединения согласно настоящему изобретению продемонстрировали действие антагонистов на человеческом mu-опиоидном рецепторе.
Тест 3: Крысиная Модель Эффективности In Vivo
В этом тесте эффективность тестируемых соединений оценивали на модели желудочно-кишечного транзита, которая позволяет оценить периферическую активность. Это исследование было одобрено Institutional Animal Care and Use Committee at Theravance, Inc. и соответствовало Guide for the Care and Use of Laboratory Animals published by the National Academy of Sciences (©1996).
a. Тест опорожнения желудка крысы
Тестируемые соединения оценивали в тесте в опорожнения желудка крысы для определения их способности обращать лоперамид-индуцированную задержку опорожнения желудка. Крыс лишали пищи в течение ночи до введения тестируемых соединений или носители внутривенным, подкожным, внутримышечным или пероральным путями введения в дозах в пределах от 0,001 до приблизительно 30 миллиграмм/килограмм (мг/кг). Введение тестируемого соединения сопровождалось подкожным введением лоперамида в дозе 1 мг/кг или носителя. Через пять минут после введения лоперамида или носителя, непитательную, нерассасывающуюся пищу на активированном угле вводили пероральным насильственным скармливанием, и животным обеспечивали свободный доступ к воде в течение шестидесяти минут эксперимента. Животных затем умерщвляли асфиксией с использованием диоксида углерода, затем подвергали торакотомии, и желудок аккуратно иссекали. Желудок лигировали по нижнему эзофагеальному сфинктеру и пилорическому сфинктеру, чтобы предотвратить дополнительное опорожнение в течение удаления ткани. Массу желудка определяли после удаления лигатур.
b. Анализ данных и результаты
Данные анализировали, используя Пакет программ GraphPad Prism Software package (GraphPad Software, Inc., San Diego, CA). Реверсивные процентные кривые выстраивали с помощью анализа нелинейной регрессии, используя сигмовидную модель доза-ответ (переменный наклон), и рассчитывали величины наилучшего приближения ID50. Минимумы и максимумы кривой фиксировали для значений лоперамидного контроля (показывающие 0% реверсирования) и контроля с носителем (показывающие 100% реверсирования) соответственно. Результаты выражали как ID50, доза, необходимая для 50%-ного реверсирования эффектов лоперамида, в миллиграммах на килограмм. Соединения Примеров 1, 9, 10-G и 12, вводимые перорально, показали значения ID50 0,09 мг/кг, 0,10 мг/кг, 0,12 мг/кг и 0,05 мг/кг соответственно, на модели опорожнения желудка.
Несмотря на то что настоящее изобретение было описано в виде его частных вариантов осуществления, специалисту должно быть понятно, что могут быть внесены различные изменения и сделаны замены эквивалентов без отхода от духа и объема изобретения. Кроме того, множество модификаций могут быть сделаны для адаптации к конкретной ситуации, материалу, композиции веществ, способу, стадии или стадиям способа, цели, духу и объему настоящего изобретения. Все такие модификации находятся в рамках приложенной формулы изобретения. Дополнительно, все публикации, патенты и патентные документы, процитированные выше, полностью включены в настоящее описание путем ссылки, как если бы каждый из них был включен индивидуальной ссылкой.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ СОЕДИНЕНИЯ 3-КАРБОКСИПРОПИЛ-АМИНОТЕТРАЛИНА | 2009 |
|
RU2512390C2 |
| СРЕДСТВА, ИНДУЦИРУЮЩИЕ АПОПТОЗ, ДЛЯ ЛЕЧЕНИЯ РАКА, ИММУННЫХ И АУТОИММУННЫХ ЗАБОЛЕВАНИЙ | 2011 |
|
RU2568611C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
| ИНГИБИТОРЫ КИНАЗЫ | 2013 |
|
RU2637944C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ ПОВРЕЖДЕНИЯ ГОЛОВНОГО МОЗГА | 2018 |
|
RU2797548C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2528046C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2556984C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2382779C2 |
| СПОСОБ МОДУЛЯЦИИ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2525115C2 |
| АГОНИСТЫ РЕЦЕПТОРА СФИНГОЗИН-1-ФОСФАТА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ В КАЧЕСТВЕ АКТИВНОГО СРЕДСТВА | 2014 |
|
RU2654483C2 |
Изобретение относится к новым соединениям формулы (I) 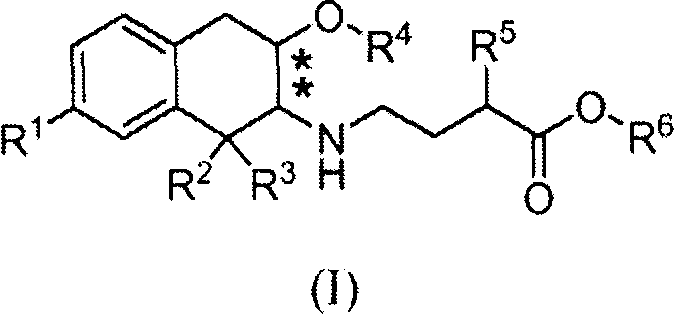 или их фармацевтически приемлемым солям, имеющим антагонистическую активность в отношении mu-опиоидного рецептора. В формуле (I) R1 обозначает -ORa или -С(О)NRbRc; R2, R3 и R4 обозначают, каждый независимо, C1-3алкил; R5 выбран из С1-6алкила, циклогексила, -(СН2)1-3-циклогексила и -(СН2)1-3-фенила; Ra, Rb и Rc обозначают, каждый независимо, водород; и R6 обозначает водород или С1-3алкил; и заместители в хиральных центрах, отмеченных звездочками, находятся в конфигурации транс. Изобретение относится также к способу получения соединений формулы (I), к промежуточным соединениям, используемым в синтезе соединений формулы (I) и способу их получения. Кроме того, изобретение относится к фармацевтической композиции, включающей соединения формулы (I), и применению таких соединений для лечения заболеваний или состояний, связанных с активностью mu-опиоидного рецептора. 9 н. и 16 з.п. ф-лы, 19 пр.
или их фармацевтически приемлемым солям, имеющим антагонистическую активность в отношении mu-опиоидного рецептора. В формуле (I) R1 обозначает -ORa или -С(О)NRbRc; R2, R3 и R4 обозначают, каждый независимо, C1-3алкил; R5 выбран из С1-6алкила, циклогексила, -(СН2)1-3-циклогексила и -(СН2)1-3-фенила; Ra, Rb и Rc обозначают, каждый независимо, водород; и R6 обозначает водород или С1-3алкил; и заместители в хиральных центрах, отмеченных звездочками, находятся в конфигурации транс. Изобретение относится также к способу получения соединений формулы (I), к промежуточным соединениям, используемым в синтезе соединений формулы (I) и способу их получения. Кроме того, изобретение относится к фармацевтической композиции, включающей соединения формулы (I), и применению таких соединений для лечения заболеваний или состояний, связанных с активностью mu-опиоидного рецептора. 9 н. и 16 з.п. ф-лы, 19 пр.
1. Соединение формулы (I):
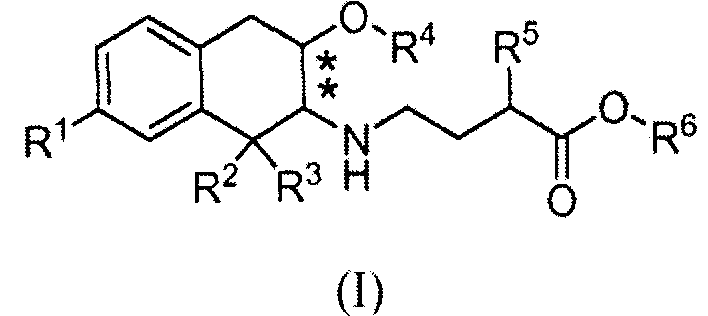
в которой
R1 обозначает -ORa или -C(O)NRbRc;
R2, R3 и R4 обозначают, каждый независимо, С1-3алкил;
R5 выбран из С1-6алкила, циклогексила, -(СН2)1-3-циклогексила и -(СН2)1-3-фенила;
Ra, Rb и Rc обозначают, каждый независимо, водород; и
R6 обозначает водород или C1-3алкил; и
в которой заместители в хиральных центрах, отмеченных звездочками, находятся в конфигурации транс;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором R1 обозначает -C(O)NH2.
3. Соединение по п.1, в котором R2 и R3 обозначают, каждый независимо, метил или этил.
4. Соединение по п.1, в котором R2 и R3 обозначают этил.
5. Соединение по п.1, в котором R4 обозначает метил.
6. Соединение по п.1, в котором R5 выбран из С3-5алкила, циклогексила, -(СН2)1-3-циклогексила и -(CH2)1-3-фенила.
7. Соединение по п.1, в котором R5 обозначает циклогексилметил.
8. Соединение по п.1, в котором R6 обозначает водород.
9. Соединение по п.1, в котором R6 обозначает метил.
10. Соединение по п.1, в котором хиральные центры имеют стереохимию (2S), (3S), как показано в формуле (Ia):

11. Соединение по п.1, выбранное из следующих соединений:
(S)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляная кислота;
метиловый эфир (S)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляной кислоты;
метиловый эфир (S)-4-((2R,3R)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляной кислоты;
метиловый эфир (S)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-4-метил-пентановой кислоты;
(S)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-4-метил-пентановая кислота;
(R)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-3-метилмасляная кислота;
(R)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-4,4-диметил-пентановая кислота;
(S)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-гептановая кислота;
(S)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-5-фенил-пентановая кислота;
(S)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-4,4-диметил-пентановая кислота;
(S)-2-Бензил-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляная кислота;
(R)-2-[2-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-этил]-гексановая кислота;
(S)-4-((2R,3R)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляная кислота;
(R)-4-((2R,3R)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляная кислота;
(R)-4-((2S,3S)-7-карбамоил-1,1-диэтил-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-2-циклогексилметил-масляная кислота;
(S)-2-циклогексилметил-4-((2S,3S)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляная кислота;
(S)-2-циклогексилметил-4-((2R,3R)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляная кислота;
(R)-2-циклогексилметил-4-((2S,3S)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляная кислота; и
(R)-2-циклогексилметил-4-((2R,3R)-1,1-диэтил-7-гидрокси-3-метокси-1,2,3,4-тетрагидро-нафталин-2-иламино)-масляная кислота
и их фармацевтическиприемлемых солей.
12. Фармацевтическая композиция, имеющая антагонистическую активность в отношении mu-опиоидного рецептора, включающая терапевтически эффективное количество соединения по любому из пп.1-11 и фармацевтически приемлемый носитель.
13. Способ получения соединения формулы (I)
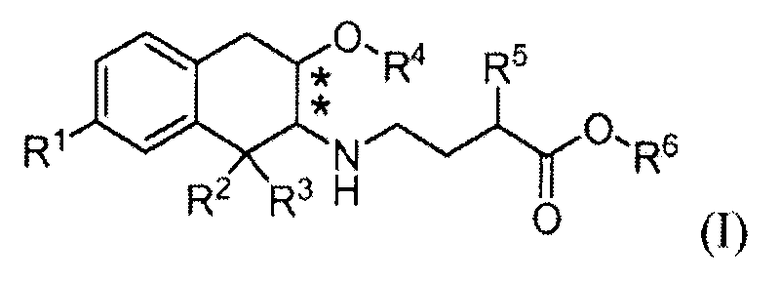
или его соли, в которой
R1 обозначает -ORa или-C(O)NRbRc;
R2, R3 и R4 обозначают, каждый независимо, С1-3алкил;
R5 выбран из C1-6алкила, циклогексила, -(СН2)1-3-циклогексила и -(СН2)1-3-фенила;
Ra, Rb и Rc обозначают, каждый, водород; и
R6 обозначает водород или C1-3алкил; и
в которой заместители в хиральных центрах, отмеченных звездочками, находятся в конфигурации транс, включающий:
(а) введение соединения формулы (II):

в реакцию с соединением формулы (III):
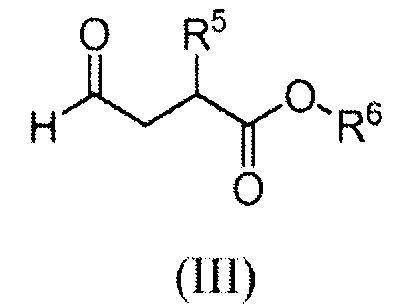
в которой R6 обозначает C1-3алкил; и
(b) когда R6 обозначает водород, введение продукта стадии (а) в контакт с избытком основания, с получением соединения формулы (I) или его соли.
14. Гидробромидная соль соединения формулы 2:

в которой R2 и R3 обозначают, каждый независимо, С1-3алкил, и заместители в хиральных центрах, отмеченных звездочками, находятся в конфигурации транс.
15. Способ получения гидробромидной соли соединения формулы 2 в твердой форме:
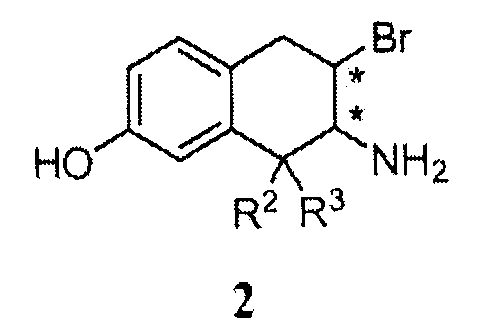
в которой R2 и R3 обозначают, каждый независимо, С1-3алкил, и заместители в хиральных центрах, отмеченных звездочками, находятся в конфигурации транс, включающий:
(а) введение рацемической смеси соединения формулы 1 и соединения, имеющего противоположную стереохимию в хиральных центрах:
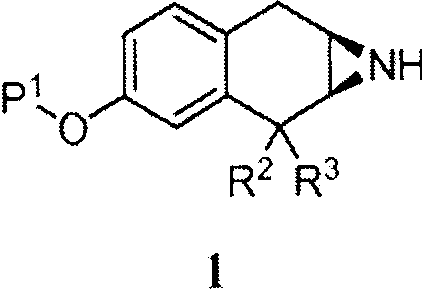
в которой Р1 обозначает C1-3алкил, в реакцию с бромидом водорода с получением гидробромидной соли соединения формулы 2; и
(b) выделение гидробромидной соли соединения формулы 2 в твердой форме.
16. Способ по п.15, в котором стадию (а) проводят в присутствии катализатора фазового переноса.
17. Соединение по любому из пп.1-11, имеющее антагонистическую активность в отношении mu-опиоидного рецептора.
18. Соединение по любому из пп.1-11 для применения в лечении заболевания или медицинского состояния у млекопитающего, которое облегчается лечением с использованием антагониста mu-опиоидного рецептора.
19. Соединение по п.18, причем заболевание или состояние представляет собой индуцируемую опиоидом дисфункцию кишечника или послеоперационную непроходимость кишечника.
20. Соединение по п.18, причем заболевание или состояние представляет собой нарушение, связанное со сниженной моторикой желудочно-кишечного тракта.
21. Способ лечения млекопитающего, имеющего медицинское состояние, которое может быть облегчено лечением антагонистом mu-опиоидного рецептора, включающий введение млекопитающему фармацевтической композиции, содержащей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по любому из пп.1-11.
22. Способ по п.21, в котором медицинское состояние представляет собой индуцируемую опиоидом дисфункцию кишечника или послеоперационную непроходимость кишечника.
23. Способ уменьшения или предотвращения побочного эффекта, связанного с использованием опиоидного средства, у млекопитающего, включающий введение млекопитающему опиоидного средства и эффективного количества соединения по любому из пп.1-11.
24. Способ изучения биологической системы или образца, включающего mu-опиоидный рецептор, включающий:
(a) введение биологической системы или образца в контакт с соединением по любому из пп.1-11; и
(b) определение эффекта, вызванного соединением на биологической системе или образце.
25. Соединение формулы (Ib), в которой R обозначает гидроксил:
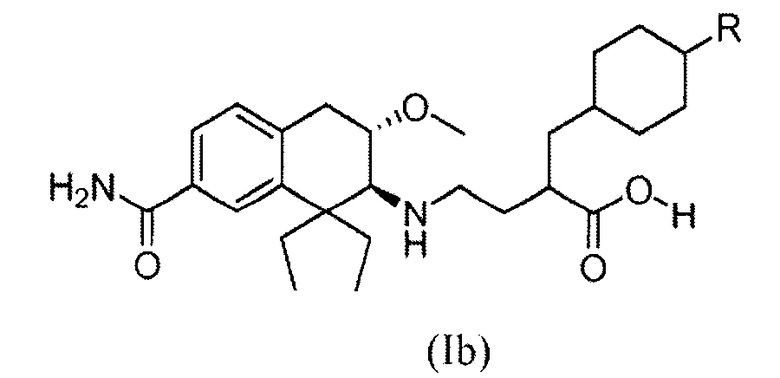
| Р.GRUNDT et al, Identification of a New Scaffold for Opioid Receptor Antagonism Based on the 2-Amino-1,1-dimethyl-7-hydroxytetralin Pharmacophore J | |||
| MED | |||
| CHEM, 2004, Vol.47, No | |||
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |
| С.ROY et al, Synthesis and Structure-Activity Relationship of Novel Aminotetralin Derivatives with High µ | |||
| Selective Opioid Affinity, BIOORG | |||
| MED | |||
| CHEM. | |||

