ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США под серийным номером 62/773,563, поданной 30 ноября 2018 г., содержание которой полностью включено в настоящий документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям, фармацевтическим композициям и способам их применения в связи с индивидуумами, инфицированными ВИЧ, ВГВ или раком.
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
Настоящая заявка содержит последовательности, перечисленные в электронном перечне последовательностей, озаглавленном PR66692_Seq_List, размером 2 KB, созданным с использованием Patent-In 3.5 12 ноября 2010 г., содержание и последовательности которого включены в настоящий документ посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
Инфицирование вирусом иммунодефицита человека 1 типа (ВИЧ-1) приводит к заболеванию синдромом приобретенного иммунодефицита (СПИД). Число случаев ВИЧ продолжает расти, и в настоящее время количество индивидуумов, страдающих от ВИЧ-инфекции в мире, оценивается в тридцать пять миллионов, см., например, http://www.sciencedirect.com/science/article/pii/S235230181630087X? via%3Dihub
В настоящее время длительное подавление репликации вируса антиретровирусными препаратами является единственным вариантом лечения инфекции ВИЧ-1. Действительно, Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило двадцать пять препаратов для шести различных классов ингибиторов, которые, как было показано, значительно увеличивают выживаемость пациентов и качество жизни. Однако все еще сохраняется потребность в дополнительных терапевтических средствах из-за ряда проблем, включающих нежелательные взаимодействия между лекарственными средствами; взаимодействия между лекарственными средствами и пищей; несоблюдение терапии; лекарственная устойчивость, обусловленная мутациями фермента-мишени и воспаление, связанное с иммунологическим повреждением, вызываемым ВИЧ-инфекцией, но не ограниченных перечисленными.
В настоящее время поста все ВИЧ-положительные пациенты получают режимы, включающие комбинации антиретровирусных препаратов, называемые высокоактивной антиретровирусной терапией ("HAART" или «ВААРТ»). Однако варианты лечения ВААРТ бывают сложными из-за необходимости вводить пациенту комбинацию различных лекарственных средств, часто ежедневно, для того чтобы избежать быстрого появления устойчивых к лекарственным средствам вариантов ВИЧ-1. Несмотря на положительное влияние ВААРТ на выживаемость пациентов, все же возможно возникновение лекарственной устойчивости, а выживаемость и качество жизни не нормализуются по сравнению с неинфицированными людьми [Lohse Ann Intern Med 2007 146; 87-95]. Действительно, частота некоторых заболеваний и смертности, не связанных со СПИДом, таких как сердечнососудистые заболевания, немощность и нейрокогнитивные нарушения, повышены у ВИЧ-инфицированных субъектов, получающих ВААРТ для супрессии вируса [Deeks Annu Rev Med 2011; 62:141-155]. Эта повышенная частота не связанных со СПИД заболеваний/смертности возникает в контексте повышенного системного воспаления, связанного с иммунологическим повреждением, вызванным ВИЧ-инфекцией, и остаточной ВИЧ-инфекцией[Нип1 J Infect Dis 2014][Byakagwa J Infect Dis 2014][Tenorio J Infect Dis 2014].
Современная антиретровирусная терапия (APT) способна эффективно подавлять репликацию ВИЧ и улучшать показатели здоровья ВИЧ-инфицированных лиц, но считается, что она не способна полностью устранить резервуары вируса ВИЧ в организме индивидуума. Геномы ВИЧ могут сохраняться в латентном состоянии в основном в иммунных клетках инфицированного индивидуума и могут реактивироваться в любое время, так что после прерывания APT репликация вируса обычно возобновляется в течение нескольких недель. У небольшого количества людей размер этого вирусного резервуара был значительно уменьшен, и после прекращения APT возобновление репликации вируса замедлялось [Henrich TJ J Infect Dis 2013][Henrich TJ Ann Intern Med 2014]. В одном случае вирусный резервуар был ликвидирован во время лечения лейкоза, и в течение нескольких лет наблюдения не наблюдалось повторного размножения вируса [Hutter G N Engl J Med 2009]. Эти примеры указывают на то, что уменьшение или устранение вирусного резервуара возможно и может привести к вирусологической ремиссии или излечению. Соответственно, разрабатываются способы устранения резервуара вируса при помощи прямых молекулярных средств, включая разрезание вирусного генома системами CRISPR/Cas9, или индукции реактивации латентного резервуара в ходе APT, что позволило бы устранить латентные клетки. Считается, что обращение латентности необходимо для того, чтобы сделать латентно инфицированные клетки доступными для клиренса.
Миметики SMACm (второго митохондриального активатора каспаз) представляют собой класс соединений, которые недавно прошли клинические испытания в качестве потенциальных средств лечения рака. Эти лекарственные средства истощают и/или ингибируют клеточный ингибитор белков апоптоза (cIAP), который действует как антиапоптотические белки, тем самым способствуя гибели раковых клеток. Антагонизм и/или истощение cIAP также приводит к активации неканонического сигнального пути NF-kB, который может индуцировать экспрессию ВИЧ и может способствовать элиминации инфицированных ВИЧ клеток. Дополнительно миметики SMAC могут селективно стимулироват гибель клеток, инфицированных ВИЧ [Campbell Cell Host Microbe 2018] или ВГВ (вируса гепатита В) [Ebert Proc Nat Acad Sci 2013], за счет антагонизма в отношении антиапоптотических белков.
Недавно появилась информация о нацеленном воздействии на неканонический путь NF-kB (ncNF-κВ) для обращения латентности в моделях на клеточных линиях. Путь ncNF-κВ обычно активируется лигированием подмножества членов семейства рецепторов TNF (ФНО -фактора некроза опухоли). В стационарном состоянии мультимолекулярный комплекс с активностью убиквитинлигазы, состоящий из фактора 2, ассоциированного с рецептором TNF (TRAF2), TRAF3 и клеточного ингибитора белков апоптоза 1 (cIAP1), ассоциироциирует с цитоплазматической частью нелигированного рецептора и конститутивно убиквитинилирует и разрушает NF-κB- индуцирующую киназу (NIK). После лигирования рецептора cIAP1 убиквитинилирует TRAF3 и аутоубиквитинилаты, что приводит к протеасомному разрушению TRAF3 и cIAP1, и, соответственно, снятию ингибирования накопления NIK. NEK. конститутивно активен и после накопления фосфорилирует ингибитор гомодимера киназы κВ-α (IKKα).
Активированный гомодимер IKKα / IKKα затем фосфорилирует неактивную форму p100 NFκB2, что приводит к убиквитинилированию убиквитинлигазой Skp1-Cull-F-box (SCFβTrCP) и протеасомному расщеплению р100, при котором высвобождается активна субъединица р52. р52 ассоциирует с RelB, и этот гетеродимер перемещается в ядро, где направляет транскрипцию с элементов промотора κВ. Помимо лигирования рецептора, ncNF-κВ может активироваться сигнальными интермедиатами каскада апоптоза. Отщепление второго митохондриального активатора каспаз (SMAC) от митохондриальной мембраны делает доступным N-концевой мотив Ala-Val-Pro-Ile, который специфически связывается с доменами промежуточных бакуловирусных повторов (BLR) белков LAP. Такое связывание BLR в cIAP1 / 2 активирует убиквитинлигазную активность комплекса TRAF2: TRAF3: cIAP, вызывая аутоубиквитинилирование и разрушение cIAP1 / 2, накопление NIK и активацию пути ncNF-κВ. Связывание SMAC с доменами BLR XIAP и ML-IAP противодействует активности ингибирования каспаз этих молекул, часто сверхэкспрессирующихся в опухолевых клетках, что приводит к усилению апоптоза. Таким образом, мотив Ala-Val-Pro-Ile SMAC стал предметом значительного внимания в онкологии, что привело к открытию класса пептидных миметиков, обладающих SMAC-подобной активностью, называемых миметиками SMAC (SMACm). SMACm мощно активирует путь ncNF-κВ и не индуцирует апоптоз в неопухолевых клетках и, соответственно, представляет интерес для обращения латентности ВИЧ. См., например, Richard Dunham et.al., The SMAC Mimetic AZD5582 представляет собой Potent HIV Latency Reversing Agent, bioRxiv, May. 2, 2018; doi: http://dx.doi.org/10.1101/312447.
Патент США №7,960,372 относится к двухвалентными диазобициклическим миметикам SMAC, которые ингибируют активность LAP.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном аспекте настоящего изобретения предложено соединение структуры, соответствующей Формуле, (I):
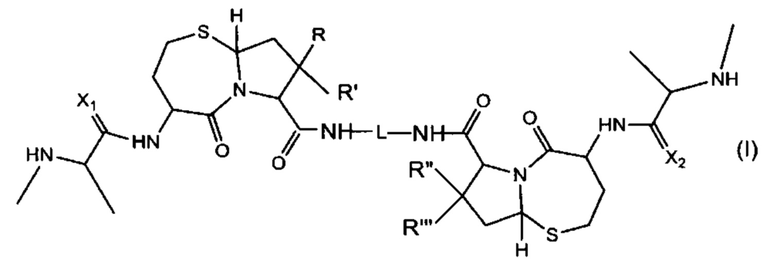
или его фармацевтически приемлемая соль или стереоизомер, где:
R, R', R'' и R''' независимо выбраны из Н и СН3;
X1 и Х2 независимо выбраны из группы, состоящей из О и S; и
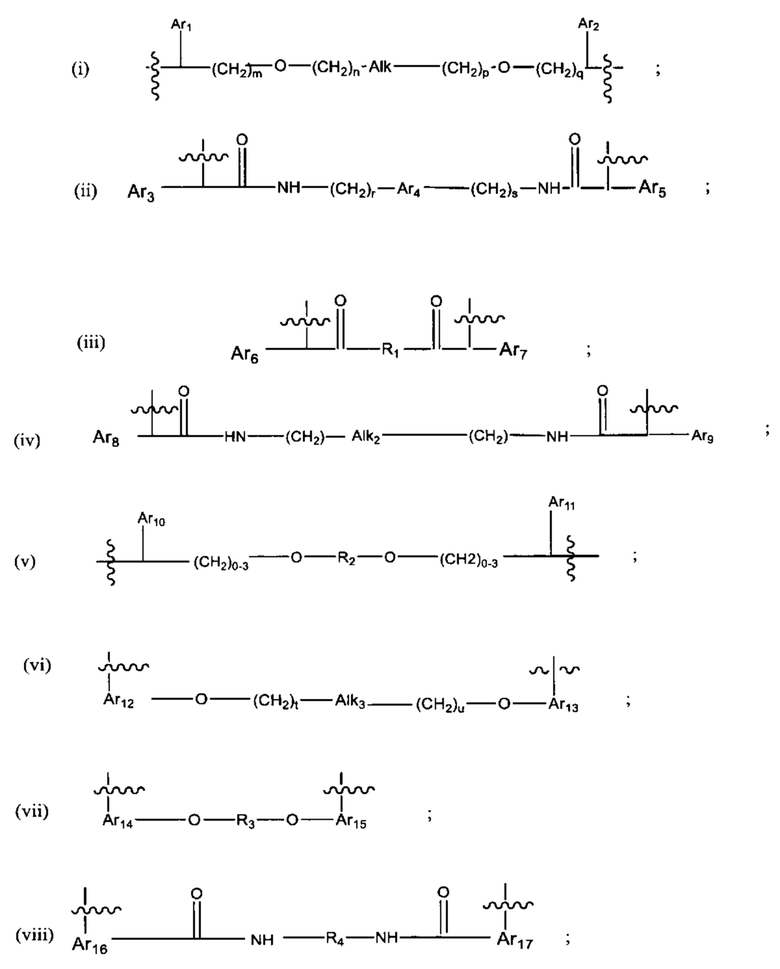
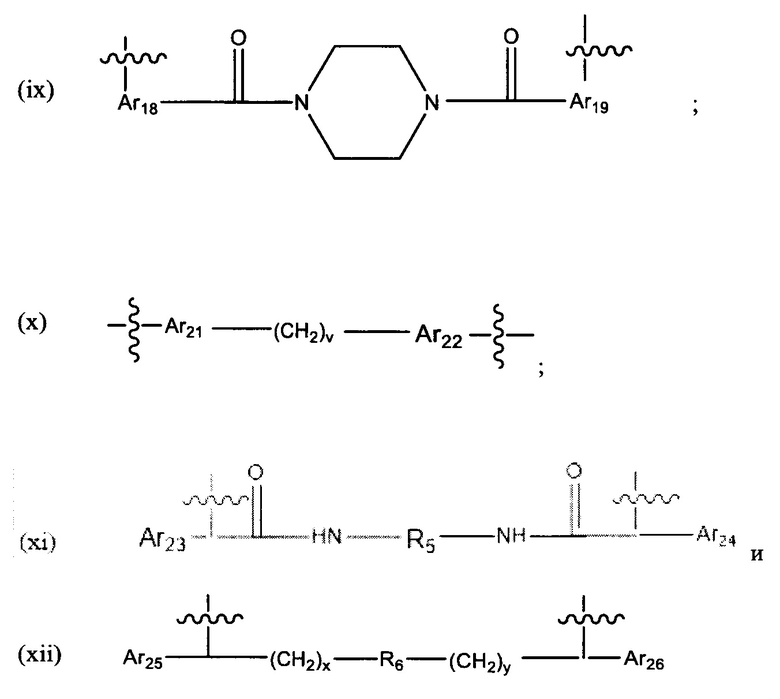
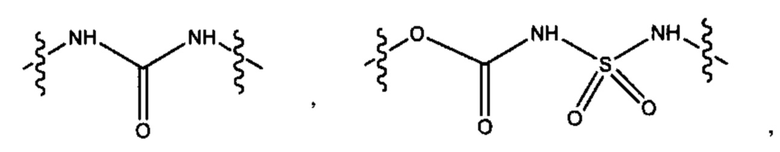
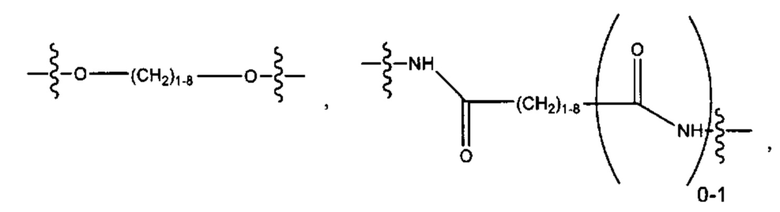
L представляет собой линкер, выбранный из группы, состоящей из:
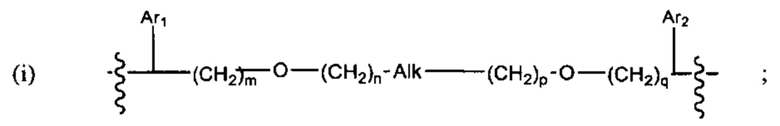
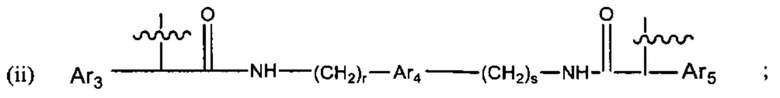
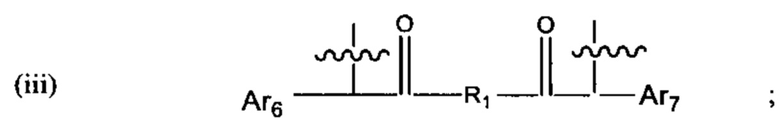
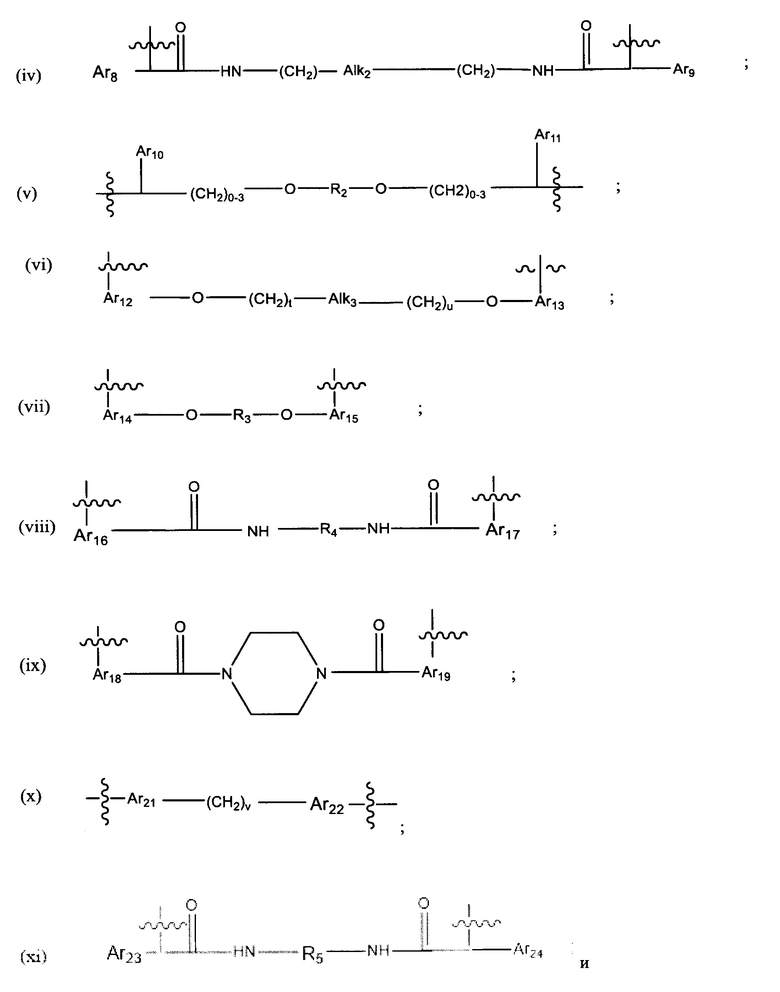
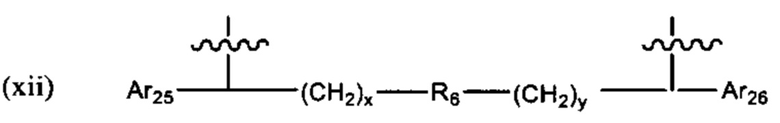
где:
Ar1, Ar2, Ar3, Ar4, Ar5, Ar6, Ar7, Ar8, Ar9, Ar10, Ar11, Ar12, Ar13, Ar14, Ar15, Ar16, Ar17, Ar18, Ar19, Ar21, Ar22, Ar23, Ar24, Ar25 и Ar26 каждый независимо выбран из (С6-С14)арила;
Alk, Alk2 и Alk3 каждый независимо выбран из:
R1, представляет собой С3-С6 циклоалкил или C1-C6 гетероцикл;
R2 выбран из группы, состоящей из -(СН2)а-, -(СН2)b-O-(СН2)с-, -(CH2)d-(C6-C14)арил-(СН2)е- и -(СН2)f-(С1-С6)гетероарил-(СН2)g-;
R3 выбран из группы, состоящей из -(СН2)h-; -(СН2)i-O-(СН2)j, -(СН2)k-(С6-С14)арил-(СН2)1- и -(СН2)m-(С1-С6)гетероарил-(СН2)m''-;
R4 представляет собой С3-С6 циклоалкил, (С6-С14)арил или (СН2)n'-(С6-С14)арил -(СН2)n'', (CH2)n'''-Alk-(CH2)n'''', где n', n'', n''' и n'''' независимо выбраны из от 1 до 8
R5 представляет собой С3-С6 циклоалкил;
R6 выбран из группы, состоящей из (CH2)z,
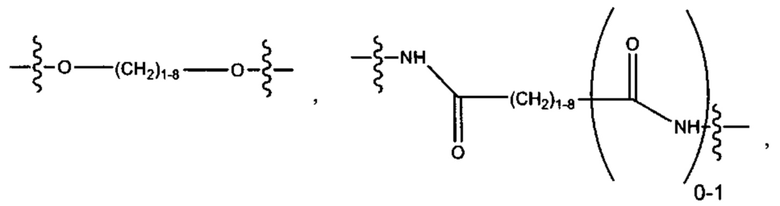
и
a, b, с, d, е, f, г, h, i, j, k, l, m, m', m'', n, p, q, r, s, t, u, v, x, у и z каждый независимо выбран из от 1 до 12.
В другом аспекте настоящее изобретение относится к соединению Формулы (Ia):
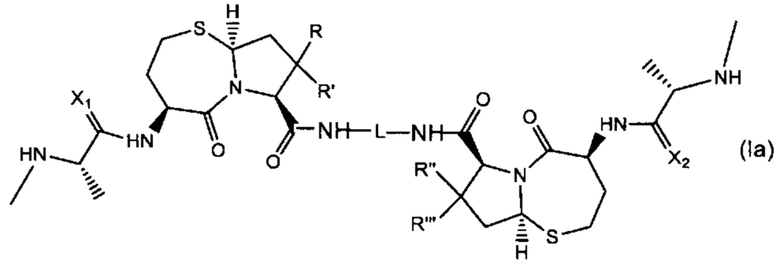
или его фармацевтически приемлемая соли или стереоизомеру, где:
R, R', R'' и R''' независимо выбраны из Н и СН3;
X1 и Х2 независимо выбраны из группы, состоящей из О и S; и
L представляет собой линкер, выбранный из группы, состоящей из:
где:
Ar1, Ar2, Ar3, Ar4, Ar5, Ar6, Ar7, Ar8, Ar9, Ar10, Ar11, Ar12, Ar13, Ar14, Ar15, Ar16, Ar17, Ar18, Ar19, Ar21, Ar22, Ar23, Ar24, Ar25 и Ar26 каждый независимо выбран из (С6-С14)арила;
Alk, Alk2 и Alk3 каждый независимо выбран из:
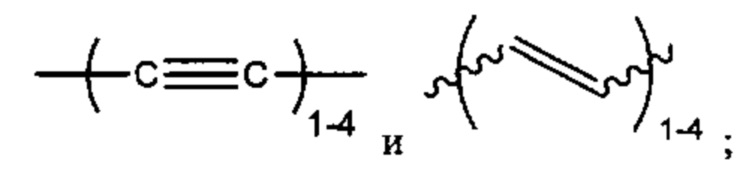
R1, представляет собой С3-С6 циклоалкил или C1-C6 гетероцикл;
R2 выбран из группы, состоящей из -(СН2)а-, -(СН2)b-O-(СН2)с-, -(CH2)d-(C6-C14)арил-(СН2)е- и -(СН2)f-(С1-С6)гетероарил-(СН2)g-;
R3 выбран из группы, состоящей из -(CH2)h-; -(СН2)i-O-(CH2)j-, -(СН2)k-(С6-С14)арил-(СН2)l- и -(СН2)m'-(С1-С6)гетероарил-(СН2)m''-;
R4 представляет собой С3-С6 циклоалкил, (С6-С14)арил или (СН2)n'-(С6-С14)арил-(СН2)n'', (CH2)n'''-Alk-(CH2)n'''', где n', n'', n''' и n'''' независимо выбраны из от 1 до 8
R5 представляет собой С3-С6 циклоалкил;
R6 выбран из группы, состоящей из (СН2)z,
и
а, b, с, d, е, f, r, h, i, j, k, 1, m, m', m'', n, p, q, r, s, t, u, v, x, у и z каждый независимо выбран из от 1 до 12.
В другом аспекте настоящее изобретение относится к соединению Формулы (Ib):
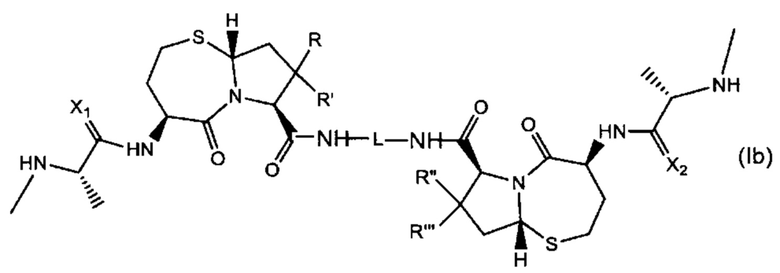
или его фармацевтически приемлемой соли или стереоизомеру, где:
R, R', R'' и R'' независимо выбраны из Н и СН3;
X1 и Х2 независимо выбраны из группы, состоящей из О и S; и
L представляет собой линкер, выбранный из группы, состоящей из:
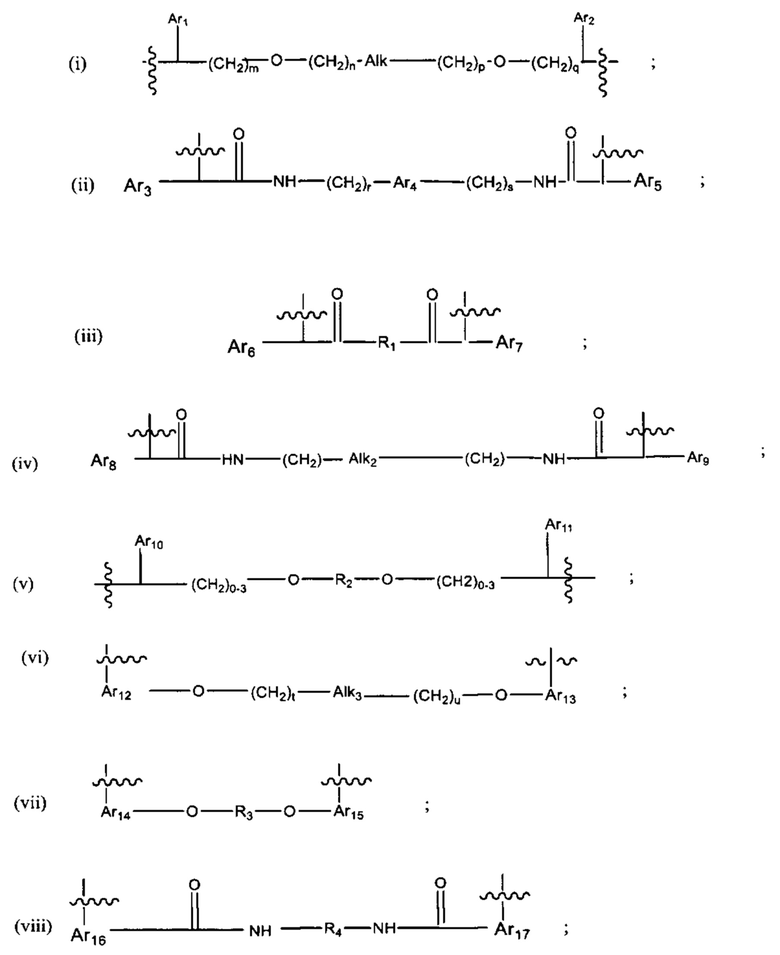
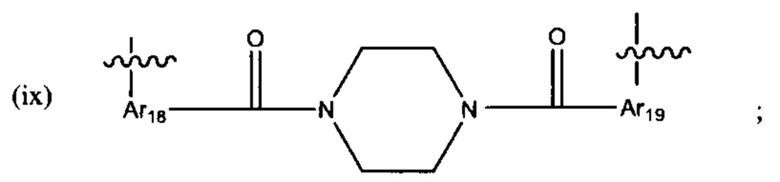
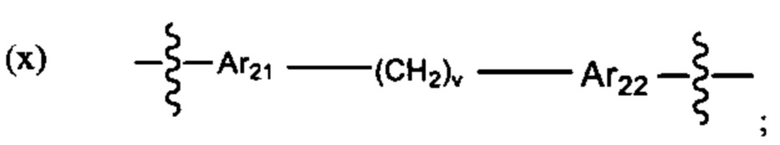
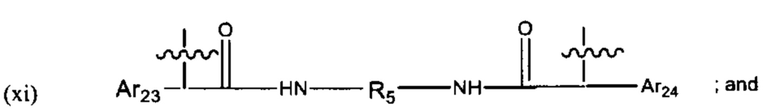
где:
Ar1, Ar2, Ar3, Ar4, Ar5, Ar6, Ar7, Ar8, Ar9, Ar10, Ar11, Ar12, Ar13, Ar14, Ar15, Ar16, Ar17, Ar18, Ar19, Ar21, Ar22, Ar23, Ar24, Ar25 и Ar26 каждый независимо выбран из (С6-С14)арила;
Alk, Alk2 а A1k3 каждый независимо выбран из:
R1, представляет собой С3-С6 циклоалкил или С1-С6 гетероцикл;
R2 выбран из группы, состоящей из -(СН2)а-, -(СН2)b-O-(СН2)с-, -(СН2)d-(С6-С14)арил-(СН2)c- и -(СН2)f-(С1-С6)гетероарил-(СН2)g-;
R3 выбран из группы, состоящей из -(СН2)h-; -(СН2)i-О-(СН2)j-, -(СН2)k-(С6-С14)арил-(СН2)l- и -(СН2)m'-(С1-С6)гетероарил-(СН2)m''-;
R4 представляет собой С3-С6 циклоалкил, (С6-С14)арил или (СН2)n'-(С6-С14)арил -(СН2)n'', (СН2)n'''-Alk-(СН2)n'''', где n', n'', n''' и n'''' независимо выбраны из от 1 до 8
R5 представляет собой С3-С6 циклоалкил;
R6 выбран из группы, состоящей из (CH2)z,
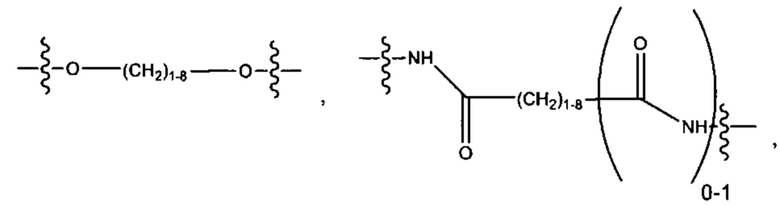
и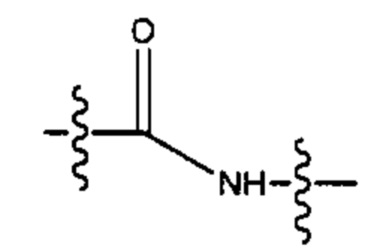
a, b, с, d, е, f, г, h, i, j, k, l, m, m', m'', n, p, q, r, s, t, u, v, x, у и z каждый независимо выбран из от 1 до 12.
В другом аспекте настоящее изобретение относится к соединению, представленному Формулой (II):
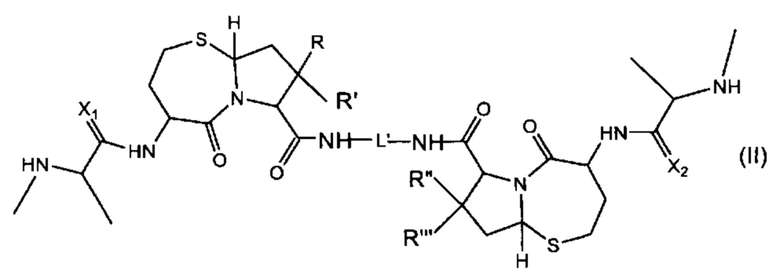
где
R, R', R'' и R''' независимо выбраны из Н и СН3;
X1 и Х2 независимо выбраны из группы, состоящей из О и S; и
L' представляет собой линкер формулы:

где:
Ar27 и Ar28 каждый независимо выбран из С6-С14 арила,
R7 выбран из группы, состоящей из
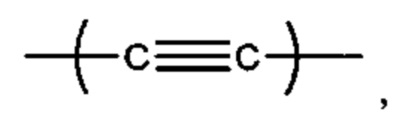 С6 арила и -(CH2)4-15-; и
С6 арила и -(CH2)4-15-; и
а' и b' независимо выбраны из от 0 до 6.
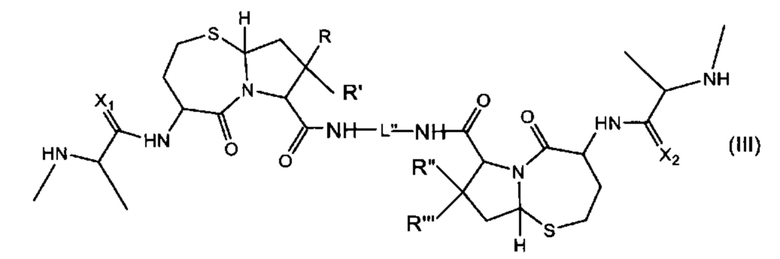
В другом аспекте настоящее изобретение относится к соединению, представленному Формулой (III):
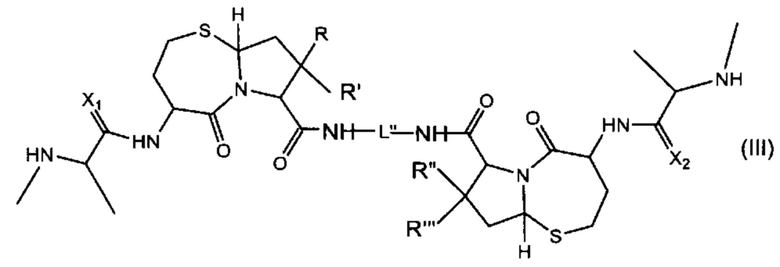
где
R, R', R'' и R''' независимо выбраны из Н и СН3;
X1 и Х2 независимо выбраны из группы, состоящей из О и S; и
L'' представляет собой линкер формулы:
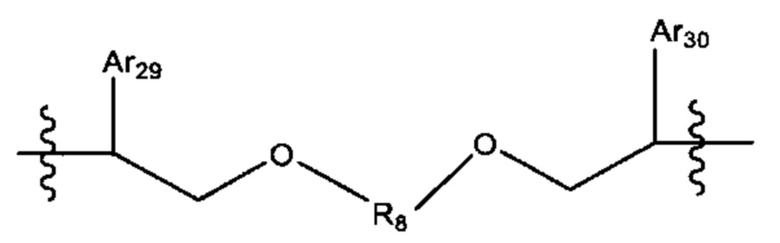
где Ar29 и Ar30 независимо выбраны из С6-С10 арила, и R8 выбран из группы, состоящей из:
 -(СН2)6-15-,- и -(СН2)d'-(С6-С10)арил-(СН2)е'-; где d' и е' выбраны независимо и лежат в диапазоне от 1 до 6.
-(СН2)6-15-,- и -(СН2)d'-(С6-С10)арил-(СН2)е'-; где d' и е' выбраны независимо и лежат в диапазоне от 1 до 6.
В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение, соответствующее Формуле (I), (Ia), (Ib), (II), (III), или его В другом аспекте настоящего изобретения предложен способ лечения или излечения ВИЧ-инфекции у субъекта, включающий введение указанному субъекту соединения Формулы (I), (Ia), (Ib), (II), (III) или его фармацевтически приемлемой соли.
В другом аспекте настоящего изобретения предложен способ истощения ВИЧ-инфицированных клеток HIV, включающий введение субъекту соединения Формулы (I), (Ia), (Ib), (II), (III) или его фармацевтически приемлемой соли.
В другом аспекте настоящего изобретения предложен истощения ВИЧ-инфицированных клеток HIV, включающий введение субъекту соединения Формулы (I), (Ia), (Ib), (II), (III) или его фармацевтически приемлемой соли и одного или более дополнительных агентов, обладающих активностью против ВИЧ. В некоторых аспектах эти агенты, обладающие активностью против ВИЧ, выбраны из группы, состоящей из антиретровирусных агентов, агентов, обращающих латентное состояние, и агентов для клиренсной терапии.
Эти и другие аспекты входят в объем изобретения, охарактеризованного в настоящем документе.
КРАТКОЕ ОПИСАНИЕ ФИГУР
ФИГ. 1 представляет собой график сравнения фармакокинетических (ФК) данных нескольких соединений Формулы I с ФК данными SMACm AZD5582.
ПОДРОБНОЕ ОПИСАНИЕ
Объект настоящего изобретения будет описан более подробно далее. Однако специалист в области техники, к которой относится объект настоящего изобретения, сможет представить многие модификации и другие варианты осуществления объекта настоящего изобретения, изложенного в данном документе, на основании идей, представленных в предшествующих описаниях. Соответственно, следует понимать, что объект настоящего изобретения не ограничивается конкретными раскрытыми вариантами осуществления, и что предполагается, что модификации и другие варианты осуществления включены в объем прилагаемой формулы изобретения. Другими словами, описанный здесь предмет охватывает все альтернативы, модификации и эквиваленты. В случае, если один или более из включенных литературных источников, патентов и подобных материалов отличаются от данной заявки или противоречат ей, включая, без ограничения перечисленным, определенные термины, использование терминов, описанные методы и т.п., приоритетной является настоящая заявка. Если не указано иное, все технические и научные термины, используемые в данном документе, имеют то же значение, которое обычно понимается специалистом в данной области. Все публикации, заявки на патенты, патенты и другие источники, упомянутые здесь, полностью включены в качестве ссылки.
Апоптоз, являющийся типом запрограммированной гибели клеток, играет важную роль в поддержании гомеостаза и регулировании количества клеток у высших организмов. Абнормальный апоптоз вовлечен в ряд заболеваний, включая аутоиммунные нарушения, дегенеративные заболевания центральной нервной системы, рак и вирусные инфекции, такие как ВИЧ. Семейство ингибиторов белков апоптоза (IAP) играет ключевую роль в подавлении передачи проапосигналов в клетках млекопитающих. Было показано, что SMACm, которые имитируют критическую тетрапептидную последовательность из второго митохондриального активатора каспазы, нарушают связывание LAP с их функциональным партнером и восстанавливают апоптотический ответ на проапоптотические стимулы в клетках. С начала 2000-х годов большие усилия были сосредоточены на разработке и получении миметиков SMAC в качестве антагонистов IAP, в частности, для стимуляции гибели опухолевых клеток, а в последнее время - для обращения латентности ВИЧ. В таких исследованиях изучали активацию неканонического пути NF-kB (ncNF-kB) в качестве потенциального механизма, по которому SMAC селективно имитирует истощение латентных клеток с ВИЧ. Примером раннего миметика SMAC является мономерный SBI-0637142, полученный исследователями из Института медицинских исследований Сэнфорда-Бернхэма. В исследованиях по истощению ВИЧ было обнаружено, что SBI-0637142 является сильнодействующим в анализах на клеточных линиях, но не проявляет активности в превращении р100-р52 или индукции каРНК ВИЧ в первичных клетках. Также была проведена большая работа по разработке двухвалентных миметиков, которые ковалентно связаны момомерными миметиками SMAC. AZD5582 от AstraZeneca и биринапант TL32711 от Medivir являются примерами димерных миметиков SMAC. В исследованиях изменения латентности ВИЧ биринапант TL32711 не проявлял активности в отношении Jurkat, превращении р100-р52 или индукции каРНК ВИЧ. Напротив, AZD5582 демонстрировал увеличение ассоциированной с клетками экспрессии РНК ВИЧ в покоящихся CD4+ Т-клетках в аналитических экспериментах с Jurkat, исследованиях превращения р100-р52 и индукции ВИЧ и каРНК (Sampey et al. BioRxiv 312447). Однако AZD5582 также может демонстрировать проблемы с переносимостью.
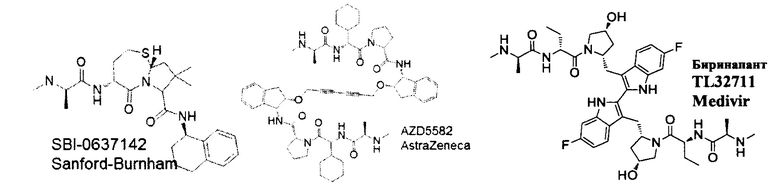
В настоящем документе раскрыты димерные SMACm, которые считаются достаточно мощными и достаточно эффективными, чтобы активировать ncNF-kB, обратить латентность ВИЧ в первичных немодифицированных человеческих клетках в качестве агентов монотерапии, и могут иметь меньшую токсичность, не связанную с мишенью, по сравнению с другими димерными SMACm, такими как AZD5582, что делает их пригодными для рассмотрения для дальнейшей разработки. В частности, димерный SMACm согласно изобретению способен индуцировать экспрессию РНК ВИЧ в нестимулированных, покоящихся первичных CD4+ Т-клетках ВИЧ-инфицированных доноров, виремия у которых полностью подавляется стандартной терапией. Считается, что другие SMACm, а именно мономерные молекулы или димерные молекулы с неоптимизированными линкерами не обладают этим эффектом в этих клетках, первичном латентном резервуаре персистирующей инфекции. Более того, димерный SMACm согласно изобретению способен к клинически измеримому изменению латентности в двух моделях на животных (SIV-инфицированные, макаки-резус с супрессией антиретровирусными и ВИЧ-инфицированные гуманизироанные мыши с супрессией препаратами APT), о чем свидетельствует периодическая виремия плазмы, которая временно возникает, несмотря на успешная продолжающуюся противовирусную терапию.
Следует понимать, что используемая в настоящем документе терминология, предназначена исключительно для цели описания конкретных вариантов осуществления и не предназаначена для ограничения объема настоящего изобретения. В этом описании и в следующей за ним формуле изобретения будет упоминаться ряд терминов, которые определены как имеющие следующие значения.
В настоящем документе, если не указано иное, "алкил" относится к одновалентной насыщенной алифатической карбильной группе, содержащей от 1 до 14 атомов углерода и, в некоторых вариантах осуществления, от 1 до 8 атомов углерода или от 1 до 6 атомов углерода. "(Сх-Су)алкил" относится к алкильным группам, содержащим от х до у атомов углерода. Термин "алкил" включает, в качестве примера, линейные или разветвленные гидрокарбильные группы, такие как метил (СН3), этил (СН3СН2), н-пропил (СН3СН2СН2), изопропил ((СН3)2СН), н-бутил (СН3СН2СН2СН2), изобутил ((СН3)2СНСН2), сек-бутил ((СН3)(СН3СН2)СН), трет-бутил ((СН3)3С), н-пентил (СН3СН2СН2СН2СН2) и неопентил ((СН3)3ССН2). Следует отметит, что упоминание, например, (C1-C12) алкила, также охватывает диапазоны внутри этой группы, например, (С1-С6)алкил.
"Алкилен" или "алкилен" относится к двухвалентным насыщенным алифатическим гидрокарбильным группам, содержащим от 1 до 10 атомов углерода и, в некоторых вариантах осуществления, от 1 до 6 атомов углерода. "(CuCv)алкилен" относится к алкиленовым группам, содержащим от u до v атомов углерода. Алкениленовые группы включают гидрокарбильные группы с разветвленной и линейной цепью. Например, подразумевается, что "(С1-С6)алкилен" включает метилен, этилен, пропилен, 2-метипропилен, диметилэтилен, пентилен и т.д. Соответственно, в качестве примера термина "пропилен" может быть приведена следующая структура:  . Аналогично, термин "диметилбутилен" можно проиллюстрировать любой из следующих трех структур или более:
. Аналогично, термин "диметилбутилен" можно проиллюстрировать любой из следующих трех структур или более: 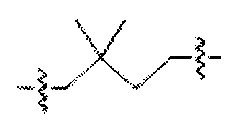 , р или
, р или 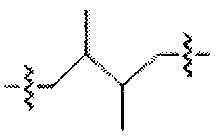 . Далее, подразумевается, что термин "(С1-С6)алкилен" включает такие разветвленные гидрокарбильные группы как циклопропилметилен, который может быть проиллюстрирован следующей структурой:
. Далее, подразумевается, что термин "(С1-С6)алкилен" включает такие разветвленные гидрокарбильные группы как циклопропилметилен, который может быть проиллюстрирован следующей структурой: 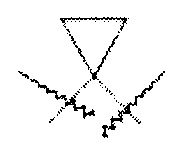 .
.
"Алкинил" или "алкин" относится к линейному одновалентному углеводородному радикалу или разветвленному одновалентному углеводородному радикалу, содержащему по меньшей мере одну тройную связь. Также подразумевается, что термин "алкинил" включает гидрокарбильные группы, содержащие одну тройную связь и одну двойную связь. Например, подразумевается, что (С2-С6) алкинил включает этинил, пропинил и т.п.
"Арил" относится к ароматической группе из от 6 до 14 атомов углерода без гетероатомов в кольце, содержащей одно кольцо (например, фенил) или множество конденсированных (сопряженных) колец (например, нафтил или антрил). В случае системы из множества колец, включая сопряженные, мостиковые и спиро-кольцевые системы, содержащей ароматические и неароматические кольца, которые не содержат гетероатомов в кольце, термин "Арил" или "Ar" применяется, когда точка присоединения находится на ароматическом атоме углерода (например, 5,6,7,8 тетрагидронафталин-2-ил представляет собой арильную группу, поскольку точка его присоединения находится в положении 2 ароматического фенильного кольца). В одном варианте реализации предпочтительная бициклическая арильная система может быть представлена формулой:
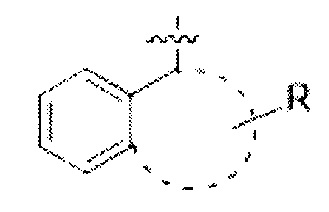
где размер кольца, конденсированного с С6 арильной группой лежит в диапазоне от 4 до 8.
В целях ясности, другие точки присоединения охватываются термином "Арил" или "Ar", например, точки присоединения на неароматическом атому углерода.
"Циклоалкил" относится к насыщенной или частично насыщенной циклической группе из от 3 до 14 атомов углерода и без гетероатомов, и имеющей единственное кольцо или множество колец, включая сопряженные, мостиковые и спиро-кольцевые системы. В случае системы из множества колец, содержащей ароматические и неароматические кольца и без гетероатомов в кольце, термин "циклоалкил" применяется, когда точка присоединения находится на неароматическом атоме углерода (например, 5,6,7,8,-тетрагидронафталин-5-ил). Термин "циклоалкил" включает циклоалкенильные группы, такие как циклогексенил.
Примеры циклоалкильных групп включают, например, адамантил, циклопропил, циклобутил, циклогексил, циклопентил, циклооктил, циклопентенил и циклогексенил. Примерами циклоалкильных групп, которые включают бициклоалкильные системы из множества колец, являются бициклогексил, бициклопентил, бициклооктил и т.п. Две такие бициклоалкильные системы из множества колец приведены в качестве примеров ниже; даны также их названия: 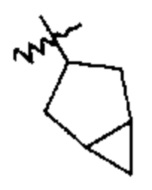 бициклогексил и
бициклогексил и 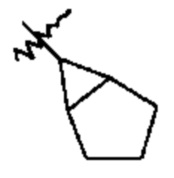 бициклогексил.
бициклогексил.
"(Cu-Cv)циклоалкил" относится к циклоалкил группе, содержащй от u до v атомов углерода.
"Спиро-циклоалкил" относится к 3-10-членному циклическому заместителю, образованному путем замены двух атомов водорода при общем атоме углерода в циклической кольцевой структуре или в алкиленовой группе, содержащей от 2 до 9 атомов углерода, примером которой является следующая структура, в которой показанная здесь группа присоединенная к связям, обозначенным волнистыми линиями, замещена спиро-циклоалкильной группой:
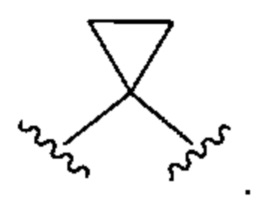
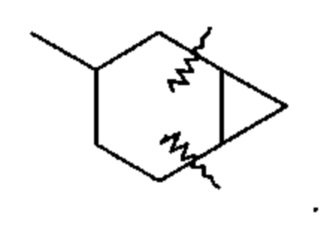
"Сопряженный циклоалкил" относится к 3-10-членному заместителю, образованному путем замены двух атомов водорода при разных атомах углерода в циклоалкильной кольцевой структуре; примером является следующая структура, в торой циклоалкильная группа, показанная здесь, содержит связи, обозначенные волнистыми линиями, которые присоединены к атомам углерода, которые замещены конденсированной циклоалкильной группой:
"AUC" относится к площади под графиком концентрации лекарственного средства в плазме (не логарифма концентрации) от времени после введения лекарственного средства.
"ЕС50" относится к концентрации лекарственного средства, которая обеспечивает половину максимального ответа. Иногда ее также преобразуют в шкалу рЕС50 (-log IC50), в соответствии в которой более высокие значения показывают экспоненциально большую эффективность.
"IC50" относится к обеспечивающей половину максимального ингибирования концентрации лекарственного средства. Иногда ее также преобразуют в шкалу pIC50 (-log IC50), в соответствии в которой более высокие значения показывают экспоненциально большую эффективность.
"Гетероарил" относится к ароматической группе из, например, и если не указано иное, от 1 до 14 атомов углерода (предпочтительно, от 1 до 12 атомов углерода, и более предпочтительно, от 2 до 12 атомов углерода) и от 1 до 6 гетероатомов (более предпочтительно от 1 до 3 гетероатомов), выбранных из кислорода, азота и серы, которая включает системы из одного кольца (например, имидазолил) и множества колец {например, бензимидазол-2-ил и бензимидазол-6-ил). В случае системы из множества колец, включая сопряженные, мостиковые и спиро-кольцевые системы, термин "гетероарил" применяется, если присутствует по меньшей мере один гитероатом в колье, и точка присоединения находится на атоме арометического кольца (например, 1,2,3,4-тетрагидрохинолин-6-ил т 5,6,7,8-тетрагидрохинолин-3-ил). В некоторых вариантах осуществления атом(ы) азота и/или серы кольца гетероарильной группы необязательно окислены с образованием фрагментов N-оксида (N→O), сульфинила или сульфонила. Префикс, указывающий число атомов углерода (например, Сх-Су) относится к общему числу атомов углерода в части гетероарильной группы, без учета числа гетероатомов. Также эта группа включает все диапазоны между х и у, например, С1-С14 включает С2-С14, С2-С9 и т.д. Более конкретно, термин «гетероарил» включает, без ограничения перечисленными: пиридил, фуранил, тиенил, тиазолил, изотиазолил, триазолил, имидазолил, имидазолинил, изоксазолил, пирролил, пиразолил, пиридазинил, пиримидинил, пуринил, фталазил, нафтилпиридил, бензофуранил, тетрагидробензофуранил, изобензофуранил, бензотиазолил, бензоизотиазолил, бензотриазолил, индолил, изоиндолил, индолизинил, дигидроиндолил, индазолил, индолинил, бензоксазолил, хинолил, изохинолил, хинолизил, хиназолил, хиноксалил, тетрагидрохинолинил, изохинолил, хиназолинонил, бензимидазолил, бензизоксазолил, бензотиенил, бензопиридазинил, птеридинил, карбазолил, карболинил, фенантридинил, акридинил, фенантролинил, феназинил, феноксазинил, фенотиазинил, фталимидил и тетразол. В одном варианте реализации например, представлена (С3-С9) гетероарильная спиро-кольцевая конденсированная система колец более предпочтительно, (С4-С6) гетероарил.
"Гетероциклический", или "гетероцикл", или "гетероциклоалкил", или "гетероциклил" относится к насыщенной или частично насыщенной группе, содержащей, например, и если не указано другое, от 1 до 14 атомов углерода (предпочтительно, от 1 до 12 атомов углерода и более предпочтительно от 2 до 12 атомов углерода) и от 1 до 6 гетероатомов (более предпочтительно, от 1 до 3 гетероатомов), выбранных из азота, серы, фосфора или кислорода, и включает системы из одного кольца или множества колец, включая конденсированные, мостиковые и спиро-кольцевые системы. Для систем из множества колец, содержащих ароматические и/или неароматические кольца, термины "гетероциклический", "гетероцикл", "гетероциклоалкил" или "гетероциклил" применяются в тех случаях, когда присутствует по меньшей мере гетероатом в кольце, и точка присоединения находится на атоме неарометического кольца (например, 1,2,3,4-тетрагидрохинолинил-3-ил, 5,6,7,8-тетрагидрохинолинил-6-ил и декагидрохинолин-6-ил). В одном варианте реализации атом(ы) азота фосфора и/или серы гетероциклической группы необязательно окислены с образованием фрагмента N-оксида, фосфаминоксида, сульфинила, сульфонила. Более конкретно, гетероциклил включает, без ограничения перечисленными: тетрагидропиранил, пиперидинил, пиперазинил, 3-пирролидинил, 2-пирролидон-1-ил, морфолинил и пирролидинил. Префикс, указывающий число атомов углерода (например, Cx-Су) относится к общему числу атомов углерода в части гетероарильной группы, без учета числа гетероатомов. Также эта группа включает все диапазоны между х и у, например, С1-С14 включает С2-С14, С2-С9 и т.д.
В одном варианте реализации присутствует, например, (С3-С9) гетероциклическая конденсированная спиро-кольцевая система, более предпочтительно, (С4-С6) гетероцикл.
Примеры гетероциклической и гетероарильной включают, без ограничения перечисленными: азетидин, пиррол, имидазол, пиразол, пиридин, пиразин, пиримидин, пиридазин, пиридон, индолизин, изоиндол, индол, дигидроиндол, индазол, пурин, хинолизин, изохинолин, хинолинил, фталазин, нафтилпиридин, хиноксалин, хиназолин, циннолин, птеридин, карбазол, карболин, фенантридин, акридин, фенантролин, изотиазол, феназин, изоксазол, феноксазин, фенотиазине, имидазолидин, имидазолин, пиперидин, пиперазин, индолин, фталимид, 1,2,3,4 тетрагидроизохинолин, 4,5,6,7 тетрагидробензо[b]тиофен, тиазол, тиазолидин, тиофен, бензо[b]тиофен, морфолин, тиоморфолин (также называемый тиаморфолином), пиперидин, пирролидин и тетрагидрофуранил.
В дополнение к вышеупомянутым вариантам осуществления, изложенным в данном документе, «конденсированный гетероциклический» или «конденсированный гетероцикл» относятся к 3-9-членному (предпочтительно, 4-6-членному) циклическому заместителю, образованному заменой двух атомов водорода у разных атомов углерода в циклоалкильном кольце, как проиллюстрировано следующей структурой, в которой циклоалкильная группа, показанная здесь, содержит связи, показанные волнистыми линиями, которые связаны с атомами углерода, которые замещены конденсированной гетероциклической группой:
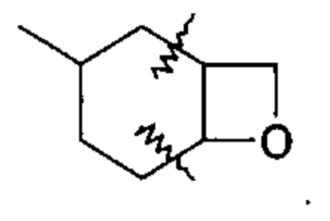
Один из вариантов осуществления спиро-кольцевой системы (например, без ограничения, образованной из R2 и R3) в формуле (I), (Ia), (Ib), (II) или (III) включает, без ограничения:
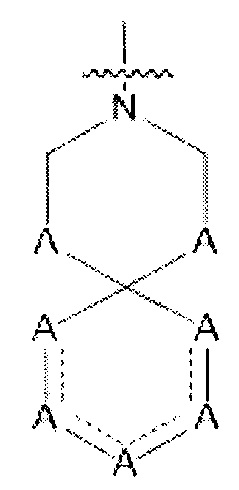 ,
,
где А может быть выбран из, например С(О), О, N, В, В(ОН), SI или Р, P(OR).
"Соединение", "соединения", "химическое соединение" и "химические соединения" в настоящем документе относится к соединению, охватываемому общими формулами, раскрытыми в настоящем документе, любыми подродами этой общей формулы, и любые формы соединения в рамках общей формулы и подкатегорий общей формулы, включая рацематы, стереоизомеры и таутомеры таких соединения или соединений.
Термин "гетероатом" обозначает, азот, кислород или серу и включает любые окисленньк формы азота, такие как N(O) {N+-О-}, и серы, такие как S(O) и S(O)2, и кватернизированные формы любого основного азота.
"Линкер" ("L") относится к веществу (например, молекуле), которая связывает две части молекулы.
"Полиморфизм" относится к ситуации, когда два или более четко различающихся фенотипа существуют в одной популяции видов, обуславливая существование более, чем одной формы или морфы. Чтобы считаться таковыми, морфы должны занимать одно обитания в одно время и относится к панмиктической популяции (со свободным скрещиванием).
"Связывание белка" относится к связыванию лекарственного средства с белками в плазме крови, мембранах ткани, эритроцитах и других компонентах крови.
"Белковый сдвиг" относится к определению сдвига связывания путем сравнения значений ЕС50, определенных в отсутствие и в присутствии сыворотки человека.
Термин "рацематы" относится к смеси энантиомеров. В одном из вариантов осуществления изобретения, соединения Формулы I, Ia, Ib, II и III, или их фармацевтически приемлемые соли, являются энантиомерно обогащенными одним энантиомером, в котором все указанные хиральные атомы углерода находятся в одной конфигурации. Обычно указание на энантиомерно обогащенное соединение или соль подразумевает, что указанный энантиомер составляет более 50% по массе от общей массы всех энантиомеров указанного соединения или соли.
"Сольват" или "сольваты" соединения относятся к соединениям, определенным выше, которые связаны со стехиометрическим или нестехиометрическим количеством растворителя. Сольваты соединения включает сольваты всех форм соединения. В некоторых вариантах осуществления растворители являются волатильными, нетоксичными и/или приемлемыми для введению человека в следовых количествах. Подходящие сольваты включают воду.
"Стереоизомер" или "стереоизомеры" относятся к соединениям, которые различаются хиральностью одного или более стереоцентров. Стереоизомеры включают энантиомеры и диастереомеры, включая соединения Формулы (I), (Ia), (Ib), (II) и (III) линкеры (L) Формул с (I) по (xiii), описанные здесь, а также линкеры, описанные в формулах (II) и (III).
"Таутомер" относятся к переходящим друг в друга формам соединения, которые различаются положением протона, таким как кето-енольные и имин-/енминовые таутомеры, или таутомерные формы гатароарильных групп, в которых один из атомов кольца присоединен как фрагменту NH кольца и фрагменту =N кольца, такие как пиразолы, имидазолы, бензимидазолы, триазолы и тетразолы.
Термин "атропизомер" относится к стереоизомеру, порожденному осью асимметрии. Это может происходить из-за затрудненного вращения вокруг одинарной связи, в тех случаях, когда барьер вращения достаточно высок для того чтобы допускать дифференциацию изомерных вариантов до и включая полное выделение стабильного, не склонного к взаимопревращениям, диастереомерного или энантиомерного варианта. Для специалиста понятно, что при введении несимметричного Rx в ядро, становится возможным образование атропоизомеров. Кроме того, при введении хирального центра в молекулу, включающую атропоизомер, два хиральных элемента совместно могут образовывать диастереомерные и энантиомерные стереохимические варианты. В зависимости от замещения вокруг оси Сх, взаимные превращения атропоизомеров могут быть возможны или невозможны, и могут зависеть от температуры. В некоторых случаях атропоизомеры могут испытывать быстрые взаимные превращения при комнатной температуре и не разделяться при обычных условиях. Другие ситуации могут допускать разделение и выделение, но взаимное превращение может происходить в течение периода продолжительностью от секунд до часов или даже дней или месяцев, так, что оптическая чистота измеримо снижается со временем. Для других молекул взаимные превращения при температуре окружающей среды и/или повышенных температурах могут быть затруднены до полной невозможности, что делает возможным разделение и выделение и дает стабильные варианты молекул. В известных случаях для присваивания названий атропоизомерам используется спиральная номенклатура. Для таких обозначений учитываются только два лиганда с максимальным приоритетом, перед осью и за ней. Если приоритетное направление поворота от переднего лиганда 1 к заднему лиганду соответствует движению по часовой стрелки, такая конфигурация называется Р, а если движению против часовой стрелки, то М.
"Фармацевтически приемлемая соль" относится к фармацевтически приемлемым солям, образованным различными органическими и неорганическими противоионами, хорошо известными в данной области, включающим, исключительно в качестве примера, натрий, калий, кальций, магний, аммоний и тетраалкиламмоний, и в случаях, когда молекула содержит основную функциональную группу, соли органических или неорганических кислот, таких как гидрохлорид, гидробромид, тартрат, мезилат, ацетат, малеат и оксалат. Подходящие соли включают соли, описанные в P. Heinrich Stahl, Camille G. Wermuth (Eds.), Handbook of Pharmaceutical Salts Properties, Selection, and Use; 2002.
"Пациент" или "субъект" относится к млекопитающим и включает людей и млекопитающих, не являющихся людьми.
"Лечение" или "процесс лечения" заболевания у пациента относится к 1) предотвращению заболевания у пациента, который предрасположен или который еще не проявляет симптомы заболевания; 2) подавлению заболевания или блокированию его развития; или 3) облегчению или обеспечению обращения заболевания.
Как указано выше, «лечение» нарушения включает предотвращение нарушения. Специалист в данной области техники поймет, что «предотвращение» не является абсолютным термином. В медицине «предотвращение» относится к профилактическому введению лекарственного средства для существенного уменьшения вероятности или тяжести нарушения или его биологического проявления или для отсрочки начала такого нарушения или его биологического проявления.
В настоящем документе термины «с перерывами» или «интервально» означают остановку и начало с регулярными или нерегулярными интервалами. В настоящем документе термин «вирусная инфекция» описывает болезненное состояние, при котором вирус проникает в здоровые клетки, использует репродуктивный аппарат клетки для размножения или репликации и, в конечном итоге, лизиса клетки, что приводит к гибели клетки, высвобождению вирусных частиц и заражению других клеток вновь образованными дочерними вирусами. Возможным результатом вирусной инфекции также может быть латентная инфекция некоторыми вирусами.
В настоящем документе термин «лечение вирусных инфекций» обозначает ингибирование репликации конкретного вируса, ингибирование передачи вируса и облегчение или ослабление симптомов заболевания, вызванного вирусной инфекцией. Лечение считается «терапевтическим», если наблюдается снижение вирусной нагрузки, снижение смертности и/или заболеваемости. «Предотвращение вирусных инфекций» означает предотвращение закрепления вируса в организме хозяина. Лечение считается «профилактическим», если субъект подвергается воздействию вируса, но не заражается вирусом в результате лечения.
В настоящем документе термин «латентность» означает понятие, описывающее 1) состояние покоя вирусной активности в популяции клеток, при котором продуцирование вируса, упаковка вируса и лизис клетки-хозяина не происходят или происходят с очень низкой частотой, или 2) понижающее регулирование или отсутствие экспрессии гена в инфицированной клетке.
В настоящем тексте "обращение латентной ВИЧ-инфекции" относится к лечению, которое повышающе регулирует экспрессию встроенных геномов ВИЧ в латентно инфицированных клетках, например, как агент, который активирует неканонический путь NF-кВ, что делает клетки чувствительными к вызываемой вирусом клеточной гибели или иммунологическому клиренсу. В настоящем тексте "истощение латентной ВИЧ-инфекции" относится к клиренсу латентно инфицированных ВИЧ клеток, который может последовать за обращением латентности ВИЧ такими реагентами, как те, которые активируют неканонический путь NF-kB.
В некоторых вариантах осуществления латентные инфицированные ВИЧ клетки представляют собой покоящиеся CD4+ Т-клетки.
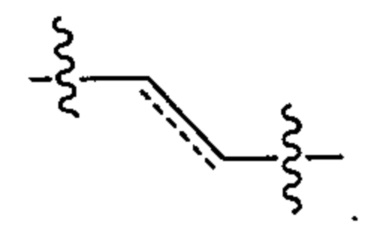
В тех случаях, где пунктирные линии встречаются рядом с одинарными связями, обозначенными сплошными линиями, пунктирная линия представляет собой необязательную двойную связь в этом положении. Аналогично, в тех случаях, где встречаются пунктирные кружки в кольцевых структурах, обозначенных сплошными линиями или сплошными кружками, пунктирные кружки представляют от одной до трех необязательных двойных связей, расположенных в соответствии с их правильной валентностью, с учетом того, есть ли в кольце какие-либо необязательные замещения вокруг кольца, как известно специалисту в данной области. Например, пунктирная линия в приведенной ниже структуре может указывать либо на двойную связь в данном положении, либо на одинарную связь в данном положении:
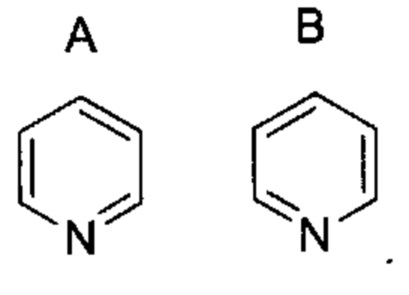
В случае, когда изображены конкретные соединения или общие формулы, содержащие ароматические кольца, такие как арильные или гетероарильные кольца, для специалиста будет понятно, что конкретное ароматическое положение любых двойных связей представляет собой комбинацию эквивалентных положений, даже если они изображены в разных положениях для разных соединений или для разных формул. Например, в двух показанных ниже пиридиновых кольцах (А и В), двойные связи изображены в разных положениях, тем не менее, известно, что они представляют одну и ту же структуру и одно и то же соединение:
Настоящее изобретение включает соединения, а также их фармацевтически приемлемые соли. Соответственно, слово "или" в контексте "соединение или его фармацевтически приемлемая соль" следует понимать, как относящееся к одному из следующих вариантов: 1) только соединение или соединение и его фармацевтически приемлемая соль (альтернативы), или 2) соединение и его фармацевтически приемлемая соль (в комбинации).
Если не указано иное, названия заместителей, которые не определены в настоящем документе в явном виде, образованы путем приведения названия концевой части функциональной группы, за которой следует соседняя функциональная группа в направлении точки присоединения. Например, заместитель "арилалкилоксикарбонил" относится к группе (арил)(алкил)ОС(O). В таких терминах как "-C(Rx)2", подразумевается, что две группы Rx могут быть одинаковыми или они могут быть разными, если Rx определен как имеющий более одной возможных форм. Дополнительно, некоторые заместители изображены как -RxRy, где "-" обозначает связь, прилежащую к основной молекуле, и Ry представляет собой концевую часть функциональной группы. Аналогично, понятно, что приведенные выше определения не включают недопустимые варианты замещения (например, метил, замещенный 5 фтор-содержащими группами). Такие недопустимые варианты замещения хорошо известны специалистам.
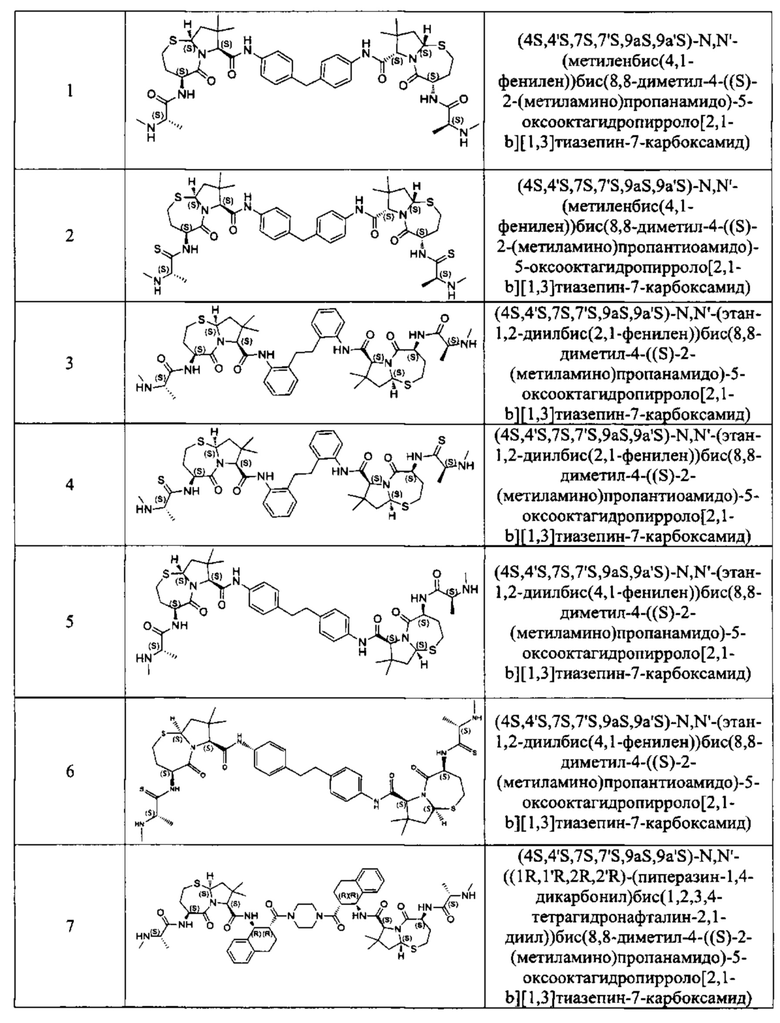
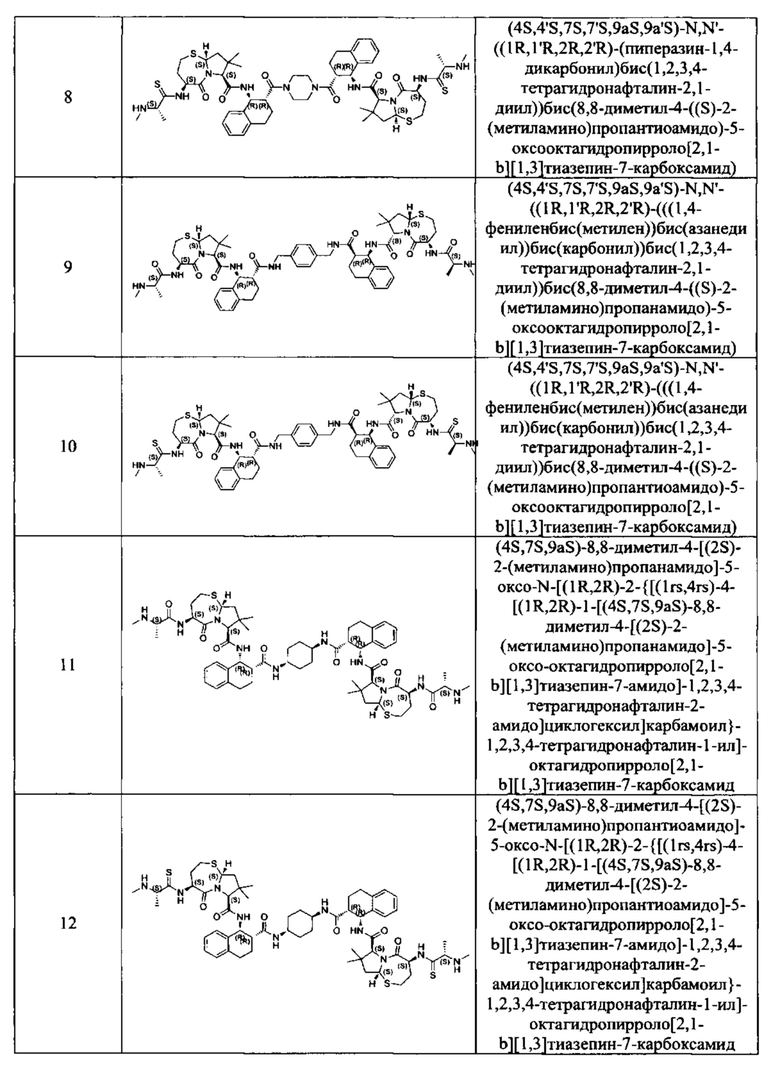
Согласно настоящему изобретению предложены соединения Формул (I), (Ia), (Ib), (II) и (III), а также в настоящем документе описаны различные формы этих соединений (например, фармацевтически приемлемые соли). Следует понимать, что подразумевается, что любое указание на соединения Формул (I), (Ia), (Ib), (II) и (III) в настоящем документе также в явном виде включает, без ограничения, соединения, представленные в Таблице 1.
В одном аспекте настоящего изобретения предложено соединение структуры, соответствующей Формуле (I):
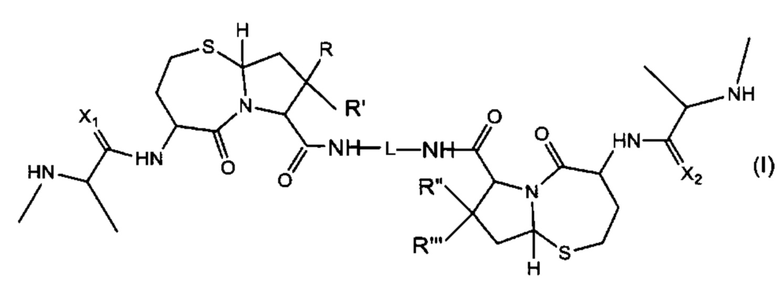
или его фармацевтически приемлемая соль или стереоизомер, где:
R, R', R'' и R''' независимо выбраны из Н и СН3;
X1 и Х2 независимо выбраны из группы, состоящей из О и S; и
L представляет собой линкер, выбранный из группы, состоящей из:
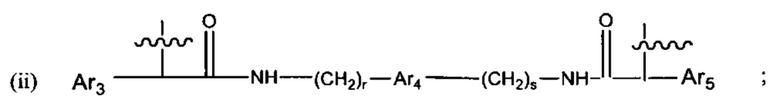
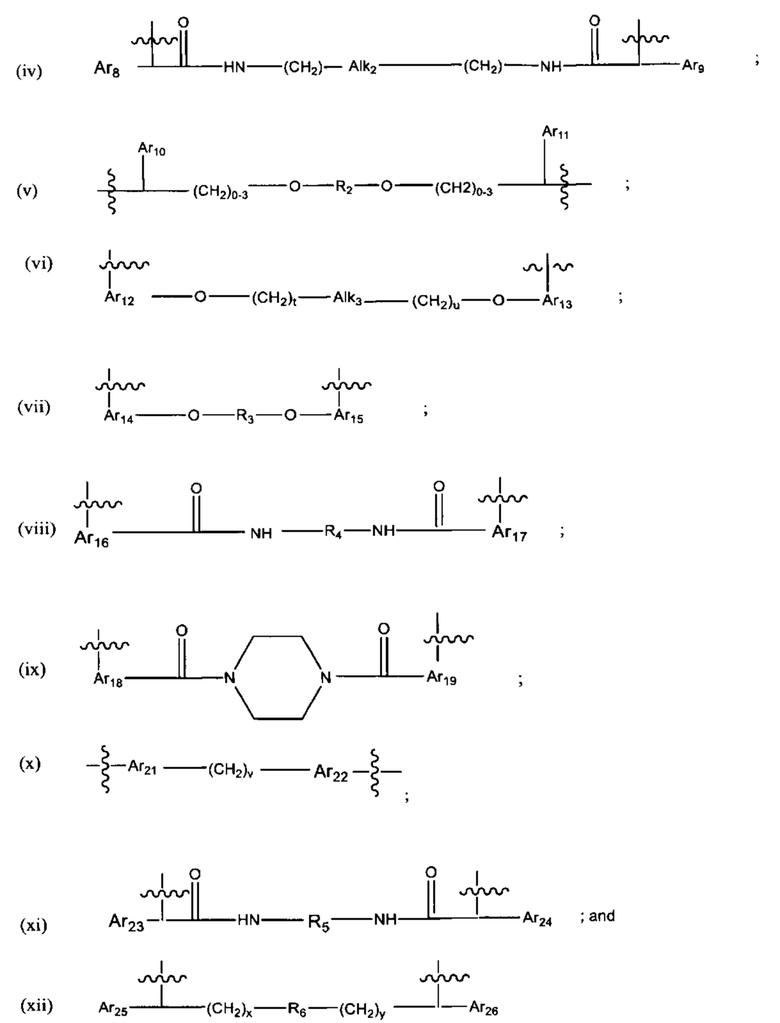
где:
Ar1, Ar2, Ar3, Ar4, Ar5, Ar6, Ar7, Ar8, Ar9, Ar10, Ar11, Ar12, Ar13, Ar14, Ar15, Ar16, Ar17, Ar18, Ar19, Ar21, Ar22, Ar23, Ar24, Ar25 и Ar26 каждый независимо выбран из (C6-C14)арила;
Alk, Alk2 и Alk3 каждый независимо выбран из:
R1, представляет собой С3-С6 циклоалкил или C1-C6 гетероцикл;
R2 выбран из группы, состоящей из -(СН2)а-, -(СН2)b-O-(СН2)с-, -(CH2)d-(C6-C14)арил-(СН2)е- и -(СН2)f-(С1-С6)гетероарил-(СН2)g-; более предпочтительно, представляет собой С2 гетероарил
R3 выбран из группы, состоящей из -(CH2)h-; -(CH2)i-O-(CH2)j-, -(СН2)k-(С6-С14)арил-(СН2)l- и -(СН2)m'-(С1-С6)гетероарил-(СН2)m''-;
R4 представляет собой С3-С6 циклоалкил, (С6-С14)арил или (СН2)n'-(С6-С14)арил-(CH2)n'', (CH2)n'''-Alk-(CH2)n'''', где n', n'', n''' и n'''' независимо выбраны из от 1 до 8
R5 представляет собой С3-С6 циклоалкил;
R6 выбран из группы, состоящей из (СН2)z,
и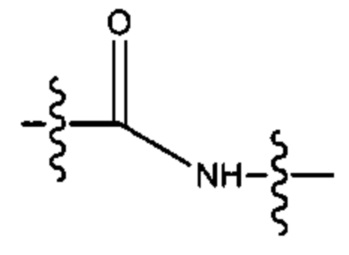
a, b, с, d, е, f, г, h, i, j, k, l, m, m', m'', n, p, q, r, s, t, u, v, x, у и z каждый независимо выбран из от 1 до 12.
В другом аспекте настоящее изобретение относится к соединению Формулы (Ia):
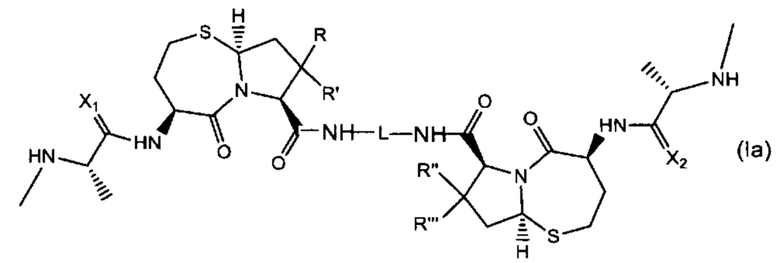
или его фармацевтически приемлемой соли или стереоизомеру, где:
R, R', R'' и R''' независимо выбраны из Н и СН3;
X1 и Х2 независимо выбраны из группы, состоящей из О и S; и
L представляет собой линкер, выбранный из группы, состоящей из:
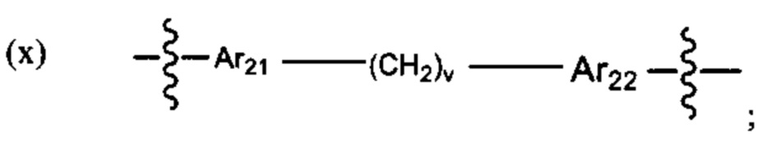
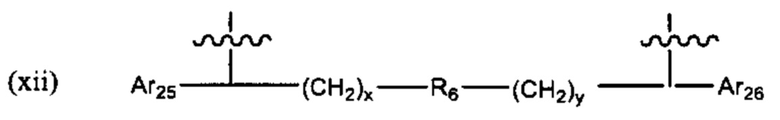
где:
Ar1, Ar2, Ar3, Ar4, Ar5, Ar6, Ar7, Ar8, Ar9, Ar10, Ar11, Ar12, Ar13, Ar14, Ar15, Ar16, Ar17, Ar18, Ar19, Ar21, Ar22, Ar23, Ar24, Ar25 и Ar26 каждый независимо выбран из (C6-C14)арила;
Alk, Alk2 and Alk3 каждый независимо выбран из:
R1, представляет собой С3-С6 циклоалкил или C1-C6 гетероцикл;
R2 выбран из группы, состоящей из -(СН2)а-, -(СН2)b-O-(СН2)с-, -(CH2)d-(C6-C14)арил-(СН2)c- и -(СН2)f-(С1-С6)гетероарил-(СН2)g-;
R3 выбран из группы, состоящей из -(СН2)h-; -(CH2)i-O-(CH2)j-, -(СН2)k-(С6-С14)арил-(CH2)l- и -(СН2)m'-(С1-С6)гетероарил-(СН2)m''-;
R4 представляет собой С3-С6 циклоалкил, (С6-С14)арил или (СН2)n'-(С6-С14)арил -(СН2)n'', (СН2)n'''-Alk-(СН2)n'''', где n', n'', n''' и n'''' независимо выбраны из от 1 до 8
R5 представляет собой С3-C6 циклоалкил;
R6 выбран из группы, состоящей из (CH2)z,

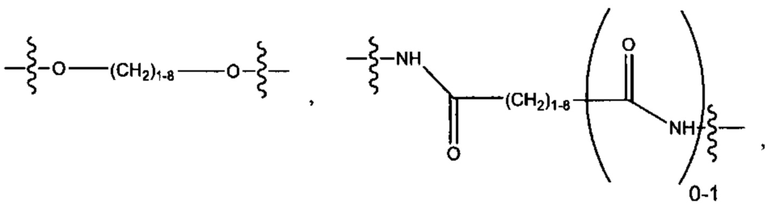
и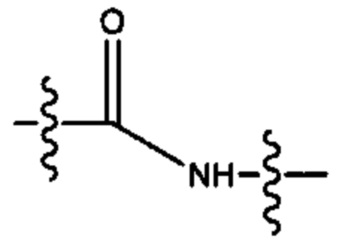
a, b, с, d, е, f, г, h, i, j, k, l, m, m', m'', n, p, q, r, s, t, u, v, x, у и z каждый независимо выбран из от 1 до 12.
В другом аспекте настоящее изобретение относится к соединению Формулы (Ib):
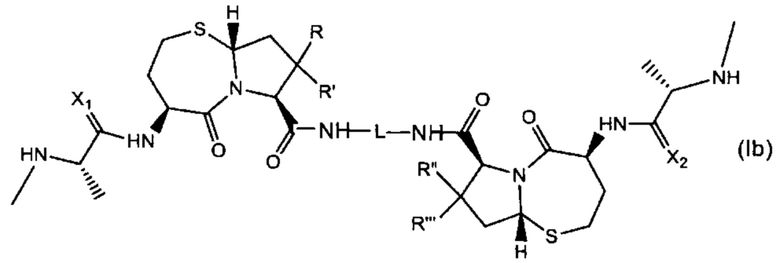
или его фармацевтически приемлемой соли или стереоизомеру, где:
R, R', R'' и R''' независимо выбраны из Н и СН3;
X1 и Х2 независимо выбраны из группы, состоящей из О и S; и
L представляет собой линкер, выбранный из группы, состоящей из:
где:
Ar1, Ar2, Ar3, Ar4, Ar5, Ar6, Ar7, Ar8, Ar9, Ar10, Ar11, Ar12, Ar13, Ar14, Ar15, Ar16, Ar17, Ar18, Ar19, Ar21, Ar22, Ar23, Ar24, Ar25 и Ar26 каждый независимо выбран из (С6-С14)арил;
Alk, Alk2 и Alk3 каждый независимо выбран из:
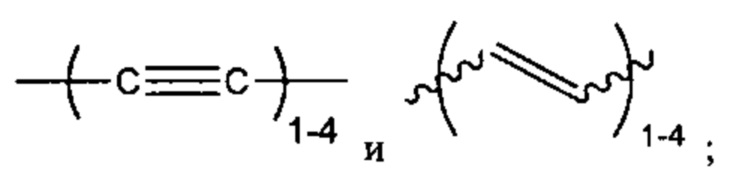
R1, представляет собой С3-С6 циклоалкил или C1-C6 гетероцикл;
R2 выбран из группы, состоящей из -(СН2)а-, -(СН2)b-O-(СН2)с-, -(СН2)d-(С6-С14)арил-(СН2)e- и -(CH2)f-(C1-C6)гетероарил-(CH2)g-;
R3 выбран из группы, состоящей из -(CH2)h-; -(CH2)i-O-(CH2)j-, -(СН2)k-(С6-С14)арил-(СН2)l- и -(СН2)m'-(С1-С6)гетероарил-(СН2)m''-;
R4 представляет собой С3-С6 циклоалкил, (С6-С14)арил или (СН2)n'-(С6-С14)арил -(СН2)n'', (CH2)n'''-Alk-(CH2)n'''', где n', n'', n''' и n'''' независимо выбраны из от 1 до 8
R5 представляет собой С3-С6 циклоалкил;
R6 выбран из группы, состоящей из (CH2)z,
и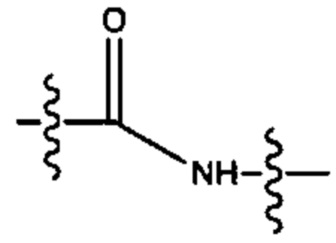
a, b, с, d, е, f, г, h, i, j, k, l, m, m', m'', n, p, q, r, s, t, u, v, x, у и z каждый независимо выбран из от 1 до 12.
В различных вариантах осуществления и в дополнение к описанному выше, каждая из переменных а, b, с, d, е, f, г, h, i, j, k, l, m, m', m'', n, p, q, r, s, t, u, v, x, у и z для Формул (I), (Ia), (Ib), (II) и (III) может быть выбрана из от 1 до 8
В различных вариантах осуществления по меньшей мере один из R'' и R''' представляет собой СН3. Предпочтительно, оба из R'' и R' каждый представляет собой СН3.
Наиболее предпочтительно, каждый из R, R', R'' и R''' представляет собой СН3.
В различных вариантах осуществления X1 и Х2 оба представляют собой О.
В различных вариантах осуществления X1 и Х2 оба представляют собой S.
В различных вариантах осуществления X1 представляет собой О, и Х2 представляет собой S.
В различных вариантах осуществления Xi представляет собой S, и Х2 представляет собой О.
В различных вариантах осуществления каждый из Alk, Alk2 и Alk3 предпочтительно представляет собой:
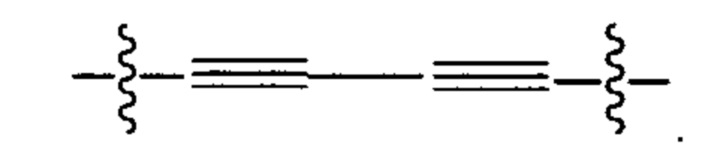
В различных вариантах осуществления каждый из Ar1, Ar2, Ar3, Ar4, Ar5, Ar6, Ar7, Ar8, Ar9, Ar10, Ar11, Ar21, Ar22, Ar23, Ar24, Ar25 и Ar26 может представлять собой С6 арил.
В различных вариантах осуществления каждый из Ar12, Ar13, Ar14, Ar15, Ar16, Ar17, Ar18 и Ar19 может представлять собой С9 арил. В предпочтительных вариантах реализации С9 арил представлен формулой
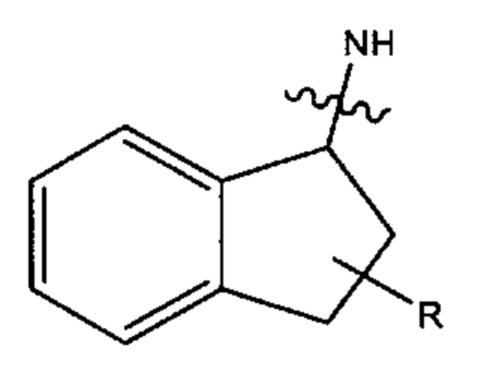
где азот в NH соединяется амидной связью с бициклической системой (системами) соединения, и R обозначает присоединение к линкеру.
В различных вариантах осуществления каждый из Ar16, Ar17, Ar18 и Ar19 представляет собойС10арил. В предпочтительных вариантах реализацииС10арил представлен формулой
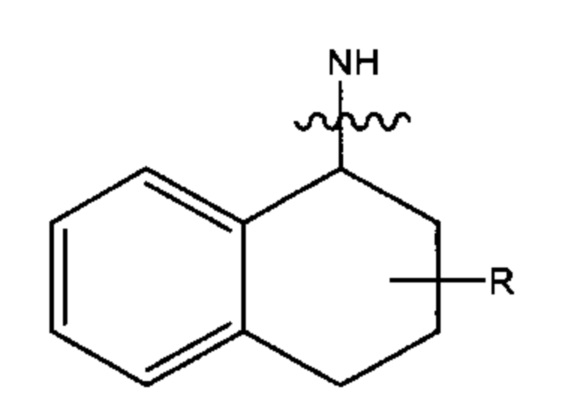
где азот в NH соединяется амидной связью с бициклической системой (системами) соединения и R обозначает присоединение к линкеру.
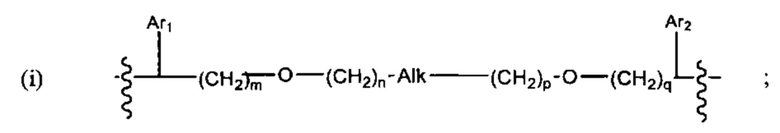
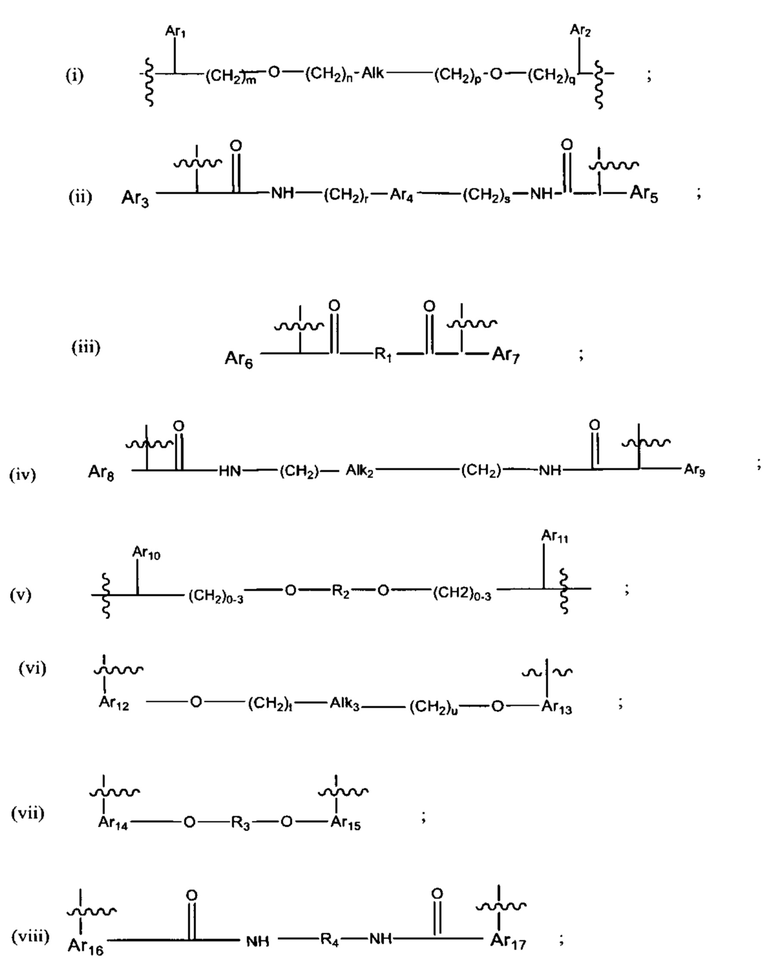
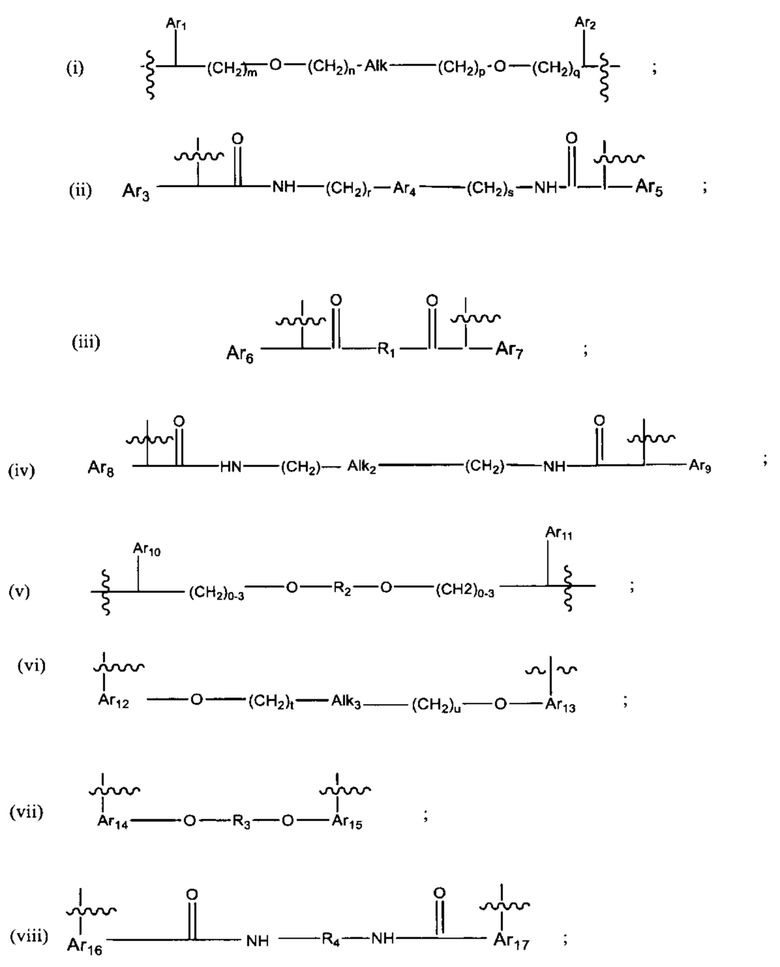
В различных вариантах осуществления линкер имеет формулу (i):
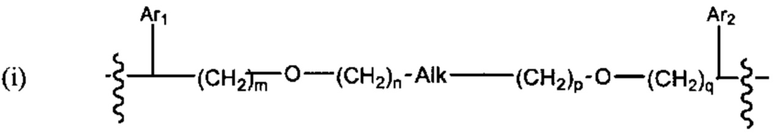
Каждый из Ar1 и Ar2 может быть независимо выбран из C6-C9 арила
Предпочтительно, в формуле (i), Ar1 представляет собой С6 арил. Alk представляет собой:
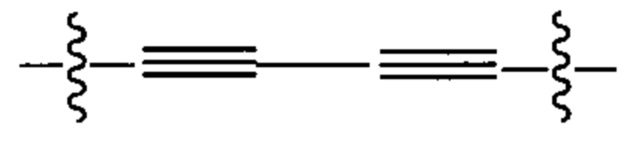
Ar2 представляет собой С6 арил, m представляет собой 1, n представляет собой 1, р представляет собой 1, и q представляет собой 1. В одном варианте реализации X1 представляет собой S, и Х2 представляет собой S. В одном варианте реализации X1 представляет собой S, и Х2 представляет собой О.
В различных вариантах осуществления линкер представляет собой формулу (ii):
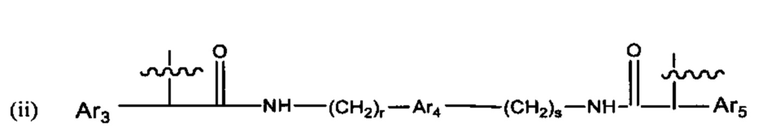
Каждый из Ar3, Ar2 и Ar5 может быть независимо выбран из С6-С9 арила Предпочтительно в формуле (ii), Ar3 представляет собой С6 арил, Ar4 представляет собой С6 арил, r представляет собой 1, Ar4 представляет собой С6 арил, s представляет собой 1, и Ar5 представляет собой С6 арил. В одном варианте реализации X1 представляет собой О, и Х2 представляет собой О. В одном варианте реализации X1 представляют собой S, и Х2 представляет собой S.
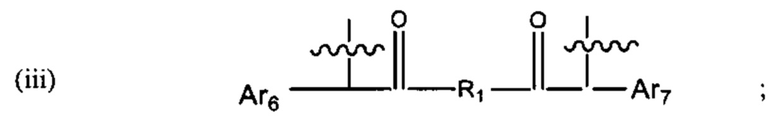
В различных вариантах осуществления линкер имеет формулу (iii):
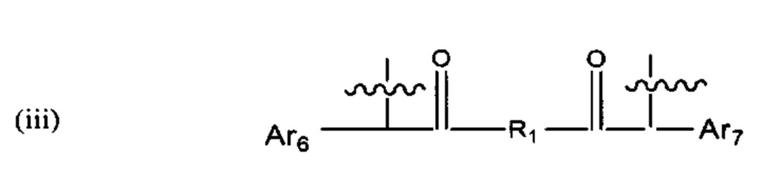
где Ar6 и Ar7 каждый независимо представляет собой С6-С9 арил, наиболее предпочтительно каждый представляет собой С6 арил;
где R1 предпочтительно представляет собой С1-С6 гетероцикл (например, С4 гетероцикл), наиболее предпочтительно:
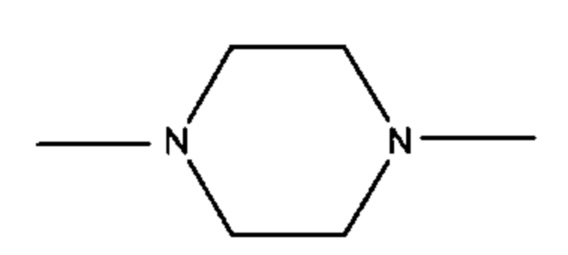
В различных вариантах осуществления линкер имеет формулу (iv):
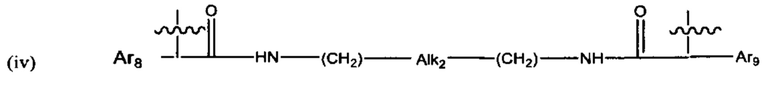
Предпочтительно в формуле (iv), Ar8 представляет собой С6-9 арил, более предпочтительно С6 арил, и Alk2 представляет собой:
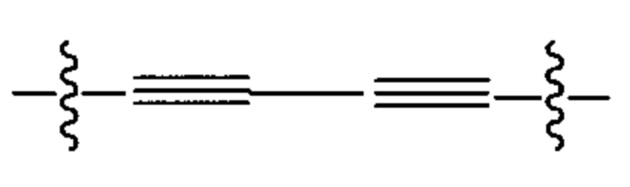
и Ar9 представляет собой С6 арил; С6-9 арил, более предпочтительно С6 арил. В различных вариантах осуществления линкер имеет формулу (v):
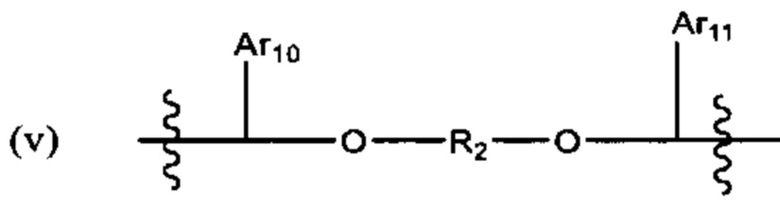
Предпочтительно в формуле (v), Ar10 представляет собой С6-9 арил, более предпочтительно С6 арил, Ar11 предпочтительно представляет собой С6-9 арил, более предпочтительно С6 арил, и R2 выбран из -(СН2)-С6-9 арил-(СН2)-, (более предпочтительно, -(СН2)-С6 арил-(СН2)-), -(CH2)1-8- (более предпочтительно -(СН2)4-, -(СН2)3-, или-(СН2)6-) и -(СН2)2-6-O-(СН2)2-6 - (более предпочтительно -(СН2)2-4-O-(СН2)2-4). В одном варианте реализации R2 представляет собой -(СН2)2-O-(СН2)2-. Если R2 представляет собой гетероарил, он предпочтительно представляет собой С2 гетероарил.
В различных вариантах осуществления линкер имеет формулу (vi):
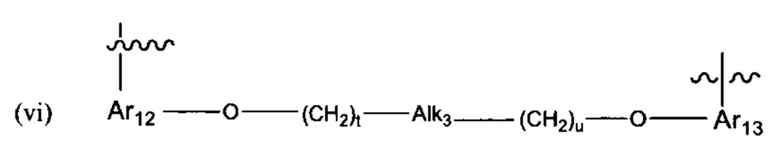
Предпочтительно в формуле (vi), Ar12 представляет собой С6-9 арил, t представляет собой 1-4 (более предпочтительно, t представляет собой 1), Alk3 представляет собой:
и представляет собой 1-4 (более предпочтительно, и представляет собой 1), и Ar13 представляет собой С6-9 арил. Наиболее предпочтительно, Ar12 и Ar13
каждый представляет собой С9арил, и наиболее предпочтительно, С9 арил представлен следующей формулой:
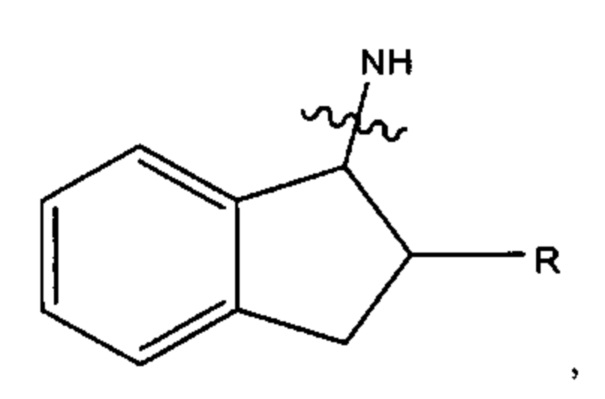
где азот в NH соединяется амидной связью с бициклической системой (системами) соединения, и R обозначает присоединение к линкеру.
В различных вариантах осуществления линкер имеет формулу (vii):
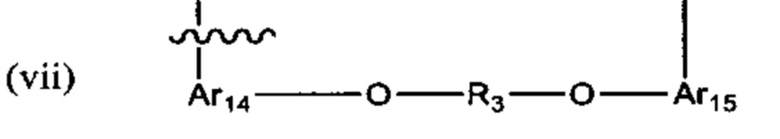
Предпочтительно в формуле (vii), Ar14 представляет собой С6-9 арил (более предпочтительно, С9 арил), R3 выбран из группы, состоящей из (СН2)1-12 (например, (СН2)4-6); -(СН2)1-6 -O-(CH2)1-6- (например, -(СН2)2-4-O-(СН2)2-4-), и Ar15 представляет собой С9 арил. Более предпочтительно, в формуле (vii), Ar14 представляет собой С9 арил, R3 выбран из группы состоящей из -(CH2)4-12-, -(СН2)2-4-O-(СН2)2-4-,
В предпочтительных вариантах реализации Ar14 и Ar15 каждый С9 следующий арил:
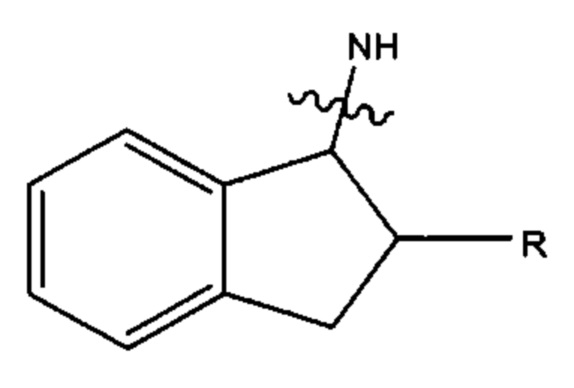
где азот в NH соединяется амидной связью с бициклической системой (системами) соединения и R обозначает присоединение к линкеру.
В различных вариантах осуществления линкер имеет формулу (viii):
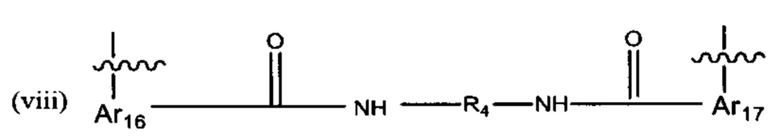
Предпочтительно, Ar16 представляет собой С6-10 арил, более предпочтительно, С9 арил или С10 арил, R4 выбран из группы, состоящей из С3-С6 циклоалкила или (С6-С14)арила и Ar17 представляет собой С6-10 арил, более предпочтительно, С9 арил или С10 арил. Более предпочтительно R4 представляет собой С6 циклоалкил, С6 арил, -СН2-С6 арил-СН2 или
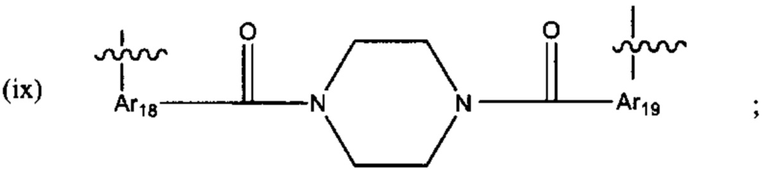
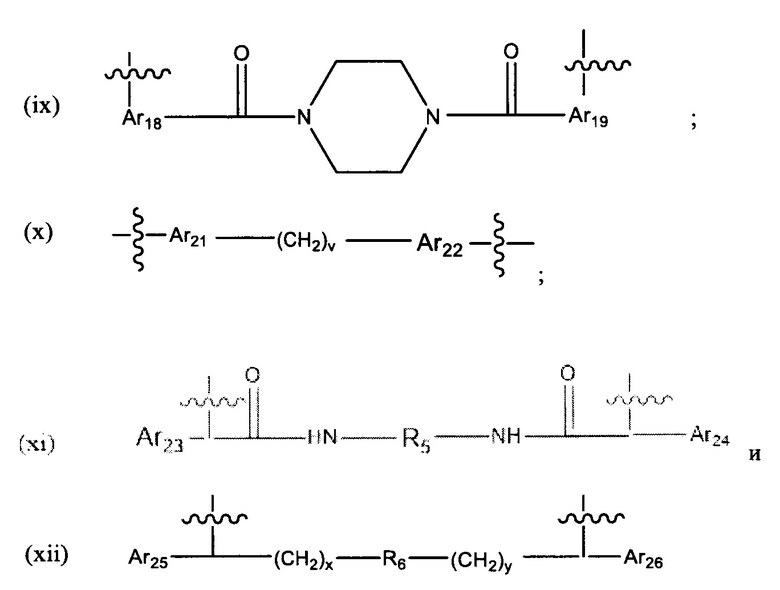
В различных вариантах осуществления линкер имеет формулу (ix):
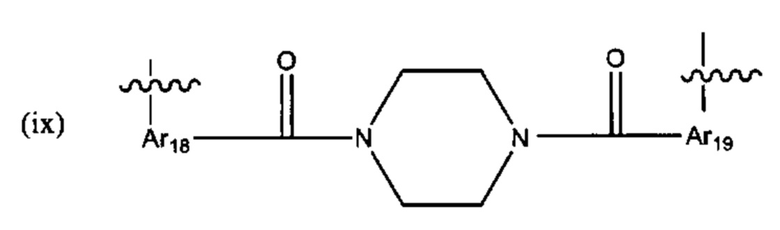
Предпочтительно, Ar18 представляет собой С6-10 арил, более предпочтительно, С9 арил или С10 арил, и Ar19 представляет собой С6-10 арил, более предпочтительно, С9 арил или С10 арил.
В различных вариантах осуществления линкер имеет формулу (х):
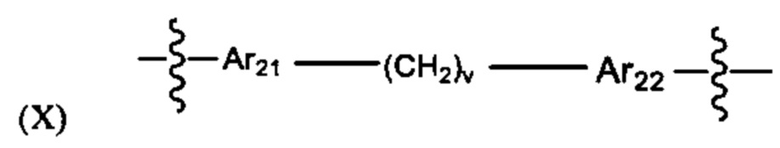
Предпочтительно A21 и А22 независимо выбраны из С6-9 арила, и v лежит в диапазоне от 1 до 6. Более предпочтительно, в одном варианте реализации Ar21 представляет собой С6 арил, v представляет собой 1, и Ar22 представляет собой С6 арил. Более предпочтительно, в одном варианте реализации Ar21 представляет собой С6 арил, v представляет собой 2, и Ar22 представляет собой С6 арил.
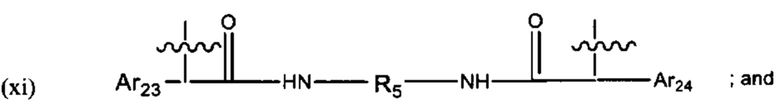
В различных вариантах осуществления линкер имеет формулу (xi):
Предпочтительно, Ar23 представляет собой С6-9 арил, более предпочтительно С6 арил, R5 представляет собой C3-C6 циклоалкил, и Ar24 предпочтительно представляет собой С6-9 арил, более предпочтительно С6 арил. Наиболее предпочтительно, R5 представляет собой С6 циклоалкил.
В другом аспекте настоящего изобретения предложено соединение Формулы (II):
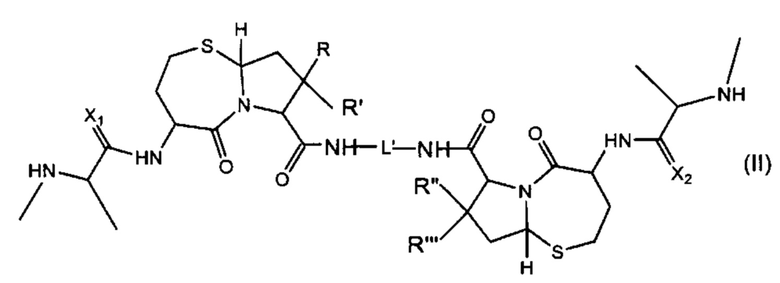
где
R, R', R'' и R''' независимо выбраны из H и СН3;
X1 и Х2 независимо выбраны из группы, состоящей из О и S; и
L' представляет собой линкер формулы:
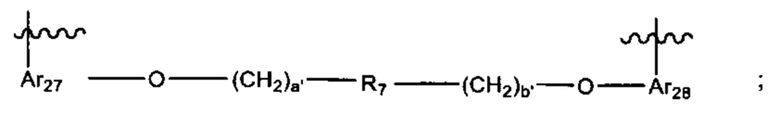
где:
Ar27 и Ar28 каждый независимо выбран из С6-С14 арила,
R7 выбран из группы, состоящей из
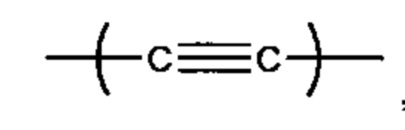 , С6 арила и -(CH2)4-15- (более предпочтительно, -(СН2)6-10-); и
, С6 арила и -(CH2)4-15- (более предпочтительно, -(СН2)6-10-); и
а' и b' независимо выбраны из от 0 до 6.
В различных вариантах осуществления R, R', R'' и R''' каждый представляет собой СН3.
В различных вариантах осуществления X1 и Х2 каждый представляет собой О;
В различных вариантах осуществления Ar27 и Ar28 каждый выбран из С6-С10 арила, и более предпочтительно, каждый представляет собой С9 арил. В различных вариантах осуществления С9 арил может быть представлен формулой
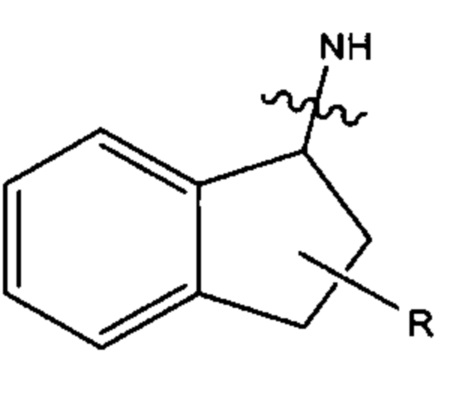
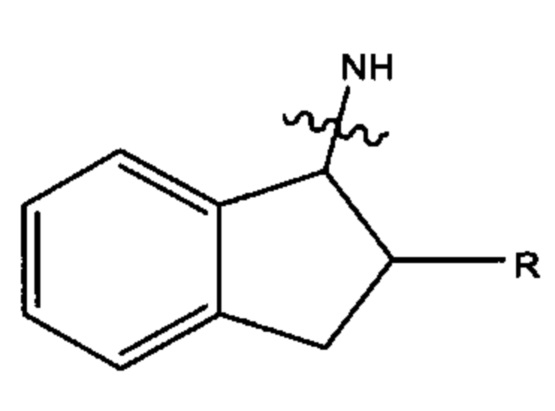
где азот в NH соединяется амидной связью с бициклической системой (системами) соединения, a R обозначает присоединение к линкеру. Наиболее предпочтительно, С9 арил имеет формулу:
В одном варианте реализации R, R', R'' и R''' каждый представляет собой СН3, X1 и Х2 каждый представляет собой О;
а' представляет собой 1, b' представляет собой 1, R7 представляет собой:
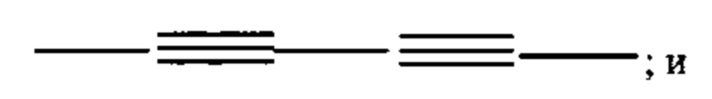
Ar27 и Ar28 каждый имеет формулу:
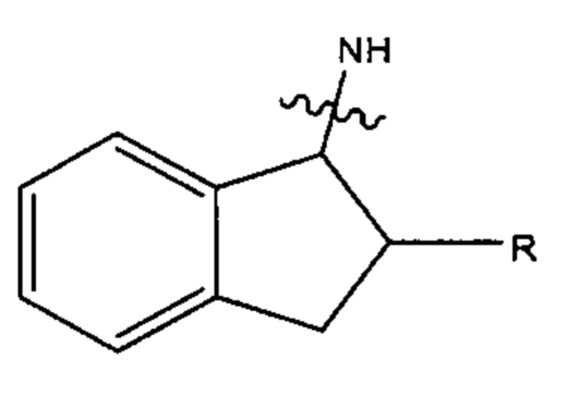
В одном варианте реализации настоящего изобретения предложено соединение Формулы (II), при условии, что если R, R', R'' и R''' каждый представляет собой СН3, X1 и Х2 каждый представляет собой О, Ar27 и Ar28 каждый имеет формулу:

и а' представляет собой 1, и b' представляет собой 1, R7 не является:
В различных вариантах осуществления предусмотрено соединение формулы (II), где а' представляет собой 0, b' представляет собой 0, и R7 представляет собой -(CH2)6-15-, (более предпочтительно -(СН2)6-10-), R, R', R'' и R''' каждый представляет собой СН3, X1 и Х2 каждый представляет собой О, и Ar27 и Ar28 каждый представляет собой С9 арил. Предпочтительно, С9 арил представлен формулой
где азот в NH соединяется амидной связью с бициклической системой (системами) соединения, и R обозначает присоединение к линкеру. Наиболее предпочтительно, С9 арил имеет формулу:
В одном варианте реализации настоящего изобретения предложено соединение Формулы (II), где а' представляет собой 0, b' представляет собой 0, R7 представляет собой -(СН2)6-, R, R', R'' и R''' каждый представляет собой СН3, X1 и Х2 каждый представляет собой О, и Ar27 и Ar28 каждый представляет собой С9 арил, представленный формулой:
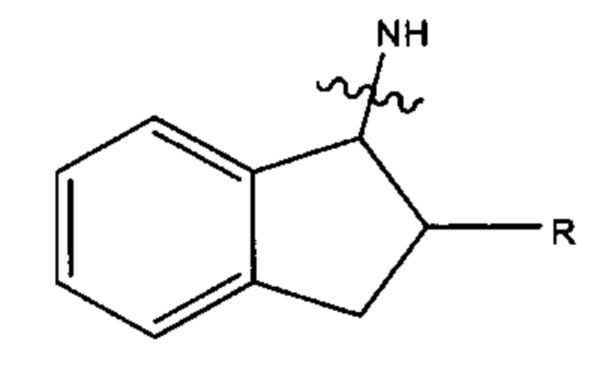
где азот в NH соединяется амидной связью с бициклической системой (системами) соединения, и R обозначает присоединение к линкеру.
В одном аспекте настоящего изобретения предложено соединение Формулы (II), с условием, что если R, R', R'' и R''' каждый представляет собой СН3, X1 и Х2 каждый представляет собой О, Ar27 и Ar28 каждый имеет формулу:
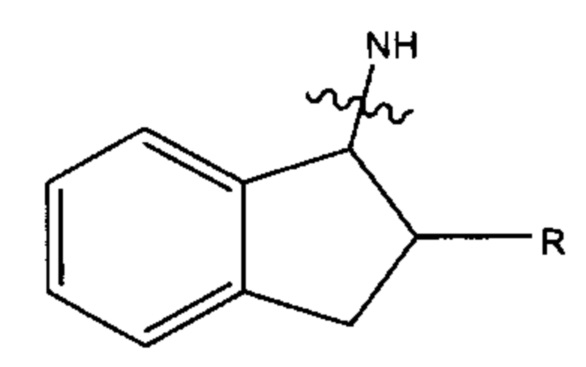
и а' представляет собой 0, b' представляет собой 0, R7 не является -(СН2)6-:
В другом аспекте настоящего изобретения предложено соединение структуры, соответствующей формуле (III):
где
R, R', R'' и R''' независимо выбраны из Н и СН3;
X1 и Х2 независимо выбраны из группы, состоящей из О и S; и
L'' представляет собой линкер формулы:
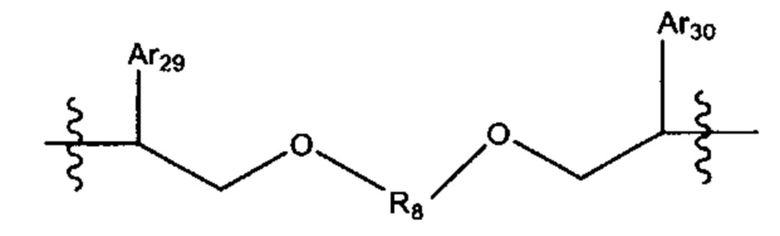
где Ar29 и Ar30 независимо выбраны из С6-С10 арила, и R8 выбран из группы, состоящей из:
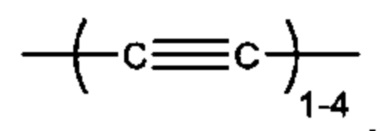 , -(CH2)6-15-,- и -(СН2)d'-(С6-С10)арил-(СН2)e'-; где d' и е' выбраны независимо и лежат в диапазоне от 1 до 6.
, -(CH2)6-15-,- и -(СН2)d'-(С6-С10)арил-(СН2)e'-; где d' и е' выбраны независимо и лежат в диапазоне от 1 до 6.
В одном варианте реализации соединения Формулы (III), R, R', R'' и R''' каждый представляет собой СН3; X1 и Х2 каждый представляет сбой О, Ar29 и Ar30 каждый представляет собой С6 арил, и R8 представляет собой:
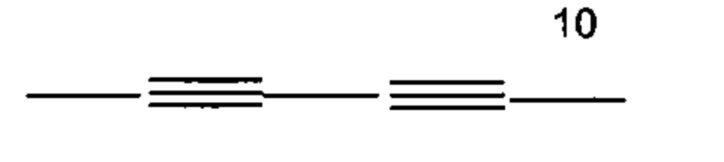
В одном варианте реализации соединения Формулы (III), R, R', R'' и R''' каждый представляет собой СН3; X1 и Х2 каждый представляет сбой О, Ar29 и Ar30 каждый представляет собой С6 арил, и R8 представляет собой -(СН2),-(С6)арил-(СН2),-.
В одном варианте реализации соединения Формулы (III), R, R', R'' и R''' каждый представляет собой СН3; X1 и Х2 каждый представляет собой О, Ar29 и Ar30 каждый представляет собой С6 арил, и R8 представляет собой -(СН2)6-.
В описанных выше формулах (II) и (III) любая из арильных может быть необязательно замещена любым из следующих заместителе, без ограничения: (С1-С6)алкил, (С1-С6)алкокси, гало, оксо, галогеналкил, бигалогеналкил, тригалогеналкил, галогеналкокси, бигалогеналкокси, тригалогеналкокси, гидроксил, амино и амид. Дополнительно, любая из арильных групп в формулах (I), (Iа) и (Ib) может также быть замещена описанными выше.
Примеры соединений, охватываемых настоящим изобретением включают, без ограничения, соединения, перечисленные в приведенной ниже Таблице 1:

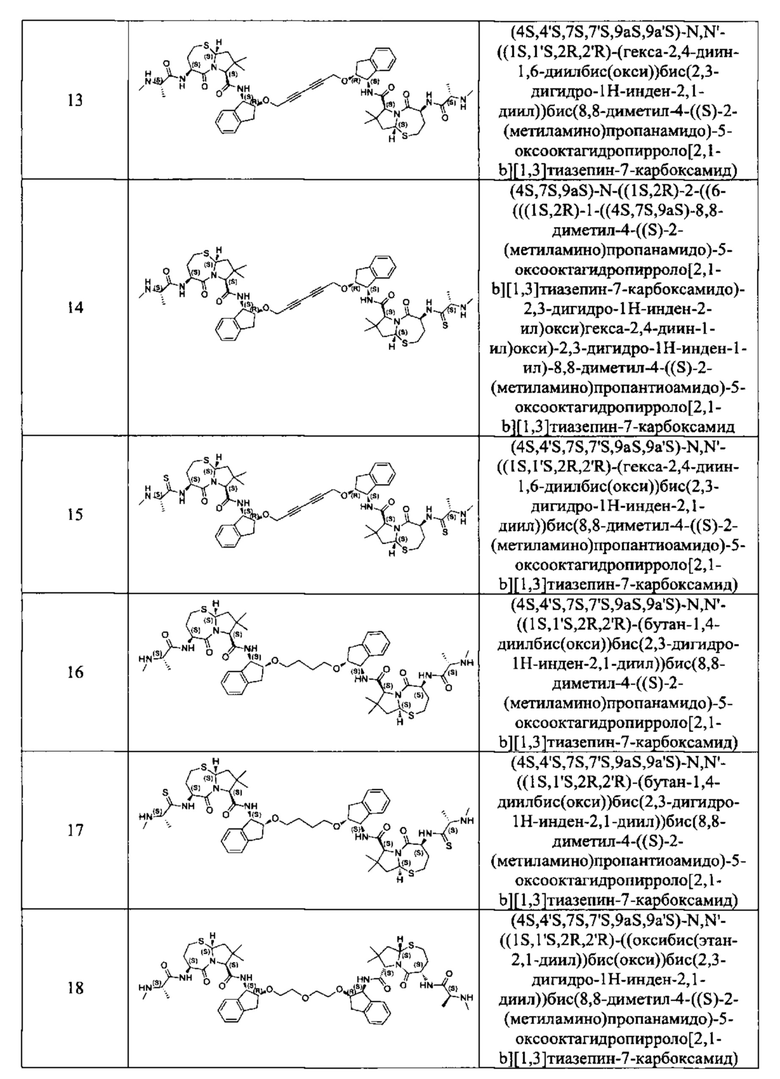
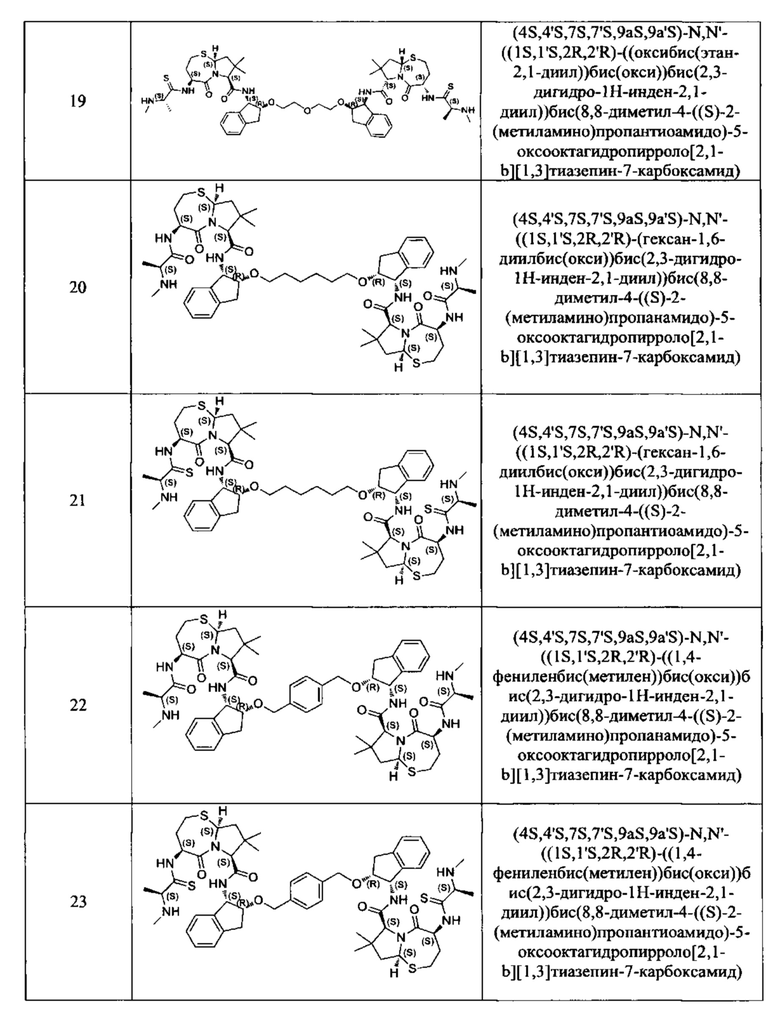
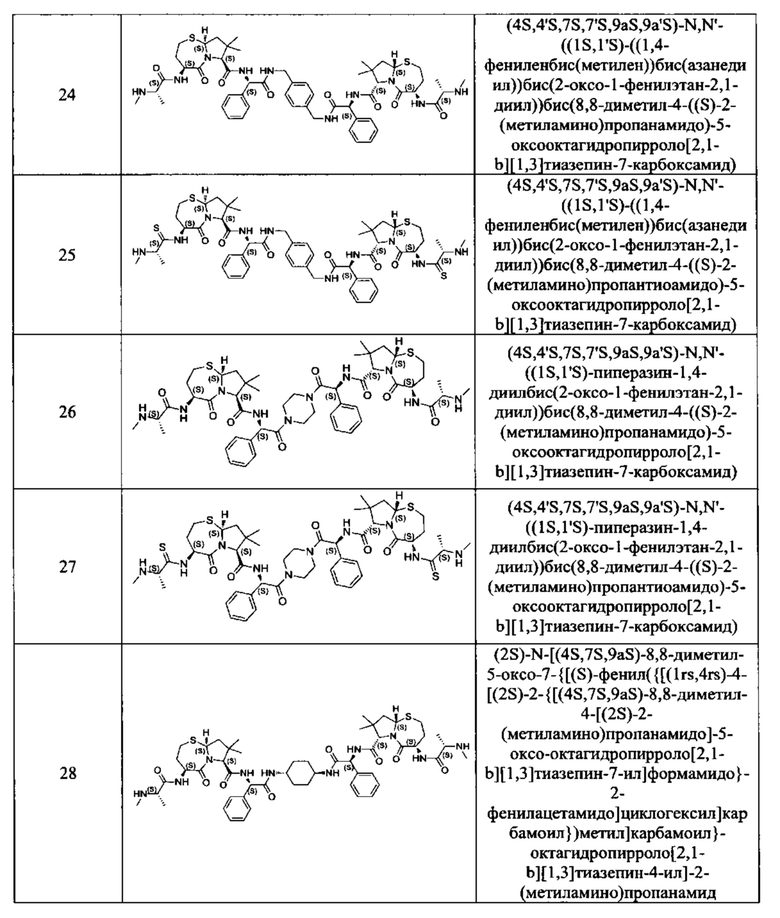
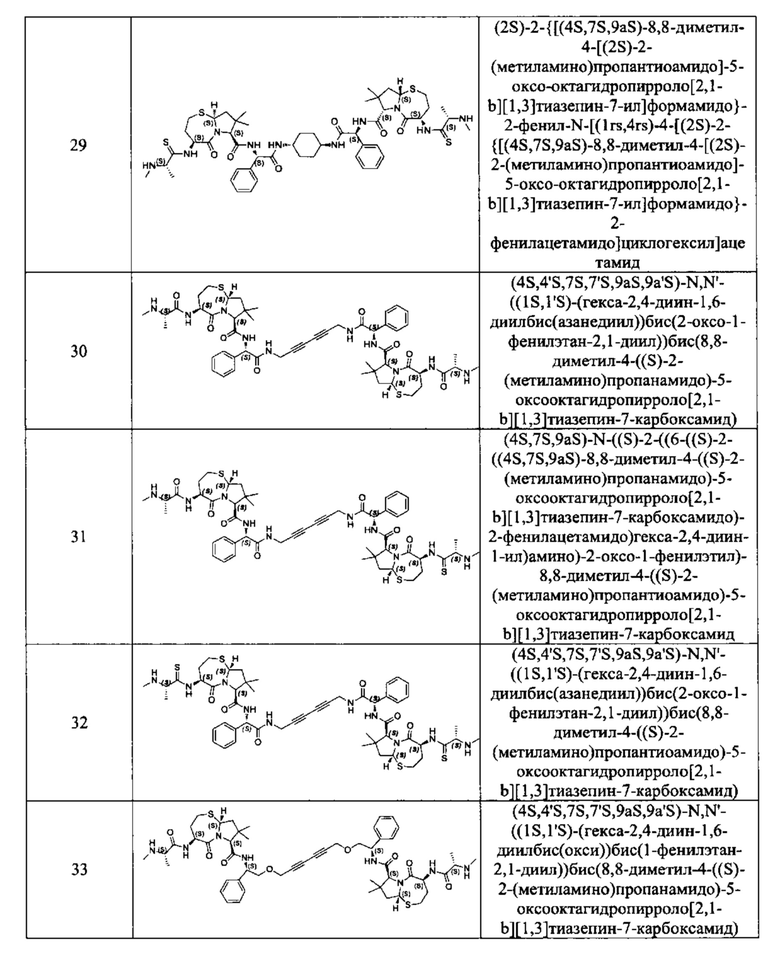
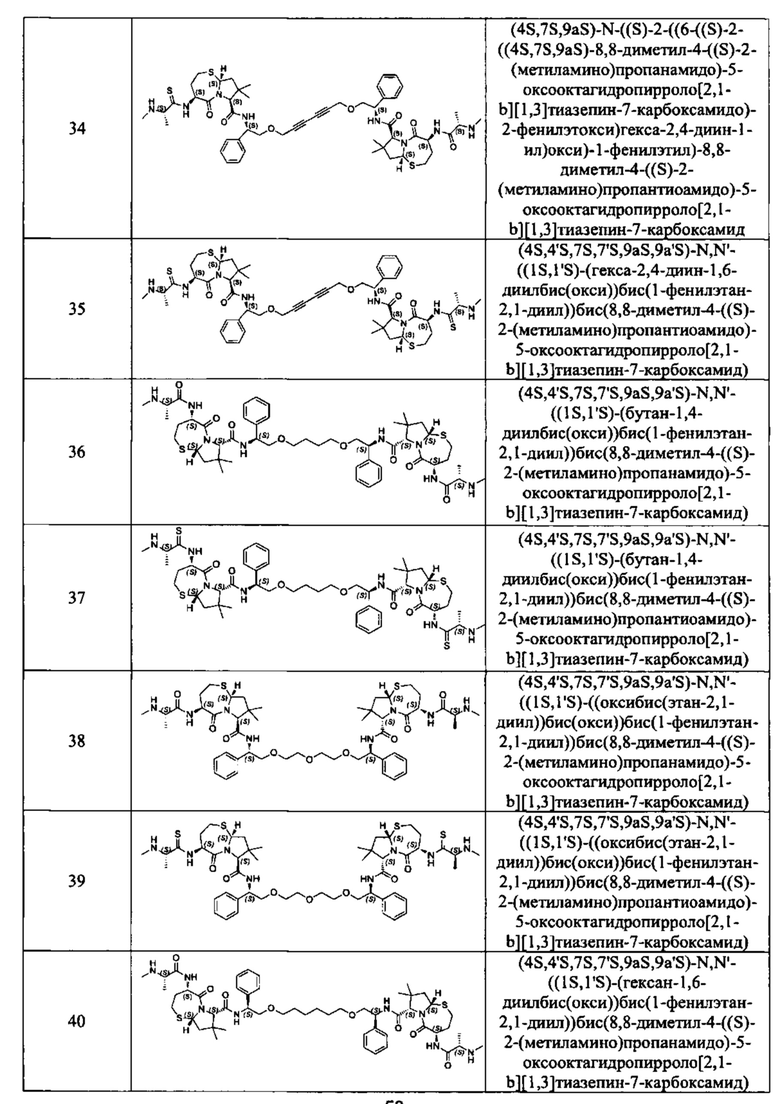
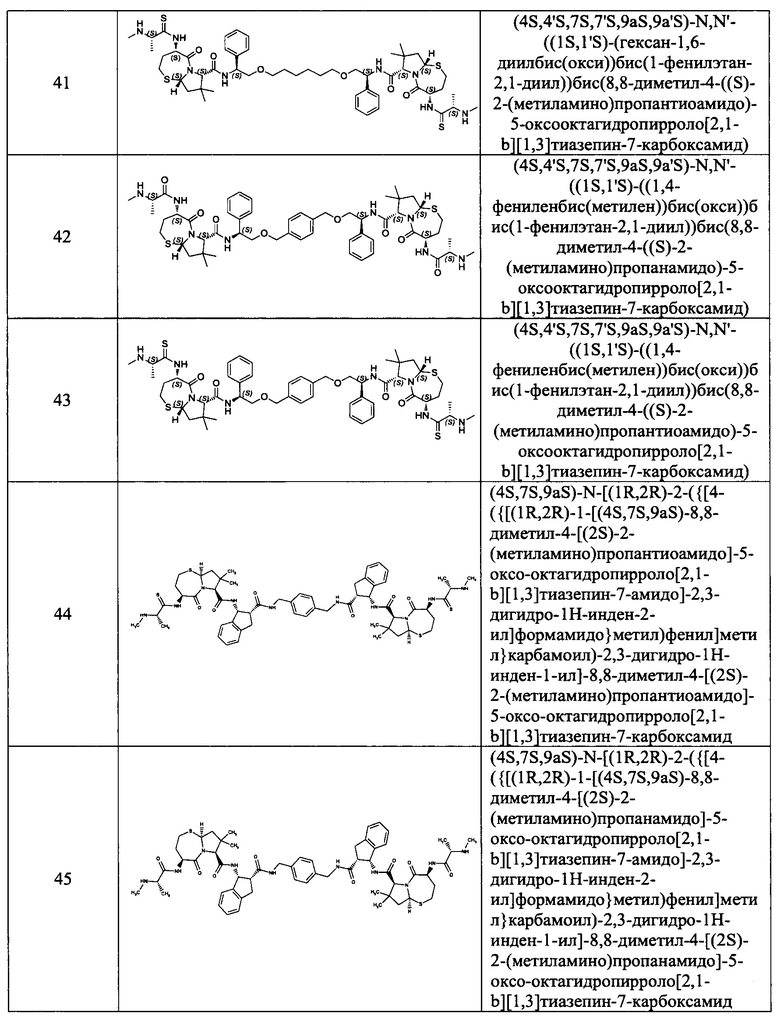
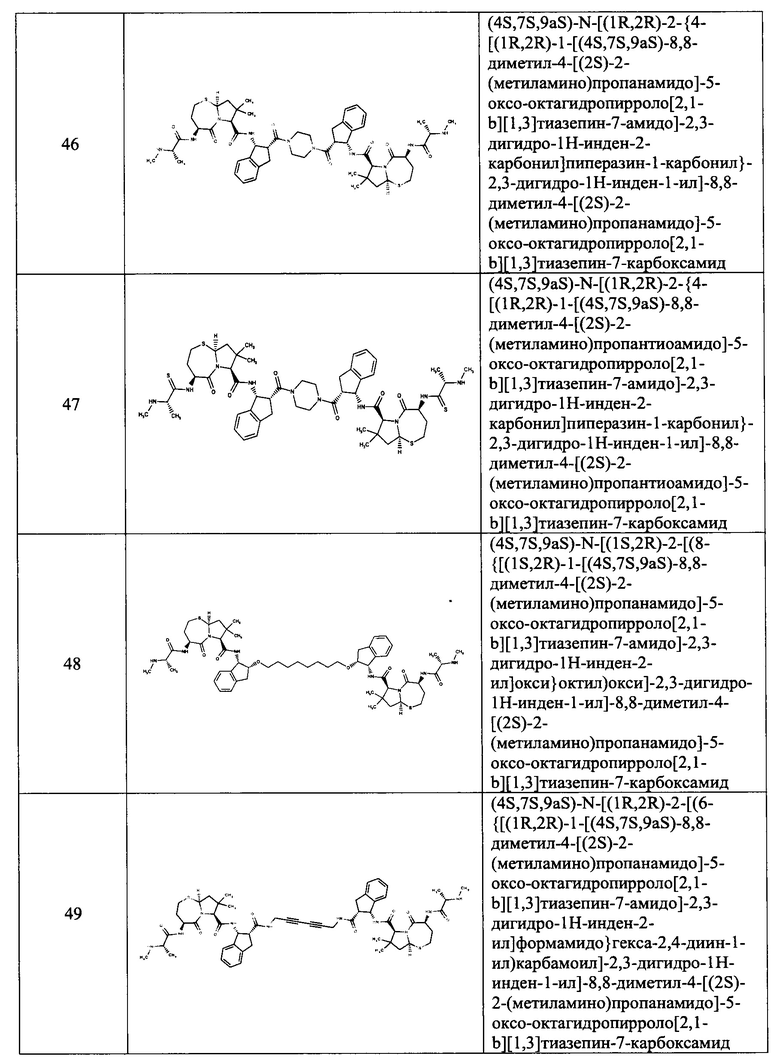
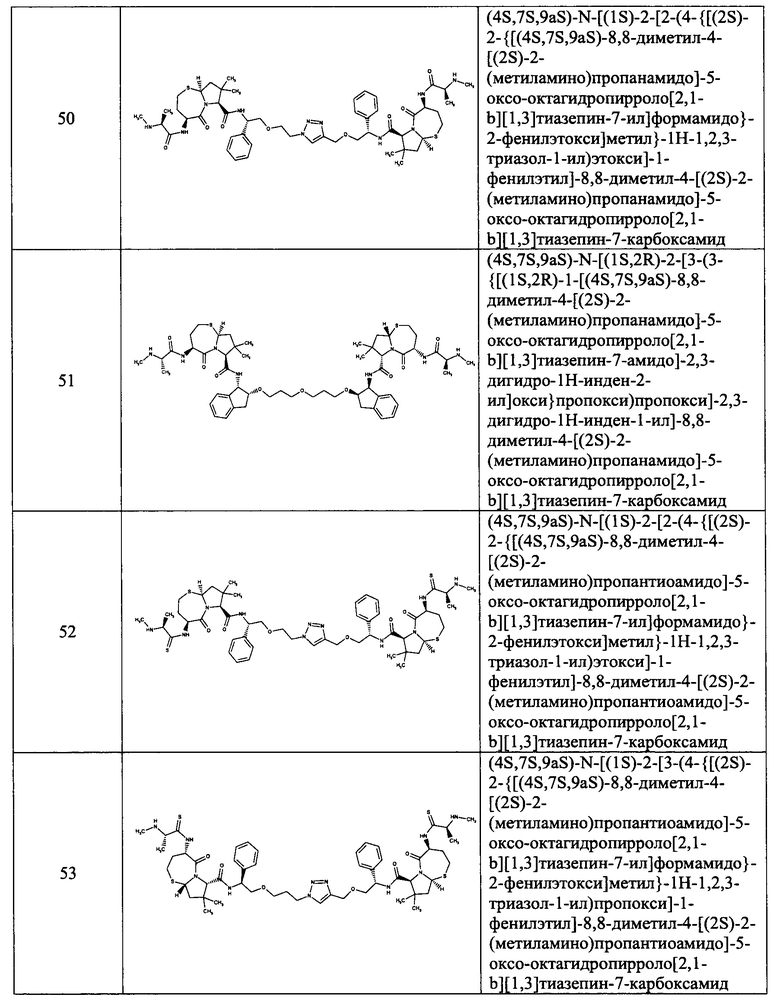
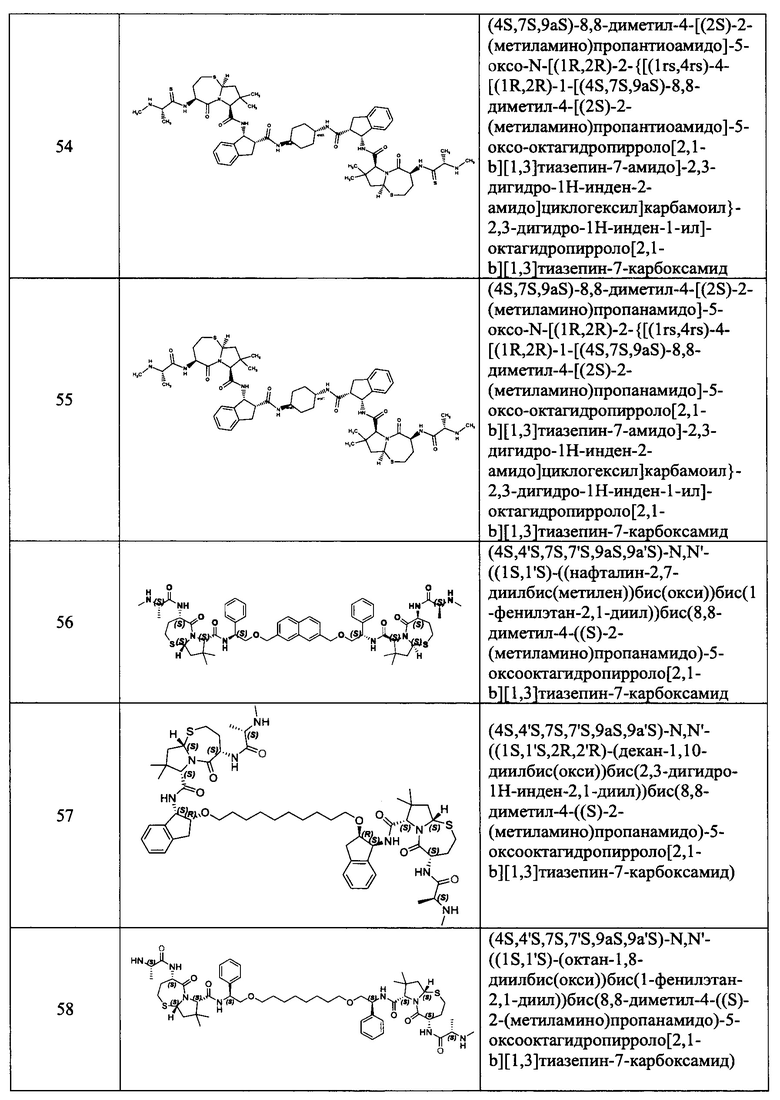
В одном аспекте настоящее изобретение относится к соединению, выбранному из группы, состоящей из:
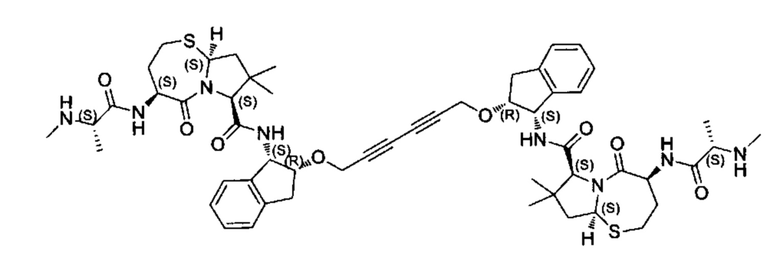
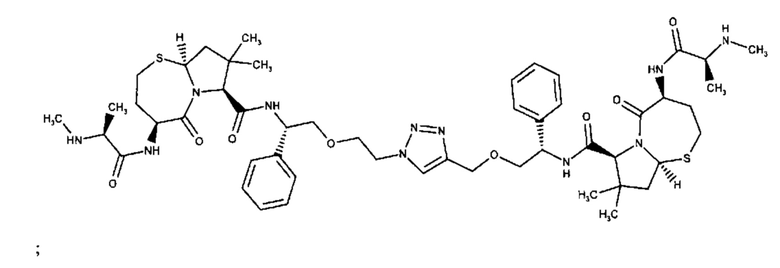
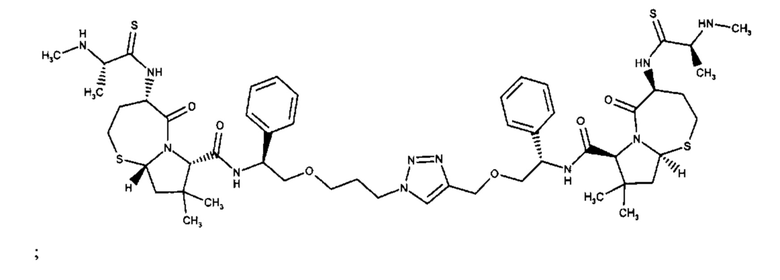
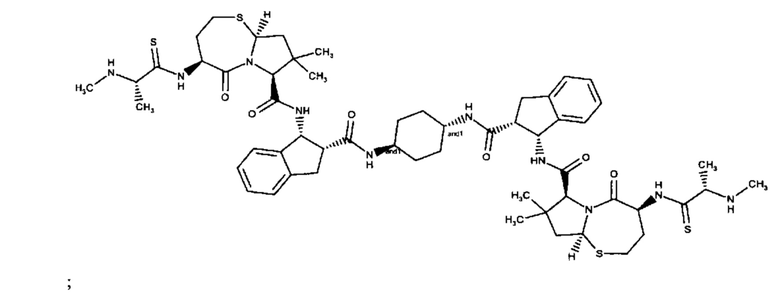
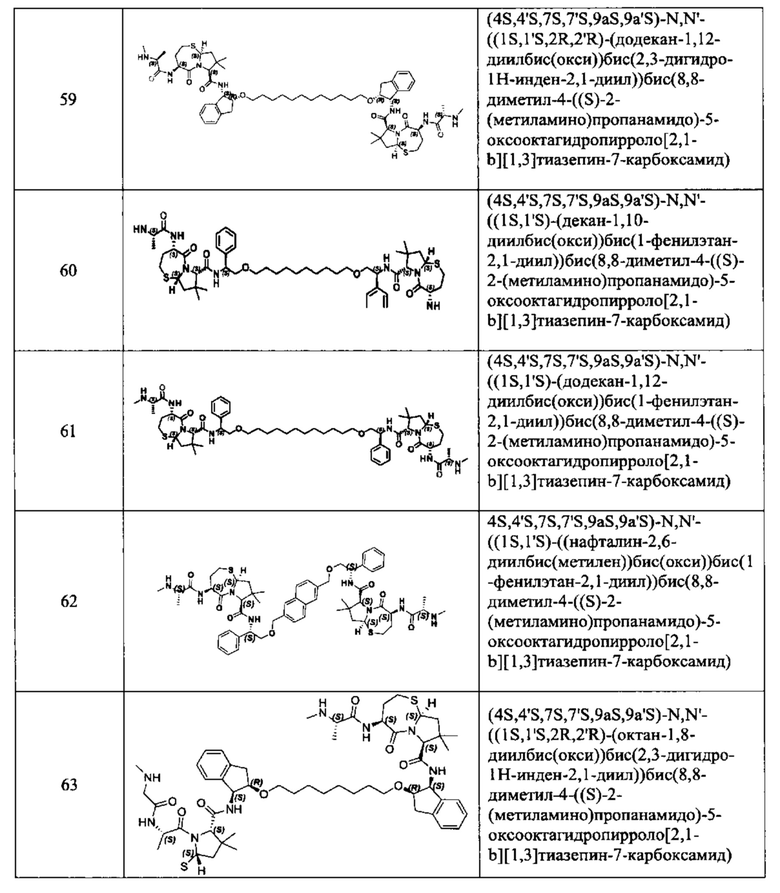
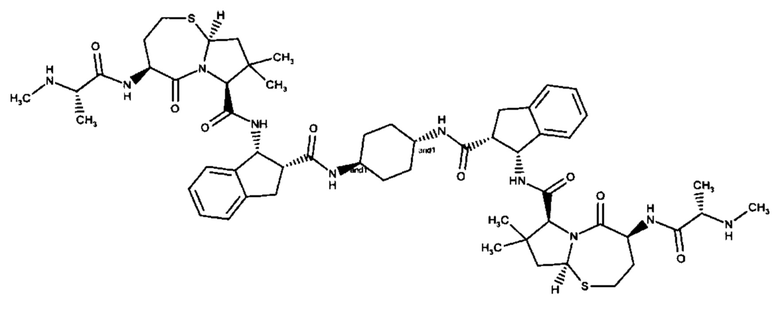 ;
;
и их фармацевтически приемлемых солей.
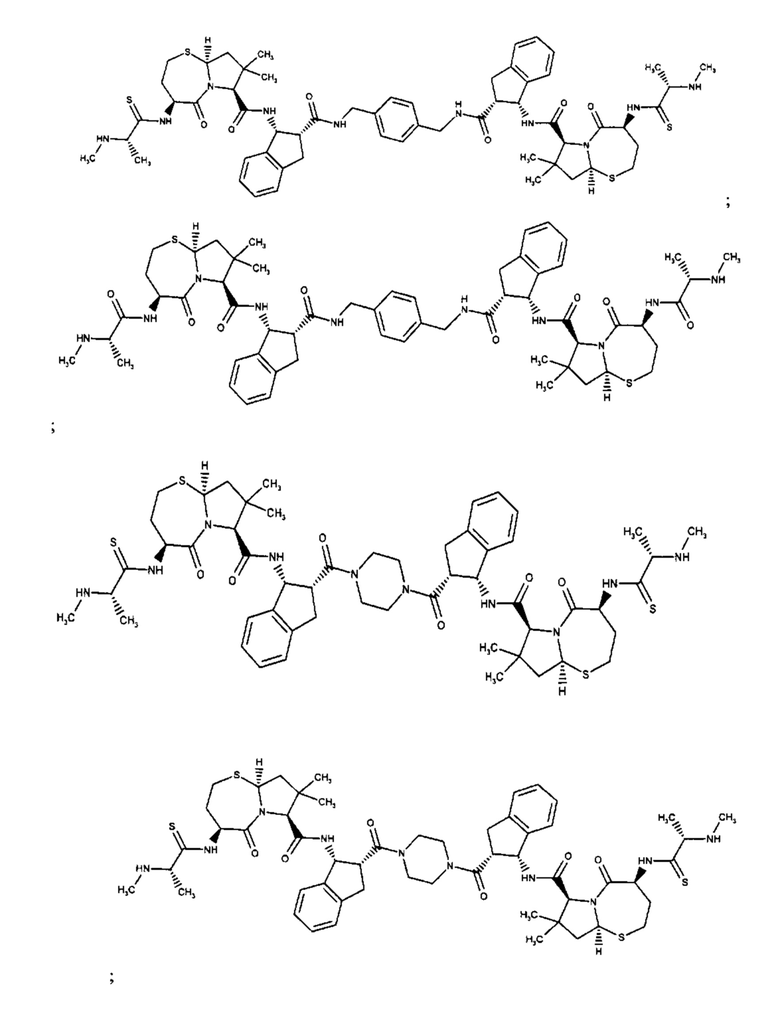
В одном аспекте настоящего изобретения предложено соединение, выбранное из группы, состоящей из:

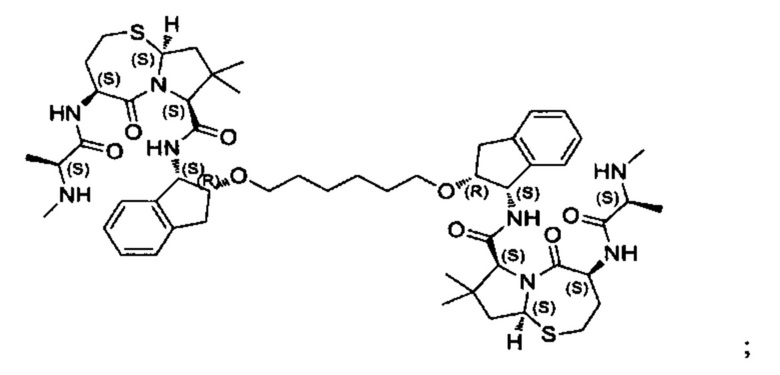
и их фармацевтически приемлемых солей.
Фармацевтически приемлемые соли также входят в объем настоящего изобретения для всех соединений 1-63, описанных в настоящем документе. Наиболее предпочтительно, каждое из соединений 1-63 может быть представлено в виде гидрохлорида (т.е., соли HCl), например, более конкретно, в виде дигидрохлорида (2 HCl). Также объем настоящего изобретения включает все соединения 1-63, представленные в виде индивидуальных молекул, включая их фармацевтически приемлемые соли, а также любые из этих соединений в форме свободных оснований.
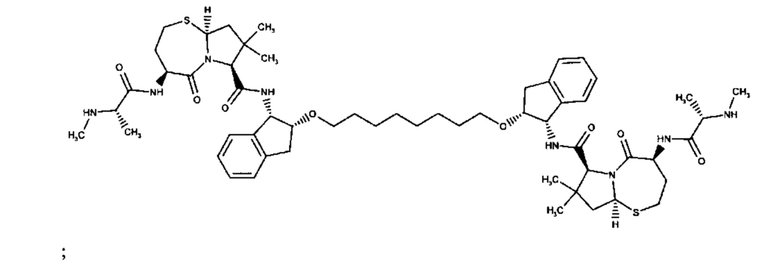
Конкретные примеры линкеров (L), которые можно применять в соответствии с настоящим изобретением включают линкеры, выбранные из группы, состоящей из:
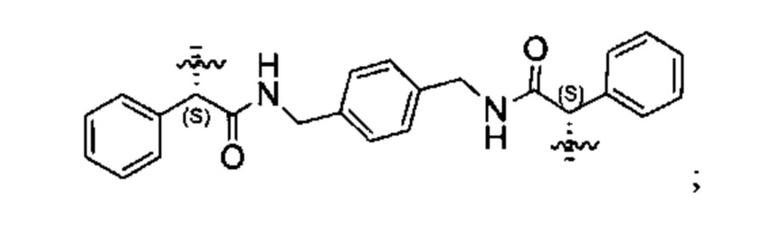
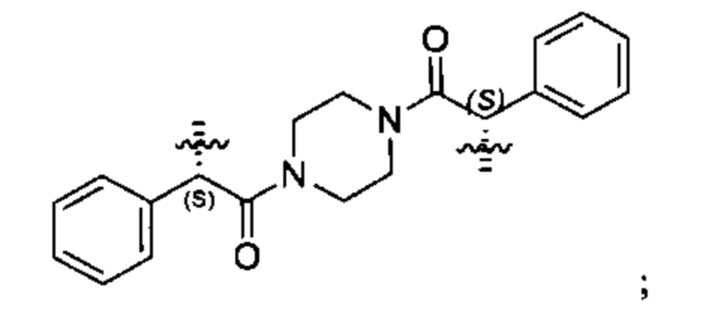
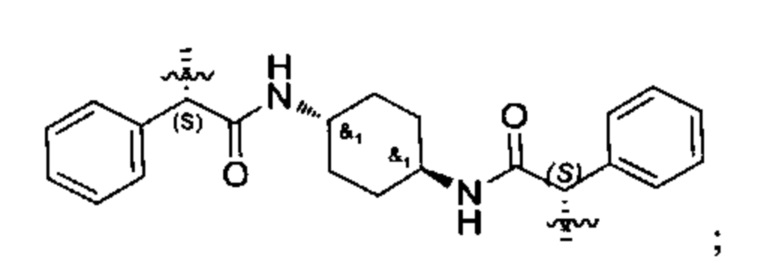
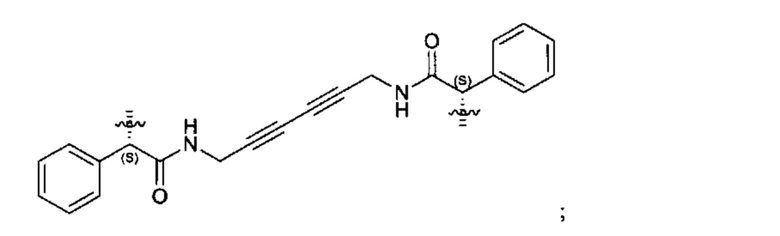
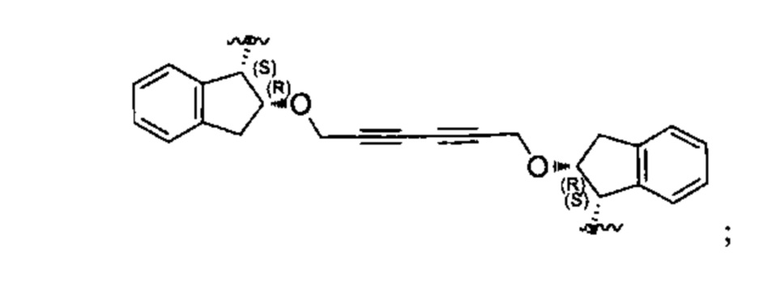
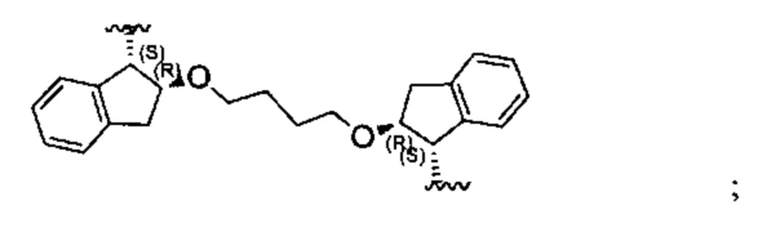
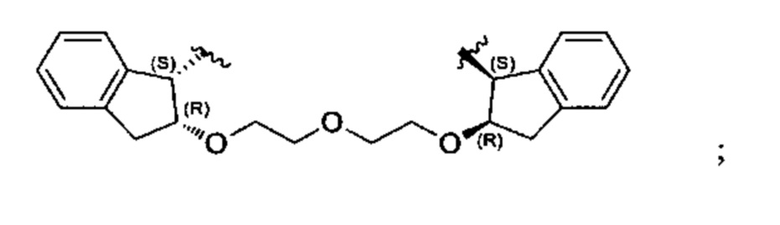
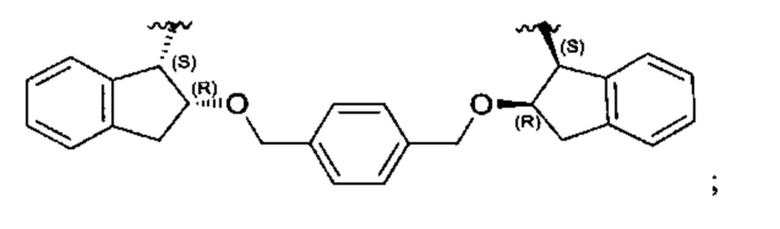
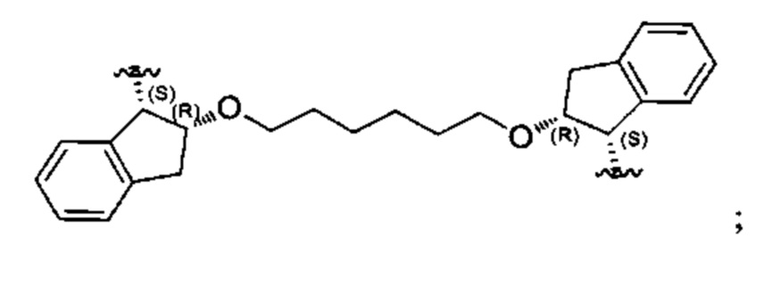
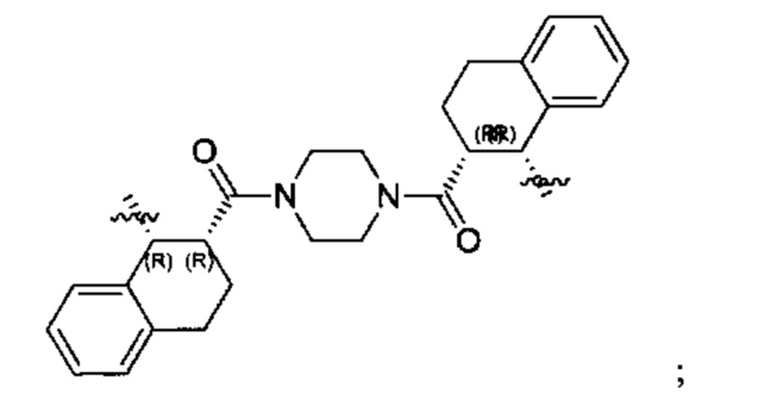
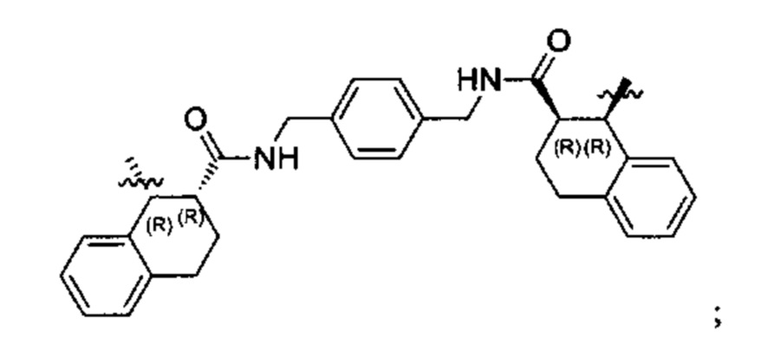
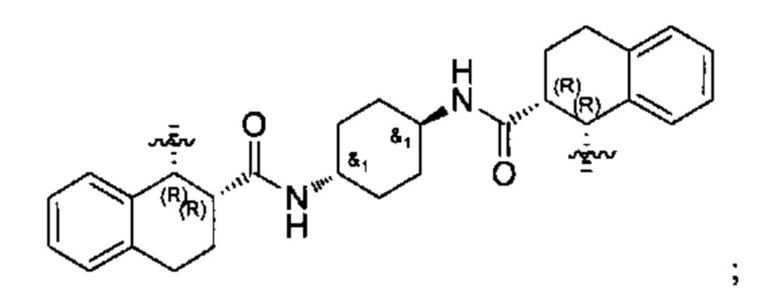
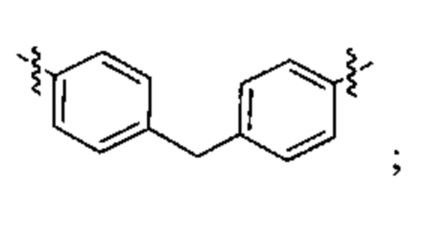
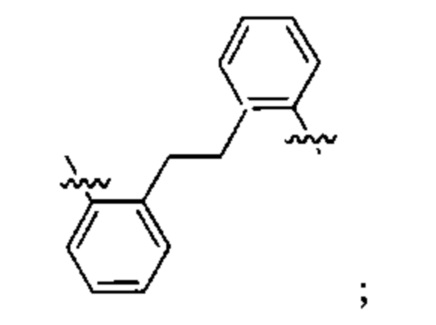
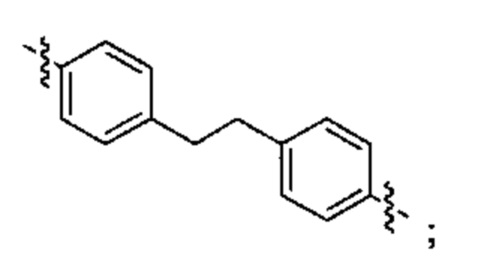
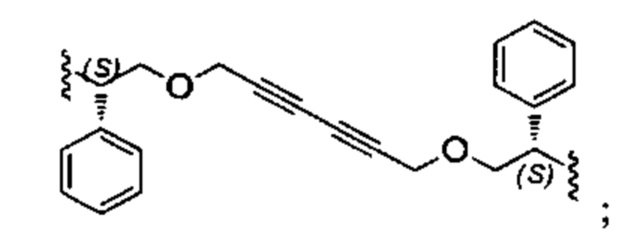
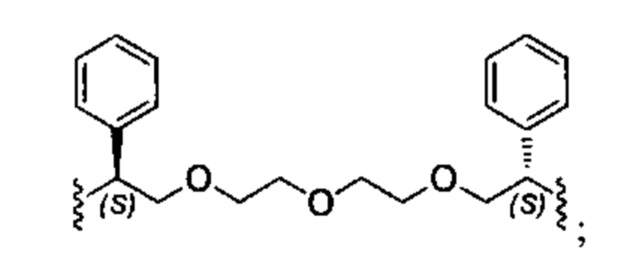
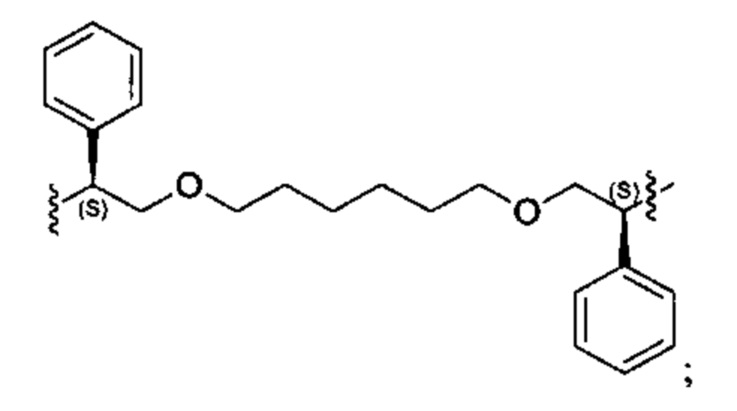
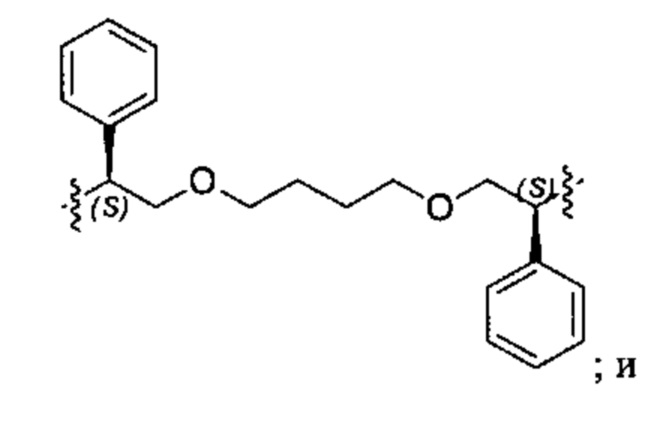
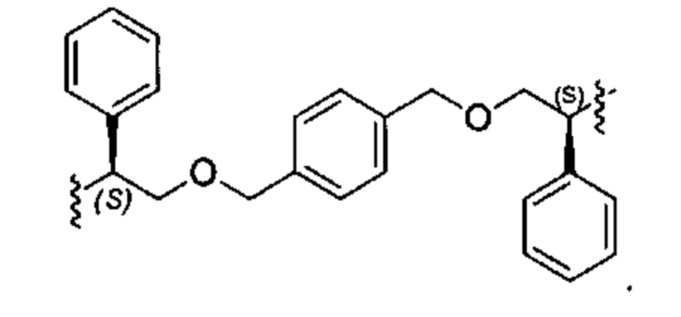
В соответствии с одним из вариантов осуществления настоящего изобретения предложена фармацевтическая композиция, содержащая соединение Формулы (I), (Ia), (Ib), (II) или (III) или его фармацевтически приемлемую соль, и фармацевтически приемлемое вспомогательное вещество. В другом варианте реализации соединение представлено в аморфной форме. В другом варианте реализации фармацевтическая композиция представлена в форме таблетки. В другом варианте реализации соединение представлено в виде дисперсии, высушенной распылением. В другом варианте реализации композиция представлена в форму наночастиц, например, частиц размером от 1 до 100 нанометров.
В соответствии с одним из вариантов осуществления настоящего изобретения предложен способ лечения ВИЧ-инфекции у субъекта, включающий введение указанному субъекту соединения Формулы (I), (Ia), (Ib), (II) или (III) или его фармацевтически приемлемой соли. В соответствии с одним из вариантов осуществления настоящего изобретения предложен способ лечения ВИЧ-инфекции у субъекта, включающий введение указанному субъекту фармацевтической в соответствии с описанием, приведенным в настоящем документе. Далее, соединения согласно настоящему изобретению, а также линкеры (L), могут существовать, в конкретных геометрических или стереоизомерных формах. Изобретение предусматривает все такие соединения, включая цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры, их рацемические смеси и другие их смеси, такие как энантиомерно или диастереомерно обогащенные смеси, которые также включены в объем настоящего изобретения. Дополнительные асимметричные атомы углерода могут присутствовать в заместителях, таких как алкильные группы. Предполагается, что все такие изомеры, а также их смеси, включены в настоящее изобретение. Оптически активные (R)- и (S)-изомеры, и d и l изомеры могут быть получены с использованием хиральных синтонов и хиральных реагентов или разделены с использованием обычных методик. Если, например, желательно получить конкретный энантиомер соединения согласно настоящему изобретению, его можно получить путем асимметричного синтеза или путем дериватизации хиральной вспомогательной группой, при этом получаемую смесь диастереомеров разделяют, и вспомогательную группу отщепляют с получением чистых целевых энантиомеров. В качестве альтернативы, в тех случаях, когда молекула содержит основную функциональную группу, такую как аминогруппа, или кислотную функциональную группу, такую как карбоксильная группа, могут быть образованы диастереомерные соли с подходящим оптических активной кислотой или основанием с последующим разделением образованных таким образом диастереомеров путем фракционной кристаллизации или с использованием хроматографических средств, известных в данной области, и с последующим выделением чистых энантиомеров. Дополнительно разделение энантиомеров и диастереомеров часто осуществляют с использованием хроматографии с применением хиральных, стационарных фаз, необязательно в комбинации с химической дериватизацией (например, образованием карбаматов из аминов).
В другом варианте реализации настоящего изобретения предложено соединение Формулы I, Ia, Ib, II или III, или его фармацевтически приемлемая соль для применения в медицинской терапии.
В другом варианте реализации настоящего изобретения предложено соединение Формулы I, Ia, Ib, II или III, или его фармацевтически приемлемая соль для применения для лечения ВИЧ-инфекции.
В другом варианте осуществления изобретения, предложено соединение Формулы I, Ia, Ib, II или III, причем указанное соединение или соль соединения применяют в изготовлении лекарственного средства для применения для лечения ВИЧ-инфекции у человека.
В одном аспекте настоящего изобретения предложен способ излечения ВИЧ-инфекции у субъекта включающий, введение указанному субъекту соединения Формул I, Ia, Ib, II или III, а также любого соединения из Таблицы 1, а также их фармацевтически приемлемых солей. «Устранение» или «излечение» болезни у пациента используется для обозначения эрадикции, остановки или окончания вируса или симптомов иммунодефицита человека, или прогрессирования симптомов вируса в течение определенного периода. В качестве примера в одном варианте реализации «устранение» или «излечение» относится к терапевтическому назначению (введению) или комбинации назначений, которые отдельно или в комбинации с одним или более другими соединениями вызывают и поддерживают устойчивый контроль вируса (уровни виремии плазмы, не определяемые, например, анализом методом полимеразной цепной реакции (ПЦР), анализом рДНК bDNA (разветвленных цепей ДНК) или анализом NASBA (амплификация на основе последовательности нуклеиновой кислоты)) вируса иммунодефицита человека после минимум двух лет без какого-либо терапевтического вмешательства. Указанные анализы ПЦР, рДНК и NASBA осуществляют с использованием методик, изветных и хорого знакомых специалисту в данной области. В качестве примера, эрадикация, прекращение, остановка или окончания вируса или симптомов иммунодефицита человека, или прогрессирования симптомов или вируса, могут поддерживаться в течение по меньшей мере двух лет.
В одном аспекте настоящего изобретения предложен способ излечения ВИЧ-инфекции у субъекта, включающий введение указанному субъекту фармацевтической композиции, содержащей соединение Формул I, Ia, Ib, II и III, равно как и его фармацевтическую соль.
В одном аспекте настоящего изобретения предложено применение соединения Формул I, Ia, Ib, II и III или его фармацевтически приемлемой соли в изготовлении лекарственного средства для применения в излечении ВИЧ-инфекции.
В одном аспекте настоящего изобретения предложено соединение Формул I, Ia, Ib, II и III, или его фармацевтически приемлемая соль для применения в излечении ВИЧ-инфекции.
Комбинации соединений Формул I, Ia, Ib, II и III и одного или более агентов, пригодных в терапии ВИЧ также могут применяться в способах излечения ВИЧ-инфекции.
В одном варианте реализации фармацевтический препарат, содержащий соединение Формулы I, Ia, Ib, II или III или его соль, представляет собой препарат, приспособленный для парентерального введения. В другом варианте реализации препарат представляет собой препарат длительного действия для парентерального введения. В другом варианте реализации препарат представляет собой препарат на основе наночастиц. Соединения согласно настоящему изобретению и их соли, сольваты или другие фармацевтически приемлемые производные, можно применять отдельно или в комбинации с другими терапевтическими агентами. Соответственно, в других вариантах осуществления способы лечения и/или предотвращения ВИЧ-инфекции у субъекта могут в дополнение к введению соединения Формулы I, Ia, Ib, II или III дополнительно включать введение одного или более дополнительных фармацевтических агентов, обладающих активность, против ВИЧ.
В таких вариантах осуществления один или более дополнительных агентов, обладающих активностью против ВИЧ, выбран из группы, состоящей из антиретровирусных агентов, агентов, обращающих латентное состояние и агентов для клиренсной терапии.
В других вариантах осуществления один или более дополнительных агентов, обладающих активностью против ВИЧ выбран из группы, состоящей из нуклеотидных ингибиторов обратной транскриптазы, ненуклеотидных ингибиторов обратной транскриптазы, ингибиторов протеазы, ингибиторов проникновения в клетку, ингибиторов прикрепления и слияния, ингибиторов интегразы, ингибиторов созревания, ингибиторов CXCR4 и/или CCR5, ингибиторов гистондеацетилазы, ингибиторов гистонкротонилтрансферазы, агонистов протеинкиназы С, ингибиторы протеасом, агонистов TLR7, ингибиторов бромодомена и нейтрализующих антител и их комбинаций.
В некоторых вариантах осуществления один или более дополнительных агентов, обладающих активностью против ВИЧ выбран из группы, состоящей из зидовудина, диданозина, ламивудина, зальцитабина, абакавира, ставудина, адефовир, адефовира дипивоксила, фозивудина, тодоксила, эмтрицитабина, аловудина, амдоксовира, элвуцитабина, невирапина, делавердина, эфавиренза, ловирида, иммунокала, олтипраза, каправирина, лерсивирина, GSK2248761, ТМС-278, ТМС-125, этравирина, саквинавира, ритонавира, индинавира, нелфинавира, ампренавира, фосампренавира, бреканавира, дуранавира, атазанавира, типранавира, палинавира, ласинавира, энфувиртида, Т-20, Т-1249, PRO-542, PROMO, TNX-355, BMS-806, BMS-663068 и BMS-626529, 5-Helix (5-хеликс), ралтегравира, элвитегравира, долутегравира, каботегравира, биктегравира, викривирока (Sch-C), Sch-D, TAK779, маравирока, TAK449, диданозина, тенофовира, лопинавира, дуранавира, вориностата, панобиностат, ромидепина, валпроиновой кислоты, моцетиностат, коротоната натрия, бриостатина, ингенола В, дисульфорама, GS-9620, JQ1, iBET151, бортезомиба, эпигаллокатехина галлата, салиноспорамида А, карфилзомиба, нейтрализующих антител широкого спектра (bNAb), eCD4-Ig, CD4-Ig и перенаправляющих антител с двойной аффинностью (DART).
Соответственно, соединения согласно настоящему изобретению Формулы (I), (Ia), (Ib), (II) или (III) и любой другой фармацевтически активный агент(ы) можно вводить совместно или раздельно, и в случае раздельного введения, введение может осуществлять одновременно или последовательно, в любом порядке. Количества соединений Формулы (I), (Ia), (Ib), (II) или (III) согласно настоящему изобретению и другого фармацевтически активного агента (агентов) и относительное время введения могут быть выбраны для обеспечения желаемого комбинированного терапевтического эффекта. Введение в комбинации соединения согласно настоящему изобретению Формулы (I), (Ia), (Ib), (II) или (III) и солей, сольватов или других фармацевтически приемлемых производных указанного соединения в другими лечебными агентами может быть комбинированным за счет введения совместно в: (1) единой фармацевтической композиции, содержащей оба соединения; или (2) раздельных фармацевтических композициях, каждая из которых содержит одно соединение. В качестве альтернативы, комбинацию можно вводить раздельно последовательным способом, в котором один агент для лечения вводят первым, а другой - вторым, или наоборот. Такое последовательное введение может быть близким или удаленным по времени. Количества соединения (соединений)Формулы I, Ia, Ib, II или III ли их солей и другого фармацевтически активного агента(агентов) и относительное время введения будут выбирать для достижения желаемого комбинированного терапевтического эффекта.
Дополнительно соединения согласно настоящему изобретению Формулы (I), (Ia), (Ib), (II) или (III) можно использовать в комбинации с одним или более другими агентами, которые могут быть полезны для лечения ВИЧ. Эти агенты могут включать антиретровирусные агенты, агенты, обращающие латентное состояние и агенты для клиренсной терапии. Ниже приведено несколько примеров антиретровирусных агентов:
нуклеотидные ингибиторы обратной транскриптазы, такие как зидовудин, диданозин, ламивудин, зальцитабин, абакавир, ставудин, адефовир, адефовир дипивоксил, фозивудин, тодоксил, эмтрицитабин, аловудин, амдоксовир, элвуцитабин и аналогичные агенты; ненуклеотидные ингибиторы обратной транскриптазы (включая агенты, обладающие противоокислительной активностью, такие как иммунокал, олтипраз и т.д.), такие как невирапин, делавердин, эфавиренз, ловирид, иммунокал, олтипраз, каправирин, лерсивирин, GSK2248761, ТМС-278, ТМС-125, этравирин и аналогичные агенты;
Ингибиторы протеазы. такие как саквинавир, ритонавир, индинавир, нелфинавир, ампренавир, фосампренавир, бреканавир, дуранавир, атазанавир, типранавир, палинавир, ласинавир и аналогичные агенты;
Ингибиторы проникновения, присоединения и слияния, такие как энфувиртид (Т-20), Т-1249, PRO-542, PRO-140, TNX-355, BMS-806, BMS-663068 и BMS-626529, 5-Helix и аналогичные агенты;
Ингибиторы интегразы, такие как ралтегравир, элвитегравир, долутегравир, каботегравира, биктегравир и аналогичные агенты;
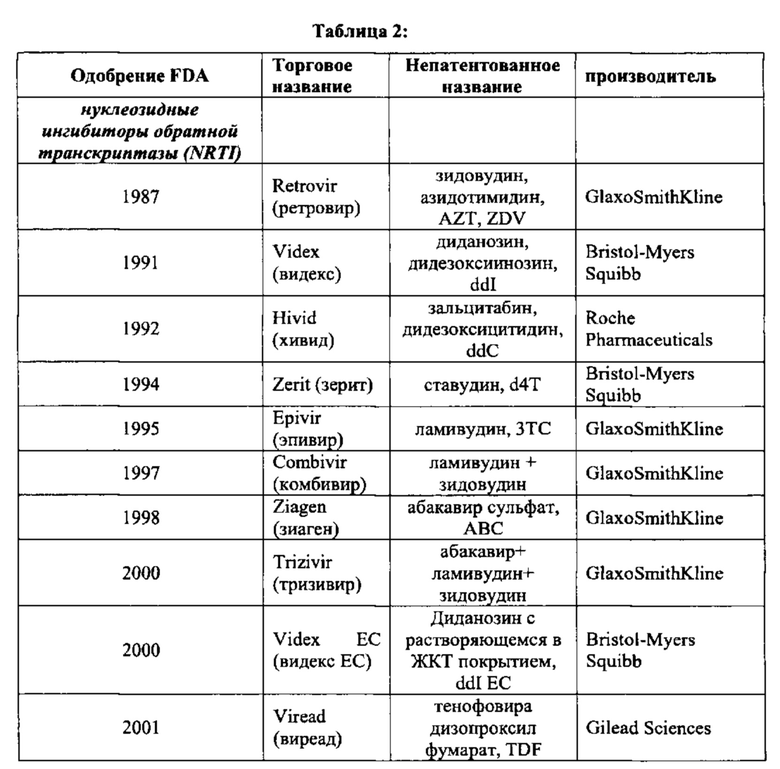
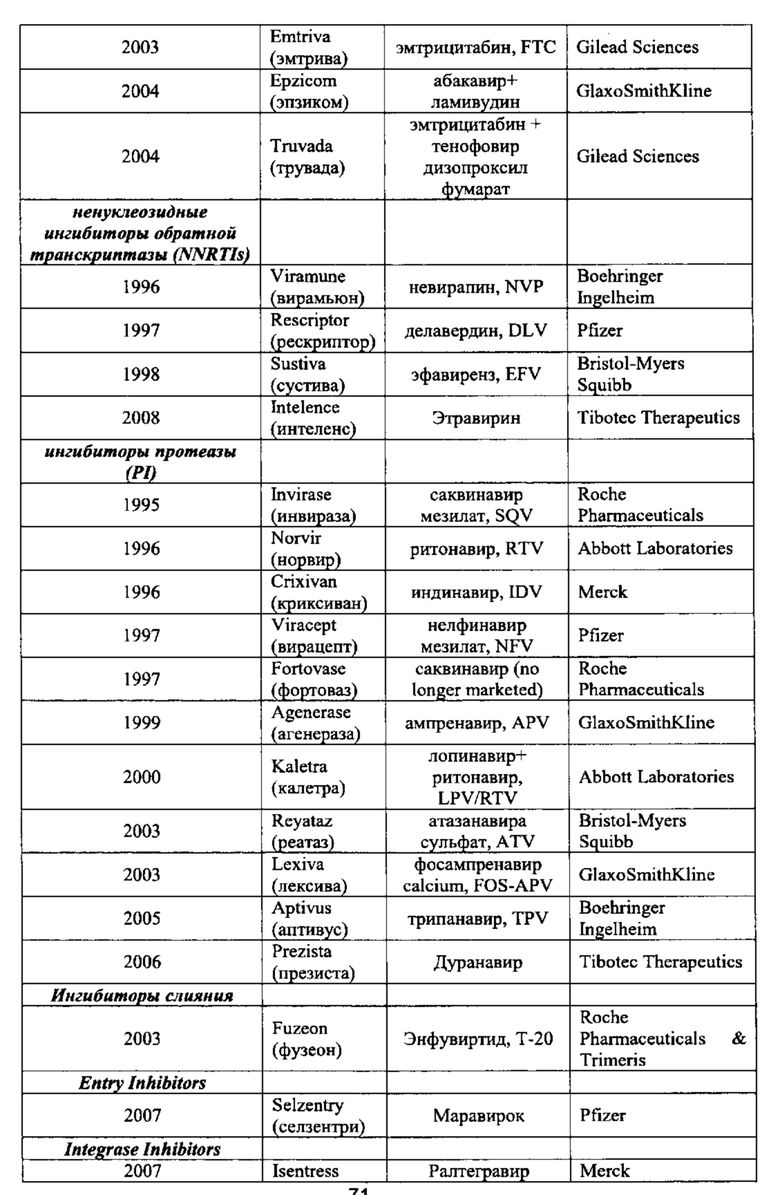
Ингибиторы созревания, такие как РА-344 и РА-457 и аналогичные агенты; и ингибиторы CXCR4 и/или CCR5. такие как викривирок (Sch-C), Sch-D, ТАК779, маравирок (UK 427,857), TAK449, а также раскрытые в WO 02/74769, PCT/US03/39644, PCT/US03/39975, PCT/US03/39619, PCT/US03/39618, PCT/US03/39740, и PCT/US03/39732 и аналогичные агенты. Дополнительные примеры, в которых_соединения согласно настоящему изобретению могут применяться в комбинации с одним или более агентами, подходящими для предотвращения или лечения ВИЧ, приведены в Таблице 2.

Настоящее изобретение можно применять в комбинации с другими агентами, которые индуцируют экспрессию ВИЧ, такими как агенты, обращающие латентное состояние. Некоторые агенты, обращающие латентное состояние включают следующие, без ограничения перечисленными: ингибиторы гистондеацетилазы (например, вориностат, панобиностат, ромидепина), ингибиторы гистонкротонилтрансферазы (коротоната натрия), агонистов протеинкиназы С (например, бриостатина, ингенола В), дисульфирам, агонисты TLR7 (например, GS-9620), ингибиторы бромодомена (например, JQ1, iBET151). Многие из этих агентов подробно описаны ниже.
Настоящее изобретение можно применять в комбинации с другими агентами, которые индуцируют экспрессию ВИЧ, такими как агенты для клиренсной терапии. Некоторые примеры агентов для клиренсной терапии или для иммунологических комбинаций для клиренса, включают следующие, без ограничения перечисленными: нейтрализующие антитела и нейтрализующие антитела широкого спектра (bNAb), eCD4-Ig, CD4-Ig и перенаправляющие белки двойной аффинности (DART).
Объем комбинаций соединений согласно настоящему изобретению с агентами против ВИЧ не ограничивается указанными выше, а включает в принципе любые комбинации с любыми фармацевтическими композициями, подходящими для лечения и/или предотвращения ВИЧ. Как отмечалось, в таких комбинациях соединения согласно настоящему изобретению и другие агенты против ВИЧ можно вводить отдельно или совместно. Кроме того, один агент можно вводить до, одновременно с или последовательно с введение другого агента (агентов). Настоящее изобретение можно применять в комбинации с одним или более агентами, которые можно применять в качестве агентов, улучшающих фармакологические свойства, а также с дополнительными соединениями для лечения или предотвращения ВИЧ или без них. Примеры таких агентов, улучшающих фармакокинетические свойства (или фармакокинетических бустеров) включают, без ограничения перечисленными: ритонавир, GS-9350 (кобицистат) и SPI-452.
Ритонавир представляет собой 10-гадрокси-2-метил-5-(1-метилэтил)-1-1[2-(1-метилэтил)-4-тиазолил]-3,6-диоксо-8,11 -бис(фенилметил)-2,4,7,12-тетраазатридекан-13-оевую кислоту, 5-тиазолилметиловый эфир, [5S-(5S*,8R*,10R*,11R*)] и доступен для приобретения в Abbott Laboratories Abbott park, Illinois, под названием Norvir (норвир). Ритонавир представляет собой ингибитор протеазы ВИЧ, назначаемый с другими антиретровирусными агентами для лечения ВИЧ-инфекции. Ритонавир также ингибирует опосредуемый Р450 метаболизм лекарственных средств, а также систему транспорта Р-гликопротеина (Pgp) клетки, обеспечивая, таким образом, повышенные концентрации активного соединения в организме.
GS-9350 (кобицистат) представляет собой соединение, разработанное компанией Gilead Sciences из Фостер-сити, Калифорния, в качестве вещества, улучшающего фармакологические свойства.
SPI-452 представляет собой соединение, разработанное компанией Sequoia Pharmaceuticals из Гейтерсберга, Мэриленд, в качестве вещества, улучшающего фармакологические свойства.
В одном варианте реализации настоящего изобретения соединение Формулы I, la, lb, или применяют в комбинации с ритонавиром. В одном варианте реализации комбинация представляет собой комбинацию с фиксированными дозами для перорального применения. В другом варианте реализации соединение формулы I, Ia, Ib, II или III выполнено в инъекционной форме продолжительного действия для парентерального введения, и ритонавир выполнен в форме композиции для перорального введения. В одном варианте реализации предложен комплект, содержащий соединение Формулы I, Ia, Ib, II или III, выполненное в инъекционной форме продолжительного действия для парентерального введения, и ритонавир, выполненный в форме композиции для перорального введения. В другом варианте реализации соединение формулы I, Ia, Ib, II или III выполнено в инъекционной форме продолжительного действия для парентерального введения, и ритонавир выполнен в форме композиции для перорального введения. В одном варианте реализации предложен набор, содержащий соединение Формулы I, Ia, Ib, II или III, выполненное в инъекционной форме продолжительного действия для парентерального введения, и ритонавир, выполненный в форме инъекционной композиции.
В другом варианте реализации настоящего изобретения соединение Формулы I, Ia, Ib, II или III применяют в комбинации с GS-9350. В одном варианте реализации комбинация представляет собой комбинацию с фиксированными дозами для перорального применения. В другом варианте реализации соединение формулы I, Ia, Ib, II или III выполнено в инъекционной форме продолжительного действия для парентерального введения, и GS-9350 выполнен в форме композиции для перорального введения. В одном варианте реализации предложен набор, содержащий соединение Формулы I, Ia, Ib, II или III, выполненное в инъекционной форме продолжительного действия для парентерального введения, и GS-9350, выполненный в форме композиции для перорального введения. В другом варианте реализации соединение Формулы I, Ia, Ib, II или III выполнено в инъекционной форме продолжительного действия для парентерального введения, и GS-9350 выполнен в форме композиции для перорального введения. В одном варианте реализации предложен набор, содержащий соединение Формулы I, Ia, Ib, II или III, выполненное в инъекционной форме продолжительного действия для парентерального введения, и GS-9350, выполненный в форме инъекционной композиции.
В одном варианте реализации настоящего изобретения соединение Формулы I, Ia, Ib, II или III применяют в комбинации с SPI-452. В одном варианте реализации комбинация представляет собой комбинацию с фиксированными дозами для перорального применения. В другом варианте реализации соединение формулы I, Ia, Ib, II или III выполнено в инъекционной форме продолжительного действия для парентерального введения, и SPI-452 выполнено в форме композиции для перорального введения. В одном варианте реализации предложен набор, содержащий соединения Формулы I, Ia, Ib, II или III, выполненное в инъекционной форме продолжительного действия для парентерального введения, и SPI-452, выполненный в форме композиции для перорального введения. В другом варианте реализации соединение Формулы I, Ia, Ib, II или III выполнено в инъекционной форме продолжительного действия для парентерального введения, и SPI-452 выполнен в форме композиции для перорального введения. В одном варианте реализации предложен набор, содержащий соединения Формулы I, Ia, Ib, II или III, выполнено в инъекционной форме продолжительного действия для парентерального введения, и SPI-452, выполненный в форме инъекционной композиции.
В одном варианте реализации настоящего изобретения соединение Формулы I, Ia, Ib, II или III применяют в комбинации с соединениями, описанными в поданной ранее заявке PCT/CN2011/0013021, которая включена в настоящий документ посредством ссылки. Приведенные выше терапевтические агенты, при использовании в комбинации с химическими соединениям, описанными в настоящем документе, могут применяться, например, в количествах, указанных в справочнике Physicians' Desk Reference (PDR, «Настольный справочник врача») или определенных иным образом специалистом в данной области.
В другом варианте реализации изобретения предложен способ лечения вирусной инфекции у млекопитающего, по меньшей мере частично опосредованной вирусом из семейства ретровирусов, причем указанный способ включает введение млекопитающему, у которого была диагностирована указанная вирусная инфекция, или у которого есть риск развития указанной вирусной инфекции, соединения Формулы I, Ia, Ib, II или III.
В другом варианте реализации изобретения предложен способ лечения вирусной инфекции у млекопитающего, по меньшей мере частично опосредованной вирусом из семейства ретровирусов, причем указанный способ включает введение млекопитающему, у которого была диагностирована указанная вирусная инфекция, или у которого есть риск развития указанной вирусной инфекции, соединения Формулы I, Ia, Ib, II или III, причем указанный вирус представляет собой вирус ВИЧ. В некоторых вариантах осуществления вирус ВИЧ представляет собой вирус ВИЧ-1.
В другом варианте реализации изобретения предложен способ лечения вирусной инфекции у млекопитающего, по меньшей мере частично опосредованной вирусом из семейства ретровирусов, причем указанный способ включает введение млекопитающему, у которого была диагностирована указанная вирусная инфекция, или у которого есть риск развития указанной вирусной инфекции, соединения Формулы I, Ia, Ib, II или III, дополнительно включающий введение терапевтически эффективного количества одного или более агентов, обладающих активностью против вируса ВИЧ.
В другом варианте реализации изобретения предложен способ лечения вирусной инфекции у млекопитающего, по меньшей мере частично опосредованной вирусом из семейства ретровирусов, причем указанный способ включает введение млекопитающему, у которого была диагностирована указанная вирусная инфекция, или у которого есть риск развития указанной вирусной инфекции, соединения Формулы I, Ia, Ib, II или III, дополнительно включающий введение терапевтически эффективного количества одного или более агентов, обладающих активностью против вируса ВИЧ, причем указанный агент, обладающий активностью против вируса ВИЧ, выбран из нуклеотидных ингибиторов обратной транскриптазы, ненуклеотидных ингибиторов обратной транскриптазы, ингибиторов протеазы, ингибиторов проникновения, присоединения и слияния, ингибиторов интегразы, ингибиторов созревания, ингибиторов CXCR4 и ингибиторов CCR5.
В другом аспекте настоящего изобретения предложен Способ истощения латентных ВИЧ-инфицированных клеток включающий введение субъекту соединения Формулы (I), (Ia), (Ib), (II) или (III) или его фармацевтически приемлемая соль.
В различных вариантах реализации указанного способа каждый из Alk, Alk2 и Alk3 представлен формулой:
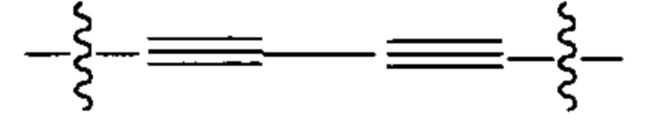
В различных вариантах реализации указанного способа каждый из Ar1, Ar2, Ar3, Ar4, Ar5, Ar6, Ar7, Ar8, Ar9, Ar10, Ar11, Ar21, Ar22, Ar23, Ar24, Ar25 и Ar26 представляет собой С6 арил. В различных вариантах реализации описанного выше изобретения каждый из Ar12, Ar13, Ar14 and Ar15, Ar16, Ar17, Ar18 и Ar19 представляет собой С9 арил.
В различных вариантах реализации описанного выше изобретения каждый из Ar16, Ar17, Ar18 и Ar19 представляет собой С10 арил.
В различных вариантах реализации описанного выше изобретения линкер (L) выбран из группы, состоящей из:
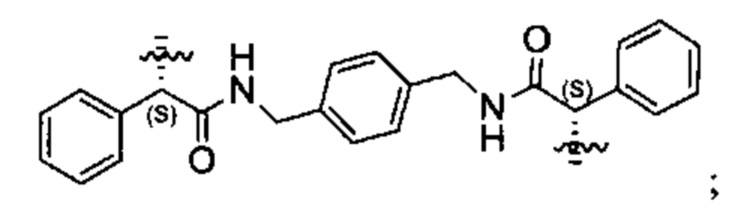
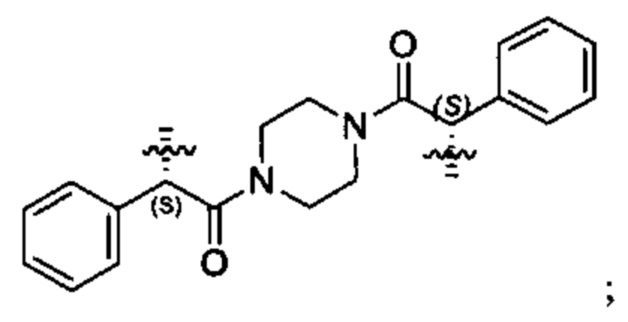
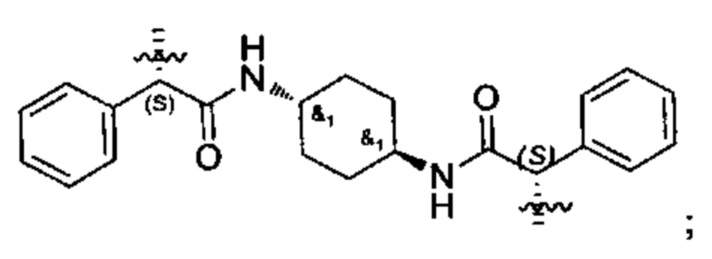
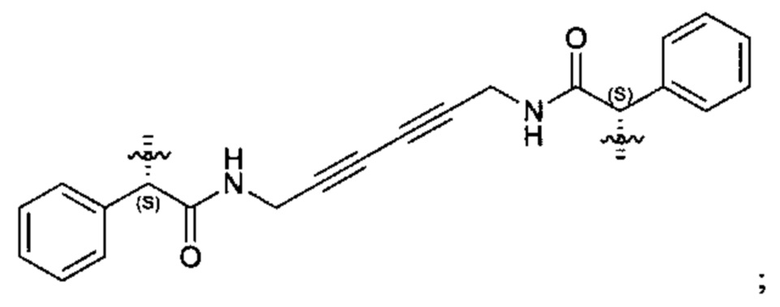
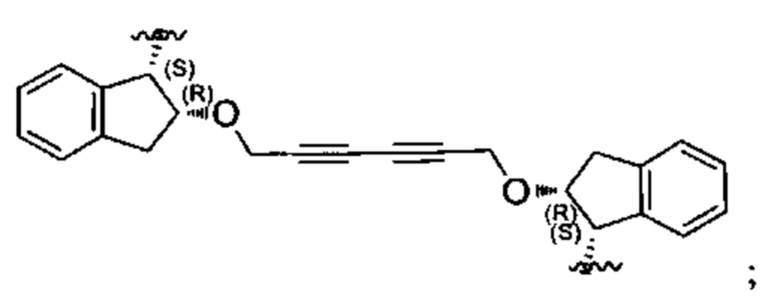
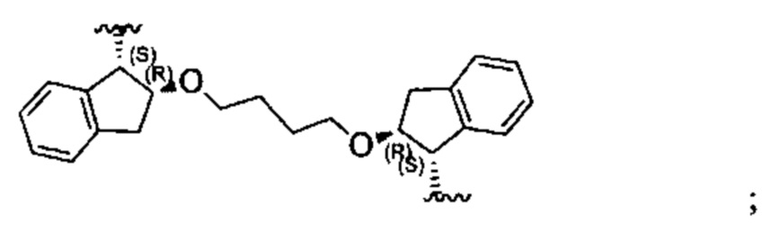
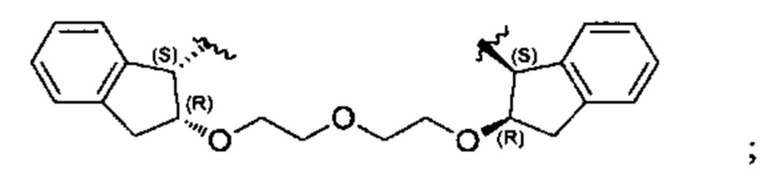
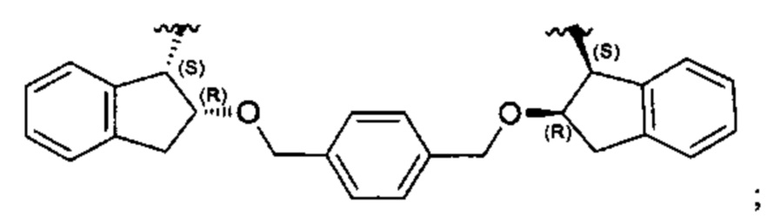
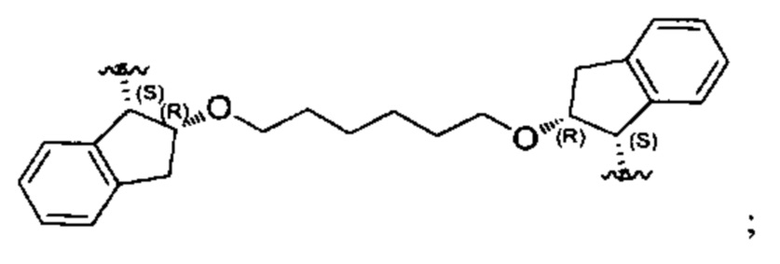
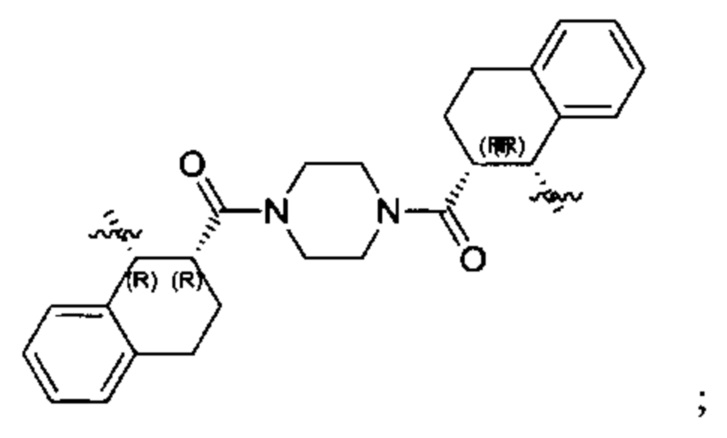
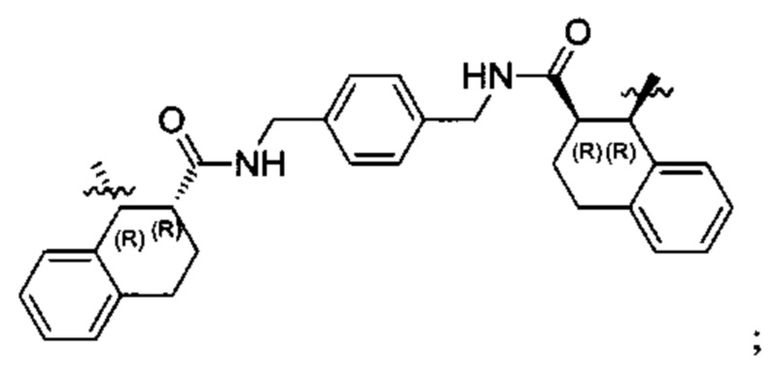
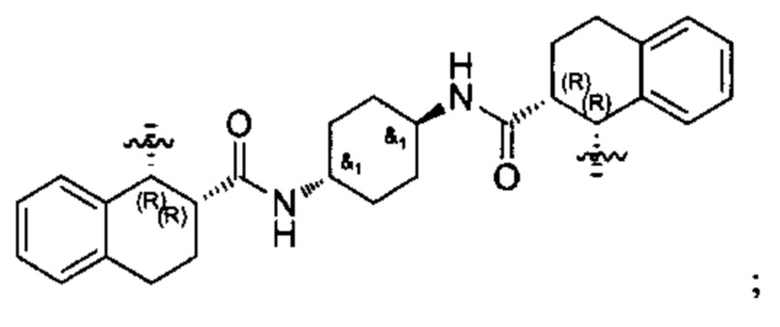
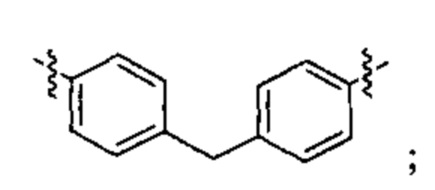
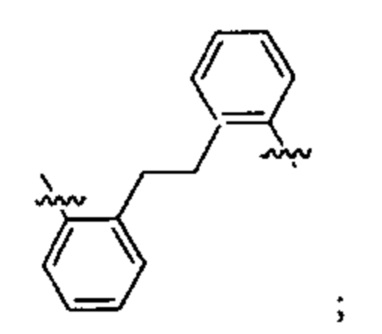
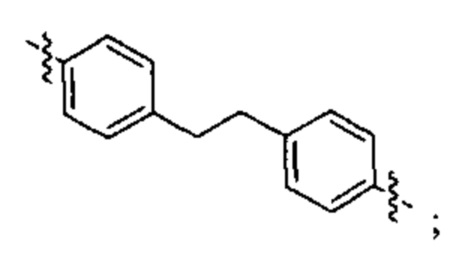
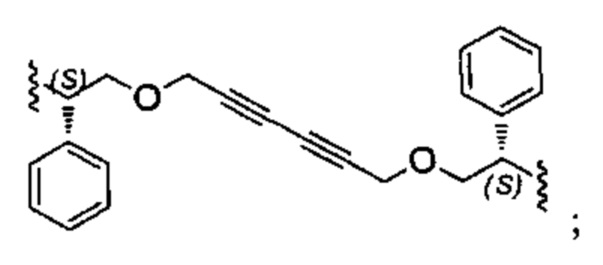
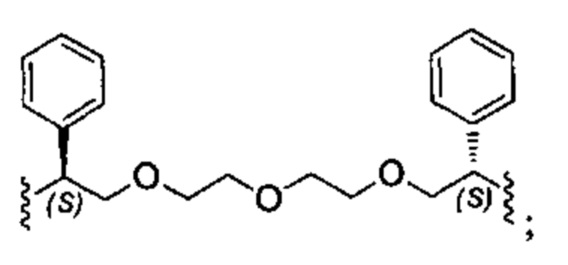
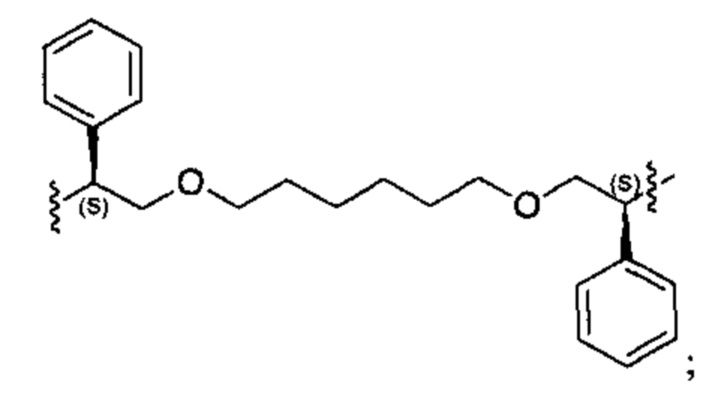
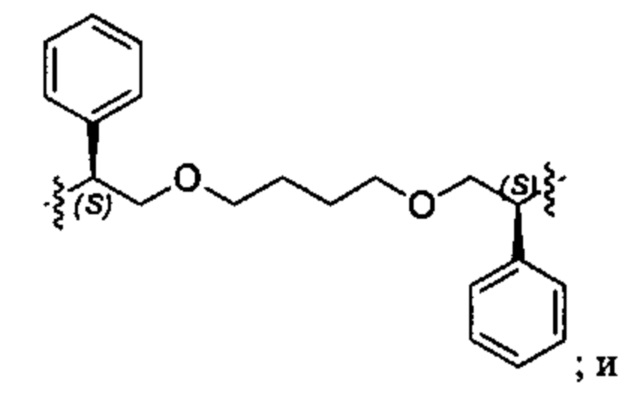
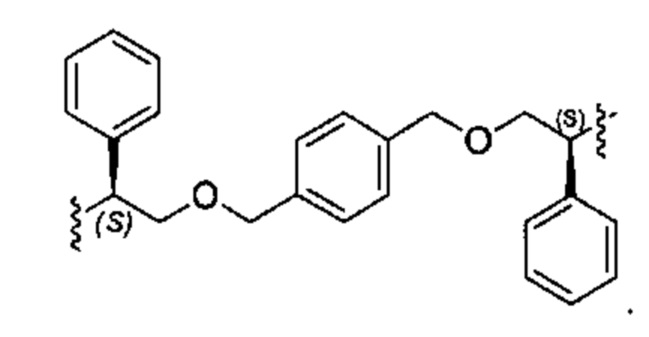
В различных вариантах реализации описанного выше изобретения, соединение выбран из группы, состоящей из следующих соединений:
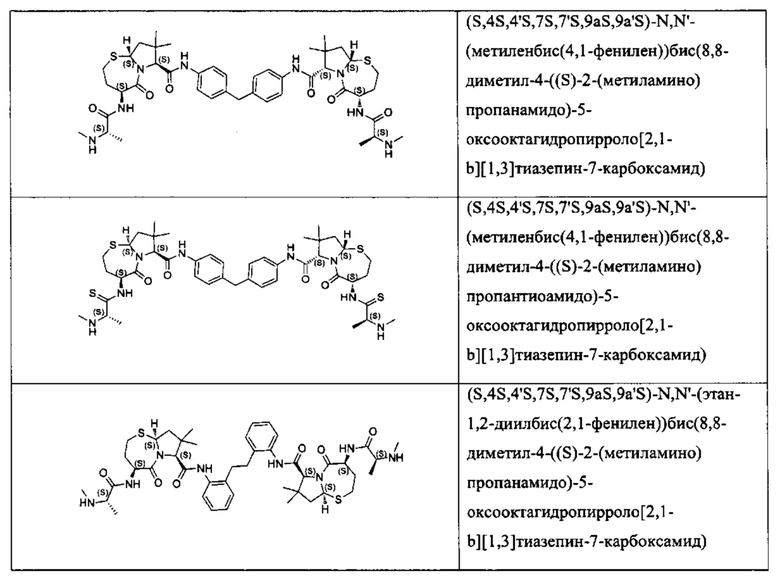
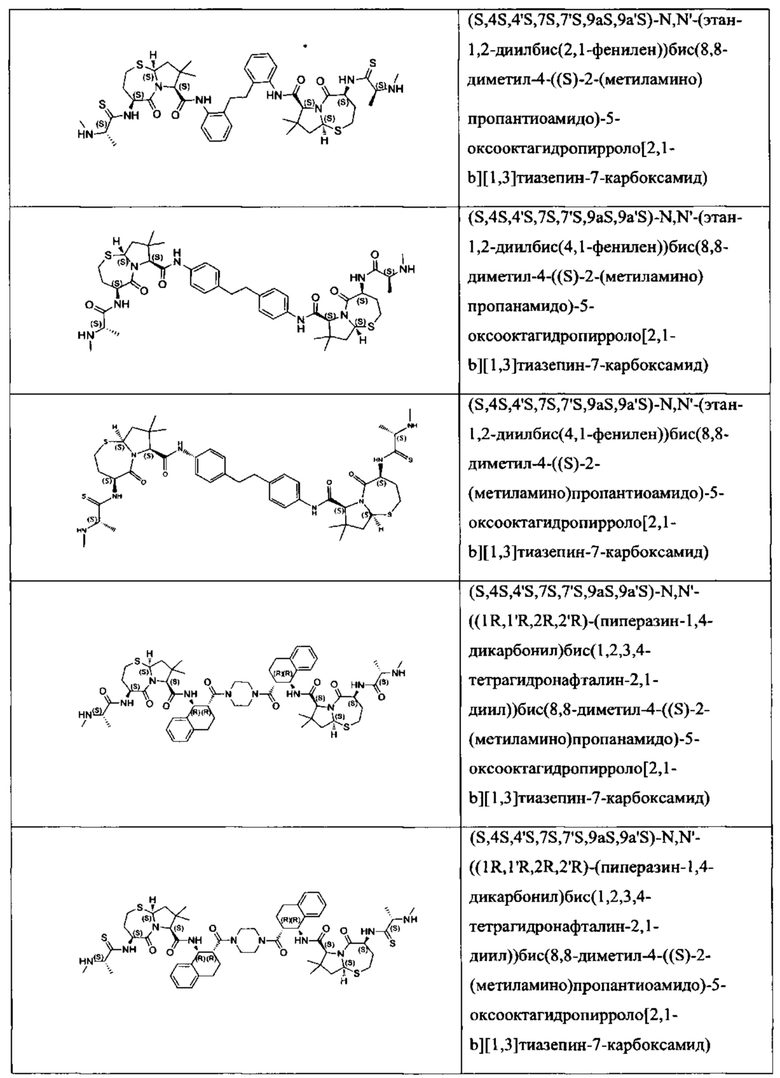
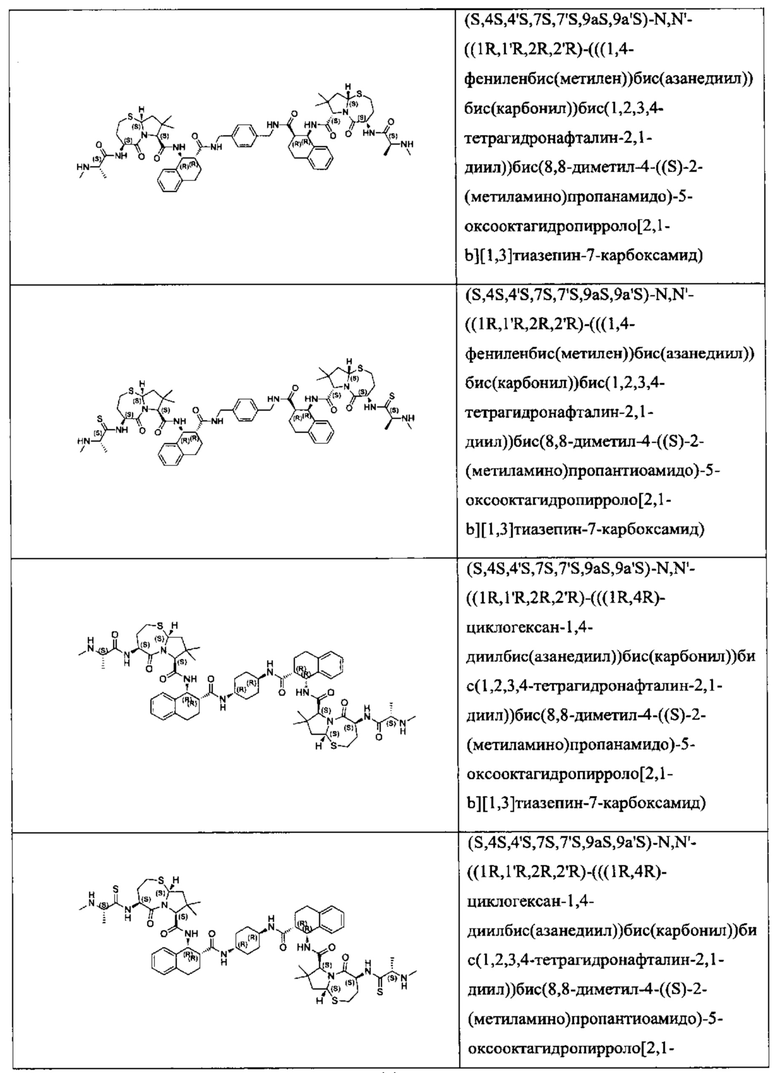
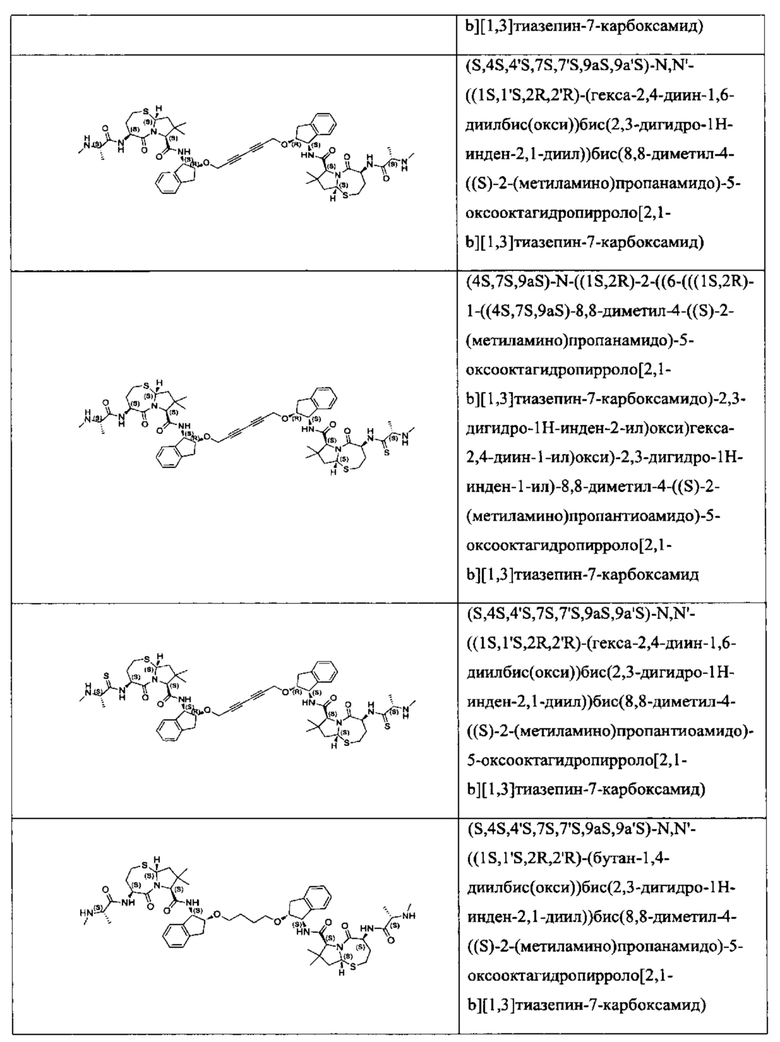
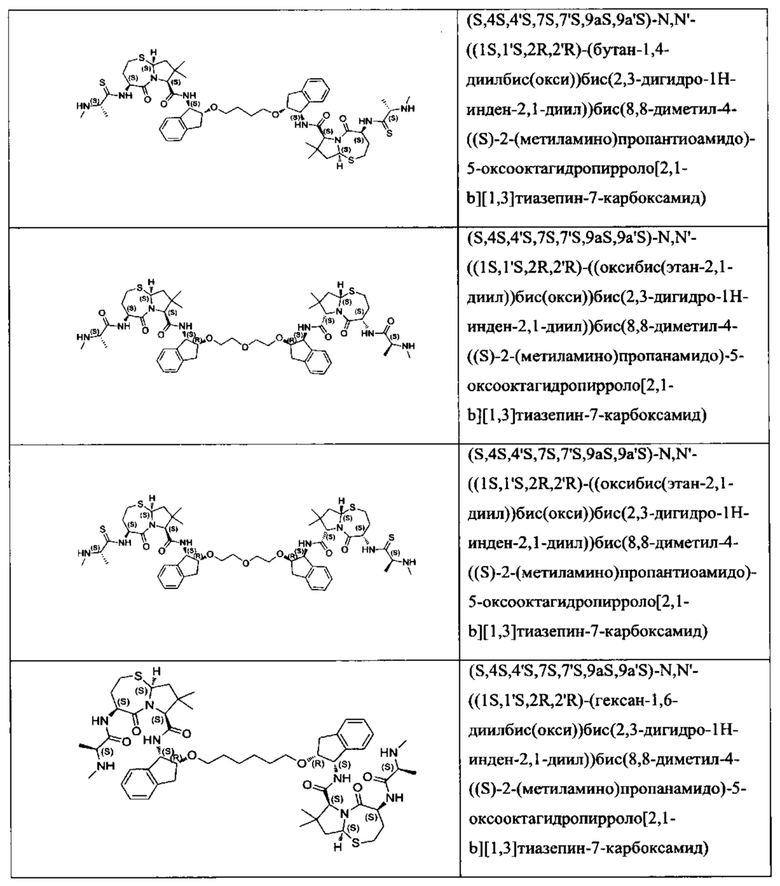
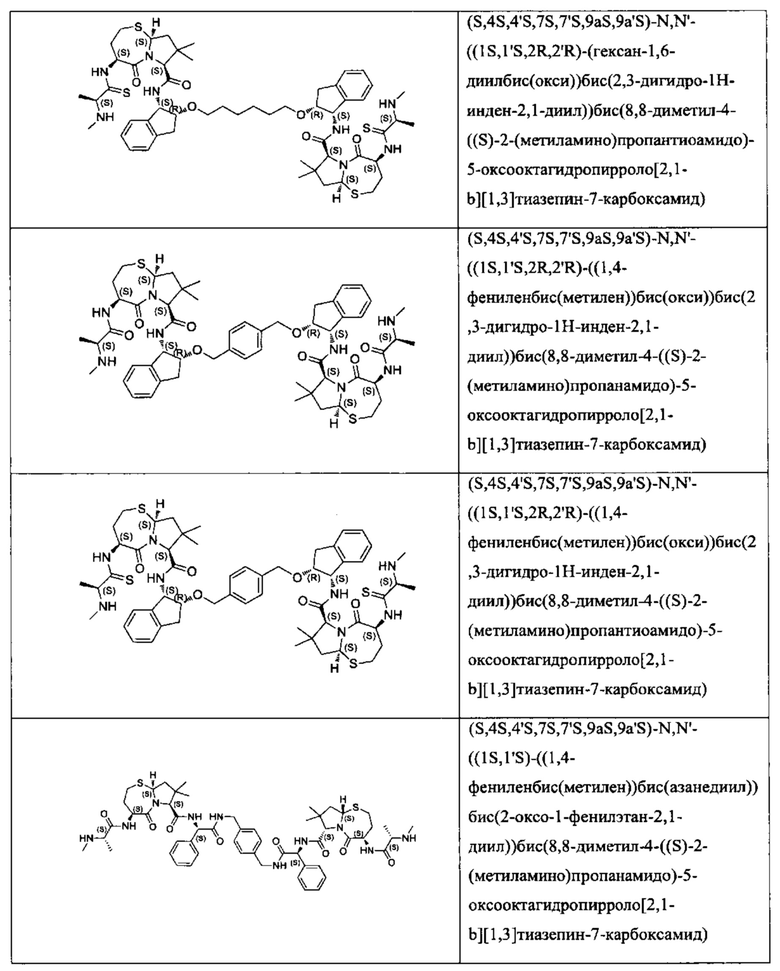
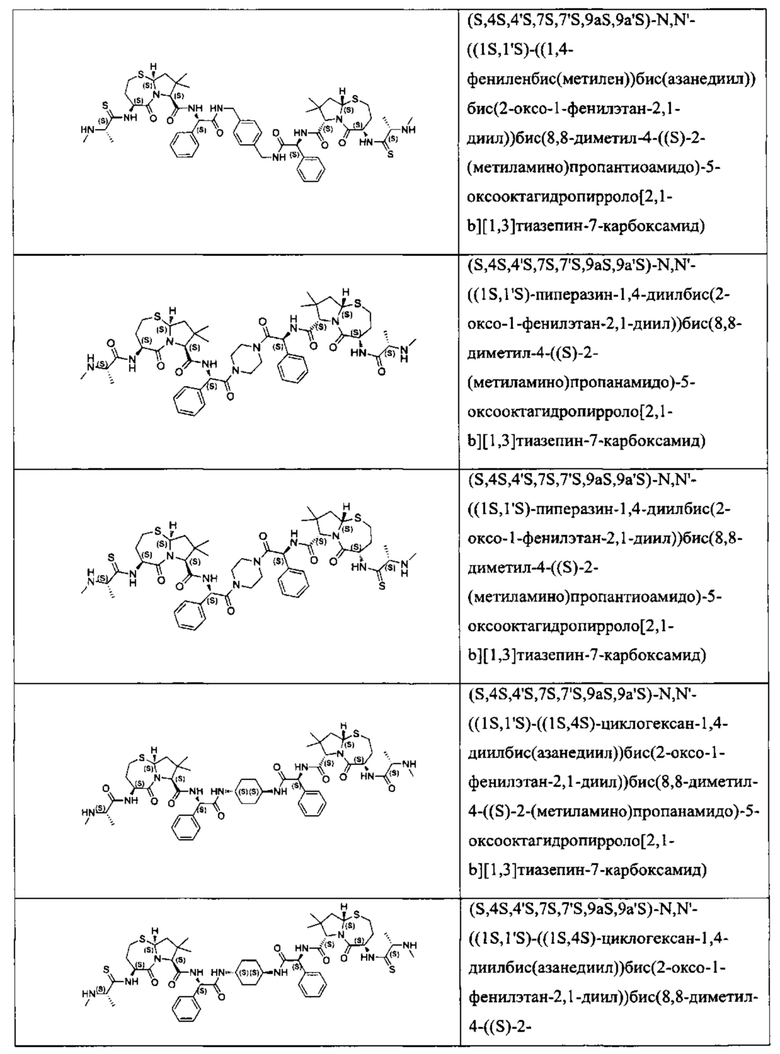
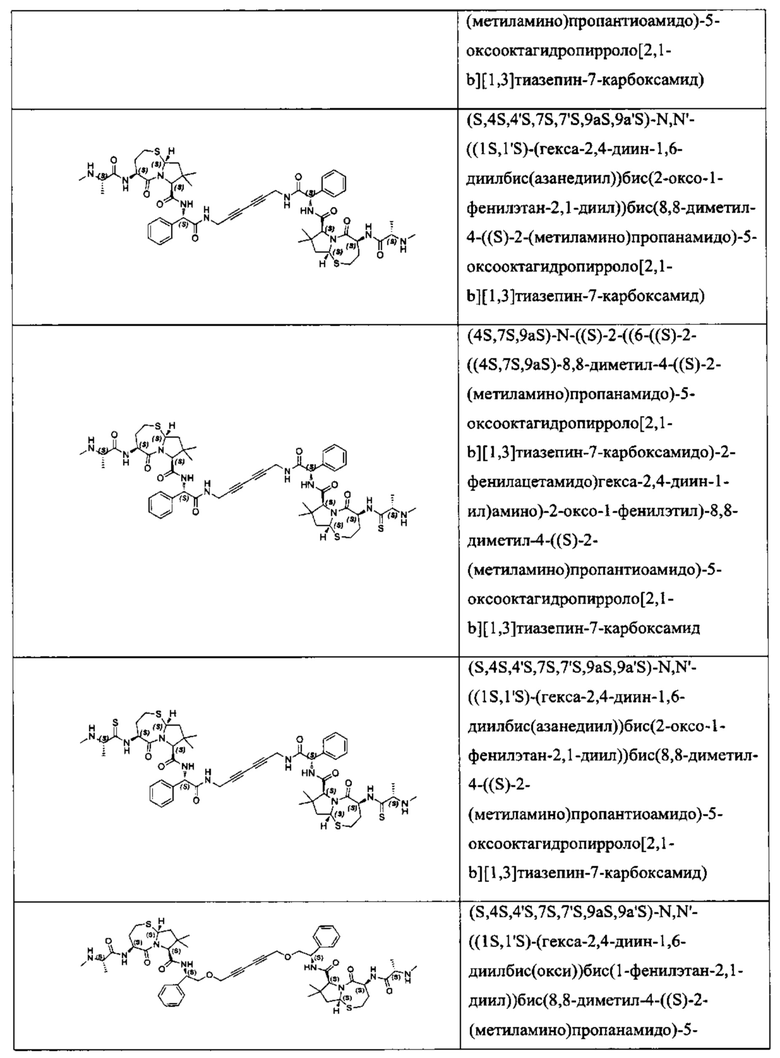
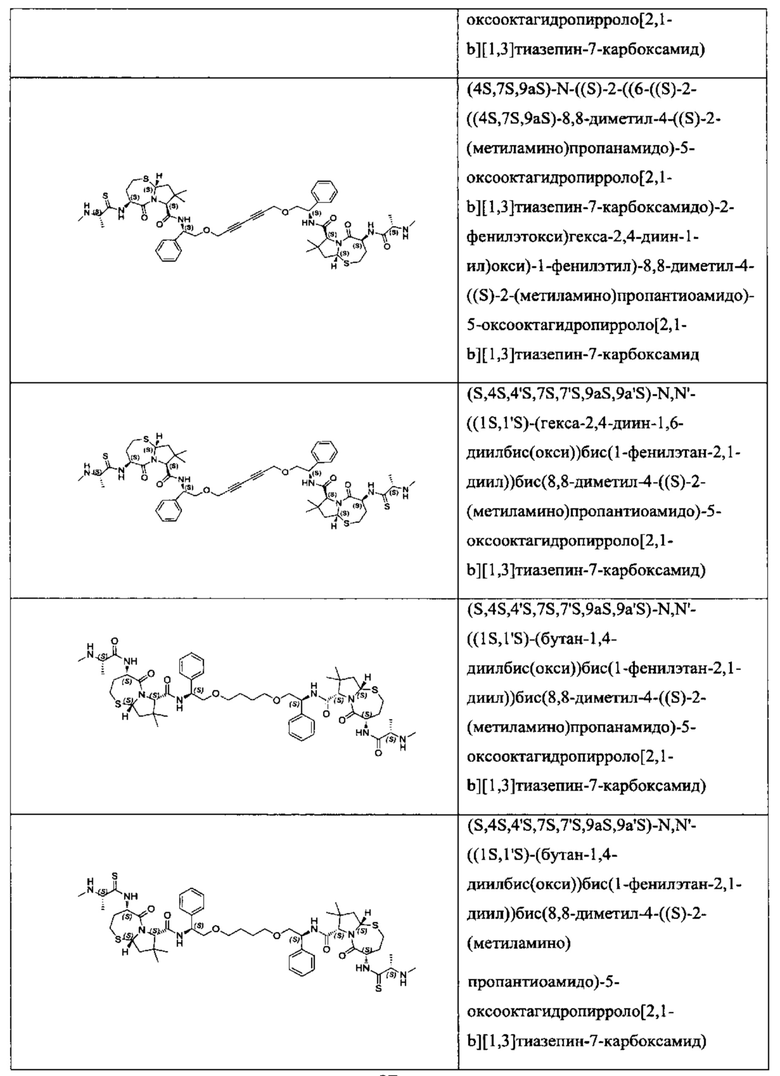
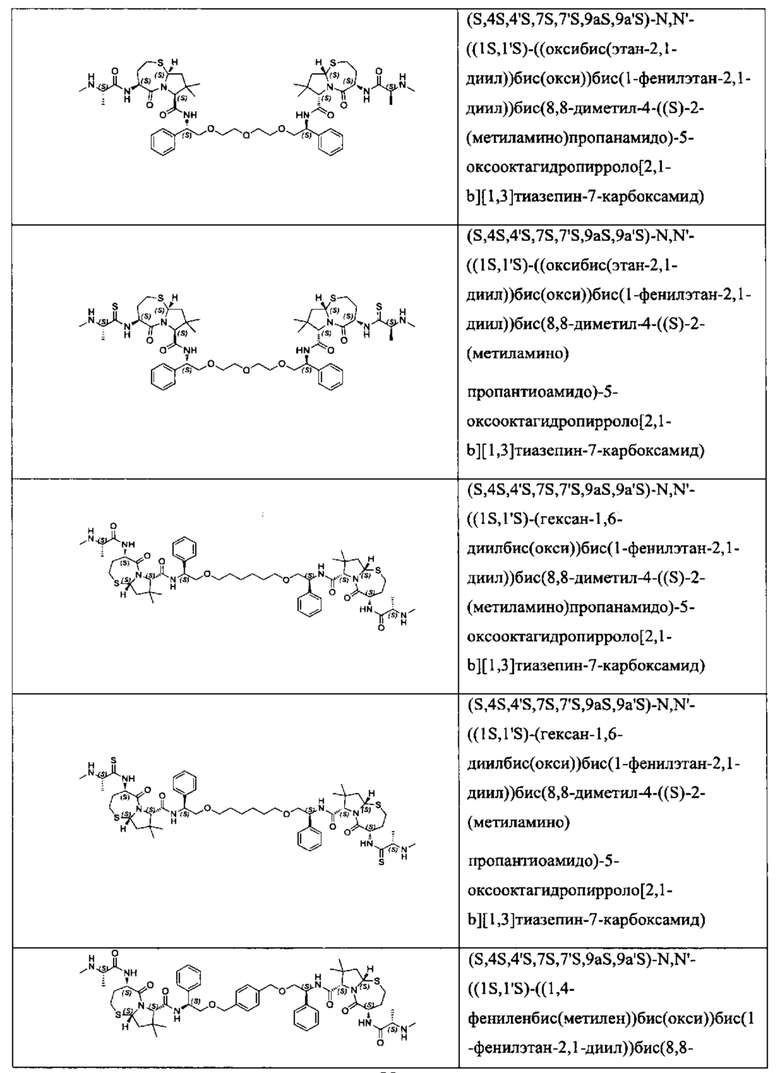
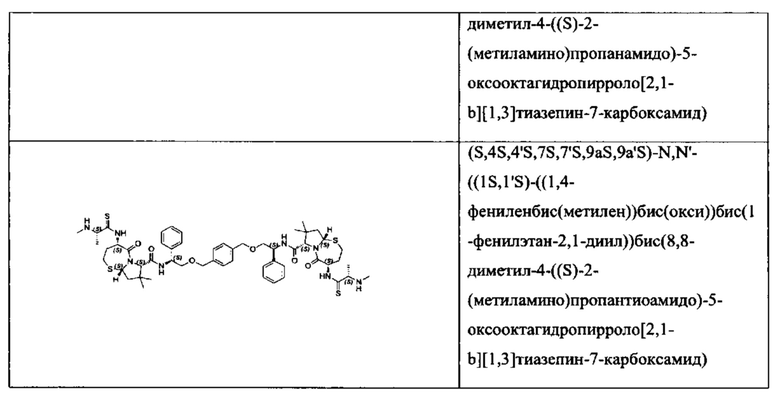
В различных вариантах реализации указанного способа Х1 Х2 каждый представляет собой О, предпочтительно для Формул (Ia) и (Ib).
В различных вариантах реализации указанного способа в линкере (L), каждый из Ar1, Ar2, Ar3, Ar4, Ar5, Ar6, Ar7, Ar8, Ar9, Ar10, Ar11, Ar20, Ar21, Ar22, Ar23 и Ar24 представляет собой С6 арил.
В различных вариантах реализации указанного способа линкер (L) выбран из группы, состоящей из (i) и (v).
В различных вариантах реализации указанного способа каждый из m, n, p, и q представляет собой 1, и каждая группа (СН2)0-3 в формуле (v) представлена (СН2).
В различных вариантах реализации указанного способа, а также соединений линкер (L) выбран из группы, состоящей из:
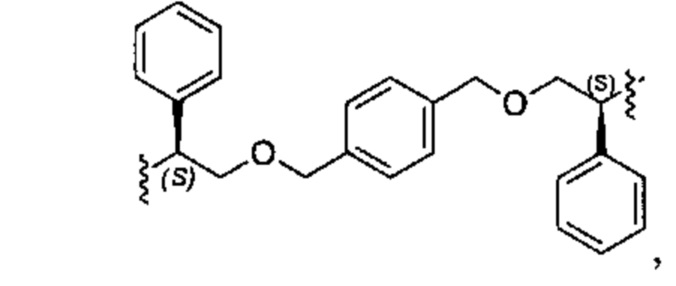
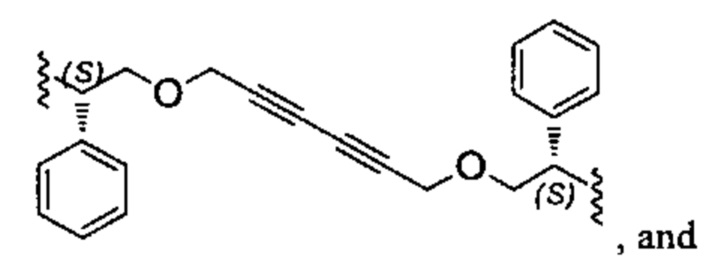

В различных вариантах реализации указанного способа линкер (L) выбран из группы, состоящей из (vi) и (vii), и каждый из Ar12, Ar13, Ar14 и Ar15 представляет собой С9 арил.
В различных вариантах реализации указанного способа каждый из Ar12 и Ar14 представляет собой 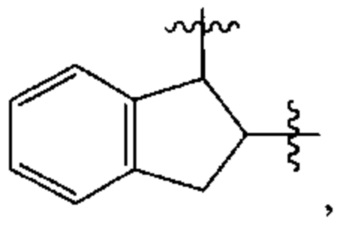
и каждый из Ar13 и Ar15 представляет собой 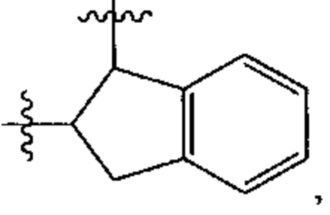 где волнистые линии представляют точки присоединения.
где волнистые линии представляют точки присоединения.
В различных вариантах реализации указанного способа линкер (L) выбран из группы, состоящей из:
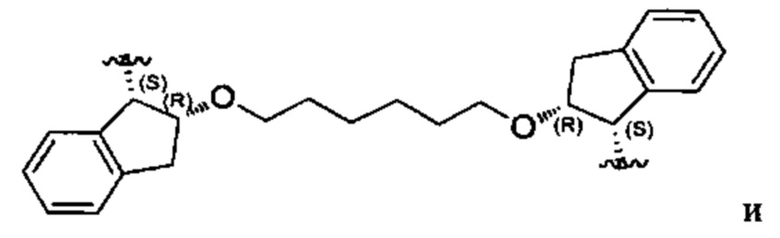

В одном варианте осуществления изобретения настоящее изобретение относится к соединению (соединение 13) формулы:
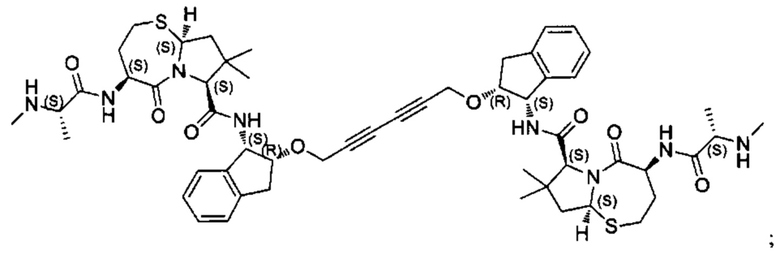
или его фармацевтически приемлемая соли. Настоящее изобретение также включает фармацевтическую композицию, содержащую это, или его фармацевтически приемлемую соль, и фармацевтически приемлемое вспомогательное вещество, включая, например, приведенные в настоящем документе. Настоящее изобретение также включает способ лечения ВИЧ-инфекции у субъекта включающий введение субъекту этого соединения или его фармацевтически приемлемой соли, а также комбинаций. Настоящее изобретение также включает это соединение, или его фармацевтически приемлемую соль, для применения для лечения ВИЧ-инфекции. Настоящее изобретение также включает применение этого соединения в изготовлении лекарственного средства для лечения ВИЧ-инфекции. Настоящее изобретение также включает способ истощения латентных ВИЧ-инфицированных клеток включающий введение субъекту этого соединения или его фармацевтически приемлемой соли, а также их комбинаций.
В одном варианте осуществления изобретения настоящее изобретение относится к соединению (соединение 20) формулы:
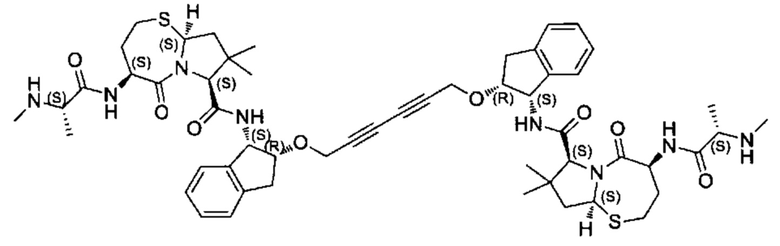
или его фармацевтически приемлемой соли. Настоящее изобретение также включает фармацевтическую композицию, содержащую это соединение, или его фармацевтически приемлемую соль, и фармацевтически приемлемое вспомогательное вещество, включая, например, приведенные в настоящем документе. Настоящее изобретение также включает способ лечения ВИЧ-инфекции у субъекта включающий введение субъекту этого соединения или его фармацевтически приемлемой соли, а также комбинаций. Настоящее изобретение также включает это соединение, или его фармацевтически приемлемую соль, для применения для лечения ВИЧ-инфекции. Настоящее изобретение также включает применение этого соединения в изготовлении лекарственного средства для лечения ВИЧ-инфекции. Настоящее изобретение также включает способ истощения латентных ВИЧ-инфицированных клеток включающий введение субъекту этого соединения или его фармацевтически приемлемой соли, а также их комбинаций.
В различных вариантах осуществления способ истощения латентной ВИЧ-инфекции дополнительно включает введение субъекту одного или более дополнительных агентов, обладающих активностью против ВИЧ, раскрытых выше в этом документе. В качестве примера, в различных вариантах осуществления один или более дополнительных агентов выбраны из группы, состоящей из нуклеотидных ингибиторов обратной транскриптазы, ненуклеотидных ингибиторов обратной транскриптазы, ингибиторов протеазы, ингибиторов проникновения в клетку, ингибиторов прикрепления и слияния, ингибиторов интегразы, ингибиторов созревания, ингибиторов CXCR4 и/или CCR5, ингибиторов гистондеацетилазы, ингибиторов гистонкротонилтрансферазы, агонистов протеинкиназы С, ингибиторы протеасом, агонистов TLR7, ингибиторов бромодомена и антител для клиренсной терапии и их комбинаций. В различных вариантах осуществления один или более дополнительных агентов, обладающих активностью против ВИЧ, выбраны из группы, состоящей из зидовудина, диданозина, ламивудина, зальцитабина, абакавира, ставудина, адефовира, адефовира дипивоксила, фозивудина, тодоксила, эмтрицитабина, аловудина, амдоксовира, элвуцитабина, невирапина, делавердина, эфавиренза, ловирида, иммунокала, олтипраза, каправирина, лерсивирина, GSK2248761, ТМС-278, ТМС-125, этравирина, саквинавира, ритонавира, индинавира, нелфинавира, ампренавира, фосампренавира, бреканавира, дуранавира, атазанавира, типранавира, палинавира, ласинавира, энфувиртида, Т-20, Т-1249, PRO-542, PRO-140, TNX-355, BMS-806, BMS-663068 и BMS-626529, 5-Helix, ралтегравира, элвитегравира, долутегравира, каботегравира, биктегравира, викривирока (Sch-C), Sch-D, TAK779, маравирока, TAK449, диданозина, тенофовира, лопинавира, дуранавира, вориностата, панобиностата, ромидепина, валпроиновой кислоты, моцетиностата, коротоната натрия, бриостатина, ингенола В, дисульфорама, GS-9620, JQ1, iBET151, бортезомида, эпигаллокатехина галлата, салиноспорамида А, карфилзомиба и нейтрализующих антител, eCD4-Ig, CD4-Ig, bNAb, DARTS и IgA.
Соединения согласно Формуле I, Ia, Ib, II и III и их фармацевтически приемлемые соли могут быть полезны для лечения рака, предраковых синдромов. В соответствующих случаях настоящее изобретение относится к способу лечения раков, выбранных из группы, состоящей из рака головного мозга (глиом), глиобластом, астроцитом, мультиформной глиобластомы, синдрома Баннайана-Зонана, болезни Коудена, болезни Лермитта-Дюкло, опухоли Вильмса, саркомы Юинга, рабдомиосаркомы, эпендимиомы, медуллобластомы, рака головы и шеи, почки, печени, меланомы, рака яичника, поджелудочной железы, аденоцарциномы, протоковой аденокарциномы, железисто-плоскоклеточной карциномы, ацинозно-клеточной карциномы, глюкагономы, инсулиномы, предстательной железы, саркомы, остеосаркома, гигантоклеточной опухоли кости, рака щитовидной железы, лимфобластного Т-клеточного лейкоза, хронического миелогенного лейкоза, хронического лейкоцитарного лейкоза, волосатоклеточного лейкоза, острого лимфобластного лейкоза, острого миелогенного лейкоза, хронического нейтрофильного лейкоза, острого лимфобластного Т-клеточного лейкоза, плазмацитомы, иммунобластного крупноклеточного лейкоза, мантийноклеточного лейкоза, множественной миеломы, мегакариобластного лейкоза, множественной миеломы, острого мегакариоцитарного лейкоза, промиелоцитарного лейкоза, эритролейкоза, злокачественной лимфомы, ходжкинской лимфомы, неходжкинской лимфомы, лимфобластной Т-клеточной лимфомы, лимфомы Беркитта, фолликулярной лимфомы, нейробластомы, рака мочевого пузыря, рака уротелия, рака вульвы, рака шейки матки, рака эндометрия, почечного рака, мезотелиома, рака пищевода, рака слюнной железы, почечноклеточного рака, рака желудка, рака носоглотки, рака щеки, рака ротовой полости, GIST (стромальная опухоль ЖКТ) и рака яичек.
В соответствующих случаях настоящее изобретение относится к способу лечения предраковых синдромов у млекопитающего, включая человека, где предраковый синдром выбран из: интраэпителиальной неоплазии шейки матки, моноклональной гаммапатии неизвестного генеза (MGUS), миелодиспластического синдрома, апластической анемии, поражений шейки матки, кожных невусов (пре-меланомы), интраэпителиальной (интрадуктальной) неоплазии предстательной железы(РГЫ), протоковой карциномы in situ (DCIS), полипов толстой кишки и тяжелого гепатита или цирроза.
Соединения Формул (I), (Ia), (Ib), (II), (III) и их фармацевтические соли можно вводить совместно с по меньшей мере одним другим агентом, о котором известно, что его можно применять для лечения или предраковых синдромов.
Под термином "совместное введение" в настоящем документе подразумевают либо одновременное введение или любой способ раздельного последовательного введения c-MYC-ингибирующего соединения в соответствии с описанием, приведенным в настоящем документе, и дополнительного активного агента или агентов, о которых известно, что они полезны для лечения рака, включая химиотерапию и лучевую терапию. Термин «дополнительный активный агент или агенты» в настоящем тексте включает любое соединение или терапевтический агент, о котором известно, или который демонстрирует полезные свойства при введении пациенту, нуждающемуся в лечении рака. Предпочтительно, если введение является не одновременным, соединения вводят близко друг к другу по времени. Кроме того, не важно, вводят ли соединения в одной лекарственной форме, например, одно соединение можно вводит путем инъекции, а другое соединение можно вводить перорально.
Примеры дополнительного активного ингредиента или ингредиентов (антинеопластического агента) для применения в комбинации или совместного введения с соединениями согласно настоящему изобретению приведены ниже. Этот список не является ограничивающим. Предусмотрены антинеопластические агенты для применения с соединениями согласно настоящему изобретению.
Обычно любой антинеопластический агент, который обладает активностью против восприимчивой опухоли, подвергаемой лечению, можно вводить совместно для лечения рака в настоящем изобретении. Примеры таких агентов можно найти в публикации Cancer Principles and Practice of Oncology V.T. Devita and S. Hellman (editors), 6е изд. (15 февраля 2001 г.), Lippincott Williams & Wilkins Publishers. Специалист в данной области сможет различить, какие комбинации агентов будут полезны, на основании конкретных характеристик лекарственных средств и рака, о котором идет речь. Типичные антинеопластические агенты, которые можно в настоящем изобретении, включают, без ограничения перечисленными, агенты, воздействующие на микротрубочки, такие как дитерпеноиды и алкалоиды барвинка; координационные комплексы платины; такие как азотистый иприт, оксазофосфорины, алкилсульфонаты, нитрозомочевины и триазены; антибиотики, такие как антрациклины, актиномицины и блеомицины; ингибиторы топоизомеразы II, такие как эпиподофиллотоксины; антиметаболиты, такие как аналоги пурина и пиримидина, и антифолатные соединения; ингибиторы топоизомеразы I, такие как камптотецины; гормоны и аналоги гормонов; ингибиторы сигнальных каскадов; не относящиеся к рецепторным тирозинкиназам ингибиторы ангиогенеза; иммунотерапевтические агенты; проапоптотические агентов; ингибиторы клеточной сигнализации; ингибиторы протеасом; и ингибиторы ракового метаболизма.
Примерами дополнительного активного ингредиента или ингредиентов (антинеопластического агента) для применения в комбинации или вводимых совместно соединениями согласно настоящему изобретения являются химиотерапевтические агенты.
Агентами, воздействующими на микротрубочки, или антимитотическими агентами являются фазоспецифические агенты, проявляющие активность против микротрубочек опухолевых клеток во время фазы М или фазы митоза клеточного цикла. Примеры агентов, воздействующих на микротрубочки включают, без ограничения перечисленными: дитерпеноиды и алкалоиды барвинка.
Дитерпеноиды, которые получают из природных источников, являются фазоспецифическими противораковыми агентами, которые действуют в G2/M фазах клеточного цикла. Считается, что дитерпеноиды стабилизируют β-тубулиновые субъединицы микротрубочек путем связывания с этим белком. Затем, по-видимому, ингибируется разборка белка, что сопровождается блокировкой митоза и последующей гибелью клеток. Примеры дитерпеноидов включают, без ограничения перечисленными, паклитаксел и его аналог доцетаксел.
Паклитаксел, 13-сложный эфир 5β,20-эпокси-1,2α,4,7β,10β,13α-гекса-гидрокситакс-11-ен-9-он-4,10-диацетат-2-бензоата с (2R,3S)-N-бензоил-3-фенилизосерином, является природным дитерпеноидным продуктом, выделенным из дерева коротколистного или американского тисса Taxus brevifolia, и коммерчески доступен в виде раствора для инъекций TAXOL® (таксол). Он является членом таксанового семейства терпенов. Впервые он был выделен в 1971 году авторами Wani et al., J. Am. Chem. Soc, 93: 2325. (1971), которые охарактеризовали его структуру с помощью химического и рентгенокристаллографического методов. Один из механизмов его активности относится к способности паклитаксела связывать тубулин, ингибируя, тем самым, рост раковых клеток. Schiff et al., Proc. Natl, Acad. Sci. USA, 77: 1561-1565 (1980); Schiff et al., Nature, 277: 665-667 (1979); Kumar, J. Biol. Chem., 256: 10435-10441 (1981). Обзор синтеза и противораковой активности некоторых производных паклитаксела, можно найти в: D.G.I. Kingston et al., Studies in Organic Chemistry vol. 26, под названием "New trends in Natural Products Chemistry 1986", Attaur-Rahman, P.W. Le Quesne, Eds, (Elsevier, Амстердам, 1986) стр. 219-235.
Паклитаксел был одобрен для клинического применения в лечении рефракторного рака яичников в Соединенных Штатах (Markman et al., Yale Journal of Biology and Medicine. 64-583, 1991; McGuire et al., Ann. Intern. Med., 111: 273, 1989) и для лечения рака груди (Holmes et al., J. Nat. Cancer Inst., 83: 1797, 1991). Он является потенциальным кандидатом для лечения новообразований на коже (Einzig et al., Proc. Am. Soc. Clin. Oncol. 20: 46) и карцином головы и шеи (Forastire et al., Sem. Oncol. 20: 56, 1990). Это соединение обнаруживает также потенциал для лечения поликистоза почек (Woo et al., Nature, 368: 750, 1994), рака легких и малярии. Лечение пациентов паклитакселом приводит к подавлению костного мозга (множественные клеточные линии, Ignoff, R.J. et al., Cancer Chemotherapy Pocket Quide, 1998), связанному с длительностью введения доз выше пороговой концентрации (50 нМ) (Kearns, С.М. et al., Seminars in Oncology, 3 (6) стр. 16-23, 1995).
Доцетаксел, 13-эфир N-трет-бутилового эфира (2R,3S)-N-карбокси-3-фенилизосерина с тригидратом 5β-20-эпокси-1,2α,4,7β,10β,13α-гексагидрокситакс-11-ен-9-он-4-ацетат-2-бензоата, коммерчески доступен в виде раствора для инъекций TAXOTER® (ТАКСОТЕР). Доцетаксел показан для лечения рака молочной железы. Доцетаксел является полусинтетическим производным паклитаксела, см. другие упомнияния, получаемым с использованием природного предшественника, 10-деацетилбаккатина III, экстрагируемого из иголок ягодного или европейского тиса. Дозоограничивающим видом токсичности доцетаксела, является нейтропения.
Алкалоиды барвинка являются фазоспецифическими антинеопластическими агентами, получаемыми из растения барвинок. Алкалоиды барвинка действуют в М-фазе (митоза) клеточного цикла за счет специфического связывания с тубулином Вследствие этого, связанная молекула тубулина не способна полимеризоваться в микротрубочки. Считается, что митоз задерживается в метафазе с последующей гибелью клеток. Примеры алкалоидов барвинка включают, без ограничения перечисленными, винбластин, винкристин и винорелбин.
Винбластин, сульфат винкалейкобластина, коммерчески доступен под маркой VELBAN® (велбан) в форме раствора для инъекций. Хотя он имеет показания в качестве возможной второй линии терапии разнообразных солидных опухолей, в первую очередь он показан при лечении рака яичек и различных лимфом, включая болезнь Ходжкина, и лимфоцитарную и гистиоцитарную лимфомы. Миелосуппрессия является дозоограничивающим побочным эффектом винбластина.
Винкристин, винкалейкобластин, 22-оксосульфат, коммерчески доступен под маркой ONCOVIN® (онковин) в форме раствора для инъекций. Винкристин показан для лечения острых лейкозов и обнаружено также, что он полезен в лечебных режимах для лечения злокачественных лимфом Ходжкина и неходжкинских лимфом. Алопеция и неврологические эффекты являются наиболее частым побочным действием винкристина, и в меньшей степени имеет место эффект миелосупрессии и воспаления слизистой желудочно-кишечного тракта.
Винорелбин, 3',4'-дидегидро-4'-дезокси-С'-норвинкалейкобластин [R-(R*,R*)-2,3-дигидроксибутандиоат (1:2)(соль)], коммерчески доступный в форме раствора для инъекций винорелбина тартрата (NAVELBINE® (навелбин)), является полусинтетическим алкалоидом барвинка. Винорелбин показан как агент монотерапии или в комбинации с другими химиотерапевтическими агентами, такими как цисплатин, в лечении различных солидных опухолей, в частности, немелкоклеточного рака легких, прогрессирующего рака молочной железы и гормональнорефракторных раков предстательной железы. Миелосупрессия является наиболее обычным дозолимитирующим побочным эффектом винорелбина.
Координационные комплексы платины являются нефазоспецифическими противораковыми агентами, которые взаимодействуют с ДНК. Комплексы платины проникают в опухолевые клетки, гидратируются и образуют внутри- и межнитевые поперечные связи в ДНК, оказывая пагубные для опухоли биологические эффекты. Примырф координационных комплексов платины включают, без ограничения перечисленными: цисплатин м карбоплатин.
Цисплатин, цис-диаминдихлорплатина, коммерчески доступен под маркой PLATINOL® (платинол) в форме раствора для инъекций. Цисплатин прежде всего показан при лечении метастатического рака яичек и рака яичников и прогрессирующего рака мочевого пузыря. Основными ограничивающими дозу побочными действиями цисплатина, являются нефротоксичность, которая может регулироваться гидратацией и диурезом, и ототоксичность.
Карбоплатин, диамин [1,1-циклобутан-дикарбоксилат(2-)-O,O'], коммерчески доступен под маркой PARAPLATIN® в форме раствора для инъекций. Карбоплатин прежде всего показан в первой и второй линии терапии распространенной карциномы яичника. Супрессия костного мозга является ограничивающей дозу токсичностью карбоплатина.
Алкилирующие агенты являются нефазовыми специфическими противораковыми агентами и сильными электрофилами. Обычно алкилирующие агенты образуют ковалентные связи путем алкилирования ДНК через нуклеофильные фрагменты молекулы ДНК, такие как фосфатная, амино-, сульфгидрильная, гидроксильная, карбоксильная и имидазольная группы. Такое алкилирование нарушает функционирование нуклеиновой кислоты, ведущее к гибели клеток. Примеры алкилирующих агентов включают, без ограничения перечисленными: азотистый иприт, такие как циклофосфамид, мелфалан и хлорамбуцил; алкилсульфонаты, такие как бусульфан; нитрозомочевины, такие как кармустин; и триазены, такие как дакарбазин.
Циклофосфамид, 2-оксида 2-бис[2-хлорэтил)амино]тетрагидро-2Н-1,3,2-оксазафосфорина моногидрат, коммерчески доступен в форме раствора для инъекций или таблеток как CYTOXAN® (цитоксан). Циклофосфамид показан в качестве агента монотерапии или в комбинации с другими химиотерапевтическими агентами при лечении злокачественных лимфом, рассеянной миеломы и лейкозов. Алопеция, тошнота, рвота и лейкопения являются наиболее частымиограничивающими дозу побочными эффектами циклофосфамида.
Мелфалан, 4-[бис(2-хлорэтил)амино]-L-фенилаланин, коммерчески доступен в форме раствора для инъекций или таблетки как ALKERAN® (алкеран). Мелфалан показан для паллиативного лечения рассеянной миеломы и неоперабельной эпителиальной карциномы яичников. Супрессия костного мозга является наиболее распространенным дозоограничивающим побочным эффектом мелфалана.
Хлорамбуцил, 4-[бис(2-хлорэтил)амино]бензолбутановая кислота, коммерчески доступен в форме таблеток как LEUKERAN® (ЛЕЙКЕРАН). Хлорамбуцил показан для паллиативного лечения хронического лимфатического лейкоза и злокачественных лимфом, таких как лимфосаркома, гигантофолликулярная лимфома и болезнь Ходжкина. Супрессия костного мозга является наиболее распространенным дозоограничивающим побочным эффектом хлорамбуцила.
Бусульфан, 1,4-бутандиолдиметансульфонат, коммерчески доступен в виде таблеток MYLERAN® (милеркн). Бусульфан показан для паллиативного лечения хронического миелогенного лейкоза. Супрессия костного мозга является наиболее распространенным дозоограничивающим побочным эффектом бусульфана.
Кармустин, 1,3-[бис(2-хлорэтил)-1-нитрозомочевина, коммерчески доступен в форме однодозовых склянок лиофилизированного материала под маркой BiCNU®. Кармустин показан для паллиативного лечения в качестве агента монотерапии в комбинации с другими агентами для лечения опухолей мозга, множественной миеломы, болезни Ходжкина и нехожжкинских лимфом. Отложенная миелосупрессия является наиболее распространенным дозоограничивающим побочным эффектом кармустина.
Дакарбазин, 5-(3,3-диметил-1-триазено)-имидазол-4-карбоксамид, коммерчески доступен в форме однодозовых склянок материала под маркой DTIC-Dome®. Дакарбазин показан для лечения метастатической злокачественной меланомы и в комбинации с другими агентами для второй линии терапии болезни Ходжкина. Тошнота, рвота и анорексия являются наиболее распространенными дозоограничивающим побочными эффектами дакарбазина.
Антибиотические антинеопластические агенты являются нефазовыми специфическими агентами, которые связываются или интеркалируются с ДНК. Обычно, такое действие приводит к образованию стабильных комплексов с ДНК или разрыву нитей, что нарушает нормальное функционирование нуклеиновых кислот, приводя к гибели клеток. Примеры антибиотических антинеопластических агентов включают, без ограничения перечисленными: дактиномицин, антроциклины, такие как даунорубицин и доксорубицин; и блеомицины.
Дактиномицин, известный также как актиномицин D, коммерчески доступен в форме для инъекций как COSMEGEN® (космеген). Дактиномицин показан для лечения опухоли Вильмса и рабдомиосаркомы. Тошнота, рвота и анорексия являются наиболее распространенными дозоограничивающим побочными эффектами дактиномицина.
Даунорубицин, (8S-цис-)-8-ацетил-10-[(3-амино-2,3,6-тридезокси-α-L-ликсогексопиранозил)окси]-7,8,9,10-тетрагидро-6,8,11-тригидрокси-1-метокси-5,12 нафтацендион гидрохлорид, коммерчески доступен в форме липосом для инъекций под маркой DAUNOXOME® (дауноксом) или в форме для инъекций под маркой CERUBIDINE®.
Даунорубицин показан для стимуляции ремиссии для лечения острого нелимфоцитарного лейкоза и распространенной ВИЧ-ассоциированной саркомой Капоши. Миелосупрессия является наиболее распространенным дозоограничивающим побочным эффектом даунорубицина.
Доксорубицин, (8S,10S)-10-[(3-амино-2,3,6-тридезокси-α-L-ликсо-гексопиранозил)окси]-8-гликолоил, 7,8,9,10-тетрагидро-6,8,11-тригидрокси-1-метокси-5,12 нафтацендиона гидрохлорид, коммерчески доступен в форме для инъекций под маркой RUBEX® (рубекс) ил ADRIAMYCIN RDF® (Адриамицин РДФ). Доксорубицин прежде всего показан для лечения острого лимфобластного лейкоза и острого миелобластного лейкоза, но он также является полезным компонентом для лечения некоторых солидных опухолей и лимфом. Миелосупрессия является наиболее распространенным дозоограничивающим побочным эффектом доксорубицина.
Блеомицин, смесь цитотоксичных гликопептидных антибиотиков, выделенных из штамма Streptomyces verticillus, коммерчески доступен под маркой BLENOXANE® (бленоксан). Блеомецин показан в качестве средства паллиативного лечения, в качестве агента монотерапии, в комбинации с другими агентами, для лечения плоскоклеточной карциномы, лимфом и карцином яичек. Легочная токсичность и кожная токсичность являются наиболее распространенными дозоограничивающим побочными эффектами блеомицина.
Ингибиторы топоизомеразы II включают, без ограничения перечисленными: эпиподофиллотоксины.
Эпиподофиллотоксины представляют собой фазоспецифические антинеопластические агенты, получаемые из растения мандрагора. Эпиподофиллотоксины обычно воздействуют на клетки в фазах S и G2 клеточного цикла за счет образования тройного комплекса с топоизомеразой II и DNA, вызывая разрывы нитей ДНК. Разрывы нитей накапливаются, приводя к гибели клеток. Примеры эпиподофиллотоксинов включают, без ограничения перечисленными: этопозид и тенипозид.
Этопозид, 4'-деметил-эпиподофиллотоксин 9[4,6-0-(R)-этилиден-β-D-глюкопиранозид], доступен для приобретения в форме раствора для инъекций или капсул под маркой VePESID®, и общеизвестен как VP-16. Этопозид показан в качестве агента монотерапии в комбинации с другими химиотерапевтическими агентами для лечения рака яичек и немелкоклеточного рака легких. Миелосупрессия является наиболее частым побочным эффектом этопозида. Случаи лейкопении имеют тенденцию быть более тяжелыми, чем тромбоцитомения.
Тенипозид, 4'-деметил-эпиподофиллотоксин 9[4,6-0-(R)-тенилиден-β-D-глюкопиранозид], доступен для приобретения в форме раствора для инъекций под маркой VUMON® (Вумон) и широко известен как VM-26. Тенипозид показан в качестве агента монотерапии в комбинации с другими химиотерапевтическими агентами для лечения острого лейкоза у детей. Миелосупрессия является наиболее распространенным дозоограничивающим побочным эффектом тенипозида. Тенипозид может вызывать как лейкопению, так и тромбоцитопению.
Антинеопластические агенты класса антиметаболитов являются фазоспецифическими антинеопластическими агентами, которые могут действовать в S фазе (синтеза ДНК) клеточного цикла путем ингибирования синтеза ДНК или путем ингибирования синтеза пуриновых или пиримидиновых оснований, тем самым, ограничивая синтез ДНК. В результате S фаза не протекает, и следует гибель клеток. Примеры антинеопластических агентов класса антиметаболитов включают, без ограничения перечисленными: фторурацил, метотрексат, цитарабин, мекаптопурин, тиогуанин и гемицитабин.
5-фторурацил, 5-фтор-2,4-(1Н,3Н) пиримидиндион, коммерчески доступен как фторурацил. Введение 5-фторурацила приводит к ингибированию синтеза тимидилата и также встраивается как в РНК, так и в ДНК. Результатом обычно является гибель клеток. 5-фторурацил показан в качестве агента монотерапии в комбинации с другими химиотерапевтическими агентами для лечения карцином молочной железы, толстого кишечника, прямой кишки, желудка и поджелудочной железы. Миелосупрессия и мукозит являются ограничивающими дозу побочными эффектами 5-фторурацила. Другие фтор-содержащие аналоги пиримидинов включают 5-фтор дезоксиуридин (флоксиридин) и 5-фтордезоксиуридин монофосфат.
Цитарабин, 4-амино-1-β-D-арабинофуранозил-2 (1H)-пиримидинон, коммерчески доступен под маркой CYTOSAR-U® (Цитозар-У) и широко известен как Ara-С. Считается, что цитарабин проявляет клеточную фазоспецифичность в S-фазе за счет ингибирования удлинения цепи ДНК путем концевого встраивания цитарабина в растущую цепь ДНК. Цитарабин показан в качестве агента монотерапии в комбинации с другими химиотерапевтическими агентами для лечения острого лейкоза. Другие аналоги цитидина включают 5-азацитидин и 2',2'-дифтордезоксицитидин (гемцитабин). Цитарабин вызывает лейкопению, тромбоцитопению и мукозит.
Меркаптопурин, 1,7-дигидро-6Н-пурин-6-тион моногидрат, коммерчески доступен под маркой PURINETHOL®. Меркаптопурин демонстрирует специфичность в отношении фазы клеточного цикла в S-фазе путем ингибирования синтеза ДНК по еще не уточненному механизму. Меркаптопурин показан в качестве агента монотерапии в комбинации с другими химиотерапевтическими агентами для лечения острого лейкоза. Миелосупрессия и желудочно-кишечный мукозит являются ожидаемыми побочными эффектами меркаптопурина в высоких дозах. Полезным аналогом меркаптопурина является азатиоприн.
Тиогуанин, 2-амино-1,7-дигидро-6Н-пурин-6-тион, коммерчески доступен под маркой TABLOID® (Таблоид). Тиогуанин демонстрирует специфичность в отношении фазы клеточного цикла в S-фазе путем ингибирования синтеза ДНК по еще не уточненному механизму. Тиогуанин показан в качестве агента монотерапии в комбинации с другими химиотерапевтическими агентами для лечения острого лейкоза. Миелосупрессия, включая тромбоцитопению и анемию, является наиболее распространенным дозоограничивающим побочным эффектом введения тиогуанина. Однако имеют место побочные эффекты со стороны желудочно-кишечного тракта, и могут быть ограничивающими дозу. Другие аналоги пурина включают пентостатин, эритрогидроксинониладенин, флударабин фосфат и кладрибин.
Гемчитабин, 2'-дезокси-2',2'-дифторцитидин топогидрохлорид (β-изомер), коммерчески доступен под маркой GEMZAR® (гезмар). Гемцитабин проявляет специфичность в отношении фазы клеточного цикла в S-фазе и путем блокирования прохождения клетки через границу G1/S. Гемцитабин показан в комбинации с цисплатином для лечения местно-распространенного немелкоклеточного рака легких и в качестве средства монотерапии для лечения местно-распространенного рака поджелудочной железы. Миелосупрессия, включая лейкопению и анемию, является наиболее распространенным дозоограничивающим побочным эффектом введения гемцитабина.
Метотрексат, N-[4[[(2,4-диамино-6-птеридинил)метил]метиламино]бензоил]-L-глутаминовая кислота, коммерчески доступен как метотрексат натрий. Метотрексат проявляет специфичность в отношении фазы клеточного цикла в S-фазе путем ингибирования синтеза, репарации и/или репликации ДНК за счет ингибирования дигидрофолатредуктазы, которая необходима для синтеза пуриновых нуклеотидов и типидилата. Метотрексат показан в качестве агента монотерапии в комбинации с другими химиотерапевтическими агентами для лечения хориокарциномы, менингеального лейкоза, неходжкинской лимфомы и карцином молочной железы, головы, шеи, яичника и мочевого пузыря. Миелосупрессия (лейкопения, тромпоцитопения и анемия)и мукозит являются ожидаемым побочным эффектом введения метотрексата.
Камптотецины, включая камптотецин и производные камптотецина, доступны или находятся в стадии разработки в качестве ингибиторов топоизомеразы I. Считается, что цитотоксическая активность камптотецина связана с его ингибирующей активностью в отношении топоизомеразы I. Примеры камптотецинов включают, но не ограничиваются перечисленными, иринотекан, топотекан и различные оптические формы 7-(4-метилпиперазинометилен)-10,11-этилендиокси-20-камптотецина, описанные ниже.
Иринотекан HCl, (4S)-4,11-диэтил-4-гидрокси-9-[(4-пиперидинопиперидино)карбонилокси]-1Н-пирано[3',4',6,7]индолизино[1,2-b]хинолинил-3,14(4Н,12Н)-дион гидрохлорид, коммерчески доступен в виде раствора для инъекций CAMPTOSAR® (камптозар).
Иринотекан представляет собой производное камптотецина, которое, как и его активный метаболит SN-38, связывается с комплексом топоизомераза I - ДНК. Считается, что цитотоксичность возникает в результате не поддающихся репарации двухцепочечных разрывов, вызванных взаимодействием тройного комплекса топоизомераза I: ДНК: иринтекан или SN-38 с ферментами репликации. Иринотекан показан для лечения метастатического рака толстой или прямой кишки. Дозоограничивающие побочные эффекты иринотекана HCl включают миелосупрессию, включая нейтропению, и эффекты со стороны желудочно-кишечного тракта, включая диарею.
Топотекан HCl, (S)-10-[(диметиламино)метил]-4-этил-4,9-дигидрокси-1Н-пирано[3',4',6,7]индолизино[1,2-b]хинолинил-3,14-(4Н,12Н)-дион моногидрохлорид, коммерчески доступен в виде раствора для инъекций HYCAMTrN(R). Топотекан представляет собой производное камптотецина, которое связывается с комплексом топоизомераза I - ДНК и предотвращает религирование разрывов одиночных цепей, вызванных топоизомеразой I в ответ на деформацию скручивания молекулы ДНК. Топотекан показан для лечения второй линии метастатического рака яичника и мелкоклеточного рака легких. Ограничивающим дозу побочным эффектом топотекана HCl является миелосупрессия, в первую очередь нейтропения.
Топотекан HCl, (S)-10-[(диметиламино)метил]-4-этил-4,9-дигидрокси-1Н-пирано[3',4',6,7]индолизино[1,2-b]хинолинил-3,14-(4Н,12Н)-дион моногидрохлорид, коммерчески доступен в виде раствора для инъекций HYCAMTEM® (хикамтин). Топотекан представляет собой производное камптотецина, которое связывается с комплексом топоизомераза I - ДНК и предотвращает религирование разрывов одиночных цепей, вызванных топоизомеразой I в ответ на торсионное напряжение в молекуле ДНК. Топотекан показан для второй линии терапии метастатического рака яичника и мелкоклеточного рака легких. Дозоограничивающим побочным эффектом топотекана HCl является миелосупрессия, в первую очередь нейтропения.
Гормоны и аналоги гормонов представляют собой соединения, которые можно применять для лечения видов рака, при которых существует взаимосвязь между гормоном (ами) и ростом и/или отсутствием роста рака. Примеры гормонов и аналогов гормонов, которые можно применять при лечении рака, включают, без ограничения перечисленными: адренокортикостероиды, такие как преднизон и преднизолон, которые можно применять для лечения злокачественной лимфомы и острого лейкоза у детей; аминоглютетимид и другие ингибиторы ароматазы, такие как анастрозол, летразол, воразол и экземестан, которые можно применять для лечения адренокортикальной карциномы и гормонозависимой карциномы молочной железы, содержащей рецепторы эстрогена; прогестрины, такие как мегестрол ацетат, которые можно применять для лечения гормонально-зависимых рака молочной железы и рака эндометрия; эстрогены, андрогены и антиандрогены, такие как флутамид, нилутамид, бикалутамид, ципротерона ацетат и 5α-редуктазы, такие как финастерид и дутастерид, которые можно применять для лечения карциномы предстательной железы и доброкачественной гипертрофии предстательной железы; антиэстрогены, такие как тамоксифен, торемифен, ралоксифен, дролоксифен, йодоксифен, а также селективные модуляторы рецепторов эстрогена (SERMS), такие как описанные в патентах США №№5681835, 5877219 и 6207716, которые можно применять для лечения гормонозависимой карциномы молочной железы и других восприимчивые виды рака; и гонадотропин-рилизинг-гормон (GnRH) и его аналоги, которые стимулируют высвобождение лютеинизирующего гормона (ЛГ) и/или фолликулостимулирующего гормона (ФСГ) для лечения карциномы предстательной железы, например, агонисты и антагонисты LHRH (рилизинг-гормона лютеинизирующего гормона), такие как гозерелин ацетат и лупролид.
Ингибиторы каскадов передачи сигнала - это те ингибиторы, которые блокируют или ингибируют химический процесс, вызывающий внутриклеточные изменения. В настоящем документе это изменение представляет собой пролиферацию или дифференцировку клеток. Ингибиторы передачи сигнала, которые можно применять в настоящем изобретении, включают ингибиторы рецепторных тирозинкиназ, нерецепторных тирозин киназы, блокаторы домена SH2 / SH3, серин/треонинкиназы, фосфотидилинозитол-3 киназы, передачи сигналов мио-инозитола и онкогенов Ras.
Некоторые протеинтирозинкиназы катализируют фосфорилирование определенных тирозильных остатков в различных белках, участвующих в регуляции роста клеток. Такие протеинтирозинкиназы можно в широком смысле классифицировать как рецепторные или нерецепторные киназы. Рецепторные тирозинкиназы представляют собой трансмембранные белки, имеющие внеклеточный лиганд-связывающий домен, трансмембранный домен и тирозин-киназный домен. Рецепторные тирозинкиназы участвуют в регуляции роста клеток и обычно называются рецепторами факторов роста. Было показано, что неадекватная или неконтролируемая активация многих из этих киназ, т.е. аберрантная активность киназы - рецептора фактора роста, например, в результате сверхэкспрессии или мутации, приводит к неконтролируемому росту клеток. Соответственно, аберрантную активность таких киназ связали со злокачественным ростом ткани. Следовательно, ингибиторы таких киназ могут обеспечить способы лечения рака. Рецепторы фактора роста включают, например, рецептор эпидермального фактора роста (EGFr), рецептор фактора роста тромбоцитов (PDGFr), erbB2, erbB4, рецептор фактора роста эндотелия сосудов (VEGFr), тирозин-киназу с иммуноглобулиноподобными доменами и доменами гомологии эпидермального фактора роста (TIE-2), рецептор фактора роста инсулина -I (IGFI), макрофагальный колониестимулирующий фактор (cfms), BTK, ckit, cmet, рецепторы фактора роста фибробластов (FGF), рецепторы Trk (TrkA, TrkB и TrkC), эфрин (eph) рецепторы и протоонкоген RET. Некоторые ингибиторы рецепторов роста находятся в стадии разработки, включая антагонисты лигандов, антитела, ингибиторы тирозинкиназы и антисмысловые олигонуклеотиды. Рецепторы факторов роста и агенты, которые ингибируют функцию рецепторов факторов роста, описаны, например, в Kath, John С., Exp. Opin. Ther. Patents (2000) 10 (6): 803-818; Shawver et al DDT Vol 2, No. 2 February 1997; и Lofts, F.J. et al, "Growth factor receptors as targets", New Molecular Targets for Cancer Chemotherapy, ed. Workman, Paul and Kerr, David, CRC press 1994, London.
В соответствующих случаях фармацевтически активные соединения согласно настоящему изобретению применяют в комбинации с ингибитором VEGFR, в соответствующих случаях, 5-[[4-[(2,3-диметил-2Н-индазол-6-ил)метиламино]-2-пиримидинил]амино]-2-метилбензолсульфонамидом или его фармацевтически приемлемой солю, в соответствующих случаях, его моногидрохлоридной солью, которая раскрыта и заявлена в международной заявке № РСТ/US 01/49367, имеющей дату международной подачи 19 декабря 2001 г., номер международной публикации WO 02/059110 и дату международной публикации 1 августа 2002 г., полное описание которой включено в настоящий документ посредством ссылки, и которая представляет собой соединение из Примера 69. 5-[[4-[(2,3-диметил-2Н-индазол-6-ил)метиламино]-2-пиримидинил]амино]-2-метилбензолсульфонамид может быть получен, как описано в международной заявке № PCT/US 01/49367.
В соответствующих случаях, 5-[[4-[(2,3-диметил-2Н-индазол-6-ил)метиламино]-2-пиримидинил]амино]-2-метилбензолсульфонамид представлен в форме моногидрохлорида. Эта солевая форма может быть получена специалистом в данной области в соответствии с описанием в международной заявке № PCT/US 01/49367, имеющей дату международной подачи 19 декабря 2001 г.
5-[[4-[(2,3-диметил-2Н-индазол-6-ил)метиламино]-2-пиримидинил]амино]-2-метилбензолсульфонамид продается в виде моногидрохлорида и известен под непатентованным названием пазопанид и торговым названием Votrient® (вотриент).
Пазопаниб назначают для лечения рака и болезней глаз/ангиогенеза. В соответствующих случаях настоящее изобретение относится к лечению рака болезни глаз/ангиогенеза, возрастной дегенерации желтого пятна, причем способы включат соединения Формулы (I) отдельно в комбинации с пазопанибом.
Тирозинкиназы, которые не являются киназами - рецепторами фактора роста, называются нерецепторными тирозинкиназами. Нерецепторные тирозинкиназы для применения в настоящем изобретении, которые являются мишенями или потенциальными мишенями противораковых препаратов, включают cSrc, Lck, Fyn, Yes, Jak, cAbl, FAK (киназу фокальной адгезии), тирозинкиназу Брутона и Bcr-Abl. Такие нерецепторные киназы и агенты, которые ингибируют функцию нерецепторных киназ тирозин, описаны в Sinh, S., Corey, S.J., (1999) Journal of Hematotherapy and Stem Cell Research 8 (5): 465-80; и Bolen, J.B., Brugge, J.S., (1997) Annual review of Immunology. 15: 371-404.
Блокаторы домена SH2 / SH3 представляют собой агенты, которые нарушают связывание домена SH2 или SH3 в различных ферментах или адаптерных белках, включая субъединицу PI3-K р85, киназы семейства Src, адаптерные молекулы (She, Crk, Nek, Grb2) и Ras-GAP. Домены SH2 / SH3 как мишени для противораковых препаратов обсуждаются в Smithgall, Т.Е. (1995), Journal of Pharmacological and Toxicological Methods. 34 (3) 125-32.
Ингибиторы серин/треонинкиназ, включая блокаторы каскада MAP-киназ, которые включают блокаторы киназ Raf (rafk), регулируемые митогенами или внеклеточными сигналами киназы (MEK) и регулируемые внеклеточными сигналами киназы (ERK); и блокаторы членов семейства протеинкиназы С, включая блокаторы PKC (альфа, бета, гамма, эпсилон, мю, лямбда, йота, дзета). Семейство киназ IkB (IKKa, IKKb), киназы семейства PKB, члены семейства киназ akt, киназы-кецеторы PDK1 и TGF-бета. Такие серин/треонинкиназы и их ингибиторы описаны в Yamamoto, Т., Taya, S., Kaibuchi, K., (1999), Journal of Biochemistry. 126 (5) 799-803; Brodt, P, Samani, A., Navab, R. (2000), Biochemical Pharmacology, 60. 1101-1107; Massague, J., Weis-Garcia, F. (1996) Cancer Surveys. 27: 41-64; Philip, P.A., Harris, A.L. (1995), Cancer Treatment and Research. 78: 3-27, Lackey, K. et al Bioorganic and Medicinal Chemistry Letters, (10), 2000, 223-226; U.S. Patent No. 6,268,391; Pearce, L.R et al. Nature Reviews Molecular Cell Biology (2010) 11, 9-22. и Martinez-Iacaci, L., et al, Int. J. Cancer (2000), 88 (1), 44-52.
В соответствующих случаях фармацевтически активные соединения согласно настоящему изобретению применяют в комбинации с ингибитором MEK. В соответствующих случаях, N-{3-[3-циклопропил-5-(2-фтор-4-йодо-фениламино)-6,8-диметил-2,4,7-триоксо-3,4,6,7-тетрагадро-2Н-пиридо[4,3-d]пиримидин-1-ил]фенил}ацетамид, или его фармацевтически приемлемая соль сольват, в соответствующих случаях, его сольват с диметилсульфоксидом, который раскрыт и заявлен в международной патентной заявке № PCT/JP 2005/011082, имеющей международную дату подачи 10 июня 2005 г.; международную публикацию № WO 2005/121142 и дату международной публикации 22 декабря 2005, которая полностью включена в настоящий документ посредством ссылки. N-{3-[3-циклопропил-5-(2-фтор-4-йодо-фениламино)-6,8-диметил-2,4,7-триоксо-3,4,6,7-тетрагидро-2Н-пиридо[4,3-d]пиримидин-1-ил]фенил}ацетамид может быть получен как описано в патентной публикации США US 2006/0014768, опубликованной 19 января 2006 г., раскрытие которой полностью включено в настоящий документ посредством ссылки.
В соответствующих случаях фармацевтически активные соединения согласно настоящему изобретению применяют в комбинации с ингибитором B-Raf. В соответствующих случаях, это N-{3-[5-(2-Амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил}-2,6-дифторбензолсульфонамид, или его фармацевтически приемлемая соль, которые раскрыты и заявлены, в международной патентной заявке № PCT/US 2009/042682, имеющей международную дату подачи 4 мая 2009 г., содержание которой полностью включено в настоящий документ посредством ссылки. N-3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил}-2,6-дифторбензолсульфонамид может быть приготовлен как описано в международной патентной заявке № PCT/US 2009/042682.
В соответствующих случаях фармацевтически активные соединения согласно настоящему изобретению применяют в комбинации с ингибитором Akt. В соответствующих случаях это N-{(1S)-2-амино-1-[(3,4-дифторфенил)метил]этил}-5-хлор-4-(4-хлор-1-метил-1Н-пиразол-5-ил)-2-фуранкарбоксамид или его фармацевтически приемлемая соль, который описан и заявлен в международной патентной заявке № PCT/US 2008/053269, имеющей международную дату подачи 7 февраля 2008 г.; международный номер публикации WO 2008/098104 и международную дату публикации 14 августа 2008 г., раскрытие которой полностью включено в настоящий документ посредством ссылки. N-{(1S)-2-амино-1-[(3,4-дифторфенил)метил]этил}-5-хлор-4-(4-хлор-1-метил-1Н-пиразол-5-ил)-2-фуранкарбоксамид представляет собой соединение из примера 224 и может быть приготовлен как описано в международной патентной заявке № PCT/US 2008/053269.
В соответствующих случаях фармацевтически активные соединения согласно настоящему изобретению применяют в комбинации с ингибитором Akt. В соответствующих случаях этот N-{(1S)-2-амино-1-[(3-фторфенил)метил]этил}-5-хлор-4-(4-хлор-1-метил-1Н-пиразол-5-ил)-2-тиофенкарбоксамид или его фармацевтически приемлемая соль, который описан и заявлен в международной патентной заявке № PCT/US 2008/053269, имеющей международную дату подачи 7 февраля 2008 г.; номер международной публикации WO 2008/098104 и дату международной публикации 14 августа 2008 г., раскрытие которой полностью включено в настоящий документ посредством ссылки. N-{(1S)-2-амино-1-[(3фторфенил)метал]этил}-5-хлор-4-(4-хлор-1-метил-1Н-пиразол-5-ил)-2-тиофенкарбоксамид представляет собой соединение из примера 96 и может быть получено как описано в международной патентной заявке № PCT/US 2008/053269. В соответствующих случаях N-{(1S)-2-амино-1-[(3-фторфенил)метил]этил}-5-хлор-4-(4-хлор-1-метил-1H-пиразол-5-ил)-2-тиофенкарбоксамид представлен в форме соли-гидрохлорида. Специалист сможет получить солевую форму на основании описания в международной патентной заявке № PCT/US 2010/022323, имеющей международную дату подачи 28 января 2010 г.
Ингибиторы членов семейства фосфотидилинозитол-3 киназы, включая блокаторы PI3-киназы, ATM, DNA-PK, и Ku, также можно применять в настоящем изобретении. Такие киназы обсуждаются в Abraham, R.T. (1996), Current Opinion in Immunology. 8 (3) 412-8; Canman, C.E., Lim, D.S. (1998), Oncogene 17 (25) 3301-3308; Jackson, S.P. (1997), International Journal of Biochemistry and Cell Biology. 29 (7): 935-8; и Zhong, H. et al, Cancer res, (2000) 60 (6), 1541-1545.
Также для настоящего изобретения представляют интерес ингибиторы передачи сигналов мио-инозитола, такие как блокаторы фосфолипазы С и аналоги миоинозита. Такие сигнальные ингибиторы описаны в Powis, G. и Kozikowski А., (1994) New Molecular Targets for Cancer Chemotherapy ed., Paul Workman, and David Kerr, CRC press 1994, London.
Другой группой ингибиторов сигнальных каскадов являются ингибиторы онкогена Ras. Такие ингибиторы включают ингибиторы фарнезилтрансферазы, геранилгеранилтрансферазы и протеаз СААХ, а также антисмысловые олигонуклеотиды, рибозимы и иммунотерапевтические средства. Было показано, что такие ингибиторы блокируют активацию ras в клетках, содержащих мутантный ras дикого типа, тем самым действуя как антипролиферативные агенты. Подавление онкогенов Ras обсуждается в Scharovsky, O.G., Rozados, V.R., Gervasoni, S.L Matar, P. (2000), Journal of Biomedical Science. 7 (4) 292-8; Ashby, M.N. (1998), Current Opinion in Lipidology. 9 (2) 99-102; и BioChim. Biophys. Acta, (19899) 1423 (3): 19-30.
Как упоминалось выше, антитела, оказывающие антагонистическое действие в отношении связывания рецепторной киназы с лигандом, также могут служить ингибиторами передачи сигнала. Эта группа ингибиторов сигнальных каскадов включает использование гуманизированных антител к внеклеточному лиганд-связывающему домену рецепторных тирозинкиназ. Например, Imclone С225 EGFR-специфическое антитело (см. Green, М.С. et al., Monoclonal Antibody Therapy for Solid Tumors, Cancer Treat. Rev., (2000), 26 (4), 269-286); Herceptin® (герцептин) - антитело к erbB2 (см. Tyrosine kinase signalling in breast cancer: ErbB family receptor tyrosine kinases, Breast Cancer Res, 2000, 2 (3), 176-183); и 2CB VEGFR2 специфическое антитело (см. Brekken, R.A. et al., Selective Inhibition of VEGFR2 Activity by a monoclonal Anti-VEGF antibody blocks tumor growth in mice, Cancer Res. (2000) 60, 5117-5124).
He относящиеся к рецепторным киназам ингибиторы ангиогенеза можно применять в настоящем изобретении. Ингибиторы VEGFR и TIE2, связанных с ангиогенезом, обсуждались выше в отношении ингибиторов передачи сигнала (оба рецептора являются рецепторными тирозин-киназами). Ангиогенез обычно связан с передачей сигналов erbB2 / EGFR, поскольку было показано, что ингибиторы erbB2 и EGFR ингибируют ангиогенез, в первую очередь экспрессию VEGF. Соответственно, не относящиеся к рецепторным киназам ингибиторы можно применять в комбинации с соединениями согласно настоящему изобретению. Например, антитела против VEGF, которые не распознают VEGFR (рецепторную тирозинкиназы), но связываются с лигандом; низкомолекулярные ингибиторы интегрина (альфа, бетаз), которые ингибируют ангиогенез; эндостатин и ангиостатин (не RTK) также могут оказаться полезными в комбинации с описанными соединениями. (См. Bruns CJ et al. (2000), Cancer Res., 60: 2926-2935; Schreiber AB, Winkler ME, Derynck R. (1986), Science, 232: 1250-1253; Yen L et al. (2000), Oncogene 19: 3460-3469).
Агенты, применяемые в иммунотерапевтических схемах, также могут применяться в комбинации с соединениями Формулы (I). Существует ряд иммунологических стратегий для создания иммунного ответа. Эти стратегии обычно относятся к сфере вакцинации против опухолей. Эффективность иммунологических подходов может быть значительно увеличена за счет комбинированного ингибирования сигнальных путей с использованием низкомолекулярного ингибитора. Обсуждение иммунологического подхода / противоопухолевой вакцины против erbB2 / EGFR можно найти в Reilly RT et al. (2000), Cancer Res. 60: 3569-3576; и Chen Y, Hu D, Eling DJ, Robbins J и Kipps TJ. (1998), Cancer Res. 58: 1965-1971.
Агенты, используемые в проапоптотических режимах (например, антисмысловые олигонуклеотиды bcl-2), также могут применяться в комбинации согласно настоящему изобретению. Члены семейства белков Bcl-2 блокируют апоптоз. Таким образом, повышающая регуляция bcl-2 связана с химиорезистентностью. Исследования показали, что эпидермальный фактор роста (EGF) стимулирует антиапоптотические члены семейства bcl-2 (т.е. mcl-1). Соответственно, стратегии, разработанные для подавления экспрессии bcl-2 в опухолях, продемонстрировали клиническую эффективность и сейчас находятся в фазе II / III испытаний, а конкретно, антисмысловой олигонуклеотид G3139 bcl-2 компании Genta. Такие проапоптотические стратегии с использованием стратегии на основе антисмысловых олигонуклеотидов для bcl-2 обсуждаются в Water JS et al. (2000), J. Clin. Oncol. 18: 1812-1823; и Китада С. и др. (1994), Antisense Res. Dev. 4: 71-79.
Ингибиторы передачи сигналов клеточного цикла ингибируют молекулы, участвующие в контроле клеточного цикла. Семейство протеинкиназ, называемых циклинзависимыми киназами (CDK), и их взаимодействие с семейством белков, называемых циклинами, контролирует прохождение цикла эукариотических клеток. Скоординированная активация и инактивация различных комплексов циклин CDK необходима для нормального прохождения клеточного цикла. Некоторые ингибиторы передачи сигналов клеточного цикла находятся в стадии разработки. Например, примеры циклинзависимых киназ, включая CDK2, CDK4 и CDK6, и их ингибиторы описаны, например, в Rosania et al, Exp.Opin. Ther. Patents (2000) 10 (2): 215-230. Кроме того, p21WAF1 / CIP1 был описан как мощный и универсальный ингибитор циклин-зависимых киназ (Cdks) (Ball et al., Progress in Cell Cycle Res., 3: 125 (1997)). Соединения, которые, как известно, индуцируют экспрессию p21WAF1 / CIP1, участвуют в подавлении пролиферации клеток и обладают опухолеподавляющей активностью (Richon et al., Proc. Nat Acad. Sci. USA 97 (18): 10014-10019 (2000).)), и включены как ингибиторы клеточной сигнализации. Ингибиторы гистондеацетилазы (HDAC) участвуют в транскрипционной активации p21WAF1 / CIP1 (Vigushin et al., Anticancer Drugs, 13 (1): 1-13 (Jan 2002)) и являются подходящими ингибиторами клеточной сигнализации для применения в комбинации согласно настоящему документу.
Примеры таких ингибиторов HDAC включают:
1. Вориностат, включая его фармацевтически приемлемые соли. Marks et al., Nature Biotechnology 25, 84 - 90 (2007); Stenger, Community Oncology 4, 384-386 (2007).
Вориностат имеет следующие химические структуру и наименование:
2. Ромидепсин, включая его фармацевтически приемлемые соли. Vinodhkumar et al., Biomedicine & Pharmacotherapy 62 (2008) 85-93. Ромидепсин имеет следующие химические структуру и наименование:
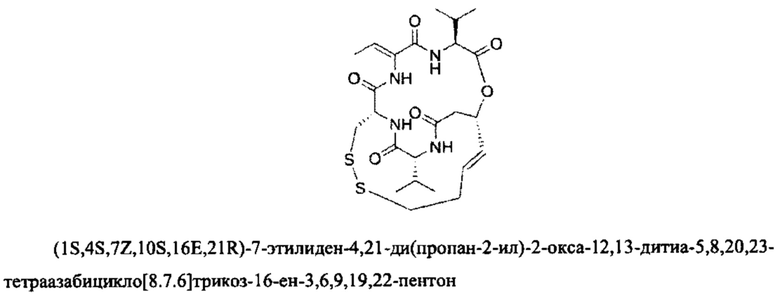
3. Панобиностат, включая его фармацевтически приемлемые соли. Drugs of the Future 32(4): 315-322 (2007).
Панобиностат имеет следующие химические структуру и наименование:
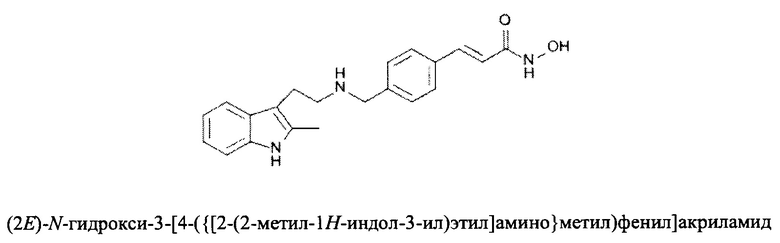
4. Вальпроевая кислота, включая ее фармацевтически приемлемые соли. Gottlicher, et al., EMBO J. 20(24): 6969-6978 (2001).
Вальпроевая кислота имеет следующие химические структуру и наименование:
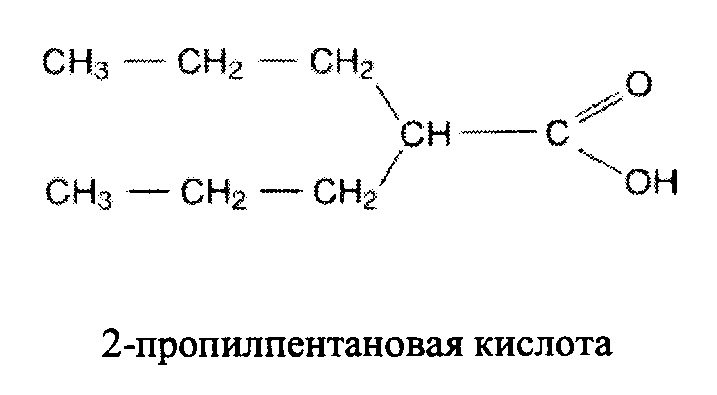
5. Моцетиностат (MGCD0103), включая его фармацевтически приемлемые соли. Balasubramanian et al., Cancer Letters 280: 211-221 (2009).
Моцетиностат имеет следующие химические структуру и наименование:

Дополнительные примеры таких ингибиторов HDAC включают приведенные в Bertrand European Journal of Medicinal Chemistry 45, (2010) 2095-2116, см., в частности, соединения, приведенные в Таблице 3 этого источника, показанные ниже.
Ингибиторы протеасом представляют собой лекарственные средства, которые блокируют действие протеасом, клеточных комплексов и разрушают белки, такие как белок р53. Несколько ингибиторов протеасом присутствуют на рынке или находятся в процессе изучения для лечения рака. Подходящие ингибиторы протеасом для применения в комбинации согласно настоящему документу включают:
1. Бортезомиб (Velcade®(велкаде)), включая его фармацевтически приемлемые соли. Adams J, Kaufftnan Μ (2004), Cancer Invest 22 (2): 304-11.
Ботрезомиб имеет следующие химические структуру и наименование.
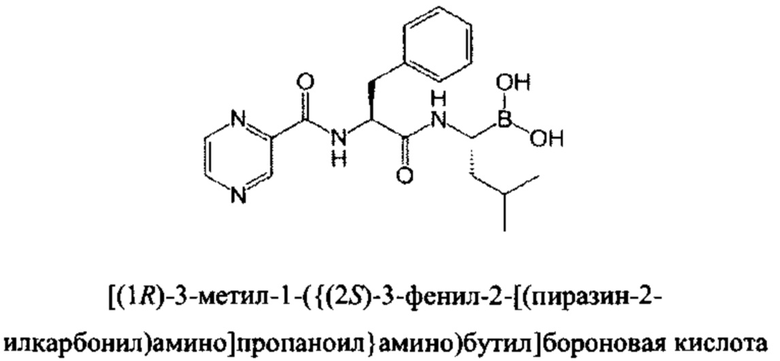
2. Дисульфирам. включая его фармацевтически приемлемые соли. Bouma et al. (1998). J. Antimicrob. Chemother. 42 (6): 817-20.
Дисульфирам имеет следующие химические структуру и наименование.
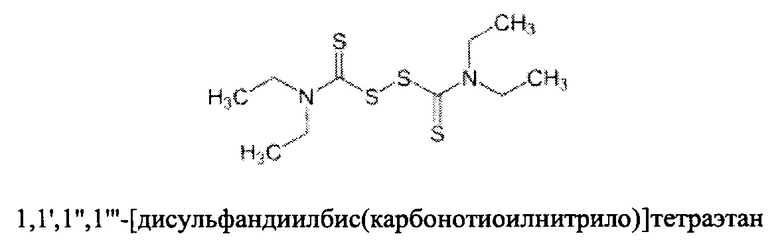
3. Эпигаллокатехин галлат (EGCG), включая его фармацевтически приемлемые соли. Williamson et al., (December 2006), The Journal of Allergy and Clinical Immunology 118 (6): 1369-74.
Эпигаллокатехин галлат имеет следующие химические структуру и наименование.
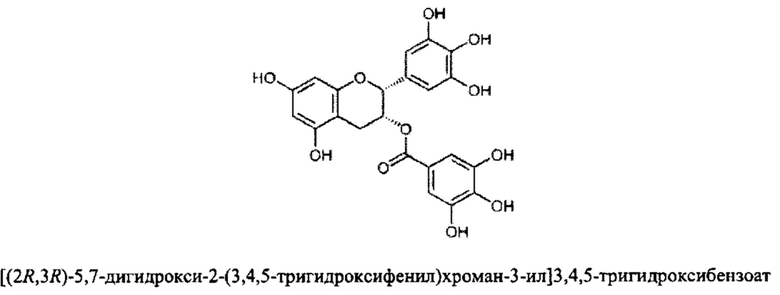
4. Салиноспорамид А, включая его фармацевтически приемлемые соли. Feling et at., (2003), Angew. Chem. Int. Ed. Engl. 42 (3): 355-7.
Салиноспорамид А имеет следующие химические структуру и наименование.
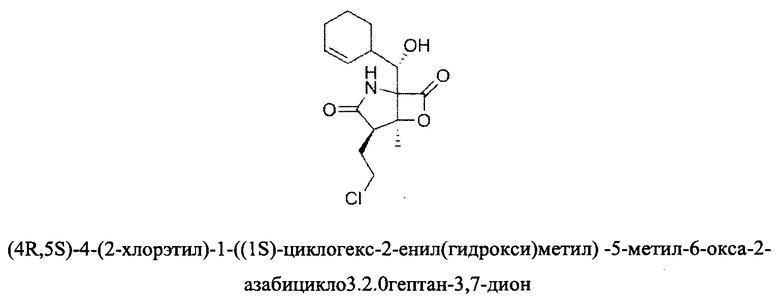
5. Карфилзомиб, включая его фармацевтически приемлемые соли. Kuhn DJ, et al, Blood, 2007, 110:3281-3290.
Карфилзомиб имеет следующие химические структуру и наименование.

Белки теплового шока массой 70 килодальтон (Hsp70) и белки теплового шока массой 90 килодальтон (Hsp90) являются семействами повсеместно экспрессируемых белков теплового шока. Экспрессия белков Hsp70 и Hsp90 повышены при некоторых типах рака. Некоторые ингибиторы Hsp70 и Hsp90 исследуются для лечения рака. Подходящие ингибиторы Hsp70 и Hsp90 для применения в комбинации согласно настоящему документу включают:
1. 17-AAG(гелданамицин), включая его фармацевтически приемлемые соли. Jia W et al. Blood. 2003 Sep 1:102(5):1824-32.
17-AAG(гелданамицин) имеет следующие химические структуру и наименование.
2. Радицикол, включая его фармацевтически приемлемые соли. (Lee et al., Mol Cell Endocrinol. 2002, 188.47-54)
Радицикол имеет следующие химические структуру и наименование.
Ингибиторы ракового метаболизма - Многие опухолевые клетки демонстрируют метаболизм, значительно отличающийся от нормальных тканей. Например, скорость гликолиза, метаболического процесса, преобразующего глюкозу в пируват, повышена, и генерируемый пируват восстанавливается до лактата вместо того чтобы окисляться в митохондриях в цикле трикарбоновых кислот (ТКК). Этот эффект частно наблюдается даже в аэробных условиях и известен как эффект Варбурга.
Лактатдегидрогеназа А (ЛДГ-А), одна из изоформ лактатдегидрогеназы, одна из изоформ лактатдегидрогеназы, экспрессируемая в мышечных клетках, играет очень важную роль в метаболизме опухолевых клеток, осуществляя восстановление пирувата до лактата, который затем может быть выведен из клетки. Было показано, что этот фермент повышающе регулируется при многих типах опухолей. Изменение метаболизма глюкозы, описанное в эффекте Варбурга, является критически важным для роста и пролиферации раковых клеток; и было показано, что блокирование ЛДГ-А с использованием РНК и приводит к уменьшению пролиферации клеток и роста опухоли в ксенографтных моделях.
D.A. Tennant et. al., Nature Reviews, 2010, 267. P. Leder, et. al., Cancer Cell, 2006, 9, 425.
Высокие уровни синтазы жирных кислот (FAS, СЖК) были обнаружены в предшественниках раковых поражений. Фармакологическое ингибирование FAS влияет на экспрессию ключевых онкогенов, участвующих как в развитии, так и в поддержании рака. Alli et al. Oncogene (2005) 24, 39-46. doi:10.1038
Ингибиторы ракового метаболизма, включая ингибиторы ЛДГ-А и ингибиторы биосинтеза жирных кислот (или ингибиторы FAS), подходят для применения в комбинации с соединениями согласно этому изобретению.
В одном варианте реализации способ лечения рака согласно заявленному изобретению включает совместное введение соединения Формулы I, Ia, Ib, II или III и/или его фармацевтически приемлемой соли и по меньшей мере одного антинеопластического агента, такого как агент, выбранный из группы, состоящей из агентов, воздействующих на микротрубочки, координационных комплексов платины, алкилирующих агентов, антибиотиков, ингибиторов топоизомеразы II, антиметаболитов, ингибиторов топоизомеразы I, гормонов и аналогов гормонов, ингибиторов сигнальных каскадов, не относящихся к рецепторным тирозинкиназам ингибиторов ангиогенеза, иммунотерапевтических агентов, проапоптотических агентов, ингибиторов сигнализации клеточного цикла; ингибиторы протеасом; и ингибиторов ракового метаболизма.
В одном варианте реализации соединение Формулы I, Ia и Ib применяют в качестве хемосенсибилизатора для повышения эффективности уничтожения опухолевых клеток.
В одном варианте реализации соединение Формулы I, Ia, Ib, II или III применяют в качестве хемосенсибилизатора для повышения эффективности уничтожения опухолевых клеток.
В одном варианте реализации соединение Формулы I, Ia, Ib, II или III применяют в комбинации с соединением, которое ингибирует активность подобной протеинкиназе R (PKR)-киназы ER, PERK (ингибитор PERK).
В соответствующих случаях соединения Формулы I, Ia, Ib, II, III и их фармацевтически приемлемые соли можно вводить совместно с по меньшей мере одним другим активным агентом, о котором известно, что он является ингибитором киназы PERK (EIF2K3), для лечения или снижения тяжести нейродегенеративного заболевания/повреждения, такого как болезнь Альцгеймера, повреждение спинного мозга, черепно-мозговая травма, ишемический инсульт, инсульт, диабет, болезнь Паркинсона, болезнь Хантингтна, Крейтцфельдта - Якоба с связанные прионные болезни, прогрессирующий супрануклеарный паралич, боковой амиотрофический склероз, инфаркт миокарда, сердечно-сосудистое заболевание, воспаление, фиброз, хронические и острые болезни печени, хронические и острые легких, хронические и легкие болезни почек, хроническая травматическая энцефалопатия (СТЕ, ХТЭ), нейродегенерация, деменция, черепно-мозговая травма, когнитивное нарушение, атеросклероз, болезни глаз, аритмии, при трансплантации органов и при транспортировки органов для трансплантации.
"Химиотерапевтический" или "химиотерапевтический агент" используется в соответствии с его прямым обычным значение и относится к химической композиции или соединению, обладающим антинеопластическими свойствами или способностью подавлять рост или пролиферацию клеток.
Дополнительно, соединения, описанные в настоящем документе, можно вводить совместно с обычными иммунотерапевтическими агентами, включая следующие, без ограничения ими: иммуностимуляторы (например, (бацилла Кальметта-Герена (БЦЖ), левамизол, интерлейкин-2, альфаинтерферон и т.д.), моноклональные антитела (например, моноклональные антитела против CD20, против HER2, против CD52, против HLA-DR и против VEGF), иммунотоксины (например,конъюгат коноклональное антитело против CD33 калихеамицин, конъюгат моноклональное антитело против CD22 - эндотоксин синегнойной палочки и т.д.), и радиоиммунотерапевтические средства (например, антиело против CD20, конъюгированное с 111In, 90Υ или 131Ι и т.д.).
В другом варианте реализации соединения, описанные в настоящем документе, можно вводить совместно с традиционными радиотерапевтическими агентами, включая, без ограничения перечисленными, радионуклиды, такие как 47Sc, 6МС 67С, 89Sr, 86Y, 87Υ и 212Bi, необязательно конъюгированные с антителами, направленными против опухолевых антигенов.
Дополнительными примерами другого активного ингредиенты или ингредиентов (антинеопластических агентов) для применения в комбинации или совместного введения с настоящими соединениями являются агенты против PD-L1.
Антител против PD-L1 и методы из получения известны в данной области. Такие антитела к PD-L1 могут быть поликлональными или моноклональными, и/или рекомбинантными, и/или гуманизированными. Примеры антител к PD-L1 раскрыты в:
патенте США №8,217,149; 12/633,339;
патенте США №8,383,796; 13/091,936;
патенте США №8,552,154; 13/120.406;
патенте США №20110280877; 13/068337;
патенте США №20130309250; 13/892671;
WO2013019906;
WO2013079174;
заявке на патент США 13/511,538 (поданной 7 августа 2012 г.), которая представляет собой заявку национальной фазы в США, основанную на международной заявке №PCT/US10/58007 (поданной в 2010 г. ;
и
заявке на патент США 13/478,511 (поданной 23 мая 2012 г.).
Дополнительные примеры антител к (называемые также CD274 или В7-Н1) и способы применения раскрыты в патенте США №7,943,743; US 20130034559, WO 2014055897, в патенте США 8,168,179 и в патенте США 7,595,048. Антитела к PD-L1 разрабатываются как иммуномодулирующие агенты для лечения рака.
В одном варианте реализации антитело к PD-L1 представляет собой антитело, раскрытое в патенте США №8,217,149. В другом варианте реализации антитело против PD-L1 содержит участки CDR антитела, раскрытого в патенте США №8,217,149.
В другом варианте реализации антитело к PD-L1 представляет собой антитело, раскрытое в заявке на патент США №13/511,538. В другом варианте реализации антитело против PD-L1 содержит участки CDR антитела, раскрытого в заявке на патент США №13/511,538.
В другом варианте реализации антитело к PD-L1 представляет собой антитело, раскрытое в заявке на патент №13/478,511. В другом варианте реализации антитело против PD-L1 содержит участки CDR антитела, раскрытого в заявке на патент США №13/478,511.
В одном варианте реализации антитело против PD-L1 представляет собой BMS-936559 (MDX-1105). В другом варианте реализации антитело против PD-L1 представляет собой MPDL3280A (RG7446). В другом варианте реализации антитело против PD-L1 представляет собой MEDI4736.
Дополнительными примерами дополнительного активного ингредиента или ингредиентов (противоопухолевого агента) для применения в комбинации или совместно с соединениями, ингибирующими путь ATF4, согласно настоящему изобретению являются антагонисты PD-1
«Антагонист PD-1» означает любое химическое соединение или биологическую молекулу, которая блокирует связывание PD-L1, экспрессируемого на раковой клетке, с PD-1, экспрессируемым на иммунной клетке (Т-клетке, В-клетке или ΝΚΤ-клетке) и предпочтительно также блокирует связывание PD-L2, экспрессируемого на раковой клетке с с PD-1, экспрессируемым иммунной клеткой. Дополнительные названия или синонимы PD-1 и его лигандов включают: PDCD1, PD1, CD279 и SLEB2 для PD-1; PDCD1L1, PDL1, В7Н1, В7-4, CD274 и В7-Н для PD-L1; и PDCD1L2, PDL2, B7-DC, Btdc и CD273 для PD-L2. В любых вариантах осуществления аспектов или вариантов реализации настоящего изобретения, в которых предполагается лечить человека, антагонист PD-1 блокирует связывание PD-L1 человека с PD-1 человека, и предпочтительно блокирует как связывание PD-L1 человека, так и связывание PD-L2 человека с PD-1 человека. Аминокислотные последовательности PD-1 человека можно найти NCBI, локус №: NP 005009. Аминокислотные последовательности PD-L1 и PD-L2 человека можно найти в NCBI, локус №: NP_054862 and NP_079515, соответственно.
Антагонисты PD-1, которые можно применять в любом из аспектов настоящего изобретения, включают моноклональное антитело (mAb, МАТ), или его антигенсвязывающий фрагмент, который специфически связывается с PD-1 или PD-L1, и предпочтительно специфически связывается с PD-1 человека или PD-L1 человека. МАТ может представлять собой человеческое антитело, гуманизированное антитело или химерное антитело, и может включают человеческую константную область. В некоторых вариантах осуществления человеческая константная область выбрана из группы, состоящей из константных областей IgG1, IgG2, IgG3 и IgG4, и в предпочтительных вариантах реализации человеческая константная область представляет собой константную область IgG1 или IgG4. В некоторых вариантах осуществления антигенсвязывающий фрагмент выбран из группы, состоящей из фрагментов Fab, Fab'-SH, F(ab')2, scFv и Fv.
Примеры MAT, которые связываются с PD-1 человека и могут применяться в различных аспектах и вариантах осуществления настоящего изобретения, описаны в US 7488802, US 7521051, US 8008449, US 8354509, US 8168757, WO 2004/004771, WO 2004/072286, WO 2004/056875 и US 2011/0271358.
Конкретные человеческие МАТ против PD-1, которые можно применять в качестве антагониста PD-1 в любом из аспектов и вариантов осуществления настоящего изобретения, включают: MK-3475, гуманизированное MAT IgG4 со структурой, описанной в WHO Drag Information, Vol.27, No. 2, 161-162 (2013) и которое содержит аминокислотные последовательности тяжелой и легкой цепей, показанные на фиг.6; ниволумаб, человеческое MAT IgG4, структура которого описана в WHO Drug Information, Vol.27, No. 1, 68-69 (2013), которое содержит аминокислотные последовательности тяжелой и легкой цепей, показанные на фиг.7; гуманизированные антитела h409Al 1, h409A16 и И409А17, которые описаны в WO 2008/156712, и АМР-514, который разрабатывается компанией Medimmune.
Другие антагонисты PD-1, которые можно применять в любом из аспектов и вариантов осуществления настоящего изобретения, включают иммуноадгезин, который специфически связывается с PD-1, и предпочтительно специфически связывается с PD-1 человека, например, слитый белок, содержащий внеклеточную или PD-1-связывающую часть PD-L1 или PD-L2, слитую с константной областью, такой как Fc-область молекулы иммуноглобулина. Примеры молекул иммуноадгезии, которые специфически связываются с PD-1, описаны в WO 2010/027827 и WO 2011/066342. Конкретные слитые белки, которые можно применять в качестве антагониста PD-1 для способа лечения, лекарственных средств и применений согласно настоящему изобретению, включают АМР-224 (также известный как B7-DCIg), который представляет собой слитый белок PD-L2-FC и связывается с PD-1 человека.
Другие примеры МАТ, которые связываются с человеческим PD-L1, и могут применяться в способах лечения, лекарственных средствах и применениях согласно настоящему изобретению, описаны в WO 2013/019906, WO 2010/077634 А1 и US 8383796. Специфические моноклональные антитела против PD-L1 человека, используемые в качестве антагониста PD-1 для способа лечения, лекарственных средств и применения согласно настоящему изобретению, включают MPDL3280A, BMS-936559, MEDI4736, MSB0010718C.
KEYTRUDA (кейтруда)/пембролизумабпредставляет собой антитело против PD-1. продаваемое для лечения рака легких компанией Merck. Аминокислотная последовательность пембролизумаба и способы его применения раскрыты в патенте США №8,168,757.
Opdivo (опдиво) / ниволумаб)представляет собой полностью человеческое моноклональное антитело, продаваемое компанией Bristol Myers Squibb, направленное против отрицательного иммунорегуляторного рецептора клеточной поверхности PD-1 (запрограммированная гибель-1 или запрограммированная гибель клеток-1 / PCD-1) человека, обладающее иммунопотенцирующей активностью. Ниволумаб связывается и блокирует активацию PD-1, трансмембранного белка суперсемейства Ig, его лигандами PD-L1 и PD-L2, что приводит к активации Т-клеток и клеточно-опосредованных иммунных ответов против опухолевых клеток или патогенов. Активированный PD-1 отрицательно регулирует активацию эффекторную функцию Т-клеток и за счет подавления активации пути Ρ13k/Akt. Другие названия ниволумаба включают: BMS-936558, MDX-1106 и ONO-4538. Последовательность аминокислот для ниволумаба, а также способы использования и получения раскрыты в патенте США №8,008.449.
Дополнительными примерами дополнительного активного ингредиента или ингредиентов (противоопухолевого агента) для применения в комбинации или совместно с соединением согласно настоящему изобретению являются иммуномодуляторы.
В настоящем документе «иммуномодуляторы» относится к любому веществу, включая моноклональные антитела, которое влияет на иммунную систему. ICOS-связывающие белки согласно настоящему изобретению можно рассматривать как иммуномодуляторы. Иммуномодуляторы можно применять в качестве противоопухолевых средств для лечения рака. Например, иммуномодуляторы включают, без ограничения перечисленными антитела против CTLA-4, такие как ипилимумаб (YERVOY), и антитела против PD-1 (Opdivo (опдиво) / ниволумаб и Keytruda (куйтруда) / пембролизумаб). Другие иммуномодуляторы включают, без ограничения перечисленными: антитела к ОХ-40, антитела в PD-L1, антитела к LAG3, антитела к ΤIM-3, антитела к 41 ВВ и антитела к GITR.
Yervoy (Ервой (ипилимумаб)) представляет собой полностью человеческое антитело к CTLA-4, продаваемое компанией Bristol Myers Squibb. Белковая структура ипилимумаба и способы его применения описаны в патентах США №№6,984,720 и 7,605,238.
В другом варианте реализации настоящего изобретения предложено соединение из Таблицы 1, описанное здесь, или Формула (I), (Ia), (Ib), (II) или (III), или его фармацевтически приемлемая соль или пролекарство для применения для лечения заболевания, состояния или нарушения, связанного с вирусом гепатита В. Настоящее изобретение предусматривает соединение из Таблицы 1, или его фармацевтически приемлемую соль или пролекарство, для применения для лечения заболевания, состояния или нарушения, связанного с вирусом гепатита В, где указанное заболевание, состояние или нарушение, связанное с вирусом гепатита В, может представлять собой желтуху, рак печени, воспаление печени, фиброз печени, цирроз печени, печеночную недостаточность, диффузное воспалительное гепатоцеллюлярное заболевание, гемофагоцитарный синдром или сывороточный гепатит).
В дальнейших вариантах осуществления соединение согласно настоящему изобретению Формулы (I), (Ia), (Ib), (II) or (III), или его фармацевтически приемлемая соль, выбраны из группы соединений, представленных в Таблице 1. Дополнительно, настоящее изобретение также охватывает каждое из этих соединений отдельно и его фармацевтически приемлемые соли.
В другом варианте реализации предложена фармацевтическая композиция, содержащая фармацевтически приемлемый разбавитель и терапевтически эффективное количество соединения Формулы I, Ia, Ib, II или III или его фармацевтически приемлемую соль.
В некоторых вариантах осуществления соединение (соединения) согласно настоящему изобретению, или фармацевтически приемлемая соль, выбраны из представленных, в Таблице 1. Соединения согласно настоящему изобретению, или фармацевтически приемлемая соль такого соединения, могут быть представлены в форме фармацевтически приемлемой соли. Термин "фрамацевтически приемлемая соль" относится к солям, полученным из фармацевтически приемлемых неорганических и органических кислот и оснований. Соответственно, слово "или" в контексте "соединение или его фармацевтически приемлемая соль" следует понимать, как относящееся либо к соединению или его фармацевтически приемлемой соли (альтернатива), или к соединению и его фармацевтически приемлемой соли (в комбинации).
В настоящем тексте термин "фармацевтически приемлемый" относится к соединениям, материалам, композициям и лекарственным формам, к соединениям, материалам, композициям и лекарственным формам, которые подходят для применения с тканями человека и животных без избыточной токсичности, раздражения или других проблем или осложнений.
Квалифицированный специалист поймет, что могут быть получены фармацевтически приемлемые соли соединений, соответствующих формулам I, Ia, Ib, II или III. Эти фармацевтически приемлемые соли могут быть получены in situ в процессе окончательного выделения и очистки соединения или путем отдельной реакции очищенного соединения в форме свободной кислоты или свободного основания с подходящим основанием или кислотой, соответственно.
Иллюстративные фармацевтически приемлемые кислотные соли соединения согласно настоящему изобретению могут быть получены из следующих кислот, включая, без ограничения, муравьиную, уксусную, пропионовую, бензойную, янтарная, гликолевую, глюконовую, молочная, малеиновая, яблочная, винная, лимонная, азотную, аскорбиновая, глюкуронвую, малеиновую, муравьиную, пировиноградную, аспарагиновую, глутаминовую, бензойную, соляную, бромоводородную, иодоводородную, изолимонную, трифторуксусную, памоевую, пропионовую, нтраниловую, метансульфоновую, оксалоуксусную, олеиновую, стеаровую, салициловую, п-гидроксибензойную, никотиновую, фенилуксусную, миндальную, эмбоновую (памоевую), метансульфоновую, фосфорную, фосфоновую, этансульфоновую, бензолсульфоновую, пантотеновую, толуолсульфоновую, 2-гидроксиэтансульфоновую, сульфаниловую, серную, салициловую, циклогексиламиносульфоновую, альгеновую, -гидроксимасляную, галактаровую и галактуровую кислоты. Предпочтительные фармацевтически приемлемые кислоты включают соли хлороводородной кислоты и трифторуксусной кислоты.
Иллюстративные фармацевтически приемлемые соли с неорганическими основаниями соединений согласно настоящему изобретению включают ионы металлов. Более предпочтительные ионы металлов включают, без ограничения, подходящие соли щелочных металлов, соли щелочноземельных металлов и другие физиологически приемлемые ионы металлов. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, магния, марганца, двухвалентного марганца, калия, натрия, цинка и т.п. и в соответствии с их обычными валентностями. Примеры солей оснований включают алюминий, кальций, литий, магний, калий, натрий и цинк. Другие примеры солей оснований включают соли аммония, кальция, магния, калия и натрия. Другие примеры солей оснований включают, например, гидроксиды, карбонаты, гидриды и алкоксиды, включая NaOH, KOH, Na2CO3, K2CO3, NaH и калий-т-бутоксид.
Соли, полученные из фармацевтически приемлемых нетоксичных органических оснований, включают соли первичных, вторичных и третичных аминов, включая, частично, триметиламин, диэтиламин, N,Ν'-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин, меглюмин (Ν-метилглюкамин) и прокаин; замещенных аминов, включая природные замещенные амины; циклических аминов; катионов четвертичного аммония; и основных ионообменных смол, таких как аргинин, бетаин, кафеин, холин, Ν,Ν-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, Ν-этилморфолин, Ν-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п.
Все вышеуказанные соли могут быть получены специалистами в данной области обычными способами из соответствующего соединения согласно настоящему изобретению. Например, фармацевтически приемлемые соли по настоящему изобретению можно синтезировать из исходного соединения, которое содержит основную или кислотную группировку, обычными химическими методами. Обычно такие соли могут быть получены путем осуществления реакции свободных таких соединений в форме свободной кислоты или основания со стехиометрическим количеством подходящего основания или кислоты в воде, в органическом растворителе или в их смесях; обычно предпочтительные неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил. Соль можно осадить из раствора и собрать фильтрацией или может быть выделена путем выпаривания растворителя. Степень ионизации соли может варьировать от полностью ионизованной до почти неионизованной. Перечни подходящих солей можно найти в Remington's Pharmaceutical Sciences. 17th ed., Mack Publishing Company, Easton, Pa., 1985, p.1418, а также в Berge, J. Pharm. Sci., 1977, 66, 1-19, or those listed in Ρ Η Stahl and С G Wermuth, editors, Handbook of Pharmaceutical Salts; Properties, Selection and Use, Second Edition Stahl/Wermuth: Wiley- VCH/VHCA, 2011 (см. http://www.wiley.com/WileyCDA/WileyTitle/productCd-3906390519.html, содержание которых полностью включено в настоящий документ посредством ссылки только в отношении перечня подходящих солей.
Соединения Формулы (I), (Ia), (Ib), (II) или (III) согласно настоящему изобретению могут существовать как в несольватированной, так и в сольватированной формах. Термин «сольват» включает соединение согласно изобретению и одну или более молекул фармацевтического приемлемого растворителя, например, этанола. Термин "гидрат" используется, когда указанный растворитель представляет собой воду. Фармацевтически приемлемые сольваты включают гидраты и другие сольваты, причем растворитель кристаллизации может быть замещен изотопом, например, D2O, d6-ацетон, d6-ДМСО.
Соединения Формулы (I), (Ia), (Ib), (II) или (III), содержащие один или более асимметричных атомов углерода, могут существовать в виде двух или более стереоизомеров. В случае, когда соединение Формулы (I), (Ia), (Ib), (II) или (III) содержит алкенильную группу или алениленовую группу, или циклоалкильную группу, возможно существование геометрических цис/транс (или Z/E) изомеров. В случае, когда соединение содержит, например, кето-группу или оксимную группу, может возникать изомерия таутомеров (таутомерия). Следовательно, одно соединение может обладать более, чем одним типом изомерии.
В объем заявленных соединений согласно настоящему изобретению включены все стереоизомеры, геометрические изомеры и таутомерные формы соединений Формулы I, Ia, Ib, II или III, включая соединения, проявляющие более одного типа изомерии, и смеси одного или более из них. Также включены соли присоединения кислоты или основания, в которых противоион обладает оптической активностью, например, D-лактат или L-лизин, или рацематы, например, DL-тартрат или DL-аргинин.
Цис/транс изомеры могут быть разделены обычными методиками, хорошо известными специалистам, например, путем хроматографии или фракционной кристаллизации.
Обычные методы получения / выделения индивидуальных энантиомеров включают хиральный синтез из подходящего оптически чистого прекурсора или разделение рацемата(или рацемата соли или производного) с использованием, например, хиральной хроматографии высокого давления (ВЭЖХ).
В качестве альтернативы, может быть проведена реакция рацемата (или рацемического прекурсора) с подходящим оптически активным соединением, например, спиртом, или, в случае, когда соединение формулы I, Ia, Ib, II или III, содержит кислотную или основную группировку, кислотой или основанием, таким как винная кислота или 1-фенилэтиламин. Полученные диастереомерные смеси могут быть разделены путем хроматограыии и/или фракционной кристаллизации, и один или оба диастереоизомера могут быть преобразованы в соответствующий чистый энантиомер(ы) средствам, хорошо известными специалисту. Хиральные соединения согласно настоящему изобретению (и их хиральные прекурсоры) могут быть получены в энантиомерно-обогащенной форме с использованием хроматографии, обычно ВЭЖХ, на смоле а асимметричной стационарной фазой и с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащего от 0 до 50% изопропанола, обычно от 2 до 20%, и от 0 до 5% алкиламина, обычно 0.1% диэтиламина. Концентрирование элюата дает обогащенную смесь.
Смеси стереоизомеров могут быть разделены любыми традиционными методиками, известными специалистам в данной области, [см., например, "Stereochemistry of Organic Compounds", Ε L Eliel (Wiley, New York, 1994).]
Настоящее изобретение включает все фармацевтически приемлемые изотопно меченные соединения Формулы (I), (Ia), (Ib), (II) или (III), в которых один или более атомов замещены атомами, имеющими такой же атомный номер, но атомная масса или массовый номер отличаются от атомной массы или массового номера, обычно обнаруживаемого в природе.
Примеры изотопов, пригодных для включения в указанные соединения согласно настоящему изобретению включают изотопы водорода, такие как 2Н и 3Н, углерода, такие как 11С, 13С и 14С, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15О, 17О и 18O, фосфора, такие как 32Р, и серы, такие как 35S. Определенные изотопно меченные соединения Формул (I), (Ia), (II) и (IIa), например, соединения, которые включают радиоактивный изотоп, пригодны для исследований распределения лекарств и/или субстратов в тканях. Радиоактивные изотопы трития, т.е., 3Н, и углерода-14, т.е., 14С, наиболее пригодны для этой цели ввиду простоты их включения и готовых средств их обнаружения. Замещение более тяжелыми изотопами, такими как дейтерий, т.е., 2Н, может дать определенные терапевтические преимущества, связанные с большей метаболической стабильностью, например, увеличенный период полувыведения in vivo или уменьшенные требования к дозировке, и, следовательно, может быть предпочтительным в некоторых обстоятельствах.
Изотопно меченные соединения формул (I), (Ia), (Ib), (II) или (III) в целом могут быть получены обычными методами, известными специалистам в данной области техники, или способами, аналогичными описанным в данном документе, с использованием подходящих изотопно меченных реагентов вместо ранее использованного немеченого реагента.
Соединения согласно настоящему изобретению можно вводить в виде пролекарств. Так, некоторые производные соединений Формулы (I), (Ia), (Ib), (II) или (III), которые могут сами обладать небольшой фармакологической активностью, или у которых фармакологическая активность может отсутствовать, при введении в организм или на организм, могут превращаться в соединения Формулы (I), (Ia), (Ib), (II) или (III) как «пролекарства».
Введение химических соединений и комбинаций таких соединений, описанных в данном документе, может осуществляться любым из приемлемых способов введения для агентов, которые служат аналогичным функциям, включая, помимо прочего, перорально, сублингвально, подкожно, внутривенно, интраназально, топически, трансдермально, внутрибрюшинно, внутримышечно, внутрилегочно, вагинально, ректально или внутрь глаза. В некоторых вариантах осуществления используется пероральное или парентеральное введение.
Фармацевтические композиции или составы включают твердые, полутвердые, жидкие и аэрозольные лекарственные формы, такие как, например, таблетки, капсулы, порошки, жидкости, суспензии, суппозитории, аэрозоли и т.п. Химические соединения также можно вводить в лекарственных формах с замедленным или контролируемым высвобождением, включая инъекции депо, осмотические насосы, пилюли, трансдермальные (включая электротранспорт) пластыри и т.п., для длительного и/или временного импульсного введения с заданной скоростью. В некоторых вариантах осуществления композиции представлены в стандартных лекарственных формах, подходящих для однократного введения точной дозы. Активный ингредиент может быть спрессован в гранулы или маленькие цилиндры и имплантирован подкожно или внутримышечно в качестве инъекционных депо-форм или имплантов. В имплантах могут применяться инертные материалы, такие как биоразлагаемые полимеры или синтетические силиконы, например, силастик или силиконовая резина.
Химические соединения также можно вводить в форме систем доставки на основе липосом, таких как небольшие однослойные везикулы, крупные однослойные везикулы и многослойные везикулы. Липосомы могут быть образованы из различных фосфолипидов, таких как холестерин, стеариламин, или фосфатидилхолины. Липосомные препараты ингибиторов тирозинкиназ также можно применять в способах согласно настоящему изобретению. Липосомные варианты ингибиторов тирозинкиназ можно применять для улучшения толерантности к ингибиторам.
Химические соединения также можно доставлять при помощи моноклональных антител в качестве индивидуальных переносчиков, с которыми связываются молекулы соединения.
Также могут быть получены химические соединения могут с растворимыми полимерами, такими как растворимые полимеры, в качестве нацеливаемых переносчиков лекарственного средства. Такие полимеры могут включать поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный остатками пальмитоила. Кроме того, могут быть получены химические соединения с биоразлагаемыми полимерами, которые можно применять до получения форм лекарственного препарата с контролируемым высвобождением, например, полимолочной кислотой, полигликолевой кислотой, сополимерами полимолочной кислоты и полигликолевой кислоты, полиэпсилонкапролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидропиранами, полицианоакрилатами или поперечно сшитыми или амфипатическими блок-сополимерами или гидрогелями.
Описанные в настоящем документе химические соединения можно вводить либо по отдельности, либо, как правило, в комбинации с обычным фармацевтическим носителем, вспомогательным веществом или т.п.(например, маннитол, лактоза, крахмал, стеарат магния, сахарин натрия, тальк, целлюлоза, кроскармеллоза натрия, глюкоза, желатин, сахароза, карбонат магния и т.п.). При желании фармацевтическая композиция может также содержать незначительные количества нетоксичных вспомогательных веществ, таких как смачивающие вещества, эмульгаторы, солюбилизаторы, буферные вещества рН и тому подобное (например, ацетат натрия, цитрат натрия, производные циклодекстрина, сорбитан монолаурат, триэтаноламин ацетат, триэтаноламин олеат и т.п.). Обычно, в зависимости от предполагаемого способа введения, фармацевтическая композиция будет содержать от около 0.005% до 95%; в некоторых вариантах осуществления примерно от 0.5% до 50% по массе химического соединения. Фактические способы приготовления таких дозированных форм известны или будут очевидны специалистам в данной области техники; например, см. Remington's Pharmaceutical Sciences, Mack Publishing Company, Истон, Пенсильвания.
В некоторых вариантах осуществления указанные композиции будут иметь форму пилюли или таблетки, и, таким образом, композиция будет содержать, наряду с активным ингредиентом, разбавитель, такой как лактоза, сахароза, дикальцийфосфат или тому подобное; смазывающее вещество, такое как стеарат магния или аналогичные вещества; и связующее вещество, такое как крахмал, гуммиарабик, поливинилпирролидин, желатин, целлюлоза, производные целлюлозы и т.п. В другой твердой лекарственной форме порошок, марум, раствор или суспензию (например, в пропиленкарбонате, растительных маслах или триглицеридах) инкапсулируют в желатиновую капсулу.
Жидкие подходящие для фармацевтического применения композиции можно, например, приготовить путем растворения, диспергирования и т.д. по меньшей мере одного химического соединения и необязательно фармацевтических вспомогательных веществ в носителе (например, воде, солевом растворе, водном растворе декстрозы, глицерине, гликолях, этаноле и т.п.) с получением раствора или суспензии. Инъекционные препараты могут быть выполнены в традиционных формах, в виде жидких растворов или суспензий, в виде эмульсий, или в твердых формах, подходящих для растворения или суспендирования в жидкости перев инъекционным введением. Процент химических соединений, содержащихся в таких парентеральных композициях, сильно зависит от их конкретной природы, а также от активности химических соединений и потребностей субъекта. Однако можно использовать процентное содержание активного ингредиента в растворе от 0.01% до 10%, и оно будет выше, если композиция представляет собой твердое вещество, которое впоследствии будет разбавлено до вышеуказанных процентов. В некоторых вариантах осуществления композиция может включать примерно от 0.2 до 2% активного агента в растворе.
Фармацевтические композиции химических соединений, описанных в настоящем документе, также можно вводить в дыхательные пути в виде аэрозоля или раствора для небулайзера или в виде мелкодисперсного порошка для инсуффляции, отдельно или в комбинации с инертным носителем, таким как лактоза. В таком случае частицы фармацевтической композиции имеют диаметр менее 50 мкм, в некоторых вариантах осуществления менее 10 мкм.
В целом, предложенные химические соединения будут вводить в терапевтически эффективном количестве любым из принятых способов введения для агентов, которые служат аналогичным целям. Режим дозирования с применением химических соединений, описанных в настоящем документе, может быть выбран в соответствии с различными факторами, включая тип, вид, возраст, массу, пол и тип заболевания, которое лечат, тяжести (т.е., стадии) заболевания, которое лечат; пути введения; функции печени и почек у пациента; и конкретных применяемых соединения или соли. Режим дозирования можно применять, например, для предотвращения, подавления (полного или частичного), или блокирования прогрессирования заболевания. Лекарственное средство можно вводить более одного раза в сутки, например, один или два раза в сутки.
Внутривенно или подкожно, пациент может получать химические соединения, описанные в настоящем документе, в терапевтически эффективных количествах, достаточных для отставки приблизительно от 0.001 до 200 мг на килограмм массы тела в сутки, например, примерно 0.005-100 мг/кг/день, например, от примерно от 0.005 до 1 мг/кг/день.
Соответственно, для введения персоне массой 70 кг, диапазон дозировки может поставлять примерно 0.35-70 мг в день. Такие количества можно вводить рядом походящих способов, например, большие объемы низких концентраций химических веществ в течение одного продолжительного периода времени или несколько раз в день. Такие количества можно вводить в течение одного или более последовательных дней, разделенных промежутками дней или комбинации того и другого в течение недели (7-дневного периода). В качестве альтернативы, низкие объемы высоких концентраций химических соединений в течение короткого периода времени, например, один раз в день в течение одного или более дней последовательно, с интервалами, или комбинация того и другого в течение недели (7-дневного периода).
В соответствии с настоящим изобретением, химические соединения, описанные в настоящем документе, можно вводить с использованием непрерывных дозирований или дозирований с интервалами. Например, введение химического соединения с интервалами может представлять собой введение от одного до шести дней в неделю или оно может означать введение в циклах (например, ежедневное введение в течение от двух до восьми последовательных недель, затем оставшийся период без введения в течение до одной недели) или он может означать введение через день. Композиции можно вводить в циклах, в которых остальные периоды между циклами (например, лечение в течение от двух до восьми недель с остатком периода до недели между лечениями).
Препараты для подкожного введения могут быть получены в соответствии с процедурами, известными в данной области, при рН в диапазоне между примерно 5 и примерно 12, такие препараты включают подходящие буферы и обеспечивающие изотоничность вещества.
В целом, химические соединения будут вводить в виде фармацевтических композиций одним из следующих способов: перорального, системного (например, трансдермального, интраназального или с применением суппозитория) или парентерального (например, внутримышечного, внутривенного или подкожного) введения. В некоторых вариантах осуществления можно использовать пероральное введения с удобной суточной дозировкой, которая может быть скорректирована в соответствии со степенью поражения. Композиции могут иметь форму таблетки, пилюли, капсулы, полутвердого вещества, порошка, состава с замедленным высвобождением, раствора, суспензии, эликсира, аэрозоля или любой другой подходящей композиции. Другим способом введения предоставленных химических веществ является ингаляция.
Выбор лекарственной формы зависит от различных факторов, таких как способ введения лекарственного средства и биодоступность лекарственного вещества. Для доставки посредством ингаляции химическое соединение может быть составлено в виде жидкого раствора, суспензии, аэрозоля или сухого порошка и загружено в подходящий дозатор для введения. Существует несколько типов фармацевтических ингаляционных аппаратов: ингаляторы-небулайзеры, ингаляторы с отмеренной дозой (MDI) и ингаляторы для сухого порошка (DPI). Распылители (небулайзеры) создают поток воздуха с высокой скоростью, который заставляет терапевтические агенты (которые сформулированы в жидкой форме) распыляться в виде тумана, который попадает в дыхательные пути пациента. MDI обычно приготовлены в виде состава со сжатым газом. При активации устройство выделяет отмеренное количество терапевтического агента с помощью сжатого газа, таким образом обеспечивая надежный способ введения установленного количества агента. DPI выделяет терапевтические агенты в форме сыпучего порошка, который может диспергироваться в потоке воздуха указанным устройством на вдохе пациента во время дыхания. Чтобы получить легкосыпучий порошок, терапевтическое средство составляют с наполнителем, таким как лактоза. Отмеренное количество терапевтического агента хранится в форме капсулы и выделяется при каждом нажатии.
Недавно были разработаны фармацевтические композиции для лекарств, которые демонстрируют низкую биодоступность, основанные на том принципе, что биодоступность может быть увеличена за счет увеличения площади поверхности, т.е. уменьшения размера частиц. Например, в патенте США №4,107,288 описан фармацевтический состав, имеющий частицы размером от 10 до 1000 нм, в котором активный материал нанесен на сшитую матрицу макромолекул. В патенте США №5,145,684 описано производство фармацевтического препарата, в котором лекарственное вещество измельчается до наночастиц (средний размер частиц 400 нм) в присутствии модификатора поверхности, а затем диспергируется в жидкой среде, в результате чего появляется фармацевтический препарат, который демонстрирует исключительно высокую биодоступность.
Указанные композиции обычно состоят из одного описанного в настоящем документе химического соединения в комбинации с одним фармацевтически приемлемым наполнителем. Приемлемые вспомогательные вещества нетоксичны, облегчают введения и не оказывают неблагоприятного воздействия на терапевтический эффект по меньшей мере одного химического соединения, описанного в настоящем документе. Такой наполнитель может быть любым твердым, жидким, полутвердым или, в случае аэрозольной композиции, газообразным наполнителем, который обычно доступен специалисту в данной области техники.
Твердые вспомогательные вещества включают крахмал, целлюлозу, тальк, глюкозу, лактозу, сахарозу, желатин, солод, рис, муку, мел, силикагель, стеарат магния, стеарат натрия, глицерин моностеарат, хлорид натрия, сушеное обезжиренное молоко и т.п. Жидкие и полутвердые вспомогательные вещества могут быть выбраны из глицерина, пропиленгликоля, воды, этанола и различных масел, включая полученные из нефтегазового сырья, животного, растительного или синтетического происхождения, например, арахисовое масло, соевое масло, минеральное масло, кунжутное масло и т.д. Жидкие носители для инъекционных растворов включают воду, волевой раствор, водный раствор декстрозы и гликоли.
Сжатые газы можно использовать для рассеивания химического соединения, описанного в настоящем документе, в форме аэрозоля. Инертные газы, подходящие для этой цели - азот, диоксид углерода и т.д. Другие подходящие фармацевтические вспомогательные вещества и их препараты описаны в Remington's Pharmaceutical Sciences, под ред. Ε.W. Martin (Mack Publishing Company, 18е изд., 1990).
Количество химического вещества может варьировать во всем диапазоне, применяемом специалистами в данной области. Обычно композиция будет содержать, в массовых процентах (масс. %), от примерно 0.01-99.99 масс. % по меньшей мере одного химического соединения, описанного в настоящем документе, в пересчете на всю композицию, где остальное количество приходится на одно или более подходящих фармацевтических вспомогательных веществ. В некоторых вариантах осуществления указанное по меньшей мере одно химическое соединение, описанное в настоящем документе, присутствует на уровне, составляющем примерно 1-80 масс. %.
В различных вариантах осуществления фармацевтические композиции согласно настоящему изобретению включают соединения Формулы (I), (Ia), (Ib), (II) или (III), их соли, и комбинации вышеперечисленного.
Различные режимы введения, дозировки и схемы введения, описанные в настоящем документе, просто указывают конкретные варианты осуществления и не должны рассматриваться как ограничивающие широкий объем изобретения. Любые перестановки, вариации и комбинации дозировок и схем введения включены в объем настоящего изобретения.
Соединения согласно настоящему изобретению могут быть получены в соответствии с различными схемами, описанными ниже.
Способы синтеза
В способах синтеза предложенных химических соединений применяются легкодоступные исходные материалы и описанные ниже общие способы и процедуры. Понятно, в тех случаях, где приведены типичные или предпочтительные условия проведения процессов (т.е., значения температуры реакции, временные значения, мольные соотношения реагентов, растворители, значения давления и т.д.), также можно использовать другие условия процесса, если не указано иное. Оптимальные условия реакций могут варьировать для конкретных используемых реагентов или растворителей, но специалист в данной области сможет установить такие условия посредством рутинных процедур оптимизации.
Кроме того, в способах согласно настоящему изобретению можно применять защитные группы, которые предотвращают участие некоторых функциональных групп в нежелательных реакциях. Подходящие защитные группы для различных функциональных групп, а также подходящие условия для защиты и удаления защиты функциональных групп хорошо известны в данной области. Например, многочисленные защитные группы описаны в Т. W. Greene and G. Μ. Wuts, Protecting Groups in Organic Synthesis, Third Edition, Wiley, New York, 1999, и цитируемых там источниках.
Кроме того, предложенные химические соединения могут содержать один или более хиральных центров, и такие соединения могут быть получены или выделены в виде чистых стереоизомеров, т.е., в виде отдельных энантиомеров или диастереомеров, или в виде стереомерно обогащенных смесей. Все такие стереоизомеры (и обогащенные смеси) включены в объем настоящего описания, если не указано иное. Чистые стереоизомеры (или обогащенные смеси) могут быть получены с использованием, например, оптически активных исходных материалов или стереоселективных реагентов, хорошо известных в данной области. В качестве альтернативы, рацемические смеси таких соединений могут быть получены с использованием, например, хиральной колоночной хроматографии, хиральных разделяющих агентов и т.п.
Исходные материалы для описанных ниже реакций представляют собой общеизвестные соединения или могут быть получены с использованием известных процедур или их очевидных модификаций. Например, многие исходные материалы можно приобрести у коммерческих поставщиков, таких как Aldrich Chemical Со. (Милуоки, Висконсин, США), Bachem (Торранс, Калифорния, США), Ernka-Chemce или Sigma (Сент-Луис, Миссури, США). Другие могут быть получены путем проведения процедур или очевидных модификаций процедур, описанных в стандартных текстах, таких как Fieser and Fieser's Reagents for Organic Synthesis, Vol.1-15 (John Wiley and Sons, 1991), Rodd's Chemistry of Carbon Compounds, Vol.1-5 and Supplemental (Elsevier Science Publishers, 1989), Organic Reactions, Vol.1-40 (John Wiley and Sons, 1991), March's Advanced Organic Chemistry, (John Wiley and Sons, 4th Edition), и Larock's Comprehensive Organic Transformations (VCH Publishers Inc., 1989).
Если не указано иное, реакции, описанные в настоящем документе, могут проходить при атмосферном давлении, обычно в диапазоне температур от -78°С до 200°С. Далее, за исключением применения в Примерах или если указано иное, указанные время и условия реакции являются приблизительными, например, реакции протекают при примерно атмосферном давлении в диапазоне температур от примерно -78°С до примерно 110°С в течение периода продолжительностью от примерно 1 до примерно 24 ч; реакции оставляют для протекания на ночь в среднем в течение примерно 16 ч.
Каждый из терминов "растворитель", "органический растворитель" и "инертный растворитель" обозначает растворитель, инертный в условиях реакции, описанной в связи с ним, включая, например, бензол, толуол, ацетонитрил, тетрагидрофуранил ("ТГФ"), диметилформамид ("ДМФА"), хлороформ, метиленхлорид (или дихлорметан), диэтиловый эфир, метанол, Ν-метилпирролидон ("ΝΜΡ"), пиридин и т.п.
Выделение и очистка химических соединений и промежуточных соединений, описанных в данном документе, при желании может быть осуществлена с помощью любой подходящей процедуры разделения или очистки, например, фильтрация, экстракция, кристаллизация, колоночная хроматография, тонкослойная хроматография или толстослойная хроматография или комбинация этих процедур. Конкретные иллюстрации подходящих процедур разделения и выделения представлены в приведенных ниже примерах. Однако также могут применяться другие эквивалентные процедуры разделения или выделения.
При желании (R)- и (S)-изомеры могут быть разделены способами, известными специалистам в данной области, например, путем образования диастереоизомерных солей или комплексов, которые могут быть разделены, например, кристаллизацией; через образование диастереоизомерных производных, которые можно разделить, например, кристаллизацией, газожидкостной или жидкостной хроматографией; за счет селективной реакции одного энантиомера с энантиомер-специфическим реагентом, например, ферментативного окисления или восстановления, с последующим разделением модифицированных и немодифицированных жнантиомеров; или газо-жидкостной или жидкостной хроматографии в хиральной среде, например, на хиральной подложке, такой как кремний со связанным хиральным лигандом, или в присутствии хирального растворителя. В качестве альтернативы, конкретный энантиомер может быть синтезирован путем асимметричного синтеза с использованием оптически активных реагентов, катализаторов или растворителей или путем превращения одного энантиомера в другой путем асимметричного преобразования.
ПРИМЕРЫ
Следующие ниже примеры служат для более полного описания способа реализации и применения вышеописанного изобретения Понятно, что это описание никоим образом не служит для ограничения истинного объема изобретения, а приведено в целях иллюстрации. В приведенных ниже примерах и приведенных выше схемах синтеза следующие сокращения имеют следующие значения. Если сокращение не определено, оно имеет общепринятое значение.
вод. - Aqueous
мкл - микролитры
мкМ - микромоль
ЯМР - ядерный магнитный резонанс
Boc - трет-бутоксикарбонил
Br - широкий
Cbz - Бензилоксикарбонил
d - дублет
Δ - химический сдвиг
°С - градусов Цельсия
ДХМ - дихлорметан
dd - дублет дублетов
DMAP - 4-(диметиламино)пиридин
DMEM - среда Игла, модифицированная по способу Дульбекко
ДМФА - Ν,Ν-диметилформамид
DMP - 2,2-диметоксипропан
ДМСО - диметилсульфоксид
EtOAc - этилацетат
ESI (электроспрей) - Ионизация методом электрораспыления
Г или г - грамм
ч или час. - часы
HCV (ВГС) - вирус гепатита С
ВЭЖХ - высокоэффективная жидкостная хроматография
Гц - Герц
IU - Международные единицы
IC50 - ингибирующая концентрация при 50% ингибировании
J - константа взаимодействия (приведенная в Гц, если не указано иное)
LHMDS - бис(триметилсилил)амид лития
Μ - мультиплет
Μ - моль
М+Н+ - исходный пик масс-спектра плюс
Mr или мг - миллиграмм
Min - Минут
мл - миллилитр
мМ - миллимолярный
Ммоль - миллимоль
MS (МС) - масс-спектр
Ть - наномоль
ppm - частей на миллион
p-TsOH - n-толуолсульфоновая кислота
q.s. - достаточное количество
S - синглет
КТ - Комнатная температуру
насыщ. - насыщенный
Τ - триплет
TBS-Cl - трет-юутилдиметилсилилхлорид
TFA (ТФУК) - трифторуксусная кислоты
Описание оборудования
1Н-ЯМР спектры записывали на спектрометре Varian. Химический сдвиги выражены в частях на миллион (ppm, единиц δ). Константы взаимодействия приведены в единицах Герц (Гц). Схема расщепления описывает кажущиеся мультиплетности как s (синглет), d (дублет), t (триплет), q (квартет), quint (квинтет), m (мультиплет), br (уширенный).
Аналитические масс-спектры (МС) низкого разрешения записывали на устройстве Waters (Acquity). Использовали следующие условия, описанные ниже.
Устройство: Agilent 1200-6100
режим сканирования: Чередующееся положительное/отрицательное электрораспыление
Диапазон сканирования: 100-1000 а.м.е.
Условия ЖХ:
ЖХМС-анализ выполняли на колонке HALO С-18, 4.6*50 мм, 2.7 мкм, С18 при 45°С.
Вводили 1.0 мкл образца
Применяли следующий градиент:
Подвижная фаза А: вода +0.1% об./об. муравьиной кислоты
Подвижная фаза В: Ацетонитрил+0.1% об./об. муравьиной кислоты
Время %А %В Скорость потока
0.00 мин 95 5 1.8 мл/мин
1.0 мин 5 95 1.8 мл/мин
2.0 мин 5 95 1.8 мл/мин
2.5 мин 95 5 1.8 мл/мин
УФ-детектирование осуществляли по суммарному поглощению сингала при сканировании на 214 нм и 254 нм
Устройство: Shimadzu ЖХМС-2020
Режим сканирования: чередующееся положительное/отрицательное электрораспыление
Диапазон сканирования: 100-2000 amu
Условиях ЖХ:
ЖХМС-анализ выполняли на колонке HALO С-18, 4.6*50 мм, 2.7 мкм, С18 при 45°С.
Вводили 1.0 мкл образца
Применяли следующий градиента
Подвижная фаза А: вода +0.1% об./об. муравьиной кислоты
Подвижная фаза В: ацетонитрил+0.1% об./об. муравьиной кислоты
Время %А %В Скорость потока
0.00 мин 95 5 1.5 мл/мин
1.0 мин 5 95 1.5 мл/мин
2.0 мин 5 95 1.5 мл/мин
2.5 мин 95 5 1.5 мл/мин
3.0 мин 95 5 1.5 мл/мин
УФ-детектирование осуществляли по суммарному поглощению сингала при сканировании на 214 нм и 254 нм
Схемы и экспериментальные процедуры
Приведенные далее схемы и процедуры показывают, как можно получить соединения согласно настоящему изобретению. Конкретные указанные растворители и условия реакции также носят иллюстративный характер и не задуманы как ограничивающие. Соединения, которые не описаны, либо доступны для приобретения, либо специалист сможет легко из приготовить с использованием доступных исходных материалов. Примеры, приведенные в настоящем документе, раскрыты исключительно в иллюстративных целях, и не предполагаются, что они будут ограничивать настоящее изобретение.
Дополнительные примеры, содержащиеся в этом документе, определены как имеющие показанную конфигурацию, методами спектроскопии, хорошо известными специалистами в данной области, включающим, без ограничения перечисленными, методы 1D и 2D ЯМП. Соединения Формулы (I), (Ia), (Ib), (II) или (III) можно, например, синтезировать в соответствии со Схемами 1-6.
В одном варианте реализации соединение формулы 1.12 можно получить как показано на Схеме 1. Бициклическая система колец в Формулах 1.5 и 1.6 может быть образована с использованием 4-компонентной реакции Уги между карбоновой кислотой 1.1, изоцианидом 1.2, альдегидом 1.3 и аммиаком с получением дипептида 1.4 с последующими удалением защиты кислотой и циклизацией. Соединения Формул 1.5 и 1.6 можно синтезировать с использованием асимметричного синтеза Уги с добавлением хиральных реагентов, таких как хиральные фосфорные кислоты (Jian Zhang, et al, Science, 2018, 361, 1087). Изоцианиды (Isocyanide Chemistry: Application in Synthesis and Material Science, V. Nenajdenko, Wiley-VCH, 1st Ed) могут быть синтезированы путем дегидрирования формамидов с использованием, например, оксихлорида фосфора, фосгена, дифосгена, толуолсульфонила хлорида и т.д.
Формамиды, в свою очередь, могут быть получены путем форматирования амина этилформиатом, смешанным ангидридом муравьиной и уксусной кислот, уксусной кислотой/карбодиимидами, активированным сложным эфиром муравьиной кислоты. Альдегид Формулы 1.3 (М. Vamos, et al, ACS Chem. Biol., 2013, 8, 725-732) может быть получен за счет ди-метилирования 4,4-диметоксибутаннитрила с последующим восстановлением при помощи DIBAL-H. Это соединение Формулы 1.8 может быть получено путем проведения реакции сочетания Boc-N-Метил-L-Ala-OH 1.7 с желаемым диастереомером 1.5 с использованием стандартных реагентов для амидного сочетания, таких как ТЗР, HATU, HBTU, EDC/HOBt, TBTU и т.п. в присутствии оснований, такого как основание Хюнига, Ν-метилморфолин и т.п., в соответствующем растворителе, таком как ДМФА, ДХМ и т.п. Алкин в формуле 1.8 можно селективно восстановить путем гидогенирования в присутствии катализатора Линдлара или альтернативным способом с получением алкена Формулы 1.9. Алкен Формулы 1.9 можно димеризовать с использованием метатезиса олефинов Граббса с получением соединения формулы 1.10. Олефин Формулы 1.10 можно восстановить с использованием гидрогенирования в присутствии металлического катализатора, такого как палладий на угле, платина, с получением соединения формулы 1.11, защитные группы Вое в котором можно удалить в кислотной среде, такой как HCl, ТФУК и т.п.
Схема 1
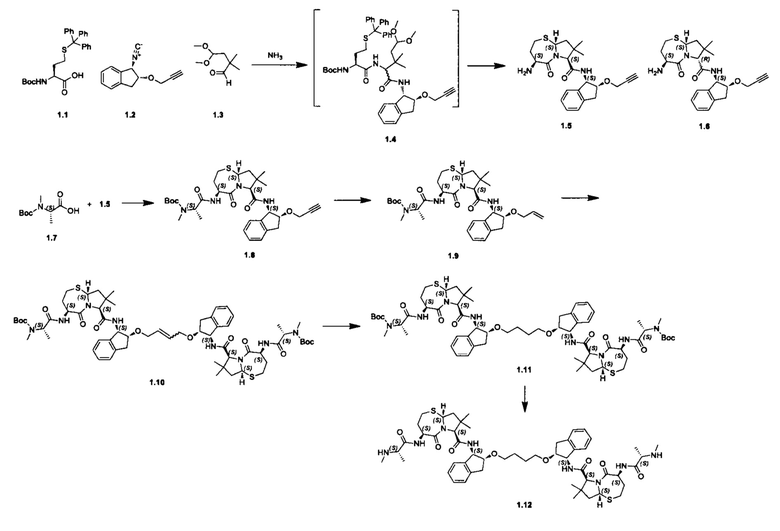
В другом варианте реализации промежуточное соединение Формулы 2.8 можно получить в соответствии со Схемой 2. промежуточное соединение 2.8 представляет собой универсальное промежуточное соединение для получения Формулы I, Ia, Ib, II или III путем сочетания с различными диаминами, известными специалисту. Бициклическое соединение Формулы 2.5 можно получить с использованием 4-компонентной реакции Уги между кислотой 2.1, изоцианидом 2.2, альдегидом 2.3 и аммиаком с получением дипептида Формулы 2.4. Соединение Формулы 2.5 можно получить путем обработки дипептида 2.4 кислотой, которая может привести к образованию бициклической кольцевой структуры, а также индоламида (О. Kreye, et al, SYNLETT, 2007, p 3188-3192). Соединение Формул 2.6 и 2.7 можно получить с использованием реакции сочетания Boc-N-Метил-L-Ala-OH 1.7 с соединением 2.5 с использованием стандартных реагентов для амидного сочетания, таких как ТЗР, HATU, HBTU, EDC/HOBt, TBTU и т.п.в присутствии основания, такого как основание Хюнига, Ν-метил морфолин и т.п., в соответствующем растворителе, таком как ДМФА, ДХМ и т.п. Кислоту Формулы 2.8 можно получить путем основного гидролиза индоламида Формулы 2.6 в присутствии гидроксид натрия или подобного соединения в растворителе, таком как, метанол.
Схема 2
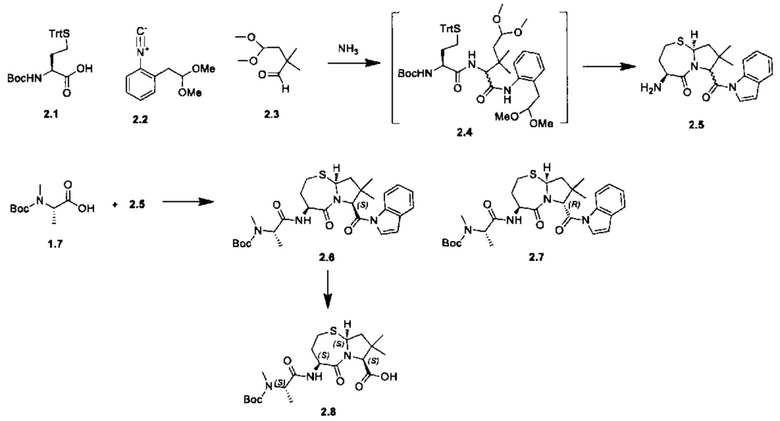
В другом варианте реализации промежуточное соединение Формулы 3.7 можно получить в соответствии со Схемой 3. Промежуточное соединение 3.7 представляет собой универсальное промежуточное соединение для получения соединений Формулы I, Ia, Ib, II или III за счет реакции сочетания с различными диаминами, известными специалисту.
Схема 3. Активированный промежуточный реагент Формулы 3.4 может быть получен следующим образом. Амидное сочетание Boc-N-метил-L-Ala-OH 1.7 с диамином Формулы 3.1 с использованием условий для сочетания на основе смешанного ангидрида или альтернативных условий для сочетания позволяет получить соединение формулы 3.2. Тионирование амида 3.2 с испольщованием пентасульфида фосфора в присутствии основания, такого как карбонат натрия или подобное ему, в растворителе, таком как ТГФ, позволяет получиь тиоамид Формулы 3.3. Нитробензотриазольный реагент Формулы 3.4 можно получить путем обработки тиоамида 3.3 нитритом натрия в уксусной кислоте в растворителях, таких как ТГФ и т.п. (М. Ashraf Shalaby, et al, J. Org. Chem. 1996, 61, 9045-9048). Соединение Формул 3.5 и 3.6 можно получить с использованием реакции индоламида 2.5 и активированного тиоацилирующего реагента Формулы 3.4 в присутствии основания, такого как основание Хюнига, триэтиламин и т.п., в растворителях, таких как ДХМ, ДМФА и т.п. Отделенный диастереоизомер Формулы 3.5 можно обработать водным основанием, таким как гидроксид натрия в спиртовом растворителе, таком как метанол, с получением промежуточного соединения Формулы 3.7.
Схема 3
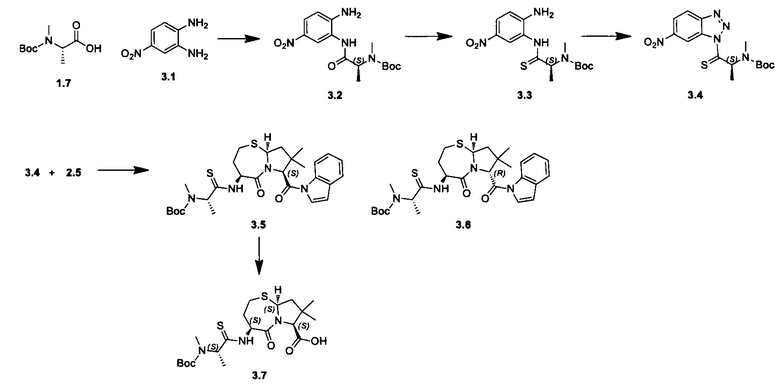
В другом варианте реализации соединения Формулы 4.3 и 4.5 можно получить в соответствии со Схемой 4. Соединения Формулы 4.2 и 4.4 могут быть получены с использованием сочетания промежуточных соединений 2.8 или 3.7 с диамином 4.1 в присутствии реагента сочетания или через активированные сложные эфиры в присутствии а основания, такого как основание Хюнига, триэтиламин и т.п., в растворителях, таких как ДМФА, ТГФ, ДХМ и т.п.. В реакциях сочетания можно использовать реагенты сочетания, такие как HATU, TBTU, ВОР. РуВОР, DEPBT, EDC/HOBt, EEDQ, но без ограничения перечисленными. Соединения Формул 4.3 и 4.5 могут быть получены путем удаления защитной группы Boc в соединениях 4.2 и 4.4 в кислотных условиях, таких как HCl, ТФУК и т.п.
Схема 4
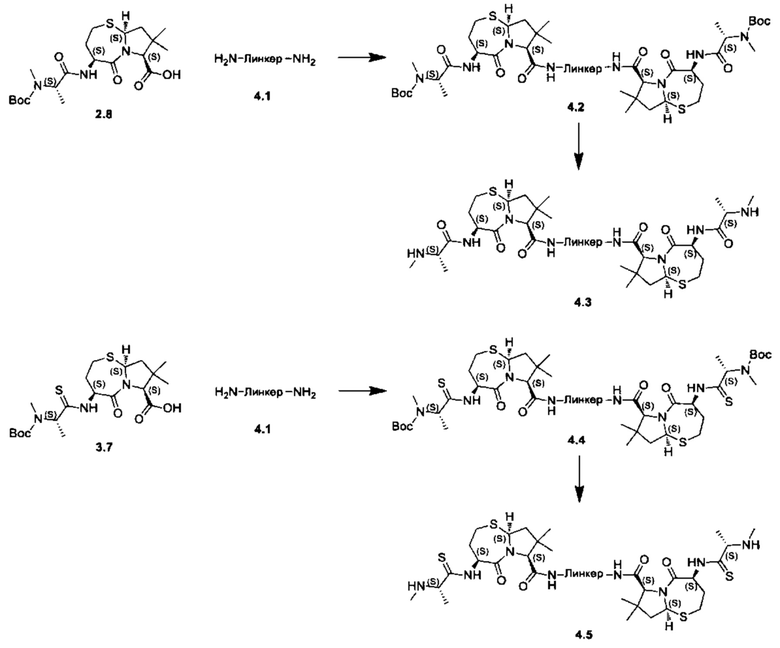
Диамины для осуществления сочетания промежуточных соединений 2.8 и 3.7 легкодоступны у коммерческих поставщиков или могут быть получены известными способами. Различные примеры показаны на Схеме 5, без ограничения типом химических компонентов или функциональными группами, используемыми для линкеров. иамины Формул 5.3 и 5.5 могут быть получены путем бис-алкилирования аминоспиртов 5.1 или 5.4, где X представляет собой уходящую группу, такую как галид, тозилат, мезилат и т.п., в присутствии основания, такого как гидрид натрия и т.п., в растворителях, таких как ДМФА, ТГФ и т.п. Соединения Формулы 5.8 и 5.10 могут быть получены с использованием реакций сочетания защищенных аминокислот 5.6 и 5.9 в условиях амидного сочетания с последующим удалением защиты кислотой.
Схема 5
Где прямоугольная рамка  представляет собой линкер.
представляет собой линкер.
В другом варианте реализации димеры Формул 6.7, 6.8, 6.9, 6.10 и 6.11 можно получить в соответствии со Схемой 6. Соединения Формул 6.3, 6.4, 6.5 и 6.6 могут быть получены путем соединения амидной связью промежуточных соединений 2.8 и 3.7 с аминами Формул 6.1 и 6.2 где X и Υ представляют собой линкеры с функциональными группами на концах, которые можно использовать для дальнейших реакций. Примеры функциональных групп включают перечисленные далее, но не ограничиваются ими: защищенный амин, защищенная карбоновая кислоты, защищенные тиолы, алкин, алкен, сульфонил хлорид, азид, гидроксил, галогениды, нитрилы, изоцианаты. Мономеры Формул 6.3 и 6.5 можно димеризовать с образованием димеров Формул 6.7 и 6.11, например, если X содержит алкин, алкен, амины или альтернативные функциональные группы, которые дополнительно могут вступать в реакцию с образованием гомодимеров по механизму опосредуемого медью сочетания алкин, метатезиса олефинов, образования мочевины или сульфамида. Мономеры Формул 6.3, 6.4, 6.5 и 6.6 моггут вступать в реакцию друг с другом с образованием гетеродимеров Формул 6.8, 6.9 и 6.10, например, если X содержит амин, a Υ содержит карбоновую кислоту, сульфонил хлорид, изоцианат, которые могут образовывать амид, сульфонамид или мочевину; или например, если X представляет собой алкин, a Υ представляет собой азид, которые могут образовывать триазол; или если X содержит гидрокси, a Υ содержит галоген, которые могут образовывать эфир. Химические реакции и реагенты не ограничены перечисленными выше, но включают также другие комбинации, известные специалистам в данной области.
Промежуточное соединение I:
(4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)метил)амино)пропанамидо)8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновая кислота
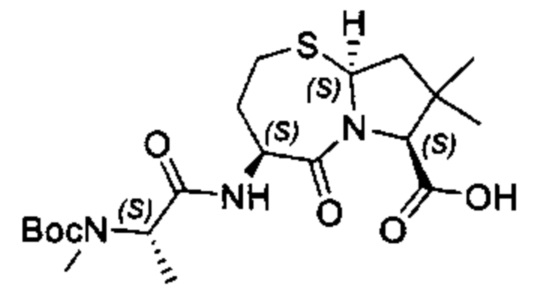
Этап 1: (E)-1-(2-нитростирил)пирролидин
К раствору 1-метил-2-нитробензола (50 г, 365 ммоль) в N,N-диметилформамиде (200 мл) добавляли пирролидин (31.1 г, 438 ммоль) и 1,1-диметокси-N,N-диметилметанамин (52.2 г, 438 ммоль). Полученную смесь перемешивали при 60°С. Через 5 ч температуру повышали до 80°С и перемешивали в течение 17 ч. После охлаждения до комнатной температуры смесь разделяли между метил-трет-бутиловым эфиром (500 мл) и водой (1 л). Водную фазу отделяли и экстрагировали метил-трет-бутиловым эфиром (500 мл). Объединенные органические слои промывали солевым раствором, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали, в результате чего получали (E)-1-(2-нитростирил)пирролидин (80 г, неочищенный) в виде темно-красного масла. Его использовали на следующем этапе без дальнейшей очистки.
Этап 2: 1-(2,2-диметоксиэтил)-2-нитробензол
К раствору (E)-1-(2-нитростирил)пирролидина (80 г, неочищенного) в метаноле (600 мл) медленно добавляли триметилхлорсилан (59.5 г, 550 ммоль). Смесь нагревали до температуры дефлегмации в течение 24 ч. После этого раствору давали остыть до комнатной температуры и концентрировали под вакуумом с получением остатка. Его разделяли между этилацетатом (500 мл) и 5% водным раствором лимонной кислоты (800 мл). Водный слой экстрагировали этилацетатом (2×300 мл). Объединенные органические слои промывали 5% водным раствором бикарбоната натрия (300 мл), а затем солевым раствором. Неочищенный продукт сушили при помощи безводного сульфата натрия, фильтровали и концентрировали, в результате чего получали 1-(2,2-диметоксиэтил)-2-нитробензол (79 г, неочищенный) в виде темно-красного масла, которое использовали на следующем этапе без дальнейшей очистки.
Этап 3: 2-(2,2-диметоксиэтил)анилин
К раствору 1-(2,2-диметоксиэтил)-2-нитробензола (79 г, 374.4 ммоль) в метаноле (1L) добавляли палладий на активированном угле (17.6 г). Смесь перемешивали в атмосфере водорода при 50 фунтов/кв. дюйм при комнатной температуре в течение 17 ч. Смесь фильтровали через диатомит и споласкивали метанолом. Фильтрат концентрировали, в результате чего получали неочищенный продукт. Неочищенный продукт растворяли в метил трет-бутиловом эфире (500 мл) и фильтровали. Фильтрат концентрировали, в результате чего получали 2-(2,2-диметоксиэтил)анилин (60 г, 331.5 ммоль, 88.5% выход) в виде темно-красного масла. Его использовали для следующего этапа без дальнейшей очистки. 1H-ЯМР (400 МГц, ДМСО-d6) δ ppm 6.98-6.89 (m, 2H), 6.63 (dd, J=7.9, 1.1 Гц, 1H), 6.51 (td, J=7.4, 1.2 Гц, 1Н), 4.80 (s, 2H), 4.55 (t, J=5.6 Гц, 1H), 3.25 (s, 6H), 2.72 (d, J=5.6 Гц, 2H).
Этап 4: N-(2-(2,2-диметоксиэтил)фенил)формамид
К раствору 2-(2,2-диметоксиэтил)анилина (60 г, 331.5 ммоль) и этилформиата (36.8 г, 497.2 ммоль) в сухом тетрагидрофуране (400 мл) медленно добавляли раствор 1М бис(триметилсилил)амида лития (597 мл, 597 ммоль). Смесь перемешивали при комнатной температуре в течение 12 ч, а затем нагревали с обратным холодильником в течение 18 ч. После этого добавляли насыщенный хлорид аммония (200 мл) и экстрагировали смесь этилацетатом (3×200 мл). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (6:1 об./об.)], в результате чего получали N-(2-(2,2 диметоксиэтил)фенил)формамид (58 г, 277.5 ммоль, 83.7% выход) в виде темно-красного масла. ЖХМС (2.5 мин, муравьиная кислота): Вр. уд. = 1.354 мин, m/z: 209.1 [M+1]+, 231.9 [M+Na]+.
Этап 5: 1-(2,2-диметоксиэтил)-2-изоцианобензол
К раствору N-(2-(2,2 диметоксиэтил)фенил)формамида (58 г, 277.5 ммоль) в дихлорметане (500 мл) при 0°С добавляли триэтиламин (143 г, 1415.3 ммоль), а затем оксихлорид фосфора (63.9 г, 416.3 ммоль). Смесь нагревали до комнатной температуры и перемешивали в течение 2 ч. После этого ее вливали в насыщенный водный бикарбонат натрия (300 мл) и экстрагировали дихлорметаном (3×200 мл). Органические слои сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (20:1 об./об.)], в результате чего получали 1-(2,2-диметоксиэтил)-2-изоцианобензол (42 г, 218.5 ммоль, 79.2% выход) в виде бледно-коричневого масла. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 7.57-7.27 (m, 4H), 4.61 (t, J=5.6 Гц, 1H), 3.27 (s, 6H), 3.00 (d, J=5.6 Гц, 2Н). ЖХМС (2.5 мин, муравьиная кислота): Вр. уд.=1.455 мин, m/z: 192.0 (M+1)+.
Этап 6: 4,4-диметокси-2,2-диметилбутаннитрил
К раствору диизопропиламинв (89.4 мл, 682 ммоль) в тетрагидрофуране (1L) при -10°С в атмосфере азота добавляли раствор 2.4 М н-бутиллития в гексане (288 мл, 682 ммоль). Через 30 мин смесь охлаждали до -78°С и добавляли раствор 4,4-диметоксибутаннитрила (40 г, 310 ммоль) в тетрагидрофуране (30 мл). Через 1 ч очень медленно добавляли метилйодид (42.4 мл, 682 ммоль). Смеси давали нагреться до комнатной температуры и перемешивали в течение ночи. После этого ее гасили насыщенным водным раствором хлорида аммония и экстрагировали этилацетатом (3×400 мл). Органические слои сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (30:1 об./об.)], в результате чего получали 4,4-диметокси-2,2-диметилбутаннитрил (35 г, 223 ммоль, 71.9% выход) в виде бледно-желтого масла. 1Н-ЯМР (400 МГц, CDCl3) δ ppm 4.60 (t, J=5.4 Гц, 1H), 3.36 (s, 6H), 1.83 (d,J=5.4 Гц, 2Н), 1.39 (s, 6H).
Этап 7: 4,4-диметокси-2,2-диметилбутаналь
К раствору 4,4-диметокси-2,2-диметилбутаннитрила (27 г, 172 ммоль) в дихлорметане (800 мл) медленно добавляли раствор 1М диизобутилалюминия гидрида в гексане (189 мл, 189 ммоль) при -78°С в течение 3.5 ч. После этого, смесь нагревали до комнатной температуры и гасили насыщенным водным хлоридом аммония (400 мл) и солью Рошели (400 мл). Водную фазу экстрагировали дихлорметаном (2×400 мл). Объединенные органические слои промывали солевым раствором (400 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (30:1 об./об.)], в результате чего получали 4,4-диметокси-2,2-диметилбутаналь (13 г, 81.1 ммоль, 47.1% выход) в виде бесцветного масла. ЖХМС (2.5 мин, муравьиная кислота): Вр. уд. = 1.453 мин, m/z: 182.9 (M+Na)+.
Этап 8: (4S,9aS)-4-амино-7-(1Н-индол-1-карбонил)-8,8-диметилгексагидропирроло[2,1-b][1,3]тиазепин-5(2Н)-он
а. Смесь N-(трет-бутоксикарбонил)-S-тритил-L-гомоцистеина (11.8 г, 24.7 ммоль), 1-(2,2-диметоксиэтил)-2-изоцианобензола (с Этапа 5) (4.5 г, 23.5 ммоль), 4,4-диметокси-2,2-диметилбутаналь (с Этапа 7) (4.5 г, 28.2 ммоль) и 7 М аммиака в метаноле (10 мл) в 2,2,2-трифторэтаноле (150 мл) перемешивали при 100°С в течение 2 ч. После этого смесь гасили 1 М вод. раствором гидроксида натрия (200 мл) и экстрагировали этилацетатом (3×150 мл). Объединенные органические фазы сушили с использованием безводного сульфата натрия, фильтровали и концентрировали, в результате чего получали неочищенный трет-бутил((2S)-1-((1-((2-(2,2-диметоксиэтил)фенил)амино)-5,5-диметокси-3,3-диметил-1-оксопентан-2-ил)амино)-1-оксо-4-(тритилтио)бутен-2-ил)карбамат (26 г, неочищенный) в виде коричневого твердого вещества. Его использовали для следующего этапа без какой-либо дополнительной очистки.
b. После этапа к раствору неочищенного трет-бутил((2S)-1-((1-((2-(2,2-диметоксиэтил)фенил)амино)-5,5-диметокси-3,3-диметил-1-оксопентан-2-ил)амино)-1-оксо-4-(тритилтио)бутен-2-ил)карбамата (26 г, неочищенный) в дихлорметане (200 мл) медленно добавляли трифторуксусную кислоту (20 мл). Смесь перемешивали при 40°С в течение 2 ч. Растворитель концентрировали, а неочищенный продукт очищали хроматографией на силикагеле [дихлорметан/метанол (25:1 об./об.)], в результате чего получали (4S,9aS)-4-амино-7-(1Н-индол-1-карбонил)-8,8-диметилгексагидропирроло[2,1-b][1,3]тиазепин-5(2Н)-он (7.5 г, 21.0 ммоль) в виде твердого вещества песочного цвета. ЖХМС (2.5 мин, муравьиная кислота): Вр. уд. = 1.361 мин, m/z: 357.9 (M+1)+.
Этап 9: трет-бутил((S)-1-(((4S,7S,9aS)-7-(1H-индол-карбонил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат
К раствору (4S,9aS)-4-амино-7-(1Н-индол-1-карбонил)-8,8-диметилгексагидропирроло[2,1-b][1,3]тиазепин-5(2Н)-он (14 г, 39.2 ммоль), N-(трет-бутоксикарбонил)-N-метил-L-аланина (8.76 г, 43.1 ммоль), 1-гидроксибензотриазола (5.82 г, 43.1 ммоль) и 4-метилморфолина (11.88 г, 117.6 ммоль) в тетрагидрофуране (400 мл) добавляли N-(3-диметиламинопропил-N'-этилкарбодиимид гидрохлорид (8.27 г, 43.1 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч. После этого ее гасили насыщенным водным раствором бикарбоната натрия (300 мл) и экстрагировали этилацетатом (3×150 мл). Объединенные органические слои промывали солевым раствором (200 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (от 2:1 об./об. до 1:1 об./об.)], в результате чего получали трет-бутил ((S)-1-(((4S,7R,9aS)-7-(1H-индол-1-карбонил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат (5.2 г, 9.6 ммоль, 24.5% выход) в виде твердого вещества песочного цветка и целевой продукт трет-бутил((S)-1-(((4S,7S,9aS)-7-(1Н-индол-1-карбонил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат (5.0 г, 9.2 ммоль, 23.5% выход) в виде бледно-желтого твердого вещества. ЖХМС (2.5 мин, муравьиная кислота): Вр. уд. = 1.740 мин, m/z: 564.8 (M+1)+.
Этап 10: (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновая кислота
К раствору трет-бутл ((S)-1-(((4S,7S,9aS)-7-(1Н-индол-1-карбонил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1 -оксопропан-2-ил)(метил)карбамата (3.9 г, 7.19 ммоль) в метаноле (60 мл) добавляли 1 М водный раствор гидроксида натрия (60 мл). Смесь перемешивали при комнатной температуре в течение 24 ч. Летучий растворитель удаляли при пониженном давлении. Оставшуюся водную фазу промывали этилацетатом (50 мл) и доводили рН водной фазу лимонной кислотой до рН 3. Водную фазу экстрагировали этилацетатом (3×60 мл). Объединенные органические фазы промывали солевым раствором (100 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (1:2 об./об.)], в результате чего получали (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновую кислоту (2 г, 4.51 ммоль, 62.7% выход) в форме белого твердого вещества. ЖХМС (2.5 мин, муравьиная кислота): Вр. уд. = 1.493 мин, m/z: 443.9 (М+1). 1Н-ЯМР (400МГц, CDCl3) δ ppm 12.57 (s, 1H), 7.75 (d, J=6.6 Гц, 1H), 5.46 (t, J=7.2 Гц. 1H), 4.64-4.60 (m, 1H), 4.43 (s, 1H), 3.98 (s, 1H), 3.17-3.11 (m, 1H), 2.88 (dd, J=14.6, 3.0 Гц, 1H), 2.74 (s, 3H), 2.27 (dd, J=12.9, 7.5 Гц, 1H), 2.12 - 2.10 (m, 1H), 1.80 (dd, J=13.0, 7.1 Гц, 1H), 1.74-1.68(m, J=11.2 Гц, 1H),1.40(s,9H), 1.25 (d, J=7.1 Гц, 3H), 1.09 (d, J=10.6 Гц, 6Н).
Промежуточное соединение II:
(4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8,9а-триметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновая кислота
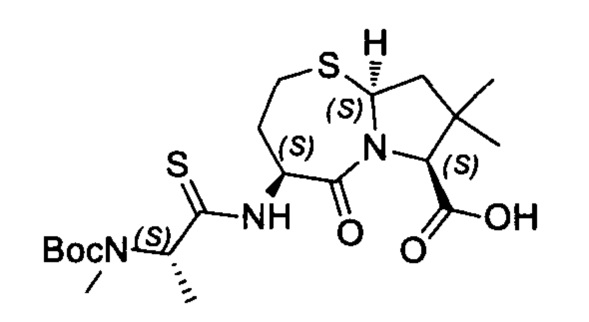
Этап 1: трет-бутил(S)-(1-((2-амино-5-нитрофенил)амино)-1-оксопропан-2-ил)(метил)карбамат
К раствору N-(трет-бутоксикарбонил)-N-метил-L-аланина (20.3 г, 100 ммоль) в сухом тетрагидрофуране (1.2 л) добавляли 4-метилморфолин (20.2 г, 200 ммоль). Смесь перемешивали при -20°С в течение 10 мин, после чего по каплям добавляли изобутилкарбонохлоридат (13.6 г, 100 ммоль). Смесь перемешивали при -15°С в течение 2 ч и нагревали до комнатной температуры в течение ночи. Смесь разбавляли этилацетатом (1.6 л) и промывали последовательно 1М вод. раствором динатрия гидрофосфата (1 л), 5% вод. раствором бикарбоната натрия (1 л) и солевым раствором (1 л). Органическую фазу сушили при помощи безводного сульфата натрия, фильтровали и концентрировали, в результате чего получали (S)-трет-бутил(1-((2-амино-5-нитрофенил)амино)-1-оксопропан-2-ил)(метил)карбамат (30 г, 88.7 ммоль, 88.7% выход) в виде темно-красного масла. ЖХМС (2.5 мин, муравьиная кислота): Вр. уд. = 1.537 мин, m/z: 360.9 (М+Na)+.
Этап 2: трет-бутил(S)-(1-((2-амино-5-нитрофенил)амино)-1-тиоксопропан-2-ил)метил)карбамат
К раствору сульфида фосфора (V) (24.4 г, 110 ммоль) в сухом тетрагидрофуране (1.6 л) добавляли карбонат натрия (5.8 г, 55 ммоль). Смесь перемешивали в течение 1 ч при комнатной температуре, которую затем снижали до 0°С. К реакционной смеси добавляли трет-бутил(S)-(1-((2-амино-5-нитрофенил)амино)-1-оксопропан-2-ил)(метил)карбамат (40 г, 110 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч. Растворитель концентрировали под вакуумом и разбавляли этилацетатом (1 л), после чего промывали 5% вод. раствором бикарбоната натрия (500 мл) и солевым раствором (500 мл). Органический слой сушили при помощи безводного сульфата натрия, фильтровали и концентрировали, в результате чего получали продукт - трет-бутил (S)-(1-((2-амино-5-нитрофенил)амино)-1-тиоксопропан-2-ил)(метил)карбамат (33 г, 93.2 ммоль, 84.7% выход) в виде оранжевого твердого вещества. ЖХМС (2.5 мин, муравьиная кислота): Вр. уд. = 1.602 мин, m/z: 376.8 (M+Na)+.
Этап 3; трет-бутил(S)-метил(1-(6-нитро-1Н-бензо[d][1,2,3]триазол-1-ил)-1-тиоксопропан-2-ил)карбамат
К раствору трет-бутил(S)-(1-((2-амино-5-нитрофенил)амино)-1-тиоксопропан-2-ил)(метил)карбамата (30 г, 84.6 ммоль) в сухом тетрагидрофуране (250 мл) и уксусной кислоте (250 мл) добавляли нитрит натрия (9 г, 130.4 ммоль). Смесь перемешивали при 0°С в течение 1.5 ч. Ее промывали последовательно водой (3×500 мл), 5% вод. раствором бикарбоната натрия (2×500 мл) и солевым раствором (500 мл). Органический слой сушили при помощи безводного сульфата натрия, фильтровали и концентрировали, в результате чего получали трет-бутил(S)-метил(1-(6-нитро-1Н-бензо[d][1,2,3]триазол-1-ил)-1-тиоксопропан-2-ил)карбамат (20 г, 54.8 ммоль, 64.8% выход) в виде коричневого масла, которое использовали для следующего этапа без дальнейшей очистки.
Этап 4: трет-бутил((S)-1-(((4S,7S,9aS)-7-(1Н-индол-1-карбонил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-тиоксопропан-2-ил)(метил)карбамат
К раствору (4S,9aS)-4-амино-7-(1Н-индол-1-карбонил)-8,8-диметилгексагидропирроло[2,1-b][1,3]тиазепин-5(2Н)-она (15 г, 42 ммоль) и трет-бутил(S)-метил(1-(6-нитро-1Н-бензо[d][1,2,3]триазол-1-ил)-1-тиоксопропан-2-ил)карбамата (20 г, 54.7 ммоль) в дихлорметане (400 мл) добавляли триэтиламин (8.6 г, 84.6 ммоль) при 0°С. Смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт очищали хроматографией на силикагеле (этилацетат /петролейный эфир=1/15), в результате чего получали трет-бутил((S)-1-(((4S,7S,9aS)-7-(1Н-индол-1-карбонил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1 -тиоксопропан-2-ил)(метил)карбамат) (3.0 г, 5.37 ммоль, 25.5% выход) в виде желтого масла и целевой диастереомер трет-бутил((S)-1-(((4S,7S,9aS)-7-(1Н-индол-1-карбонил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-тиоксопропан-2-ил)(метил)карбамат (3.9 г, 6.98 ммоль, 29.4% выход) в виде желтого масла. ЖХМС (2.5 мин, муравьиная кислота): Вр. уд. = 1.905 мин, m/z: 580.8 (M+Na)+.
Этап 5: (4S,7S,9aS)-4-(2-((трет-6утоксикарбонил)(метил)амино)этантиоамидо)-8,8,9a-триметил-5-оксооктагидропирроло[2,1-b] [1,3]тиазепин-7-карбоновая кислота
К раствору трет-бутил(2-(((4S,7S,9aS)-7-(1Н-индол-1-карбонил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-2-тиоксоэтил)(метил)карбамата (1.5 г, 2.69 ммоль) в метаноле медленно добавляли 1М вод. раствор гидроксида натрия (60 мл). Смесь перемешивали при комнатной температуре в течение ночи. Летучий растворитель удаляли при пониженном давлении, а оставшийся раствор экстрагировали этилацетатом (50 мл). рН водного слов доводили 20% водным раствором лимонной кислоты до рН 4, а затем экстрагировали этилацетатом (3×100 мл). Органические слои объединяли, промывали солевым раствором (100 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали, в результате чего получали (4S,7S,9aS)-4-(2-((трет-бутоксикарбонил)(метил)амино)этантиоамидо)-8,8,9а-триметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновую кислоту (1 г, 21.8 ммоль, 81% выход) в виде коричневого масла. ЖХМС (2.5 мин, муравьиная кислота): Вр. уд. = 1.632 мин, m/z: 481.8 (М+Na)+.
Пример 1
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(метиленбис(4,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
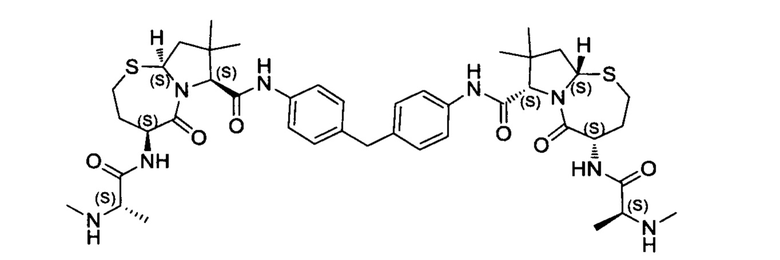
Этап 1: N, N'-(метиленбис(4,1-фенилен)) диформамид
К раствору 4,4'-метилендианилина (5 г, 25.2 ммоль) в толуоле (40 мл) добавляли муравьиную кислоту (4.64 г, 100.8 ммоль). Смесь перемешивали при 110°С в течение 2 часов. Реакционную смесь концентрировали. Неочищенный продукт промывали дихлорметаном и фильтровали с получением целевого продукта N,N'-(метиленбис(4,1-фенилена))диформамид (4.7 г, 18,5 ммоль, выход 73.4%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.165 минуты, m/z: 255.1 (М+1)+.
Этап 2: бис(4-изоцианофенил)метан
К раствору N,N'-(метиленбис(4,1-фенилена))диформамида (4.7 г, 18.5 ммоль) в дихлорметане (150 мл) при 0°С добавляли триэтиламин (19 г, 188.7 ммоль), после чего добавляли оксихлорид фосфора (8.5 г, 55.4 ммоль). Смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. Реакционную смесь вливали в насыщенный водный раствор бикарбоната натрия (100 мл) и экстрагировали дихлорметаном (3×60 мл). Органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [дихлорметан/метанол (25:1 об./об.)] в результате чего получали бис(4-изоцианофенил)метан (1.6 г, 7.3 ммоль, выход 39.6%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.715 минуты, m/z: 218.9 (M+1)+.
Этап 3: (4S,4S,9aS,9a'S)-N,N'-(метиленбис(4,1-фенилен))бис(4-амино-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
Смесь N-(трет-бутоксикарбонил)-S-тритил-L-гомоцистеина (2.4 г, 5.04 ммоль), бис(4-изоцианофенил)метана (500 мг, 2.29 ммоль), 4,4-диметокси-2,2-диметилбутаналя (954 мг, 5,95 ммоль) и 7 М аммония в метаноле (2 мл, 14 ммоль) в 2,2,2-трифторэтаноле (10 мл) перемешивали при 80°С в течение 30 минут в микроволновых условиях. Смесь гасили 1 М водного гидроксида натрия (20 мл) и экстрагировали этилацетатом (3×15 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, в результате чего получали неочищенный продукт ди-трет-бутил((2S,2'S)-((((метиленбис(4,1-фенилен))бис(азандиил))бис(5,5-диметокси-3,3-диметил-1-оксопентан-1,2-диил))бис(азандиил))бис(1-оксо-4-(тритилтио)бутан-1,2-диил))дикарбамат (2.0 г, неочищенный) в виде коричневого твердого вещества. Его использовали на следующем этапе без дополнительной очистки. К раствору неочищенного ди-трет-бутил((2S,2'S)-((((метиленбис(4,1-фенилен))бис(азандиил))бис(5,5-диметокси-3,3-диметил-1-оксопентан-1,2-диил))бис(азандиил))бис(1-оксо-4-(тритилтио)бутан-1,2-диил))дикарбамата (2 г, неочищенный) в дихлорметане (20 мл) медленно добавляли трифторуксусную кислоту (5 мл). Смесь перемешивали при 40°С в течение 2 часов. Реакционную смесь концентрировали и очищали хроматографией на силикагеле [дихлорметан/метанол (6:1 об./об.)], в результате чего получали (4S,4'S,9aS,9a'S)-N,N'-(метиленбис(4,1-фенилен))бис(4-амино-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) (500 мг, 0.74 ммоль) в виде песчаного твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.263 минуты, m/z: 678.8 (М+1)+.
Этап 4: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((метиленбис(4,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
К раствору (4S,4'S,9aS,9a'S)-N,N'-(метиленбис(4,1-фенилен))бис(4-амино-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) (550 мг, 0.810 ммоль), N-(mpem-бутоксикарбонил)-N-метил-L-аланина (362 мг, 1.782 ммоль), 1-гидроксибензотриазола (241 мг, 1.782 ммоль) и 4-метилморфолина (491 мг, 4.860 ммоль) в тетрагидрофуране (30 мл) добавляли N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид (342 мг, 1.782 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов. Смесь гасили насыщенным вод. раствором бикарбоната натрия (25 мл) и экстрагировали этилацетатом (3×15 мл). Объединенные органические фазы промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [дихлорметан/метанол (25:1 об./об.)], в результате чего получали нечистый продукт, который дополнительно очищали с помощью хиральной-ВЭЖХ, в результате чего получали целевой продукт ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((метиленбис(4,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (55 мг, 0.052 ммоль, выход 6.4%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.828 минуты, m/z: 425 [(М - 2 Вос)+2]+/2,
Этап 5: (4S,4'S,7S,7'S,9aS,9a'S)-N.N'-(метиленбис(4,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((метиленбис(4,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (55 мг, 0.052 ммоль) в дихлорметане (6 мл) добавляли 4 н. хлорид водорода в 1,4-диоксане (1.5 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли при пониженном давлении и твердое вещество сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(метиленбис(4,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид (43 мг, 0.046 ммоль, выход 88.5%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 10.08 (s, 2H), 9.27-9.24 (m, 2H), 8.87-8.84 (m, 2H), 8.80(d, 3=6.8 Гц, 2Н), 7.52(d, J=8.0 Гц, 2H), 7.14(d, J=8.0 Гц, 2H), 5,47 (m, J=7.8 Гц, 2H), 4.71(t, J=9.4 Гц, 2H), 4.26 (s, 2H), 3.84 (s, 2H), 3.71-3.65 (m, 2H), 3.17(t, J=12.4 Гц, 2Н), 2.95-2.91 (m, 2H), 2.48-2.47 (m, 6H), 2.23-2.18 (m, 2H), 2.14-2.10 (m, 2H), 1.96-1.90 (m, 2H), 1.85-1.76 (m, 2H), 1.42 (s, 4H), 1.39(d, J=7.2 Гц, 6H), 1.09 (m, 6H), 1.02 (m, 6H). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.268 минуты, т/г: 848.9 (М+1)+.
Пример 2:
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(метиленбис(4,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид
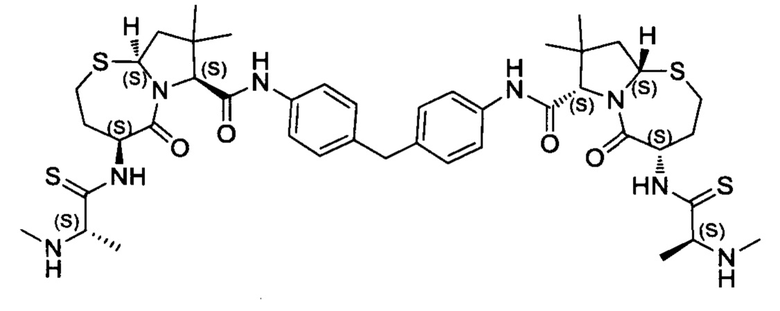
Этап 1: ди-трет-бутил((2S,2'S)-(4S,4'S,7S,7'S,9aS,9a'S)-(((метиленбис(4,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2диил))бис(метилкарбамат)
К раствору (4S,4'S,9aS,9a'S)-N,N'-(метиленбис(4,1-фенилен))бис(4-амино-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) (Пример 1, Этап 3) (260 мг, 0.38 ммоль) в дихлорметане (10 мл) при 0 °С добавляли триэтиламин (116.3 мг, 1,15 ммоль). Через 10 минут добавляли раствор трет-бутил(S)-метил(1-(6-нитро-1Н-бензо[d][1,2,3]триазол-1-ил)-1-тиоксопропан-2-ил)карбамата (321.9 мг, 0,88 ммоль) в дихлорметане (5 мл). Смесь перемешивали при комнатной температуре в течение еще 2 часов. Растворитель удаляли при пониженном давлении, в результате чего получали желтый сироп. Остаток очищали с помощью тонкослойной хроматографии [этилацетат /петролейный эфир (1:4 об./об.)], в результате чего получали желтое масло (200 мг, 0.18 ммоль), которое дополнительно очищали с помощью хиральной-ВЭЖХ, в результате чего получали титульный продукт (45 мг, 0.04 ммоль, выход 10.9%). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.998 минуты, m/z: 980.6 (M - Вос+1)+.
Этап 2: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(метиленбис(4,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((метиленбис(4,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (45 мг, 0.042 ммоль) в диэтиловом эфире (2 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Смесь концентрировали при пониженном давлении и сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(метиленбис(4,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (22 мг, 0.025 ммоль, выход 55%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 11.08-11.96 (m, 2H), 10.15 (s, 1H), 9.89 (s, 1H), 9.64 (s, 1H), 8.66 (s, 1H), 8.42 (s, 1H), 7.52(d, J=8.0 Гц, 2H), 7.14(d, J=8.0 Гц, 2Н), 5.43(t, J=7.6 Гц, 2H), 5.16-5.08 (m, 2H), 4.29-4.20 (m, 4Н), 3.84 (s, 2H), 3.72-3.67 (m. 2H), 3.22-3.16 (m, 2H), 3.01-2.98 (m, 2H), 2.49 (s, 6H), 2.25-2.21 (m, 4H), 2.00-1.92 (m, 4H), 1.46-1.43 (m, 6H), 1.11 (m, 6H), 1.02 (m, 6H). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.298 минуты, m/z: 880.6 (M+1)+.
Пример 3
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(2,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид

Этап 1: 2,2'-(этан-1,2-диил)дианилин
К раствору 1,2-бис(2-нитрофенил)этана (9.5 г, 34.9 ммоль) в этаноле (200 мл) добавляли палладий на активированный уголь (1 г). Смесь перемешивали в атмосфере водорода (1 атм) при комнатной температуре в течение 5 часов. Катализатор фильтровали и фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (1:1 об./об.)], в результате чего получали 2,2'-(этан-1,2-диил)дианилин (5 г, 23.6 ммоль, выход 67.6%) в виде желтого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.336 минут, 212.9 (M+1)+.
Этап 2: N.N'-(этан-1,2-диилбис(2,1-фенилен))диформамид
К раствору 2,2'-(этан-1,2-диил)дианилина (5 г, 23.6 ммоль) в толуоле (100 мл) добавляли муравьиную кислоту (4.3 г, 94,4 ммоль). Смесь перемешивали при кипячении с обратным холодильником в течение ночи. Смесь концентрировали при пониженном давлении, в результате чего получали неочищенный продукт, который промывали петролейным эфиром, в результате чего получали N,N'-(этан-1,2-диилбис(2,1-фенилен))диформамид (5 г, 18.7 ммоль, выход 79.2%) в виде белого твердого вещества.
Этап 3: 1,2-бис(2-изоцианофенил)этан
К раствору N,N'-(этан-1,2-диилбис(2,1-фенилен))диформамида (2 г, 7.46 ммоль) в дихлорметане 50 мл) при 0°С добавляли триэтиламин (7.5 г, 74.6 ммоль), после чего добавляли оксихлорид фосфора (3.4 г, 22.4 ммоль). Смесь нагревали до комнатной температуры и перемешивали в течение 4 часов. Затем реакционную смесь выливали в насыщенный вод. раствор бикарбоната натрия (50 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (4:1 об./об.)], в результате чего получали 1,2-бис(2-изоцианофенил)этан (1.5 г, 6.47 ммоль, выход 86.7%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.763 минуты, m/z: 233.0 (M+1)+.
Этап 4: (4S,4'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(2,1-фенилен))бис(4-амино-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)
Смесь N-(трет-бутоксикарбонил)-S-тритил-L-гомоцистеина (4,1 г, 8.6 ммоль), бис(4-изоцианофенил)метана (1 г, 4.3 ммоль), 4,4-диметокси-2,2-диметилбутаналя (1.45 г, 9.0 ммоль) и 7М аммиака в метаноле (2.4 мл, 16.8 ммоль) в 2,2,2-трифторэтаноле (20 мл) перемешивали при 80°С в течение 30 минут в микроволновых условиях. Реакцию гасили 1М раствором гидроксида натрия (40 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного ди-трет-бутил((2S,2'S)-((((этан-1,2-диилбис(2,1-фенилен))бис(азандиил))бис(5.5-диметокси-3,3-диметил-1-оксопентан-1,2-диил))бис(азандиил))бис(1-оксо-4-(тритилтио)бутан-1,2-диил))дикарбамата (4.0 г, неочищенный) в виде коричневого твердого вещества. К раствору неочищенного ди-трет-бутил((2S,2'S)-((((этан-1,2-диилбис(2,1-фенилен))бис(азандиил))бис(5,5-диметокси-3,3-диметил-1-оксопентан-1,2-диил))бис(азандиил))бис(1-оксо-4-(тритилтио)бутан-1,2-диил))дикарбамата (4 г, неочищенный) в дихлорметане (50 мл) медленно добавляли трифторуксусную кислоту (10 мл). Смесь перемешивали при 40 °С в течение 2 часов. Реакционную смесь концентрировали и очищали хроматографией на силикагеле [дихлорметан/метанол (6:1 об./об.)], в результате чего получали (4S,4'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(2,1-фенилен))бис(4-амино-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) (2.3 г, 3.32 ммоль, выход 77.2%) в виде желтого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.317 минуты, m/z: 692.9 (M+1)+.
Этап 5: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((этан-1,2-диилбис(2,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
К раствору(4S,4'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(2,1-фенилен))бис(4-амино-8,8-диметил-5-оксооктагадропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) (1 г, 1.44 ммоль), N-(трет-бутоксикарбонил)-N-метил-L-аланина (580 мг, 2.88 ммоль), 1-гидроксибензотриазола (430 мг, 3.17 ммоль) и 4-метилморфолина (580 мг, 5.76 ммоль) в тетрагидрофуране (30 мл) добавляли N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид (580 мг, 3.04 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов. Реакцию гасили насыщенным вод. раствором бикарбоната натрия (25 мл) и экстрагировали этилацетатом (3×15 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (1:1 об./об.)], в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((этан-1,2-диилбис(2,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (1.2 г, 1.13 ммоль, выход 78.5%) в виде бежевого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.910 минуты, m/z: 431.9[(M-2Boc)+2]+/2.
Этап 6: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(2,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((этан-1,2-диилбис(2,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (90 мг, 0.085 ммоль) в метаноле (10 мл) добавляли 4 н. хлорид водорода в 1,4-диоксане (5 мл). Смесь перемешивали при комнатной температуре в течение 4 часов. Затем смесь концентрировали и кристаллизовали из эфира и метанола, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(2,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктапндропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (15 мг, 0.016 ммоль, выход 18.8%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d3) δ ppm 9.51 (s, 2H), 9.15 (s, 2H), 8.83-8.77 (m, 4H), 7.39-7.53 (m, 4H), 7.21-7.12 (m, 4H), 5.50(t, J=8.0 Гц, 2H), 4.74(t, J=9.6 Гц, 2H), 4.38 (s, 2H), 3.90-3.82 (m, 2H), 3.17-3.11 (m, 2H), 2.99-2.82 (m, 6H), 2.48 (m, 6H), 2.26-2.21 (m, 2H), 2.04-1.95 (m, 4H), 1.72-1.63 (m, 2H), 1.36(d, J=6.4 Гц, 6H), 1.14 (m, 6H), 1.12 (m, 6H). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.293 минуты, m/z: 862.9 (M+1)+.
Пример 4
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(2,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
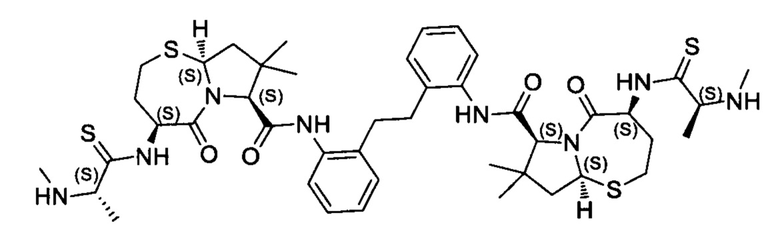
Этап 1: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((этан-1,2-диилбис(2,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат)
К раствору (4S,4'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(2,1-фенилен))бис(4-амино-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) (300 мг, 0.44 ммоль) в дихлорметане (20 мл) при 0 °С добавляли триэтиламин (132 мг, 1.31 ммоль) и трет-бутил(S)-метил(1-(6-нитро-1Н-бензо[d][1,2,3]триазол-1-ил)-1-тиоксопропан-2-ил)карбамат (400 мг, 1.09 ммоль). Смесь нагревали до комнатной температуры и перемешивали в течение ночи. Смесь промывали насыщенным вод. раствором бикарбоната натрия (15 мл) и солевым раствором (15 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали препаративной ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((этан-1,2-диилбис(2,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (50 мг, 0.046 ммоль, выход 10.5%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 2.173 минуты, m/z: 1116.6 (M+Na)+.
Этап 2: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(2,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((этан-1,2-диилбис(2,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (50 мг, 0.046 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлорид водорода в 1,4-диоксане (4 мл). Смесь перемешивали при комнатной температуре в течение 4 часов. Смесь концентрировали и сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(2,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид (35 мг, 0.036 ммоль, выход 78.3%) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 10.85(d, J=6.8 Гц, 2Н), 9.59 (s, 2Н), 9.49 (s, 2H), 8.59 (s, 2H), 7.39-7.33 (m, 4H), 7.21-7.11 (m, 4H), 5.45(t, J=8.0 Гц, 2H), 5.18(t, J=8.2 Гц, 2H), 4.41 (s, 2H), 4.25-4.23 (m, 2H), 3.72-3.65 (m, 2H), 3.51-3.47 (m, 2H), 3.16(t, J=12.4 Гц, 2H), 2.93-2.88 (m, 2H), 2.50 (s, 6H), 2.28-2.16 (m, 4H), 2.06-2.00 (m, 2H), 1.84-1.75 (m, 2H), 1.41(d, J=6.4 Гц, 6Н), 1.14 (m, 12H). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.389 минуты, m/z: 894.8 (М+1)+.
Пример 5
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(4,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b](1,3]тиазепин-7-карбоксамида) дитрифторацетат
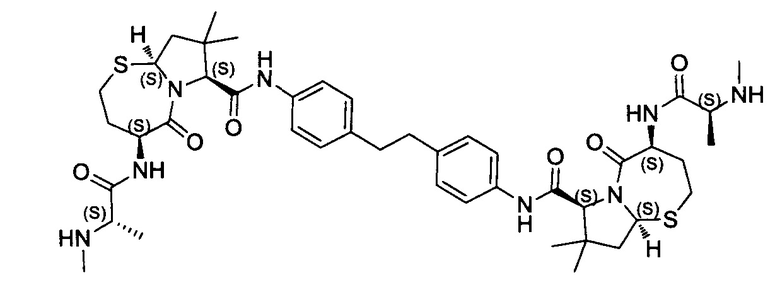
Этап 1: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((этан-1,2-диилбис(4,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1 -оксопропан-1,2-диил))бис(метилкарбамат)
К раствору (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение I) (230 мг, 0.519 ммоль) в 1,2-дихлорэтане (20 мл) добавляли N-этоксикарбонил-2-этокси-1,2-дигидрохинолинил (EEDQ) (175 мг, 0.709 ммоль), N,N-диизопропилэтиламин (120 мг, 0.930 ммоль) и 4,4'-(этан-1,2-диил)дианилин (50 мг, 0.236 ммоль). Смесь перемешивали при 50°С в течение ночи. Растворитель удаляли при пониженном давлении с последующей хроматографией на силикагеле [петролейный эфир/этилацетат (2:1 об./об.)], в результате чего получали целевой продукт ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((этан-1,2-диилбис(4,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (100 мг, 0.094 ммоль, выход 39.8%) в виде желтого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.828 минуты, m/z: 432.0 {[(М-2 Вос)+2]/2}+.
Этап 2: (4S,4'S,7S,7'S,9aS,9а'S)-N,N'-(этан-1,2-диилбис(4,1-фенилен))бис(8,8-диметил-4-((8)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дитрифторацетат
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((этан-1,2-диилбис(4,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (100 мг, 0.094 ммоль) в метаноле (5 мл) добавляли 4 н. хлорид водорода в метаноле (5 мл). Смесь перемешивали при комнатной температуре в течение 6 часов. Смесь концентрировали и очищали препаративной-ВЭЖХ, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(4,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)дитрифторацетат (64 мг, 0.059 ммоль, выход 62.8%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 9.99 (s, 2H), 8.94 (s, 2H), 8.80-8.78 (m, 4H), 7.50(d, J=8.0 Гц, 4H), 7.17(d, J=8.0 Гц, 4Н), 5.49(t, J=7.6 Гц, 2H), 4.73(t, J=9.0 Гц, 2H), 4.24 (s, 2H), 3.89-3.83 (m, 2H), 3.21-3.15 (m, 2H), 3.12-3.09 (m, 2H), 2.80 (m, 2H), 2.50 (s, 6H), 2.24-2.20 (m, 2H), 2.13-2.11 (m, 2H), 1.98-1.93 (m, 2H), 1.85-1.76 (m, 2H), 1.38(d. J=6.8 Гц, 6H), 1.10 (m, 6H), 1.03 (m, 6H). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания=1.284 минуты, m/z: 862.9 (M+1)+.
Пример 6
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(4,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид
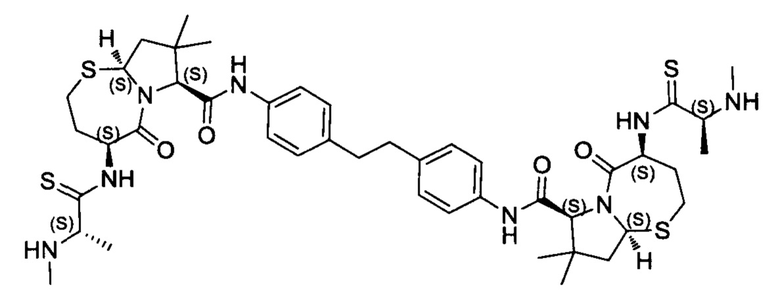
Этап 1: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((этан-1,2-диилбис(4,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандии))бис(1 -тиоксопропан-1,2-диил))бис(метилкарбамат)
К раствору (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение II) (141 мг, 0.312 ммоль) в 1,2-дихлорэтане (20 мл) добавляли N-этоксикарбонил-2-этокси-1,2-дигидрохинолинил (EEDQ) (105 мг, 0.423 ммоль), N,N-диизопропилэтиламин (72 мг, 0.564 ммоль) и 4,4'-(этан-1,2-диил)дианилин (30 мг, 0.141 ммоль). Смесь перемешивали при 50°С в течение ночи. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (2:1 об./об.)] с последующей препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((этан-1,2-диилбис(4,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (60 мг, 0.055 ммоль, выход 39.0%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 2.059 минут.
Этап 2: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(4,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-6утил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((этан-1,2-диилбис(4,1-фенилен))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (70 мг, 0.064 ммоль) в метаноле (5 мл) добавляли 4 н. хлорид водорода в метаноле (3 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(этан-1,2-диилбис(4,1-фенилен))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)дигидрохлорид (43 мг, 0.044 ммоль, выход 68.8%) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 11.04(d, J=6.0 Гц, 2H), 10.95(d, J=6.8 Гц, 2Н), 10.11-10.10 (m, 2Н), 9.80 (s, 1H), 9.56 (s, 1H), 8.64 (s, 1H), 8.43 (s, 1H), 7.51(d, J=8.0 Гц, 4H), 7.15(d, J=8.4 Гц, 4H), 5.45 (кв, J=5.6 Гц, 2Н), 5.18-5.09 (m, 2Н), 4.29 (s, 2Н), 4.24-4.21 (m, 2Н), 3.22-3.17 (m, 2Н), 3.02-2.99 (m, 2Н), 2.80 (s, 2Н), 2.50 (s, 6H), 2.26-2.22 (m, 4H), 2.02-1.91 (m, 4H), 1.46-1.43 (m, 6H), 1.12 (s, 6H), 1.04 (m, 12H). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.326 минуты, m/z: 895.1 (M+1)+.
Пример 7
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(1R,1'R,2R,2'R)-(пиперазин-1,4-дикарбонил)6ис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
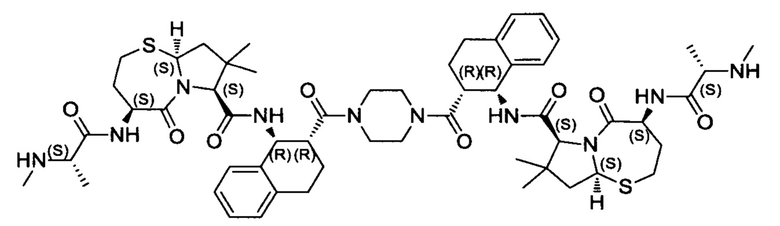
Этап 1: (1R,2R)-1-((трет-бутоксикарбонил(амино)-1,2,3,4-тетрагидронафталин-2-карбоновая кислота
Карбонат натрия добавляли к раствору гидрохлорида (1R,2R)-1-амино-1,2,3,4-тетрагидронафталин-2-карбоновой кислоты (1,3 г, 5.71 ммоль) в диоксане/воде (1:1 об./об., 100 мл), чтобы довести рН до 8. Затем добавляли ди-трет-бутил дикарбонат (1,62 г, 7,42 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. По завершении реакции рН смеси доводили до 4 с помощью 20%-го раствора лимонной кислоты. Смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (3/2 об./об.)], в результате чего получали (1R,2R)-1-((трет-бутоксикарбонил)амино)-1,2,3,4-тетрагидронафталин-2-карбоновую кислоту (1.3 г, 4,46 ммоль, выход 78.1%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.519 минуты, m/z: 289.9 (M-1)-.
Этап 2: ди-трет-бутил((1R,1'R,2R,2'R-(пиперазин-1,4-дикарбонил)бис(1,2,3,4-тетрагидронафталин-2,1-диил))дикарбамат
К раствору (1R,2R)-1-((трет-бутоксикарбонил)амино)-1,2,3,4-тетрагидронафталин-2-карбоновой кислоты (270 мг, 0.90 ммоль) и пиперазина (39.9 мг, 0.46 ммоль) в N,N-диметилформамиде (12 мл) при -10°С медленно добавляли триэтиламин (187.5 мг, 1.85 ммоль) и диэтил цианофосфонат (226.7 мг, 1.38 ммоль). Смесь перемешивали в течение 20 минут. Смеси давали нагреться до комнатной температуры и перемешивали в течение 3 часов. Реакцию гасили ледяной водой (50 мл), в результате чего получали осадок, который фильтровали и сушили, в результате чего получали ди-трет-бутил((1R,1'R,2R,2'R)-(пиперазин-1,4-дикарбонил)бис(1,2,3,4-тетрагидронафталин-2,1-диил))дикарбамат (435 мг, 0.69 ммоль, выход 68.7%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.74 минуты, m/z: 532.7 (М - Вос+1)+.
Этап 3: пиперазин-1,4-диилбис(((1R,2R)-1-амино-1,2,3,4-тетрагидронафталин-2-ил)метанона) дигидрохлорид
К раствору ди-трет-бутил((1R,1'R,2R,2'R)-(пиперазин-1,4-дикарбонил)бис(1,2,3,4-тетрагидронафталин-2,1-диил))дикарбамата (435 мг, 1.0 ммоль) в дихлорметане (20 мл) медленно добавляли 4 н. хлорид водорода в 1,4-диоксане (2 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель концентрировали при пониженном давлении, в результате чего получали неочищенный пиперазин-1,4-диилбис(((1R,2R)-1-амино-1,2,3,4-тетрагидронафталин-2-ил)метанона) дигидрохлорид (402 мг) в виде белого твердого вещества. Его использовали в реакции без дальнейшей очистки. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.07 минуты, m/z: 432.9 (M+1)+.
Этап 4: ди-трет-бутил((2R,2'S)-(((4S,4'R,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R-(пиперазин-1,4-дикарбонил)бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
Раствор пиперазин-1,4-диилбис(((1R,2R)-1-амино-1,2,3,4-тетрагидронафталин-2-ил)метанона) дигидрохлорида (150 мг, 0,297 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение I) (319 мг, 0.68 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимид метиодида (200 мг, 1.02 ммоль), 1-гидроксибензотриазола (187 мг, 1.36 ммоль) и N,N-диизопропилэтиламина (269 мг, 2.04 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (30 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали нечистый продукт (93 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-(пиперазин-1,4-дикарбонил)бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (64 мг, 0.05 ммоль, выход 13.9%) в виде белого твердого вещества. ЖХМС (3.0 минуты, муравьиная кислота): Время удерживания = 2.51 минуты, m/z: 1282.6 (М+1)+, 642.5 {[(М - 2Boc)+2]/2}+.
Этап 5: (4S,4'S,7S,7'S,9aS,9a'S)-N.N'-((1R,1'R,2R,2'R)-пиперазин-1,4-дикарбонил)бис(1,2,3,4-тетрагадронафталин-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-(пиперазин-1,4-дикарбонил)бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (64 мг, 0.05 ммоль) в дихлорметане (5 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли под вакуумом, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1R,1'R,2R,2'R)-(пиперазин-1,4-дюсарбонил)бис(1,2,3,4-тетрагидронафталин-2,1 -диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид (50 мг, 0.043 ммоль, выход 84.3%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 8.64 (s, 2H), 8.00(d, J=8.8 Гц, 1Н), 7.87(d, J=8.4 Гц, 1Н), 7.31-7.25 (m, 2H), 7.19-7.07 (m, 6H), 5.50-5.45 (m, 2H), 5.40-5.34 (m, 2H), 4.70-4.64 (m, 2H), 3.78-3.65 (m, 6H), 3.62-3.59 (m, 2H), 3.49-3.47 (m, 2H), 3.22-3.11 (m, 4H), 3.08-3.03 (m, 4H), 2.93-2.91 (m, 2H), 2.76 (s, 4H), 2.43 (m, 6H), 2.17-2.20 (m, 2H), 2.10-2.01 (m, 4H), 1.94-1.88 (m, 2H), 1.76-1.58 (m, 2H), 1.35-1.32 (m, 6H), 1.03-1.00 (m, 12H). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.407 минуты, m/z: 1082.8 (M+1)+.
Пример 8
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1R,1'R,2R,2'R)-(пиперазин-1,4-дикарбонил)бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
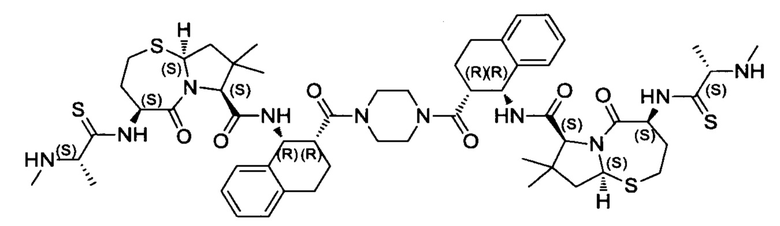
Этап 1: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-(пиперазин-1,4-дикарбонил)бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат)
Раствор пиперазин-1,4-диилбис(((1R,2R)-1-амино-1,2,3,4-тетрагидронафталин-2-ил)метанона) дигидрохлорида (Пример 7, Этап 3) (150 мг, 0.297 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение II) (272.8 мг, 0.593 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимид метиодида (170.7 мг, 0.89 ммоль), 1-гидроксибензотриазола (160.5 мг, 1.188 ммоль) и N,N-диизопропилэтиламина (230.2 мг, 1.782 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (30 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали нечистый продукт (170 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-(пиперазин-1,4-дикарбонил)бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (90 мг, 0.07 ммоль, выход 23.6%) в виде белого твердого вещества. ЖХМС (3.0 минуты, муравьиная кислота): Время удерживания = 2.75 минуты, m/z: 1315.6 (M+1)+.
Этап 2: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1R,1'R,2R,2'R)-(пиперазин-1,4-дикарбонил)бис(1,2,3,4-тетрагадронафталин-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-(пиперазин-1,4-дикарбонил)бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (96 мг, 0.073 ммоль) в дихлорметане (10 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1R,1'R,2R,2'R)-(пиперазин-1,4-дикарбонил)бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (60 мг, 0.05 ммоль, выход 68.8%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 11.12-10.98 (m, 1H), 10.89-10.87 (m, 1H), 9.81 (s, 1H), 9.62 (s, 1H), 8.67 (s, 1H), 8.44-8.36 (m, 1H), 8.15-8.07 (m, 1H), 7.95-7.88 (m, 1H), 7.26-7.25 (m, 2H), 7.15-7.09 (m, 6H), 5.47-5.41 (m, 2H), 5.36-5.35 (m, 2H), 5.17-5.03 (m, 2H), 4.41-4.32 (m, 2H), 3.78-3.65 (m, 4H), 3.60-3.47 (m, 4H), 3.39 (s, 2H), 3.25-3.09 (m, 4H), 3.03-2.96 (m, 2H), 2.75 (s, 4H), 2.51 (s, 6H), 2.30-2.19 (m, 6H), 2.02 (s, 2H), 1.91 (s, 2H), 1.66-1.62 (m, 2H), 1.45-1.41 (m, 6H), 1.04 (s, 6H), 1.00 (s, 6H). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.246 минуты, m/z: 1114.4 (M+1)+.
Пример 9
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
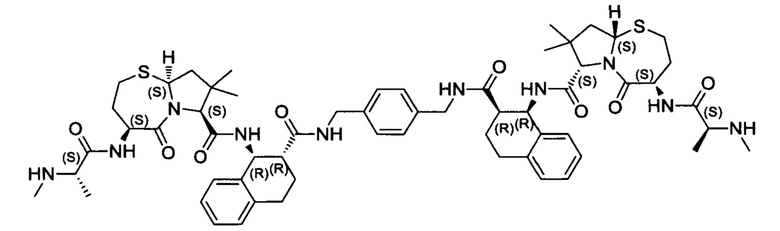
Этап 1: ди-трет-бугил((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))дикарбамат
К раствору (1R,2R)-1-((трет-бутоксикарбонил)амино)-1,2,3,4-тетрагидронафталин-2-карбоновой кислоты (114 мг, 0.39 ммоль) и 1,4-фенилендиметанамина (26 мг, 0.19 ммоль) в N,N-диметилформамиде (5 мл) при -10°С медленно добавляли триэтиламин (79 мг, 0.78 ммоль) и диэтилцианофосфонат (96 мг, 0.59 ммоль). Смесь перемешивали в течение 20 минут.Смеси давали нагреться до комнатной температуры и перемешивали в течение 3 часов. Реакцию гасили ледяной водой (50 мл), в результате чего получали осадок, который фильтровали и сушили, в результате чего получали ди-трет-бутил((1R,1'R,2R,2')-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))дикарбамат (180 мг, 0.26 ммоль, выход 67.4%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота); Время удерживания = 1.736 минуты, га/г: 704.8 (M+Na)+.
Этап 2: (1R,1'R,2R,2'R)-N,N'-(1,4-фениленбис(метилен))бис(1-амино-1,2,3,4-тетрагидронафталин-2-карбоксамида)дигидрохлорид
К раствору ди-трет-бутил((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1 -диил))дикарбамата (180 мг, 0.26 ммоль) в дихлорметане (10 мл) медленно добавляли 4 н. хлорид водорода в 1,4-диоксане (1 мл). Смесь перемешивали при комнатной температуре в течение 4 часов и растворитель удаляли при пониженном давлении, в результате чего получали (1R,1'R,2R,2'R)-N,N'-(1,4-фениленбис(метилен))бис(1-амино-1,2,3,4-тетрагидронафталин-2-карбоксамида) дигидрохлорид (150 мг) в виде белого твердого вещества. Его использовали на следующем этапе без дополнительной очистки. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.085 минуты, m/z: 482.9 (M+1)+.
Этап 3: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-тил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
Раствор (1R,1'R,2R,2'R)-N,N'-(1,4-фениленбис(метилен))бис(1-амино-1,2,3,4-тетрагидронафталин-2-карбоксамида)дигидрохлорида (150 мг, 0.27 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение I) (240 мг, 0.54 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимид метиодида (155.3 мг, 0.81 ммоль), 1-гидроксибензотриазола (146 мг, 1.08 ммоль) и N,N-диизопропилэтиламина (209.3 мг, 1.62 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (30 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2:1 об./об.)], в результате чего получали неочищенный продукт (170 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (76 мг, 0.057 ммоль, выход 21.1%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.80 минуты, m/z: 566.9 {[M-2Boc)=2]/2}+.
Этап 4: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(8,8-диметил-4-((S)-2-(метидамино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7S,9aS,9а'S)-((((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (80 мг, 0.06 ммоль) в дихлорметане (6 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали и сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(8,8-диметил-4-((8)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид (46.2 мг, 0.038 ммоль, выход 60.5%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 9.24 (s, 2H), 8.86-8.85 (m, 2H), 8.78(d, J=6.8 Гц, 2Н), 8.36(t, J=5.2 Гц, 2Н), 7.94(d, J=9.2 Гц, 2Н), 7.31-7.29 (m, 2H), 7.22 (s, 4H), 7.14-7.12 (m, 4H), 7.09-7.06 (m, 3H), 5.48(t, J=7.8 Гц, 2H), 5.43-5.30 (m, 2H), 4.72-4.67 (m, 2H), 4.38-4.33 (m, 2H), 4.14 (s, 2H), 4.08-4.03 (m, 2H), 3.91-3.86 (m, 2H), 3.21-3.15 (m, 2H), 2.96-2.87 (m, 4H), 2.82-2.68 (s, 4H), 2.47(t, J=4.4 Гц, 6Н), 2.21-2.16 (m, 2H), 2.09-1.99 (m, 6Н), 2.00-1.96 (m, 2H), 1.75-1.70 (m, 2H), 1.38(d, J=6.8 Гц, 6Н), 1.03 (s, 6Н), 1.00 (s, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.453 минуты, m/z: 1133.7 (M+1)+, 1155.6 (М+Na)+.
Пример 10
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)

Этап 1: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат)
Раствор (1R,1'R,2R,2'R)-N,N'-(1,4-фениленбис(метилен))бис(1-амино-1,2,3,4-тетрагидронафталин-2-карбоксамида) дигидрохлорида (Пример 9, Этап 2) (116 мг, 0.21 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение II) (201 мг, 0.44 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимид метиодида (120 мг, 0.63 ммоль), 1-гидроксибензотриазола (113 мг, 0.84 ммоль) и N,N-диизопропилэтиламина (279 мг, 2.16 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (30 мл) и экстрагировали дихлорметаном (3×30 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали нечистый продукт (110 мг) в виде желтого масла, который дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-6утил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (54 мг, 0.039 ммоль, выход 18.9%) в виде белого твердого вещества. ЖХМС (3.0 минуты, муравьиная кислота): Время удерживания = 2.614 минуты, m/z: 1388 (M+Na)+.
Этап 2: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (54 мг, 0.04 ммоль) в дихлорметане (5 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и разбавляли водой. Ph доводили до 8~9 с помощью бикарбоната натрия и экстрагировали дихлорметаном (3×10 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью тонкослойной хроматографии [дихлорметан/метанол (8/1 об./об.)], в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагадронафталин-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) (6 мг, 0.005 ммоль, выход 13%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 8.43-8.37 (m, 2H), 7.85-7.82 (m, 2H), 7.33-7.27 (m, 2H), 7.20 (s, 4H), 7.15-7.13 (m, 4H), 7.08-7.06 (m, 2H), 5.51-5.46 (m, 2H), 5.40-5.37 (m, 2H), 5.10-5.06 (m, 2H), 4.40-4.34 (m, 2H), 4.16 (s, 2H), 4.04-4,00 (m, 2H), 3.49-3.42 (m, 2H), 3.22-3.14 (m, 2H), 2.98-2.91 (m, 4H), 2.79-2.69 (m, 4H), 2.42-2.31 (m, 2H), 2.27-2.20 (m, 2H), 2.16 (m, 6H), 2.07-1.98 (m, 4H), 1.74-1.65 (m, 2H), 1.22-1.21 (m, 6H), 1.07-1.06 (m, 6H), 1.03-1.02 (m, 6H). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.52 минуты, 1164.7 (M+1)+.
Пример 11
(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-N-[(1R,2R)-2-{1(1rs,4rs)-4-[(1R,2R)-1-[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-амидо]-1,2,3,4-тетрагидронафталин-2-амидо]циклогексил]карбамоил}-1,2,3,4-тетрагидронафталин-1-ил]-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида дигидрохлорид
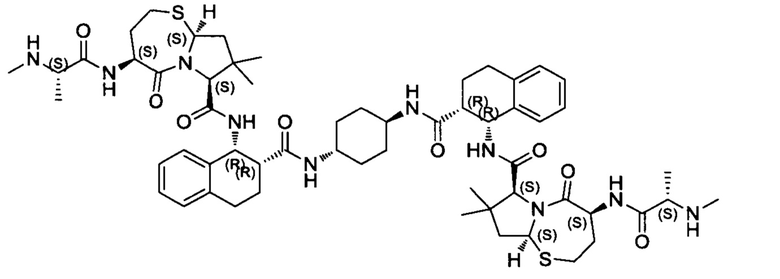
Этап 1: ди-трет-бутил((1R,1'R,2,2'R)-((((1R,4R)-циклогексан-1,4-диил)бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))дикарбамат
К раствору (1R,2R)-1-((трет-бутоксикарбонил)амино)-1,2,3,4-тетрагидронафталин-2-карбоновой кислоты (250 мг, 0.858 ммоль) и (1r,4r)-циклогексан-1,4-диамина (49 мг, 0.429 ммоль) в N,N-диметилформамиде (10 мл) при -10°С медленно добавляли N,N-диизопропилэтиламин (221 мг, 1.716 ммоль) и диэтилцианофосфонат (210 мг, 1.287 ммоль). Смесь перемешивали в течение 20 минут. Смеси давали нагреться до комнатной температуры и перемешивали в течение 3 часов. Реакцию гасили ледяной водой (100 мл), в результате чего получали осадок, который фильтровали, в результате чего получали ди-трет-бутил((1R,1'R,2R,2'R)-((((1R,4R)-циклогексан-1,4-диил)бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))дикарбамат (240 мг, 0.363 ммоль, выход 84.6%) в виде оранжевого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.724 минуты, m/z: 628.8 (М+Na)+.
Этап 2: (1R,1'R,2R,2'R)-N,N'-((1R,4R)-циклогексан-1,4-диил)бис(1-амино-1,2,3,4-тетрагидронафталин-2-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((1R,1'R,2R,2'R)-((((1R,4R)-циклогексан-1,4-диил)бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))дикарбамата (150 мг, 0,23 ммоль) в дихлорметане (5 мл) медленно добавляли 4 н. хлорид водорода в 1,4-диоксане (0.5 мл). Смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении, в результате чего получали (1R,1'R,2R,2'R)-N,N'-((1R,4R)-циклогексан-1,4-диил)бис(1-амино-1,2,3,4-тетрагидронафталин-2-карбоксамида) дигидрохлорид (130 мг, неочищенный) в виде желтого сиропа. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.122 минуты, m/z: 461.1 (M+1)+
Этап 3: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-циклогексан-1,4-диил)бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1 -диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
Раствор (1R,1'R,2R,2'R)-N,N'-((1R,4R)-циклогексан-1,4-диил)бис(1-амино-1,2,3,4-тетрагидронафталин-2-карбоксамида) дигидрохлорида (130 мг, 0.24 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение 1) (216.2 мг, 0.49 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимид метиодида (140.1 мг, 0.73 ммоль), 1-гидроксибензотриазола (131.7 мг, 0.97 ммоль) и N,N-диизопропилэтиламина (189 мг, 1.46 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (50 мл) и экстрагировали дихлорметаном (3×50 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали неочищенный продукт (170 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-6утил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-((((1R,4R)-циклогексан-1,4-диил)бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (86 мг, 0.06 ммоль, выход 26.8%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.96 минуты, m/z: 555.9 {[М-2Boc)+2]/2}+.
Этап 4: (4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-N-{(1R,2R)-2-{[(1rs,4rs)-4-[(1R,2R)-1-[(4S,7S,9aS)-8,8-даметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-амидо]-1,2,3,4-тетрагидронафталин-2-амидо]циклогексил]карбамоил} -1,2,3,4-тетрагидронафталин-1-ил]-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-((((1R,4R)-циклогексан-1,4-диил)бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (80 мг, 0.073 ммоль) в дихлорметане (10 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель концентрировали при пониженном давлении и твердое вещество сушили в глубоком вакууме, в результате чего получали указанное в заголовке соединение(62 мг, 0.052 ммоль, выход 79.8%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 9.29 (s, 1H), 8.89-8.88 (m, 1H), 8.78(d, J=6.8 Гц, 2Н), 7,90(d, J=9.2 Гц, 2Н), 7.74(d, J=7.6 Гц, 2Н), 7.28-7.26 (m, 2Н), 7.12-7.05 (m, 6H), 5,46(t, J=7.6 Гц, 2Н), 5.36-5.33 (m, 2Н), 4.71-4.67 (m, 2Н), 4.15 (s, 2Н), 3,90 (s, 2Н), 3.41-3.39 (m, 2Н), 3.16(t, J=11.8 Гц, 2Н), 2,96-2.92 (m, 2Н), 2.79-2.64 (m, 6H), 2.48 (s, 6H), 2.19-2.11 (m, 4H), 2.01-1.94 (m, 6H), 1.78-1.68 (m, 6H), 1.40(d, J=6.8 Гц, 6H), 1.20-1.12 (m, 4H), 1.02 (s, 6H), 1.00 (s, 6H). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.280 минуты, m/z: 1110.8 (M+1)+.
Пример 12
(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-N-[(1R,2R)-2-{[(1rs,4rs)-4-[(1R,2R)-1-[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-амидо]-1,2,3,4-тетрагидронафталин-2-амидо]циклогексил]карбамоил}-1,2,3,4-тетрагидронафталин-1-ил]-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида дигидрохлорид
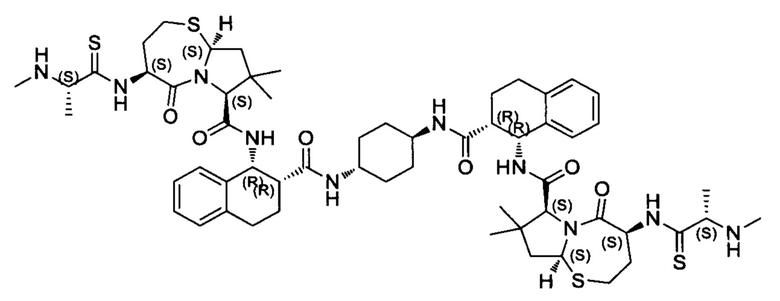
Этап 1: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-((((1R,4R)-циклогексан-1,4-диил)бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат)
Смесь (1R,1'R,2R,2'R)-N,N'-((1R,4R)-циклогексан-1,4-диил)бис(1-амино-1,2,3,4-тетрагидронафталин-2-карбоксамида) дигидрохлорида (Пример 11, Этап 2) (200 мг, 0.375 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение II) (413 мг, 0.901 ммоль), N-(3-диметиламинопропил)-N-этилкарбодиимида гидрохлорида (216 мг, 1.125 ммоль), 1-гидроксибензотриазола (203 мг, 1.5 ммоль) и N,N-диизопропилэтиламина (290 мг, 2.25 ммоль) в дихлорметане (15 мл) перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь вливали в воду (15 мл) и экстрагировали дихлорметаном (2×10 мл). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали. Затем неочищенный продукт очищали препаративной-ТСХ [петролейный эфир/этилацетат (1:2 об./об.)] с последующей препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-((((1R,4R)-иклогексан-1,4-диил)бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1 -диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (140 мг, 0.104 ммоль, выход 28.0%) в виде бежевого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 2.115 минуты, m/z: 572.1 {[(M-2Boc)+2]/2}+.
Этап 2: (4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-N-[(1R,2R)-2-{[(1rs,4rs)-4-[(1R,2R)-1-[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-амидо]-1,2,3,4-тетрагидронафталин-2-амидо]циклогексил]карбамоил}-1,2,3,4-тетрагидронафталин-1-ил]-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидадигидрохдорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-((((1R,4R)-циклогексан-1,4-диил)бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (140 мг, 0.104 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлорид водорода в 1,4-диоксане (0.5 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и сушили в глубоком вакууме, в результате чего получали указанное в заголовке соединение(70 мг, 0.058 ммоль, выход 55.3%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 11.04-11.00 (m, 2H), 10.91 (s, 2H), 8.67 (s, 1H), 8.43 (s, 1H), 7.97(t, J=8.4 Гц, 2H), 7.76-7.75 (m, 2H), 7.27-7.25 (m, 2H), 7.15-7.06 (m, 6H), 5.45-5.33 (m, 4H), 5.15-5.09 (m, 2H), 4.32 (s, 2H), 4.18(d, J=9.2 Гц, 2Н), 3.51-3.47 (m, 2H), 3.22-3.17 (m, 2H), 3.07-3.00 (m, 2H), 2.79-2.66 (m, 6H), 2.48 (s, 6H), 2.33-2.18 (m, 4H), 2.05-1.94 (m, 6H), 1.79-1.74 (m, 6H), 1.45 (s, 6H), 1.24-1.14 (m, 4H), 1.04-1.03 (m, 6H), 1.00 (m, 6H). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.242 минуты, m/z: 1144.4 (M+1)+, 572.3 [(М+2)/2]+.
Пример 13
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(гекса-2,4-диин-1,6-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламиио)пропанамидо)-5-оксооктагидропирроло [2,1-b][1,31 тиазепин-7-карбоксамида) дигидрохлорид
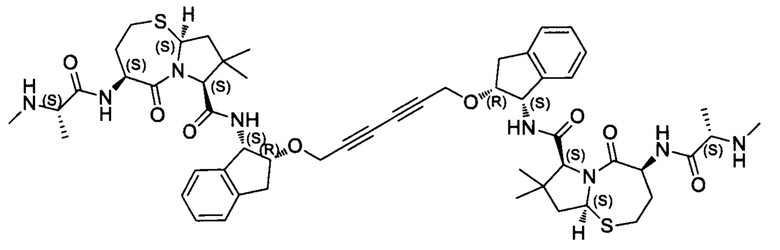
Этап 1: (1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден-1 -амин
К раствору (1S,2R)-1-амино-2,3-дигидро-1Н-инден-2-ола (2 г, 13.4 ммоль) в тетрагидрофуране (50 мл) при 0°С порциями добавляли гидрид натрия (60%, дисперсия в Парафиновом Масле) (0.59 г, 14.7 ммоль). Смеси давали нагреться до комнатной температуры. Затем добавляли 3-бромпроп-1-ин (1.75 г, 14.7 ммоль) и полученную смесь нагревали до 70°С. Ее перемешивали при 70°С в течение ночи. Смесь гасили ледяной водой (50 мл) и экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (3:2 об./об.)], в результате чего получали (1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден-1-амин (1.2 г, 6.4 ммоль, выход 47.8%) в виде черного масла. ЖХМС (электроспрей, m/z): 187.1, 188.1 [M+H]+, время удерживания 0.942 минут. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 7.36-7.29 (m, 1H), 7.22-7.13 (m, 3H), 4.25(dd, J=4.2, 2.4 Гц, 2Н), 4.22-4.15 (m, 2H), 3.43(t, J=2.4 Гц, 1Н), 2.93 (квд, J=16.1,3.5 Гц, 2H), 1.75 (s, 2H).
Этап 2: трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден-1-ил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат
К раствору (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (260 мг, 0.587 ммоль), 2-(7-азабензотриазол)-N,N,N',N'-тетраметилурония гексафторфосфата (305 мг, 0.801 ммоль) и N,N-диизопропилэтиламина (138 мг, 1.068 ммоль) в 1,2-дихлорэтане (10 мл) добавляли (1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден-1-амин (100 мг, 0.534 ммоль). Смесь перемешивали при 50°С в течение ночи. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (1:1 об./об.)] и дополнительно очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (3/2 об./об.)], в результате чего получали трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден-1-ил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат (160 мг, 0.261 ммоль, выход 48.9%). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.662 минуты, m/z: 634.8 (M+Na)+.
Этап 3: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(гекса-2,4-диин-1,6-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
а. К раствору трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден-1-ил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата (160 мг, 0.261 ммоль) и пиридина (124 мг, 1.566 ммоль) в ацетонитриле (7 мл) добавляли ацетат меди (57 мг, 0.313 ммоль). Смесь перемешивали при 85°С в течение 1 часа. Смесь охлаждали, концентрировали и разбавляли этилацетатом (15 мл). Добавляли водный аммиак (20-кратное разведение, 15 мл). Водную фазу экстрагировали этилацетатом (2×10 мл). Объединенные органические слои промывали солевым раствором (15 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью тонкослойной хроматографии [этилацетат /петролейный эфир (2/1 об./об)] с последующей препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(гекса-2,4-диин-1,6-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (80 мг, 0.065 ммоль, выход 50.2%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.853 минуты, m/z: 511.8 {[(M-2Boc)+2]/2}+.
b. К раствору трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден-1-ил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата (1.0 г, 1.63 ммоль) и пиридина (775 мг, 9.79 ммоль) в ацетонитриле (20 мл) добавляли ацетат меди (366 мг, 1.96 ммоль). Смесь перемешивали при 85°С в течение 1 часа. Реакционную смесь охлаждали, концентрировали и разбавляли этилацетатом (100 мл). Добавляли раствор 20-кратно разведенного аммиака (50 мл). Водную фазу экстрагировали этилацетатом (2×50 мл). Органический слой объединяли и промывали солевым раствором. Сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Остаток очищали хроматографией на колонке с силикагелем, используя для элюирования этилацетат /петролейный эфир = 1/1, в результате чего получали неочищенный продукт. Затем неочищенный продукт очищали препаративной-ВЭЖХ с получением ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(гекса-2,4-диин-1,6-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (800 мг, 0.65 ммоль, выход 80.2%) в виде белого твердого вещества. ЖХМС (электроспрей, m/z): 1222.6, 512.2 [М/2-Вос+Н]+, время удерживания 1.735 минут. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 7.96 (d, J=8.8 Гц, 2Н), 7.77(d, J=6.5 Гц, 2Н), 7.24 (ддд, J=14.9, 8.9, 2.8 Гц, 8Н), 5.48(t, J=7.9 Гц, 2Н), 5.38(dd, J=8.7, 5.3 Гц, 2Н), 4.67-4.62 (m, 2Н), 4.30 (m, 6Н), 4.23 (s, 2Н), 3.15(t, J=11.9 Гц, 2Н), 3.03(d, J=4.4 Гц, 2Н), 2.88(d, J=16.4 Гц, 2Н), 2.73 (s,6H), 2.26-2.17 (m, 2Н), 2.16-2.05 (m, 6Н), 1.76 (dt, J=23.3, 10.1 Гц, 4Н), 1.40 (s, 18Н). 1.24(d, J=7.1 Гц, 6Н), 1.07(d, J=14.9 Гц, 12Н).
Этап 4: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R-(гекса-2,4-диин-1,6-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидро хлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(гекса-2,4-диин-1,6-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (80 мг, 0.065 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлорид водорода в 1,4-диоксане (1.5 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и твердое вещество сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(гекса-2,4-диин-1,6-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (55 мг, 0.050 ммоль, выход 76.9%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц. ДМСО-d6) δ ppm 9.53-9.50 (m, 2Н), 8.95-8.91 (m, 2Н), 8,88(d, J=7.2 Гц, 2Н), 8.00(d, J=8.8 Гц, 2Н), 7.28-7.20 (m, 8Н), 5.51(t, J=7.8 Гц, 2Н), 5.38(dd, J=8.6, 5.4 Гц, 2Н), 4.76-4.71 (m, 2Н), 4.24-4.28 (m, 6Н), 4.23 (s, 2Н), 3.93-3.85 (m, 2Н), 3.22-3.16 (m. 2Н), 3.10-3.00 (m, 4Н), 2.94-2.90 (m, 2H), 2.47(t, J=5.2 Гц, 6Н), 2.29-2.14 (m, 4Н), 1.87-1.79 (m, 4H), 1.42(d, J=6.8 Гц, 6Н), 1.10 (s, 6Н), 1.06 (s, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.328 минуты, m/z: 512.0 [(М+2)/2]+.
Пример 14
(4S,7S,9aS)-N-((1S,2R)-2-((6-(((1S,2R)-1-((4S,7S,9aS)-8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2,3-дигидро-1Н-инден-2-ил)окси)гекса-2,4-диин-1-ил)окси)-2,3-дигидро-1Н-индеи-1-ил)-8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
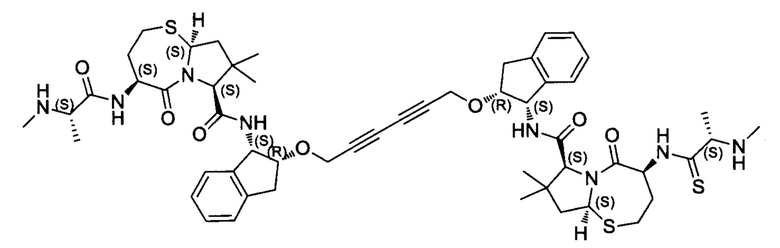
Этап 1: трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден-1-ил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-тиоксопропан-2-ил)(метил)карбамат
К раствору (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (500 мг, 1.088 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (621 мг, 1.632 ммоль) и N,N-диизопропилэтиламина (309 мг, 2.393 ммоль) в 1,2-дихлорэтане (15 мл) добавляли (1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден-1-амин (224 мг, 1.197 ммоль). Смесь перемешивали при 50°С в течение ночи. Смесь концентрировали и очищали хроматографией на силикагеле [петролейный эфир/этилацетат (3:2 об./об.)] с последующей препаративной-ТСХ [петролейный эфир/этилацетат (2:3 об./об.)], в результате чего получали трет-бутил((8)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1H-инден-1-ил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-тиоксопропан-2-ил)(метил)карбамат (400 мг, 0.636 ммоль, выход 58.5%). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.812 минуты, т/г: 650.8 (M+Na)+.
Этап 2: трет-бутил((S)-1-(((4S,7S,9aS)-7-(((1S,2R)-2-((6-(((1S,2R)-1-((4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2.3-дигидро-1Н-инден-2-ил)окси)гекса-2,4-диин-1 -ил)окси)-2,3-дигидро-1Н-инден-1-ил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1 -оксопропан-2-ил)(метил)карбамат
К раствору трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден-1-ил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата (390 мг, 0.636 ммоль), трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден-1-ил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-тиоксопропан-2-ил)(метил)карбамата (400 мг, 0.636 ммоль) и пиридина (301 мг, 3.816 ммоль) в ацетонитриле (15 мл) добавляли ацетат меди (277 мг, 1.526 ммоль). Смесь перемешивали при 85°С в течение 40 минут. После охлаждения растворитель удаляли при пониженном давлении. Неочищенный продукт разбавляли этилацетатом (20 мл) и водным аммиаком (20-кратное разведение, 20 мл). Водную фазу экстрагировали этилацетатом (2×15 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали препаративной-ТСХ [петролейный эфир/этилацетат (2:3 об./об.)] и препаративной-ВЭЖХ, в результате чего получали трет-бутил((S)-1-(((4S,7S,9aS)-7-(((1S,2R)-2-((6-(((1S,2R)-1-((4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2,3-дигидро-1Н-инден-2-ил)окси)гекса-2,4-диин-1-ил)окси)-2,3-дигидро-1Н-инден-1-ил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат (50 мг, 0.040 ммоль, выход 6.29%) и ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9a8S9a'S)-((((1S,1'S,2R,2'R)-(гекса-2,4-диин-1,6-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (30 мг, 0.024 ммоль, выход 3.77%), оба в виде белых твердых веществ.
Этап 3: (4S,7S,9aS)-N-((1S,2R)-2-((6-(((1S,2R)-1-((4S,7S,9aS)-8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2,3-дигидро-1Н-инден-2-ил)окси)гекса-2.4-диин-1-ил)окси)-2.3-дигидро-1Н-инден-1-ил)-8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида дигидрохлорид
К раствору трет-бутил((S)-1-(((4S,7S,9aS)-7-(((1S,2R)-2-((6-(((1S,2R)-1-((4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагадропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2,3-дигидро-1Н-инден-2-ил)окси)гекса-2,4-диин-1-ил)окси)-2,3-дигидро-1Н-инден-1-ил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата (50 мг, 0.040 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлорид водорода в 1,4-диоксане (0.5 мл). Смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали и сушили, в результате чего получали (4S,7S,9aS)-N-((1S,2R)-2-((6-(((1S,2R)-1-((4S,7S,9aS)-8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2,3-дигидро-1Н-инден-2-ил)окси)гекса-2,4-диин-1-ил)окси)-2,3-Дигидро-1Н-инден-1-ил)-8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида дигидрохлорид (20 мг, 0.018 ммоль, выход 45%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 11.03-10.93 (m, 2Н), 9.74-9.58 (m, 1Н), 9.28 (s, 1Н), 8,86-8,84 (m, 2Н), 8.65-8.45 (m, 1Н), 8.11-8.05 (m, 1Н), 8.02-8.00 (m, 1Н), 7.27-7.18 (m, 8Н), 5.52-5.43 (m, 2Н), 5.40-5.37 (m, 2Н), 5.17 (кв, J=6.4 Гц, 1Н), 4.76-4.65 (m, 1Н), 4.38-4.23 (m, 8Н), 3.91-3.88 (m, 1Н), 3.73-3.65 (m, 1Н), 3.51-3.46 (m, 1Н), 3.22-3.16 (m, 2Н), 3.10-2.97 (m, 4Н), 2.95-2.90 (m, 1Н), 2.49 (s, 6Н), 2.33-2,12 (m, 4Н), 1.96-1.76 (m, 4Н), 1.45(d, J=6.4 Н, 3H), 1.41(d, J=6.8 Н, 3H), 1.10-1.06 (m, 12Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.643 минуты, m/z: 520.0 [(М+2)/2]+.
Пример 15
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(гекса2,4-диин-l,6-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид
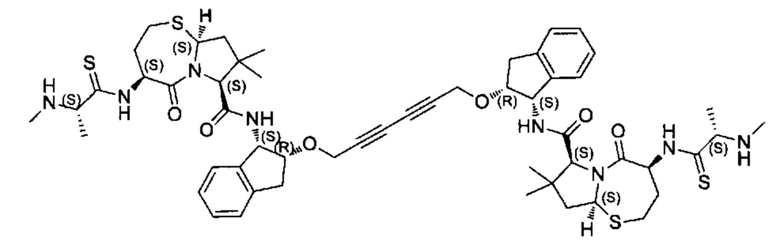
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(гекса-2,4-диин-1,6-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметал-5-оксооктагидропирроло[2,1-b][1,3]таазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (Пример 14, Этап 2) (35 мг, 0.028 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлорид водорода в 1,4-диоксане (0.5 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(гекса2,4-диин-1,6-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (12 мг, 0.011 ммоль, выход 39.3%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 11.00-10.91 (m, 2Н), 9.66-9.51 (m, 2Н), 8.62-8.44 (m, 2Н), 8.08(dd, J=13.9, 9.0 Гц, 2Н), 7.28-7.18 (m, 8Н), 5.46 (кв, J=9.3 Гц, 2Н), 5.38(dd, J=8.6, 5.4 Гц, 2Н), 5.17(t, J=10.2 Гц, 2Н), 4.38-4.26 (m, 10H), 3.23-3.17 (m, 2Н), 3.06-2.99 (m, 6Н), 2.50 (s, 6Н), 2.33-2,17 (m, 4Н), 1.95-1.84 (m, 4Н), 1.44(d, J=6.4Н, 6Н), 1.10 (s, 6Н), 1.08 (s, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания=1.505 минуты, m/z: 1055.6 (М+1)+.
Пример 16
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S,2R,2'R)-(бутан-1,4-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)
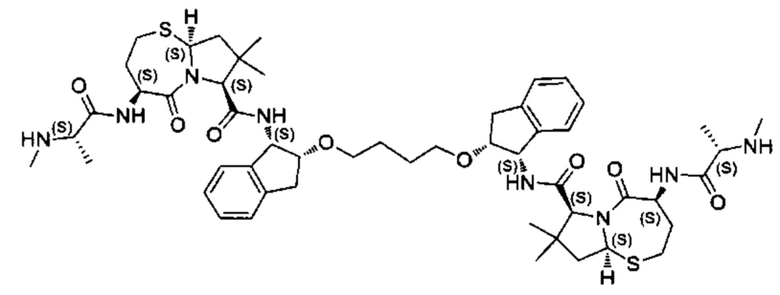
Этап 1: (1S,2R)-1-изоциано-2-(проп-2-ин-1-илокси)-2.3-дигидро-1Н-инден
Суспензию (1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1H-инден-1-амина (3.31 г, 17.68 ммоль) и нагревали в этилформиате (2S,8 мл, 354 ммоль) при 70°С в течение 21 часа. После охлаждения растворитель удаляли при пониженном давлении, в результате чего получали неочищенный формамид (3.71 г). К суспензии неочищенного формамида в дихлорметане (ДХМ) (35 мл) при 0°С добавляли Et3N (12.32 мл, 88 ммоль), а затем POCl3 (2.471 мл, 26.5 ммоль). Через 2 часа неочищенный продукт вливали в смесь ДХМ (150 мл) и нас.вод. NaHCO3 (60 мл). Водную фазу отделяли и экстрагировали дихлорметаном (50 мл). Объединенные органические слои промывали солевым раствором (40 мл), сушили (Na2SO4), фильтровали и концентрировали. Неочищенный продукт абсорбировали на Целите® и очищали хроматографией на силикагеле [1-20% EtOAc в гексанах], в результате чего получали (1S,2R)-1-изоциано-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден, 0.15 этилацетат (сольват) (1.88 г, 8.93 ммоль, выход 50.5%) в виде светло-желтого масла. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 2.99(dd, J=16.4, 5.9 Гц, 1Н), 3.11(dd, J=16.0, 6.2 Гц, 1Н), 3.53(t, J=2.3 Гц, 1Н), 4.26-4.38 (m, 2H), 4,44 (кв, J=5.6 Гц, 1Н), 5.34-5.45 (m, 1Н), 7.28-7.38 (m, 3Н), 7,44(d, J=7.0 Гц, 1Н); ЖХМС(электроспрей) m/z: 171.0 (M-(NC))+.
Этап 2: (4S,7S,9aS)-4-амино-8,8-диметил-5-оксо-N-((1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден-1-ил)октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид и (4S,7R,9aS)-4-амино-8,8-диметил-5-оксо-N-((1S,2R)-2-(проп-2-ин-1-илокси)-2.3-дигидро-1Н-инден-1-ил)октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
Смесь N-(трет-бутоксикарбонил)-S-тритил-1-гомоцистеина (2.05 г, 4.29 ммоль), (1S,2R)-1-изоциано-2-(проп-2-ин-1-илокси)-2,3-дигидро-1H-индена, 0.15 этилацетата (сольват) (0.903 г, 4.29 ммоль), 4,4-диметокси-2,2-диметилбутаналя (1.2 г, 7,49 ммоль) и аммиака, 7 М в МеОН (1.226 мл, 8.58 ммоль) в трифторэтаноле (16 мл) нагревали при 80°С. Через 1 час растворитель удаляли при пониженном давлении. Неочищенный продукт разбавляли дихлорметаном (100 мл) и промывали 1H. NaOH (15 мл). Водный слой отделяли и экстрагировали этилацетатом (75 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили (MgSO4), фильтровали и концентрировали, в результате чего получали пену. К этому материалу добавляли дихлорметан (ДХМ) (20 мл) и ТФУК (4 мл, 51.9 ммоль). Смесь нагревали при 35-40°С. Через 1 час к смеси добавляли ТФУК (2 мл) и нагревали в течение 1.5 часа. Смесь оставляли перемешиваться при к.т. в течение ночи. Добавляли ТФУК (2 мл) и нагревали смесь при 50°С в течение 1 часа. Неочищенный продукт абсорбировали на Целите® и очищали С18 с помощью устройства ISCO [20-30% градиент ACN, 0.1% муравьиной кислоты], в результате чего получали 2 фракции. Лиофилизированные фракции по отдельности разбавляли этилацетатом (150 мл) и промывали нас.вод. NaHCO3 (25 мл), сушили (Na2SO4), фильтровали и концентрировали. Получали два диастереоизомера - (4S,7S,9aS)-4-амино-8,8-диметил-5-оксо-N-((1S,2R)-2-(проп-2-ил-1-илокси)-2,3-дигидро-1H-инден-1-ил)октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид (290.1 мг, выход 15.8%) и (4S,7R,9aS)-4-амино-8,8-диметил-5-оксо-N-((1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1H-инден-1-ил)октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид (283.3 мг, 15.4%), оба выделены в виде пены.
Данные диастереоизомера 7S: 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 1.06 (s, 3H), 1.09 (s, 3H), 1.67- 1.82 (m, 2H), 1.96-2.05 (m, 1H), 2.23(dd, J=12.9, 7,4 Гц, 1H),2.81 (dt, J=11.5,2.6 Гц, 1H), 2.96 - 3.17 (m, 3H), 3.43(t, J=2.3 Гц, 1Н), 3.68(d, J=9.8 Гц, 1Н), 4.13(t, J=2.5 Гц, 2H), 4.16 -4.19 (m, 1H), 4.28 - 4.35 (m, 1H), 5.27 - 5.43 (m, 2Н), 7.13 - 7.32 (m, 4Н), 7.81(d, J=8.6 Гц, 1H); ЖХМС(электроспрей) m/z: 42S,3 (М+1)+.
Данные диастереоизомера 7R: 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 1.05 (s, 3H), 1,31 (s, 3H), 1.55 - 1.67 (m, 1H), 1.69(d, J=13.3 Гц, 1H), 1.94 (br. s., 1H), 2.00-2.06 (m, 1H), 2.77 (dt, J=l 1.6, 2.8 Гц, 1H), 2.97(dd, J=16.4, 3.1 Гц, 1H), 3.08(dd, J=16.4, 5.5 Гц, 1H), 3.14 - 3.24 (m, 1H), 3.39(t, J=2,1 Гц, 1H), 3.69(d, J=9.8 Гц, 1H), 4.16 - 4.23 (m, 1H), 4.26 - 4.37 (m, 3H), 5.33(dd, J=8.6, 5.5 Гц, 1H), 5.48(d, J=9.0 Гц, 1H), 7.11- 7.27 (m, 4 Н), 8.35 (d, J=9.0 Гц, 1H); ЖХМС(электроспрей) m/z: 42S,7 (М+1)+.
Этап 3: трет-бутил((S)-1-(((4S,75.9а5)-8,8-диметил-5-оксо-7-(((1S,2R)-2-(проп-2-ин-1-илокси)-2.3-дигидро-1H-инден-1-ил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат
К раствору (4S,7S,9aS)-4-амино-8,8-диметил-5-оксо-N-((1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1H-инден-1-ил)октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида (287 мг, 0.671 ммоль) и Boc-N-Me-Ala-OH (205 мг, 1.007 ммоль) в N,N-диметилформамиде (ДМФА) (3 мл) при 0°С добавляли основание Хунига (0.586 мл, 3.36 ммоль) с последующим добавлением по каплям T3P, 50 мас. % в EtOAc (0.699 мл, 1.175 ммоль). Через 45 минут смесь разбавляли этилацетатом (100 мл) и промывали нас.вод. NaHCO3 (25 мл), затем солевым раствором (25 мл), сушили (Na2SO4), фильтровали и концентрировали. Неочищенный продукт абсорбировали на Целите® и очищали хроматографией на силикагеле [20-80% EtOAc в гексанах], в результате чего получали трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1H-инден-1-ил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат (314.5 мг, 0.513 ммоль, выход 76%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1.13 (s, 3H), 1.20 (s, 3H), 1,35(d, J=7.0 Гц, 3H), 1.48 (s, 9Н), 1.79 - 1.90 (m, 1H), 1.97 - 2,12 (m, 1H), 2.29(dd, J=13.3, 7.0 Гц, 2H), 2.50 (br. s., 1H), 2.77 - 2.84 (m, 4Н), 3.04 - 3.16 (m, 2H), 3.25 - 3.36 (m, 1H), 4.10(dd, J=15.6, 2.3 Гц, 1H), 4.17(dd, J=15.6,2.3 Гц, 1H), 4.32 (s, 1H), 4.49 (кв, J=5.1 Гц, 1H), 4.55(dd, J=10.0, 6.1 Гц, 1H), 5.17(dd, J=9.2, 7.6 Гц, 1H), 5.54(dd, J=8.2, 5.9 Гц, 1H), 7.17 - 7.25 (m, 3H), 7.31(d, J=6.2 Гц, 1H), 7,42(d, J=8.6 Гц, 2H); ЖХМС(электроспрей) m/z: 613.4 (М+1)+.
Этап 4: трет-бутил((S)-1-(((4S,7S,9aS)-7-(((1S,2R)-2-(аллилокси)-2,3-дигидро-1H-инден-1-ил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,31 тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат
Суспензию трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден-1-ил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата (51.5 мг, 0.084 ммоль) и катализатор Линдлара (12.8 мг, 6.01 мкмоль) перемешивали в атмосфере водорода (1 атм) в МеОН (10 мл) в течение 2.5 часа. Суспензию фильтровали через 0.45 мкм диск и фильтрат выпаривали при пониженном давлении, в результате чего получали трет-бутил((S)-1-(((4S,7S,9aS)-7-(((1S,2R)-2-(аллилокси)-2,3-дигидро-1H-инден-1-ил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат (51.4 мг, 0.071 ммоль, выход 85%, чистота 85%) в виде пленки. ЖХМС(электроспрей) m/z: 615.4, 617.6 (М+1) смесь олефина и О-пропила (85:15 соотношение согласно протонному ЯМР).
Этап 5: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2')-(((E/Z)-бутил-2-ен-1,4-диил)бис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
Смесь трет-бутил((S)-1-(((4S,7S,9aS)-7-(((1S,2R)-2-(аллилокси)-2,3-дигидро-1H-инден-1-ил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата (51.4 мг, 0.084 ммоль) и катализатор Граббса 2-го поколения (6.39 мг, 7.52 мкмоль) перемешивали в дихлорметане (ДХМ) (350 мкл) при к.т.в атмосфере азота. Через 1 день к этой смеси добавляли вторую партию катализатора Граббса 2-го поколения (6.39 мг, 7.52 мкмоль) Через 4 дня неочищенную смесь очищали хроматографией на силикагеле [20-100% EtOAC в гексанах], в результате чего получали аддукт в виде смеси цис- и транс- изомеров (10.9 мг, 21.8%). ЖХМС(электроспрей) m/z: 1199.7 (М-1).
Этап 6: ди-трет-бутил((2S,2'S,)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(бутин-1,4-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
Суспензию ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(((E/Z)-бут-2-ен-1,4-диил)бис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (13 мг, 10.82 мкмоль) и палладия, 10 мас. % на активированном угле, Degussa, тип Е101 (1.151 мг, 1.082 мкмоль) перемешивали в атмосфере водорода (1 атм) в метаноле (4 мл). Через 1 день смесь фильтровали через 0.45 мкм диск и повторно подвергали гидрированию при 30 фунт./кв. дюйм в МеОН. Добавляли новую партию Pd/C и смесь перемешивали в атмосфере водорода при 50 фунт./кв. дюйм. Через 4 дня суспензию фильтровали через 0.45 мкм диск и растворитель выпаривали при пониженном давлении. Неочищенный продукт очищали С18 с помощью устройства ISCO [50-90% градиент ACN, 0.1% муравьиной кислоты], в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(бутан-1,4-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (4.5 мг, 3.74 мкмоль, выход 34.6%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1.08 (s, 3H), 1.17 (s, 3H), 1,35(d, J=7.0 Гц, 3H), 1.41-1.51 (m, 12H), 1.75-1.86 (m, 1H), 1.91(dd, J=13.3, 9.4 Гц, 1H),2,13 - 2.29 (m, 2H), 2.56 - 2.68 (m, 1H), 2.80 (s, 3H), 2.90 - 3.03 (m, 2H), 3.14(t, J=12.7 Гц, 1H), 3.31-3.51 (m,2Н),4.14(кв,1=4.3 Гц, 1H),4.28 (s, 1H), 4.51(dd, J=10.2, 5.9 Гц, 1H), 5.12(t, J=8.4 Гц, 1H), 5.46(dd, J=8.2, 5.9 Гц, 1H), 7.14 - 7.25 (m, 4 Н), 7.29(d, J=7.0 Гц, 1H), 7.35(d, J=4.3 Гц, 1H); ЖХМС(электроспрей) m/z: 502.4 {[(М - 2 Вос)+2]/2}+.
Этап 7: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(бутан-1,4-диилбис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(бутан-1,4-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (4.5 мг, 3.74 мкмоль) в дихлорметане (ДХМ) (1 мл) при к.т.добавляли хлорид водорода (4Mb 1,4-диоксане) (0.2 мл, 0.800 ммоль). Через 5 часов растворитель удаляли при пониженном давлении. Неочищенный продукт очищали С18 с помощью устройства ISCO [10-100% градиент ACN, 0.1% муравьиной кислоты], в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(бутан-1,4-диилбис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) (2.8 мг, 2.73 мкмоль, выход 73.1%) в виде белого твердого вещества. 1H-ЯМР (400 МГц, ХЛОРОФОРМ-d) δ ppm 1.11 (s, 3H), 1.17 (s, 3H), 1.29(d, J=7.0 Гц, 3H), 1.53 (br. s., 2H), 1.79 - 1.98 (m, 2H), 2,16 - 2.32 (m, 2H), 2.43 (s, 3H), 2.72(dd, J=14.8, 2.3 Гц, 1H), 2.99(d, J=4.7 Гц, 2H), 3.14 (кв, J=7.0 Гц, 1H), 3.24(t, J=12.9 Гц, 1H), 3.33 - 3.43 (m, 1H), 3.50(d, J=9.0 Гц, 1H), 4.18 (кв, J=4.7 Гц, 1H), 4.30 (s, 1H), 4.59(dd, J=10.1, 7.0 Гц, 1H), 5.17(t, J=8.4 Гц, 1H), 5.45(dd, J=8.4, 5.7 Гц, 1H), 7.15 - 7.25 (m, 3H), 7.29 -7.39 (m, 2H), 8.34(d,. J=6.6 Гц, 1H); ЖХМС(электроспрей) m/z: 1003.5 (М+1)+.
Пример 17
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(бутан-1,4-диилбис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
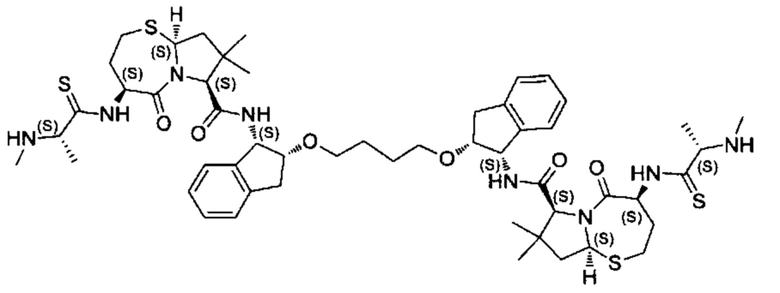
Этап 1: бутан-1,4-диилбис(4-метилбензолсульфонат)
К раствору бутан-1,4-диола (2 г, 22,19 ммоль) и триэтиламина (8.98 г, 48,82 ммоль) в дихлорметане (100 мл) добавляли 4-диметиламинопиридин (0.54 г, 4.44 ммоль) с последующим добавлением п-тозил хлорида (9.31 г, 48,82 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов. Смесь концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (3:1 об./об.)], в результате чего получали бутан-1,4-диилбис(4-метилбензолсульфонат) (4 г, 10.04 ммоль, выход 45.2%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.731 минуты, m/z: 420.8 (M+Na)+.
Этап 2: (1S,1'S,2R,2'R)-2,2'-(бутан-1,4-диилбис(окси))бис(2.3-дигидро-1Н-инден-1-амин)
К раствору (1S,2R)-1-амино-2,3-дигидро-1Н-инден-2-ола (749 мг, 5.02 ммоль) в тетрагидрофуране (80 мл) при 0°С порциями добавляли гидрид натрия (60%, дисперсия в Парафиновом Масле) (221 мг, 5.52 ммоль). Смеси давали нагреться до комнатной температуры. Затем добавляли бутан-1,4-диилбис(4-метилбензолсульфонат) (1 г, 2.51 ммоль). Полученную смесь перемешивали при 70°С в течение ночи. Реакцию гасили ледяной водой (50 мл) и экстрагировали этилацетатом (3 × 30 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [дихлорметан /метанол (10:1 об./об.), в результате чего получали (1S,1'S,2R,2'R)-2,2'-(бутан-1,4-диилбис(окси))бис(2,3-дигидро-1Н-инден-1-амин) (220 мг, 0.62 ммоль, выход 24.7%) в виде коричневого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания - 1.206 минуты, m/z: (М+1)+.
Этап 3: ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(бутан-1,4-диилбис(окси))бис(2.3-дигидро-1H-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат)
К смеси (1S,1'S,2R,2'R)-2,2'-(бутан-1,4-диилбис(окси))бис(2,3-дигидро-1H-инден-1-амина) (130 мг, 0.369 ммоль) и (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение II) (373 мг, 0.812 ммоль) в 1,2-дихлорэтане (15 мл) добавляли N-этоксикарбонил-2-этокси-1,2-дигидрохинолинил (274 мг, 1.107 ммоль) и N,N-диизопропилэтиламин (190 мг, 1.476 ммоль). Полученную смесь перемешивали при 50°С в течение ночи. Реакцию гасили водой (50 мл) и экстрагировали дихлорметаном (3 × 50 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (1/1 об./об.)] с последующей препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(бутан-1,4-диилбис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (60 мг, 0.049 ммоль, выход 13.3%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =2,154 минуты, m/z: 517.9 {[(М-2 Вос)+2]/2}+.
Этап 4: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(бутан-1,4-диилбис(окси))бис(2,3 -дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(бутан-1,4-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (60 мг, 0.049 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлорид водорода в 1,4-диоксане (3 мл). Смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель концентрировали при пониженном давлении и твердое вещество сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(бутан-1,4-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (30 мг, 0.027 ммоль, выход 57.7%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 11.01 (s, 1Н), 10.93 (s, 1Н), 9.63 (s, 1Н), 9.48 (s, 1Н), 8.64 (s, 2Н), 8.47 (s, 2Н), 7.98-7.93 (m, 2Н), 7.24-7.20 (m, 8Н), 5.46 (кв, J=8,8 Гц, 2Н), 5.35-5.32 (m, 2Н), 5.19-5.14 (m, 2Н), 4.28-4.26 (m, 4Н), 4.10-4.09 (m, 2Н), 3.73-3.65 (m, 2Н), 3.51-3.47 (m, 2Н), 3.39-3.38 (m, 2Н), 3.20-3.14 (m, 2Н), 2.96-2.95 (m, 4Н), 2.50 (s, 6Н), 2.27-2,13 (m, 4Н), 1.94-1.82 (m, 4Н), 1.45 (s, 10H), 1.07 (m, 12Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.408 минуты, m/z: 518.0 [(М+2)/2]+.
Пример 18
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-((оксибис(этан-2,1-диил))бис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид
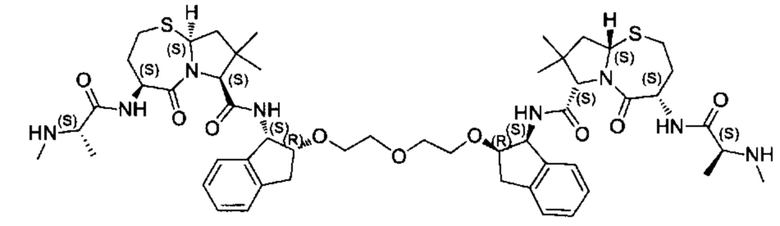
Этап 1: (1S,1'S,2R,2'R)-2,2'-((оксибис(этан-2,1-диил))бис(окси))бис(2,3-дигидро-1H-инден-1-амин)
К раствору (1S,2R)-1-амино-2,3-дигидро-1H-инден-2-ола (800 мг, 5.36 ммоль) в сухом тетрагидрофуране (20 мл) порциями медленно добавляли гидрид натрия (60%, дисперсия в Парафиновом Масле) (241 мг, 10.04 ммоль) при 0°С в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 30 минут и затем добавляли оксибис(этан-2,1-диил)бис(4-метилбензолсульфонат) (1.0 г, 2.44 ммоль). Смесь кипятили с обратным холодильником и перемешивали в течение 5 часов. Смесь осторожно гасили водой (150 мл) и экстрагировали этилацетатом (3 × 50 мл). Объединенные органические слои промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии (дихлорметан /метанол (8:1 об./об.)], в результате чего получали (1S,1'S,2R,2'R)-2,2'-((оксибис(этан-2,1-диил))бис(окси))бис(2,3-дигидро-1H-инден-1-амин) (410 мг, 1.11 ммоль, выход 53%) в виде желтого масла. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.107 минуты, m/z: 368.9 (М+1)+.
Этап 2: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((((1S,1'S,2R,2'R)-((оксибис(этан-2,1-диил))бис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b[1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
Раствор (1S,1'S,2R,2'R)-2,2'-((оксибис(этан-2,1-диил))бис(окси))бис(2,3-дигидро-1Н-инден-1-амина) (100 мг, 0.27 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение 1) (252.8 мг, 0.57 ммоль), N-этоксикарбонил-2-этокси-1,2-дигидрохинолинила (201,3 мг, 0.81 ммоль) N,N-диизопропилэтиламина (140.3 мг, 1.09 ммоль) в 1,2-дихлорэтане (6 мл) перемешивали при 50°С в течение ночи. Растворитель удаляли при пониженном давлении и остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (1/1 об./об.)], в результате чего получали продукт (260 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((((1S,1'S,2R,2'R)-((оксибис(этан-2,1-диил))бис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (180 мг, 0.15 ммоль, выход 54.4%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1,85 минут, [1/2(М-Вос)+Н]+=509.9 {[(М-2 Вос)+2]/2}.
Этап 3: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-((оксибис(этан-2,1-диил))бис(окси))бис(2.3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-((оксибис(этан-2,1-диил))бис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (180 мг, 0.12 ммоль) в дихлорметане (5 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи.
Растворитель концентрировали при пониженном давлении и твердое вещество сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-((оксибис(зтан-2,1-диил))бис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (12S,4 мг, 0.12 ммоль, выход 80%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 9.56 (m, 2Н), 8.95-8.90 (m, 4Н), 7.89(d, J=8.4 Гц, 2Н), 7.23-7.20 (m, 8Н), 5.51(t, J=7.6 Гц, 2Н), 5.34-5.30 (m, 2Н), 4.73(t, J=8.4 Гц, 2Н), 4.22-4.18 (m, 4Н), 3.91-3.89 (m, 2Н), 3.54-3.53 (m, 4Н), 3.46 (s, 4Н), 3.19(t, J=12.2 Гц, 2Н), 2.98-2.90 (s, 6Н), 2.46 (s, 6Н), 2.24-2,14 (m, 4Н), 1.86-1.77 (m, 4Н), 1.42-1.40 (m, 6Н), 1.06-1.05 (m, 12Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания=1,318 минуты, m/z: 1018.7 (М+1)+.
Пример 19
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S,2R,2'R)-((оксибис(этан-2,1-диил))бис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b|[1,3]тиазепин-7-карбоксамида) дигидрохлорид
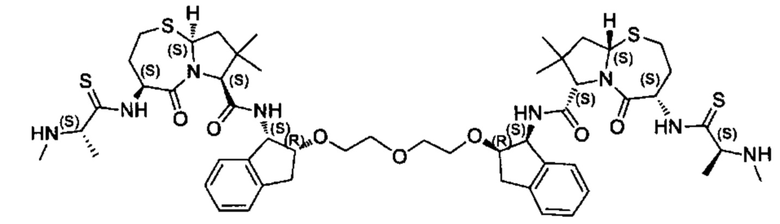
Этап 1: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-((оксибис(этан-2,1-диил))бис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1.2-диил))бис(метилкарбамат)
Раствор (1S,1'S,2R,2'R)-2,2'-((оксибис(этан-2,1-диил))бис(окси))бис(2,3-дигидро-1Н-инден-1-амина) (100 мг, 0.27 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение II) (262 мг, 0.57 ммоль), N-этоксикарбонил-2-этокси-1,2-дигидрохинолинила (201,3 мг, 0.81 ммоль) и N,N-диизопропилэтиламина (140.3 мг, 1.09 ммоль) в 1,2-дихлорэтане (6 мл) перемешивали при 50°С в течение ночи. Растворитель удаляли при пониженном давлении и неочищенный продукт очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (1/1 об./об.)], в результате чего получали продукт (200 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-((оксибис(этан-2,1-диил))бис(окси))бис(2,3-дигадро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктапцфопирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (150 мг, 0.13 ммоль, выход 44.1%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =2,11 минуты, m/z: 526.1 {[(М-2 Вос)+2]/2}+.
Этап 2: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-((оксибис(этан-2,1-диил))бис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-((оксибис(этан-2,1-диил))бис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (150 мг, 0,122 ммоль) в дихлорметане (5 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель концентрировали при пониженном давлении и твердое вещество сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-((оксибис(этан-2,1-диил))бис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (80.1 мг, 0.071 ммоль, выход 60%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 11.06-10.93 (m, 2Н), 9.76 (s, 1Н), 9.58 (s, 1Н), 8.65 (s, 1Н), 8.45 (s, 1Н), 7.98(t, J=10.0 Гц, 2Н), 7.23-7.20 (m, 8Н), 5.46 (кв, J=7.6 Гц, 2Н), 5.34-5.31 (m, 2Н), 5.19-5.13 (m, 2Н), 4.30-4.25 (m, 4Н), 4.18-4.17 (m, 2Н), 3.54-3.53 (m, 4Н), 3.47-3.46 (m, 4Н), 3.20(t, J=12.2 Гц, 2Н), 3.00-2.98 (m, 6Н), 2.50(s, 6Н), 2.33-2.21 (m, 4Н), 1.96-1.82 (m, 4Н), 1.45-1.43 (m, 6Н), 1.07 (s, 12Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.404 минуты, m/z: 1072.7 (M+Na)+.
Пример 20
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S,2R,2'R)-(гексан-1,6-диилбис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
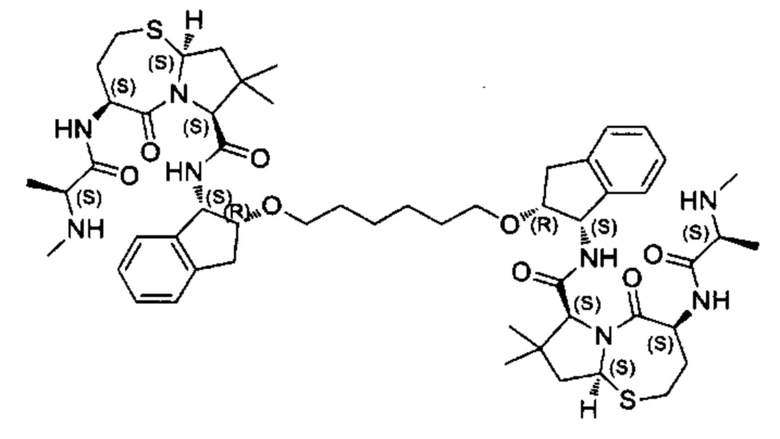
Этап 1: (1S,1'S,2R,2'R)-2,2'-(гексан-l,6-диилбис(окси))бис(2,3-дигидро-1H-инден-1-амин)
К раствору (1S,2R)-1-амино-2,3-дигидро-1Н-инден-2-ола (1,35 г, 9.10 ммоль) в тетрагидрофуране (80 мл) при 0°С порциями добавляли гидрид натрия (60%, дисперсия в Парафиновом Масле) (428 мг, 10.70 ммоль). Смеси давали нагреться до комнатной температуры. Затем добавляли 1,6-дибромгексан (1 г, 4.12 ммоль). Полученную смесь перемешивали при 70°С в течение ночи. Смесь гасили ледяной водой (50 мл) и экстрагировали этилацетатом (3 × 30 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [этилацетат/метанол (10:1 об./об.)], в результате чего получали (170 мг, 0.45 ммоль, выход 10.9%) в виде коричневого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.284 минуты, m/z: 381.0 (М+1)+.
Этап 2: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((((1S,1'S,2R,2'R)-(гексан-1.6-диилбис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
К смеси (1S,1'S,2R,2'R)-2,2'-(гексан-1,6-диилбис(окси)бис(2,3-дигидро-1H-инден-1-амина) (120 мг, 0.315 ммоль) и (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение I) (307 мг, 0,694 ммоль) в 1,2-дихлорэтане (8 мл) добавляли N-этоксикарбонил-2-этокси-1,2-дигидрохинолинил (233 мг, 0.945 ммоль) и N,N-диизопропилэтиламин (162 мг, 1.26 ммоль). Полученную смесь перемешивали при 50°С в течение ночи. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (3/2 об./об.)] с последующей препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(гексан-1,6-диилбис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (60 мг, 0.049 ммоль, выход 15.6%) в виде бесцветного масла. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =2.218 минуты, m/z: 515.9 {[(М-2 Вос)+2]/2}+.
Этап 3: (4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S,2R,2'R)-Ггексан-1,6-диилбис(окси))бис(2.3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(гексан-1,6-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (60 мг, 0.049 ммоль) в метаноле (5 мл) добавляли 4 н. хлорид водорода в 1,4-диоксане (4 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли при пониженном давлении и твердое вещество сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(гексан-1,6-диилбис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (30 мг, 0.027 ммоль, выход 55.1%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 9.33 (s, 2Н), 8,87-8,85 (m, 4Н), 7.91(d, J=8.4 Гц, 2Н), 7.24-7.21 (m, 8Н), 5.50(t, J=7.8 Гц, 2Н), 5.35-5.31 (m, 2Н), 4,76-4.72 (m, 2Н), 4.24 (s, 2Н), 4.11-4.10 (m, 2Н), 3.91-3.87 (m, 2Н), 3.72-3.65 (m, 2Н), 3.51-3,47 (m, 2Н), 3.39-3.37 (m, 4Н), 3.19(t, J=12.4 Гц, 2Н), 2.98-2.97 (m, 4Н), 2.93-2.89 (m, 2Н), 2.48 (s, 6Н), 2.24-2,13 (m, 4Н), 1.84-1.79 (m, 4Н), 1.41-1,39 (m, 10H), 1.08 (m, 6Н), 1.05 (m, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1,365 минуты, m/z: 1030.8 (М+1)+.
Пример 21
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(гексан-1,6-диилбис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
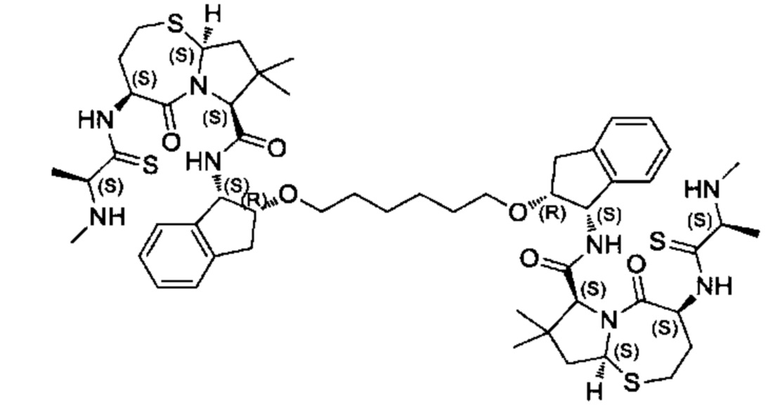
Этап 1: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(гексан-1,6-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазегшн-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат)
К смеси (1S,1'S,2R,2'R)-2,2'-(гексан-1,6-диилбис(окси)бис(2,3-дигидро-1H-инден-1-амина) (60 мг, 0,158 ммоль) и (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение II) (159 мг, 0.347 ммоль) в 1,2-дихлорэтане (5 мл) добавляли N-этоксикарбонил-2-этокси-1,2-дигидрохинолинил (117 мг, 0.474 ммоль) и N,N-диизопропилэтиламин (81 мг, 0.632 ммоль). Смесь перемешивали при 50°С в течение ночи. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали с помощью тонкослойной хроматографии [этилацетат /петролейный эфир (1/1 об./об.)] с последующей препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2,S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(гексан-1,6-диилбис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (30 мг, 0.024 ммоль, выход 15.2%) в виде бесцветного масла. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =2.218 минуты, m/z: 532.8 {[(М-2 Вос)+2]/2}+.
Этап 2: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(гексан-1,6-диилбис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(гексан-1,6-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (30 мг, 0.024 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлорид водорода в 1,4-диоксане (2 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и твердое вещество сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(гексан-1,6-диилбис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-даметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (13 мг, 0.011 ммоль, выход 48.3%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 11.02(d, J=6.4 Гц, 2Н), 10.94(d, J=6.4 Гц, 2Н), 9.71 (s, 1Н), 9.54 (s, 1H), 8.64 (s, 1Н), 8.44 (s, 1Н), 8.01-7.95 (m, 2Н), 7.24-7.21 (m, 8Н), 5.46 (кв, J=8.0 Гц, 2Н), 5.35-5.32 (m, 2Н), 5.21-5.13 (m, 2Н), 4.29-4.27 (m, 4Н), 4.11-4.07 (m, 2Н), 3.72-3.65 (m, 2Н), 3.41-3.36 (m, 6Н), 3.23-3.17 (m, 2Н), 3.01-2.97 (m, 6Н), 2.51 (s, 6Н), 2.33-2.23 (m, 4Н), 1.96-1.83 (m, 4Н), 1.45-1.43 (m, 10H), 1.08 (m, 12Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания=1.449 минуты, m/z: 532.0 [(М+2)/2]+.
Пример 22
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-((1,4-фениленбис(метилен))бис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)

Этап 1: (1S,1'S,2R,2'R)-2,2'-((1,4-фениленбис(метилен))бис(окси))бис(2,3-дигидро-1H-инден-1-амин)
К раствору (1S,2R)-1-амино-2,3-дигидро-1Н-инден-2-ола (800 мг, 5.36 ммоль) в сухом тетрагидрофуране (20 мл) при 0°С порциями медленно добавляли гидрид натрия (60%, дисперсия в Парафиновом Масле) (244 мг, 10.04 ммоль) в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 30 минутут, после чего добавляли 1,4-бис(бромметил)бензол (644 мг, 2.43 ммоль). Смесь кипятили с обратным холодильником в течение 5 часов. Смесь осторожно гасили водой (150 мл) и экстрагировали дихлорметаном (3 × 50 мл). Объединенные органические слои промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью тонкослойной хроматографии [дихлорметан/метанол (7:1 об./об.)], в результате чего получали (1S,1'S,2R,2'R)-2,2'-((1,4-фениленбис(метилен))бис(окси))бис(2,3-дигидро-1H-инден-1-амин) (200 мг, 0.53 ммоль, выход 22%) в виде желтого масла. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.208 минуты, m/z: 400.9 (М+1)+.
Этап 2: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-((1,4-фениленбис(метилен))бис(окси)')бис(2.3-дигидро-1H-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][3,1]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1.2-диил)1бис(метилкарбамат)
Раствор (1S,1'S,2R,2'R)-2,2'-((1,4-фениленбис(метилен))бис(окси))бис(2,3-дигидро-1Н-инден-1-амина) (80 мг, 0.21 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение 1) (198 мг, 0.45 ммоль), N-этоксикарбонил-2-этокси-1,2-дигидрохинолинила (157.6 мг, 0.64 ммоль) и N,N-диизопропилэтиламина (110 мг, 0.85 ммоль) в 1,2-дихлорэтане (5 мл) перемешивали при 50°С в течение ночи. Растворитель концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (1/1 об./об.)], в результате чего получали продукт (110 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-((1,4-фениленбис(метилен))бис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (68 мг, 0.045 ммоль, выход 26.1%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.86 минуты, m/z: 526.2 {[(М-2 Вос)+2]/2}+.
Этап 3: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-(1S,1'S,2R,2'R)-((1,4-фениленбис(метилен))бис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-((1,4-фениленбис(метилен))бис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (68 мг, 0.055 ммоль) в дихлорметане (5 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Неочищенный продукт разбавляли водой и доводили рН до 8~9 с помощью бикарбоната натрия и экстрагировали дихлорметаном (3 × 20 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [дихлорметан/метанол (9/1 об./об.)], в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-((1,4-фениленбис(метилен))бис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) (38,8 мг, 0.037 ммоль, выход 67.3%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 8.63-8.62 (m, 2Н), 7.98(d, J=9.2 Гц, 2Н), 7.26-7.23 (m, 12Н), 5.50(t, J=7.8 Гц, 2Н), 5.41-5.37 (m, 2Н), 4.67-4.72 (m, 2Н), 4.51 (s, 4Н), 4.25-4.20 (m, 4Н), 3.58-3.56 (m, 2Н), 3.16-2.95 (m, 6Н), 2.74-2.61 (s, 2Н), 2.40 (s, 6Н), 2.34-2,19 (m, 2Н), 2.05-2.02 (m, 2Н), 1.81-1.76 (m, 2Н), 1.73-1.68 (m, 2Н), 1,30(d, J=6.4 Гц, 6Н), 1.04 (m, 12Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1,397 минуты, m/z: 1051.7 (М+1)+.
Пример 23
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-((1,4-фениленбис(метилен))бис(окси))бис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)
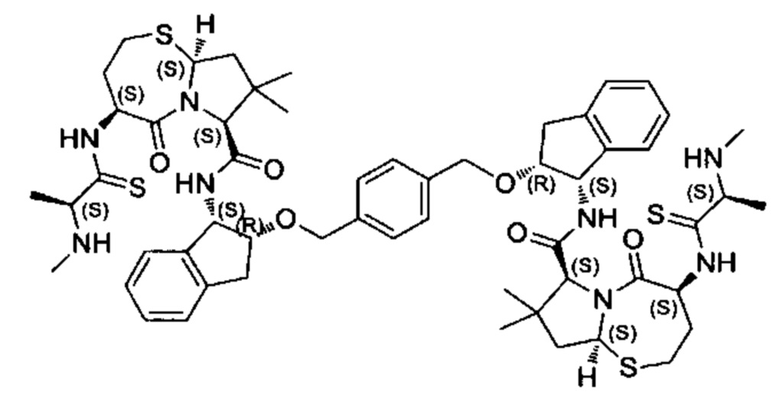
Этап 1: трет-бутил((S)-1-(((4S,7S,9aS)-7-(((1S,2R)-2-((4-((((1S,2R)-1-((4S,7S,9aS)-4-((S)-2-трет-бутоксикарбонил)метил)амино)пропантиоамидо)8,8-диметил-5-оксооктагадропирроло[2,1-b][1,3]тиазепин-7-карботиоамидо)-2,3-дигидро-1H-инден-2-ил)окси)метил)бензил)окси)2,3-дигадро-1Н-инден-1-ил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-тиоксопропан-2-ил)(метил)карбамат
Раствор (1S,1'S,2R,2'R)-2,2'-((1,4-фениленбис(метилен))бис(окси))бис(2,3-дигидро-1H-инден-1-амина) (80 мг, 0.21 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение II) (205.1 мг, 0.45 ммоль), К-этоксикарбонил-2-этокси-1,2-дигидрохинолинила (157.6 мг, 0.64 ммоль) и N,N-диизопропилэтиламина (110 мг, 0.85 ммоль) в 1,2-дихлорэтане (5 мл) перемешивали при 50°С в течение ночи. Растворитель удаляли при пониженном давлении и неочищенный продукт очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (1/1 об./об.)], в результате чего получали продукт (120 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали трет-бутил((S)-1-(((4S,7S,9aS)-7-(((1S,2R)-2-((4-((((1S,2R)-1-((4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карботиоамидо)-2,3-дигидро-1H-инден-2-ил)окси)метил)бензил)окси)-2,3 -дигидро-1H-инден-1-ил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-тиоксопропан-2-ил)(метил)карбамат (60 мг, 0.047 ммоль, выход 22.4%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =2,144 минуты, m/z: 541.9 {[(М-2 Вос)+2]/2}+.
Этап 2: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-((1,4-фениленбис(метилен))бис(окси))бис(2.3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-((1,4-фениленбис(метилен))бис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (60 мг, 0.047 ммоль) в дихлорметане (5 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении. Неочищенный продукт разбавляли водой и доводили рН до 8~9 с помощью бикарбоната натрия и экстрагировали дихлорметаном (3*20 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [дихлорметан/метанол (10/1 об./об.)], в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-((1,4-фениленбис(метилен))бис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) (21.9 мг, 0.020 ммоль, выход 42.6%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 7.98(d, J=8,8 Гц, 2Н), 7.24 (m, 12Н), 5.51-5.46 (m, 2Н), 5.42-5.39 (m, 2Н), 5.11 (d, J=10.4 Гц, 2Н), 4.51 (s, 4Н), 4.29(d, J=10.4 Гц, 2Н), 4.24-4.21 (m, 2Н), 3.31 (s, 4Н), 3.17-3.09 (m, 2Н), 3.05-2.97 (s, 4Н), 2.79-2.73 (m, 2Н), 2.26-2.26 (m, 10H), 1.81-1.73 (m, 2Н), 1.68-1.66 (m, 2Н), 1.29-1.28 (m, 6Н), 1.06-1.05 (m, 12Н). ЖХМС (2,5 минуты, муравьиная кислота): Время удерживания =1.443 минуты, m/z: 1083.7 (М+1)+.
Пример 24
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-((1,4-фениленбис(метилен))бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)
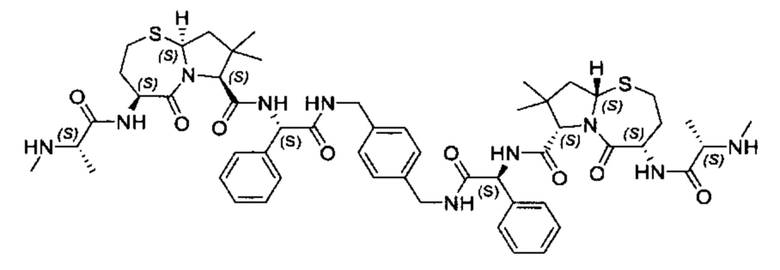
Этап 1: (S)-2-((трет-бутоксикарбонил)амино)-2-фенилуксусная кислота
К раствору (S)-2-амино-2-фенилуксусной кислоты (3.0 г, 19.8 ммоль) в 1 М раствора гидроксида натрия (28 мл) добавляли раствор ди-трет-бутил дикарбоната (4.75 г, 21.78 ммоль) в трет-бутиловом спирте (16 мл). Полученную суспензию перемешивали при комнатной температуре в течение 1 часа. Летучий растворитель удаляли при пониженном давлении и рН оставшегося раствора доводили до 3 с помощью лимонной кислоты. Твердое вещество фильтровали и сушили, в результате чего получали (S)-2-((трет-бутоксикарбонил)амино)-2-фенилуксусную кислоту (4.1 г, 16.3 ммоль, выход 82%). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.48 минут, [М+Н]+=273.9,
Этап 2: ди-трет-бутил((1S,1'S)-((1,4-фениленбис(метилен))бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))дикарбамат
К раствору (S)-2-((трет-бутоксикарбонил)амино)-2-фенилуксусной кислоты (700 мг, 2.78 ммоль) в N,N-диметилформамиде (15 мл) при -15°С добавляли 1,4-фенилендиметанамин (189 мг, 1,39 ммоль) в N,N-диметилформамиде (5 мл). Затем последовательно добавляли диэтил цианофосфонат (680 мг, 4.17 ммоль) и триэтиламин (562 мг, 5.56 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (400 мл), в результате чего получали осадок, который фильтровали и сушили в глубоком вакууме, в результате чего получали ди-трет-бутил((1S,1'S)-((1,4-фениленбис(метилен))бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))дикарбамат (1.1 г, 1.83 ммоль, выход 59.6%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.56 минуты, m/z: 502.7 [(М-Вос)+1]+.
Этап 3: (2S,2'S)-N,N'-(1,4-фениленбис(метилен))бис(2-амино-2-фенилацетамида) дигидрохлорид
К раствору ди-трет-бутил((1S,1'S)-((1,4-фениленбис(метилен))бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))дикарбамата (1.1 г, 1.83 ммоль) в дихлорметане (10 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 2 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи и растворитель удаляли при пониженном давлении, в результате чего получали (2S,2'S)-N,N'-(1,4-фениленбис(метилен))бис(2-амино-2-фенилацетамида) дигидрохлорид (810 мг, неочищенный) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =0.38 минуты, m/z: 403.0 (М+1)+.
Этап 4: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-(((1S,1'S)-((1,4-фениленбис(метилен))бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
Раствор (2S,2'S)-N,N'-(1,4-фениленбис(метилен))бис(2-амино-2-фенилацетамида) дигидрохлорида (200 мг, 0.43 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (379 мг, 0.86 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимид метиодида (246 мг, 1.28 ммоль), 1-гидроксибензотриазола(231 мг, 1.71 ммоль) и N,N'-диизопропилэтиламина (331 мг, 2.56 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (50 мл) и экстрагировали дихлорметаном (3 × 50 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали неочищенный продукт (96 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-((1,4-фениленбис(метилен))бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (67 мг, 0.053 ммоль, выход 12.6%) в виде белого твердого вещества. ЖХМС (3.0 минуты, муравьиная кислота): Время удерживания =2.55 минуты, m/z: 526.5 {[(М-2 Вос)+2]/2}+, 1153.4 (М-Вос+1)+.
Этап 5: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-((1,4-фениленбис(метилен))бис(азандиил))бис(2-оксо-1-фенил этан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (67 мг, 0.05 ммоль) в дихлорметане (10 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении, в результате чего получали неочищенный продукт. Неочищенный продукт разбавляли водой, рН доводили до 8~9 с помощью бикарбоната натрия и экстрагировали дихлорметаном (3 × 20 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [дихлорметан / метанол (8/1 об./об.)], в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-((1,4-фениленбис(метилен))бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) (47.5 мг, 0.045 ммоль, выход 84%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 8.79 (t, J=5.8 Гц, 2H), 8.52(d, J=8.0 Гц, 2H), 8.21(d, J=6.8 Гц, 2H), 7,46-7,44 (m, 4Н), 7.38-7.29 (m, 6Н), 6.96 (s, 4Н), 5.55-5.50 (m, 4Н), 4.69-4.65 (m, 2Н), 4.23 (s, 6Н), 3.22-3.16 (m, 2Н), 2.96-2.88 (m, 4Н), 2.22 (s, 8Н), 2,14-2.09 (m, 2Н), 1.84-1.71 (m, 4Н), 1.12(d, J=7.2 Гц, 6H), 1.05 (s, 6Н), 0.90 (s, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.248 минуты, m/z: 1052.7 (М+1)+.
Пример 25
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-((1,4-фенилембис(метилен))бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид
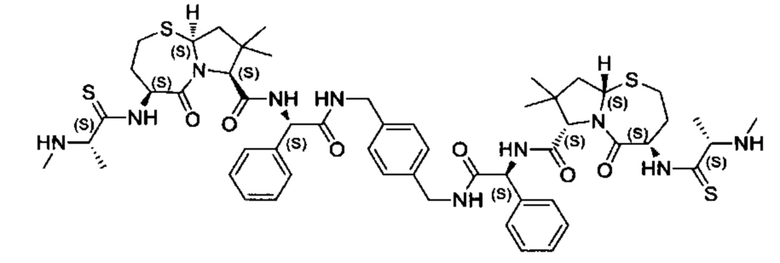
Этап 1: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-((1,4-фениленбис(метилен))бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1.2-диил))бис(метилкарбамат)
Раствор (2S,2'S)-N,N'-(1,4-фениленбис(метилен))бис(2-амино-2-фенилацетамида) дигидрохлорида (200 мг, 0.42 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение II) (391 мг, 0.85 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимид метиодида (243 мг, 1.27 ммоль), 1-гидроксибензотриазола (228 мг, 1.69 ммоль) и N,N-диизопропилэтиламина (327 мг, 2.53 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение ночи. Смесь гасили водой (50 мл) и экстрагировали дихлорметаном (3 × 50 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали неочищенный продукт (96 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-6ynui((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-((1,4-фениленбис(метилен))бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидрогшрроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (63 мг, 0.049 ммоль, выход 11.7%) в виде белого твердого вещества. ЖХМС (3.0 минуты, муравьиная кислота): Время удерживания =2.76 минуты, m/z: 542.9 {[(М-2 Вос)+2]/2}+, 1185.6 (М-Вос+1)+.
Этап 2: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(1,4-фениленбис(метилен))бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1R,1'R,2R,2'R)-(((1,4-фениленбис(метилен))бис(азандиил))бис(карбонил))бис(1,2,3,4-тетрагидронафталин-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (63 мг, 0.049 ммоль) в дихлорметане (5 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-((1,4-фениленбис(метилен))бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (33,2 мг, 0.029 ммоль, выход 57.8%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 8,83(t, J=6.0 Гц, 2Н), 8.61(d, J=6.4 Гц, 2Н), 7,46-7,44 (m, 4Н), 7.38-7.29 (m, 6Н), 6.95 (s, 4Н), 5.56-5.53 (m, 2Н), 5.51-5.46 (m, 2Н), 5.17-5.13 (m, 2Н), 4.27-4.20 (m, 4Н), 4.12-4.08 (m, 2Н), 3.72-3.65 (m, 1Н), 3.52-3.47 (m, 1Н), 3.25-3.18 (m, 2Н), 3.01-2.97 (m, 2Н), 2.42(d, J=4.8 Гц, 6Н), 2.34-2.26 (m, 4Н), 2.01-1.90 (m, 2Н), 1.81-1.76 (m, 2Н), 1,39(d, J=5.4 Гц, 6Н), 1.08 (s, 6Н), 0.93 (s, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1,312 минуты, m/z: 1085.7 (М+1)+.
Пример 26
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-пиперазин-1,4-диилбис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорид
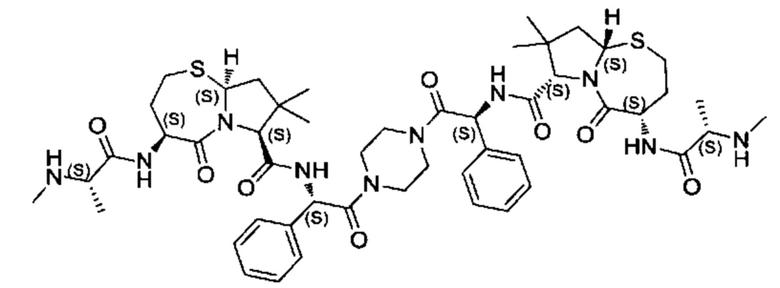
Этап 1: ди-трет-бутил((1S,1'S)пиперазин-1,4-диилбис(2-оксо-1-фенилэтан-2,1-диил))дикарбамат
К раствору (S)-2-((трет-бутоксикарбонил)амино)-2-фенилуксусной кислоты (500 мг, 2,1 ммоль) в N,N-диметилформамиде (15 мл) при -15°С последовательно добавляли пиперазин (85.6 мг, 1.0 ммоль) в N,N-диметилформамиде (5 мл), диэтилцианофосфонат (488 мг, 3.0 ммоль) и триэтиламин (402.7 мг, 4.0 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Смесь вливали в воду (300 мл), в результате чего получали осадок, который фильтровали и сушили, в результате чего получали ди-трет-бутил((1S,1'S)-пиперазин-1,4-диилбис(2-оксо-1-фенилэтан-2,1-диил))дикарбамат (640 мг, 1.2 ммоль, выход 58.2%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.57 минуты, m/z: 575.8 (M+Na)+.
Этап 2: (2S,2'S)-1,1'-(пиперазин-1,4-диил)бис(2-амино-2-фенилэтан-1-он) дигидрохлорид
К раствору ди-трет-бутил((1S,1'S)-пиперазин-1,4-диилбис(2-оксо-1-фенилэтан-2,1-диил))дикарбамата (200 мг, 0.32 ммоль) в дихлорметане (10 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении и сушили в глубоком вакууме, в результате чего получали неочищенный (2S,2'S)-1,1'-(пиперазин-1,4-диил)бис(2-амино-2-фенилэтан-1-она) дигидрохлорид (150 мг) в виде белого твердого вещества, которое использовали на следующем этапе без дальнейшей очистки. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =0.39 минуты, m/z: 352.9 (М+1)+.
Этап 3: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-пиперазин-1,4-диилбис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
Раствор (2S,2'S)-1,1'-(пиперазин-1,4-диил)бис(2-амино-2-фенилэтан-1-она) дигидрохлорида (300 мг, 0.85 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (750 мг, 1.69 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимид метиодида (490 мг, 2.56 ммоль), 1-гидроксибензотриазола (460 мг, 3.40 ммоль) и N,N-диизопропилэтиламина (660 мг, 5.11 ммоль) в дихлорметане (20 мл) перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (100 мл) и экстрагировали дихлорметаном (3 × 10 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали неочищенный продукт (170 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-пиперазин-1,4-диилбис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (80 мг, 0.067 ммоль, выход 13.5%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.73 минуты, m/z: 502.0 {[(М-2 Вос)+2]/2}+, [М-Вос+Н]+=1104.8 (М-Вос+1)+.
Этап 3: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-пиперазин-1,4-диилбис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-пиперазин-1,4-диилбис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (80 мг, 0.067 ммоль) в дихлорметане (5 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-пиперазин-1,4-диилбис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (36 мг, 0.033 ммоль, выход 49.3%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 8.56-8.51 (m, 2Н), 8.47(d, J=7.6 Гц, 2Н), 7.37-7.33 (m, 10H), 5.88-5.81 (m, 2Н), 5.50(t, J=7.6 Гц, 2Н), 4.68(t, J=7,4 Гц, 2Н), 4.10-4.08 (m, 2Н), 3.55-3.37 (m, 4Н), 3.25-3.13 (m, 6Н), 2.97-2.90 (m, 4Н), 2.32 (s, 6Н), 2,18-2,13 (m, 4Н), 1.97-1.87 (m, 2Н), 1.57-1.49 (m, 2Н), 1.24(d, J=5.6 Гц, 6Н), 1.00 (s, 6Н), 0.81-0.78 (m, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1,368 минуты, m/z: 1003.8 (М+1)+.
Пример 27
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-пиперазин-1,4-диилбис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
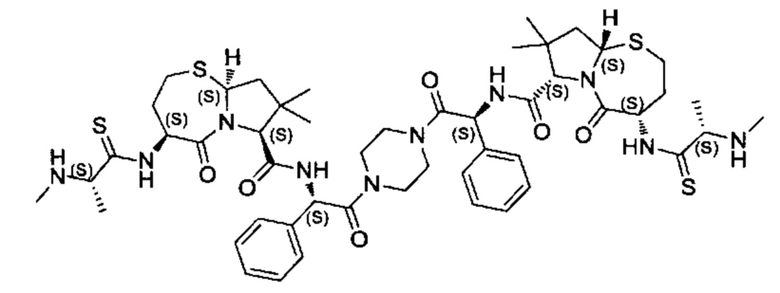
Этап 1: ди-трет-бутил(2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-пиперазин-1,4-диилбис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат)
Раствор (2S,2'S)-1,1'-(пиперазин-1,4-диил)бис(2-амино-2-фенилэтан-1-она) дигидрохлорида (200 мг, 0.47 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение II) (432.2 мг, 0.94 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида метиодида (270.4 мг, 1.41 ммоль), 1-гидроксибензотриазола (254.1 мг, 1,88 ммоль) и N,N-диизопропилэтиламина (364.5 мг, 2.82 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (80 мл) и экстрагировали дихлорметаном (3 × 80 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали неочищенный продукт (170 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ с получением целевого продукта (90 мг, 0.073 ммоль, выход 15,5%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =2.04 минуты, m/z: 641.0 [M+2Na]/2,
Этап 2: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-пиперазин-1,4-диилбис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-пиперазин-1,4-диилбис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (90 мг, 0.073 ммоль) в дихлорметане (5 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении и сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-пиперазин-1,4-диилбис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (52.3 мг, 0.047 ммоль, выход 64.2%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 8.53(d, J=7.6 Гц, 2Н), 7,41-7.34 (m, 12Н), 5.89-5.82 (m, 2Н), 5.49-5.43 (m, 2Н), 5.14(t, J=9.6 Гц, 2Н), 4.17-4.09 (m, 4Н), 3.57 (s, 2Н), 3.41 (s, 2Н), 3.23-3.07 (m, 5Н), 3.01-2.96 (m, 3H), 2.44 (s, 6Н), 2.33-2.26 (m, 2Н), 2.20-2,17 (m, 2Н), 2.08-1.99 (m, 2Н), 1.60-1.57 (m, 2Н), 1.40(t, J=7.6 Гц, 6Н), 1.02 (s, 6Н), 0.86-0.82 (m, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1,341 минуты, m/z: 1035.7 (М+1)+.
Пример 28
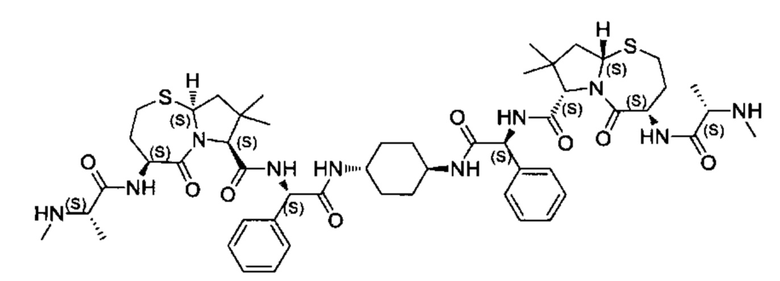
(2S)-N-[(4S,7S,9aS)-8,8-диметил-5-оксо-7-{[(S)-фенил({[(1rs,4rs)-4-[(2S)-2-{[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-ил]формамидо}-2-фенилацетамидо]циклогексил]карбамоил})метил]карбамоил}-октагидропирроло[2,1-b][1,3]тиазепин-4-ил]-2-(метиламино)пропанамида дигидрохлорид
Этап 1: ди-трет-бутил((1S,1'S)-(((1S,4S)-циклогексан-1,4-диил)бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))дикарбамат
К раствору (S)-2-((трет-бутоксикарбонил)амино)-2-фенилуксусной кислоты (600 мг, 2,1 ммоль) в N,N-диметилформамиде (20 мл) при -15°С последовательно добавляли (1 г,4 г)-циклогексан-1,4-диамин (136.3 мг, 1.0 ммоль), диэтил цианофосфонат (584 мг, 3.0 ммоль) и триэтиламин (483 мг, 4.0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь вливали в воду (300 мл), в результате чего получали осадок, который фильтровали и сушили, в результате чего получали ди-трет-бутил((1S,1'S)-(((1S,4S)-циклогексан-1,4-диил)бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))дикарбамат (640 мг, 1.1 ммоль, выход 46.2%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.61 минуты, m/z: 480.8 (М-Вос+1)+.
Этап 2: (2S,2'S)-N,N'-((1S,4S)-циклогексан-1,4-диил)бис(2-амино-2-фенилацетамида) дигидрохлорид
К раствору ди-трет-бутил((1S,1'S)-(((1S,4S)-циклогексан-1,4-диил)бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))дикарбамата (460 мг, 0.32 ммоль) в дихлорметане (10 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 2 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и сушили под вакуумом, в результате чего получали (2S,2'S)-N,N'-((1S,4S)-циклогексан-1,4-диил)бис(2-амино-2-фенилацетамида) дигидрохлорид (неочищенный, 400 мг) в виде белого твердого вещества. Этот материал использовали на следующем этапе без дальнейшей очистки. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =0.37 минуты, m/z: 381.0 (М+1).
Этап 3: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-(((1R,4R)-циклогексан-1,4-диил)бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
Раствор (2S,2'S)-N,N'-((1S,4S)-циклогексан-1,4-диил)бис(2-амино-2-фенилацетамида) дигидрохлорида (200 мг, 0.44 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (387 мг, 0,87 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимид метиодида (239 мг, 1.25 ммоль), 1-гидроксибензотриазола (225 мг, 1.67 ммоль) и N,N-диизопропилэтиламина (323 мг, 2.50 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (50 мл) и экстрагировали дихлорметаном (3 × 50 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали неочищенный продукт (110 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил ди-трет-6ymu((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-(((1R,4R)-циклогексан-1,4-диил)бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (61 мг, 0.049 ммоль, выход 11.2%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания=1.68 минуты, m/z: 1130.7 (М-Вос+1)+.
Этап 4: (2S)-N-[(4S,7S,9aS)-8,8-диметил-5-оксо-7-{[(S)(фенил({[(1rs,4rs)-4-[(2S)-2-{[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-ил]формамидо}-2-фенилацетамидо]циклогексил]карбамоил})метил]карбамоил}-октагидропирроло[2,1-b][1,3]тиазепин-4-ил]-2-(метиламино)пропанамида дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-(((1R,4R)-циклогексан-1,4-диил)бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (61 мг, 0.05 ммоль) в дихлорметане (5 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель концентрировали при пониженном давлении и сушили в глубоком вакууме, в результате чего получали указанное в заголовке соединение(36.6 мг, 0.033 ммоль, выход 66.5%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 9.25 (s, 2Н), 8.91-8,82 (m, 4Н), 8.49(d, J=8.4 Гц, 2Н), 8.23(d, J=7.6 Гц, 2Н), 7,42-7,40 (m, 4Н), 7.35-7.31 (m, 4Н), 7.28-7.24 (m, 2Н), 5.52(t, J=8.0 Гц, 2Н), 5.46(d, J=8.0 Гц, 2Н), 4.75-4.70 (m, 2Н), 4.19 (m, 2Н), 3.87 (m, 2Н), 3.72-3.65 (m, 1Н), 3.50-3.39 (m, 3H), 3.25-3.19 (m, 2Н), 2.96-2.92 (m, 2Н), 2.48 (s, 6Н), 2.24-2,13 (m, 4Н), 1.92-1.69 (m, 6Н), 1.58-1.56 (m, 2Н), 1,39(d, J=6.8 Гц, 6Н), 1.04 (s, 6Н), 0.94 (s, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1.404 минуты, m/z: 1031.7 (М+1)+.
Пример 29
(2S)-2-{[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-ил]формамидо}-2-фенил-N-[(1rs,4rs)-4-[(2S)-2-{[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-ил|формамидо}-2-фенилацетамидо]циклогексил]ацетамида дигидрохлорид
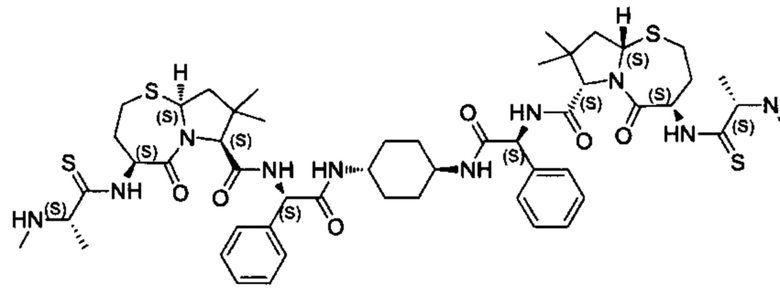
Этап 1: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-(((1R,4R)-циклогексан-1,4-диил)бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидрошфроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат)
Раствор (2S,2'S)-N,N'-((1S,4S)-циклогексан-1,4-диил)бис(2-амино-2-фенилацетамида) дигидрохлорида (170 мг, 0.45 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение II) (362 мг, 0.79 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимид метиодида (216 мг, 1.13 ммоль), 1-гидроксибензотриазола (203 мг, 1.5 ммоль) и N,N-диизопропилэтиламина (291 мг, 2.25 ммоль) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (50 мл) и экстрагировали дихлорметаном (3 × 50 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат /петролейный эфир (2/1 об./об.)], в результате чего получали продукт (210 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-(((1R,4R)-циклогексан-1,4-диил)бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (127 мг, 1.0 ммоль, выход 26.8%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =2,182 минуты, m/z: 532,1 {[(М-2 Вос)+2]/2}+.
Этап 2: (2S)-2-{[(4S,7S,9aS)-8,8-диметил-4-((2S)-2-(метиламино)пропантиоамидо 1-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-ил]формамидо}-2-фенил-N-[(1rs,4rs)-4-[(2S)-2-{[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-ил]формамидо}-2-фенилацетамидо]иклогексил]ацетамида дигидрохлорид
К раствору ди-трет-бутил(2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-(((1R,4R)-циклогексан-1,4-диил)бис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(азандиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (127 мг, 0.10 ммоль) в дихлорметане (10 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении и твердое вещество сушили в глубоком вакууме, в результате чего получали указанное в заголовке соединение(100 мг, 0.08 ммоль, выход 87,4%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 11.02-10.90 (m, 2Н), 9.75 (s, 1Н), 9.58 (s, 1Н), 8.63-8.44 (m, 4Н), 8.26(d, J=7.6 Гц, 2Н), 7,42-7,40 (m, 4Н), 7.35-7.31 (m, 4Н), 7.28-7.25 (m, 2Н), 5.50-5.45 (m, 4Н), 5.18-5.12 (m, 2Н), 4.27-4.21 (m, 4Н), 3.48-3.93 (m, 2Н), 3.26-3.17 (m, 2Н), 3.10-2.98 (m, 4Н), 2.48 (s, 6Н), 2.33-2.23 (m, 4Н), 2.03-1.95 (m, 2Н), 1.81-1.74 (m, 4Н), 1.58 (d, J=7.2 Гц, 2Н), 1.45-1.43 (m, 6Н), 1.07 (s, 8Н), 0.92 (s, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1,351 минуты, m/z: 1063.6 (М+1)+.
Пример 30
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S)-(гекса-2,4-диин-1,6-диилбис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
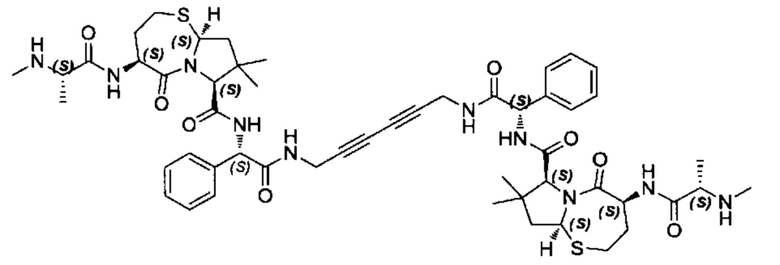
Этап 1: трет-бутил (S)-(2-оксо-1-фенил-2-(проп-2-ин-1-иламино)этил)карбамат
К раствору (S)-2-((трет-бутоксикарбонил)амино)-2-фенилуксусной кислоты (600 мг, 2.3 ммоль) в дихлорметане (18 мл) добавляли проп-2-ин-1-амин (131.5 мг, 2.3 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (1.82 г, 4.8 ммоль) и N,N-диизопропилэтиламин (925.8 мг, 7.2 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Смесь гасили водой (300 мл) и экстрагировали дихлорметаном (3 × 200 мл). Объединенные органические слои промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (1/3 об./об.)], в результате чего получали трет-бутил (S)-(2-оксо-1-фенил-2-(проп-2-ин-1-иламино)этил)карбамат (неочищенный, 300 мг) в виде желтого масла. ЖХМС (3.0 минуты, муравьиная кислота): Время удерживания =1.677 минуты, m/z: 289 (М+1)+.
Этап 2: (S)-2-амино-2-фенил-N-(проп-2-ин-1-ил)ацетамид гидрохлорид
К раствору трет-бутил(S)-(2-оксо-l -фенил-2-(проп-2-ин-1-иламино)этил)карбамата (300 мг, 1.04 ммоль) в дихлорметане (12 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении, в результате чего получали трет-бутил(S)-(2-оксо-1-фенил-2-(проп-2-ин-1-иламино)этил)карбамат (300 мг) в виде желтого твердого вещества, которое использовали на следующем этапе без дальнейшей очистки. ЖХМС (3.0 минуты, муравьиная кислота): Время удерживания =0.801 минуты, m/z: 189 (М+1)+.
Этап 3: трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((S)-2-оксо-1-фенил-2-(проп-2-ин-1-иламино)этил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат
К раствору (S)-2-амино-2-фенил-N-(проп-2-ин-1-ил)ацетамида гидрохлорида (250 мг, 1.1 ммоль) в 1,2-дихлорэтане (15 мл) добавляли (4S,7S,9aS)-4-((S)-3-((трет-бутоксикарбонил)(метил)амино)-2-оксобутил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновую кислоту (Промежуточное соединение I) (589 мг, 1,3 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,'N'-тетраметалурония гексафторфосфат (808 мг, 2,1 ммоль) и N,N-диизопропилэтиламин (412 мг, 3.2 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (300 мл) и экстрагировали дихлорметаном (3 × 60 мл). Объединенные органические слои промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((S)-2-оксо-1-фенил-2-(проп-2-ин-1-иламино)этил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат (578 мг, 0.919 ммоль, выход 83.5%) в виде желтого масла. ЖХМС (3.0 минуты, муравьиная кислота): Время удерживания - 1.794 минуты, m/z: 636 (M+Na)+.
Этап 4 ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-7,7'-(3S,14S)-4,13-диоксо-3,14-дифенил-2,5,12,15-тетраазагексадека-7,9-диин-1,16-диоии)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-2,1-диил))бис(метилкарбамат)
К смеси трет-бутил((S)4-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((S)-2-оксо-1-фенил-2-(проп-2-ин-1-иламино)этил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата (170 мг, 0.27 ммоль) в ацетонитриле (8 мл) и пиридина (131,6 мг, 1,62 ммоль) добавляли ацетат меди (II) (60.4 мг, 0.33 ммоль). Полученную смесь перемешивали при 85°С в течение 1 часа в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, гасили водой (50 мл) и экстрагировали дихлорметаном (3 × 50 мл). Объединенные органические слои промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали неочищенный продукт (86 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-7,7'-((3S,14S)-4,13-диоксо-3,14-дифенил-2,5,12,15-тетраазагексадека-7,9-диин-1,16-диоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-2,1-диил))бис(метилкарбамат) (58 мг, 0.047 ммоль, выход 17.1%) в виде белого твердого вещества. ЖХМС (3.0 минуты, муравьиная кислота): Время удерживания =2.044 минуты, m/z: 1247 (M+Na)+.
Этап 5: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,l'S)-(гекса2,4-диин-1,6-диилбис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-7,7'-((3S,14S)-4,13-диоксо-3,14-дифенил-2,5,12,15-тетраазагексадека-7,9-диин-1,16-диоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-2,1-диил))бис(метилкарбамата) (38 мг, 0.03 ммоль) в дихлорметане (7 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Растворитель удаляли при пониженном давлении и продукт сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(гекса-2,4-диин-1,6-диилбис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (30 мг, 0.027 ммоль, выход 88.3%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 9.08-8,81 (m, 8Н), 8.52(d, J=8.0 Гц, 2Н), 7,42-7,40 (m, 3H), 7.37-7.28 (m, 5Н), 5.54-5.47 (m, 4Н), 4.76-4.71 (m, 2Н), 4.19 (s, 2Н), 4.0(d, J=5.2 Гц, 4Н), 3.89-3.84 (m, 2Н), 3.25-3.18 (m, 2Н), 2.97-2.91 (m, 2Н), 2.49 (s, 6Н), 2.24-2,19 (m, 2Н), 2,17-2,12 (m, 2Н), 1.93-1.83 (m, 2Н), 1.75-1.70 (m, 2Н), 1,38(d, J=6.4 Гц, 6Н), 1.05 (s, 6Н), 0.91 (s, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания =1,309 минуты, m/z: 1025.7 (М+1)+.
Пример 31
(4S,7S,9aS)-N-((S)-2-((6-((S)-2-((4S,7S,9aS)-8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2-фенилацетамидо)гекса-2,4-диин-1-ил)амино)-2-оксо-1-фенилэтил)-8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида дигидрохлорид
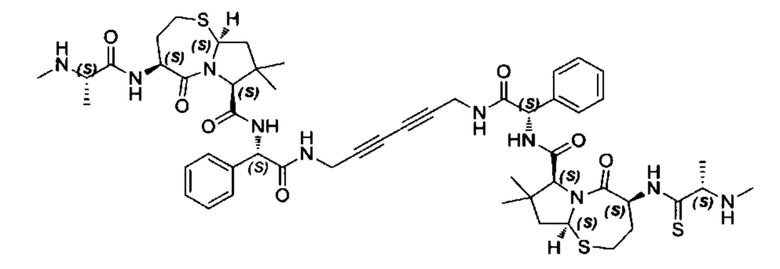
Этап 1: трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((S)-2-оксо-1-фенил-2-(проп-2-ин-1-иламино)этил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-тиоксопропан-2-ил)(метил)карбамат
К раствору (S)-2-амино-2-фенил-N-(проп-2-ин-1-ил)ацетамида гидрохлорида (216 мг, 0.96 ммоль) в 1,2-дихлорэтане (10 мл) добавляли (4S,7S,9aS)-4-((S)-3-((трет-бутоксикарбонил)(метил)амино)-2-оксобутил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновую кислоту (527 мг, 1.15 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N'N'-тетраметилурония гексафторфосфат (700 мг, 1.84 ммоль) и N,N-диизопропилэтиламин (356 мг, 2.76 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Смесь гасили водой (100 мл) и экстрагировали дихлорметаном (3×100 мл). Объединенные органические слои промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((S)-2оксо-1-фенил-2-(проп-2-ин-1-иламино)этил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-тиоксопропан-2-ил)(метил)карбамат (500 мг, неочищенный) в виде желтого масла. ЖХМС (3.0 минуты, муравьиная кислота): Время удерживания = 2.004 минуты, m/z: 652 (M+Na)+.
Этап 2: трет-бутил((S)-1-(((4S,7S,9aS)-7-(((S)-2-((6-((S)-2-((4S,7S,9aS)-4-((S)2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2-фенилацетамидо)гекса-2,4-диин-1-ил)амино)-2-оксо-1-фенилэтил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат и ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-7,7'-((3S,14S)-4,13-диоксо-3,14-дифенил-2,5,12,15-тетраазагексадека-7,9-диин-1,16-диоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,31тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-2,1-диил))бис(метилкарбамат)
К смеси трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((S)-2-оксо-1-фенил-2-(проп-2-ин-1-иламино)этил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата (300 мг, 0.5 ммоль) и трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((S)-2-оксо-1-фенил-2-(проп-2-ин-1-иламино)этил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-тиоксопропан-2-ил)(метил)карбамата (308 мг, 0.5 ммоль) в ацетонитриле (8 мл) и пиридине (232 мг, 2.9 ммоль) добавляли ацетат меди (II) (107 мг, 0.6 ммоль). Смесь нагревали до 85°С и перемешивали в течение 1 часа. Смесь охлаждали до комнатной температуры, разбавляли водой (50 мл) и экстрагировали дихлорметаном (3×50 мл). Органический слой объединяли, промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали трет-бутил((S)-1-(((4S,7S,9aS)-7-(((S)-2-((6-((S)-2-((4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2-фенилацетамидо)гекса-2,4-диин-1-ил)амино)-2-оксо-1-фенилэтил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат (20 мг, 0.016 ммоль, выход 3.3%) [ЖХМС (3.0 минуты, муравьиная кислота): Время удерживания = 2.100 минуты, m/z: 1263 (M+Na)+] и ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-7,7-((3S,14S)-4,13-диоксо-3,14-дифенил-2,5,12,15-тетраазагексадека-7,9-диин-1,16-диоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-2,1-диил))бис(метилкарбамат) (8 мг, 0.006 ммоль, выход 1,4%) [ЖХМС (3.0 минуты, муравьиная кислота): Время удерживания = 2.367 минуты, m/z: 1279 (M+Na)+] в виде белых твердых веществ. Другой диастереоизомер не собирали.
Этап 3: (4S,7S,9aS)-N-((S)2-((6-((S)-2-((4S,7S,9aS)8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2-фенилацетамидо)гекса-2,4-диин-1-ил)амино)-2-оксо-1-фенилэтил)-8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида дигидрохлорид
К раствору трет-бутил((S)-1-(((4S,7S,9aS)-7-(((S)-2-((6-((S)-2-((4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)иропантиоами до)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2-фенилацетамидо)гекса-2,4-диин-1-ил)амино)-2-оксо-1-фенилэтил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата (20 мг, 0.016 ммоль) в дихлорметане (8 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 0.5 мл).Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Растворитель концентрировали при пониженном давлении и твердое вещество сушили в глубоком вакууме, в результате чего получали (4S,7S,9aS)-N-((S)-2-((6-((S)-2-((4S,7S,9aS)-8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2-фенилацетамидо)гекса-2,4-диин-1-ил)амино)-2-оксо-1-фенилэтил)-8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида дигидрохлорид (13 мг, 0.012 ммоль, выход 72.2%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 8.84-8.81 (m, 3Н), 8.57-8.48 (m, 4Н), 8.27-8.24 (m, 2Н), 7.43-7.30 (m, 11Н), 5.53-5.46 (m, 5Н), 5.12-5.09 (m, 1Н), 4.69-4.64 (s, 2Н), 4.26(d, J=5.6 Гц, 1Н), 4.21 (m, 1Н), 4.00(d, J=5.2 Гц, 4Н), 3.24-3.16 (m, 3Н), 3.01-2.88 (m, 4Н), 2.23 (s, 3Н), 2.16(d, J=4.0 Гц, 2Н), 1.83-1.69 (m, 6Н), 1.24-1.20 (m, 6Н), 1.05(d, J=8.0 Гц, 6Н), 0.92-0.90 (m, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.425 минуты, m/z: 1040.6 (М+1)+.
Пример 32
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(гекса-2,4-диин-1,6-диилбис(азандиил))бис(2-оксо-1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)дигидрохлорид
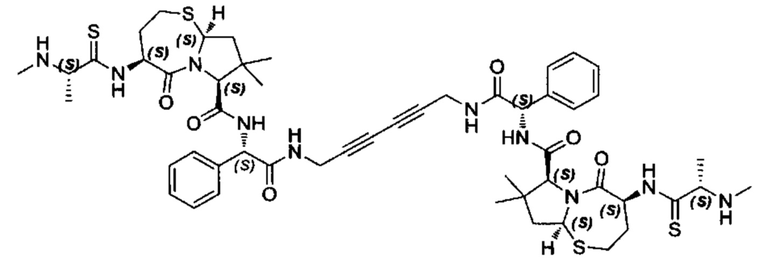
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-7,7'-((3S,14S)-4,13-диоксо-3,14-дифенил-2,5,12,15-тетраазагексадека-7,9-диин-1,16-диоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-2,1-диил))бис(метилкарбамата) (Пример 31, Этап 2) (8 мг, 0.006 ммоль) в дихлорметане (8 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 0.5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. Растворитель концентрировали при пониженном давлении и сушили в глубоком вакууме, в результате чего получали указанное в заголовке соединение (2 мг, 0.002 ммоль, выход 27.8%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD-d4) δ 7.47-7.46 (m, 4Н), 7.40-7.34 (m, 6Н), 5.58-5.53 (m, 2Н), 5.50 (m, 2Н), 5.28-5.25 (m, 2Н), 4.23(d, J=3.2 Гц, 2Н), 4.03 (s, 4Н), 3.79-3.71 (m, 2Н), 3.47-3.41 (m, 2Н), 3.05-3.00 (m, 2Н), 2.44(d, J=9.6 Гц, 6Н), 2.39-2.34 (m, 2Н), 2.28-2.21 (m, 2Н), 1.83-1.77 (m, 2Н), 1.62-1.55 (m, 2Н), 1.31 (s, 6Н), 1.61 (s, 6Н), 0.96 (s, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.546 минуты, m/z: 1056.6 (М+1)+.
Пример 33
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S)-(гекса-2,4-диин-1,6-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
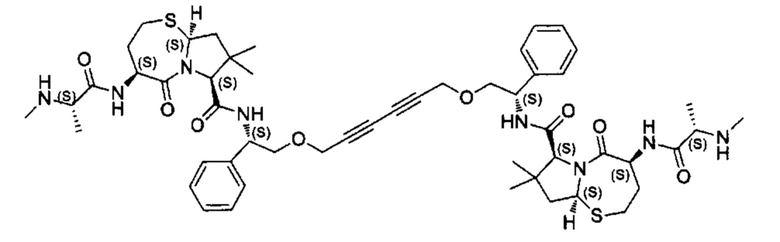
Этап 1: (S)-1-фенил-2-(проп-2-ин-1-илокси)этан-1-амин
К раствору (S)-2-амино-2-фенилэтан-1-ола (6 г, 43.7 ммоль) в сухом тетрагидрофуране (200 мл) при 0°С добавляли гидрид натрия (60%, дисперсия в Парафиновом Масле) (1.9 г, 48.1 ммоль). Смесь перемешивали при 0°С в течение 20 минут. Затем добавляли 3-бромпроп-1-ин (5.72 г, 48.1 ммоль). Смесь нагревали при 70°С в течение ночи. После охлаждения, к этой смеси добавляли ледяную воду (200 мл) и смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические слои промывали солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле [петролейный эфир/этилацетат (1:1 об./об.)], в результате чего получали (S)-1-фенил-2-(проп-2-ин-1-илокси)этан-1-амин (2.5 г, 14.3 ммоль, выход 32.65%) в виде оранжевого масла. 1Н-ЯМР 1 (400 МГц, ДМСО-d6) δ ppm 7.44-7.14 (m, 5Н), 4.18-4.12 (m, 2Н), 4.08-4.00 (m, 1Н), 3.55-3.47 (m, 1Н), 3.44-3.37 (m, 2Н), 1.84 (s, 2Н).
Этап 2: трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((S)-1-фенил-2-(проп-2-ин-1-илокси)этил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат
К раствору (S)-1-фенил-2-(проп-2-ин-1-илокси)этан-1-амина (308 мг, 1.76 ммоль) в дихлорметане (10 мл) добавляли (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновую кислоту (650 мг, 1.47 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (948 мг, 2.49 ммоль) и N,N-диизопропилэтиламин (454 мг, 3.52 ммоль) при комнатной температуре. Смесь перемешивали в течение ночи. Реакцию гасили водой (300 мл) и экстрагировали дихлорметаном (3×20 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле [этилацетат/петролейный эфир (1/5 об./об.)], в результате чего получали трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((S)-1-фенил-2-(проп-2-ин-1-илокси)этил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат (194 мг, 0.32 ммоль, выход 22.63%) в виде желтого масла. ЖХМС (3.0 минуты, муравьиная кислота): Время удерживания = 1.70 минуты, m/z: 601 (М+1)+.
Этап 3: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,14S)-3,14-дифенил-5,12-диокса-2,15-диазагексадека-7,9-дииндиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
Смесь трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((S)-1-фенил-2-(проп-2-ин-1-илокси)этил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата (194 мг, 0,32 ммоль), ацетата меди (II) (70.4 мг, 0.39 ммоль) и пиридина (50.6 мг, 0.64 ммоль) в ацетонитриле (10 мл) перемешивали при 85°С в течение 1 часа. К этой смеси добавляли водный аммиак (20-кратное разведение) до тех пор, пока она не становилась синей, и смесь экстрагировали дихлорметаном (3×30 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали неочищенный продукт (130 мг). Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((3S,14S)-3,14-дифенил-5,12-диокса-2,15-диазагексадека-7,9-дииндиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (70 мг, 0.058 ммоль) в виде белого порошка. ЖХМС (3.0 минуты, муравьиная кислота): Время удерживания = 2.10 минуты, m/z: 1199.7 (М+1)+.
Этап 4: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(гекса-2,4-диин-1,6-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил-((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((3S,14S)-3,14-дифенил-5,12-диокса-2,15-диазагексадека-7,9-дииндиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (70 мг, 0.058 ммоль) в дихлорметане (15 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель концентрировали при пониженном давлении и сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(гекса-2,4-диин-1,6-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]таазепин-7-карбоксамида) дигидрохлорид (50 мг, 0.047 ммоль, выход 79.36%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 9.24 (s, 2Н), 8.86(d, J=6.4 Гц, 2Н), 8.81(d, J=7.2 Гц, 2Н), 8.38(d, J=8.0 Гц, 2Н), 7.39-7.32 (m, 8Н), 7.29-7.25 (m, 2Н), 5.47(t, J=8.0 Гц, 2Н), 5.04 (кв, J=6.8 Гц, 2Н), 4.73-4.69 (m, 2Н), 4.30 (s, 4Н), 4.17 (s, 2Н), 3.86 (кв, J=6.9 Гц, 2Н), 3.62(d, J=6.0 Гц, 4Н), 3.19-3.13 (m, 2Н), 2.92-2.87 (m, 2Н), 2.47 (s, 6Н), 2.22-2.17 (m, 2Н), 2.13-2.09 (m, 2Н), 1.87(dd, J=12.4, 8.8 Гц, 2Н), 1.80-1.72 (m, 2Н), 1.38(d, J=6.8 Гц, 6Н), 1.06 (s, 6Н), 1.01 (s, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.28 минуты, m/z: 999.7 (М+1)+.
Пример 34
(4S,7S,9aS)-N-((S)-2-((6-((S)-2-((4S,7S,9aS)-8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2-фенилэтокси)гекса-2,4-диин-1-ил)окси)-1-фенилэтил)-8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
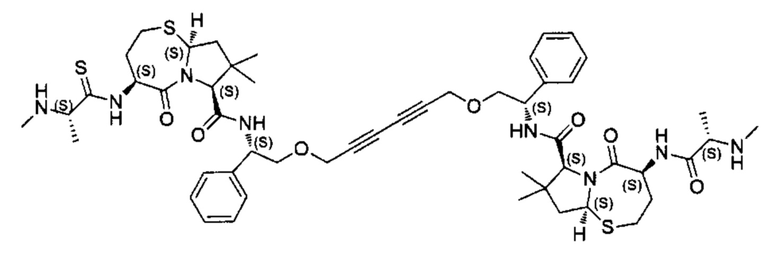
Этап 1: трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((S)-1-фенил-2-(проп-2-ин-1-илокси)этил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-тиоксопропан-2-ил)(метил)карбамат
К раствору (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (500 мг, 1.088 ммоль), 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (624 мг, 1.64 ммоль) и N,N-диизопропилэтиламина (281 мг, 2.18 ммоль) в 1,2-дихлорэтане (15 мл) добавляли (S)-1-фенил-2-(проп-2-ин-1-илокси)этан-1-амин (210 мг, 1.20 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Смесь очищали хроматографией на силикагеле [петролейный эфир/этилацетат (2:1 об./об.)] и препаративной-ТСХ [петролейный эфир/этилацетат (1:1 об./об.)], в результате чего получали трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((S)-1-фенил-2-(проп-2-ин-1-илокси)этил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-тиоксопропан-2-ил)(метил)карбамат (400 мг, 0.65 ммоль, выход 59.1%). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.988 минуты, m/z: 638.8 (M+Na)+.
Этап 2: ди-тркт-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((3S,14S)-3,14-дифенил-5,12-диокса-2,15-диазагексадека-7,9-дииндиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат), трет-бутил((S)-1-(((4S,7S,9aS)-7-(((S)-2-((6-((S)-2-((4S,7S,9aS)-4-((S)2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2-фенилэтокси)гекса-2,4-диин-1-ил)окси-)-1-фенилэтил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамат и ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S, 14)-3,14-дифенил-5,12-диокса-2,15-диазагексадека-7,9-дииндиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b1[1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат)
К раствору трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((1S,2R)-2-(проп-2-ин-1-илокси)-2,3-дигидро-1Н-инден-1-ил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-тиоксопропан-2-ил)(метил)карбамата (378 мг, 0.616 ммоль), трет-бутил((S)-1-(((4S,7S,9aS)-8,8-диметил-5-оксо-7-(((S)-1-фенил-2-(проп-2-ин-1-илокси)этил)карбамоил)октагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата (370 мг, 0.616 ммоль) и пиридина (292 мг, 3.696 ммоль) в ацетонитриле (15 мл) добавляли ацетат меди (II) (269 мг, 1.478 ммоль). Смесь перемешивали при 85°С в течение 40 минут. Реакционнную смесь концентрировали и разбавляли этилацетатом (20 мл) и добавляли водный аммиак (20-кратное разведение, 20 мл). Водную фазу отделяли и экстрагировали этилацетатом (2×15 мл). Объединенную органическую фазу промывали солевым раствором (20 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали препаративной-ТСХ [петролейный эфир/этилацетат (2:3 об./об.)] и препаративной-ВЭЖХ с получением целевых продуктов ди-трет-бутил((2S,2'S)-(((4S,4,S)7S,7'S,9aS,9а'S)-((3S,14S)-3,14-дифенил-5,12-диокса-2,15-диазагексадека-7,9-дииндиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (100 мг, 0.083 ммоль, выход 13.5%) [ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.914 минуты, m/z: 499.8 {[(М-2Вос)+2]/2}+], трет-бутил((S)-1-(((4S,7S,9aS)-7-(((S)-2-((6-((S)-2-((4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)1фопантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2-фенилэтокси)гекса-2,4-диин-1-ил)окси)-1-фенилэтил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата (66 мг, 0.054 ммоль, выход 8.77%) [ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 2.042 минуты, m/z: 507.8 {[(М-2Вос)+2]/2}+] и ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,14S)-3,14-дифенил-5,12-диокса-2,15-диазагексадека-7,9-дииндиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (40 мг, 0.032 ммоль, выход 5.19%) [ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 2.186 минуты, m/z: 516.1 {[(М-2Вос)+2]/2}+].
Этап 3: (4S,7S,9aS)-N-((S)-2-((6-((S)-2-((4S,7S,9aS)-8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2-фенилэтокси)гекса-2,4-диин-1-ил)окси)-1-фенилэтил)-8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
К раствору трет-бутил((S)-1-(((4S,7S,9aS)-7-(((S)-2-((6-((S)-2-((4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2-фенилэтокси)гекса-2,4-диин-1-ил)окси)-1-фенилэтил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата (60 мг, 0.049 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлорид водорода в 1,4-диоксане (1 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель концентрировали и неочищенный продукт очищали препаративной-ТСХ [дихлорметан/метанол (5:1 об./об.)], в результате чего получали (4S,7S,9aS)-N-((S)-2((6-((S)-2-((4S,7S,9aS)-8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2-фенил этокси)гекса-2,4-диин-1-ил)окси)-1-фенилэтил)-8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид (30 мг, 0.029 ммоль, выход 59.2%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 8.36-8.33 (m, 1Н), 8.29(d, J=8.0 Гц, 1Н), 8.18(d, J=6.8 Гц, 1Н), 7.39-7.33 (m, 8Н), 7.29-7.26 (m, 2Н), 5.50-5.44 (m, 2Н), 5.11-5.01 (m, 3Н), 4.67-4.63 (m, 1Н), 4.30 (s, 4Н), 4.23-4.18 (m, 2Н), 3.62(d, J=6.0 Гц, 4Н), 3.40-3.35 (m, 2Н), 3.20-3.12 (m, 2Н), 2.97-2.85 (m, 3Н), 2.38-2.33 (m, 1Н), 2.22 (s, 6Н), 2.16-2.15 (m, 2Н), 1.91-1.83 (m, 2Н), 1.78-1.65 (m, 2Н), 1.24 (s, 3Н), 1.1 l(d, J=6.8 Гц, 3Н), 1.07(d, J=8.4 Гц, 6Н), 1.00(d, J=6.8 Гц, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.251 минуты, m/z: 1015.4 (М+1)+.
Пример 35
(4S,4'S,7S,7'S,9aS,9a'S)-N,N,-((1S,1'S)-(гекса-2,4-диин-1,6-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)
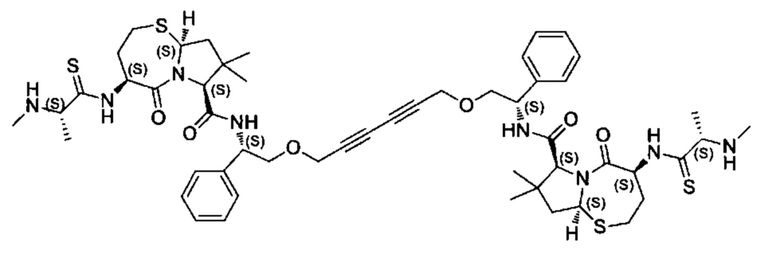
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,14S)-3,14-дифенил-5,12-диокса-2,15-диазагексадека-7,9-дииндиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (Пример 34, Этап 2) (40 мг, 0.032 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлорид водорода в 1,4-диоксане (1 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель концентрировали и неочищенный продукт очищали препаративной-ТСХ (дихлорметан/метанол = 5:1), в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(гекса-2,4-диин-1,6-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) (19 мг, 0.018 ммоль, выход 56.2%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 8.39-8.36 (m, 2Н), 7.73-7.67 (m, 2Н), 7.39-7.33 (m, 8Н), 7.30-7.25 (m, 2Н), 5.49-5.44 (m, 2Н), 5.11-5.02 (m, 4Н), 4.30 (m, 3Н), 4.14(dd, J=5.6, 3.6 Гц, 1Н), 3.62(d, J=6.0 Гц, 4Н), 3.56 (s, 2Н), 3.20-3.13 (m, 2Н), 2.96-2.89 (m, 2Н), 2.35-2.31 (m, 2Н), 2.24-2.17 (m, 8Н), 1.91-1.86 (m, 2Н), 1.79-1.61 (m, 4Н), 1.26-1.21 (m, 8Н), 1.08 (s, 6Н), 1.01 (s, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.314 минуты, m/z: 1031.4 (М+1)+.
Пример 36
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(бутан-1,4-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
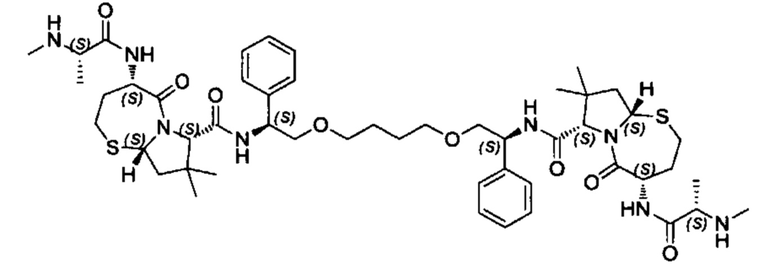
Этап 1: (1S,1'S)-2,2'-(бутан-1,4-диилбис(окси))бис(1-фенилэтан-1-амин)
К раствору (S)-2-амино-2-фенилэтан-1-ола (1 г, 7.29 ммоль) в тетрагидрофуране (80 мл) при 0°С медленно добавляли гидрид натрия (60%, дисперсия в Парафиновом Масле) (321 мг, 8.02 ммоль). Смеси давали нагреться до комнатной температуры. Затем добавляли 1,4-Дибромбутан (787 мг, 3.65 ммоль). Полученную смесь нагревали до 70°С и перемешивали в течение ночи. После охлаждения добавляли ледяную воду (50 мл) и смесь экстрагировали этилацетатом (3×30 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью тонкослойной хроматографии [дихлорметан/метанол (10:1 об./об.)], в результате чего получали продукт (1S,1'S)-2,2'-(бутан-1,4-диилбис(окси))бис(1-фенилэтан-1-амин) (190 мг, 0.58 ммоль, выход 15.9%) виде черного масла. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.162 минуты, m/z: 329.0 (М+1)+.
Этап 2: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,12S)-3,12-дифенил-5,10-диокса-2,13-диазатетрадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1.2-диил))бис(метилкарбамат)
К смеси (1S,1'S)-2,2'-(бутан-1,4-диилбис(окси))бис(1-фенилэтан-1-амина) (90 мг, 0.274 ммоль) и (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение I) (267 мг, 0.603 ммоль) в 1,2-дихлорэтане (12 мл) добавляли N-этоксикарбонил-2-этокси-1,2-дигидрохинолинил (203 мг, 0.822 ммоль) и N,N-диизопропилэтиламин (141 мг, 1.096 ммоль). Смесь перемешивали при 50°С в течение ночи. Растворитель удаляли при пониженном давлении. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)] с последующей препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2,S)-(((4S,4'S,7S,7'S,9aS)9a'S)-((3S,12S)-3,12-дифенил-5,10-диокса-2,13-диазатетрадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (48 мг, 0.041 ммоль, выход 14.6%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.835 минуты, m/z: 489.9 {[(М-2Вос)+2]/2}+.
Этап 3: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(бутан-1,4-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((3S,12S)-3,12-дифенил-5,10-диокса-2,13-диазатетрадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (48 мг, 0.041 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлорид водорода в 1,4-диоксане (1 мл). Смесь перемешивали при комнатной температуре в течение 3 часов. Смесь концентрировали и сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(бутан-1,4-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (34 мг, 0.032 ммоль, выход 79.2%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 9.34 (s, 1Н), 8.62 (s, 1Н), 8.81(d, J=7.2 Гц, 2Н), 8.34(d, J=8.0 Гц, 2Н), 7.37-7.30 (m, 8Н), 7.27-7.23 (m, 2Н), 5.47(t, J=8.0 Гц, 2Н), 4.99 (кв, J=6.4 Гц, 2Н), 4.71(t, J=8.4 Гц, 2Н), 4.17 (s, 2Н), 3.88-3.84 (m, 2Н), 3.49-3.48 (m, 4Н), 3.41-3.29 (m, 4Н), 3.16(t, J=12.4 Гц, 2Н), 2.90-2.87 (m, 2Н), 2.46 (s, 6Н), 2.22-2.19 (m, 2Н), 2.12-2.10 (m, 2Н), 1.89-1.84 (m, 2Н), 1.79-1.70 (m, 2Н), 1.42 (s, 4Н), 1.39(d, J=6.8 Гц, 6Н), 1.06 (m, 6Н), 1.00 (m, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.323 минуты, m/z: 978.8 (М+1)+.
Пример 37
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S)-(бутан-1,4-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
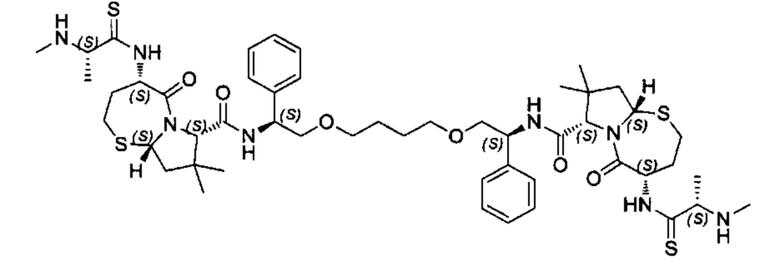
Этап 1: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,12S)-3,12-дифенил-5,10-диокса-2,13-диазатетрадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат)
К смеси (1S,1'S)-2,2'-(бутан-1,4-диилбис(окси))бис(1-фенилэтан-1-амина) (100 мг, 0.304 ммоль) и (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение II) (308 мг, 0.670 ммоль) в 1,2-дихлорэтане (12 мл) добавляли N-этоксикарбонил-2-этокси-1,2-дигидрохинолинил (226 мг, 0.912 ммоль) и N,N-диизопропилэтиламин (157 мг, 1.216 ммоль). Смесь перемешивали при 50°С в течение ночи. Растворитель удаляли при пониженном давлении. Неочищенный продукт очищали с помощью тонкослойной хроматографии (этилацетат /петролейный эфир = 1/1) с последующей препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((3S,12S)-3,12-дифенил-5,10-диокса-2,13-диазатетрадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (55 мг, 0.045 ммоль, выход 14.8%) в виде бесцветного масла. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 2.044 минуты, m/z: 506 {[(М-2Вос)+2]/2}+.
Этап 2: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(бутан-1,4-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((3S,12S)-3,12-дифенил-5,10-диокса-2,13-диазатетрадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (55 мг, 0.045 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлорид водорода в 1,4-диоксане (1 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли при пониженном давлении и продукт сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(бутан-1,4-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (35 мг, 0.032 ммоль, выход 71.4%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 10.96 (s, 1Н), 10.89 (s, 1Н), 9.71 (s, 1Н), 9.55 (s, 1Н), 8.61 (s, 1Н), 8.40(t, J=8.2 Гц, 2Н), 7.37-7.30 (m, 8Н), 7.27-7.23 (m, 2Н), 5.42 (кв, J=8.0 Гц, 2Н), 5.16-5.11 (m, 2Н), 5.02-4.98 (m, 2Н), 4.26-4.23 (m, 2Н), 4.21-4.20 (m, 2Н), 3.49-3.48 (m, 4Н), 3.33 (s, 4Н), 3.19-3.13 (m, 2Н), 2.96-2.92 (m, 2Н), 2.48 (s, 6Н), 2.24-2.21 (m, 4Н), 1.93-1.88 (m, 4Н), 1.43-1.42 (m, 6Н), 1.08 (m, 6Н), 1.02 (m, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.326 минуты, m/z: 506.0 [(М+2)/2]+.
Пример 38
(4S,4'S,7S,7'S,9aS,9a'S)-N,N,-((1S,1'S)-((оксибис(этан-2,1-диил))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
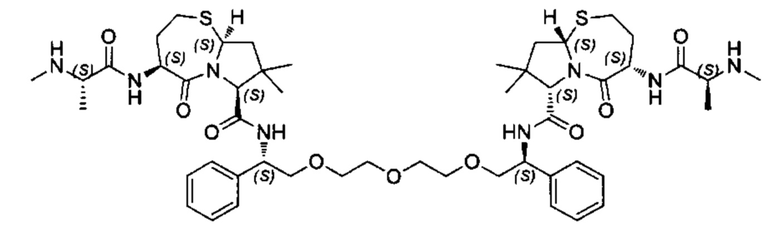
Этап 1: оксибис(этан-2,1-диил) бис(4-метилбензолсульфонат)
К раствору 4-тозилхлорида (11.86 г, 62.2 ммоль) и 2,2'-оксибис(этан-1-ола) (3.0 г, 28.30 ммоль) в дихлорметане (200 мл) при 0°С добавляли KOH (14.27 г, 25.4 ммоль). Смесь оставляли перемешиваться в течение ночи. К этой смеси добавляли ледяную воду (150 мл) с последующим экстрагированием дихлорметаном (3×200 мл). Объединенные органические слои сушили с использованием безводного сульфата магния, фильтровали и концентрировали. Остаток кристаллизовали из безводного метанола, в результате чего получали оксибис(этан-2,1-диил)бис(4-метилбензолсульфонат) (9.3 г, 22.4 ммоль, 79%) в виде белого твердого вещества. 1Н-ЯМР 1 (400 МГц, ДМСО-d6) δ ppm 7.78(d, J=8.0 Гц, 4Н), 7.47(d, J=8.0 Гц, 4Н), 4.07- 4.05 (m, 4Н), 3.53-3.51 (m, 4Н), 2.42 (s, 6Н); ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.675 минуты, m/z: 414.5 (М+1)+, 436.8 (M+Na)+.
Этап 2: (1S,1'S)-2,2'-((оксибис(этан-2,1-диил))бис(окси))бис(1-фенилэтан-1-амин)
К раствору (S)-2-амино-2-фенилэтан-1-ола (364.1 мг, 2.65 ммоль) в сухом тетрагидрофуране (10 мл) при 0°С порциями добавляли суспензию гидрида натрия (60%, дисперсия в Парафиновом Масле) (144.8 мг, 6.03 ммоль) в сухом тетрагидрофуране (10 мл) в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 2 часов. К этой смеси добавляли оксибис(этан-2,1-диил)бис(4-метилбензолсульфонат) (500 мг, 1.21 ммоль) и смесь перемешивали при комнатной температуре в течение 12 часов. Смесь гасили водой (50 мл) и экстрагировали этилацетатом (3×50 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (1/1 об./об.)], в результате чего получали (1S,1'S)-2,2'-((оксибис(этан-2,1-диил))бис(окси))бис(1-фенилэтан-1-амин) (260 мг, 0.76 ммоль, выход 62%) в виде желтого масла. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.085 минуты, m/z: 345.0 (М+1)+.
Этап 3: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,13S)-3,13-дифенил-5,8,11-триокса-2,14-диазапентадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1.2-диил))бис(метилкарбамат)
Раствор (1S,1'S)-2,2'-((оксибис(этан-2,1-диил))бис(окси))бис(1-фенилэтан-1-амина) (150 мг, 0.44 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (405.6 мг, 0.91 ммоль), N-этоксикарбонил-2-этокси-1,2-дигидрохинолинила (323 мг, 1.31 ммоль) и N,N-диизопропилэтиламина (225.1 мг, 1.74 ммоль) в 1,2-дихлорэтане (8 мл) перемешивали при 50°С в течение ночи. Смесь концентрировали при пониженном давлении и очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (2/1 об./об.)], в результате чего получали неочищенный продукт (360 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,13S)-3,13-дифенил-5,8,11-триокса-2,14-диазапентадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (180 мг, 0.15 ммоль, выход 33.5%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.79 минуты, m/z: 498.1 {[(М-2Вос)+2]/2}+.
Этап 4: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-((оксибис(этан-2,1-диил))бис(1-окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((3S,13S)-3,13-дифенил-5,8,11-триокса-2,14-диазапентадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (180 мг, 0.15 ммоль) в дихлорметане (6 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 2 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении и сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-((оксибис(этан-2,1-диил))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (130 мг, 0.131 ммоль, 76%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 9.50 (s, 2Н), 8.91 (s, 2Н), 8.83(d, J=6.4 Гц, 2Н), 8.37(d, J=7.6 Гц, 2Н), 7.38-7.31 (m, 8Н), 7.27-7.23 (m, 2Н), 5.47(t, J=7,8 Гц, 2Н), 5.00 (кв, J=7.2 Гц, 2Н), 4.70(t, J=8,8 Гц, 2Н), 4.17 (s, 2Н), 3,88-3,82 (m, 2Н), 3.56-3.40 (m, 12Н), 3.15(t, J=12.2 Гц, 2Н), 2.91-2,87 (m, 2Н), 2.45 (s, 6Н), 2.22-2.09 (m, 4Н), 1.90-1.71 (m, 4Н), 1.39(d, J=6,8 Гц, 6Н), 1.06 (m, 6Н), 1.01 (m, 6Н); ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.284 минуты, m/z: 995,8 (М+1)+.
Пример 39
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S)-((оксибис(этан-2,1-диил))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
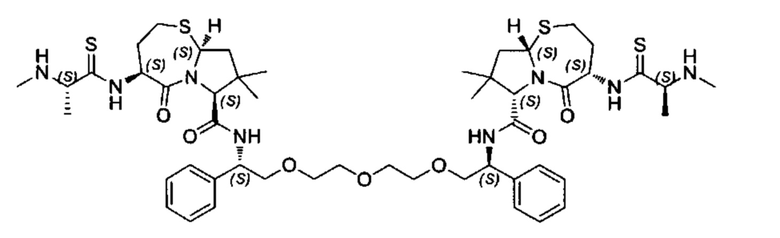
Этап 1: ди-трет-бутил((2S,2'S)-((4S,4'S,7S,7'S',9aS,9а'S)-((3S,13S)-3,13-дифенил-5,8,11-триокса-2,14-диазапентадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат)
Раствор (1S,1'S)-2,2'-((оксибис(этан-2,1-диил))бис(окси))бис(1-фенилэтан-1-амина) (50 мг, 0.15 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (Промежуточное соединение II) (140.1 мг, 0.30 ммоль), N-этоксикарбонил-2-этокси-1,2-дигидрохинолинила (107.7 мг, 0.45 ммоль) и N,N-диизопропилэтиламина (75 мг, 0.60 ммоль) в 1,2-дихлорэтане (5 мл) перемешивали при 50°С в течение ночи. Растворитель удаляли при пониженном давлении. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (1/1 об./об.)], в результате чего получали неочищенный продукт (360 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((3S,13S)-3,13-дифенил-5,8,11-триокса-2,14-диазапентадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (100 мг, 0.082 ммоль, выход 60%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 2.02 минуты, m/z: 514.0 {[(М-2Вос)+2]/2}+.
Этап 2: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-((оксибис(этан-2,1-диил))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1.3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,13S)-3,13-дифенил-5,8,11-триокса-2,14-диазапентадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (100 мг, 0.082 ммоль) в дихлорметане (5 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-((оксибис(этан-2,1-диил))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (50 мг, 0.05 ммоль, выход 60%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 10.95(d, J=6.4 Гц, 1Н), 10.88(d, J=6.4 Гц, 1Н), 9.70 (s, 1Н), 9.53 (s, 1Н), 8.61 (s, 1Н), 8.44-8.40 (m, 3Н), 7.37-7.31 (m, 8Н), 7.27-7.24 (m, 2Н), 5.42 (кв, J=8.0 Гц, 2Н), 5.13 (кв, J=7.6 Гц, 2Н), 5.03-4.98 (m, 2Н), 4.25-4.20 (m, 4Н), 3.55-3.54 (m, 4Н), 3.51-3.47 (m, 4Н), 3.44 (s, 4H), 3.16(t, J=12 Гц, 2H), 2.48 (s, 6H), 2.24-2.21 (m, 4H), 1.94-1.89 (m, 4H), 1.43(d, J=6.4 Гц, 6H), 1.08 (m, 6H), 1.03 (m, 6H). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.355 минуты, m/z: 1026.8 (М+1)+.
Пример 40
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S)-(гексан-1,6-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
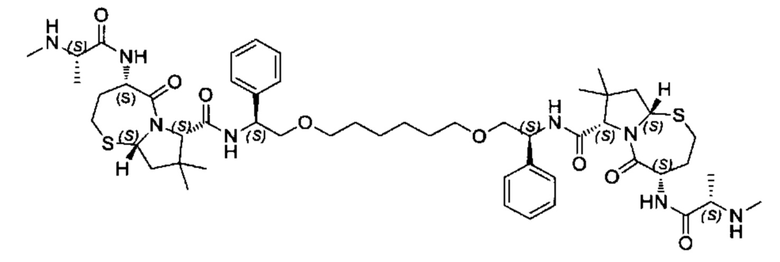
Этап 1: (1S,1'S)-2,2'-(гексан-1,6-диилбис(окси))бис(1-фенилэтан-1-амин)
К смеси (S)-2-амино-2-фенилэтан-1-ола (1.0 г, 7.20 ммоль) в сухом тетрагидрофуране (20 мл) при 0°С порциями медленно добавляли гидрид натрия (60%, дисперсия в Парафиновом Масле) (330 мг, 13.8 ммоль) в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 30 минут и добавляли раствор 1,6-дибромгексана (810 мг, 3.31 ммоль) в сухом тетрагидрофуране (10 мл). Смесь кипятили с обратным холодильником и перемешивали в течение еще 5 часов. После охлаждения к этой смеси осторожно добавляли воду (300 мл) с последующим экстрагированием дихлорметаном (3×100 мл). Объединенные органические слои промывали солевым раствором, сушили, фильтровали и концентрировали. Остаток очищали с помощью тонкослойной хроматографии (дихлорметан/метанол (7:1 об./об.)], в результате чего получали (1S,1'S)-2,2'-(гексан-1,6-диилбис(окси))бис(1-фенилэтан-1-амин) (410 мг, 1.15 ммоль, выход 25.6%) в виде желтого масла. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.24 минуты, m/z: 357.0 (М+1)+.
Этап 2: ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,14S)-3,14-дифенил-5,12-диокса-2,15-диазагексадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат)
Раствор (1S,1'S)-2,2'-(гексан-1,6-диилбис(окси))бис(1-фенилэтан-1-амина) (90 мг, 0.25 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (235.2 мг, 0.53 ммоль), N-этоксикарбонил-2-этокси-1,2-дигидрохинолинила (187.3 мг, 0.76 ммоль) и диизопропилэтиламина (130.5 мг, 1.01 ммоль) в 1,2-дихлорэтане (5 мл) перемешивали при 50°С в течение ночи. Растворитель удаляли при пониженном давлении. Остаток очищали с помощью тонкослойной хроматографии [этилацетат/петролейный эфир (1:1 об./об.)], в результате чего получали неочищенный продукт (120 мг) в виде желтого масла. Этот материал дополнительно очищали препаративной-ВЭЖХ, в результате чего получали ди-трет-бутил((2S,2'S)-(((4S,4,S,7S,7'S,9aS,9а,S)-((3S,14S)-3,14-дифенил-5,12-диокса-2,15-диазагексадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (50 мг, 0.041 ммоль, выход 16.4%) в виде белого твердого вещества. ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.89 минуты, m/z: 503.9 {[(М-2Вос)+2]/2}+.
Этап 3: (4S,4'S,7S,7'S,9aS,9a'S')-N,N'-((1S,1'S)-(гексан-1,6-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид
К раствору ди-трет-бутил((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,14S)-3,14-дифенил-5,12-диокса-2,15-диазагексадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азандиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (100 мг, 0.083 ммоль) в дихлорметане (5 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении и сушили в глубоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(гексан-1,6-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида) дигидрохлорид (41.9 мг, 0.038 ммоль, выход 45.8%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 9.34 (s, 2Н), 8.88-8.86 (m, 2Н), 8.82(d, J=7.8 Гц, 2Н), 8.36(d, J=8.4 Гц, 2Н), 7.38-7.31 (m, 8Н), 7.27-7.23 (m, 2Н), 5.47(t, J=7.8 Гц, 2Н), 5.48-5.02 (m, 2Н), 4.73-4.68 (m, 2Н), 4.17 (s, 2Н), 3.88-3.83 (m, 2Н), 3.72-3.65 (m, 2Н), 3.51-3.47 (m, 6Н), 3.32-3.28 (m, 4Н), 3.19-3.13 (m, 2Н), 2.90-2.87 (m, 2Н), 2.46(t, J=4.4Н, 6Н), 2.22-2.17 (m, 2Н), 2.13-2.09 (m, 2Н), 1.89-1.84 (m, 2Н), 1.79-1.34 (m, 2Н), 1.39-1.38 (m, 10Н), 1.06 (s, 6Н), 1.01 (m, 6Н). ЖХМС (2.5 минуты, муравьиная кислота): Время удерживания = 1.258 минуты, m/z: 1006.8 (М+1)+.
Пример 41
(4S,4'S,7S,7'S,9aS,9a'S)-N,N',-((1S,1'S)-(гексан-1,6-дилбис(окси))бис(1фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) дигидрохлорид
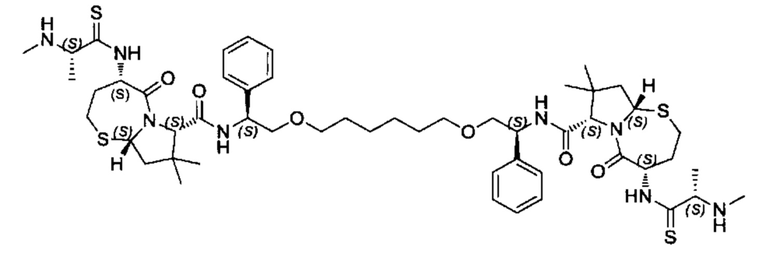
Этап 1: ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((3S,14S)-3,14-дифенил-5,12-диокса-2,15-диазагексадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат)
Раствор (1S,1'S)-2,2'-(гексан-1,6-диилбис(окси))бис(1-фенилэтан-1-амина) (90 мг, 0.25 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (243.7 мг, 0.53 ммоль), N-этоксикарбонил-2-этокси-1,2-дигидрохинолинила (187.3 мг, 0.76 ммоль) и N,N-диизопропилэтиламина (130.5 мг, 1.01 ммоль) в 1,2-дихлорэтана (5 мл) перемешивали при 50°С в течение ночи. Растворитель удаляли при пониженном давлении. Остаток очищали тонкослойной хроматографией [этилацетат/петролейный эфир (1/1 об./об.)], в результате чего получали неочищенный продукт (120 мг) в виде желтого масла. Затем этот материал очищали препаративной ВЭЖХ, в результате чего получали ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((3S,14S)-3,14-дифенил-5,12-диокса-2,15-диазагексадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (130 мг, 0.104 ммоль, 41.4% выход) в виде белого твердого вещества. ЖХМС (2.5 мин, муравьиная кислота): Bp. уд. = 2.094 мин, m/z: 520.0 {[(М-2Вос)+2]/2}+.
Этап 2: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(гексан-1,6-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) дигидрохлорид
К раствору ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,14S)-3,14-дифенил-5,12-диокса-2,15-диазагексадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (130 мг, 0.10 ммоль) в дихлорметане (5 мл) добавляли хлорид водорода (4 н. в 1,4-диоксане, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении и сушили в высоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(гексан-1,6-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) дигидрохлорид (78.4 мг, 0.07 ммоль, 70.0% выход) в форме белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 10.96-10.88 (m, 2Н), 9.62 (s, 2Н), 8.61 (s, 1Н), 8.42 (t, J=8.4 Гц, 3Н), 7.37-7.31 (m, 8Н), 7.27-7.23 (m, 2Н), 5.42 (q, J=8.0 Гц, 2Н), 5.17-5.10 (m, 2Н), 5.03-4.98 (m, 2Н), 4.24-4.20 (m, 4Н), 3.57 (s, 2Н), 3.54-3.47 (m, 4Н), 3.40-3.38 (m, 2Н), 3.32-3.28 (m, 4Н), 3.19-3.13 (m, 2Н), 2.96-2.93 (m, 2Н), 2.48-2.47 (m, 6Н), 2.24-2.21 (m, 4Н), 1.94-1.81 (m, 4Н), 1.44-1.42 (m, 10Н), 1.09 (s, 6Н), 1.03 (m, 6Н). ЖХМС (2.5 мин, муравьиная кислота): Bp. уд. = 1.300 мин, m/z: 1038.7 (М+1)+.
Пример 42
(4S,4'S,7S,7,S,9aS,9a'S)-N,N,-((1S,1'S)-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) дигидрохлорид
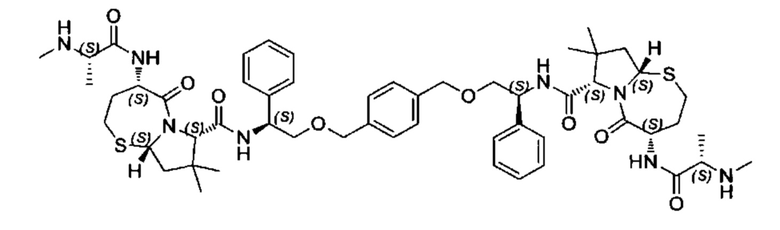
Этап 1: (1S,1'S)-2,2'-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-1-амин)
К раствору (S)-2-амино-2-фенилэтан-1-ол (2.2 г, 16.0 ммоль) в сухом тетрагидрофурана (30 мл) добавляли гидрид натрия (60%, дисперсия в жидком парафине) (760 мг, 31.6 ммоль). Через 30 минут добавляли 1,4-бис-бромметилбензол (2.0 г, 7.6 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Смесь гасили ледяной водой (50 мл) и экстрагировали этилацетатом (3×60 мл). Объединенные органические слои промывали солевым раствором, сушили (Na2SO4), фильтровали и концентрировали. Неочищенный продукт очищали хроматографией на силикагеле (дихлорметан/метанол (20:1 об./об.)], в результате чего получали (1S,1'S)-2,2'-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-1-амин) (1.2 г, 3.2 ммоль, 31% выход) в виде желтого масла. ЖХМС (2.5 мин, муравьиная кислота): Bp. уд. = 1.14 мин, m/z: 377.0 (M+1)+.
Этап 2: трет-бутил-ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(азанедиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) Раствор (1S,1'S)-2,2'-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-1-амин) (100 мг, 0.266 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (247.5 мг, 0.56 ммоль), N-этоксикарбонил-2-этокси-1,2-дигидрохинолинила (197.1 мг, 0.80 ммоль) и N,N-диизопропилэтиламина (137.3 мг, 0.29 ммоль) в 1,2-дихлорэтане (6 мл) перемешивали при 50°С в течение ночи. Смесь концентрировали при пониженном давлении и очищали тонкослойной хроматографией [этилацетат/петролейный эфир (2:1 об./об.)], в результате чего получали неочищенный продукт (200 мг)в виде желтого масла. Затем этот материал очищали препаративной ВЭЖХ, в результате чего получали трет-бутил-ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(азанедиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамат) (160 мг, 0.130 ммоль, 49.1% выход) в виде белого твердого вещества. ЖХМС (2.5 мин, муравьиная кислота): Bp. уд. = 1.86 мин, m/z: 514.1 {[(М-2Вос)+2]/2}+.
Этап 3: (4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S)-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) дигидрохлорид
К раствору ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(азанедиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (160 мг, 0.13 ммоль) в дихлорметане (15 мл) добавляли хлорид водорода (4 N в 1,4-диоксане, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении и сушили в высоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) дигидрохлорид (110 мг, 0.11 ммоль, 78% выход) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 9.39 (s, 2Н), 8.88-8.86 (m, 2Н), 8.81 (d, J=6.8 Гц, 2Н), 8.42 (d, J=8.0 Гц, 2Н), 7.39-7.32 (m,8Н), 7.28-7.25 (m, 2Н), 7.20 (s, 4Н), 5.47 (t, J=7.6 Гц, 2Н), 5.08 (q, J=6.0 Гц, 2Н), 4.70 (t, J=9.0 Гц, 2Н), 4.49-4.43 (m, 4Н), 4.18 (s, 2Н), 3.86 (s, 2Н), 3.72-3.63 (m, 2Н), 3.55-3.47 (m, 2Н), 3.13 (t, J=12.2 Гц, 2Н), 2.82-2.78 (m, 2Н), 2.46 (s, 6Н), 2.21-2.16 (m, 2Н), 2.09-2.06 (m, 2Н), 1.88-1.83 (m, 2Н), 1.77-1.68 (m, 2Н), 1.39 (d, J=6.8 Гц, 6Н), 1.05 (m, 6Н), 0.97 (m, 6Н). ЖХМС (2.5 мин, муравьиная кислота): Bp. уд. = 1.334 мин, m/z: 1027.8 (М+1)+.
Пример 43
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1,S)-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) дигидрохлорид
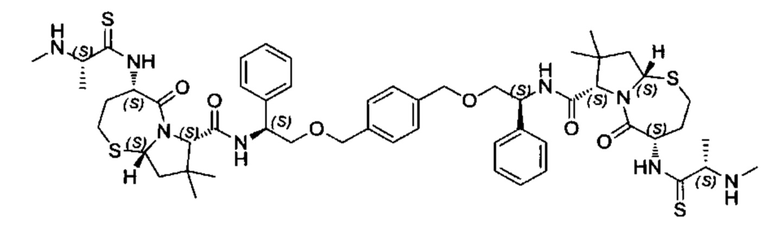
Этап 1: ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-((1,4-фениленбис(метилен))бис(окси))бис(1-фенил этан-2,1-диил))бис(азанедиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат)
К раствору (1S,1'S)-2,2'-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-1-амин) (100 мг, 0.266 ммоль), (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропантиоамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (промежуточного соединения II) (256.4 мг, 0.56 ммоль),-N-этоксикарбонил-2-этокси-1,2-дигидрохинолинила (197.1 мг, 0.80 ммоль) и N,N-диизопропилэтиламина (137.3 мг, 0.29 ммоль) в 1,2-дихлорэтане (6 мл) перемешивали при 50°С в течение ночи. Смесь концентрировали и очищали тонкослойной хроматографией [этилацетат/петролейный эфир (1:1 об./об.)], в результате чего получали неочищенный продукт (200 мг)в виде желтого масла. Затем этот материал очищали препаративной ВЭЖХ, в результате чего получали ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(азанедиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамат) (100 мг, 0.08 ммоль, 30% выход) в виде белого твердого вещества. ЖХМС (2.5 мин, муравьиная кислота): Bp. уд. = 2.08 мин, m/z: 530.6 {[(М-2Вос)+2]/2}+.
Этап 2: (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) дигидрохлорид
К раствору ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(азанедиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-тиоксопропан-1,2-диил))бис(метилкарбамата) (100 мг, 0.08 ммоль) в дихлорметане (5 мл) добавляли хлорид водорода (4 N в 1,4-диоксане, 1 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали при пониженном давлении и сушили в высоком вакууме, в результате чего получали (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-((1,4-фениленбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропантиоамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) дигидрохлорид (53 мг, 0.05 ммоль, 63% выход) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 10.97 (d, J=6.0 Гц, 1Н), 10.89 (d, J=6.0 Гц, 1Н), 9.74 (s, 1Н), 9.57 (s, 1Н), 8.62 (s, 1Н), 8.49-8.45 (m, 3Н), 7.39-7.32 (m, 8Н), 7.28-7.25 (m, 2Н), 7.20 (s, 4Н), 5.42 (q, J=7.6 Гц, 2Н), 5.16-5.06 (m, 4Н), 4.49-4.43 (m, 4Н), 4.27-4.21 (m, 4Н), 3.65-3.59 (m, 2Н), 3.42-3.38 (m, 2Н), 3.14 (t, J=12 Гц, 2Н), 2.89-2.86 (m, 2Н), 2.48-2.47 (m, 6Н), 2.24-2.18 (m, 4Н), 1.92-1.76 (m, 4Н), 1.43 (d, J=6.4 Гц, 6Н), 1.07 (m, 6Н), 0.99 (m, 6Н). ЖХМС (2.5 мин, муравьиная кислота): Bp. уд. = 1.397 мин, m/z: 1058.7 (М+1)+.
Примеры 44-55
Примеры 44-55 получали аналогично другим соединениям, описанным в настоящем документе. Было обнаружено, что эти соединения характеризуются данными, представленными ниже.
Пример 44
(4S,7S,9aS)-N-[(1R,2R)-2-({[4-({[(1R,2R)-1-[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-амидо]-2,3-дигидро-1Н-инден-2-ил]формамидо}метил)фенил]метил}карбамоил)-2,3-дигидро-1Н-инден-1-ил]-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
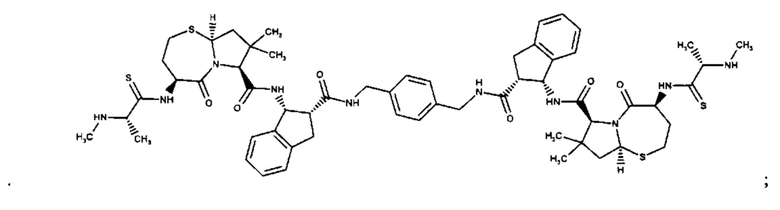
ЖХМС (электроспрей, m/z): 1137.6 [М+Н]+, время удерживания 1.265 мин.
1Н-ЯМР (400МГц, ДМСО-d6): δ ppm 10.90 (d, J=34.6 Гц, 2Н), 9.81-9.43 (m, 2Н), 8.62 (s, 1Н), 8.52-8.36 (m, 3Н), 7.94 (dd, J=16.9, 9.3 Гц, 2Н), 7.34-7.12 (m, 12Н), 5.67 (t, J=8.6 Гц, 2Н), 5.45 (q, J=8.3 Гц, 2Н), 5.20-5.05 (m, 2Н), 4.43 (d, J=14.8 Гц, 2Н), 4.30 (s, 2Н), 4.13 (d, J=13.3 Гц, 2Н), 3.95 (dd, J=15.1, 4.3 Гц, 2Н), 3.48-3.39 (m, 2Н), 3.28-3.16 (m, 4Н), 3.11-3.01 (m, 2Н), 3.01-2.91 (m, 2Н), 2.48 (s, 6Н), 2.39-2.16 (m, 6Н), 1.83-1.65 (m, 2Н), 1.43 (d, J=6.0 Гц, 6Н), 1.19-0.90 (m, 12Н).
Пример 45
(4S,7S,9aS)-N-[(1R,2R)-2-({[4-({[(1R,2R)-1-[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-амидо]-2,3-дигидро-1Н-инден-2-ил]формамидо}метил)фенил]метил}карбамоил)-2,3-дигидро-1Н-инден-1-ил]-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
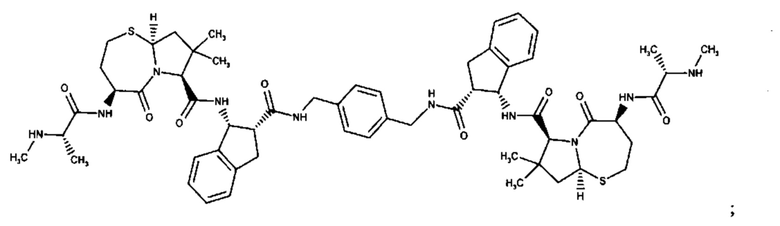
ЖХМС (электроспрей, m/z): 1105.7 [М+Н]+, время удерживания 1.268 мин.
1Н-ЯМР (400 МГц, ДМСО-d6): δ ppm 9.07 (s, 2Н), 8.92-8.72 (m, 4Н), 8.48-8.38 (m, 2Н), 7.88 (d, J=8.9 Гц, 2Н), 7.41-6.98 (m, 12Н), 5.71-5.62 (m, 2Н), 5.57-5.43 (m, 2Н), 4.76-4.65 (m, 2Н), 4.44 (dd,J=14.7, 6.7 Гц, 2Н), 4.11 (s, 2Н), 4.00-3.91 (m, 2Н), 3.91-3.82 (m, 2Н), 3.47-3.41 (m, 2Н), 3.25-3.12 (m, 4Н), 3.05 (dd, J=16.2, 8.6 Гц, 2Н), 2.90 (d, J=13.0 Гц, 2Н), 2.50-2.42 (m, 6Н), 2.28-2.15 (m, 4Н), 2.13-2.02 (m, 2Н), 1.76-1.63 (m, 2Н), 1.43-1.32 (m, 6Н), 0.98 (s, 12Н).
Пример 46
(4S,7S,9aS)-N-[(1R,2R)-2-{4-[(1R,2R)-1-[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-амидо]-2,3-дигидро-1Н-инден-2-карбонил]пиперазин-1-карбонил}-2,3-дигидро-1H-инден-1-ил]-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
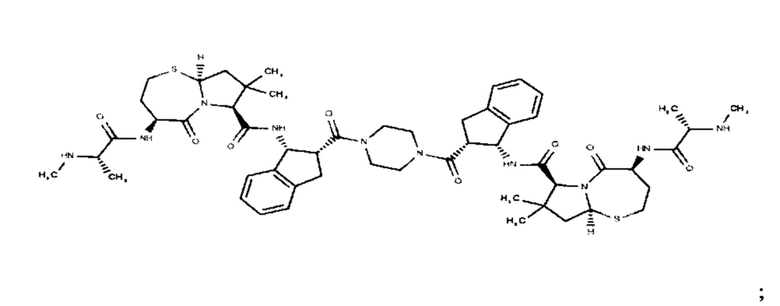
ЖХМС (электроспрей, m/z): 1055.3 [М+Н]+, время удерживания 1.185 мин.
1Н-ЯМР (400 МГц, ДМСО-d6): δ ppm 9.66-9.32 (m, 2Н), 9.06-8.71 (m, 4Н), 7.98-7.85 (m, 1Н), 7.67 (d, J=9.5 Гц, 1Н), 7.38-7.11 (m, 8Н), 5.78-5.59 (m, 2Н), 5.55- 5.40 (m, 2Н), 4.72-4.58 (m, 2Н), 4.09-3.80 (m, 6Н), 3.77-3.47 (m, 10Н), 3.46-3.32 (m, 4Н), 3.29-3.12 (m, 2Н), 3.02-2.77 (m, 4Н), 2.50-2.43 (m, 6Н), 2.28-2.09 (m, 4Н), 1.46-1.37 (m, 6Н), 1.07-0.83 (m, 12Н).
Пример 47
(4S,7S,9aS)-N-[(1R,2R)-2-{4-[(1R,2R)-1-[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-амидо]-2,3-дигидро-1Н-инден-2-карбонил]пиперазин-1-карбонил}-2,3-Дигидро-1Н-инден-1-ил]-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
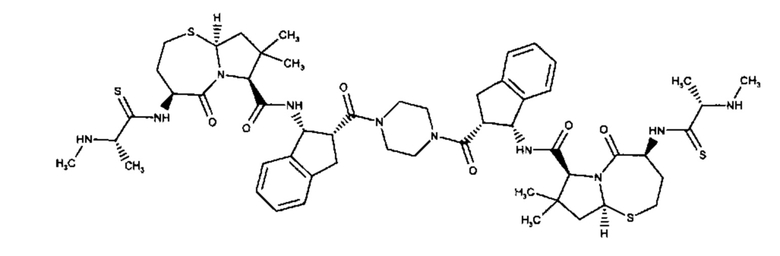
ЖХМС (электроспрей, m/z): 1086.6 [М+Н]+, время удерживания 1.337 мин.
1Н-ЯМР (400МГц, ДМСО-d6): δ ppm 11.37-10.79 (m, 2Н), 9.95-9.38 (m, 2Н), 8.80-8.22 (m, 2Н), 7.97 (dd, J=28.6, 9.8 Гц, 1H), 7.72 (d, J=9.4 Гц, 1Н), 7.38-7.09(m, 8Н), 5.79-5.59 (m, 2Н), 5.44 (dd, J=16.3, 8.2 Гц, 2Н), 5.22-4.91 (m, 2Н), 4.71-4.31 (m, 2Н), 4.15- 3.79 (m, 4Н), 3.80-3.38 (m, 10Н), 3.33-3.12 (m, 4Н), 3.04-2.83 (m, 4Н), 2.51-2.49 (m, 6Н), 2.37-2.00 (m, 6Н), 1.48-1.38 (m, 6Н), 1.01 (d, J=9.6 Гц, 12Н).
Пример 48
(4S,7S,9aS)-N-[(1S,2R)-2-[(8-{[(1S,2R)-1-[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-амидо]-2,3-дигидро-1Н-инден-2-ил|окси}октил)окси]-2,3-дигидро-1Н-инден-1-ил]-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
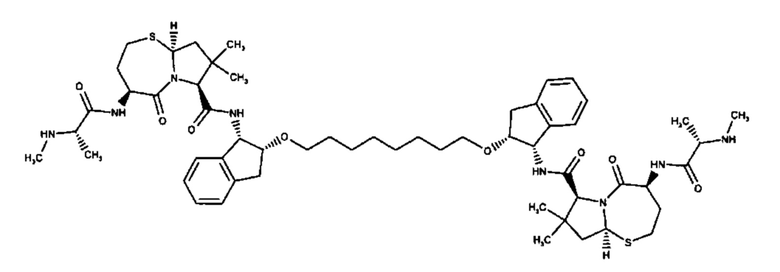
ЖХМС (электроспрей, m/z): 1059.7 [М+Н]+, время удерживания 1.348 мин.
1Н-ЯМР (400 МГц, ДМСО-d6) d ppm δ 9.50 (s, 2Н), 8.93 (s, 2Н), 8.87 (d, J=6.6 Гц, 2Н), 7.91 (d, J=8.7 Гц, 2Н), 7.27-7.19 (m, 8Н), 5.51 (t, J=7.9 Гц, 2Н), 5.33 (dd, J=8.6, 5.4 Гц, 2Н), 4.78-4.70 (m, 2Н), 4.24 (s, 2Н), 4.11 (dd, J=9.3, 4.1 Гц, 2Н), 3.89 (dd, J=10.5, 6.1 Гц, 2Н), 3.44-3.38 (m, 4Н), 3.20 (t, J=11.8 Гц, 2Н), 2.98 (d, J=3.7 Гц, 4Н), 2.95-2.85 (m, 2Н), 2.47 (s, 6Н), 2.27-2.11 (m, 4Н), 1.89-1.76 (m, 4Н), 1.47-1.36 (m, 10Н), 1.24-1.17 (m, 8Н), 1.08 (s, 6Н), 1.06 (s, 6Н).
Пример 49
(4S,7S,9aS)-N-[(1R,2R)-2-[(6-{[(1R,2R)-1-[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-амидо]-2,3-дигидро-1H-инден-2-ил]формамидо}гекса-2,4-диин-1-ил)карбамоил1-2,3-дигидро-1Н-инден-1-ил]-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
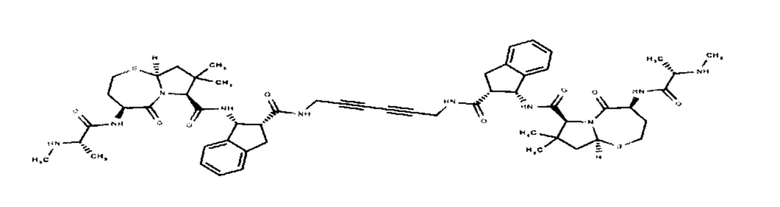
ЖХМС (электроспрей, m/z): 1105.7 [М+Н]+, время удерживания 1.370 мин.
1Н-ЯМР (400МГц, ДМСО-d6) d ppm δ 9.05 (s, 2Н), 8.77 (d, J=7.0 Гц, 4Н), 8.45 (s, 2Н), 7.87 (d, J=9.3 Гц, 2Н), 7.29 (d, J=8.2 Гц, 2Н), 7.17-7.05 (m, 6Н), 5.46 (t, J=8.0 Гц, 2Н), 5.37 (dd, J=9.1, 5.4 Гц, 2Н), 4.72-4.63 (m, 2Н), 4.12-4.01 (m, 4Н), 3.89 (dd, J=12.1, 6.5 Гц, 2Н), 3.82 (d, J=4.3 Гц, 1Н), 3.77 (d, J=4.8 Гц, 1Н), 3.65 (s, 1Н), 3.57 (s, 1Н), 3.21-3.11 (m, 2Н), 2.96-2.88 (m, 2Н), 2.87-2.80 (m, 2Н), 2.78-2.64 (m, 4Н), 2.18 (dd, J=12.9, 7.3 Гц, 2Н), 2.12-1.90 (m, 8Н), 1.77-1.67 (m, 2Н), 1.37(d, J=6.8 Гц, 6Н), 1.24 (s, 4Н), 0.99 (d, J=13.7 Гц, 12Н).
Пример 50
(4S,7S,9aS)-N-[(1S)-2-[2-(4-{[(2S)-2-{[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-ил]формамидо}-2-фенилэтокси]метил}-1Н-1,2,3-триазол-1-ил)этокси]-1-фенилэтил]-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
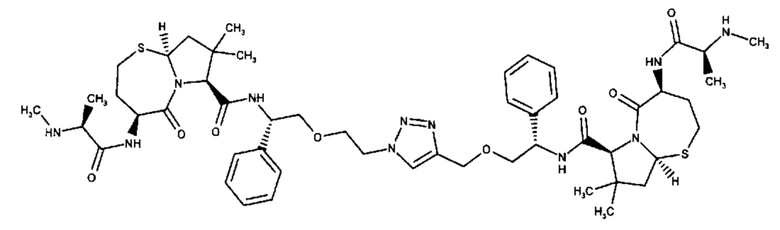
ЖХМС (электроспрей, m/z): 1032.7 [М+Н]+, время удерживания 1.224 мин.
1Н-ЯМР (400МГц, ДМСО-d6) d ppm δ 9.74-9.45 (m, 2Н), 8.92 (d, J=5.8 Гц, 2Н), 8.85 (d, J=7.0 Гц, 2Н), 8.39 (d, J=8.0 Гц, 2Н), 7.79(s, 1H), 7.42-7.15 (m, 10Н), 5.46 (t, J=7.5 Гц, 2Н), 5.01 (td, J=14.0, 6.5 Гц, 2Н), 4.73-4.65 (m, 2Н), 4.49- 4.45 (m, 4Н), 4.17 (d, J=3.4 Гц, 2Н), 3.92-3.81 (m, 2Н), 3.87 (t, J=4.7 Гц, 2H), 3.63-3.50 (m, 4H), 3.23-3.06 (m, 2H), 2.95-2.80 (m, 2H), 2.45 (t, J=5.0 Гц, 6H), 2.26-2.03 (m, 4H), 1.92-1.66 (m, 4H), 1.39 (d, J=6.8T4, 6H), 1.04 (s, 6H), 0.96 (s, 6H).
Пример 51
(4S,7S,9aS)-N-[(1S,2R)-2-[3-(3-{[(1S,2R)-1-[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3)тиазепин-7-амидо]-2,3-дигидро-1H-инден-2-ил]окси}пропокси)пропокси]-2,3-дигидро-1Н-инден-1-ил]-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо)-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
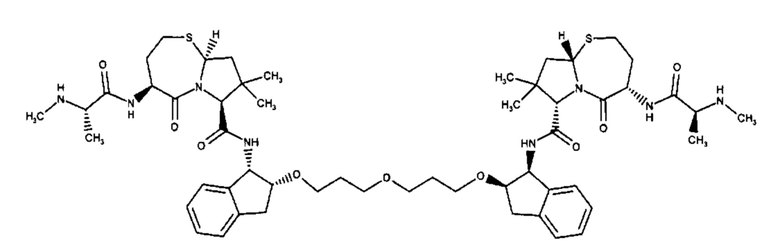
ЖХМС (электроспрей, m/z): 1047.7 [М+Н]+, время удерживания 1.147 мин.
1Н-ЯМР (400 МГц, ДМСО-d6) d ppm δ 9.47 (s, 2Н), 8.99-8.81 (m, 4Н), 7.91 (d, J=8.7 Гц, 2Н), 7.28-7.18 (m, 8Н), 5.51 (t, J=7.9 Гц, 2Н), 5.34 (dd, J=8.6, 5.4 Гц, 2Н), 4.74 (dd, J=9.9, 7.7 Гц, 2Н), 4.24 (s, 2Н), 4.12 (dd, J=9.1, 4.0 Гц, 2Н), 3.89 (dd, J=11.1, 6.4 Гц, 2Н), 3.52-3.39 (m, 6Н), 3.32-3.30 (m, 2Н), 3.25-3.15 (m, 2Н), 2.98 (d, J=3.5 Гц, 4Н), 2.95-2.87 (m, 2Н), 2.47 (s, 6Н), 2.30-2.10 (m, 4Н), 1.88-1.75 (m, 4Н), 1.73-1.58 (m, 4Н), 1.41 (d, J=6.9 Гц, 6Н), 1.07 (d, J=9.5 Гц, 12Н).
Пример 52
(4S,7S,9aS)-N-[(1S)-2-[2-(4-{[(2S)-2-{[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо)-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-ил]формамидо}-2-фенилэтокси]метил}-1Н-1,2,3-триазол-1-ил)этокси1-1-фенилэтил]-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
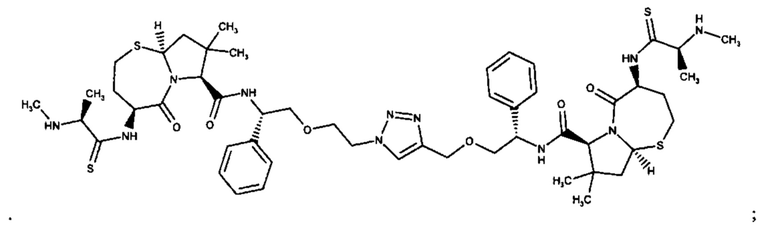
ЖХМС (электроспрей, m/z): 1064.6 [М+Н]+, время удерживания 1.267 мин.
1Н-ЯМР (400 МГц, ДМСО-d6) d ppm δ 11.02-10.87 (m, 2Н), 9.65 (d, J=70.2 Гц, 2Н), 8.62 (s, 1Н), 8.43 (t, J=7.0 Гц, 3Н), 7.78 (s, 1Н), 7.37-7.23 (m, 10Н), 5.42 (q, J=7.6 Гц, 2Н), 5.17-5.07 (m, 2Н), 5.07-4.95 (m, 2Н), 4.53-4.42 (m, 4Н), 4.30-4.16 (m, 4Н), 3.61-3.54 (m, 4Н), 3.21-3.09 (m, 2Н), 2.99-2.88 (m, 2Н), 2.49-2.44 (m, 6Н), 2.29-2.14 (m, 4Н), 1.95-1.77 (m, 4Н), 1.43 (d, J=6.3 Гц, 6Н), 1.24 (s, 2Н), 1.07 (s, 6Н), 0.98 (s, 6Н).
Пример 53
(4S,7S,9aS)-N-[(1S)-2-[3-(4-{[(2S)-2-{[(4S,7S,9aS)-8,8-дметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-ил]формамндо}-2-фенилэтокси]метил}-1Н-1,2,3-триазол-1-ил)пропокси)-1-фенилэтил]-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
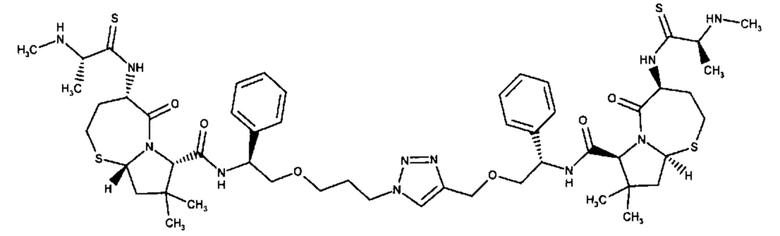
ЖХМС (электроспрей, m/z): 1079.7 [М+Н]+, время удерживания 1.148 мин.
1Н-ЯМР (400 МГц, ДМСО-d6): d ppm δ 10.99 (d, J=3.8 Гц, 1Н), 10.90 (d, J=6.0 Гц, 1H), 9.75 (s, 1H), 9.57 (s, 1H), 8.63 (s, 1H), 8.51-8.31 (m, 3H), 7.88 (s, 1H), 7.40-7.23 (m, 10H), 5.49-5.35 (m, 2H).5.13 (dd, J=16.0, 9.1 Гц, 2H), 5.07-4.98 (m, 2H), 4.50 (s, 2H), 4.36-4.15 (m, 6Н), 3.61-3.55 (m, 4H), 3.39-3.28 (m, 2Н), 3.21-3.10 (m, 2H), 3.01-2.88 (m, 2H), 2.49-2.40 (m, 6H), 2.30-2.14 (m, 4H), 2.02-1.81 (m, 6H), 1.43 (d, J=6.4 Гц, 6H), 1.11-1.04 (m, 6H), 1.03 (s, 3H), 0.96 (s, 3H).
Пример 54
(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-N-[(1R,2R)-2-{[(1rs,4rs)-4-[(1R,2R)-1-[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропантиоамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-амидо]-2,3-дигидро-1Н-инден-2-амидо]циклогексил]карбамоил}-2,3-дигидро-1Н-инден-1-ил]-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
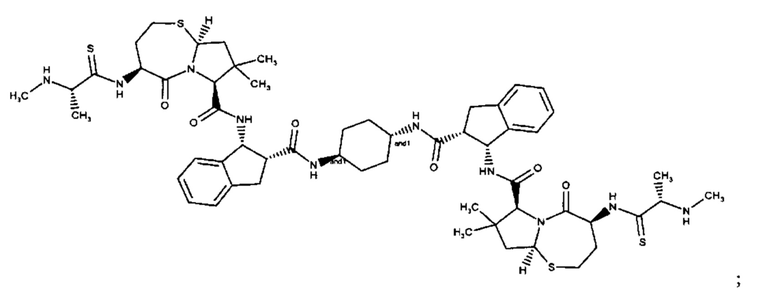
ЖХМС (электроспрей, m/z): 1115.6 [М+Н]+, время удерживания 1.136 мин.
1Н-ЯМР (400 МГц, ДМСО-d6): d ppm δ 11.00-10.80 (m, 2Н), 9.56 (s, 1H), 9.41 (s, 1Н), 8.64 (s, 1H), 8.46 (s, 1H), 8.00-7.82 (m, 4H), 7.45-7.12 (m, 8H), 5.60 (t, J=16.0 Гц, 2H), 5.50-5.37 (m, 2H), 5.17-5.07 (m, 2H), 4.41-4.26 (m, 2H), 4.14(d, J=8.0 Гц, 2H), 3.75-3.65 (m, 2H), 3.35-3.27 (m, 2H), 3.25-3.07 (m, 4H), 3.05-2.91 (m, 4H), 2.50 (s, 6H), 2.30-2.02 (m, 6H), 1.85-1.67 (m, 6H), 1.44 (d, J=4.0 Гц, 6H), 1.25-1.10 (m, 4H), 1.03 (d, J=8.0 Гц, 12H).
Пример 55
(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-N-[(1R,2R)-2-{[(1rs,4rs)-4-[(1R,2R)-1-[(4S,7S,9aS)-8,8-диметил-4-[(2S)-2-(метиламино)пропанамидо]-5-оксо-октагидропирроло[2,1-b][1,3]тиазепин-7-амидо]-2,3-дигидро-1Н-инден-2-амидо]циклогексил1карбамоил}-2,3-дигидро-1Н-инден-1-ил]-октагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид
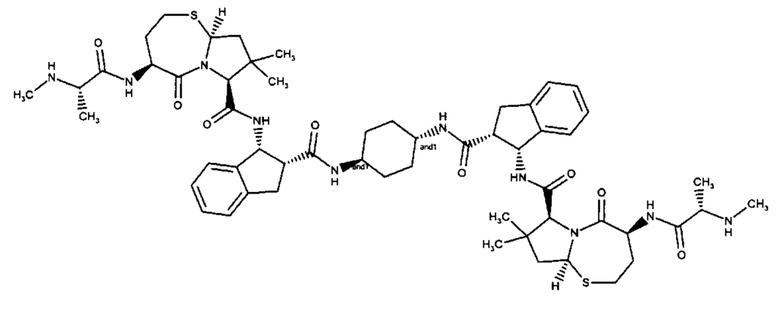
ЖХМС (электроспрей, m/z): 1083.7 [М+Н]+, время удерживания 1.091 мин.
1Н-ЯМР (400 МГц, ДМСО-d6): d ppm δ 9.17 (s, 2Н), 8.92 (s, 2Н), 8.75 (d, J=4.0 Гц, 2Н), 7.90 (s, 2Н), 7.84 (d, J=8.0 Гц, 2Н), 7.30-7.05 (m, 8Н), 5.60 (t, J=16.0 Гц, 2Н), 5.48 (t, J=16.0 Гц, 2Н), 4.75-4.60(m, 2Н), 4.11 (s, 2Н), 3.98-3.85 (m, 2Н), 3.38-3.28 (m, 2Н), 3.24-3.10 (m, 4Н), 3.07-2.97 (m, 2Н), 2.95-2.85 (m, 2Н), 2.50-2.43 (m, 6Н), 2.25-2.00 (m, 6Н), 1.87-1.73 (m, 4Н), 1.73-1.62 (m, 2Н), 1.40 (d, J=8.0 Гц, 6Н), 1.25-1.17 (m, 2Н), 1.17-1.10 (m, 2Н), 1.02 (s, 12Н).
Пример 56
(4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-((нафталин-2,7-диилбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)
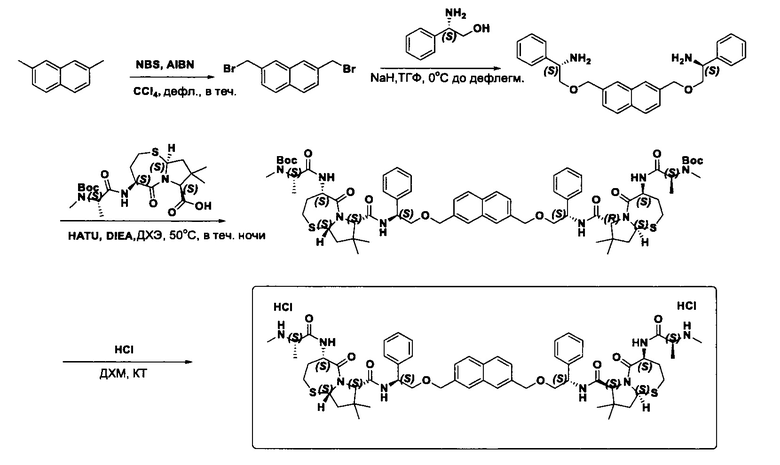
Синтез 2,7-бис(бромметил)нафталина
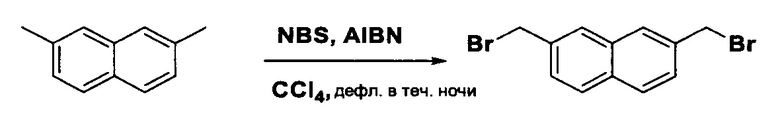
К раствору 2,7-диметилнафталина (1 г, 6.40 ммоль) и 1-бромпирролидин-2,5-диона (2.5 г, 14.08 ммоль) в перхлорметане (40 мл) добавляли 2,2'-азобис-(2-метилпропаннитрил) (105 мг, 0.64 ммоль). Смесь перемешивали при 80°С в течение ночи. Нерастворимое твердое вещество удаляли фильтрацией через слой целита. Фильтрат промывали насыщенным раствором бикарбоната натрия (20 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта. Его очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 100:1), в результате чего получали 2,7-бис(бромметил)нафталин (600 мг, 1.91 ммоль, 29.9% выход) в форме белого твердого вещества.
Синтез (1S,1'S)-2,2'-((нафталин-2,7-диилбис(метилен))бис(окси))бис(1-фенилэтан-1-амина)
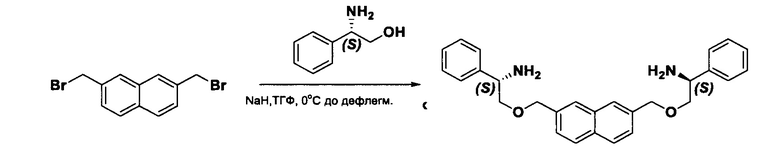
К раствору (S)-2-амино-2-фенилэтан-1-ола (585 мг, 3.92 ммоль) в тетрагидрофуране (20 мл) при 0°С добавляли порциями гидрид натрия (60%, дисперсия в жидком парафине) (171 г, 4.27 ммоль). Смесь нагревали до комнатной температуры естественным путем. Затем капали 2,7-бис(бромметил)нафталин (560 мг, 1.78 ммоль) и нагревали полученную смесь до 70°С. Ее перемешивали при 70°С в течение ночи. Реакционную смесь гасили ледяной водой (30 мл) и экстрагировали этилацетатом (3×20 мл). Органические слои объединяли, промывали солевым раствором (20 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта. Его очищали колоночной хроматографией на силикагеле (дихлорметан /метанол = 15:1), в результате чего получали (1S, 1'S)-2,2'-((нафталин-2,7-диилбис(метилен))бис(окси))бис(1-фенилэтан-1-амин) (200 мг, 0.469 ммоль, 26.3% выход) в виде черного масла.
ЖХМС (электроспрей, m/z): 426.8 [М+Н]+, время удерживания 1.047 мин.
Синтез трет-бутил((S)-1-(((4S,7R,9aS)-7-(((S)-2-((7-(((S)-2-((4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2-фенилэтокси)метил)нафталин-2-ил)метокси)-1-фенилэтил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата
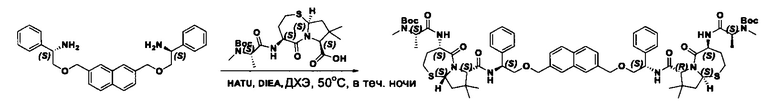
К раствору (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (499 мг, 1.13 ммоль), этил-2-этоксихинолинил-1(2Н)-карбоксилата (325 мг, 1.31 ммоль) и N,N-диизопропилэтиламина (243 мг, 1.88 ммоль) в 1,2-дихлорэтане (10 мл) добавляли (1S,1'S)-2,2'-((нафталин-2,7-диилбис(метилен))бис(окси))бис(1-фенилэтан-1-амин) (200 мг, 0.47 ммоль). Смесь перемешивали при 50°С в течение ночи. Реакционную смесь концентрировали с получением неочищенного продукта. Его очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1:4), в результате чего получали неочищенный продукт. Затем неочищенный продукт очищали препаративной ЖХВД с получением продукта (110 мг, 0.086 ммоль, 18.3% выход) в форме белого твердого вещества.
ЖХМС (электроспрей, m/z): 539.3 [М/2-Вос+Н]+, время удерживания 1.770 мин.

Синтез (4S,4,S,7S,7'S,9aS,9а'S)-N,N'-((1S,1,S)-((нафталин-2,7-диилбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)
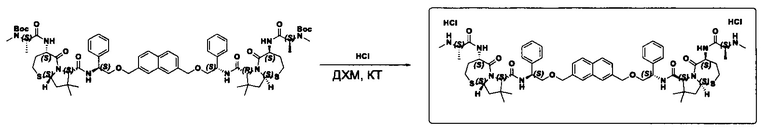
К раствору трет-бутил ((S)-1-(((4S,7R,9aS)-7-(((S)-2-((7-(((S)-2-((4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамидо)-2-фенилэтокси)метил)нафталин-2-ил)метокси)-1-фенилэтил)карбамоил)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-4-ил)амино)-1-оксопропан-2-ил)(метил)карбамата (110 мг, 0.086 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлороводородную кислоту в диоксане (0.5 мл). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали с получением целевого продукта (90 мг, 0.078 ммоль, 90.9% выход) в форме белого твердого вещества. Затем это соединение лиофилизировали для удаления остатка растворителя.
ЖХМС (электроспрей, m/z): 1077.4 [М+Н]+, время удерживания 1.253 мин.
1Н-ЯМР (400 МГц, ДМСО-di) δ ppm 9.48 (s, 2Н), 9.01 - 8.77 (m, 4Н), 8.47 (d, J = 8.1 Гц, 2Н), 7.85 (d, J = 8.4 Гц, 2Н), 7.68 (s, 2Н), 7.45 - 7.33 (m, 10Н), 7.31 - 7.25 (m, 2Н), 5.46 (t, J = 7.9 Гц, 2Н), 5.14 (dd, J = 13.1, 6.6 Гц, 2Н), 4.74 - 4.59 (m, 6Н), 4.20 (s, 2Н), 3.85 (s, 2Н), 3.72 - 3.60 (m, 4Н), 3.13 (t, J = 12.3 Гц, 2Н), 2.84 - 2.73 (m, 2Н), 2.45 (s, 6Н), 2.18 (dd, J = 12.6, 7.0 Гц, 2Н), 2.11 - 2.02 (m, 2Н), 1.87 (dd, J = 12.5, 9.0 Гц, 2Н), 1.73 (dd, J = 22.6, 11.5 Гц, 2Н), 1.40 (d, J = 6.9 Гц, 6Н), 1.05 (s, 6Н), 0.99 (s, 6Н).
Пример 57
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S,2R,2'R)-(декан-1,10-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)дигидрохлорид
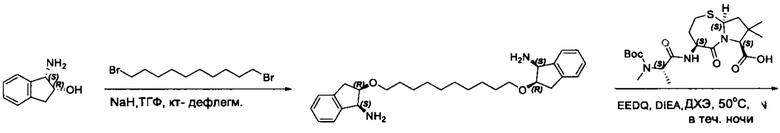
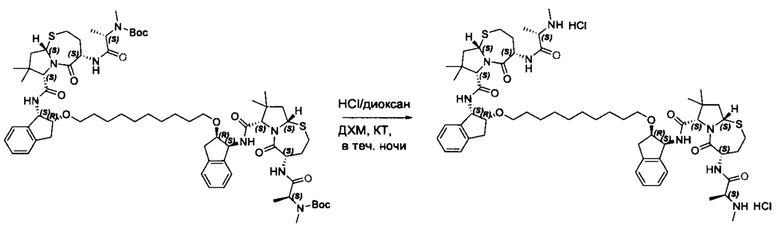
Синтез (1S,1'S,2R,2'R)-2,2'-(декан-1,10-диилбис(окси))бис(2,3-дигидро-1H-инден-1-амина)
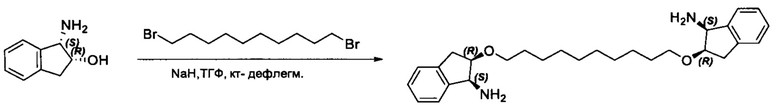
К раствору (1S,2R)-1-амино-2,3-дигидро-1Н-инден-2-ола (1 г, 6.71 ммоль) в тетрагидрофуране (60 мл) при 0°С добавляли порциями гидрид натрия (60%, дисперсия в жидком парафине) (293 мг, 7.32 ммоль). Смесь нагревали до комнатной температуры естественным путем. Затем добавляли 1,10-дибромдекан (1.0 г, 3.05 ммоль). Полученную смесь перемешивали при 70°С в течение ночи. Реакционную смесь гасили ледяной водой (30 мл) и экстрагировали этилацетатом (3 × 30 мл). Органический слой объединяли, промывали солевым раствором (50 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта. Его очищали хроматографией в режиме Combi-Flash, используя для элюирования дихлорметан/метанол = 20:1 и тонкостенной хроматографией с использованием для проявки этилацетатом/петролейный эфир = 3/1, в результате чего получали целевой продукт (100 мг, 0.23 ммоль, 7.5% выход) в виде темного масла.
ЖХМС (электроспрей, m/z): 437.0 [М+Н]+, время удерживания 1.117 мин.
Синтез ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(декан-1,10-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азанедиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата)
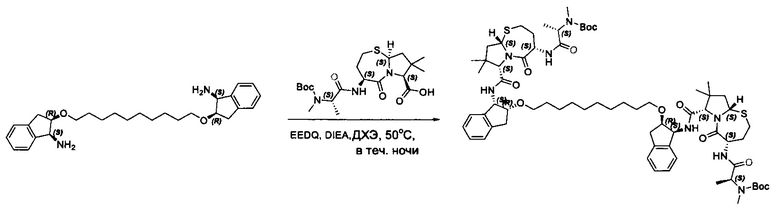
Смесь (1S,1'S,2R,2'R)-2,2'-(декан-1,10-диилбис(окси))бис(2,3-дигидро-1Н-инден-1-амина) (100 мг, 0.229 ммоль) и (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (220 мг, 0.505 ммоль) в 1,2-дихлорэтане (8 мл) добавляли N-этоксикарбонил-2-этокси-1,2-дигидрохинолинил (170 мг, 0.687 ммоль) и N,N-диизопропилэтиламин (118 мг, 0.916 ммоль). Полученную смесь перемешивали при 50°С в течение ночи. Реакционную смесь концентрировали с получением неочищенного продукта. Его очищали тонкослойной хроматографией, с использованием для проявки этилацетат/петролейный эфир = 2/1, в результате чего получали неочищенный продукт. Затем неочищенный продукт очищали препаративной ЖХВД с получением продукта (100 мг, 0.077 ммоль, 33.9% выход) в виде бежевого твердого вещества.
ЖХМС (электроспрей, m/z): 543.6 [M/2-Boc+H]+, время удерживания 2.072 мин.

Синтез (4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S,2R,2'R)-(декан-1,10-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло|2,1-b][1,3]тиазепин-7-карбоксамид)дигидрохлорида
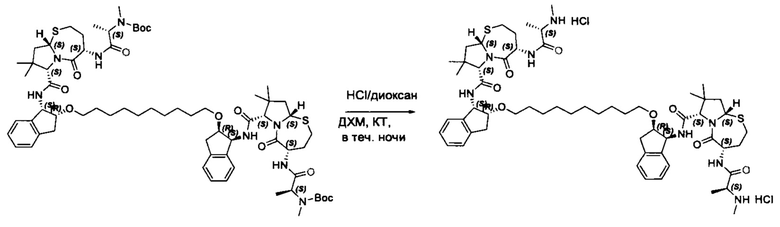
К раствору ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((((1S,1'S,2R,2'R)-(декан-1,10-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азанедиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (100 мг, 0.078 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлороводородную кислоту в диоксане (0.7 мл). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали с получением целевого продукта (62 мг, 0.053 ммоль, 67.9% выход) в форме белого твердого вещества.
ЖХМС (электроспрей, m/z): 1087.8 [М-2HCl+Н]+ (Расч. М-2HCl+Н=1087.6), время удерживания 1.254 мин.
1Н-ЯМР (400 МГц, ДМСО) δ ppm 9.43 (s, 2Н), 8.99 - 8.76 (m, 4Н), 7.90 (d, J = 8.7 Гц, 2Н), 7.34 - 7.09 (m, 8Н), 5.51 (t, J = 7.7 Гц, 2Н), 5.34 (dd, J = 8.3, 5.5 Гц, 2Н), 4.82 - 4.67 (m, 2Н), 4.24 (s, 2Н), 4.12 (dd, J = 8.4, 3.7 Гц, 2Н), 3.89 (dd, J = 11.0, 6.0 Гц, 2Н), 3.42 - 3.36 (m, 4Н), 3.20 (t, J = 12.1 Гц, 2Н), 2.98 (d, J = 3.2 Гц, 4Н), 2.95 - 2.86 (m, 2Н), 2.50 - 2.44 (m, 6Н), 2.29 - 2.09 (m, 4Н), 1.90 - 1.74 (m, 4Н), 1.54 - 1.35 (m, 10Н), 1.28 - 1.15 (m, 12Н), 1.09 (s, 6Н), 1.06 (s, 6Н).
Пример 58
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-(1S,1'S)-(октан-1,8-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)дигидрохлорид
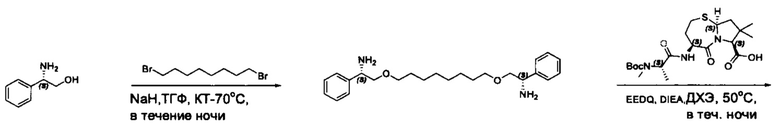
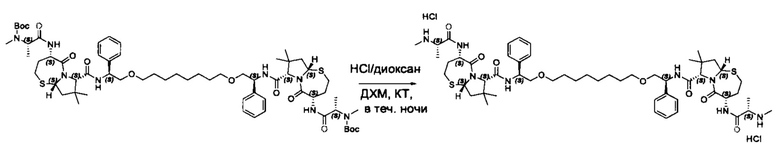
Синтез (1S,1'S)-2,2'-(октан-1,8-диилбис(окси))бис(1-фенилэтан-1-амина)
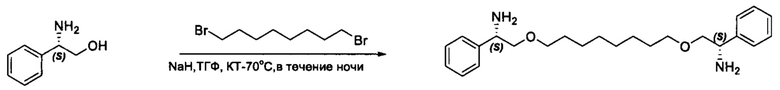
К раствору (S)-2-амино-2-фенилэтан-1-ола (1 г, 7.29 ммоль) в тетрагидрофуране (60 мл) при 0°С добавляли порциями гидрид натрия (60%, дисперсия в жидком парафине) (318 мг, 7.94 ммоль). Смесь нагревали до комнатной температуры естественным путем. Затем добавляли 1,8-дибромоктан (901 мг, 3.31 ммоль). Полученную смесь перемешивали при 70°С в течение ночи. Реакционную смесь гасили ледяной водой (30 мл) и экстрагировали этилацетатом (3 × 30 мл). Органический слой объединяли, промывали солевым раствором (50 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта. Его очищали на колонке Combi-Flash, используя для элюирования дихлорметан/метанол = 25:1, а затем тонкослойной хроматографией, используя для проявки этилацетат/петролейный эфир = 3/1, в результате чего получали целевой продукт (400 мг, 1.04 ммоль, 31.4% выход) в виде желтого масла.
ЖХМС (электроспрей, m/z): 385.0 [M+H]+, время удерживания 1.037 мин.
Синтеза ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((3S,16S)-3,16-дифенил-5,14-диокса-2,17-диазаоктадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата)
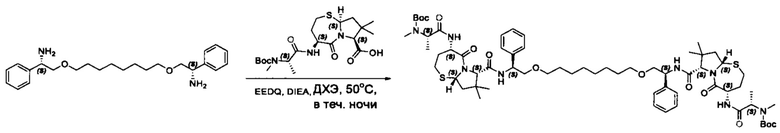
Смесь (1S,1'S)-2,2'-(октан-1,8-диилбис(окси))бис(1-фенилэтан-1-амина) (100 мг, 0.26 ммоль) и (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (252 мг, 0.57 ммоль) в 1,2-дихлорэтане (10 мл) добавляли в N-этоксикарбонил-2-этокси-1,2-дигидрохинолинил (193 мг, 0.78 ммоль) и N,N-ииизопропилэтиламин (134 мг, 1.04 ммоль). Полученную смесь перемешивали при 50°С в течение ночи. Реакционную смесь концентрировали с получением неочищенного продукта. Его очищали тонкослойной хроматографией, с использованием для проявки этилацетат/петролейный эфир = 3/1, в результате чего получали неочищенный продукт. Then неочищенный продукт очищали препаративной ЖХВД с получением продукта (100 мг, 0.081 ммоль, 31.2% выход) в форме белого твердого вещества.
ЖХМС (электроспрей, m/z): 517.8 [1/2М-Вос+Н]+, время удерживания 1.859 мин.
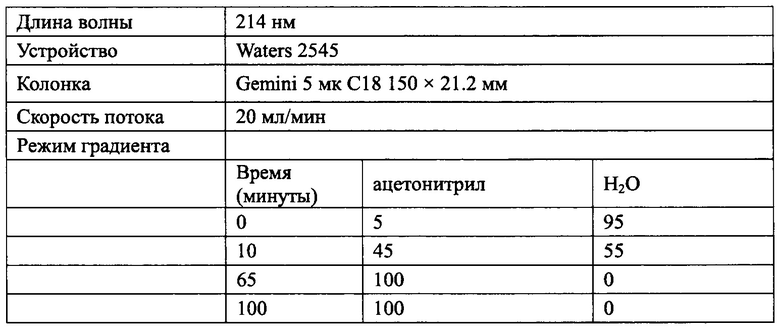
Синтез (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S)-(октан-1,8-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)дигидрохлорида
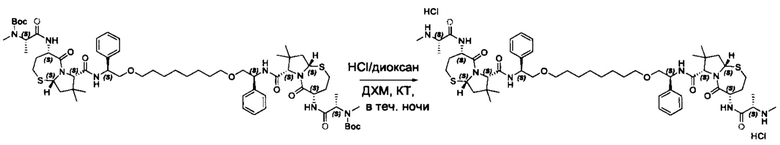
К раствору ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,16S)-3,16-дифенил-5,14-диокса-2,17-диазаоктадекандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (100 мг, 0.081 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлороводородную кислоту в диоксане (0.7 мл). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали с получением целевого продукта (60 мг, 0.054 ммоль, 66.7% выход) в форме белого твердого вещества.
ЖХМС (электроспрей, m/z): 1035.7 [М-2HCl+Н]+ (Расч. М-2НС1+Н=1035.6), время удерживания 1.211 мин.
1Н-ЯМР (400 МГц, ДМСО) δ ppm 9.45 (s, 2Н), 8.89 (d, J = 6.1 Гц, 2Н), 8.82 (d, J = 7.0 Гц, 2Н), 8.37 (d, J = 8.1 Гц, 2Н), 7.40 - 7.30 (m, 8Н), 7.28 - 7.21 (m, 2Н), 5.46 (t, J = 7.8 Гц, 2Н), 5.00 (dd, J = 13.4, 6.6 Гц, 2Н), 4.75 - 4.65 (m, 2Н), 4.17 (s, 2Н), 3.85 (d, J = 5.6 Гц, 2Н), 3.55 - 3.47 (m, 4Н), 3.39 - 3.33 (m, 4Н), 3.16 (t, J = 12.0 Гц, 2Н), 2.94 - 2.82 (m, 2Н), 2.45 (s, 6Н), 2.19 (dd, J = 12.6, 7.0 Гц, 2Н), 2.14 - 2.06 (m, 2Н), 1.87 (dd, J = 12.4, 8.8 Гц, 2Н), 1.75 (dd, J = 22.3, 11.4 Гц, 2Н), 1.47 - 1.35 (m, 10Н), 1.24 - 1.16 (m, 8Н), 1.06 (s, 6Н), 1.01 (s, 6Н).
Пример 59
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S,2R,2'R)-(додекан-1,12-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)дигидрохлорид
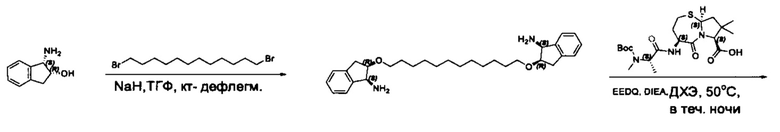
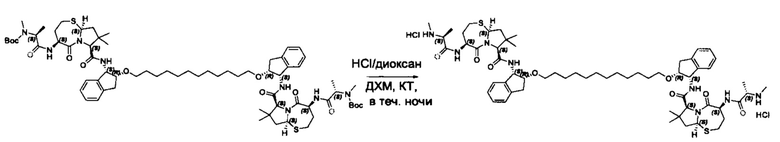
Синтез (1S,1'S,2R,2'R)-2,2'-(додекан-1,12-диилбис(окси))бис(2,3-дигидро-1Н-инден-1-амина)
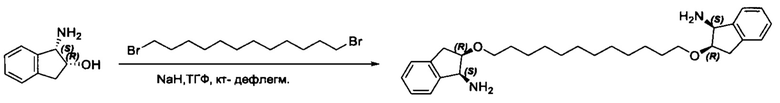
К раствору (1S,2R)-1-амино-2,3-дигидро-1Н-инден-2-ола (1 г, 5.70 ммоль) в тетрагидрофуране (60 мл) при 0°С добавляли порциями гидрид натрия (60%, дисперсия в жидком парафине) (249 мг, 6.22 ммоль). Смесь нагревали до комнатной температуры естественным путем. Затем добавляли 1,12-дибромдодекан (0.85 г, 2.59 ммоль). Полученную смесь перемешивали при 70°С в течение ночи. Реакционную смесь гасили ледяной водой (30 мл) и экстрагировали этилацетатом (3 × 30 мл). Органический слой объединяли, промывали солевым раствором (50 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта. Его очищали тонкослойной хроматографией, с использованием для проявки этилацетат/петролейный эфир = 3/1, в результате чего получали целевой продукт (160 мг, 0.34 ммоль, 13.3% выход) в виде темного масла.
ЖХМС (электроспрей, т/г): 465.0 [М+Н]+, время удерживания 1.149 мин.
Синтез ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(додекан-1,12-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азанедиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло(2,1-b][1,3)тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата)
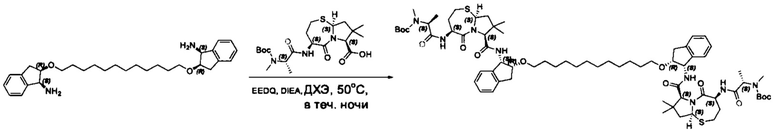
Смесь (1S,1'S,2R,2'R)-2,2'-(додекан-1,12-диилбис(окси))бис(2,3-дигидро-1Н-инден-1-амина) (110 мг, 0.237 ммоль) и (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (231 мг, 0.521 ммоль) в 1,2-дихлорэтане (8 мл) добавляли к N-этоксикарбонил-2-этокси-1,2-дигидрохинолинилу (176 мг, 0.711 ммоль) и N,N-диизопропилэтиламину (122 мг, 0.948 ммоль). Полученную смесь перемешивали при 50°С в течение ночи. Реакционную смесь концентрировали с получением неочищенного продукта. Его очищали тонкослойной хроматографией, с использованием для проявки этилацетат/петролейный эфир = 3/1, в результате чего получали неочищенный продукт. Затем неочищенный продукт очищали препаративной ЖХВД с получением продукта (105 мг, 0.080 ммоль, 33.7% выход) в виде бежевого твердого вещества.
ЖХМС (электроспрей, m/z): 557.9 [1/2М-Вос+Н]+, время удерживания 2.312 мин.
Синтез (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(додекан-1,12-диилбис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)дигидрохлорида
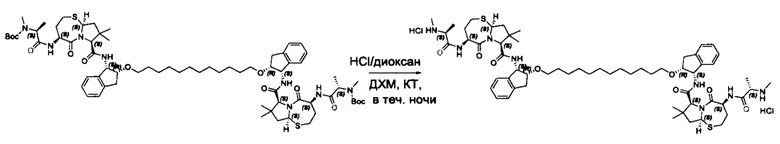
К раствору ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(додекан-1,12-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азанедиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (105 мг, 0.080 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлороводородную кислоту в диоксане (0.7 мл). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали с получением целевого продукта (65 мг, 0.055 ммоль, 68.6% выход) в форме белого твердого вещества.
ЖХМС (электроспрей, m/z): 558.0 [1/2М-HCl+Н]+, время удерживания 1.448 мин.
1Н-ЯМР (400 МГц, ДМСО) δ ppm 9.45 (s, 2Н), 9.03 - 8.77 (m, 4Н), 7.90 (d, J = 8.7 Гц, 2Н), 7.29 - 7.14 (m, 8Н), 5.51 (t, J = 7.8 Гц, 2Н), 5.34 (dd, J = 8.4, 5.5 Гц, 2Н), 4.78 - 4.69 (m, 2Н), 4.24 (s, 2Н), 4.11 (dd, J = 8.8, 4.0 Гц, 2Н), 3.89 (dd, J = 12.9, 6.1 Гц, 2Н), 3.47 - 3.36 (m, 4Н), 3.20 (t, J = 12.5 Гц, 2Н), 2.98 (d, J = 3.3 Гц, 4Н), 2.95 - 2.86 (m, 2Н), 2.47 (s, 6Н), 2.26 - 2.07 (m, 4Н), 1.90 - 1.73 (m, 4Н), 1.49 - 1.36 (m, 10Н), 1.25 - 1.17 (s, 16Н), 1.08(s, 6Н), 1.06(s, 6Н).
Пример 60
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S)-(декан-1,10-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)дигидрохлорид
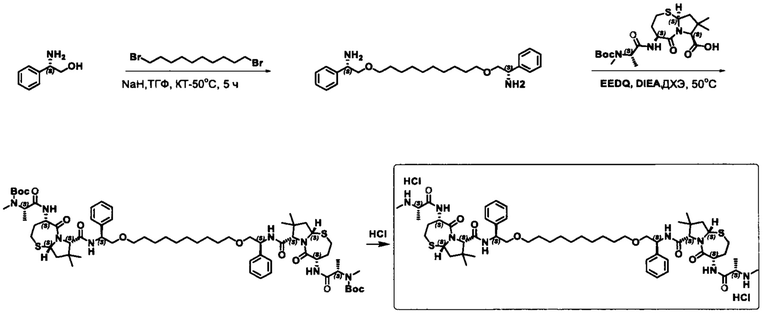
Синтез (1S,1'S)-2,2'-(декан-1,10-диилбис(окси))бис(1-фенилэтан-1-амина)
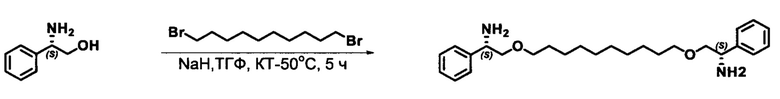
К раствору (S)-2-амино-2-фенилэтан-1-ола (2.1 г, 15.4 ммоль) в тетрагидрофуране (40 мл) партиями добавляли гидрид натрия (0.672 г, 16.8 ммоль) при 0°С. Смесь перемешивали при комнатной температуре в течение 40 мин. Затем добавляли 1,10-дибромдекан (2.1 г, 7 ммоль) и перемешивали смесь при комнатной температуре в течение 10 мин и нагревали до 50°С в течение 3 ч. Реакцию гасили путем добавления воды (100 мл). Затем реакционную смесь экстрагировали этилацетатом (3*100 мл). Объединенные органические слои промывали солевым раствором, сушили с использованием безводного сульфата натрия и концентрировали под вакуумом с получением неочищенного продукта. Его очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = 15:1) и ТСХ (петролейный эфир/этилацетат = 1:3), в результате чего получали (1S,1'S)-2,2'-(декан-1,10-диилбис(окси))бис(1-фенилэтан-1-амин) (0.5 г, 1.21 ммоль, 7.9% выход) в виде желтого масла.
ЖХМС (электроспрей, m/z): 413.0 [М+Н]+, время удерживания 1.073 мин.
Синтез ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,18S)-3,18-дифенил-5,16-диокса-2,19-диазаэйкозандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата)
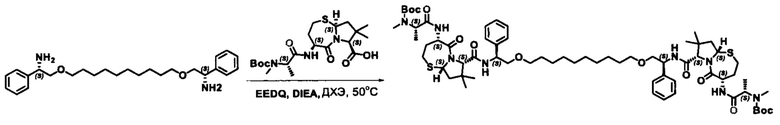
К раствору (1S,1'S)-2,2'-(декан-1,10-диилбис(окси))бис(1-фенилэтан-1-амина) (150 мг, 0.364 ммоль) и (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (355 мг, 0.800 ммоль) в 1,2-дихлорэтане (10 мл) добавляли 2-этокси-1-этоксикарбонил-1,2-дигидрохинолинил (270 мг, 1.091 ммоль) и N,N-диизопропилэтиламин (188 мг, 1.454 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. В это время ее разбавляли водой (10 мл), затем ее экстрагировали дихлорметаном (3*20 мл). Органические слои объединяли, промывали солевым раствором, сушили с использованием безводного сульфата натрия и концентрировали с получением неочищенного продукта. Его очищали препаративной ТСХ (петролейный эфир/этилацетат = 1:3) с получением неочищенного продукта. Затем неочищенный продукт очищали препаративной ВЭЖХ с получением ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,18S)-3,18-дифенил-5,16-диокса-2,19-диазаэйкозандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (70 мг, 0.055 ммоль, выход 15.2%) в виде коричневого твердого вещества.
ЖХМС (электроспрей, m/z): 532.1 [1/2М-Вос+Н]+, время удерживания 1.994 мин.


Синтез (4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S)-(декан-1,10-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)дигидрохлорида
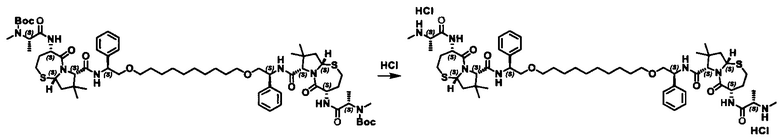
К раствору ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,18S)-3,18-дифенил-5,16-диокса-2,19-диазаэйкозандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (70 мг, 0.055 ммоль) в дихлорметане (5 мл) добавляли 4М хлороводородную кислоту (0.7 мл) в диоксане. Полученную смесь перемешивали в течение 4 ч при комнатной температуре. После этого ее концентрировали с получением (4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S)-(декан-1,10-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)дигидрохлорида (60 мг, 0.053 ммоль, выход 96.0%) в форме светло-желтого твердого вещества.
ЖХМС (электроспрей, m/z): 1062.8 [М-2HCl+Н]+ (Расч. М-2HCl+Н=1063.6), время удерживания 1.213 мин.
1Н-ЯМР (400 МГц, ДМСО-d6): δ ppm 9.42 (s, 2Н), 8.99 - 8.77 (m, 4Н), 8.38 (d, J = 8.0 Гц, 2Н), 7.43 - 7.21 (m, 10Н), 5.56 - 5.42 (t, 2Н), 5.05 - 4.98 (t, 2Н), 4.79 - 4.65 (t, 2Н), 4.19 (s, 2Н), 3.87 (s, 2Н), 3.57 - 3.46 (t, 4Н), 3.42 - 3.35 (m, 4Н), 3.23 - 3.11 (t, 2Н), 2.90 (d, J = 12.0 Гц, 2Н), 2.47 (m, 6Н), 2.26 - 2.17 (m, 2Н), 2.16 - 2.07 (m, 2Н), 1.93 - 1.83 (dd, J = 12.0, 8.0 Гц, 2Н), 1.83 - 1.70 (m, 2Н), 1.50 - 1.35 (m, 10Н), 1.21 (d, J = 16.0 Гц, 12Н), 1.06 (d, J = 20.0 Гц, 12Н).
Пример 61
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S)-(додекан-1,12-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)дигидрохлорид
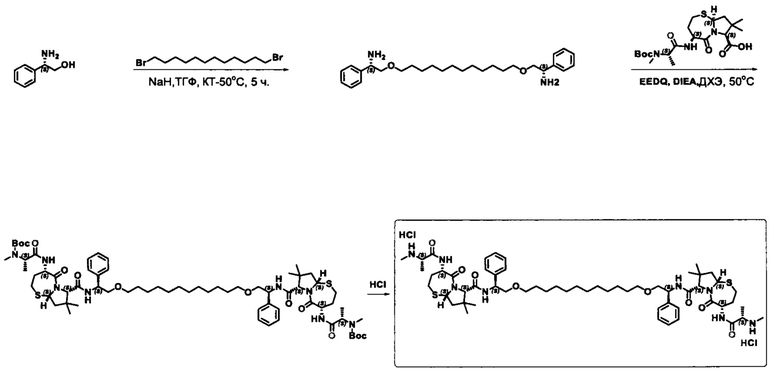
Синтез (1S,1'S)-2,2'-(додекан-1,12-диилбис(окси))бис(1-фенилэтан-1-амина)
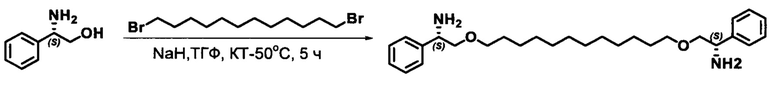
К раствору (S)-2-амино-2-фенилэтан-1-ола (0.920 г, 6.71 ммоль) в тетрагидрофуране (50 мл) партиями добавляли гидрид натрия (0.293 г, 7.31 ммоль) при 0°С. Смесь перемешивали при комнатной температуре в течение 40 мин. Затем добавляли 1,12-дибромдодекан (1 г, 3.05 ммоль) и перемешивали смесь при комнатной температуре в течение 10 мин и нагревали до 50°С в течение 3 ч. После этого реакцию гасили путем добавления воды (50 мл). Затем реакционную смесь экстрагировали этилацетатом (3*50 мл). Объединенные органические слои промывали солевым раствором, сушили с использованием безводного сульфата натрия и концентрировали под вакуумом с получением неочищенного продукта. Его очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = 15:1) и препаративной ТСХ (петролейный эфир/этилацетат = 1:3), в результате чего получали (1S,1'S)-2,2'-(додекан-1,12-диилбис(окси))бис(1-фенилэтан-1-амин) (70 мг, 0.159 ммоль, выход 5.2%) в виде коричневого масла.
ЖХМС (электроспрей, m/z): 441.0 [М+Н]+, время удерживания 1.162 мин.
Синтез ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,20S)-3,20-дифенил-5,18-диокса-2,21-диазадокозандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата)
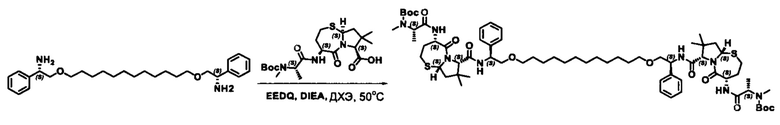
К раствору (1S,1'S)-2,2'-(додекан-1,12-диилбис(окси))бис(1-фенилэтан-1-амина) (70 мг, 0.159 ммоль) и (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (211.4 мг, 0.477 ммоль) в 1,2-дихлорэтане (5 мл) добавляли 2-этокси-1-этоксикарбонил-1,2-дигидрохинолинил (157 мг, 0.635 ммоль) и N,N-диизопропилэтиламин (123 мг, 0.953 ммоль). Полученную смесь перемешивали при 50°С в течение ночи. После этого ее разбавляли водой (10 мл), затем ее экстрагировали дихлорметаном (3*20 мл). Органические слои объединяли, промывали солевым раствором, сушили с использованием безводного сульфата натрия и концентрировали с получением неочищенного продукта. Его очищали препаративной ТСХ (петролейный эфир/этилацетат = 1:3) с получением неочищенного продукта. Затем неочищенный продукт очищали препаративной ЖХВД с получением ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((3S,20S)-3,20-дифенил-5,18-диокса-2,21-диазадокозандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (20 мг, 0.016 ммоль, выход 9.7%) в виде коричневого твердого вещества.
ЖХМС (электроспрей, m/z): 546.1 [1/2М-Вос+Н]+, время удерживания 2.192 мин.

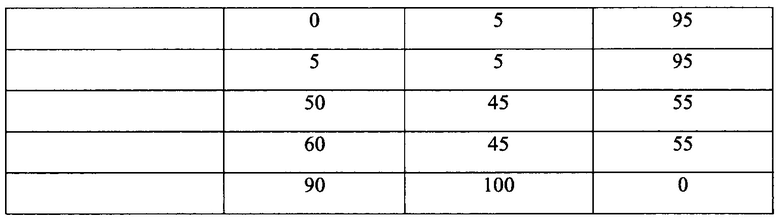
Синтез (4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S)-{додекан-1,12-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид) дигидрохлорида
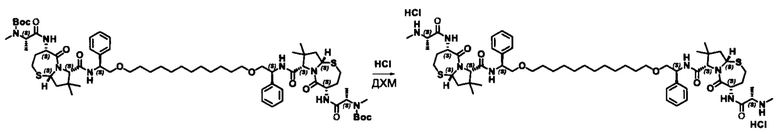
К раствору ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((3S,20S)-3,20-дифенил-5,18-диокса-2,21-диазадокозандиоил)бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (20 мг, 0.016 ммоль) в дихлорметане (5 мл) добавляли 4М хлороводородную кислоту (0.5 мл) в диоксане. Полученную смесь перемешивали в течение 4 ч при комнатной температуре. После этого концентрировали с получением (4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S)-(додекан-1,12-диилбис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)дигидрохлорида (11 мг, 0.009 ммоль, выход 60.9%) в форме белого твердого вещества.
ЖХМС (электроспрей, m/z): 546.1 [М/2-HCl+Н]+ (Расч. М/2-HCl+Н=546.3), время удерживания 1.397 мин.
1Н-ЯМР (400 МГц, MeOD-d6): δ ppm 8.36 (d, J = 8.0 Гц, 2Н), 7.39 (d, J = 8.0 Гц, 4Н), 7.34 - 7.27 (t, 4Н), 7.27 - 7.21 (t, 2Н), 5.51 - 5.43 (t, 2Н), 5.12 - 5.05 (dd, J = 4.0, 4.0 Гц, 2Н), 4.75 (d, J = 8.0 Гц, 2Н), 4.22 (s, 2Н), 3.89 (d, J = 8.0 Гц, 2Н), 3.66 - 3.59 (m, 4Н), 3.50 - 3.43 (m, 2Н), 3.40 - 3.33 (m, 2Н), 2.96 - 2.87 (m, 2Н), 2.67 (d, J = 4.0 Гц, 6Н), 2.38 - 2.29 (m, 2Н), 2.29 - 2.20 (d, J = 16.0 Гц, 2Н), 2.06 - 1.99 (d, J = 12.0 Гц, 2Н), 1.99 - 1.91 (m, 2Н), 1.57 - 1.46 (m, 10Н), 1.26 (s, 18Н), 1.15 (s, 6Н), 1.04 (s, 6Н).
Пример 62
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S)-((нафталин-2,6-диилбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)
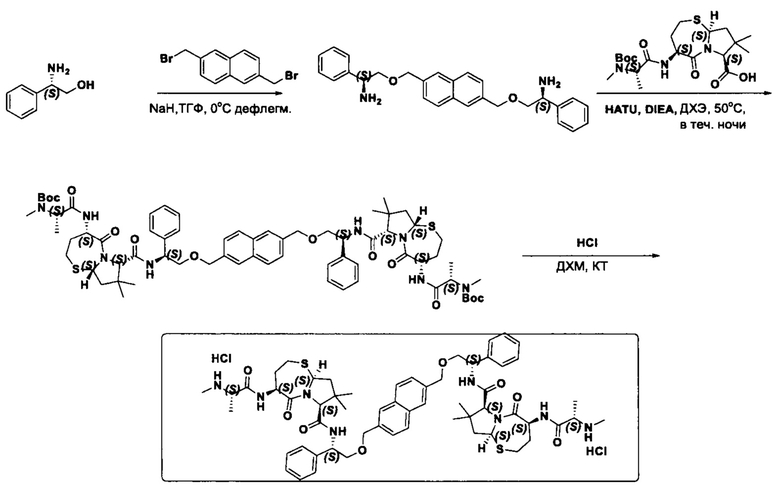
Синтез (1S,1'S)-2,2'-((нафталин-2,6-диилбис(метилен))бис(окси))бис(1-фенилэтан-1-амина)
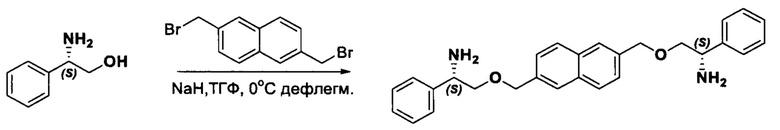
К раствору (S)-2-амино-2-фенилэтан-1-ола (480 мг, 3.5 ммоль) в тетрагидрофуране (20 мл) при 0°С добавляли порциями гидрид натрия (60%, дисперсия в жидком парафине) (166 г, 4.1 ммоль). Смесь нагревали до комнатной температуры естественным путем. Затем капали 2,6-бис(бромметил)нафталин (500 мг, 1.6 ммоль) и нагревали полученную смесь до 70°С. Ее перемешивали при 70°С в течение ночи. Реакционную смесь гасили ледяной водой (30 мл) и экстрагировали этилацетатом (3 × 20 мл). Органические слои объединяли, промывали солевым раствором (20 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта. Его очищали колоночной хроматографией на силикагеле (дихлорметан/метанол = 15:1), в результате чего получали (1S,1'S)-2,2'-((нафталин-2,6-диилбис(метилен))бис(окси))бис(1-фенилэтан-1-амин) (400 мг, 0.938 ммоль, 58.9% выход) в виде черного масла.
ЖХМС (электроспрей, m/z): 427.2 [М+Н]+, время удерживания 1.037 мин.
Синтез ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S)-((нафталин-2,6-диилбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(азанедиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата)

К раствору (4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновой кислоты (259 мг, 0.59 ммоль), этил 2-этоксихинолинил-1(2Н)-карбоксилата (162 мг, 0.66 ммоль) и N,N-диизопропилэтиламина (121 мг, 0.94 ммоль) в 1,2-дихлорэтане (5 мл) добавляли (1S,1'S)-2,2'-((нафталин-2,6-диилбис(метилен))бис(окси))бис(1-фенилэтан-1-амин) (100 мг, 0.23 ммоль). Смесь перемешивали при 50°С в течение ночи. Реакционную смесь концентрировали с получением неочищенного продукта. Его очищали колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 1:4), в результате чего получали неочищенный продукт. Затем неочищенный продукт очищали препаративной ЖХВД с получением продукта (100 мг, 0.078 ммоль, 33.4% выход) в форме белого твердого вещества.
ЖХМС (электроспрей, m/z): 539.3 [М/2-Вос+Н]+, время удерживания 1.778 мин.
Синтез (4S,4'S,7S,7'S,9aS,9aS)-N,N'-((1S,1'S)-((нафталин-2,6-диилбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамида)
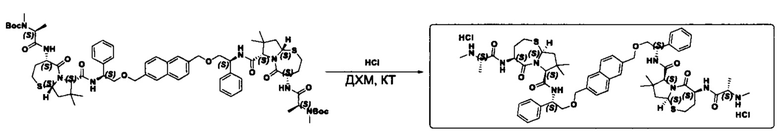
К раствору ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9а'S)-((((1S,1'S)-((нафталин-2,6-диилбис(метилен))бис(окси))бис(1-фенилэтан-2,1-диил))бис(азанедиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (92 мг, 0.072 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлороводородную кислоту в диоксане (0.5 мл). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали с получением целевого продукта (20 мг, 0.017 ммоль, 24.2% выход) в форме белого твердого вещества. Затем это соединение лиофилизировали для удаления остатка растворителя.
ЖХМС (электроспрей, m/z): 1077.7 [М-2HCl+Н]+, время удерживания 1.426 мин.
1Н-ЯМР (400 МГц, ДМСО-d6) δ ppm 9.30 (s, 2Н), 8.91 - 8.77 (m, 4Н), 8.45 (d, J = 8.2 Гц, 2Н), 7.82 - 7.72 (m, 4Н), 7.45 - 7.32 (m, 10Н), 7.28 (t, J = 7.2 Гц, 2Н), 5.46 (t, J = 7.9 Гц, 2Н), 5.13 (dd, J = 13.3, 6.6 Гц, 2Н), 4.74 - 4.59 (m, 6Н), 4.20 (s, 2Н), 3.90 - 3.80 (m, 2Н), 3.72 - 3.61 (m, 4Н), 3.13 (t, J = 12.4 Гц, 2Н), 2.83 - 2.73 (m, 2Н), 2.49 - 3.42 (m, 6Н), 2.18 (dd, J = 12.5, 6.8 Гц, 2Н), 2.10 - 2.02 (m, 2Н), 1.86 (dd, J = 12.7, 8.8 Гц, 2Н), 1.73 (dd, J = 22.5, 11.2 Гц, 2Н), 1.40 - 1.33 (m, 6Н), 1.05 (s, 6Н), 0.98 (s, 6Н).
Пример 63
(4S,4'S,7S,7'S,9aS,9а'S)-N,N'-((1S,1'S,2R,2'R)-{октан-1,8-диилбис(окси))бис(2,3-дигидро-1H-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)дигидрохлорид
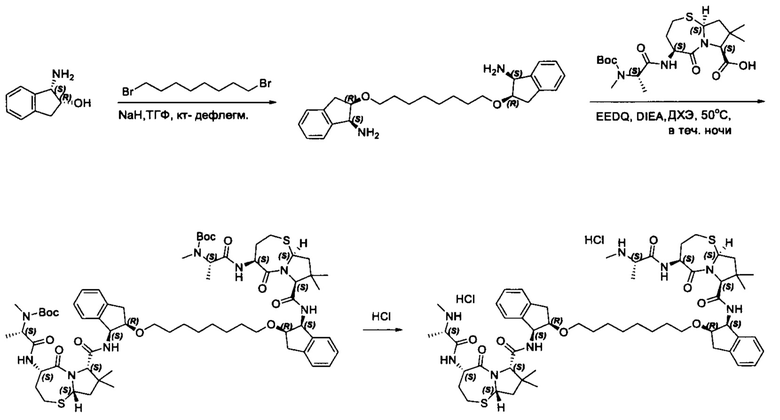
Синтез (1S,1'S,2R,2'R)-2,2'-(октан-1,8-диилбис(окси))бис(2,3-дигидро-1Н-инден-1-амина)
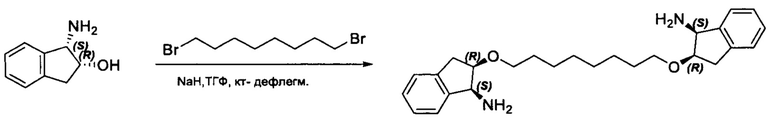
К раствору (1S,2R)-1-амино-2,3-дигидро-1Н-инден-2-ола (1.21 г, 8.09 ммоль) в тетрагидрофуране (50 мл) при 0°С добавляли порциями гидрид натрия (60%, дисперсия в жидком парафине) (380 мг, 9.57 ммоль). Смесь нагревали до комнатной температуры естественным путем. Затем добавляли 1,8-дибромоктан (1.0 г, 3.68 ммоль). Полученную смесь перемешивали при 70°С в течение ночи. Реакционную смесь гасили ледяной водой (30 мл) и экстрагировали этилацетатом (3 × 30 мл). Органический слой объединяли, промывали солевым раствором (50 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта. Его очищали с использованием устройства Combi-Flash, используя для элюирования дихлорметан/метанол = 10:1 а затем тонкослойной хроматографией, используя для проявки этилдихлорметан/метанол = 10/1, в результате чего получали целевой продукт (170 мг, 0.42 ммоль, 11.4% выход) в виде темного масла.
Синтез ди-трет-бутил ((2S,2'S)-((((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(октан-1,8-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азанедиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата)
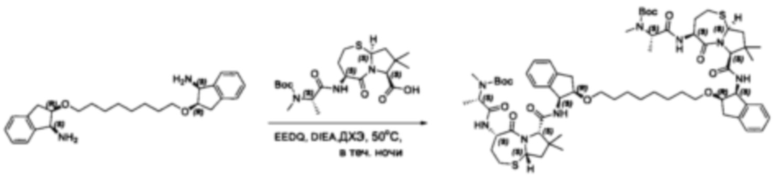
(1S,1S,2R,2'R)-2,2'-(октан-1,8-диилбис(окси))бис(2,3-дигидро-1Н-инден-1-амин) (150 мг, 0.367 ммоль) и ((4S,7S,9aS)-4-((S)-2-((трет-бутоксикарбонил)(метил)амино)пропанамидо)-8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоновую кислоту (358 мг, 0.808 ммоль) в 1,2-дихлорэтане (10 мл) добавляли к N-этоксикарбонил-2-этокси-1,2-дигидрохинолинилу (272 мг, 1.101 ммоль) и N,N-диизопропилэтиламину (189 мг, 1.468 ммоль). Полученную смесь перемешивали при 50°С в течение ночи. Реакционную смесь концентрировали с получением неочищенного продукта. Его очищали тонкослойной хроматографией, с использованием для проявки этилацетат/петролейный эфир = 3/1, в результате чего получали неочищенный продукт. Затем неочищенный продукт очищали препаративной ЖХВД с получением продукта (150 мг, 0.119 ммоль, 32.4% выход) в виде бежевого твердого вещества.
ЖХМС (электроспрей, m/z): 529.9 [1/2М-Вос+Н]+, время удерживания 2.027 мин.
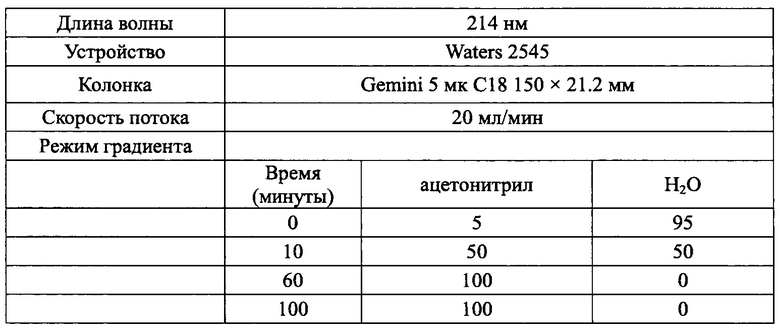
Синтез (4S,4'S,7S,7'S,9aS,9a'S)-N,N'-((1S,1'S,2R,2'R)-(октан-1,8-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(8,8-диметил-4-((S)-2-(метиламино)пропанамидо)-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7-карбоксамид)дигидрохлорида
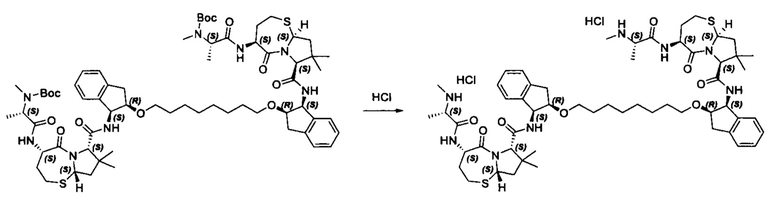
К раствору ди-трет-бутил ((2S,2'S)-(((4S,4'S,7S,7'S,9aS,9a'S)-((((1S,1'S,2R,2'R)-(октан-1,8-диилбис(окси))бис(2,3-дигидро-1Н-инден-2,1-диил))бис(азанедиил))бис(карбонил))бис(8,8-диметил-5-оксооктагидропирроло[2,1-b][1,3]тиазепин-7,4-диил))бис(азанедиил))бис(1-оксопропан-1,2-диил))бис(метилкарбамата) (150 мг, 0.119 ммоль) в дихлорметане (5 мл) добавляли 4 н. хлороводородную кислоту в диоксане (0.5 мл). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали с получением целевого продукта (85 мг, 0.075 ммоль, 63.1% выход) в форме белого твердого вещества.
ЖХМС (электроспрей, m/z): 1058.8 [М-2HCl+Н]+ (Расч. М-2HCl+Н=1059.6), время удерживания 1.348 мин.
1Н-ЯМР (400 МГц, ДМСО) δ ppm 9.50 (s, 2Н), 8.93 (s, 2Н), 8.87 (d, J = 6.6 Гц, 2Н), 7.91 (d, J = 8.7 Гц, 2Н), 7.27 - 7.19 (m, 8Н), 5.51 (t, J = 7.9 Гц, 2Н), 5.33 (dd, J = 8.6, 5.4 Гц, 2Н), 4.78 - 4.70 (m, 2Н), 4.24 (s, 2Н), 4.11 (dd, J = 9.3, 4.1 Гц, 2Н), 3.89 (dd, J = 10.5, 6.1 Гц, 2Н), 3.44 - 3.38 (m, 4Н), 3.20 (t, J = 11.8 Гц, 2Н), 2.98 (d, J = 3.7 Гц, 4Н), 2.95 - 2.85 (m, 2Н), 2.47 (s, 6Н), 2.27 - 2.11 (m, 4Н), 1.89 - 1.76 (m, 4Н), 1.47 - 1.36 (m, 10Н), 1.24 - 1.17 (m, 8Н), 1.08 (s, 6Н), 1.06 (s, 6Н).
• Анализ обращения латентности в клетках Jurkat.
• Анализ обращения латентности на клетках Jurkat.
Клоны Jurkat с ВИЧ-люциферазой поддерживали на среде RPMI 1640 (Gibco от Life Technologies), содержащей 10% (по объему) фетальной бычьей сыворотки (SAFC/Sigma-Aldrich) и 25 ед/мл пенициллина, 25 ед/мл стрептомицина (Gibco от Life Technologies), и разделяли 1:4 каждые 3-4 дня с сохранением плотности клеток ~0.3-1 млн клеток/мл. Клоны Jurkat поддерживали на среде с добавлением 500 нМ EFV. Три клона клеток Jurkat (С16, I15, и N6), каждый из который нес один или два интегрированных провируса ВИЧ, экспрессирующих репортерный ген люциферазы, добавляли в равных количествах в количестве, составляющем в общей сложности 5000 клеток на лунку, в 384-луночные планшеты, содержащие титрованные разведения соединения. Тестирование зависимости «доза-ответ» проводили с растворенными в диметилсульфоксиде (ДМСО; Fisher Scientific, Мерелбеке, Бельгия) соединениями, распределенными по серийным 3-кратным 14-точечным титрованным разведениям, полученным в двух повторностях, используя цифровой капельный дозатор D300e Digital Droplet Dispenser (Hewlett-Packard), с получением конечных концентраций в анализе от 10 мкМ до 2.1 пМ в 50 мкл среды с конечной концентрацией 0.5% ДМСО (по объему). Клетки и соединение инкубировали при 37°С в течение 48 часов, если не указано иное, с последующим добавлением 20 мкл люциферазы Steady-Glo® (Promega). Люминесценцию в результате индукции экспрессируемой вирусом люциферазы измеряли с применением многоканального планшет-ридера EnVision 2102 (Perkin Elmer). Зависимость «доза-ответ» анализировали с помощью GraphPad PRISM 6 с применением четырехпараметрической логистической регрессионной модели для вычисления концентрации соединения, обеспечивающей полумаксимальный ответ (ЕС50) и максимальный процент активации по сравнению с контролем-основой.
Результаты вышеописанного анализа приведены в таблице 3.
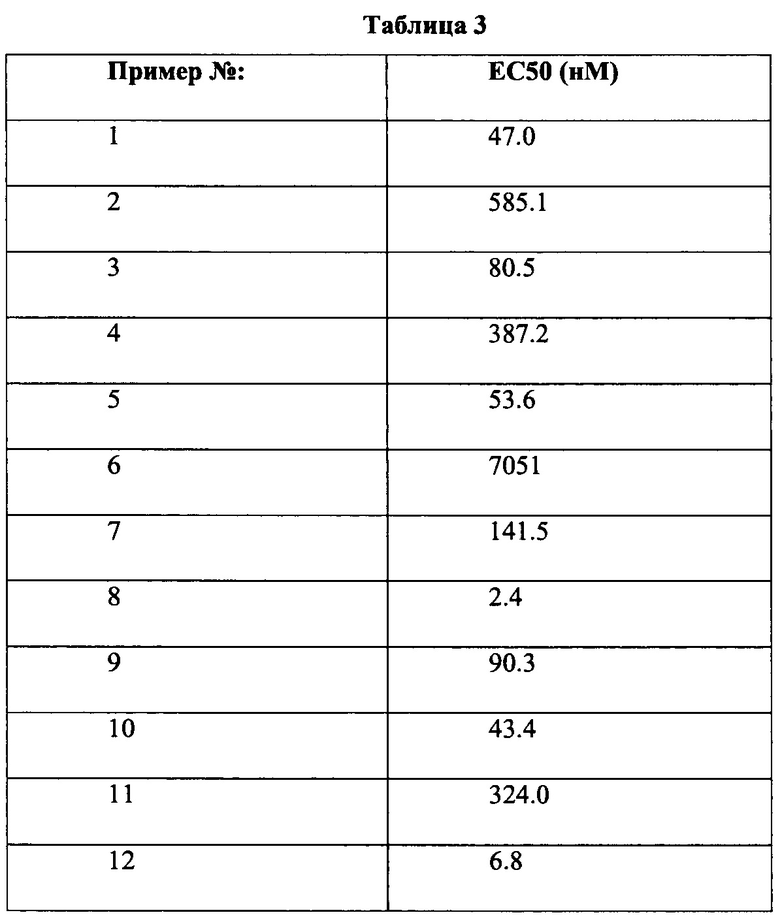
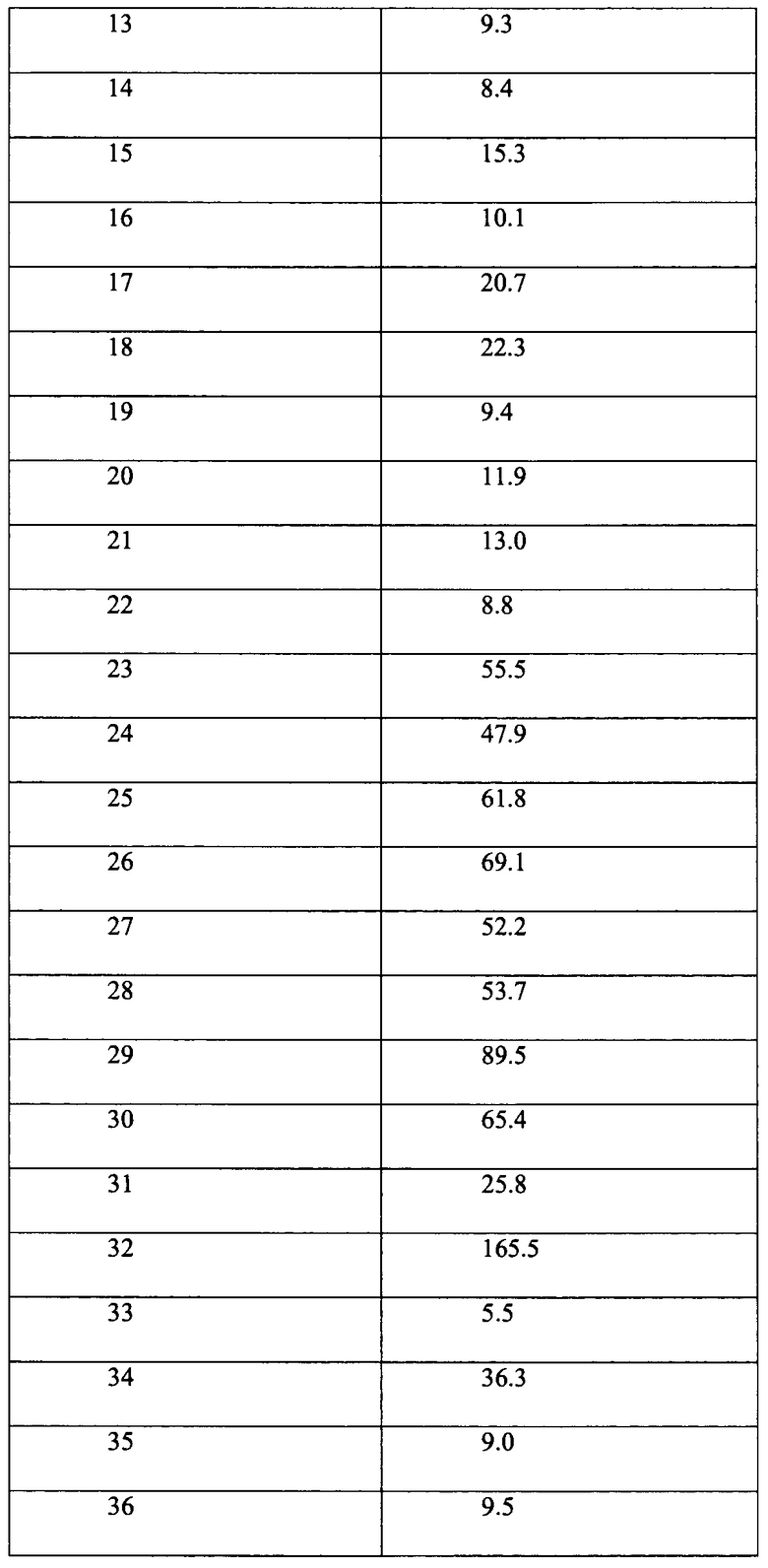
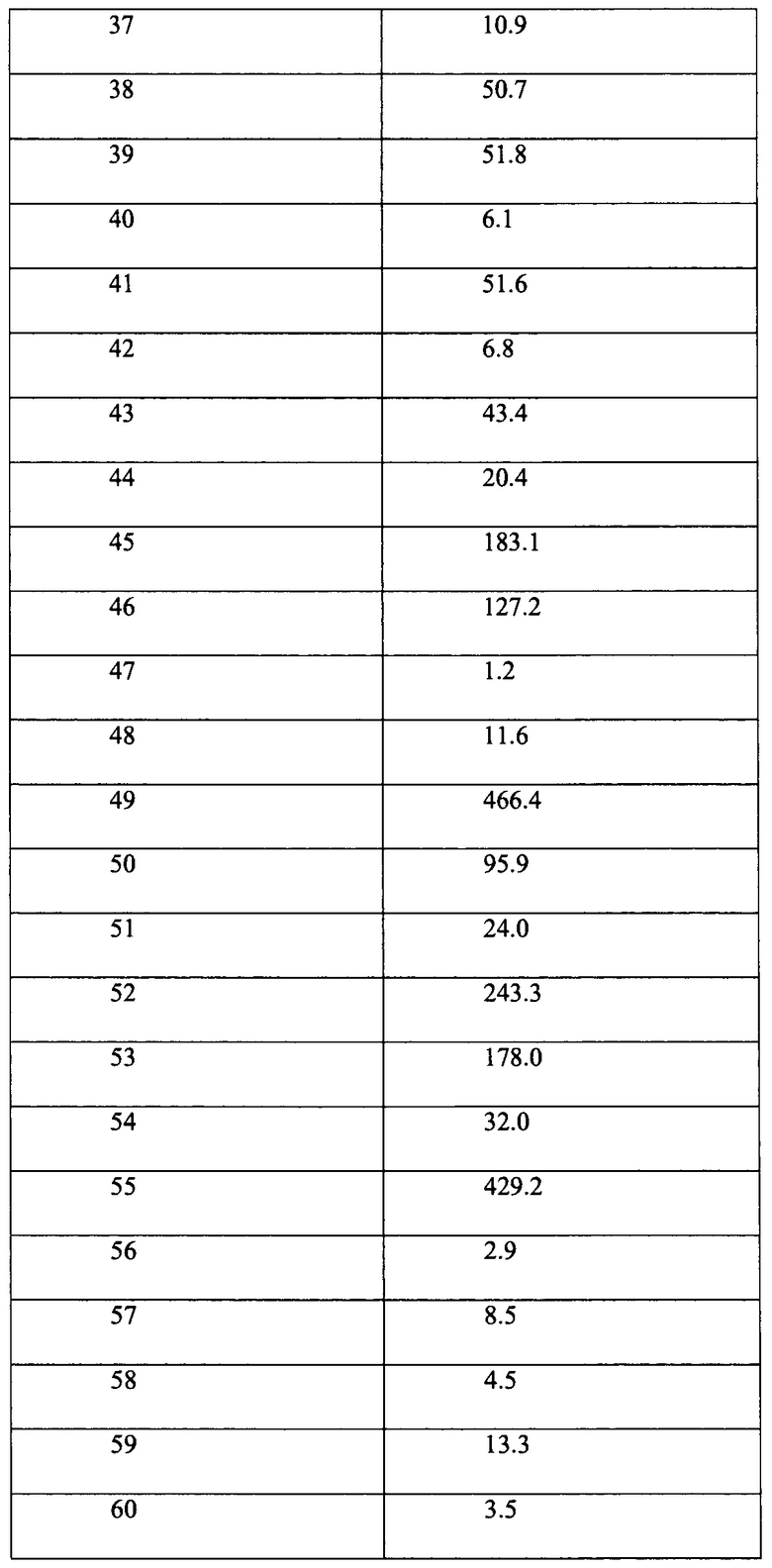
Фармакокинетические данные
Фармакокинетические (ФК) данные нескольких соединений-миметиков SMAC формулы I для грызунов сравнивали с данными ФК SMACm AZD5582 (фиг. 1). Как видно на фиг. 1, кривые зависимости концентрации лекарственного средства в плазме от времени для соединений формулы I демонстрируют зависимость ФК, сопоставимую с наблюдаемой для AZD5582.
Индукция гена NFkB2
5×105 CD4 Т-клеток от здоровых доноров обрабатывали в течение 24 ч 6-кратными 7-точечными серийными разведениями соединений, начиная с 1.0 мкМ. Тотальную РНК выделяли с применением набора RNEasy Mini Kit (Qiagen), следуя инструкциям изготовителя. Получали следующие наборы праймерных зондов TaqMan от Applied Biosystems: Hs00174517_m1 (NFKB2) и №02800695 (HPRT1). Использовали ПЦР в реальном времени на основе TaqMan (мастер-микс Fast Virus 1-Step Master Mix, Applied Biosystems) для амплификации генов хозяина, представляющих интерес. Генную экспрессию нормировали по HPRT1 и использовали сравнительный метод пороговых циклов (СТ) (ΔΔСТ) для измерения относительной кратности изменения генной экспрессии по сравнению с необработанными клетками. Данные анализировали в системе для ПЦР в реальном времени QuantStudio3. Результаты приведены в таблице 4.
Линия р100/р52 WB
Культивирование и обработка клеток
Получали культуры клеток для оценки нескольких соединений SMACm. Замороженные клетки МКПК от здоровых доноров размораживали и высевали в планшеты с глубокими лунками по 2 мл с плотностью 2×106 клеток/мл. Получали серийные разведения каждого соединения путем 7-кратного разведения 1:6, начиная со стартовой конечной дозы 1000 нМ. Затем клетки объединяли с соединением и держали при 37° в течение 24 ч. На следующий день клетки осаждали и помещали на хранение при -80° до лизиса.
Сбор лизированных целых клеток
При подготовке лизиса клеток замороженные осажденные клетки и аликвоту клеточного буфера для лизиса NP40 (Invitrogen, кат. № FNN0021), а также аликвоту 10х коктейля ингибиторов протеаз (Sigma, кат. № Р-2714) размораживают на льду. После размораживания готовят буфер для полного лизиса, добавляя 10% от итогового объема коктейля ингибиторов протеаз к буферу NP40, а затем достаточный объем 0.1 М ПМСФ (Sigma, кат. №93482) и 1 М ДТТ (Sigma, кат. №43816) с получением конечной концентрации 1 мМ каждого из них в буфере для полного лизиса. Затем проводят лизис клеток путем ресуспендирования каждого клеточного осадка в 30 мкл на 10^6 клеток с последующей инкубацией на льду в течение 30 минут с интенсивным перемешиванием на вортексе каждые 10 минут. Затем лизированные клетки центрифугируют при 13000 RPM в течение 10 минут при 4°С в охлаждаемой микроцентрифуге. Затем супернатанты (лизаты) переносили в новые микроцентрифужные пробирки и либо помещали на хранение при -80°, либо немедленно использовали для анализа концентрации белка и вестерн-блоттинга.
Процедура анализа в микропланшетах по методу Брэдфорда
Концентрацию белка в каждом лизате определяют с применением детергент-совместимого анализа по методу Брэдфорда (Pierce, кат. №23246), следуя инструкциям изготовителя микропланшетов.
Разделение и иммунодетекция белков
Капиллярный вестерн-блоттинг проводили с применением системы ProteinSimple Jess. В общей сложности 1 мкг клеточного лизата получали в соответствии с предложенным протоколом от ProteinSimple. Первичное антитело к р100/р52 (Cell, Signaling #3017) разводят 1:10 для применения в системе Jess. Модуль для разделения 12-230 кДа Jess Separation Module (SM-W004) применяют в сочетании с вторичным конъюгированным с красителем для ближнего ИК-спектра антителом против кролика (043-819). Модуль для разделения включает предварительно заполненные планшеты, куда загружают образцы, первичные и вторичные антитела, а также реагент для нормирования белка и разбавители, для проведения разделения и детекции. После проведения анализа данные анализируют, используя программное обеспечение Compass, предоставляемое ProteinSimple.
Анализ методом иммуноблоттинга
В качестве альтернативы капиллярного электрофореза проводили традиционные анализы методом иммуноблоттинга, 10 мкг клеточного лизата загружали в лунку на 4-20% Трис-глициновые ДСН-ПААГ-гели. Белок с ДСН-ПААГ-гелей переносили на ПВДФ-комплекты для переноса Turbo Midi PVDF Transfer Packs (BioRad) с применением протокола для «смешанных молекулярных масс» для одного геля среднего формата (постоянный ток 2.5 А до 25 В, в течение 7 минут) системы Trans-Blot Turbo Transfer System (Bio-Rad) с готовым модулем Trans-Blot, следуя инструкциям изготовителя. После переноса ПВДФ-мембраны блокировали в 5% бычьем сывороточном альбумине (БСА) в 1x TBS (BioRad) с 0.1% Tween-20 в течение 1 часа при комнатной температуре при аккуратном встряхивании. Первичные антитела добавляли и инкубировали в течение ночи при 4°С (антитело против р100/р52, Cell Signaling Technology #3017, 1:1000; HRP конъюгированное антитело против актина, Abcam #49900, 1:30000). После первичного окрашивания мембрану три раза промывали 1x Tris-забуференным солевым раствором (TBS)+0.1% TWEEN® 20, каждый раз в течение 10 минут. После промывания мембрану инкубировали в 5% БСА в 1x TBS+0.1% TWEEN® 20 с подходящим вторичным антителом в течение 2 часов при комнатной температуре. После вторичного окрашивания мембрану двукратно промывали в течение 10 минут 1x TBS+0.1% TWEEN® 20 с последующим 10-минутным промыванием 1x TBS. Затем мембрану осушали фильтровальной бумагой и захватывали изображение непроявленной мембраны в системе визуализации ChemiDoc MP с применением программного обеспечения Image Lab (BioRad). Использовали достаточное количество реагента ECL (GE Healthcare) для покрытия мембраны и получали ряд изображений, начиная с 0.001 секунды и удваивая/утраивая время экспозиции до тех пор, пока люминесценция от проявленной мембраны не насыщала изображение. Затем проявленную мембрану промывали три раза 1x TBS в течение 5 минут для удаления остаточного реагента ECL и хранили при 4°С в достаточном количестве 1x TBS, чтобы мембрана была полностью погружена. Затем проводили денситометрию изображений проявленной мембраны с применением программного обеспечения Image Lab. Некоторые мембраны очищали в течение 1 минуты стриппинг-буфером для вестерн-блоттинга One Minute Plus (GM Biosciences), после чего промывали три раза в течение 10 минут 1x TBS. Затем очищенные мембраны блокировали в 5% БСА в 1x TBS+0.1% TWEEN® 20 в течение часа и повторно зондировали в течение ночи новым первичным антителом. Результаты р100/р52 WB-анализа линии приведены в таблице 4.
Клеточно-ассоциированная РНК ВИЧ(каРНК)
Во время продуктивной фазы жизненного цикла вируса ВИЧ продуцирует значительное число транскриптов, образующихся путем дифференцированного сплайсинга, в совокупности называемых клеточно-ассоциированной РНК ВИЧ-(каРНК ВИЧ), в некоторых из инфицированных клеток. У инфицированных ВИЧ индивидуумов, получающих супрессивную антиретровирусную терапию (ART), изменение уровней каРНК ВИЧ представляет собой признанный косвенный показатель эффективности обращения латентности ВИЧ.
Для измерения каРНК ВИЧ мононуклеарные клетки периферической крови (МКПК) выделяют из лейкоцитов, полученных путем проточного лейкофереза. Тотальные CD4+ Т-клетки выделяли из МКПК путем отрицательного отбора с применением набора для обогащения по CD4+ Т-клеткам человека EasySep Human CD4+ Т cell Enrichment kit (StemCell, Ванкувер, Канада), следуя рекомендациям изготовителя. Покоящиеся CD4+ Т-клетки выделяют путем отрицательного отбора с использованием иммуномагнитной колонки, как описано ранее (25), и либо немедленно криоконсервируют, либо поддерживают в течение двух дней на среде IMDM (Gibco) с 10% ФБС, 2 мкг/мл ИЛ-2 (Peprotech) и антиретровирусными средствами для предотвращения размножения вируса.
Для обработки SMACm 3-5 репликатов 2-5×106 CD4+ Т-клеток обрабатывали в течение 48 часов при 37°С в 1 мл среды RPMI 1640 с 10% ФБС, 10 мкг/мл энфувиртида (Sigma) и 200 нМ рилпивирина. 10 нМ форбол-12-миристат-13-ацетата (РМА; Sigma) с 1 мкМ иономицина (Sigma) использовали в качестве положительного контроля для активации LRA (изменяющих латентность агентов) и 0.2% основу - ДМСО применяли в качестве отрицательного контроля. После обработки клетки лизировали, и РНК и ДНК совместно экстрагировали с применением набора AllPrep 96 RNA/DNA Kit (Qiagen, Валенсия, Калифорния), следуя инструкциям изготовителя, с доведением объема буфера для лизиса до 0.6 мл, добавляя обработку ДНКазой I на колонке (Qiagen) и элюируя РНК в 50 мкл воды.
ОТ-кПЦР выполняли в трех повторностях для каждой из трех лунок-репликатов с применением мастер-микса для одностадийной ОТ-кПЦР TaqMan Fast Virus 1-step RT-qPCR Master Mix (Applied Biosciences) с 5 мкл выделенной РНК и 900 нМ праймеров для капсида ВИЧ HIV-gag (5'-ATCAAGCAGCTATGCAAATGTT-3' (SEQ ID NO: 1)) и обратного gag (5'-CTGAAGGGTACTAGTAGTTCCTGCTATGTC-3' (SEQ ID NO: 2)), и 250 нМ зонда FAM/ZEN/IABFQ HIV-gag (5'-ACCATCAATGAGGAAGCTGCAGAATGGGA-3' (SEQ ID NO: 3)). Образцы амплифицировали и данные собирали с помощью системы для ПЦР в реальном времени QuantStudio™ 3 (Applied Biosystems) со следующими условиями проведения циклов: один цикл при 50°С в течение 5 минут (обратная транскрипция), один цикл при 95°С в течение 20 секунд (инактивация обратной транскриптазы); 50 циклов при 95°С в течение 3 секунд; и 60°С в течение 20 секунд (денатурация и отжиг/достройка).
Абсолютное число копий РНК ВИЧ HIV gag на реакцию определяли с применением стандарта для кПЦР HIV gag gBlock qPCR, соответствующего последовательности ДНК продукта кПЦР (5'-ATCAAGCAGCCATGCAAATGTTAAAAGAGACCATCAATGAGGAAGCTGCAGAATGGGATAGATTGCATCCAGTGCATGCAGGGCCTATTGCACCAGGCCAGATGAGAGAACCAAGGGGAAGTGACATAGCAGGAACTACTAGTACCCTTCAG-3' (SEQ ID NO: 4); Integrated DNA Technologies), и число копий нормировали по числу клеток, подсчитанному с применением светлопольной микроскопии. Эффективность ОТ-кПЦР должна была составлять от 90% до 110%. Аналитические значения с положительным сигналом, которые были меньше нижнего предела детекции (LLOD), равного 7 копиям HIV gag на реакцию, приравнивали к LLOD. Анализ выполняли с применением программного обеспечения QuantStudio™ Design and Analysis Software (Applied Biosystems) и пакета программного обеспечения R (подробно описаны ниже).
В таблице 4 приведены сравнительные данные анализов Jurkat ЕС50, р100/р52 WB, индукции гена NFkB2 и каРНК для нескольких миметиков SMAC согласно описанию в настоящем документе и миметиков-конкурентов.
Соединения согласно настоящему изобретению (а именно, примеры 33, 13, 42, 40 и 20) представляют собой мощные димерные SMACm, которые способны активировать путь ncNF-kB и индуцировать экспрессию ВИЧ. Указанные молекулы эффективно истощают cIAP1 и cIAP2, приводят к расщеплению р100 с высвобождением р52, активируют путь ncNF-kB и индуцируют экспрессию ВИЧ. Кроме того, они обладают свойствами, которые задействуют путь ncNF-kB in vivo. В частности, считается, что примеры 13 и 20 обладают наилучшей комбинацией неожиданных и удивительных свойств.
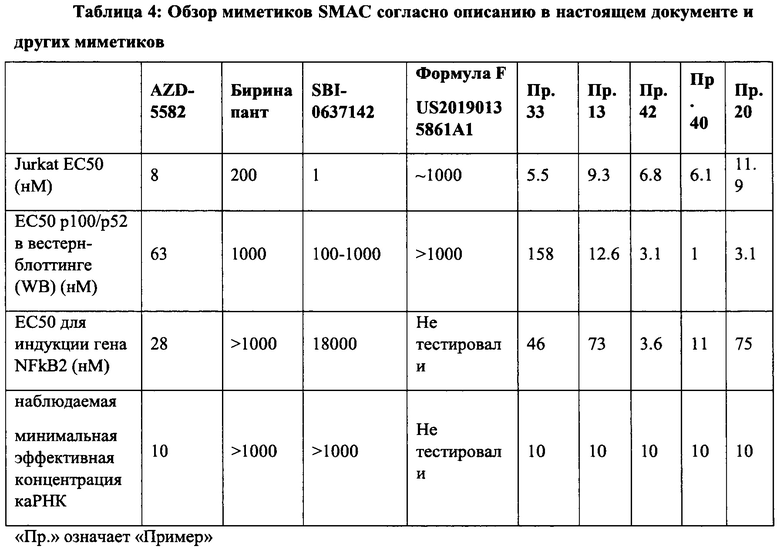
где:
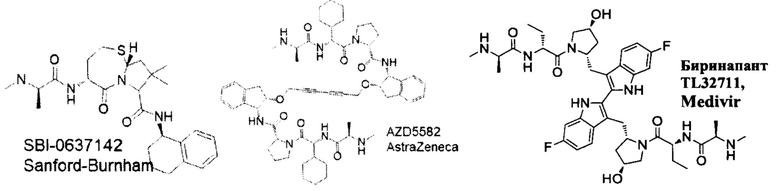
и описанный димер SBI формулы F: 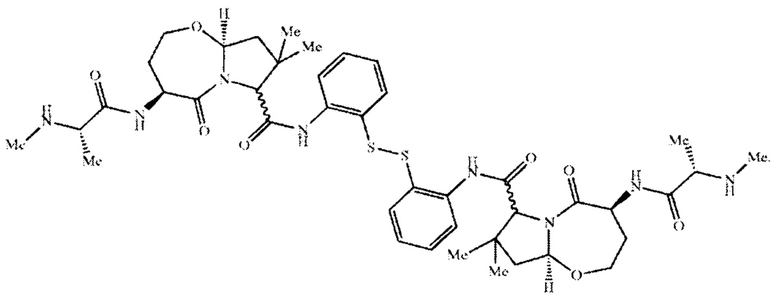
Цитотоксичность
Жизнеспособность клеток Jurkat определяли с применением люминесцентного анализа жизнеспособности клеток CellTiter-Glo® (Promega), однородного способа определения уровней АТФ в культуральной лунке, которые соответствуют наличию метаболически активных клеток в культуре. Клетки культивировали согласно имеющимся описаниям для анализов на клетках Jurkat. Для оценки SMACm-опосредованной токсичности в контексте ФНО-а, в культуру добавляли 10 нг/мл ФНО-а (R&D Systems). Часть клеток удаляли и добавляли в каждую лунку, содержащую клетки, 30 мкл реагента Promega CellTiter-Glo® и измеряли люминесценцию с применением планшет-ридера Perkin Elmer EnVision. Зависимость «доза-ответ» анализировали с помощью GraphPad PRISM 6, с применением четырехпараметрической логистической регрессионной модели для вычисления концентрации соединения, которая уменьшает жизнеспособность клеток на 50% по сравнению с необработанными контролями (СС50).
Были предприняты усилия для обеспечения точности используемых численных значений (например, количеств, температуры и т.п.), однако следует учитывать возможность экспериментальных погрешностей и отклонений..
Специалисту в данной области техники будут понятны многие способы и материалы, аналогичные или эквивалентные описанным в настоящем документе, подходящие для применения при практическом осуществлении изобретения, описанного в настоящем документе. Настоящее раскрытие ни в коем случае не ограничено только описанными способами и материалами.
Если не указано иное, используемые в настоящем документе технические и научные термины имеют значения, соответствующие общеизвестным значениям для специалистов в области техники, к которой принадлежит указанный предмет изобретения, и согласуются с источниками: Singleton et al (1994) Dictionary of Microbiology and Molecular Biology, 2nd Ed., J. Wiley & Sons, New York, NY; и Janeway, C, Travers, P., Walport, M., Shlomchik (2001) Immunobiology, 5th Ed., Garland Publishing, New York.
В тексте настоящего описания и в формуле изобретения термины «содержать», «содержит» и «содержащий» используются в неисключительном смысле, кроме случаев, когда контекст предполагает иное. Следует понимать, что варианты реализации, описанные в настоящем документе, включают варианты реализации, включающие термины «состоящие из» и/или «состоящие по существу из».
В настоящем документе термин «приблизительно» в отношении значения охватывает, согласно некоторым вариантам реализации, отклонения, составляющие ±50%, согласно некоторым вариантам реализации - ±20%, согласно некоторым вариантам реализации - ±10%, согласно некоторым вариантам реализации - ±5%, согласно некоторым вариантам реализации - ±1%, согласно некоторым вариантам реализации - ±0.5%; и согласно некоторым вариантам реализации - ±0.1% от указанного количества, если такие отклонения подходят для осуществления описанных способов или применения описанных композиций.
Если приведен диапазон значений, подразумевается, что включено каждое промежуточное значение, вплоть до десятых долей единицы от нижнего предела значений, если контекстом явно не продиктовано иное, между верхним и нижним пределом диапазона, а также любое другое указанное или промежуточное значение в указанном диапазоне. Верхний и нижний пределы этих малых диапазонов, которые могут независимо быть включены в меньшие диапазоны, также включены, с учетом того, что какой-либо предел в указанном диапазоне может быть специально исключен. Если указанный диапазон включает один или оба предела, также включены диапазоны, исключающие любой один или оба этих включенных предела.
Специалист в области техники, к которой относится предмет настоящего изобретения, сможет представить многие модификации и другие варианты осуществления описанного в настоящем документе изобретения, учитывая преимущества идей, изложенных в описаниях выше и на связанных с ними чертежах. Таким образом, следует понимать, что предмет изобретения не ограничен конкретными раскрытыми вариантами осуществления, и что модификации и другие варианты осуществления включены в объем прилагаемой формулы изобретения. Хотя в настоящем описании используются конкретные термины, они имеют исключительно общий и описательный смысл, и не предназначены для ограничения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ АЗОЛЫ, ПРОТИВОВИРУСНЫЙ АКТИВНЫЙ КОМПОНЕНТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2010 |
|
RU2452735C1 |
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
| БИЦИКЛИЧЕСКИЕ КЕТОНЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2797922C2 |
| Аналоги сплицеостатина | 2013 |
|
RU2618523C2 |
| КОМБИНИРОВАННОЕ ЛЕЧЕНИЕ АГОНИСТОМ ТОЛЛ-ПОДОБНОГО РЕЦЕПТОРА (TLR7) И ИНГИБИТОРОМ СБОРКИ КАПСИДА ВИРУСА ГЕПАТИТА В | 2016 |
|
RU2718917C2 |
| РЕАКЦИИ МАКРОЦИКЛИЗАЦИИ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ДРУГИЕ ФРАГМЕНТЫ, ПРИГОДНЫЕ В ПОЛУЧЕНИИ АНАЛОГОВ ХАЛИХОНДРИНА B | 2014 |
|
RU2710545C2 |
| ФЕНОКСИПИРИДИНИЛАМИДНЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ PDE4-ОПОСРЕДОВАННЫХ БОЛЕЗНЕННЫХ СОСТОЯНИЙ | 2009 |
|
RU2509077C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА ИЛИ ТЕТРАГИДРОПИРАНА, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ИЛИ ГИДРАТЫ, ИЛИ СОЛЬВАТЫ ЭТИХ СОЕДИНЕНИЙ, ИЛИ ИХ СОЛЕЙ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ И ФУНГИЦИДНОЕ СРЕДСТВО | 1992 |
|
RU2084439C1 |
| РЕГУЛЯТОРЫ-ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2797408C2 |
| ПРОИЗВОДНЫЕ АДАМАНТИЛА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ JNK-ОПОСРЕДОВАННОГО РАССТРОЙСТВА | 2012 |
|
RU2626890C2 |
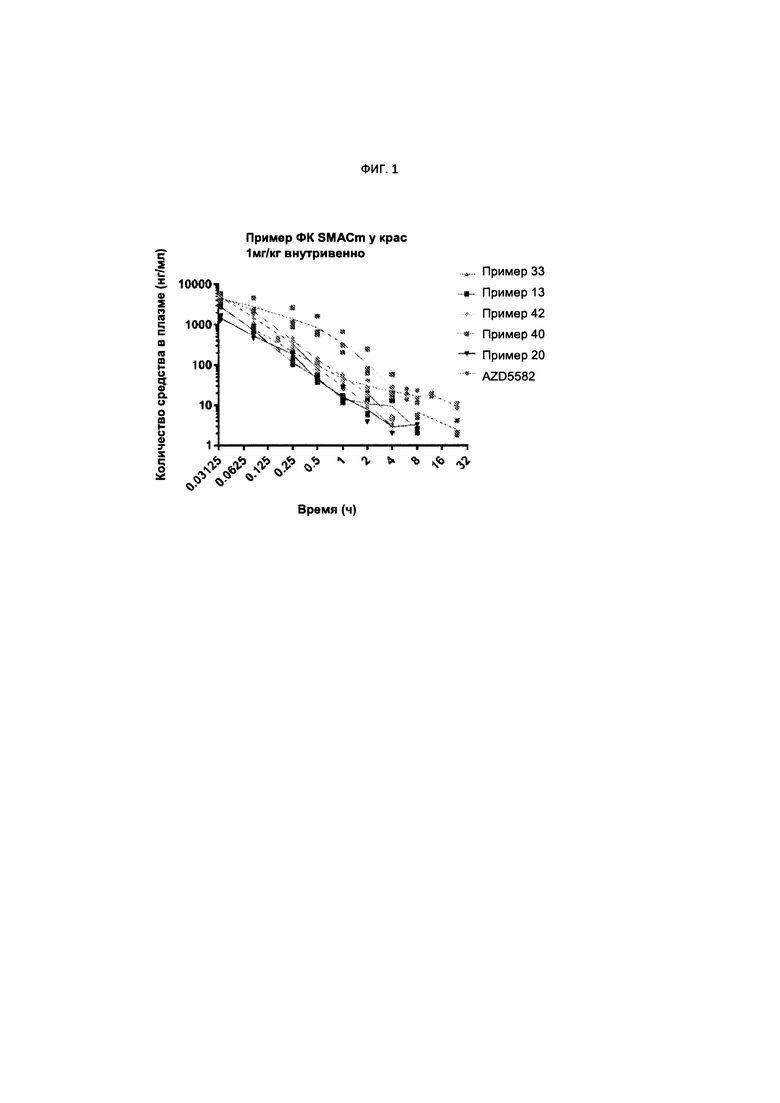
Группа изобретений относится к области органической химии и фармацевтики и направлена на обеспечение соединения, обладающего активностью в отношении ВИЧ. Раскрывается соединение, имеющее структуру, представленную ниже, или его фармацевтически приемлемая соль. Кроме того, раскрываются фармацевтические композиции на основе указанного соединения или его фармацевтически приемлемой соли. Группа изобретений обеспечивает эффективное подавление ВИЧ-инфекции. 4 н. и 4 з.п. ф-лы, 1 ил., 4 табл., 63 пр.
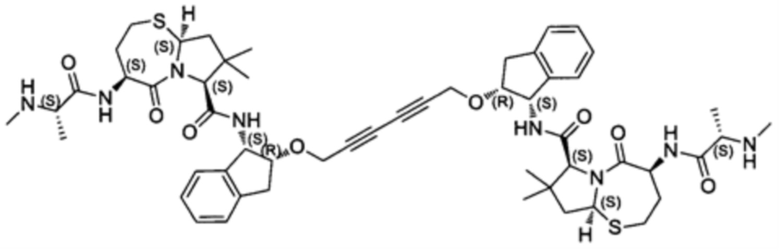
1. Соединение, имеющее структуру:
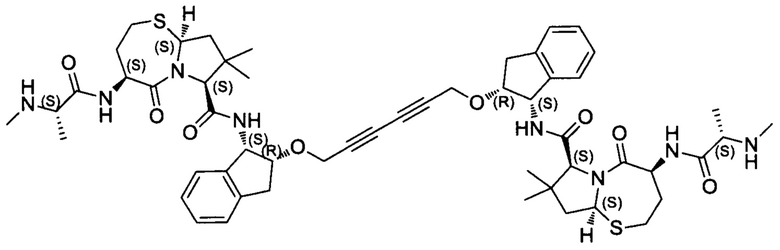
или его фармацевтически приемлемая соль.
2. Соль по п. 1, отличающаяся тем, что указанной солью является гидрохлорид.
3. Соль по п. 2, отличающаяся тем, что указанным гидрохлоридом является дигидрохлорид.
4. Фармацевтическая композиция для лечения ВИЧ-инфекции, содержащая от 0,5% до 50% по массе соединения по п. 1 или его фармацевтически приемлемой соли и фармацевтически приемлемое вспомогательное вещество.
5. Композиция по п. 4, отличающаяся тем, что указанная композиция приспособлена для парентерального введения.
6. Соединение, имеющее структуру:
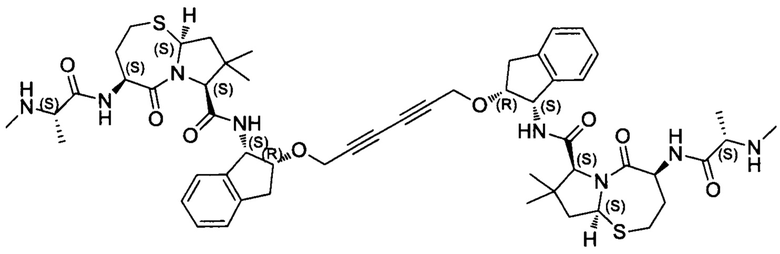
7. Фармацевтическая композиция для лечения ВИЧ-инфекции, содержащая от 0,5% до 50% по массе соединения по п. 6 или его фармацевтически приемлемой соли и фармацевтически приемлемое вспомогательное вещество.
8. Композиция по п. 7, отличающаяся тем, что указанная композиция приспособлена для парентерального введения.
| WO 2007130626 A2, 15.11.2007 | |||
| WO 2015187998 A2, 10.12.2015 | |||
| WO 2014085489 A1, 05.06.2014 | |||
| WO 2011050068 A2, 28.04.2011 | |||
| EP 2058312 A1, 13.05.2009 | |||
| MITCHELL VAMOS et al | |||
| Expedient Synthesis of Highly Potent Antagonists of Inhibitor of Apoptosis Proteins (IAPs) with Unique Selectivity for ML-IAP | |||
| Acs Chemical Biology, 19.04.2013, vol | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |

