Настоящее изобретение касается 4-оксо-1-(3-замещенный фенил)-1,4-дигидро-1,8-нафтиридин-3-карбоксамидов, пригодных в качестве ингибиторов фосфодиэстеразы-4.
Гормоны представляют собой соединения, оказывающие различное влияние на клеточную активность. Во многих отношениях гормоны действуют в качестве мессенжеров для запуска специфических клеточных ответов и активности. Многие эффекты, производимые гормонами, однако, вызваны не только отдельным эффектом именно этого гормона. Напротив, гормон сначала связывается с рецептором, вызывая, таким образом, высвобождение второго соединения, которое продолжает влиять на клеточную активность. В этом случае гормон называют первым мессенжером, тогда как соединение называют вторым мессенжером. Циклический аденозинмонофосфат (аденозин 3',5'-циклический монофосфат, "cAMP" или "циклический АМР") известен как второй мессенжер для гормонов, включающих эпинефрин, глюкагон, кальцитонин, кортикотрофин, липотропин, лютеинизирующий гормон, норэпинефрин, паратиреоидный гормон, тироидстимулирующий гормон и вазопрессин. Таким образом, cAMP опосредует клеточные ответы на гормоны. Циклический AMP также опосредует клеточные ответы на различные нейротрансмиттеры.
Фосфодиэстеразы ("PDE") представляют собой семейство ферментов, которые метаболизируют 3',5'-циклические нуклеотиды до 5'-нуклеозидмонофосфатов, прекращая, таким образом, активность cAMP в качестве второго месасенжера. Конкретная фосфодиэстераза, фосфодиэстераза-4 ("PDE4", также известная как "PDE-IV"), которая представляет собой PDE типа IV и обладает высоким сродством и специфичностью к cAMP, вызвала интерес как потенциальная мишень для разработки новых антиастматических и противовоспалительных соединений. Известно, что PDE4 существует в виде, по меньшей мере, четырех изоферментов, каждый из которых кодируется отдельным геном. Каждый из четырех известных генных продуктов PDE4, как полагают, играет разную роль в аллергических и/или воспалительных ответах. Поэтому считается, что ингибирование PDE4, в частности конкретных специфических изоформ PDE4, производящих неблагоприятные ответы, может благоприятно действовать на аллергические и воспалительные симптомы. Было бы желательно получить новые соединения и композиции, ингибирующие активность PDE4.
Основным недостатком применения ингибиторов PDE4 является побочный рвотный эффект, наблюдаемый в случае некоторых соединений-кандидатов, как описано в C.Burnouf et al., ("Burnouf"), Ann.Rep.In.Med.Chem., 33:91-109(1998). B.Hughes et al., Br.J.Pharmacol., 118:1183-1191(1996); M.J.Perry et al., Cell Biochem. Biophys., 29:113-132(1998); S.B.Christensen et al., J.Med.Chem., 41:821-835(1998); и Burnouf описывает различную степень тяжести нежелательных побочных эффектов, ингибируемых различными соединениями. Как описано в M.D.Houslay et al., Adv.In Pharmacol., 44:225-342(1998) and D.Spina et al., Adv.In Pharmacol., 44:33-89(1998), существует значительный интерес, и проводятся исследования терапевтических ингибиторов PDE4.
Международная патентная публикация WO9422852 описывает хинолины в качестве ингибиторов PDE4. Международная патентная публикация WO9907704 описывает производные 1-арил-1,8-нафтилидин-4-она в качестве ингибиторов PDE4.
A.H.Cook, и др., J.Chem.Soc., 413-417(1943) описывает гамма-пиридилхинолины. Другие хинолиновые соединения описаны в Kei Manabe и др, J.Org.Chem., 58(24):6692-6700(1993); Kei Manabe и др., J.Am.Chem.Soc., 115(12):5324-5325(1993); Kei Manabe и др., J.Am.Chem.Soc., 114(17):6940-6941(1992).
Соединения, включающие кольцевые системы, описаны различными исследователями как эффективные для различных терапевтических целей и применений. Например, Международная патентная публикация WO98/25883 описывает кетобензамиды в качестве ингибиторов калпаина, Европейская патентная публикация № ЕР 811610 и патенты США №№ 5679712, 5693672 и 5747541 описывают замещенные бензоилгуанидины в качестве блокаторов натриевых каналов, патент США № 5736297 описывает кольцевые системы, полезные в качестве светочувствительной композиции.
Патенты США №№ 5491147, 5608070, 5622977, 5739144, 5776958, 5780477, 5786354, 5798373, 5849770, 5859034, 5866593, 5891896 и Международная патентная публикация WO 95/35283 описывают ингибиторы PDE4, являющиеся тризамещенными производными арил- или гетероарилфенилов. Патент США № 5580888 описывает ингибиторы PDE4, являющиеся производными стирола. Патент США № 5550137 описывает ингибиторы PDE4, являющиеся производными фениламинокарбонилов. Патент США № 5340827 описывает ингибиторы PDE4, являющиеся производными фенилкарбоксамидов. Патент США № 5780478 описывает ингибиторы PDE4, являющиеся тетразамещенными фенильными производными. Международная патентная публикация WO 96/00215 описывает производные оксима в качестве ингибиторов PDE4. Патент США № 5633257 описывает ингибиторы PDE4, являющиеся цикло(алкил и алкенил)фенилалкенил(арил и гетероарил)производными.
Однако остается потребность в новых соединениях и композициях, которые терапевтически ингибируют PDE4 с минимальными побочными эффектами.
Настоящее изобретение направлено на биарилзамещенные 1,8-нафтиридин-4(1Н)-оны, представленные общей формулой (I):
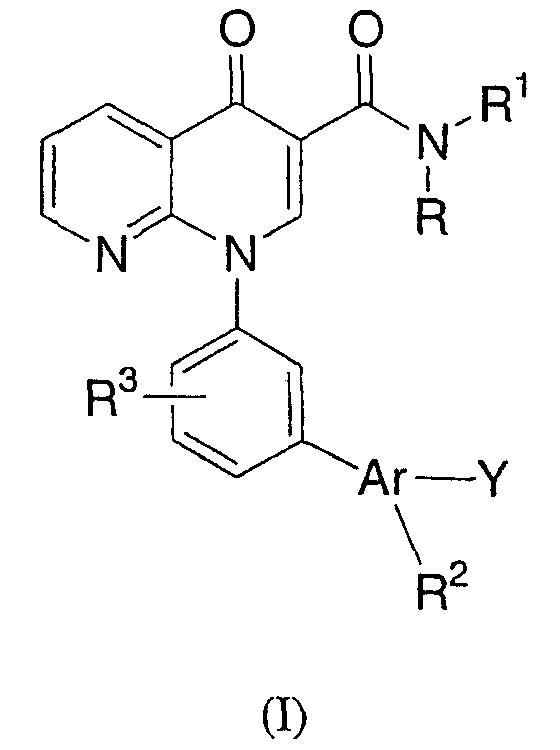
или их фармацевтически приемлемые соли, которые являются ингибиторами фосфодиэстеразы-4.
Настоящее изобретение также касается фармацевтической композиции, включающей эффективное количество новых биарилзамещенных 1,8-нафтиридин-4(1Н)-онов и фармацевтически приемлемый носитель. Настоящее изобретение также предлагает способ лечения у млекопитающих, например, астмы, хронического бронхита, хронического обструктивного заболевания легких (COPD), эозинофильной гранулемы, псориаза и других доброкачественных или злокачественных пролиферативных кожных заболеваний, эндотоксического шока (и связанных с этим состояний, таких как ламинит и колика у лошадей), септического шока, язвенного колита, болезни Крона, реперфузионного поражения миокарда и мозга, воспалительного артрита, остеопороза, хронического гломерулонефрита, атопического дерматита, крапивницы, респираторного дистресс-синдрома у взрослых, респираторного дистресс-синдрома у детей, хронического обструктивного заболевания легких у животных, несахарного диабета, аллергического ринита, аллергического конъюнктивита, весеннего конъюнктивита, рестеноза артерий, атеросклероза, нейрогенного воспаления, боли, кашля, ревматоидного артрита, анкилозирующего спондилита, отторжения трансплантата и заболевания "трансплантат против хозяина", повышенной секреции желудочной кислоты, сепсиса или септического шока бактериального, грибкового или вирусного типа, воспаления и опосредованной цитокинами хронической дегенерации ткани остеоартрита, рака, кахексии, мышечной гипотрофии, депрессии, ухудшения памяти и монополярной депрессии, острого и хронического нейродегенеративного заболевания с воспалительными компонентами, болезни Паркинсона, болезни Альцгеймера, травмы спинного мозга, травмы головы, рассеянного склероза, роста опухоли и инвазии раковой опухоли в нормальные ткани путем введения эффективного количества соединений формулы (I) или соединения-предшественника, образующего в in vivo условиях соединения формулы (I), которые являются ингибиторами фосфодиэстеразы-4. Настоящее изобретение, кроме того, предлагает способ улучшения познавательной способности у здоровых людей.
Подробное описание изобретения
В одном варианте осуществления настоящее изобретение направлено на соединения, представленные формулой (I):
и их фармацевтически приемлемые соли,
где Ar представляет собой фенил, пиридил, пиримидил, индолил, хинолинил, тиенил, пиридонил, оксазолил, оксадиазолил, тиадиазолил или имидазолил; или их оксиды, когда Ar представляет собой гетероарил;
Y представляет собой -СООН, С1-6алкил(С1-4алкил)n-СООН, -С3-4циклоалкил(С1-4алкил)m-СООН, где -С1-6алкил и С3-4циклоалкил необязательно замещены галогеном, алкокси, гидрокси или нитрилом, и (С1-4алкил) заместители необязательно связаны с образованием С3-4циклоалкила; где n имеет значение 0, 1, 2, 3 или 4, m имеет значение 0, 1 или 2;
R представляет собой Н или -С1-6алкил;
R1 представляет собой Н или -С1-6алкил, -С3-6циклоалкил, -С1-6алкокси, -С2-6алкенил, С3-6алкинил, гетероарил или гетероцикл, необязательно замещенный независимо 1-3 заместителями, выбранными из галогенС1-6алкила, -С1-6алкила, -С1-6алкокси, ОН, амино, -(С0-6алкил)-SOp-(С1-6алкил), нитро, CN, =N-O-С1-6алкила, -O-N=С1-6алкила или галогена, где р имеет значение 0, 1 или 2;
R2 представляет собой Н, галоген, -CN, -NO2, -С1-6алкил, -С3-6циклоалкил, -О-С3-6циклоалкил, О-С1-6алкил, О-С3-6циклоалкил-С1-6алкил(С3-6циклоалкил)(С3-6циклоалкил), -С1-6алкокси, фенил, гетероарил, гетероцикл, амино, -С(О)-С1-6алкил, -С(О)-О-С1-6алкил, -С1-6алкил(=N-OH), -C(N=NOH)С1-6алкил, -С0-6алкил(окси)С1-6алкилфенил, SOkNH(С0-6алкил) или -(С0-6алкил)-SOk-(С1-6алкил), где фенил, гетероарил или гетероцикл необязательно замещены галогеном, -С1-6алкилом, -С1-6алкокси, гидрокси, амино или -С(О)-О-С1-6алкилом, и где алкил или циклоалкил необязательно замещены 1-6 заместителями, независимо выбранными из атомов галогенов или -ОН, и где k имеет значение 0, 1 или 2;
R3 выбран из Н, галогена, CN, -С1-6алкила, -С3-6циклоалкила, нитро, -С(О)-С1-6алкила, -С(О)-О-С0-6алкила, -SOn'NH(С0-6алкил) или -(С0-6алкил)SOn'-(С1-6алкил), О-С1-6алкила, -О-С3-6циклоалкила, где n' имеет значение 0, 1 или 2, и где алкил и циклоалкил необязательно замещены 1-6 заместителями, независимо выбранными из галогена или ОН.
В альтернативном варианте группа Y:
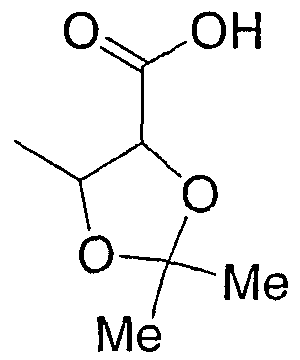
или -С3-6циклоалкил(С1-4алкил)m-СООН, где С3-6циклоалкил необязательно замещен галогеном, алкокси, гидрокси или нитрилом, и (С1-4алкил) заместители необязательно связаны с образованием С3-6циклоалкила; где n имеет значение 0, 1, 2, 3 или 4; m имеет значение 0, 1.
В объем этого варианта изобретения входит группа соединений или их фармацевтически приемлемые соли, где:
Y представляет собой -С3-4циклоалкил(С1-4алкил)m-СООН, где С3-4циклоалкил необязательно замещен галогеном, алкокси, гидрокси или нитрилом, и (С1-4алкил) заместители необязательно связаны с образованием С3-4циклоалкила; где n имеет значение 0, 1, 2, 3 или 4, m имеет значение 0, 1 или 2.
В объем этого варианта изобретения входит также еще одна группа соединений или их фармацевтически приемлемые соли, где:
Y представляет собой циклопропил-СООН;
Ar представляет собой фенил.
Эта группа охватывает также подгруппу соединений или их фармацевтически приемлемые соли, где:
R1 представляет собой -С1-6алкил, необязательно замещенный 1-3 заместителями, независимо выбранными из -С1-6алкила, -С1-6алкокси, ОН, амино, -(С0-6алкил)-SOp-(С1-6алкил), нитро, CN, =N-O-С1-6алкила, -O-N=С1-6алкила или галогена.
Эта группа охватывает также подгруппу соединений или их фармацевтически приемлемые соли, где:
R1 представляет собой -С3-6циклоалкил, необязательно замещенный 1-3 заместителями, независимо выбранными из -С1-6алкила, -С1-6алкокси, ОН, амино, -(С0-6алкил)-SOp-(С1-6алкил), нитро, CN, =N-O-С1-6алкила, -O-N=С1-6алкила или галогена.
Эта группа охватывает еще одну подгруппу соединений или их фармацевтически приемлемые соли, где:
R представляет собой водород.
Эта группа охватывает также еще одну подгруппу соединений или их фармацевтически приемлемые соли, где:
R2 представляет собой водород, или -С1-3алкил, или галоген;
R3 представляет собой водород или галоген.
Эта группа охватывает также еще одну подгруппу соединений или их фармацевтически приемлемые соли, где:
R1 представляет собой -С3-6циклоалкил, необязательно замещенный метилом или галогеном;
R представляет собой водород.
Эта группа охватывает также еще одну подгруппу соединений или их фармацевтически приемлемые соли, где:
R1 представляет собой циклопропил, необязательно замещенный метилом или галогеном;
R и R2 представляют собой водород;
R3 представляет собой водород или галоген.
В другом аспекте в объем варианта изобретения, описанного выше, входит еще одна группа соединений или их фармацевтически приемлемые соли, где:
Y представляет собой циклопропил-СООН;
Ar представляет собой пиридил, пиримидил или их оксид.
Эта группа охватывает подгруппу соединений или их фармацевтически приемлемые соли, где:
R1 представляет собой -С1-6алкил, необязательно замещенный 1-3 заместителями, независимо выбранными из -С1-6алкила, -С1-6алкокси, ОН, амино, -(С0-6алкил)-SOp-(С1-6алкил), нитро, CN, =N-O-С1-6алкила, -O-N=С1-6алкила или галогена.
Эта группа охватывает другую подгруппа соединений или их фармацевтически приемлемые соли, где:
R1 представляет собой -С3-6циклоалкил, необязательно замещенный 1-3 заместителями, независимо выбранными из -С1-6алкила, -С1-6алкокси, ОН, амино, -(С0-6алкил)-SOp-(С1-6алкил), нитро, CN, =N-O-С1-6алкила, -O-N=С1-6алкила или галогена.
Эта группа охватывает еще одну подгруппу соединений или их фармацевтически приемлемые соли, где:
R представляет собой водород.
Эта группа охватывает также еще одну подгруппу соединений или их фармацевтически приемлемые соли, где:
R2 представляет собой водород или -С1-3алкил;
R3 представляет собой водород или галоген.
Эта группа охватывает также еще одну подгруппу соединений или их фармацевтически приемлемые соли, где:
R1 представляет собой -С3-6циклоалкил, необязательно замещенный метилом или галогеном;
R представляет собой водород.
Эта группа охватывает также еще одну подгруппу соединений или их фармацевтически приемлемые соли, где:
R1 представляет собой циклопропил, необязательно замещенный метилом или галогеном;
R и R2 представляют собой водород;
R3 представляет собой водород или галоген.
Примерами соединений согласно настоящему изобретению являются следующие:
2-(транс)-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-3-ил}метилпропановая кислота;
2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}метилпропановая кислота;
3-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-3-метилбутановая кислота;
{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}(гидрокси)уксусная кислота;
1-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
2-(цис)-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
5-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-2,2-диметил-1,3-диоксолан-4-карбоновая кислота;
1-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-3-ил}циклопропанкарбоновая кислота;
1-циано-3-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-2,2-диметилциклопропанкарбоновая кислота;
2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-3-фтор-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
(цис)-2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-3-ил}циклопропанкарбоновая кислота;
2-{3'-бром-5'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-3-метил-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-2-метил-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
2-{3-хлор-3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
2-(цис)-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-3-фтор-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-карбоновая кислота;
2-{3'-[3-(морфолин-4-илкарбонил)-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
2-{3'-[4-оксо-3-({[5-(трифторметил)-1,3,4-тиадиазол-2-ил]амино}карбонил)-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
2-{3'-[3-({[2-(метилтио)этил]амино}карбонил)-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
2-{3'-[3-({[2-(метилсульфонил)этил]амино}карбонил)-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
2-{3'-[4-оксо-3-{[2,2,2-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
2-(5-{3-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]фенил}тиен-2-ил)циклопропанкарбоновая кислота;
2-{3'-[3-{[(циклопропилметил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
2-{3'-[3-{[(1-цианоциклопропил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота;
3-{3'-[3-[(изопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-3-метилбутановая кислота и их фармацевтически приемлемые соли.
В другом варианте осуществления настоящее изобретение касается соединений, представленных формулой (I):
и их фармацевтически приемлемых солей, где
Ar представляет собой фенил, пиридил, пиримидил, индолил, хинолинил, тиенил, пиридонил, оксазолил, оксадиазолил, тиадиазолил или имидазолил; или их оксиды, если Ar представляет собой гетероарил;
Y представляет собой -COOR4, С1-6алкил(С1-4алкил)n-COOR4, -С3-4циклоалкил(С1-4алкил)m-COOR4, где -С1-6алкил и С3-4циклоалкил необязательно замещены галогеном, алкокси, гидрокси или нитрилом, и (С1-4алкил) заместители необязательно связаны с образованием С3-4циклоалкила; где n имеет значение 0, 1, 2, 3 или 4, m имеет значение 0, 1 или 2;
R и R4 каждый независимо выбран из Н или -С1-6алкила;
R1 представляет собой Н или -С1-6алкил, -С3-6циклоалкил, -С1-6алкокси, -С2-6алкенил, С3-6алкинил, гетероарил или гетероцикл, необязательно замещенный 1-3 заместителями, независимо выбранными из галогенС1-6алкила, -С1-6алкила, -С1-6алкокси, ОН, амино, -(С0-6алкил)-SOp-(С1-6алкил), нитро, CN, =N-O-С1-6алкила, -O-N=С1-6алкила или галогена, где р имеет значение 0, 1 или 2, или R1 представляет собой -С1-6алкил, моно- или ди-замещенный заместителями, выбранными из фенила и -С3-6циклоалкила;
R2 представляет собой Н, галоген, -CN, -NO2, -С1-6алкил, -С3-6циклоалкил, -О-С3-6циклоалкил, О-С1-6алкил, О-С3-6циклоалкил-С1-6алкил(С3-6циклоалкил)(С3-6циклоалкил), -С1-6алкокси, фенил, гетероарил, гетероцикл, амино, -С(О)-С1-6алкил, -С(О)-О-С1-6алкил, -С1-6алкил(=N-OH), -C(N=NOH)С1-6алкил, -С0-6алкил(окси)С1-6алкилфенил, SOkNH(С0-6алкил) или -(С0-6алкил)-SOk-(С1-6алкил), где фенил, гетероарил или гетероцикл необязательно замещены галогеном, -С1-6алкилом, -С1-6алкокси, гидрокси, амино или -С(О)-О-С1-6алкилом, и где алкил или циклоалкил необязательно замещены 1-6 заместителями, независимо выбранными из атомов галогенов или -ОН, и где k имеет значение 0, 1 или 2;
R3 выбран из Н, галогена, CN, -С1-6алкила, -С3-6циклоалкила, нитро, -С(О)-С1-6алкила, -С(О)-О-С0-6алкила, -SOn'NH(С0-6алкил) или -(С0-6алкил)SOn'-(С1-6алкил), О-С1-6алкила, -О-С3-6циклоалкила, где n' имеет значение 0, 1 или 2, и где алкил и циклоалкил необязательно замещены 1-6 заместителями, независимо выбранными из галогена или ОН.
В одном аспекте соединения по настоящему изобретению являются полезными для лечения дефицита познавательной способности (такого как ухудшение памяти, как указано в настоящем описании) из-за психологической дисфункции, неврологического расстройства (такого как удар) или психиатрической дисфункции.
В другом аспекте настоящее изобретение направлено на способ улучшения познавательной способности у здорового субъекта, включающий введение безопасного, улучшающего познавательную способность количества ингибитора фосфодиэстеразы-4. В частности, настоящее изобретение направлено на способ улучшения памяти, запоминания, удерживания в памяти, способности вспоминать, уверенности и рассудительности у здоровых субъектов, включающий введение безопасного и эффективного количества ингибитора фосфодиэстеразы-4 формулы I.
В объеме настоящей заявки в качестве субъекта имеется в виду субъект, обладающий познавательной способностью в нормальных пределах для субъектов этого возраста или другой классификации. Познавательная способность здорового человека, а также улучшение его познавательной способности показана с приведением испытаний соединений в водном лабиринте Морриса (Morris), описанном McNamara and Skelton, Psychobiology, 1993, 21, 101-108. Более подробно соответствующая методика описана в WO 96/25948. Другие методы определения улучшения познавательной способности включают, но не ограничиваются этим, "T" критерий Maze; тест вытянутой руки Maze; замедленный непарный и замедленный парный критерий; процедуру пассивного избегания; 5-выборочное испытание, раскрытые в WO 01/87281 А2, опубликованной 22 ноября 2001 г.
В настоящем описании классы здоровых субъектов включают подростков, взрослых и людей старшего возраста со средней познавательной способностью; подростков, взрослых и людей старшего возраста с познавательной способностью выше средней и подростков, взрослых и людей старшего возраста с познавательной способностью ниже средней.
В настоящем описании подростки определяются как люди младше 18 лет. В описании взрослые определяются как люди возраста 18 лет и старше. Этот класс охватывает людей от 18 до 40 лет. В настоящем описании люди старшего возраста определяются как люди возраста 40 лет и старше. Этот класс охватывает людей возраста от 55 лет и старше, возраста от 65 лет и старше, возраста от 70 лет и старше.
Как должно быть понятно специалистам, начиная от 25 лет познавательная способность у здоровых людей снижается со скоростью, которую можно измерить и воспроизвести, например, следующими описанными методами: Cambridge Neurophysiological Test Automated Battery (CANTAB, de Jager CA, Milwain E, Budge M. Early detection of isolated memory deficits in the eldery: the need for more sensitive neurophysiological tests. Psycol Med 2002 Apr; 32(3):483-91); Cognitive Drug research Battery (CDR, Barker A, Jones R, Simpson P, Wesnes K. (1995). Scopolamine induced cognitive impairment as a predictor of cognitive decline in healthy elderly volunteers. International Journal of Geriatric Psychiatry 10:1059-1062). Таким образом, по достижении возраста 40 лет познавательная способность существенно снижается, и ее можно улучшить способом, улучшающим познавательную способность.
В настоящем описании "алкил", а также другие группы, имеющие префикс "алк", например, такие как алкокси, алканоил, алкенил, алкинил и т.п., означают углеродные цепи, которые могут быть линейными или разветвленными, или их сочетания. Примеры алкильных групп включают метил, этил, пропил, бутил, втор- и трет-бутил, пентил, гексил, гептил и т.п. "Алкенил", "алкнинил" и подобные термины включают углеродные цепи, содержащие, по меньшей мере, одну ненасыщенную С-С связь.
Термин "галогеналкил", например "галогенС1-6алкил", означает алкил, замещенный одной или несколькими группами галогенов.
Термин "циклоалкил" означает карбоциклы, не содержащие гетероатомов, и включает моно-, би- и трициклические насыщенные карбоциклы, а также конденсированные кольцевые системы. Такие конденсированные кольцевые системы могут включать одно кольцо, являющееся частично или полностью ненасыщенным, такое как бензольное кольцо, для образования конденсированной кольцевой системы, такой как бензоконденсированные карбоциклы. Циклоалкил включает такие конденсированные кольцевые системы, как спироконденсированные кольцевые системы. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, декагидронафталин, адамантан, инданил, инденил, флуоренил, 1,2,3,4-тетрагидронафталин и т.п. Аналогичным образом, "циклоалкенил" означает карбоциклы, не содержащие гетероатомов и содержащие, по меньшей мере, одну неароматическую двойную С-С связь, и включает моно-, би- и трициклические частично насыщенные карбоциклы, а также бензоконденсированные циклоалканы. Примеры циклоалкенила включают циклогексенил, инденил и т.п.
Термин "циклоалкокси", если конкретно не указано иное, включает циклоалкильную группу, соединенную со связывающим атомом окси.
Термин "алкокси", если конкретно не указано иное, включает алкильную группу, соединенную со связывающим атомом окси.
Термин "арил", если конкретно не указано иное, включает многокольцевые системы, а также системы с одним кольцом, например, такие как фенил или нафтил.
Термин "арилокси", если конкретно не указано иное, включает многокольцевые системы, а также системы с одним кольцом, например, такие как фенил или нафтил, соединенные в месте присоединения через связывающий атом окси.
Термин "С0-6алкил" включает алкилы, содержащие 6, 5, 4, 3, 2, 1 или 0 атомов углерода. Алкил, не содержащий атомов углерода, представляет собой водородный заместитель или простую связь - в зависимости от того, является ли алкил концевой группой или мостиковой группой.
Термин "гетеро", если конкретно не указано иное, включает один или несколько атомов О, S или N. Например, гетероциклоалкил и гетероарил включают кольцевые системы, содержащие один или несколько атомов О, S или N в кольце, включая комбинации таких атомов. Гетроатомы замещают кольцевые атомы углерода. Так, например, гетероциклоС5алкил представляет собой пятичленное кольцо, содержащее от 5 до 0 атомов углерода.
Примеры гетероарила включают, например, пиридинил, хинолинил, изохинолинил, пиридазинил, пиримидинил, пиразинил, хиноксалинил, фурил, бензофурил, дибензофурил, тиенил, бензотиенил, пирролил, индолил, пиразолил, индазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, имидазолил, бензимидазолил, оксадиазолил, тиадиазолил,триазолил, тетразолил.
Термин "гетероарилокси", если конкретно не указано иное, включает гетероарильную группу, соединенную через связывающий атом окси с местом присоединения.
Примеры гетероарил(С1-6)алкила включают, например, фурилметил, фурилэтил, тиенилметил, тиенилэтил, пиразолилметил, оксазолилметил, оксазолилэтил, изоксазолилметил, тиазолилметил, тиазолилэтил, имидазолилметил, имидазолилэтил, бензимидазолилметил, оксадиазолилметил, оксадиазолилэтил, тиадиазолилметил, тиадиазолилэтил, триазолилметил, триазолилэтил, тетразолилметил, тетразолилэтил, придинилметил, пиридинилэтил, пиридазинилметил, пиримидинилметил, пиразинилметил, хинолинилметил, изохинолинилметил и хиноксалинилметил.
Примеры гетероциклоС3-7алкила включают, например, азетидинил, пирролидинил, пиперидинил, пиперазинил, морфолинил, тетрагидрофуранил, имидазолинил, пирролидин-2-он, пиперидин-2-он и тиоморфолинил.
Примеры арил(С1-6)алкила включают, например, фенил(С1-6)алкил и нафтил(С1-6)алкил.
Примеры гетероциклоС3-7алкилкарбонил(С1-6)алкила включают, например, азетидинил карбонил(С1-6)алкил, пирролидинил карбонил(С1-6)алкил, пиперидинил карбонил(С1-6)алкил, пиперазинил карбонил(С1-6)алкил, морфолинил карбонил(С1-6)алкил и тиоморфолинил карбонил(С1-6)алкил.
Термин "амин", если конкретно не указано иное, включает первичные, вторичные и третичные амины.
Термин "карбамоил", если не указано иное, включает -NHC(O)OC1-C4алкил и -ОС(О)NHC1-C4алкил.
Термин "галоген" включает атомы фтора, хлора, брома и иода.
Термин "необязательно замещенный" означает и замещенный, и незамещенный. Так, например, необязательно замещенный арил может представлять собой пентафторфенил или фенильное кольцо. Кроме того, замещение может быть произведено по любой из групп. Например, замещенный арил(С1-6)алкил включает замещение по арильной группе, а также замещение по алкильной группе.
Термин "оксид" гетероарильных групп используется в его обычном, широко известном химическом смысле и включает, например, N-оксиды гетероатомов азота.
Описанные в настоящей заявке соединения содержат одну или несколько двойных связей и могут, таким образом, образовывать цис/транс изомеры, а также другие конформационные изомеры. Настоящее изобретение включает все такие возможные изомеры, а также смеси таких изомеров.
Описанные в настоящей заявке соединения могут содержать один или несколько центров асимметрии и могут, таким образом, образовывать диастереомеры, а также оптические изомеры. Настоящее изобретение включает все такие возможные диастереомеры, а также их рацемические смеси, их по существу чистые разделенные энантиомеры, все возможные геометрические изомеры и их фармацевтически приемлемые соли. Приведенная выше формула I показана без определенной стереохимии в определенных положениях. Настоящее изобретение включает все стереоизомеры формулы I и их фармацевтически приемлемые соли. Кроме того, включены смеси стереоизомеров, а также выделенные конкретные стереоизомеры. В ходе стадий синтеза, используемых для получения таких соединений, или при использовании стадий рацемизации или эпимеризации, которые известны специалистам в данной области, продуктами таких стадий могут быть смеси стереоизомеров.
Термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот. Если соединение по настоящему изобретению является кислотным, его соответствующую соль можно получить из фармацевтически приемлемых нетоксичных оснований, включая неорганические основания и органические основания. Соли, полученные из таких неорганических оснований, включают соли алюминия, аммония, кальция, меди (одновалентной и двухвалентной), двухвалентного железа, одновалентного железа, лития, магния, марганца (одновалентного и двухвалентного), калия, натрия, цинка и т.п. Особенно предпочтительны соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых нетоксичных органических оснований, включают соли первичных, вторичных и третичных аминов, а также циклических аминов и замещенных аминов, таких как природные и синтетически полученные замещенные амины. Другие фармацевтически приемлемые нетоксичные органические основания, из которых могут быть образованы соли, включают ионообменные смолы, такие как, например, аргинин, бетаин, кафеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п.
Если соединение по настоящему изобретению является основным, его соответствующую соль можно получить из фармацевтически приемлемых нетоксичных кислот, включая органические и неорганические кислоты. Такие кислоты включают, например, уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромистоводородную, хлористоводородную, изетионовую, молочную, малеиновую, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую кислоту и т.п. Особенно предпочтительными являются бензолсульфоновая, лимонная, бромистоводородная, хлористоводородная, малеиновая, фосфорная, серная и винная кислоты.
Фармацевтические композиции по настоящему изобретению включают соединение, представленное формулой I (или его фармацевтически приемлемые соли), в качестве активного ингредиента, фармацевтически приемлемый носитель и, необязательно, другие терапевтические ингредиенты или адъюванты. Такие дополнительные терапевтические ингредиенты включают, например, i) антагонисты рецепторов лейкотриена, ii) ингибиторы биосинтеза лейкотриена, iii) кортикостероиды, iv) антагонисты рецепторов Н1, v) агонисты бета-2-адренорецептора, vi) селективные ингибиторы СОХ-2, vii) статины, viii) нестероидные противовоспалительные лекарственные средства ("NSAID") и ix) антагонисты М2/М3. Композиции включают композиции, подходящие для перорального, ректального, местного и парентерального (включая подкожное, внутримышечное и внутривенное) введения, хотя наиболее подходящее введение для любого конкретного случая зависит от конкретного пациента, природы и тяжести состояния, для лечения которого вводят данный активный ингредиент.
Фармацевтические композиции можно получить в виде стандартной лекарственной формы, и они могут быть получены любым способом, хорошо известным в области фармацевтики.
Для местного применения можно использовать кремы, мази, желе, растворы или суспензии, содержащие соединение формулы I. Местное применение в соответствии с настоящим изобретением охватывает полоскание для полости рта и полоскание для горла.
Для лечения таких состояний, как i) легочных заболеваний, например астмы, хронического бронхита, хронического обструктивного заболевания легких (COPD), респираторного дистресс-синдрома у взрослых, респираторного дистресс-синдрома у детей, кашля, хронического обструктивного заболевания легких у животных, ii) желудочно-кишечных расстройств, например язвенного колита, болезни Крона и повышенной секреции желудочной кислоты, iii) инфекционных заболеваний, например сепсиса или септического шока бактериального, грибкового или вирусного типа, эндотоксического шока (и связанных с этим состояний, таких как ламинит и колика у лошадей), и септического шока, iv) неврологических заболеваний, например травмы спинного мозга, травмы головы, нейрогенного воспаления, боли и реперфузионного поражения головного мозга, v) воспалительных заболеваний, например псориатического артрита, ревматоидного артрита, анкилоизирующего спондилита, остеоартрита, воспаления и опосредованной цитокинами хронической дегенерации ткани, vi) аллергических заболеваний, например аллергического ринита, аллергического конъюнктивита и эозинофильной гранулемы, vii) психиатрических заболеваний, например депрессии, ухудшения памяти и монополярной депрессии, vii) нейродегенеративных заболеваний, например болезни Паркинсона, болезни Альцгеймера, острого и хронического рассеянного склероза, ix) дерматологических заболеваний, например псориаза и других доброкачественных или злокачественных пролиферативных кожных заболеваний, атопического дерматита и крапивницы, x) онкологических заболеваний, например рака, роста опухоли и инвазии раковой опухоли в нормальные ткани, xi) метаболических расстройств, например несахарного диабета, xii) костных заболеваний, например остеопороза, xiii) сердечно-сосудистых расстройств, например рестеноза артерий, атеросклероза, реперфузионного поражения миокарда, xiv) других заболеваний, например хронического гломерулонефрита, весеннего конъюнктивита, отторжения трансплантата и заболевания "трансплантат против хозяина" и кахексии, являющихся чувствительными к ингибированию PDE4, полезными являются дозы в пределах от около 0,001 мг/кг до около 140 мг/кг массы тела или, альтернативно, от около 0,5 мг до около 2,7 г на пациента в день. Кроме того, должно быть понятно, что соединения по настоящему изобретению, ингибирующие PDE4, можно вводить в профилактически эффективных дозах для профилактики вышеперечисленных состояний.
Количество активного ингредиента, которое можно сочетать с материалом носителя для получения единицы лекарственной формы, варьируется в зависимости от субъекта, подлежащего лечению, и от конкретного способа введения. Например, композиция, предназначенная для перорального введения человеку, может содержать от около 0,5 мг до около 5 г активного вещества, соединенного с подходящим и удобным количеством материала носителя, которое может варьироваться от около 5 до около 95 процентов от общей массы композиции. Стандартные лекарственные формы обычно содержат от около 0,01 мг до около 1000 мг активного ингредиента, типично, 0,01 мг, 0,05 мг, 0,25 мг, 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг, 500 мг, 600 мг, 800 мг или 1000 мг.
Однако должно быть понятно, что конкретный уровень дозирования для любого конкретного пациента зависит от различных факторов, включая возраст, массу тела, общее состояние здоровья, пол, рацион питания, время введения, путь введения, скорость выведения из организма, комбинацию лекарственных средств и тяжесть конкретного заболевания, которое лечат.
На практике соединения, представленные формулой I, или их фармацевтически приемлемые соли по настоящему изобретению можно объединять в качестве активного ингредиента в смеси с фармацевтическим носителем в соответствии с традиционными методами получения фармацевтических композиций. Носитель может быть в разнообразных формах в зависимости от формы препарата, которая желательна для введения, например пероральной или парентеральной (включая внутривенную). Так, фармацевтические композиции по настоящему изобретению могут предоставляться в виде отдельных дозированных единиц, подходящих для перорального введения, таких как капсулы, саше или таблетки, каждая содержащая предварительно определенное количество активного ингредиента. Кроме того, композиции могут быть приготовлены в форме порошка, гранул, раствора, суспензии в водной жидкости, в виде неводной жидкости, в виде эмульсии масло-в-воде или эмульсии вода-в-масле. Помимо обычных лекарственных форм, описанных выше, соединение, представленное формулой I, или его фармацевтически приемлемые соли также можно вводить при помощи средств и/или устройств доставки с контролируемым высвобождением. Композиции могут быть получены любым из способов, применяемых в фармацевтике. Как правило, такие способы включают стадию сочетания активного ингредиента с носителем, состоящим из одного или нескольких необходимых ингредиентов. Как правило, композиции получают однородным и равномерным смешиванием активного ингредиента с жидкими носителями, или тонкоизмельченными твердыми носителями, или и тем, и другим. Затем продукту придают удобную форму с получением желаемого внешнего вида препарата.
Таким образом, фармацевтические композиции по настоящему изобретению могут включать фармацевтически приемлемый носитель и соединение или фармацевтически приемлемую соль формулы I. Соединения формулы I или их фармацевтически приемлемые соли можно также включать в фармацевтические композиции в сочетании с одним или несколькими другими терапевтически активными соединениями.
Фармацевтический носитель, который используют, может представлять собой, например, твердое вещество, жидкость или газ. Примеры твердых носителей включают лактозу, белую глину, сахарозу, тальк, желатин, агар, пектин, аравийскую камедь, стеарат магния и стеариновую кислоту. Примерами жидких носителей являются сахарный сироп, арахисовое масло, оливковое масло и вода. Примеры газообразных носителей включают диоксид углерода и азот.
При получении композиций для пероральной лекарственной формы можно использовать любые удобные фармацевтические среды. Например, воду, гликоли, масла, спирты, отдушки, консерванты, красители и т.п. можно использовать для получения пероральных жидких препаратов, таких как суспензии, эликсиры и растворы; тогда как носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие вещества, смазки, связующие, разрыхлители и т.п. можно использовать для получения пероральных твердых препаратов, таких как порошки, капсулы и таблетки. Из-за простоты введения таблетки и капсулы являются предпочтительными разовыми дозами пероральной лекарственной формы, при этом используют твердые фармацевтические носители. Таблетки необязательно могут иметь покрытие, нанесенное стандартным водным или неводным способом.
Таблетку, содержащую композицию по настоящему изобретению, можно получить прессованием или формованием, необязательно с одним или несколькими вспомогательными ингредиентами или адъювантами. Прессованные таблетки можно получить прессованием в подходящей машине активного ингредиента в свободнотекучей форме, такой как порошок или гранулы, необязательно смешанный со связующим, смазывающим веществом, инертным разбавителем, поверхностно-активным веществом или диспергирующим агентом. Формованные таблетки можно получить формованием в подходящей машине смеси порошкообразного соединения, смоченного жидким инертным разбавителем. Каждая таблетка предпочтительно содержит от около 0,1 мг до около 500 мг активного ингредиента, а каждые саше или капсула предпочтительно содержит от около 0,1 мг до около 500 мг активного ингредиента.
Фармацевтические композиции по настоящему изобретению, подходящие для парентерального введения, могут быть приготовлены в виде растворов или суспензий активных соединений в воде. Может быть добавлено подходящее поверхностно-активное вещество, такое как, например, гидроксипропилцеллюлоза. Дисперсии также могут быть приготовлены в глицерине, жидких полиэтиленгликолях и их смесях в маслах. Кроме того, может быть добавлен консервант для предотвращения вредного роста микроорганизмов.
Фармацевтические композиции по настоящему изобретению, подходящие для введения путем инъекции, включают стерильные водные растворы или дисперсии. Кроме того, композиции могут находиться в форме стерильных порошков для приготовления таких растворов или дисперсий для немедленного введения. Во всех случаях готовая форма для инъекции должна быть стерильной и достаточно жидкой для обеспечения легкого введения через шприц. Фармацевтические композиции должны быть стабильными в условиях изготовления и хранения; поэтому они, предпочтительно, должны быть защищены от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может представлять собой растворитель или дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), растительные масла и подходящие смеси этих веществ.
Фармацевтические композиции по настоящему изобретению могут находиться в форме, подходящей для местного применения, такой как, например, аэрозоль, крем, мазь, лосьон, тонкоизмельченный порошок и т.п. Кроме того, композиции могут находиться в форме, подходящей для чрескожного использования. Такие композиции могут быть получены с использованием соединения формулы I по настоящему изобретению или его фармацевтически приемлемых солей с применением традиционных способов переработки. В качестве примера, крем или мазь получают смешиванием гидрофильного вещества и воды вместе с 5-10 мас.% соединения с получением крема или мази, имеющих желаемую консистенцию.
Фармацевтические композиции по настоящему изобретению могут находиться в форме, подходящей для ректального введения, где носитель представляет собой твердое вещество. Предпочтительно, чтобы смесь образовывала стандартные единицы лекарственной формы, такой как суппозитории. Подходящие носители включают масло какао и другие вещества, широко известные из области техники. Суппозитории можно удобным образом получать сначала смешиванием композиции с размягченным или расплавленным номителем(ями), с последующим охлаждением и формованием в специальных формах.
Помимо вышеперечисленных ингредиентов носителя, фармацевтические композиции, описанные выше, могут включать, если это является подходящим, один или несколько дополнительных ингредиентов носителя, например разбавители, буферы, отдушки, связующие, поверхностно-активные вещества, загустители, смазывающие вещества, консерванты (включая антиоксиданты) и т.п. Кроме того, могут быть включены другие адъюванты для придания композиции изотоничности с кровью предполагаемого реципиента. Композиции, содержащие соединение формулы I или его фармацевтически приемлемые соли, можно получать в форме порошка или жидкого концентрата.
Было обнаружено, что соединения и фармацевтические композиции по настоящему изобретению проявляют биологическую активность как ингибиторы PDE4. Соответственно, следующий аспект настоящего изобретения включает лечение у млекопитающих, например, i) легочных заболеваний, например астмы, хронического бронхита, хронического обструктивного заболевания легких (COPD), респираторного дистресс-синдрома у взрослых, респираторного дистресс-синдрома у детей, кашля, хронического обструктивного заболевания легких у млекопитающих, ii) желудочно-кишечных расстройств, например язвенного колита, болезни Крона и повышенной секреции желудочной кислоты, iii) инфекционных заболеваний, например сепсиса или септического шока бактериального, грибкового или вирусного типа, эндотоксического шока (и связанных с этим состояний, таких как ламинит и колика у лошадей) и септического шока, iv) неврологических заболеваний, например травмы спинного мозга, травмы головы, нейрогенного воспаления, боли и реперфузионного поражения головного мозга, v) воспалительных заболеваний, например псориатического артрита, ревматоидного артрита, анкилоизирующего спондилита, остеоартрита, воспаления и опосредованной цитокинами хронической дегенерации ткани, vi) аллергических заболеваний, например аллергического ринита, аллергического конъюнктивита и эозинофильной гранулемы, vii) психиатрических заболеваний, например депрессии, ухудшения памяти и монополярной депрессии, vii) нейродегенеративных заболеваний, например болезни Паркинсона, болезни Альцгеймера, острого и хронического рассеянного склероза, ix) дерматологических заболеваний, например псориаза и других доброкачественных или злокачественных пролиферативных кожных заболеваний, атопического дерматита и крапивницы, x) онкологических заболеваний, например рака, роста опухоли и инвазии раковой опухоли в нормальные ткани, xi) метаболических расстройств, например несахарного диабета, xii) костных заболеваний, например остеопороза, xiii) сердечно-сосудистых расстройств, например рестеноза артерий, атеросклероза, реперфузионного поражения миокарда, и xiv) других заболеваний, например хронического гломерулонефрита, весеннего конъюнктивита, отторжения трансплантата и заболевания "трансплантат против хозяина" и кахексии - заболеваний, которые поддаются лечению путем ингибирования изофермента PDE4 и повышения, в результате этого, уровней сАМР - путем введения эффективного количества соединений по настоящему изобретению. Термин "млекопитающие" включает человека, а также других млекопитающих, таких как, например, собаки, кошки, лошади, свиньи и коровы. Соответственно, должно быть понятно, что лечение млекопитающих, отличных от человека, представляет собой лечение заболеваний, клинически соответствующих тем, которые приводились в качестве примера выше как относящиеся к заболеваниям человека.
Кроме того, как описано выше, соединение по настоящему изобретению можно использовать в сочетании с другими терапевтическими соединениями. В частности, можно использовать комбинации соединения-ингибитора PDE4 с i) антагонистами рецепторов лейкотриена, ii) ингибиторами биосинтеза лейкотриена, iii) селективными ингибиторами СОХ-2, iv) статинами, v) NSAID, vi) антагонистами М2/М3, vii) кортикостероидами, viii) антагонистами рецепторов Н1 (гистамина) и ix) агонистами бета-2-адренорецептора.
Так, например, легочные заболевания, такие как астма, хронический бронхит, хроническое обструктивное заболевание легких (COPD), респираторный дистресс-синдром у взрослых, респираторный дистресс-синдром у детей, кашель, хроническое обструктивное заболевание легких у млекопитающих, удобно лечить с использованием капсул, саше или таблеток, каждая из которых содержит 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг активного ингредиента - соединения по настоящему изобретению или его фармацевтически приемлемой соли, которые вводят один, два или три раза в день.
Желудочно-кишечные расстройства, такие как язвенный колит, болезнь Крона и повышенная секреция желудочной кислоты, удобно лечить с использованием капсул, саше или таблеток, каждая из которых содержит 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг активного ингредиента - соединения по настоящему изобретению или его фармацевтически приемлемой соли, которые вводят один, два или три раза в день.
Инфекционные заболевания, такие как сепсис или септический шок бактериального, грибкового или вирусного типа, эндотоксический шок (и связанные с этим состояния, такие как ламинит и колика у лошадей) и септический шок, удобно лечить с использованием капсул, саше или таблеток, каждая из которых содержит 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг активного ингредиента - соединения по настоящему изобретению или его фармацевтически приемлемой соли, которые вводят один, два или три раза в день.
Неврологические заболевания, такие как травма спинного мозга, травма головы, нейрогенное воспаления, боль и реперфузионное поражение головного мозга, удобно лечить с использованием капсул, саше или таблеток, каждая из которых содержит 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг активного ингредиента - соединения по настоящему изобретению или его фармацевтически приемлемой соли, которые вводят один, два или три раза в день.
Воспалительные заболевания, такие как псориатический артрит, ревматоидный артрит, анкилоизирующий спондилит, остеоартрит, воспаление и опосредованная цитокинами хроническая дегенерация ткани, удобно лечить с использованием капсул, саше или таблеток, каждая из которых содержит 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг активного ингредиента - соединения по настоящему изобретению или его фармацевтически приемлемой соли, которые вводят один, два или три раза в день.
Аллергические заболевания, такие как аллергический ринит, аллергический конъюнктивит и эозинофильная гранулема, удобно лечить с использованием капсул, саше или таблеток, каждая из которых содержит 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг активного ингредиента - соединения по настоящему изобретению или его фармацевтически приемлемой соли, которые вводят один, два или три раза в день.
Психиатрические заболевания, такие как депрессия, ухудшение памяти и монополярная депрессия, удобно лечить с использованием капсул, саше или таблеток, каждая из которых содержит 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг активного ингредиента - соединения по настоящему изобретению или его фармацевтически приемлемой соли, которые вводят один, два или три раза в день.
Нейродегенеративные заболевания, такие как болезнь Паркинсона, болезнь Альцгеймера, острый и хронический рассеянный склероз, удобно лечить с использованием капсул, саше или таблеток, каждая из которых содержит 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг активного ингредиента - соединения по настоящему изобретению или его фармацевтически приемлемой соли, которые вводят один, два или три раза в день.
Дерматологические заболевания, такие как псориаз и другие доброкачественные или злокачественные пролиферативные кожные заболевания, атопический дерматит и крапивница, удобно лечить с использованием капсул, саше или таблеток, каждая из которых содержит 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг активного ингредиента - соединения по настоящему изобретению или его фармацевтически приемлемой соли, которые вводят один, два или три раза в день.
Онкологические заболевания, такие как рак, рост опухоли и инвазия раковой опухоли в нормальные ткани, удобно лечить с использованием капсул, саше или таблеток, каждая из которых содержит 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг активного ингредиента - соединения по настоящему изобретению или его фармацевтически приемлемой соли, которые вводят один, два или три раза в день.
Метаболические расстройства, такие как несахарный диабет, удобно лечить с использованием капсул, саше или таблеток, каждая из которых содержит 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг активного ингредиента - соединения по настоящему изобретению или его фармацевтически приемлемой соли, которые вводят один, два или три раза в день.
Костные заболевания, такие как остеопороз, сердечно-сосудистые расстройства, такие как рестеноз артерий, атеросклероз, реперфузионное поражение миокарда, и другие заболевания, такие как хронический гломерулонефрит, весенний конъюнктивит, отторжение трансплантата и заболевание "трансплантат против хозяина" и кахексия, удобно лечить с использованием капсул, саше или таблеток, каждая из которых содержит 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг или 500 мг активного ингредиента - соединения по настоящему изобретению или его фармацевтически приемлемой соли, которые вводят один, два или три раза в день.
Для улучшения познавательной способности (например, улучшения памяти, способности к запоминанию, удерживанию в памяти, способности вспоминать, уверенности и рассудительности) полезными являются дозы от около 0,0001 мг/кг до около 50 мг/кг массы тела в день или от около 0,005 мг до около 2,5 г на пациента в день. Альтернативно, дозы от около 0,001 мг до 10 мг соединения на килограмм массы тела в день или, альтернативно, от около 0,05 мг до около 500 мг на пациента в день.
Количество активного ингредиента, которое можно объединять с материалом носителя с получением единицы лекарственной формы, варьируется в зависимости от подлежащего лечению хозяина и конкретного пути введения. Например, удобно, если композиция, предназначенная для перорального введения человеку, содержит от около 0,005 мг до около 2,5 г активного вещества в соединении с подходящим и удобным количеством материала носителя. Стандартные лекарственные формы обычно содержат от около 0,005 мг до около 1000 мг активного ингредиента, обычно 0,005, 0,01 мг, 0,05 мг, 0,25 мг, 1 мг, 5 мг, 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг, 500 мг, 600 мг, 800 мг или 1000 мг, при введении один, два или три раза в день.
Используемые в настоящей заявке аббревиатуры имеют значения, указанные в таблице ниже. Аббревиатуры, которые не указаны в таблице, имеют общепринятые значения, если не указано иное.
Аббревиатуры алкильных групп
Анализы, демонстрирующие биологическую активность LPS, и FMLP-индуцированные TNF-α и LTB4 анализы с использованием цельной крови человека
Из цельной крови получают белок и обогащенную клетками среду, подходящие для исследования биохимической эффективности противовоспалительных соединений, таких как селективные ингибиторы PDE4. Нормальная, нестимулированная кровь человека не содержит детектируемые уровни TNF-α и LTB4. При стимуляции при помощи LPS активированные моноциты экспрессируют и секретируют TNF-α вплоть до 8 часов, и уровни содержания в плазме остаются стабильными в течение 24 часов. Опубликованные результаты исследований показали, что ингибирование TNF-α путем повышения внутриклеточного сАМР в результате ингибирования PDE4 и/или повышенной активности аденилилциклазы происходит на уровне транскрипции. Синтез LTB4 также чувствителен к уровням внутриклеточного сАМР и может быть полностью ингибирован селективными ингибиторами PDE4. Поскольку в течение 24 часов LPS-стимуляции цельной крови продуцируется небольшое количество LTB4, для синтеза LTB4 активированными нейтрофилами необходима дополнительная LPS-стимуляция с последующей fMLP-стимуляцией цельной крови. Таким образом, используя одну и ту же пробу крови, можно определить активность соединения на двух суррогатных маркерах активности PDE4 в цельной крови с использованием методики, описанной ниже.
Свежую кровь брали из вены здоровых людей (мужчин и женщин), добровольно участвующих в испытании, и собирали в гепаринизированные пробирки. У этих добровольцев не было никаких явно выраженных воспалительных состояний, и они не принимали никаких NSAID в течение, по меньшей мере, 4 дней до взятия крови. 500-мкл аликвоты крови предварительно инкубировали либо с 2 мкл носителя (DMSO), либо с 2 мкл испытываемого соединения в различных концентрациях в течение 15 минут при 37°С. Затем добавляли либо 10 мкл носителя (PBS) в виде контрольного раствора, либо 10 мкл LPS (конечная концентрация 1 мкг/мл, #L-2630 (Sigma Chemical Co., St.Louis, MO) из E.coli, серотип 0111:В4; разбавленный в 0,1% мас./об. BSA (в PBS)). После 24 часов инкубации при 37°С к крови добавляли еще 10 мкл PBS (контроль) или 10 мкл LPS (конечная концентрация 1 мкг/мл) и инкубировали в течение 30 минут при 37°С. Затем кровь стимулировали либо 10 мкл PBS (контроль), либо 10 мкл fMLP (конечная концентрация 1 мкМ, #F-3560 (Sigma); разбавленный в 0,1% мас./об. BSA (в PBS)) в течение 15 минут при 37°С. Образцы крови центрифугировали при 1500хg в течение 10 минут при 4°С с получением плазмы. 50-мкл аликвоту плазмы смешивали с 200 мкл метанола для осаждения белка и центрифугировали, как описано выше. Супернатант анализировали на LTB4 с использованием набора для иммуноферментного анализа (#520111 от Cyaman Chemical Co., Ann Arbor, MI) в соответствии с процедурой изготовителя. TNF-α анализировали в разбавленной плазме (в PBS), используя набор ELISA (Cistron Biotechnology, Pine Brook, NJ), в соответствии с процедурой изготовителя. Полученные в примерах значения ИК50 обычно находились в пределах от 0,075 мкМ до 25 мкМ.
Противоаллергическая активность in vivo
Соединения по настоящему изобретению были испытаны на их действие в отношении IgE-опосредованного аллергического легочного воспаления, индуцированного у сенсибилизированных морских свинок вдыханием антигена. Морских свинок сначала сенсибилизировали к овальбумину при слабой циклофосфамидиндуцированной иммуносупрессии путем внутрибрюшинной инъекции антигена в сочетании с гидроксидом алюминия и коклюшевой вакциной. Через две и четыре недели вводили бустерные дозы антигена. По прошествии шести недель животных стимулировали овальбумином в форме аэрозоля под защитой антигистаминного средства (мепирамина), который вводили внутрибрюшинно. Еще через 48 часов осуществляли бронхиальный альвеолярный лаваж (BAL) и подсчитывали количество эозинофилов и других лейкоцитов в BAL жидкостях. Также удаляли легкие для гистологического исследования на воспалительное поражение. Введение соединений настоящего изобретения (0,001-10 мг/кг в/б или п/о) до трех раз в течение 48 часов с последующей стимуляцией антигеном приводило к значительному снижению эозинофилии и накоплению других воспалительных лейкоцитов. Также наблюдалось менее воспалительное поражение в легких животных, обработанных соединениями согласно настоящему изобретению.
Протокол анализа активности PDE с использованием SPA
Осуществляли отбор соединений, ингибирующих гидролиз сАМР до АМР посредством сАМР-специфической фосфодиэстеразы типа IV, в формате 96-луночного планшета следующим образом:
В 96-луночный планшет при 30°С добавляли испытываемое соединение (растворенное в 2 мкл DMSO), 188 мл субстратного буфера, содержащего [2,8-3H]аденозин 3',5'-циклофосфат (сАМР, от 100 нМ до 5 мкМ), 10 мМ MgCl2, 1 мМ EDTA, 50 мМ Трис, рН 7,5. Реакцию инициировали добавлением 10 мл человеческой рекомбинантной PDE4 (количество контролировали так, чтобы за 10 мин образовалось ˜10% продукта). Через 10 минут реакцию останавливали добавлением 1 мг PDE-SPA гранул (Amersham, Pharmacia Biotech, Inc., Piscataway, NJ). Определяли количество образованного продукта АМР на счетчике для 96-луночных планшетов Wallac Microbeta® (FG&G Wallac Co., Gaithersburg, MD). Сигнал отсутствия фермента определяли как фоновый. 100% активность определяли по детектируемому сигналу в присутствии фермента и DMSO за вычетом фонового сигнала. Соответственно рассчитывали процент ингибирования. Значение ИК50 аппроксимировали методом нелинейной регрессии с использованием уравнения стандартные 4 параметра/множественные участки связывания на основе данных титрования по десяти точкам.
Значения ИК50 примеров, раскрытые ниже, определяли при 100 нМ сАМР, используя очищенный слитый белок GST человеческой рекомбинантной фосфодиэстеразы IVb (met-248), полученный из системы экспрессии бакуловирус/Sf-9. Значения ИК50 примеров, раскрытые ниже, находятся в пределах от 0,01 нМ до 2300 нМ.
Приведенные ниже примеры предназначены для иллюстрации некоторых предпочтительных вариантов выполнения изобретения и не предназначены для какого-либо ограничения настоящего изобретения.
Если конкретно не указано иное, эксперименты осуществляли в следующих условиях. Все операции осуществляли при комнатной температуре или при температуре окружающей среды, т.е. при температуре в пределах 18-25°С. Выпаривание растворителя осуществляли на роторном испарителе при пониженном давлении (600-4000 Па: 4,5-30 мм рт.ст.) при температуре бани до 60°С. Ход реакции отслеживали методом тонкослойной хроматографии (ТСХ), и время реакции приводится только в целях иллюстрации. Точки плавления нескорректированы, и "d" означает разложение. Указанные точки плавления были определены для веществ, полученных, как описано ниже. В некоторых примерах получения при выделении веществ с разными точками плавления может иметь место полиморфизм. Структуру и чистоту всех конечных продуктов подтверждали, по меньшей мере, одним из следующих методов: ТСХ, масс-спектрометрия, ядерный магнитный резонанс (ЯМР) или микроаналитические данные. Если указан выход продукта, эти данные представлены исключительно в иллюстративных целях. Если представлены данные ЯМР, они представлены в форме дельта (δ) единиц для основных диагностических протонов, выражены в миллионных долях (ppm) относительно тетраметилсилана (TMS) в качестве внутреннего стандарта, получены при 300 МГц, 400 МГц или 500 МГц с использованием указанного растворителя. Общепринятые аббревиатуры, используемые для обозначения формы сигнала, следующие: s - синглет; d - дублет; t -триплет; m - мультиплет; br - уширенный; и т.д. Кроме того, "Ar" означает ароматический сигнал. Химические символы имеют их обычные значения; также использовали следующие аббревиатуры: об. (объем), мас. (масса), b.p. (точка кипения), m.p. (точка плавления), л (литр(ы)), мл (миллилитры), г (грамм(ы)), мг (миллиграмм(ы)), моль (моли), ммоль (миллимоли), экв. (эквивалент(ы)).
Способы синтеза
Соединения по настоящему изобретению можно получить в соответствии со следующими способами. Заместители - те же, что и в Формуле I, за исключением случаев, если указано иное.
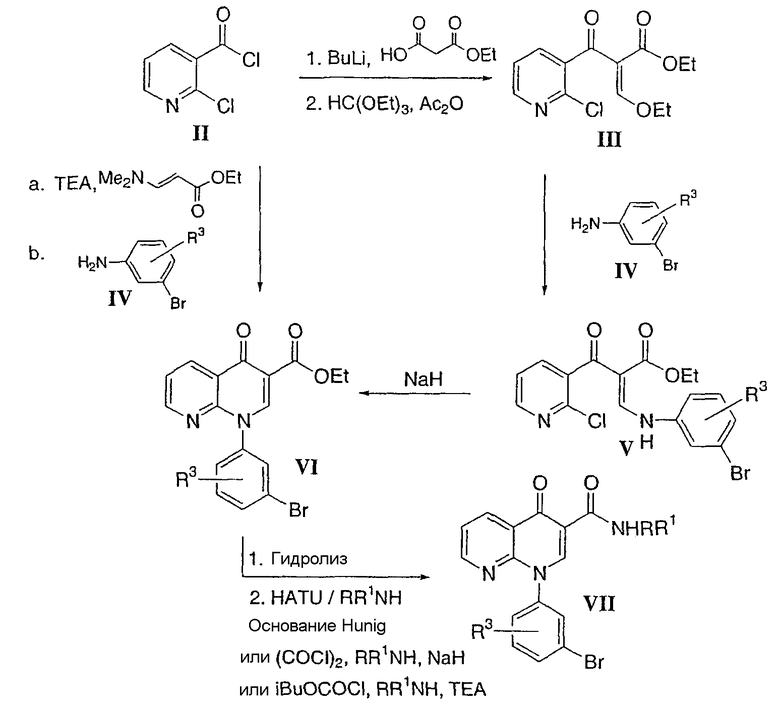
1-(3-Бромфенил)-1,4-дигидро-[1,8]нафтиридин-4-он-3-карбоксамид общей формулы VII получали в соответствии с общим способом, представленным на Схеме 1. Сначала этил 2-хлорникотиноилацетат (II) конденсировали с триэтилортоформиатом в присутствии уксусного ангидрида с получением 2-хлорникотиноилакрилата (III). После добавления подходяще замещенного галогенариламина формулы IV получали 3-ариламиноакрилат типа V. Циклизацию соединения V до 1-галогенарил-1,4-дигидро-[1,8]нафтиридин-4-он-карбоксилата формулы VI осуществляли в присутствии избыточного количества сильного основания при комнатной температуре. Альтернативно, промежуточное соединение VI можно получить способом, осуществляемом в одном сосуде, с использованием, например, 2-хлорникотиноилхлорида и этил-N,N-диметиламиноакрилата и галогенариламина IV в присутствии основания, такого как триэтиламин, в растворителе, таком как ацетонитрил. Гидролиз VI и последующее сочетание полученной карбоновой кислоты с амином (RR1NH2) с использованием HATU и основания Hunig дает желаемое ключевое промежуточное соединение - арилбромид VII.
Схема 1
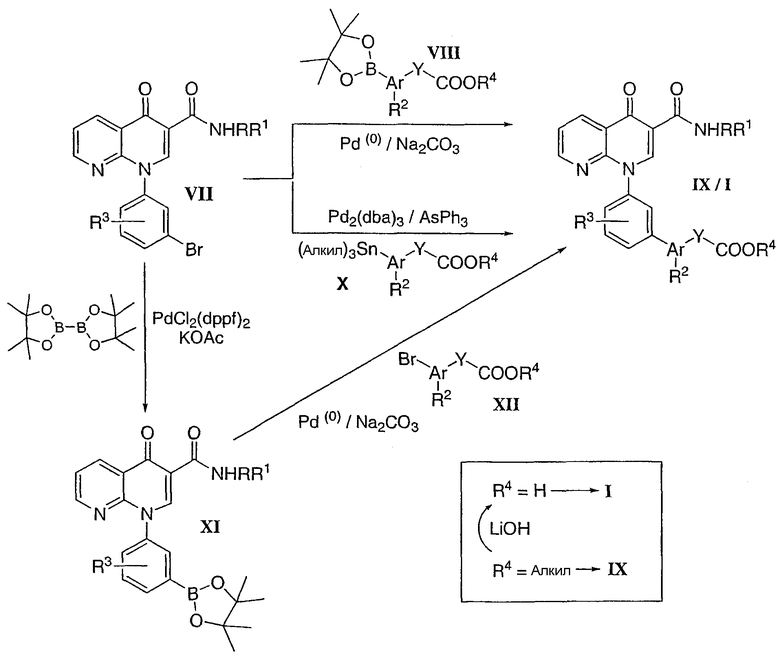
Соединения формулы I получали, используя любой из описанных ниже общих подходов. Катализируемое палладием сочетание Suzuki-Miaura между арилбромидом типа VII и соответствующе замещенным арилпинаколборонатом типа VIII может привести к образованию желаемого соединения I или соответствующего сложного алкилового эфира IX. Сложноэфирное соединение IX может быть гидролизовано при помощи LiOH в ТГФ/МеОН с получением желаемой кислоты I. Альтернативно, арилбромид VII можно преобразовать в пинаколборонат XI путем катализируемого палладием сочетания с пинаколдибораном. Описанная выше реакция Suzuki-Miyaura между соединением XI и арилбромидом типа XII может привести к образованию кислоты I или соответствующего сложного алкилового эфира IX. В заключение, реакция сочетания Stille между арилбромидом типа VII и соответствующе замещенным арилстаннаном типа X также приводит к получению желаемой кислоты формулы I.
Схема 2
И сложный эфир бороната типа VIII, и станнан типа X (Схема 3) можно получить из промежуточного соединения арилбромида XII путем катализируемого палладием сочетания с использованием, соответственно, пинаколдиборана и гексаалкилдиолова.
Схема 3
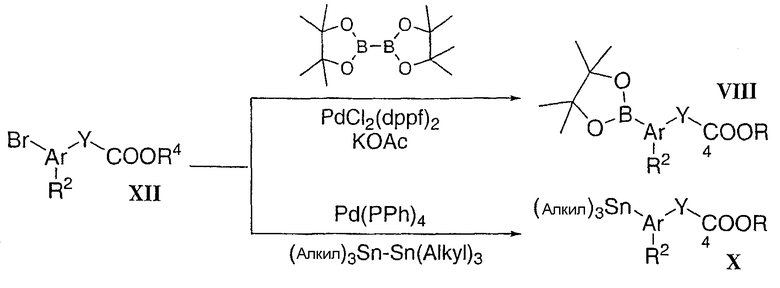
Промежуточные соединения типа XII, в которых Y представляет собой циклопропан, как в соединении формулы XIV, можно получить с использованием любой из следующих четырех общих процедур (Схема 4). Сложный эфир коричной кислоты типа XIII, коммерчески доступный или полученный реакцией Horner-Emmons из соответствующего альдегида XV, можно преобразовать в циклопропановое соединение с использованием диазометана в присутствии каталитического количества диацетата палладия. Полученный транс-циклопропановый сложный эфир XIV можно разделить методом ВЭЖХ на колонке Chiral-Pak с получением двух отдельных энантиомеров. Во втором подходе альдегид XV можно преобразовать в соответствующий стирол XVI при помощи реакции Wittig с последующим энантиоселективным циклопропанированием, используя комплекс бис-оксазолиновый хиральный лиганд/медь и диазоацетат (Evans et al. J.Am.Chem.Soc. 1991, 113, 726). Полученную смесь транс-XIV- и цис-XVII-циклопропана можно разделить методом селективного гидролиза в щелочных условиях (тип гидролиза: транс>цис). Индивидуальные энантиомеры (XIV или XVII) можно получить, используя либо R, либо S хиральный лиганд. Цис-циклопропановый сложный эфир XIV также можно получить из цис-сложного эфира коричной кислоты XVIII, используя описанную выше процедуру диазометан/Pd(Ас)2. Цис-сложный эфир коричной кислоты XVIII можно получить модифицированным способом олефинирования Horner-Emmons с использованием бис(трифторэтил)фосфонового эфира и сильного основания (Still et al. Tetrahedron Lett, 1983, 24, 4405).
Схема 4
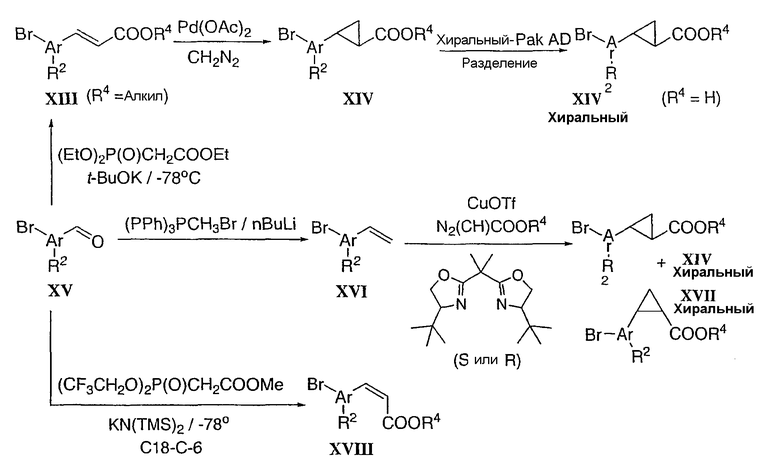
Цис- и транс-хиральные циклопропановые промежуточные соединения типа XIV и XVII, соответственно, также можно получить следующими двумя способами. Коричную кислоту XIII в присутствии CDI преобразовывают в ацилимидазол XIX, который в щелочных условиях взаимодействует с оптически чистым оксазолидиноном XX, с образованием хирального циннаматоксазолидинона XXI. Циклопропанирование соединения XXI в соответствии с описанным выше способом дает смесь (>5 к 1) диастереоизомеров XXII, которую можно разделить (кристаллизация, SiO2). В результате гидролиза получают желаемый хиральный циклопропил XIV. Аналогичным образом, рацемическую смесь циклопропила типа XIV (XVII) можно преобразовать в две стадии в 1:1 диастереоизомерную смесь описанного выше циннаматоксазолидинона XXI.
Схема 5
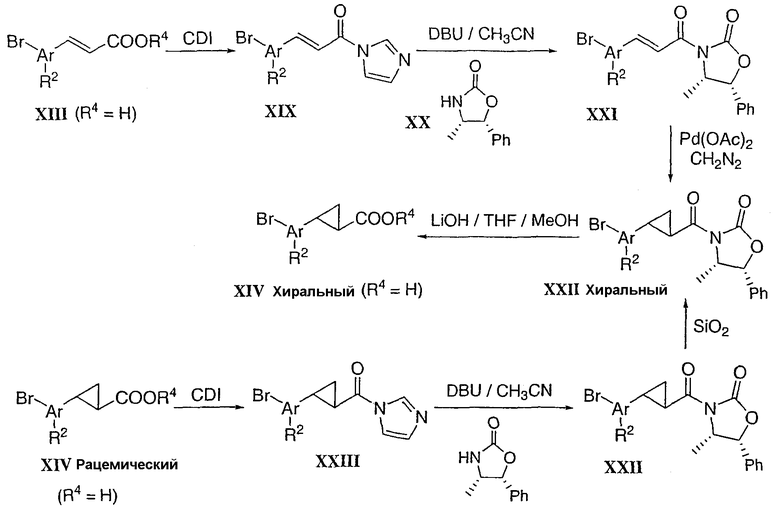
Промежуточные соединения типа XXV (Схема 6) можно получить депротонированием сложного эфира типа XXIV с использованием избыточного количества сильного основания в присутствии алкилирующего агента, такого как R5I. Количество основания и алкилирующего агента по отношению к исходному сложному эфиру соотносится с уровнем замещения (моно vs. бис). Для пропионовых аналогов типа XXVII трет-бутиловый сложный эфир XXVI можно депротонировать с использованием пространственно затрудненного основания. Добавление алкилирующего агента приводит к смеси моно- и бис-алкилированного соединения. При повторе этой процедуры получают, в основном, сложный эфир типа XXVII. Циклопропиловый аналог типа XXX можно получить в две стадии из соответствующего арилацетонитрила XXVIII. Реакция фазового переноса с использованием нитрила XXVIII и 2-хлорбромэтана в водном растворе сильного основания дает циклопропилнитрил XXIX (Org. Prep. & Proc. 1995, 27, 355). Гидролиз с использованием NaOH в кипящем этаноле дает желаемую кислоту XXX. (1,1-Диметил)этиларил типа XXXIII можно получить в три стадии. (2-Хлор-1,1-диметилэтил)арил типа XXXI можно преобразовать в соответствующий сложный эфир XXXII сначала путем гашения соли Гриньяра XXXI диоксидом углерода с последующей этерификацией полученной кислоты диазометаном. Электрофильное замещение сложного арилового эфира XXXII в среде, генерирующей катион иодония, дает желаемый иодарил XXXIII (J. Am. Chem. Soc. 1948, 70, 370).
Схема 6
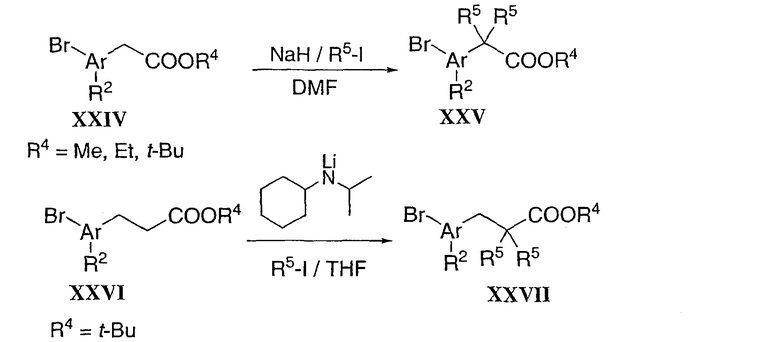
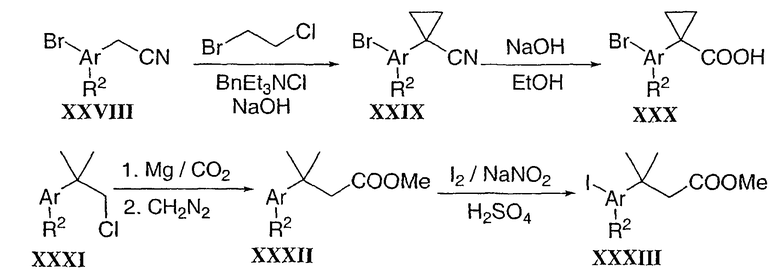
Замещенный циклопропил типа XXXV можно получить в две стадии (Схема 7). Аддукт Knoevenagel XXXIV, полученный конденсацией альдегида XV и этилцианоацетата, можно преобразовать в циклопропил XXXV в присутствии 2-нитропропана и основания в кипящем этаноле (Tetrahedron Lett. 1985, 26, 1923). В завершение, промежуточные циклические ацеталевые соединения типа XXXVII можно получить в две стадии из циннамата XIII. Бис-гидроксилирование циннамата XIII с использованием условий, разработанных Sharpless et al. (AD-mix. J. Org. Chem. 1992, 57, 2768), после конденсации с ацетоном в присутствии каталитического количества кислоты диола XXXVI, дает желаемый хиральный ацеталь XXXVII.
Схема 7
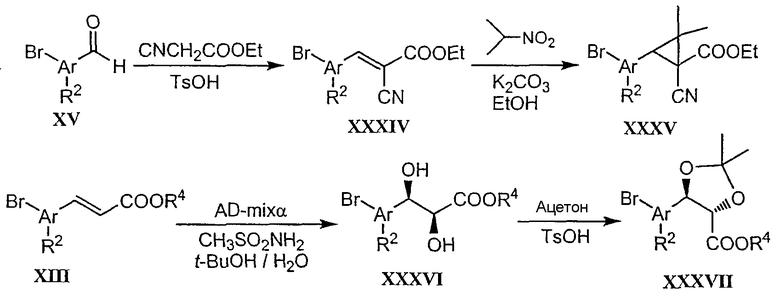
НАФТИРИДИНОН 1
N-Изопропил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид
Стадия 1: Этил 3-(3-броманилино)-2-(2-хлорникотиноил)акрилат
Смесь этил 2-хлорникотиноилацетата (полученного в соответствии со способом, описанным в J. het. Chem., 30, 855. 1993) (1 экв.), триэтилортоформиата (1,5 экв.) и уксусного ангидрида (5 экв.) нагревали при 130°С в течение 2,5 часов. Летучие компоненты отгоняли и остаток дважды выпаривали вместе с ксилолом. Маслянистый остаток растворяли в метиленхлориде и медленно добавляли 3-броманилин (1,2 экв.). Полученный раствор перемешивали при комнатной температуре в течение 18 часов и выпаривали растворитель. Полученное неочищенное соединение использовали без очистки на следующей стадии.
Стадия 2: Этил 1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксилат
Неочищенное соединение со стадии 1 растворяли в тетрагидрофуране (0,3 М), раствор охлаждали до 0°С и по порциям добавляли гидрид натрия (60% дисперсия в масле, 1,3 экв.). После перемешивания при 0°С в течение 1 часа смеси давали нагреться до комнатной температуры. Через 2 часа к суспензии добавляли воду и нерастворимое твердое вещество отфильтровывали и промывали водой в обильном количестве. После высыхания твердое вещество перемешивали в простом эфире при комнатной температуре в течение 24 часов и фильтровали с получением указанного в заголовке соединения в виде твердого вещества кремового цвета.
1Н ЯМР (Ацетон-d6) δ 1,32 (т, 3H), 4,29 (кв, 2H), 7,54-7,63 (м, 2H), 7,69 (дд, 1H), 7,78 (дд, 1H), 7,93 (с, 1H), 8,66-8,71 (м, 3H).
Альтернативно, для стадий 1-2 можно использовать следующий способ.
Смесь 2-хлорникотиноилхлорида (1 экв.), триэтиламина (4 экв.) и этил 3,3-диметиламиноакрилата (1,5 экв.) в ацетонитриле (0,5 М) нагревали до температуры кипения с обратным холодильником в течение 3 часов, охлаждали до 40-50°С и добавляли 3-броманилин (1 экв.). Реакционную смесь нагревали до температуры кипения с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли водой (2 объема). Продукт выделяли фильтрованием и промывали водой, простым эфиром или смесью ацетонитрил-вода (1:1).
Стадия 3: 1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоновая кислота
Суспензию этил 1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксилата со стадии 2 (1 экв.) в смеси тетрагидрофуран-метанол (0,15 М) и 1 н. водный раствор гидроксида натрия (2 экв.) нагревали примерно при 50°С в течение 20 минут при перемешивании. После охлаждения смесь разбавляли водой и подкисляли 1 н. водным раствором HCl. После перемешивания в течение 45 минут осадок отфильтровывали, тщательно промывали водой и сушили с получением указанной в заголовке кислоты в виде твердого вещества кремового цвета.
1Н ЯМР (Ацетон-d6) δ 7,65 (т, 1Н), 7,76 (м, 2Н), 7,84 (д, 1Н), 7,99 (с, 1Н), 8,87 (м, 2Н), 9,01 (с, 1Н).
Стадия 4: N-изопропил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид
К суспензии 1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоновой кислоты со стадии 3 (1 экв.) и триэтиламина (3 экв.) в тетрагидрофуране (0,08 М) при 0°С добавляли изобутилхлорформиат (1,8 экв.). После перемешивания при 0°С в течение 2 часов добавляли изопропиламин (5 экв.) и смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. Затем смесь распределяли между этилацетатом и водой, органическую фазу сушили и упаривали до получения твердого вещества, которое перемешивали в простом эфире при комнатной температуре в течение 3 часов и фильтровали с получением N-изопропил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамида в виде белого твердого вещества.
1Н ЯМР (Ацетон-d6) δ 1,25 (д, 6H), 4,17 (м, 1H), 7,59-7,63 (м, 2H), 7,70 (д, 1H), 7,80 (д, 1H), 7,94 (с, 1H), 8,73 (м, 1H), 8,78 (д, 1H), 8,85 (с, 1H), 9,61 (ушир, NH).
НАФТИРИДИНОН 2
N-Циклопропил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамид
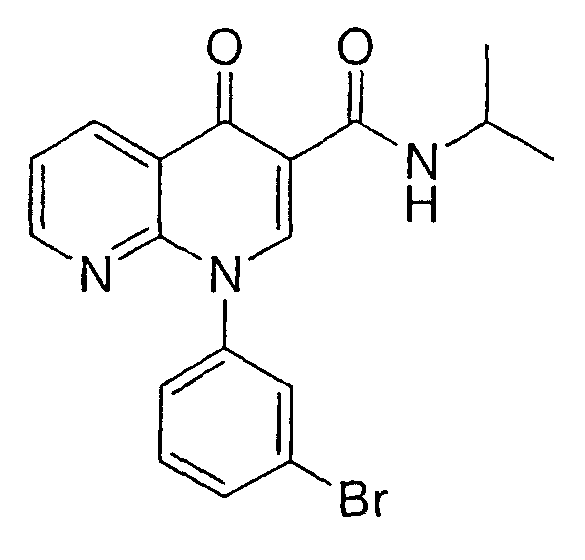
Следуя способу получения НАФТИРИДИНОНА 1, но заменяя на стадии 4 изопропиламин циклопропиламином, получали указанное в заголовке соединение в виде рыхлого белого твердого вещества.
1Н ЯМР (Ацетон-d6) δ 0,59 (м, 2H), 0,80 (м, 2H), 2,96 (м, 1Н), 7,59-7,68 (м, 2H), 7,72 (дд, 1H), 7,82 (дд, 1H), 7,97 (с, 1H), 8,72-8,81 (м, 2H), 8,89 (с, 1H), 9,70 (ушир., NH).
НАФТИРИДИНОН 3
N-Циклопропил-4-оксо-1-[3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1,4-дигидро-1,8-нафтиридин-3-карбоксамид
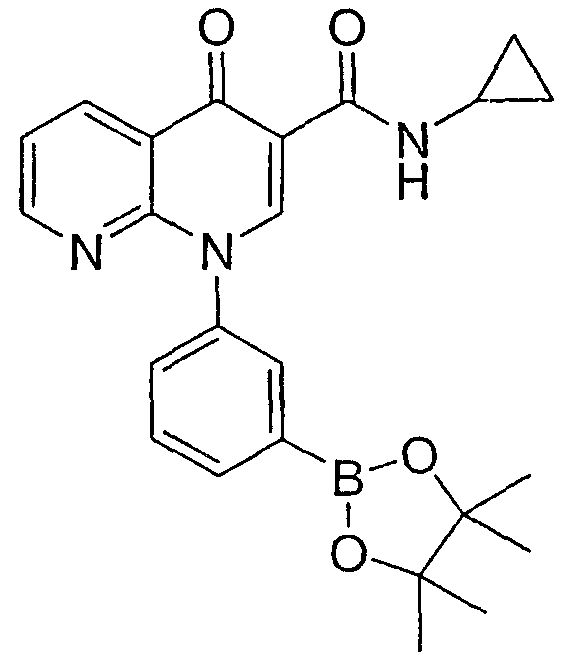
Смесь НАФТИРИДИНОНА 2 (1,0 экв.), пинаколдиборана (1,5 экв.), КОАс (4 экв.) и PdCl2(dppf) (0,05 экв.) в DMF (0,2 М) перемешивали при 70-80°С в течение 3 часов. Смесь охлаждали до комнатной температуры, разбавляли EtOAc и раствором NH4Cl. Органические экстракты промывали Н2О, насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. После кристаллизации из простого эфира и флэш-хроматографии (CH2Cl2:EtOAc, 50:50) маточного раствора получали указанное в заголовке соединение в виде белого твердого вещества.
1Н ЯМР (500 МГц, ацетон-d6): 9,78 (с, 1H), 8,90 (с, 1H), 8,79 (дд, 1H), 8,72 (дд, 1H), 7,94 (д, 1H), 7,91 (с, 1H), 7,80 (д, 1H), 7,69 (т, 1Н), 7,62 (дд, 1H), 2,9 (м, 1H), 1,38 (с, 12H), 0,80 (м, 2H), 0,60 (м, 2H).
НАФТИРИДИНОН 4
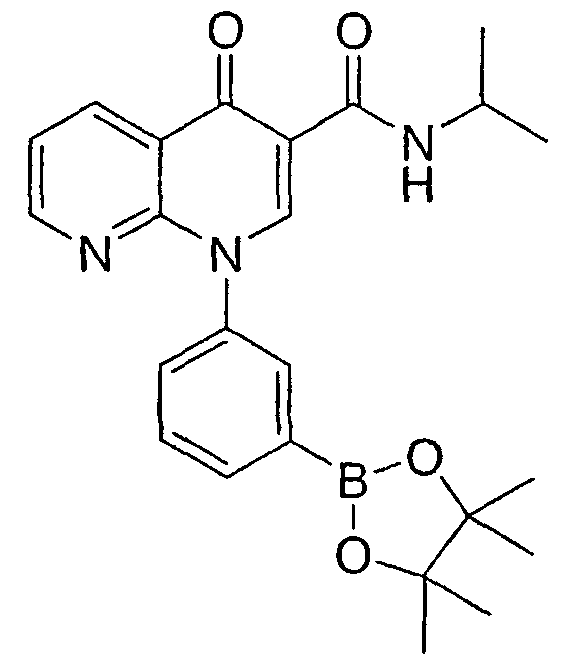
Получали в соответствии со способом, описанным для получения НАФТИРИДИНОНА 3, но используя НАФТИРИДИНОН 1 в качестве исходного вещества.
ПРИМЕР 1
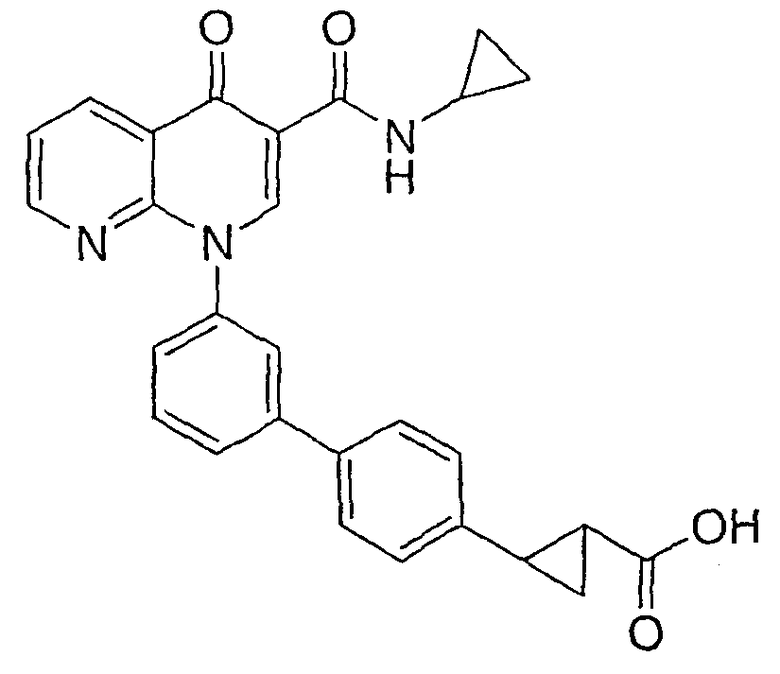
2-(транс)-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
Стадия 1: Этил 2-(транс)-(4-бромфенил)циклопропанкарбоксилат
К смеси этил 4-бромциннамата и Pd(OAc)2 (0,05 экв.) в метиленхлориде (1М) при 0°С добавляли по каплям раствор CH2N2 в простом эфире до тех пор, пока по данным ЯМР реакция не завершалась. Смесь фильтровали через слой силикагеля и концентрировали с получением указанного в заголовке соединения в виде масла.
Стадия 2: Этил-2-(транс)-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]циклопропанкарбоксилат
Смесь бромида со стадии 1 (1,0 экв.), пинаколдиборанового эфира (1.4 экв.), КОАс (3,5 экв.) и PdCl2(dppf) (0,03 экв.) в DMF (0,14 М) перемешивали при 60°С в течение 24 часов. Полученную смесь охлаждали до комнатной температуры, разбавляли EtOAc:гексаном (1:1). Органическую фазу промывали водой, насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Флэш-хроматография (гексан:EtOAc; 90:10) давала указанное в заголовке соединение.
Стадия 3: 2-(транс)-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]циклопропанкарбоновая кислота
Смесь сложного эфира со стадии 2 и NaOH (20%, 30 мл) нагревали до 100°С в течение 1,5 час, охлаждали до комнатной температуры, подкисляли 10% HCl и экстрагировали EtOAc. Органический экстракт сушили над Na2SO4 и выпаривали растворитель с получением указанного в заголовке соединения.
Стадия 4: 2-(транс)-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
Смесь НАФТИРИДИНОНА 2 (1,0 экв.), кислоты со стадии 3 (1,5 экв.), Na2СО3 (3,5 экв., 2М в Н2О), Pd(ОАс)2 (0,05 экв.) и PPh3 (0,15 экв.) или PdCl2dppf (0,05 экв.) в н-пропаноле-DMF (1:1, 0,1 М) перемешивали при 70°С в течение 2 часов. Смесь охлаждали до комнатной температуры, гасили АсОН и разбавляли EtOAc. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Флэш-хроматография (CH2Cl2:EtOAc; 60:40, 2% АсОН) давала указанное в заголовке соединение в виде белого твердого вещества.
1H ЯМР (500 МГц, CDCl3): δ 9,9 (д, 1H), 9,08 (с, 1H), 8,77 (дд, 1H), 8,69 (дд, 1H), 7,71 (д, 1H), 7,60 (м, 2H), 7,52 (д, 2H), 7,45 (м, 1H), 7,38 (д, 1H), 7,13 (д, 2H), 2,97 (м, 1H), 2,54 (м, 1H), 1,87 (м, 1Н), 1,60 (м, 1H), 1,35 (м, 1H), 0,85 (м, 2H), 0,65 (м, 2H). МС (Н-): 464,2.
Оптически активные изомеры примера 1 могут быть выделены отдельно методом хроматографии с использованием хиральной колонки; например, Chiral Pak AD, элюируя гексаном:EtOH или гексаном:iPrOH, содержащим 0,2% TFA.
Альтернативно, разделение можно осуществить на промежуточном соединении (транс)-2-(4-бромфенил)циклопропанкарбоновой кислоте.
(транс)-2-(4-Бромфенил)циклопропанкарбоновая кислота
К раствору сложного эфира со стадии 1 в ТГФ-МеОН (4:1, 0,5 М) добавляли LiOH (3 экв., 2М) и смесь перемешивали при 50°С в течение 1 часа. Органический растворитель выпаривали, водную фазу подкисляли 1 н. раствором HCl и кислоту экстрагировали EtOAc (3х). Органическую фазу промывали насыщенным солевым раствором, сушили и выпаривали растворитель с получением 2-(4-бромфенил)циклопропанкарбоновой кислоты.
Оптически активные предшественники получали разделением на хиральной колонке (Chiral Pak AD), элюируя гексаном:EtOH или гексаном:iPrOH, содержащим 0,2% TFA.
Следующая альтернатива представляет собой использование хирального вспомогательного соединения, как описано ниже.
Стадия 1: (транс)-3-(4-Бромфенил)-1-имидазол-1-илпропенон
К раствору (транс)-3-(4-бромфенил)акриловой кислоты (1,0 экв.) в толуоле (0,2 М) добавляли CDI (1,5 экв.). Смесь перемешивали при комнатной температуре в течение 3 часов. Полученный остаток выделяли фильтрованием с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 2: (транс)-3-[3-(4-бромфенил)-акрилоил]-4-метил-5-фенилоксазолидин-2-он
Смесь 3-(4-бромфенил)-1-имидазол-1-илпропенона (1,05 экв.) со стадии 1, (4R,5S)-(+)-4-метил-5-фенил-2-оксазолидинона (1,0 экв.) или (-)-изомера и Et3N (1,2 экв.) в CH3CN (0,2 М) кипятили с обратным холодильником в течение ночи. Полученную смесь охлаждали до комнатной температуры, фильтровали через слой силикагеля и концентрировали. Кристаллизация в гексане:Et2O давала указанное в заголовке соединение в виде белого твердого вещества.
Стадия 3: (транс)-3-[2-(4-бромфенил)циклопропанкарбонил]-4-метил-5-фенилоксазолидин-2-он
К раствору (транс)-3-[3-(4-бромфенил)акрилоил]-4-метил-5-фенилоксазолидин-2-она со Стадии 2 и Pd(ОАс)2 (0,05 экв.) в ТГФ (0,2 М) по порциям добавляли CH2N2 до завершения реакции. Реакцию отслеживали путем анализа аликвот методом ЯМР. Полученную смесь концентрировали и подвергали флэш-хроматографии (гексан:EtOAc; 3:2) с получением двух отдельных диастереоизомеров. Каждый диастереоизомер отдельно подвергали описанным ниже процедурам с получением (+) и (-) энантиомеров примера 1.
Стадия 4: 2-(транс)-(4-бромфенил)циклопропанкарбоновая кислота
К раствору амида со стадии 3 в ТГФ-EtOH-Н2О (4:1:1, 0,1 М) добавляли LiOH (2,4 экв., 2М) и смесь перемешивали при комнатной температуре в течение 2 часов. Смесь нейтрализовали до рН 7 при помощи 1 н. раствора HCl, органический растворитель выпаривали и полученный остаток растворяли в простом эфире. Органическую фазу промывали 1 н. раствором NaOH (2Х). Объединенные водные слои подкисляли и экстрагировали простым эфиром (3Х). Органический экстракт промывали насыщенным солевым раствором, сушили и выпаривали растворитель с получением (+) или (-)-(транс)-2-(4-бромфенил)циклопропанкарбоновой кислоты в виде белого твердого вещества.
Стадия 5: 2-(транс)-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
Смесь НАФТИРИДИНОНА 3 (1,0 экв.), 2-(транс)-(4-бромфенил)циклопропанкарбоновой кислоты (1,2 экв.) со стадии 4, Na2СО3 (3,5 экв., 2М в Н2О), Pd(ОАс)2 (0,05 экв.) и PPh3 (0,15 экв.) в н-пропаноле (0,1М) перемешивали при 70°С в течение 4 часов. Смесь охлаждали до комнатной температуры, выливали в воду и экстрагировали EtOAc (2х). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Флэш-хроматография (CH2Cl2:EtOAc; 50:50, 2% АсОН) давала указанное в заголовке соединение в виде белого твердого вещества.
Еще одна альтернатива представляет собой энантиоселективное циклопропанирование с использованием, например, комплекса бис-оксазолиновый хиральный лиганд/медь и диазоацетата (Evans et al., J. Am. Chem. Soc. 1991, 113, 726) для получения оптически активного этил 2-(транс)-(4-бромфенил)циклопропанкарбоксилата из 4-бромстирола. Селективный гидролиз смеси цис- и транс-изомера с использованием LiOH (1 экв. в расчете на транс-эфир) давал 2-(транс)-(4-бромфенил)циклопропанкарбоновую кислоту и этил-2-(цис)-(4-бромфенил)циклопропанкарбоксилат, который можно использовать в примере 8.
ПРИМЕР 2
2-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-3-ил}циклопропанкарбоновая кислота
Стадия 1: Метил-2-(транс)-(3-бромфенил)циклопропанкарбоксилат
К смеси 3-бромкоричной кислоты и Pd(OAc)2 (0,05 экв.) в метиленхлориде (1М) при 0°С добавляли по каплям раствор CH2N2 в простом эфире до тех пор, пока по данным ЯМР реакция не завершалась. Смесь фильтровали через слой силикагеля и концентрировали с получением указанного в заголовке соединения.
Стадия 2: 2-(3-бромфенил)циклопропанкарбоновая кислота
К раствору сложного эфира со стадии 1 в ТГФ-МеОН (4:1, 0,5 М) добавляли LiOH (3 экв., 2М) и смесь перемешивали при 50°С в течение 1 часа. Органический растворитель выпаривали, водную фазу подкисляли 1 н. раствором HCl и кислоту экстрагировали EtOAc (3Х). Органическую фазу промывали насыщенным солевым раствором, сушили и выпаривали растворитель с получением 2-(3-бромфенил)циклопропанкарбоновой кислоты.
Стадия 3: 2-(транс)-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-3-ил}циклопропанкарбоновая кислота
Смесь НАФТИРИДИНОНА 3 (1,0 экв.), кислоты со стадии 2 (1,2 экв.), Na2СО3 (3,5 экв., 2М в Н2О), PdCl2(dppf) (0,05 экв.) в н-пропаноле (0,1 М) перемешивали при 90°С в течение 3 часов. Смесь охлаждали до комнатной температуры, гасили при помощи HCl и разбавляли EtOAc. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Флэш-хроматография (CH2Cl2:EtOAc:NH4OH; 75:25:2,5) давала указанное в заголовке соединение в виде белого твердого вещества.
1Н ЯМР (500 МГц, ацетон-d6): δ м.д. 0,61 (м, 2 H), 0,80 (м, 2H), 1,45 (м, 1H), 1,52 (м, 1H), 2,01 (м, 1H), 2,54 (м, 1H), 2,97 (дт, J=11,35, 3,47 Гц, 1H), 7,25 (д, J=8,20 Гц, 1H), 7,43 (т, J=7,57 Гц, 1H), 7,63 (м, 4H), 7,75 (т, J=7,88 Гц, 1H), 7,95 (д, J=8,20 Гц, 1H), 8,04 (с, 1H), 8,75 (дд, J=4,41, 1,89 Гц, 1H), 8,80 (дд, 7=8,20, 1,89 Гц, 1H), 8,96 (с, 1H), 9,76 (с, 1H).
ПРИМЕР 3
2-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-3-ил}-2-метилпропановая кислота
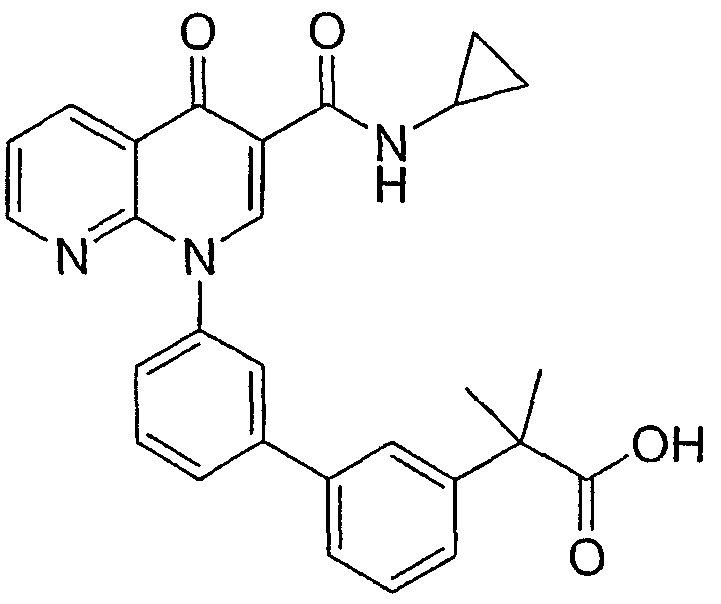
Стадия 1: Метил 2-(3-бромфенил)-2-метилпропаноат
К раствору LiHMDS (1 М, ТГФ, 2,1 экв.) в ТГФ (0,06 М) добавляли метил 3-бромфенилацетат (1 экв.). Через 15 минут добавляли MeI (4 экв.) и реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 18 часов. Смесь гасили 10% раствором HCl, разбавляли EtOAc, промывали 10% раствором HCl, насыщенным солевым раствором, сушили и выпаривали растворитель. Флэш-хроматография (гексан:EtOAc; 95:5) давала указанное в заголовке соединение.
Стадия 2: 2-(3-бромфенил) метилпропановая кислота
К раствору сложного эфира со стадии 1 в ТГФ-МеОН (4:1, 0,5 М) добавляли LiOH (3 экв., 2М) и смесь перемешивали при 50°С в течение 1 часа. Органический растворитель выпаривали, водную фазу подкисляли 1 н. раствором HCl и кислоту экстрагировали EtOAc (3х). Органическую фазу промывали насыщенным солевым раствором, сушили и выпаривали растворитель с получением указанной кислоты.
Стадия 3: 2-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-3-ил}-2-метилпропановая кислота
Смесь НАФТИРИДИНОНА 3 (1,0 экв.), кислоты со стадии 2 (1,5 экв.), Na2СО3 (3,5 экв., 2М в Н2О) и Pd(Ph3)4, или PdCl2(dppf), или Pd(ОАс)2(Ph3P)3 (0,05 экв.) в н-пропаноле (0,1 М) перемешивали при 70-90°С в течение 3 часов. Смесь охлаждали до комнатной температуры, гасили при помощи HCl и разбавляли EtOAc. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Флэш-хроматография (CH2Cl2:MeOH; 99:1) давала указанное в заголовке соединение в виде белого твердого вещества.
1Н ЯМР (500 МГц, Ацетон-d6): δ 11,0 (с, OH), 9,76 (с, NH), 8,95 (с, 1H), 8,74 (дд, 1H), 8,71 (дд, 1H), 8,02 (с, 1H), 7,88 (д, 1H), 7,81 (с, 1H), 7,72 (т, 1H), 7,65 (м, 2H), 7,58 (дд, 1H), 7,46 (д, 1H), 2,94 (м, 1H), 1,61 (с, 6H), 0,77 (м, 2H), 0,58 (м, 2H). МС +ECI Q1 (М+1) 468,3.
ПРИМЕР 4
2-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-2-метилпропановая кислота
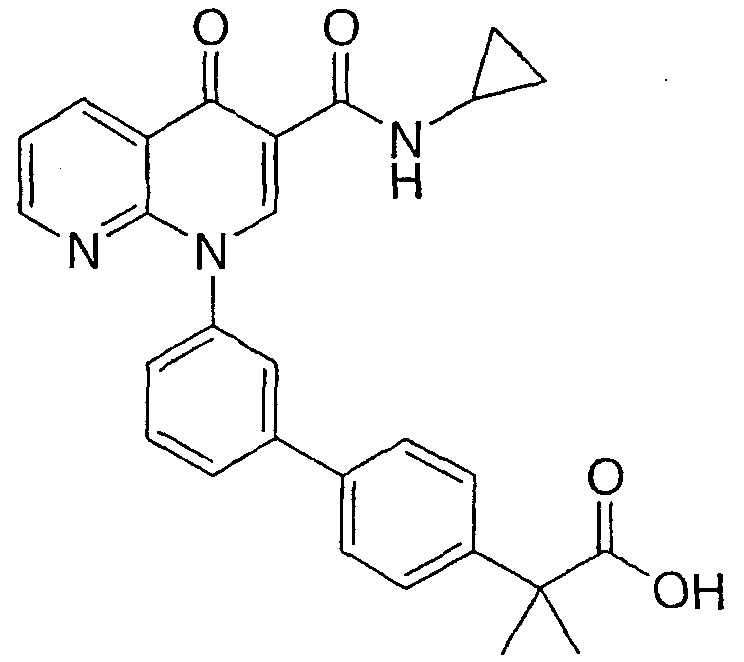
Получали в соответствии со способом, описанным в примере 3, но используя в качестве исходного вещества 4-бромфенилацетат. Флэш-хроматография (CH2Cl2:EtOAc, 40:60, 2% АсОН) давала указанное в заголовке соединение в виде белого твердого вещества.
1Н ЯМР (500 МГц, ДМСО-d6): δ 9,75 (д, 1H), 8,81 (с, 1H), 8,77 (м, 1H), 8,71 (д, 1H), 7,92 (с, 1H), 7,86 (д, 1H), 7,70 (д, 2H), 7,70-7,55 (м, 3H), 7,45 (д, 2H), 2,88 (м, 1H), 1,50 (с, 6H), 0,77 (м, 2H), 0,55 (м, 2H).
ПРИМЕР 5
3-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-3-метилбутановая кислота
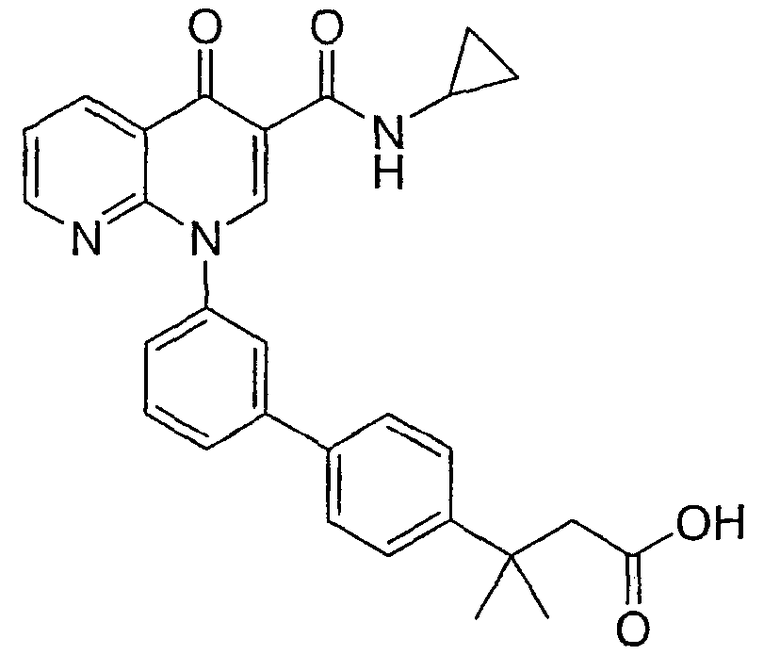
Получали в соответствии со способом, описанным в примере 3, стадия 2 и 3, но используя в качестве исходного вещества метиловый эфир 3-(4-иодфенил)-3-метилмасляной кислоты (полученный в соответствии со способом, описанным в J. Am. Chem. Soc. 1948, 70, 370). Флэш-хроматография (CH2Cl2:EtOAc, 40:60, 2% АсОН) давала указанное в заголовке соединение в виде белого твердого вещества.
1Н ЯМР (500 МГц, ДМСО-d6): δ 9,75 (д, 1H), 8,82 (с, 1H), 8,78 (дд, 1H), 8,72 (дд, 1H), 7,94 (с, 1H), 7,86 (д, 1H), 7,68-7,60 (м, 2H), 7,66 (д, 2H), 7,57 (д, 1H), 7,49 (д, 2H), 2,88 (м, 1H), 2,60 (с, 2H), 1,40 (с, 6H), 0,76 (м, 2H), 0,55 (м, 2H).
ПРИМЕР 6
{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}(гидрокси)уксусная кислота
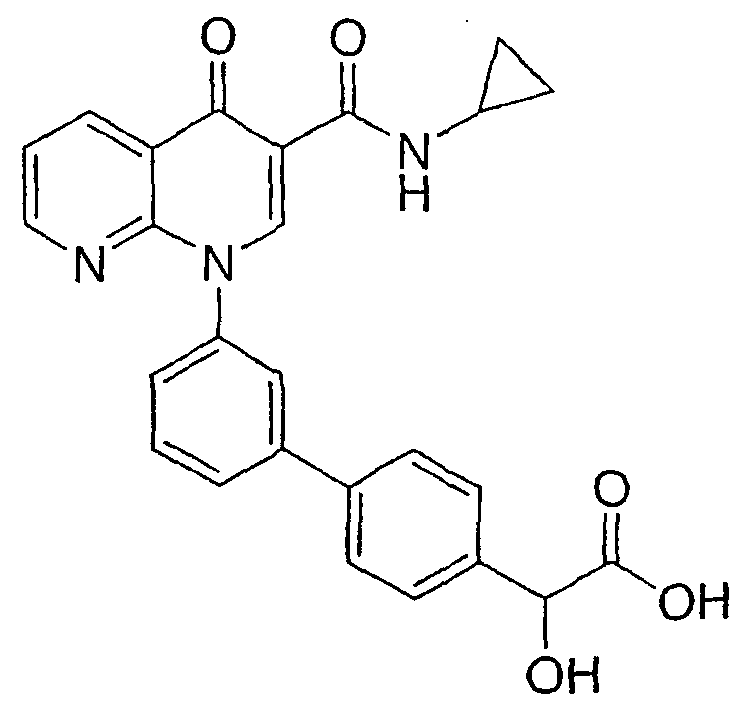
Получали в соответствии со способом, описанным в примере 3, стадия 3, но используя в качестве исходного вещества 4-бромминдальную кислоту. Флэш-хроматография (CH2Cl2:MeOH, от 9:1 до 8:2) с последующим интенсивном перемешиванием остатка в CH2Cl2/эфире и выделением путем фильтрования давала указанное в заголовке соединение в виде белого твердого вещества.
1Н ЯМР (500 МГц, ацетон-d6): δ 9,75 (д, 1H), 8,83 (с, 1Н), 8,79 (дд, 1Н), 8,74 (дд, 1Н), 7,97 (с, 1H), 7,89 (д, 1H), 7,73 (д, 2H), 7,70 (т, 1H), 7,65 (дд, 1H), 7,61 (д, 1Н), 7,52 (д, 2H), 5,03 (с, 1H), 2,92-2,90 (м, 1Н), 0,80-0,77 (м, 2H), 0,59-0,56 (м, 2H).
ПРИМЕР 7
1-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-циклопропанкарбоновая кислота
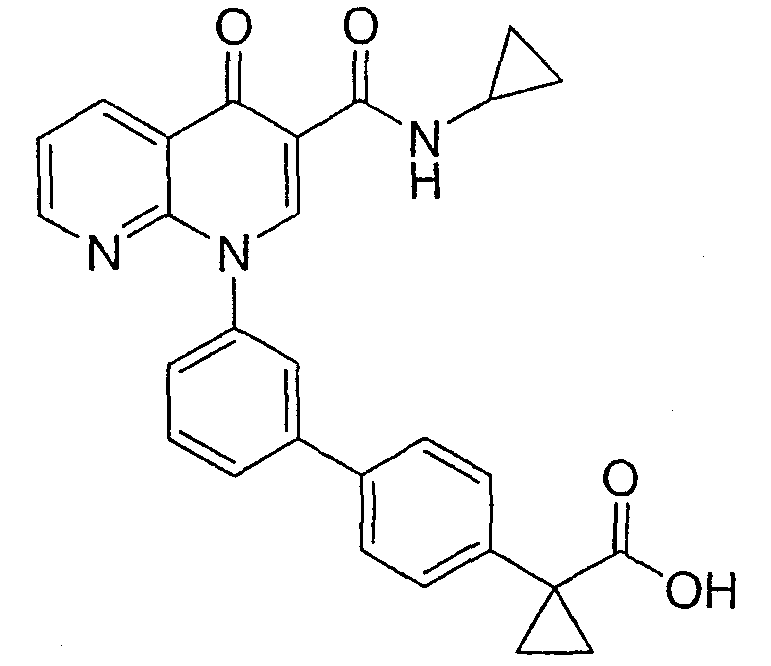
Стадия 1: 1-(4-бромфенил)циклопропанкарбонитрил
Смесь 4-бромфенилацетонитрила (1 экв.), 1,2-бромхлорэтана (1,5 экв.), бензилтриэтиламмонийхлорида (0,14 экв.) в 50% растворе NaOH (4М) нагревали до 50°С в течение 18 часов. Смесь охлаждали до комнатной температуры, гасили 5% раствором HCl и разбавляли простым эфиром. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Флэш-хроматография (гексан:EtOAc; 95:5) давала указанное в заголовке соединение.
Стадия 2: 1-(4-бромфенил)циклопропанкарбоновая кислота
Смесь нитрила (1 экв.) со стадии 1, 25% раствора NaOH (12 экв.) в EtOH (0,5М) нагревали до 100°С в течение 5 часов. Смесь охлаждали до комнатной температуры, гасили АсОН и разбавляли простым эфиром. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Флэш-хроматография (гексан:EtOAc, от 90:10 до 50:50) давала указанное в заголовке соединение.
Стадия 3: 1-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
Получали в соответствии со способом, описанным в примере 3, стадия 3, но используя в качестве исходного вещества кислоту со стадии 2. Флэш-хроматография (CH2Cl2:MeOH, 98:2) давала указанное в заголовке соединение в виде белого твердого вещества.
1Н ЯМР (500 МГц, ацетон-d6): δ 9,92 (д, 1H), 9,77 (д, 1Н), 8,96 (с, 1H), 8,79 (дд, 1H), 8,74 (дд, 1H), 8,00 (с, 1H), 7,76-7,72 (м, 3H), 7,65 (дд, 1Н), 7,61 (дд, 1H), 7,52 (д, 2H), 2,98-2,94 (м, 1H), 1,60-1,58 (м, 2H), 1,25-1,22 (м, 2H), 0,82-0,78 (м, 2H), 0,62-0,59 (м, 2H).
ПРИМЕР 8
2-(цис)-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
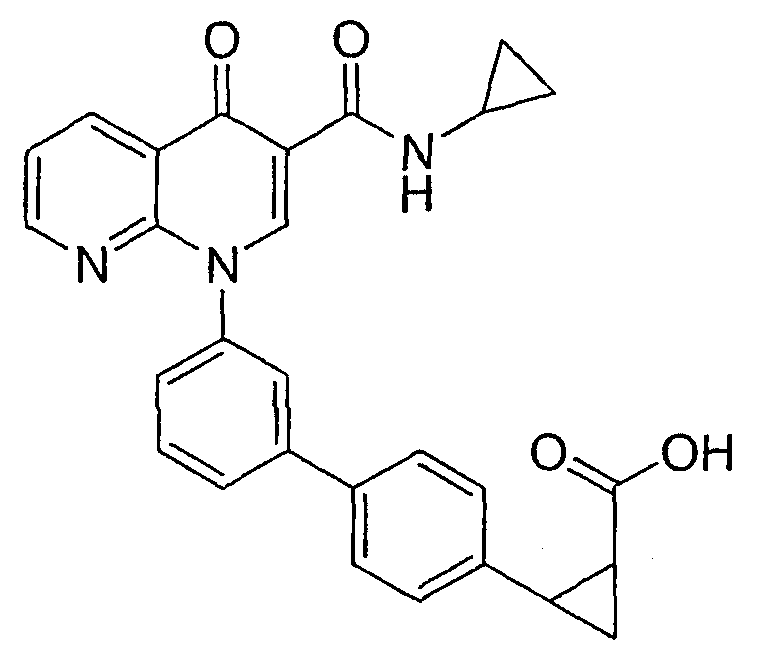
Стадия 1: Метил-2-(цис)-3-(4-бромфенил)проп-2-еноат
К раствору бис(трифторэтил)(метоксикарбонилметил)фосфоната и 18-краун-6 (5 экв.) в ТГФ (0,05 М) при -78°С добавляли KHMDS (1 экв., 0,5 М, толуол), а затем 4-бромбензальдегид (1 экв.). Реакционную смесь перемешивали при -78°С в течение 1 часа, гасили насыщенным раствором хлорида аммония и разбавляли простым эфиром. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Флэш-хроматография (гексан:EtOAc, от 9:1 до 7:3) давала указанное в заголовке соединение.
Стадия 2: Метил-2-(цис)-(4-бромфенил)циклопропанкарбоксилат
Смесь сложного эфира со стадии 1 и Pd(OAc)2 (0,05 экв.) в метиленхлориде (1 М) при 0°С добавляли по каплям раствор CH2N2 в простом эфире до тех пор, пока по данным ЯМР анализа реакция не завершалась. Флэш-хроматография (гексан:EtOAc, от 100:0 до 90:10) давала указанное в заголовке соединение.
Стадия 3: Метил-2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}- циклопропанкарбоксилат
Получали в соответствии со способом, описанным в примере 3, стадия 3, но используя в качестве исходного вещества сложный эфир из примера 8, стадия 2. Продукт очищали флэш-хроматографией (гексан:EtOAc, 60:40) с последующим интенсивном перемешиванием остатка в гексане/эфире и выделением путем фильтрования с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 4: 2-(цис)-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}- циклопропанкарбоновая кислота
К раствору сложного эфира в ТГФ-EtOH (1:1, 0,05 М) добавляли LiOH (5 экв., 2М) и смесь перемешивали при 60°С в течение 4 часов. Смесь охлаждали до комнатной температуры, экстрагировали простым эфиром. Водную фазу подкисляли 1 н. раствором HCl и кислоту экстрагировали EtOAc (3Х). Органическую фазу промывали насыщенным солевым раствором, сушили и выпаривали растворитель. Остаток интенсивно перемешивали в смеси CH2Cl2/гексан/ацетон и выделяли фильтрованием с получением указанного в заголовке соединения в виде не совсем белого твердого вещества.
1Н ЯМР (500 МГц, ДМСО-d6): δ 11,88 (ушир. с, 1Н), 9,75 (д, 1H), 8,80 (с, 1H), 8,79 (дд, 1H), 8,74 (дд, 1H), 7,95 (с, 1H), 7,87 (д, 1Н), 7,63-7,70 (м, 4H), 7,59 (д, 1H), 7,35 (д, 2H), 2,89-2,93 (м, 1H), 2,62 (дд, 1H), 2,04-2,09 (м, 1H), 1,31-1,35 (м, 1H), 0,77-0,87 (м, 2H), 0,56-0,59 (м, 2H).
Оптически активные диастереоизомеры примера 8 можно выделить по отдельности методом хроматографии с использованием хиральной колонки; например, Chiral Pak AD, элюируя гексаном:EtOH или гексаном:iPrOH, содержащим 0,2% TFA.
Альтернативно, оптически активное промежуточное соединение может быть получено, как описано ниже.
(цис)-2-(4-Бромфенил)циклопропанкарбоновая кислота
К раствору сложного эфира со стадии 2 в ТГФ-МеОН (4:1, 0,05 М) добавляли LiOH (3 экв., 2М) и смесь перемешивали при 50°С в течение 4 часа. Органический растворитель выпаривали, водный слой подкисляли 1 н. раствором HCl и кислоту экстрагировали EtOAc (3Х). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили и упаривали с получением 2-(4-бромфенил)циклопропанкарбоновой кислоты. Оптически активные промежуточные соединения получали разделением на хиральной колонке (Chiral Pak AD) с использованием гексана:EtOH (90:10 с 0,2% TFA).
Другая альтернатива представляет собой использование хирального вспомогательного соединения, как описано ниже.
Стадия 1: (цис)-1-{[2-(4-Бромфенил)циклопропил]карбонил}-1Н-имидазол
К раствору (цис)-2-(4-бромфенил)циклопропанкарбоновой кислоты (1,0 экв.) в толуоле (0,2 М) добавляли CDI (1,5 экв.). Смесь перемешивали при 50°С в течение 18 часов. Растворитель выпаривали и остаток интенсивно перемешивали в смеси гексан/CH2Cl2, фильтровали и фильтрат упаривали с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 2: (цис)-3-{[2-(4-Бромфенил)циклопропил]карбонил}-4-метил-5-фенил-1,3-оксазолидин-2-он
Смесь (цис)-1-{[2-(4-бромфенил)циклопропил]карбонил}-1Н-имидазола (1 экв.) со стадии 1, (4R,5S)-(+)-4-метил-5-фенил-2-оксазолидинона (1,2 экв.) или (-)-изомера и DBU (добавляемого при 0°С, 1.2 экв.) в CH3CN (0,2М) перемешивали при 0°С в течение 4 часов. Растворитель выпаривали и остаток очищали флэш-хроматографией (гексан:EtOAc, от 100:0 до 80:20) с получением каждого диастереоизомера.
Стадия 3: (цис)-2-(4-бромфенил)циклопропанкарбоновая кислота
К раствору амида (либо (+), либо (-) изомер) со стадии 2, описанной выше) в ТГФ-Н2О (4:1, 0,5М) при 0°С добавляли LiOH (1,6 экв., 2М) и Н2О2 (35%, 4 экв.). Смесь перемешивали при 0°С в течение 4 часов. Органический растворитель выпаривали и смесь экстрагировали CH2Cl2, водную фазу подкисляли 1 н. раствором HCl и кислоту экстрагировали EtOAc (3х). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили и растворитель выпаривали с получением либо (+), либо (-) оптически активной (цис)-2-(4-бромфенил)циклопропанкарбоновой кислоты.
Еще одна альтернатива представляет собой энантиоселективное циклопропанирование с использованием, например, комплекса бис-оксазолиновый хиральный лиганд/медь и диазоацетата (Evans et al., J. Am. Chem. Soc. 1991, 113, 726) для получения оптически активного этил-2-(цис)-(4-бромфенил)циклопропанкарбоксилата из 4-бромстирола. Селективный гидролиз смеси цис- и транс-изомера с использованием LiOH (1 экв. в расчете на транс-изомер) давал 2-(транс)-(4-бромфенил)циклопропанкарбоновую кислоту (используемую в Примере 1) и желаемый этил-2-(цис)-(4-бромфенил)циклопропанкарбоксилат.
ПРИМЕР 9
5-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-2,2-диметил-1,3-диоксолан-4-карбоновая кислота
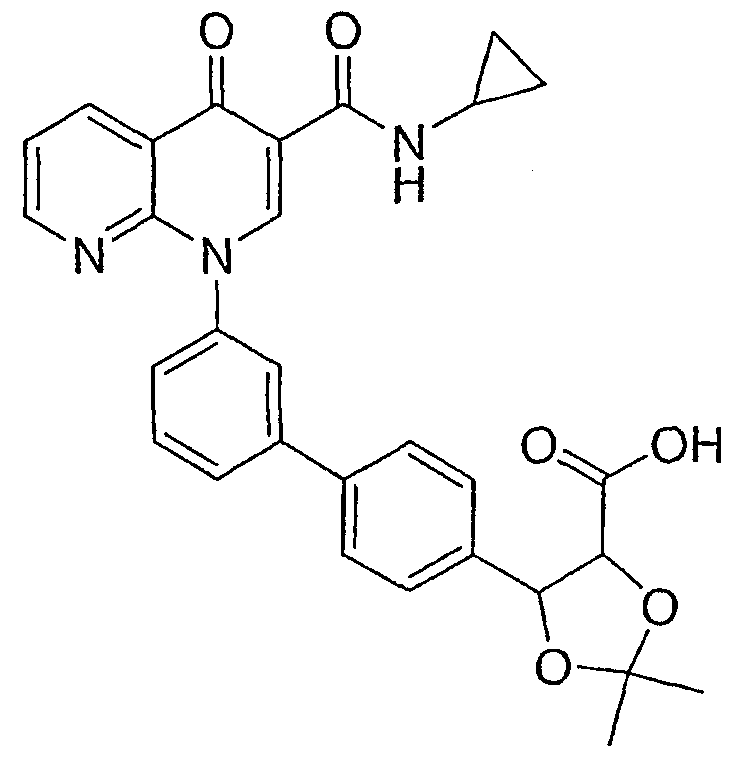
Стадия 1: Этил 3-(4-бромфенил)-2,3-дигидроксипропаноат
Следовали методике, описанной в J. Org. Chem., 1992, 57, 2768, с использованием этил-4-бромциннамата в качестве исходного вещества. Остаток очищали флэш-хроматографией (гексан:EtOAc, 50:50) с получением указанного в заголовке соединения.
Стадия 2: Этил 5-(4-бромфенил)-2,2-диметил-1,3-диоксолан-4-карбоксилат
Раствор диола со стадии 1 (1 экв.) в ацетоне (0,2М), диметоксипропана (8 экв.) и pTsOH (0,05 экв.) перемешивали при комнатной температуре в течение 4 часов. Растворитель выпаривали, остаток растворяли в EtOAc и промывали водным раствором NaHCO3. Органический экстракт промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Флэш-хроматография (гексан:EtOAc, от 95:5 до 85:15) давала указанное в заголовке соединение.
Стадия 3: Этил 5-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-2,2-диметил-1,3-диоксолан-4-карбоксилат
Смесь НАФТИРИДИНОНА 3 (1,0 экв.), сложного эфира со стадии 2 (1,2 экв.), Na2СО3 (2,5 экв., 2М в Н2О) и Pd(ОАс)2(Ph3P)3 (0,05 экв.) в DMF-H2O (0,3М) перемешивали при 80°С в течение 4 часов. Смесь охлаждали до комнатной температуры, разбавляли EtOAc и фильтровали через целит (промывали EtOAc). Органические экстракты промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Флэш-хроматография (гексан:EtOAc, 30:70) давала указанное в заголовке соединение в виде белого твердого вещества.
Стадия 4: 5-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-2,2-диметил-1,3-диоксолан-4-карбоновая кислота
К раствору сложного эфира со стадии 3 в ТГФ-МеОН (1:1, 0,05 М) добавляли LiOH (3 экв., 2М) и смесь перемешивали при 50°С в течение 15 минут. Органический растворитель выпаривали, водную фазу подкисляли 1 н. раствором HCl и кислоту экстрагировали EtOAc (3х). Объединенный органический экстракт промывали насыщенным солевым раствором, сушили и выпаривали растворитель, остаток растирали в порошок в смеси гексан/простой эфир и выделяли фильтрованием с получением указанного в заголовке соединения.
1Н ЯМР (500 МГц, ацетон-4): δ 11,4 (ушир. с, 1H), 9,76 (д, 1Н), 8,97 (с, 1H), 8,79 (дд, 1H), 8,75 (дд, 1Н), 8,02 (т, 1H), 7,94 (д, 1H), 7,83 (д, 2H), 7,76 (т, 1H), 7,60-7,68 (м, 4H), 5,27 (д, 1H), 4,40 (д, 1H), 2,95-2,99 (м, 1H), 1,58 (с, 3H), 1,53 (с, 3H), 0,79-0,82 (м, 2H), 0,59-0,63 (м, 2H).
ПРИМЕР 10
1-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-3-ил}-циклопропанкарбоновая кислота
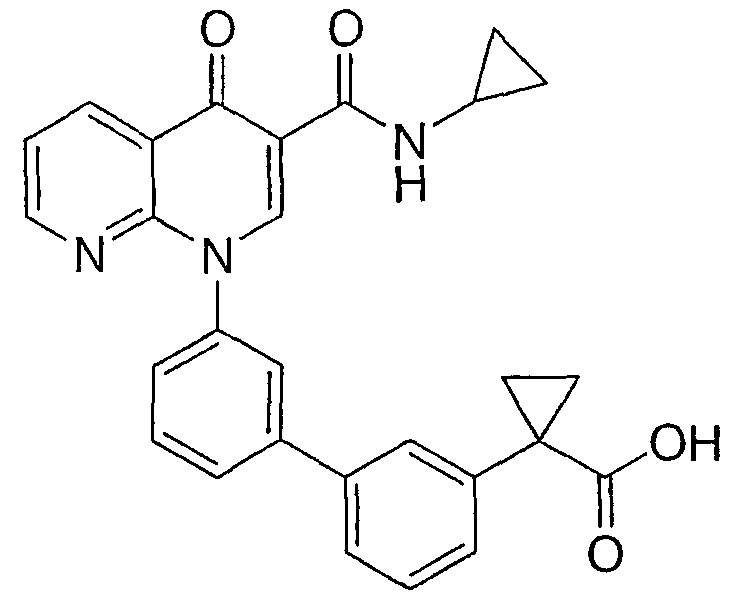
Получали в соответствии со способом, описанным в примере 7, но используя в качестве исходного вещества 3-бромфенилацетонитрил. Остаток очищали флэш-хроматографией (CH2Cl2:МеОН, 96:4), растирали в порошок в гексане/CH2Cl2 и затем выделяли путем фильтрования с получением указанного в заголовке соединения в виде белого твердого вещества.
1Н ЯМР (500 МГц, CDCl3): δ 9,88 (д, 1H), 9,06 (с, 1Н), 8,79 (дд, 1H), 8,69 (дд, 1Н), 7,70 (д, 1H), 7,60-7,52 (м, 3H), 7,49 (д, 1H), 7,44 (дд, 1Н), 7,39-7,30 (м, 3H), 2,95 (м, 1H), 1,65 (м, 2H), 1,25 (м, 2H), 0,82 (м, 2H), 0,65 (м, 2H).
ПРИМЕР 11
1-Циано-3-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-2,2-диметилциклопропанкарбоновая кислота
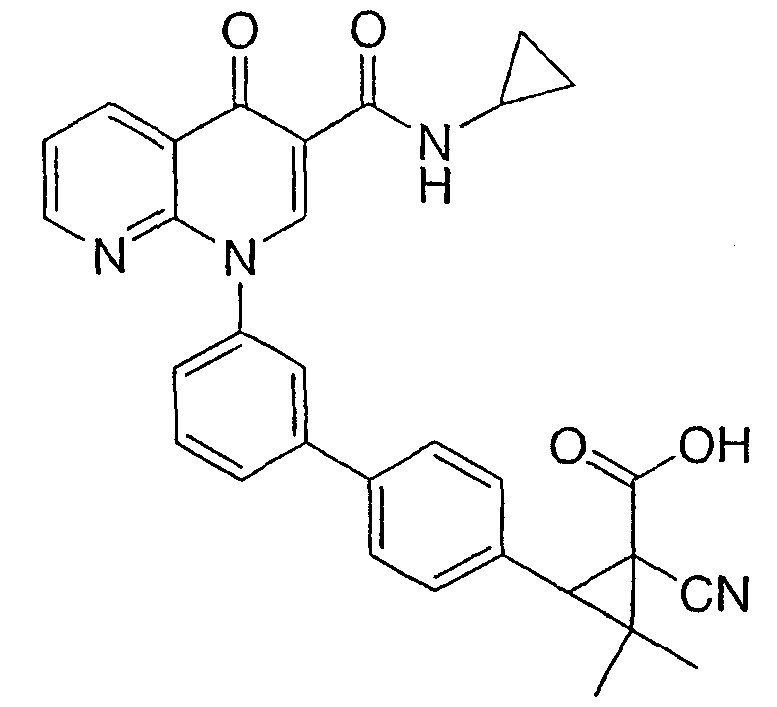
Стадия 1:трет-бутил (2Е)-3-(4-бромфенил)-2-цианопроп-2-еноат
Смесь 4-бромбензальдегида (1 экв.), трет-бутилцианоацетата (1 экв.) и NH4OAc (1 экв.) в бензоле (1М) кипятили с обратным холодильником в течение 4 часов, удаляя при этом воду при помощи ловушки Дина-Старка. Смесь охлаждали до комнатной температуры, разбавляли гексаном и растирали в порошок. Указанное в заголовке соединение выделяли после фильтрования в виде белого твердого вещества.
Стадия 2: трет-Бутил-3-(4-бромфенил)-1-циано-2,2-диметилциклопропанкарбоксилат
Следуя методике, описанной в Tetrahedron Lett. 1985, 1923, смесь 2-нитропропана (1 экв.), К2СО3 (1 экв.) и сложного эфира со стадии 1 (1 экв.) в EtOH (1,2 М) перемешивали при 100°С в закрытой бутыли в течение 3 часов. Смесь охлаждали до комнатной температуры, разбавляли EtOAc, промывали раствором NH4Cl, насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Флэш-хроматография (гексан:EtOAc, 95:5) давала указанное в заголовке соединение в виде масла.
Стадия 3: трет-Бутил-1-циано-3-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-2,2-диметилциклопропанкарбоксилат
Получали в соответствии со способом, описанным в примере 7, стадия 3, но используя в качестве исходного вещества сложный эфир со стадии 2. Остаток очищали флэш-хроматографией (гексан:EtOAc, от 60:40 до 30:70).
Стадия 4: 1-Циано-3-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-2,2-диметилциклопропанкарбоновая кислота
Сложный эфир со стадии 3 (1 экв.) в СН2Cl2- TFA-DMS (3:2:1, 0,02 М) перемешивали при комнатной температуре в течение 48 часов. Летучие вещества выпаривали, остаток растирали с эфиром и затем выделяли фильтрованием с получением указанного в заголовке соединения в виде белого твердого вещества.
1Н ЯМР (500 МГц, ацетон-d6): δ 9,73 (с, 1Н), 8,94 (с, 1H), 8,77 (дд, 1H), 8,72 (дд, 1H), 8,01 (т, 1H), 7,92 (д, 1H), 7,81 (д, 2H), 7,73 (т, 1H), 7,64 (дд, 1H), 7,59 (дд, 1H), 7,51 (д, 2H), 2,93 (м, 1Н), 2,9 (ушир. с, OH), 1,52 (с, 3H), 1,36 (с, 3H), 0,78 (м, 2H), 0,58 (м, 2H).
ПРИМЕР 12
2-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-3-фтор-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
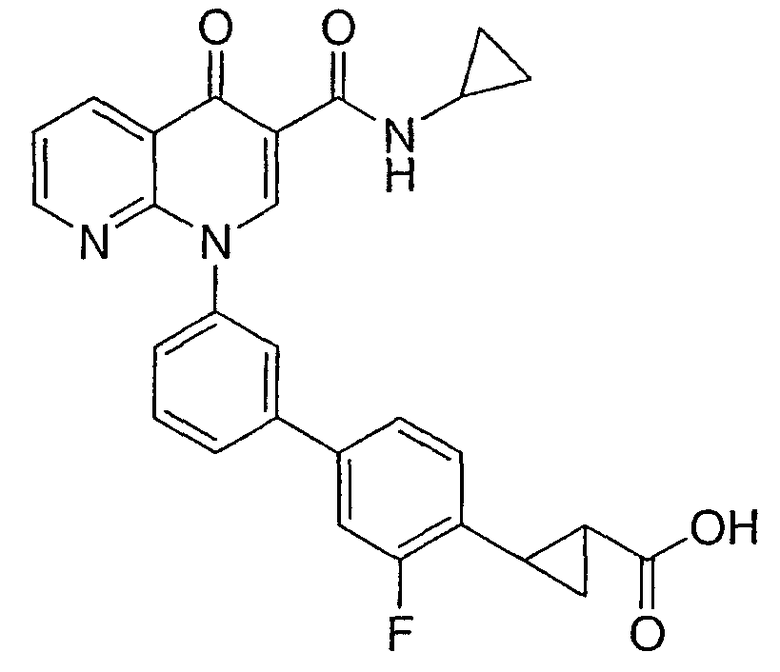
Стадия 1: Метил-2-(4-бром-2-фторфенил)циклопропанкарбоксилат
К смеси 4-бром-2-фторкоричной кислоты и Pd(OAc)2 (0,05 экв.) в метиленхлориде (1М) при 0°С добавляли по каплям раствор CH2N2 в простом эфире до тех пор, пока по данным ЯМР реакция не завершалась. Остаток очищали флэш-хроматографией (гексан:EtOAc, от 100:0 до 70:30).
Стадия 2: Метил-2-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-3-фтор-1,1'-бифенил-4-ил}циклопропанкарбоксилат
Смесь НАФТИРИДИНОНА 3 (1,0 экв.), сложного эфира со стадии 1 (1,3 экв.), Na2СО3 (3,5 экв., 2М в Н2О), Pd(ОАс)2 (0,05 экв.) и PPh3 (0,15 экв.) или PdCl2dppf (0,05 экв.) в н-пропаноле (0,1М) перемешивали при 60-80°С в течение 1-3 часов. Смесь охлаждали до комнатной температуры, выливали в воду и экстрагировали EtOAc (2х). Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Флэш-хроматография (CH2Cl2:EtOAc; 90:10) давала указанное в заголовке соединение.
Стадия 3: 2-(транс)-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-3-фтор-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
К раствору сложного эфира со стадии 2 в ТГФ-МеОН (2:1, 0,2М) добавляли LiOH (5 экв., 2М) и смесь перемешивали при комнатной температуре в течение 6 часов. Органический растворитель выпаривали, водную фазу подкисляли АсОН и кислоту экстрагировали EtOAc (3Х). Органический слой промывали насыщенным солевым раствором, сушили и выпаривали растворитель. Остаток растирали в порошок в CH2Cl2/эфире и затем выделяли фильтрованием с получением указанного в заголовке соединения в виде твердого вещества.
1Н ЯМР (500 МГц, CDCl3): δ 9,85 (д, 1H), 9,08 (с, 1Н), 8,80 (дд, 1H), 8,70 (дд, 1H), 7,71 (д, 1H), 7,63 (т, 1H), 7,60 (с, 1H), 7,46 (дд, 1Н), 7,40 (д, 1Н), 7,28 (м, 2H), 7,01 (т, 1H), 2,97 (м, 1H), 2,66 (м, 1H), 1,93 (м, 1H), 1,62 (м, 1Н), 1,40 (м, 1H), 0,84 (м, 2H), 0,66 (м, 2H).
Оптически активную (+) или (-)-(транс)-2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-3-фтор-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту можно получить, используя оптически активный (+) или (-)-(транс)-метил 2-(4-бром-2-фторфенил)циклопропанкарбоксилат, полученный в соответствии со способом, описанным в примере 29, стадии 5-8.
ПРИМЕР 13
(цис)2-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-3-ил}циклопропанкарбоновая кислота
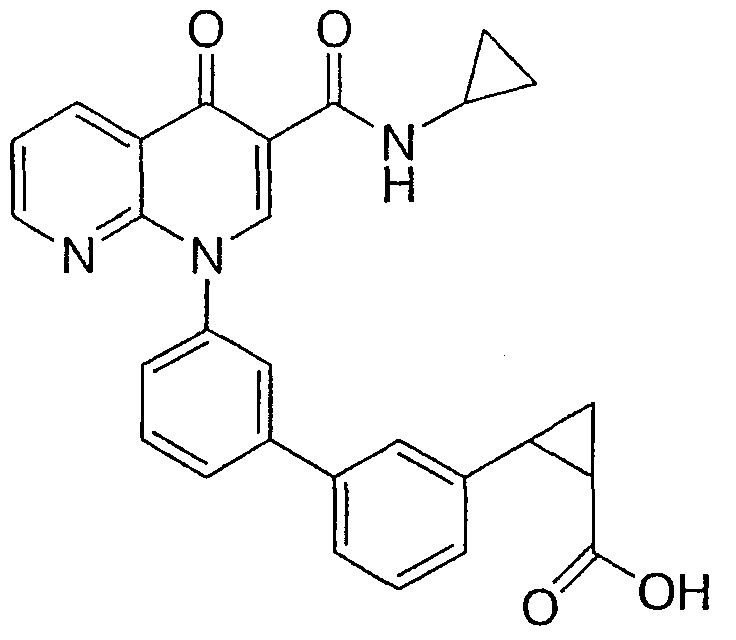
Получали в соответствии со способом, описанным в примере 8, но используя 3-бромбензальдегид в качестве исходного вещества.
1Н ЯМР (500 МГц, ацетон-d6): δ 9,78 (ушир. с, 1H), 8,96 (с, 1H), 8,79 (д, 1H), 8,76 (д, 1H), 7,97 (с, 1H), 7,91 (д, 1H), 7,75 (т, 1H), 7,7 (с, 1Н), 7,59-7,65 (м, 3H), 7,34-7,40 (м, 2H), 2,69 (дд, 1H), 2,12-2,15 (м, 1Н), 1,67 (дд, 1H), 1,35-1,39 (м, 1H), 0,79-0,83 (м, 2H), 0,61 (ушир. с, 2H).
ПРИМЕР 14
2-{3'-Бром-5'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
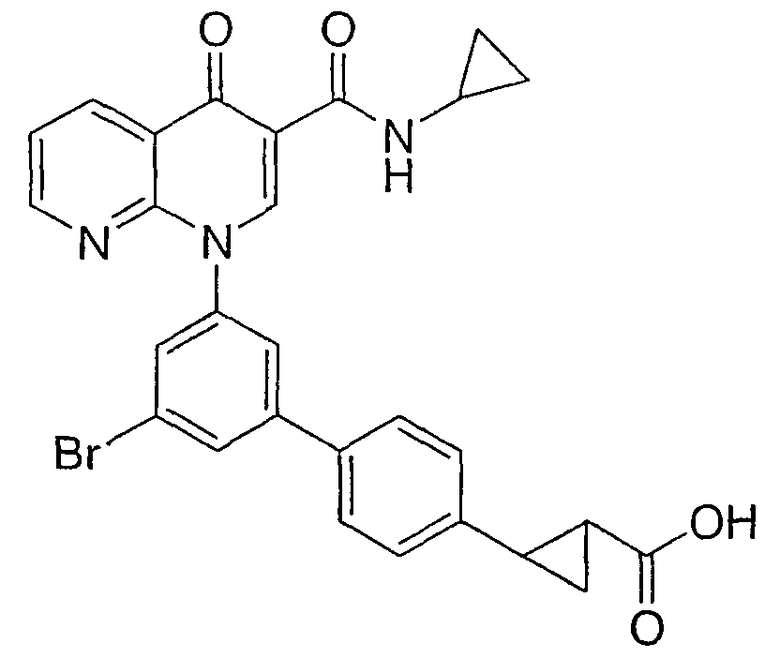
Стадия 1: N-Циклопропил-1-(3,5-дибромфенил)-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид
Получали в соответствии со способом, описанным для получения НАФТИРИДИНОНА 2, но используя 3,5-диброманилин в качестве исходного вещества.
Стадия 2: Этил-2-[4-(триметилстаннил)фенил]циклопропанкарбоксилат
Смесь этил-2-(транс)-(4-бромфенил)циклопропанкарбоксилата из примера 1, стадия 1, гексаметилдиолова (3,5 экв.) в 1,4-диоксане (0,2М) перемешивали при 60°С в течение 18 часов, растворитель выпаривали и остаток очищали флэш-хроматографией (гексан:EtOAc; 95:5) с получением указанного в заголовке соединения.
Стадия 3: Этил-2-{3'-бром-5'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоксилат
Смесь сложного эфира со стадии 2 (1,7 экв.), дибром соединения со стадии 1 (1 экв.), Pd2(dba)3 (0,05 экв.) и трифениларсина (0,25 экв.) в DMF (0,05М) нагревали при 80°С в течение 2 часов. Смесь охлаждали до комнатной температуры, разбавляли EtOAc и водой, органический слой промывали насыщенным солевым раствором, сушили и выпаривали растворитель. Остаток очищали флэш-хроматографией (CH2Cl2:МеОН, от 99:1 до 90:10) с получением указанного в заголовке соединения.
Стадия 4: 2-(транс)-{3'-Бром-5'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
К раствору сложного эфира со стадии 3 в ТГФ-МеОН-Н2О (2:1:0,5, 0,1М) добавляли LiOH (5 экв., 2М) и смесь перемешивали при 50°С в течение 4 часов. Органический растворитель выпаривали, водную фазу подкисляли АсОН и кислоту экстрагировали EtOAc (3х). Органическую фазу промывали насыщенным солевым раствором, сушили и выпаривали растворитель. Остаток очищали флэш-хроматографией (CH2Cl2:МеОН, от 99:1 до 90:10) с получением указанного в заголовке соединения.
1Н ЯМР (500 МГц, ДМСО-d6):δ 9,71 (д, 1H), 8,84 (с, 1Н), 8,80 (дд, 1H), 8,72 (дд, 1H), 8,08 (с, 1H), 8,01 (с, 1H), 7,91 (с, 1H), 7,71 (д, 2H), 7,65 (дд, 1H), 7,29 (д, 2H), 2,91-2,89 (м, 1H), 2,46-2,43 (м, 1H), 1,88-1,85 (м, 1H), 1,47-1,45 (м, 1Н), 1,43-1,38 (м, 1H), 0,81-0,77 (м, 2H), 0,57-0,56 (м, 2H).
Оптически активные предшественники получали разделением на хиральной колонке (Chiral Pak AD), элюируя гексаном:EtOH (35:65), содержащим 0,2% TFA.
ПРИМЕР 15
2-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-3-метил-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
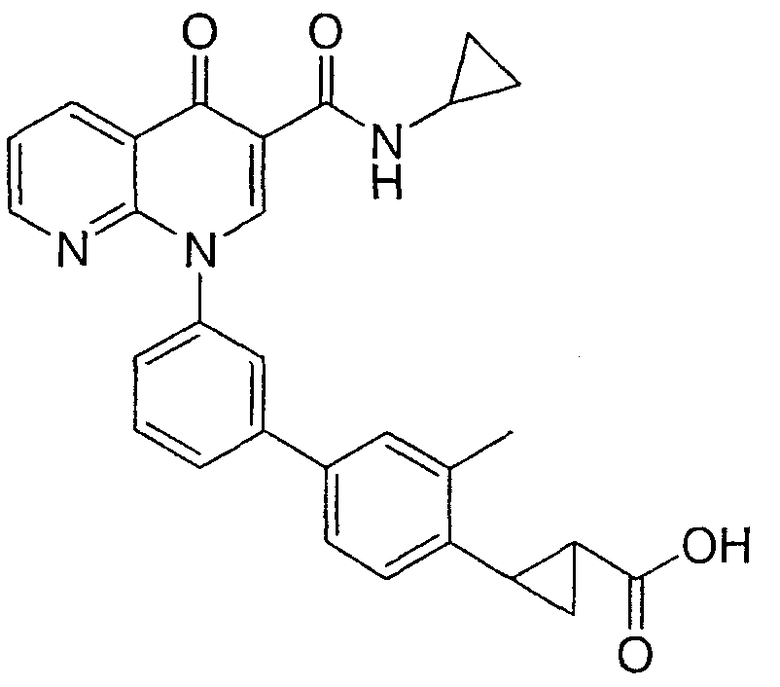
Стадия 1: Этил-(2Е)-3-(4-бром-2-метилфенил)проп-2-еноат
К раствору 4-бром-2-метилбензальдегида (1 экв.) и триэтилфосфоноацетата (1,1 экв.) в ТГФ (0,3 М) при комнатной температуре добавляли по каплям трет-бутоксид калия (1,1 экв., 1 М, ТГФ). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, гасили 10% раствором HCl, разбавляли простым эфиром, промывали раствором NaHCO3, насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Флэш-хроматография (гексан:EtOAc, от 90:10 до 70:30) давала указанное в заголовке соединение.
Стадия 2: 2-(транс)-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-3-метил-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
Получали в соответствии со способом, описанным в примере 12, но используя в качестве исходного вещества сложный эфир со стадии 1.
1Н ЯМР (500 МГц, CDCl3): δ 9,82 (д, 1H), 9,10 (с, 1H), 8,80 (дд, 1H), 8,70 (дд, 1H), 7,60 (м, 1H), 7,48 (м, 2H), 7,40 (м, 2H), 7,22 (д, 1H), 7,02 (с, 1H), 6,97 (м, 1H), 2,98 (м, 1H), 2,55 (м, 1Н), 2,30 (с, 3H), 1,95 (м, 1Н), 1,65 (м, 1H), 1,40 (м, 1H), 0,85 (м, 2H), 0,65 (м, 2H).
ПРИМЕР 16
2-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-2-метил-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
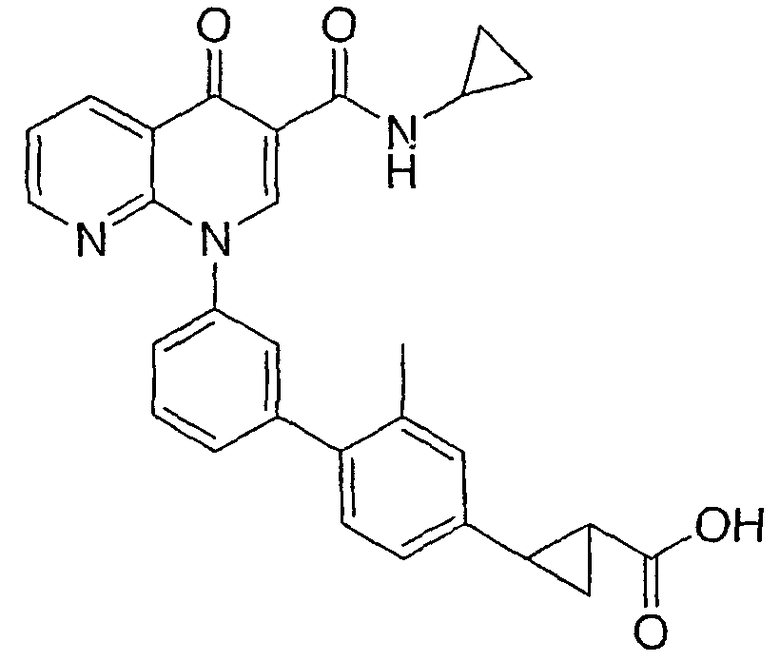
Получали в соответствии со способом, описанным в примере 15, но используя 4-бром-3-метилбензальдегид в качестве исходного вещества.
Получали указанное в заголовке соединение 2-(транс)-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-2-метил-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту.
1Н ЯМР (500 МГц, CDCl3): δ 9,88 (с, 1Н), 9,10 (с, 1H), 8,80 (д, 1Н), 8,70 (д, 1Н), 7,72 (д, 1H), 7,60 (м, 2H), 7,45 (дд, 1H), 7,40 (м, 3H), 7,05 (д, 1H), 2,98 (м, 1Н), 2,55 (м, 1H), 2,45 (с, 3H), 1,78 (м, 1Н), 1,60 (м, 1H), 1,40 (м, 1Н), 0,85 (м, 2H), 0,75 (м, 2H).
ПРИМЕР 17
2-{3-Хлор-3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
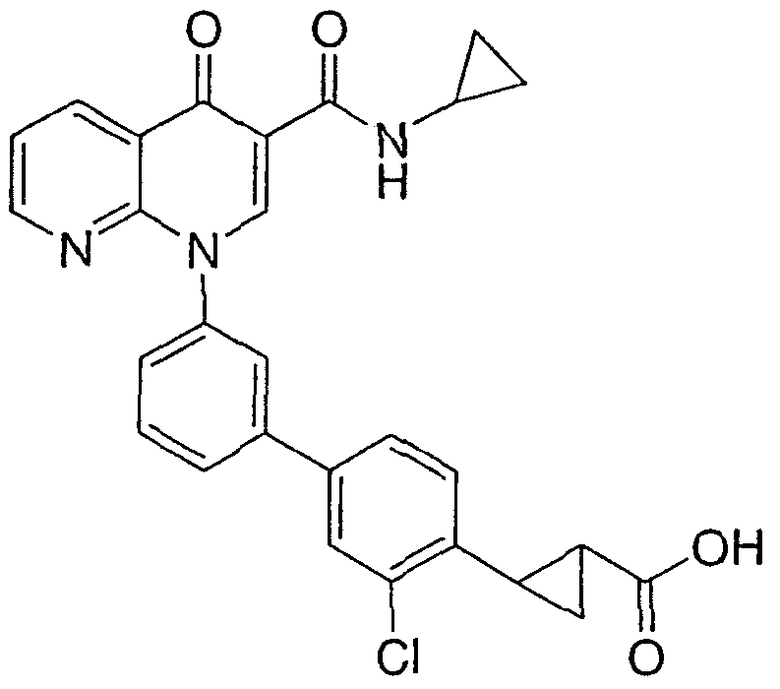
Получали в соответствии со способом, описанным в примере 15, но используя 4-бром-2-хлорбензальдегид в качестве исходного вещества.
1Н ЯМР (500 МГц, CDCl3): δ 9,88 (с, 1H), 9,10 (с, 1H), 8,80 (д, 1H), 8,70 (д, 1Н), 7,72 (д, 1H), 7,65 (м, 3H), 7,45 (м, 3H), 7,05 (д, 1H), 2,95 (м, 1Н), 2,70 (м, 1H), 1,80 (м, 1H), 1,65 (м, 1H), 1,35 (м, 1H), 0,85 (м, 2H), 0,65 (м, 2H).
Получали указанное в заголовке соединение - 2-(транс)-{3-хлор-3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту.
ПРИМЕР 18
2-(цис)-{3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-3-фтор-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
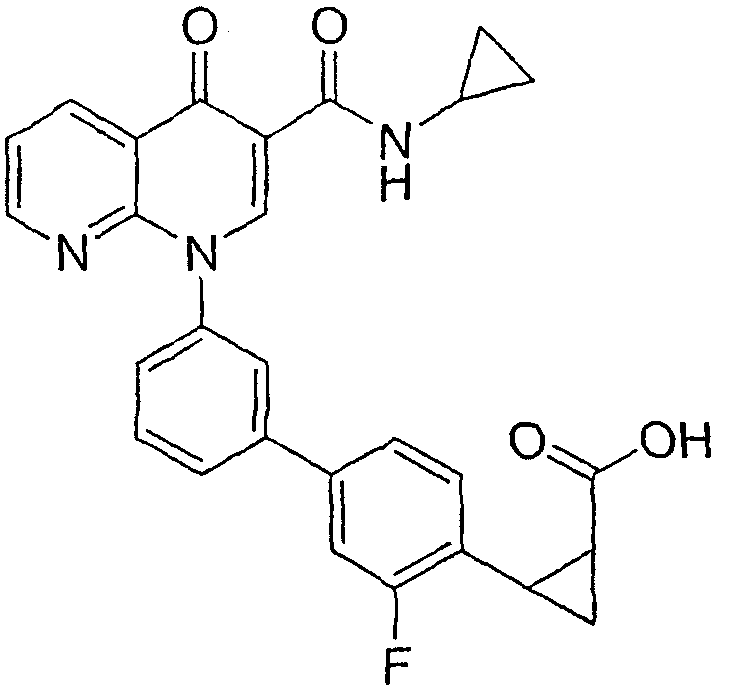
Получали в соответствии со способом, описанным в примере 8, но используя 4-бром-2-фторбензальдегид в качестве исходного вещества.
1Н ЯМР (500 МГц, ДМСО-d6): δ 11,91 (ушир. с, 1H), 9,75 (д, 1H), 8,83 (с, 1H), 8,79 (дд, 1H), 8,74 (дд, 1H), 8,02 (с, 1H), 7,93 (д, 1H), 7,70 (т, 1H), 7,62-7,66 (м, 2H), 7,57 (д, 1Н), 7,54 (д, 1H), 7,36 (т, 1Н), 2,88-2,94 (м, 1H), 2,55 (кв, 1H), 2,08-2,13 (м, 1H), 1,53 (дд, 1H), 1,36-1,40 (м, 1H), 0,77-0,81 (м, 2H), 0,56-0,59 (м, 2H).
ПРИМЕР 19
3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-карбоновая кислота
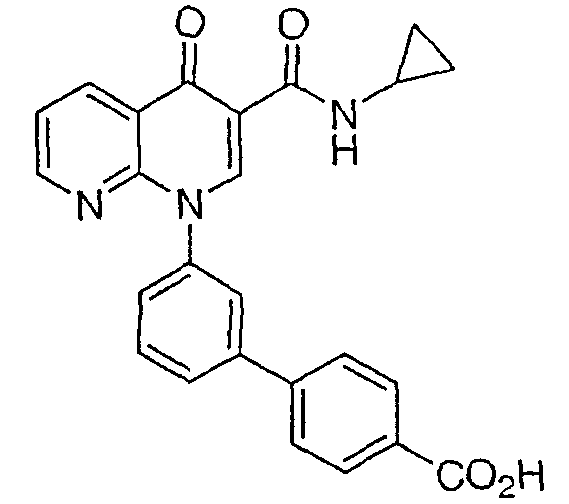
Стадия 1: Этил-3-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-карбоксилат
Смесь НАФТИРИДИНОНА 3 (1 экв.), этил-4-бромбензоата (3 экв.), Na2СО3 (3,5 экв., 2М в Н2О), Pd(PPh3)4 (0,15 экв.) в н-пропаноле-DMF (1:1, 0,1 М) перемешивали при 80°С в течение 2 часов. Смесь охлаждали до комнатной температуры, гасили насыщенным солевым раствором и разбавляли EtOAc. Органические экстракты промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Остаток растирали в порошок в простом эфире/EtOAc и затем выделяли фильтрованием с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 2: 3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-карбоновая кислота
К раствору сложного эфира со стадии 2 в ТГФ-МеОН-СН2Cl2 (2:2:0,5, 0,2 М) добавляли LiOH (5 экв., 2М) и смесь перемешивали при комнатной температуре в течение 6 часов. Органический растворитель выпаривали, водную фазу подкисляли 1 н. раствором HCl и кислоту экстрагировали EtOAc (3Х). Органический слой промывали насыщенным солевым раствором, сушили и выпаривали растворитель. Флэш-хроматография (СН2Cl2:МеОН, 80:20 с NH3) давала указанное в заголовке соединение.
1Н ЯМР (500 МГц, ДМСО-d6): δ 11,2 (с, OH), 9,75 (с, NH), 8,77 (дд, 1Н), 8,73 (дд, 1H), 8,13 (д, 2H), 8,07 (с, 1H), 7,98 (д, 1H), 7,91 (д, 2H), 7,77 (т, 2H), 7,70 (д, 1H), 7,60 (дд, 1H), 2,94 (м, 1H), 0,78 (м, 2H), 0,57 (м, 2H). МС +ESI Q1 (М+1) 426,2.
ПРИМЕР 20
2-{3'-[3-[(Морфолин-4-илкарбонил)-4-оксо-1,8-нафтиридин-1(4Н)]-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
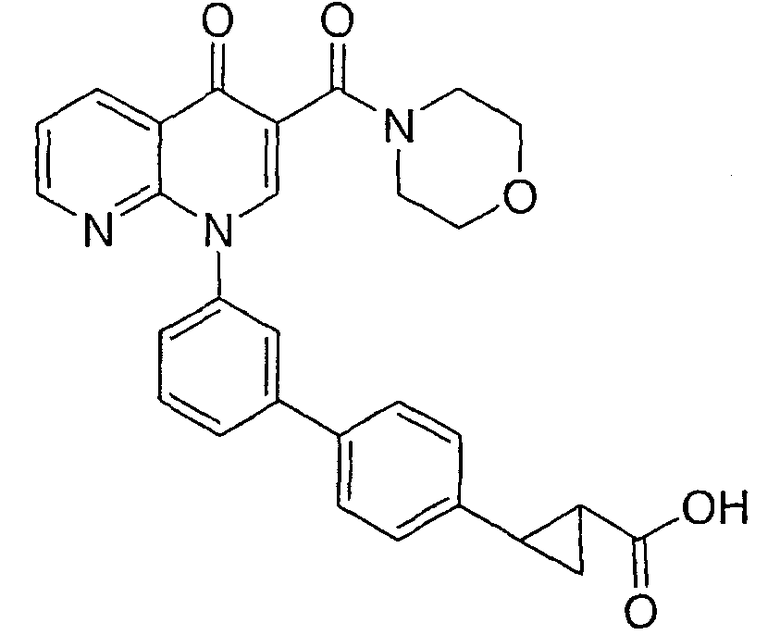
Стадия 1: 1-(3-Бромфенил)-3-(морфолин-4-илкарбонил]-1,8-нафтиридин-1(4Н)-он
К раствору кислоты примера получения НАФТИРИДИНОНА 1, стадия 3 (1 экв.), в ТГФ (0,1М) добавляли оксалилхлорид (1,5 экв.) и 2 капли DMF. Смесь нагревали до температуры кипения с обратным холодильником в течение 1 часа, охлаждали до комнатной температуры и переносили в раствор морфолина (3 экв., депротонированный при помощи NaH в ТГФ при 0°С). Смесь перемешивали при комнатной температуре в течение 18 часов, гасили раствором NH4Cl и разбавляли EtOAc. Органический слой промывали насыщенным солевым раствором, сушили и выпаривали растворитель. Остаток растирали в порошок в простом эфире/EtOAc и затем выделяли фильтрованием с получением указанного в заголовке соединения.
Стадия 2: Этил-2-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]циклопропанкарбоксилат
Получали в соответствии с методикой, описанной для получения НАФТИРИДИНОНА 3, но используя в качестве исходного вещества этил-2-(транс)-(4-бромфенил)циклопропанкарбоксилат из примера 1, стадия 1.
Стадия 3: Этил-2-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]циклопропанкарбоновая кислота
К раствору сложного эфира со стадии 2 в ТГФ-Н2О (0,2М) добавляли LiOH (3 экв., 2М) и смесь перемешивали при комнатной температуре в течение 18 часов. Органический растворитель выпаривали, водную фазу подкисляли 1 н. раствором HCl и кислоту экстрагировали EtOAc (3х). Органический слой промывали насыщенным солевым раствором, сушили и выпаривали растворитель с получением указанной в заголовке карбоновой кислоты.
Стадия 4: 2-(транс)-{3'-[3-[(Морфолин-4-илкарбонил)-4-оксо-1,8-нафтиридин-1(4Н)]-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
Смесь кислоты со стадии 2 (1 экв.), амида со стадии 1 (1 экв.), Na2CO3 (3,5 экв.; 2М в Н2О), Pd(PPh3)4 (0,15 экв.) в н-пропаноле-DMF (1:1, 0,1М) перемешивали при 80°С в течение 2 часов. Смесь охлаждали до комнатной температуры, гасили насыщенным солевым раствором и разбавляли EtOAc. Органический экстракт промывали насыщенным солевым раствором, сушили над Na2SO, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (CHCl3:ТГФ, 50:50), растирали в порошок в простом эфире/ацетоне и затем выделяли фильтрованием с получением указанного в заголовке соединения в виде твердого вещества.
1Н ЯМР (500 МГц, CDCl3): δ 10,5 (с, NH), 8,80 (дд, 1H), 8,71 (дд, 1H), 8,37 (с, 1H), 7,73 (д, 1H), 7,68 (с, 1H), 7,63 (т, 1H), 7,55 (д, 2H), 7,45 (м, 2H), 7,17 (д, 2H), 3,83 (ушир. с, 6H), 3,53 (ушир. с, 2H), 2,59 (м, 1H), 1,93 (м, 1H), 1,65 (м, 1H), 1,38 (м, 1H). МС +ESI Q1 (М+1) 496,1.
ПРИМЕР 21
2-{3'-[4-Оксо-3-({[5-(трифторметил)-1,3,4-тиадиазол-2-ил]амино}карбонил)-1,8-нафтиридин-1(4Н)]-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
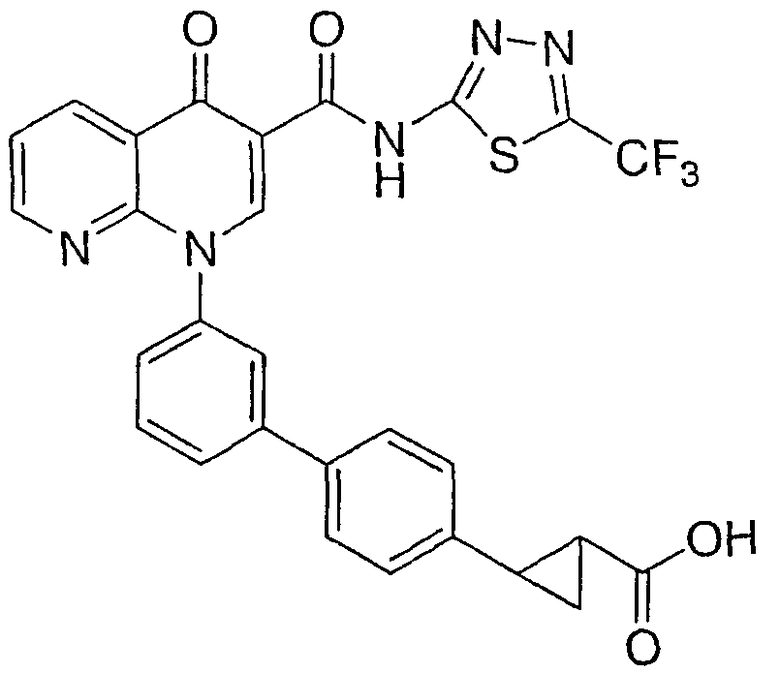
Стадия 1: 1-(3-Бромфенил)-4-оксо-N-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]-1,4-дигидро-1,8-нафтиридин-3-карбоксамид
К раствору кислоты примера получения НАФТИРИДИНОНА 1, стадия 3 (1 экв.) в DMF (0,1М) добавляли HATU (4 экв.), диизопропилэтиламин (8 экв.), а затем 5-(трифторметил)-1,3,4-тиадиазол-2-амин (4 экв.). Смесь перемешивали при комнатной температуре в течение 12 часов и затем нагревали до 80°С в течение 2 часов, охлаждали до комнатной температуры и разбавляли водой. Остаток затем выделяли фильтрованием, промывали простым эфиром, растирали в порошок в ацетоне с получением указанного в заголовке соединения.
Стадия 2: 2-(транс)-{3'-[4-Оксо-3-({[5-(трифторметил)-1,3,4-тиадиазол-2-ил]амино}карбонил)-1,8-нафтиридин-1(4Н)]-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
Получали в соответствии со способом, описанным в примере 20, стадии 2, 3 и 4, но используя амид со стадии 1 в качестве исходного вещества. Очищали с помощью флэш-хроматографии (CHCl3:МеОН, 80:20 с NH3).
1Н ЯМР (500 МГц, ДМСО-d6): δ 13,8 (с, OH), 9,95 (с, NH), 9,10 (с, 1Н), 8,83 (м, 2H), 7,98 (с, 1H), 7,88 (д, 1H), 7,71 (д, 2H), 7,69 (м, 2H), 7,64 (д, 1Н), 7,28 (д, 2H), 2,43 (м, 1H), 1,86 (м, 1H), 1,46 (м, 1Н), 1,38 (м, 1H). МС +ESI Q1 (М+1) 578,2.
ПРИМЕР 22
2-{3'-[3-({[2-(Метилтио)этил]амино}карбонил)-4-оксо-1,8-нафтиридин-1(4Н)]-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
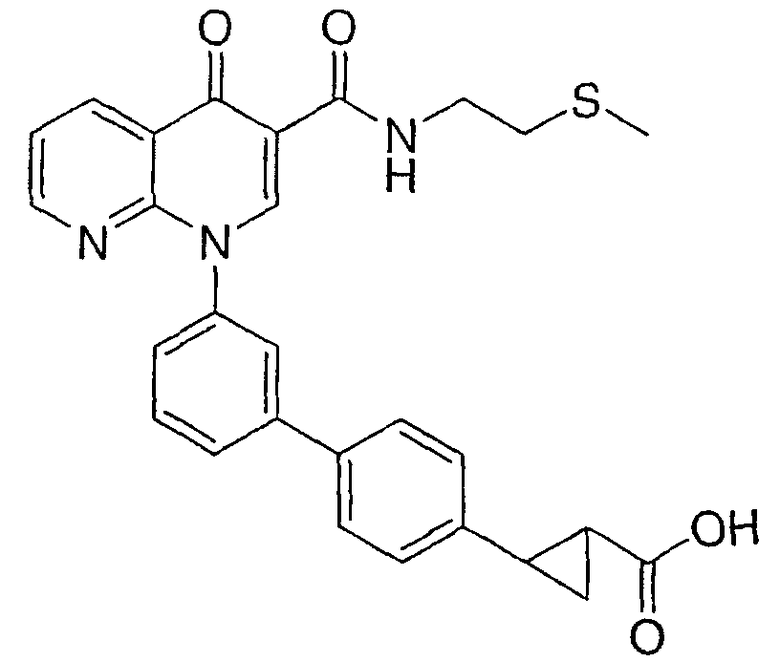
Стадия 1: 1-(3-Бромфенил)-N-[2-(метилтио)этил]-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоксамид
Получали в соответствии со способом, описанным в примере 21, стадия 1, но используя в качестве исходного вещества 2-(метилтио)этанамид.
Стадия 2: Этил-2-{3'-[3-({[2-(метилтио)этил]амино}карбонил)-4-оксо-1,8-нафтиридин-1(4Н)]-ил]-1,1'-бифенил-4-ил}циклопропанкарбоксилат
Смесь амида со стадии 1 (1 экв.), этил-2-(транс)-(4-бромфенил)циклопропанкарбоксилата из примера 1 стадия 1 (1 экв.), Na2CO3 (3,5 экв.; 2М в Н2О), Pd(PPh3)4 (0,05 экв.) в н-пропаноле (0,1М) перемешивали при 80°С в течение 3 часов. Смесь охлаждали до комнатной температуры, гасили насыщенным солевым раствором и разбавляли EtOAc. Органические экстракты промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Остаток растирали в порошок в простом эфире/МеОН, выделяли фильтрованием и промывали простым эфиром с получением указанного в заголовке соединения в виде твердого вещества.
Стадия 3: 2-(транс)-{3'-[3-({[2-(Метилтио)этил]амино}карбонил)-4-оксо-1,8-нафтиридин-1(4Н)]-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
К раствору сложного эфира со стадии 2 в ТГФ-МеОН-Н2О (2:1:0,5, 0,1 М) добавляли LiOH (5 экв., 2М) и смесь перемешивали при комнатной температуре в течение 18 часов. Органический растворитель выпаривали, водную фазу подкисляли 1 н. раствором HCl и кислоту экстрагировали EtOAc (3Х). Органический слой промывали насыщенным солевым раствором, сушили и выпаривали растворитель. Остаток растирали в порошок с простым эфиром/МеОН и затем выделяли фильтрованием с получением указанного в заголовке соединения в виде твердого вещества.
1Н ЯМР (500 МГц, CDCl3): δ 10,21 (с, OH), 9,16 (с, NH), 8,89 (дд, 1H), 8,76 (дд, 1H), 7,78 (д, 1H), 7,65 (т, 1H), 7,63 (с, 1H), 7,53 (д, 2H), 7,51 (дд, 1H), 7,43 (д, 1H), 7,20 (д, 2H), 3,76 (тд, 2H), 2,82 (т, 2H), 2,60 (м, 1H), 2,21 (с, 3H), 1,96 (м, 1Н), 1,66 (м, 1Н), 1,42 (м, 1H). МС +ESI Q1 (М+1) 500,2.
ПРИМЕР 23
2-{3'-[3-({[2-(Метилсульфонил)этил]амино}карбонил)-4-оксо-1,8-нафтиридин-1(4Н)]-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
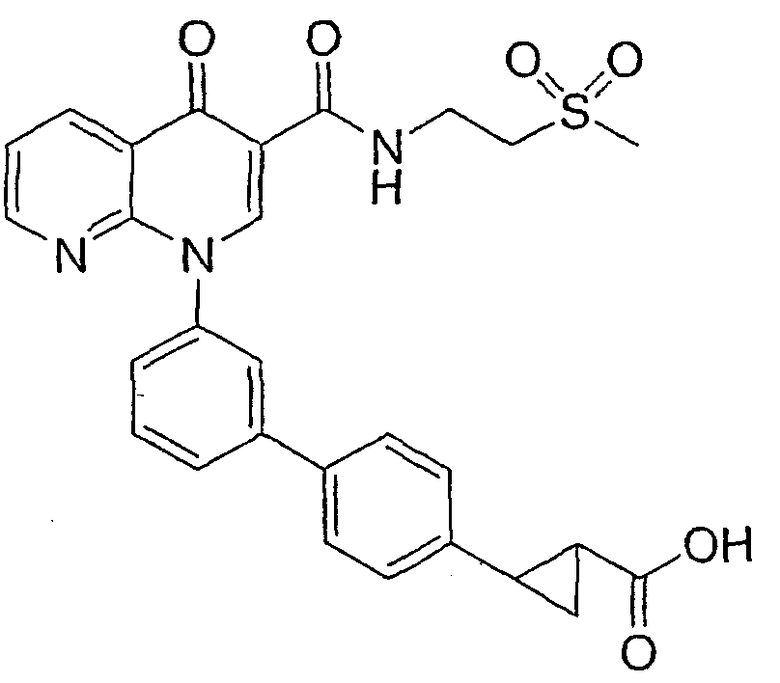
К раствору соединения примера 22 (1 экв.) в ТГФ-МеОН-Н2О (2:1:1, 0,1М) добавляли оксон (2 экв.). Смесь перемешивали при комнатной температуре в течение 18 часов и разбавляли водой. Остаток выделяли фильтрованием, промывали простым эфиром с получением указанного в заголовке соединения в виде твердого вещества.
1Н ЯМР (500 МГц, ДМСО-d6): δ 12,4 (с, OH), 9,97 (с, NH), 8,83 (с, 1H), 8,78 (дд, 1H), 8,75 (дд, 1H), 7,95 (с, 1Н), 7,87 (д, 1H), 7,66 (д, 2H), 7,64 (м, 2H), 7,59 (д, 1H), 7,28 (д, 2H), 3,82 (тд, 2H), 3,42 (т, 2H), 3,05 (с, 3Н), 2,44 (м, 1Н), 1,87 (м, 1Н), 1,46 (м, 1Н), 1,40 (с, 1Н). МС +ESI Q1 (М+1) 532,1.
Получали указанное в заголовке соединение 2-(транс)-{3'-[3-({[2-(метилсульфонил)этил]амино}карбонил)-4-оксо-1,8-нафтиридин-1(4Н)]-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
ПРИМЕР 24
2-{3'-[4-Оксо-3-{[2,2,2-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
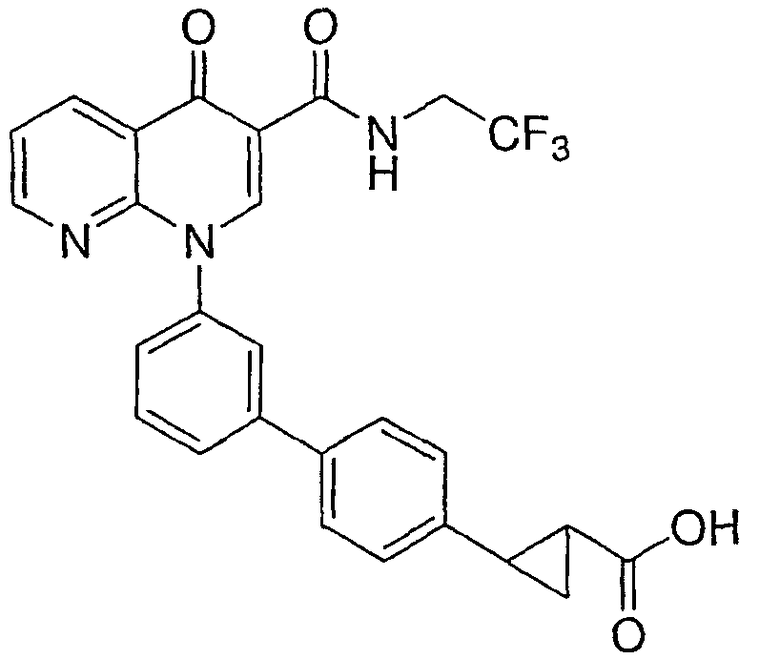
Получали в соответствии со способом, описанным в примере 22, но используя в качестве исходного вещества 2,2,2-трифторэтанамид.
1Н ЯМР (500 МГц, CDCl3): δ 11,3 (с, OH), 10,34 (с, NH), 9,11 (с, 1H), 8,88 (дд, 1Н), 8,77 (дд, 1H), 7,78 (д, 1H), 7,68 (т, 1H), 7,65 (с, 1H), 7,56 (д, 2H), 7,52 (дд, 1H), 7,44 (дд, 1H), 7,22 (д, 2H), 4,21 (ушир. квт, 2H), 2,62 (м, 1H), 1,97 (м, 1H), 1,70 (м, 1H), 1,45 (м, 1H). МС +ESI Q1 (М+1) 508,1.
Получали указанное в заголовке соединение 2-(транс)-{3'-[4-оксо-3-{[2,2,2-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту.
ПРИМЕР 25
2-(5-{3-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]фенил}тиен-2-ил)циклопропанкарбоновая кислота
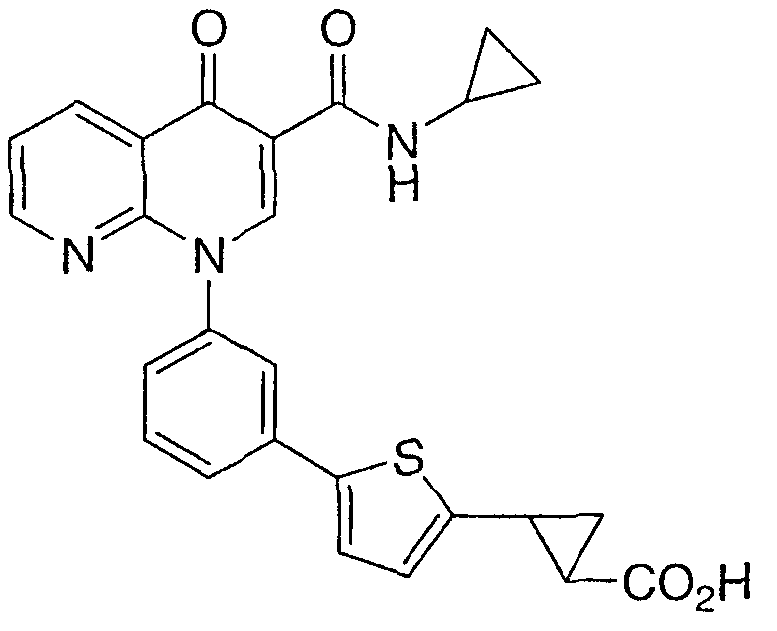
Получали в соответствии со способом, описанным в примере 15, но используя в качестве исходного вещества 5-бромтиофен-2-карбальдегид.
1Н ЯМР (500 МГц, CDCl3): δ 10,21 (с, OH), 9,77 (с, NH), 9,04 (с, 1H), 8,79 (дд, 1H), 8,69 (дд, 1H), 7,66 (д, 1Н), 7,54 (м, 2H), 7,45 (дд, 1H), 7,28 (д, 1H), 7,13 (дд, 1Н), 6,77 (дд, 1H), 2,97 (м, 1H), 2,68 (м, 1H), 1,93 (м, 1Н), 1,65 (м, 1H), 1,36 (м, 1H), 0,83 (м, 2H), 0,66 (м, 2H). МС +ESI Q1 (М+1) 472,2.
Получали указанное в заголовке соединение 2-(транс)-(5-{3-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]фенил}тиен-2-ил)циклопропанкарбоновую кислоту.
ПРИМЕР 26
2-{3'-[3-{[(Циклопропилметил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
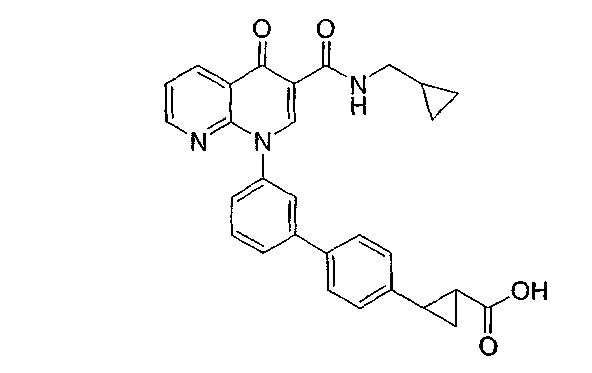
Получали в соответствии со способом, описанным в примере 22, но используя в качестве исходного вещества 1-циклопропилметанамин.
1Н ЯМР (500 МГц, ДМСО-d6): δ 9,83 (с, NH), 8,82 (с, 1H), 8,79 (дд, 1H), 8,75 (дд, 1H), 7,96 (с, 1H), 7,87 (д, 1Н), 7,65 (м, 3H), 7,57 (д, 2H), 7,28 (д, 2H), 3,38 (т, 2H), 2,45 (м, 1H), 1,87 (м, 1H), 1,47 (м, 1H), 1,40 (м, 1Н), 1,08 (м, 1H), 0,49 (м, 2H), 0,26 (м, 2H). МС +ESI Q1 (М+1) 480,1.
Получали указанное в заголовке соединение 2-(транс)-{3'-[3-{[(циклопропилметил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту.
ПРИМЕР 27
2-{3'-[3-{[(1-Цианоциклопропил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновая кислота
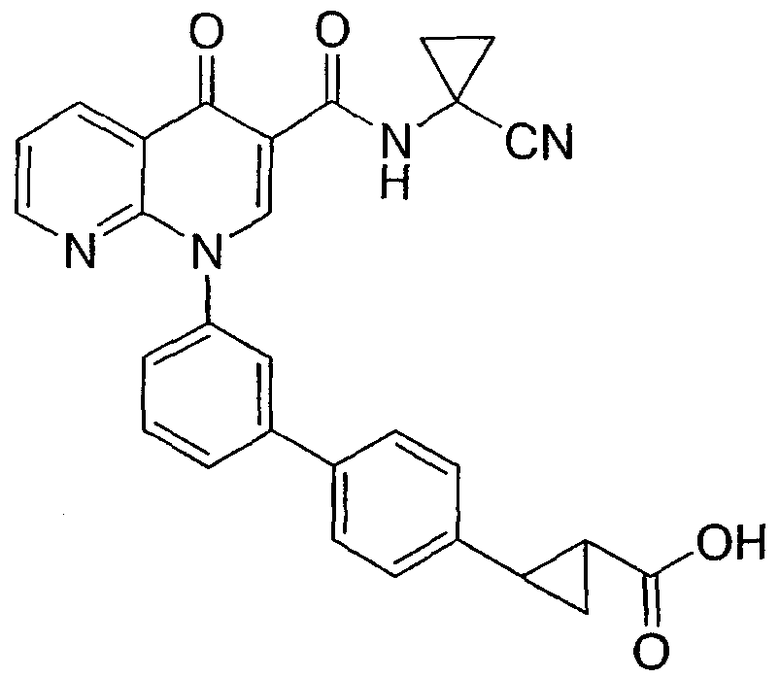
Получали в соответствии со способом, описанным в примере 21, но используя в качестве исходного вещества 1-аминоциклопропанкарбонитрил.
1Н ЯМР (500 МГц, CDCl3): δ 11,51 (с, OH), 10,36 (с, NH), 9,11 (с, 1Н), 8,84 (дд, 1H), 8,76 (дд, 1H), 7,78 (д, 1H), 7,67 (т, 1H), 7,63 (с, 1H), 7,57 (д, 2H), 7,53 (дд, 1H), 7,41 (д, 1H), 7,22 (д, 2H), 2,67 (м, 1H), 1,98 (м, 1H), 1,74 (м, 1H), 1,66 (м, 2H), 1,45 (м, 1H), 1,40 (м, 2H). МС +ESI Q1 (М+1) 519,2.
Получали указанное в заголовке соединение 2-(транс)-{3'-[3-{[(1-Цианоциклопропил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту.
ПРИМЕР 28
3-{3'-[3-[(Изопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-3-метилбутановая кислота
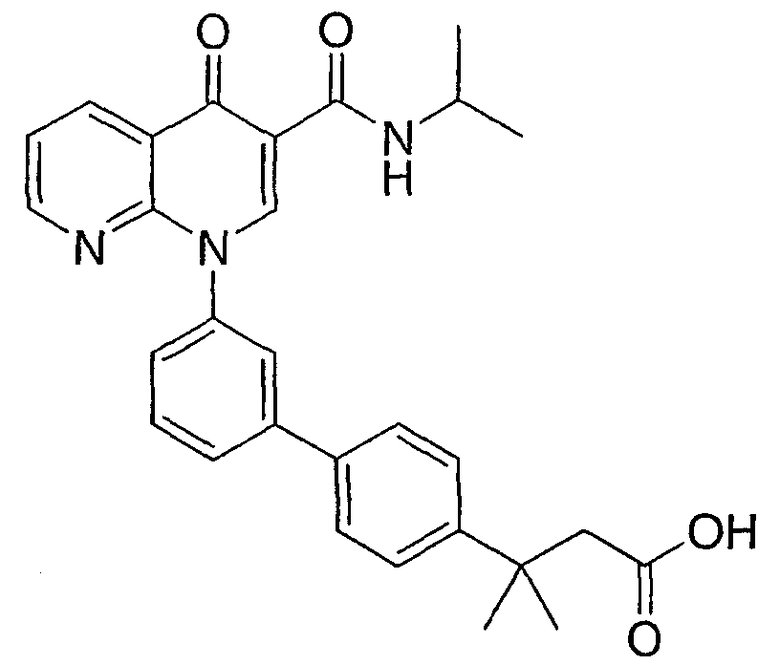
Получали в соответствии со способом, описанным в примере 5, но используя НАФТИРИДИНОН 4 в качестве исходного вещества.
1Н ЯМР (500 МГЦ, CDCl3): δ 9,76 (д, 1H), 9,11 (с, 1H), 8,85 (д, 1H), 8,73 (д, 1H), 7,77 (д, 1H), 7,65 (м, 2H), 7,58 (д, 2H), 7,47 (м, 3H), 7,41 (д, 1H), 4,33 (м, 1H), 2,71 (с, 2H), 1,51 (с, 6H), 1,33 (дд, 6H).
ПРИМЕР 29
(+)-(транс)-2-{3-Фтор-3'-[4-оксо-3-{[2,2,2-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}циклопропанкарбоновая кислота
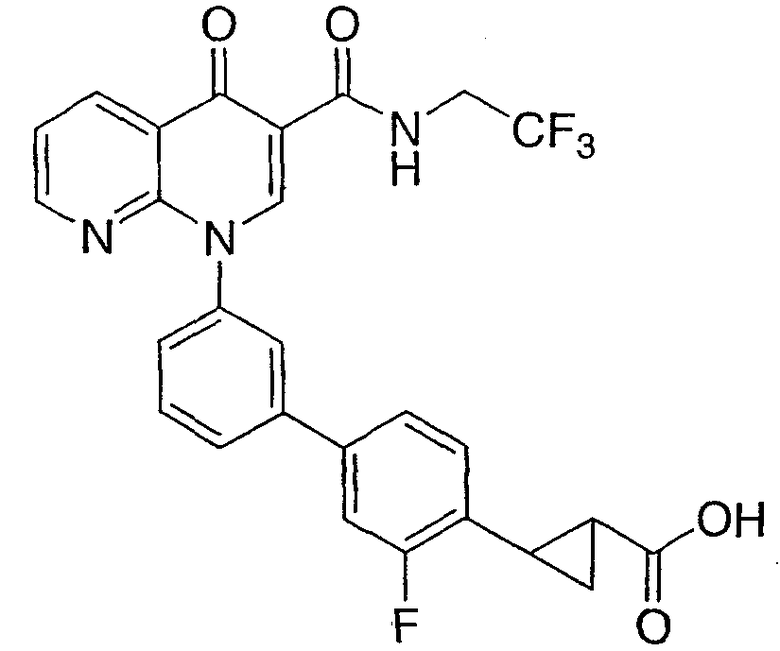
Стадия 1: Этил-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксилат
Смесь этил-2-хлорникотиноилацетата (коммерчески доступного или полученного в соответствии со способом, описанным в J. Het. Chem., 30, 855, 1993) (1 экв.), триэтиламина (4 экв.) и этил-3,3-диметиламиноакрилата (1,5 экв.) в ацетонитриле (0,5 М) нагревали до температуры кипения с обратным холодильником в течение 3 часов, охлаждали до 40-50°С и добавляли 3-броманилин (1 экв.). Реакционную смесь нагревали до температуры кипения с обратным холодильником в течение ночи, охлаждали до комнатной температуры, разбавляли водой (2 объема). Продукт выделяли фильтрованием и промывали водой, простым эфиром или смесью ацетонитрил-вода (1:1).
1Н ЯМР (Ацетон-d6) δ 1,32 (т, 3H), 4,29 (кв, 2H), 7,54-7,63 (м, 2H), 7,69 (дд, 1Н), 7,78 (дд, 1H), 7,93 (с, 1Н), 8,66-8,71 (м, 3Н).
Стадия 2: 1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоновая кислота
Суспензию этил 1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксилата со стадии 1 (1 экв.) в смеси тетрагидрофуран-метанол (0,15М) и 1 н. водный раствор гидроксида натрия (2 экв.) нагревали примерно при 50°С в течение 20 минут при перемешивании. После охлаждения смесь разбавляли водой и подкисляли 1 н. водным раствором HCl. После перемешивания в течение 45 минут осадок отфильтровывали, тщательно промывали водой и сушили с получением указанной в заголовке кислоты в виде твердого вещества кремового цвета.
1Н ЯМР (Ацетон-d6): δ 7,65 (т, 1H), 7,76 (м, 2H), 7,84 (д, 1H), 7,99 (с, 1Н), 8,87 (м, 2H), 9,01 (с, 1Н).
Стадия 3: 1-(3-бромфенил)-N-(2,2,2-трифторэтил)-1,4-дигидро-1,8-нафтиридин-4-он-3-карбоксамид
К суспензии 1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоновой кислоты со стадии 2 (1 экв.) и триэтиламина (3 экв.) в тетрагидрофуране (0,08 М) при 0°С добавляли изобутилхлорформиат (1,8 экв.). После перемешивания при 0°С в течение 2 часов добавляли 2,2,2,-трифторэтиламин (5 экв.) и смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. Затем смесь распределяли между этилацетатом и водой, органическую фазу сушили и упаривали до получения твердого вещества, которое перемешивали в простом эфире при комнатной температуре в течение 3 часов и фильтровали с получением N-(2,2,2,-трифторэтил)-1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоксамида в виде белого твердого вещества.
Альтернативно:
К суспензии 1-(3-бромфенил)-1,4-дигидро[1,8]нафтиридин-4-он-3-карбоновой кислоты со стадии 2 (1 экв.) в DMF (0,1М) добавляли HATU (4 экв.), диизопропилэтиламин (8 экв.), а затем 2,2,2-трифторэтиламин (4 экв.). Смесь перемешивали при комнатной температуре в течение 12 часов, затем нагревали при 80°С в течение 2 часов, охлаждали до комнатной температуры и разбавляли водой. Остаток затем выделяли фильтрованием, промывали простым эфиром и растирали в порошок с ацетоном с получением указанного в заголовке соединения.
Стадия 4: N-(2,2,2,-трифторэтил)-1-[3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1,4-дигидро-1,8-нафтиридин-4-он-3-карбоксамид
Смесь 1-(3-бромфенил)-N-(2,2,2,-трифторэтил)-1,4-дигидро-1,8-нафтиридин-4-он-3-карбоксамида (1,0 экв.), пинаколдиборана (1,5 экв.), КОАс (4 экв.) и PdCl2(dppf) (0,05 экв.) в DMF (0,2 М) перемешивали при 70-80°С в течение 3 часов. Смесь охлаждали до комнатной температуры, разбавляли EtOAc и раствором NH4Cl. Органические экстракты промывали Н2О, насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. После кристаллизации из EtOAc-простого эфира-гексана (1:1:2) и флэш-хроматографии (CH2Cl2:EtOAc, 50:50) маточного раствора получали указанное в заголовке соединение в виде белого твердого вещества.
Стадия 5: 4-Бром-2-фтор-1-винилбензол
К суспензии бромида метилтрифенилфосфония (1,1 экв.) в тетрагидрофуране (0,13 М) при -78°С добавляли по каплям в течение 20 минут н-бутиллитий (2,5 М в гексане, 1 экв.). Реакционную смесь перемешивали при -78°С в течение 15 минут, нагревали до 0°С, перемешивали в течение 15 минут и снова охлаждали до -78°С. Добавляли по каплям в течение 30 минут раствор 4-бром-2-фторбензальдегида (1 экв.) в 100 мл ТГФ. Полученной смеси давали медленно нагреться до комнатной температуры и перемешивали в течение 1 часа. Полученную смесь гасили насыщенным раствором NH4Cl и разбавляли 2 объемами гексана. Органические экстракты промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали. Остаток разбавляли гексаном:EtOAc (9:1) и фильтровали через слой силикагеля. Фракции объединяли и концентрировали с получением желаемого вещества.
Стадия 6: (+)-этил 2-(4-бром-2-фторфенил)циклопропанкарбоксилат
К суспензии комплекса меди (I) и бензолтрифторметансульфоната (0,01 экв.) в хлороформе (100 мл) при комнатной температуре добавляли (R,R)-2,2'-изопропилиден-бис(4-трет-бутил-2-оксазолин) (0,01 экв.). Смесь перемешивали в течение 1 часа и затем переносили через трубку со стекловатой в раствор стирола со стадии 5 (1,2 экв.) в хлороформе (0,1М) при 4°С. К этому раствору по каплям в течение 3 часов добавляли раствор этилдиазоацетата (0,8 экв.) в хлороформе (0,1М). Полученную смесь перемешивали в течение ночи при 4°С. Смесь концентрировали. Флэш-хроматография (гексан:EtOAc; 95:5) давала указанное в заголовке соединение. (84:16, транс:цис-изомеры, методика, описанная в Evans и др.. J. Am. Chem. Soc. 1991, 113, 726).
Стадия 7: (+)-2-(4-бром-2-фторфенил)циклопропанкарбоновая кислота
К раствору (+)-этил 2-(4-бром-2-фторфенил)циклопропанкарбоксилата (смесь сложных эфиров, 1 экв.) в тетрагидрофуране-метаноле (2:1) добавляли гидроксид лития (0,84 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 2 дней. Полученную смесь концентрировали, разбавляли водой, экстрагировали простым эфиром (2х) с получением цис-эфира. Водную фазу подкисляли 10% раствором HCl, экстрагировали простым эфиром (2х) с получением (+)-транс-кислоты. Органические экстракты, содержащие транс-кислоту, объединяли и промывали насыщенным солевым раствором, сушили над MgSO4, фильтровали и концентрировали.
Стадия 8: (+)-метил 2-(4-бром-2-фторфенил)циклопропанкарбоксилат
К раствору (+)-2-(4-бром-2-фторфенил)циклопропанкарбоновой кислоты со стадии 7 (1 экв.) в метиленхлориде добавляли эфирный раствор диазометана до тех пор, пока по данным ТСХ реакция не завершилась. Полученную смесь концентрировали. Неочищенный сложный эфир использовали без очистки на следующей стадии.
Стадия 9: (+)-метил 2-{3-фтор-3'-[4-оксо-3-{[(2,2,2,-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]бифенил-4-ил}циклопропанкарбоксилат
К раствору (+)-метил 2-(4-бром-2-фторфенил)циклопропанкарбоксилата со стадии 8 (1,2 экв.) и боронатного сложного эфира со стадии 4 (1 экв.) в DMF-iPrOH (1:1, 0,1 М) добавляли трис(дибензилиденацетон)дипалладий(0) (0,055 экв.), 2,2-(диметиламино)-2'-(дициклогексилфосфино)бифенил (0,13 экв.) и раствор карбоната натрия (2 М, 4 экв.). Реакционную смесь нагревали при 78°С в течение 2 часов. Реакционную смесь фильтровали через целит и силикагель (1:1) и промывали EtOAc. Фильтрат концентрировали в вакууме и оставшийся растворитель отгоняли в вакууме. Полученное желтое твердое вещество интенсивно перемешивали в простом эфире. Остаток затем выделяли фильтрованием и промывали эфиром. Маточный раствор дополнительно очищали флэш-хроматографией (толуол:EtOAc, от 100:0 до 70:30).
Стадия 10: (+)-2-{3-фтор-3'-[4-оксо-3-{[(2,2,2,-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]бифенил-4-ил}циклопропанкарбоновая кислота
К раствору сложного эфира со стадии 9 в ТГФ-МеОН-Н2О (7:3:1, 0,05М) добавляли LiOH (1,1 экв., 1М) и смесь перемешивали при 50°С в течение 3 часов, а затем при комнатной температуре в течение 24 часов. Органический растворитель выпаривали, водную фазу подкисляли 1 н. раствором HCl и кислоту экстрагировали EtOAc (3Х). Органическую фазу промывали насыщенным солевым раствором, сушили и выпаривали растворитель с получением 2-(3-бромфенил)циклопропанкарбоновой кислоты. Полученное твердое вещество интенсивно перемешивали в гексане-простом эфире-ацетоне в течение 1 часа и затем собирали фильтрованием.
Энантиомер: Пример 65, (-)-2-{3-фтор-3'-[4-оксо-3-{[(2,2,2,-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]бифенил-4-ил}циклопропанкарбоновую кислоту можно получить с использованием (S,S)-2,2'-изопропилиден-бис(4-трет-бутил-2-оксазолина) на стадии 6.
1Н ЯМР (500 МГц, CDCl3): δ 10,31 (с, NH), 9,10 (с, 1H), 8,9 (дд, 1Н), 8,78 (м, 1Н), 7,78 (дд, 1H), 7,71 (т, 1H), 7,64 (ушир. с, 1H), 7,53 (м, 1Н), 7,47 (дд, 1H), 7,35 (м, 2H), 7,09 (т, 1H), 4,16 (м, 2H), 3,77 (м, 1Н), 2,00 (м, 1H), 1,71 (м, 1H), 1,52 (м, 1H). МС +ESI Q1 (М+1) 525,9.
ПРИМЕР 30
1-({3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]бифенил-4-ил}метил)циклобутанкарбоновая кислота
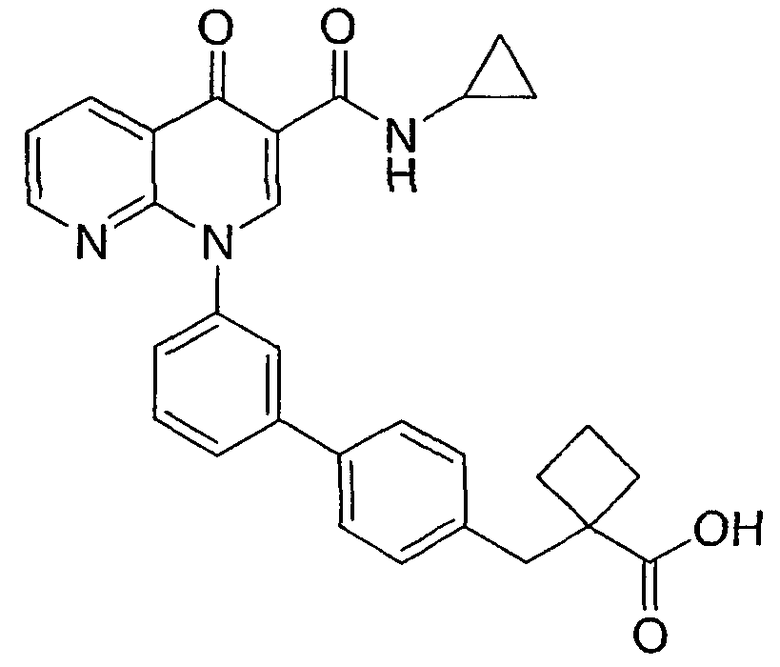
Стадия 1: этил 1-(4-бромбензил)циклобутанкарбоксилат
К раствору LDA (1,2 экв.) в ТГФ при -78°С добавляли этилциклобутанкарбоксилат (1 экв.) и реакционную смесь нагревали до 40°С в течение 10 минут. Реакционную смесь снова охлаждали до -78°С в течение 1 часа, затем добавляли к смеси 4-бромбензилбромид (1,1 экв.) в виде твердого вещества. Реакционную смесь нагревали до 0°С на ледяной бане. Реакционную смесь гасили насыщенным раствором NH4Cl и экстрагировали этилацетатом 3х, сушили над MgSO4 и упаривали досуха. Флэш-хроматография с использованием смеси 95:5 гексан:этилацетат давала этил 1-(4-бромбензил)циклобутанкарбоксилат.
Стадия 2: Этил 1-({3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]бифенил-4-ил}метил)циклобутанкарбоксилат
Смесь НАФТИРИДИНОНА 3 (1,0 экв.), сложного эфира со стадии 1 (1,5 экв.), Na2СО3 (3,5 экв., 2М в Н2О), Pd(ОАс)2 (0,05 экв.) и PPh3 (0,15 экв.) или PdCl2dppf (0,05 экв.) в н-пропаноле-DMF (1:1, 0,1М) перемешивали при 70°С в течение 2 часов. Смесь охлаждали до комнатной температуры, гасили при помощи АсОН и разбавляли EtOAc. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Флэш-хроматография (CH2Cl2/MeOH; 99:1) давала указанное в заголовке соединение.
Стадия 3: 1-({3'-[3-[(Циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]бифенил-4-ил}метил)циклобутанкарбоновая кислота
К раствору сложного эфира со стадии 2 в ТГФ-МеОН-Н2О (2:1:0,5, 0,1М) добавляли LiOH (5 экв., 2М) и смесь перемешивали при 50°С в течение 4 часов. Органический растворитель выпаривали, водную фазу подкисляли при помощи АсОН и кислоту экстрагировали EtOAc (3х). Органический слой промывали насыщенным солевым раствором, сушили и выпаривали растворитель. Остаток очищали флэш-хроматографией (CH2Cl2/МеОН, 10% NH4OH, 85:15) с получением указанного в заголовке соединения в виде белого твердого вещества.
1Н ЯМР (500 МГц, ацетон-d6): δ 9,87 (с, NH), 9,1 (с, 1н), 8,84 (дд, 1H), 8,72 (дд, 1H), 7,72 (д, 1н), 7,61 (м, 2H), 7,49 (м, 3H), 7,39 (д, 1H), 7,27 (м, 1H), 3,18 (с, 2H), 3,05 (м, 1H), 2,5 (м, 2H), 2,12 (м, 2H), 1,95 (м, 2H), 0,90 (м, 2H), 0,70 (м, 2H). МС 492,0 (-).
Следующие соединения получали в соответствии со способами, описанными выше. В таблице указаны их (М+1)+ значения, полученные методом масс-спектрометрии низкого разрешении в условиях электронного распыления или химической ионизации. * Указывают значение (М-1)-.
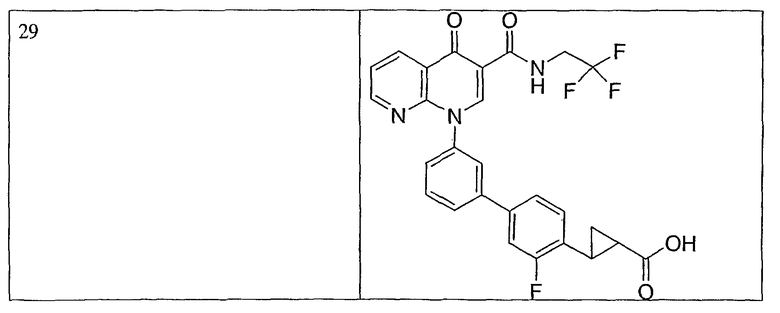
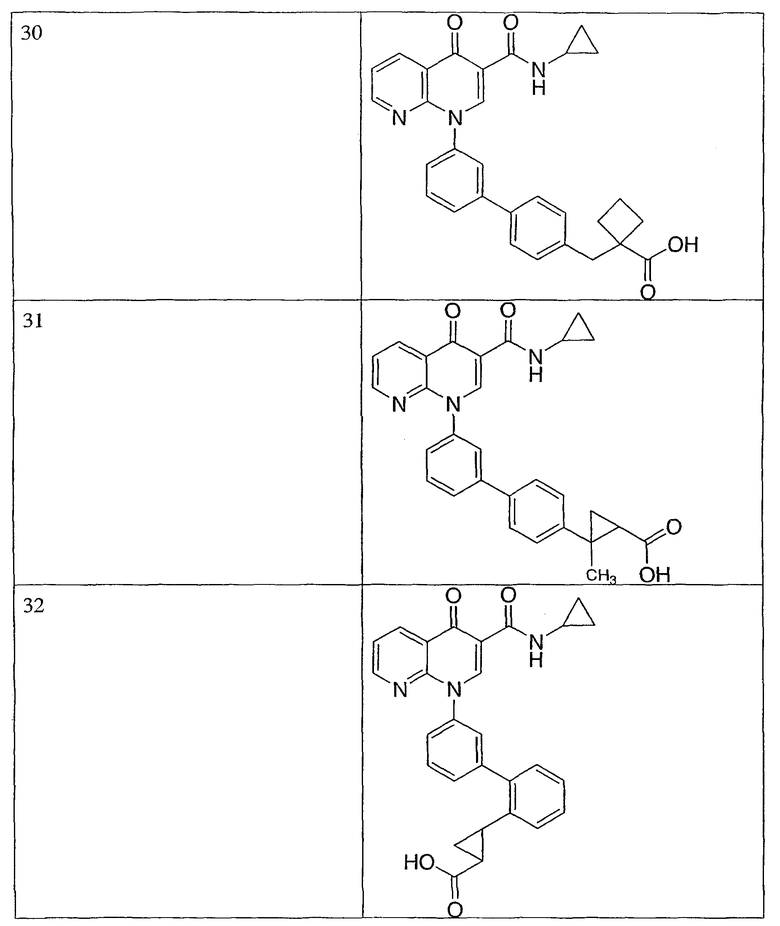
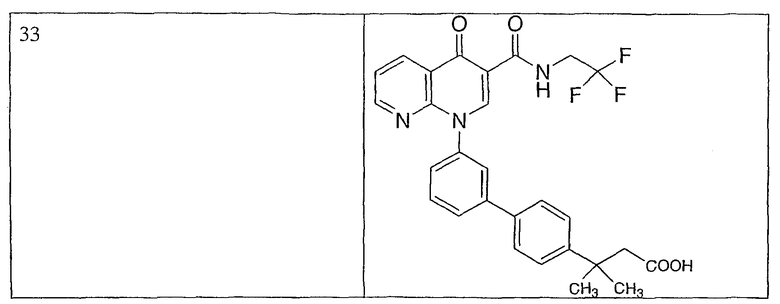
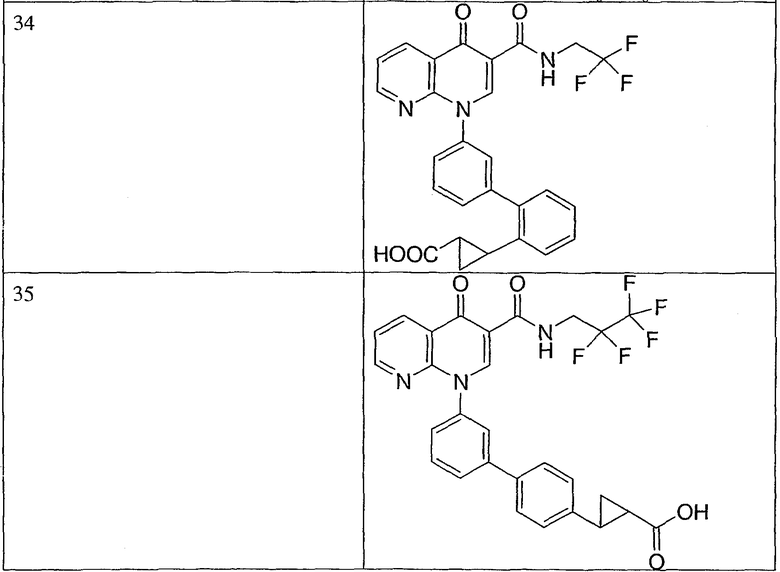
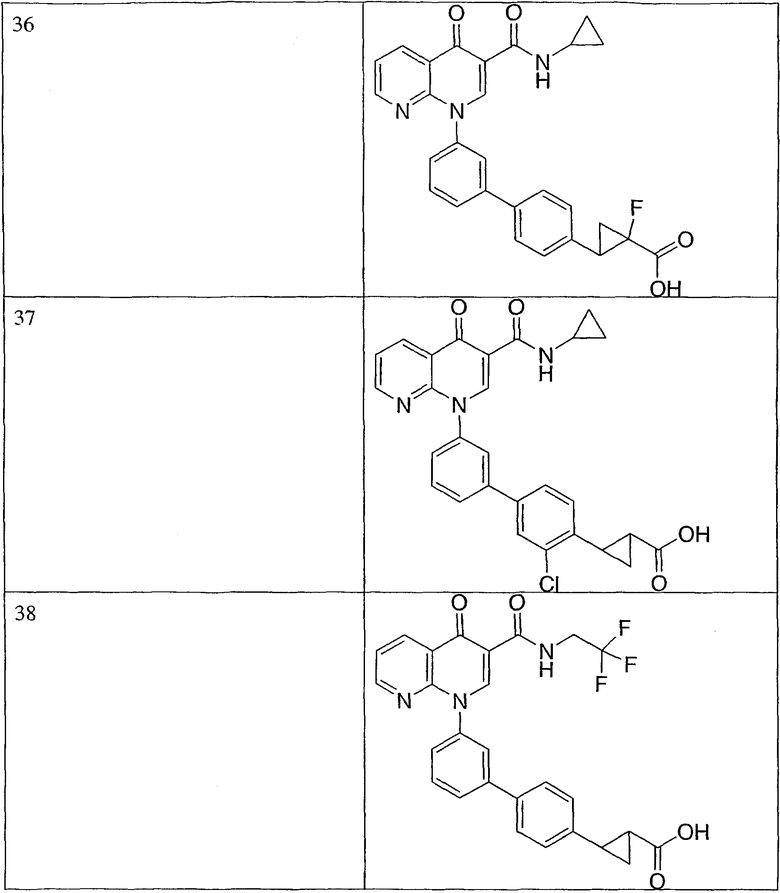
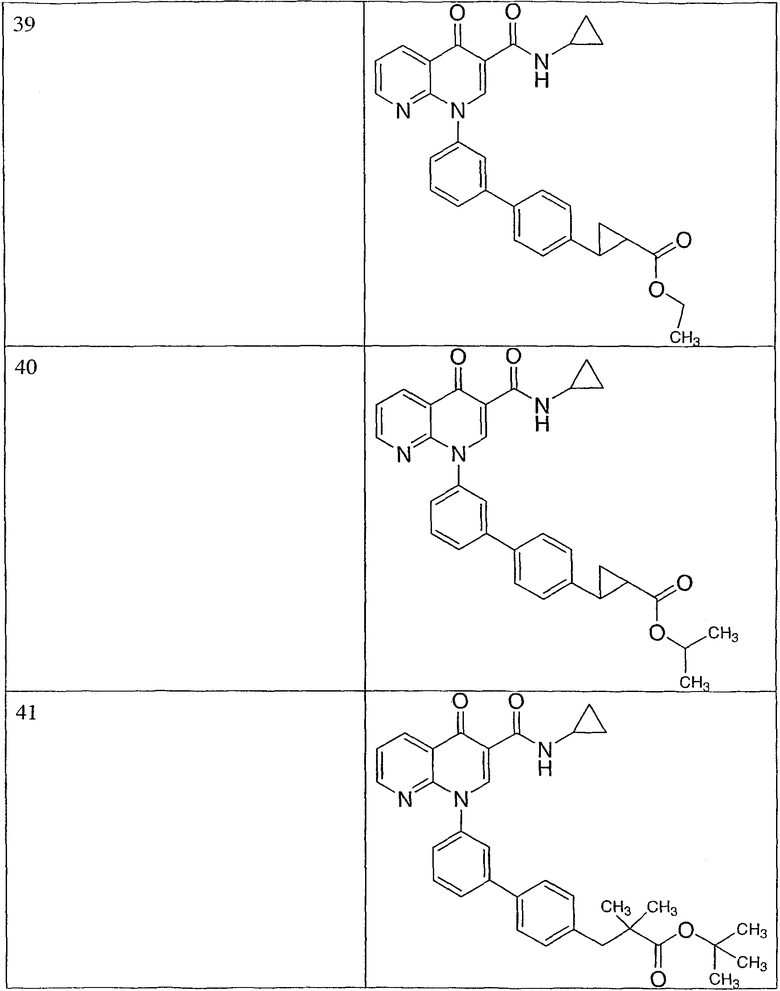

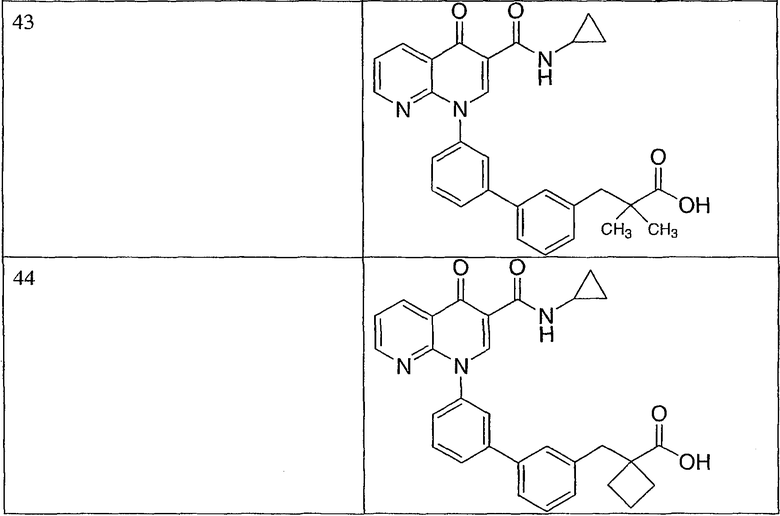
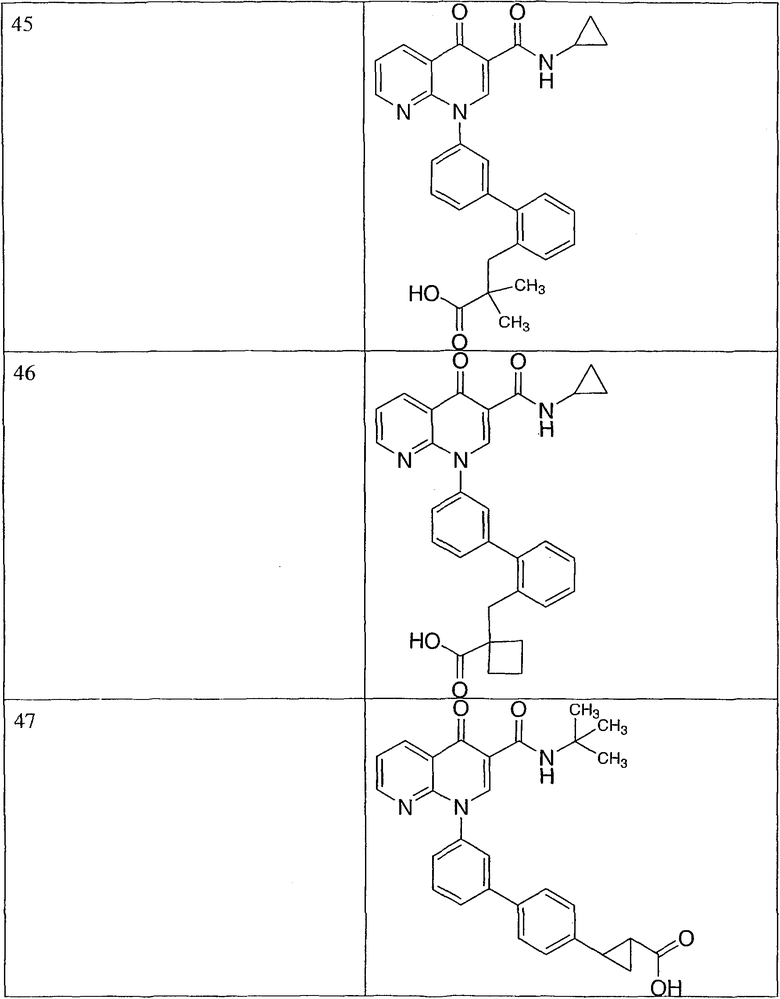
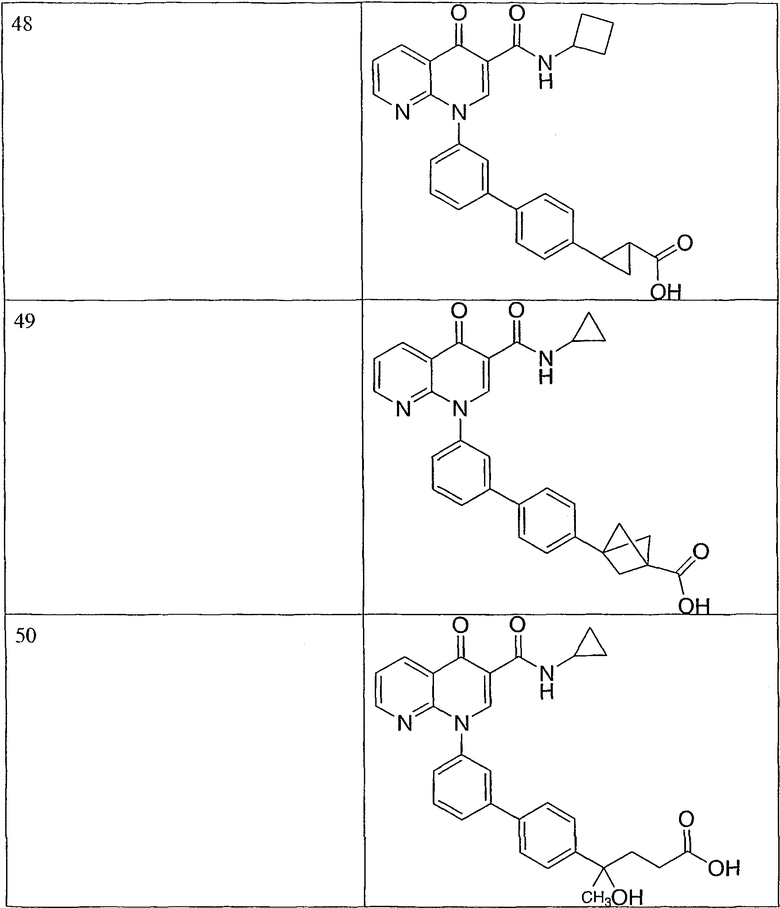
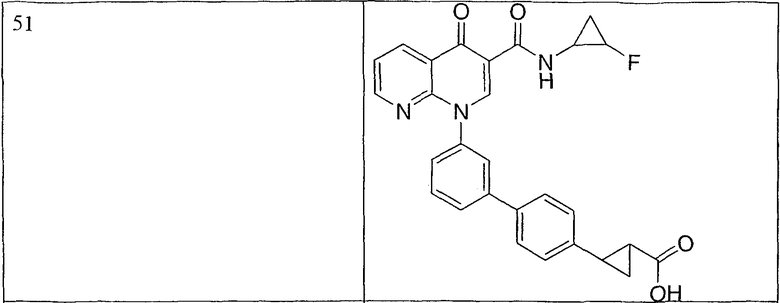
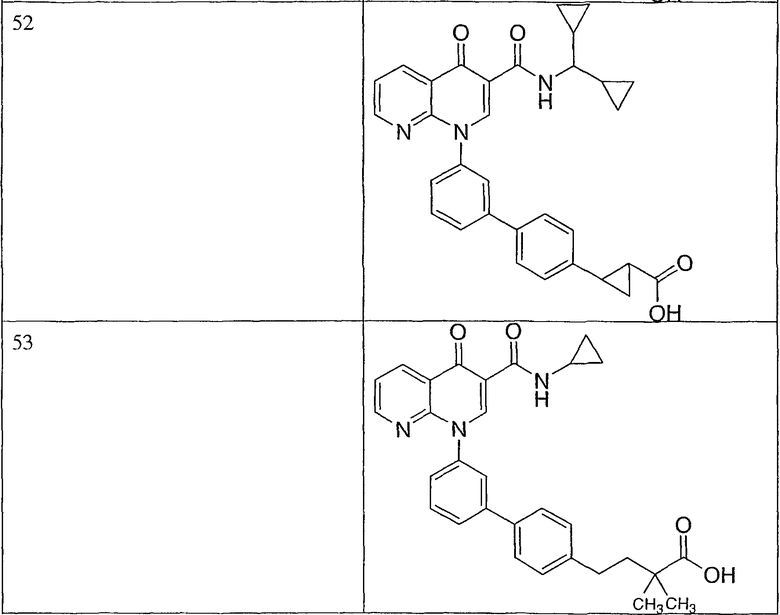
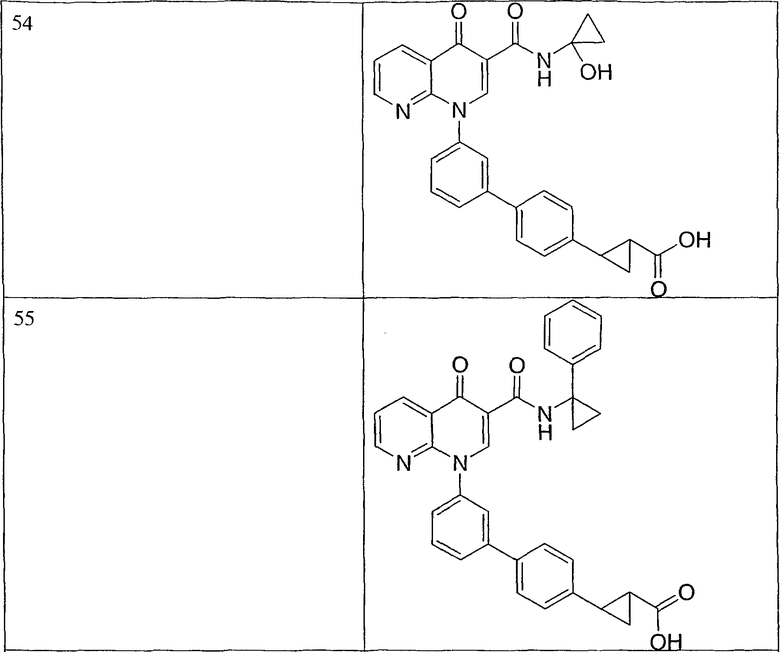
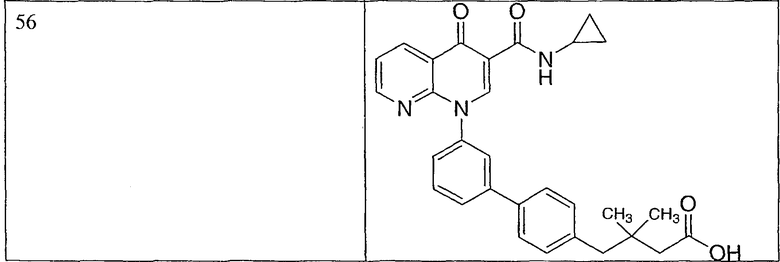
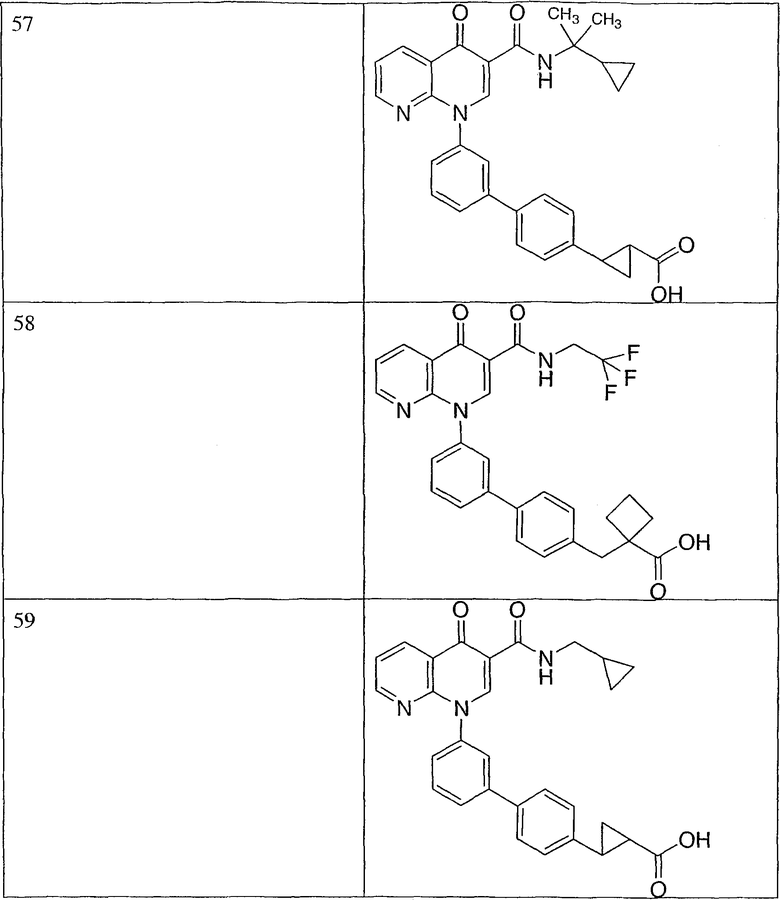
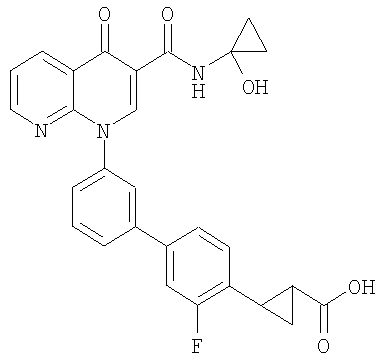
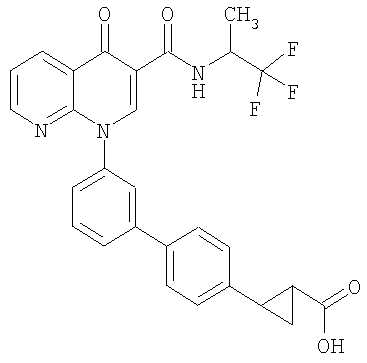
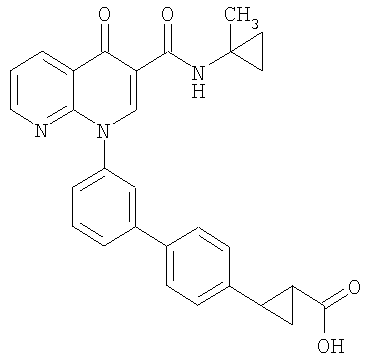
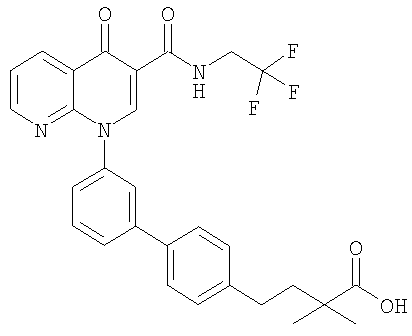
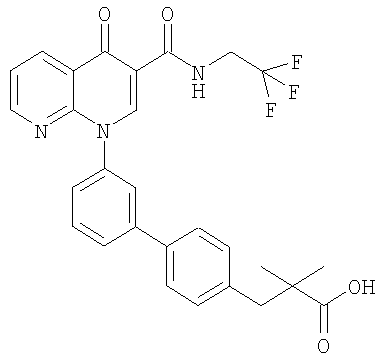
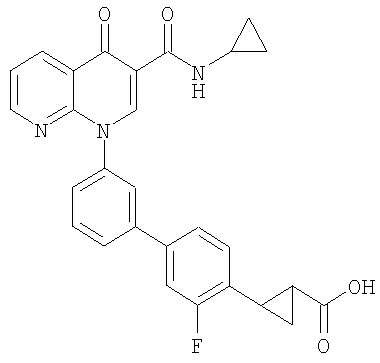
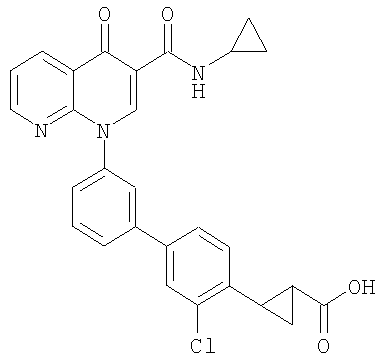
Изобретением также охватываются другие варианты или модификации, являющиеся очевидными для специалистов в данной области. Настоящее изобретение не ограничивается чем-либо, за исключением представленной ниже формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 3-ОКСО-1-ЦИКЛОБУТЕНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2296753C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ-4 | 2003 |
|
RU2323938C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ СРЕДСТВА ПРОТИВ ВИРУСОВ СЕМЕЙСТВА FLAVIVIRIDAE | 2004 |
|
RU2355687C2 |
| ИНГИБИТОРЫ КАТЕХОЛ-О-МЕТИЛТРАНСФЕРАЗЫ И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2011 |
|
RU2586974C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОПРОПИЛАМИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ H-ГИСТАМИНОВОГО РЕЦЕПТОРА | 2007 |
|
RU2449989C2 |
| ИНГИБИТОРЫ ПОЛИ(ADP-РИБОЗО)ПОЛИМЕРАЗЫ | 2007 |
|
RU2455286C2 |
| ИНГИБИТОРЫ MAGL НА ОСНОВЕ ПИРАЗОЛА | 2018 |
|
RU2789157C2 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ ТЕРАПЕВТИЧЕСКОГО ЭФФЕКТА ИНГИБИТОРА LSD1 НА ОСНОВЕ ЭКСПРЕССИИ INSM1 | 2018 |
|
RU2789449C2 |
| НОВОЕ СОЕДИНЕНИЕ БИФЕНИЛА ИЛИ ЕГО СОЛЬ | 2016 |
|
RU2726622C2 |
| ГАЛОГЕНЗАМЕЩЕННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2013 |
|
RU2756506C2 |
Описываются новые 4-Оксо-1-(3-замещенный фенил)-1,4-дигидро-1,8-нафтиридин-3-карбоксамиды общей формулы (I):
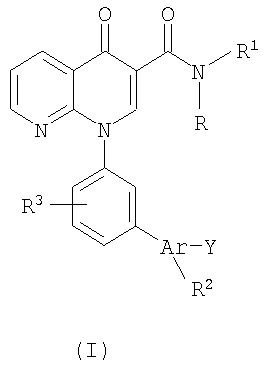
или их фармацевтически приемлемые соли, где
Ar - фенил или тиенил;
Y означает -СООН, C1-6алкил (С1-4алкил)n-СООН, С3-4циклоалкил-СООН, где C1-6алкил и С3-4циклоалкил необязательно замещены галогеном, гидрокси или CN, и (С1-4алкил) заместители необязательно связаны с образованием С3-4циклоалкила; где n имеет значение 0, 1, 2, 3 или 4;
R - Н или -C1-6алкил;
R1 - Н, С1-6алкил, необязательно замещенный галогеном; С3-6циклоалкил, необязательно замещенный галогеном, С1-6алкилом, ОН, CN или фенилом; (C1-6алкил)-SOр-(С1-6алкил), где р имеет значение 0, 1 или 2; или 1,3,4-тиадиазолил, замещенный галогенC1-С6алкилом; или R1 означает метил, замещенный циклопропилом;
R2 - Н, галоген или C1-6алкил;
R3 - Н или галоген, фармацевтические композиции на их основе. Соединения обладают ингибирующей активностью и могут быть использованы для лечения астмы и воспалительных заболеваний. 7 н. и 15 з.п. ф-лы, 4 табл.
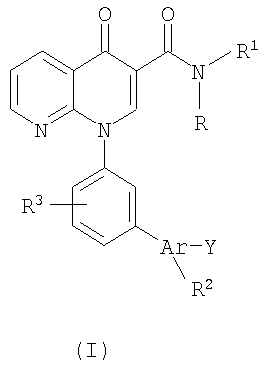
или их фармацевтически приемлемые соли,
где Ar представляет собой фенил или тиенил;
Y представляет собой -СООН, С1-6алкил(С1-4алкил)n-СООН, -С3-4циклоалкил-СООН, где -С1-6алкил и С3-4циклоалкил необязательно замещены галогеном, гидрокси или CN, и (С1-4алкил) заместители необязательно связаны с образованием С3-4циклоалкила; где n имеет значение 0, 1, 2, 3 или 4;
R представляет собой Н или -C1-6алкил;
R1 представляет собой Н, -С1-6алкил, необязательно замещенный галогеном; -С3-6циклоалкил, необязательно замещенный галогеном, -C1-6алкилом, ОН, CN или фенилом; -(С1-6алкил) -SOp- (С1-6алкил), где р имеет значение 0, 1 или 2; или 1,3,4-тиадиазолил, замещенный галогенС1-6алкилом; или R1 представляет собой метил, замещенный циклопропилом;
R2 представляет собой Н, галоген или -С1-6алкил;
R3 выбран из Н или галогена.
где n имеет значение 0, 1, 2, 3 или 4.
где R1 представляет собой -С3-6алкил, необязательно замещенный галогеном.
где R1 представляет собой -С3-6циклоалкил, необязательно замещенный заместителями, выбранными из -С3-6алкила, ОН, CN или галогена.
где R представляет собой водород.
где R1 представляет собой -С3-6циклоалкил, необязательно замещенный метилом или галогеном; и
R представляет собой водород.
R и R2 представляют собой водород.
где R и R3 представляют собой водород;
R1 представляет собой -С3-6циклоалкил, необязательно замещенный метилом или галогеном, или -C1-6алкил, необязательно замещенный 1-3 галогенами; и
Ar представляет собой фенил.
Y представляет собой -СН2-С3-4циклоалкил-СООН или -С3-4циклоалкил-СООН.
2-(транс)-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
2-(транс)-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-3-ил}циклопропанкарбоновую кислоту;
2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-3-ил}-2-метилпропановую кислоту;
2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-2-метилпропановую кислоту;
3-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-3-метилбутановую кислоту;
{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}(гидрокси)уксусную кислоту;
1-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
2-(цис)-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
5-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-2,2-диметил-1,3-диоксолан-4-карбоновую кислоту;
1-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-3-ил}циклопропанкарбоновую кислоту;
1-циано-3-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-2,2-диметилциклопропанкарбоновую кислоту;
2-(транс)-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-3-фтор-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
(цис)-2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-3-ил}циклопропанкарбоновую кислоту;
2-(транс)-{3'-бром-5'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
2-(транс)-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-3-метил-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
2-(транс)-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-2-метил-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
2-(транс)-{3-хлор-3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
2-(цис)-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-3-фтор-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-карбоновую кислоту;
2-(транс)-{3'-[3-(морфолин-4-илкарбонил)-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
2-(транс)-3'-[4-оксо-3-({[5-(трифторметил)-1,3,4-тиадиазол-2-ил]амино}карбонил)-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
2-(транс)-{3'-[3-({[2-(метилтио)этил]амино}карбонил)-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
2-(транс)-{3'-[3-({[2-(метилсульфонил)этил]амино}карбонил)-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
2-(транс)-{3'-[4-оксо-3-{[2,2,2-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
2-(транс)-(5-{3-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил] фенил}тиен-2-ил)циклопропанкарбоновую кислоту;
2-(транс)-{3'-[3-{[(циклопропилметил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту;
2-(транс)-{3'-[3-{[(1-цианоциклопропил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}циклопропанкарбоновую кислоту; или
3-{3'-[3-[(изопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-1,1'-бифенил-4-ил}-3-метилбутановую кислоту; или фармацевтически приемлемую соль перечисленных соединений.
(+)-(транс)-2-{3-фтор-3'-[4-оксо-3-{[(2,2,2-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]бифенил-4-ил}циклопропанкарбоновую кислоту;
1-({3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}метил)циклобутанкарбоновую кислоту;
(транс)-2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}-2-метилциклопропанкарбоновую кислоту;
(транс)-2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-2-ил}циклопропанкарбоновую кислоту;
3-метил-3-{3'-[4-оксо-3-{[(2,2,2-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]бифенил-4-ил}бутановую кислоту;
(транс}-2-{3'-[4-оксо-3-{[(2,2,2-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]бифенил-2-ил}циклопропанкарбоновую кислоту;
(транс)-2-{3'-[4-оксо-3-{[(2,2,3,3,3-пентафторпропил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]бифенил-4-ил} циклопропанкарбоновую кислоту;
(транс)-2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}-1-фторциклопропанкарбоновую кислоту;
(+)-(транс)-2-{3-хлор-3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}циклопропанкарбоновую кислоту;
(-)-(транс)-2-{3'-[4-оксо-3-{[(2,2,2-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]бифенил-4-ил}циклопропанкарбоновую кислоту;
(+)-(транс)-этил 2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}циклопропанкарбоксилат;
(+)-(транс)-изопропил 2-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}циклопропанкарбоксилат;
трет-бутил 3-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}-2,2-диметилпропаноат;
3-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}-2,2,-диметилпропановую кислоту;
3-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-3-ил}-2,2-диметилпропановую кислоту;
1-({3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-3-ил}метил)циклобутанкарбоновую кислоту;
3-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-2-ил}-2,2-диметипропановую кислоту;
1-({3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-2-ил}метил)циклобутанкарбоновую кислоту;
(+)-(транс)-2-{3'-[3-[(трет-бутиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}циклопропанкарбоновую кислоту;
(+)-(транс)-2-{3'-[3-[(никлопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}циклопропанкарбоновую кислоту;
3-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}бицикло[1.1.1] пентан-1-карбоновую кислоту;
4-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}-4-гидроксипентановую кислоту;
(транс)-2-{3'-[3-{[(±)-цис-(2-фторциклопропил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-(+)-бифенил-4-ил}циклопропанкарбоновую кислоту;
(+)-(транс)-2-{3'-[3-[(дициклопропилметил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}-циклопропанкарбоновую кислоту;
4-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}-2,2-диметилбутановую кислоту;
(+)-(транс)-2-{3'-[3-{[(1-гидроксициклопропил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}циклопропанкарбоновую кислоту;
(+)-(транс)-2-{3'-[4-оско-3-{[(1-фенилциклопропил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}циклопропанкарбоновую кислоту;
4-{3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}-3,3-диметилбутановую кислоту;
(+)-(транс)-2-{3'-[3-{[(1-циклопропил-1-метилэтил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}циклопропанкарбоновую кислоту;
1-({3'-[4-оксо-3-{[(2,2,2-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]бифенил-4-ил}метил)циклобутанкарбоновую кислоту;
(+)-(транс)-2-{3'-[3-[(циклопропилметил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}-циклопропанкарбоновую кислоту;
(-)-(транс)-2-{3-фтор-3'-[3-{[(1-гидроксициклопропил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}циклопропанкарбоновую кислоту;
(транс)-2-{3'-[4-оксо-3-{[((±)-2,2,2-трифтор-1-метилэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]-(+)-бифенил-4-ил}циклопропанкарбоновую кислоту;
(+)-(транс)-2-{3'-[3-{[(1-метилциклопропил)амино]карбонил}-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}-циклопропанкарбоновую кислоту;
2,2-диметил-4-{3'-[4-оксо-3-{[(2,2,2-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]бифенил-4-ил)бутановую кислоту;
2,2-диметил-3-{3'-[4-оксо-3-{[(2,2,2-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил] бифенил-4-ил}пропановую кислоту;
(-)-(транс)-2-{3-хлор-3'-[3-[(циклопропиламино)карбонил]-4-оксо-1,8-нафтиридин-1(4Н)-ил]-бифенил-4-ил}циклопропанкарбоновую кислоту; или
(+)-(транс)-2-{3'-[4-оксо-3-{[(2,2,2-трифторэтил)амино]карбонил}-1,8-нафтиридин-1(4Н)-ил]бифенил-4-ил}циклопропанкарбоновую кислоту;
или фармацевтически приемлемую соль перечисленных соединений.
фармацевтически приемлемый носитель.
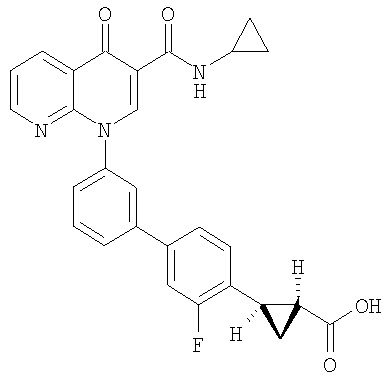
| WO 9907704 А, 18.02.1999 | |||
| ПРОИЗВОДНЫЕ НАФТИРИДИНА И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2240322C2 |
| ПРОИЗВОДНЫЕ ПИРИДОНКАРБОНОВОЙ КИСЛОТЫ ИЛИ ИХ СОЛИ И АНТИБАКТЕРИАЛЬНОЕ СРЕДСТВО | 1996 |
|
RU2167873C2 |

