Для настоящей заявки испрашивается приоритет согласно разделу 35 Кодекса законов США, статья 119(е), по предварительной заявке США 60/081310, поданной 10 апреля 1998 г., текст которой включен далее полностью посредством ссылки. Текст и формула изобретения заявки США, озаглавленной "Process for Alkylating Hindered Sulfonamides", поданной 9 апреля 1999 г., которой был присвоен номер экспресс-почты EE645346913US, также включен полностью посредством ссылки.
Предпосылки изобретения
Настоящее изобретение относится к способу алкилирования затрудненных сульфонамидов посредством присоединения Михаэля (Michael) к пропиолатам, а также к новым промежуточным соединениям, получаемым в указанном способе. Продукты вышеупомянутой реакции могут быть превращены в ингибиторы матриксных металлопротеиназ.
Ингибиторы матриксных металлопротеиназ (matrix metalloproteinase, ММР), как известно, полезны при лечении состояния, выбранного из группы, состоящей из артрита (включая остеоартрит и ревматоидный артрит), воспалительного заболевания кишечника, болезни Крона, эмфиземы, острого респираторного дистресс-синдрома, астмы, хронического обструктивного заболевания легких, болезни Альцгеймера, токсичности при трансплантации органов, кахексии, аллергических реакций, контактной аллергии, рака, изъязвления тканей, рестеноза, заболевания периодонта, врожденного буллезного эпидермолиза, остеопороза, расшатывания искуственных суставных протезов, атеросклероза (включая разрыв атеросклеротической бляшки), аневризмы аорты (включая аневризмы брюшной аорты и аорты головного мозга), застойной сердечной недостаточности, инфаркта миокарда, удара, церебральной ишемии, травмы головы, повреждения спинного мозга, нервно-дегенеративных нарушений (острых и хронических), аутоиммунных нарушений, болезни Хантингтона, болезни Паркинсона, мигрени, депрессии, периферической невропатии, боли, церебральной амилоидной ангиопатии, ноотропной или познавательной активации, бокового амиотрофического склероза, рассеянного склероза, развития глазных кровеносных сосудов, повреждения роговицы, дегенерации желтого пятна, аномального заживления раны, ожогов, диабета, инвазии опухоли, роста опухоли, опухолевого метастазирования, рубцевания роговицы, склерита, СПИДа, сепсиса, септического шока и других заболеваний, характеризующихся ингибированием экспрессии металлопротеиназ или ADAM (включая TNF-α). Кроме того, продукты, которые могут быть получены из соединений и способами по данному изобретению, могут быть использованы в комбинационной терапии со стандартными нестероидными противовоспалительными лекарствами (в дальнейшем НПВЛ), ингибиторами ЦОГ-2 и аналгетиками для лечения артрита, а также в сочетании с цитотоксическими лекарствами, такими как адриамицин, дауномицин, цисплатин, этопозид, таксол, таксотер, и алкалоидами, такими как винкристин, при лечении рака.
Алкилсульфонамиды, которые могут быть получены способами по настоящему изобретению, описаны в литературе. РСТ публикации WO 96/27583 и WO 98/07697, опубликованные 7 марта 1996 г. и 26 февраля 1998 г. соответственно, относятся к арилсульфонилгидроксамовым кислотам. Вышеуказанные ссылки относятся к способам получения сульфонамидов с использованием способов, отличных от описанных в данном изобретении. Каждая из вышеупомянутых публикаций включена в данное изобретение во всей полноте посредством ссылки.
Краткое изложение сущности изобретения
Настоящее изобретение относится к соединению формулы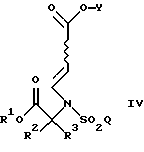
где R1 является (С1-С6)алкилом или возможно замещенным бензилом;
R2 и R3 являются независимо (С1-С6)алкилом, или R2 и R3 вместе образуют 3-7-членный циклоалкил, пиран-4-ильное кольцо или бициклокольцо формулы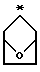
где звездочка обозначает атом углерода, общий для R2 и R3;
Q является (С1-С6)алкилом, (C6-C10)арилом, (С2-С9)гетероарилом, (C6-C10)арил(С1-С6)алкилом, (С2-С9)гетероарил(С1-С6)алкилом, (C6-C10)арилокси(С1-С6)алкилом, (C6-C10)арилокси(C6-C10)арилом, (C6-C10)арилокси(С2-С9)гетероарилом, (C6-C10)арил(C6-C10)арилом, (C6-C10)арил(С2-С9)гетероарилом, (C6-C10)арил(C6-C10)арил(С1-С6)алкилом,
(C6-C10)арил(C6-C10)арил(C6-C10)арилом, (C6-C10)арил(C6-C10)арил(С2-С9)гетероарилом, (С2-С9)гетероарил(C6-C10)арилом, (С2-С9)гетероарил(С2-С9)гетероарилом,
(C6-C10)арил(С1-С6)алкокси(С1-С6)алкилом, (C6-C10)арил(С1-С6)алкокси(C6-C10)арилом, (C6-C10)арил(С1-С6)алкокси(С2-С9)гетероарилом, (С2-С9)гетероарилокси(С1-С6)алкилом, (С2-С9)гетероарилокси(C6-C10)арилом,
(С2-С9)гетероарилокси(С2-С9)гетероарилом, (С2-С9)гетероарил(С1-С6)алкокси(С1-С6)алкилом, (С2-С9)гетероарил(С1-С6)алкокси(C6-C10)арилом или (С2-С9)гетероарил(С1-С6)алкокси(С2-С9)гетероарилом;
где каждая (C6-C10)арильная или (С2-С9)гетероарильная группировка указанных (C6-C10)арила, (С2-С9)гетероарила, (C6-C10)арил(С1-С6)алкила, (С2-С9)гетероарил(С1-С6)алкила, (C6-C10)арилокси(С1-С6)алкила, (C6-C10)арилокси(C6-C10)арила, (C6-C10)арилокси(С2-С9)гетероарила, (C6-C10)арил(C6-C10)арила, (C6-C10)арил(С2-С9)гетероарила, (C6-C10)арил(C6-C10)арил(С1-С6)алкила, (C6-C10)арил(C6-C10)арил(C6-C10)арила, (C6-C10)арил(C6-C10)арил(С2-С9)гетероарила, (С2-С9)гетероарил(C6-C10)арила,
(С2-С9)гетероарил(С2-С9)гетероарила, (C6-C10)арил(С1-С6)алкокси(С1-С6)алкила, (С6-С10)арил(С1-С6)алкокси(C6-C10)арила, (C6-C10)арил(С1-С6)алкокси(С2-С9)гетероарила, (С2-С9)гетероарилокси(С1-С6)алкила, (С2-С9)гетероарилокси(C6-C10)арила,
(С2-С9)гетероарилокси(С2-С9)гетероарила, (С2-С9)гетероарил(С1-С6)алкокси(С1-С6)алкила, (С2-С9)гетероарил(С1-С6)алкокси(C6-C10)арила или (С2-С9)гетероарил(С1-С6)алкокси(С2-С9)гетероарила возможно замещена по любому из кольцевых атомов углерода, способных к образованию дополнительной связи, одним или более чем одним заместителем на кольцо, независимо выбранным из фтора, хлора, брома, (С1-С6)алкила, (С1-С6)алкокси, пepфтоp(C1-С3)алкила, перфтор(С1-С3)алкокси и (С6-С10)арилокси;
a Y является водородом, (С1-С6)алкилом или приемлемой защитной группой.
Предпочтительными являются те соединения формулы IV, где R2 и R3 вместе образуют циклобутильное, циклопентильное, пиран-4-ильное кольцо или бициклокольцо формулы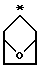
где звездочка обозначает атом углерода, общий для R2 и R3;
и где Q является 4-(4-фторфенокси)фенилом.
Настоящее изобретение также относится к способу получения соединения формулы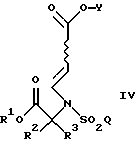
где R1, R2, R3, Q и Y такие, как определено выше;
при котором соединение формулы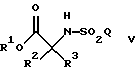
где R1 является возможно замещенным бензилом; a R2, R3 и Q такие, как определено выше;
подвергают взаимодействию с соединением формулы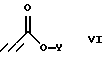
где Y является (С1-С6)алкилом;
в присутствии основания, такого как тетрабутиламмонийфторид, карбонат калия, третичные амины и карбонат цезия, предпочтительно тетрабутиламмонийфторида, и полярного растворителя, такого как тетрагидрофуран, ацетонитрил, трет-бутанол, трет-амиловые спирты и N,N-диметилформамид, предпочтительно тетрагидрофурана.
Настоящее изобретение также относится к способу, при котором указанное соединение формулы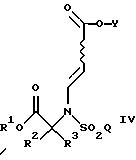
где R1, R2, R3, Q и Y такие, как определено выше;
восстанавливают с помощью восстановителя, такого как палладиевые катализаторы, и источника водорода, предпочтительно водорода над палладием на углероде, в растворителе, таком как спирты или тетрагидрофуран, предпочтительно этаноле, с образованием соединения формулы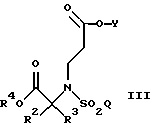
где R4 является водородом; и
R2, R3, Q и Y такие, как определено выше.
Настоящее изобретение также относится к способу, при котором дополнительно указанное соединение формулы III, где R4 является водородом, подвергают взаимодействию с аминами, такими как дициклогексиламин, с образованием солей аминов, таких как дициклогексиламмониевая соль соединения формулы III.
Термин "защитная группа" в качестве заместителя для Y соответствует описанному в Greene and Wuts, Protective Groups in Organic Synthesis (John Wiley & Sons, Inc., Wiley Interscience Second Edition, 1991).
Термин "алкил", используемый в тексте, если не оговорено особо, включает в себя насыщенные одновалентные углеводородные радикалы с прямыми, разветвленными или циклическими группировками или их комбинации.
Термин "алкокси", используемый в тексте, включает в себя О-алкильные группы, где "алкил" определен выше.
Термин "арил", используемый в тексте, если не оговорено особо, включает в себя органический радикал, полученный из ароматического углеводорода путем удаления одного водорода, такой как фенил или нафтил.
Термин "гетероарил", используемый в тексте, если не оговорено особо, включает в себя органический радикал, полученный из ароматического гетероциклического соединения путем удаления одного водорода, такой как пиридил, фурил, пироил, тиенил, изотиазолил, имидазолил, бензимидазолил, тетразолил, пиразинил, пиримидил, хинолил, изохинолил, бензофурил, изобензофурил, бензотиенил, пиразолил, индолил, изоиндолил, пуринил, карбазолил, изоксазолил, тиазолил, оксазолил, бензтиазолил или бензоксазолил. Предпочтительные гетероарилы включают в себя пиридил, фурил, тиенил, изотиазолил, пиразинил, пиримидил, пиразолил, изоксазолил, тиазолил или оксазолил. Наиболее предпочтительные гетероарилы включают в себя пиридил, фурил или тиенил.
Термин "ацил", используемый в тексте, если не оговорено особо, включает в себя радикал общей формулы R-(C=О)-, где R является алкилом, алкокси, арилом, арилалкилом или арилалкокси, а термины "алкил" или "арил" такие, как определено выше.
Термин "ацилокси", используемый в тексте, включает в себя О-ацильные группы, где "ацил" определен выше.
Волнистая линия (то есть 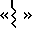 ) в формуле IV означает, что карбоксигруппа может существовать в цис- или транс-конфигурации.
) в формуле IV означает, что карбоксигруппа может существовать в цис- или транс-конфигурации.
Соединения формул I-V могут иметь хиральные центры и поэтому существуют в различных диастереомерных или энантиомерных формах. Данное изобретение относится ко всем оптическим изомерам, таутомерам и стереоизомерам соединений формул I-V, а также их смесям.
Предпочтительно соединения формулы I' существуют в виде экзоизомера формулы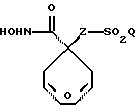 I'
I'
Усовершенствованные пути синтеза с повышенным выходом
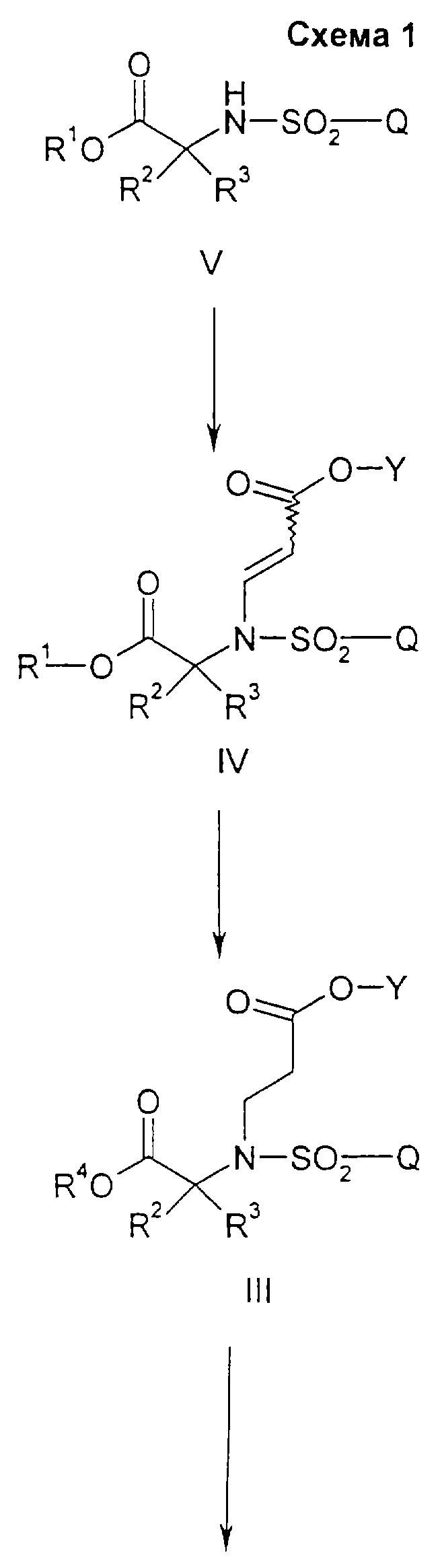
Настоящее изобретение касается также усовершенствованной методологии для получения соединений структуры (формулы) I на схеме 1 (смотри раздел Подробное описание изобретения)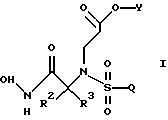
а также новых промежуточных соединений, полезных в данном отношении. Соединения структуры I обладают ценными фармакологическими свойствами. Соответственно, предложены предпочтительные промежуточные соединения согласно структуре IV, как упомянуто выше,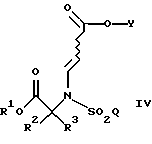
где R1 является [(А1)СН2]с[(А2)СН2]b[(А3)СH2]аС-, причем каждый из a, b и с равен 1; каждый из A1, A2 и А3 независимо выбран из группы, состоящей из Н, (С1-С5)алкила, фенила или замещенного фенила. В предпочтительном примере R1 является трет-бутилом, а каждый из A1, A2 и А3 является водородом.
Как подробно описано ниже, обеспечение таких промежуточных соединений способствует синтезу фармацевтических соединений по изобретению с высоким выходом. Было установлено, что в реакции присоединения Михаэля (смотри ниже, использование данной реакции на предварительных стадиях синтеза), при которой соединение формулы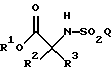
подвергают взаимодействию с соединением формулы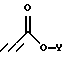
достигается существенное и неожиданное преимущество, если группа R1 определена согласно данному конкретному примеру настоящего изобретения (R1 является [(А1)СН2]с[(А2)СН2]b[(А3)СН2]аС-, причем каждый из a, b и с равнен 1; и каждый из A1, A2 и А3 независимо выбран из группы, состоящей из Н, (C1-C5)алкила, фенила или замещенного фенила), по сравнению с другими примерами, где R1 такая, как определено в настоящем описании, включая, например, бензильную группу.
Таким образом, в настоящем изобретении предложены способы получения таких соединений, как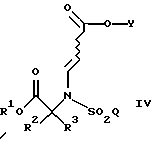
в которых R1 является [(А1)СН2]с[(А2)СН2]b[(А3)СН2]аС-, причем каждый из а, b и с равен 1; и каждый из A1, A2 и А3 независимо выбран из группы, состоящей из Н, (C1-C5)aлкилa и фенила или замещенного фенила, а также способы их дальнейшего использования.
Такие дополнительные способы включают в себя востановление указанного соединения IV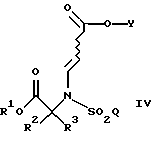
где R1 является [(А1)СН2]с[(А2)СН2]b[(А3)СН2]аС-, причем каждый из a, b и с равен 1; и каждый из A1, A2 и А3 независимо выбран из группы, состоящей из Н, (С1-С5)алкила и фенила или замещенного фенила, а R2, R3, Y и Q такие, как определено выше,
восстановителем с образованием соединения формулы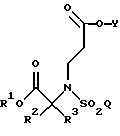
В еще одном из аспектов данного изобретения указанная методология дополнительно включает в себя гидролиз данного соединения, где R1, R2, R3, Y и Q такие, как определено выше, в кислых условиях с образованием соединения формулы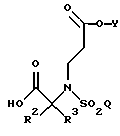
где группы R2, R3, Y и Q такие, как определено выше.
В другом воплощении настоящего изобретения соединение формулы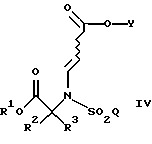
где R1, R2, R3, Y и Q такие, как описано выше, сначала подвергают гидролизу в кислых условиях с образованием соединения формулы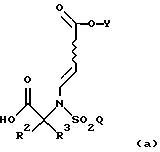
где R2, R3, Y и Q соответствуют описанным выше; а затем на второй стадии соединение (а) обрабатывают восстановителем с образованием соединения формулы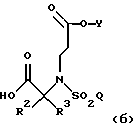
где R2, R3, Y и Q такие, как определено выше.
Подробное описание
Следующие ниже реакционные схемы иллюстрируют получение соединений по настоящему изобретению. Если не оговорено особо, n, R1, R2, R3, Q и Z на данных реакционных схемах и в дальнейшем обсуждении определены, как выше.
Схема 1 относится к получению ингибирующих матриксные металлопротеиназы соединений формулы I.
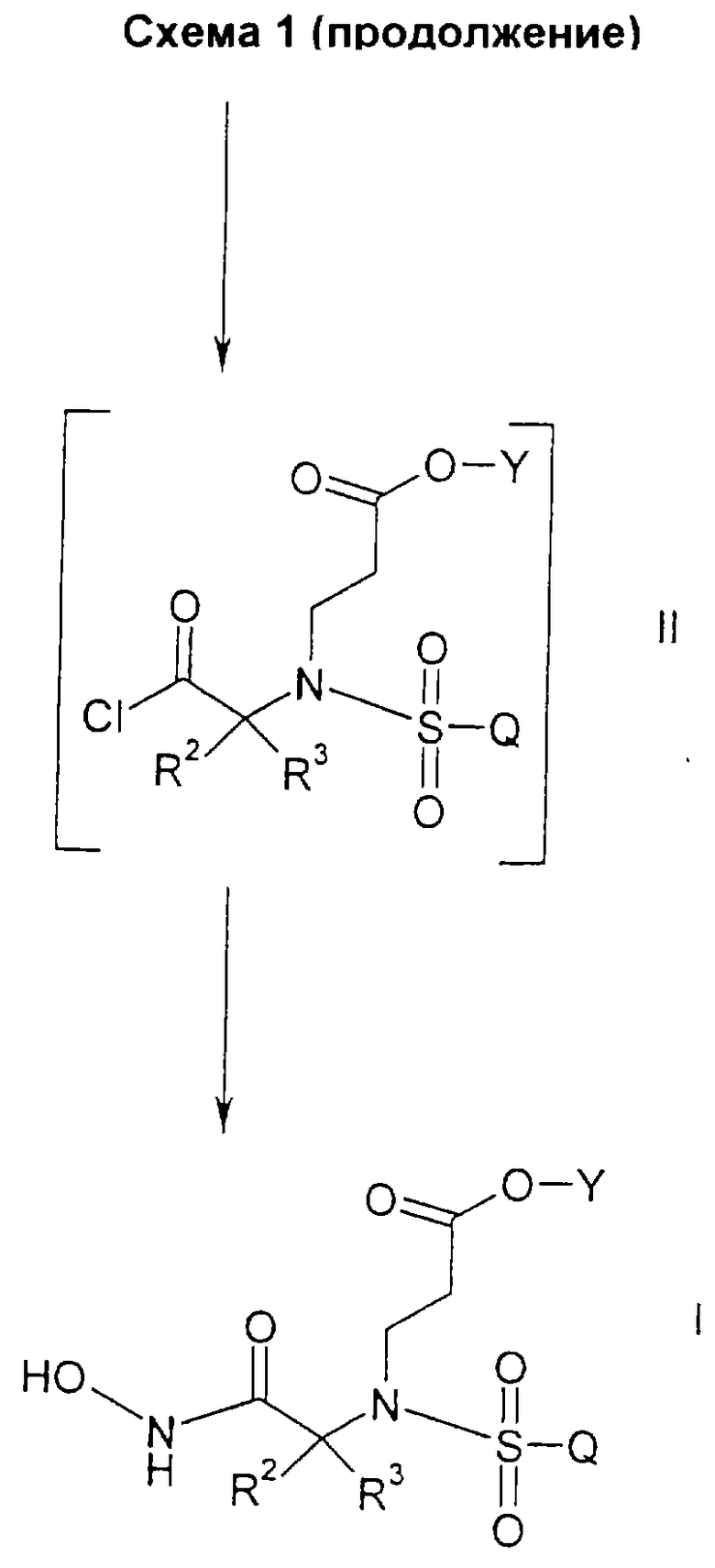
Согласно схеме 1 соединения указанной формулы I получают из соединений формулы II путем взаимодействия с образованным in situ силилированным гидроксиламином с последующей обработкой кислотой. Образованные in situ силилированные гидроксиламиновые соединения получают путем взаимодействия гидроксиламина гидрохлорида или гидроксиламина сульфата, предпочтительно гидроксиламина гидрохлорида, с ((С1-С4)алкил)3cилилгалогенидом в присутствии основания с образованием О-триметилсилилгидроксиламина, N,О-бистриметилсилилгидроксиламина или их комбинаций. Приемлемыми основаниями являются пиридин, 2,6-лутидин или диизопропилэтиламин, предпочтительно пиридин. Реакцию проводят при температуре от примерно 0oС до примерно 22oС (то есть при комнатной температуре) в течение от примерно 1 до примерно 12 часов, предпочтительно в течение примерно 1 часа. Приемлемыми кислотами являются соляная или серная кислоты, предпочтительно соляная.
Соединения указанной формулы II, предпочтительно в невыделенном виде, получают из соединений формулы III, где R4 является водородом, путем взаимодействия с оксалилхлоридом или тионилхлоридом, предпочтительно оксалилхлоридом, и катализатором, предпочтительно примерно 2% N,N-диметилформамида, в инертном растворителе, таком как метиленхлорид или толуол. Реакцию проводят при температуре от примерно 0oС до примерно 22oС (то есть при комнатной температуре) в течение от примерно 1 до примерно 12 часов, предпочтительно в течение примерно 1 часа.
Соединения формулы III, где R4 является водородом, могут быть получены из соединений формулы IV, где R1 является возможно замещенным бензилом, в результате восстановления в полярном растворителе. Приемлемые восстановители включают в себя палладиевый катализатор с источником водорода, такой как водород над палладием, водород над палладием на углероде или гидроксидом палладия на углероде, предпочтительно водород над палладием на углероде. Приемлемые растворители включают в себя тетрагидрофуран, метанол, этанол и изопропанол и их смеси, предпочтительно этанол. Данную реакцию проводят при температуре примерно 22oС (то есть при комнатной температуре) в течение периода времени от 1 до 7 дней, предпочтительно в течение примерно 2 дней.
Соединения формулы III, где R5 не является водородом, например, является протонированным амином (таким как протонированный первичный, вторичный или третичный амин), щелочным металлом или щелочноземельным металлом, могут быть получены из соединений формулы III, где R5 является водородом, путем обработки водным или спиртовым раствором, содержащим приемлемый катион (например натрий, калий, дициклогексиламин, кальций и магний, предпочтительно дициклогексиламин) с последующим выпариванием полученного раствора досуха, предпочтительно при пониженом давлении, или фильтрованием осадка, предпочтительно осадка соли дициклогексиламина.
Соединения формулы IV, где R1 является (С1-С6)алкилом или возможно замещенным бензилом, могут быть получены из соединений формулы V, где R1 является возможно замещенным бензилом, в результате присоединения Михаэля к эфиру пропиоловой кислоты в присутствии основания в полярном растворителе. Приемлемыми пропиолатами являются соединения формулы 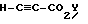 где Y является (С1-С6)алкилом. Соединения формулы H-C≡C-CO2Y имеются в продаже или могут быть получены хорошо известными специалистам методами. Приемлемые основания включают в себя тетрабутиламмонийфторид, карбонат калия, третичные амины и карбонат цезия, предпочтительно тетрабутиламмонийфторид. Приемлемые растворители включают в себя тетрагидрофуран, ацетонитрил, трет-бутанол, трет-амиловые спирты и N,N-диметилформамид, предпочтительно тетрагидрофуран. Вышеуказанную реакцию проводят при температуре от примерно -10oС до примерно 60oС, предпочтительно в пределах от 0oС до примерно 22oС (то есть при комнатной температуре). Соединения формулы IV получают в виде смесей геометрических изомеров по двойной олефиновой связи (то есть цис- и транс-изомеров); разделение данных изомеров не является обязательным.
где Y является (С1-С6)алкилом. Соединения формулы H-C≡C-CO2Y имеются в продаже или могут быть получены хорошо известными специалистам методами. Приемлемые основания включают в себя тетрабутиламмонийфторид, карбонат калия, третичные амины и карбонат цезия, предпочтительно тетрабутиламмонийфторид. Приемлемые растворители включают в себя тетрагидрофуран, ацетонитрил, трет-бутанол, трет-амиловые спирты и N,N-диметилформамид, предпочтительно тетрагидрофуран. Вышеуказанную реакцию проводят при температуре от примерно -10oС до примерно 60oС, предпочтительно в пределах от 0oС до примерно 22oС (то есть при комнатной температуре). Соединения формулы IV получают в виде смесей геометрических изомеров по двойной олефиновой связи (то есть цис- и транс-изомеров); разделение данных изомеров не является обязательным.
Соединения указанной формулы I, где Y является (С1-С6)алкилом, могут быть подвергнуты омылению до свободной кислоты (где Y является водородом) с использованием такого основания, как гидроксид натрия, в протонном растворителе, таком как этанол, метанол, вода или смесь воды с этанолом, воды с толуолом или воды с тетерагидрофураном. Предпочтительной системой растворителей является смесь воды и толуола. Реакцию проводят в течение периода времени от 30 минут до 24 часов, предпочтительно в течение примерно 2 часов.
Соединения формулы V, где R1 является возможно замещенным бензилом, могут быть получены известными методами. Алкилсульфонамиды, которые могут быть получены способами по данному изобретению, и исходные вещества формулы V также описаны в литературе. РСТ публикации WO 96/27583 и WO 98/07697, опубликованные 7 марта 1996 г. и 26 февраля 1998 г. соответственно, относятся к арилсульфонилгидроксамовым кислотам. Каждая из упомянутых публикаций включена полностью посредством ссылки.
Соединения формулы V, где R2 и R3 являются тетрагидропиран-4-илом или бициклокольцом формулы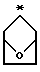
где звездочка обозначает атом углерода, общий для R2 и R3, могут быть получены способами, аналогичными изложенным в примерах 2 и 3.
Соединения формулы I, которые являются основными по своей природе, способны к образованию большого количества различных солей с разнообразными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемыми для введения животным, на практике часто желательно сначала выделить соединение формулы I из реакционной смеси в виде фармацевтически неприемлемой соли, затем просто превратить последнюю обратно в свободное основание обработкой щелочным реагентом, а затем превратить это свободное основание в фармацевтически приемлемую соль присоединения кислоты. Соли присоединения кислот основных соединений по данному изобретению могут быть легко получены путем обработки основного соединения по существу эквивалентным количеством выбранной органической или минеральной кислоты в водном растворителе или приемлемом органическом растворителе, таком как метанол или этанол. При осторожном выпаривании растворителя получается желаемая соль в твердом виде.
Кислотами, используемыми для получения фармацевтически приемлемых солей присоединения кислот основных соединений по данному изобретению, являются кислоты, которые образуют нетоксичные соли присоединения кислот, то есть соли, содержащие фармакологически приемлемые анионы, такие как соли гидрохлорид, гидробромид, гидроиодид, нитрат, сульфат или бисульфат, фосфат или кислый фосфат, ацетат, лактат, цитрат или кислый цитрат, тартрат или битартрат, сукцинат, малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат и памоат [то есть 1,1'-метиленбис-(2-гидрокси-3-нафтоат)].
Те соединения формулы I, которые являются также кислыми по своей природе, способны к образованию солей оснований с различными фармакологически приемлемыми катионами. Примеры таких солей включают в себя соли щелочных или щелочно-земельных металлов, в частности соли натрия и калия. Такие соли получают общепринятыми методами. Химическими основаниями, используемыми в качестве реагентов для получения фармацевтически приемлемых солей оснований по данному изобретению, являются те основания, которые образуют нетоксичные соли оснований с описанными здесь кислыми соединениями формулы I. Такими нетоксичными солями оснований являются соли, полученные из фармакологически приемлемых катионов, например натрия, калия, кальция и магния и так далее. Такие соли могут быть легко получены обработкой соответствующего кислого соединения водным раствором, содержащим желаемые фармакологически приемлемые катионы, с последующим выпариванием полученного раствора досуха, предпочтительно при пониженном давлении. С другой стороны, они также могут быть получены путем смешивания вместе растворов кислых соединений в низших спиртах и алкоксида желаемого щелочного металла с последующим выпариванием полученного раствора досуха описанным выше способом. В обоих случаях предпочтительно использовать стехиометрические количества реагентов с тем, чтобы обеспечить их полное взаимодействие и максимальные выходы продуктов.
Способность соединений формулы I или их фармацевтически приемлемых солей (в дальнейшем именуемых активными соединениями) ингибировать матриксные металлопротеиназы или ADAMs (например ингибировать продукцию фактора некроза опухоли (tumor necrosis factor, TNF)) и, следовательно, проявлять свою эффективность в лечении заболеваний, в которые вовлечены матриксная металлопротеиназа или ADAM (например продуцирование фактора некроза опухоли), может быть определена с помощью тест-анализов in vitro, хорошо известных специалистам. Тест на ингибирование человеческой коллагеназы является примером теста, подтверждающего истинность полученных конечных продуктов, получаемых способами данного изобретения.
Дополнительные предпочтительные примеры изобретения
Настоящее изобретение также касается улучшенной методологии получения соединений со структурой (формулой) I на схеме 1 (смотри раздел Подробное описание изобретения ниже)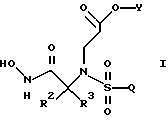
и новых промежуточных продуктов, полезных в данном отношении. Соединения структуры I обладают ценными фармакологическими свойствами. Соответственно, предложены предпочтительные промежуточные соединения согласно структуре IV, упомянутой выше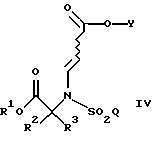
в которых R1 является [(А1)СН2]с[(А2)СН2]b[(А3)СН2]аС-, причем каждый из а, b и с равен 1; каждый из A1, A2 и А3 независимо выбран из группы, состоящей из Н, (С1-С5)алкила и фенила или замещенного фенила. В предпочтительном примере R1 является трет-бутилом, а каждый из A1, A2 и А3 является водородом.
Обеспечение таких промежуточных соединений способствует синтезу фармацевтических соединений по данному изобретению с высоким выходом. Было установлено, что в реакции присоединения Михаэля (смотри ниже, использование данной реакции на стадиях предварительного синтеза), при которой соединение формулы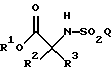
подвергают взаимодействию с соединением формулы,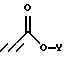
достигается существенное и неожиданное преимущество, если группа R1 определена согласно данному конкретному примеру настоящего изобретения (то есть R1 является [(А1)СН2] с[(А2)СН2] b[(А3)СН2] аС-, причем каждый из а, b и с равен 1; каждый из A1, A2 и А3 независимо выбран из группы, состоящей из Н, (C1-C5)алкила и фенила или замещенного фенила). Это заметно при сравнении с другими примерами, где R1 такой, как определено в настоящем описании, включая, например, бензильную или возможно замещенную бензильную группу. Поэтому использование, например, трет-бутила в качестве R1 является предпочтительным по сравнению с тем не менее очень полезными группами, такими, например, как бензил.
Согласно практике данного изобретения бензильная или замещенная бензильная группы являются очень полезными в качестве R1. Например, в отношении структуры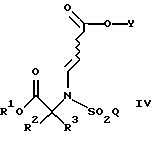
условия реакции могут быть выбраны таким образом (смотри выше), что на одной стадии происходит не только восстановление двойной углерод-углеродной связи, но и отщепление бензила от карбоксильной группы. Хотя это могло бы показаться очень выгодным, оказывается, что присутствие бензила или замещенного бензила в положении R1 благоприятствует побочным реакциям, которые могут снизить общую эффективность реакции присоединения Михаэля. Хотя практика данного изобретения не ограничена никакой теорией, оказывается, что повышение эффективности реакции Михаэля даже за счет простоты следующих стадий может иметь большое значение в определении эффективности полной синтетической схемы. Таким образом, настоящий пример предусматривает альтернативу другой полезной технологии изобретения.
Опять же, не ограничиваясь теорией, возможно, что R1 группы, такие как трет-бутил, мешают побочным реакциям (например, вследствие пространственных затруднений) в большей степени, чем другие группы R1, такие как бензил, в ходе присоединения Михаэля. Данный эффект может быть более важен для успешного протекания всей реакции, чем эффективность прямого сочетания. Поэтому практика настоящего изобретения включает в себя альтернативные эффективные средства для получения промежуточных соединений, необходимых для успешного производства активных фармацевтических средств.
Настоящее изобретение, таким образом, предлагает способы получения таких соединений, как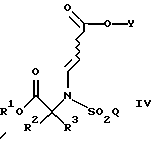
в которых R1 является [(A1)CH2]c[(A2)CH2]b[(A3)CH2]aC-, причем каждый из a, b и с равен 1; каждый из A1, A2 и А3 независимо выбран из группы, состоящей из Н, (С1-С5)алкила и фенила или замещенного фенила, а также способы их дальнейшего использования.
Что касается выбора групп R1, то, как ожидают, могут быть использованы дополнительные группы с эффектом трет-бутила, или могут быть выявлены другие на основании указаний в данном изобретении. Таким образом, опытный практик понимает, что данное изобретение подразумевает использование в качестве R1 любой группы, которая относительно бензила уменьшает скорость побочных реакций в ходе присоединения Михаэля.
Такие дополнительные способы включают в себя восстановление указанного соединения IV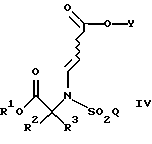
где R1 является [(А1)СН2]с[(А2)СH2]b[(А3)СН2]аС-, причем каждый из а, b и с равен 1; каждый из A1, A2 и А3 независимо выбран из группы, состоящей из Н, (С1-С5)алкила и фенила или замещенного фенила, а R2, R3, Y и Q такие, как описано выше,
при помощи восстановителя с образованием соединения формулы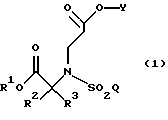
В другом аспекте данного изобретения указанная методология дополнительно включает в себя гидролиз вышеназванного соединения, где R1, R2, R3, Y и Q такие, как описано выше, в кислых условиях с образованием соединения формулы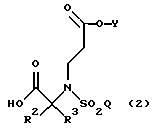
где R2, R3, Y и Q такие, как описано выше.
В связи с выбором групп Y для практики данного изобретения нужно отметить, что в соединениях по данному изобретению Y предпочтительно является водородом или (С1-С6)алкилом. Что касается вышеописанных способов (превращение структуры (1) в структуру (2) непосредственно выше), то особенно предпочтительно, чтобы Y являлся (С1-С6)алкилом. Эта (C1-С6)алкильная группа обладает особенно ценным свойством, заключающимся в том, что хотя она является неустойчивой при гидролизе в щелочных условиях, она достаточно устойчива при гидролизе в кислых условиях, используемом в практике данного изобретения. Таким образом, когда R1 является трет-бутилом, например, то предпочтительно проводить гидролиз в мягких кислых условиях (смотри, например, пример 4), удаляя трет-бутильную группу и оставляя на месте группировку Y в качестве функциональной группы. Так как (С1-С6)алкильная группа в качестве Y по сравнению с оставшейся незащищенной карбоксильной группой является устойчивой как к образованию кислого хлорида, так и к последующему введению гидроксамовой кислоты, то окончательные химические превращения по данному изобретению могут быть направлены на соответствующую карбонильную группу. В практике данного изобретения используются другие группировки, помимо (С1-С6)алкильной группы, для достижения того же самого функционального результата.
В дополнительном воплощении данного изобретения соединение формулы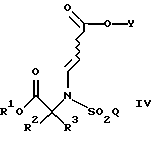
где R1, R2, R3, Y и Q такие, как определено выше, сначала подвергают гидролизу в кислых условиях с образованием соединения формулы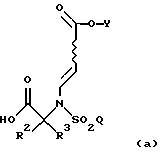
где R2, R3, Y и Q остаются такими, как определено выше; а затем на второй стадии соединение формулы (а) обрабатывают восстановителем с образованием соединения формулы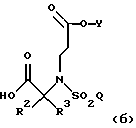
где R2, R3, Y и Q определены как выше.
В связи с вышеописанными реакциями следует отметить, что для гидролиза в кислых условиях могут быть использованы различные кислоты. Среди минеральных кислот можно упомянуть НсС, НВr и H2SO4. Также можно использовать приемлемые карбоновые кислоты, например муравьиную или трифторуксусную кислоту. Без ограничения дополнительный класс пригодных кислот включает в себя сульфоновые кислоты, например п-толуолсульфоновую кислоту и метансульфоновую кислоту.
Что касается условий восстановления, упомянутых в качестве приемлемых согласно практике данного аспекта изобретения, то нужно отметить следующее. Подходящими каталитическими условиями являются такие, при которых восстановителем является водород над катализатором, выбранным из группы, состоящей из оксида платины или никеля Рэнея, или нанесенного катализатора, который выбран из группы, состоящей из палладия на углероде или платины на углероде. Опять же, следует понимать, что подбор эквивалентно эффективных агентов и условий оставляется за исполнителем.
Биологический тест
Ингибирование человеческой коллагеназы (ММР-1)
Человеческую рекомбинантную коллагеназу активируют трипсином в следующем соотношении: 10 мкг трипсина на 100 мкг коллагеназы. Трипсин и коллагеназу инкубируют при комнатной температуре в течение 10 минут, затем добавляют пятикратный избыток (50 мкг/10 мкг трипсина) соевого ингибитора трипсина.
Готовят 10 мМ исходного раствора ингибитора в диметилсульфоксиде, а затем разбавляют по следующей схеме:
10 мМ --->120 мкМ ---> 12 мкМ ---> 1,2 мкМ ---> 0,12 мкМ
Затем в трех повторностях вносят по 25 микролитров каждой концентрации в соответствующие лунки 96-луночного микрофлуорометрического планшета. Конечная концентрация ингибитора имеет разведение 1:4 после добавления фермента и субстрата. Положительный контроль (фермент без ингибитора) помещают в лунки D1-D6, а пустой контроль (ни фермента, ни ингибитора) помещают в лунки D7-D12.
Коллагеназу разбавляют до 400 нг/мл и затем добавляют по 25 мкл в соответсвующие лунки микрофлуорометрического планшета. Конечная концентрация коллагеназы в тесте составляет 100 нг/мл.
Субстрат (DNP-Pro-Cha-Gly-Cys(Me)-His-Ala-Lys(NMA)-NH2) готовят в виде 5 мМ исходного раствора в диметилсульфоксиде, а затем разбавляют до 20 мМ в буфере для анализа. Анализ начинают добавлением 50 мкл субстрата на лунку микрофлуорометрического планшета для достижения конечной концентрации 10 мкМ.
Показания флуоресценции (360 нм возбуждение, 460 нм испускание) были сняты в момент времени 0, а затем с 20-минутными интервалами. Анализ проводят при комнатной температуре в течение 3 часов.
Показания флуоресценции в зависимости от времени наносят на график для пустого контроля и проб с коллагеназой (средние данные по трем измерениям). Значение времени, при котором достигается хороший сигнал (пустой контроль) и которое находится на линейной части кривой (обычно около 120 мин), выбирают для определения значений IC50. Время ноль используют как пустой контроль для каждого соединения при каждой концентрации, и эти значения вычитают из данных при 120 мин. По полученным данным строят график зависимости концентрации ингибитора от % контроля (флуоресценция ингибитора, деленная на флуоресценцию одной коллагеназы х100). Значения IC50 определяют по концентрации ингибитора, которая дает сигнал, составляющий 50% от контроля.
Если значения IC50 составляют менее 0,03 мкМ, ингибитор испытывают в концентрациях 0,3 мкМ, 0,03 мкМ, 0,03 мкМ и 0,003 мкМ.
Следующие примеры иллюстрируют получение соединений по настоящему изобретению. Значения точек плавления не корректировали. Данные ЯМР приведены в миллионных долях (δ) и относятся к сигналу дейтерия растворителя образца (дейтериохлороформ, если не оговорено особо). Коммерческие реагенты использованы без дополнительной очистки. ТГФ обозначает тетрагидрофуран. ДМФ обозначает N,N-диметилформамид. Термин "хроматография" относится к колоночной хроматографии, проводимой с использованием 32-63 мм силикагеля и под давлением азота (флэш-хроматография). Комнатная температура или температура окружающей среды означает 20-25oС. Все реакции в неводной среде проводились в атмосфере азота с целью повышения выхода. Концентрирование при пониженном давлении означает, что использовался роторный испаритель.
Пример 1
3-[[4-(4-Фторфенокси)бензолсульфонил] -(1-гидроксикарбамоилциклопентил)амино]пропионовая кислота
А) Бензиловый эфир 1-[4-(4-фторфенокси)бензолсульфониламино]-циклопентанкарбоновой кислоты
К смеси 12,41 г (0,032 моль) соли толуол-4-сульфоновой кислоты бензилового эфира 1-аминоциклопентанкарбоновой кислоты (может быть получена описанными в литературе методами, например согласно патенту США 4745124) и 10,0 г (0,035 моль, 1,1 эквивалента) 4-(4-фторфенокси)бензолсульфонилхлорида (получен согласно примеру получения 3) в 113 мл толуола добавили 11,0 мл (0,079 моль, 2,5 эквивалента) триэтиламина. Полученную в результате смесь перемешивали в течение ночи при температуре окружающей среды, затем промыли 2 н. соляной кислотой (2х100 мл) и рассолом (100 мл), высушили над сульфатом натрия и сконцентрировали до 30 мл. 149 мл гексана добавили по каплям в течение 3 часов, в результате чего образовался твердый осадок, который был гранулирован при 0oС в течение часа и отфильтрован с получением 12,59 г (85%) бензилового эфира 1-[4-(4-фторфенокси)бензолсульфониламино]-циклопентанкарбоновой кислоты.
1Н ЯМР (CDCl3) 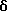 7.78-7.82 (m, 2H), 7.30-7.39 (m, 5H), 7.06-7.12 (m, 2H), 6.99-7.04 (m, 2H), 6.93-6.97 (m, 2H), 5.15 (s, 1H), 5.02 (s, 2H), 2.04-2.13 (m, 2H), 1.92-1.98 (m, 2H),1.62-1.69 (m, 4H).
7.78-7.82 (m, 2H), 7.30-7.39 (m, 5H), 7.06-7.12 (m, 2H), 6.99-7.04 (m, 2H), 6.93-6.97 (m, 2H), 5.15 (s, 1H), 5.02 (s, 2H), 2.04-2.13 (m, 2H), 1.92-1.98 (m, 2H),1.62-1.69 (m, 4H).
Образец массой 4,0 г гранулировали в смеси 4 мл этилацетата и 40 мл гексанов в течение ночи с получением 3,72 г (93% извлечение) бензилового эфира 1-[4-(4-фторфенокси)бензолсульфониламино]циклопентанкарбоновой кислоты в виде светлых желтовато-коричневых твердых частиц, т. пл. 97.0-97,5oС.
Б) Бензиловый эфир 1-{(2-этоксикарбонилвинил)-[4-(4-фторфенокси)бензолсульфонил]-амино}циклопентанкарбоновой кислоты
Раствор 25,0 г (53,2 ммоль) бензилового эфира 1-[4-(4-фторфенокси)бензолсульфониламино] циклопентанкарбоновой кислоты и 10,8 мл (106 ммоль, 2 эквивалента) этилпропиолата в 200 мл безводного тетрагидрофурана при 1oС обработали 53,2 мл (53,2 ммоль, 1 эквивалент) раствора тетрабутиламмонийфторида в тетрагидрофуране (1 М) в течение 45 минут. Полученный раствор оставили медленно нагреваться до температуры окружающей среды при перемешивании в течение ночи. Тетрагидрофуран заменили толуолом при пониженном давлении, а толуольный раствор промыли водой и рассолом, разбавили толуолом до 600 мл, перемешивали с 90 г силикагеля в течение 3 часов, отфильтровали и сконцентрировали до 25,14 г (83%) бензилового эфира 1-{(2-этоксикарбонилвинил)-[4-(4-фторфенокси)бензолсульфонил]амино}циклопентанкарбоновой кислоты в виде оранжевого масла. 1H ЯМР (СDCl3) показал соотношение транс/цис 1,5:1.
Транс δ 7.74-7.78 (m, 2H), 7.72 (d, J=14 Гц, 1Н), 7.26-7.36 (m, 5H), 6.96-7.12 (m, 4Н), 6.78-6.84 (m, 2H), 5.44 (d, J=14 Гц, 1H), 5.11 (s, 2H), 4.12 (q, J=7.1 Гц, 2Н), 2.08-2.43 (m, 4H), 1.63-1.80 (m, 4H), 1.24 (t, J=7.1 Гц, 3Н). Цис δ 7.68-7.72 (m, 2H), 7.26-7.36 (m, 5H), 6.96-7.12 (m, 4H), 6.86-6.91 (m, 2H), 6.47 (d, J=8.1 Гц, 1H), 5.90 (d, J=8.1 Гц, 1H), 5.11 (s, 2H), 3.93 (q, J=7.2 Гц, 2H), 2.08-2.43 (m, 4H), 1.63-1.80 (m, 4H), 1.17 (t, J=7.2 Гц, 3Н).
В) 1-{ (2-Этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил]-амино} -циклопентанкарбоновая кислота
Раствор 2,50 г (4,4 ммоль) бензилового эфира 1-{(2-этоксикарбонилвинил)-[4-(4-фторфенокси)бензолсульфонил]амино}циклопентанкарбоновой кислоты в 25 мл этанола обработали 2,5 г катализатора, представляющего собой 10%-ный палладий на углероде с 50%-ной влажностью, и встряхивали под давлением водорода, равным 53 фунт-сила на кв. дюйм (365,42 кПа) в течение 21 часа. Катализатор удалили фильтрацией и промыли этанолом (4х25 мл). Фильтрат и промывные воды объединили и сконцентрировали под вакуумом до 1,74 г (82%) сырой 1-{ (2-этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил] амино} циклопентанкарбоновой кислоты в виде вязкого масла.
1Н ЯМР (CDCl3) δ 7.78-7.82 (m, 2H), 6.94-7.09 (m, 6Н), 4.09 (q, J=7.2 Гц, 2H), 3.56-3.60 (m, 2H), 2.75-2.79 (m, 2H), 2.33-2.39 (m, 2H), 1.93-2.03 (m, 2H), 1.69-1.76 (m, 2H), 1.56-1.63 (m, 2H), 1.22 (t, J=7.2 Гц, 3Н).
Г) Дициклогексиламиниевая соль 1-{(2-этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил]-амино}-циклопентанкарбоновой кислоты
Раствор 3,10 г (6,5 ммоль) сырой 1-{(2-этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил] амино}циклопентанкарбоновой кислоты в 30 мл этанола обработали 1,28 мл (6,5 ммоль, 1 эквивалент) дициклогексиламина при температуре окружающей среды с образованием твердых частиц в течение 5 минут. Данную смесь перемешивали в течение ночи при температуре окружающей среды, а затем в течение 5 часов при 0oС. Белые твердые частицы выделили фильтрацией, промыли 10 мл холодного этанола, а затем подвергли воздушной сушке с получением 2,89 г (67%) дициклогексиламиниевой соли 1-{(2-этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил]амино}циклопентанкарбоновой кислоты.
1Н ЯМР (CDCl3) δ 7.86-7.91 (m, 2H), 6.99-7.09 (m, 4H), 6.90-6.94 (m, 2H), 5.3 (br s, 2H), 4.07 (q, J=7.1 Гц, 2H), 3.54-3.59 (m, 2H), 2.88-2.95 (m, 4H), 2.31-2.38 (m, 2H), 1.95-2.22 (m, 6H), 1.68-1.77 (m, 6H), 1.53-1,60 (m, 4H), 1.40-1.50 (m, 4H), 1.21 (t, J=7.1 Гц, 3Н), 1.14-1.22 (m, 6H). Т. пл. 164,5-165,9oС.
Д) 1-{ (2-Этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил]-амино} -циклопентанкарбоновая кислота
Раствор 3,0 г (4,5 ммоль) дициклогексиламиниевой соли 1-{(2-этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил]амино}циклопентанкарбоновой кислоты в 30 мл дихлорметана обработали 30 мл 2 н. соляной кислоты при температуре окружающей среды, при этом сразу образовался осадок твердых частиц. Данную смесь перемешивали при температуре окружающей среды в течение 3 часов. Твердые частицы отфильтровали, водную фазу экстрагировали дихлорметаном, объединенные органические фазы промыли водой, высушили над сульфатом натрия и сконцентрировали под вакуумом до 2,2 г (100%) 1-{(2-этоксикарбонилэтил)-[4-(4-
фторфенокси)бензолсульфонил] амино} циклопентанкарбоновой кислоты в виде прозрачного масла.
1Н ЯМР (DMSO-d6) δ 12.68 (bs, 1H), 7.76-7.80 (m, 2H), 7.25-7.31 (m, 2H), 7.16-7.21 (m, 2H), 7.03-7.08 (m, 2H), 4.01 (q, J=7.1 Гц, 2H), 3.48-3.54 (m, 2H), 2.64-2.70 (m, 2H), 2.13-2.21 (m, 2H), 1.90-1.98 (m, 2H), 1.52-1.59 (m, 4H), 1.14 (t, J=7.1 Гц, 3Н).
Е) Этиловый эфир 3-{ (1-хлоркарбонилциклопентил-[4-(4-фторфенокси)бензолсульфонил]амино}пропионовой кислоты
Раствор 7,26 г (15,1 ммоль) 1-{(2-этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил] амино}циклопентанкарбоновой кислоты в 73 мл дихлорметана обработали 1,4 мл (17 ммоль, 1,1 эквивалента) оксалилхлорида и 0,02 мл (0,3 ммоль, 0,02 эквивалента) диметилформамида при температуре окружающей среды, что привело к выделению пузырьков, и перемешивали в течение ночи. Полученный раствор этилового эфира 3-{ (1-хлоркарбонилциклопентил)-[4-(4-фторфенокси)бензолсульфонил] амино} пропионовой кислоты был использован для получения этилового эфира 3-[[4-(4-фторфенокси)бензолсульфонил] -(1-гидроксикарбамоилциклопентил)амино]пропионовой кислоты без выделения.
Полученный также раствор этилового эфира 3-{(1-хлоркарбонилциклопентил)-[4-(4-фторфенокси)бензолсульфонил] амино} пропионовой кислоты был сконцентрирован под вакуумом до масла.
1Н ЯМР (CDCl3) δ 7.84-7.87 (m, 2H), 6.97-7.12 (m, 6Н), 4.10 (q, J=7.2 Гц, 2H), 3.55-3.59 (m, 2H), 2.68-2.72 (m, 2H), 2.47-2.53 (m, 2H), 1.95-2.02 (m, 2H), 1.71-1.76 (m, 4Н), 1.24 (t, J=7.2 Гц, 3Н).
Ж) Этиловый эфир 3-[[4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклопентил)амино]пропионовой кислоты
Раствор 1,37 г (19,7 ммоль, 1,3 эквивалента) гидрохлорида гидроксиламина в 9,2 мл (114 ммоль, 7,5 эквивалентов) безводного пиридина при 0oС обработали 5,8 мл (45 ммоль, 3,0 эквивалента) триметилсилилхлорида, что вызвало выпадение в осадок твердых частиц белого цвета. Смесь оставили нагреваться до температуры окружающей среды в течение ночи. Затем данную смесь охладили до 0oС и обработали раствором 7,54 г (15,1 ммоль) этилового эфира 3-{(1-хлоркарбонилциклопентил)-[4-(4-фторфенокси)бензолсульфонил] амино} пропионовой кислоты в 73 мл дихлорметана, полученного как описано выше, без выделения, что вызвало экзотерму до примерно 8oС. Данную смесь перемешивали при 0oС в течение 30 минут и при температуре окружающей среды в течение примерно одного часа. Реакционную смесь затем обработали 50 мл 2 н. водной соляной кислоты и перемешивали при температуре окружающей среды в течение часа. Водную фазу экстрагировали дихлорметаном, объединенные органические фазы промыли 2 н. водной соляной кислотой (2х50 мл) и водой (50 мл). Полученный раствор этилового эфира 3-[[4-(4-фторфенокси)бензолсульфонил] -(1-гидроксикарбамоилциклопентил)амино] пропионовой кислоты в дихлорметане использовали для получения 3-[[4-(4-фторфенокси)бензолсульфонил] -(1-гидроксикарбамоилциклопентил)амино] пропионовой кислоты без выделения. Аликвота была сконцентрирована до пены.
1Н ЯМР (DMSO-d6) δ 10.37 (s, 1H), 8.76 (s, 1H), 7.74-7.79 (m, 2H), 7.24-7.30 (m, 2H), 7.14-7.20 (m, 2H), 7.01-7.05 (m, 2H), 3.99 (q, J=7.1 Гц, 2H), 3.42-3.47 (m, 2H), 2.62-2.67 (m, 2H), 2.16-2.23 (m, 2H), 1.77-1.85 (m, 2H), 1.43-1.52 (m, 4H), 1.13 (t, J=7.1 Гц, 3Н).
Полученный также раствор был сконцентрирован под вакуумом до 6,71 г (89%) этилового эфира 3-[[4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоилциклопентил)амино]пропионовой кислоты в виде стойкой сухой пены.
З) 3-[[4-(4-Фторфенокси)бензолсульфонил] -(1-гидроксикарбамоилциклопентил)амино]пропионовая кислота
Раствор 7,48 г (15,1 ммоль) этилового эфира 3-[[4-(4-фторфенокси)бензолсульфонил] -(1-гидроксикарбамоилциклопентил)амино] пропионовой кислоты в дихлорметане сконцентрировали в роторном испарителе с добавлением 75 мл толуола. Данный раствор обработали 75 мл воды, охладили до 0oС и обработали 6,05 г (151 ммоль, 10 эквивалентов) гидроксида натрия в гранулах в течение 10 минут при интенсивном перемешивании. Данную смесь перемешивали в течение 15 минут при 0oС и нагрели до температуры окружающей среды в течение часа. Водную фазу отделили, разбавили 7,5 мл тетрагидрофурана, охладили до 0oС и обработали 33 мл 6 н. водной соляной кислоты в течение 20 минут. Смесь перемешивали с 75 мл этилацетата при температуре от 0oС до температуры окружающей среды, затем этилацетатную фазу отделили и промыли водой. Раствор в этилацетате медленно обработали 150 мл гексанов при температуре окружающей среды, что вызвало выпадение в осадок твердых частиц, и перемешивали в течение ночи. После фильтрации получили 5,01 г 3-[[4-(4-фторфенокси)бензолсульфонил] -(1-гидроксикарбамоилциклопентил)амино] пропионовой кислоты в виде белого твердого вещества (выход 71% от 1-{(2-этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил]амино}циклопентанкарбоновой кислоты).
1Н ЯМР (DMSO-d6) δ 12.32 (s, 1H), 10.43 (s, 1H), 8.80 (s, 1H), 7.82 (d, J=8.6 Гц, 2Н), 7.28-7.35 (m, 2H), 7.20-7.26 (m, 2Н), 7.08 (d, J=8.9 Гц, 2Н), 3.44-3.49 (m, 2H), 2.61-2,66 (m, 2H), 2.24-2.29 (m, 2H), 1.86-1.90 (m, 2H), 1.54-1.55 (m, 4H). Т. пл. 162,9-163,5oС (разложение).
Пример 2
3-[[4-(4-Фторфенокси)-бензолсульфонил] -(4-гидроксикарбамоилтетрагидропиран-4-ил)-амино]-пропионовая кислота
А) Бензиловый эфир 4-[N-(дифенилметилен)амино]тетрагидропиран-4-карбоновой кислоты
К суспензии гидрида натрия (6,56 г, 0,164 моль) в диметиловом эфире этиленгликоля (150 мл) при 0oС добавляют раствор бензилового эфира N-(дифенилметилен)глицина (0,07398 моль) в диметиловом эфире этиленгликоля (50 мл) по каплям через капельную воронку. Затем к этому раствору в диметиловом эфире этиленгликоля порциями по 10 мл в течение приблизительно 5 минут добавляют раствор 2-бромэтилового эфира (23,21 г, 0,090 моль) в диметиловом эфире этиленгликоля (50 мл). Ледяную баню удаляют, после чего реакционную смесь перемешивают при комнатной тепературе в течение 16 часов. Смесь разбавляют диэтиловым эфиром и промывают водой. Водный слой экстрагируют диэтиловым эфиром. Объединенные органические экстракты промывают рассолом, сушат над сульфатом магния и концентрируют до получения сырого продукта. Посредством хроматографии на силикагеле с элюцией сначала 4 л смеси 5% этилацетат/гексан, а затем 4 л смеси 10% этилацетат/гексан получают бензиловый эфир 4-[N-(дифенилметилен)амино] тетрагидропиран-4-карбоновой кислоты в виде прозрачного желтого масла.
Б) Бензиловый эфир 4-аминотетрагидропиран-4-карбоновой кислоты
К раствору бензилового эфира 4-[N-(дифенилметилен)амино]тетрагидропиран-4-карбоновой кислоты (0,047 моль) в диэтиловом эфире (120 мл) добавляют 1 М водный раствор соляной кислоты (100 мл). Смесь интенсивно перемешивают в течение 16 часов при комнатной температуре. Слои разделяют, водный слой промывают диэтиловым эфиром. Затем рН водного слоя доводят до 10 разбавленным водным раствором гидроксида аммония и экстрагируют дихлорметаном. Органический экстракт сушат над сульфатом натрия и концентрируют с получением бензилового эфира 4-аминотетрагидропиран-4-карбоновой кислоты.
В) Бензиловый эфир 4-[4-(4-фторфенокси)бензолсульфониламино]тетрагидропиран-4-карбоновой кислоты
К раствору бензилового эфира 4-аминотетрагидропиран-4-карбоновой кислоты (0,0404 моль) в N,N-диметилформамиде (40 мл) добавляют триэтиламин (5,94 мл, 0,043 моль). К указанному раствору порциями добавляют твердый 4-(4-фторфенокси)бензолсульфонилхлорид (12,165 г, 0,0424 моль). Полученную смесь перемешивают при комнатной температуре в течение 16 часов, а затем большую часть растворителя удаляют выпариванием под вакуумом. Остаток распределяют между насыщенным раствором бикарбоната натрия и дихлорметаном. Водный слой отделяют и экстрагируют дихлорметаном. Объединенные органические слои промывают рассолом и сушат над сульфатом натрия. После выпаривания растворителя под вакуумом получают сырой бензиловый эфир 4-[4-(4-фторфенокси)бензолсульфониламино] тетрагидропиран-4-карбоновой кислоты. Посредством флэш-хроматографии на силикагеле с элюцией сначала смесью 25% этилацетат/гексан, затем смесью 50% этилацетат/гексан получают бензиловый эфир 4-[4-(4-фторфенокси)бензолсульфониламино]тетрагидропиран-4-карбоновой кислоты.
Г) Бензиловый эфир 4-{(2-этоксикарбонилвинил)-[4-(4-фторфенокси)-бензолсульфонил]-амино}-тетрагидропиран-4-карбоновой кислоты
Раствор продукта с предыдущей стадии (53,2 ммоль) и 10,8 мл (106 ммоль, 2 эквивалента) этилпропиолата в 200 мл безводного тетрагидрофурана при температуре 1oС обрабатывают 53,2 мл (53,2 ммоль, 1 эквивалент) раствора тетрабутиламмонийфторида в тетрагидрофуране (1 М) в течение 45 минут. Полученный раствор оставляют медленно нагреваться до температуры окружающей среды и перемешивают в течение ночи. Тетрагидрофуран заменяют толуолом при пониженном давлении, а толуольный раствор промывают водой и рассолом, разбавляют толуолом до 600 мл, перемешивают с 90 г силикагеля в течение 3 часов, фильтруют и концентрируют до соединения, указанного в заголовке.
Д) 4-{(2-Этоксикарбонилэтил)-[4-(4-фторфенокси)-бензолсульфонил]-амино} тетрагидропиран-4-карбоновая кислота
Раствор продукта со стадии Г (4,4 ммоль) в 25 мл этанола обрабатывают 2,5 г катализатора, представляющего собой 10%-ный палладий на угле с 50%-ной влажностью, и встряхивают под давлением водорода 53 фунт-сила на кв. дюйм (365,42 кПа) в течение 21 часа. Катализатор удаляют фильтрацией и промывают этанолом (4х25 мл). Фильтрат и промывные воды объединяют и концентрируют под вакуумом до сырого продукта.
Е) Этиловый эфир 3-{(4-хлоркарбонилтетрагидропиран-4-ил)-[4-(4-фторфенокси)-бензолсульфонил]-амино}-пропионовой кислоты
Раствор продукта реакции со стадии Д (15,1 ммоль) в 73 мл дихлорметана обрабатывают 1,4 мл (17 ммоль, 1,1 эквивалента) оксалилхлорида и 0,02 мл (0,3 ммоль, 0,02 эквивалента) диметилформамида при температуре окружающей среды, что вызывает образование пузырьков, а затем перемешивают в течение ночи. Полученный в результате раствор указанного в заголовке соединения используют на стадии Ж без выделения.
Ж) Этиловый эфир 3-[[4-(4-фторфенокси)-бензолсульфонил]-(4-гидроксикарбамоилтетрагидропиран-4-ил)-амино]-пропионовой кислоты
Раствор гидрохлорида гидроксиламина (19,7 ммоль, 1,3 эквивалента) в 9,2 мл (114 ммоль, 7,5 эквивалентов) безводного пиридина при 0oС обрабатывают 5,8 мл (45 ммоль, 3,0 эквивалента) триметилсилилхлорида, что вызывает выпадение в осадок твердых частиц белого цвета. Смесь оставляют нагреваться до температуры окружающей среды в течение ночи. Затем данную смесь охлаждают до 0oС и обрабатывают раствором продукта со стадии Е (15,1 ммоль) в 73 мл дихлорметана, что вызывает экзотерму до примерно 8oС. Затем смесь перемешивают при 0oС в течение 30 минут и при температуре окружающей среды в течение примерно одного часа. Далее реакционную смесь обрабатывают 50 мл водной 2 н. соляной кислоты и перемешивают при температуре окружающей среды в течение часа. Водную фазу экстрагируют дихлорметаном и объединенные органические фазы промывают 2 н. водной соляной кислотой (2х50 мл) и водой (50 мл). Полученный раствор указанного в заголовке соединения в дихлорметане используют на следующей стадии.
З) 3-[[4-(4-Фторфенокси)-бензолсульфонил] -(4-гидроксикарбамоилтетрагидропиран-4-ил)-амино]-пропионовая кислота
Раствор 15,1 ммоль продукта со стадии Ж в дихлорметане концентрируют с использованием роторного испарителя с добавлением 75 мл толуола. Данный раствор обрабатывают 75 мл воды, охлаждают до 0oС и обрабатывают 6,05 г (151 ммоль, 10 эквивалентов) гидроксида натрия в гранулах в течение 10 минут при интенсивном перемешивании. Полученную смесь перемешивают в течение 15 минут при 0oС и нагревают до температуры окружающей среды в течение часа. Водную фазу отделяют, разбавляют 7,5 мл тетрагидрофурана, охлаждают до 0oС, а затем обрабатывают 33 мл 6 н. водной соляной кислоты в течение 20 минут. Полученную смесь перемешивают с 75 мл этилацетета при температуре от 0oС до температуры окружающей среды, затем этилацетатную фазу отделяют и промывают водой. Этилацетатный раствор концентрируют с получением соединения, указанного в заголовке.
Пример 3
3-[[4-(4-Фторфенокси)-бензолсульфонил] -(3-гидроксикарбамоил-8-оксабицикло[3.2.1]окт-3-ил)-амино]-пропионовая кислота
A) Бензиловый эфир 3-(бензгидрилиденамино)-8-оксабицикло[3.2.1]октан-3-карбоновой кислоты
К суспензии гидрида натрия (0,41 г, 17,1 ммоль) в N,N-диметилформамиде (50 мл) при 0oС добавляют по каплям раствор бензилового эфира N-дифенилметиленглицина (7,8 ммоль) в N,N-диметилформамиде (50 мл). После перемешивания в течение 30 минут при комнатной температуре добавляют по каплям раствор цис-2,5-бис(гидроксиметил)-тетрагидрофурана дитозилата (4,1 г, 9,3 ммоль) (получен известными из литературы способами, например как описано в JOC, 47, 2429-2435 (1982)) в N,N-диметилформамиде (50 мл). Реакционную смесь постепенно нагревают до 100oС на масляной бане и перемешивают при данной температуре в течение ночи. Растворитель выпаривают под вакуумом, а остаток переносят в воду и экстрагируют дважды диэтиловым эфиром. Объединенные органические экстракты промывают рассолом, сушат над сульфатом магния и концентрируют с получением сырого продукта.
Б) Гидрохлорид бензилового эфира 3-амино-8-оксабицикло[3.2.1]октан-3-карбоновой кислоты
Двухфазную смесь бензилового эфира 3-(бензгидрилиденамино)-8-оксабицикло[3.2.1] октан-3-карбоновой кислоты (3,9 ммоль) в водном растворе 1 н. соляной кислоты (100 мл) и диэтилового эфира (100 мл) перемешивают при комнатной температуре в течение ночи. Затем водный слой концентрируют для получения указанного в заголовке соединения.
B) Бензиловый эфир 3-экзо-[4-(4-фторфенокси)бензолсульфониламино]-8-оксабицикло[3.2.1]-октан-3-карбоновой кислоты
Раствор гидрохлорида бензилового эфира 3-амино-8-оксабицикло[3.2.1]октан-3-карбоновой кислоты (2,9 ммоль), 4-(4-фторфенокси)бензолсульфонилхлорида (923 мг, 3,2 ммоль) и триэтиламина (0,9 мл, 6,5 ммоль) в N,N-диметилформамиде (45 мл) перемешивают при комнатной температуре в течение ночи. Растворитель удаляют под вакуумом и остаток переносят в насыщенный водный раствор бикарбоната натрия. После двукратного экстрагирования метиленхлоридом объединенные органические слои промывают рассолом, сушат над сульфатом магния и концентрируют до коричневого масла. Указанное в заголовке соединение выделяют с помощью хроматографии на диоксиде кремния, используя в качестве элюента 1%-ный метанол в метиленхлориде.
Г) Бензиловый эфир 3-{(2-этоксикарбонилвинил)-[4-(4-фторфенокси)-бензолсульфонил]-амино}-8-оксабицикло[3.2.1]октан-3-карбоновой кислоты
Раствор продукта с предыдущей стадии (53,2 ммоль) и 10,8 мл (106 ммоль, 2 эквивалента) этилпропиолата в 200 мл безводного тетрагидрофурана при 1oС обрабатывают 53,2 мл (53,2 ммоль, 1 эквивалент) раствора тетрабутиламмонийфторида в тетрагидрофуране (1 М) в течение 45 минут. Полученный раствор оставляют медленно нагреваться до температуры окружающей среды и перемешивают в течение ночи. Тетрагидрофуран замещают толуолом при пониженном давлении, толуольный раствор промывают водой и рассолом, разбавляют до 600 мл толуолом, перемешивают с 90 г силикагеля в течение 3 часов, фильтруют и концентрируют до указанного в заголовке соединения.
Д) 3-{(2-Этоксикарбонилэтил)-[4-(4-фторфенокси)-бензолсульфонил]-амино} -8-оксабицикло[3.2.1]октан-3-карбоновая кислота
Раствор продукта со стадии Г (4,4 ммоль) в 25 мл этанола обрабатывают 2,5 г катализатора, представляющего собой 10%-ный палладий на углероде с 50%-ной влажностью, и встряхивают под давлением водорода 53 фунт-сила на кв. дюйм (365,42 кПа) в течение 48 часов. Катализатор удаляют фильтрованием и промывают этанолом (4х25 мл). Фильтрат и промывные воды объединяют и концентрируют под вакуумом до сырого продукта.
Е) Этиловый эфир 3-{(3-хлоркарбонил-8-оксабицикло[3.2.1]окт-3-ил)-[4-(4-фторфенокси)-бензолсульфонил]-амино}-пропионовой кислоты
Раствор 15,1 ммоль продукта со стадии Д в 73 мл дихлорметана обрабатывают 1,4 мл (17 ммоль, 1,1 эквивалента) оксалилхлорида и 0,02 мл (0,3 ммоль, 0,02 эквивалента) диметилформамида при температуре окружающей среды, что вызывает выделение пузырьков, и перемешивают в течение ночи. Полученный раствор указанного в заголовке соединения используют на стадии Ж без выделения.
Ж) Этиловый эфир 3-[[4-(4-фторфенокси)-бензолсульфонил] -(3-гидроксикарбамоил-8-оксабицикло[3.2.1]окт-3-ил)-амино]-пропионовой кислоты
Раствор гидрохлорида гидроксиламина (19,7 ммоль, 1,3 эквивалента) в 9,2 мл (114 ммоль, 7,5 эквивалентов) безводного пиридина при 0oС обрабатывают 5,8 мл (45 ммоль, 3,0 эквивалента) триметилсилилхлорида, что вызывает выпадение в осадок твердых частиц белого цвета. Смесь оставляют нагреваться до температуры окружающей среды в течение ночи. Затем ее охлаждают до 0oС и обрабатывают раствором продукта со стадии Е (15,1 ммоль) в 73 мл дихлорметана, что вызывает экзотерму до примерно 8oС. Смесь перемешивают при 0oС в течение 30 минут и при температуре окружающей среды в течение часа. Затем реакционную смесь обрабатывают 50 мл 2 н. водной соляной кислоты и перемешивают при температуре окружающей среды в течение одного часа. Водную фазу экстрагируют дихлорметаном, объединенные органические фазы промывают 2 н. водной соляной кислотой (2х50 мл) и водой (50 мл). Раствор указанного в заголовке соединения в дихлорметане используют на следующей стадии.
З) 3-[[4-(4-Фторфенокси)-бензолсульфонил] -(3-гидроксикарбамоил-8-оксабицикло[3.2.1]окт-3-ил)-амино]-пропионовая кислота
Раствор 15,1 ммоль продукта со стадии Ж в дихлорметане концентрируют в роторном испарителе с добавлением 75 мл толуола. Данный раствор обрабатывают 75 мл воды, охлаждают до 0oС, обрабатывают 6,05 г (151 ммоль, 10 эквивалентов) гидроксида натрия в гранулах в течение 10 минут при интенсивном перемешивании. Затем данную смесь перемешивают в течение 15 минут при 0oС и нагревают до температуры окружающей среды в течение часа. Водную фазу отделяют, разбавляют 7,5 мл тетрагидрофурана, охлаждают до 0oС и обрабатывают 33 мл 6 н. водной соляной кислоты в течение 20 минут. Данную смесь перемешивают с 75 мл этилацетата при температуре от 0oС до температуры окружающей среды, этилацетатную фазу отделяют и промывают водой. Этилацетатный раствор концентрируют до указанного в заголовке соединения.
Получение 1
4-Фторфениловый эфир 4-(4-фторфенокси)бензолсульфоновой кислоты
Раствор 14,68 г (0,131 моль, 2,0 эквивалента) трет-бутилата калия в 27 мл безводного N-метилпирролидинона обработали раствором 15,39 г (0,137 моль, 2,1 эквивалента) 4-фторфенола в 27 мл безводного N-метилпирролидинона при температуре окружающей среды, что вызвало мягкую экзотерму до 45oС. Раствор 13,81 г (0,065 моль) 4-хлорбензолсульфонилхлорида в 27 мл безводного N-метилпирролидинона медленно добавили к темной реакционной смеси, что вызвало мягкую экзотерму до 44oС. Полученную смесь перемешивали при комнатной температуре в течение часа, а затем при 130oС в течение 11 часов. Охлажденную реакционную смесь обработали 162 мл воды, внесли небольшое количество 4-фторфенилового эфира 4-(4-фторфенокси)бензолсульфоновой кислоты в качестве затравки и проводили гранулирование при комнатной температуре в течение ночи. Полученные твердые частицы отфильтровали с получением 20,24 г (85%) 4-фторфенилового эфира 4-(4-фторфенокси)бензолсульфоновой кислоты.
1Н ЯМР (CDCl3) δ 7.74 (dd, J=7.0, 2.0 Гц, 2Н), 7.14-6.97 (m, 10Н). Т. пл. 78-83oС.
Получение 2
Натриевая соль 4-(4-фторфенокси)бензолсульфоновой кислоты
К взвеси 47,43 г (0,131 моль) 4-фторфенилового эфира 4-(4-фторфенокси)бензолсульфоновой кислоты в 475 мл этанола добавили 13,09 г (0,327 моль, 2,5 эквивалента) гидроксида натрия в гранулах. Данную смесь нагревали с обратным холодильником в течение 3 часов и перемешивали в течение ночи при комнатной температуре. Полученные твердые частицы отфильтровали с получением 37,16 г (98%) натриевой соли 4-(4-фторфенокси)бензолсульфоновой кислоты.
1Н ЯМР (СD3ОD) δ 7.73-7.78 (m, 2Н), 7.05-7.13 (m, 2Н), 6.99-7.05 (m, 2H), 6.90-6.95 (m, 2H).
Получение 3
4-(4-Фторфенокси)бензолсульфонилхлорид
К взвеси 15,0 г (0,052 моль) натриевой соли 4-(4-фторфенокси)бензолсульфоновой кислоты в 150 мл безводного толуола добавили 11,3 мл (0,155 моль, 3 эквивалента) тионилхлорида и 0,04 мл (0,5 ммоль, 0,01 эквивалента) диметилформамида. Полученную смесь перемешивали при комнатной температуре в течение 48 часов, профильтровали через кизельгур и сконцентрировали при пониженном давлении до 40 мл. Данный раствор был использован без дальнейшей очистки для получения бензилового эфира 1-[4-(4-фторфенокси)бензолсульфониламино]циклопентанкарбоновой кислоты.
Порцию в 5,0 мл данного раствора сконцентрировали до 1,77 г 4-(4-фторфенокси)бензолсульфонилхлорида в виде масла, что соответствовало выходу 96%.
1Н ЯМР (СDСl3) δ 7.92-7.97 (m, 2H), 7.01-7.13 (m, 6H). Часть полученного таким же образом масла кристаллизовали из гексана, т. пл. 80oС.
Пример 4
Получение 1
1-[4-(4-Фторфенокси)-бензолсульфониламино]циклопентанкарбоновая кислота
Бензиловый эфир 1-[4-(4-фторфенокси)-бензолсульфониламино]циклопентанкарбоновой кислоты (15 г, 32 ммоль) в 75 мл ТГФ соединили с 75 мл (150 ммоль) 2 н. водного гидроксида натрия и перемешивали при нагревании с обратным холодильником в течение часа. Реакционную смесь охладили до температуры окружающей среды и разбавили 100 мл воды и 100 мл этилацетата. рН водной фазы довели до значения 1,2 и отделили этилацетатный слой. Этилацетатный слой промыли 100 мл воды и высушили над сульфатом магния. Этилацетат упарили в вакууме и заменили 75 мл метил-трет-бутилового эфира. Полученный продукт отфильтровали и высушили с получением 11,16 г (92%) 1-[4-(4-фторфенокси)бензолсульфониламино]циклопентанкарбоновой кислоты.
1Н ЯМР (СDСl3) δ 7.71-7.78 (m, 2H), 6.88-7.04 (m, 6H), 5.04 (s, 1H), 2.01-2.13 (m, 2H), 1.92-1.98 (m, 2H), 1.44-1.68 (m, 4H).
Получение 2
Трет-бутиловый эфир 1-[4-(4-фторфенокси)-бензолсульфониламино]-циклопентанкарбоновой кислоты
К раствору 1-[4-(4-фторфенокси)бензолсульфониламино] циклопентанкарбоновой кислоты (10,22 г, 27 ммоль) в 100 мл метиленхлорида при -78oС было сконденсировано 40 мл изобутилена. Добавили концентрированную серную кислоту (0,3 мл) и оставили смесь нагреваться до температуры окружающей среды и перемешивали в течение 22 часов. Затем смесь промыли 2 н. NaOH (3х50 мл), органический слой высушили над сульфатом магния и выпарили с получением 11,17 г (95%) трет-бутилового эфира 1-[4-(4-фторфенокси)бензолсульфониламино]-циклопентанкарбоновой кислоты.
1Н ЯМР (СDСl3) δ 7.74-7.77 (m, 2Н), 6.85-7.13 (m, 6H), 4.95 (s, 1H), 1.92-2.02 (m, 2Н), 1.78-1.88 (m, 2H), 1.50-1.65 (m, 4Н), 1.35 (s, 9H).
Опытный практик может найдет другие многочисленные стратегии для синтеза промежуточных продуктов реакций, описанных здесь. Например, может быть осуществлена этерификация изобутилена с такой молекулой, как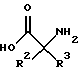
с последующим сульфированием с помощью, например, QSO2Cl группировки. Также следует отметить, что, например, трет-бутиловые эфиры вышеуказанных структур могут быть легко получены или могут быть куплены.
Получение 3
Трет-бутиловый эфир 1-{ (2-этоксикарбонилвинил)-[4-(4-фторфенокси)бензолсульфонил]-амино}-циклопентанкарбоновой кислоты
К смеси трет-бутилового эфира 1-[4-(4-фторфенокси)-бензолсульфониламино] циклопентанкарбоновой кислоты (1,0 г, 2,3 ммоль) в 10 мл ТГФ и 2,3 мл (2,3 ммоль) 1 М тетрабутиламмонийфторида в ТГФ добавили 0,23 мл (2,3 ммоль) этилпропиолата при температуре окружающей среды. После перемешивания в течение часа реакционную смесь подвергли ВЭЖХ (высокоэффективная жидкостная хроматография) и упарили досуха в вакууме. Остаток растворили в 20 мл этилацетата и промыли водой (2х10 мл), органический раствор упарили до масла. Это масло хроматографировали на силикагеле, элюируя смесью 10% этилацетат/гексан, получив 0,95 г (выход 77%) трет-бутилового эфира 1-{(2-этоксикарбонилвинил)-[4-(4-фторфенокси)бензолсульфонил]-амино}-циклопентанкарбоновой кислоты в виде бесцветного масла. 1H ЯМР (СDСl3) показал соотношение 1,5:1 транс/цис.
Транс: δ 5 7.79-7.83 (m, 2H), 7.63 (d, J=14 Гц, 1Н), 6.89-7.05 (m, 4H), 5.44 (d, J=14 Гц, 1H), 4.08 (q, J=7.1 Гц, 1Н), 2.08-2.43 (m, 4H), 1.63-1.80 (m, 4H), 1.39 (s, 9H), 1.22 (t, J=7.1 Гц, 3Н). Цис: 7.62-7.69 (m, 2H), 6.91-6.85 (m, 2H), 6.55 (d, J=8.1 Гц, 1H), 5.85 (d, J=8.1 Гц, 1H), 3.81 (q, J= 7.2 Гц, 2H), 2.08-2.43 (m, 4H), 1.19-1.25 (m, 4H), 1.49 (s, 9H), 1.11 (q, J=7.2 Гц, 3Н).
Получение 4
Трет-бутиловый эфир 1-{(2-этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил]-амино}-циклопентанкарбоновой кислоты
Раствор трет-бутилового эфира 1-{(2-этоксикарбонилвинил)-[4-(4-фторфенокси)бензолсульфонил] -амино} -циклопентанкарбоновой кислоты (1,23 г, 2,3 ммоль) в 50 мл этанола с 723 мг 5% Pd/C катализатора гидрировали при температуре окружающей среды до тех пор, пока ВЭЖХ не показала завершения реакции. Катализатор отфильтровали, фильтрат упарили до получения масла, которое хроматографировали на силикагеле, элюируя 105 этилацетата в гексане. Трет-бутиловый эфир 1-{(2-этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил] -амино}-циклопентанкарбоновой кислоты был выделен в виде бесцветного масла (875 мг, выход 71%).
1Н ЯМР (CDCl3) δ 7.75-7.80 (m, 2H), 6.86-7.01 (m, 6H), 4.09 (q, J=7.2 Гц, 2H), 3.44-3.48 (m, 2H), 2,66-2.72 (m, 2H), 2.09-2.15 (m, 2H), 1.52-1.74 (m, 4H), 1.43 (s, 9H), 1.21 (t, J=7.2 Гц, 3Н).
Получение 5
Дициклогексиламиниевая соль 1-{(2-этоксикарбонилэтил)-[4-(4-фторфенокси)-бензолсульфонил]-амино}-циклопентанкарбоновой кислоты
Раствор трет-бутилового эфира 1-{(2-этоксикарбонилэтил)-[4-(4-фторфенокси)бензолсульфонил] -амино} -циклопентанкарбоновой кислоты (0,225 г, 0,42 ммоль) в 4 мл толуола обработали метансульфоновой кислотой (0,06 мл, 0,84 ммоль) при температуре окружающей среды в течение 18 часов. Полученный раствор промыли водным раствором бикарбоната натрия и упарили до бесцветного масла. Это масло растворили в 2 мл этанола и обработали дициклогексиламином (0,084 мл, 0,42 ммоль). Полученный продукт, дициклогексиламиниевую соль 1-{ (2-этоксикарбонилэтил)-[4-(4-фторфенокси)-бензолсульфонил] -амино}-циклопентанкарбоновой кислоты, отфильтровали и высушили с получением 223 мг (выход 80%) белого твердого вещества. Данные ЯМР и время удерживания на ВЭЖХ аналогичны образцу, полученному по пути с использованием бензилового эфира.
Изобретение относится к новым производным сульфонамидов общей формулы I, где R1 - [(A1)CH2] c[(А2)СН2]b[(А3)СН2]аС-, причем каждый из a, b и с равен 1; каждый из A1, А2 и А3 независимо выбран из Н, (С1-С5)алкила и фенил или замещенного фенила; R2 и R3 независимо (С1-С6)алкил, или R2 и R3 вместе образуют 3-7-членный циклоалкил, тетрагидропиран-4-ильное кольцо или бициклокольцо формулы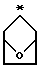
где звездочка обозначает атом углерода, общий для R2 и R3; Q - (C1-С6)алкил, (С6-С10)арил, (С6-С10)арил(С1-С6)алкил, (С6-С10)арилокси(С1-С6)алкил, (С6-С10)арилокси(С6-С10)арил, (С6-С10)арил(С6-С10)арил, (С6-С10)арил(С6-С10)арил(С1-С6)алкил, (С6-С10)арил(С6-С10)арил(С6-С10)арил, (С6-С10)арил(С1-С6)алкил или (С6-С10)арил(С1-С6)алкокси(С6-С10)арил, где каждая (С6-С10)арильная группировка указанных (С6-С10)арила, (С6-С10)арил(С1-С6)алкила, (С6-С10)арилокси(С1-С6)алкила, (С6-С10)арилокси(С6-С10)арила, (С6-С10)арил(С6-С10)арила, (С6-С10)арил(С6-С10)арил(С1-С6)алкила,
(С6-С10)арил(С6-С10)арил(С6-С10)арила, (С6-С10)арил(С1-С6)алкила или (С6-С10)арил(С1-С6)алкокси(С6-С10)арила возможно замещена по любому из кольцевых атомов углерода, способных к образованию дополнительной связи, одним или более чем одним заместителем на кольцо, независимо выбранным из фтора, хлора, брома, (С1-С6)алкила, (С1-С6)алкокси, перфтор (С1-С3)алкила, перфтор(С1-С3)алкокси и (С6-С10)арилокси; и Y - водород или (С1-С6)алкил. Соединения I - промежуточные продукты в синтезе ингибиторов матриксных металлопротеиназ. 2 с. и 10 з.п. ф-лы.
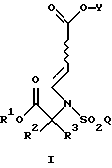
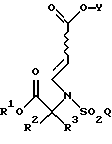
где R1 является [(А1)СН2] с[(А2)СН2] b[(А3)СН2] аС-, причем каждый из а, b и с равен 1; каждый из A1, A2 и А3 независимо выбран из группы, состоящей из Н, (С1-С5)алкила и фенила или замещенного фенила;
R2 и R3 являются независимо (С1-С6)алкилом, или R2 и R3 вместе образуют 3-7-членный циклоалкил, тетрагидропиран-4-ильное кольцо или бициклокольцо формулы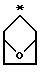
где звездочка обозначает атом углерода, общий для R2 и R3;
Q является (С1-С6)алкилом, (С6-С10)арилом, (С6-С10)арил(С1-С6)алкилом,
(С6-С10)арилокси(С1-С6)алкилом, (С6-С10)арилокси(С6-С10)арилом, (С6-С10)арил(С6-С10)арилом, (С6-С10)арил(С6-С10)арил(С1-С6)алкилом,
(С6-С10)арил(С6-С10)арил(С6-С10)арилом, (С6-С10)арил(С1-С6)алкокси(С1-С6)алкилом или (С6-С10)арил(С1-С6)алкокси(С6-С10)арилом;
где каждая (С6-С10)арильная группировка указанных (С6-С10)арила, (С6-С10)арил(С1-С6)алкила, (С6-С10)арилокси(С1-С6)алкила, (С6-С10)арилокси(С6-С10)арила, (С6-С10)арил(С6-С10)арила, (С6-С10)арил(С6-С10)арил(С1-С6)алкила, (С6-С10)арил(С6-С10)арил(С6-С10)арила, (С6-С10)арил(С1-С6)алкокси(С1-С6)алкила или (С6-С10)арил(С1-С6)алкокси(С6-С10)арила возможно замещена по любому из кольцевых атомов углерода, способных к образованию дополнительной связи, одним или более чем одним заместителем на кольцо, независимо выбранным из фтора, хлора, брома, (С1-С6)алкила, (С1-С6)алкокси, перфтор(С1-С3)алкила, перфтор(С1-С3)алкокси и (С6-С10)арилокси;
Y является водородом или (С1-С6)алкилом.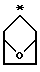
где звездочка обозначает атом углерода, общий для R2 и R3.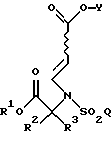
где R1 является [(А1)СН2] с[(А2)СН2] b[(А3)СН2] аС-, причем каждый из a, b и с равен 1; каждый из A1, A2 и А3 независимо выбран из группы, состоящей из Н, (С1-С5)алкила и фенила или замещенного фенила;
R2 и R3 являются независимо (С1-С6)алкилом, или R2 и R3 вместе образуют 3-7-членный циклоалкил, тетрагидропиран-4-ильное кольцо или бициклокольцо формулы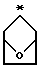
где звездочка обозначает атом углерода, общий для R2 и R3;
Q является (С1-С6)алкилом, (С6-С10)арилом, (С6-С10)арилокси(С1-С6)алкилом, (С6-С10)арилокси(С6-С10)арилом, (С6-С10)арил(С1-С6)алкилом, (С6-С10)арил(С6-С10)арилом, (С6-С10)арил(С6-С10)арил(С1-С6)алкилом, (С6-С10)арил(С6-С10)арил(С6-С10)арилом, (С6-С10)арил(С1-С6)алкокси(С1-С6)алкилом или (С6-С10)арил(С1-С6)алкокси(С6-С10)арилом,
где каждая (С6-С10)арильная группировка указанных (С6-С10)арила, (С6-С10)арилокси(С1-С6)алкила, (С6-С10)арилокси(С6-С10)арила, (С6-С10)арил(С1-С6)алкила, (С6-С10)арил(С6-С10)арила, (С6-С10)арил(С6-С10)арил(С1-С6)алкила, (С6-С10)арил(С6-С10)арил(С6-С10)арила, (С6-С10)арил(С1-С6)алкокси(С1-С6)алкила или (С6-С10)арил(С1-С6)алкокси(С6-С10)арила возможно замещена по любому из кольцевых атомов углерода, способных к образованию дополнительной связи, одним или более чем одним заместителем на кольцо, независимо выбранным из фтора, хлора, брома, (С1-С6)алкила, (С1-С6)алкокси, перфтор(С1-С3)алкила, перфтор(С1-С3)алкокси и (С6-С10)арилокси;
Y является (С1-С6)алкилом,
отличающийся тем, что соединение формулы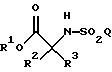
где R1, R2, R3 и Q определены выше,
подвергают взаимодействию с соединением формулы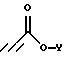
где Y является(С1-С6)алкилом,
в присутствии основания и полярного растворителя.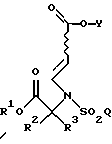
где R1, R2, R3, Y и Q такие, как определено в п. 4,
восстанавливают восстановителем с образованием соединения формулы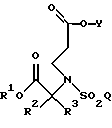
где R1, R2, R3, Y и Q определены выше.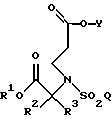
где R1, R2, R3, Y и Q такие, как определено в п. 7,
дополнительно гидролизуют в кислых условиях с образованием соединения формулы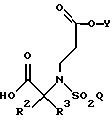
где R2, R3, Y и Q такие, как определено выше.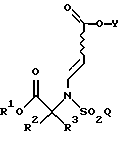
где R1, R2, R3, Y и Q такие, как определено в п. 4,
гидролизуют в кислых условиях с образованием соединения формулы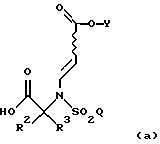
где R2, R3, Y и Q определены выше; и
(б) вторую дополнительную стадию, на которой указанное соединение (а) восстанавливают восстановителем с образованием соединения формулы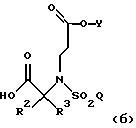
где R2, R3, Y и Q определены выше.
| Способ получения сульфоксидных или сульфоновых производных тиокарбаматов | 1973 |
|
SU523636A3 |
| WO 9627583, 12.09.1996 | |||
| WO 9807697, 26.02.1998 | |||
| TETPAHEDRON LETTERS, v.36, № 3, 1995, р.417-420. | |||

