Объектами настоящего изобретения являются рацемические или энантиомерно чистые производные 4-пирролидинов, способ их получения, фармацевтические композиции, включающие упомянутые производные, и их использование для профилактики и лечения заболеваний.
В частности, объектом настоящего изобретения являются соединения формулы I
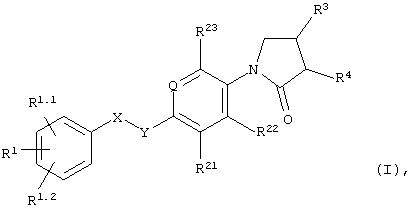
в которых
Q обозначает=N- или=C(R24)-;
X-Y обозначает -СН2-СН2-, -СН=СН- или -СН2-О-;
R1, R1.1 и R1.2 независимо друг от друга выбраны из группы, включающей водород, галоид, галоид(С1-С6)алкил, цианогруппу, (С1-С6)алкоксигруппу или галоид-(С1-С6)алкоксигруппу;
R21, R22 и R23 независимо друг от друга выбраны из группы, включающей водород и галоид;
R24 обозначает водород, галоид или метил;
R3 обозначает -NHR6;
R4 обозначает водород; и
R6 обозначает -С(О)Н, -С(О)-(С1-С3)алкил, С(О)-галоид(С1-С3)алкил, -С(О)O(С1-С3)алкил, -С(О)NH2 или -SO2-(С1-С3)алкил;
а также их индивидуальные изомеры и рацемические или нерацемические смеси.
В особенности, объектом настоящего изобретения являются соединения формулы I*
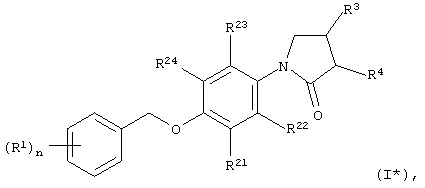
в которых
R1 обозначает галоид, галоид(С1-С6)алкил, цианогруппу, (C1-С6)алкоксигруппу или галоид(С1-С6)алкоксигруппу;
R21, R22, R23 и R24 независимо друг от друга выбраны из группы, включающей водород и галоид;
R3 обозначает -NHR6;
R4 обозначает водород;
R6 обозначает -СО-(С1-С3)алкил или -SO2-(С1-С3)алкил; и
n принимает значения 0, 1, 2 или 3;
а также их индивидуальные изомеры и рацемические или нерацемические смеси.
Было обнаружено, что соединения общей формулы I и I*, а также их индивидуальные изомеры и рацемические или нерацемические смеси (здесь и далее: активные соединения), являются селективными ингибиторами моноаминоксидазы В.
Моноаминоксидаза (МАО, ЕС 1.4.3.4) является флавинсодержащим ферментом, отвечающим за окислительное дезаминирование эндогенных моноаминных нейромедиаторов, таких как допамин, серотонин, адреналин или норадреналин, а также следовых аминов, например, фенилэтиламина, равно как и ряда аминосодержащих ксенобиотиков. Фермент существует в двух формах, МАО-А и МАО-В, кодируемых разными генами (A.W.Bach и др., Proc. Natl. Acad. Sci. USA, 1988, т.85, cc.4934-4938) и различающихся распределением в тканях, структурой и субстратной специфичностью. МАО-А обладает большим сродством к серотонину, октопамину, адреналину и норадреналину, тогда как естественными субстратами для МАО-В являются фенилэтиламин и тирамин. Допамин, как полагают, окисляется обеими изоформами. МАО-В широко распространена в некоторых органах, включая мозг (А.М.Cesura и A.Pletscher, Prog. Drug Research, 1992, т.38, cc.171-297). Активность МАО-В в мозге, как оказывается, увеличивается с возрастом. Это увеличение приписывают связанному со старением глиозу (C.J.Fowler и др., J. Neural Transm., 1980, т.49, cc.1-20). Кроме того, активность МАО-В существенно выше в мозге пациентов с болезнью Альцгеймера (Р.Dostert и др., Biochem. Pharmacol. 1989, т.38, cc.555-561) и, как обнаружилось, высоко выражена в астроцитах вокруг старческих бляшек (Saura и др., Neuroscience, 1994, т.70, cc.755-774). В связи с этим, поскольку окислительное дезаминирование первичных моноаминов с помощью МАО приводит к образованию аммиака, альдегидов и перекиси водорода, веществ с установленной или потенциальной токсичностью, предполагается, что имеются логические основания для применения селективных ингибиторов МАО-В для лечения слабоумия и болезни Паркинсона. Ингибирование МАО-В вызывает снижение ферментативной инактивации допамина и, вследствие этого, пролонгирование доступности нейромедиатора в допаминовых нейронах. Дегенеративные процессы, связанные с возрастом и болезнями Альцгеймера и Паркинсона, могут быть также отнесены на счет окислительной нагрузки в результате увеличенной активности МАО и обусловленного ею повышения выработки перекиси водорода Н2O2 под воздействием МАО-В. Поэтому действие ингибиторов МАО-В может проявляться как в снижении выработки кислородных радикалов, так и в подъеме уровней моноаминов в мозгу.
Учитывая описанное выше значение МАО-В при неврологических расстройствах, существует значительный интерес к получению мощных и селективных ингибиторов, которые позволили бы контролировать эту ферментативную активность. Фармакология некоторых известных ингибиторов МАО-В обсуждается, например, D. Bentué-Ferrer и др. в CNS Drugs, 1996, т.6, сс.217-236. Тогда как главным ограничением активности необратимого и неселективного ингибитора МАО является необходимость соблюдения диетических предосторожностей вследствие риска вызвать гипертонический криз при приеме диетического тирамина, равно как и возможность взаимодействия с другими лекарственными средствами (D.M.Gardiner и др., J. Clin. Psychiatry, 1996, т.57, сс.99-104), эти побочные явления менее значимы для обратимых и селективных ингибиторов МАО, в частности МАО-В. Таким образом, существует потребность в ингибиторах МАО-В, обладающих высокой селективностью и не имеющих вредных побочных эффектов, типичных для необратимых ингибиторов МАО с низкой селективностью по отношению к ферменту.
Нижеследующие определения общих терминов, используемых в контексте, действительны вне зависимости от того, употреблены ли рассматриваемые термины по отдельности или в определенном сочетании. Следует учитывать, что употребленные в описании и пунктах формулы изобретения формы единственного числа подразумевают также формы множественного числа, если только из контекста однозначно не следует обратное.
Термин «их индивидуальные изомеры и рацемические или нерацемические смеси» означает Е- и Z-изомеры, их смеси, равно как и индивидуальные конфигурационные изомеры и их смеси, при этом термин "рацемические смеси" означает смеси двух энантиомеров в соотношении 1:1, а «нерацемические смеси» означает смеси двух энантиомеров в соотношении, отличном от 1:1.
Термин «(C1-C6)алкил», используемый в настоящей заявке, означает насыщенные углеводородные остатки с прямой или разветвленной цепью, содержащие от 1 до 6 атомов углерода, такие как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил и им подобные, предпочтительно содержащие от 1 до 3 атомов углерода. Соответственно, термин «(C1-C3)алкил» означает насыщенный углеводородный остаток с прямой или разветвленной цепью, содержащий от 1 до 3 атомов углерода.
Термин «галоген» («галоид») означает фтор, хлор, бром и иод.
«Галоид(С1-С6)алкил» или «галоид(С1-С6)алкокси» означает (низш.)алкильный остаток или (низш.)алкоксильный остаток соответственно, как это определено в контексте, замещенный по любому положению одним или несколькими атомами галогена, как это определено в контексте. Примеры галоидалкильных остатков включают 1,2-дифторпропил, 1,2-дихлорпропил, трифторметил, 2,2,2-трифторэтил, 2,2,2-трихлорэтил и 3,3,3-трифторпропил и подобные им, но не ограничиваются перечисленными. «Галоидалкокси» включает трифторметоксигруппу.
«(С1-С6)алкокси» означает остаток -О-R, в котором R является низшим алкильным остатком, как это определено в контексте. Примеры алкоксильных радикалов включают метоксигруппу, этоксигруппу, изопропоксигруппу и подобные им, но не ограничиваются перечисленными.
«Фармацевтически приемлемые соли» соединения означают соли, являющиеся приемлемыми фармацевтически, то есть в общем смысле безопасными, не токсичными, не являющиеся нежелательными ни в биологическом, ни в каком-либо ином смысле, обладающие желаемой фармакологической активностью исходного соединения. Эти соли являются производными неорганической или органической кислоты или основания. Если это возможно, соединения формулы I могут быть преобразованы в фармацевтически приемлемые соли. Следует иметь в виду, что фармацевтически приемлемые соли включены в настоящее изобретение.
Еще в одном варианте осуществления настоящего изобретения его объектами являются соединения формулы I*, в которых R3 обозначает -NHR6, R6 обозначает -СО-(С1-С6)алкил или -SO2-(С1-С6)алкил, а R4 обозначает водород. Примером подобного соединения является (RS)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид.
Соединения формулы I* могут быть замещены n заместителями R1, выбранными из группы, включающей галоид, галоид(С1-С6)алкил, цианогруппу, (С1-С6)алкоксигруппу или галоид(С1-С6)алкоксигруппу, где n является целым числом, выбранным из 0, 1, 2 и 3. Предпочтительно, n принимает значения 1 или 2. Предпочтительными соединениями формулы I* являются такие, в которых R1 обозначает галоид или галоид(С1-С6)алкил. Особенно предпочтительными являются такие соединения формулы I*, в которых R1 обозначает фтор, хлор или трифторметильную группу. Еще в одном варианте осуществления настоящего изобретения его объектами являются соединения формулы I*, в которых n принимает значения 0 или 1. Еще в одном варианте осуществления настоящего изобретения его объектами являются соединения формулы I*, в которых n принимает значение 1. В том случае, когда соединения замещены двумя или тремя заместителями R1, каждый R1 может быть тем же самым или иным. В одном из вариантов осуществления настоящего изобретения его объектами являются соединения формулы I, в которых Q обозначает =C(R24)-, где R24 обозначает водород, галоид или метил. В другом варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых Q обозначает =СН-, =CF- или =С(СН3)-. Еще в одном варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых Q обозначает =N-.
В одном из вариантов осуществления настоящего изобретения его объектами являются соединения формулы I, в которых -X-Y- обозначает -СН2-O-. В другом варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых -X-Y- обозначает -СН2-СН2- или -СН=СН-.
В одном из вариантов осуществления настоящего изобретения его объектами являются соединения формулы I, в которых R1, R1.1 и R1.2 независимо друг от друга выбраны из группы, включающей водород, галоид, метил, галоидметил, цианогруппу, метоксигруппу или галоидметоксигруппу. В другом варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых R1, R1.1 и R1.2 обозначают галоид, например, фтор, например, 2,4,6-трифтор-, 2,4,5-трифтор-, 2,3,6-трифтор-, 2,3,4-трифтор-или 3,4,5-трифтор-. Еще в одном варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых R1.2 обозначает водород, a R1 и R1.1 независимо друг от друга выбраны из группы, включающей водород, галоид, (С1-С6)алкил, галоид(С1-С6)алкил, цианогруппу, (С1-С6)алкоксигруппу или галоид(С1-С6)алкоксигруппу. Еще в одном варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых R1.2 обозначает водород, а R1 и R1.1 независимо друг от друга выбраны из группы, включающей галоид и (С1-С6)алкил. Еще в одном варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых R1.2 обозначает водород, R1.1 обозначает галоид, а R1 обозначает галоид или (С1-С6)алкил. Еще в одном варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых R1.1 и R1.2 обозначают водород, а R1 обозначает галоид, (С1-С6)алкил, галоид(С1-С6)алкил, цианогруппу, (С1-С6)алкоксигруппу или галоид(С1-С6)алкоксигруппу. Еще в одном варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых R1.1 и R1.2 обозначают водород, а R1 обозначает галоид, метил, галоидметил, цианогруппу, метоксигруппу или галоидметоксигруппу. Еще в одном варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых R1.1 и R1.2 обозначают водород, а R1 обозначает фтор, например, 2-фтор-, 3-фтор- или 4-фтор-, хлор, например, 3-хлор-, метил, например, 4-метил-, галоидметил, например, 3-трифторметил-, цианогруппу, метоксигруппу, например, 2-метокси-, 3-метокси- или 4-метокси-, или галоидметоксигруппу, например, 3-трифторметокси-. В другом варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых R1, R1.1 и R1.2 обозначают водород.
В другом варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых R21, R22 и R23 обозначают водород.
Еще в одном варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых R24 обозначает водород.
Еще в одном варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых R24 обозначает -NHR6, где R6 обозначает -С(О)Н, -С(О)-СН3, -С(О)-CH2F, -С(О)-CHF2, -С(О)-CF3, -С(О)O-СН3, -С(О)-NH2 или -SO2-СН3.
В одном из вариантов осуществления настоящего изобретения его объектами являются соединения формулы I, имеющие (S)-конфигурацию.
В другом варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых Q обозначает =C(R24)-, где R24 обозначает водород, X-Y обозначает -СН2-O-, R1.1 и R1.2 обозначают водород, R1 обозначает водород или галоид, R21, R22 и R23 обозначают водород, R3 обозначает -NHR6, R4 обозначает водород, а R6 обозначает -С(О)Н, -С(О)-(C1-С3)алкил, С(О)-галоид(С1-С3)алкил, -С(О)O(С1-С3)алкил, -С(О)NH2 или -SO2-(С1-С3)алкил.
Еще в одном варианте осуществления настоящего изобретения его объектами являются соединения формулы I, в которых Q обозначает =C(R24)-, где R24 обозначает водород, X-Y обозначает -СН2-O-, R1.1 и R1.2 обозначают водород, R1 обозначает водород или галоид, R21, R22 и R23 обозначают водород, R3 обозначает -NHR6, R4 обозначает водород, а R6 обозначает -С(О)Н, -С(О)-СН3, -С(О)-CH2F, -С(О)-CHF2, -С(О)-CF3, -С(О)O-СН3, -С(О)-NH2 или -SO2-СН3.
Примеры соединений формулы I включают соединения, выбранные среди нижеследующих:
(RS)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(S)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(R)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(RS)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}формамид,
(S)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}формамид,
(R)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}формамид,
метиловый эфир (RS)-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}-карбаминовой кислоты,
(RS)-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}мочевина,
(RS)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}метансульфамид,
(S)-2-фтор-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(S)-2,2-дифтор-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(S)-2,2,2-трифтор-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(RS)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(R)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(S)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(RS)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}формамид,
(RS)-N-[1-(4-бензилоксифенил)-5-оксопирролидин-3-ил]ацетамид,
(RS)-N-{1-[4-(2-фторбензилоксифенил]-5-оксопирролидин-3-ил}ацетамид,
(RS)-(Е)-N-(1-{4-[2-(3-фторфенил)винил]фенил}-5-оксопирролидин-3-ил)ацетамид,
(RS)-N-(1-{4-[2-(3-фторфенил)этил]фенил}-5-оксопирролидин-3-ил)ацетамид,
(RS)-N-{1-[6-(4-фторбензилокси)пиридин-3-ил]-5-оксопирролидин-3-ил}ацетамид,
(S)-N-{1-[4-(3-хлорбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(S)-N-{1-[4-(2,6-дифторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(S)-N-{5-оксо-1-[4-(2,4,6-трифторбензилокси)фенил]пирролидин-3-ил}ацетамид,
(S)-N-{1-[4-(3-метоксибензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(S)-N-{5-оксо-1-[4-(4-трифторметилбензилокси)фенил]пирролидин-3-ил}ацетамид,
(S)-N-{1-[4-(4-метилбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид и
(S)-N-{1-[4-(3-цианобензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид.
Еще в одном варианте осуществления настоящего изобретения его объектом является способ получения соединений формулы I, состоящий во введении соединения формулы II
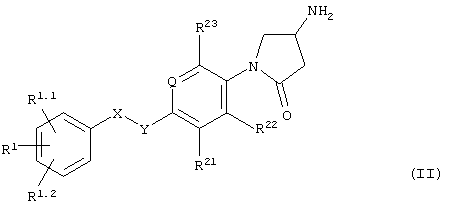
в реакцию с изоцианатом или с ацилирующим агентом формулы Z-С(О)-(C1-С3)алкил, Z-С(О)-галоид(С1-С3)алкил, Z-С(О)O(С1-С3)алкил или Z-SO2-(C1-С3)алкил, где Z обозначает активирующую группу, например, галоид или ангидрид.
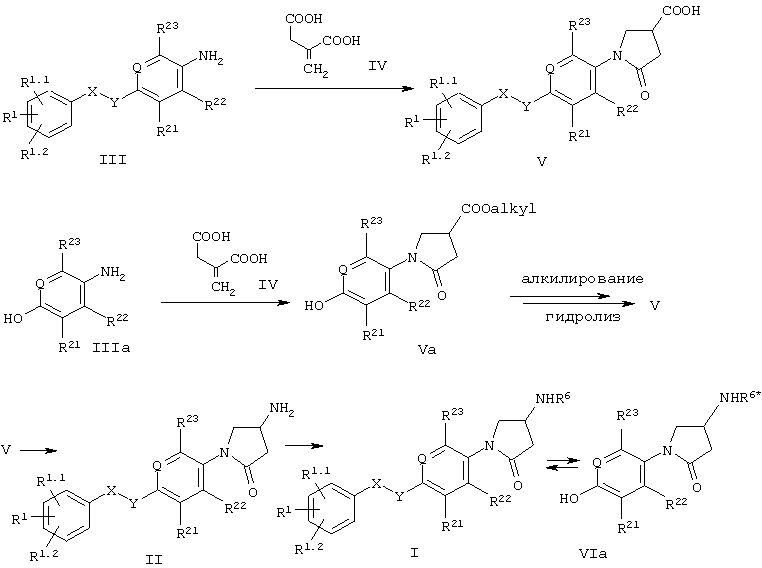
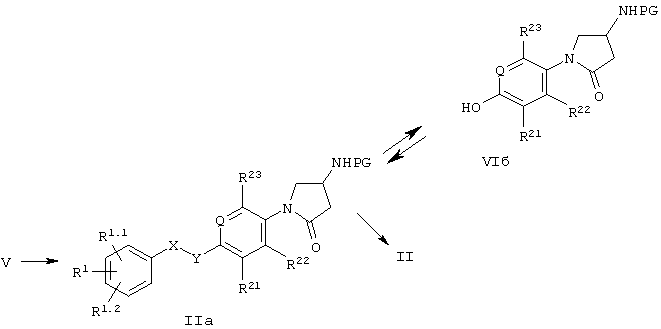
На схеме 1 показаны основные пути получения соединений формулы I. Промежуточные соединения III и IIIa могут быть введены в реакцию с неразбавленной итаконовой кислотой IV в диапазоне температур от 80°С до 200°С.
Соединения формулы Va могут быть подвергнуты алкилированию посредством синтеза простых эфиров по Вильямсону с использованием незамещенного или замещенного бензильного производного, выбранного из бензилгалогенидов, тозилатов, метансульфонатов (мезилатов) и трифторметансульфонатов (трифлатов). Используемыми при этом основаниями могут быть, например, алкоголяты или карбонаты, такие как карбонаты натрия, калия или цезия. Предпочтительными растворителями являются низшие спирты, ацетонитрил или низшие кетоны в температурном диапазоне от 20°С до температуры кипения с обратным холодильником.
Другим подходом является конденсация по Мицунобу: необязательно замещенный бензиловый спирт вводят в реакцию с соединением формулы Va в инертном растворителе, например, диэтиловом эфире или тетрагидрофуране, добавляя диалкилазодикарбоксилаты в присутствии фосфинов, например, трибутил- или трифенилфосфина. Гидролиз соединений формулы Va может быть осуществлен общепринятыми способами, такими как гидролиз в кислотных условиях, например с помощью соляной кислоты, или в основных условиях, например с помощью гидроксидов лития, натрия или калия в смесях со спиртами и водой в качестве растворителя.
Соединения формулы II и IIa могут быть получены исходя из кислотных производных формулы V путем нуклеофильных миграций от атома углерода к атому азота, таких как, например, перегруппировка Гофмана или Курциуса, через образование соответствующего изоцианата. Последующая обработка изоцианата водными растворами кислот приводит непосредственно к аминам формулы II. Обработка образующегося на промежуточной стадии изоцианата соответствующими спиртами приводит к защищенным аминопроизводным формулы IIa. Для обработки изоцианата выбирают такие спирты, которые приводят к типичным карбаматам, используемым в качестве защитных групп для аминов, например, трет-бутоксикарбонильной, бензилоксикарбонильной или флуоренилметоксикарбонильной группе. Снятие защиты с аминогрупп с целью получения соединений формулы II производят согласно хорошо известным из литературы методикам.
Дальнейшее преобразование полученных соединений в соединения формулы II может быть осуществлено с помощью стандартных способов, таких как, например, реакции с активированными ацильными производными, например, ацилгалогенидами или ангидридами, или реакций конденсации кислот с использованием, например, карбодиимидов в качестве конденсирующего агента, или реакции с изоцианатами.
В тех соединениях формулы I или IIa, где -X-Y- обозначает -СН2-О-, необязательно замещенный бензильный остаток может выступать в качестве промежуточной группы, которая может быть расщеплена посредством гидрогенолиза. Получаемые в результате соединения формулы VIa или VIб могут быть снова подвергнуты алкилированию с использованием другой бензильной группы в упомянутых выше условиях. Как известно специалистам в данной области, этот процесс возможен только при том условии, что R6* и PG (защитная группа) являются группами, устойчивыми в упомянутых выше условиях реакции по отношению к гидрогенолизу и реакции алкилирования.
Схема 1
Другой способ получения соединений формулы I включает реакции кросс-сочетания арилстаннанов [Lam и др.. Tetrahedron Lett., т.43, с.3091 (2002)], арилборонатов [Lam и др., Synlett, т.5, с.674 (2000); Chan и др., Tetrahedron Lett., т.39, с.2933 (1998)] или арилгалогенидов [Buchwald и др., J. Amer. Chem. Soc., т.118, с.7215 (1996)] с соответствущими пирролидонами (схема 2)
Схема 2
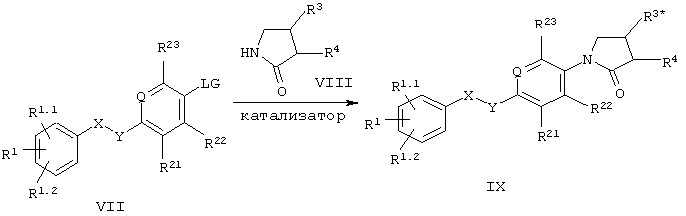

где LG обозначает уходящую группу, например, галоид, например, хлор, бром или иод, или SnR3 или В(ОН)2, a R3* обозначает -NHR6 или алкоксикарбонильную группу.
Согласно настоящему изобретению, возможность получения соединений общей формулы III, где -X-Y- является -СН2-О-, т.е. соединений формулы IIIб, представлена на схеме 3. Промежуточные соединения формулы XII могут быть получены путем нуклеофильного замещения исходя из ароматических нитросоединений формулы XI, содержащих уходящие группы в параположении, с помощью бензиловых спиртов формулы X. Примерами уходящих групп в параположении являются галоиды (F, Cl, Br, I), тозилаты, мезилаты или трифлаты. Эти реакции замещения могут быть осуществлены как без растворителя, так и в инертных растворителях, подобных, например, толуолу или ксилолу. Предпочтительная температура проведения реакции лежит в диапазоне от 50°С до 150°С. Альтернативно, соединения формулы XII могут быть получены путем синтеза простых эфиров по Вильямсону исходя из n-нитрофенолов формулы XIV и бензилгалогенидов, тозилатов, мезилатов или трифлатов формулы XIII. Используемыми при этом основаниями могут быть, например, алкоголяты или карбонаты (карбонат натрия, калия или цезия). Предпочтительными растворителями являются низшие спирты, ацетонитрил или низшие кетоны при температуре в диапазоне от 20°С до температуры кипения с обратным холодильником. Другим подходом является конденсация по Мицунобу бензиловых спиртов с n-нитрофенолами формулы XIV. Реакцию, как обычно, проводят в инертных растворителях, таких как, например, диэтиловый эфир или тетрагидрофуран, используя диалкилазодикарбоксилаты в присутствии фосфинов, например, трибутил- или трифенилфосфина.
Ключевые промежуточные соединения формулы XII восстанавливают до аминосоединений IIIб путем каталитического гидрирования, например, при использовании в качестве катализатора платины на угле в низших спиртах, этилацетате или тетрагидрофуране. Альтернативным способом является восстановление нитрогруппы металлами, такими как железо, олово или цинк, в кислой среде, такой как разбавленная соляная кислота или уксусная кислота. Металлы также могут быть заменены солями металлов, например, хлоридом олова (II).
Схема 3
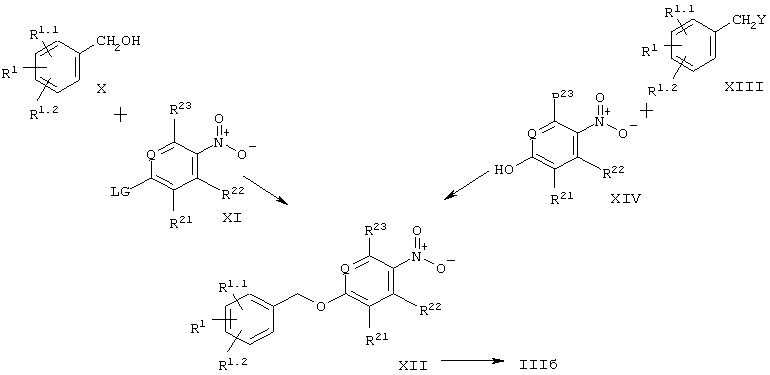 где LG обозначает уходящую группу, например, галоид, OTf и т.д., a Y также обозначает уходящую группу, например, галоид, OTf и т.д. или ОН (в случае конденсации по Мицунобу).
где LG обозначает уходящую группу, например, галоид, OTf и т.д., a Y также обозначает уходящую группу, например, галоид, OTf и т.д. или ОН (в случае конденсации по Мицунобу).
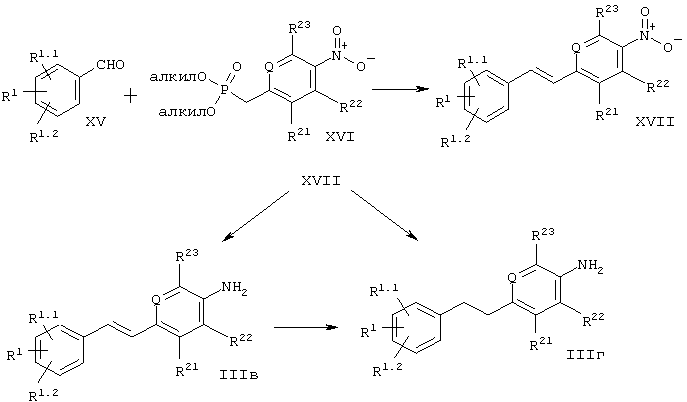
Промежуточные соединения формулы III, где -X-Y- является -СН=СН-, т.е. соединение формулы IIIв, или где -X-Y- является -СН2-СН2-, т.е. соединение формулы IIIг, могут быть получены с помощью способа, представленного на схеме 4. Промежуточные соединения формулы XVII могут быть получены по реакции олефинизации необязательно замещенных ароматических альдегидов формулы XV с помощью диалкил-(4-нитробензил)фосфонатов формулы XVI в присутствии основания, например, гидрида натрия, приводящей к соответствующим нитроолефинам формулы XVII.
Ключевые промежуточные соединения формулы XVII могут быть селективно восстановлены до аминоолефина формулы IIIв с помощью металлов или солей металлов, таких как, например, хлорид олова (II), или посредством каталитического гидрирования, такого как, например, гидрирование с использованием платины на угле в качестве катализатора и низших спиртов, этилацетата или тетрагидрофурана в качестве растворителя. Аминопроизводные формулы IIIг могут быть получены из нитропроизводных формулы XVII или аминоолефинов формулы IIIв путем гидрирования с использованием палладия на угле в качестве катализатора и низших спиртов, этилацетата или тетрагидрофурана в качестве растворителя.
Схема 4
Соединения общей формулы I могут также существовать в оптически чистой форме. Разделение на энантиомеры может быть осуществлено с помощью общепринятых способов: или на ранней стадии синтеза, исходя из соединений формулы V, путем образования соли с оптически активным амином, таким как, например, (+)- или (-)-1-фенилэтиламин или (+)- или (-)-1-нафтилэтиламин, и разделения диастереомерных солей посредством фракционной кристаллизации; или путем образования производного с хиральным вспомогательным соединением, таким как, например, (+)- или (-)-2-бутанол, (+)- или (-)-1-фенилэтанол или (+)- или (-)-ментол, и разделения диастереомерных продуктов с помощью хроматографии и/или кристаллизации с последующим разрывом связи со вспомогательным хиральным соединением; или на самой последней стадии синтеза путем разделения энантиомеров формулы I с помощью хиральной хроматографии. Помимо этого, соединения формулы I также могут быть получены исходя из энантиомерно чистых интермедиатов, полученных с помощью биотрансформации, например, путем гидролиза сложных эфиров формулы Va ферментами, такими, как, например, холестераза, получаемая из Candida cylindracea. Определение абсолютной конфигурации получаемого производного пирролидинона можно осуществить посредством анализа чистых диастереомерных солей или производных общепринятыми спектроскопическими методами, среди которых наиболее подходящим является рентгеновская спектроскопия монокристаллов.
Соединения формулы I являются, как уже было упомянуто, ингибиторами моноаминоксидазы В и могут быть применены для лечения или профилактики заболеваний в тех случаях, когда ингибиторы МАО-В могли бы быть полезны. Подобные случаи включают острые и хронические неврологические расстройства, расстройства познавательной функции и расстройства памяти. К показанным к лечению неврологическим расстройствам относятся, например, травматические или хронические дегенеративные процессы в нервной системе, такие как болезнь Альцгеймера, другие виды слабоумия, снижение познавательной функции до минимума или болезнь Паркинсона. Другие показания включают психиатрические заболевания, такие как депрессия, беспокойство, беспричинное беспокойство, социофобия, шизофрения, расстройства питания и метаболические расстройства, такие как ожирение, а также профилактика и лечение абстинентных синдромов, вызванных злоупотреблением алкоголем, никотином и другими вырабатывающими привыкание средствами. Среди других показаний к лечению могут быть периферийная невропатия, вызванная химиотерапией рака (WO 97/33572), синдром недостатка оценки (WO 01/34172) или лечение рассеянного склероза (WO 96/40095) и других воспалительных неврологических заболеваний.
Соединения формулы I особенно полезны при лечении и профилактике болезни Альцгеймера и старческого слабоумия.
Фармакологическую активность соединений исследовали с помощью следующей методики.
Молекулы комплементарной ДНК, кодирующие МАО-А и МАО-В, временно трансфектировали в клетки ядерного антигена вируса Эпштейна-Барра (EBNA) согласно процедуре, описанной Schlaeger и Christensen (Cytotechnology, т.15, cc.1-13, (1998). После трансфекции клетки гомогенизировали с помощью гомогенизатора Polytron в 20 мМ буферном растворе трис-HCl, рН 8,0, содержащем 0,5 мМ этиленгликольтетрауксусной кислоты (ЭГТК) и 0,5 мМ фторангидрида фенилметансульфокислоты. Клеточные мембраны отделяли центрифугированием при 45000 xg, и после двух стадий промывания с помощью 20 мМ буферного раствора трис-HCl, рН 8,0, содержащего 0,5 мМ ЭГТК, мембраны снова суспендировали в вышеуказанном буферном растворе и сохраняли в виде аликвот при -80°С до дальнейшего использования.
Ферментативную активность МАО-А и МАО-В анализировали в 96-ячеечных планшетах посредством спектрофотометрического анализа, заимствованного из способа, описанного М.Zhou и N.Panchuk-Voloshina (Analytical Biochemistry, т.253, cc.159-164, (1997). В кратком изложении, аликвоты мембран инкубировали в буферном растворе, содержащем 0,1 М фосфата калия, рН 7,4, и различные концентрации соединений в течение 30 минут при 37°С. По истечении этого периода инициировали ферментативную реакцию путем добавления тирамина в качестве субстрата МАО совместно с 1 Ед./мл пероксидазы хрена (Roche Biochemicals) и 80 мкМ N-ацетил-3,7-дигидроксифеноксазина (Amplex Red, Molecular Probes). Образцы инкубировали еще в течение 30 минут при 37°С и окончательном объеме 200 мкл, после чего определяли поглощение при длине волны 570 нм с помощью считывающего устройства для планшетов SpectraMax (Molecular Devices). Фоновое (неспецифическое) поглощение определяли в присутствии 10 мкМ клоргилина в случае МАО-А или 10 мкМ L-депренила в случае МАО-В. Величины IC50 (концентрации вещества, обеспечивающей 50% ингибирование) определяли по кривым ингибирования, полученным исходя из девяти концентраций ингибитора в параллельных опытах, путем аппроксимации данных четырехпараметрическим логистическим уравнением с помощью компьютерной программы.
Соединения по настоящему изобретению являются специфическими ингибиторами МАО-В. Значения IC50 предпочтительных соединений формулы I, измеренные в ходе вышеописанного анализа, находятся в пределах 1 мкМ или меньшей величины, как правило, в пределах 0,1 мкМ или меньшей величины, в идеальном случае 0,02 мкМ или меньшей величины.
Соединения формулы I могут применяться в качестве лекарственных средств, например в виде фармацевтических препаратов. Фармацевтические препараты могут быть введены перорально, например в форме таблеток, таблеток в оболочке, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Однако введение может также быть осуществлено через прямую кишку, например в форме суппозиториев, или парентерально, например в форме растворов для инъекций.
Соединения формулы I могут обрабатываться совместно с фармацевтически инертными неорганическими или органическими носителями для производства фармацевтических препаратов. Лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли и другие подобные вещества могут применяться, например, в качестве подобных носителей для таблеток, таблеток в оболочке, драже и твердых желатиновых капсул. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и другие подобные вещества; однако, в зависимости от природы активной субстанции в случае мягких желатиновых капсул обычно не требуется никаких носителей. Подходящими носителями для производства растворов и сиропов являются, например, вода, полиолы, сахароза, инвертный сахар, глюкоза и другие подобные вещества. Адъюванты, такие как спирты, полиолы, глицерин, растительные масла и другие подобные соединения, могут быть использованы для приготовления водных растворов для инъекций водорастворимых солей соединений формулы I, но, как правило, не являются необходимыми. Подходящими носителями для суппозиториев являются, например, натуральные или отвержденные масла, воски, жиры, полужидкие или жидкие полиолы и другие подобные вещества.
Кроме того, фармацевтические препараты могут содержать консерванты, солюбилизаторы, стабилизаторы, смачивающие средства, эмульгаторы, подсластители, красители, ароматизаторы, соли для варьирования осмотического давления, буферные растворы, маскирующие средства или антиоксиданты. Они могут также содержать другие терапевтически полезные вещества.
Как было упомянуто ранее, лекарственные средства, содержащие соединение формулы I и терапевтически инертный наполнитель, также являются объектом настоящего изобретения, равно как и способ производства подобных лекарственных средств, включающий превращение одного или нескольких соединений формулы I и, если это требуется, одного или нескольких других терапевтически полезных веществ в дозированную галеновую форму совместно с одним или несколькими терапевтически инертными носителями.
Дозировка может варьироваться в широких пределах и будет, конечно, приспосабливаться к индивидуальным требованиям в каждом конкретном случае. В общем, эффективная дозировка для перорального или парентерального введения находится в пределах 0,01-20 мг/(кг·день), причем дозировка в пределах 0,1-10 мг/(кг·день) является предпочтительной для всех описанных показаний. Суточная дозировка для взрослого человека с массой тела 70 кг находится, соответственно, в пределах 0,7-1400 мг в сутки, предпочтительно от 7 до 700 мг в сутки.
Следующие примеры даны для иллюстрации настоящего изобретения. Их следует рассматривать не как ограничивающие объем изобретения, а как представляющие изобретение. Аббревиатура «КТ» означает «комнатная температура».
Пример 1: (RS)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
а) (RS)-1-(4-бензилоксифенил)-5-оксопирролидин-3-карбоновая кислота
Смешивают 18,8 г (94,4 ммоль) 4-бензилоксианилина с 12,28 г (94,4 ммоль) итаконовой кислоты. Твердую смесь нагревают до температуры 130°С. Через 20 минут расплавленный материал затвердевает. После охлаждения полученное твердое вещество растирают с этилацетатом, что приводит к 28,26 г (96% от теоретического) (RS)-1-(4-бензилоксифенил)-5-оксопирролидин-3-карбоновой кислоты в виде сероватого твердого вещества. Масс-спектр (МС): m/e=311 (М)+.
б) (RS)-1-[4-бензилоксифенил]-5-оксопирролидинкарбонилхлорид
Суспензию 9,50 г (30,5 ммоль) (RS)-1-[4-(3-бензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты в 100 мл дихлорметана обрабатывают 13,3 мл (183 ммоль) тионилхлорида при КТ в течение 18 часов. Для получения конечного продукта реакционную смесь упаривают досуха при пониженном давлении, затем диспергируют остаток в толуоле и снова упаривают досуха, что приводит к количественному выходу (RS)-1-[4-бензилоксифенил]-5-оксопирролидин-карбонилхлорида в виде желтоватого твердого вещества, которое используют на следующей стадии без дополнительной очистки и характеризации.
в) Трет-бутиловый эфир (RS)-[1-(4-бензилоксифенил)-5-оксопирролидин-3-ил]карбаминовой кислоты
Раствор 0,20 г (0,6 ммоль) (RS)-1-(4-бензилоксифенил)-5-оксопирролидин-3-карбонилхлорида в 12 мл толуола охлаждают до 0°С и добавляют 0,058 г (0,9 ммоль) азида натрия. Реакционной смеси дают нагреться до КТ и продолжают перемешивание в течение 1 часа. После этого смесь нагревают до температуры 80°С, добавляют 1,88 мл (20 ммоль) трет-бутанола и продолжают перемешивание еще в течение 1 часа. Для получения конечного продукта смесь охлаждают, разбавляют этилацетатом и последовательно экстрагируют насыщенным раствором бикарбоната натрия, водой и соляным раствором. Органическую фазу сушат над сульфатом натрия и упаривают при пониженном давлении, что приводит к неочищенному соединению в виде коричневатого твердого вещества. Полученный материал подвергают хроматографической очистке на силикагеле, используя смесь 2:1 н-гексана и этилацетата в качестве элюента. Получают 0,13 г (55% от теоретического) трет-бутилового эфира (RS)-[1-(4-бензилоксифенил)-5-оксопирролидин-3-ил]карбаминовой кислоты в виде твердого вещества белого цвета. МС: m/e=400 (М+NH4)+.
г) Трет-бутиловый эфир (RS)-[1-(4-гидроксифенил)-5-оксопирролидин-3-ил]карбаминовой кислоты
Раствор 82 мг (0,2 ммоль) трет-бутилового эфира (RS)-[1-(4-бензилоксифенил)-5-оксопирролидин-3-ил]карбаминовой кислоты в 2 мл тетрагидрофурана (ТГФ) гидрируют в присутствии 7 мг палладия на угле (10%) при давлении окружающей среды и КТ в течение 18 часов. Для получения конечного продукта реакционную смесь фильтруют над дикалитом, а затем упаривают при пониженном давлении. Неочищенный трет-бутиловый эфир (RS)-[1-(4-гидроксифенил)-5-оксопирролидин-3-ил]карбаминовой кислоты получают в виде бесцветного маслянистого вещества, которое непосредственно используют на следующей стадии без дополнительной очистки и характеризации.
д) Трет-бутиловый эфир (RS)-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}карбаминовой кислоты
Раствор 62 мг (0,21 ммоль) неочищенного трет-бутилового эфира (RS)-[1-(4-гидроксифенил)-5-оксопирролидин-3-ил]карбаминовой кислоты в 3 мл 2-бутанона обрабатывают 0,031 мл (0,23 ммоль) 3-фторбензилбромида и 59 мг (0,42 ммоль) карбоната калия и перемешивают смесь при температуре 50°С в течение 18 часов. Для получения конечного продукта реакционную смесь разбавляют этилацетатом и экстрагируют водой. Органическую фазу сушат над сульфатом натрия и упаривают при пониженном давлении. Полученный материал подвергают хроматографической очистке на силикагеле, используя смесь 2:1 смесь н-гексана и этилацетата в качестве элюента. Получают 61 мг (72% от теоретического) трет-бутилового эфира (RS)-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}-карбаминовой кислоты в виде твердого вещества белого цвета. МС: m/e=401 (М+Н)+.
е) Гидрохлорид (RS)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она
Раствор 49 мг (0,12 ммоль) трет-бутиловото эфира (RS)-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}карбаминовой кислоты в 1 мл диоксана обрабатывают 0,10 мл соляной кислоты (37%). Желтоватый раствор нагревают до температуры 45°С в течение 1 часа. Для получения конечного продукта реакционную смесь упаривают при пониженном давлении и растирают твердый остаток с диэтиловым эфиром. После фильтрования и высушивания получают 33 мг (79% от теоретического) гидрохлорида (RS)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она в виде твердого вещества белого цвета. МС: m/e=301 (М+Н)+.
ж) (RS)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
Раствор 25 мг (0,07 ммоль) гидрохлорида (RS)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она в 1 мл хлористого метилена обрабатывают 22 мкл (0,16 ммоль) триэтиламина и охлаждают до температуры 0°С. К получаемому раствору добавляют 6 мкл (0,08 ммоль) ацетилхлорида и продолжают перемешивание при температуре 0°С в течение 30 минут. Для получения конечного продукта реакционную смесь обрабатывают 2 мл раствора гидроксида аммония, органическую фазу отделяют, а затем сушат над сульфатом натрия и упаривают при пониженном давлении. Полученный материал подвергают хроматографической очистке на силикагеле, используя смесь 95:5 хлористого метилена и метанола в качестве элюента. Получают 20 мг (78% от теоретического) (RS)-14-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамида в виде твердого вещества белого цвета. МС: m/e=343 (М+Н)+.
Пример 2: (S)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
a) (RS)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновая кислота
В металлической чашечке смешивают в твердом виде 257,0 г (2,355 моль) 4-аминофенола и 301,75 г (2,32 моль) итаконовой кислоты. Смесь осторожно нагревают на плитке при перемешивании металлическим шпателем. При температуре 110-120°С при кипении начинается эндотермическая реакция, а при подъеме температуры до 150°С реакционная масса превращается в бежевое твердое вещество. Продукт песочного цвета оставляют охлаждаться до КТ в течение 1-2 часов. Неочищенную (RS)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновую кислоту используют на следующей стадии без дополнительной очистки и характеристики.
б) Метиловый эфир (RS)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновой кислоты
В десятилитровой четырехгорлой колбе, оборудованной дефлегматором, термометром и механической мешалкой, растворяют неочищенную (RS)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновую кислоту в смеси 5000 мл метанола, 24 мл концентрированной серной кислоты и 400 мл 2,2-диметоксипропана и перемешивают при кипячении с обратным холодильником в течение 2 часов. Для получения конечного продукта реакционный раствор доводят до половины исходного объема посредством перегонки, а затем переносят в двадцатилитровый сосуд. Добавляют при перемешивании при температуре 40°С 2500 мл смеси льда с водой (1:1). Немедленно начинается кристаллизация, образующиеся в результате которой мелкие белые кристаллы собирают на фильтровальной воронке. Их промывают холодной водой общим количеством 2000 мл до обесцвечивания фильтрата и приобретения им нейтральной реакции. Хорошо спрессованный и предварительно высушенный продукт на фильтровальной воронке сушат при пониженном давлении, что приводит к 980 г (84% от теоретического, 2 стадии) метилового эфира (RS)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновой кислоты в виде твердого вещества белого цвета; МС: m/e=234 (М+Н)+.
в) Метиловый эфир (R)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновой кислоты и (S)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновая кислота
Аккуратно перемешивают суспензию 50,22 г (213,5 ммоль) метилового эфира (RS)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновой кислоты (98% HPLC) в 500 мл циклогексана. При добавлении 2,0 л 0,1М раствора хлорида натрия, 50 мМ раствора сульфата магния, 3 мМ буферного раствора фосфата калия рН 6,0 образуется эмульсия/суспензия, рН которой доводят до 6,0. Доводят температуру до 30°С. Гидролиз инициируют путем добавления 201 мг холестеразы из Candida cylindracea (Roche Applied Science, Industrial Products, Enzyme Projects, Sandhofer Str. 116, D-68305, Mannheim, Germany, заказ №10129046103) и выдерживают постоянное значение рН 6,0 путем контролируемого добавления 0,1 н. раствора NaOH (постоянный рН) при аккуратном перемешивании. После добавления титрующего агента общим количеством 1016 мл (в течение ночи; степень превращения 48,6%) реакционную смесь эктрагируют 3,5 л и 2×2,5 л хлористого метилена, а затем 3,5 л этилацетата. Объединенные растворы в хлористом метилене сушат над сульфатом натрия, упаривают и сушат под высоким вакуумом, что приводит к 22,5 г (95,6 ммоль; 44,8%) метилового эфира (R)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновой кислоты в виде белых кристаллов; чистота: ВЭЖХ: >99%; оптическая чистота: 96,3%, исключая ошибки; [α]D=-27,7° (с=1,02; этанол); МС: 235,1.
Водную фазу доводят до рН 2,2 с помощью 32% соляной кислоты и экстрагируют 3×3,5 л этилацетата. Объединенные органические фазы сушат над сульфатом натрия, упаривают и сушат под высоким вакуумом, что приводит к 21,9 г (99,0 ммоль; 46,4%) (S)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновой кислоты; чистота: ВЭЖХ: >99%; оптическая чистота 99,1%, исключая ошибки; [α]D=25,4° (с=1,05; этанол); МС: 221,1.
г) Метиловый эфир (S)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновой кислоты
Смесь 26 г (117,5 ммоль) (S)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновой кислоты, 0,66 мл серной кислоты и 100 мл диметоксипропана в 700 мл метанола нагревают до кипения с обратным холодильником в течение 3 часов. Для получения конечного продукта реакционную смесь уменьшают в объеме до 4/5 исходного объема, а затем добавляют остаток при перемешивании к смеси льда и воды. Выпавший в осадок продукт собирают на фильтровальной воронке, промывают холодной водой и сушат под высоким вакуумом, что приводит к 23,7 г (86% от теоретического) метилового эфира (S)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновой кислоты в виде твердого вещества белого цвета; МС: m/e=234 (M-H)+; оптическая чистота 97,4%, исключая ошибки.
д) Метиловый эфир (S)-1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты
Раствор 14,23 г (110,6 ммоль) 3-фторбензилового спирта и 27,19 г (108,8 ммоль) трифенилфосфина в 150 мл тетрагидрофурана добавляют по каплям в течение 50 минут в атмосфере азота при температуре 0°С к раствору 23,65 г (100,5 ммоль) метилового эфира (S)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновой кислоты и 21,62 г (100,5 ммоль) диизопропилазодикарбоксилата в 200 мл тетрагидрофурана. Смесь оставляют нагреваться до КТ и продолжают перемешивание в течение 18 часов. Для получения конечного продукта смесь упаривают при пониженном давлении. Твердый остаток растирают в 400 мл диэтилового эфира, после чего остается твердое вещество белого цвета, состоящее в основном из желаемого продукта и трифенилфосфиноксида. После фильтрования твердый материал растирают в 100 мл холодного метанола, что приводит к 23,5 г (68% от теоретического) метилового эфира (S)-1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты в виде твердого вещества белого цвета [МС: m/e=344 (M+H)+] совместно со следовыми количествами трифенилфосфина и диизопропилгидразодикарбоксилата.
е) (S)-1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновая кислота
Раствор 25,61 г (74,6 ммоль) метилового эфира (S)-1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты в 650 мл диоксана обрабатывают 175 мл соляной кислоты (37%). Смесь нагревают до 50°С в течение 18 часов в закрытой колбе. Для получения конечного продукта раствор упаривают при пониженном давлении, что приводит к неочищенной кислоте в виде твердого вещества желтого цвета. Для очистки неочищенную кислоту растирают при температуре 0°С в 50 мл этилацетата. Твердое вещество собирают на фильтровальной воронке, а затем сушат под высоким вакуумом, что приводит к 20,3 г (82% от теоретического) (S)-1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты в виде желтоватого твердого вещества; МС: m/e=330 (М+Н)+.
ж) Гидрохлорид (S)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она
Раствор 20,0 г (61 ммоль) (S)-1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты в 300 мл диоксана обрабатывают 6,7 мл (61 ммоль) N-метилморфолина. После этого охлаждают реакционную смесь до температуры -8°С и добавляют 8,14 мл (61 ммоль) изобутилового эфира хлормуравьиной кислоты. После перемешивания в течение 5 минут добавляют раствор 7,98 г (121 ммоль) азида натрия в 40 мл воды при повышении температуры до 0°С. После перемешивания в течение 70 минут при температуре 0°С суспензию фильтруют над дикалитом. Фильтрат разбавляют 700 мл толуола и переносят в делительную воронку. Органический слой отделяют, затем дважды промывают 250 мл насыщенного раствора бикарбоната натрия и дважды - 200 мл насыщенного раствора хлорида натрия. После этого органический слой сушат над сульфатом натрия и после добавления 400 мл толуола растворитель и остаточный изобутиловый спирт упаривают до достижения объема 350 мл. Раствор постепенно нагревают до температуры 80°С и выдерживают при этой температуре в течение 70 минут. После охлаждения раствор образующегося на промежуточной стадии изоцианата концентрируют до достижения объема около 300 мл и прибавляют по каплям к раствору 25,4 мл соляной кислоты (37%) в 100 мл диоксана при нагревании до температуры 45°С. После окончания прибавления температуру повышают до 60°С в течение 1 часа, причем гидрохлорид уже начинает осаждаться. Смесь охлаждают до температуры 0°С и собирают образующийся твердый материал на фильтровальной воронке. После промывания трет-бутилметиловым эфиром продукт сушат под высоким вакуумом. Получают 14,6 г (71% от теоретического) гидрохлорида (S)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она в виде твердого вещества белого цвета. МС: m/e=301 (М+Н)+.
з) (S)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
Аналогично тому, как это было описано в примере 1ж), ацетилирование гидрохлорида (S)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она приводит к образованию (S)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамида в виде кристаллического твердого вещества белого цвета. МС: m/e=343 (М+Н)+.
Пример 3: (R)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
а) Метиловый эфир (R)-1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты
Аналогично тому, как это описано в примере 2д), алкилирование метилового эфира (R)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновой кислоты [пример 2в)] 3-фторбензиловым спиртом приводит к метиловому эфиру (R)-1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты в виде твердого вещества белого цвета. МС: m/e=344 (М+Н)+.
б) (R)-1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновая кислота
Аналогично тому, как это описано в примере 2е), гидролиз метилового эфира (R)-1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты соляной кислотой (37%) в диоксане приводит к (R)-1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоте в виде твердого вещества белого цвета. МС: m/e=330 (М+Н)+.
в) Гидрохлорид (R)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она
Аналогично тому, как это описано в примере 2ж), перегруппировка Курциуса (R)-1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты приводит к гидрохлориду (R)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она в виде твердого вещества белого цвета. МС: m/e=301 (M+H)+.
г) (R)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил} ацетамид
Аналогично тому, как это описано в примере 1ж), ацетилирование гидрохлорида (R)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она приводит к (R)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамиду в виде кристаллического твердого вещества белого цвета. МС: m/e=343 (М+Н)+.
Пример 4: (RS)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил)формамид
Приготовляют смесь 190 мг (1,9 ммоль) уксусного ангидрида и 108 мг (2,3 ммоль) муравьиной кислоты при температуре 0°С, затем нагревают ее до температуры 60°С в течение 2 часов. После охлаждения до КТ раствор разбавляют 1 мл безводного тетрагидрофурана, а затем добавляют раствор 215 мг (0,7 ммоль) (RS)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она [пример 1е)] в 2 мл хлористого метилена (амин получают из соответствующего гидрохлорида посредством обработки триэтиламином и экстракции из смеси хлористого метилена и воды). Образующуюся суспензию перемешивают в течение 1 часа. Для получения конечного продукта реакционную смесь разбавляют хлористым метиленом и дважды промывают водой. Органический слой отделяют, сушат над сульфатом натрия и упаривают. Получают 126 мг (54% от теоретического) (RS)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}формамида в виде твердого вещества белого цвета. МС: m/e=329 (М+Н)+.
Пример 5: (S)-N-{1-[4-(3-Фторбензилокси)фенил]-5-оксопирролидин-3-ил}формамид
Аналогично тому, как это описано в примере 4, ацилирование (S)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она [пример 2ж)] приводит к (S)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}формамиду в виде полутвердого вещества белого цвета (выход 81% от теоретического). МС: m/e=329 (М+Н)+.
Пример 6: (R)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}формамид
Аналогично тому, как это описано в примере 4, ацилирование (R)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она [пример 3в)] приводит к (R)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}формамиду в виде твердого вещества светло-желтого цвета (выход 94% от теоретического). МС: m/e=329 (М+Н)+.
Пример 7: Метиловый эфир (RS)-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил)карбаминовой кислоты
Раствор 250 мг (0,74 ммоль) гидрохлорида (RS)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она [Пример 1е)] в 12 мл хлористого метилена охлаждают до температуры 0°С и последовательно обрабатывают 226 мкл (1,6 ммоль) триэтиламина и 64 мкл (0,8 ммоль) метилового эфира хлормуравьиной кислоты. Смесь оставляют нагреваться до КТ и перемешивают в течение 1 часа. Для получения конечного продукта к реакционной смеси добавляют хлористый метилен и воду. Органический слой отделяют, сушат над сульфатом натрия и упаривают. Получают 203 мг (76% от теоретического) метилового эфира (RS)-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}карбаминовой кислоты в виде твердого вещества белого цвета. МС: m/e=359 (M+H)+.
Пример 8: (RS)-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}мочевина
Раствор 250 мг (0,74 ммоль) гидрохлорида (RS)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она [пример 1е)] в 2 мл N,N-диметилформамида охлаждают до температуры 0°С и последовательно обрабатывают 386 мкл (2,2 ммоль) N-этилдиизопропиламина и 307 мкл (2,2 ммоль) триметилсилилизоцианата. Смесь оставляют нагреваться до КТ и перемешивают в течение 4 часов. Для получения конечного продукта реакционную смесь упаривают при пониженном давлении. Красный остаток растворяют в хлористом метилене и промывают органическую фазу водой. После отделения органического слоя и высушивания его над сульфатом натрия его упаривают, что приводит к маслянистому веществу красного цвета. Неочищенный продукт подвергают хроматографической очистке на силикагеле с градиентом смеси от 95:5 к 90:10 хлористого метилена и метанола в качестве элюента. После хроматографической очистки продукт дополнительно растирают в этилацетате и бикарбонате натрия при КТ. Получают 153 мг (60% от теоретического) (RS)-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}мочевины в виде твердого вещества белого цвета. МС: m/e=344 (М+Н)+.
Пример 9: (RS)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}метансульфонамид
Раствор 250 мг (0,74 ммоль) гидрохлорида (RS)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она [пример 1е)] в 8 мл хлористого метилена охлаждают до температуры 0°С и последовательно обрабатывают 226 мкл (2,2 ммоль) триэтиламина и 64 мкл (2,2 ммоль) метансульфохлорида. Смесь перемешивают в течение 30 минут при температуре 0°С. Для получения конечного продукта реакционную смесь дважды промывают водой, отделяют органический слой и сушат его над сульфатом натрия. После выпаривания растворителя неочищенный материал подвергают хроматографической очистке на силикагеле, используя смесь 98:2 хлористого метилена и метанола в качестве элюента. Получают 235 мг (84% от теоретического) (RS)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}метансульфонамида в виде твердого вещества белого цвета. МС: m/e=377 (M-Н)+.
Пример 10: (S)-2-фтор-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
Раствор 100 мг (0,3 ммоль) гидрохлорида (S)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она [пример 2ж)] в 0,3 мл N,N-диметилформамида последовательно обрабатывают 100 мкл (0,6 ммоль) N-этилдиизопропиламина и 55 мкл (0,6 ммоль) метил фторацетата. Образующуюся в результате бежевую суспензию нагревают до температуры 50°С в течение 18 часов. Для получения конечного продукта реакционную смесь упаривают, после чего остаток растворяют в хлористом метилене и промывают раствор 1 мл соляной кислоты (1 н.). Органический слой отделяют, сушат над сульфатом натрия и упаривают. Неочищенный продукт подвергают хроматографической очистке на силикагеле, используя смесь 98:2 хлористого метилена и метанола в качестве элюента. Получают 30 мг (28% от теоретического) (S)-2-фтор-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамида в виде твердого вещества белого цвета. МС: m/e=378 (M+NH4)+.
Пример 11: (S)-2,2-дифтор-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
Раствор 103 мг (0,3 ммоль) гидрохлорида (S)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она [Пример 2ж)] в 0,5 мл N,N-диметилформамида последовательно обрабатывают 180 мкл (1,0 ммоль) N-этилдиизопропиламина, 20 мкл (0,3 ммоль) дифторуксусной кислоты и 102 мг (0,3 ммоль) тетрафторбората O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU) при КТ, после чего перемешивают в течение 6 часов. Для получения конечного продукта реакционную смесь упаривают при пониженном давлении. Получаемый в результате остаток растворяют в 3 мл хлористого метилена и промывают раствор 1,5 мл насыщенного раствора бикарбоната натрия и 1,5 мл соляной кислоты (0,1 н.). Органическую фазу сушат над сульфатом натрия и упаривают. Неочищенный материал подвергают хроматографической очистке на силикагеле, используя смесь 98:2 хлористого метилена и метанола в качестве элюента. Получают 21 мг (18% от теоретического) (S)-2,2-дифтор-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамида в виде твердого вещества белого цвета. МС: m/e=396 (M+NH4)+.
Пример 12: (S)-2,2,2-трифтор-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
Раствор 10 мг (0,3 ммоль) гидрохлорида (S)-4-амино-1-[4-(3-фторбензилокси)фенил]пирролидин-2-она [Пример 2ж)] в 2,5 мл хлористого метилена охлаждают до температуры 0°С и последовательно обрабатывают 90 мкл (0,6 ммоль) триэтиламина и 50 мкл (0,33 ммоль) трифторуксусного ангидрида. Реакционную смесь оставляют нагреваться до КТ и перемешивают всего в течение 3,5 часов. Для получения конечного продукта реакционную смесь разбавляют 2 мл хлористого метилена. Получаемый в результате раствор дважды промывают 2 мл воды, органический слой отделяют, сушат над сульфатом натрия и упаривают. Неочищенный материал подвергают хроматографической очистке на силикагеле, используя смесь 98:2 хлористого метилена и метанола в качестве элюента. Получают 60 мг (51% от теоретического) (S)-2,2,2-трифтор-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамида в виде твердого вещества белого цвета. МС: m/e=414 (М+NH4)+.
Пример 13: (RS)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
а) Метиловый эфир (RS)-1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты
Раствор 5,0 г (21,3 ммоль) метилового эфира (RS)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновой кислоты [пример 26)] в 200 мл 2-бутанона обрабатывают 3,55 мл (27,6 ммоль) 4-фторбензилбромида и 5,88 г (42,5 ммоль) карбоната калия и перемешивают смесь при температуре 90°С в течение 3 часов. Для получения конечного продукта реакционную смесь разбавляют этилацетатом и экстрагируют водой. Органическую фазу отделяют, сушат над сульфатом натрия и упаривают при пониженном давлении. Полученный материал подвергают хроматографической очистке на силикагеле, используя смесь 98:2 хлористого метилена и метанола в качестве элюента. Получают 7,18 г (98% от теоретического) метилового эфира (RS)-1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты в виде твердого вещества белого цвета. МС: m/e=344 (М+Н)+.
б) (RS)-1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновая кислота
Приготавливают суспензию 7,12 г (20,7 ммоль) метилового эфира (RS)-1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты в 10,3 мл раствора гидроксида натрия (1 н.) и добавляют тетрагидрофуран до получения прозрачного раствора. Смесь нагревают до температуры 50°С в течение 1 часа. Для получения конечного продукта тетрагидрофуран упаривают при пониженном давлении. Получаемую белую суспензию разбавляют водой, а затем фильтруют. Белый продукт обрабатывают толуолом и упаривают при пониженном давлении для удаления основной массы воды. Азеотропную перегонку повторяют троекратно. Получают 5,78 г (85% от теоретического) (RS)-1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты в виде твердого вещества белого цвета.
в) Трет-бутиловый эфир (RS)-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}карбаминовой кислоты
Раствор 5,16 г (15,7 ммоль) (RS)-1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты в 70 мл тетрагидрофурана обрабатывают 1,76 мл (15,7 ммоль) N-метилморфолина. После этого реакционную смесь охлаждают до температуры -10°С и добавляют 2,08 мл (15,7 ммоль) изобутилового эфира хлормуравьиной кислоты. После перемешивания в течение 3 минут добавляют раствор 2,06 г (31,3 ммоль) азида натрия в 10 мл воды при повышении температуры до 0°С. После перемешивания в течение 45 минут при температуре 0°С суспензию разбавляют 200 мл толуола и переносят в делительную воронку. Органический слой дважды промывают 1000 мл насыщенного раствора бикарбоната натрия и дважды - 100 мл насыщенного раствора хлорида натрия. После этого органический слой сушат над сульфатом натрия и упаривают растворитель до достижения объема около 80 мл. Раствор постепенно нагревают до температуры 80°С и выдерживают при этой температуре в течение 30 минут. После этого добавляют 35,3 мл (376 ммоль) трет-бутанола и перемешивают смесь при температуре 80°С в течение 18 часов. Затем отгоняют растворитель при пониженном давлении и подвергают остаток хроматографической очистке на силикагеле, используя смесь 98:2 хлористого метилена и метанола в качестве элюента. Получают 4,82 г (77% от теоретического) трет-бутилового эфира (RS)-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}карбаминовой кислоты в виде твердого вещества белого цвета. МС: m/e=401 (М+Н)+.
г) Гидрохлорид (RS)-4-амино-1-[4-(4-фторбензилокси)фенил]пирролидин-2-она
Аналогично тому, как это описано в примере 1е), расщепление трет-бутоксикарбонильной группы в трет-бутиловом эфире (RS)-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}карбаминовой кислоты в кислой среде приводит к гидрохлориду (RS)-4-амино-1-[4-(4-фторбензилокси)фенил]пирролидин-2-она в виде твердого вещества белого цвета (выход 80% от теоретического). МС: m/e=301 (М+Н)+.
д) (RS)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
Аналогично тому, как это описано в примере 1ж), ацетилирование (R)-4-амино-1-[4-(4-фторбензилокси)фенил]пирролидин-2-она приводит к (RS)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамиду в виде твердого вещества белого цвета (выход 98% от теоретического). МС: m/e=343 (М+Н)+.
Пример 14: (R)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид и (S)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
Разделение 0,25 г (0,7 ммоль) двух энантиомеров (RS)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамида (пример 13) производят на препаративной колонке для хиральной ВЭЖХ (CHIRALPAK® AD, давление: 17 бар, объемная скорость: 35 мл/минуту), используя смесь 4:1 н-гептана и этанола в качестве элюента. Получают 100 мг (39% от теоретического) элюируемого первым (R)-(+)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамида [МС: m/e=343 (M++Н)] и 90 мг (35% от теоретического) элюируемого позднее (S)-(-)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамида [МС: m/e=343 (М+Н)+], каждый в виде твердого вещества белого цвета.
Пример 15: (RS)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}формамид
Аналогично тому, как это описано в примере 4а), ацилирование гидрохлорида (RS)-4-амино-1-[4-(4-фторбензилокси)фенил]пирролидин-2-она [пример 13г)] приводит к (RS)-К-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}формамиду в виде твердого вещества белого цвета (выход 77,5% от теоретического). МС: m/e=328 (М+Н)+.
Пример 16: (RS)-N-[1-(4-бензилоксифенил)-5-оксопирролидин-3-ил]ацетамид
а) Гидрохлорид (RS)-4-амино-1-(4-бензилоксифенил)пирролидин-2-она
Аналогично тому, как это описано в примере 1е), расщепление трет-бутоксикарбонильной группы в трет-бутиловом эфире (RS)-[1-(4-бензилоксифенил)-5-оксопирролидин-3-ил]карбаминовой кислоты [пример 1в)] приводит к гидрохлориду (RS)-4-амино-1-(4-бензилоксифенил)пирролидин-2-она в виде твердого вещества белого цвета (выход 84% от теоретического).
б) (RS)-N-[1-(4-бензилоксифенил)-5-оксопирролидин-3-ил]ацетамид
Аналогично тому, как это описано в примере 1ж), ацетилирование гидрохлорида (RS)-4-амино-1-(4-бензилоксифенил)пирролидин-2-она приводит к (RS)-N-[1-(4-бензилоксифенил)-5-оксопирролидин-3-ил]ацетамиду в виде твердого вещества белого цвета (выход 21% от теоретического). МС: m/e=325 (М+Н)+.
Пример 17: (RS)-N-{1-[4-(2-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
а) Метиловый эфир (RS)-1-[4-(2-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты
Аналогично тому, как это описано в примере 13а), алкилирование метилового эфира (RS)-1-(4-гидроксифенил)-5-оксопирролидин-3-карбоновой кислоты [пример 2б)] с помощью 2-фторбензилбромида при КТ с использованием карбоната цезия в качестве основания приводит к метиловому эфиру (RS)-1-[4-(2-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты в виде твердого вещества светло-желтого цвета (выход 82% от теоретического).
б) (RS)-1-[4-(2-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновая кислота
Аналогично тому, как это описано в примере 13б), гидролиз метилового эфира (RS)-1-[4-(2-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты приводит к (RS)-1-[4-(2-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоте в виде твердого вещества грязно-белого цвета (выход 82% от теоретического).
в) Гидрохлорид (RS)-4-амино-1-[4-(2-фторбензилокси)фенил]пирролидин-2-она
Аналогично тому, как это описано в примере 2ж), перегруппировка Курциуса (RS)-1-[4-(2-фторбензилокси)фенил]-5-оксопирролидин-3-карбоновой кислоты и гидролиз образующегося на промежуточной стадии изоцианата приводит к гидрохлориду (RS)-4-амино-1-[4-(2-фторбензилокси)фенил]пирролидин-2-она в виде твердого вещества белого цвета (выход 85% от теоретического). МС: m/e=301 (М+Н)+.
г) (RS)-N-{1-[4-(2-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
Аналогично тому, как это описано в примере 1ж), ацетилирование (R)-4-амино-1-[4-(2-фторбензилокси)фенил]пирролидин-2-она приводит к (RS)-N-{1-[4-(2-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамиду в виде твердого вещества белого цвета (выход 98% от теоретического). МС: m/e=343 (М+Н)+.
Пример 18: (RS)-(Е)-К-(1-N-[2-(3-фторфенил)винил]фенил}-5-оксопирролидин-3-ил)ацетамид
а) (Е)-1-фтор-3-[2-(4-нитрофенил)этенил]бензол
Суспензию 677 мг гидрида натрия (55% дисперсия в масле) в 10 мл N,N-диметилформамида охлаждают до температуры 0°С. После этого прибавляют по частям 5,61 г (20,5 ммоль) диэтил(4-нитробензил)фосфоната. Реакционную смесь оставляют нагреваться до КТ и перемешивают в течение 1,5 часов. После этого смесь охлаждают до температуры -10°С и прибавляют по каплям раствор 1,5 г (12,1 ммоль) 3-фторбензальдегида в 5 мл N,N-диметилформамида. Перемешивание продолжают в течение 30 минут при температуре 0°С, а затем при КТ. Для получения конечного продукта в реакционную смесь добавляют лед и этилацетат. Органический слой отделяют, сушат над сульфатом магния и упаривают при пониженном давлении, что приводит к неочищенному кристаллическому продукту, перекристаллизация которого из смеси диэтилового эфира и гептана приводит к 2,41 г (82% от теоретического) (Е)-1-фтор-3-[2-(4-нитрофенил)этенил]бензола в виде твердого вещества желтого цвета; МС: m/e=243 (M)+.
б) (Е)-4-[2-(3-фторфенил)винил]фениламин
Раствор 2,41 г (10 ммоль) (Е)-1-фтор-3-[2-(4-нитрофенил)этенил]бензола в 25 мл этилацетата продувают аргоном, после чего гидрируют при КТ и атмосферном давлении в течение 4 часов, используя 0,241 г палладия на угле (5%) в качестве катализатора. Для получения конечного продукта катализатор отфильтровывают над дикалитом и упаривают получаемый в результате раствор при пониженном давлении. Получаемый твердый материал кристаллизуют из смеси диэтилового эфира и гептана, что приводит к 1,32 г (62,5% от теоретического) (Е)-4-[2-(3-фторфенил)винил]фениламина в виде твердого вещества оранжевого цвета; МС: m/e=213 (М)+.
в) (RS)-(Е)-1-{4-[2-(3-фторфенил)винил]фенил}-5-оксопирролидин-3-карбоновая кислота
Смесь 600 мг (2,8 ммоль) (Е)-4-[2-(3-фторфенил)винил]фениламина и 366 мг (2,8 ммоль) итаконовой кислоты нагревают до температуры 130°С. По прошествии 1 часа расплавленный материал охлаждают до КТ, после чего получаемое в результате твердое вещество растирают с этилацетатом, что приводит к 568 мг (62% от теоретического) (RS)-(E)-1-{4-[2-(3-фторфенил)винил]фенил}-5-оксопирролидин-3-карбоновой кислоты в виде мелкого порошка желтого цвета; МС: m/e=324 (M-Н)+.
г) Трет-бутиловый эфир (RS)-(Е)-(1-{4-[2-(3-фторфенил)винил]фенил}-5-оксопирролидин-3-ил)карбаминовой кислоты
Раствор 150 мг (0,46 ммоль) (RS)-(Е)-1-{4-[2-(3-фторфенил)винил]фенил}-5-оксопирролидин-З-карбоновой кислоты в 2 мл тетрагидрофурана охлаждают до температуры -15°С и прибавляют по каплям 63 мг (0,46 ммоль) изобутилового эфира хлормуравьиной кислоты. Через 5 минут добавляют раствор 60 мг (0,92 ммоль) азида натрия в 0,5 мл воды. Смесь перемешивают при температуре 0°С в течение 45 минут, а затем оставляют нагреваться до КТ. Добавляют толуол и промывают разбавленный раствор насыщенным раствором бикарбоната натрия. Органический слой отделяют, сушат над сульфатом магния и упаривают при пониженном давлении. Остаток растворяют в 5 мл толуола и нагревают раствор до температуры 80°С. Через 30 минут добавляют 1,1 мл (1,2 ммоль) трет-бутанола и продолжают нагревание в течение 18 часов. Для получения конечного продукта реакционную смесь упаривают, и неочищенный продукт непосредственно подвергают хроматографической очистке на силикагеле, используя смесь 95:5 хлористого метилена и метанола в качестве элюента. После кристаллизации из диэтилового эфира получают 104 мг (57% от теоретического) трет-бутилового эфира (RS)-(E)-(1-{4-[2-(3-фторфенил)винил]фенил}-5-оксопирролидин-3-ил)карбаминовой кислоты в виде твердого вещества светло-желтого цвета; МС: m/e=397 (М+Н)+.
д) Гидрохлорид (RS)-(Е)-4-амино-1-{4-[2-(3-фторфенил)винил]фенил}-пирролидин-2-она
Раствор 104 мг (0,26 ммоль) трет-бутилового эфира (RS)-(E)-(1-{4-[2-(3-фторфенил)винил]фенил}-5-оксопирролидин-3-ил)карбаминовой кислоты в 2,5 мл тетрагидрофурана обрабатывают 192 мг соляной кислоты (37%). Смесь нагревают до температуры 45°С в течение 2 часов, после чего оставляют перемешиваться в течение 18 часов при КТ. Продукт частично осаждается из реакционной смеси, которую упаривают для получения неочищенного гидрохлорида. Последний перекристаллизовывают из диэтилового эфира, что приводит к 74 мг (85% от теоретического) гидрохлорида (RS)-(Е)-4-амино-1-{4-[2-(3-фторфенил)винил]фенил}пирролидин-2-она в виде твердого вещества белого цвета; МС: m/e=297 (М+Н)+.
е) (RS)-(Е)-N-(1-{4-[2-(3-фторфенил)винил]фенил)-5-оксопирролидин-3-ил)ацетамид
Суспензию 61 мг (0,18 ммоль) гидрохлорида (RS)-(Е)-4-амино-1-{4-[2-(3-фторфенил)винил]фенил}пирролидин-2-она в 2,5 мл хлористого метилена обрабатывают 45 мг (0,44 ммоль) триэтиламина. Смесь охлаждают до температуры 0°С, после чего добавляют 20 мг (0,26 ммоль) ацетилхлорида. После выдерживания в течение 1 часа при температуре 0°С смесь оставляют нагреваться до КТ и разбавляют хлористым метиленом. После промывания водой органический слой сушат над сульфатом магния и упаривают. Неочищенный продукт кристаллизуют из диэтилового эфира, что приводит к 49 мг (78% от теоретического) (RS)-(Е)-N-(1-{4-[2-(3-фторфенил)винил]фенил}-5-оксопирролидин-3-ил)ацетамида в виде твердого вещества светло-коричневого цвета; МС: m/e=339 (М+Н)+.
Пример 19: (RS)-N-(1-{4-[2-(3-фторфенил)этил]фенил}-5-оксопирролидин-3-ил)ацетамид
а) 4-[2-(3-фторфенил)этил]фениламин
Аналогично тому, как это описано в примере 18б), гидрирование (Е)-1-фтор-3-[2-(4-нитрофенил)этенил]бензола [пример 18а)] с использованием палладия на угле (10%) в течение 5 часов с одновременным восстановлением двойной связи приводит с количественным выходом к 4-[2-(3-фторфенил)этил]фениламину в виде твердого вещества желтого цвета. МС: m/e=215 (М)+.
б) (RS)-1-{4-[2-(3-фторфенил)этил]фенил}-5-оксопирролидин-3-карбоновая кислота
Аналогично тому, как это описано в примере 18в), реакция 4-[2-(3-фторфенил)этил]фениламина с итаконовой кислотой приводит к (RS)-1-{4-[2-(3-фторфенил)этил]фенил}-5-оксопирролидин-3-карбоновой кислоте в виде твердого вещества свето-коричневого цвета; МС: m/e=326 (M-Н)+.
в) Трет-бутиловый эфир (RS)-(1-{4-[2-(3-фторфенил)этил]фенил}-5-оксопирролидин-3-ил)карбаминовой кислоты
Аналогично тому, как это описано в примере 18г), перегруппировка Курциуса (RS)-1-{4-[2-(3-фторфенил)этил]фенил}-5-оксопирролидин-3-карбоновой кислоты и обработка образующегося на промежуточной стадии изоцианата трет-бутанолом приводят к трет-бутиловому эфиру (1-{4-[2-(3-фторфенил)этил]фенил}-5-оксопирролидин-3-ил)карбаминовой кислоты в виде твердого вещества грязно-белого цвета (выход 36% от теоретического); МС: m/e=399 (М+Н)+.
г) Гидрохлорид (RS)-4-амино-1-N-[2-(3-фторфенил)этил]фенил}пирролидин-2-она
Аналогично тому, как это описано в примере 18д), расщепление трет-бутоксикарбонильной группы соляной кислотой приводит к гидрохлориду (RS)-4-амино-1-{4-[2-(3-фторфенил)этил]фенил}пирролидин-2-она в виде твердого вещества грязно-белого цвета (выход 67,5% от теоретического). МС: m/e=299 (М+Н)+.
д) (RS)-N-(1-N-[2-(3-фторфенил)этил]фенил}-5-оксопирролидин-3-ил)ацетамид
Аналогично тому, как это описано в примере 18е), ацетилирование (RS)-4-амино-1-{4-[2-(3-фторфенил)этил]фенил}пирролидин-2-она приводит к (RS)-N-(1-{4-[2-(3-фторфенил)этил]фенил}-5-оксопирролидин-3-ил)ацетамиду в виде твердого вещества белого цвета после кристаллизации из диэтилового эфира (выход 85,6% от теоретического). МС: m/e=341 (М+Н)+.
Пример 20: (RS)-N-{1-[6-(4-фторбензилокси)пиридин-3-ил]-5-оксопирролидин-3-ил}ацетамид
а) 2-(4-фторбензилокси)-5-нитропиридин
Аналогично тому, как это описано в Journal of Medicinal Chemistry, т.33, cc.2087-2093, (1990), реакция 4-фторбензилового спирта вместо бензилового спирта с 2-хлор-5-нитропиридином приводит к 2-(4-фторбензилокси)-5-нитропиридину в виде твердого вещества желтого цвета.
б) 6-(4-фторбензилокси)пиридин-3-иламин
Смесь 0,70 г (2,8 ммоль) 2-(4-фторбензилокси)-5-нитропиридина и 2,36 г (4,2 ммоль) порошка железа в 35 мл воды и 0,7 мл уксусной кислоты нагревают при кипячении с обратным холодильником в течение 4 часов. Для получения конечного продукта реакционную смесь обрабатывают водой и этилацетатом при интенсивном перемешивании, после чего фильтруют над слоем дикалита. Органический слой отделяют, сушат над сульфатом натрия и упаривают при пониженном давлении. Получают 0,28 г (45% от теоретического) 6-(4-фторбензилокси)пиридин-3-иламина в виде зеленоватого твердого вещества, которое используют на следующей стадии без дополнительной очистки.
в) (RS)-1-[6-(4-фторбензилокси)пиридин-3-ил]-5-оксопирролидин-3-карбоновая кислота
Аналогично тому, как это описано в примере 1а), реакция 6-(4-фторбензилокси)пиридин-3-иламина с итаконовой кислотой приводит к (RS)-1-[6-(4-фторбензилокси)пиридин-3-ил]-5-оксопирролидин-3-карбоновой кислоте в виде твердого вещества зеленого цвета (выход 47% от теоретического).
г) Дигидрохлорид (RS)-4-амино-1-[6-(4-фторбензилокси)пиридин-3-ил]пирролидин-2-она
Аналогично тому, как это описано в примере 2ж), перегруппировка Курциуса (RS)-1-[6-(4-фторбензилокси)пиридин-3-ил]-5-оксопирролидин-3-карбоновой кислоты и обработка получаемого на промежуточной стадии изоцианата приводит к дигидрохлориду (RS)-4-амино-1-[6-(4-фторбензилокси)пиридин-3-ил]пирролидин-2-она в виде твердого вещества светло-желтого цвета.
д) (RS)-N-{1-[6-(4-фторбензилокси)пиридин-3-ил]-5-оксопирролидин-3-ил}ацетамид
Аналогично тому, как это описано в примере 1ж), ацетилирование дигидрохлорида (RS)-4-амино-1-[6-(4-фторбензилокси)пиридин-3-ил]пирролидин-2-она приводит к (RS)-N-{1-[6-(4-фторбензилокси)пиридин-3-ил]-5-оксопирролидин-3-ил}ацетамиду в виде твердого вещества белого цвета (выход 37% от теоретического). МС: m/e=344 (М+Н)+.
Пример 21: (S)-N-{1-[4-(3-хлорбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
а) (S)-N-[1-(4-гидроксифенил)-5-оксопирролидин-3-ил]ацетамид
Раствор 4,67 г (13,6 ммоль) (S)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамида в 500 мл тетрагидрофурана гидрируют в присутствии 726 мг палладия на угле (10%) при атмосферном давлении и КТ в течение 18 часов. Если реакция протекает не до конца, катализатор отфильтровывают над дикалитом и добавляют еще 726 мг палладия на угле (10%). Для получения конечного продукта реакционную смесь фильтруют над дикалитом, а затем упаривают при пониженном давлении. Неочищенный (S)-N-[1-(4-гидроксифенил)-5-оксопирролидин-3-ил]ацетамид получают в виде твердого вещества грязно-белого цвета, которое непосредственно используют на следующей стадии без дальнейшей очистки. МС: m/e=235 (М+Н)+.
б) (S)-N-{1-[4-(3-хлорбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
Раствор 15 мг (0,064 ммоль) (S)-N-[1-(4-гидроксифенил)-5-оксопирролидин-3-ил]ацетамида в 30 мл ацетона обрабатывают 0,01 мл (0,074 ммоль) 2-хлорбензилбромида и 22 мг (0,067 ммоль) карбоната цезия и перемешивают смесь при температуре 40°С в течение 4 часов. Для получения конечного продукта реакционную смесь фильтруют и упаривают досуха. Остаток подвергают хроматографической очистке на силикагеле, используя смесь 19:1 хлористого метилена и метанола в качестве элюента. Получают 17 мг (72% от теоретического) (S)-N-{1-[4-(3-хлорбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамида в виде твердого вещества белого цвета. МС: m/e=359,3 (М+Н)+.
Пример 22: (S)-N-{1-[4-(2,6-дифторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
Аналогично тому, как это описано в примере 21б), исходя из (S)-N-[1-(4-гидроксифенил)-5-оксопирролидин-3-ил]ацетамида [пример 21а] получают указанное в заглавии соединение путем алкилирования с помощью 2,6-дифторбензилбромида и карбоната цезия при температуре 40°С в течение ночи. Выход 85% от теоретического в виде твердого вещества белого цвета. МС: m/e=361,3 (М+Н)+.
Пример 23: (S)-N-{5-оксо-1-[4-(2,4,6-трифторбензилокси)фенил]пирролидин-3-ил}ацетамид
Аналогично тому, как это описано в примере 21б), исходя из (S)-N-[1-(4-гидроксифенил)-5-оксопирролидин-3-ил]ацетамида [пример 21а] получают указанное в заглавии соединение путем алкилирования с помощью 2,4,6-трифторбензилбромида и карбоната цезия при температуре 40°С в течение ночи. Выход 53% от теоретического в виде твердого вещества белого цвета. МС: m/e=379,4 (М+Н)+.
Пример 24: (S)-N-{1-[4-(3-метоксибензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
Аналогично тому, как это описано в примере 21б), исходя из (S)-N-[1-(4-гидроксифенил)-5-оксопирролидин-3-ил]ацетамида [пример 21а] получают указанное в заглавии соединение путем алкилирования с помощью 3-метоксибензилбромида и карбоната цезия при температуре 40°С в течение ночи. Выход 58% от теоретического в виде твердого вещества белого цвета. МС: m/e=355,2 (М+Н)+.
Пример 25: (S)-N-{5-оксо-1-[4-(4-трифторметил-бензилокси)фенил]пирролидин-3-ил}ацетамид
Аналогично тому, как это описано в примере 21б), исходя из (S)-N-[1-(4-гидроксифенил)-5-оксопирролидин-3-ил]ацетамида [пример 21а] получают указанное в заглавии соединение путем алкилирования с помощью 4-(трифторметил)бензилбромида и карбоната цезия при температуре 40°С в течение ночи. Выход 55% от теоретического в виде твердого вещества белого цвета. МС: m/e=393,3(M+H)+.
Пример 26: (S)-N-{1-[4-(4-метилбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
Аналогично тому, как это описано в примере 21б), исходя из (S)-N-[1-(4-гидроксифенил)-5-оксопирролидин-3-ил]ацетамида [пример 21а] получают указанное в заглавии соединение путем алкилирования с помощью 4-метилбензилбромида и карбоната цезия при температуре 40°С в течение ночи. Выход 83% от теоретического в виде твердого вещества белого цвета. МС: m/e=339,1 (M+H)+.
Пример 27: (S)-N-{1-[4-(3-цианобензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид
Аналогично тому, как это описано в примере 21б), исходя из (S)-N-[1-(4-гидроксифенил)-5-оксопирролидин-3-ил]ацетамида [пример 21а] получают указанное в заглавии соединение путем алкилирования с помощью 3-(бромметил)бензонитрила и карбоната цезия при температуре 40°С в течение ночи. Выход 91% от теоретического в виде твердого вещества светло-желтого цвета. МС: m/e=350,3 (M+H)+.
Пример А: Таблетки
Таблетки следующего состава изготавливают общепринятым способом:
Пример Б: Таблетки
Таблетки следующего состава изготавливают общепринятым способом:
Пример В: Капсулы
Изготавливают капсулы следующего состава:
Активный ингредиент, имеющий подходящий размер частиц, кристаллическую лактозу и микрокристаллическую целлюлозу смешивают друг с другом до образования гомогенной смеси и просеивают, после чего добавляют тальк и стеарат магния. Получаемой в результате смесью заполняют твердые желатиновые капсулы подходящего размера.
Пример Г: Раствор для инъекций
Раствор для инъекций может иметь следующий состав и приготавливается обычным способом:
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 4-ПИРРОЛИДИНОФЕНИЛБЕНЗИЛОВОГО ЭФИРА | 2003 |
|
RU2336268C2 |
| ЗАМЕЩЕННЫЕ МЕТИЛФЕНИЛКЕТОНЫ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2007 |
|
RU2493149C2 |
| ПРИМЕНЕНИЕ ОКСАЗОЛИДИНОН-ХИНОЛИНОВЫХ ГИБРИДНЫХ АНТИБИОТИКОВ ДЛЯ ЛЕЧЕНИЯ СИБИРСКОЙ ЯЗВЫ И ДРУГИХ ИНФЕКЦИЙ | 2004 |
|
RU2351335C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДОНА В КАЧЕСТВЕ ИНГИБИТОРОВ МАОВ | 2003 |
|
RU2336267C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ТЕТРАЗОЛОНА | 2014 |
|
RU2664644C2 |
| ПРОИЗВОДНЫЕ СУЛЬФОНИЛПИРРОЛИДИНА | 2001 |
|
RU2272026C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2001 |
|
RU2419608C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛИДИН-2-КАРБОКСАМИДЫ | 2011 |
|
RU2564022C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ ПИРРОЛИДИНИЛГИДРОКСАМОВОЙ КИСЛОТЫ | 1999 |
|
RU2163235C1 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2298550C2 |
Изобретение относится к новым производным пирролидона формулы I
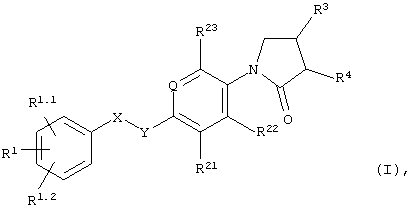
где: Q означает =N, =C(R24)-; X-Y означает -СН2-СН2-, -СН=СН-, -СН2-О-; R1, R1.1 и R1.2 независимо друг от друга выбраны из группы, включающей Н, галоид, галоид(C1-C6)алкил, CN, (C1-C6)алкоксигруппу; R21, R22 и R23 независимо друг от друга выбраны из группы, включающей Н, галоид; R24 означает Н; R3 означает -NHR6; R4 означает Н; R6 означает -С(O)Н, -С(O)-(C1-C3)алкил, С(O)-галоид(C1-C3)алкил, -С(O)O(C1-C3)алкил, -C(O)NH2, -SO2-(C1-C3)алкил; а также его индивидуальные изомеры, рацемические и нерацемические смеси. Соединения ингибируют моноаминоксидазу В, что позволяет использовать их в фармацевтической композиции, предназначенной для профилактики и лечения заболевания, опосредованного ингибитором моноаминоксидазы В, в частности болезни Альцгеймера и старческого слабоумия. 3 н. и 12 з.п. ф-лы.
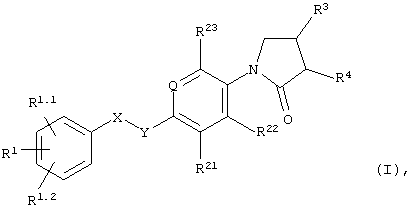
в которой Q обозначает=N- или=C(R24)-;
X-Y обозначает -СН2-CH2-, -СН=СН- или -СН2-О-;
R1, R1.1 и R1.2 независимо друг от друга выбраны из группы, включающей водород, галоид, галоид(C1-C6)алкил, цианогруппу или (C1-C6)алкоксигруппу;
R21, R22 и R23 независимо друг от друга выбраны из группы, включающей водород и галоид;
R24 обозначает водород;
R3 обозначает -NHR6;
R4 обозначает водород; и
R6 обозначает -С(O)Н, -С(O)-(C1-C3)алкил, С(O)-галоид(C1-C3)алкил, -С(O)O(C1-C3)алкил, -C(O)NH2 или -SO2-(C1-C3)алкил;
а также его индивидуальные изомеры и рацемические или нерацемические смеси.
(RS)-N-{l-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(S)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(R)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(RS)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}формамид,
(S)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}формамид,
(R)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}формамид, метиловый эфир (RS)-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}-карбаминовой кислоты,
(RS)-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}мочевину,
(RS)-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}метансульфонамид,
(S)-2-фтор-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(S)-2,2-дифтор-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(S)-2,2,2-трифтор-N-{1-[4-(3-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(RS)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(R)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(S)-N-{1-[4-(4-фторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(RS)-N-{1-[4-(4-фторбензилоксифенил]-5-оксопирролидин-3-ил}формамид,
(RS)-N-[1-(4-бензилокси)фенил)-5-оксопирролидин-3-ил]ацетамид,
(RS)-N-{1-[4-(2-фторбензилоксифенил]-5-оксопирролидин-3-ил}ацетамид,
(RS)-(Е)-N-(1-{4-[2-(3-фторфенил)винил]фенил}-5-оксопирролидин-3-ил)ацетамид,
(RS)-N-(1-{4-[2-(3-фторфенил)этил]фенил}-5-оксопирролидин-3-ил)ацетамид,
(RS)-N-{1-[6-(4-фторбензилокси)пиридин-3-ил]-5-оксопирролидин-3-ил}ацетамид,
(S)-N-{1-[4-(3-хлорбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(S)-N-{1-[4-(2,6-дифторбензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(S)-N-{5-оксо-1-[4-(2,4,6-трифторбензилокси)фенил]пирролидин-3-ил}ацетамид,
(S)-N-{1-[4-(3-метоксибензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид,
(S)-N-{5-оксо-1-[4-(4-трифторметилбензилокси)фенил]пирролидин-3-ил}ацетамид и
(S)-N-{1-[4-(3-цианобензилокси)фенил]-5-оксопирролидин-3-ил}ацетамид.
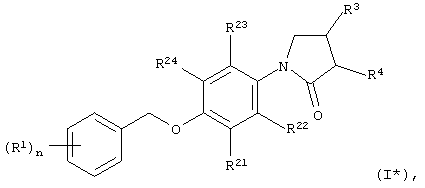
в котором R1 обозначает галоид, галоид(C1-C6)алкил, цианогруппу или (C1-C6)алкоксигруппу;
R21, R22, R23 независимо друг от друга выбраны из группы, включающей водород и галоид;
R3 обозначает -NHR6;
R4 обозначает водород; и
R6 обозначает -С(O)-(C1-C3)алкил или -SO2-(C1-C3)алкил; и
n принимает значения 0,1,2 или 3;
а также его индивидуальные изомеры и рацемические или нерацемические смеси.
| US 4287351, 01.09.1981 | |||
| СПОСОБ ИЗГОТОВЛЕНИЯ МЕТАЛЛИЧЕСКОЙ ДЕТАЛИ, СОДЕРЖАЩЕЙ ВНУТРЕННИЕ ЭЛЕМЕНТЫ ЖЕСТКОСТИ, ОБРАЗОВАННЫЕ КЕРАМИЧЕСКИМИ ВОЛОКНАМИ | 2009 |
|
RU2500831C2 |
| ПРОИЗВОДНЫЕ ИЗОКСАЗОЛА, ИХ ПРИМЕНЕНИЕ В ПРОИЗВОДСТВЕ ИНГИБИРУЮЩЕГО МОНОАМИНООКСИДАЗУ АГЕНТА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ МОНОАМИНООКСИДАЗЫ ТИПА В | 1995 |
|
RU2140414C1 |

