ОБЛАСТЬ ТЕХНИКИ
Данное изобретение относится к производным нафтиридина, используемым в качестве лекарственного средства, в частности в качестве ингибитора фосфодиэстеразы типа IV.
Астма является респираторным заболеванием, которое вызывает хрипы и приступы при спазмах дыхательных путей. Количество пациентов постоянно увеличивается и, как предсказывают, будет увеличиваться дальше.
В настоящее время для лечения астмы в качестве бронхолитических средств в основном используются производные ксантина, такие как аминофиллин и теофиллин, и β-стимуляторы, такие как прокатерол.
Функциональный механизм этих соединений направлен на облегчение спазмов гладкой мускулатуры дыхательных путей посредством увеличения концентрации внутриклеточного циклического аденозин-3',5'-монофосфата (цАМФ) через активацию фермента аденилатциклазы, вырабатывающего внутриклеточный цАМФ, или ингибирование фермента фосфодиэстеразы (ФДЭ), гидролизующего цАМФ, в гладкой мускулатуре дыхательных путей (Internal Medicine, 69, 207-214 (1992)).
Известно, что повышенная концентрация цАМФ вызывает ингибирование спазмов гладкой мускулатуры дыхательных путей (Clin. Exp. Allergy, 22, 337-344 (1992), Drugs of the Future, 17, 799-807 (1992)), что способствует улучшению состояния больных астмой.
Однако известно, что производные ксантина вызывают системное побочное действие, такое как гипотензия и кардиотоническое действие (J. Cyclic Nucleotide and Protein Phosphorylation Res., 10, 551-564 (1985), J. Pharmacol. Exp. Ther., 257, 741-747 (1991)), и β-стимуляторы могут явиться причиной гипочувствительности и, при повышении дозы, вызвать такие побочные эффекты, как дрожание пальцев и учащенное сердцебиение.
С другой стороны, было обнаружено, что ФДЭ делится, по крайней мере, на пять различных типов от I до V, и каждый из них имеет различное распространение или функции (Pharmacol. Ther., 51, 13-33 (1991)). В частности, ФДЭ типа IV не действует на циклический гуанозин-3',5'-монофосфат (цГМФ), но осуществляет специфический гидролиз цАМФ среди нуклеотидов, и ее присутствие распознается как в гладкой мускулатуре дыхательных путей, так и в инфильтрующих клетках.
Также было описано, что ингибиторы ФДЭ типа IV демонстрируют ингибирующее действие на инфильтрацию эозинофилов за счет антигенов и факторов агрегации тромбоцитов у морских свинок (Eur. J. Pharmacol., 255, 253-256 (1994)) и ингибируют высвобождение вредных белков (базальный белок миелина (МВР), катионный белок эозинофилов (ЕСР)) из эозинофилов (Br. J. Pharmacol., 115, 39-47 (1995)). Также было описано, что они оказывают ингибирующее действие на спазмы гладкой мускулатуры посредством сокращающих веществ (гистамин, метахолин, LTD4) (Br. J. Pharmacol., 113, 1423-1431 (1994)), ингибируют вырабатывание ИЛ-4 - цитокина, который, как известно, в значительной степени участвует в астме (J. Invest., Dermatol., 100, 681-684 (1993)), оказывают ингибирующее действие на увеличение проницаемости сосудов дыхательных путей (Fundam. Clin. Pharmacol., 6, 247-249 (1992)) и оказывают ингибирующее действие на гиперчувствительность дыхательных путей (Eur. J. Pharmacol., 275, 75-82 (1995)). Таким образом, ожидается, что ингибитор ФДЭ типа IV может стать агентом для лечения астмы, имеющим меньшее количество побочных эффектов.
В качестве ингибиторов ФДЭ типа IV известно множество соединений, включая производные нафтиридина. Авторы данного изобретения ранее описали производное нафтиридина, представленное следующей формулой, в которой в положении 4 (R6) находится циклический заместитель, такой как арил, гетероарил или циклоалкил, и в положении 3 (R5) находится незамещенная или замещенная низшая алкильная группа
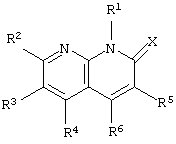
где R5 является атомом водорода или низшей алкильной группой и R6 является арильной группой, имеющей заместитель, гетероарильной группой, имеющей заместитель, циклоалкильной группой или адамантильной группой.
Для получения дополнительной информации см. ссылку (WO 96/06843).
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторы данного изобретения провели исследования с целью получения нового соединения, которое эффективно и селективно ингибировало бы ФДЭ типа IV и было бы полезным для профилактики и лечения респираторных заболеваний, таких как бронхиальная астма, при этом имело бы меньшее количество побочных эффектов, а также лекарственного средства, содержащего указанное соединение.
Авторы данного изобретения также провели обширные исследования соединений, оказывающих ингибирующее действие на ФДЭ типа IV, и в результате обнаружили, что соединение, в котором определенный заместитель (-X-R6) введен в положение 3 ранее описанного соединения (WO 96/06843), является новым соединением и оказывает сильное ингибирующее действие на ФДЭ типа IV, а также имеет превосходную пероральную абсорбируемость и метаболически стабильно. Следовательно, было обнаружено, что данное соединение крайне полезно как ингибитор ФДЭ типа IV, что и составило данное изобретение.
Соответственно, данное изобретение относится к новому производному нафтиридина, представленному следующей общей формулой (I), или его фармацевтически приемлемой соли и лекарственному препарату, содержащему указанное соединение в качестве активного ингредиента.
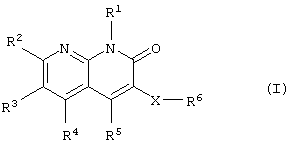
где R1: -R0, низший алкиленциклоалкил или циклоалкил;
R0: низший алкил;
R2, R3 и R4: -Н, -R0, галоген, низший алкилен-ОН,
- низший алкилен-SH, -низший алкилен-O-R0, -низший алкилен-3-R0,- низший алкилен-O-CO-R0, -низший алкилен-S-CO-R0, -ОН, -О-R0, -S-R0, -SO-R0, -SO2-R0, -NH2, -NHR0, -NR
R9: -Н, -R0 или -низший алкилен-арил;
R5: циклоалкил, который может быть замещен группой, выбранной из R10, циклоалкенил, который может быть замещен группой, выбранной из R10, гетероциклическая группа, которая может быть замещена группой, выбранной из R10, или фенил, который может быть замещен группой, выбранной из R10;
R6: -ОН, -OR7, -COOH, -COOR7, -CONH2, -CONHR7, -CON(R7)2,
-O-COR7, -O-COOR7, -CHO, -COR7, -NH2, -NHR7, -N(R7)2, -NHCOR7,
-N(R7)COR7, -NHSO2R7, -N(R7)SO2R7, -CN, -NHCOOR7, -N(R7)COOR7,
-C(NH)NH2, -NHC(NH)NH2 или -N(R7)С(NH)NH2, или группа формулы
-Y-R8;
R7: низший алкил, который может быть замещен группой, выбранной из группы, включающей -ОН, -фенил, -галоген, -OR0, -CO2H, -CO2R0, -NH2, -NHR0, -NR
R8: циклоалкил, который может быть замещен группой, выбранной из R10, арил, который может быть замещен группой, выбранной из R10, или гетероциклическая группа, которая может быть замещена группой, выбранной из R10;
R10: -ОН, -фенил, -галоген, -OR0, -СО2Н, -CO2R0, -NH2,
-NHR0, -NR
Y: связь, -О-, -COO-, -CONH-, -CON(R7)-, -O-СО-, -O-СОО-, -СО-, -NH-, -N(R7)-, -NHCO-, -N(R7)CO-, -NHCOO-, -N(R7)COO-, -NHSO2- или -N(R7)SO2-, и
X: связь или низший алкилен, или низший алкенилен (далее принимаются те же значения).
Также, в соответствии с данным изобретением, представлено лекарственное средство, в частности ингибитор ФДЭ типа IV, которое содержит производное нафтиридина или его соль.
Далее данное изобретение описывается более подробно.
В контексте данного описания термин "низший" означает прямую или разветвленную углеводородную цепь, имеющую от 1 до 6 атомов углерода. Примеры "низшего алкила" включают метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, изопентил, неопентил, гексил и подобные. Предпочтительным являются алкил, имеющий от 1 до 4 атомов углерода, и особенно предпочтительным является метил или этил. Термин "низший алкилен" означает двухвалентную группу, образованную удалением любого из атомов водорода из описанного выше "низшего алкила", и предпочтительным является алкилен, имеющий от 1 до 4 атомов углерода, особенно предпочтительным является метилен, этилен или пропилен. "Низший алкенилен" означает группу, имеющую одну или более двойных связей в любом положении "низшего алкилена", имеющего два или более атомов углерода, и предпочтительно является алкениленом, имеющим от 2 до 4 атомов углерода.
"Циклоалкил" предпочтительно является циклоалкилом, имеющим от 3 до 8 атомов углерода, в частности предпочтительно циклопропилом или циклогексилом. "Циклоалкенил" предпочтительно является циалоалкенилом, имеющим от 5 до 8 атомов углерода, особенно предпочтительно циклогексенилом. Термин "арил" означает ароматическую углеводородную группу, имеющую от 6 до 14 атомов углерода, предпочтительно фенил. "Гетероциклическая группа" представляет собой моноциклическую - трициклическую гетероциклическую группу, имеющую от 1 до 4 гетероатомов, выбранных из группы, включающей атом азота, атом кислорода и атом серы, которая может образовывать мостиковое кольцо или сконденсированное кольцо с бензольным кольцом. Такой гетероцикл предпочтительно является пяти - семичленной насыщенной или ненасыщенной моноциклической гетероциклической группой, особенно предпочтительно пиридином, пиперидином, морфолином, тиофеном, тиазолом, имидазолом, тетразолом, пиразином или пиперазином.
Термин "галоген" означает F, Cl, Вr или I.
Фраза "который может быть замещен" означает "незамещенный" или "имеющий от 1 до 5 заместителей, которые могут быть одинаковыми или отличаться друг от друга".
Заместитель в "циклоалкиле, который может быть замещен", "циклоалкениле, который может быть замещен", "гетероциклической группе, которая мажет быть замещена", "фениле, который может быть замещен" и "ариле, который может быть замещен", специально не ограничен до тех пор, пока он может быть использован как заместитель в кольцах указанных соединений, которые могут быть использованы для получения лекарственных средств, в частности ингибитора ФДЭ типа IV, но предпочтительно он является -ОН, -фенилом, -галогеном, -OR0, -CO2H, -CO2R0, -NH2, -NHR0, -NR
Группа X-R6 в положении 3 нафтиридина предпочтительно является группой более гидрофильной, чем алкильная группа, имеющая такое же количество атомов углерода. Например, Х предпочтительно является связью или низшим алкиленом и R6 предпочтительно является -ОН, -СООН, -COOR7, -O-COR7, -NH2, -NHR7, -N(R7)2, -C(NH)NH2, -NHC(NH)NH2 или -N(R7)С(NH)NH2 или группой, представленной формулой -Y-R8. R8 предпочтительно является арилом или гетероциклической группой. Эти группы могут быть замещены группой, выбранной из группы, включающей -ОН, -фенил, -галоген, -OR0, -CO2H, -CO2R0, -NH2, -NHR0, -NR
Группа R5 в положении 4 нафтиридина предпочтительно является циклоалкилом, фенилом, который может быть замещен в положении 3, или подобными. Заместитель предпочтительно является галогеном, низшим алкилом или подобными. Группы R3 и R4 в положениях 5 и 6 нафтиридина каждая предпочтительно является низшим алкилом или атомом водорода, более предпочтительно атомом водорода. Группа R2 в положении 7 нафтиридина предпочтительно является низшим алкилом, галогеном, низшим алкилен-ОН или группой, представленной формулой -CH=N-OH.
Среди соединений в соответствии с данным изобретением особенно предпочтительными являются следующие соединения:
3-(2-амидиноэтил)-4-(3-хлорфенил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-он,
4-(3-хлорфенил)-1-этил-3-(2-гуанидиноэтил)-7-метил-1,8-нафтиридин-2(1Н)-он,
4-циклогексил-1-этил-7-метил-3-[2-(1Н-тетразол-5-ил)этил]-1,8-нафтиридин-2(1Н)-он,
4-(3-хлорфенил)-1-этил-7-метил-3-[3-(1Н-тетразол-5-ил)пропил]-1,8-нафтиридин-2(1Н)-он,
4-(3-бромфенил)-1-этил-7-метил-3-[2-(1Н-тетразол-5-ил)этил]-1,8-нафтиридин-2(1Н)-он,
3-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропановая кислота,
3-(4-циклогексил-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропановая кислота,
3-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]бензойная кислота,
3-[4-(3-хлорфенил)-1-этил-7-(гидроксииминометил)-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропановая кислота,
3-[7-хлор-4-(3-хлорфенил)-1-этил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропановая кислота,
3-[1-этил-7-метил-4-(3-метилфенил)-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропановая кислота,
4-(3-хлорфенил)-1-этил-7-метил-3-(пиперидин-4-ил)-1,8-нафтиридин-2(1Н)-он и
1-{2-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]этил}пиперидин-4-карбоновая кислота и их соли.
В зависимости от видов заместителей соединения в соответствии с данным изобретением могут существовать в виде геометрических изомеров и таутомеров, их выделенные формы и смеси включены в объем данного изобретения.
Также соединения в соответствии с данным изобретением в некоторых случаях могут иметь асимметричные атомы углерода и на основе этих атомов могут существовать (R)- и (S)-формы оптических изомеров. Данное изобретение включает все такие оптические изомеры в смешанной и разделенной форме.
В соединения в соответствии с данным изобретением также включены фармакологически приемлемые пролекарства. Фармакологически приемлемые пролекарства представляют собой соединения, имеющие группы, которые могут быть превращены в определенные группы в соответствии с данным изобретением, такие как NН2, ОН и СО2Н, при растворении или при определенных физиологических условиях. Примеры групп, которые могут образовывать пролекарства, включают те, которые описаны в "Prog. Med.", 5, 2157-2161 (1985) и "Pharmaceutical Research and Development" (Hirokawa Publishing Co., 1990), том 7, Drug Design, 163-198.
Соединения в соответствии с данным изобретением могут образовывать кислотно-аддитивные соли или, в зависимости от видов заместителей, соли с основаниями. Такие соли являются фармацевтически приемлемыми солями, и их иллюстративные примеры включают кислотно-аддитивные соли с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота, и органическими кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфокислота, этансульфокислота, аспарагиновая кислота и глутаминовая кислота, соли с неорганическими основаниями, как, например, соли с натрием, калием, магнием, кальцием и алюминием, и органическими основаниями, такими как метиламин, этиламин, этаноламин, лизин и орнитин, и аммониевые соли.
Кроме того, данное изобретение также включает различные гидраты, сольваты и полиморфные вещества соединения (I) в соответствии с данным изобретением и их соли.
Способ получения
Соединение в соответствии с данным изобретением и его фармацевтически приемлемые соли могут быть получены различными известными методами синтеза с учетом свойств соединения, основанных на базовой (основной) структуре соединения или видах заместителей. В этом случае, в зависимости от вида функциональной группы, иногда бывает эффективна, с производственной точки зрения, замена функциональной группы в исходном соединении или на промежуточной стадии на подходящую защитную группу, а именно группу, которая может быть легко превращена в функциональную группу. Затем целевое соединение может быть получено удалением защитной группы, как того требует каждый конкретный случай. Примерами таких функциональных групп могут быть гидроксильная и карбоксильная группы, а указанными защитными группами, которые необязательно используют в зависимости от условий реакции, могут быть защитные группы описанные, например, в "Protective Groups in Organic Synthesis" (2-е изд.), изданной Green и Wuts.
(1) Первый способ получения
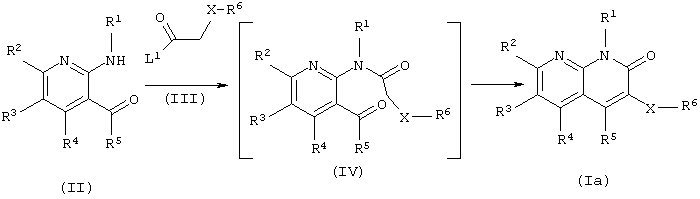
где L1 является уходящей группой (это же значение применяется и далее).
В данном способе получения соединения (Iа) в соответствии с данным изобретением получают взаимодействием производного аминопиридина (II) с ацилирующим агентом, представленным обшей формулой (III), с получением производного амида (IV) и затем непосредственно реакцией замыкания кольца.
Предпочтительные примеры уходящей группы, представленной L1, включают атомы галогена, ацилокси, карбонаты, такие как алкилоксикарбонилокси, и остатки органической сульфокислоты, такие как метансульфонилокси и п-толуолсульфонилокси. Также, через объединение заместителя на XR6 с L1, соединение общей формулы (III) может образовывать внутримолекулярный или межмолекулярный ангидрид кислоты (например, глутаровый ангидрид).
Реакцию проводят в органическом растворителе, инертном к реакции, выбранном из галогенированных углеводородов, таких как дихлорметан, дихлорэтан и хлороформ; ароматических углеводородов, таких как бензол, толуол и ксилол; простых эфиров, таких как диэтиловый эфир, тетрагидрофуран (ТГФ) или диоксан; N,N-диметилформамида (ДМФА) или без растворителя, при температуре от охлаждения до нагревания. При проведении реакции производное аминопиридина (II) и ацилирующий агент (III) могут быть использованы в эквивалентных количествах или одно из них может быть использовано в избытке, и иногда, для более гладкого прохождения реакции, полезно проводить реакцию в присутствии органического основания (предпочтительно, триэтиламина, пиридина или 4-(N,N-диметиламино)пиридина), неорганического основания (предпочтительно, гидроксида натрия или карбоната калия) или металлического основания (предпочтительно, гидрида натрия, метилата натрия или трет-бутилата калия).
В данном способе получения выделение производного амида (IV) и реакция замыкания кольца могут быть проведены постадийно. В этом случае, учитывая растворитель, температуру, основание и т.д., указанные выше условия могут применяться в каждой реакции.
(2) Второй способ получения
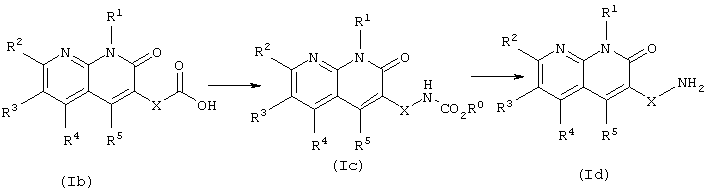
В данном способе получения соединение (Id) в соответствии с данным изобретением, имеющее аминогруппу, получают из соединения (Ib) в соответствии с данным изобретением, имеющего карбоксильную группу. Соединение карбамата (Ic), получаемое в качестве промежуточного соединения, также является соединением в соответствии с данным изобретением.
Соединение (Ic) в соответствии с данным изобретением может быть получено взаимодействием соединения изоцианата, которое получают перегруппировкой Курциуса кислотного азида, полученного взаимодействием реакционноспособного производного карбоксильной группы, полученной из соединения (Ib), такого как ангидрид кислоты, с солью азида, такой как азид натрия, или методом взаимодействия с дифенилфосфорилазидом (ДФФА), или которое получают перегруппировкой Хоффмана первичного амида, полученного из соединения (Ib) обычной реакцией амидирования, с соединением спирта.
Реакцию проводят в органическом растворителе, инертном к реакции, выбранном из галогенированных углеводородов, ароматических углеводородов, простых эфиров и ДМФА, или без растворителя, при температуре от охлаждения до нагревания. При проведении реакции соединение спирта может быть использовано в эквивалентном количестве или в избытке по отношению к соединению (Ib).
Соединение (Id) в соответствии с данным изобретением получают, удаляя из соединения (Iс) в соответствии с данным изобретением защитную группу аминогруппы карбаматного типа, описано в указанной ранее "Protective Groups in Organic Synthesis" (2-е изд.). Данная реакция может проводиться последовательно, без выделения соединения (Iс), следуя указанной выше реакции.
(3) Третий способ получения
Соединение, имеющее карбоксильную группу на R8 соединения (I), может быть получено гидролизом трифторметильной группы на R8.
Реакцию проводят в органическом растворителе, инертном к реакции, выбранном из галогенированных углеводородов, ароматических углеводородов, простых эфиров и ДМФА, или без растворителя, в присутствии кислоты (хлористоводородной кислоты, серной кислоты или подобных) или основания (гидроксида натрия, метилата натрия или подобных) при температуре от охлаждения до нагревания.
(4) Четвертый способ получения
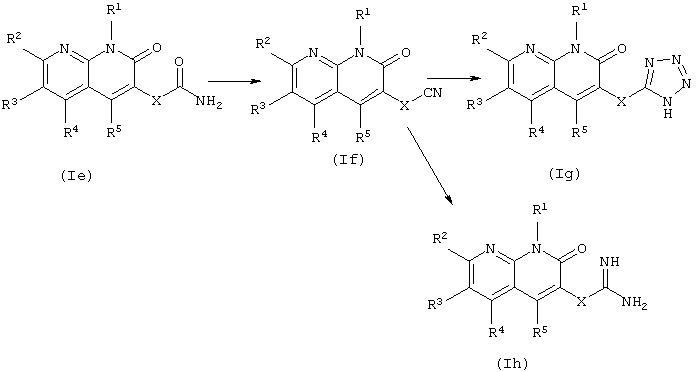
В данном способе получения соединения (If), (Ig) и (Ih) в соответствии с данным изобретением получают из соединения (Ie) в соответствии с данным изобретением по указанному выше пути синтеза.
Соединение (If) в соответствии с данным изобретением может быть получено дегидратацией соединения (Ie) в соответствии с данным изобретением. Может быть использован обычный метод дегидратации, такой как описан, например, в "JIKKEN KAGAKU KOZA" (4-е изд.), изданном Химическим обществом Японии, том 20, (1992) (Maruzen).
Соединение (Ig) в соответствии с данным изобретением может быть получено взаимодействием соединения (If) в соответствии с данным изобретением с солью азида, такой как азид натрия. Реакцию проводят в органическом растворителе, инертном к реакции, выбранном из галогенированных углеводородов, ароматических углеводородов, простых эфиров, спиртов, таких как метанол и этанол, ДМФА и воды, или без растворителя при температуре от охлаждения до нагревания.
При проведении реакции соль азида может быть использована в эквивалентном количестве или в избытке по отношению к соединению (If), и в некоторых случаях для более гладкого проведения реакции полезно проводить реакцию в присутствии кислоты (уксусной кислоты, трифторуксусной кислоты, гидрохлорида триэтиламина, хлористоводородной кислоты, хлорида алюминия или подобных) или основания (пиридина, триэтиламина, гидроксида натрия, гидроксида калия, метилата натрия, трет-бутилата калия или подобных).
Соединение (Ih) в соответствии с данным изобретением может быть получено взаимодействием соединения (If) в соответствии с данным изобретением с аммиаком, аммониевой солью, такой как хлорид аммония, или амидом металла, таким как амид натрия. Также оно может быть получено взаимодействием имидоилхлорида, полученного взаимодействием соединения (If) с хлороводородом, с аммониевой солью, такой как хлорид аммония. Реакцию проводят в органическом растворителе, инертном к реакции, выбранном из галогенированных углеводородов, ароматических углеводородов, простых эфиров, спиртов, ДМФА и воды, или без растворителя при температуре от охлаждения до нагревания и при давлении от обычного до повышенного. При проведении реакции аминирующий агент может быть использован в эквивалентном количестве или в избытке по отношению к соединению (If).
(5) Пятый способ получения
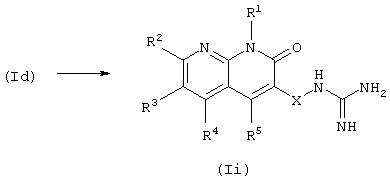
В данном способе получают соединение (Ii) из соединения (Id) в соответствии с данным изобретением реакцией получения гуанидина.
Примеры гуанидинобразующих агентов, используемых в данной реакции, включают цианамид, амидиносульфат, 1-амидинопиразол и S-метилизотиомочевину. Реакцию проводят в органическом растворителе, инертном к реакции, выбранном из галогенированных углеводородов, ароматических углеводородов, простых эфиров, спиртов, ДМФА и воды, или без растворителя при температуре от охлаждения до нагревания. При проведении реакции гуанидинобразующий агент может быть использован в эквивалентном количестве или в избытке по отношению к соединению (Id), и в некоторых случаях, для более гладкого проведения реакции, полезно проводить реакцию в присутствии кислоты (уксусной кислоты, трифторуксусной кислоты, хлористоводородной кислоты, серной кислоты или подобных) или основания (пиридина, диметиламинопиридина, триэтиламина, гидроксида натрия, гидроксида калия, метилата натрия, трет-бутилата калия или подобных).
(6) Шестой способ получения
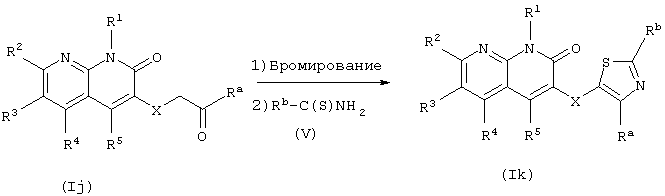
где Ra и Rb могут быть одинаковыми или различными и каждый является Н или группой, представленной R7 или R8 (те же значения применяются ниже).
В данном способе получения производное тиазола (Ik) получают из соединения (Ij) в соответствии с данным изобретением.
Целевое соединение может быть получено взаимодействием с тиоамидом (V) соединения брома, которое получают взаимодействием соединения (Ij) с бромирующим агентом, таким как бром, N-бромсукцинимид или бензилтриметиламмонийтрибромид, после выделения или без выделения. Реакцию проводят в органическом растворителе, инертном к реакции, выбранном из галогенированных углеводородов, ароматических углеводородов, простых эфиров, спиртов, уксусной кислоты, ДМФА и воды, или без растворителя при температуре от охлаждения до нагревания. При проведении реакции соединение (Ij) и бромирующий агент или соединение брома и тиоамид (V) могут быть использованы в эквивалентных количествах или один из них может быть использован в избытке, и в некоторых случаях, для более гладкого проведения реакции, полезно проводить реакцию в присутствии кислоты или основания.
(7) Седьмой способ получения
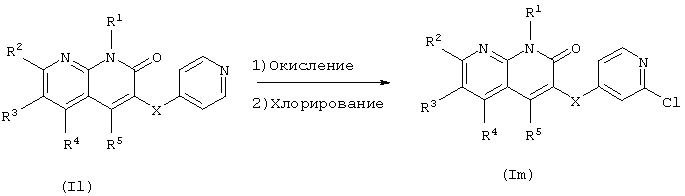
В данном способе получения атом хлора вводят в пиридиновое кольцо соединения (II) по настоящему изобретению.
Целевое соединение может быть получено взаимодействием соединения оксида пиридина, полученного взаимодействием соединения (Il) с окисляющим агентом, таким как м-хлорпербензойная кислота, перуксусная кислота или перекись водорода, с хлорирующим агентом, таким как оксихлорид фосфора, пентахлорид фосфора или тионилхлорид, после выделения или без выделения. Реакцию проводят в органическом растворителе, инертном к реакции, выбранном из галогенированных углеводородов, ароматических углеводородов, простых эфиров, спиртов, уксусной кислоты, ДМФА и воды, или без растворителя при температуре от охлаждения до нагревания. При проведении реакции соединение (Il) и окисляющий агент или соединение оксида пиридина и хлорирующий агент могут быть использованы в эквивалентных количествах или один из них может быть использован в избытке, и в некоторых случаях, для более гладкого проведения реакции, полезно проводить реакцию в присутствии кислоты или основания.
Атом хлора может быть превращен в различные заместители проведением обычного замещения в соединении (Im) по изобретению, полученном согласно данному способу получения, как описано в публикации WO 97/19078 и т.д.
(8) Синтез исходных материалов
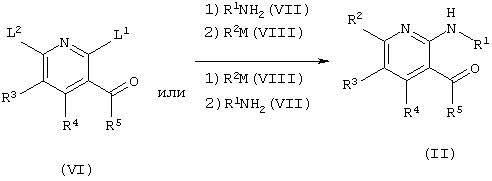
где L2 является уходящей группой, такой же как L1, и М является Н или солью металла (те же значения применяются ниже).
Исходное соединение (II), в котором заместитель R2 и пиридиновое кольцо объединены со связью углерод-углерод, и исходное соединение (VI), имеющее уходящие группы в положении 2 и в положении 6 пиридинового кольца, могут быть синтезированы методом, описанным на страницах 19-21 публикации WO 97/19078.
Исходное соединение (II), в котором заместитель R2 и пиридиновое кольцо не объединены со связью углерод-углерод, может быть синтезировано проведением реакции замещения исходного соединения (VI) с соединением амина (VII), имеющим R1-группу, и нуклеофильным реагентом R2M (VIII) последовательно. Порядок проведения замещения подходит для заместителей R1NH и R2 и уходящих групп (L1 и L2). Реакцию проводят в органическом растворителе, инертном к реакции, выбранном из воды, ароматических углеводородов, простых эфиров и ДМФА, или без растворителя при температуре от охлаждения до нагревания. В некоторых случаях, для более гладкого проведения реакции, полезно проводить реакцию в присутствии органического основания, неорганического основания (предпочтительно, гидроксида натрия или карбоната калия) или основания металла.
Соединение в соответствии с данным изобретением, полученное каждым из представленных выше способов, может быть далее превращено в различные соединения в соответствии с данным изобретением, посредством реакций амидирования, сульфонамидирования, этерификации, гидролиза, алкилирования, восстановления сложного эфира или нуклеофильного замещения. Амидирование, сульфонамидирование и этерификация может проводиться согласно методам, описанным в "JIKKEN KAGAKU KOZA" (4-е издание), изданном Химическим обществом Японии, том 22 (1992) (Maruzen), гидролиз может проводиться согласно методу, описанному в параграфе, посвященном снятию защиты карбоксильной группы, в указанном выше "Protective Groups in Organic Synthesis" (2-е издание), алкилирование может проводиться согласно методу, описанному в "JIKKEN KAGAKU KOZA" (4-е издание), изданном Химическим обществом Японии, том 20 (1992) (Maruzen), и восстановление сложного эфира может проводиться согласно методу, описанному, например, в "JIKKEN KAGAKU KOZA" (4-е издание), изданном Химическим обществом Японии, том 20 (1992) (Maruzen). Нуклеофильное замещение может осуществляться взаимодействием соединения, имеющего алкильную группу, замещенную ОН, с тионилхлоридом или подобными соединениями с получением производного алкилхлорида, или с метансульфонилхлоридом или п-толуолсульфонилхлоридом с получением сложного эфира органического сульфоната, с последующим взаимодействием с нуклеофилом. Альтернативно указанное замещение может быть достигнуто проведением реакции Мицунобу. Реакцию проводят в органическом растворителе, инертном к реакции, выбранном из галогенированных углеводородов, ароматических углеводородов, простых эфиров и ДМФА, или без растворителя при температуре от охлаждения до нагревания. В некоторых случаях для более гладкого проведения реакции полезно проводить реакцию в присутствии основания.
Продукт реакции, полученный в каждом из указанных выше способов получения, выделяют и очищают в виде свободного соединения, соли или различных сольвагов, таких как гидрат. Соль может быть получена проведением обычных реакций получения соли.
Выделение и очистка проводятся с помощью обычно используемых химических методов, таких как экстракция, концентрирование, упаривание, кристаллизация, фильтрация, перекристаллизация и различные виды хроматографии.
Различные изомеры могут быть выделены обычным методом, используя различие физико-химических свойств соответствующих изомеров. Например, оптические изомеры могут быть разделены обычным методом оптического разделения, таким как фракционная кристаллизация или хроматография. Также оптический изомер может быть получен из подходящего оптически активного исходного соединения.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Что касается ингибирования ФДЭ, в настоящее время известно, по крайней мере, пять типов от I до V, и соединение в соответствии с данным изобретением оказывает особенно превосходное действие в отношении ингибирования ФДЭ типа IV и, следовательно, может быть использовано в качестве агента для профилактики и/или лечения респираторных заболеваний (например, бронхиальной астмы (включая неинфекционно-аллергическую бронхиальную астму), хронического бронхита, легочных заболеваний и респираторного дистресс-синдрома у взрослых (РДСВ)), в которых участвует ФДЭ типа IV. В частности, соединение в соответствии с данным изобретением может рассматриваться как агент для профилактики и/или лечения бронхиальной астмы.
Кроме того, соединение в соответствии с данным изобретением также может быть использовано в качестве агента для профилактики и/или лечения других заболеваний, в которых известно участие ФДЭ типа IV, таких как заболевания, в которые вовлечены цитокин (ИЛ-1, ИЛ-4, ИЛ-6 и ФНО (фактор некроза опухоли)) или подобные (например, ревматоидный артрит, язвенный колит, болезнь Крона, сепсис, септический шок, эндотоксический шок, грамотрицательный бактериальный сепсис, синдром токсического шока, нефрит, гепатит, инфекции (бактериальные и вирусные) и недостаточность кровообращения (сердечная недостаточность, атеросклероз, инфаркт миокарда, удар и подобные)). Также, так как соединение в соответствии с данным изобретением хорошо метаболизируется ферментом Р450, метаболизирующим лекарственные средства, находящимся в микросоме печени, и имеет хорошую пероральную абсорбируемость и продолжительность действия, оно может быть использовано в качестве лекарственного средства длительного действия, имеющего хороший фармакокинетический профиль.
Доступность соединения в соответствии с данным изобретением подтверждена следующими тестами.
Экспериментальный пример 1. Ингибирующее действие на ФДЭ типа IV.
1) Раствор, содержащий ФДЭ типа IV, выделяют из мышечной ткани желудочка сердца крысы следующим образом. Сердце, вырезанное у самца крысы Wistar под анестезией эфиром, промывают физиологическим раствором и затем отделяют желудочек. Отделенный желудочек тонко нарезают ножницами и суспендируют в буфере А (20 мМ Бис-Трис, 50 мМ ацетата натрия, 2 мМ ЭДТК, 5 мМ 2-меркаптоэтанола, 2 мМ бензамидена, 0,05 мМ фенилметилсульфонилфторида, рН 6,5), содержащего 1% PROTEASE INHIBITOR COCKTAIL For Mammalian Cell Extracts (Sigraa). Затем клетки разрушают, используя Polytron, и подвергают ультрацентрифугированию (100000 G, 60 минут, 4°С) с получением растворимой фракции.
2) Полученную растворимую фракцию вносят в колонку (2,6×10 см) с Q-сефарозой, уравновешенную буфером А. Затем колонку промывают 1200 мл буфера А для удаления неприсоединенного белка. Белок, присоединенный к колонке, элюируют 750 мл буфера А, содержащего линейный градиент раствора ацетата натрия от 0,05 до 1,00 М, и разделяют на 110 пробирок, содержащих по 7 мл фракции. Исследуют действие ФДЭ, направленное на метаболизацию цАМФ, в каждой фракции, полученной в присутствии или в отсутствие цГМФ и кальция/кальмодулина. Каждую фракцию, которая демонстрирует цАМФ-метаболизирующее действие и подвержена влиянию присутствия цГМФ или кальция/кальмодулина на цАМФ-метаболизирующее действие, используют в качестве исходного раствора для проверки ингибирующего действия в отношении ФДЭ типа IV.
3) Каждое тестируемое соединение в предварительно установленной концентрации подвергают 10-минутному взаимодействию при температуре 30°С в реакционной смеси, содержащей 40 мМ Трис-НСl (рН 8,0), 5 мМ хлорида магния, 4 мМ 2-меркаптоэтанола, 1 мкМ цАМФ, 1 мкКи/мл [3Н]цАМФ и исходный раствор ФДЭ типа IV. Реакцию останавливают добавлением 1/2 объема 20 мг/мл суспензии SPA-шариков силиката иттрия, покрытых полилизином (Amersham), содержащей 18 мМ сульфата цинка и 5 мкМ 3-изобутил-1-метилксантина (ИБМК), к реакционному раствору и измеряют радиоактивность.
Концентрацию тестируемого соединения при ингибировании 50% метаболической активности ФДЭ типа IV определяют как IС50 и рассчитывают для каждого соединения.
С помощью указанного выше теста и метода, описанного в WO 97/19078, таким же образом измеряют ингибирующее действие на ФДЭ типов I, II, III и V.
В результате проведения указанного выше тестирования ингибирующего действия было подтверждено, что соединения примеров 2, 7, 16, 21, 28, 38, 39, 40, 41, 43, 47, 58, 65, 66, 67 и 68 имеют IC50 11 нМ или менее для ФДЭ типа IV, включая соединения, имеющие мощное действие при 0,002 нМ.
Экспериментальный пример 2. Тестирование метаболизма лекарственного средства in vitro с использованием микросомы печени.
1) Суспензию микросомы печени человека и крысы (микросома человека: Xenotech, микросома крысы: Charles River) разбавляют 100 мМ Na-K фосфатным буфером (рН 7,4) до концентрации белка 0,5 мг/мл. К порции 100 мкл полученной суспензии добавляют 2 мкл раствора тестируемого соединения (10 мкг/мл раствор в ацетонитриле), 500 мкл 200 мМ Na-K фосфатного буфера (рН 7,4), 50 мкл 1 мМ ЭДТК-NaOH (рН 7,4) и 200 мкл очищенной воды, получая раствор субстрата (концентрация в реакционном растворе: микросома печени (в качестве белковой составляющей) 0,05 мг/мл, тестируемое соединение 20 нг/мл, 100 мМ Na-K фосфатного буфера, 0,1 мМ ЭДТК-NaOH).
2) Рабочую среду восстановленного НДДФ получают смешиванием 42 мг НАДФ, 5 мл 100 мМ глюкоза-6-фосфатазы (Г6Ф) и 5 мл 100 мМ MgCl2 и добавлением 57 мкл Г6Ф-дегидрогеназы (около 1750 Ед/5 мг/мл) к смеси. Полученную смесь нагревают при температуре 37°С в течение 5 минут и затем охлаждают на льду до использования.
3) 900 мкл порцию раствора субстрата предварительно инкубируют при температуре 37°С в течение 5 минут и затем к ней добавляют 100 мкл рабочей системы восстановленного НАДФ с последующим проведением реакции при температуре 37°С в течение 10, 20 и 30 минут. После прерывания реакции добавлением 2 мл этилацетата всю смесь охлаждают на льду.
Контрольный образец готовят добавлением 100 мкл рабочей системы восстановленного НАДФ после добавления 2 мл этилацетата (время протекания реакции 0 минут).
4) В реакционный раствор добавляют 100 мкл внутреннего стандарта, имеющего предварительно определенную концентрацию (раствор ацетонитрила), 1 мл 0,5 М фосфорной кислоты и 2 мл этилацетата с последующим встряхиванием в течение 10 минут. Через 10 минут центрифугирования при 2500 об/мин слой этилацетата отделяют и выпаривают досуха, а остаток растворяют в 100 мкл растворителя ВЭЖХ с подвижной фазой. Тестируемое соединение элюируют примерно через 12 минут, а внутренний стандарт примерно через 16 минут при следующих условиях. Условия ВЭЖХ с подвижной фазой: ацетонитрил/20 мМ ацетат аммония=2:3 (об./об.), колонка: Discovery RP Amide С16, 4,6×35 мм (SUPELCO), скорость потока 0,8 мл/мин, определение: УФ 286 нм).
5) Рассчитывают отношение (остаточное отношение) высоты пика через 10, 20 или 30 минут реакции к высоте пика каждого тестируемого соединения в контрольной группе (отношение высоты пика к внутреннему стандарту).
В результате проведения указанного выше теста подтвердилось, что соединения примеров 2, 21, 28, 41, 43, 47, 65 и 67 хорошо метаболизируются ферментом Р450, метаболизирующим лекарственные средства, присутствующим в микросоме печени.
Экспериментальный пример 3. Оценка пероральной абсорбируемости и фармакокинетического профиля с использованием ингибирующего действия ФДЭ типа IV в качестве показателя.
Данное исследование проводят для оценки пероральной абсорбируемости и фармакокинетических профилей соединений, ингибирующих ФДЭ типа IV в соответствии с данным изобретением.
1) Каждое тестируемое соединение, суспендированное в очищенной воде, содержащей 0,5% метилцеллюлозы, перорально вводят семинедельным самцам крыс Fisher в дозе 3 мг/кг. В контрольной группе тем же образом вводят растворитель (0,5% метилцеллюлозу в очищенной воде, 3 мл/кг). После перорального введения периодически отбирают образцы крови в присутствии гепарина из хвостовой вены под эфирной анестезией и готовят плазму обычным методом.
2) Плазму, полученную от каждой крысы, которой вводили тестируемое соединение или растворитель, добавляют к измерительной системе ФДЭ типа IV, описанной выше в экспериментальном примере 1, таким образом, чтобы конечная концентрация составляла 0,1%, и измеряют ингибирующее действие в отношении ФДЭ типа IV.
В результате данного теста было обнаружено, что соединения примеров 2, 21, 28, 41, 43, 47, 65 и 67 имеют хорошую пероральную абсорбируемость и метаболическую стабильность по сравнению с соединением сравнения (соединение сравнения: 4-(3-хлорфенил)-1-этил-7-метил-2-оксо- 1,2-дигидро-1,8-нафтиридин).
Экспериментальный пример 4. Ингибирование антиген-индуцированной инфильтрации эозинофилов в дыхательные пути.
В экспериментах по ингибированию антиген-индуцированной инфильтрации эозинофилов в дыхательные пути in vivo, которые служат показателем активности соединений против астмы, использовали модифицированную методику, описанную в работе J.Pharmacol.Exp.Ther., 2000 Dec; 295(3): 1149-55.
У сенсибилизированных самок крыс BN вызывали воспаление дыхательных путей посредством ингаляции 1% аэрозоля альбумина в течение 20 мин. Каждое соединение (в дозе 3 мг/кг) вводили перорально за 1 ч перед началом ингаляции антигена. В качестве нормальной контрольной группы использовали группу особей, подвергшихся ингаляции физиологическим раствором. Величину ингибирования инфильтрации эозинофилов в дыхательные пути для контрольной группы и нормальной контрольной группы принимали за 0 и 100% соответственно и посредством этого оценивали активность испытываемых соединений в качестве ингибиторов.
Полученные результаты для некоторых соединений по изобретению, показывающие их применимость для лечения астмы, приведены ниже.
Соединение по примеру № Ингибирование инфильтрации
эозинофилов (%)
2 90
28 72
43 62
47 85
65 77
67 80
На основе результатов экспериментальных примеров 1-4 можно сказать, что соединения в соответствии с данным изобретением обладают ингибируюшим действием в отношении ФДЭ типа IV и, следовательно, могут быть использованы в качестве агента для профилактики и лечения заболеваний, в которых участвует ФДЭ типа IV.
Фармацевтические композиции, содержащие одно, два или более соединений в соответствии с данным изобретением или их соли в качестве активного ингредиента, получают с использованием носителей, наполнителей и других добавок, которые обычно используют при получении лекарственных средств.
Введение может быть либо пероральным, в виде, например, таблеток, пилюль, капсул, гранул, порошков или жидкостей, либо парентеральным, в виде, например, внутривенных или внутримышечных инъекций, суппозиториев, чрескожных препаративных форм, назальных препаративных форм или ингаляций. Оптимальная доза выбирается в каждом конкретном случае, например, в зависимости от симптомов, возраста и пола каждого пациента, но обычно составляет от 0,001 до 100 мг/кг в день для взрослого пациента при пероральном введении, и данная доза может вводиться один раз в день или быть разделена на 2-4 приема в день. Также, если в соответствии с симптомами осуществляется внутривенное введение, дозу вводят один или несколько раз в день обычно в количестве от 0,001 до 10 мг/кг в день для взрослого пациента. Ингаляции делаются один или несколько раз в день обычно в дозе от 0,0001 до 1 мг/кг в день для взрослого пациента, чрескожные формы наносят один или несколько раз в день в количестве от 0,0001 до 1 мг/кг в день для взрослого пациента.
Твердые композиции для перорального введения в соответствии с данным изобретением используют, например, в виде таблеток, порошков или гранул. В таких твердых композициях одно или несколько активных веществ смешивают, по крайней мере, с одним инертным наполнителем, таким как лактоза, маннит, глюкоза, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза, крахмал, поливинилпирролидон или алюмосиликат магния. Обычно композиция может содержать инертные добавки, включая смазочные агенты, такие как стеарат магния, и разрыхлители, такие как карбоксиметилкрахмал-натрий или солюбилизирующий агент. При необходимости таблетки или пилюли могут быть покрыты пленкой из сахара или соединением, растворимым в желудке или кишечном тракте.
Жидкие композиции для перорального введения содержат, например, фармацевтически приемлемые эмульсии, жидкости, суспензии, сиропы и эликсиры, и содержат обычные инертные растворители, такие как очищенная вода или этанол. В добавление к инертному растворителю данные композиции также могут содержать вспомогательные агенты, такие как солюбилизирующий агент, увлажняющий агент и суспендирующий агент, а также подсластители, вкусовые добавки, ароматизаторы и антисептики.
Инъекции для парентерального введения включают асептические водные или неводные жидкости, суспензии и эмульсии. Примеры водных растворителей включают дистиллированную воду для инъекций и физиологический раствор. Примеры неводных растворителей включают пропиленгликоль, полиэтиленгликоль, растительное масло, такое как оливковое масло, спирт, такой как этанол, и полисорбат 80 (торговая марка). Такие композиции также могут содержать агент, регулирующий тоничность (осмотическое давление), антисептик, увлажняющий агент, эмульгирующий агент, диспергирующий агент, стабилизирующий агент и солюбилизирующий агент. Эти композиции стерилизуют, например, фильтрацией через улавливающий бактерии фильтр, примешиванием антимикробного препарата или облучением. Кроме того, они могут быть в виде первоначальных стерильных твердых композиций, растворяемых в стерильной воде или стерильном растворителе для инъекций непосредственно перед введением.
ОПТИМАЛЬНЫЙ ВАРИАНТ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
Данное изобретение иллюстрируется следующими примерами, которые тем не менее не ограничивают объем данного изобретения. Методы получения исходных соединений показаны в ссылочных примерах. В данном случае 3-(3-хлорбензоил)-2-этиламино-6-диметоксиметилпиридин получают согласно методике ссылочного примера 44 публикации WO 97/19078, а 3-замещенные производные 2-этиламино-6-метилпиридина, такие как 3-(3-хлорбензоил)-2-этиламино-6-метилпиридин и 3-циклогексанкарбонил-2-этиламино-6-метилпиридин получают согласно методикам, описанным в ссылочных примерах 45, 48 и 51 публикации WO 97/19078 соответственно.
В ссылочных примерах и таблицах используются следующие аббревиатуры. Пр.: номер примера, СПр.: номер ссылочного примера, №: номер соединения. Данные: физико-химические данные (МС: масс-спектрометрия с бомбардировкой быстрыми атомами (М+H)+, Т. пл.: температура плавления (°С), разл.: разложение, ЯМР1: δ (м.д.) характеристических пиков 1H-ЯМР в СDСl3, ЯМР2: δ (м.д.) характеристических пиков 1H-ЯМР в ДМСО-d6), соль: соли и содержащее растворитель (окc: оксалат, фум: фумарат, чистая колонка: свободное соединение, цифра перед компонентом, например 1 НСl, означает моногидрохлорид), син.: способ получения (каждая цифра означает номер примера или ссылочного примера с подобным способом получения), Me: метил, Et: этил, сРr: циклопропил, сНех: циклогексил, Ph: фенил. Ас: ацетил, Рy2: пиридин-2-ил и Рy4: пиридин-4-ил. Кроме того, цифра перед каждым заместителем показывает положение замещения, например 2-Сl-Ру4 означает 2-хлорпиридин-4-ил и 3Сl-Рh означает 3-хлорфенил.
Ссылочный пример 1
Раствор 3-(3-хлорбензоил)-2-этиламино-6-метилпиридина в ДМФА обрабатывают 60% гидридом натрия и затем подвергают взаимодействию с моноэтилхлорглутаратом при нагревании. Затем раствор обрабатывают и очищают обычным методом с получением этил-4-{N-[3-(3-хлорбензоил)-6-метилпиридин-2-ил]-N-этил-карбамоил}бутаноата. Полученное соединение подвергают взаимодействию с метилатом натрия в этаноле при нагревании, затем к реакционной смеси добавляют концентрированную серную кислоту с последующим взаимодействием при нагревании в течение 2 дней. Затем реакционную смесь обрабатывают и очищают обычным методом с получением этил-3-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропаноата. МС: 399.
Ссылочный пример 2
По методике ссылочного примера 1 получают этил-[4-(3- хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]ацетат. ЯМР1: 6,91 (1Н, д, J=8,1 Гц), 4,13 (2Н, кв, J=7,1 Гц), 3,43 (2Н, с).
Ссылочный пример 3
По методике ссылочного примера 1 получают метил-4-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]бутаноат. ЯМР1: 6,88 (1Н, д, J=7,8 Гц), 4,67 (2Н, кв, J=7,0 Гц), 2,27 (2Н, т, J=7,6 Гц).
Ссылочный пример 4
После взаимодействия 3-(3-хлорбензоил)-2-этиламино-6-метилпиридина с изохроман-1,3-дионом (75%) при нагревании соединение, полученное после обычной обработки, подвергают взаимодействию с метилиодидом в 2-бутаноне в присутствии карбоната калия. Затем реакционную смесь обрабатывают и очищают обычным методом с получением метил-2-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]бензоата в виде желтого твердого вещества. МС: 433.
Ссылочный пример 5
После обработки 4-цианобутановой кислоты метилатом натрия в метаноле обработанное соединение подвергают взаимодействию с пивалоилхлоридом в ТГФ. Полученное таким образом соединение подвергают взаимодействию с 3-(3-хлорбензоил)-2-этиламино-6-метилпиридином при нагревании. Затем соединение, полученное обычной обработкой, обрабатывают в этаноле в присутствии метилата натрия при нагревании. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 4-(3-хлорфенил)-3-(2-цианоэтил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она в виде бледно-желтого твердого вещества. МС: 352.
Ссылочный пример 6
Соединение, полученное взаимодействием 3'-трифторметилфенилуксусной кислоты с пивалоилхлоридом в ТГФ в присутствии триэтиламина перемешивают при добавлении 3-(3-хлорбензоил)-2-этиламино-6-метилпиридина при температуре 150°С в течение 15 часов. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 4-(3-хлорфенил)-1-этил-7-метил-3-(3-трифторметилфенил)-1,8-нафтиридин-2(1Н)-она в виде бесцветного твердого вещества. МС: 443.
Ссылочный пример 7
По методике ссылочного примера 6 получают 4-(3-хлорфенил)-1-этил-7-метил-3-(4-трифторметилфенил)-1,8-нафтиридин-2(1Н)-он. МС: 443.
Ссылочный пример 8
Этил-2-(2-аминотиазол-4-ил)ацетат подвергают взаимодействию с ацетилхлоридом в дихлорэтане в присутствии триэтиламина и затем реакционную смесь обрабатывают и очищают обычным методом с получением этил-2-(2-ацетиламинотиазол-4-ил)ацетата в виде бесцветного твердого вещества. МС: 229.
Ссылочный пример 9
Этил-2-(2-ацетиламинотиазол-4-ил)ацетат подвергают взаимодействию со смесью этанол -1 М водный раствор гидроксида натрия (1:1) при комнатной температуре, затем реакционную смесь обрабатывают и очищают обычным методом с получением 2-(2-ацетиламинотиазол-4-ил)уксусной кислоты в виде бесцветного твердого вещества. МС: 201.
Ссылочный пример 10
Используя 2-(2-ацетиламинотиазол-4-ил)уксусную кислоту, по методике ссылочного примера 6 получают N-{4-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]тиазол-2-ил}ацетамид. МС: 439.
Ссылочный пример 11
Этилизонипекотат подвергают взаимодействию с бензилбромацетатом в ацетонитриле в присутствии карбоната цезия и затем реакционную смесь обрабатывают и очищают обычным методом с получением этил-1-(бензилоксикарбонилметил)изонипекотата в виде бесцветного маслянистого вещества. МС: 306.
Ссылочный пример 12
Этил-1-(бензилоксикарбонилметил)изонипекотат подвергают каталитическому восстановлению в атмосфере водорода 1 атм в этаноле в присутствии 10% палладия на угле. Полученное соединение обрабатывают метилатом натрия в этаноле и затем подвергают взаимодействию с пивалоилхлоридом в ТГФ.
Полученное соединение подвергают взаимодействию с 3-(3-хлорбензоил)-2-этиламино-6-метилпиридином при нагревании и затем обрабатывают метилатом натрия в этаноле. Затем реакционную смесь обрабатывают и очищают обычным методом с получением этил-1-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пиперидин-4-карбоксилата в виде желтого маслянистого вещества. МС: 454.
Ссылочный пример 13
Раствор диизопропиламина в ТГФ обрабатывают 1,6 М раствором бутиллития/гексана. Затем к смеси по каплям добавляют 2,6-дихлорпиридин вместе с ТГФ и оставляют взаимодействовать, затем по каплям добавляют 3-хлорбензальдегид. Реакционную смесь обрабатывают и очищают обычным методом и полученное соединение подвергают взаимодействию с диоксидом марганца в толуоле при нагревании. Реакционную смесь обрабатывают и очищают обычным методом с получением 3-(3-хлорбензоил)-2,6-дихлорпиридина. МС: 286.
Ссылочный пример 14
К раствору 2,6-дихлор-3-(3-хлорбензоил)пиридина в ТГФ добавляют 70% водный раствор этиламина с последующим взаимодействием при комнатной температуре. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 6-хлор-3-(3-хлорбензоил)-2-этиламинопиридина. ЯМР1: 8,95 (1Н, шир.с), 6,48 (1Н, д, J=7,8 Гц), 1,31 (3Н, т, J=7,1 Гц).
Ссылочный пример 15
Глутаровый ангидрид и 6-хлор-3-(3-хлорбензоил)-2-этиламинопиридин подвергают взаимодействию при нагревании при температуре 150°С и затем обрабатывают и очищают обычным методом. Полученное соединение подвергают взаимодействию с метилиодидом в 2-бутаноне в присутствии карбоната калия при температуре 60°С. Затем реакционную смесь обрабатывают и очищают обычным методом с получением метил-3-[7-хлор-4-(3-хлорфенил)-1-этил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропаноата в виде бесцветного твердого вещества. МС: 405.
Ссылочный пример 16
3-(3-Хлорбензоил)-2-этиламино-6-диметоксиметилпиридин в ДМФА обрабатывают 60% гидридом натрия с последующим взаимодействием с моноэтилхлорглутаратом. Затем реакционную смесь обрабатывают и очищают обычным методом. Полученное соединение подвергают взаимодействию с этанолом в присутствии метилата натрия при нагревании и реакционную смесь обрабатывают и очищают обычным методом. Затем полученное соединение обрабатывают раствором 6 М хлористоводородная кислота - диоксан (1:1) при нагревании, затем реакционную смесь обрабатывают и очищают обычным методом. Полученное соединение далее подвергают взаимодействию с метилиодидом в ДМФА в присутствии карбоната калия, затем реакционную смесь обрабатывают и очищают обычным методом с получением метил-3-[4-(3-хлорфенил)-1-этил-7-формил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропаноата. ЯМР1: 10,11 (1Н, д, J=0,6 Гц), 2,45-2,95 (4Н, м), 1,43 (3Н, т, J=7,1 Гц).
Ссылочный пример 17
15% водный раствор тиометилата натрия по каплям добавляют к раствору 2,6-дихлор-3-(3-хлорбензоил)пиридина в ДМФА при охлаждении на льду. После завершения реакции реакционную смесь обрабатывают и очищают обычным методом. Полученное соединение подвергают взаимодействию с 70% водным раствором этиламина в загерметизированной пробирке при нагревании до температуры 110°С. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 3-(3-хлорбензоил)-2-этиламино-6-метилсульфанилпиридина. ЯМР1: 6,36 (1Н, дд, J=8,2, 0,7 Гц), 2,58 (3Н, с), 1,32 (3Н, т, J=7,1 Гц).
Ссылочный пример 18
К раствору 4-(3-хлорфенил)-1-этил-7-метил-3-(пиридин-4- ил)-1,8-нафтиридина-2(1Н)-она в дихлорметане добавляют м-хлорпербензойную кислоту при комнатной температуре с последующим перемешиванием в течение 5 часов. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 4-(3-хлорфенил)-1-этил-7-метил-3-(1-оксипиридин-4-ил)-1,8-нафтиридина-2(1Н)-она в виде бесцветного твердого вещества. МС: 392.
Ссылочный пример 19
К раствору 3-(3-хлорбензоил)-2-этиламино-6-метилпиридина в ДМФА добавляют хлорацетилхлорид и пиридин с последующим взаимодействием при комнатной температуре. Затем реакционную смесь обрабатывают и очищают обычным методом с получением N-[3-(3-хлорбензоил)-6-метилпиридин-2-ил]-N-этилхлорацетамида. К раствору соединения в ацетонитриле добавляют N-трет-бутоксикарбонилпиперазин и карбонат калия с последующим взаимодействием при нагревании. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 2-[4-трет-бутоксикарбонилпиперазин-1-ил]-N-[3-(3-хлорбензоил)-6-метилпиридин-2-ил]-N-этилацетамида. Полученное соединение подвергают взаимодействию с метилатом натрия в метаноле при нагревании. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 3-(1-трет-бутоксикарбонил-пиперазин-4-ил)-4-(3-хлорфенил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она. ЯМР1: 4,73 (2Н, кв, J=7,3 Гц), 3,15-3,35 (2Н, м), 1,38 (9Н, с).
Ссылочный пример 20
К раствору 4-(3-хлорфенил)-1-этил-3-(3-гидроксипропил)-7-метил-1,8-нафтиридин-2(1Н)-она в ТГФ добавляют триэтиламин и метансульфонилхлорид с последующим взаимодействием при нагревании. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 4-(3-хлорфенил)-1-этил-3-(3-метансульфонилоксипропил)-7-метил-1,8-нафтиридин-2(1Н)-она. ЯМР1: 4,67 (2Н, кв, J=7,1 Гц), 4,18 (2Н, т, J=6,3 Гц), 2,93 (3Н, с).
Ссылочный пример 21
К раствору 4-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]бутановой кислоты в ТГФ добавляют оксалилхлорид и одну каплю ДМФА с последующим перемешиванием при комнатной температуре. Реакционную смесь по каплям добавляют к охлажденному на льду раствору концентрированного водного аммиака в ТГФ и затем смесь перемешивают в течение 30 минут. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 4-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]бутанамида. К раствору полученного соединения в дихлорэтане добавляют пиридин, оксихлорид фосфора и одну каплю ДМФА с последующим перемешиванием при комнатной температуре. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 4-(3-хлорфенил)-3-(3-цианопропил)-1-этил-7-метил-1,8- нафтиридин-2(1Н)-она. ЯМР1: 6,90 (1Н, д, J=8,2 Гц), 4,67 (2Н, кв, J=7,1 Гц), 1,70-2,10 (2Н, м).
Ссылочный пример 22
К суспензии магния в диэтиловом эфире по каплям добавляют 2-бромтиофен при комнатной температуре и полученной смеси дают прореагировать. После охлаждения до температуры 0°С в реакционную смесь добавляют 2-хлор-6-метилникотиновую кислоту и, после нагревания до комнатной температуры смесь перемешивают в течение 12 часов. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 2-хлор-6-метил-3-(тиофен-2-карбонил)пиридина. ЯМР1: 7,79 (1Н, дд, J=5,0; 1,1 Гц), 7,78 (1Н, д, J=7,7 Гц), 2,63 (3Н, с).
Ссылочный пример 23
В загерметизированной пробирке нагревают и перемешивают 2-хлор-6-метил-3-(тиофен-2-карбонил)пиридин и 70% водный раствор этиламина. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 2-этиламино-6-метил-3-(тиофен-2-карбонил)пиридина. ЯМР1: 7,97 (1Н, д, J=8,1 Гц), 7,62 (1Н, дд, J=5,0; 1,1 Гц), 1,28 (3Н, т, J=7,3 Гц).
Ссылочный пример 24
Раствор 3-(3-хлорбензоил)-6-диметоксиметил-2-этиламинопиридина в ДМФА обрабатывают гидридом натрия при температуре 0°С и затем добавляют моноэтилхлорглутарат с последующим нагреванием при температуре 80°С при перемешивании. Затем реакционную смесь обрабатывают и очищают обычным методом. Полученное соединение растворяют в этаноле и в полученный раствор добавляют метилат натрия при температуре 0°С с последующим нагреванием с обратным холодильником в течение 1 часа. Реакционную смесь охлаждают до температуры 0°С и затем добавляют концентрированную серную кислоту с последующим нагреванием с обратным холодильником в течение 1 часа. Затем реакционную смесь обрабатывают обычным методом. Полученное соединение растворяют в диоксане и в полученный раствор добавляют 6 М хлористоводородную кислоту при температуре 0°С, после нагревания до комнатной температуры смесь перемешивают в течение 3 часов. Затем реакционную смесь обрабатывают обычным методом с получением 3-[4-(3-хлорфенил)-1-этил-7-формил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропановой кислоты. ЯМР1: 10,12 (1Н, с), 7,70 (1Н, д, J=8,1 Гц), 1,44 (3Н, т, J=6,9 Гц).
Ссылочный пример 25
К раствору 3-(3-хлорбензоил)-6-диметоксиметил-2-этиламинопиридина в ацетоне добавляют 6 М хлористоводородную кислоту и смесь оставляют взаимодействовать при комнатной температуре в течение 5 часов. После удаления растворителя выпариванием пропиленоксид и хлорацетилхлорид добавляют к продукту, полученному обычным жидкостным разделением в метил-трет-бутиловом эфире, и смесь перемешивают при температуре 60°С в течение 14 часов. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 2-xлop-N-[3-(3-хлорбензоил)-6-формилпиридин-2-ил]-N-этилацетамида в виде желтого маслянистого вещества. ЯМР1: 10,1 (1Н, д, J=0,6 Гц), 7,95 (2Н, шир.с), 1,0-1,5 (3Н, м).
Ссылочный пример 26
К раствору 2-хлор-N-[3-(3-хлорбензоил)-6-формилпиридин- 2-ил]-N-этилацетамида в ацетонитриле добавляют карбонат цезия и этилизонипекотат с последующим перемешиванием при температуре 60°С в течение 3 часов. После удаления неорганического вещества фильтрацией и выпаривания растворителя полученный остаток растворяют в этаноле, добавляют метилат натрия и нагревают с обратным холодильником в течение 15 минут. Затем реакционную смесь обрабатывают и очищают обычным методом с получением этил-1-[4-(3-хлорфенил)-1-этил-7-формил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пиперидин-4-карбоксилата в виде желтого маслянистого вещества. МС: 468.
Ссылочный пример 27
К раствору этил-1-[4-(3-хлорфенил)-1-этил-7-формил-2- оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пиперидин-4-карбоксилата в этаноле добавляют борогидрид натрия при охлаждении на льду и перемешивают в течение 15 минут. Затем реакционную смесь обрабатывают и очищают обычным методом с получением этил-1-[4-(3-хлорфенил)-1-этил-7-гидроксиметил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пиперидин-4-карбоксилата в виде желтого маслянистого вещества. МС: 470.
Ссылочный пример 28
К раствору (1-ацетилпиперидин-4-ил)уксусной кислоты в ТГФ при комнатной температуре добавляют триэтиламин и пивалоилхлорид и перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь фильтруют и концентрируют. Затем к остатку добавляют 3-циклогексанкарбонил-2-этиламино-6-метилпиридин с последующим перемешиванием при температуре 150°С в течение 14 часов. К продукту, полученному обработкой реакционной смеси, добавляют этанол и метилат натрия и полученную смесь нагревают с обратным холодильником в течение 1 часа. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 3-(1-ацетилпиридин-4-ил)-4-циклогексил-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она в виде бесцветного твердого вещества. МС: 396.
Ссылочный пример 29
К раствору 3-(3-бромбензоил)-2-этиламино-6-метилпиридина в дихлорэтане добавляют п-толуолсульфонилхлорид, (1-этоксикарбонилпиперидин-4-ил)уксусную кислоту и 4-диметиламинопиридин с последующим перемешиванием при температуре 80°С в течение 12 часов. К продукту, полученному обработкой реакционной смеси, добавляют этанол и метилат натрия, полученную смесь нагревают с обратным холодильником в течение 1 часа. Затем реакционную смесь обрабатывают и очищают обычным методом с получением 4-(3-бромфенил)-3-(1-этоксикарбонилпиперидин-4-ил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она в виде бледно-коричневого твердого вещества. МС: 498.
Ссылочный пример 30
4-(3-Бромфенил)-3-(2-цианоэтил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-он синтезируют по методике ссылочного примера 5. МС: 396.
Ссылочный пример 31
4-Циклогексил-3-(2-цианоэтил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-он синтезируют по методике ссылочного примера 5. МС: 324.
Ссылочный пример 32
4-(3-Хлорфенил)-1-этил-3-(2-гидроксиэтил)-7-метил-1,8-нафтиридин-2(1Н)-он синтезируют по методике представленного ниже примера 16. ЯМР1: 6,92 (1Н, д, J=8,2 Гц), 4,67 (2Н, кв, J=7,0 Гц), 3,74 (2Н, т, J=5,8 Гц).
Ссылочный пример 33
5-(3-Хлорфенил)-8-этил-6-[3-(морфолин-4-ил)-3-оксопропил]-7-оксо-7,8-дигидро-1,8-нафтиридин-2-карбальдегид синтезируют по методике представленного ниже примера 6. ЯМР1: 7,69 (1Н, д, J=8,1 Гц), 3,51-3,67 (8Н, м), 1,44 (3Н, т, J=7,0 Гц).
Ссылочный пример 34
К 50 мл раствора дихлорэтана, содержащего 7,5 г 3-(3- хлорбензоил)-2-этиламино-6-метилпиридина, добавляют 5 мл моноэтилхлормалоната и 6,5 г 4-диметиламинопиридина. Смесь перемешивают при комнатной температуре в течение 30 минут и затем при нагревании на масляной бане при температуре 80°С в течение 30 минут. Реакционную смесь охлаждают до комнатной температуры и после добавления 1 М хлористоводородной кислоты экстрагируют хлороформом. Органический слой промывают насыщенным раствором соли и затем растворитель выпаривают. Полученный остаток очищают хроматографией на колонке с силикагелем (от гексан-этилацетат до хлороформ-этилацетат) с получением 7,10 г этил-4-(3-хлорфенил)-1-этил- 7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-карбоксилата в виде бесцветных кристаллов.
Пример 2
15 мл 1 М водного раствора гидроксида натрия добавляют к 15 мл раствора ТГФ-метанол (1;1), содержащего 2,70 г этил-3-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропаноата с последующим перемешиванием при нагревании на масляной бане при температуре 80°С в течение 2 часов. После охлаждения до комнатной температуры смесь доводят до рН 3 добавлением 1 М хлористоводородной кислоты и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и затем растворитель выпаривают. Полученный остаток очищают хроматографией на колонке с силикагелем (хлороформ-метанол) и далее перекристаллизовывают из диизопропилового эфира-этилацетата с получением 1,27 г 3-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропановой кислоты в виде бесцветных кристаллов.
Пример 3
Смесь 1,00 г 3-(3-хлорбензоил)-2-этиламино-6-метилпиридина и 5,17 г 2,2-диметилглутарового ангидрида перемешивают при температуре 200°С в течение 1,5 дней. Реакционную смесь охлаждают до комнатной температуры и после добавления 0,5 М хлористоводородной кислоты нагревают с обратным холодильником в течение 2 часов. Затем реакционную смесь охлаждают до комнатной температуры и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и затем растворитель выпаривают. Полученный остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат) и затем перекристаллизовывают из этанола-воды с получением 198 мг 3-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]-2,2-диметилпропановой кислоты в виде оранжевых кристаллов.
Ссылочный пример 35
10 мл полифосфорной кислоты добавляют к 320 мг 4-(3-хлорфенил)-3-циано-1-этил-1,8-нафтиридин-2(1Н)-она с последующим перемешиванием при нагревании на масляной бане при температуре 130°С в течение 2 часов. Реакционную смесь выливают в ледяную воду, доводят до рН 6 добавлением 1 М водного раствора гидроксида натрия и экстрагируют хлороформом. Органический слой промывают насыщенным раствором соли и затем растворитель выпаривают. Полученный остаток очищают хроматографией на колонке с силикагелем (хлороформ-этилацетат) и затем перекристаллизовывают из этанола с получением 180 мг 4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-карбоксамида в виде бесцветных кристаллов.
Пример 5
0,3 мл оксалилхлорида и одну каплю ДМФА добавляют к 20 мл раствора ТТФ, содержащего 1,00 г 3-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропановой кислоты с последующим перемешиванием при комнатной температуре в течение еще 30 минут. Реакционную смесь по каплям добавляют к 10 мл охлажденного на льду раствора ТГФ, содержащего 1,0 мл морфолина, с последующим перемешиванием в течение 30 минут. К реакционной смеси добавляют 1 М хлористоводородную кислоту и смесь экстрагируют этилацетатом. Органический слой промывают водой и насыщенным раствором соли и затем растворитель выпаривают. Полученный остаток очищают хроматографией на колонке с силикагелем (хлороформ-метанол) и затем перекристаллизовывают из диизопропилового эфира-этилацетата с получением 780 мг 4-(3-хлорфенил)-1-этил-7-метил-3-[3-(морфолин-4-ил)-3-оксопропил]-1,8-нафтиридин-2(1Н)-она в виде бесцветных кристаллов.
Пример 6
При охлаждении на льду 630 мг 1-этил-3-(3-диметил- аминопропил)карбодиимида гидрохлорида, 154 мг диметиламина гидрохлорида и 0,53 мл триэтиламина последовательно добавляют к 10 мл раствора ДМФА, содержащего 700 мг 3-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропановой кислоты. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 2 часов. Реакционную смесь разбавляют водой и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и затем растворитель выпаривают. Полученный остаток очищают хроматографией на колонке с силикагелем (хлороформ-этилацетат) и затем перекристаллизовывают из диизопропилового эфира с получением 262 мг 3-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]-N,N-диметилпропанамида в виде бесцветных кристаллов.
Ссылочный пример 38
К 10 мл раствора толуола, содержащего 1,00 г 4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-карбоновой кислоты добавляют 900 мг дифенилфосфорилазида (ДФФА) и 0,5 мл триэтиламина с последующим нагреванием на масляной бане при температуре 100°С в течение 1 часа. Затем в реакционную смесь добавляют 10 мл этанола и смесь перемешивают при нагревании в течение 30 минут. После охлаждения до комнатной температуры реакционную смесь разбавляют водой и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и затем растворитель выпаривают. Полученный остаток очищают хроматографией на колонке с силикагелем (толуол-этилацетат) и затем перекристаллизовывают из этилацетата с получением 590 мг этил-N-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]карбамата в виде бесцветных кристаллов.
Ссылочный пример 39
720 мг этил-N-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо- 1,2-дигидро-1,8-нафтиридин-3-ил]карбамата перемешивают в 40 мл этанола - 3 М водного раствора гидроксида натрия (1:1) при нагревании на масляной бане при температуре 100°С в течение 4 часов. После охлаждения до комнатной температуры реакционную смесь разбавляют водой и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и затем растворитель выпаривают. Полученный остаток очищают хроматографией на колонке с силикагелем (толуол-этилацетат) и затем перекристаллизовывают из диизопропилового эфира-этилацетата с получением 298 мг 3-амино-4-(3-хлорфенил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она в виде бесцветных кристаллов.
Пример 12
К 15 мл раствора дихлорэтана, содержащего 1,38 г 3-(2-аминоэтил)-4-(3-хлорфенил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она, добавляют 720 мг 37% водного раствора формалина и 0,55 мл уксусной кислоты при охлаждении при температуре 0°С с последующим перемешиванием в течение 30 минут. Затем в смесь добавляют 2,05 г триацетоксиборогидрида натрия и смесь нагревают до комнатной температуры, затем перемешивают в течение 2 часов. Реакционную смесь охлаждают до температуры 0°С, доводят до рН 8 добавлением 1 М водного раствора гидроксида натрия и экстрагируют хлороформом. Из органического слоя выпаривают растворитель и остаток очищают хроматографией на колонке с силикагелем (хлороформ-метанол). К полученному маслянистому веществу в 5 мл метанола добавляют 194 мг щавелевой кислоты и затем растворитель выпаривают. Полученные неочищенные кристаллы перекристаллизовывают из ацетонитрила с получением 300 мг 4-(3-хлорфенил)-3-(2-диметиламиноэтил)-1-этил-7-метилнафтиридин-2(1Н)-она монооксалата полугидрата в виде бесцветных кристаллов.
Ссылочный пример 40
К 5 мл раствора ДМФА, содержащего 1,00 г 3-амино-4-(3- хлорфенил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она добавляют 191 мг 60% гидрида натрия с последующим нагреванием до температуры 60°С. К реакционной смеси добавляют 551 мг N-(2-хлорэтил)диметиламина гидрохлорида и 1,08 мл триэтиламина вместе с 5 мл ДМФА и реакционную смесь перемешивают в течение 1 часа. После охлаждения до температуры 0°С реакционную смесь разбавляют 5 мл воды и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и растворитель выпаривают. Остаток очищают хроматографией на колонке с силикагелем (хлороформ-метанол). К полученному маслянистому веществу в 5 мл метанола добавляют 83 мг щавелевой кислоты и затем растворитель выпаривают. Полученные неочищенные кристаллы перекристаллизовывают из метанола с получением 93 мг 4-(3-хлорфенил)-3-(2-диметиламиноэтиламино)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она монооксалата полугидрата в виде бесцветных кристаллов.
Пример 14
К 10 мл раствора дихлорэтана, содержащего 630 мг 3-(3-аминопропил)-4-(3-хлорфенил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она добавляют 243 мг метансульфонилхлорида и 0,30 мл триэтиламина при охлаждении на льду с последующим перемешиванием в течение 1 часа при нагревании до комнатной температуры. Реакционную смесь разбавляют этилацетатом и промывают насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. Из органического слоя выпаривают растворитель, полученный остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат) и затем перекристаллизовывают из этанола-воды с получением 204 мг N-{3-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропил}метансульфонамида в виде бесцветных кристаллов.
Пример 16
Смесь 1,00 г 3-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропановой кислоты, 20 мл метанола и 0,5 мл концентрированной серной кислоты нагревают с обратным холодильником в течение целого дня и ночи. После охлаждения до комнатной температуры в смесь добавляют насыщенный водный раствор бикарбоната натрия и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и затем растворитель выпаривают. Остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат). Полученное соединение растворяют в 20 мл ТГФ и добавляют 500 мг борогидрида натрия. При нагревании с обратным холодильником по каплям добавляют 3 мл метанола и смесь затем продолжают нагревать с обратным холодильником в течение 3 часов. После охлаждения до комнатной температуры добавляют 1 М хлористоводородную кислоту и смесь экстрагируют этилацетатом. Органический слой промывают водой и насыщенным раствором соли, растворитель выпаривают. Остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат) и затем перекристаллизовывают из диизопропилового эфира с получением 497 мг 4-(3-хлорфенил)-1-этил-3-(3-гидроксипропил)-7-метил-1,8-нафтиридин-2(1Н)-она в виде бесцветных кристаллов.
Пример 17
При охлаждении на льду 5,60 мл триэтиламина и 4,90 мл пивалоилхлорида добавляют в 80 мл раствора ТГФ, содержащего 5,08 г 5-кетогексановой кислоты, с последующим перемешиванием при комнатной температуре в течение 1 часа. Реакционную смесь фильтруют и растворитель выпаривают. К остатку, полученному выпариванием растворителя, добавляют 2,00 г 3-(3-хлор-бензоил)-2-этиламино-6-метилпиридина и смесь перемешивают при нагревании при температуре 150°С в течение 2 дней. После охлаждения до комнатной температуры реакционную смесь разбавляют этилацетатом и промывают 1 М водным раствором гидроксида натрия и насыщенным раствором соли. Растворитель органического слоя выпаривают, полученный остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат) и затем перекристаллизовывают из этанола с получением 774 мг 4-(3-хлорфенил)-1-этил-7-метил-3-(3-оксобутил)-1,8-нафтиридин-2(1Н)-она в виде желтых кристаллов.
Пример 18
Смесь 700 мг 4-(3-хлорфенил)-3-(2-цианоэтил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она, 388 мг азида натрия, 411 мг триэтиламина гидрохлорида и 10 мл 1-метилпирролидин-2-она перемешивают при температуре 130°С в течение 20 часов. Реакционную смесь охлаждают до комнатной температуры, подкисляют добавлением 1 М хлористоводородной кислоты и затем экстрагируют этилацетатом. Органический слой промывают водой и насыщенным раствором соли и растворитель выпаривают. Остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат) и затем перекристаллизовывают из этилацетата-диизопропилового эфира с получением 130 мг 4-(3-хлорфенил)-1-этил-7-метил-3-[2-(1Н-тетразол-5-ил)этил]-1,8-нафтиридин-2(1Н)-она в виде бледно-желтых кристаллов.
Пример 19
К 10 мл раствора ТГФ, содержащего 500 мг 4-(3-хлорфенил)-1-этил-3-(3-гидроксипропил)-7-метил-1,8-нафтиридин-2(1Н)-она добавляют 300 мг п-толуолсульфонилхлорида, 0,15 мл триэтиламина и каталитическое количество 4-диметиламинопиридина с последующим нагреванием с обратным холодильником в течение 2 часов. Затем добавляют 300 мг п-толуолсульфонилхлорида, 0,15 мл триэтиламина и каталитическое количество 4-диметиламинопиридина с последующим нагреванием с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры добавляют воду и смесь экстрагируют этилацетатом. Органический слой промывают водой и насыщенным раствором соли и растворитель выпаривают при пониженном давлении. Остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат) и полученное соединение перемешивают с 300 мл имидазола, 250 мг карбоната калия и 10 мл ДМФА при нагревании на масляной бане при температуре 80°С в течение 2 часов. Затем добавляют 300 мг иодида калия с последующим перемешиванием при нагревании на масляной бане при температуре 80°С в течение еще 2 часов. После охлаждения до комнатной температуры реакционную смесь разбавляют водой и затем экстрагируют этилацетатом. Органический слой промывают водой и насыщенным раствором соли и затем растворитель выпаривают. Полученный остаток очищают хроматографией на колонке с силикагелем (хлороформ-метанол-водный аммиак) и затем растворяют в этилацетате. К раствору добавляют 4 М раствор хлороводорода в этилацетате и затем растворитель выпаривают при пониженном давлении. Остаток перекристаллизовывают из ацетонитрила-этилацетата с получением 447 мг 4-(3-хлорфенил)-1-этил-3-[3-(имидазол-1-ил)пропил]-7-метил-1,8-нафтиридин-2(1Н)-она моногидрохлорида 0,2 гидрата в виде бесцветного кристаллического твердого вещества.
Пример 20
1,40 г метил-2-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо- 1,2-дигидро-1,8-нафтиридин-3-ил]бензоата перемешивают в 20 мл метанола и 10 мл 1 М водного раствора гидроксида натрия при температуре 60°С в течение 15 часов. Реакционную смесь охлаждают до комнатной температуры и добавляют 10 мл 1 М хлористоводородной кислоты. Полученные осадки собирают фильтрацией и перекристаллизовывают из этилацетата-диизопропилового эфира с получением 540 мг 2-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]бензойной кислоты 0,6 гидрата в виде бесцветного кристаллического твердого вещества.
Пример 21
Смесь 700 мг 4-(3-хлорфенил)-1-этил-7-метил-3-(3-три- фторметилфенил)-1,8-нафтиридин-2(1Н)-она и 5 мл концентрированной серной кислоты перемешивают при температуре 120°С в течение 2 часов. Реакционную смесь выливают в ледяную воду и экстрагируют этилацетатом. Органический слой промывают водой и насыщенным раствором соли и затем растворитель выпаривают. Полученные неочищенные кристаллы перекристаллизовывают из ацетонитрила с получением 459 мг 3-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]бензойной кислоты в виде бледно-желтых кристаллов.
Пример 22
При охлаждении на льду 897 мг метилата натрия добавляют к 40 мл раствора метанола, содержащего 3,45 г 3-(4-метоксикарбонилфенил)пропановой кислоты с последующим перемешиванием в течение 30 минут. После концентрации реакционной смеси полученный остаток разбавляют 50 мл ТГФ. После охлаждения на льду к смеси добавляют 3,07 мл пивалоилхлорида с последующим перемешиванием при комнатной температуре в течение 1 часа. К остатку, полученному концентрацией реакционной смеси после фильтрации, добавляют 800 мг 3-(3-хлорбензоил)-2-этиламино-6-метилпиридина с последующим перемешиванием при температуре 150°С в течение 14 часов. После охлаждения до комнатной температуры реакционную смесь разбавляют этилацетатом и промывают 1 М водным раствором гидроксида натрия и насыщенным раствором соли. Растворитель выпаривают из органического слоя и к остатку добавляют 50 мл метанола и 900 мг метилата натрия с последующим нагреванием с обратным холодильником в течение 3 часов. Затем добавляют 40 мл 1 М водного раствора гидроксида натрия и смесь перемешивают при температуре 60°С в течение 16 часов. Затем к реакционной смеси добавляют 50 мл 1 М хлористоводородной кислоты. После концентрации при пониженном давлении полученный остаток разбавляют этилацетатом и промывают насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. После выпаривания растворителя полученные неочищенные кристаллы перекристаллизовывают из этанола с получением 764 мг 4-{[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]метил}бензойной кислоты в виде бледно-желтых кристаллов.
Пример 23
К 20 мл раствора диоксана, содержащего 783 мг 3-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]бензойной кислоты, добавляют 0,48 мл ДФФА, 0,31 мл триэтиламина и 0,89 мл трет-бутанола с последующим нагреванием с обратным холодильником в течение 18 часов. После охлаждения до комнатной температуры и концентрации реакционной смеси при пониженном давлении к полученному остатку добавляют этилацетат с последующим промыванием насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. Органический слой концентрируют при пониженном давлении и полученный остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат). К 680 мг из 680 мг полученного соединения в 5 мл этилацетата добавляют 5 мл раствора 4 М хлороводород-этилацетат при охлаждении на льду с последующим перемешиванием при комнатной температуре в течение 2 часов. Реакционную смесь концентрируют и полученные неочищенные кристаллы перекристаллизовывают из этанола-этилацетата с получением 486 мг 3-(3-аминофенил)-4-(3-хлорфенил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она моногидрохлорида в виде бледно-коричневых кристаллов.
Пример 26
К 50 мл раствора метанола, содержащего 3,16 г 4-пиридилуксусной кислоты моногидрохлорида, добавляют 1,97 г метилата натрия при охлаждении на льду и смесь перемешивают в течение 1 часа. Реакционную смесь концентрируют и к остатку добавляют 50 мл ТГФ. При охлаждении на льду к смеси добавляют 2,24 мл пивалоилхлорида. Затем полученную смесь обрабатывают по методике примера 17 и затем обрабатывают по методике получения соли с получением 98 мг 4-(3-хлорфенил)-1-этил-7-метил-3-(пиридин-4-ил)-1,8-нафтиридин-2(1Н)-она моногидрохлорида в виде бледно-желтых кристаллов.
Пример 28
Смешанный раствор, содержащий 400 мг 3-(1-ацетилпиперидин-4-ил)-4-(3-хлорфенил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-он, 5 мл этанола и 5 мл 6 М хлористоводородной кислоты нагревают с обратным холодильником в течение 15 часов. Реакционную смесь концентрируют и после добавления насыщенного водного раствора бикарбоната натрия экстрагируют хлороформом. Органический слой промывают водой и насыщенным раствором соли, затем растворитель выпаривают. Остаток очищают хроматографией на колонке с силикагелем (хлороформ-метанол-водный аммиак) и полученное желтое маслянистое вещество растворяют в 5 мл метанола, к которому последовательно добавляют 1 мл раствора метанола, содержащего 45 мг фумаровой кислоты. Растворитель выпаривают и полученный остаток перекристаллизовывают из этанола-этилацетата с получением 187 мг 4-(3-хлорфенил)-1-этил-7-метил-3-(пиперидин-4-ил)-1,8-нафтиридин-2(1Н)-она монофумарата в виде бледно-желтых кристаллов.
Пример 29
К 20 мл раствора хлороформа, содержащего 510 мг 4-(3- хлорфенил)-1-этил-3-(3-оксобутил)-7-метил-1,8-нафтиридин-2(1Н)-она, по каплям добавляют 1,03 мл брома при охлаждении на льду. После добавления к реакционной смеси добавляют насыщенный водный раствор тиосульфата натрия, смесь экстрагируют хлороформом и органический слой промывают насыщенным раствором соли. Органический слой сушат над безводным сульфатом магния и затем растворитель выпаривают. Полученное соединение растворяют в 10 мл этанола, добавляют 104 мг тиоацетамида и перемешивают при температуре 70°С в течение 2 часов. Растворитель реакционной смеси выпаривают и к полученному остатку добавляют хлороформ. Раствор промывают водой и насыщенным раствором соли и сушат над безводным сульфатом магния. Растворитель органического слоя выпаривают, остаток очищают хроматографией на колонке с силикагелем (этилацетат-гексан) и затем перекристаллизовывают из ацетонитрила с получением 301 мг 4-(3-хлорфенил)-3-[(2,4-диметилтиазол-5-ил)метил]-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она в виде бесцветных кристаллов.
Пример 30
К 670 мг 4-(3-хлорфенил)-1-этил-7-метил-3-(1-оксипиридин-4-ил)-1,6-нафтиридин-2(1Н)-она добавляют 1,6 мл оксихлорида фосфора и 2,4 мл триэтиламина с последующим перемешиванием при температуре 60°С в течение 1 часа. Растворитель выпаривают и к полученному остатку добавляют воду. Смесь экстрагируют этилацетатом, органический слой промывают насыщенным раствором соли и сушат над безводным сульфатом магния. Растворитель выпаривают, полученный остаток очищают хроматографией на колонке с силикагелем (этилацетат-гексан) и затем перекристаллизовывают из этанола-воды с получением 175 мг 4-(3-хлорфенил)-3-(2-хлорпиридин-4-ил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она в виде бесцветных кристаллов.
Пример 32
К 10 мл раствора этилацетата, содержащего 570 мг 3-(1-трет-бутоксикарбонилпиперазин-4-ил)-4-(3-хлорфенил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она, добавляют 10 мл раствора 4 М хлороводород-этилацетат с последующим перемешиванием при комнатной температуре в течение 1 часа. После добавления воды реакционную смесь нейтрализуют 1 М водным раствором гидроксида натрия и затем экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и сушат над безводным сульфатом магния. Растворитель выпаривают, полученный остаток очищают хроматографией на колонке с силикагелем (хлороформ-метанол-водный аммиак) и затем перекристаллизовывают из диизопропилового эфира с получением 115 мг 4-(3-хлорфенил)-1-этил-7-метил-3-(пиперазин-1-ил)-1,8-нафтиридин-2(1Н)-она в виде бледно-желтых кристаллов.
Пример 34
К 10 мл раствора ДМФА, содержащего 500 мг 4-(3-хлорфенил)-1-этил-3-(3-гидроксипропил)-7-метил-1,8-нафтиридин-2(1Н)-она добавляют 500 мг карбоната калия и 0,5 мл метилиодида с последующим перемешиванием при нагревании на масляной бане при температуре 80°С в течение 2 часов. Затем добавляют 1,0 г карбоната калия и 1,0 мл метилиодида с последующим перемешиванием при нагревании на масляной бане при температуре 80°С в течение целого дня и ночи. После охлаждения реакционной смеси до комнатной температуры в нее добавляют воду и экстрагируют этилацетатом. Органический слой промывают водой и насыщенным раствором соли и сушат над безводным сульфатом магния. Растворитель выпаривают, полученный остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат) и затем перекристаллизовывают из гексана-диизопропилового эфира с получением 160 мг 4-(3-хлорфенил)-1-этил-3-[3-(метоксикарбонилокси)пропил]-7-метил-1,8-нафтиридин-2(1Н)-она в виде бесцветных кристаллов.
Пример 35
К 10 мл раствора трет-бутанола, содержащего 500 мг 4-(3-хлорфенил)-1-этил-3-(3-гидроксипропил)-7-метил-1,8-нафтиридин-2(1Н)-она, последовательно добавляют 300 мг трет-бутилата натрия и 0,2 мл метилиодида с последующим перемешиванием при нагревании на масляной бане при температуре 60°С в течение 2 часов. После охлаждения реакционной смеси до комнатной температуры в нее добавляют воду и 1 М хлористоводородную кислоту и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и сушат над безводным сульфатом магния. Растворитель выпаривают, полученный остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат) и затем перекристаллизовывают из диизопропилового эфира с получением 180 мг 4-(3-хлорфенил)-1-этил-3-(3-метоксипропил)-7-метил-1,8-нафтиридин-2(1Н)-она в виде бесцветных кристаллов.
Пример 36
К 60 мл раствора этанола, содержащего 3,54 г этил- (8-метил-8-азабицикло[3.2.1]октан-3-ил)ацетата, добавляют 30 мл 1 М водного раствора гидроксида натрия с последующим перемешиванием при комнатной температуре в течение 14 часов. Затем к реакционной смеси добавляют 30 мл 1 М хлористоводородной кислоты и растворитель выпаривают. К полученному остатку добавляют этанол и после удаления нерастворимого вещества фильтрацией растворитель снова выпаривают с получением 3,55 г неочищенного продукта. 3,50 г соединения растворяют в 40 мл метанола и 837 мг метилата натрия с получением натриевой соли. После выпаривания метанола полученный остаток суспендируют в 40 мл ТГФ и 2,87 мл пивалоилхлорида с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционной смеси добавляют диэтиловый эфир и после удаления нерастворимого вещества фильтрацией растворитель выпаривают. К полученному остатку добавляют 800 мг 3-(3-бромбензоил)-2-этиламино-6-метилпиридина с последующим перемешиванием при температуре 150°С в течение 14 часов. К реакционной смеси добавляют этилацетат и раствор промывают 1 М водным раствором гидроксида натрия, водой и насыщенным раствором соли и затем сушат над безводным сульфатом магния. После выпаривания растворителя к полученному остатку добавляют 50 мл этанола и 837 мг метилата натрия с последующим нагреванием с обратным холодильником в течение 1 часа. После выпаривания растворителя добавляют этилацетат, раствор промывают водой и насыщенным раствором соли и затем сушат над безводным сульфатом магния. Растворитель выпаривают и полученный остаток очищают хроматографией на колонке с силикагелем (хлороформ-метанол-29% водный аммиак) с получением 150 мг продукта. Соединение обрабатывают 4 М хлороводородом с получением гидрохлорида, который затем перекристаллизовывают из этанола-этилацетата с получением 69 мг 4-(3-хлорфенил)-1-этил-7-метил-3-(8-метил-8-азабицикло[3.2.1]октан-3-ил)-1,8-нафтиридин-2(1Н)-она моногидрохлорида в виде бесцветных кристаллов.
Пример 37
К 10 мл раствора ТГФ, содержащего 700 мг 4-(3-хлорфенил)-1-этил-3-(3-метансульфонилоксипропил)-7-метил-1,8-нафтиридин-2(1Н)-она, добавляют 0,5 мл пирролидина и 300 мг иодида калия с последующим перемешиванием при нагревании на масляной бане при температуре 60°С в течение 1,5 часов. После охлаждения до комнатной температуры к смеси добавляют воду и 1 М хлористоводородную кислоту и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и сушат над безводным сульфатом магния. Затем растворитель выпаривают и остаток очищают хроматографией на колонке с силикагелем (хлороформ-метанол-водный аммиак). Полученное соединение растворяют в этилацетате и добавляют 4 М раствор хлороводород-этилацетат. Затем остаток, полученный выпариванием растворителя, перекристаллизовывают из этилацетата-ацетонитрила с получением 133 мг 4-(3-хлорфенил)-1-этил-7-метил-3-[3-(пирролидин-1-ил)пропил]-1,8-нафтиридин-2(1Н)-она моногидрохлорида в виде бесцветных кристаллов.
Пример 38
К смеси 2,00 г 4-(3-хлорфенил)-1-этил-3-(2-гидроксиэтил)-7-метил-1,8-нафтиридин-2(1Н)-она, 1,0 мл триэтиламина и 20 мл ТГФ по каплям добавляют 0,5 мл метансульфонилхлорида с последующим перемешиванием при комнатной температуре в течение 30 минут. К реакционной смеси добавляют воду и смесь экстрагируют этилацетатом. Органический слой последовательно промывают 1 М хлористоводородной кислотой, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и затем сушат над безводным сульфатом магния. К 20 мл раствора остатка, полученного выпариванием растворителя, в ТГФ добавляют 1,0 мл триэтиламина, 1,0 мл этилизонипекотата и 500 мг иодида калия с последующим перемешиванием при нагревании на масляной бане при температуре 60°С в течение целого для и ночи. После охлаждения реакционной смеси до комнатной температуры в нее добавляют воду и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и сушат над безводным сульфатом магния. После выпаривания растворителя полученный остаток очищают хроматографией на колонке с силикагелем (хлороформ-метанол) с получением 1,70 г соединения сложного эфира. Затем по методике примера 2 получают 593 мг 1-{2-[4-(3-хлорфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]этил}пиперидин-4-карбоновой кислоты в виде бесцветных кристаллов.
Пример 39
К 15 мл раствора 1,4-диоксана, содержащего 314 мг 3-(2-аминоэтил)-4-(3-хлорфенил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она добавляют 404 мг 1-амидинопиразола моногидрохлорида и 0,48 мл диизопропилэтиламина с последующим перемешиванием в течение 122 часов. Реакционный раствор упаривают, полученное твердое вещество удаляют фильтрацией и твердое вещество далее промывают хлороформом. Объединенный фильтрат и промывки концентрируют и остаток очищают хроматографией на колонке с силикагелем (хлороформ-метанол-водный аммиак). Полученное маслянистое вещество растворяют в 5 мл этанола и туда последовательно добавляют 4 М хлороводород-этилацетат и ацетонитрил. Растворитель выпаривают с получением 347 мг 4-(3-хлорфенил)-1-этил-3-(2-гуанидиноэтил)-7-метил-1,8-нафтиридин-2(1Н)-она моногидрохлорида моногидрата в виде бесцветного твердого вещества.
Пример 40
Хлороводород барботируют в 20 мл раствора этанол-хлороформ (1:1), содержащего 461 мг 4-(3-хлорфенил)-3-(2- цианоэтил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она, при температуре -78°С в течение 30 минут, раствор перемешивают при температуре 5°С в течение 18 часов и затем реакционный раствор упаривают. К полученному твердому веществу добавляют 15 мл этанола и 505 мг ацетата аммония с последующим перемешиванием в течение 90 часов. Реакционный раствор упаривают и остаток очищают хроматографией на колонке с силикагелем (хлороформ-метанол-водный аммиак). 0,4 мл 4 М хлороводорода-этилацетата добавляют к полученному маслянистому веществу в 5 мл этанола. После выпаривания растворителя полученный остаток перекристаллизовывают из этанола-этилацетата с получением 268 мг 3-(2-амидиноэтил)-4-(3-хлорфенил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она моногидрохлорида в виде бесцветных кристаллов.
Пример 42
К 7 мл раствора ТГФ-этанол (5:2), содержащего 300 мг метил-3-[4-(3-хлорфенил)-1-этил-7-формил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропионата, добавляют 9 мг борогидрида натрия при охлаждении на льду. После перемешивания в течение 1 часа к реакционной смеси добавляют воду и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и затем растворитель выпаривают. Остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат). Используя 200 мг из 210 мг полученного соединения, соединение обрабатывают по методике примера 2 с получением 130 мг 3-[4-(3-хлорфенил)-1-этил-7-гидроксиметил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропановой кислоты в виде бледно-желтых кристаллов.
Пример 43
К 33 мл раствора метанол-пиридин (10:1), содержащего 1,50 г метил-3-[4-(3-хлорфенил)-1-этил-7-формил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропаноата, добавляют 300 мг гидроксиламина гидрохлорида при охлаждении на льду. После перемешивания в течение 1 часа реакционную смесь разбавляют водой и экстрагируют этилацетатом. Органический слой промывают водой и насыщенным раствором соли и затем растворитель упаривают. Полученный остаток обрабатывают по методике примера 2 с получением 72 мг 3-[4-(3-хлорфенил)-1-этил-7-гидроксииминометил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропановой кислоты в виде бесцветных кристаллов.
Пример 44
В загерметизированной пробирке смесь 4,4 г 6-хлор-3-(3-хлорбензоил)-2-этиламинопиридина, 5 мл ТГФ и 10 мл 40% водного раствора метиламина перемешивают при нагревании на масляной бане при температуре 100°С в течение 2 часов. После охлаждения до комнатной температуры добавляют хлороформ. Органический слой промывают водой и насыщенным раствором соли и затем растворитель упаривают. К остатку добавляют 2 мл бензоилхлорида, 2 г 4-диметиламинопиридина и 100 мл дихлорэтана с последующим нагреванием на масляной бане при температуре 80°С в течение 2 часов. Затем добавляют 1 мл бензоилхлорида и 1 г 4-диметиламинопиридина и смесь перемешивают при нагревании на масляной бане при температуре 100°С в течение 1 часа. После охлаждения до комнатной температуры к смеси добавляют воду и экстрагируют хлороформом. Органический слой промывают водой и насыщенным раствором соли и затем растворитель выпаривают. Остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат). К 50 мл раствора полученного соединения в ДМФА добавляют 700 мг 60% гидрида натрия при охлаждении на льду с последующим перемешиванием в течение 30 минут. Затем туда добавляют 2,7 мл моноэтилхлорглутарата с последующим перемешиванием при нагревании на масляной бане при температуре 80°С в течение 1 часа. Затем к смеси добавляют 2,7 мл моноэтилхлорглутарата с последующим перемешиванием при нагревании на масляной бане при температуре 80°С в течение еще 1 часа. После охлаждения до комнатной температуры смесь разбавляют водой и экстрагируют этилацетатом. Органический слой промывают водой и насыщенным раствором соли и затем растворитель выпаривают. Остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат). Полученное соединение нагревают с обратным холодильником вместе с 1,5 г метилата натрия и 50 мл этанола в течение 1 часа. После охлаждения до комнатной температуры к реакционной смеси добавляют 2 мл концентрированной серной кислоты и смесь нагревают с обратным холодильником в течение целого дня и ночи. Реакционную смесь охлаждают до комнатной температуры и после добавления насыщенного водного раствора бикарбоната натрия экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и затем растворитель выпаривают. Остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат). Полученное соединение перемешивают с 10 мл ТГФ-метанола (1:1) и 20 мл 1 М водного раствора гидроксида натрия при нагревании на масляной бане при температуре 80°С в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь доводят до рН 2 добавлением 1 М хлористоводородной кислоты и затем экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и затем растворитель выпаривают. Полученный остаток перекристаллизовывают из ацетонитрила с получением 1,43 г 3-[4-(3-хлорфенил)-1-этил-7-метиламино-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропановой кислоты в виде бледно-желтых кристаллов.
Пример 46
Смесь 1,2 г 3-(3-бромбензоил)-2-этиламино-6-метилпиридина и 2,1 г глутарового ангидрида перемешивают при нагревании при температуре 150°С в течение 15 часов. После охлаждения до комнатной температуры добавляют 10 мл 1 М хлористоводородной кислоты и нагревают с обратным холодильником в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь разбавляют водой и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и затем растворитель выпаривают. К остатку добавляют 50 мл этанола и 0,5 мл концентрированной серной кислоты с последующим нагреванием с обратным холодильником в течение 1 часа. Реакционную смесь охлаждают до комнатной температуры, разбавляют водой и экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и затем растворитель выпаривают. Остаток очищают хроматографией на колонке с силикагелем (хлороформ). Затем соединение обрабатывают по методике примера 2 с получением 1,00 г 3-[4-(3-бромфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пропановой кислоты в виде бледно-желтых кристаллов.
Пример 47
К 20 мл раствора ТГФ, содержащего 2,34 г 3-циклогексанкарбонил-2-этиламино-6-метилпиридина, добавляют 3 мл моноэтилхлорглутарата и 2,8 мл 2,6-лутидина с последующим перемешиванием при нагревании на масляной бане при температуре 60°С в течение 1 часа. После охлаждения реакционной смеси до комнатной температуры в нее добавляют воду и экстрагируют этилацетатом. Органический слой промывают 3 М хлористоводородной кислотой, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли и сушат над безводным сульфатом магния. Растворитель выпаривают и 1,00 г метилата натрия добавляют к 20 мл раствора полученного остатка в этаноле с последующим нагреванием с обратным холодильником в течение 1 часа. Затем к реакционному раствору добавляют 20 мл 1 М водного раствора гидроксида натрия с последующим нагреванием с обратным холодильником в течение еще 1 часа. После охлаждения смеси до комнатной температуры в нее добавляют воду, смесь нейтрализуют 1 М хлористоводородной кислотой и затем экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и сушат над безводным сульфатом магния. Растворитель выпаривают и остаток перекристаллизовывают из этанола-воды с получением 764 мг 3-(4-циклогексил-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил)пропионовой кислоты в виде бесцветных кристаллов.
Пример 52
К 370 мг этил-{4-[4-(3-бромфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пиперидин-1-ил}ацетата добавляют 5 мл 6 М хлористоводородной кислоты с последующим перемешиванием при температуре 100°С в течение 15 часов. После удаления растворителя полученные неочищенные кристаллы перекристаллизовывают из этанола-ацетонитрила с получением 185 мг {4-[4-(3-бромфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пиперидин-1-ил}уксусной кислоты моногидрохлорида в виде бесцветных кристаллов.
Пример 55
К 1,28 г 4-(3-бромфенил)-3-(1-этоксикарбонилпиперидин-4- ил)-1-этил-7-метил-1,8-нафтиридин-2(1Н)-она добавляют 20 мл концентрированной хлористоводородной кислоты с последующим перемешиванием при температуре 100°С в течение 3 часов. Растворитель выпаривают и остаток подщелачивают добавлением концентрированного водного аммиака, смесь экстрагируют этилацетатом. Органический слой промывают насыщенным раствором соли и затем сушат над безводным сульфатом натрия. После удаления растворителя из органического слоя полученные неочищенные кристаллы перекристаллизовывают из ацетонитрила и затем перекристаллизовывают из этанола-воды с получением 357 мг 4-(3-бромфенил)-1-этил-7-метил-3-(пиперидин-4-ил)-1,8-нафтиридин-2(1Н)-она в виде бесцветных кристаллов.
Пример 56
К 30 мл раствора метанола, содержащего 2,56 г имидазо[1,2-а]пиридин-3-илуксусной кислоты, добавляют 783 мг метилата натрия с последующим перемешиванием при комнатной температуре в течение 30 минут. Растворитель выпаривают и к полученному остатку добавляют 15 мл N-метилпирролидона и 2,15 мл пивалоилхлорида с последующим перемешиванием при комнатной температуре в течение 2 часов. Затем к смеси добавляют 900 мг 3-(3-бромбензоил)-2-этиламино-6-метилпиридина и смесь перемешивают при температуре 150°С в течение 16 часов. К реакционному раствору добавляют воду и смесь экстрагируют этилацетатом. Органический слой промывают водой и насыщенным раствором соли и сушат над безводным сульфатом магния. После удаления растворителя остаток очищают хроматографией на колонке с силикагелем (хлороформ-метанол) и затем перекристаллизовывают из ацетонитрила с получением 240 мг 4-(3-бромфенил)-1-этил-3-(имидазо[1,2-а]пиридин-3-ил)-7-метил-1,8-нафтиридин-2(1Н)-она в виде желтых кристаллов.
Пример 57
К 5 мл раствора ацетонитрила, содержащего 400 мг 4-(3-бромфенил)-1-этил-7-метил-3-(пиперидин-4-ил)-1,8-нафтиридин-2(1Н)-она добавляют 0,11 мл этилбромацетата и 0,13 мл триэтиламина с последующим перемешиванием при комнатной температуре в течение 13 часов. К реакционному раствору добавляют этилацетат, раствор промывают водным раствором бикарбоната натрия и насыщенным раствором соли и сушат над безводным сульфатом магния. После удаления из органического слоя растворителя полученный остаток очищают хроматографией на колонке с силикагелем (гексан-этилацетат) и затем перекристаллизовывают из этилацетата-диизопропилового эфира с получением 120 мг этил-{4-[4-(3-бромфенил)-1-этил-7-метил-2-оксо-1,2-дигидро-1,8-нафтиридин-3-ил]пиперидин-1-ил}ацетата в виде бесцветных кристаллов.
По методике указанных выше примеров или ссылочных примеров получают соединения по примерам и ссылочным примерам, указанные в таблицах 1-4 соответственно. Структуры и физико-химические данные соединений ссылочных примеров 34 и 45 и примеров 1-69 показаны в таблицах 1-4.
Кроме того, структуры других соединений данного изобретения показаны в таблицах 5 и 6. Они могут быть легко синтезированы по указанным выше способам получения, методикам, описанным в примерах, и методикам, очевидным для специалистов в данной области техники, или по модифицированным методикам.
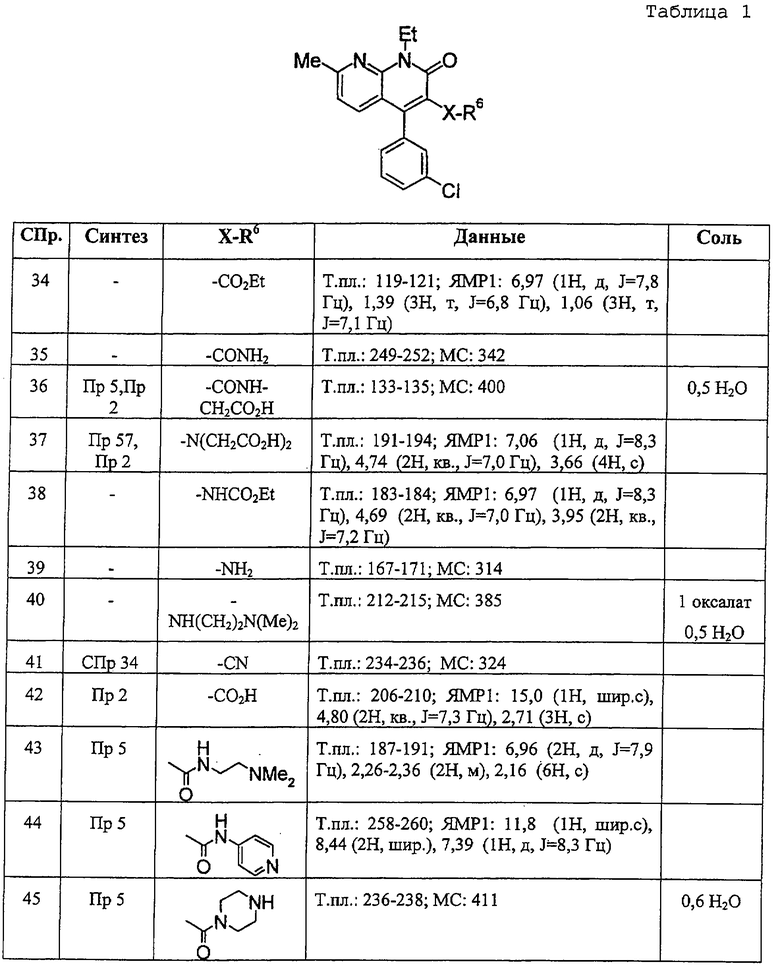
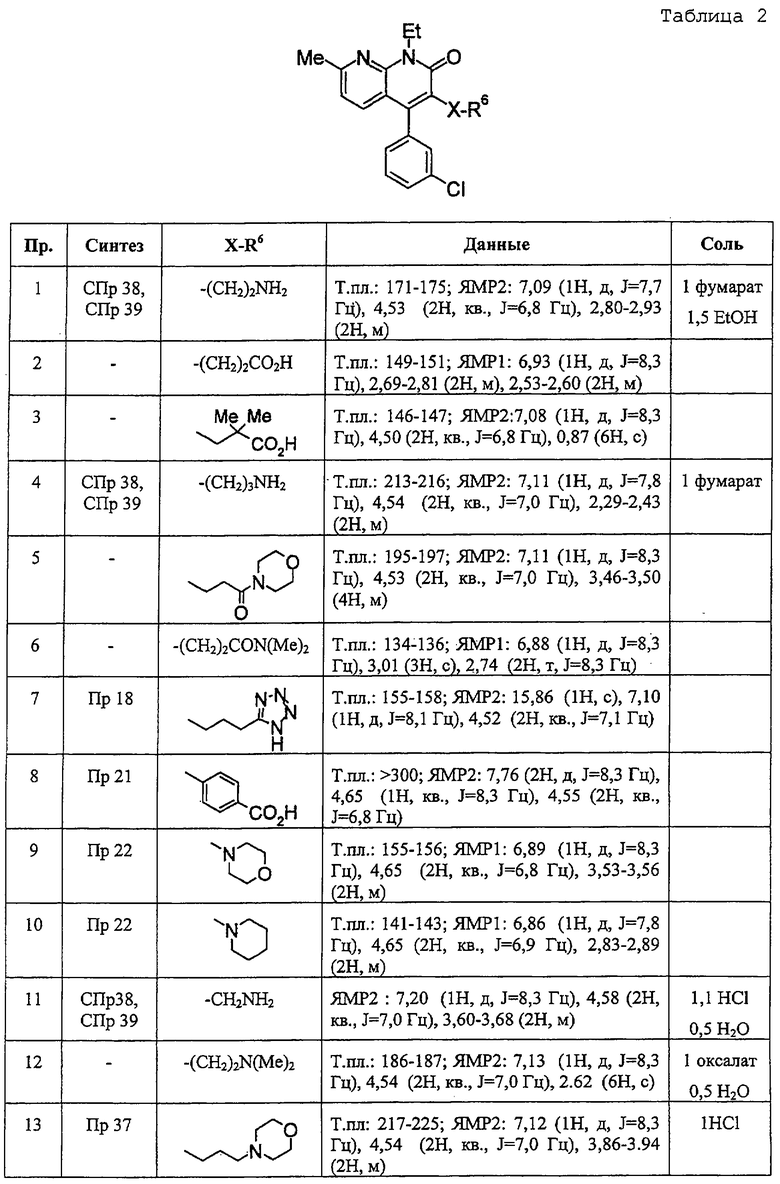
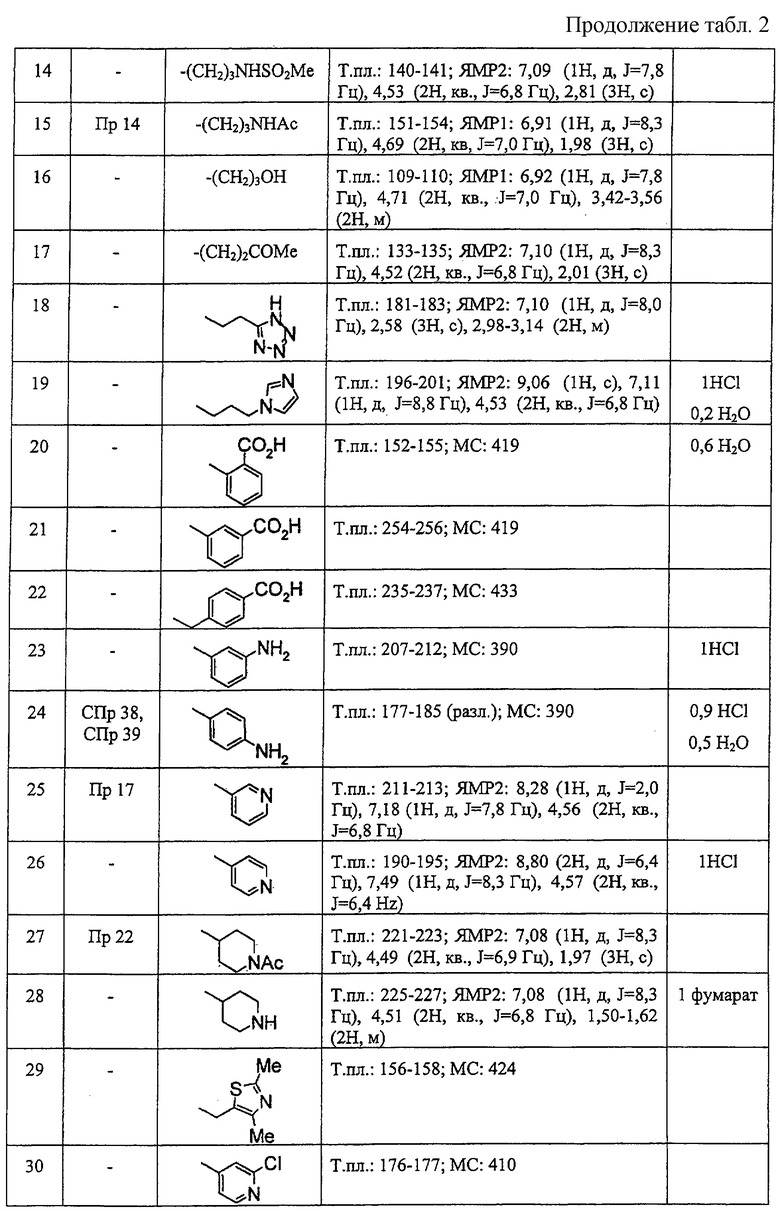
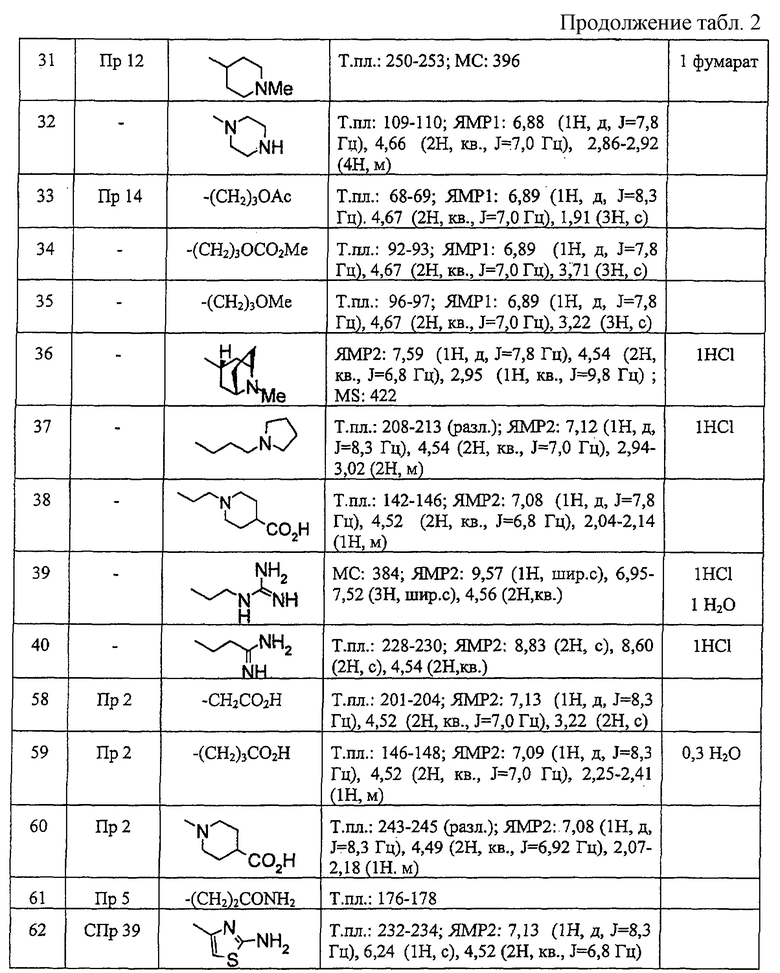
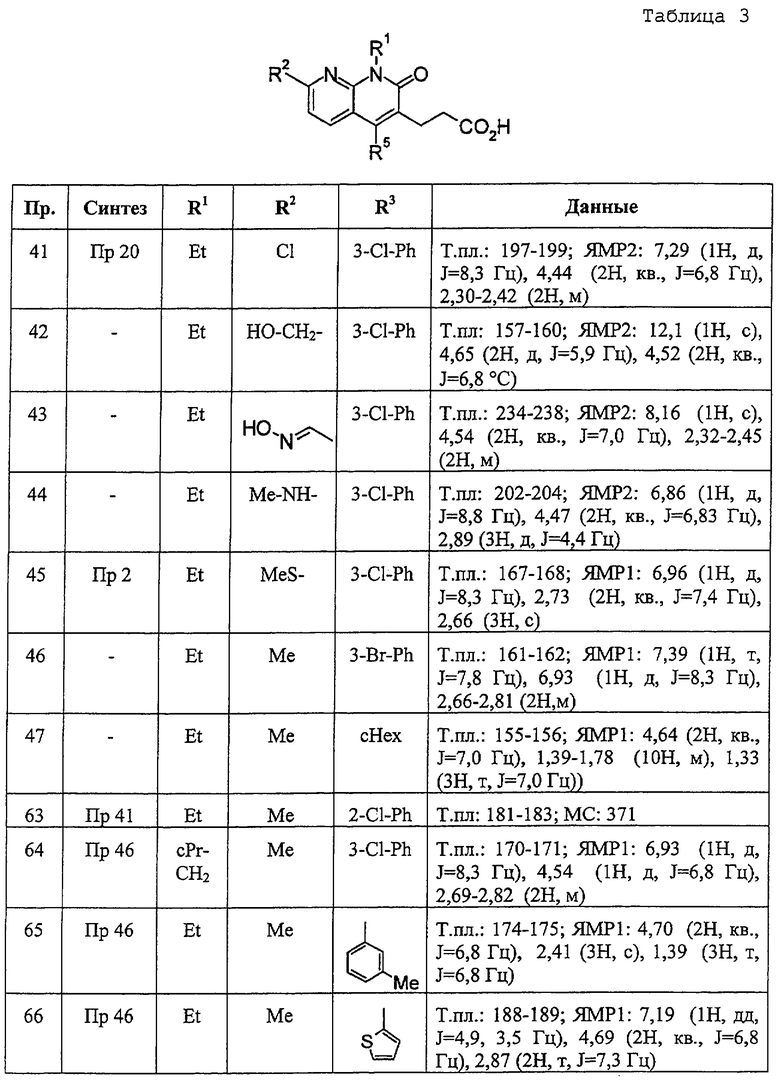
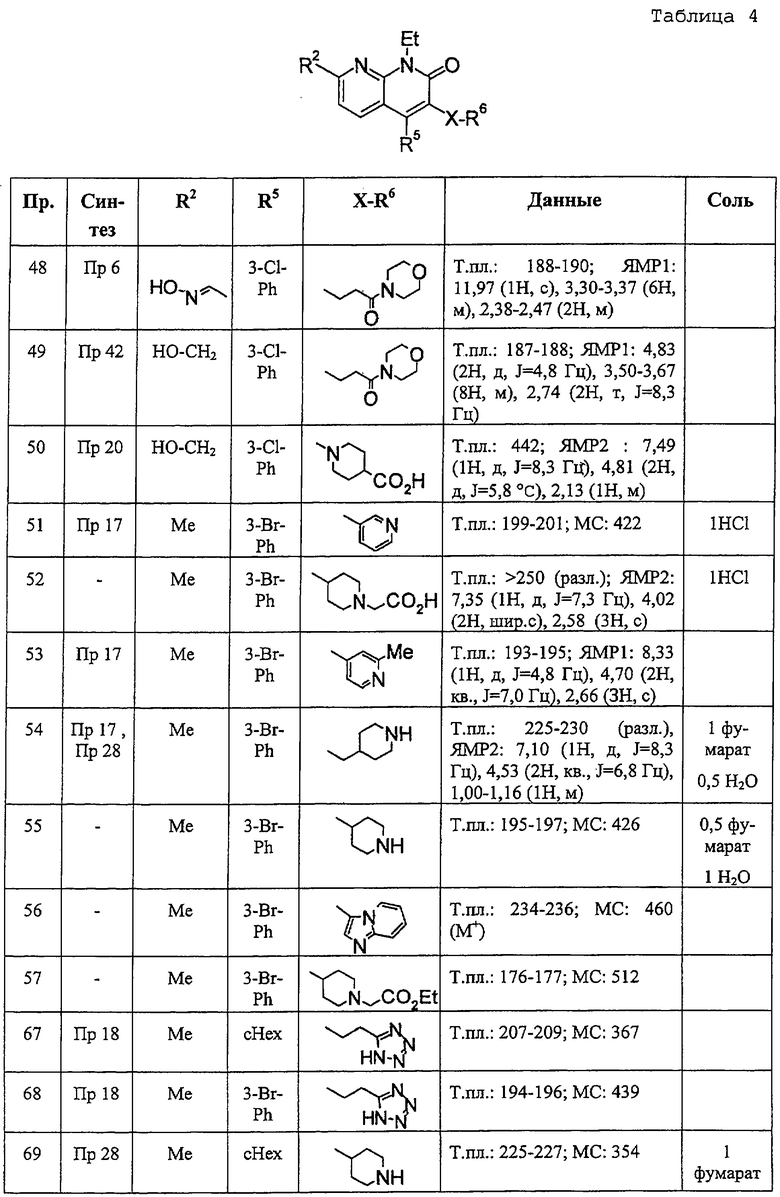
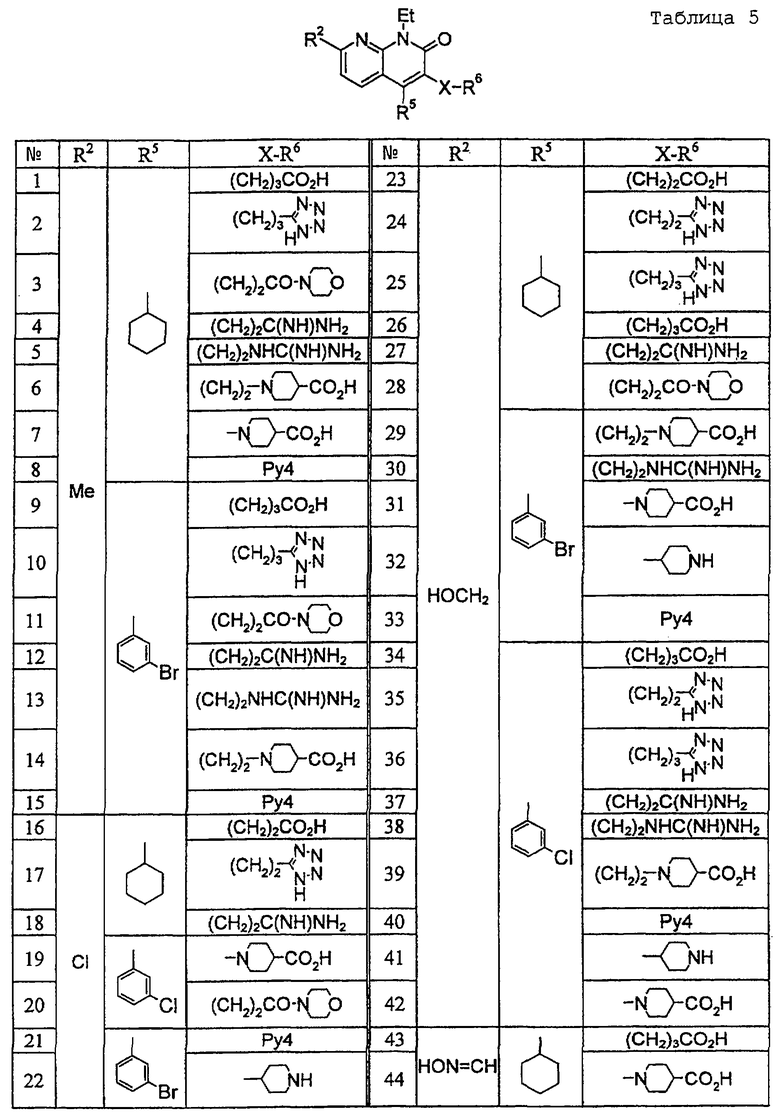
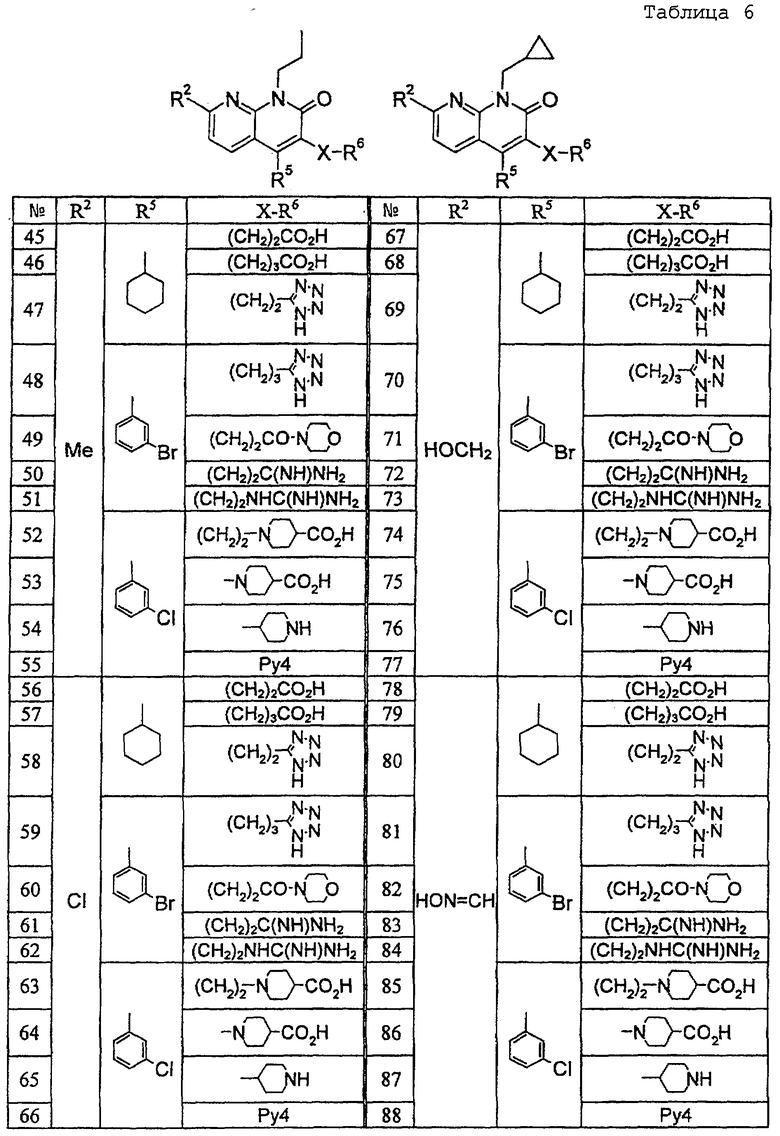
Описываются производные нафтиридина общей формулы (I) или их фармацевтически приемлемые соли формулы
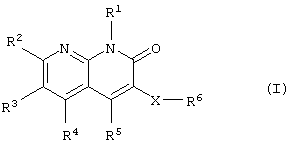
где R1: -R0 или низший алкиленциклоалкил; R0: низший алкил; R2: -R0, галоген, -низший алкилен-ОН, -S-R0, -NHR0 или -CH=N-OR9, которые могут быть одинаковыми или отличаться друг от друга; R3 и R4: -H, R9: -H;
R5: циклогексил; тиенил или фенил, который может быть замещен группой, выбранной из низшего алкила и галогена;
R6: -ОН, -OR7, -COOH, -CONH2, -CON(R7)2, -O-COR7, -O-COOR7, -COR7, -NH2, -N(R7)2, -N(R7)COR7, -N(R7)SO2R7, -C(NH)NH2, -NHC(NH)NH2 или группа формулы -Y-R8;
R7: низший алкил, который может быть замещен группой -CO2R0;
R8: фенил, который может быть замещен группой, выбранной из R10, или гетероциклическая группа, выбранная из морфолинила, тетразолила, имидазолила, пиридила, пиперидила, тиазолила, пиперазила, пирролидила или имидазо[1,2-а]пиридила, которая может быть замещена группой, выбранной из R10;
R10: -галоген, -CO2Н, -NH2 или -COR0, или группа, описанная как R7; Y: связь или -CO- и X: связь или низший алкилен. Данные соединения могут быть использованы в качестве профилактических или лечебных агентов для респираторных заболеваний, связанных с ингибированием ФДЭ типа IV. 2 с. и 5 з.п. ф-лы, 6 табл.
где R1 - R0 или -низший алкилен-циклоалкил;
R0 - низший алкил;
R2 - R0, -галоген, -низший алкилен-ОН, -S-R0, -NHR0 или -CH=N-OR9, которые могут быть одинаковыми или отличаться друг от друга;
R3 и R4 - Н
R9 - Н;
R5 - циклогексил, тиенил или фенил, который может быть замещен группой, выбранной из низшего алкила и галогена;
R6 - ОН, -OR7, -СООН, -CONH2, -CON(R7)2, -O-COR7, -O-COOR7, -COR7, -NH2, -N(R7)2, -N(R7)COR7, -N(R7)SO2R7, -C(NH)NH2, -NHC(NH)NH2 или группа формулы -Y-R8;
R7 - низший алкил, который может быть замещен группой -CO2R0;
R8 - фенил, который может быть замещен группой, выбранной из R10, или гетероциклическая группа, выбранная из морфолинила, тетразолила, имидазолила, пиридила, пиперидила, тиазолила, пиперазила, пирролидила или имидазо[1,2-а]пиридила, которая может быть замещена группой, выбранной из R10;
R10 - галоген, -СO2Н, -NH2 или -COR0, или группа, описанная как R7;
Y - связь или -СО-;
X - связь или низший алкилен.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

