Данное изобретение относится к соединениям, которые полезны для лечения воспаления и которые содержат пиперазиновые или пиперидиновые группы, связанные с положением 5 или 6 индола, бензимидазола или бензотриазола. Более конкретно, данное изобретение относится к новым ортозамещенным индолам и N-замещенным индолам, а также к способам лечения сердечных и почечных нарушений с использованием этих соединений и их производных.
Уровень техники
Было установлено, что большое число хронических и острых состояний связано с нарушением нормального хода (пертурбацией) воспалительной реакции. В этой реакции участвует большое количество цитокинов, в том числе IL-1, IL-6, IL-8 и TNF. По-видимому, активность этих цитокинов в регулировании воспаления основывается по меньшей мере частично на активации фермента в пути передачи клеточных сигналов, члена семейства МАР-киназ, обычно известного как р38 и альтернативно известного как CSBP и RK. Эта киназа активируется двойным фосфорилированием после стимуляции физико-химическим стрессом, обработкой липополисахаридами или провоспалительными цитокинами, такими как IL-1 и TNF. Таким образом, ингибиторы киназной активности р38 являются полезными противовоспалительными агентами.
В заявках РСТ WO98/28292, WO98/06715, WO98/07425 и WO96/40143, которые включены сюда в качестве ссылок, описана взаимосвязь ингибиторов киназы р38 с различными патологическими состояниями. Как упоминается в этих заявках, ингибиторы киназы р38 полезны для лечения различных заболеваний, связанных с хроническим воспалением. Эти заявки перечисляют ревматоидный артрит, ревматоидный спондилит, остеоартрит, подагрический артрит и другие относящиеся к артриту состояния, сепсис, септический шок, эндотоксиновый бактериально-токсический шок, сепсис, вызываемый грамотрицательными бактериями, синдром токсического шока, астму, респираторный дистресс-синдром взрослых, удар (сердечную и мозговую недостаточность), реперфузионное повреждение, повреждения ЦНС, такие как невральная травма и ишемия, псориаз, рестеноз, церебральную малярию, хроническое воспалительное заболевание легких, силикоз, легочный саркоз (патологическое разрастание мягких тканей легких), заболевания с резорбций (рассасыванием) костей, такие как остеопороз, реакцию "трансплантат против хозяина", болезнь Крона, язвенный колит, в том числе воспалительное заболевание пищеварительного тракта (IBD) и pyresis*.
В вышеуказанных заявках РСТ описаны соединения, которые являются ингибиторами киназы р38, которые, как указывается, полезны для лечения этих патологических состояний. Эти соединения являются либо имидазолами, либо индолами, замещенными в положении 3 или 4 пиперазиновым или пиперидиновым кольцом, связанным через карбоксамидную связь. Дополнительные соединения, которые являются конъюгатами пиперазинов с индолами, описаны в качестве инсектицидов в WO97/26252, также включенной сюда в качестве ссылки.
Описание изобретения
Данное изобретение относится к соединениям, полезным для лечения воспаления в общем, в том числе специфических состояний, таких как состояния, описанные в разделе "Уровень техники" выше. Было обнаружено, что некоторые новые соединения ингибируют киназу р38, в частности киназу р38 α, и, следовательно, полезны для лечения заболеваний, опосредованных этим ферментом. Соединения данного изобретения являются соединениями формулы:

предпочтительно соединениями формул:


и их фармацевтически приемлемыми солями,
где каждый из Z1 и Z2 обозначает независимо CR4 или N;
каждый R4 обозначает независимо Н или C1-С6алкил или арил, причем каждый указанный алкил и арил необязательно включает один или несколько гетероатомов, выбранных из О, S и N, и необязательно замещен одним или несколькими заместителями, выбранными из галогена, OR, SR, NR2, RCO, COOR, CONR2, OOCR или NROCR, где R обозначает Н или C1-С6алкил, или одним или несколькими CN или =O, или одним или несколькими алифатическими или ароматическими 5- или 6-членными кольцами, необязательно содержащими 1-2 гетероатома;
R1 обозначает

где X1 обозначает СО или его изостер;
m равно 0 или 1;
Y обозначает необязательно замещенный алкил, необязательно замещенный арил или необязательно замещенный арилалкил или два Y, взятые вместе, могут образовывать С2-С3алкиленовый мостик;
n равно 0, 1 или 2;
Z3 обозначает СН или N;
X2 обозначает СН, СН2 или их изостер и
Ar состоит из одной или двух фенильных групп, связанных непосредственно с X2, необязательно замещенных галогеном, нитро, C1-С6алкилом, С2-С6алкенилом, С2-С6алкинилом, CN или CF3 или RCO, COOR, CONR2, NR2, OR, SR, OOCR или NROCR, где R обозначает Н или C1-С6алкил,
или фенилом, который необязательно замещен вышеуказанными заместителями;
R2 обозначает Н или обозначает C1-С6алкил или арил, причем каждый указанный алкил или арил необязательно включает один или несколько гетероатомов, которые представляют собой О, S или N, и необязательно замещен одним или несколькими заместителями, выбранными из галогена, OR, SR, NR2, RCO, COOR, CONR2, OOCR или NROCR, где R обозначает Н или C1-С6алкил, или одним или несколькими CN или =O, или одним или несколькими алифатическими или ароматическими 5- или 6-членными кольцами, необязательно содержащими 1-2 гетероатома;
R3 обозначает Н, галоген, NO2, C1-С6алкил, С2-С6алкенил, С2-С6алкинил, CN, OR, SR, NR2, RCO, COOR, CONR2, OOCR или NROCR, где R обозначает Н или C1-С6алкил.
Таким образом, в одном аспекте, данное изобретение относится к соединениям формул, представленных выше. В других аспектах, данное изобретение относится к способам получения этих соединений, к содержащим их фармацевтическим композициям и к способам лечения воспаления с использованием этих соединений. Данное изобретение относится также к лечению состояний, связанных с сердечной недостаточностью, с использованием соединений данного изобретения и других описанных здесь соединений.
Способы осуществления данного изобретения
Соединения формул 1-4 полезны в различных физиологических применениях, дополнительно описанных ниже. Предпочтительные варианты соединений включают соединения, в которых оба Z1 и Z2 представляют собой СН или в которых Z1 представляет собой CR4, а Z2 представляет собой СН; таким образом, предпочтительные соединения данного изобретения включают производные индола. Особенно предпочтительными заместителями в положении 3 являются заместители, связанные через карбоксамидные связи. Таким образом, некоторыми предпочтительными вариантами R4 является формула RNHCO-, где R представляет собой алкил или замещенный алкил.
Обычно, заместители на азотсодержащей части индольно-го, бензимидазольного или бензотриазольного колец предназначены для повышения растворимости. Таким образом, в типичном случае, заместители R2 и R4 являются полярными или содержат полярные группы.
В других предпочтительных вариантах, заместители соединений данного изобретения, являются заместителями, приведенными ниже.
Для R1:
X1 обозначает СО или его изостер. Таким образом, кроме СО, X1 может являться СН2, SO, SO2 или СНОН. Предпочтительным является СО.
Z3 обозначает СН или N; Z3=СН является предпочтительным.
Обычно m равно 1; однако, в некоторых соединениях данного изобретения m может быть равно 0; таким образом, этот заместитель является пятичленным кольцом.
X2 обозначает СН2, если Ar состоит из одной фенильной группы, или СН, если Ar состоит из двух фенильных групп, или может быть их изостером. Таким образом, для подходящих вариантов Ar, X2 может быть любой из альтернатив, описанных выше для X1.
Фенильные группы, представленные Ar, могут быть необязательно замещены заместителями, включающими C1-С6алкил, галоген, RCO, COOR, CONR2, OR, SR, NR2, OOCR, NROCR, NO2, CN или CF3, где R обозначает Н или С1-С6алкил. Фенильные группы могут быть также замещены дополнительным фенильным остатком, предпочтительно в положении 4. Дополнительный фенильный остаток может быть замещен заместителями, указанными выше. Дополнительный фенил может быть замещен во всех пяти положениях, но предпочтительно менее, предпочтительно в 1-2 положениях, или вообще является незамещенным. Предпочтительные заместители включают С1-С6алкил, OR, NR2 и галоген, в частности, галоген и ОСН3. Заместители могут занимать все пять положений фенильного заместителя, предпочтительно 1-2 положения, или этот фенил может быть незамещенным.
n может быть равно 0, 1 или 2 и предпочтительно равно 0. Однако, когда n равно 1, Y присутствует и может быть алкилом, арилалкилом или арилом, которые могут быть необязательно замещены заместителями, представленными выше для Ar. Когда n равно 2, обе группы Y вместе могут составлять алкиленовый мостик. Предпочтительным является этиленовый мостик. Предпочтительные значения Y, когда n равно 1, включают незамещенный алкил и незамещенный арилалкил.
Для R2:
R2 обозначает предпочтительно Н, но может быть также подходящим заместителем. Такие заместители представляют собой в типичном случае и предпочтительно алкил или замещенный алкил. Алкил или замещенный алкил могут необязательно включать один или несколько гетероатомов, которыми могут быть О, N или S, предпочтительно N или О. Возможные заместители алкильной группы приведены выше; предпочтительные заместители включают OR, где R обозначает Н или C1-С6алкил и =O. Предпочтительными заместителями алкильной группы являются также циклические группы, такие как пиперазин, пиридин, пиперидин, фенил и т.п. Предпочтительно, алкильные группы R2 содержат 0, 1 или 2 заместителя. Предпочтительными значениями R2 являются -(CO)O-Y', где Y' обозначает, например, -(CH2)nNR2, где n является целым числом от 0 до 6, а R имеет указанные выше значения; или Y' обозначает, например, алифатическую или ароматическую циклическую систему, такую как

Дополнительные иллюстративные значения R2 включают в себя никотиноил и его изомеры, акрилоил и заместители общей формулы Y'(CH2)nNH(CH2)nCHOH(CH2)n-, где Y' обозначает характерный для данного типа радикалов заместитель, такой как необязательно замещенный алкил, пиперазинил, пиперидинил, циклогексил, фенил или метокси, и подобные и, где каждое n независимо является целым числом от 1 до 3. Y' является очень вариабельным и может в общем включать любую невлияющую часть молекулы. Дополнительные значения включают Y'NH(CH2)n-CO, где Y' и n имеют указанные выше значения; а также Y'(CH2)nNH(CH2)nCO, где Y' и n имеют указанные выше значения; и Y'(СН2)nСО и Y'(CH2)nNHCO, где Y' и n имеют указанные выше значения; и R2N(CH2)n-, где R представляет собой C1-С6алкил и n является целым числом 1-3.
Для R3:
Хотя R3 может быть Н, включены и другие значения, и они могут быть предпочтительными. Они включают галоген, OR, NR2 и C1-С6алкил, как особенно желательные.
В вариантах, где Z1 или Z2, предпочтительно Z1, представляет собой CR4, где R4 является отличным от Н, предпочтительные значения R4 включают R2N(СН2)n-, где каждый R независимо обозначает C1-С6алкил или Н и n является целым числом 1-6; или Y'(-CH2)n-, где Y' имеет указанные выше значения и n является целым числом 1-6; или Y'NHCO; или R2NCO, где заместители R2, взятые вместе, образуют кольцо, которое может быть замещено, предпочтительно алкилом, арилалкилом и подобными. Когда R4 обозначает Y'(СН2)n-, например, Y' может быть

Дополнительные иллюстративные значения R4 включают 2-, 3- и 4-пиридил, 2-, 3- и 4-пиперидил.
Соединения формул (1)-(4) могут быть получены в форме их фармацевтически приемлемых кислотно-аддитивных солей, включая соли неорганических кислот, таких как хлористоводородная, серная, бромистоводородная или фосфорная кислота, или соли органических кислот, таких как уксусная, винная, янтарная, бензойная, салициловая кислота и подобные. Если присутствует карбоксильная часть, эти соединения могут быть получены в виде соли с фармацевтически приемлемым основанием, в том числе с неорганическими основаниями, такими как гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид аммония и т.п., или соли с органическим основанием, таким как кофеин.
Особенно предпочтительными соединениями данного изобретения являются соединения формул (5) и (6):

В этих соединениях R1 имеет приведенную формулу, где каждый X3 независимо обозначает галоген, C1-С6алкил, OR или NR2, где R обозначает Н или C1-С6алкил, а р является целым числом 0-3. R2, R3 и R4 имеют указанные выше значения.
Предпочтительными являются также подобные соединения, где положения заместителей R3 и показанного заместителя R1 являются обращенными; т.е. R3 находится в положении 5, а R1 находится в положении 6.
Синтез соединений данного изобретения
Соединения данного изобретения могут быть синтезированы различными способами, большинство которых известны в данной области per se. Индольная, бензимидазольная или бензотриазольная части могут быть полезны per se, а заместитель R1 может быть присоединен к ним. R1 может быть полезен как таковой или его синтез может быть завершен, когда пиперазильный или пиперидильный остаток уже связан с индольной, бензимидазольной или бензотриазольной частью. Альтернативно, в частности, в вариантах, в которых R3 обозначает заместитель, отличный от водорода, подходящим образом замещенное производное п-аминобензойной кислоты может быть циклизовано и затем замещено пиперазином или пиперидином.
Так, например, как показано на схеме реакций 1, пиперазин, защищенный трет-бутоксикарбонилом (ВОС), связывают с 5-карбоксибензимидазолом (или 5-карбоксииндолом или 5-карбоксибензотриазолом) в реакционной смеси, содержащей связующий агент, такой как EDAC, в инертном апротонном растворителе с получением связанного карбоксамида, который затем освобождают от защитных групп и обрабатывают замещенными или незамещенными бензилгалогенидами или бензоилгалогенидами.
Схема 1

R4 представляет собой, например, 2,6-дифторфенил; 3,4-дифторфенил, 2,3-дифторфенил; 3,5-дифторфенил; 3-хлорфенил; 4-хлорфенил; 4-карбоксиметилфенил; 4-метоксифенил; 4-трифторметилоксифенил; 4-метилфенил; 6-хлорпиперонил; трет-бутилкарбоксифенил; 3-трифторфенил; 2,4-дихлорфенил; 3,4-дихлорфенил; фенил; метоксифенил или п-толуил.
Альтернативно, как показано на схеме реакций 2, 2,5-карбоксилированный бензимидазол (или индол или бензотриазол) взаимодействует с пиперазиновой или пиперидиновой частью, уже замещенной Х2-Ar. В этой реакции пиперазил- или пиперидилпроизводное непосредственно взаимодействует с карбоксилированным бициклом, содержащим гетероатом в присутствии связующего агента, такого как EDAC, в присутствии инертного растворителя, как описано выше.
Схема 2

Для получения замещенного пиперазина, требующегося для схемы 1, пиперазин сначала превращают в ВОС-производное и затем он взаимодействует с ArCHO в присутствии боргидрида при кислых условиях с образованием замещенного пиперазина, как показано на схеме реакций 3.
Схема 3

Альтернатива связывания производного пиперазина или пиперидина с индолом, бензимидазолом или бензотриазолом показана на схеме реакций 4. В этой реакции получают производное пиперазинового или пиперидинового кольца с подходящей уходящей группой, как показано, и затем обрабатывают основанием, таким как NaH, в инертном растворителе с получением целевого конъюгата.
Схема 4
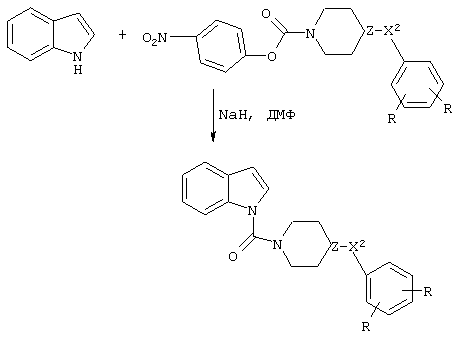
Другая альтернатива показана на схеме реакций 5. По этой методике защищенный пиперидон взаимодействует в присутствии основания, такого как NaH, с подходящим фосфонатным эфиром с образованием защищенного бензиленпиперидина. Затем этот продукт освобождают от защитных групп и подвергают реакции с карбоксилатом индола, бензимидазола или бензотриазола с использованием соответствующего дегидратирующего агента. Затем продукт восстанавливают с получением целевого арилалкилированного производного пиперидина.
Схема 5

Схема реакций 6 иллюстрирует способ получения соединений данного изобретения, в котором индол замещен в 6-членном кольце. В схеме реакций 6, подходящим образом замещенный анилин взаимодействует с 1-метилмеркаптил-2,2-диалкоксиэтаном в присутствии трет-бутирилхлорида и основания с получением целевого индола. В зависимости от характера замещения исходного анилина, может образоваться более чем один изомер, как показано. Метилмеркаптильную группу, остающуюся на 5-членном кольце, восстанавливают никелем Ренея и обязательную метильную группу, включенную в исходную анилиновую часть, гидролизуют с получением соответствующей карбоновой кислоты. Затем полученная кислота взаимодействует с желаемым производным пиперидина или пиперазина в присутствии связующего агента, такого как EDC.
Схема 6

Алкилирование атомов азота на индольном, бензимидазольном или бензотриазольном кольце в соединениях per se проводят общепринятыми способами реакцией галогена заместителя, который должен быть присоединен, в присутствии основания и ацетона, как показано на иллюстративной альтернативной схеме 7.
Схема 7

или

где Х=Н, ОМе, Cl;
каждый R представляет собой Н или алкил;
n является целым числом; или

где Х=Н, ОНС3, Cl, СН3 и т.д.;
каждый R представляет собой Н, алкил, арил
или вместе оба R образуют

Заместители в положении 3 индола могут быть модифицированы с использованием методик, показанных на схеме 8:
Схема 8

где Х=ОМе, Cl, СН3;
каждый R обозначает Н, алкил, арил или вместе группы R обозначают пиперазинил, 4-бензилпиперазинил и подобные.
Для синтеза соединений, в которых n равно 1 --, т.е. в которых пиперидиновое кольцо содержит один дополнительный заместитель, иной, чем заместители, которые являются обязательными в соединениях данного изобретения, 4-замещенный пиперидин сначала защищают при помощи ВОС2О в ТГФ или другом апротонном растворителе и затем подвергают взаимодействию, например, с алкилиодидом, в присутствии смеси S-бутил-литий/TMEDA с использованием, например, эфира в качестве растворителя с получением алкилированного пиперидина. Затем алкилированный пиперидин превращают в соединение данного изобретения удалением защитных групп с последующим образованием карбоксамидной связи с индоильным остатком. Это проиллюстрировано примером ниже.
Для соединений данного изобретения, которые являются индолами, замещенными в положении 3, может быть успешно использована схема реакций, показанная в примере 23. Обычно, карбоксамидный исходный материал обрабатывают трифторуксусным ангидридом с получением трифторацетилсодержащего промежуточного продукта, который также является соединением данного изобретения. При обработке основанием, образуется 3-карбоновая кислота, которая может затем взаимодействовать с подходящим амином с образованием дополнительных соединений данного изобретения.
Введение и применение
Соединения данного изобретения полезны для лечении состояний, связанных с воспалением. Таким образом, соединения формул (1)-(4) или их фармацевтически приемлемые соли используют для приготовления лекарственного средства для профилактики или терапии млекопитающих, в том числе человека, по поводу состояний, характеризующихся избыточным продуцированием цитокинов и/или неприемлемой или нерегулируемой активностью цитокинов на таких клетках, как кардиомиоциты, кардиофибробласты и макрофаги.
Соединения данного изобретения ингибируют продуцирование цитокинов, таких как TNF, IL-1, IL-6 и IL-8, которые являются важными провоспалительными составляющими во многих различных патологических состояниях и синдромах. Таким образом, ингибирование этих цитокинов является выгодным в регулировании и облегчении многих заболеваний. Показано, что соединения данного изобретения ингибируют член семейства МАР-киназ, называемый р38 МАРК (или р38), CSBP или SAPK-2. Показано, что активация этого белка регулирует продуцирование простаноидов, таких как PGE2, и металлопротеиназ матрикса (межклеточного вещества тканей), таких как коллагеназа-3, и сопровождает обострение заболеваний в ответ на стресс, вызванный, например, обработкой липополисахаридами или цитокинами, такими как TNF и IL-1. Следовательно, ингибирование активности р38 позволяет предсказать способность лекарственного средства обеспечивать полезное действие при лечении заболеваний, таких как болезнь коронарных артерий, застойная сердечная недостаточность, кардиомиопатия, миокардит, васкулит, рестеноз, которые встречаются после пластической операции на сосудах сердца, атеросклероз, ревматоидный артрит, ревматоидный спондилит, остеоартрит, подагрический артрит и другие артритные состояния, рассеянный склероз, острый респираторный дистресс-синдром у взрослых (ARDS), астма, хроническая обструктивная болезнь легких (ХОБЛ), силикоз, легочный саркоз (патологическое разрастание тканей легких), сепсис, септический шок, эндотоксиновый бактериально-токсический шок, синдром токсического шока, сердечная и мозговая недостаточности (удар), которые характеризуются ишемией, и реперфузионное повреждение, в хирургических процедурах, таких как процедуры при трансплантации и отторжениях трансплантата, искусственном (экстракорпоральном) кровообращении, обходном шунтировании кардиолегочной артерии, обходном шунтировании коронарных артерий, повреждениях ЦНС, в том числе открытой и закрытой травме головы, в лечении воспалительных глазных состояний, таких как конъюнктивит и увеит, острой почечной недостаточности, гломерулонефрита, воспалительных заболеваний пищеварительного тракта, таких как болезнь Крона или язвенный колит, реакции "трансплантат против хозяина", заболеваний с резорбцией костей, таких как остеопороз, диабета типа II, pyresis*, псориаза, кахексии, вирусных заболеваний, таких как вызываемые ВИЧ, CMV, вирусом герпеса, и церебральной малярии, опухолевых метастазов и острой боли, такой как боль, сопровождающая зубную хирургию, дисменорею и постортопедическую хирургию.
В последние несколько лет было показано, что р38 содержит группу МАР-киназ, обозначенных р38α, р38β, р38γ и р38δ. Jiang, Y. et al. J. Biol. Chem (1996) 271:17920-17926 первые описали характеристику р38β как белка из 372 аминокислот, близкородственного р38α. Kumar, S. et al. Biochem. Biophys. Res. Comm.(1997) 235:533-538 и Stein, В. et al. J. Biol. Chem. (1997) 272:19509-19517 сообщили о второй изоформе р38β, р38β2, содержащей 364 аминокислоты с 73% идентичностью к р38α. Все эти сообщения дают доказательства того, что р38β активируется провоспалительными цитокинами и стрессовыми условиями окружающей среды, хотя вторая описанная изоформа р38β, р38β2, по-видимому, является преимущественно экспрессированной в ЦНС, сердечную и скелетную мышцы в сравнении с более распространенной экспрессией в ткани р38α. Кроме того, наблюдали, что активированный фактор транскрипции 2 (ATF-2) является лучшим субстратом для р38β2, чем для р38α, что предполагает, что с этими формами могут быть связаны разные механизмы действия. Физиологическая роль p38β1 была поставлена под вопрос последними в двух последних сообщениях, так как киназа р38β1 не могла быть обнаружена в ткани человека и не проявляла поддающейся определению киназной активности с субстратами р38α.
Об идентификации р38γ было сообщено в Li, Z. et al. Biochem. Biophys. Res. Comm. (1996) 228:334-340 и об идентификации р38δ было сообщено в Wang, X., et ai., J. Biol. Chem. (1997) 272:23668-23674 и Kumar, S. et al. Biochem. Biophys. Res. Comm. (1997) 235:533-538. Эти данные предполагают, что эти две изоформы р38 (γ и δ) представляют уникальную субпопуляцию семейства МАРК на основе распределений их экспрессии в тканях, утилизации субстрата, ответной реакции на прямые и опосредованные стимулы и чувствительности к ингибиторам киназ.
Различные результаты в отношении ответной реакции на лекарственные средства, нацеленные на семейство р38, как среди р38α, так и для предполагаемых p38β1 или р38β2 или для обоих, были опубликованы в Jiang, Kumar and Stein, указанных выше, а также в Eyers, P.A. et al. Chem. and Biol. (1995) 5:321-328. В еще одной статье Wang, Y. et al. J. Biol. Chem. (1998) 273:2161-2168 высказывается предположение о значении таких дифференциальных эффектов. Как указывается в Wang, ряд стимулов, таких как инфаркт миокарда, гипертензия, клапанные заболевания, вирусный миокардит и кардиомиопатия с дилатацией сердца, приводят к увеличению рабочей нагрузки сердца и повышенному механическому стрессу в отношении кардиомиоцитов. Говорится, что это приводит к адаптивной гипертрофической реакции, которая, если ее не регулировать, имеет несомненно негативные последствия. Wang цитирует прежние исследования, которые показали, что в подвергнутых реперфузии ишемических сердцах активности р38 МАРК повышаются в связи с гипертрофией и запрограммированным некрозом клеток. В указанной статье Wang показывает, что активация активности р38β ведет к гипертрофии, тогда как активация активности р38α ведет к апоптозу миоцитов. Таким образом, избирательное ингибирование активности p38α в сравнении с активностью p38β будет выгодным для лечения состояний, связанных с сердечной недостаточностью. Эти состояния включают застойную сердечную недостаточность, кардиомиопатию, миокардит, васкулит, рестеноз сосудов, клапанное заболевание, состояния, связанные с искусственным (экстракорпоральным) кровообращением, шунтированием коронарных артерий, трансплантатами и васкулярными трансплантатами.
Кроме того, с учетом того, что α-изоформа является токсичной в других типах мышечных клеток, α-селективные ингибиторы были бы полезными для состояний, связанных с кахексией, приписываемой TNF, или других состояний, таких как рак, инфекция или аутоиммунное заболевание.
Соединения, описанные здесь, которые селективно ингибируют активность изоформы p38α, полезны для лечения состояний, связанных с активацией р38α, в частности, состояний, связанных с гипертрофией сердца, ишемией или другим стрессом окружающей среды, таким как окислительное повреждение, гиперосмолярность или другие агенты или факторы, которые активируют киназу р38α, или сердечной недостаточности, например застойной сердечной недостаточности, кардиомиопатии и миокардита.
Соединения, которые проявляют эту активность, являются соединениями формулы

где R1, R2, R3, Z1 и Z2 имеют указанные в п.1 значения. Способ введения и формулирования описанных здесь соединений будет зависеть от природы состояния, тяжести состояния, конкретного проходящего лечение субъекта и оценки лечащего врача; формулирование будет зависеть от способа введения. Так как эти соединения являются небольшими молекулами, их удобно вводить пероральным путем, формулируя их с подходящими фармацевтическими наполнителями, пригодными для получения таблеток, капсул, сиропов и т.п. Подходящие препаративные формы для перорального введения могут также включать незначительное количество компонентов, таких как буферы, улучшающие вкус и запах агенты и т.п. Обычно количество активного ингредиента в этих препаративных формах находится в диапазоне 5-95% от количества всей композиции, но допускается широкое варьирование в зависимости от носителя. Подходящие носители включают сахарозу, пектин, стеарат магния, лактозу, арахисовое масло, оливковое масло, воду и т.п.
Соединения, применимые в данном изобретении, могут также вводиться в качестве суппозиториев или других трансмукозных носителей. В типичном случае, такие препаративные формы будут включать наполнители, которые облегчают прохождение соединения через слизистую оболочку, такие как фармацевтически приемлемые детергенты.
Эти соединения могут также вводиться местно, для локальных состояний, таких как псориаз, или в виде препаративных форм, предназначенных для проникновения через кожу. Такие композиции включают лосьоны, кремы, мази и т.п., которые могут быть приготовлены известными способами.
Эти соединения могут также вводиться инъекцией, в том числе внутривенной, внутримышечной, подкожной или внутрибрюшинной инъекции. Типичными препаративными формами для такого применения являются жидкие композиции в изотонических носителях, таких как раствор Хенка или раствор Рингера.
Альтернативные препаративные формы включают назальные спреи, липосомные композиции, композиции замедленного высвобождения и т.п., которые известны в данной области.
Может быть использована любая подходящая препаративная форма. Обзор известных в данной области композиций можно найти в Remington's Pharmaceutical Sciences, latest edition. Mack Publishing Company, Easton, PA. Ссылка на этот справочник является обычной в данной области.
Дозы соединений данного изобретения будут зависеть от ряда факторов, которые будут меняться от пациента к пациенту. Однако считается, что обычная суточная пероральная доза будет составлять 0,001-100 мг/кг от общего веса тела, предпочтительно от 0,01 до 50 мг/кг и более предпочтительно около 0,01 мг/кг-10 мг/кг. Схема приема лекарственного средства будет, однако, варьироваться в зависимости от состояний, которые должны быть подвергнуты лечению, и оценки практикующего врача.
Как упоминалось выше, хотя соединения данного изобретения могут быть использованы для человека, они также пригодны для применения в ветеринарии для лечения животных.
Следующие ниже примеры предназначены для иллюстрации, но не для ограничения данного изобретения.
Примеры 1-3 иллюстрируют схему реакции 1:
Пример 1
Получение 4-ВОС-пиперазинилбензимидазол-5-карбоксамида

Вензимидазол-5-карбоновую кислоту (3,25 г, 20 ммоль) подвергали взаимодействию с 2,52 г (20 ммоль) диизопропил-карбодиимида в сухом ДМФ при комнатной температуре в течение 15 минут. К этой реакционной смеси добавляли 3,75 г (20 ммоль) трет-бутил-1-пиперазинкарбоксилата и смесь перемешивали в течение 18 часов. Смесь выливали в воду и экстрагировали метиленхлоридом (3×100 мл). Органические экстракты промывали опять водой, солевым раствором и сушили над MgSO4. После удаления растворителя в вакууме остаток хроматографировали на колонке силикагеля с элюированием смесью CHCl3-метанол (градиент, метанол 0-5%) с получением 5,69 г (86%) продукта. 1Н-ЯМР (ДМСО-d6): с 8,3 (1Н), м 7,7-7,6 (2Н), м 7,2-7,3 (1Н), м 3,6-3,3 (8Н), с 1,4 (9Н); MS (ESI) m/e 330 (М+).
Пример 2
Получение пиперазинилбензимидазол-5-карбоксамида
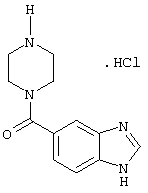
N-ВОС-пиперазинилбензимидазол-5-карбоксамид (5,6 г) перемешивали в 20 мл смеси 4 М HCl-диоксан в течение 1 часа. Диоксан удаляли при пониженном давлении с получением гидрохлоридной соли с количественным выходом. Ее использовали для алкилирований без какой-либо дополнительной очистки.
Пример 3
Получение 4-(2,6-дифторбензил)-пиперазинилбензимидазол-5-карбоксамида

А. Пиперазинилбензимидазол-5-карбоксамид (0,186 г, 0,5 ммоль) помещали в 5 мл ДМФ и добавляли 0,101 г (1 ммоль) триэтиламина и перемешивали в течение 15 минут при комнатной температуре. К этой реакционной смеси добавляли 0,104 г 2,6-дифторбензилбромида и смесь перемешивали в течение 20 часов. Смесь выливали в воду и экстрагировали метиленхлоридом (3×50 мл). Объединенный экстракт дополнительно промывали солевым раствором, водой и сушили над MgSO4. Растворитель удаляли в вакууме и остаток хроматографировали на колонке силикагеля с элюированием смесью хлороформ-метанол (0-5% метанол, градиент). Упаривание целевой фракции давало 48,9 мг целевого продукта; MS(ESI) m/e 356 (М+).
В. С использованием методики, описанной в части А, получали следующие соединения:
MS(ESI) m/e 356 (М+)
MS(ESI) m/e 356 (М+)
MS(ESI) m/e 354 (М+)
MS(ESI) m/e 378 (М+)
MS(ESI) m/e 350 (M+)
MS(ESI) m/e 404 (М+)
MS(ESI) m/e 334 (М+)
MS(ESI) m/e 403 (M+)
MS(ESI) m/e 403 (M+)
MS(ESI) m/e 428 (М+)
циннамоил-хлоридом
MS(ESI) m/e 369 (М+)
MS(ESI) m/e 334 (М+)
MS(ESI) m/e 402 (М+)
MS(ESI) m/e 364 (M+)
Пример 4 иллюстрирует схему реакций 2:
Пример 4
Получение 4-(3,4-дихлорфенил)пиперазинилбензимидазол5-карбоксамида
А. Бензимидазол-5-карбоновую кислоту (1 ммоль, 162 мг) растворяли в 5 мл сухого ДМФ и подвергали взаимодействию с гидрохлоридом 1-этил-3-(3-диметиламинопропил)карбодиимида в течение 15 минут. Добавляли 1-(3,4-дихлорфенил)пиперазин, 1 ммоль (231 мг) с последующим добавлением 10 мг DMAP. Эту смесь перемешивали в течение 20 часов при комнатной температуре. Реакционную смесь выливали в воду и экстрагировали метиленхлоридом (3×50 мл). Экстракты объединяли, промывали солевым раствором, водой и сушили над MgSO4. После выпаривания растворителя остаток хроматографировали на колонке силикагеля со смесью хлороформ-метанол (0-5% метанол, градиент) в качестве элюента. Упаривание желательных фракций давало 150 мг (40%) указанного в заголовке соединения; MS(ESI) m/e 375 (M+).
В. С использованием методики части А получали следующие соединения:
MS(ESI) m/e 431 (М+)
MS(ESI) m/e 346 (М+)
MS(ESI) m/e 432 (М+)
MS(ESI) m/e 354 (M+)
MS(ESI) m/e 354 (M+)
MS(ESI) m/e 320 (M+)
Пример 5 иллюстрирует схему реакций 3:
Пример 5
А. Получение 4-(4-метилтиобензил)пиперазинилбензимидазол-5-карбоксамида

Смесь 4-(метилтио)бензальдегида, 305 мг (2 ммоль), и N-BOC-пиперазина, 372 мг (1 ммоль), перемешивали в сухом метаноле в течение 30 минут. К этой смеси добавляли 1,6 г боргидрида на полимерном носителе (2,5 ммоль)/г, на Амберлите, IRA-400, Aldrich) и смесь перемешивали в течение 24 часов. Полимер удаляли фильтрованием и выпаривание растворителя давало 4-ВОС-1-(4-метилтио)бензилпиперазина с количественным выходом. MS(ESI) m/e 322 (М+).
4-ВОС-1-(4-метилтио)бензилпиперазин помещали в 10 мл смеси 1:1 ТФУ/метиленхлорид и перемешивали в течение 1 часа при комнатной температуре. Растворители удаляли в вакууме и остаток использовали без очистки для связывания с бензимидазол-5-карбоновой кислотой.
Бензимидазол-5-карбоновую кислоту (2 ммоль, 324 мг) помещали в 15 мл сухого ДМФ и подвергали реакции с 2 ммоль (382 мг) EDAC при комнатной температуре в течение 15 минут. Добавляли вышеописанный 1-(4-метилтио)бензилпиперазин в виде раствора в ДМФ с последующим добавлением 505 мг (5 ммоль) ТФУ. Смесь перемешивали в течение 20 часов. Смесь выливали в воду и экстрагировали метиленхлоридом (3×50 мл). Объединенные экстракты промывали солевым раствором, водой и сушили над MgSO4. Растворитель удаляли в вакууме и остаток хроматографировали. Упаривание желательных фракций давало указанное в заголовке соединение; MS(ESI) m/e 366 (М+).
С использованием этой методики получали следующие соединения:
MS(ESI) m/e 410(М+)
MS(ESI) m/e 391 (М+)
MS(ESI) m/e 396 (М+)
MS(ESI) m/e 412 (М+)
В. Получение 4-бензилпиперидинилбензимидазол-5-карбоксамида

Бензимидазол-5-карбоновую кислоту (1,62 г, 10 ммоль) подвергали взаимодействию с EDAC (1,92 г, 10 ммоль) в 40 мл сухого ДМФ при комнатной температуре в течение 15 минут. К реакционной смеси добавляли 4-бензилпиперидин (1,75 г, 10 ммоль) и DMAP (˜20 мг, катализатор) и эту смесь перемешивали при комнатной температуре в течение 20 часов. Смесь выливали в воду и экстрагировали метиленхлоридом (3×100 мл). Объединенный экстракт промывали водой, солевым раствором и опять водой. Экстракт сушили над MgSO4 и упаривали. Остаток хроматографировали на колонке силикагеля со смесью хлороформ-метанол (0-5% метанол) в качестве элюента. Упаривание желательных фракций Мавало 1,5 г (47%) продукта после перекристаллизации из смеси этилацетатгексан. 1Н-ЯМР (CDCl3) δ 7,8 (с, 1Н), 7,1-7,3 (м, 8Н), 4,8-4,7 (шм, 1H), 3,7-3,9 (шм, 1Н), 3,1-2,7 (шм, 2Н), 2,55 (д, 2Н), 2,0-1,1 (м, 5Н). MS(ESI) m/e 319 (М+), 318 (М+-Н).
Пример 6
Получение дополнительных пиперидинилсодержащих производных бензимидазола
Схема реакций в данном примере является в общем следующей:
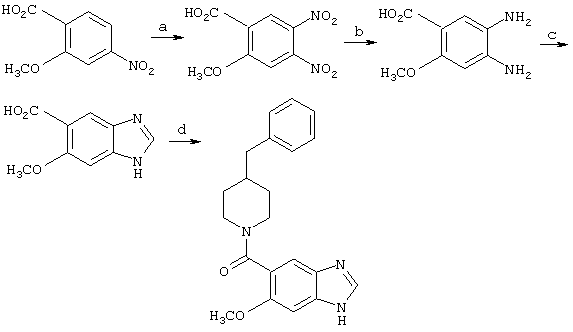
a) Азотная кислота/серная кислота, 100°С, 1 час.
b) Метанол, 10% Pd/C, муравьиная кислота, 1-3 часа.
c) 90% водная муравьиная кислота, кипячение с обратным холодильником, 1,5 часа.
d) Бензилпиперидин, EDAC•HCl, DMAP, ДМФ.
2-метокси-3,4-динитробензойная кислота
4-нитро-2-метоксибензойную кислоту, 3,09 г, добавляли к 20 мл смеси азотная кислота:серная кислота 1:1 при 0°С. После завершения добавления реакционную смесь нагревали при 100°С в течение 30 минут. Охлаждали до комнатной температуры и выливали в 200 мл охлажденной льдом воды. Водный слой экстрагировали этилацетатом и промывали насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали с получением желтого твердого вещества. Этот материал очищали хроматографией на силикагеле с использованием смеси этилацетат/гексан/метанол/уксусная кислота 5/5/1/0,1 в качестве элюента. Полученное желтое твердое вещество использовали на следующей стадии. EIMS М+ 242 (Эксп.242). ЯМР, d6 DMSO: 8,5 (с, 1Н), 7,95 (с, 1Н), 4,05 (с, 3Н).
2-метокси-3,4-диаминобензойная кислота
2-метокси-3,4-динитробензойную кислоту (1,0 г) растворяли в метаноле (50 мл) и обрабатывали 100 мг 10% палладия на угле. Реакционную смесь барботировали азотом и помещали на баню со льдом. При обработке 5 мл муравьиной кислоты было отмечено сильное выделение пузырьков газа из жидкости, которое прекращалось при последующем охлаждении. Реакционную смесь фильтровали через целит и концентрировали с получением рыжевато-коричневого твердого вещества. (При хранении быстро происходит обесцвечивание). EIMS М+ 182, Эксп.182.
6-метокси-5-бензимидазолкарбоновая кислота
2-метокси-3,4-диаминобензойную кислоту (0,5 г) растворяли в 10 мл 90% водной муравьиной кислоты. Смесь доводили до кипячения с обратным холодильником и поддерживали в течение 90 минут. Охлаждали до комнатной температуры и растворитель удаляли при пониженном давлении с получением темного твердого вещества. EIMS M+ 192 (Эксп.192).
6-метокси-(4-бензилпиперидинил)бензимидазол-5-карбоксамид

6-метокси-5-бензимидазолкарбоновую кислоту (1 эквивалент) обрабатывали 1,1 эквивалента EDAC•HCl и 1 эквивалентом 4-бензилпиперидина в присутствии каталитического количества DMAP в смеси ДМФ/ДХМ 1:1 в течение 3-6 часов. Затем реакционную смесь концентрировали и помещали в этилацетат. После промывания 5% водным карбонатом натрия и раствором насыщенного хлорида натрия органический слой сушили над безводным сульфатом натрия и концентрировали с получением неочищенного материала. Этот неочищенный материал очищали хроматографией на диоксиде кремния. М+ 349, Эксп.349.
6-хлор-(4-бензилпиперидинил)бензимидазол-5-карбоксамид

получали таким же образом. МН+ 353, Эксп.353.
Пример 7
N-пропилирование 4-бензилпиперидинилбензимидазол-5-карбоксамида

4-(4-бензил)пиперидинилбензимидазол-5-карбоксамид (318 мг, 1 ммоль) помещали в 20 мл ацетона. Добавляли КОН (твердый, 280 мг, 5 ммоль) с последующим добавлением 2-иод-пропана (1 г, ˜6 ммоль) и эту смесь кипятили с обратным холодильником в течение 20 часов. Ацетон удаляли в вакууме и остаток экстрагировали из воды метиленхлоридом (3×50 мл). Экстракт сушили, упаривали и остаток хроматографировали на силикагеле, используя смесь CHCl3-метанол (0-3% метанол) в качестве элюента. MS(ESI) m/e 360 (М+). ВЭЖХ: (колонка Vydac С18, смесь 5-40% ацетонитрил/вода, содержащая 0,1% ТФУ), два пика, обнаруживающие оба изомера.
Пример 8
Получение 4-бензилпиперидинилиндол-5-карбоксамида

Индол-5-карбоновую кислоту (1,61 г, 10 ммоль) вводили во взаимодействие с EDAC (1/92 г, 10 ммоль) в 40 мл сухого ДМФ в течение 15 минут. Добавляли 4-бензилпиперидин (1,75 г, 10 ммоль) с последующим добавлением DMAP (20 мг, катализатор) и реакционную смесь перемешивали в течение 20 часов. Смесь выливали в воду и экстрагировали метиленхлоридом (3×100 мл). Объединенный экстракт промывали разбавленной хлористоводородной кислотой, насыщенным раствором бикарбоната натрия и водой и сушили над MgSO4. После выпаривания растворителя остаток хроматографировали, используя смесь метиленхлорид/метанол (0-2% метанол, градиент) с получением 1,60 г (50%) продукта после перекристаллизации из смеси эфир-гексан. MS(ESI) m/e 318 (М+), (317+-Н). 1H-ЯМР (CDCl3) δ 8,5 (с, 1Н), 7,7 (с, 1Н), 7,4-7,15 (м, 8Н), 6,8 (с, 1Н), 4,8-4,6 (шм, 1Н), 4,1-3,99 (шм, 1Н), 3,1-2,7 (шм, 2Н), 2,6 (д, 2Н), 1,9-1,7 (шм, 3Н), 1,4-1,2 (шС, 2Н).
Пример 9
Получение 4-бензилпиперидинил-1-(2-пропил)индол-5-карбоксамида

Смесь 4-бензилпиперидинилбензимидазол-5-карбоксамида (318 мг, 1 ммоль), твердого КОН (280 мг, 5 ммоль) и 2-иод-пропана (1 г, 6 ммоль) кипятили с обратным холодильником в 20 мл ацетона в течение 20 часов. После удаления ацетона в вакууме остаток экстрагировали из воды метиленхлоридом (3×50 мл). Объединенный экстракт сушили, упаривали и хроматографировали с получением 180 мг (50%) целевого продукта. 1H-ЯМР (CDCl3) δ 7,7 (с, 1Н), 7,4-7,1 (м, 7Н), 4,8-4,6 (м, 1Н), 3,0-2,7 (шм, 4Н), 2,6 (д, 2Н), 1,8-1,45 (м, 3Н), 1,5 (д, 6Н), 1,3-1,1 (м, 2Н). MS(ESI) m/e 360 (М).
Пример 10
Получение 4-(4-хлорбензил)пиперазинил-1-(2-пропил)-индол-5-карбоксамида

4-(4-хлорбензил)пиперазинилиндол-5-карбоксамид (420 мг, 1,32 ммоль) помещали в ацетон. Добавляли твердый КОН (280 мг, 5 ммоль) с последующим добавлением 2-иодпропана (1 г, 6 ммоль) и смесь кипятили с обратным холодильником при перемешивании в течение 20 часов. Ацетон удаляли в вакууме и остаток экстрагировали из воды метиленхлоридом. Экстракт сушили и упаривали и остаток хроматографировали на силикагеле с использованием смеси этилацетат/гексан (этил-ацетат 0-25%, градиент) и перекристаллизовывали из смеси эфир-гексан с получением 300 мг продукта. 1Н-ЯМР (CDCl3): δ 7,6 (с, 1Н), 7,3-7,1 (м, 6Н), 6,5 (с, 1Н), 4,65-4,55 (м, 1H), 3,8-3,5 (м, 4Н), 3,4 (с, 2Н), 2,4-2,5 (с, 4Н), 1,5 (д, 6Н). MS(ESI) m/e 395 (М+).
Пример 11
Получение других аналогов
С использованием методики примера 8 получают следующие соединения:
MS(ESI) m/e 318 (М+), (317+-Н)
MS(ESI) m/e 320 (М+), (319+-H)
С использованием методики примера 10 эти соединения алкилировали, например,
MS(ESI) m/e 360 (М+)
Пример 12
Получение 3-хлорбензилпиперазинил-N-бензилбензимидазол-5- и -6-карбоксамидов
А. На этой стадии описана методика образования N-бензилпроизводных соединений данного изобретения; в последующих стадиях описано алкилирование другими частями молекул.
3-хлорбензилпиперазинилбензимидазол-5-карбоксамид (0,12 г, 0,33 ммоль) и бензилбромид (0,058 г, 0,33 ммоль) в 15 мл ДМФ объединяли с К2СО3 (0,09 г, 0,66 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, затем нагревали при 45°С в течение 3 часов. Добавляли EtOAc и промывали водой. Органический слой выпаривали и изомеры разделяли колоночной хроматографией на силикагеле с использованием 5% МеОН в EtOAc с получением изомера а (70 мг, 48%), MS(ESI) m/e 444 (М+) и изомера b (40 мг, 27%), MS(ESI) m/e 444 (M+).
Такая же обработка 6-карбоксамида дает соответствующее соединение, где R2 представляет собой бензил.
В. 3-хлорбензилпиперазинил-N-(2-пропил)бензимидазол-5- и 6-карбоксамиды
3-хлорбензилпиперазинилбензимидазол-5-карбоксамид алкилировали с заменой бензилбромида на стадии А 2-иодпропаном. Изомеры разделяли с использованием тех же условий для хроматографии. Изомер a, MS(ESI) m/e 396 (M+); изомер b, MS(ESI) m/e 396 (M+).
Аналогичная обработка 6-карбоксамида дает соответствующее соединение, где R2 представляет собой 2-пропил.
С. 3-хлорбензилпиперазинил-N-метилбензимидазол-5- и 6-карбоксамиды
3-хлорбензилпиперазинилбензимидазол-5-карбоксамид алкилировали с заменой бензилбромида в методике стадии А иодметаном. Измеры разделяли с использованием колоночной хроматографии с 50% ацетоном в ацетонитриле в качестве элюирующего растворителя. Изомер a, MS(ESI) m/e 368 (М+), изомер b, MS(ESI) m/e 368 (М+).
Подобная обработка 6-карбоксамида дает соответствующее соединение, где R2 представляет собой метил.
Подобным образом, 4-бензилпиперидинил-(1-метил)индол-5-карбоксамид (MS(ESI) m/e 332 (М+)) получали из 4-бензилпиперидинилиндол-5-карбоксамида.
Аналогичная обработка 6-карбоксамида дает соответствующее соединение, где R2 представляет собой метил.
D. 3-хлорбензилпиперазинил-N-этилбензимидазол-5- и 6-карбоксамиды
3-хлорбензилпиперазинилбензимидазол-5-карбоксамид алкилировали с заменой бензилбромида на стадии А иодэтаном. Изомер a, MS(ESI) m/e 382 (М+); изомер b, MS(ESI) m/e 382 (М+).
Аналогичная обработка 6-карбоксамида дает соответствующее соединение, где R2 представляет собой этил.
Подобным образом, 4-бензилпиперидинил-(1-этил)индол-5-карбоксамид (MS(ESI) m/e 346 (М+)) получали из 4-бензилпиперидинилиндол-5-карбоксамида.
Аналогичная обработка 6-карбоксамида дает соответствующее соединение, где R2 представляет собой этил.
Пример 13
Получение 4-(4-хлорбензил)пиперидинилиндол-5-карбоксамида

Этот пример иллюстрирует схему реакций 5.
А. Получение N-BOC-4-(4-хлорбензилен)пиперидина

N-BOC-пиперидон (2,0 г, 10 ммоль) объединяли с диэтил-4-хлорбензилфосфонатом (2,6 г, 10 ммоль) в сухом ТГФ. Добавляли гидрид натрия (400 мг, 60% дисперсия в минеральном масле, 10 ммоль) и смесь кипятили с обратным холодильником в течение трех часов. ТГФ удаляли в вакууме и остаток экстрагировали из воды метиленхлоридом. Экстракт сушили над MgSO4, упаривали и остаток хроматографировали на силикагеле с получением 0,615 г целевого продукта. 1H-ЯМР (CDCl3): δ 7,3 (д, 2Н), 7,1 (д, 2Н), 6,3 (с, 1Н), 3,55-3,50 (м, 2Н), 3,45-3,35 (м, 2Н), 2,45-2,35 (м, 2Н), 2,30-2,25 (м, 2Н), 1,25 (с, 9Н). EIMS 307 (М+), 251 (М+-С3Н8).
В. Связывание 4-хлорбензиленпиперидина с индол-5-карбоновой кислотой
N-BOC-4-(4-хлорбензилен)пиперидин, описанный выше, освобождали от защитных групп перемешиванием в 20 мл смеси 1:1 дихлорметан-трифторуксусная кислота в течение 1 часа, упаривали и сушили в вакууме в течение 1 часа, удаляя все остатки трифторуксусной кислоты. Его повторно растворяли в 15 мл дихлорметана и ТФУ-соль нейтрализовали добавлением небольшого избытка триэтиламина. Раствор А.
Индол-5-карбоновая кислота (0,32 г, 2 ммоль) взаимодействовала с 0,383 г EDAC в 30 мл сухого дихлорметана в течение 15 минут. К этому раствору добавляли раствор в метиленхлориде 4-хлорбензиленпиперидина (раствор А) с последующим добавлением 10 мг DMAP. Смесь перемешивали в течение 20 часов. Смесь промывали водой, 2 н. HCl, 5% карбонатом натрия и затем водой. Органический раствор сушили, упаривали и остаток хроматографировали на силикагеле с элюированием смесью этилацетат-гексан (1:4). Выход: 260 мг (37%). EIMS 350 (М+), 315 (M+-Cl). 1H-ЯМР (CDCl3): δ 8,4 (с, 1Н), 7,7 (с, 1Н), 7,3-7,0 (м, 7Н), 6,5 (с, 1Н), 6,25 (с, 1Н), 3,8-3,0 (шм, 4Н), 2,6-2,20 (шм, 4Н).
С. Гидрирование 4-(4-хлорбензилен)пиперидининдол-5-карбоксамида
4-(4-хлорбензилен)пиперидининдол-5-карбоксамид (240 мг, 0,68 ммоль) растворяли в 40 мл ТГФ. Добавляли Pd/C (25 мг) и смесь гидрировали (1 атм) в течение 20 часов с быстрым перемешиванием. Катализатор удаляли фильтрованием через целит, органический раствор выпаривали и остаток перекристаллизовывали из смеси метиленхлорид/гексан. Выход количественный. EIMS 352 (M+), 351 (М+-Н).
Пример 14
С использованием общей методики, описанной в примере 13, получали следующие соединения:
MS(ESI) m/e 353 (М+)
MS(ESI) m/e 353 (M+)
Пример 15
Синтез цис-2-метил-4-бензилпиперидин-1-илиндол-5-карбоксамида

А. Смесь 4-бензилпиперидина (3,52 мл, 10/0 ммоль) и ди-трет-бутилдикарбоната (5,45 г, 25,0 ммоль) в 100 мл ТГФ кипятили с обратным холодильником в течение 20 часов. После охлаждения до комнатной температуры реакционную смесь выливали в воду и экстрагировали этилацетатом (2×100 мл). Объединенный органический экстракт промывали водой и солевым раствором. Экстракт сушили над Na2SO4 и упаривали. Остаток хроматографировали на колонке силикагеля с использованием смеси 10% этилацетат-гексан. Упаривание желательных фракций давало 5,02 г (91%) продукта в виде масла. MS(ESI) m/e 275 (М+).
В. Смесь 1-ВОС-4-бензилпиперидина (0,825 г, 3,0 ммоль) и N,N,N',N'-тетраметилэтилендиамина (TMEDA) (0,59 мл, 3,9 ммоль) в 6 мл Et2O охлаждали до -78°С в атмосфере аргона. Добавляли по каплям 1,3 М раствор втор-BuLi в циклогексане (3,0 мл, 3,9 ммоль). После завершения добавления реакционную смесь перемешивали при -20°С в течение 30 минут и охлаждали опять до -78°С. Добавляли метилиодид (0,28 мл, 4,5 ммоль) и реакционную смесь перемешивали при -78°С в течение 5 минут, охлаждающую баню удаляли и перемешивание продолжали еще в течение 3 минут. Реакционную смесь выливали в воду и экстрагировали этилацетатом (2×25 мл). Объединенный органический экстракт промывали водой и солевым раствором. Экстракт сушили над Na2SO4 и упаривали с получением 0,58 г (67%) масла, которое давало одно пятно по ТСХ (силикагель, смесь 10% этилацетат-гексан). Этот материал использовали непосредственно на следующей стадии. MS(ESI) m/e 289 (M+).
С. К раствору 1-ВОС-2-метил-4-бензилпиперидина (0,29 г, 1,0 ммоль) в 5 мл дихлорметана добавляли трифторуксусную кислоту (ТФУ) (0,5 мл). После перемешивания при комнатной температуре в течение 10 часов реакционную смесь упаривали в вакууме и азеотропно перегоняли дважды с дихлорметаном и дважды с гексаном. Остаток растворяли в 5 мл дихлорметана и добавляли диизопропилэтиламин (1,6 мл, 10 ммоль). В отдельной колбе смесь 5-индолкарбоновой кислоты (0,19 г, 1,2 ммоль) и EDAC (0,23 г, 1,2 ммоль) растворяли в 15 мл дихлорметана и перемешивали при комнатной температуре в течение 5 минут. К этой реакционной смеси добавляли первый раствор и полученную смесь перемешивали при комнатной температуре в течение 20 часов. Реакционную смесь выливали в воду и экстрагировали этилацетатом (2×50 мл). Объединенный органический экстракт промывали водой и солевым раствором. Экстракт сушили над Na2SO4 и упаривали. Остаток хроматографировали на колонке силикагеля со смесью 1% МеОН-дихлорметан. Упаривание желательных фракций давало 0,18 г (54%) продукта в виде масла.
При испытании, описанном ниже, указанное в заголовке соединение имело IC50=280 нМ.
Пример 16
Получение 4-хлор-(4-бензилпиперидинил)индол-5-карбоксамида и 6-хлор-(4-бензилпиперидинил)индол-5-карбоксамида

A. Синтез этого индола выполняли по способу Gassman, P.G. J. Am. Chem. Soc. (1974) 96:5495-5507. К раствору 2,0 г (10,8 ммоль) 4-амино-2-хлорметилбензоата в 30 мл CH2Cl2 при -60°С добавляли 1,2 г (10,8 ммоль) трет-бутилгипохлорита в 20 мл CH2Cl2. Спустя 10 минут добавляли 10,8 ммоль диметилацеталя метилтиоацетальдегида в 10 мл СН2Cl2, перемешивание продолжали при -60°С в течение 1 часа. Затем добавляли 10,8 ммоль Et3N в 10 мл CH2Cl2 и раствору давали нагреться до комнатной температуры. Растворитель выпаривали и остаток растворяли в 30 мл CCl4, добавляли 5 мл Et3N и смесь кипятили с обратным холодильником в течение 4 часов. Растворитель удаляли и остаток растворяли в 50 мл эфира. Циклизацию ацеталя с индолом проводили перемешиванием этого раствора в течение 3 часов с 20 мл 2 н. HCl. Эфирный слой промывали насыщенным NaHCO3, сушили, фильтровали и упаривали. Изомерные индолы разделяли колоночной хроматографией на силикагеле. Структуру изомеров идентифицировали ЯМР-спектроскопией. Изомер а: 5-карбоксиметил-4-хлор-3-тиометилиндол, 1Н-ЯМР (CDCl2): δ 2,35 (с, 3Н), 3,95 (с, 3Н), 7,32 (с, 1Н), 7,42 (с, 1Н), 8,33 (с, 1Н), 8,61 (с, 1Н). Изомер b: 5-карбоксиметил-6-хлор-3-тиометилиндол, 1H-ЯМР (CDCl3): δ 2,42 (с, 3Н), 3,97 (с, 3Н), 7,20 (с, 1Н), 7,25 (д, 1Н), 7,71 (д, 1Н), 8,58 (с, 1Н).
Раствор 100 мг 5-карбоксиметил-4-хлор-3-тиометилиндола (изомера а) в 10 мл этанола обрабатывали никелем Ренея W-2 до завершения детиометилирования. Выделенный эфир индола обрабатывали NaOH в смеси метанол:вода (1:1) и получали в результате 60 мг 4-хлориндол-5-карбоновой кислоты в виде белого твердого вещества, 1Н-ЯМР (ДМСО-d6) δ 6,61 (с, 1Н), 7,41 (д, 1Н), 7,52 (с, 1Н), 7,62 (д, 1Н), 11,62 (с, 1Н).
К раствору 50 мг (0,25 ммоль) вышеуказанной индолкарбоновой кислоты в 10 мл ДМФ добавляли 50 мг (0,28 ммоль) 4-бензилпиперидина и 60 мг (0,28 ммоль) EDAC. Реакционную смесь перемешивали в течение ночи, разбавляли этилацетатом и промывали водой, органический слой сушили, фильтровали и упаривали с получением белого твердого вещества. Его очищали хроматографией на силикагеле с последующей кристаллизацией с получением 50 мг 4-хлор-5-(4-бензилпиперидинил)индолкарбоксамида в виде белого твердого вещества, MS (M+ 352).
Изомер b превращали в 6-хлориндол-5-карбоновую кислоту с использованием такой же последовательности реакций, как описанная выше, и связывали с 4-бензилпиперидином с получением 6-хлор-5-(4-бензилпиперидинил)индолкарбоксамида в виде белого твердого вещества, MS (M+ 352).
В. С использованием способа стадии А получали 4-хлор-(4-{4-фторбензил)пиперидинил)индол-5-карбоксамид и 6-хлор-(4-(4-фторбензил)пиперидинил)индол-5-карбоксамид.
Пример 17
Соответствующие 6-пиперидинилпроизводные индола
Соединения, сходные с соединениями, описанными на стадиях А и В примера 16, но в которых пиперидинильный заместитель находится в положении 6, синтезируют следующим образом:

а) метанол/тионилхлорид, кипячение с обратным холодильником;
b)
i) N-хлорсукцинимид, ДХМ, диметилацеталь метилтио-ацетальдегида, триэтиламин, кипячение с обратным холодильником;
ii) CHCl3, кипячение с обратным холодильником;
iii) HCl
c) никель Ренея, EtOH
d) метанол, гидроксид натрия, кипячение с обратным холодильником;
e) бензилпиперидин, EDAC•HCl, DMA.P, ДМФ/ДХМ
f) ацетон, гидроксид калия, никотиноилхлорид.
Следующие соединения получали в соответствии с этим способом:
4-бензилпиперидинил-5-хлориндол-6-карбоксамид:

МН+ 351, эксп.352;
4-бензилпиперидинил-7-хлориндол-6-карбоксамид:

МН+ 351, эксп.352;
1-никотиноил-4-бензилпиперидинил-7-хлориндол-6-карбоксамид:

MH+ 457, эксп.457;
1-никотиноил-3-(2-диметиламино)этиламинокарбонил-4-бензилпиперидинил-7-хлориндол-6-карбоксамид:

МН+ 571, эксп.571.
(См. примеры 19-21 в отношении присоединения заместителей в положениях 1 и 3).
Пример 18
Получение 4-метокси-(4-бензилпиперидинил)индол-5-карбоксамида и 6-метокси-(4-бензилпиперидинил)индол-5-карбоксамида

А. Получение 4-метоксииндол-и 6-метоксииндол-5-карбоновых кислот

Соответствующие метиловые эфиры индолкарбоновых кислот получали модифицированным способом схемы 6 в соответствии с Inoue, S. Heterocycles, (1992) 34:1017-1029, в котором эти две изомерные индолкарбоновые кислоты получали в соотношении 3:2. 5-карбоксиметил-4-метоксииндол. 1H-ЯМР (CDCl3) δ 2,42 (с, 3Н), 3,92 (с, 3Н), 4,13 (с, 3Н), 7,14 (д, 1Н), 7,18 (д, 1Н), 7,55 (д, 1Н), 9,41 (с, 1Н). 5-карбоксиметил-6-метоксииндол. 1H-ЯМР (CDCl3) δ 2,38 (с, 3Н), 3,81 (с, 3Н), 3,95 (с, 3Н), 6,83 (с, 1Н), 7,21 (с, 1Н), 8,22 (с, 1Н), 8,50 (с, 1H).
В. Превращение в указанное в заголовке соединение
4-метокси-или 6-метоксииндол-5-карбоновую кислоту связывали с 4-бензилпиперидином с получением указанных в заголовке соединений, MS (М+ 349).
Кроме того, 4-метоксииндол-5-карбоновую кислоту связывали с 4-(4-фторбензил)пиперидином с получением 4-метокси-(4-(4-фторбензил)пиперидинил)индол-5-карбоксамида, MS (M+ 367) и 6-метоксииндол-5-карбоновую кислоту связывали с 4-(4-фторбензил)пиперидином с получением 6-метокси-(4-(4-фторбензил)пиперидинил)индол-5-карбоксамида, MS (M+ 367).
Пример 19
Получение N-(3-циклогексилметиламино-2-гидроксипропил)-4-бензилпиперидинилиндол-5-карбоксамида

А. Указанное в заголовке соединение получали в соответствии со схемой 7. К охлажденному на льду раствору 1,0 г (3,0 ммоль) 4-бензилпиперидинилиндол-5-карбоксамида в ацетоне добавляли 15 ммоль порошкообразного КОН с последующим добавлением 3,0 ммоль эпибромгидрина и смесь перемешивали в течение 30 минут. Смесь фильтровали и раствор упаривали. Остаток растворяли в этилацетате, промывали водой, сушили и упаривали. После очистки колоночной хроматографией на силикагеле получали 435 мг эпоксида. MS (М+ 373).
К раствору 200 мг (0,54 ммоль) вышеуказанного эпоксида индола в 5 мл МеОН добавляли 121 мг (1,1 ммоль) циклогексилметиламина и смесь кипятили с обратным холодильником в течение 1 часа. Неочищенный продукт очищали на колонке силикагеля. Затем это аминосоединение превращали в его HCl-соль обработкой раствором HCl в этаноле. MS (M+ 487).
В. Согласно методике стадии А, но с заменой циклогексилметиламина пиперазином получали N-(3-N-метилпиперазинил-2-гидроксипропил)-4-бензилпиперидинилиндол-5-карбоксамид:

MS (M+ 473);
при замене циклогексилметиламина бензиламином получали N-(3-бензиламино-2-гидроксипропил)-4-бензилпиперидинил-индол-5-карбоксамид:

MS (M+ 481);
при замене циклогексилметиламина п-метоксибензиламином получали N-[3-{(4-метоксибензил)амино}-2-гидроксипропил]-5-(4-бензилпиперидинилиндол)-5-карбоксамид:

MS (M+ 511);
при замене циклогексилметиламина пропиламином получали N-{3-н-пропиламино-2-гидроксипропил}-(4-бензилпиперидинилиндол)-5-карбоксамид:

MS (М+ 433).
Пример 20
Получение других 1-замещенных производных
А. Получение N-(4-пиридоил)-4-бензилпиперидинилиндол-5-карбоксамида

0,318 мг (1 ммоль) 4-бензилпиперидинилиндол-5-карбоксамида растворяли в 15 мл сухого ДМФ, добавляли 80 мг (60% суспензии в масле) гидрида натрия и смесь перемешивали в течение 30 минут в атмосфере азота. Смесь охлаждали до 0°С и добавляли 200 мг (1,1 ммоль) гидрохлорида изоникотинилхлорида и смесь перемешивали в течение 20 часов при комнатной температуре. Реакцию останавливали добавлением насыщенного раствора хлорида аммония, разбавляли водой и продукт экстрагировали дихлорметаном. Экстракт сушили, упаривали и остаток хроматографировали на силикагеле (этилацетат-гексан, градиент 50-75% этилацетат) с получением 150 мг чистого продукта. ESI MS (M+ 423, М+-Н, 422).
С использованием методики предыдущего абзаца, но с заменой гидрохлорида изоникотинилхлорида гидрохлоридом 4-пиколилхлорида, получали N-(4-пиридилметил)-4-бензилпиперидинилиндол-5-карбоксамид:

MS (М+ 409).
В. Получение 1-никотиноил-(4-бензилпиперидинил)индол-6-карбоксамида:
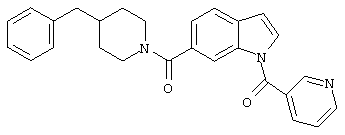
Это соединение получали таким же образом, М+ 423.
С. Получение 1-никотиноил-6-метокси-(4-бензилпиперидинил)индол-5-карбоксамида:

Это соединение получали таким же образом. М+ 490.
D. Получение N-метилацетил-4-бензилпиперидинилиндол-5-карбоксамида и его свободной кислоты:

1,95 г (6,13 ммоль) 4-бензилпиперидинилиндол-5-карбоксамида растворяли в 30 мл сухого ДМФ и обрабатывали 320 мг (8 ммоль, 60% суспензии в масле) гидрида натрия в течение 30 минут. Реакционную смесь охлаждали до 0°С и добавляли 1,225 г (8 ммоль) бромметилацетата и перемешивание продолжали в течение 1 часа при 0°С. Баню со льдом удаляли и перемешивание продолжали еще в течение 6 часов при комнатной температуре. Реакцию останавливали добавлением насыщенного раствора хлорида аммония, разбавляли водой и продукт экстрагировали дихлорметаном. Экстракт сушили, упаривали и остаток очищали колоночной хроматографией на силикагеле с элюированием смесью этилацетат-гексан (25-35% этилацетат) с получением 2,2 г (92%) целевого продукта. MS: M+, 390; М+-1, 389. 1H-ЯМР (CDCl3): δ 7,7 (с, 1Н), 7,35-7,1 (м, 8Н), 6,6 (с, 1Н), 5,1 (с, 2Н), 3,75 (с, 3Н), 3,0-2,7 (шм, 4Н), 2,6 (д, 2Н), 1,9-1,2 (м, 5Н).
2,15 г (5,5 ммоль) 4-бензилпиперидинилиндол-5-карбоксамид-1-метилацетата с предыдущей стадии помещали в 20 мл этанола. Добавляли раствор 2,0 г К2СО3 в 20 мл воды и смесь кипятили с обратным холодильником в течение 2 часов. Этанол удаляли при пониженном давлении, оставшийся раствор разбавляли водой и подкисляли концентрированной HCl. Осажденный продукт собирали фильтрованием, промывали водой и сушили с получением 1,9 г продукта. MS: M+, 376; M+-H, 375.
Е. Получение 1-акрилоил-(4-бензилпиперидинил)индол-5-карбоксамида
0,318 г (1 ммоль) (4-бензилпиперидинил)индол-5-карбоксамида помещали в 15 мл сухого ацетона и подвергали реакции с 0,2 г (5 ммоль) порошкообразного КОН в течение 15 минут. Смесь охлаждали на льду и добавляли 0,225 мг (2,5 ммоль) акрилоилхлорида одной порцией. Перемешивание продолжали при 0°С в течение 20 минут, после чего реакционную массу дополнительно перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли в вакууме и остаток экстрагировали этилацетатом из воды. Экстракт сушили и упаривали. ТСХ (этилацетат-гексан) и масс-спектр (M+ при 372) подтверждала образование целевого продукта. Этот продукт использовали без дополнительной очистки на следующей стадии.
F. 1-[3-(2-пропиламино)пропионил]-(4-бензилпиперидинил)-5-индолкарбоксамид:

Полученный, как описано выше, продукт растворяли в 20 мл дихлорметана и подвергали реакции с 0,1 мл изопропиламина при комнатной температуре в течение 18 часов. Растворитель удаляли и продукт очищали колоночной хроматографией на силикагеле с элюированием смесью хлороформ-метанол (95:5). Выход: 180 мг, М+, 431.
G. 1-(3-пиперазинилпропионил)-(4-бензил)пиперидинил-индол-5-карбоксамид:

1-Акрилоил-(4-бензил)пиперидинилиндол-5-карбоксамид с предыдущей стадии подвергали взаимодействию с трет-бутил-1-пиперазинкарбоксилатом, как описано выше. Продукт освобождали от защитных групп, используя раствор HCl в метаноле. М+ 458.
Н. 1-(3-бензиламинопропионил)-(4-бензилпиперидинил)индол-5-карбоксамид:

получали реакцией 1-акрилоил-(4-бензилпиперидинил)ин-дол-5-карбоксамида с бензиламином. М+ 479.
I. 1-(3-морфолинилпропионил)-4-(4-бензилпиперидинил)индол-5-карбоксамид

получали реакцией 1-акрилоил-(4-бензилпиперидинил)индол-5-карбоксамида с морфолином. M+ 459.
J. Получение н-пропиламида 4-бензилпиперидинилиндол-5-карбоксамид-1-уксусной кислоты:

4-бензилпиперидинилиндол-5-карбоксамид-1-уксусную кислоту (200 мг, 0,53 ммоль) примера 21 подвергали взаимодействию со 120 мг (0,61 ммоль) EDC в 10 мл сухого дихлорметана в течение 30 минут. Добавляли н-пропиламин (100 мкл, избыток) и смесь перемешивали в течение 20 часов. Раствор разбавляли дихлорметаном, промывали водой и 5% раствором карбоната натрия. Органический раствор сушили и упаривали и остаток очищали хроматографией на силикагеле, используя смесь этилацетат-гексан (3:2) с получением 100 мг продукта. MS (М+ 417).
К. Получение (4-метоксибензил)амида 4-бензилпиперидинил-индол-5-карбоксамид-1-уксусной кислоты:

Согласно методике, описанной для предыдущей стадии, но с заменой н-пропиламина п-метоксибензиламином, получали (4-метоксибензил)амид 4-бензилпиперидинилиндол-5-карбоксамид-1-уксусной кислоты. MS, ESI: М+Н, 496.
L. Получение 1-(диэтиламиноэтил)-6-метокси-(4-бензилпиперидинил)индол-5-карбоксамида:

0,3 г (0,862 ммоль) 6-метокси-(4-бензилпиперидинил)индол-5-карбоксамида растворяли в 20 мл сухого ДМФ. Раствор охлаждали на бане со льдом и подвергали взаимодействию с 0,12 г NaH (3 ммоль, 60% суспензия). Добавляли 0,172 мг (1 ммоль) гидрохлорида 2-(диэтиламино)этилхлорида, и смесь перемешивали в течение 18 часов. Реакционную смесь выливали в воду и экстрагировали дихлорметаном (3×75 мл). Объединенный экстракт повторно промывали водой, сушили над безводным MgSO4, упаривали и очищали хроматографией на силикагеле на хроматотроне с использованием смеси СН2Cl2/метанол (95:5) с получением 0,22 г целевого продукта, который превращали в HCl-соль и лиофилизировали, М+ 448.
М. Согласно методике предыдущей стадии, но с заменой гидрохлорида 1-(диэтиламино)этилхлорида гидрохлоридом 1-(диэтиламино)-н-пропилхлорида, получали 1-(диэтиламино)-н-пропил-(4-бензилпиперидинил)индол-5-карбоксамид,
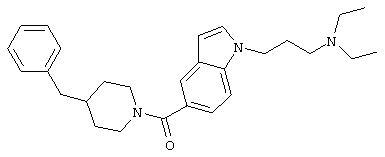
M+ 468.
Подобным образом, получали 1-(диэтиламино)-этил-(4-бензилпиперидинил)индол-5-карбоксамид,

M+ 454.
Подобным образом, получали 1-(диэтиламино)-н-пропил-6-хлор-(4-бензилпиперидинил)индол-5-карбоксамид,

M+ 502.
Подобным образом получали 1-(диэтиламино)-этил-(4'-фтор-4-бензилпиперидинил)индол-5-карбоксамид,

М+, 472.
Аналогично получали 1-(диэтиламино)-н-пропил-6-метокси-(4-бензилпиперидинил)индол-5-карбоксамид,

М+, 498.
Пример 21
Получение 3-замещенных индолов
Общая методика для синтеза 3-замещенных индолов описана следующим образом:
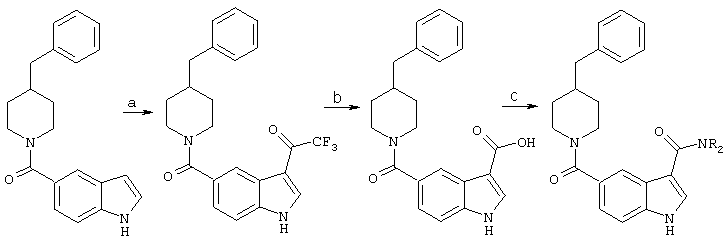
a) Трифторуксусный ангидрид, ТГФ, 0°С, 3 часа.
b) вод. NaOH, кипячение с обратным холодильником, 3-6 часов.
c) R2NH, EDAC·HCl, DMAP, ДХМ/ДМФ, 3-6 часов.
А. 3-трифторацетил-4-бензилпиперидинилиндол-5-карбоксамид: 4-бензилпиперидинилиндол-5-карбоксамид (1 экв.) растворяли в безводном ТГФ. Реакционный сосуд продували азотом и помещали на баню со льдом. Через шприц добавляли трифторуксусный ангидрид (1,2-1,3 экв.). Реакции давали продолжаться при 0°С, пока не переставал обнаруживаться исходный материал согласно анализу тонкослойной хроматографии. В некоторых случаях требовалось добавление дополнительного количества трифторуксусного ангидрида для облегчения завершения реакции. После завершения реакции реакционную смесь концентрировали и повторно растворяли в минимальном количестве этилацетата для хроматографии с использованием диоксида кремния. Неочищенный материал хроматографировали с использованием этилацетата и гексанов (1:1). Идентичность продукта 3-трифторацетил-4-бензилпиперидинилиндол-5-карбоксамида определяли масс-спектроскопией с бомбардировкой электронами. (МН+ 413 (эксп.414), основной пик 240).
Подобным образом, с использованием в качестве исходного материала 4-бензилпиперидинилиндол-6-карбоксамида или 6-метокси-(4-бензилпиперидинил)индол-5-карбоксамида, получали соответствующие 3-трифторацетилпроизводные.
В. 4-бензилпиперидинилиндол-5-карбоксамид-3-карбоно-вая кислота: Производное трифторацетилиндола со стадии А суспендировали в водном гидроксиде натрия (10 н., 5-6 экв.) и кипятили с обратным холодильником. В начале дефлегмации добавляли минимальное количество метанола для облегчения растворения. Реакционную смесь поддерживали при кипячении в течение 3-6 часов. По завершении реакционную смесь охлаждали до комнатной температуры, разбавляли водой и промывали эфиром. Затем водный слой подкисляли до рН 4 концентрированной HCl, поместив на баню со льдом. Затем кислоту экстрагировали этилацетатом и промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и концентрировали с получением твердого вещества. Твердое вещество очищали хроматографией на диоксиде кремния с использованием смеси этилацетат:гексаны:метанол:уксусная кислота, 5:5:1:0,1. Идентичность продукта определяли масс-спектроскопией с бомбардировкой электронами. (МН+ 361 (эксп.361), 317, основной пик 144).
Подобным образом, другие 3-фторацетилпроизводные, полученные на стадии А, превращали в соответствующие 3-карбоновые кислоты.
С. 3-(2-диметиламино)этиламинокарбоксамидил-(4-бензилпиперидинил)индол-5-карбоксамид: Карбоновую кислоту стадии В (1 экв.) обрабатывали 1,1 эквивалентом EDAC•HCl и 1 экв. диметиламиноэтилендиамина в присутствии каталитического количества DMAP в ДМФ/ДХМ 1:1 в течение 3-6 часов. Затем реакционную смесь концентрировали и помещали в этил-ацетат. После промывания 5% водным карбонатом натрия и раствором насыщенного хлорида натрия органический слой сушили над безводным сульфатом натрия и концентрировали с получением неочищенного материала. Неочищенный материал очищали хроматографией на диоксиде кремния. Идентичность продукта, показанного ниже, определяли масс-спектроскопией с бомбардировкой электронами. (МН+ 432 (эксп.432)).

D. Подобным образом, производя подходящие замены карбоксамида и участвующего в реакции амина, получали следующие соединения данного изобретения в соответствии со схемой реакций, приведенной в начале этого примера; все эти соединения являются соединениями приведенной выше формулы, но с другими заместителями на карбонильной части молекулы в положении 3 индольной части молекулы, как указано.
i. 3-(2-метоксиэтиламинокарбоксамидил)-(4-бензилпиперидинил)индол-5-карбоксамид (3-карбонильный заместитель представляет собой 2-метоксиэтиламино, МН+ 418, эксп.418);
ii. 3-(2-метиламиноэтиламинокарбоксамидил)-(4-бензилпиперидинил)индол-5-карбоксамид (3-карбонильный заместитель представляет собой 2-метиламиноэтиламино, МН+ 418, эксп.418);
iii. 3-(N-метил-2-аминоэтиламинокарбоксамидил)-(4-бензилпиперидинил)индол-5-карбоксамид (3-карбонильный заместитель представляет собой 2-аминоэтил(метил)амино, МН+ 418, эксп.418);
iv. 3-(4-бензилпиперидинилкарбоксамидил)-(4-бензилпиперидинил)индол-5-карбоксамид (3-карбонильный заместитель представляет собой 4-бензилпиперидинил, MH+ 519, эксп.519);
v. 3-(4-бензилпиперидинилкарбоксамидил)-(4-бензилпиперидинил)индол-6-карбоксамид (3-карбонильный заместитель представляет собой 4-бензилпиперидинил, МН+ 519, эксп.519);
vi. 3-(4-фторбензиламинокарбоксамидил)-(4-бензилпиперидинил)индол-5-карбоксамид (3-карбонильный заместитель представляет собой 4-фторбензиламино, МН+ 469, эксп.469);
vii. 3-2-(3,4-диметоксифенил)этиламинокарбоксамидил-(4-бензилпиперидинил)индол-5-карбоксамид (3-карбонильный заместитель представляет собой 2-(3,4-диметоксифенил)этиламино, МН+ 525, эксп.525);
viii. 3-трифторацетил-(4-бензилпиперидинил)индол-5-карбоксамид (3-карбонильный заместитель представляет собой трифторметил, МН+ 413, эксп.414);
ix. 3-трифторацетил-(4-бензилпиперидинил)индол-6-карбоксамид (3-карбонильный заместитель представляет собой трифторметил, МН+ 413, эксп.414);
х. 6-метокси-3-(2-диметиламиноэтиламино)карбоксамидил-(4-бензилпиперидинил)индол-5-карбоксамид (3-карбонильный заместитель представляет собой 2-диметиламиноэтил, включающий также заместитель 6-метокси в положении 6, МН+ 462, эксп.462).
Формулы соединений i-x, описанных выше, показаны ниже.


Альтернативно, соединения данного изобретения, которые являются производными индола с заместителями в положении 3, могут быть получены с использованием схемы реакций 8, приведенной ранее.
Е. Получение 3-морфолинометил-(4-бензилпиперидинил)индол-5-карбоксамида:
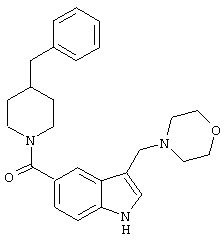
По схеме 8 0,318 г (1 ммоль) 4-бензилпиперидинилиндол-5-карбоксамида, 0,1 г параформальдегида (3,3 ммоль) и 0,1 мл морфолина помещали в 25 мл абсолютного этанола и подкисляли добавлением 1 мл этанольного раствора HCl. Смесь кипятили с обратным холодильником в течение 18 часов. Растворитель удаляли и остаток экстрагировали из 5% раствора в карбонате натрия дихлорметаном. Экстракт сушили, упаривали и остаток очищали колоночной хроматографией с использованием смеси этилацетат-метанол (95:5) с получением 0,15 г целевого продукта, который превращали в HCl-соль и лиофилизировали. М+ 454.
F. Получение диэтиламинометил-(4-бензилпиперидинил)индол-5-карбоксамида:

По схеме 8 это соединение получали следующим образом. К суспензии 0,41 г (1,28 ммоль) 4-бензилпиперидинилиндол-5-карбоксамида в 5 мл ледяной уксусной кислоты добавляли охлажденную на льду смесь 1,2 мл водного формальдегида (37%) и 0,16 мл этиламина (1,5 ммоль). Реакционную смесь перемешивали в течение 30 минут при 0°С и затем перемешивание продолжали при комнатной температуре в течение 18 часов. Смесь выливали в воду, подщелачивали добавлением 20% раствора гидроксида натрия и экстрагировали этилацетатом. Экстракт сушили и упаривали. Остаток очищали хроматографией на силикагеле с элюированием смесью хлороформ-метанол-триэтиламин (95:5:0,5) с получением 0,22 г указанного в заголовке соединения. MS: 403, М+; 331, M+-Net2.
Пример 22
Тест на ингибирование киназы р38
Тестируемые соединения солюбилизировали в ДМСО и растворяли в воде до желаемых концентраций. Киназу р38 разбавляли до 10 мкг/мл в буфере, содержащем 20 мМ MOPS, pH 7,0, 25 мМ бета-глицерофосфат, 2 мг/мл желатина, 0,5 мМ ЭДТА и 4 мМ ДТТ.
Реакцию проводили смешиванием 20 мкл тестируемого соединения с 10 мкл субстратной смеси, содержащей 500 мкг/мл пептидного субстрата и 0,2 мМ АТФ (+ 200 мкКи/мл гамма-32P-АТФ) в 4× тест-буфере. Реакцию инициировали добавлением 10 мкл киназы р38. Конечные условия теста были следующими: 25 мМ MOPS, рН 7,0, 26,25 мМ бета-глицерофосфат, 80 мМ KCl, 22 мМ MgCl2, 3 мМ MgSO4, 1 мг/мл желатина, 0,625 мМ ЭДТА, 1 мМ ДТТ, 125 мкг/мл пептидного субстрата, 50 мкМ АТФ и 2,5 мкг/мл фермента. Через 40 минут инкубации при комнатной температуре реакцию останавливали добавлением 10 мкл на одну реакционную смесь 0,25 М фосфорной кислоты.
Часть реакционной смеси наносили в виде пятна на диск фосфоцеллюлозной бумаги Р81, фильтры сушили в течение 2 минут и затем промывали 4× в 75 мМ Н3PO4. Фильтры промывали быстро в 95% этаноле, сушили, затем помещали в сцинтилляционные кюветы с жидкой сцинтилляционной смесью.
Альтернативно, субстрат сначала биотинилировали и полученные реакционные смеси наносили в виде пятен на содержащие стрептавидин SAM2™ фильтры-квадраты (Promega). Эти фильтры промывали 4× в 2 М NaCl, 4× в 2 М NaCl с 1% фосфорной кислотой, 2× в воде и быстро в 95% этаноле. Фильтры-квадраты сушили и помещали в сцинтилляционные кюветы с жидкой сцинтилляционной смесью.
Включение в имп/мин определяли на сцинтилляционном счетчике. Относительную активность фермента рассчитывали вычитанием имп/мин фона (имп/мин, измеренные в отсутствие фермента) из каждого результата и сравнением полученных результатов с результатами, полученными в отсутствие ингибитора. Величины IC50 определяли вычерчиванием графиков с использованием общепринятых пакетов программного обеспечения. Приблизительные величины IC50 рассчитывали по формуле
IC50 (прибл.)=(А×i)/(1-А)
где А = дробная активность, a i = общая концентрация ингибитора.
Пример 23
Активность соединений данного изобретения
Активность соединений данного изобретения определяли, как описано выше. Испытуемыми соединениями были 4-бензилпиперидинил- или 4-бензилпиперазинилиндол-5- или 6-карбоксамиды. Обычно, пиперидинилпроизводное превосходило соответствующее пиперазинилпроизводное. IC50 для ингибирования р38α показана в таблице 1.
Видно также, что нахождение заместителей пиперидинила или пиперазинила в положении 3, 4, 5 и 6 приводит к более высокой активности, чем нахождение этих заместителей в положении 2 или 7.
Те же самые соединения тестировали на их специфичность в отношении р38α в сравнении с р38β. Эти результаты показаны в таблице 2.
Активность в отношении р38β также определяли для соединений данного изобретения для определения влияния положения заместителя пиперидинила или пиперазинила. Испытывали также влияние замещения на бензильной части, присоединенной к положению 4 пиперазина или пиперидина. Эти результаты показаны в таблице 3 как процентное ингибирование активности р38β при концентрации 50 мкМ соединения.
Замена индольной части на бензимидазольную часть молекулы в соединениях данного изобретения также приводила к значительному ингибированию р38β при тестировании этих соединений при 50 мкМ. 4-бензилпиперидинилбензимидазол-5-карбоксамид дал 85%-ное ингибирование; 4-(3-хлорбензил)пиперазинилбензимидазол-5-карбоксамид дал 66%-ное ингибирование.
Соединения данного изобретения обычно являются специфическими в отношении p38α в сравнении с p38β. Видно, что специфичность в отношении α в сравнении с β является специфичностью десятикратного порядка.
Специфичность соединений данного изобретения определяли также в отношении других киназ, в том числе р38γ, ERK-2, РКА, РКС, cdc-2, EGF-R и DNA-PK, как показано в таблице 4. Тестируемыми соединениями были 4-бензилпиперидинилиндол-5- и 6-карбоксамиды с числом, указывающим положение кольца карбоксамида.
5
6
Эти результаты даны в виде приблизительных величин IC50 (мкМ), полученных, при испытании этих соединений при 50 мкМ, и рассчитаны по формуле в примера 22. Исключением являются величины для р38α, в этом случае величины IC50 были определены с помощью кривых зависимости от концентрации, используемой в тесте.
Как показано, все тестируемые соединения являются высокоспецифичными для указанных других киназ.
В таблице 5 показано ингибирование р38α соединениями данного изобретения, которые являются 4-(бензилпиперидинил)индол-5-карбоксамидами или 4-[(4-фторбензил)пиперидинил]индол-5-карбоксамидами, т.е. соединениями формул (1) или (2);

где R1 имеет формулу (11) или (12):

Приведенные величины представляют собой IC50 в мкМ.
0,577


Некоторые другие соединения данного изобретения также тестировали. Соединение формулы (3), т.е. соединение, в котором карбоксамид находится в положении 6, R1 имеет формулу (11), R3 представляет собой Н, R2 представляет собой Н, Z1 представляет собой CCOCF3 и Z2 представляет собой СН, показало 41%-ное ингибирование при 1 мкМ. Подобным образом соединение, в котором R1 имеет формулу (11) и которое замещено в положении 6 индолом, R2 представляет собой 
и оба Z1 и Z2 обозначают СН, имеет IC50 0,505 мкМ.
Испытывали также два соединения, в которых R1 имеет формулу:

Одно из этих соединений, соединение формулы (2), в котором R3 представляет собой МеО, R2 представляет собой Н и оба Z1 и Z2 представляют собой СН, дало 63%-ное ингибирование при 0,2 мкМ; другое соединение, соединение формулы (3), в котором R3 представляет собой Н, R2 представляет собой Н, Z1 представляет собой N и Z2 представляет собой СН, имеет IC50 2,15 мкМ. Наконец, тестировали одно соединение, в котором R1 был заместителем формулы:

и которое является соединением формулы (3), в котором R3 представляет собой Н, R2 представляет собой Н и оба Z1 и Z2 представляют собой СН, и это соединение дало 51%-ное ингибирование при 1 мкМ.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ИНДОЛЬНОГО РЯДА В КАЧЕСТВЕ ИНГИБИТОРОВ p38 КИНАЗЫ | 2000 |
|
RU2278115C2 |
| НОВЫЕ СОЕДИНЕНИЯ, КОТОРЫЕ ЯВЛЯЮТСЯ ИНГИБИТОРАМИ ERK | 2013 |
|
RU2660429C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА МИНЕРАЛОКОРТИКОИДОВ | 2012 |
|
RU2598842C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА ЭСТРОГЕНОВЫХ РЕЦЕПТОРОВ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2018 |
|
RU2797808C2 |
| PROTAC, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА ТАУ-БЕЛОК, И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2805523C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОНАФТАЛИНА И ТЕТРАГИДРОИЗОХИНОЛИНА В КАЧЕСТВЕ РАЗРУШИТЕЛЕЙ ЭСТРОГЕНОВОГО РЕЦЕПТОРА | 2017 |
|
RU2797244C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2382779C2 |
Изобретение относится к новым ортозамещенным и N-замещенным индолам формулы (α) или (β)

или их фармацевтически приемлемым солям, где Z1-CR4 или N, R4-H, C1-С6алкил, необязательно включающий атом 0 или N, возможно замещенный галогеном, кето, 5-6-членным циклоалифатическим радикалом, возможно содержащим 1-2 атома 0 или N, Z2-CH или CR, где R-C1-С6-алкил, R1-

где X1-CO, или его изостер, m=0, 1, Y - алкил, возможно замещенный, или два Y вместе образуют С2-С3-алкилен, n=0, 1 или 2, Z3-CH или N, Х2-СН, СН2 или их изостер, Ar - одна или две фенильные группы, связанные с X2, где фенил может быть замещен, R2-H, C1-С6алкил, или арил, где каждый арил возможно включает атом 0 или N и может быть замещен. Соединения являются селективными ингибиторами р 38α-киназы. 1 н. и 33 з.п. ф-лы, 5 табл.

или его фармацевтически приемлемые соли,
где Z1 представляет собой CR4 или N, где R4 представляет собой Н или C1-С6алкил необязательно включающий один гетероатом, выбранный из О и N, и необязательно замещенный одним или несколькими заместителями, выбранными из группы, состоящей из галогена, =O и одного или нескольких 5- или 6-членных алифатических колец, необязательно содержащих 1-2 гетероатома, выбранных из О и N;
Z2 представляет собой СН или CR, где R представляет собой C1-С6алкил;
R1 представляет собой

где X1 представляет собой СО или его изостер;
m равно 0 или 1;
Y представляет собой необязательно замещенный алкил или два Y, взятые вместе, могут образовать С2-С3алкиленовый мостик;
n равно 0, 1 или 2;
Z3 представляет собой СН или N;
Х2 представляет собой СН, СН2 или их изостер и
Ar состоит из одной или двух фенильных групп, связанных непосредственно с X2, причем указанные одна или две фенильные группы необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, C1-С6алкила, С2-С6алкенила, CN, CF3, RCO, COOR, NR2, OR и SR и фенила, который необязательно замещен одним или несколькими вышеуказанными заместителями; где R в вышеуказанных необязательных заместителях представляет собой Н или С1-С6алкил; R2 представляет собой Н, C1-С6алкил или арил, причем каждый алкил или арил необязательно включает один гетероатом, выбранный из О и N, и необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, OR, NR2, CN или =O (где R2 представляет собой алкил), и одним или несколькими алифатическими или ароматическими 5- или 6-членными кольцами, необязательно содержащими 1-2 гетероатома, где R в указанных необязательных заместителях представляет собой Н или C1-С6алкил;
R3 выбран из группы, состоящей из Н, галогена и O-C1-С6алкила.



где каждый X3 представляет собой независимо C1-С6алкил, галоген, OR или NR2 и р равно 0, 1, 2 или 3.
где каждый Х3 представляет собой независимо C1-С6алкил, галоген, OR или NR2 и р равно 0, 1, 2 или 3.

или имеет структуру формулы (5) или (6), где положения на индольном или бензимидазольном кольце, занимаемые R3 и заместителем, иллюстрированным как R1, являются обращенными, где R2, R3 и R4 имеют определенные в п.1 значения и каждый X3 представляет собой независимо галоген, C1-С6алкил, OR или NR2, где R представляет собой Н или C1-С6алкил, и р равно 0, 1, 2 или 3.

где R2, R4, X3 и р имеют указанные в п.19 значения.

где R2, X3 и р имеют определенные в п.19 значения.
4-хлор-4-бензилпиперидинилиндол-5-карбоксамид;
6-хлор-4-бензилпиперидинилиндол-5-карбоксамид,
4-хлор-(4-(4-фторбензил)пиперидинил)индол-5-карбоксамид;
6-хлор-(4-(4-фторбензил)пиперидинил)индол-5-карбоксамид;
4-метокси-(4-бензилпиперидинил)индол-5-карбоксамид;
6-метокси-(4-бензилпиперидинил)индол-5-карбоксамид;
4-метокси-(4-(4-фторбензил)пиперидинил)индол-5-карбоксамид;
6-метокси-(4-(4-фторбензил)пиперидинил)индол-5-карбоксамид;
N-(3-циклогексилметиламино-2-гидроксипропил)-(4-бензилпиперидинил)индол-5-карбоксамид;
N-(3-N-метилпиперазинил-2-гидроксипропил)-(4-бензилпиперидинил)индол-5-карбоксамид;
N-(3-бензиламино-2-гидроксипропил)-(4-бензилпиперидинил)индол-5-карбоксамид;
N-[3-{(4-метоксибензил)амино}-2-гидроксипропил]-(4-бензилпиперидинил)индол-5-карбоксамид;
N-{3-н-пропиламино-2-гидроксипропил}-(4-бензилпиперидинил)индол-5-карбоксамид;
N-(4-пиридоил)-(4-бензилпиперидинил)индол-5-карбоксамид;
N-(4-пиридилметил)-(4-бензилпиперидинил)индол-5-карбоксамид;
N-метилацетил-(4-бензилпиперидинил)индол-5-карбоксамид;
N-ацетил-4-бензилпиперидинилиндол-5-карбоксамид;
N-(н-пропиламид)ацетил-4-бензилпиперидинилиндол-5-карбоксамид;
н-бутиламид 4-бензилпиперидинилиндол-5-карбоксамид-1-уксусной кислоты;
4-метоксибензиламид 4-бензилпиперидинилиндол-5-карбоксамид-1-уксусной кислоты;
3-(2-метоксиэтиламинокарбоксамидил)-(4-бензилпиперидинил)индол-5-карбоксамид;
3-(2-метиламиноэтиламинокарбоксамидил)-(4-бензилпиперидинил)индол-5-карбоксамид;
3-(2-аминоэтиламинокарбоксамидил)-(4-бензилпиперидинил)индол-5-карбоксамид;
3-(4-бензилпиперидинилкарбоксамидил)-(4-бензилпиперидинил)индол-5-карбоксамид;
3-(4-бензилпиперидинилкарбоксамидил)-(4-бензилпиперидинил)индол-6-карбоксамид;
3-(4-фторбензилкарбоксамидил)-(4-бензилпиперидинил)индол-5-карбоксамид;
3-[2-(3,5-диметоксифенил)этилкарбоксамидил]-(4-бензилпиперидинил)индол-5-карбоксамид;
6-метокси-(4-бензилпиперидинил)индол-5-карбоксамид;
3-трифторацетил-(4-бензилпиперидинил)индол-5-карбоксамид;
6-метокси-3-(2-диметиламиноэтиламино)карбоксамидил-(4-бензилпиперидинил)индол-5-карбоксамид;
3-трифторацетил-4-бензилпиперидинилиндол-5-карбоксамид;
4-бензилпиперидинилиндол-5-карбоксамид-3-карбоновая кислота;
3-(2-диметиламино)этиламинокарбоксамидил-(4-бензилпиперидинил)индол-5-карбоксамид;
или соединения общей формулы (1) или (2)


3-[2-диметиламиноэтиламинокарбонил]-4-бензилпиперидинил-6-метоксииндол-5-карбоксамид; или
4-бензилпиперидинил-6-метоксибензимидазол-5-карбоксамид.
Приоритеты: от 22.05.1998, по заявке США 60/086531.
соединения формулы (α) или (β) по пп. 1-34 или их фармацевтически приемлемые соли, в которых
Z1 представляет собой CR4 или N; где
R4 представляет собой Н или C1-С6алкил;
Z2 представляет собой СН или CR, где R представляет собой C1-С6алкил;
R1 представляет собой

где X1 представляет собой СО или его изостер (CH2, SO или SO2);
m равно 1;
n равно 0;
Z3 представляет собой СН или N;
X2 представляет собой СН, СН2 или СО; и
Ar состоит из одной или двух фенильных групп, связанных непосредственно с X2, причем указанные одна или две фенильные группы необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, C1-С6алкила, CN, CF3, COOR, NR2, OR и SCH3 и фенила; где R в вышеуказанных необязательных заместителях представляет собой Н или метил или этил;
R2 представляет собой Н, C1-С6алкил или бензил; R3 выбран из Н.
от 03.08.1998, по заявке США №09/128137
соединения формулы (α) или (β) по пп. 1-34 или их фармацевтически приемлемые соли, в которых
Z1 представляет собой CR4 или N; где
R4 представляет собой Н или С1-С4алкил необязательно включающий один гетероатом, выбранный из О;
Z2 представляет собой СН или CR, где R представляет собой C1-С4алкил;
R1 представляет собой
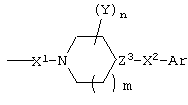
где X1 представляет собой СО или его изостер;
m равно 0 или 1;
Y представляет собой необязательно замещенный алкил;
n равно 0, 1;
Z3 представляет собой СН или N;
Х2 представляет собой СН, СН2 или их изостер; и
Ar состоит из одной или двух фенильных групп, связанных непосредственно с X2, причем указанные одна или две фенильные группы необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, C1-С6алкила, CN, CF3, RCO, COOR, NR2, OR и SR и фенила, который необязательно замещен одним или несколькими вышеуказанными заместителями; где R в вышеуказанных необязательных заместителях представляет собой Н или C1-С6алкил;
R2 представляет собой Н, C1-С6алкил, причем каждый алкил необязательно включает один гетероатом, выбранный из О;
R3 выбран из группы, состоящей из Н, галогена и O-C1-С6алкила.
соединения от 24.03.1999, по заявке №09/275176
формулы (α) или (β) по пп. 1-34 или их фармацевтически приемлемые соли, в которых
Z1 представляет собой CR4 или N; где
R4 представляет собой Н или C1-С6алкил необязательно включающий один гетероатом, выбранный из О и N, и необязательно замещенный одним или несколькими заместителями, выбранными из группы, состоящей из галогена, =O и одного 5- или 6-членного алифатического кольца, необязательно содержащего 1-2 гетероатома, выбранных из N;
Z2 представляет собой СН или CR, где R представляет собой C1-С6алкил;
R1 представляет собой

где X1 представляет собой СО или его изостер;
m равно 0 или 1;
Y представляет собой необязательно замещенный алкил, или два Y, взятые вместе, могут образовать С2-С3алкиленовый мостик;
n равно 0, 1 или 2;
Z3 представляет собой СН или N;
X2 представляет собой СН, СН2 или их изостер; и
Ar состоит из одной или двух фенильных групп, связанных непосредственно с Х2, причем указанные одна или две фенильные группы необязательно замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена, C1-С6алкила, С2-С6алкенила, CN, CF3, RCO, COOR, NR2, OR и SR и фенила, который необязательно замещен одним или несколькими вышеуказанными заместителями; где R в вышеуказанных необязательных заместителях представляет собой Н или C1-С6алкил;
R2 представляет собой Н, C1-С6алкил, причем каждый алкил необязательно включает один гетероатом, выбранный из О и N, и необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, OR, NR2, CN или =O (где R2 представляет собой алкил), и одним алифатическим или ароматическим 5- или 6-членным кольцом, необязательно содержащим 1-2 гетероатома, где R в указанных необязательных заместителях представляет собой Н или C1-С6алкил;
R3 выбран из группы, состоящей из Н, галогена и O-C1-С6алкила.
от 21.05.1999, по заявке PCT/US 99/11222 - соединения формулы (α) или (β) по пп.1-34 или их фармацевтически приемлемые соли, где R4 представляет собой C1-С6алкил необязательно включающий один гетероатом, выбранный из О и N, и необязательно замещенный одним или несколькими заместителями, выбранными из группы, состоящей из одного 5- или 6-членного алифатического кольца, необязательно содержащего 1-2 гетероатома, выбранных из О или нескольких 5- или 6-членных алифатических колец, необязательно содержащих 1-2 гетероатома, выбранных из О и N;
R2 представляет собой Н, C1-С6алкил, причем каждый алкил необязательно включает один гетероатом, выбранный из О и N, и необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из галогена, OR, NR2, CN или =O (где R2 представляет собой алкил), и одним алифатическим или ароматическим 5- или 6-членным кольцом, необязательно содержащим 1-2 гетероатома, где R в указанных необязательных заместителях представляет собой Н или C1-С6алкил; R3 выбран из группы, состоящей из Н, галогена и O-C1-С6алкила.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| ЕР 0431945 А2 12.06.1991 | |||
| Устройство для продвижения длинногогибКОгО пРОВОдА чЕРЕз пОлыЕ пРОХОды | 1976 |
|
SU831090A3 |

