Настоящее изобретение относится к некоторым гетероциклическим соединениям в качестве ингибиторов протеинкиназы р38 ("р38"). В частности, настоящее изобретение относится к имидазозамещенным гетеробициклическим соединениям, способу их получения, содержащим их фармацевтическим препаратам и способам их применения.
Митоген-активируемые протеинкиназы (MAP) представляют собой семейство пролин-содержащих серин/треонинкиназ, которые активируют их субстраты двойным фосфорилированием. Киназы активируются различными сигналами, включая питательный и осмотической шок, УФ-облучение, факторы роста, эндотоксин и воспалительные цитокины. Одна группа MAP киназ представляет собой группу киназы р38, которая включает различные изоформы (например, р38α, р38β, р38δ и р38γ). Киназы р38 ответственны за фосфорилирование и активацию факторов транскрипции, а также других киназ, и сами активируются физическими и химическими факторами, предвоспалительными цитокинами и бактериальным липополисахаридом.
Более важно, показано, что продукты фосфорилирования р38 медиируют выработку воспалительных цитокинов, включая TNF, IL-1, IL-6 и циклокигеназу-2. Каждый из этих цитокинов включен в многочисленные заболевания и состояния. Например, TNF-α является цитокином, изначально вырабатываемым активированными моноцитами и макрофагами. Его избыточная или нерегулируемая выработка играет роль в патогенезе ревматоидного артрита. Недавно было показано, что ингибирование выработки TNF находит широкое применение для лечения воспаления, синдрома раздраженного кишечника, болезни Альцгеймера, болезни Крона, рассеянного склероза и астмы.
TNF также включен в вирусные инфекции, такие как ВИЧ, вирус энфлюенции и вирус герпеса, включая простой вирус герпеса типа-1 (HSV-1), простой вирус герпеса типа-2 (HSV-2), цитомегаловирус (CMV), варицелла-зостер вирус (VZV), вирус Эпштейна-Барра, вирус герпеса человека 6 (HHV-6), вирус герпеса человека 7 (HHV-7), вирус герпеса человека 8 (HHV-8), среди прочего ложное бешенство и ринотрахеиты.
Обычно IL-1 вырабатывается активированными моноцитами и макрофагами и играет роль во многих патофизиологических состояниях, включая ревматоидный артрит, лихорадку и снижение резорбции кости.
Ингибирование этих цитокинов ингибированием киназы р38 полезно для контроля, уменьшения и излечения многих этих заболеваний.
В одном варианте осуществления (i), настоящее изобретение относится к соединениям формулы I:
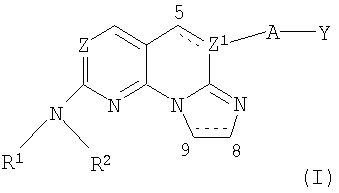
где:
Z представляет собой N или СН;
Z1 выбран из N и СН, когда связь между атомами С5 и Z1 является простой связью, и Z1 представляет собой С, когда связь между С5 и Z1 является двойной связью;
R1 представляет собой водород или алкил;
R2 представляет собой алкил, аралкил, циклоалкил, гетероалкил, гетероциклил, арил или гетероарил;
А отсутствует или представляет собой -О-, -CHR'-, -C(=O)-, -S(O)n- или -NR3-, где n обозначает 0, 1 или 2, R' представляет собой водород или алкил и R3 представляет собой водород, алкил, арил, гетероарил или циклоалкил;
связь между атомами С5 и Z1 является одинарной или двойной связью;
связь между атомами С8 и С9 является одинарной или двойной связью и Y представляет собой алкил, гетероалкил, циклоалкил, арил или гетероарил;
или его изомеры, фармацевтически приемлемые соли, эфиры или пролекарства.
В другом варианте осуществления настоящее изобретение предполагает:
(ii) Соединение в соответствии с (i) формулы:
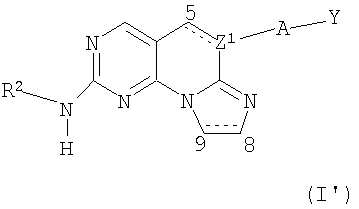
где:
R2 выбран из необязательно замещенного аралкила, необязательно замещенного циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного арила и С1-4алкила, необязательно замещенного одним заместителем, выбранным из гидрокси, O(С1-4алкила) или S(O)2(C1-4алкила);
А отсутствует или представляет собой -О-;
Y представляет собой фенил, необязательно замещенный одной или двумя группами, выбранными из галогена, гидрокси, амино, алкила или гетероалкила;
или его изомеры, фармацевтически приемлемые соли, эфиры или пролекарства; или
(iii) Соединение в соответствии с (ii), где R2 выбран из
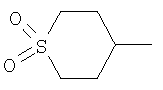
циклопентила, циклогексила, тетрагидропиранила, пиперидинила, тианонила, тетрагидро-1,1-диоксид-2-Н-тиопиранила, фенила и бензила, где каждая из указанных R2 групп необязательно замещена одним заместителем, выбранным из гидрокси, галогена, O(С1-4алкила) или S(O)2(С1-4алкила); или R2 выбран из С1-4алкила, необязательно замещенного одним заместителем, выбранным из гидрокси, O(С1-4алкила) или S(O)2(С1-4алкила); или
(iv) Соединение в соответствии с (ii) формулы
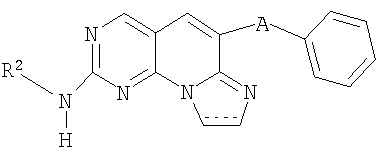
где R2 определен в (ii); или
(v) Соединение в соответствии с (iv), где R2 выбран из циклопентила, циклогексила, тетрагидропиранила, пиперидинила, тианонила, тетрагидро-1,1-диоксид-2-Н-тиопиранила, фенила и бензила, где каждая из указанных R2 групп необязательно замещена заместителем, выбранным из гидрокси, галогена, О(СН3) или S(O)2(СН3); или R выбран из С1-4алкила, необязательно замещенного одним заместителем, выбранным из гидрокси, О(СН3) или S(O)2(СН3); или
(vi) Соединение в соответствии с (i), где Z представляет собой -N-; или
(vii) Соединение в соответствии с (i) или (vi), где Z1 представляет собой -С- и связь между Z1 и С5 является двойной связью; или
(viii) Соединение в соответствии с любым одним из (i), (vi) или (vii), где А отсутствует или -О-; или
(ix) Соединение в соответствии с любым одним из (i), (vi)-(viii), где R1 представляет собой водород; или
(х) Соединение в соответствии с любым одним из (i), (vi)-(ix), где R2 выбран из циклопентила, циклогексила, тетрагидропиранила, пиперидинила, тианонила, тетрагидро-1,1-диоксид-2-Н-тиопиранила, фенила и бензила, где каждая из указанных R2 групп необязательно замещена одним из гидрокси, галогена, O(С1-4алкила) или S(O)2(С1-4алкила); или R2 выбран из С1-4алкила, необязательно замещенного одним заместителем, выбранным из гидрокси, O(C1-4алкила) или S(O)2(С1-4алкила); или
(xi) Соединение в соответствии с любым одним из (i), (vi)-(x), где Y представляет собой фенил, необязательно замещенный галогеном, гидрокси, амино, алкилом или гетероарилом; или
(xii) Соединение формулы Ia,
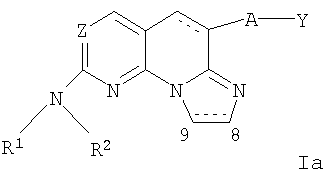
где
Y представляет собой арил;
Z представляет собой N или СН;
А отсутствует или представляет собой О;
R1 представляет собой водород;
R2 представляет собой алкил, гидроксиалкил, необязательно замещенный циклоалкил, необязательно замещенный гетероциклил, необязательно замещенный арил или необязательно замещенный аралкил; и связь между С8 и С9 является одинарной или двойной связью; или
(xiii) Соединение в соответствии с (xii), где Z представляет собой N, А представляет собой -О- и связь между С8 и С9 является двойной связью; или
(xiv) Соединение в соответствии с (xiii), где:
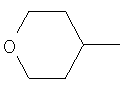
R2 представляет собой 4-тетрагидропиранил и Y представляет собой 4-фторфенил; или
R2 представляет собой 4-фторфенил и Y представляет собой 4-фторфенил; или
(xv) Соединение в соответствии с (xii), где Z представляет собой N, А отсутствует и связь между С8 и С9 является двойной связью; или
(xvi) Соединение в соответствии с (xv), где:
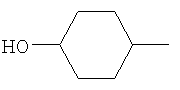
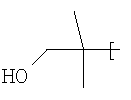
R2 представляет собой 4-гидроксициклогексил и Y представляет собой 2-хлорфенил;
R2 представляет собой 4-тетрагидропиранил и Y представляет собой 2-хлорфенил;
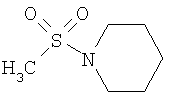
R2 представляет собой 4-(N-метилсульфонилпиперидинил) и Y представляет собой 2-хлорфенил;
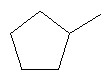
R2 представляет собой циклопентил и Y представляет собой is 2-хлорфенил;
R2 представляет собой 4-(N-метилсульфонилпиперидинил) и Y представляет собой 2-хлорфенил;
R2 представляет собой 4-тетрагидро,1,1-диоксид-2-Н-тиопиранил и Y представляет собой 2-хлорфенил;
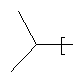
R2 представляет собой изопропил и Y представляет собой 2-хлорфенил; или
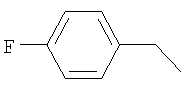
R2 представляет собой 4-фторбензил и Y представляет собой 2-хлорфенил; или
(xvii) Соединение в соответствии с (i) формулы
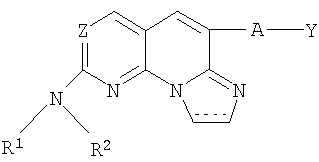
(xviii) Соединение в соответствии с (xvii), где Z представляет собой N, А отсутствует, R1 представляет собой водород, Y представляет собой 2-хлорфенил и R представляет собой 4-гидроксициклогексил; или
(xix) Соединение в соответствии с (i) формулы I(с):
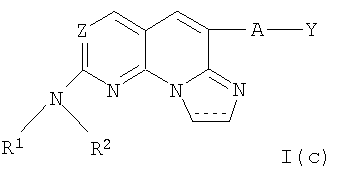 или
или
(хх) Соединение в соответствии с (xix), где Z представляет собой N, А отсутствует, R1 представляет собой водород, Y представляет собой 2-хлорфенил и R2 представляет собой 4-гидроксициклогексил.
Соединения формулы I и их вышеупомянутые соли являются ингибиторами протеинкиназ и проявляют эффективную активность против р38 in vivo. Следовательно, соединения могут использоваться для лечения заболеваний, опосредованных предвоспалительными цитокинами, такими как TNF и IL-1.
В другом варианте осуществления настоящее изобретение относится к способам лечения заболеваний или состояний, опосредованных р38, в которых терапевтически эффективное количество соединения формулы I вводится пациенту, нуждающемуся в таком лечении.
В другом варианте осуществления настоящее изобретение относится к способам получения описанных выше соединений.
В другом варианте осуществления настоящее изобретение относится к способам получения лекарственных средств, пригодных для лечения заболеваний и состояний, опосредованных р38.
Как здесь используется, термин "алкил" обозначает линейный насыщенный моновалентный углеводородный радикал или разветвленный насыщенный моновалентный углеводородный радикал с одним - шестью атомами углерода, например, метил, этил, н-пропил, 2-пропил, трет-бутил, пентил и т.д.
Термин "арил" обозначает моновалентный моноциклический или бициклический ароматический углеводородный радикал, предпочтительно фенил, который необязательно замещен независимо одним или несколькими заместителями. Когда арильная группа замещена предпочтительно одним, двумя или тремя заместителями, заместители предпочтительно выбраны из группы, состоящей из алкила, галогеналкила, галогена, гидрокси, нитро, циано, амино, галогеналкокси, гетероалкил, метилендиокси, этилендиокси, Y-арила, Y-гетероарила, Y-циклоалкила, Y-гетероциклила, Y-ORp-Y-NRpRq, -Y-C(O)-Rp, -YS(O)0-2Rp, -Y-N-S(O)2Rp, -Y-S(O)2NRpRq, -Y-N-C(O)NRpRq, где Y отсутствует или представляет собой C1-С3 алкиленовую группу, и Rp и Rq каждый независимо выбран из водорода, алкила, галогеналкила, гидрокси, алкокси, арила, гетероарила, циклоалкила и гетероциклила. Более конкретно термин арил включает, но не ограничивается ими, фенил, хлорфенил, метоксифенил, 1-нафтил, 2-нафтил и их производные.
"Аралкил" обозначает радикал -RxRy, где Rx представляет собой алкиленовую группу и Ry представляет собой арильную группу как определено выше, например, бензил, фенилэтилен и т.д.
Используемый здесь термин "циклоалкил" обозначает насыщенный моновалентный циклический углеводородный радикал с тремя-семью атомами углерода в кольце, например, циклопентил, циклобутил, циклогексил и т.д. Насыщенный моновалентный циклический углеводородный радикал, как определено выше, может быть необязательно замещен одним, двумя или тремя заместителями, которые не являются водородом. Предпочтительно, заместители выбраны из группы, состоящей из алкила, гидрокси, алкокси, амино, монозамещенного амино, дизамещенного амино, галогеналкила, галогена, цианоалкила, оксо (т.е., кислород карбонила), гетероалкила, гетероциклила, гидроксиалкила и -(X)n-C(O)R' (где Х представляет собой О или NR'', n обозначает 0 или 1, R'' представляет собой водород, алкил, галогеналкил, амино, монозамещенный амино, дизамещенный амино, гидрокси, алкокси, алкил или необязательно замещенный фенил, и R' представляет собой Н или алкил), и -S(O)nR' (где n обозначает 0-2). Более конкретно, термин замещенный циклоалкил включает, например, замещенный циклопентил, замещенный циклогексил и т.д.
Термин "гало" или "галоген", относящийся к заместителю, обозначает фтор, хлор, бром или йод, предпочтительно хлор.
Термин "галогеналкил" обозначает алкил, замещенный одним или несколькими одинаковыми или различными атомами галогена, например, -CH2Cl, -CF3, -СН2CF3, -СН2CCl3 и т.д., а также включает алкильные группы, такие как перфторалкил, где все водородные атомы алкила замещены атомами фтора.
Используемый здесь термин "гетероалкил" обозначает определенный выше алкильный радикал, где один, два или три атома водорода замещены заместителем, независимо выбранным из группы, состоящей из -ORa -NRbRc, и S(O)nRd (где n имеет значение от 0 до 2), где гетерорадикал присоединен через атом углерода, где Ra представляет собой водород, ацил, алкил, циклоалкил или циклоалкилалкил; или Rb и Rc независимо друг от друга представляют собой водород, ацил, алкил, циклоалкил или циклоалкилалкил, или Rb и Rc вместе с атомом азота, к которым они присоединены, образует гетероцикло или гетероарил; и когда n обозначает 0, Rd представляет собой водород, алкил, циклоалкил или циклоалкилалкил, и когда n обозначает 1 или 2, Rd представляет собой алкил, циклоалкил, циклоалкилалкил, амино, ациламино, моноалкиламино или диалкиламино. Примеры включают, но не ограничиваются ими, 2-гидроксиэтил, 3-гидроксипропил, 2-гидроксиметилэтил, 2,3-дигидроксипропил, 1-гидроксиметилэтил, 3-гидроксибутил, 2,3-дигидроксибутил, 2-гидрокси-1-метилпропил, 2-аминоэтил, 3-аминопропил, 2-метилсульфонилэтил, аминосульфонилметил, аминосульфонилэтил, метиламиносульфонилметил, метиламиносульфонилэтил, метиламиносульфонилпропил и т.д. Когда Ra представляет собой водород, радикал -ORa также представляет собой "гидроксиалкил" и включает, но не ограничивается ими, 2-гидроксиэтил, 3-гидроксипропил, 2-гидроксиметилэтил, 2,3-дигидроксипропил, 1-гидроксиметилэтил, 3-гидроксибутил, 2,3-дигидроксибутил и 2-гидрокси-1-метилпропил.
"Гетероарил" обозначает моновалентный моноциклический или бициклический радикал с 5-12 атомами в кольце, имеющий по крайней мере одно ароматическое кольцо, содержащее один, два или три гетероатома в кольце, выбранных из N, О или S, причем остальные атомы в кольце являются атомами С, где когда гетероарил представляет собой бициклическую систему с одним карбоциклическим и/или неароматическим кольцом, гетероарильный радикал присоединен через гетероарильное кольцо. Гетероарильное кольцо необязательно замещено одним или несколькими заместителями, предпочтительно одним или двумя заместителями, независимо друг от друга выбранными из алкила, галогеналкила, галоген, нитро, циано, амино, метилендиокси, Y-арила, Y-гетероарила, Y-циклоалкила, -Y-гетероциклила, -Y-OR', -YNR'R'', -Y-C(O)R', -Y-O-C(O)-R', -Y-S(O)0-2-R', -Y-N-SO2-R', -Y-SO2-NR'R'', и -Y-N-C(O)-NR'R'', где Y отсутствует или представляет собой C1-С3 алкиленовую группу и R' и R'' независимо друг от друга представляют собой водород, алкил, галогеналкил, гидрокси, алкокси, арил, гетероарил, циклоалкил или гетероциклил. Более конкретно, термин гетероарил включает, но не ограничивается ими, пиридил, фуранил, тиенил, тиазолил, изотиазолил, триазолил, имидазолил, изоксазолил, пирролил, пиразолил, пиримидинил, бензофуранил, тетрагидробензофуранил, изобензофуранил, бензотиазолил, бензоизотиазолил, бензотриазолил, индолил, изоиндолил, бензоксазолил, хинолил, тетрагидрохинолинил, изохинолил, бензимидазолил, бензизоксазолил или бензотиенил, имидазо[1,2-а]-пиридинил, имидазо [2,1-b] тиазолил и их производные.
"Монозамещенный амино" обозначает радикал -NHRе, где Rе представляет собой алкил, гетероалкил, галогеналкил, циклоалкил, циклоалкилалкил, гидроксиалкил, арил, аралкил, аралкенил, гетероарил, гетероаралкил, гетероаралкенил, гетероциклил или гетероциклилалкил, например, метиламино, этиламино, фениламин, бензиламин и т.д. Аналогично, термин "дизамещенный амино" обозначает радикал -RgRh, где Rg и Rh представляют собой, независимо друг от друга, алкил, гетероалкил, галогеналкил, циклоалкил, циклоалкилалкил, гидроксиалкил, арил, аралкил, аралкенил, гетероарил, гетероаралкил, гетероаралкенил, гетероциклил или гетероциклилалкил, или Rg и Rh вместе с атомом азота, к которым они присоединены, образуют гетероциклильное кольцо. Примеры включают, но не ограничиваются ими, диметиламино, метилэтиламино, ди(1-метилэтил)амино, пиперазинил и т.д.
"Гетероциклил" обозначает насыщенный циклический радикал, где один или два атома в кольце являются гетероатомами, выбранными из N, О или S(O)n, (где n имеет значение от 0 до 2), причем остальные атомы в кольце являются атомами С, где один или два атома С могут необязательно содержать группу кислорода карбонила, например, один или два атома в кольце могут быть группами формулы -С(=O)-. Гетероциклическое кольцо может быть необязательно замещено независимо одним, двумя или тремя заместителями, выбранными из алкила, гидрокси, гидроксиалкила, алкокси, гетероалкила, галогеналкила и -(X)n-C(O)R (где Х представляет собой О или NR', n обозначает 0 или 1, R представляет собой водород, алкил, галогеналкил, амино, монозамещенный амино, дизамещенный амино, гидрокси, алкокси или необязательно замещенный фенил, и R' представляет собой Н или алкил), Y-арил, Y-гетероарил, Y-циклоалкил, Y-гетероциклил, Y-ORp, -Y-NRpRq, -Y-C(O)-Rp, -YS(O)0-2Rp, -Y-N-S(O)2Rp, -Y-S(O)2NRpRq, -Y-N-C(O)NRpRq, где Y отсутствует или представляет собой C1-С3 алкиленовую группу, и Rp и Rq каждый независимо друг от друга выбраны из водорода, алкила, галогеналкила, гидрокси, алкокси, арила, гетероарила, циклоалкила и гетероциклила. Более конкретно, термин гетероциклил включает, но не ограничивается ими, тетрагидропиранил, пиперидинил, пиперазинил, морфолинил и их производные.
Термин "ацил" обозначает группу -C(O)Rr, где Rr представляет собой алкил, галогеналкил, гетероалкил, арил, гетероарил, аралкил или гетероаралкил.
"Алкокси", "арилокси", "аралкилокси" или "гетероаралкилокси" обозначает радикал -OR, где R представляет собой алкил, арил, аралкил или гетероаралкил, соответственно, как определено выше, например, метокси, фенокси, пиридин-2-илметилокси, бензилокси и т.д.
Когда связь в формуле является двойной связью, где одна из двух связей обозначена пунктирной линией, как в 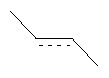 , если конкретно не указано иное, это обозначает, что связь необязательно может быть простой связью или двойной связью, с соответствующим выбором соседних атомов. Так, когда указано "пунктирная линия представляет собой связь", это означает, что присутствует двойная связь; и когда указано "пунктирная линия не представляет собой связь", это означает, что присутствует простая связь.
, если конкретно не указано иное, это обозначает, что связь необязательно может быть простой связью или двойной связью, с соответствующим выбором соседних атомов. Так, когда указано "пунктирная линия представляет собой связь", это означает, что присутствует двойная связь; и когда указано "пунктирная линия не представляет собой связь", это означает, что присутствует простая связь.
"Уходящая группа" имеет значение, обычно используемое в синтетической органической химии, т.е. атом или группу, которая может быть замещена нуклеофилом и включает галоген (такой как хлор, бром и йод), алкансульфонилокси, аренесульфонилокси, алкилуглеродилокси (например, ацетокси), арилуглеродилокси, мезилокси, тозилокси, трифторметансульфонилокси, арилокси (например, 2,4-динитрофенокси), метокси, N,O-диметилгидроксиламино и т.д.
"Фармацевтически приемлемый эксципиент" обозначает эксципиент, который полезен для получения фармацевтической композиции, который обычно является безопасным, нетоксичным и биологически и иначе приемлемым, и включает эксципиенты, которые являются приемлемыми для ветеринарного применения и для фармацевтического применения для человека.
"Фармацевтически приемлемый эксципиент" как используется в описании и формуле изобретения включает один и несколько таких эксципиентов.
"Фармацевтически приемлемая соль" соединения обозначает соль, которая является фармацевтически приемлемой и которая обладает желаемой фармакологической активностью исходного соединения. Такие соли включают: (1) кислотные аддитивные соли, полученные с неорганическими кислотами, такими как соляная кислота, бромоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.д.; или полученные с органическими кислотами, такими как уксусная кислота, пропановая кислота, гексановая кислота, циклопентанпропановая кислота, гликолевая кислота, пирувиновая кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метан сульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталенсульфоновая кислота, 4-толуолсульфоновая кислота, камфорсульфоновая кислота, 4-метилбицикло[2,2,2]-окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 4,4'-метиленбис-(3-гидрокси-2-ен-1-карбоновая кислота), 3-фенилпропановая кислота, триметилуксусная кислота, трет-бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и т.д.; или (2) соли, полученные замещением кислотного протона в исходном соединении ионом металла, например, ионом щелочного металла, ионом щелочно-земельного металла или ионом аллюминия; или связыванием с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, триметамин, N-метилглюкамин и т.д.
Используемый здесь термин "пролекарство" обозначает любое соединение, которое высвобождает активное исходного лекарственное средство Формулы I in vivo, когда такое пролекарство вводится млекопитающему. Пролекарства соединения формулы I получают модификацией одной или нескольких функциональных групп в соединении формулы I таким образом, что модификация(и) может быть снята in vivo для высвобождения исходного соединения. Пролекарства включают соединения формулы I, где гидрокси, амино или сульфгидрильная группа в соединении формулы I связана с любой группой, которая может отщепляться in vivo с получением свободной гидроксильной, амино или сульфгидрильной группы, соответственно. Примеры пролекарств включают, но не ограничиваются ими, эфиры (например, ацетатные, форматные и бензоатные производные), карбаматы (например, N,N-диметиламинокарбонил) гидроксифункциональных групп соединений формулы I и т.д.
"Защитная группа" обозначает группу атомов, которые при присоединении к реакционной группе в молекуле защищают, уменьшают или предотвращают ее реактивность. Примеры защитных групп приведены в T.W.Green и P.G.Futs, Protective Groups in Organic Chemistry, (Wiley, 2nd ed. 1991) и Harrison и Harrison и др., Compendium of Synthetic Organic Methods, Vols.1-8 (John Wiley and Sons, 1971-1996). Примерные аминозащитные группы включают формильные, ацетильные, трифторацетильные, бензильные, бензилоксикарбонильные (CBZ), трет-бутоксикарбонильные (Boc), триметилсилильные (TMS), 2-триметилсилилэтансульфонильные (SES), тритильные и замещенные тритильные группы, аллилоксикарбонил, 9-флуоренилметилоксикарбонил (FMOC), нитровератрилоксикарбонил (NVOC) и т.д. Примерные гидроксизащитные группы включают группы, где гидроксигруппа ацилирована или алкилирована, такие как бензиловые и тритильные эфиры, а также алкиловые эфиры, тетрагидропираниловые эфиры, триалкилсилильные эфиры и аллильные эфиры.
"Обработка" или "лечение" заболевания включает: (1) предотвращение заболевания, т.е. предотвращение развития клинических симптомов заболевания у млекопитающего, которое может быть подвергнуто или предрасположено к заболеванию, но симптомы заболевания еще не присутствуют или не проявляются; (2) ингибирование заболевания, т.е. остановка или уменьшение развития заболевания или его клинических симптомов; или (3) облегчение заболевания, т.е. начало регрессии заболевания или его клинических симптомов.
"Терапевтически эффективное количество" обозначает количество соединения, которое при введении млекопитающему для лечения заболевания является достаточным для воздействия такого лечения на болезнь. "Терапевтически эффективное количество" зависит от соединения, заболевания и его серьезности и возраста, веса и других факторов излечиваемого млекопитающего.
В одном варианте осуществления настоящее изобретение относится к соединениям формулы:
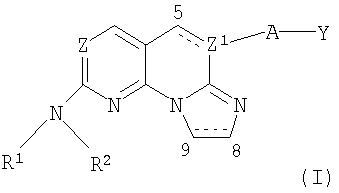
где Z, Z1, R1, R2, A, Y и связи между С5 и Z1 и С8 и С9 определены выше.
Предпочтительные соединения формулы I включают соединения, в которых А отсутствует или -О-, оба Z и Z1 представляют собой N, Y представляет собой арил, R1 представляет собой водород и R2 определен выше для соединений формулы (I). Более предпочтительными соединениями являются соединения формулы I, где Z представляет собой N, Z1 представляет собой С так, что связь между С5 и Z1 является двойной связью, А отсутствует или представляет собой -О, Y представляет собой арил (более предпочтительно необязательно замещенный фенил), R1 представляет собой водород и R2 определен выше для соединений формулы I.
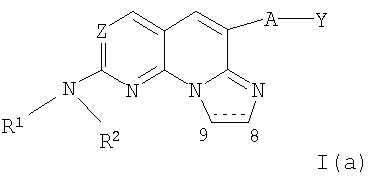
Еще более предпочтительными соединениями являются соединения формулы I(а):
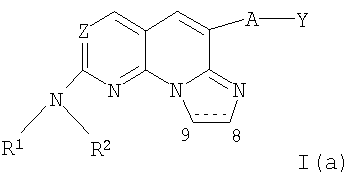
где Z представляет собой N, А отсутствует или представляют собой -О-, Y представляет собой необязательно замещенный фенил, R1 представляет собой водород и R2 определен выше.
Наиболее предпочтительными соединениями являются соединения формулы I(а), где Z представляет собой N, А отсутствует, Y представляет собой фенил, замещенный галогеном, гидрокси, амино, алкилом или гетероалкилом, R1 представляет собой водород и R2 представляет собой алкил, гетероалкил, необязательно замещенный циклоалкил, необязательно замещенный гетероциклил или замещенный фенил или бензил.
Особенно предпочтительными соединениями являются соединения формулы I(а), где А отсутствует, Y представляет собой фенил, замещенный галогеном, пунктирная линия обозначает двойную связь, R1 представляет собой водород и R2 представляет собой алкил (более предпочтительно низший алкил, необязательно замещенный гидрокси, метокси или метилсульфонилом), гетероалкил, необязательно замещенный циклоалкил (более предпочтительно циклопентил или циклогексил, необязательно замещенные метокси), необязательно замещенный гетероциклил (более предпочтительно необязательно замещенный пиперидинил), или замещенный фенил или бензил (более предпочтительно фенил или бензил, замещенные галогеном). В таблице I ниже представлены некоторые примерные соединения формулы I(а).
Другими примерными соединениями изобретения являются соединения формулы I(b).
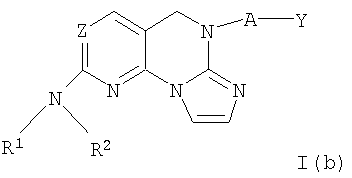
где R1, Z, R2, А и Y определены выше для соединений формулы (I) и I(а).
Другими примерными соединениями изобретения являются соединения формулы I(с)
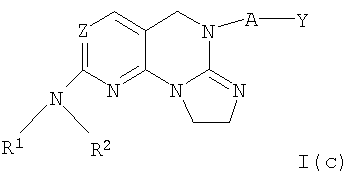
где Z, R1, R2, А и Y определены выше для соединений формулы (I) и I(а).
Предпочтительным соединением (12) формулы I(с) является соединение, в котором Z представляет собой N, А отсутствует, R1 представляет собой водород, Y представляет собой 2-хлорфенил и R2 представляет собой 4-гидроксициклогексил. Масс-спектр МН+ 399.
Таблица 1
Примерными соединениями формулы I(а) являются соединения, где пунктирная линия обозначает связь, Z представляет собой N, R1 представляет собой водород и значения R2, А и Y указаны ниже:
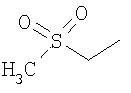
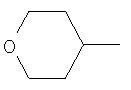
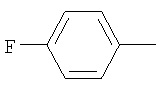
Предпочтительным соединением (13) формулы I(а) является соединение, в котором Z представляет собой N, пунктирная линия не является связью, R1 представляет собой водород, R2 представляет собой 4-гидроксициклогексил и Y представляет собой 2-хлорфенил. МН+ 396, tпл от 169,3°С до 175,8°С.
Особенно предпочтительными соединениями формулы I(а) являются соединения (3) и (7) Таблицы 1 выше.
Соединения настоящего изобретения могут существовать в свободных формах, а также в формах сольватов, включая гидратированные формы, и включены в объем настоящего изобретения. Кроме того, как указано выше, настоящее изобретение также включает все фармацевтически приемлемые соли соединений наряду с пролекарственными формами соединений и все стереоизомеры в чистой хиральной форме или смеси рацематов других форм в смеси.
Соединения формулы I способны также образовывать фармацевтически приемлемые кислотные аддитивные соли. Все эти формы также включены в объем изобретения.
Фармацевтически приемлемые кислотный аддитивные соли соединений формулы I включают соли, полученные с неорганическими кислотами, такими как соляной, азотной, фосфорной, серной, бромоводородной, йодостоводородной, фосфористой и т.д., а также соли, полученные с органическими кислотами, такими как алифатическими моно- и дикарбоновыми кислотами, фенилзамещенными алкановыми кислотами, гидроксиалкановыми кислотами, алкандионовыми кислотами, ароматическими кислотами, алифатическими и ароматическими сульфоновыми кислотами и т.д. Такие соли включают сульфат, пиросульфат, бисульфат, сульфит, бисульфит, нитрат, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид, йодид, ацетат, пропионат, каприлат, изобутират, оксалат, малонат, сукцинат, суберат, себакат, фумарат, малеат, манделат, бензоат, хлорбензоат, метилбензоат, динитробензоат, фталат, бензолсульфонат, толуолсульфонат, фенилацетат, цитрат, лактат, малеат, тартрат, метансульфонат и т.д. Также включены соли с аминокислотами, такие как аргинат и т.д. и глюконат, галактуронат (см., например, Berge и др., "Pharmaceutical Salts," J. of Pharmaceutical Science, 1977, 66, 1-19).
Кислотные аддитивные соли основных соединений могут быть получены взаимодействием формы свободного основания с достаточным количеством нужной кислоты с получением соли обычным способом. Форма свободного основания может быть восстановлена взаимодействием формы соли с основанием и выделением свободного основания обычным способом. Формы свободных оснований могут отличаться от их соответствующих форм солей определенными физическими свойствами, такими как растворимость в полярных растворителях, но в любом случае соли являются эквивалентными соответствующему свободному основанию по цели настоящего изобретения.
Фармацевтически приемлемые основные аддитивные соли могут быть получены с ионами металлов или аминами, такими как ионы щелочных и щелочно-земельного металла или органические амины. Примеры ионов металлов, которые используются в качестве катионов, включают натрий, калий, магний, кальций и т.д. Примерами подходящих аминов являются N,N'-дибензилэтилендиамин, хлоророкаин, холин, диэтаноламин, этилендиамин, N-метилглюкамин и прокаин (см., например, Berge и др., выше).
Основные аддитивные соли кислотных соединений могут быть получены взаимодействием формы свободной кислоты с достаточным количеством нужного основания с получением соли обычным способом. Форма свободной кислоты может быть получена взаимодействием формы соли с кислотой и выделением свободной кислоты обычным способом. Формы свободных кислот могут отличаться от их соответствующих форм солей определенными физическими свойствами, такими как растворимость в полярных растворителях, но в любом случае соли являются эквивалентными соответствующей свободной кислоты по цели настоящего изобретения.
Соединения настоящего изобретения могут быть получены различными способами, используя методики, хорошо известные среднему специалисту в данной области техники. На следующих схемах представлены способы получения соединений изобретения, где используемые сокращения имеют следующее значение:
МСРВА: м-хлорпербензойная кислота.
NMP: 1-метил-2-пирролидинон.
ТГФ: тетрагидрофуран.
TLC: тонкослойная хроматография.
EtOAc: этилацетат.
LAH: литийалюмогидрид.
ДМФА: диметилформамид.
DMPU: 1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидинон.
На схеме 1 показан общий способ получения соединений формулы I(а) (1) (соединения формулы I(а), где пунктирная линия не является связью) исходя из защищенного тиопиридин-5-карбоксилата 1, где Rp и Rpa представляют собой защитные группы, такие как низший алкил и бензил; и RL представляет собой уходящую группу, такую как галоген. Защитные и уходящие группы описаны на стр.6, строка 30 - стр.7, строка 9 и стр.5, строки 22-26, соответственно. Общий способ получения соединения 5, из карбоксилата 1 на схеме описан в WO 0129042 и WO 0129041.
Соединение 1 обрабатывают триэтиламином и водным аммиаком с получением соединения 2. Соединение 2 восстанавливают литийалюмогидридом или другими восстанавливающими агентами, хорошо известными в уровне техники, с получением соединения 3. Соединение 3 окисляют до альдегида 4 обработкой порошком активированного оксида марганца или другими способами окисления, известными в уровне техники. Соединение 4 реагирует с эфиром общей формулы -A-Y-C(O)ORz, где А и Y определены выше и Rz представляет собой низший алкил, арил или циклоалкил, с получением пиримидинона 5.
Соединение 5 реагирует с N-(2-гидроксиэтил)фталимидом с получением фталимида 6. Фталимид 6 перемешивают с гидразинмоногидратом и соединение 7 выделяют в чистой форме. Соединение 7 циклюзуют в трициклический сульфид 8 реакцией с триметилаллюминием.
Трициклический сульфид 8 может непосредственно реагировать с амином общей формулы -NH2R1R2 с получением соединения общей формулы I(а)(1).
Схема 1
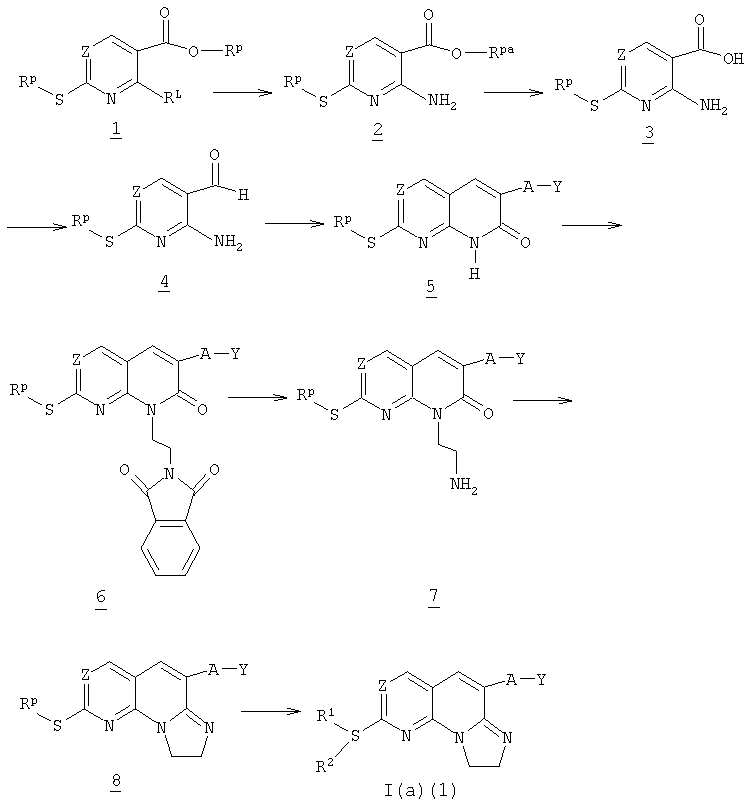
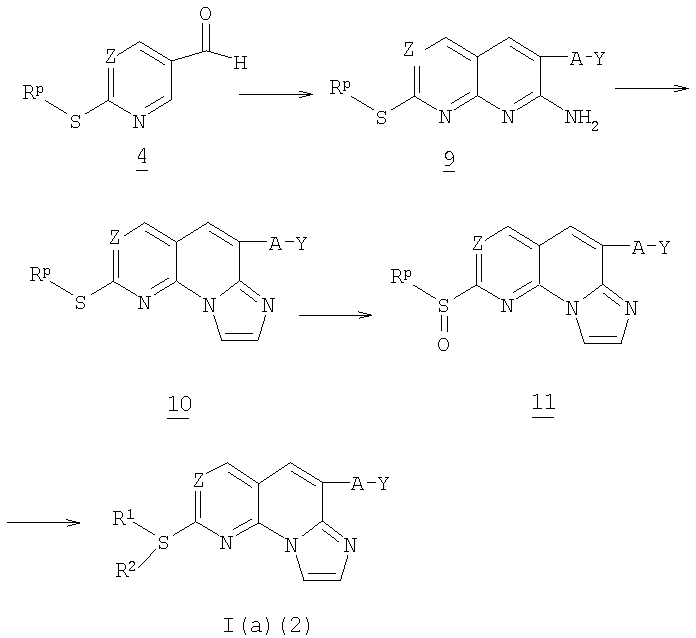
На схеме 2 представлен общий способ получения соединения формулы I(а)(2) (соединения формулы I(а), в которых пунктирная линия обозначает связь), исходя из альдегида 4 из схемы 1 выше и нагревая его с цианидом общей формулы CN-CH2-A-Y, где А и Y указаны выше, и карбонатом калия в DPMU с получением аминопиридопиримидина 9. Соединение 9 обрабатывают триэтиламином и 1,2-дихлорэтилэтиловым эфиром с получением сульфида формулы 10.
Соединение 10 затем окисляют до соответствующего сульфоксида 11 способом, описанным в схеме 1. Сульфоксид 11 затем реагирует с амином формулы -NH2R1R2 с получением соединения общей формулы I(а).
Альтернативно, сульфид 10 может реагировать непосредственно с -NH2R1R2 с получением соединения общей формулы I(а)(2).
Схема 2
На схеме 3 представлен общий способ получения соединения формулы I(с) исходя из соединения 12. Соединение 12 получают в соответствии со способом, описанным в WO 0129042 и WO 0129041.
Схема 3
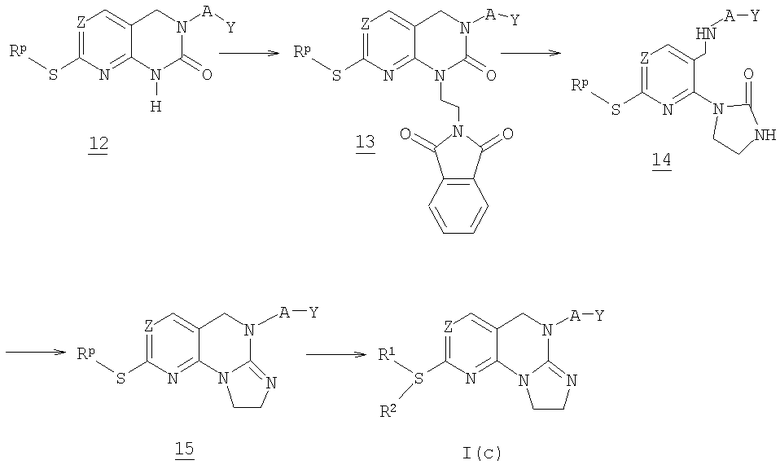
Соединение 12 обрабатывают трифенилфосфином, диэтилазодикарбоксилатом и N-(2-гидроксиэтил)фталимидом с получением соединения 13. Соединение 13 затем реагирует с гидразингидратом с получением соединения 14, которое затем реагирует с раствором триметилаллюминия с получением соединения 15. Соединение 15 реагирует с амином формулы -H2NR1R2 с получением соединения формулы I(с). Альтернативно, соединение 15 может окисляться 3-хлорпербензойной кислотой с получением соответствующего сульфоксида (не показано), который затем реагирует с амином -NR1R2 с получением нужного соединения формулы I(с).
Эти способы получения соединений настоящего изобретения также являются объектом настоящего изобретения, включая следующие способы:
(i) Способ получения соединения по п.1, включающий:
а) реакцию соединения формулы А с цианидом формулы CN-CH2-A-Y, где A, Z и Y описаны в п.1 формулы изобретения, и Rp выбран из низшего алкила и бензила, с получением соединения формулы В;
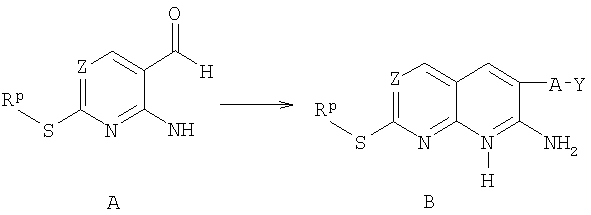
б) обработку соединения В реагентом, выбранным из триэтиламина, триалкиламинового основания, 1,4-диазабицикло[2,2,2]октаном или диизопропилэтиламином, и 1,2-дихлорэтиловым эфиром с получением соединения С;
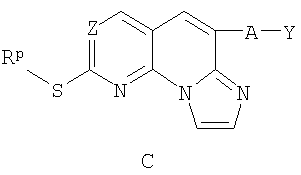
в) окисление соединения С реагентом, выбранным из 3-хлорпербензойной кислоты, перекиси водорода /муравьиной кислоты, перекиси водорода/ метилтриоксорения VII или OXONE, с получением соединения D; и
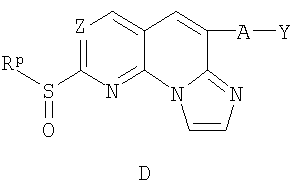
г) взаимодействие соединения D с амином формулы -H2N-R1R2, где R1 и R2 определены в п.1 формулы изобретения, в течение достаточного периода времени, с получением соединения по п.1.
(ii) Способ получения соединения по п.1, включающий:
а) реакцию соединения формулы Е, где Z, А и Y определены в п.1 формулы изобретения и Rp представляет собой низший алкил или бензил, с реагентом, выбранным из трифенилфосфина, диэтилазодикарбоксилата и N-(2-гидроксиэтил)фталимида с получением соединения F;
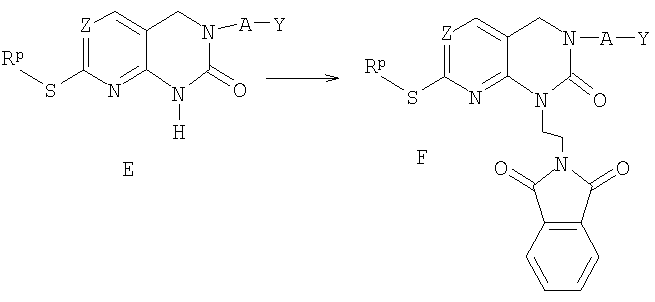
б) реакцию соединения F с гидразином с получением соединения G;
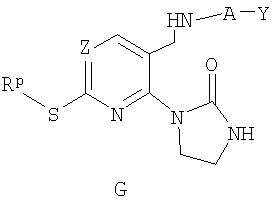
в) реакцию соединения G с триметилаллюминием с получением соединения Н;
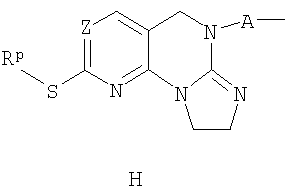
г) необязательно окисление соединения Н реагентом, выбранным из 3-хлорпербензойной кислоты, перекиси водорода /муравьиной кислоты, перекиси водорода/ метилтриоксорения VII или OXONE, с получением соответствующего сульфоксида, и взаимодействие сульфоксида с амином формулы -H2N-R1R2, где R1 и R2 определены в п.1 формулы изобретения, в течение достаточного периода времени, с получением соединения по п.1.
Соединения формулы I и фармацевтически приемлемые соли основных соединений формулы I с кислотами могут использоваться в качестве лекарственных средств, например, в форме фармацевтических препаратов. Фармацевтические препараты могут вводиться энтерально, например, орально в форме таблеток, покрытых таблеток, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий, назально, например, в форме назальных спреев, или ректально, например, в форме свечей. Однако они также могут вводиться парентерально, например, в форме инъекционных растворов.
Соединения формулы I и их вышеупомянутые фармацевтически приемлемые соли могут находиться в фармацевтически инертных, органических или неорганических носителях для получения фармацевтических препаратов. Например, в качестве таких носителей для таблеток, покрытых таблеток, драже и твердых желатиновых капсул могут использоваться лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли и т.д. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и т.д.; однако в зависимости от природы активного ингредиента носитель может не использоваться для мягких желатиновых капсул. Подходящими носителями для получения растворов и сиропов являются, например, вода, полиолы, сахароза, инвертированный сахар, глюкоза и т.д. Подходящими носителями для свечей являются, например, природные или отвержденные масла, воски, жиры, полутвердые и жидкие полиолы и т.д.
Фармацевтические препараты также могут содержать консерванты, солюбилизаторы, стабилизаторы, увлажняющие агенты, эмульсификаторы, подсластители, красители, отдушки, соли для регулирования осмотического давления, буферы, маскирующие агенты или антиоксиданты. Они также могут содержать терапевтически ценные вещества, отличные от соединений формулы I и их вышеупомянутых фармацевтически приемлемых солей.
Лекарственные средства, которые содержат соединение формулы I или фармацевтически приемлемую соль основного соединения формулы I с кислотой в смеси с подходящим фармацевтическим носителем, также являются объектом настоящего изобретения, а также способ получения таких лекарственных средств, который включает введение одного или нескольких таких соединений или солей и, при необходимости, одного или нескольких других терапевтически ценных веществ в галеновую форму для введения вместе с подходящим фармацевтическим носителем.
Как указано выше, соединения формулы I и их вышеупомянутые фармацевтически приемлемые соли могут использоваться согласно изобретению в качестве терапевтически активных веществ, особенно в качестве противовоспалительных агентов или для предотвращения отторжения после введения трансплантата. Дозировка может изменяться в широких пределах и будет зависеть от индивидуальных требований в каждом конкретном случае. Обычно, в случае введения взрослому пациенту подходящая ежедневная дозировка составляет от около 0,1 мг/кг до около 100 мг/кг, предпочтительно от около 0,5 мг/кг до около 5 мг/кг. Ежедневная дозировка может вводиться одной дозой или в раздельных дозах и, кроме того, верхний предел первоначальной дозировки может быть повышен, когда это необходимо.
Наконец, применение соединений формулы I и их вышеупомянутых фармацевтически приемлемых солей для получения лекарственных средств, особенно для лечения или профилактики воспалительных, иммунологических, онкологических, бронхолегочных, дерматологических и сердечно-сосудистых заболеваний, для лечения астмы, заболеваний центральной нервной системы или диабетических осложнений или для предотвращения отторжения после введения трансплантата, также является объектом изобретения.
Соединения формулы I могут использоваться, но не ограничиваясь ими, для лечения любого заболевания или болезненного состояния человека или другого млекопитающего, которое возникает или вызывается избыточной или нерегулируемой выработкой TNF или киназы р38 у такого млекопитающего. Соответственно, настоящее изобретение относится к способу лечения цитокин-медиируемого заболевания, который включает введение эффективного цитокин-модулирующего количества соединения формулы I или его фармацевтически приемлемой соли или таутомера.
Соединения формулы I могут быть полезными, но не ограничиваясь ими, для лечения воспаления у субъекта и для применения в качестве антипиретиков для лечения лихорадки. Соединения по изобретению могут быть полезными для лечения артрита, включая, но не ограничиваясь ими, ревматоидный артрит, спондилоартропатии, подагрический артрит, остеоартрит, системную красную волчанку и ювенильный артрит, и других артритных состояний. Такие соединения могут быть полезными для лечения легочных заболеваний или воспаления легких, включая респираторный дистрессовый синдром у взрослых, легочный саркоидоз, астму, силикоз и хроническую легочную воспалительную болезнь. Соединения также являются полезными для лечения вирусных и бактериальных инфекций, включая сепсис, септический шок, грам-отрицательный сепсис, малярию, менингит, вторичную к инфекции катексию или злокачественность, вторичную к острому синдрому иммунодефицита катексию (СПИД), СПИД, ARC (комплекс, связанный со СПИДом), пневмонию и вирус герпеса. Соединения также являются полезными для лечения заболеваний резорбции кости, такого как остеопороз, эндотоксического шока, синдрома токсического шока, реперфузионной травмы, аутоиммунного заболевания, включающего реакцию хозяина против трансплантата и отторжение трансплантата, сердечно-сосудистых заболеваний, включая атеросклероз, тромбоз, острую сердечную недостаточность и сердечные реперфузионные травмы, почечных реперфузионных травм, заболевания печени и нефрита и миалгий после инфекции.
Соединения также являются полезными для лечения гриппа, рассеянного склероза, рака, диабетов, системной красной волчанки (SLE), кожных состояний, таких как псориаз, экзема, ожоги, дерматит, келоидное образование и образование рубцовой ткани. Соединения по изобретению также могут быть полезны для лечения желудочно-кишечных состояний, таких как болезнь воспаленного кишечника, болезнь Крона, гастрит, синдром раздраженного кишечника и язвенный колит. Соединения также могут быть полезны для лечения офтальмологических заболеваний, таких как ретинит, ретинопатия, ювеит, окулярная фотофобия и острых травм тканей глаза. Соединения по изобретению также могут быть полезны для лечения ангиогенеза, включая неоплазию; метастаз; офтальмологических состояний, таких как отторжение трансплантата роговицы, окулярная неоваскуляризация, ретинальная неоваскуляризация, включая неоваскуляризация после травмы или инфекции, диабетическая ретинопатия, ретролентальная фиброплазия и неососудистая глаукома; язвенных заболеваний, таких как язва желудка; патологических, но незлокачественных состояний, таких как гемангиомы, включая инфантильные гемангиомы, ангиофиброму носоглотки и асосудистый некроз кости; диабетической нефропатии и кардиомиопатии; и заболеваний женской половой системы, таких как эндометриоз. Соединения по изобретение также могут быть полезными для предотвращения выработки циклооксигеназы-2.
Кроме применения для лечения человека, эти соединения также могут использоваться для ветеринарного лечения домашних животных, экзотических животных и животных фермы, включая млекопитающих, грызунов и т.д. Наиболее предпочтительные животные включают лошадей, собак и кошек.
Соединения настоящего изобретения могут также использоваться для совместного лечения, частично или полностью, вместо других обычных противовоспалительных средств, вместе со стероидами, ингибиторами циклооксигеназы-2, NSAID, DMARD, иммуносуппрессорами, ингибиторами 5-липоксигеназы, антагонистами LTB4 и ингибиторами LTA4 гидролазы.
Используемый здесь термин "TNF медиируемое заболевание" обозначает любое и все заболевания и болезненные состояния, в которых играет роль TNF, контролируя TNF или вызывая высвобождение других монокинов с помощью TNF, таких как, но не ограничиваясь ими, IL-1, IL-6 или IL-8. Болезненное состояние, в котором, например, IL-1 является главным компонентом и выработка или действие которого вызывается или начинается в ответ на TNF, следовательно, понимаются под заболеванием, опосредованным TNF.
Используемый здесь термин "р38 медиируемое заболевание" обозначает любое и все заболевания и болезненные состояния, в которых играет роль р38, контролируя р38 или вызывая высвобождение других факторов с помощью р38, таких как, но не ограничиваясь ими, IL-1, IL-6 или IL-8. Болезненное состояние, в котором, например, IL-1 является главным компонентом и выработка или действие которого вызывается или начинается в ответ на р38, следовательно, понимаются под заболеванием, опосредованным р38.
Поскольку TNF-β имеет близкую структурную гомологию с TNF-α (также известным как катектин), и так как они индуцируют похожие биологические отклики и связываются с одним клеточным рецептором, синтез TNF-α, и TNF-β ингибируется соединениями настоящего изобретения, и, следовательно, они вместе здесь обозначены "TNF", если описание не предполагает иное.
ПРИМЕРЫ
Пример 1 (Соединение #12)
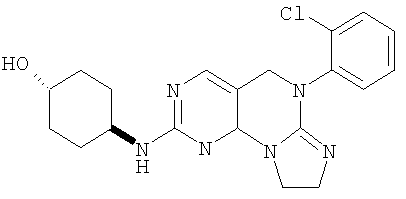
Этот пример иллюстрирует получение соединения формулы I(с).
Стадия 1
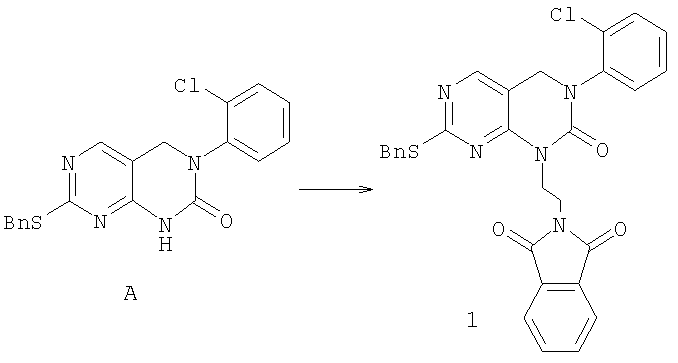
К суспензии 3,0 г (7,84 ммоль) пиримидинона А, 4,12 г (15,7 ммоль) трифенилфосфина и 4,12 г (15,7 ммоль) N-(2-гидроксиэтил)фталимида в 45 мл 1,4-диоксана при 5°С по каплям добавляли раствор 2,73 г (2,5 мл, 15,7 ммоль) диэтилазодикарбоксилата в 10 мл 1,4-диоксана в течение 20 мин через дополнительное горлышко. Суспензию was перемешивали при 5°С в течение 1 часа и затем при комнатной температуре в течение ночи, после чего суспензия превращалась в желтый раствор. Раствор упаривали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя 7,5% этилацетатом в дихлорметане, с получением 3,92 г фталимида 1 (90% выход).
Стадия 2
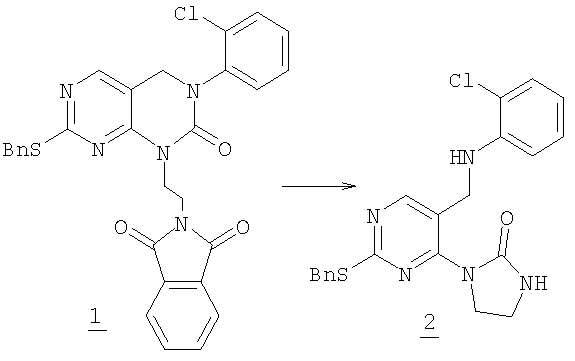
Смесь 1,5 г (2,7 ммоль) фталимида 1 со стадии 1 перемешивали с 2,0 мл гидразингидрата в 100 мл метанола в течение ночи при комнатной температуре. Раствор упаривали в вакууме и остаток разделяли между хлороформом и водой. Раствор хлороформа промывали соляным раствором, высушивали (MgSO4) и упаривали. Продукт очищали колоночной хроматографией на силикагеле, элюируя 10% метанолом в дихлорметане с получением 1,0 г промежуточного соединения 2 (87% выход).
Стадия 3

К раствору 1,56 г (3,7 ммоль) соединения 2 в 30 мл толуола добавляли по каплям 2,3 мл раствора триметилаллюминия (2М в толуоле, 4,6 ммоль). Реакционную смесь кипятили с обратным холодильником в течение одного часа до завершения. Толуол удаляли в вакууме и остаток гасили 100 мл насыщенного водного NH4Cl и 100 г льда. Добавляли 200 мл этилацетата и смесь отфильтровывали. Твердое вещество собирали опять и смешивали с 200 мл этилацетата и 200 мл воды. Его перемешивали в течение 15 минут и отфильтровывали. Эту операцию повторяли еще один раз. Все три фильтрата объединяли и переносили в отдельный сосуд и слои разделяли. Водный слой экстрагировали еще раз этилацетатом. Объединенный раствор этилацетата (около 800 мл) промывали соляным раствором и высушивали (MgSO4) и удаляли в вакууме с получением 1,4 г трициклического соединения 3. (светло-желтое твердое вещество, 94% выход).
Стадия 4
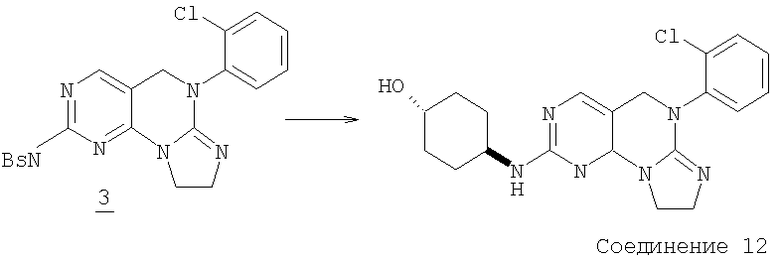
Раствор 0,171 г (0,42 ммоль) трициклического сульфида 3 в 20 мл дихлорметана охлаждали до 5°С. Добавляли 0,094 г (0,42 ммоль) 3-хлорпербензойной кислоты (77% макс.). Смесь перемешивали в течение 30 минут до завершения и раствор выливали в насыщенный водный Na2SO3, экстрагировали дихлорметаном (2×75 мл). Дихлорметан затем промывали холодным насыщенным водным NaHCO3, соляным раствором и высушивали (MgSO4). Растворитель удаляли в вакууме, получая 0,17 г сульфоксида (96% выход). Этот сульфоксид смешивали с 0,21 г (1,8 ммоль) транс-4-аминоциклогексанола и 1 мл NMP нагревали при 120°С в масляной бане в течение 45 минут до окончания реакции. Смесь разбавляли водой и экстрагировали этилацетатом (3×30 мл). Объединенный раствор этилацетата затем промывали водой (3×30), соляным раствором и высушивали (MgSO4). Растворитель удаляли в вакууме и полученный остаток очищали колоночной хроматографией на силикагеле, элюируя МеОН/СН2Cl2/Et3N (1/9/0,4), с получением 19 мг соединения 13. (масс-спектр М+1=399)
Пример 2 (Соединение 13)
Этот пример иллюстрирует получение соединения формулы I(а) следуя методике, описанной на схеме 1.
Стадия 1
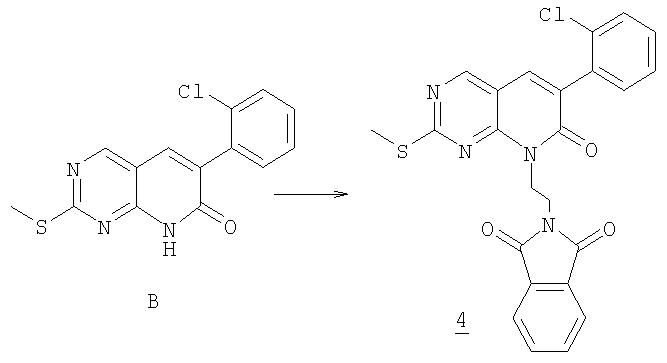
К суспензии 3,0 г (9,88 ммоль) пиридона В, 5,18 г (19,8 ммоль) трифенилфосфина и 3,78 г (19,8 ммоль) N-(2-гидроксиэтил)фталимида в 45 мл 1,4-диоксана при 5°С по каплям добавляли раствор 2,73 г (2,5 мл, 15,7 ммоль) диэтилазодикарбоксилата в 10 мл 1,4-диоксана в течение 20 мин через дополнительное горлышко. Суспензию затем перемешивали при 5°С в течение 1 часа и затем при комнатной температуре в течение ночи. Суспензию отфильтровывали и твердое вещество промывали метанолом, затем эфиром. Получали белое твердое вещество, 2,17 г желаемого продукта. Фильтрат упаривали и очищали у колоночной хроматографией на силикагеле, элюируя 5% этилацетатом в гексане с получением еще 1,2 г желаемого продукта. Общий выход 4 составлял 71%.
Стадия 2
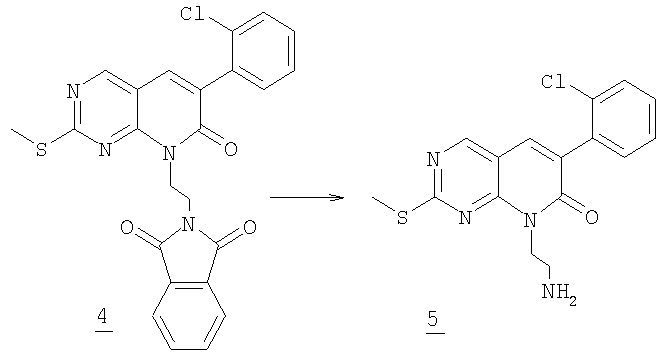
К суспензии 1,2 г (2,5 ммоль) фталимида 4 в 100 мл метанола добавляли 6 мл гидразинмоногидрата. Ее перемешивали при комнатной температуре в течение ночи до превращения суспензии в чистый желтый раствор. Смесь упаривали и остаток разбавляли водой и экстрагировали хлороформом. Органический раствор промывали соляным раствором и высушивали (MgSO4) и остаток очищали колоночной хроматографией на силикагеле, элюируя 10% метанолом в дихлорметане с получением 0,48 г аминоэтилпиридона 5 (55% выход).
Стадия 3
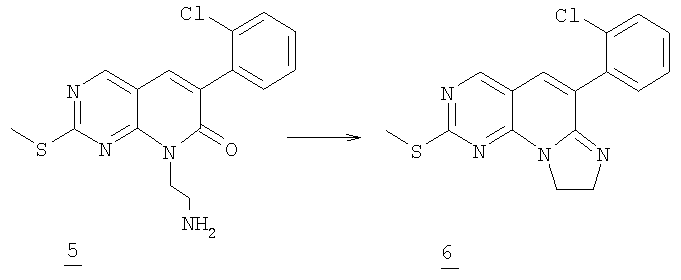
К раствору 0,48 г (1,4 ммоль) соединения 5, в 10 мл толуола по каплям добавляли 0,87 мл раствора триметилаллюминия (2М в толуоле, 1,76 ммоль). Реакционную смесь кипятили с обратным холодильником в течение двух часов до завершения. Толуол удаляли в вакууме и остаток гасили 50 мл насыщенного водного NH4Cl и экстрагировали этилацетатом (3×75 мл). Объединенный органический раствор промывали соляным раствором и высушивали (MgSO4) и упаривали в вакууме с получением 0,36 г трициклического соединения 6 (80% выход).
Стадия 4
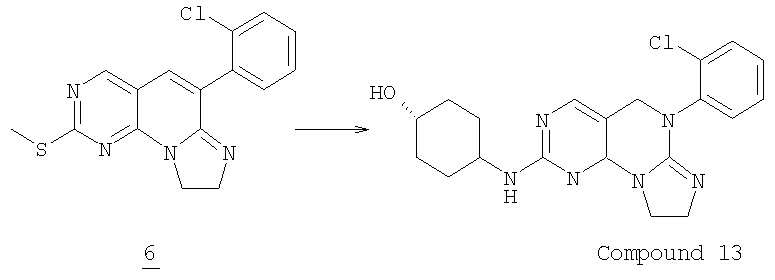
Смесь 0,1 г (0,3 ммоль) соединения 6 и 0,325 г (1 ммоль) транс-А-аминоциклогексанола в 0,5 мл NMP нагревали при 200°С в бане с песком (нагреваемое покрытие) в течение 14 часов. После охлаждения смесь разбавляли водой и экстрагировали этилацетатом (3x50 мл). Объединенный органический раствор промывали водой (3х50 мл), соляным раствором и высушивали (MgSO4). Растворитель удаляли в вакууме и продукт очищали колоночной хроматографией на силикагеле, элюируя градиентом от 5% до 20% метанола в дихлорметане с получением 0,102 г соединения 13. (масс-спектр М+1=396, tпл=169,3-175,8°С).
Пример 3: (Соединение 1)
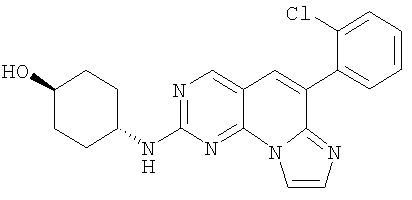
Стадия 1
4-Амино-2-бензилсульфанилпиримидин-5-карбоксилат
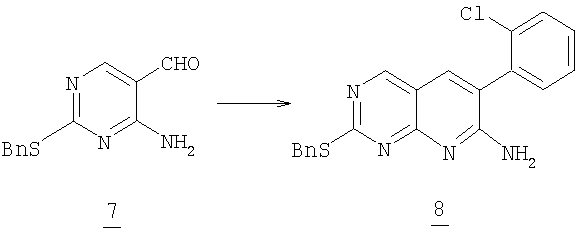
Смесь 5 г (20,4 ммоль) альдегида 7, 3,71 г (24,4 ммоль) 2-хлорбензилцианида и 18 г (130 ммоль) карбоната калия в 50 мл DMPU нагревали при 110°С в масляной бане в течение 3 часов до окончания реакции. Смесь выливали в 600 мл воды и экстрагировали 300 мл этилацетата. Органический растворитель промывали водой (3x250 мл), разбавляли 200 мл гексана, промывали соляным раствором, высушивали (MgSO4) и отфильтровывали через слой силикагеля. Растворитель удаляли в вакууме. Оставшееся твердое вещество перемешивали в 50 мл этилацетата в течение 30 минут. Твердое вещество отфильтровывали и промывали небольшим количеством этилацетата, получая 2,23 г аминопиридопиримидина 8. Фильтрат упаривали и остаток очищали колоночной хроматографией на силикагеле, элюируя 50% этилацетатом в гексане, выделяя дополнительные 0,62 г 2-бензилсульфанил-6-(2-хлорфенил)пиридо[2,3-d]пиримидин-7-иламина 8 (общий выход: 37%).
Стадия 2
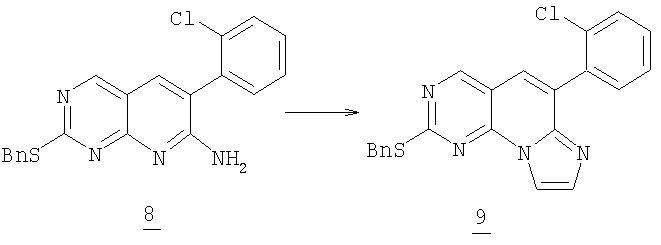
К смеси 3 г (7,9 ммоль) амина 8, 4,8 г (6,6 мл, 47,6 ммоль) триэтиламина, 5 мл воды и 50 мл ацетонитрила добавляли общее количество 4,0 мл (32 ммоль) 1,2-дихлорэтилэтилового эфира в течение 6 часов (1 эквивалент/час в первые два часа и 0,5 эквивалент/час в следующие четыре часа) до окончания реакции. Смесь выдерживали в течение ночи при комнатной температуре, разбавляли водой и экстрагировали этилацетатом (2x100 мл). Органический раствор промывали соляным раствором и высушивали (MgSO4) и упаривали в вакууме. Очистка колоночной хроматографией на силикагеле, элюируя 30% этилацетатом в гексане с получением 2 г сульфида 9 (63% выход).
Стадия 3
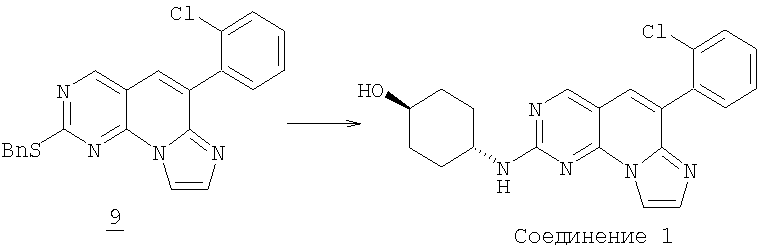
Смесь 0,056 г (0,14 ммоль) соединения 9 и 0,053 г (0,46 ммоль) транс-4-аминоциклогексанола в 1 мл NMP нагревали при 205°С в бане с песком (нагреваемая оболочка) в течение 14 часов. После охлаждения смесь разбавляли водой и экстрагировали этилацетатом (3x50 мл). Объединенный органический раствор промывали водой (3x50 мл), соляным раствором и высушивали (MgSO4). Растворитель удаляли в вакууме с получением 0,39 г желаемого соединения 1 (Соединение 1). (масс-спектр М+1=394,tпл=116-119°С).
Стадия 4
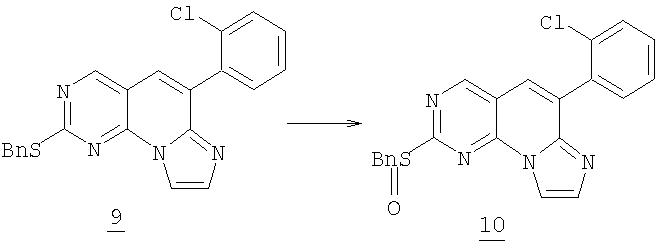
К охлажденному раствору 2 г (5 ммоль) сульфида 9 в 60 мл дихлорметана при 5°С добавляли 1,11 г (5 ммоль) 3-хлорпербензойной кислоты (77% макс.). Реакция заканчивалась через 3 часа и смесь выливали в насыщенный водный NaHCO3 и экстрагировали дихлорметаном. Органический раствор затем промывали 10% водным NaHSO3, соляным раствором и высушивали (MgSO4). После удаления растворителя в вакууме получали 2 г сульфоксида 10. (96% выход). Соответствующий метилсульфоксид (заместитель Bn для Me) получали аналогичным образом. Сульфоксид 10 затем превращали в соединение №1 как описано на стадии 4 выше.
Пример 4 (Соединение 3)
Получение 1-(метилсульфонил)пиперидин-4-амина (15)
Стадия 1
Бензил 1-бензилпиперидин-4-илкарбамат
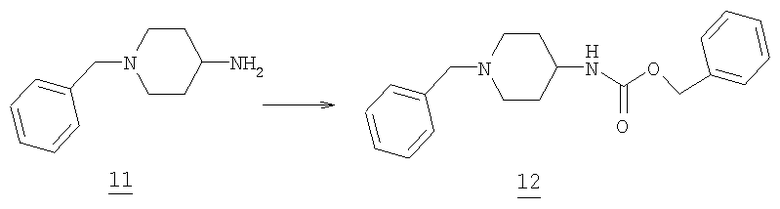
К 0°С раствору 4-амино-1-бензилпиперидина 11 (41,2 г, 216,5 ммоль) и триэтиламина (51,3 мл, 369 ммоль) в 600 мл тетрагидрофурана по каплям добавляли бензилхлорформат (31 мл, 217 ммоль) в течение от 30 до 45 мин с такой скоростью, чтобы температура реакции поддерживалась между 5°С и 10°С. После окончания добавления реакционную смесь нагревали до комнатной температуры и перемешивали в течение 12 часов. Растворитель и примеси удаляли при пониженном давлении. Затем добавляли воду (500 мл) и этилацетат (1,2 L) и реакцию разделяли между двумя фазами. Органический слой промывали насыщенным водного раствором бикарбоната натрия (2Х, 150 мл) и затем высушивали (соляным раствором, MgSO4). Упаривание растворителя приводило к получению желто-коричневой жидкости, которую очищали колоночной хроматографией (SiO2, EtOAc/гексан - от 30/70 до EtOAc - 100) с получением 27,8 г амина 12 в виде белого твердого вещества (масс-спектр М+=324,tпл=79,1-79,6°C).
Стадия 2
Бензилпиперидин-4-илкарбамат
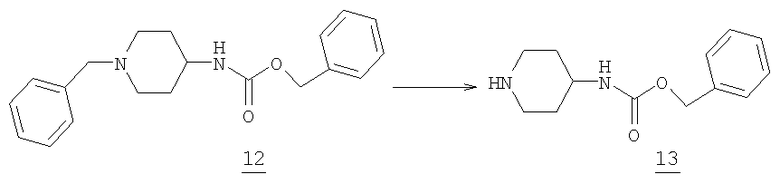
Бензиламин 12 (27,8 г, 85,7 ммоль) растворяли в 400 мл метиленхлорида при комнатной температуре и по каплям добавляли 1-хлорэтилхлорформат (25,4 г, 178 ммоль) в 50 мл метиленхлорида через дополнительное горлышко. После окончания добавления реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель и примеси удаляли при пониженном давлении и добавляли метанол (500 мл). Реакцию кипятили с обратным холодильником при перемешивании в течение 1 часа и затем охлаждали до комнатной температуры. Удаление растворителя реакции упариванием приводило к получению 26,3 г пиперидина 13 в виде белого твердого вещества (масс-спектр М+1=235, tпл=190,7-192,2°С).
Стадия 3
Бензил 1-(метилсульфонил)пиперидин-4-илкарбамат
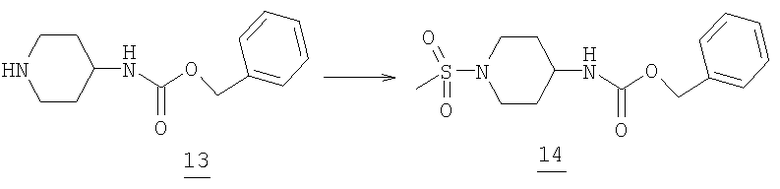
Защищенный пиперидин 13 (10 г, 42,7 ммоль) и триэтиламин (12 мл, 86,7 ммоль) растворяли в 500 мл метиленхлорида при комнатной температуре. По каплям добавляли метансульфонилхлорид (4,3 мл, 55,5 ммоль) в 20 мл метиленхлорида через дополнительное горлышко. После окончания добавления реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель и примеси удаляли при пониженном давлении. Добавляли этилацетат (500 мл) и водный раствор соляной кислоты (0,5М, 350 мл). Реакцию разделяли между двумя фазами и водный слой удаляли. Органический слой промывали опять водным раствором соляной кислоты (0,5М, 2Х, 100 мл) и затем насыщенным водным раствором бикарбоната натрия (3Х, 100 мл). Растворитель реакции затем высушивали (соляным раствором, MgSO4) и упаривали при пониженном давлении с получением 9,2 г метансульфонамида 14 (tпл=148,6-152,8°С).
Стадия 4
1-(Метилсульфонил)пиперидин-4-амин
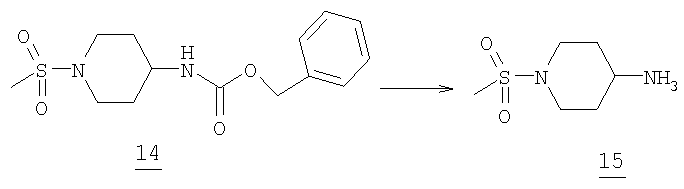
Метансульфонамид 14 (9,2 г, 29,5 ммоль) растворяли в 200 мл тетрагидрофурана при комнатной температуре в 500 мл круглодонной колбе в атмосфере азота. Затем добавляли палладий на угле (10%, 2-3 г) и реакционный сосуд наполняли водородом (3Х). Баллон водорода вводили в реакционную колбу и раствор перемешивали в течение 15 часов (добавляли еще катализатор и баллон с водородом наполняли при необходимости). К реакции добавляли метиленхлорид (100 мл) и отфильтровывали через целит. Упаривание растворителей при пониженном давлении приводило к получению 4,63 г желаемого амина 15 (масс-спектр М+1=179, tпл=65,3-65,7°С).
Стадия 5
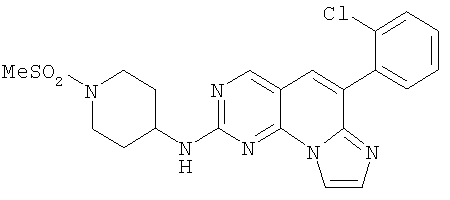
Смесь 0,6 г (1,43 ммоль) сульфоксида 10 из примера 3 и 0,51 г (2,86 ммоль) 1-(метилсульфонил)пиперидин-4-амина 15 в 1 мл NMP нагревали при от 70 до 80°С в течение 1 часа. Охлаждали и разбавляли водой и экстрагировали этилацетатом (2×75 мл). [Исходный реагент 15 и продукт имеют одинаковое значение Rf на ТСХ (5% МеОН/СН2Cl2), масс-спектр продукта реакции: М+1=457)]. Органический раствор промывали водой (2×75 мл), соляным раствором и высушивали (MgSO4) и упаривали. Остаток очищали колоночной хроматографией, элюируя 30% этилацетатом в дихлорметане с получением 0,473 г (73% выход) продукта. Его растворяли в 1 мл дихлорметана и добавляли 1,24 мл 1М HCl/эфира. Суспензию перемешивали в течение 30 минут, отфильтровывали и промывали эфиром с получением 0,47 г соединения 3. (масс-спектр М+1=457, tпл=188-194°С)
Пример 5 (Соединение 2)
Смесь 0,7 г (1,67 ммоль) сульфоксида 10 из примера 3 и 0,84 г (8,4 ммоль) 4-аминотетрагидропирана в 1 мл NMP перемешивали в течение 10 минут. Реакция является экзотермической и заканчивалась через 10 минут. Смесь разбавляли водой и экстрагировали этилацетатом. Органический раствор промывали соляным раствором и высушивали (MgSO4) и упаривали в вакууме. Очистка колоночной хроматографией на силикагеле, элюируя 60% этилацетатом в гексане, приводила к получению 0,46 г (73% выход) продукта. Это твердое вещество в 1 мл дихлорметана смешивали с 1,45 мл 1М HCl/эфира и смесь перемешивали в течение 1 часа. Растворитель удаляли в вакууме с получением 0,48 г соединения 2. (масс-спектр М+1=380, tпл=211-214°С)
Пример 6 (Соединение 5)
Получение 4-аминотетрагидротиопирандиоксида
Стадия 1
4-аминотетрагидротиопиран

Суспензионную смесь 5 г (0,043 моль) тетрагидротиопиран-4-она 16, 29,26 г (0,215 моль) тригидрата ацетата натрия и 14,9 г (0,215 моль) гидрохлорида гидроксиламина в 200 мл этанола кипятили с обратным холодильником в течение шести часов до окончания реакции. Реакционную смесь охлаждали и этанол удаляли при пониженном давлении. Остаток разбавляли 400 мл ледяной воды и экстрагировали этилацетатом, 3×150 мл. Органический раствор промывали соляным раствором, высушивали и отфильтровывали. Фильтрат упаривали при пониженном давлении с получением 5,6 г (количественный выход) тианоноксима 17 (белое твердое вещество).
Стадия 2
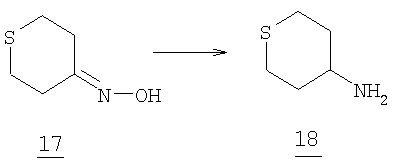
К 76 мл 1М LAH/ТГФ (0,076 моль) по каплям добавляли раствор 2 г (0,015 моль) тианоноксима 17 в 30 мл ТГФ. После окончания добавления смесь перемешивали при кипении с обратным холодильником в течение 7 часов и затем при комнатной температуре в течение ночи. Суспензию охлаждали и по каплям осторожно добавляли 2,9 мл воды, затем 2,9 мл 15% водного гидроксида натрия и затем 8,7 мл воды. Суспензию перемешивали в течение 30 мин, отфильтровывали через целит и промывали 200 мл этилацетата. Фильтрат высушивали, отфильтровывали и упаривали досуха при пониженном давлении (<50°С) с получением 1,62 г (92,3% выход) 4-аминотетрагидротиопирана 18.
Стадия 3
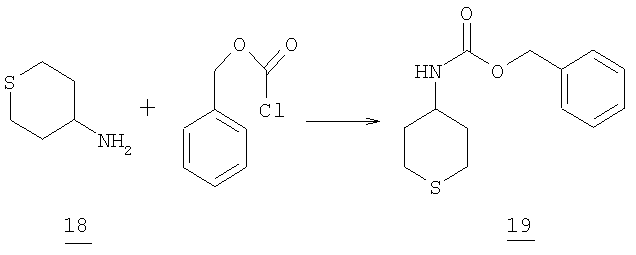
К суспензии of 2,3 г (0,0197 моль) 4-аминотетрагидротиопирана 18 в 25 мл 1N NaOH добавляли по каплям при 5°С 3,14 мл (3,75 г, 0,022 моль) бензилхлорформата. Твердое вещество образовывалось немедленно и после окончания добавления смесь перемешивали при комнатной температуре в течение 1 часа, отфильтровывали и промывали водой. Затем промывали гексаном (50 мл). После высушивания (высушивание на воздухе) получали 4,4 г (88% выход) продукта 19. (масс-спектр М+1=252).
Стадия 4
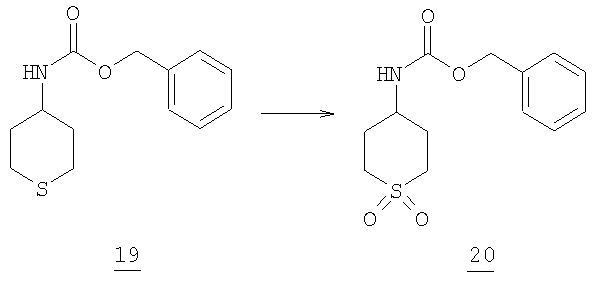
К раствору 40 г (0,159 моль) сульфида 19 в 200 мл дихлорметана при комнатной температуре добавляли небольшими порциями 75 г (0,33 моль) 3-хлорпербензойной кислоты. Реакция является экзотермической. Реакционную смесь перемешивали в течение ночи. Смесь промывали водным насыщенным Na2SO3 (2×150 мл) и затем NaHCO3 (3×150 мл). Органический раствор затем промывали соляным раствором и высушивали над MgSO4. Смесь отфильтровывали и фильтрат упаривали в вакууме с получением 37,5 г сульфона 20 (83% выход).
Стадия 5
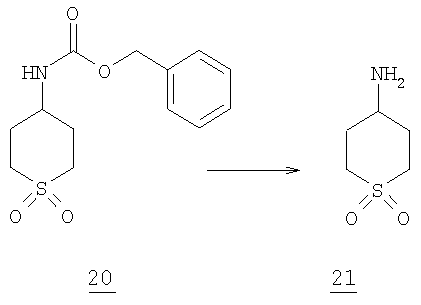
К смеси 10 г (0,026 моль) N-бензоилоксикарбонилсульфона 20 в 150 мл этанола в колбе Парра добавляли 0,5 г гидроксида палладия (20% на угле, катализатор Пильмана). Ее гидрировали в гидрогенизаторе Парра при 50 фунт/дюум2 в течение 15 часов до завершения. Отфильтровывали через целит и упаривали в вакууме с получением 5,0 г свободного амина 21 (68% выход).
Стадия 6
Смесь 1,4 г (0,41 ммоль) сульфоксида 10 и 1,22 г (8,2 ммоль) 4-аминотетрагидротиопирандиоксида 21 в 3 мл NMP нагревали при 70°С в течение 2 часов. Смесь охлаждали, разбавляли водой и экстрагировали этилацетатом (2×75 мл). Объединенный раствор этилацетата промывали водой (3×75 мл), соляным раствором и высушивали (MgSO4) и сырой продукт очищали колоночной хроматографией на силикагеле, элюируя 4% метанолом в дихлорметане с получением 1,5 г желаемого продукта. Его растворяли в 3 мл дихлорметана и по каплям добавляли 4,5 мл 1М HCl/эфира. Суспензию перемешивали в течение 30 минут и твердое вещество отфильтровывали и промывали эфиром с получением 1,86 г (98% выход) соединения 5. (масс-спектр М+1=428, tпл=221,8-226,8°С).
Пример 7 (Соединение 6)
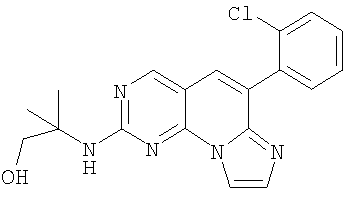
Смесь 0,4 г (1,2 моль) сульфоксида 10 и 0,52 г (5,8 ммоль) 2-амино-2-метил-1-пропанола в 10 мл хлороформа и 0,8 мл NMP нагревали при 70°С в течение 3 часов. Хлороформ удаляли при пониженном давлении. Добавляли 40 мл смеси метанол/эфир (1:1). Суспензию перемешивали в течение 10 минут и отфильтровывали. Твердое вещество промывали эфиром с получением 0,258 г (58,5% выход) желаемого продукта. Его растворяли в 15 мл 10% метанола в дихлорметане и добавляли 0,85 мл 1М HCl/эфира. Смесь перемешивали в течение 30 минут и упаривали. Добавляли эфир и твердое вещество отфильтровывали и промывали эфиром с получением 0,295 г соединения 6. (масс-спектр М+1=368, tпл=190,8-194,3°С)
Пример 8 (Соединение 7)
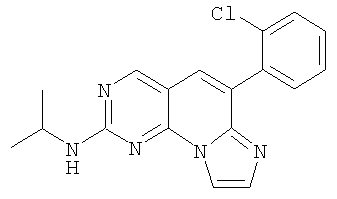
Стеклянный закрывающийся (тефлоновая крышка) реактор загружали 0,4 г (1,2 ммоль) сульфоксида 10. Его насыщали азотом в течение 2 минут, пока реакционный сосуд охлаждали ледяной водной баней. Добавляли 5 мл изопропиламина и сосуд закрывали. Красный раствор нагревали при 64°С в масляной бане в течение 2 часов до окончания реакции. Смесь охлаждали и реакционный сосуд промывали дихлорметаном. Растворитель удаляли в вакууме и образец очищали колоночной хроматографией на силикагеле, элюируя градиентом от 5 до 10% этилацетата в дихлорметане с получением 0,34 г (87% выход) продукта. Это светло-желтое твердое вещество растворяли в минимальном количестве дихлорметана и добавляли 1,2 мл 1М HCl/эфира, затем 5 мл эфира. Желтую суспензию перемешивали в течение 1 часа и отфильтровывали и промывали эфиром с получением 0,34 г соединения 7. (Масс-спектр М+1=338, tпл=210-212,5°С)
Пример 9 (Соединение 8)
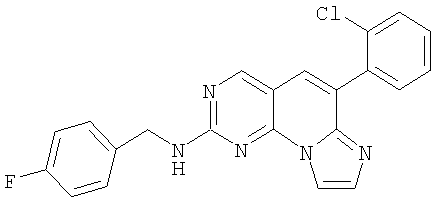
Смесь 0,4 г (1,2 ммоль) сульфоксида 10 и 0,29 г (2,33 ммоль) 4-фторбензиламина в 5 мл хлороформа нагревали при 60°С в масляной бане в течение 1,5 часов до окончания реакции. Продукт очищали непосредственно колоночной хроматографией, элюируя градиентом от 5 до 10% этилацетата в дихлорметане с получением 0,42 г (89% выход) продукта. Желтое твердое вещество растворяли в минимальном количестве дихлорметана и добавляли 1,25 мл 1М HCl/эфира. Смесь перемешивали в течение 30 минут и растворитель удаляли в вакууме. Добавляли эфир и твердое вещество отфильтровывали и промывали эфиром с получением 0,469 г соединения 8. (масс-спектр М+1=404, tпл=178-180°С)
ПРИМЕР 10 (Соединение 9)

Смесь 0,4 г (1,2 ммоль) сульфоксида 10 и 0,36 г (2,4 ммоль) 2-аминоэтилметилсульфона в 3 мл NMP нагревали при 65°С в течение 30 минут. [2-аминометилсульфон получали реакцией 4-метоксибензиламина с метилвинилсульфоном в присутствии триэтиламина в ТГФ при комнатной температуре, N-бензильную группу удаляли при перемешивании в метаноле в присутствии формата аммония и 10% Pd-C]. Смесь охлаждали, разбавляли водой и этилацетатом. Полученное твердое вещество отфильтровывали, промывали этилацетатом и затем 40 мл метанола с получением 0,34 г (72,6% выход) продукта. Его растворяли в 5 мл 10% метанола в дихлорметане и добавляли 1 мл 1М HCl/эфира. Перемешивание продолжали в течение 30 минут и растворитель удаляли в вакууме. Добавляли эфир для растирания и полученное твердое вещество отфильтровывали и промывали дополнительными 10 мл эфира с получением 0,374 г соединения 9. (масс-спектр М+1=402, tпл=172,2-176°С)
Пример 11 (Соединение 10)
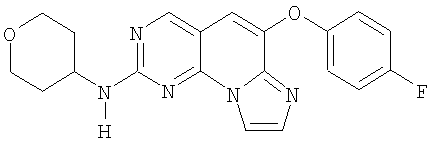
Стадия 1
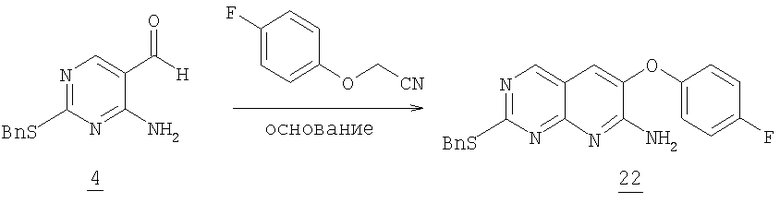
Смесь 2,48 г (10,1 ммоль) альдегида 4 из примера 2, 1,83 г (12 ммоль) 4-фторфеноксиацетонитрила (полученного из 4-фторфенола и йодацетонитрила в K2CO3/ДМФА) и 7 г (50,6 ммоль) карбоната калия в 30 мл NMP нагревали при 110°С в течение 4 часов до окончания реакции. Смесь выливали в 200 мл воды и экстрагировали этилацетатом (3×125 мл). Органический растворитель промывали водой (3×150 мл), соляным раствором, разбавляли 200 мл гексана, высушивали (MgSO4) и отфильтровывали через слой силикагеля. Растворитель удаляли в вакууме. Полученное твердое вещество очищали колоночной хроматографией на силикагеле, элюируя градиентом от 5% до 20% этилацетата в дихлорметане с получением 1,3 г аминопиридопиримидина 22, (37% выход). (масс-спектр М+1=379, tпл=186,2-192,9°С)
Стадия 2
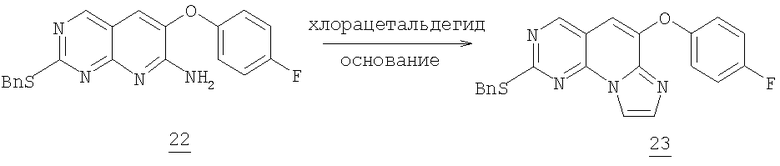 К смеси 1,3 г (3,4 ммоль) амина 22, 1,54 г (2,1 мл, 15,3 ммоль) триэтиламина, 3 мл воды и 30 мл ацетонитрила добавляли общее количество 2,4 мл (19,6 ммоль, 5,8 екв.) 1,2-дихлорэтилэтилового эфира в течение 6 часов (1 эквивалент/час первые два часа и 0,5 эквивалент/час следующие четыре часа) до окончания реакции. Смесь оставляли в течение ночи при комнатной температуре, разбавляли водой и экстрагировали этилацетатом (2×100 мл). Органический раствор промывали соляным раствором и высушивали (MgSO4) и упаривали в вакууме. Очистка колоночной хроматографией на силикагеле, элюируя 10% этилацетатом в гексане с получением 0,78 г (57% выход) фенокситрициклического имидазола 23 (57% выход).
К смеси 1,3 г (3,4 ммоль) амина 22, 1,54 г (2,1 мл, 15,3 ммоль) триэтиламина, 3 мл воды и 30 мл ацетонитрила добавляли общее количество 2,4 мл (19,6 ммоль, 5,8 екв.) 1,2-дихлорэтилэтилового эфира в течение 6 часов (1 эквивалент/час первые два часа и 0,5 эквивалент/час следующие четыре часа) до окончания реакции. Смесь оставляли в течение ночи при комнатной температуре, разбавляли водой и экстрагировали этилацетатом (2×100 мл). Органический раствор промывали соляным раствором и высушивали (MgSO4) и упаривали в вакууме. Очистка колоночной хроматографией на силикагеле, элюируя 10% этилацетатом в гексане с получением 0,78 г (57% выход) фенокситрициклического имидазола 23 (57% выход).
Стадия 3
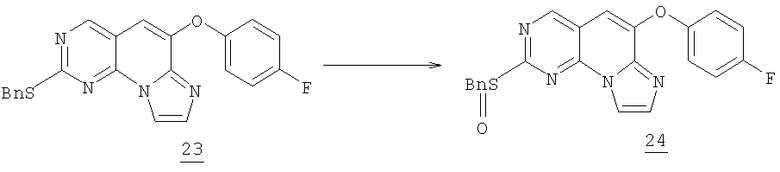
К охлажденному раствору 0,77 г (1,92 ммоль) сульфида 23 в 30 мл дихлорметана при 5°С добавляли 0,43 г (1,92 ммоль) 3-хлорпербензойной кислоты (77% макс.). Реакция заканчивалась через 1 час и смесь выливали в холодный насыщенный водный NaHCO3 и экстрагировали дихлорметаном. Органический раствор промывали соляным раствором и высушивали (MgSO4). После удаления растворителя в вакууме получали 0,763 г сульфоксида (95% выход).
Стадия 4
Смесь 0,4 г (0,96 ммоль) сульфоксида 24 и 0,48 г (4,78 ммоль) 4-аминотетрагидропирана в 1 мл NMP нагревали при от 60 до 70°С в течение 30 минут. Смесь разбавляли водой и экстрагировали этилацетатом. Органический раствор промывали соляным раствором и высушивали (MgSO4) и упаривали в вакууме. Очистка колоночной хроматографией на силикагеле, элюируя градиентом от 10 до 30% этилацетата в дихлорметане для элюирования неполярных примесей и в конце 2,5% метанолом в дихлорметане для элюирования продукта приводила к получению 0,210 г (58% выход). Это твердое вещество в 1 мл дихлорметана смешивали с 0,83 мл 1М HCl/эфира и смесь перемешивали в течение 1 часа, разбавляли эфиром и отфильтровывали с получением 0,21 г соединения 10. (масс-спектр М+1=380, tпл=198-206°С)
Пример 12 (Соединение 11)
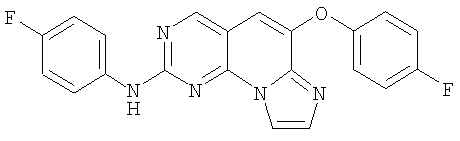
К 0,11 г (1,0 ммоль) 4-фторанилина в 5 мл дихлорметана при 5°С по каплям добавляли 0,5 мл (1,0 ммоль) реагента триметилаллюминия (2М/толуол). После добавления перемешивание продолжали в течение 10 минут. Этот раствор затем добавляли пипеткой к раствору 0,355 г (0,85 ммоль) сульфоксида из примера 10 в 10 мл дихлорметана при 5°С. Смесь затем перемешивали в течение 17 часов и затем кипятили с обратным холодильником в течение 3 часов. Его выливали в насыщенный водный раствор NH4Cl. Смесь отфильтровывали и фильтрат переносили в отдельный сосуд, где слои разделяли. Водный слой экстрагировали еще раз дихлорметаном и объединенный органический раствор промывали соляным раствором и высушивали (соляным раствором, MgSO4). После удаления растворителя в вакууме остаток очищали колоночной хроматографией на силикагеле, элюируя 10% этилацетатом в дихлорметане с получением 0,16 г желтого твердого вещества. Его смешивали с 5 мл 20% метанола в дихлорметане и добавляли 0,49 мл 1М HCl/эфира. Перемешивали в течение 30 минут и растворитель упаривали досуха. Добавляли 10 мл эфира и желтую суспензию перемешивали в течение 10 минут, отфильтровывали и промывали дополнительным количеством эфира с получением 0,145 г соединения 11. (масс-спектр М+1=390, tпл=289,3-291,1°С)
Среднему специалисту в данной области техники ясно, что к вышеприведенным схемам применимы некоторые модификации и они входят в объем настоящего изобретения. Например, некоторые стадии могут включать применение защитных групп для функциональных групп, которые не совместимы с конкретными условиями реакции.
Пример 13
Далее представлены примерные фармацевтические составы, содержащие соединение формулы (I).
Состав таблетки
Следующие ингредиенты смешивали до однородности и спрессовывали в таблетки с одним делением.
Состав капсулы
Следующие ингредиенты смешивали до однородности и помещали в твердую желатиновую капсулу.
Состав суспензии
Следующие ингредиенты смешивали с получением суспензии для орального введения.
Инъекционный состав
Следующие ингредиенты смешивали с получением инъекционного состава.
Все указанные выше ингредиенты, за исключением воды, объединяли и нагревали при 60-70°С с перемешиванием. Затем добавляли достаточное количество воды при 60°С при активном перемешивании с получением эмульсии ингредиентов и затем добавляли воду до 100 г.
Состав свечей
Свечи общим весом 2,5 г получали смешиванием соединения по изобретению с Witepsol® H-15 (триглицериды насыщенных растительных жирных кислот; Riches-Nelson, Inc., New York), и имеют следующий состав:
Пример 14
Ингибирование р-38 (MAP) киназы. Анализ In Vitro
Ингибирующая активность р-38 MAP киназы соединений настоящего изобретения in vitro показана измерением переноса γ-фосфата из γ-33Р-АТФ р-38 киназой к основному белку миелина (МВР), используя небольшие модификации способа, описанного в Ahn, N. G.; и др. J. of Biol. Chem. Vol.266 (7), 4220-4227, (1991).
Фосфорилированную форму рекомбинантной р38 MAP киназы экспрессировали с SEK-1 и МЕКК в Е. Coli (см., Khokhlatchev, А. и др. J. of Biol. Chem. Vol.272 (17), 11057-11062, (1997) и затем очищали аффинной хроматографией, используя колонку Nickel.
Фосфорилированную р38 MAP киназу разбавляли в киназном буфере (20 мМ 3-(N-морфолино)пропансульфоновая кислота, рН 7,2, 25 мМ β-глицеринфосфат, 5 мМ этиленгликольбис(бета-аминоэтиловый эфир)-N,N,N',N'-тетрауксусная кислота, 1 мМ ванадата натрия, 1 мМ дитиотреитол, 40 мМ хлорид магния). Тестируемое соединение растворяли в ДМСО или только ДМСО (контроль) добавляли и образцы инкубировали в течение 10 мин при 30°С. Киназную реакцию инициировали добавлением смеси субстрата, содержащей МВР и γ-33Р-АТФ. После инкубирования в течение дополнительных 20 мин при 30°С реакцию останавливали добавлением 0,75% фосфорной кислоты. Фосфорилированный МВР затем отделяли от оставшейся γ-33Р-АТФ с помощью фосфоцеллюлозной мембраны (Millipore, Bedfrod, MA) и определяли количество с помощью стинтилляционного счетчика (Packard, Meriden, CT).
р-38 ингибирующие активности (выраженные как IC50, концентрация, вызывающая 50% ингибирование анализируемого р-38 фермента) соединений, представленные в таблице 1 в описании, составляют между 0,001 мкМ и 10 мкМ. Например, в таблице 2 ниже показаны некоторые значения ингибирования р38, измеренные с помощью анализа примера 14:
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ IAP | 2008 |
|
RU2491276C2 |
| БИЦИКЛИЧЕСКИЕ ПИРИДИНЫ И ПИРИМИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ Р38 | 2003 |
|
RU2301233C2 |
| 6-АЛКОКСИПИРИДОПИРИМИДИНЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АКТИВНОСТЬЮ ИНГИБИТОРОВ КИНАЗЫ МАР р38 | 2003 |
|
RU2324695C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 1999 |
|
RU2270833C2 |
| ГЕТЕРОАЛКИЛАМИНОЗАМЕЩЕННЫЕ БИЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ р38 | 2000 |
|
RU2265606C2 |
| ЗАМЕЩЕННЫЕ ПИПЕРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ | 2002 |
|
RU2300533C2 |
| ИНГИБИТОРЫ IAP | 2006 |
|
RU2451025C2 |
| ПРОИЗВОДНЫЕ 1, 2, 4-ТРИАЗИН-6-ОНА, ИНГИБИРУЮЩИЕ ВИЧ | 2005 |
|
RU2401833C2 |
| ПРОИЗВОДНЫЕ ПИРИДОПИРИМИДИНОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2269527C2 |
| БЕНЗОКСАЗИНИЛ-АМИДОЦИКЛОПЕНТИЛ-ГЕТЕРОЦИКЛИЧЕСКИЕ МОДУЛЯТОРЫ ХЕМОКИНОВЫХ РЕЦЕПТОРОВ | 2004 |
|
RU2301802C2 |
Описываются новые имидазоконденсированные соединения общей формулы I
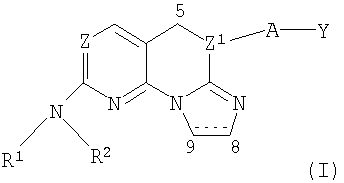
где Z представляет собой N;
Z1 представляет собой N, когда связь между атомами С5 и Z1 является простой связью, и Z1 представляет собой С, когда связь между С5 и Z1 является двойной связью;
R1 представляет собой водород;
R2 представляет собой С1-6алкил, С1-6гидроксиалкил, фенил-С1-4алкил, замещенный галогеном, (С1-4алкил)SO2 С1-6алкил, С5-6циклоалкил, возможно замещенный гидрокси, фенил, замещенный галогеном, гетероциклил, возможно замещенный и выбранный из группы, состоящей из тетрагидропиранила, (N-метилсульфонил)пиперидинила или тетрагидро-1,1-диоксид-2-Н-тиопиранила;
А отсутствует или представляет собой -O-; связь между атомами С5 и Z1 является одинарной или двойной связью; связь между атомами С8 и С9 является одинарной или двойной связью, и Y представляет собой фенил, замещенный галогеном; или их фармацевтически приемлемые соли, обладающие ингибирующей активностью в отношении р-38 MAP киназы, и фармацевтическая композиция, их содержащая. Описываемые соединения могут применяться, например, при лечении и/или профилактике таких заболеваний как ревматоидный артрит, лихорадка или снижение резорбции кости. 2 н. и 14 з.п. ф-лы, 2 табл.
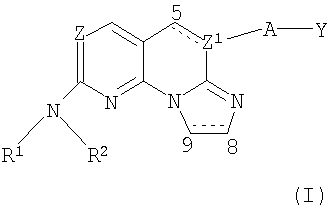
где Z представляет собой N;
Z1 представляет собой N, когда связь между атомами С5 и Z1 является простой связью, и Z1 представляет собой С, когда связь между С5 и Z1 является двойной связью;
R1 представляет собой водород;
R2 представляет собой С1-6алкил, С1-6гидроксиалкил, фенил-С1-4алкил, замещенный галогеном, (С1-4алкил)SO2 С1-6алкил, С5-6циклоалкил, возможно замещенный гидрокси, фенил, замещенный галогеном, гетероциклил, возможно замещенный и выбранный из группы, состоящей из тетрагидропиранила, (N-метилсульфонил)пиперидинила или тетрагидро-1,1-диоксид-2-Н-тиопиранила;
А отсутствует или представляет собой -O-;
связь между атомами С5 и Z1 является одинарной или двойной связью;
связь между атомами С8 и С9 является одинарной или двойной связью, и
Y представляет собой фенил, замещенный галогеном; или
их фармацевтически приемлемые соли.
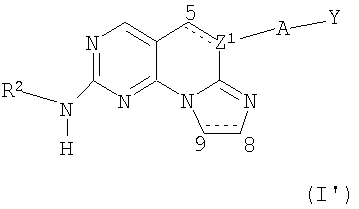
где Z1 представляет собой N, когда связь между атомами С5 и Z1 является простой связью, и Z1 представляет собой С, когда связь между С5 и Z1 является двойной связью;
R2 выбран из фенил-С1-4алкила, замещенного галогеном, C5-6циклоалкила, возможно замещенного гидрокси, гетероциклила, возможно замещенного и выбранного из группы, состоящей из тетрагидропиранила, (N-метилсульфонил)пиперидинила или тетрагидро-1,1-диоксид-2-Н-тиопиранила, фенила, замещенного галогеном, и С1-4алкила, С1-4гидроксиалкила или (С1-4алкил)SO2 С1-4алкила;
А отсутствует или представляет собой -O-;
Y представляет собой фенил, замещенный галогеном; или
их фармацевтически приемлемые соли.

где Y представляет собой фенил, замещенный галогеном;
Z представляет собой N;
А отсутствует или представляет собой -O-;
R1 представляет собой водород;
R2 представляет собой С1-6алкил, С1-6гидроксиалкил, C5-6циклоалкил, возможно замещенный гидрокси, гетероциклил, возможно замещенный и выбранный из группы, состоящей из тетрагидропиранила, (N-метилсульфонил)пиперидинила или тетрагидро-1,1-диоксид-2-Н-тиопиранила, фенил, замещенный галогеном, или фенил-С1-4алкил, замещенный галогеном; и
связь между С8 и С9 является одинарной или двойной связью.
R2 представляет собой 4-тетрагидропиранил и Y представляет собой 4-фторфенил; или
R2 представляет собой 4-фторфенил и Y представляет собой 4-фторфенил.
R2 представляет собой 4-гидроксициклогексил и Y представляет собой 2-хлорфенил;
R2 представляет собой 4-тетрагидропиранил и Y представляет собой 2-хлорфенил;
R2 представляет собой 4-(N-метилсульфонилпиперидинил) и Y представляет собой 2-хлорфенил;
R2 представляет собой циклопентил и Y представляет собой 2-хлорфенил;
R2 представляет собой 4-тетрагидро-1,1-диоксид-2-Н-тиопиранил и Y представляет собой 2-хлорфенил;
R2 представляет собой изопропил и Y представляет собой 2-хлорфенил, или
R2 представляет собой 4-фторбензил и Y представляет собой 2-хлорфенил.
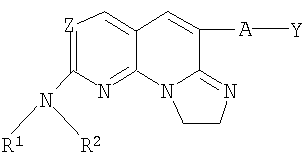
где Z, R1, R2, А и Y имеют значения, определенные в п.1.
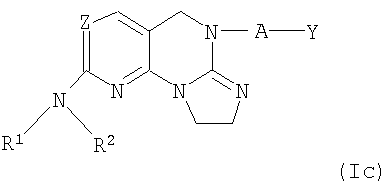
где Z, R1, R2, А и Y имеют значения, определенные в п.1.
| Сорбционный датчик сопротивления к влагомеру | 1959 |
|
SU129042A1 |
| ОПТИЧЕСКОЕ СТЕКЛО | 0 |
|
SU218380A1 |
| ПИРИДО[3,2-Е]ПИРАЗИНОНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРЕПАРАТ НА ИХ ОСНОВЕ И СПОСОБ ПОЛУЧЕНИЯ ПРЕПАРАТА | 1996 |
|
RU2143433C1 |

