Данное изобретение относится к способам получения хиральных 1,4-дизамещенных пиперазинов и промежуточным соединениям, пригодным для этих способов.
Пиперазины формулы А
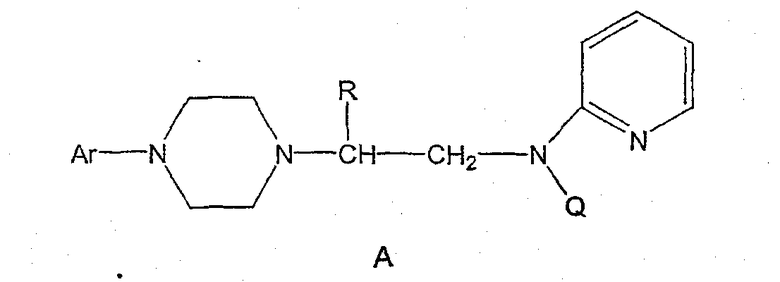
где R представляет собой низший алкил, Ar представляет собой незамещенный или замещенный арил или гетероарил, Q представляет собой водород, СО-(низший)алкил, СО-циклоалкил или СО-арил, являются высокоэффективными средствами, связывающими 5НТ1А-рецептор. В патенте США № 6127357 описываются такие производные пиперазина, которые пригодны для лечения расстройств центральной нервной системы (CNS). Производные пиперазина формулы А содержат асимметричный атом углерода, поэтому они могут находиться в двух оптически активных формах. В настоящее время хорошо известно, что энантиомеры связываются с рецепторами с разной эффективностью и селективностью, они могут иметь разный метаболический путь и вызывать разные побочные эффекты. В заявке WO 9703982 указывается, что предпочтительные энантиомеры пиперазинов формулы А проявляют улучшенное сродство к связыванию 5НТ1А-рецепторов и биологическую доступность. Поэтому существует потребность в эффективном, простом, дешевом и безопасном способе получения оптически предпочтительных пиперазинов.
В заявке WO 9533725 описывается способ синтеза некоторых хиральных пиперазинов формулы А алкилированием соответствующего 1-арилпиперазина энантиомерно чистым 2-(5-метил-2,2-диоксидо-1,2,3-оксатиазолидин-3-ил)пиридином.
Одним из традиционных подходов к получению 1,4-дизамещенных пиперазинов является бис-алкилирование первичных аминов бис(2-хлорэтил)аминами, так называемыми азотистыми ипритами. Несколько оптически активных пиперазинов, структурно отличающихся от соединений формулы А, были получены конденсацией N-замещенного бис(2-хлорэтил)амина с выбранным хиральным амином в соответствии с методиками Natsuka et al., J. Med. Chem. 1987, 1779 и WO 9424115, а также с природной аминокислотой в соответствии с методиками Acta Pol. Pharm. 1999, 56, p. 41; CA 131:157745. Однако сохраняется потребность в способе получения синтетически полезных молекул азотистых ипритов. В публикации Chem.&Pharm.Bulletin Japan 1954, 275 описывается превращение бис(2-хлорэтил)амина в N-бис(2-хлорэтил)аминоацетонитрил, в публикации Chem.&Pharm. Bulletin Japan 1957, 487 описывается безуспешная попытка разделить соответствующий рацемический N-бис(2-хлорэтил)аланин, и сложное разделение 2-[N-бис(2-хлорэтил)амино]пропанамида.
Полифункциональные хиральные амины могут быть получены несколькими многостадийными способами, но прямое замещение реакционноспособной функциональной группы обычно приводит к получению рацемических аминов.
В публикации Effenberger et al., Angew. Chem. 1983, 95 [1], 50 сообщалось, что трифлаты реагируют с простыми вторичными аминами в условиях инверсии Вальдена (Walden). Данный способ применим к синтезу сложных эфиров как (R)-, так и (S)-α-аминокислот. Он позволяет получать асимметричную С(α),N-связь с высокой степенью стереоселективности посредством единственной реакции, и иногда использовался с очень незначительными видоизменениями (Quadri et al., Biorg.&Med.Chem.Letters 2, 1661, 1992; Taylor et al., Tetrahedron Letters 37, 1297, 1996). В публикации Hoffman and Hwa-Ok Kim, Tetrahedron Letters 31, 2953, 1990 описана замена трифлатов сложными (4-нитробензол)сульфонилоксиэфирами в реакции с гидразинами.
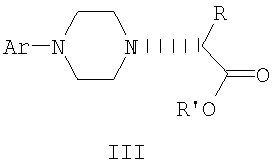
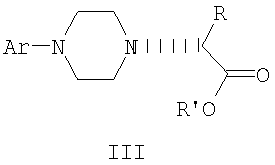
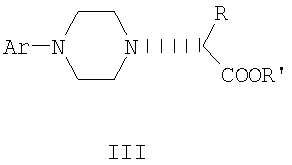
Данное изобретение включает способ получения соединения формулы III
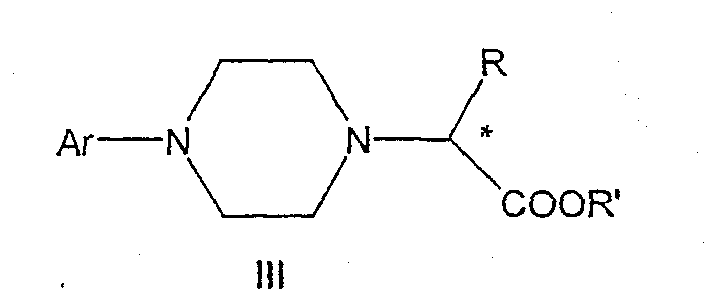
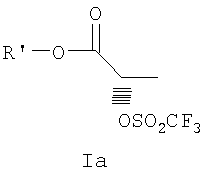
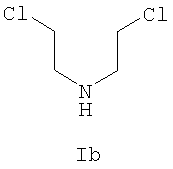
где R и R' каждый независимо представляет собой С1-С3 алкильную группу; Ar представляет собой дигидробензодиоксинил, или бензодиоксинил, или фенил, необязательно замещенный до трехкратно заместителями, независимо друг от друга выбранными из галогена, метокси, галогенметила, дигалогенметила и тригалогенметила; причем указанный способ включает взаимодействие соединения формулы Ia и соединения формулы Ib с получением соединения формулы II
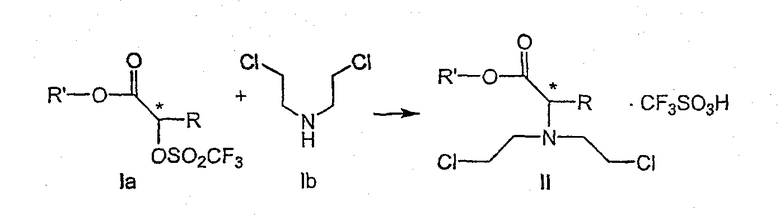
и дальнейшее взаимодействие соединения формулы II с ариламином Ar-NH2, где Ar принимает значения, определенные выше, с получением соединения формулы III. Предпочтительно, данные стадии осуществляются каскадным методом с получением соединения III без выделения промежуточного соединения II.
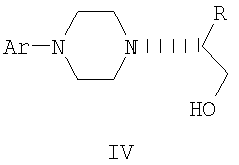
В предпочтительной форме выполнения изобретения соединение формулы Ia представляет собой единственный (S)- или (R)-энантиомер, который приводит к получению единственного энантиомера соединения формулы II, имеющего обращенную конфигурацию, т.е. (R) или (S). Гидридное восстановление соединения формулы III далее протекает с сохранением конфигурации с получением промежуточного соединения формулы IV.
Изобретение включает также взаимодействие соединения формулы IV с получением промежуточных продуктов формулы V:
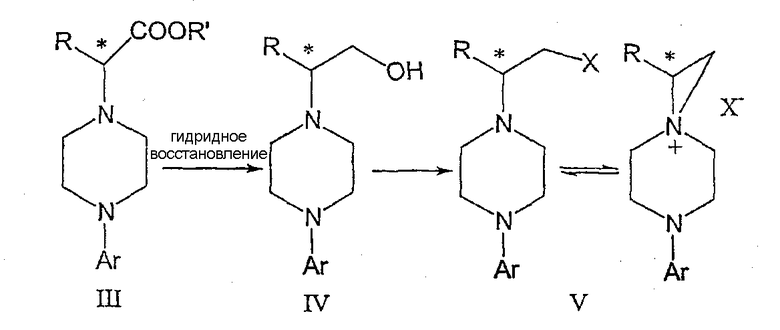
где Х представляет собой удаляемую группу, такую как галоген (в частности, хлор или бром), метансульфонилокси, п-толуолсульфонилокси или п-бромфенилсульфонилокси.
Изобретение включает также новые соединения, представленные формулами II, III, IV и V, и их оптические изомеры.
Изобретение также включает следующие стадии способа, в которых соединение V используется для получения соединений VII, VIII и IX:
обработка соединения формулы V соединением формулы VI в апротонном растворителе
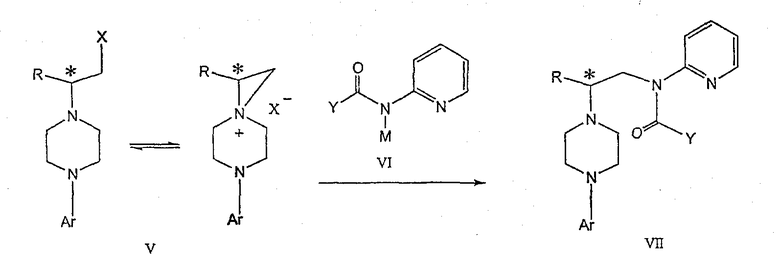
где М представляет собой щелочной металл (например, Na, Li, K), и Y представляет собой фрагмент, выбранный из группы, включающей С1-С6-алкокси, С1-С6-алкил, С3-С7-циклоалкил и С3-С7-циклоалкокси;
обработка соединения формулы VII протонной кислотой с получением соединения формулы VIII
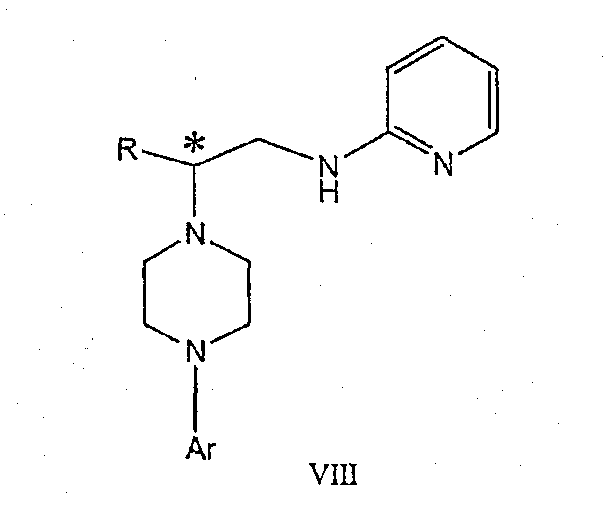
обработка соединения формулы VIII ароильным соединением, выбранным из ароилхлорида, ароилбромида и ароилангидрида, в присутствии основания с получением соединения формулы IX
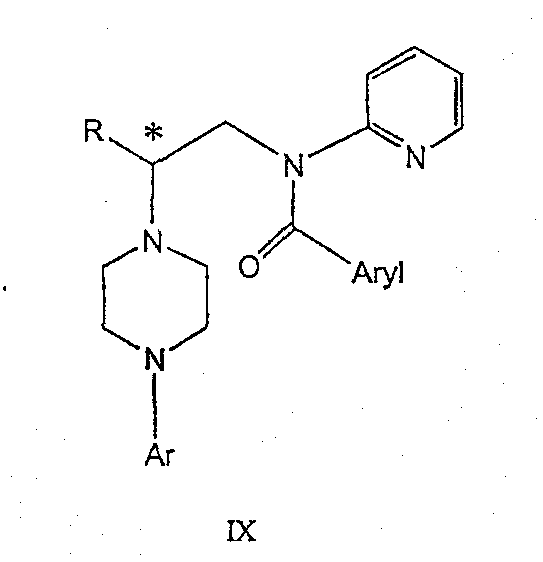
где Ar представляет собой С6-С12-ароматическую группу, необязательно замещенную до трехкратно заместителями, независимо друг от друга выбранными из группы, включающей атомы галогенов, алкил, алкокси, алкоксикарбонил, нитро, амино, алкиламино, диалкиламино, галогеналкил, дигалогеналкил, тригалогеналкил, нитрил и амидогруппу, каждый из которых содержит не более шести атомов углерода.
Данное изобретение раскрывает способ получения специфических энантиомерных соединений в качестве промежуточных соединений для получении 1,4-дизамещенных пиперазинов, которые пригодны в качестве средства, связывающего серотониновый 1А рецептор. В качестве основных реагентов служат хиральные производные азотистых ипритов. Данный способ обеспечивает более простую последовательность реакций, чем последовательность, известная из уровня техники. Новый синтез хиральных 1,4-дизамещенных пиперазинов приводит к получению стабильных при хранении промежуточных продуктов синтеза соединений формулы IX, которая описана выше.
Различные аспекты предпочтительного осуществления данного изобретения представлены на схеме 1:
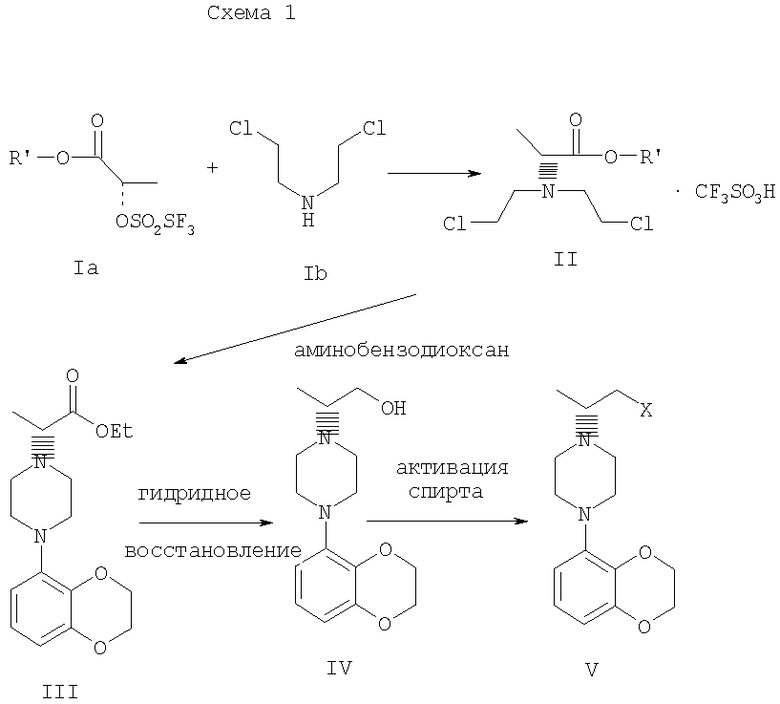
(S)-2-[(трифторметилсульфонил)окси]пропионат, представленный на схеме 1, является коммерчески доступным, или такие лактаттрифлаты легко могут быть получены из соответствующих алкиллактатов, например, в соответствии с методиками, описанными в публикациях Prasad et al., J. Chem. Soc. Perkin Trans I, 1991, 3331, и Wang and Xu, Tetrahedron 54, 12597, 1998. Бис(2-хлорэтил)амин выделяется в виде свободного основания из его гидрохлоридной соли. Реакция первой стадии, показанная на схеме 1, проводится в инертном органическом растворителе, в котором растворимы исходные вещества, таком как тетрагидрофуран, диоксан, 1,2-диметоксиэтан, диэтиловый эфир, трет-бутилметиловый эфир, метиленхлорид, хлорбензол, трифторметилбензол или толуол. Температура не является определяющим фактором, и подходящей может быть температура в интервале от 0°С до примерно 50°С, предпочтительно в интервале от температуры ледяной бани до комнатной температуры. Более высокие температуры ускоряют нежелательный процесс отщепления. Реакция обычно проводится в течение 4-6 часов, хотя может использоваться и более длительное перемешивание в течение 18-24 часов. Выходы соответствующего соединения формулы II могут высокими, например, 83%, но, как правило, выходы находятся в интервале 50-65%. Тетрагидрофуран является оптимальным растворителем, однако он очень чувствителен к присутствию следов трифторметансульфокислоты или ангидрида трифторметансульфокислоты, которые могут инициировать частичную полимеризацию тетрагидрофурана и приводить к получению гелеобразного вещества, затрудняющего выделение продукта.
Предпочтительное осуществление данного изобретения включает одностадийный процесс, в котором соединение II, полученное в хлорбензоле в виде кристаллической соли трифторметансульфокислоты, используется для алкилирования 2,3-дигидро-1,4-бензодиоксин-5-амина в хлорбензоле для получения соединения III. Соединение формулы II может подвергаться взаимодействию с 2,3-дигидро-1,4-бензодиоксин-5-амином в кипящем хлорбензоле в течение от примерно 8 до примерно 18 часов. Таким образом, получение соединения формулы III может осуществляться в виде последовательного процесса с использованием хлорбензольного растворителя без необходимости промежуточного выделения соединения формулы II.
Сложный аминоэфир соединения формулы III может выделяться в виде свободного основания или превращаться в стабильную гидрохлоридную соль. Альтернативно, соединение формулы III получают конденсацией 2,3-дигидро-1,4-бензодиоксин-5-амина со свободным основанием соединения формулы II в аналогичных условиях, и оба промежуточных соединения II и III без дополнительной очистки используются на последующих стадиях.
Предпочтительное осуществление получения соединения формулы III из соединения формулы II включает взаимодействие аминобензодиоксина, как показано на схеме I. В другом варианте данного изобретения аминофенил используется вместо аминобензодиоксана, где фенил может быть замещенным до трехкратно заместителями, независимо друг от друга выбранными из галогена, метокси, галогенметила, дигалогенметила и тригалогенметила.
Промежуточные соединения формулы III могут подвергаться восстановлению до спирта формулы IV с помощью восстановителей. Реакция проводится стандартными методами, известными специалисту, например, применением комплексного гидрида металла или борсодержащего восстановителя в условиях, обеспечивающих отсутствие эпимерности.
В предпочтительном осуществлении способа данного изобретения восстановление проводится при кипячении с обратным холодильником в простом эфире или в тетрагидрофуране при 20-40оС с использованием алюмогидрида лития. Энантиомерная чистота выделяемого спирта IV составляет 98% или более, как определено на хиральной колонке с использованием образца рацемического IV в качестве эталона.
В еще одном аспекте данного изобретения спирт соединения формулы IV может обрабатываться метансульфонилхлоридом в присутствии органического основания в метиленхлориде для получения промежуточного соединения формулы V. В альтернативном варианте спирт формулы V или его гидрохлоридная соль нагревается с тионилхлоридом в кипящем хлороформе с получением гидрохлоридной соли соединения формулы V.
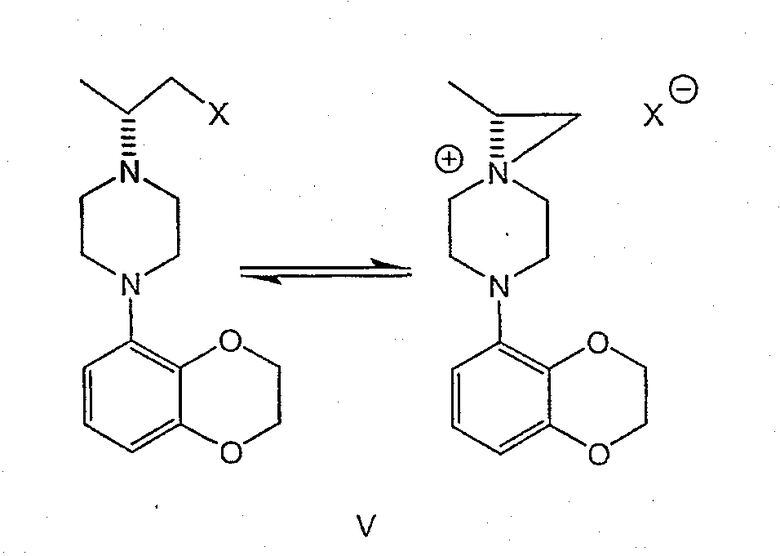
В зависимости от природы удаляемой группы Х, кислотности среды, концентрации или полярности растворителя указанные пиперазины могут находиться в равновесии с 6-аза-3-азониаспиро[2,5]октановыми производными.
Данное изобретение включает также новые соединения формулы II, III и IV. Предпочтительные соединения включают следующие:
метиловый эфир N,N-бис(2-хлорэтил)-(R)-аланина, трифторметансульфонат;
этиловый эфир N,N-бис(2-хлорэтил)-(R)-аланина, трифторметансульфонат;
этиловый эфир (R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-α-метил-1-пиперазинуксусной кислоты;
этиловый эфир (R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-α-этил-1-пиперазинуксусной кислоты;
метиловый эфир (R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-α-метил-1-пиперазинуксусной кислоты;
метиловый эфир (R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-α-этил-1-пиперазинуксусной кислоты;
(R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-β-метил-1-пиперазинэтанол; и
(R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-β-этил-1-пиперазинэтанол.
Соединение V может подвергаться взаимодействию с соединением формулы VI для получения соединения формулы VII. Y представляет фрагмент, выбранный из группы, включающей С1-С6-алкокси, С1-С6-алкил, С3-С7-циклоалкил и С3-С7-циклоалкокси.
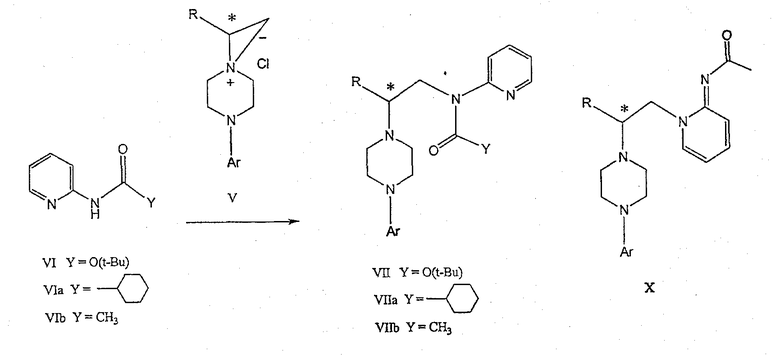
Аминопиридильный фрагмент вводится путем замещения. Из предшествующего уровня техники не ясно, насколько серьезно побочные реакции, описанные выше, могут влиять на проведение такого замещения. Многое зависит от конкретного алкилирующего агента. В соответствии с заявкой WO 9703982, аминопиридин VIa в конкретных условиях может подвергаться обработке соединениями формулы Va, где Х представляет собой удаляемую группу, с получением соединения VIIa. В ходе работы над данным изобретением авторами было установлено, что анион N-алканоильных соединений (т.е. VIb) взаимодействует с соединением V (Х=Cl) со значительным (˜20%) нежелательным алкилированием по азоту пиридильного кольца, результатом которого является соединение Х. В предпочтительном варианте данного изобретения Y представляет собой алкоксигруппу, более предпочтительно С1-С6алкокси.
Данное изобретение обеспечивает практическое осуществление синтеза N-арилпиперазинов, где хиральность вводится на стадии образования пиперазинового кольца, а 2-аминопиридильное замещение осуществляется реакцией замещения.
Применение t-Boc-2-аминопиридина, VI, как описано в данном изобретении, приводит к значительному снижению количества (<7%) образующегося побочного продукта, повышая долю желательного соединения VII. Как указано выше, защитная t-Boc-группа легко удаляется, и после этого свободный амин может подвергаться ацилированию.
В данном описании и в прилагаемой формуле изобретения, если не указано иное, термин «галоген» относится к F, Cl и Br, термины «алкил», «алкан», «алканол» и «алкокси» включают алкильные группы с прямой и разветвленной цепью.
Приведенные далее примеры представлены для иллюстрации некоторых форм осуществления данного изобретения, но не должны рассматриваться как ограничивающие объем притязаний данного изобретения.
ПРИМЕР I
Этиловый эфир N,N-бис(2-хлорэтил)-(R)-аланина, трифторметансульфонат
Суспензию бис(2-хлорэтил)амина гидрохлорида (0,392 г; 2,1 ммоль) в 5 N водном растворе гидроксида натрия (3 мл) экстрагируют эфиром (2x10 мл) и объединенные экстракты промывают минимальным количеством воды и насыщенным раствором соли. Эфирный раствор быстро сушат над сульфатом магния и фильтруют. К фильтрату добавляют тетрагидрофуран (2 мл) и эфир осторожно удаляют при пониженном давлении на роторном испарителе без нагрева. Остаток смешивают с раствором этилового эфира (S)-2-[(трифторметилсульфонил)окси]пропионовой кислоты (0,5 г; 2 ммоль) в тетрагидрофуране (1 мл). Реакционную смесь перемешивают в течение 24 часов при комнатной температуре, при этом не наблюдается выпадение осадка. Летучие растворители удаляют при пониженном давлении, оставшееся вязкое масло растворяют в эфире (8 мл) и по истечении 60 минут слегка помутневший раствор фильтруют. Фильтрат обрабатывают н-гептаном, который добавляют по каплям для индуцирования кристаллизации; конечное соотношение н-гептан/эфир составляет 1:3. Кристаллическую соль собирают фильтрованием и быстро промывают небольшим количеством эфира. Получают 0,653 г (выход 83,3%) этилового эфира N,N-бис(2-хлорэтил)-(R)-аланина трифторметансульфоната; т.пл. 73-74,5°С; 1Н ЯМР (300 МГц, CDCl3) δ 1,35 (т, J=7,1 Гц, 3Н), 1,76 (д, J=7,2 Гц, 3Н), 3,87 (м, 2Н), 4,00 (м, 2Н), 4,35 (кв., J=7,1 Гц, 2Н), 4,57 (кв., J=7,2 Гц, 1Н), 9,02 (ушир. 1Н).
ПРИМЕР II
Этиловый эфир (R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-α-метил-1-пиперазинуксусной кислоты
Раствор 2,3-дигидро-1,4-бензодиоксин-5-амина (0,327 г; 2,16 ммоль) в хлорбензоле (2 мл) добавляют к раствору этилового эфира N,N-бис(2-хлорэтил)-(R)-аланила (соль трифторметансульфоновой кислоты; 0,850 г; 2,16 ммоль) в таком же растворителе (2 мл). Реакционную смесь нагревают до 130°С и выдерживают при этой температуре в течение 15 часов, затем летучие растворители удаляют на роторном испарителе и полутвердый остаток распределяют между 10% раствором бикарбоната натрия (15 мл) и эфиром. Органические экстракты промывают раствором соли, сушат над сульфатом магния и фильтруют. ТСХ (хлороформ) показывает получение этилового эфира (R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-α-метил-1-пиперазинуксусной кислоты в качестве нового продукта с Rf 0,15. Добавление 1 N HCl в эфирном растворе приводит к превращению этилового эфира (R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-α-метил-1-пиперазинуксусной кислоты в его гидрохлоридную соль, которую собирают фильтрованием; 0,615 г (80%), т.пл. 168-171оС. Соль может быть перекристаллизована из смеси этанол-эфир или ацетон-эфир. 1Н ЯМР (300 МГц, ДМСО-d6) δ 1,25 (т, J=7,1 Гц, 3Н), 1,58 (д, J=7,2 Гц, 3Н), 3,16 (м, 2Н), 3,36 (м, 2Н), 4,23 (м, 4Н), 4,26-4,38 (м, 3Н), 4,48 (уш., 4Н), 6,52 (д, J=7,9 Гц, 1Н), 6,57 (д, J=8 Гц, 1Н), 6,76 (т, J=8 Гц, 1Н), 11,3 (уш., <1Н).
ПРИМЕР III
(R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-β-метил-1-пиперазинэтанол
Гидрохлоридную соль, полученную в примере II (1,07 г; 3 ммоль), суспендируют в 5% водном растворе бикарбоната натрия (6
мл) и экстрагируют эфиром. Органическую фазу отделяют, промывают раствором соли, быстро сушат над сульфатом магния и фильтруют. Фильтрат при перемешивании добавляют к суспензии алюмогидрида лития (0,34 г; 9 экв.) и смесь кипятят при умеренном кипячении с обратным холодильником в течение 3 часов. После охлаждения смесь разлагают водой (1 мл) и 0,5 N соляной кислотой (7 мл). Водный слой отделяют, подщелачивают 10% раствором бикарбоната натрия и снова экстрагируют эфиром. Объединенные экстракты промывают небольшими количествами воды и раствора соли, сушат над сульфатом магния, фильтруют и выпаривают. Маслянистый продукт (0,69 г; выход 82%) медленно кристаллизуется из неподвижного раствора, и продукт перекристаллизовают из смеси н-бутанол/н-гептан; т.пл. 92°С, энантиомерная чистота 98%; 1H ЯМР (300 МГц, CDCl3) δ 1,03 (д, J=7 Гц, 3Н), 2,74 (м, 2Н), 2,97 (м, 3Н), 3,14 (м, 4Н), 3,42 (т, J=11 Гц, 1Н), 3,57 (дд, J=11 Гц, J1=5 Гц, 1Н), 4,35 (сим. м, 4Н), 6,53 (д, J=7,9 Гц, 1Н), 6,61 (д, J=7,9 Гц), 6,75 (т, J=7,9 Гц, 1Н).
ПРИМЕР IV
Этиловый эфир (R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-α-метил-1-пиперазинуксусной кислоты
Свободное основание бис(2-хлорэтил)амина выделяют распределением его гидрохлоридной соли между 5 N водным раствором гидроксида натрия и метиленхлоридом в соответствии с методикой примера I. Выделенный бис(2-хлорэтил)амин (0,94 г; 6,56 ммоль) добавляют двумя порциями в течение 1 часа при перемешивании к раствору (S)-2-[(трифторметилсульфонил)окси]пропионата (0,82 г; 3,28 ммоль) в хлорбензоле (10 мл) при комнатной температуре. Реакционную смесь перемешивают в течение дополнительных 2 часов, твердый осадок отфильтровывают и промывают небольшим количеством хлорбензола. Фильтрат смешивают с раствором 2,3-дигидро-1,4-бензодиоксин-5-амина (0,46 г; 3 ммоль) и реакционную смесь кипятят с обратным холодильником в течение 18 часов. После охлаждения продукт подщелачивают 5% водным раствором бикарбоната натрия (20 мл) и дважды экстрагируют эфиром (50 мл). Объединенные экстракты промывают водой, раствором соли, сушат над сульфатом магния и фильтруют. Фильтрат концентрируют в вакууме, в результате получают неочищенный продукт, который может непосредственно подвергаться восстановлению или пропускается через рыхлый слой силикагеля в хлороформе с получением соединения III (0,49 г; общий выход 50%). Продукт идентичен продукту, описанному в примере II.
ПРИМЕР V
(R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-β-метил-1-пиперазинэтанол
Свободное основание бис(2-хлорэтил)амина (28,4 г; 0,2 моль) выделяют из его гидрохлоридной соли, как описано в примере IV, и смешивают с раствором (S)-2-[(трифторметилсульфонил)окси] пропионата (20 г; 0,08 моль) в хлорбензоле (150 мл). Смесь перемешивают в течение 3 часов при комнатной температуре и полученную густую суспензию промывают водой (100 мл) и 10% раствором бикарбоната натрия (100 мл). Органическую фазу переносят в колбу, содержащую 2,3-дигидро-1,4-бензодиоксин-5-амина (9,6 г; 0,064 мол) и реакционную смесь при перемешивании кипятят с обратным холодильником в течение 18 часов. Из раствора выпадает небольшое количество желтого осадка. Смесь охлаждают до комнатной температуры, добавляют 10% водный раствор бикарбоната натрия (55 мл) и перемешивают в течение 1 часа. Органический слой отделяют, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток растворяют в тетрагидрофуране (50 мл) и по каплям при перемешивании добавляют к суспензии алюмогидрида лития (9,1 г; 0,24 моль) в тетрагидрофуране (50 мл). Смесь выдерживают при 40°С в течение 90 минут, охлаждают и разлагают, добавляя по каплям этилацетат (200 мл). После этого продукт экстрагируют 2 N соляной кислотой (500 мл), водную часть промывают три раза этилацетатом (150 мл), подщелачивают 10 N гидроксидом натрия и повторно экстрагируют продукт этилацетатом (2x200 мл). Объединенные экстракты промывают раствором соли, сушат над сульфатом натрия, фильтруют и выпаривают в вакууме. Остаток в виде масла самопроизвольно кристаллизуется, и анализ ТСХ продукта (этилацетат-гексан 3:2) показывает общую область со спиртом примера III. Данные спектроскопии и энантиомерная чистота идентичны данным спектроскопии и чистоте спирта, полученного в примере III. Общий выход 9,1 г (51%) из расчета на 2,3-дигидро-1,4-бензодиоксин-5-амин.
ПРИМЕР VI
6-(2,3-Дигидро-1,4-бензодиоксин-5-ил)-1-метил-6-аза-3-азаспиро[2,5]октан хлорид
Раствор спирта, полученного согласно методике примера III (0,5 г; 1,8 ммоль), в метиленхлориде (15 мл) обрабатывают триэтиламином (0,2 г; 1,98 ммоль). Смесь перемешивают при охлаждении на ледяной бане и по каплям добавляют раствор метансульфонилхлорида (0,24 г; 2,1 ммоль) в метиленхлориде (2 мл). Спустя 20 минут ледяную баню удаляют и реакционную смесь оставляют на ночь при комнатной температуре. Полученный раствор промывают последовательно небольшим количеством воды, 5% водным раствором бикарбоната натрия и раствором соли, затем сушат над сульфатом магния и фильтруют. Летучие растворители удаляют на роторном испарителе, получая маслянистый продукт (0,5 г). 1Н ЯМР (300 МГц, CDCl3) δ 1,55 (д, J=7,2 Гц, 3Н), 2,54 (дд, J=15 Гц, J1= 7,5 Гц, 1Н), 2,64-2,81 (м, 5Н), 3,11 (м, 4Н), 4,11 (сим.м, 1Н), 4,27 (м, 4Н), 6,52 (д, J=7,8 Гц, 1Н), 6,57 (д, J=8 Гц, 1Н), 6,76 (т, J=7,8 Гц, 1Н).
ПРИМЕР VII
(R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-1-(2-хлор-1-метилэтил)пиперазин
Раствор спирта, полученного в соответствии с методикой примера III (0,3 г; 1,08 ммоль), в метиленхлориде (5 мл) подкисляют эфирной HCl, полученный раствор выпаривают и полукристаллический остаток растирают с эфиром. После декантирования продукт кристаллизуют из смеси ацетонитрил-эфир, т.пл. 207-210°С. Полученную соль (0,35 г) суспендируют в хлороформе (6 мл), добавляют тионилхлорид (0,2 г) и полученную смесь кипятят с обратным холодильником в течение 8 часов. Полученному раствору дают охладиться, летучие растворители удаляют в вакууме, остаток освобождают от легких компонентов с помощью толуола и сушат. ТСХ (этилацетат-гексан 3:2) показывает отсутствие исходного спирта. 1Н ЯМР (300 МГц, ДМСО-d6) δ 1,56 (д, J=7 Гц, 3Н), 3,45 (м, 6Н), 4,64 (м, 2Н), 4,75 (м, 1Н); спектр также показывает присутствие азиридинового продукта. Продукт может использоваться непосредственно для алкилирования 2-(трет-бутоксикарбониламино)пиридина.
Множество вариантов данного изобретения, не проиллюстрированных в данном описании, понятно квалифицированному специалисту. Данное изобретение не ограничивается представленными в описании иллюстративными примерами, но включает все объекты, составляющие объем притязаний прилагаемой формулы изобретения, и их эквиваленты.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИПЕРАЗИНА И ПРОМЕЖУТОЧНОЕ ВЕЩЕСТВО | 2003 |
|
RU2314294C2 |
| СПОСОБ СИНТЕЗА ХИРАЛЬНЫХ N-АРИЛПИПЕРАЗИНОВ | 2003 |
|
RU2315762C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2125566C1 |
| 2,3-ДИГИДРО-1,4-БЕНЗОДИОКСИ-5-ИЛ-ПИПЕРАЗИНОВЫЕ ПРОИЗВОДНЫЕ ИЛИ ИХ СОЛИ | 1994 |
|
RU2118322C1 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ТЕРАПИИ ПРОТИВ ВИЧ | 2019 |
|
RU2806030C2 |
| ПРОИЗВОДНЫЕ БЕНЗОДИОКСОЛА, БЕНЗОФУРАНА, ДИГИДРОБЕНЗОФУРАНА И БЕНЗОДИОКСАНА И СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ | 1997 |
|
RU2190609C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИН-4-ОНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СЕРОТОНИНАНТАГОНИСТИЧЕСКАЯ, ДОПАМИНАНТАГОНИСТИЧЕСКАЯ И АНТИГИСТАМИННАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1990 |
|
RU2028297C1 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1992 |
|
RU2193561C2 |
| ЗАМЕЩЕННЫЕ БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ПРИМЕНЕНИЕ В КАЧЕСТВЕ СРЕДСТВ ПРОТИВ ОЖИРЕНИЯ И ГИПЕРХОЛЕСТЕРИНЕМИИ | 2000 |
|
RU2278114C2 |
| СПОСОБЫ СИНТЕЗА СПИРО-ОКСИНДОЛЬНЫХ СОЕДИНЕНИЙ | 2010 |
|
RU2544852C2 |
Изобретение относится к промежуточным соединениям формулы II, III, IV,
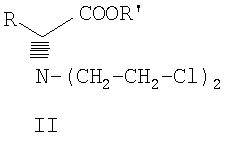
где R и R', каждый независимо друг от друга, представляет собой C1-С3-низший алкил, и его оптические изомеры и соли.
где R и R', каждый независимо друг от друга, представляет собой C1-С3-низший алкил, Ar представляет собой дигидробензодиоксинил, бензодиоксинил или фенил, необязательно замещенный до трехкратно заместителями, независимо друг от друга выбранными из галогена, метокси, галогенметила, дигалогенметила и тригалогенметила, и его оптические изомеры.
где Ar представляет собой дигидробензодиоксинил, или бензодиоксинил, или фенил, необязательно замещенный до трехкратно заместителями, независимо друг от друга выбранными из галогена, метокси, галогенметила, дигалогенметила и тригалогенметила, и его оптические изомеры. Изобретение также относится к способу получения соединений формулы III, IV, V, которые являются промежуточными продуктами для получения средства, связывающего 5НТ1А-рецепторы, и пригодно для лечения расстройств центральной нервной системы (CNS). Технический результат - упрощение процесса и получение оптически предпочтительных пиперазинов. 8 н. и 5 з.п. ф-лы.
где R и R', каждый независимо друг от друга, представляет собой С1-С3-алкильную группу; Ar представляет собой дигидробензодиоксинил, бензодиоксинил или фенил, необязательно замещенный до трехкратно заместителями, независимо друг от друга выбранными из галогена, метокси, галогенметила, дигалогенметила, тригалогенметила;
включающий взаимодействие соединения формулы Ia
где R' имеет определенные выше значения,
и соединения формулы Ib
с получением соединения формулы II
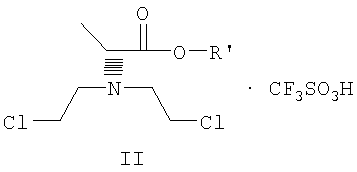
где R' имеет определенные выше значения,
и дальнейшее взаимодействие соединения формулы II с ариламином Ar-NH2, где Ar имеет определенные выше значения, с получением соединения формулы III.
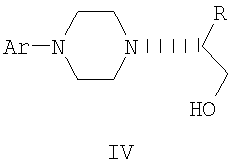
где Ar и R имеют значения, определенные в п.1,
включающий восстановление соединения формулы III
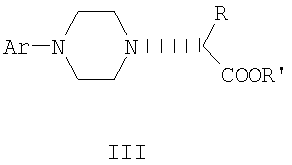
где R, R' и Ar имеют значения, определенные в п.1.
где Ar и R имеют значения, определенные в п.1,
включающий восстановление соединения формулы III, представляющего собой, по существу, чистый R-энантиомер
где R, R' и Ar имеют значения, определенные в п.1.
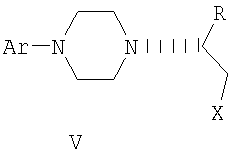
где Х представляет собой пригодную удаляемую группу, независимо выбранную из группы, включающей бром, хлор, метансульфонилокси, п-толуолсульфонилокси и п-бромфенилсульфонилокси, и Ar и R имеют значения, определенные в п.1,
включающий превращение соединения формулы IV
в целевой продукт.
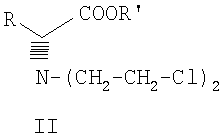
где R и R', каждый независимо друг от друга, представляет собой C1-С3-низший алкил, и его оптические изомеры и соли.
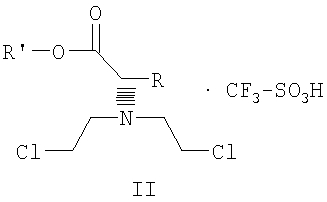 .
.
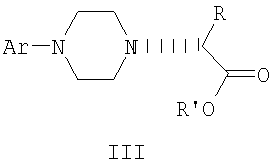
где R и R', каждый независимо друг от друга, представляет собой C1-С3-низший алкил, Ar представляет собой дигидробензодиоксинил, бензодиоксинил или фенил, необязательно замещенный до трехкратно заместителями, независимо друг от друга выбранными из галогена, метокси, галогенметила, дигалогенметила и тригалогенметила,
и его оптические изомеры.
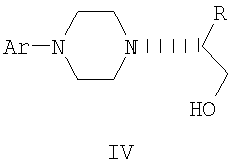
где Ar представляет собой дигидробензодиоксинил, или бензодиоксинил, или фенил, необязательно замещенный до трехкратно заместителями, независимо друг от друга выбранными из галогена, метокси, галогенметила, дигалогенметила и тригалогенметила, и его оптические изомеры.
метиловый эфир N,N-бис(2-хлорэтил)-(R)-аланина, трифторметансульфонат;
этиловый эфир N,N-бис(2-хлорэтил)-(R)-аланина, трифторметансульфонат;
этиловый эфир (R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-α-метил-1-пиперазинуксусной кислоты;
этиловый эфир (R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-α-этил-1-пиперазинуксусной кислоты;
метиловый эфир (R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-α-метил-1-пиперазинуксусной кислоты;
метиловый эфир (R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-α-этил-1-пиперазинуксусной кислоты;
(R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-β-метил-1-пиперазинэтанол; и
(R)-4-(2,3-дигидро-1,4-бензодиоксин-5-ил)-β-этил-1-пиперазинэтанол, и их оптические изомеры.
| DATABASE CROSSFIRE BEILSTEIN, vol | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| DATABASE CROSSFIRE BEILSTEIN, no.2, 1986, р.314-333 | |||
| Резиновая смесь на основе ненасыщенного каучука | 1981 |
|
SU960203A1 |
| S.J.STACHEL ET AL., TETRAHEDON LETTERS, vol.40, 1999, p.5811-5812 | |||
| J.M.GOLEC ET AL, BIORG | |||
| MED.CHEM.LETT, vol.7, no.17, 1997, p.2181-2186 | |||
| D.MULLER ET AL, J.ORG.CHEM, vol.62, no.2, 1997, p.411-416 | |||
| G.M.TAYLOR ET AL, | |||

