Данное изобретение относится к новому способу получения производных гидроксамовой кислоты и их фармацевтически приемлемых солей. Эти соединения и композиции полезны в качестве аналгезирующих, противовоспалительных, диуретических, анестезирующих или нейропротекторных средств или средств для лечения острых или функциональных кишечных болезней, таких как боль в животе, для лечения млекопитающих, особенно человека.
Предпосылки изобретения
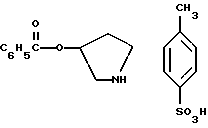
Опиоидные анальгетики, такие как морфин, являются терапевтически полезными, но их применение сильно ограничено из-за их побочных эффектов, таких как лекарственная зависимость. Поэтому требуются анальгетики с высокой эффективностью и пониженной тенденцией вызывать лекарственную зависимость. Важные фармакологические и биохимические исследования были проведены, чтобы открыть опиоидные пептиды и опиоидные рецепторы, и открытие подтипов опиоидных рецепторов, таких как μ,δ,κ в периферических нервах у различных видов, включая человека, положило начало к созданию новых анальгетиков. Так как считается, что опиоидные анальгетики, такие как морфин, действуют как агонист μ - рецептора, то изучалось отличие действия, основанного на агонизме к κ -рецептору, от действия, основанного на агонизме к μ - рецептору. Недавно в свете вышеупомянутой проблемы поступило сообщение о κ -селективных агонистах, например EMD-60400: A. Barber et al. , Naunyn-Schmled. Arch. Pharmacol. , 345 (Suppl.): Abst456. Причем некоторые из них были изучены в клинических испытаниях (Med. Res. Rev., 12, 525 (1992)).
WO 96/30339 описывает соединение формулы: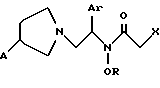
и его соль, где
A обозначает водород, гидрокси или OY, где Y обозначает гидрокси-защитную группу;
Ar обозначает фенил, возможно замещенный одним или более (предпочтительно до 3-х) заместителями, выбранными из гало, гидрокси, C1-C4алкил, C1-C4алкокси, CF3, C1-C4алкокси-C1-C4алкилокси, и карбокси-C1-C4алкилокси;
X обозначает фенил, нафтил, бифенил, инданил, бензофуранил, бензотиофенил, 1-тетралон-6-ил, C1-C4алкилендиокси, пиридил, фурил и тиенил, причем эти группы возможно замещены 1-3 заместителями, выбранными из гало, C1-C4алкил, C1-C4алкокси, гидрокси, NO2, CF3 и SO2CH3; и
R обозначает водород, C1-C4алкил или гидрокси-защитную группу.
Производные гидроксамовой кислоты формулы (I), в которой A обозначает водород или гидрокси и R обозначает водород или C1-C4 алкил, проявляют значительную агонистическую активность по отношению к опиоидному κ -рецептору. Поэтому эти κ -агонисты особенно полезны в качестве аналгезирующих средств у млекопитающих, особенно у людей. Они также полезны в качестве противовоспалительных, диуретических, анестезирующих или нейропротекторных средств, или средств для лечения острых или функциональных кишечных заболеваний, таких боль в животе, для лечения млекопитающих, особенно человека.
Краткое описание изобретения
Согласно данному изобретению предложен выгодный синтетический способ получения соединений формулы I, указанной выше, в которой A обозначает гидрокси, Ar обозначает фенил или фенил, замещенный 1-3 заместителями, выбранными из хлора, метила и CF3, более предпочтителен 3,4-дихлорфенил, и R обозначает водород. Предпочтительная конфигурация атома углерода, к которому присоединяется группа Ar, (S).
Предпочтительными индивидуальными соединениями, которые могут быть получены способом по изобретению являются:
2-(3,4-дихлорфенил)-N-гидрокси-N-[2-(3-(S)-гидроксипирролидин-1-ил)-1-(S)-фенилэтил]ацетамид;
2-(4-бромфенил)-N-гидрокси-N-[2-(3-(S)-гидроксипирролидин-1- ил)-1-(S)-фенилэтил]ацетамид;
N-гидрокси-N-[2-(3-(S)-гидроксипирролидин-1-ил)-1-(S)-фенилэтил] -2-(4-трифторметилфенил]ацетамид;
2-(4-хлорфенил)-N-гидрокси-N-[2-(3-(S)-гидроксипирролидин-1-ил)-1-(S)-фенилэтил]ацетамид;
2-(2,3-дихлорфенил)-N-гидрокси-N-[2-(3-(S)-гидроксипирролидин-1- ил)-1-(S)-фенилэтил]ацетамид;
2-(2,4-дихлорфенил)-N-гидрокси-N-[2-(3-(S)-гидроксипирролидин-1- ил)-1-(S)-фенилэтил]ацетамид;
2-(2,5-дихлорфенил)-N-гидрокси-N-[2-(3-(S)-гидроксипирролидин-1- ил)-1-(S)-фенилэтил]ацетамид;
2-(2,6-дихлорфенил)-N-гидрокси-N-[2-(3-(S)-гидроксипирролидин-1- ил)-1-(S)-фенилэтил]ацетамид;
N-гидрокси-N-[2-(3-(S)-гидроксипирролидин-1-ил)-1-(S)-фенилэтил] - 2-(2,3,6-трихлорфенил)ацетамид;
2-(3,4-дихлорфенил)-N-[2-(3-(S)-гидроксипирролидин-1-ил)-1-(S)- фенилэтил]ацетамид; и
2-(3,4-диметилфенил)-N-гидрокси-N-[2-[(3-(S)-гидроксипирролидин- 1-ил)-1-(S)-фенилэтил]ацетамид.
Согласно данному изобретению также предложены новые промежуточные соединения, которые полезны для получения соединений формулы 1; эти промежуточные соединения включают:
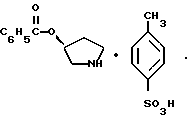
пирролидин-3-илового эфира бензойной кислоты п-метилфенилсульфонат структуры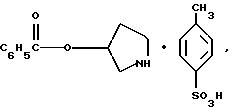
композицию, включающую 1-(2-гидрокси-2-фенил-этил)-пирролидин-3-иловый эфир бензойной кислоты структуры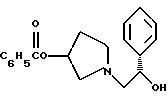
и его региоизомер структуры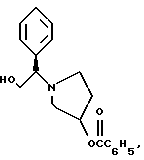
1-(2-хлор-2-фенил-этил)-пирролидин-3-иловый эфир бензойной кислоты структуры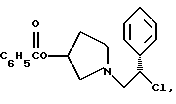
1-(2-бензилоксиамино-2-фенил-этил)-пирролидин-3-иловый эфир бензойной кислоты структуры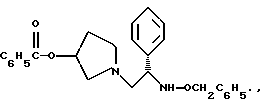
оксалат 1-(2-бензилоксиамино-2-фенил-этил)-пирролидин-3-илового эфира бензойной кислоты структуры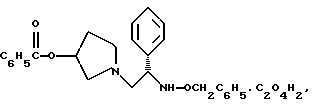
и N-бенэилокси-2-(3,4-дихлор-фенил)-N-[2-(3-гидрокси-пирролидин-1- ил)-1-фенил-этил]-ацетамида бензойной кислоты эфир структуры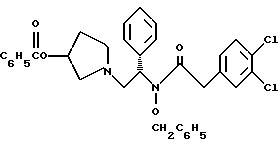
и N-бензилокси-2-(3,4-дихлор-фенил)-N-[2-(3-гидрокси-пирролидин-1- ил)-1-фенил-этил]-ацетамид структуры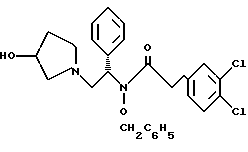
Подробное описание изобретения
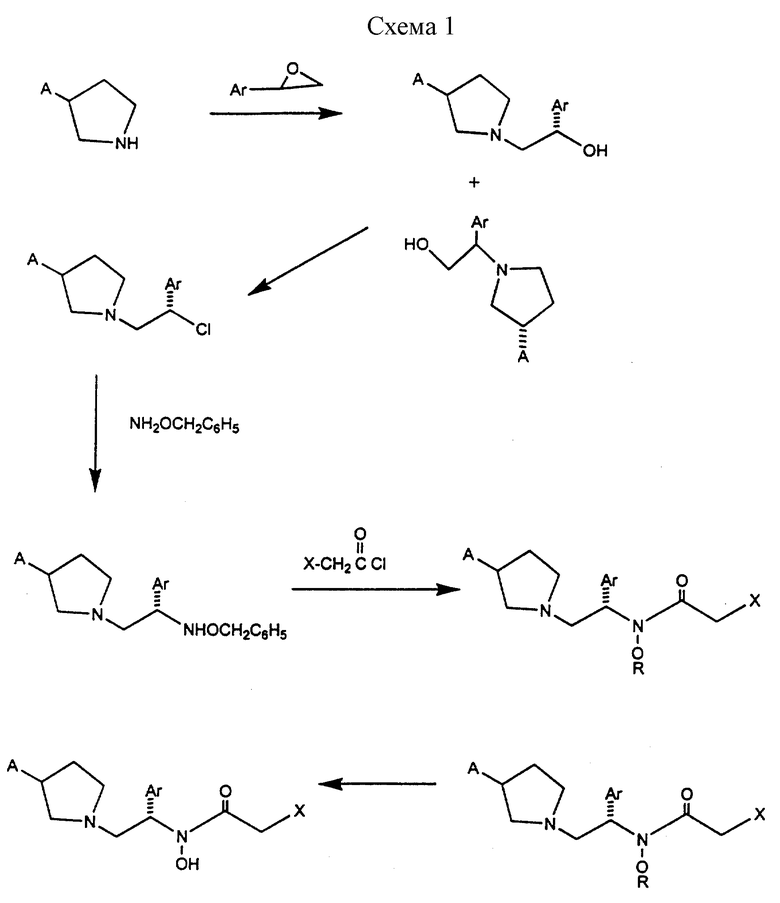
Соединения формулы I могут быть выгодно получены с помощью реакционной схемы 1 (схему 1 см. в конце текста),
где A обозначает гидрокси или OY, где Y обозначает гидрокси-защитную группу;
Ar обозначает фенил, возможно замещенный одним или более (предпочтительно до 3-х) заместителями, выбранными из гало, гидрокси, C1-C4алкил, C1-C4алкокси, CF3, C1-C4алкокси-C1-C4алкилокси, и карбокси-C1-C4алкилокси;
X обозначает фенил, нафтил, бифенил, инданил, бензофуранил, бензотиофенил, 1-тетралон-6-ил, C1-C4алкилендиокси, пиридил, фурил и тиенил, причем эти группы возможно замещены 1-3 заместителями, выбранными из гало, C1-C4алкил, C1-C4алкокси, гидрокси, NO2, CF3 и SO2CH3, и
R обозначает бензильную группу.
Авторами данного изобретения найдена схема внедрения и удаления защитных групп, которые превращают промежуточные соединения, которые ранее считались нестабильными, в соединения, с которыми можно работать. Было обнаружено, что бензоильная группа является особенно полезной в качестве защитной группы Y. Селективное расщепление бензил-защищенной гидроксамовой кислоты требует выбора подходящего катализатора.
Соединение, с которого снимают защиту в примере девять (стадия 9), содержит дополнительную функциональность, которая не инертна в условиях гидрогенизации. А именно, 3,4-дихлорзамещенное ароматическое кольцо склонно к дегалогенизации, и связь азот - кислород в группировке гидроксамовой кислоты может подвергаться гидрогенолизу до вторичного амида. Эти нежелательные реакции контролируются выбором подходящего катализатора и содержанием кислоты. Отбор проводили среди большого круга катализаторов гидрогенизации, которые доводят до минимума возможность этих двух побочных реакций (Предпочтительным является Johnson Matthey тип A 11190A - 5). Кроме того, содержание кислоты значительно уменьшает степень дегалогенизации. Хотя в присутствии кислоты наблюдается дополнительное дезоксигенирование, этот побочный продукт удаляется на последующих стадиях. Образование специфических солей соединений, показанных на следующей схеме, делает ключевые промежуточные соединения кристаллическими, позволяя провести очистку.
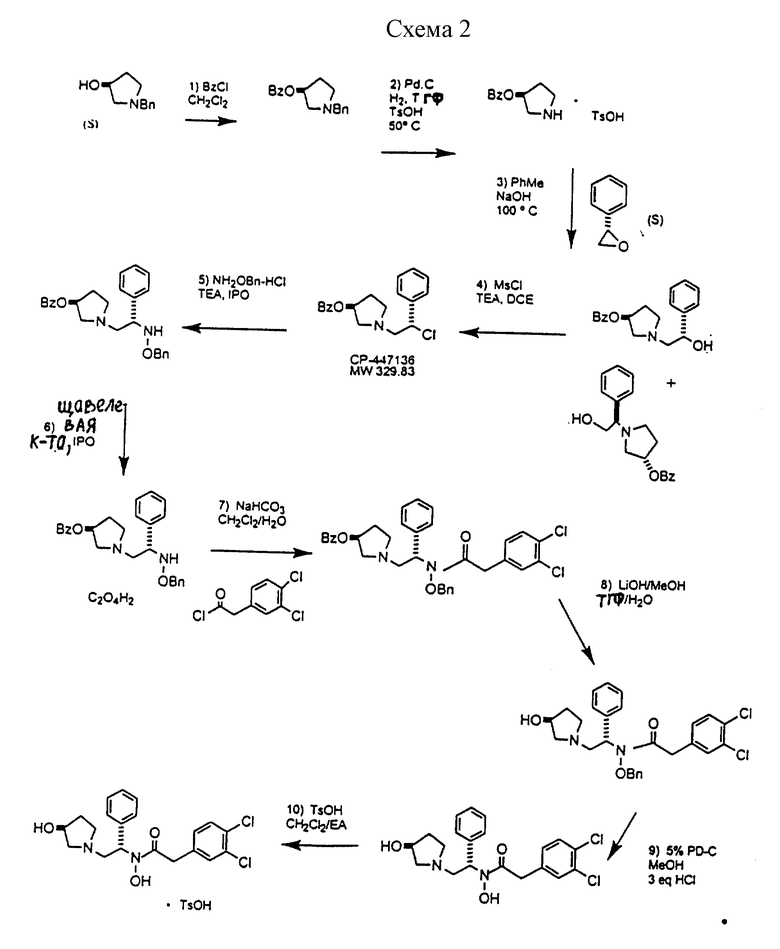
Подробная схема 2 получения 2-(3,4-дихлорфенил)-N-гидрокси-N-[1-(S)- фенил-2-(3-(S)-гидрокси-пирролидин-1-ил)этил] ацетамида п-метилфенилсульфоната показана ниже (схему 2 см. в конце текста) и описывается подробно в примерах 1-10.
Примеры
Данное изобретение иллюстрируется примерами, приведенными ниже. Следует понимать, что данное изобретение не ограничивается конкретными подробностями этих примеров.
Пример 1
1-бензил-пирролидин-3-иловый эфир бензойной кислоты
К раствору 100,0 г S-N-бензил-3-гидроксипирролидина (0,56 моль, 1,0 экв. ) в 500 мл метиленхлорида добавляют 65,0 мл бензоилхлорида (0,56 моль, 1,0 экв. ) при 0oC в течение 15 мин. Реакция протекает при перемешивании в течение дополнительного часа. ВЭЖХ анализ показал, что остаются только следы исходных веществ. К полученной желтой суспензии при 0oC добавляют раствор 59,4 г карбоната натрия (0,56 моль, 1,0 экв.), растворенного в 500 мл воды. Слои разделяют и водный слой экстрагируют 500 мл метиленхлорида. Летучие примеси удаляют при атмосферном давлении с получением 155,1 г (98%) соединения, указанного в заголовке, в виде масла, которое используют на следующей стадии без дополнительной очистки.
1H ЯМР (CDCl3) □ 7,99 (m, 2H), 7.63-7.19 (m, 8H), 3.98 (m, 1H), 3.72 (m, 2H), 3.08-2.92 (m, 1H), 2.89-2.70 (m, 2H), 2.63 (m, 1H), 2.38 (m, 1H), 2.10-1.93 (m, 1H).
ПБМС (масс-спектрометрия с использованием протонной бомбардировки) (M+1)+ = 282
ВЭЖХ tr (время удерживания) = 12,39 мин (Zorbax C8, 4,6 х 150 мм, 220 нм, 1 мл/мин, 600:400:2:1 H2O:TEA (триэтаноламин):OHAc (уксусная кислота).
Пример 2
Пирролидин-3-илового эфира бензойной кислоты п-метилфенилсульфонат
К раствору 25,0 г соединения примера 1 (89 ммоль, 1,0 экв.) в 250 мл ТГФ добавляют 7,5 г 10% палладия на углероде (50% увлажнение) и 16,9 г (89 ммоль, 1,0 экв.) моногидрата пара-толуолсульфокислоты. Затем смесь гидрогенизируют на шейкере Парра при 50 фунт/кв.дюйм (344,75 кПа) избыточное и 50oC на протяжении всей ночи. В первой половине дня водород удаляют и смесь фильтруют через броунмиллерит, чтобы удалить катализатор. ВЭЖХ анализ показал, что остаются только следы исходных веществ. Осадок с фильтра промывают ТГФ, и летучие примеси удаляют под вакуумом, чтобы получить суспензию. Далее замещают ТГФ изопропиловым эфиром, проводят фильтрацию и высушивание под вакуумом, чтобы получить 30,2 г (89%) соединения, указанного в заголовке, в виде белого твердого осадка.
1H ЯМР (d6-ДМСО) □ 8,97 (шир. s, 2H), 8.00 (m, 2H), 7.66 (m, 1H), 7.57-7.42 (m, 4H), 7.08 (m, 2H), 5.51 (m, 1H), 3.40-3.24 (m, 4H), 2.30-2.12 (m, 5H).
ХИАД/МС (химическая ионизация при атмосферном давлении/масс-спектрометрия) (M+1)+ = 192 (свободное основание)
ВЭЖХ tr = 2,29 мин (Zorbax C8, 4,6 х 150 мм, 220 нм, 1 мл/мин, 600:400: 2:1 H2O:MeCN:TEA:HOAc)
Пример 3
1-(2-гидрокси-2-фенил-этил)-пирролидин-3-иловый эфир бензойной кислоты
К двухфазной смеси 25,0 г соединения примера 2 (69 ммоль, 1,0 экв.) в 125 мл толуола добавляют 2,75 г гидроксида натрия (69 ммоль, 1,0 экв.), растворенного в 20 мл воды, а затем 8,27 г (69 ммоль, 1 экв.) оксида (S)-стирола. Реакционную смесь нагревают с дефлегмацией в течение ночи, после чего ВЭЖХ анализ показал, что определяются только следы исходных веществ. После охлаждения до комнатной температуры слои разделяют. Органический слой промывают дополнительными 1,4 г (35 ммоль, 0,5 экв.) гидроксида натрия, растворенного в 20 мл воды, затем 20 мл воды. Растворитель толуол удаляют под вакуумом с получением 19,77 г (92%) густого масла, которое затвердевает при стоянии. Сырой продукт содержит смесь региоизомеров в соотношении ~1,2:1,0 и используется без дополнительной очистки в следующей стадии.
ЯМР двух региоизомеров
□ - открытый региоизомер: 1H ЯМР (CDCl3) □ 8,06 (m, 2H), 7.63-7.23 (m, 8H), 5.45 (m, 1H), 4.75 (m, 1H), 3.27-3.03 (m, 2H), 2.94-2.75 (m, 2H), 2.67-2.51 (m, 2H), 2.40 (m, 1H), 2.16-1.99 (m, 1H).
□ - открытый региоизомер: 1H ЯМР (CDCl3) □ 8,00 (m, 2H), 7.61-7.22 (m, 8H), 5.38 (m, 1H), 3.87 (m, 2H), 3.61 (m, 1H), 3.00-2.75 (m, 3H), 2.58 (m, 1H), 2.31 (m, 1H), 2.20-1.92 (m, 2H).
ХИАД/МС (M+1)+ = 312
ВЭЖХ tr = 11,65 и 12,19 мин (Zorbax C8, 4,6 х 150 мм, 220 нм, 1 мл/мин, 600:400:2:1 H2O:MeCN:TEA:HOAc)
Пример 4
1-(2-хлор-2-фенил-этил)-пирролидин-3-иловый эфир бензойной кислоты
К раствору смеси 50,0 г (161 ммоль) соединения примера 3 в 500 мл дихлорэтана добавляют 24,7 мл (177 ммоль, 1,1 экв.) триэтиламина. При 0oC 13,7 мл (177 ммоль, 1,1 экв.) метансульфонилхлорида добавляют по каплям в течение 20 минут, чтобы поддержать температуру <5oC. Смесь оставляют нагреваться до комнатной температуры, и через 2,5 часа ТСХ анализ (силикагель, 254 нм, 60: 40 гексаны/этилацетат) показал, что исходные вещества израсходованы. Раствор соединения, указанного в заголовке (CP-447136), используют непосредственно в следующей реакции.
Для аналитических оценочных целей образец реакционной смеси промывают водным раствором бикарбоната натрия и летучие примеси удаляют под вакуумом с получением соединения, указанного в заголовке (CP-447135), в виде масла.
1H ЯМР (CDCl3) □ 8,03 (d, 2H), 7.64-7.22 (m, 8H), 5.40 (m, 1H), 4.97 (t, 1H), 3.30-2.98 (m, 2H), 2.90-2.63 (m, 2H), 2.31 (m, 1H), 2.09-1.89 (m, 1H).
ПБМС (M+1)+ = 330
Пример 5
1-(2-бензилоксиамино-2-фенил-этил)-пирролидин-3-иловый эфир бензойной кислоты
Раствор соединения примера 4 обрабатывают дополнительными 25,4 мл триэтиламина (354 ммоль, 2,2 экв.) и 30,8 г (193 ммоль, 1,2 экв.) O-бензилгидроксиламина гидрохлорида. Реакционную смесь доводят до 50oC и затем добавляют 100 мл изопропанола, чтобы растворить O-бензилгидроксиламина гидрохлорид. Реакционной смеси дают возможность в течение ночи перемешиваться при дефлегмации в атмосфере азота. В первой половине дня ТСХ анализ (силикагель, 254 нм, 60: 40 гексаны/этилацетат) показал, что исходные вещества израсходованы. Реакционную смесь охлаждают до комнатной температуры и затем быстро нейтрализуют добавлением 400 мл 1н. NaOH (pH реакционной смеси 11). После разделения слоев органическую фазу промывают 250 мл воды. Органический слой отделяют и удаляют летучие примеси под вакуумом с получением сырого продукта в виде масла.
Пример 6
Оксалат 1-(2-бензилоксиамино-2-фенил-этил)-пирролидин-3-илового эфира бензойной кислоты
Сырое масло из примера 5 растворяют в 500 мл изопропанола и обрабатывают 20,3 г (161 моль, 1,0 экв.) щавелевой кислоты · 2H2O. Полученную суспензию перемешивают в течение ночи и затем охлаждают до 0oC и фильтруют. Затем сырой осадок с фильтра разжижают 300 мл горячего изопропанола. Суспензии дают возможность остыть до комнатной температуры в течение ночи. В первой половине дня твердое вещество отфильтровывают и полученный осадок на фильтре промывают сначала изопропанолом, затем изопропиловым эфиром. Твердый осадок высушивают под вакуумом с получением 48,1 г (59%) соединения, указанного в заголовке, в виде тусклого белого твердого вещества.
1H ЯМР (CDCl3) □ 7,97 (m, 2H), 7.66 (m, 1H), 7.56-7.17 (m, 12H), 5.38 (m, 1H), 4.58 (s, 2H), 4.52 (s, 2H), 4.30 (m, 1H), 3.38-3.13 (m, 4H), 3.09-2.92 (m, 2H), 2.39-2.26 (m, 1H), 2.09-1.96 (m, 1H).
ХИАД/МС (M+1)+ = 417
ВЭЖХ tr = 4,26 мин (Zorbax C8, 4,6 х 150 мм, 225 нм, 2 мл/мин, 1:1 [600: 400:2:1 H2O:MeCN:TEA:HOAc]:MeCN)
Пример 7
N-бензилокси-2-(3,4-дихлор-фенил)-N-[2-(3-гидрокси-пирролидин-1- ил)-1-фенил-этил]-ацетамида бензойной кислоты эфир
К раствору 899 г (4,37 ммоль) 3,4-дихлорфенилуксусной кислоты в 10,5 л метиленхлорида добавляют 586 г (4,62 моль, 1,05 экв.) оксалилхлорида при комнатной температуре. За этим следует осторожное добавление 31 г (0,42 моль, 0,10 экв. ) диметилформамида (остерегаться выделения газа). После прекращения выделения газа аликвоту быстро гасят в метаноле, чтобы гарантировать завершение реакции путем превращения в соответствующий метиловый эфир. ВЭЖХ анализ показал, что определяются только следы исходных веществ. Раствор (3,4-дихлор-фенил)-ацетилхлорида (UK-279292) используется в следующей реакции.
1H ЯМР (CDCl3) □ 7,39-7,46 (m, 1H), 7.34-7.36 (m, 1H), 7.07-7.11 (m, 1H), 4.11 (s, 2H).
ВЭЖХ (сложного метилового эфира) tr = 1,72 мин (Zorbax C8, 4,6 х 150 мм, 225 нм, 2 мл/мин, 1:1 [600:400:2:1 H2O:MeCN:TEA:HOAc]:MeCN)
К суспензии 2118 г (4,18 моль) продукта из примера 6 в 10,5 л метиленхлорида добавляют суспензию 1,780 г (21,1 моль, 5,0 экв.) натрия бикарбоната в 21 л воды (остерегаться выделения газа). Двухфазную смесь охлаждают до 0oC и добавляют раствор (3,4-дихлор-фенил)-ацетилхлорида (UK-279292) (4,37 моль, 1,05 экв.) в метиленхлориде с такой скоростью, чтобы температура оставалась менее чем 10oC. pH контролируют и поддерживают между 8 и 9. После завершения добавления ВЭЖХ анализ показал, что исходные вещества израсходованы. Добавляют дополнительные 10,5 л воды и реакция протекает при перемешивании в течение ночи при комнатной температуре. В первой половине дня помешивание прекращают и позволяют слоям разделиться. Органический слой собирают и концентрируют под вакуумом до получения масла, которое используют без дополнительной очистки на следующей стадии (чистота сырого продукта 93,9%).
1H ЯМР (CD3OH) □ 7,92 (d, 2H), 7.56-7.17 (m, 15H), 7.01-6.95 (m, 1H), 5.75-5.64 (m, 1H), 5.32 (m, 1H), 4.95-4.87 (m, 1H), 4.56-4.48 (m, 1H), 3.88-3.78 (m, 1H), 3.74-3.61 (m, 2H), 3.14-2,96 (m, 2H), 2.92-2.86 (m, 1H), 2.72 (m, 1H), 2.45 (m, 1H), 2.32 (m, 1H), 2.00-1.90 (m, 1H).
ХИАД/МС (M+1)+ = 603
ВЭЖХ tг = 10,4 мин (Zorbax C8, 4,6 х 150 мм, 225 нм, 2 мл/мин, 1:1 [600: 400:2:1 H2O:MeCN:TEA:HOAc]:MeCN)
Пример 8
N-бензилокси-2-(3,4-дихлор-фенил)-N-[2-(3-гидрокси-пирролидин-1- ил)-1-фенил-этил]-ацетамид
К раствору сырого продукта из примера 7 (4,18 моль теоретически) в 26 л в 1: 1 (по объему) смеси ТГФ и метанола добавляют раствор 356 г (8,28 моль, 2,0 экв. ) гидроксида лития · H2O, растворенного в 6,5 л воды. Реакционную смесь перемешивают в течение ночи при комнатной температуре. В первой половине дня pH было более 13, и ВЭЖХ анализ показал, что исходные вещества израсходованы. Затем летучие примеси удаляют под вакуумом, выдерживая тигель при температуре менее 40oC. К неочищенному продукту добавляют 13 л метиленхлорида и 13 л воды. Слои разделяют и органическую фазу промывают дополнительными 13 л воды. Растворитель удаляют под вакуумом с получением сырого продукта (1,990 г, 95% теоретически за две стадии), который переносят непосредственно в следующую реакцию (чистота сырого продукта 84,1%).
1H ЯМР (CD3OH) □ 7,49-7,23 (m, 12H), 7.10-7.02 (m, 1H), 5.72-5.60 (m, 1H), 4.98-4.90 (m, 1H), 4.50-4.42 (m, 1H), 4.25 (m, 1H), 3.92-3.83 (m, 1H), 3.76-3.67 (m, 1H), 3.62-3.52 (m, 1H), 2.97 (m, 1H), 2.87 (m, 1H), 2.63 (m, 1H), 2.52 (m, 1H), 2.03 (m, 1H), 1.70-1.60 (m, 1H).
ХИАД/МС (M+1)+ = 499
ВЭЖХ tr = 3,13 мин (Zorbax C8, 4,6 х 150 мм, 225 нм, 2 мл/мин, 1:1 [600: 400:2:1 H2O:MeCN:TEA:HOAc]:MeCN)
Пример 9
2-(3,4-дихлор-фенил)-N-гидрокси-N-[2-(3-гидрокси-пирролидин-1- ил)-1-S-фенил-этил]-ацетамид
Раствор продукта из примера 8 (3,98 моль теоретически) в 40 л метанола обрабатывают 995 мл (12 моль, 3.0 экв.) концентрированной соляной кислоты и 400 r 5% Pd · С (50% увлажнение, Johnson Matthey тип A 11190A-5). После откачки паров и трехкратной продувки азотом подают водород, чтобы создать незначительное повышенное давление. Дополнительный водород добавляют, чтобы поддерживать незначительное повышенное давление. Протекание реакции контролируют ТСХ (силикагель, 90:10 метиленхлорид:метанол с добавкой гидроксида аммония, Rf исходного вещества 0,65, Rf продукта 0,30), исходное вещество расходуется за ~5 часов. Из системы откачивают пары и трижды продувают азотом. Катализатор удаляют фильтрацией через броунмиллерит, далее 30 л метанола промывают осадок катализатора. Затем HCl/MeOH нейтрализуют осторожным добавлением продукта, содержащего раствор 1350 г (16 моль, 4,0 экв.) натрия бикарбоната, растворенного в 10 л воды. Затем удаляют метанол под вакуумом и добавляют 4 л метиленхлорида и 2 л воды. После разделения слоев органическую фазу промывают дополнительными 10 л воды, снова разделяют и переводят на стадию получения соли без дополнительной очистки.
Пример 10
2-(3,4-дихлорфенил)-N-гидрокси-N-[1-(S)-фенил-2-(3-(S)-гидрокси- пирролидин-1-ил)этил]ацетамида п-метилфенилсульфонат
Раствор 2-(3,4-дихлорфенил)-N-гидрокси-N-[1-(S)-фенил-2-(1- пирролидинил)этил] ацетамида (3,98 моль теоретически) в метиленхлориде из предыдущего примера обрабатывают 757 г (3,98 моль, 1,0 экв.) пара-толуолсульфоновой кислоты · H2O и перемешивают до растворения. За этим следует фильтрация через фильтр с размером пор 0,2 микрона, чтобы удалить частицы. Затем метиленхлорид замещают этилацетатом до конечного объема 6 л. После охлаждения при комнатной температуре продукт осаждается и его оставляют для перемешивания в течение ночи. В первой половине дня суспензию охлаждают до 0oC в течение 90 минут и фильтруют. Осадок с фильтра промывают 2 раза по 500 мл холодного этилацетата. После высушивания масса стала 1,529 г, 66% от теоретического за две стадии. Чистота по ВЭЖХ на этот момент составляла 96,5%.
1,514 г твердого вещества, полученного выше, обрабатывают 7,5 л воды и суспензию перемешивают в течение ночи при комнатной температуре. Твердые вещества отфильтровывают и осадок с фильтра промывают 2 л изопропилового эфира. После высушивания масса стала 1,440 г (95,1%, чистота по ВЭЖХ 97,3%).
1,429 г твердого вещества, полученного выше, обрабатывают 5 л смеси этилацетат: метанол в соотношении 6:1. Суспензию нагревают до растворения и затем раствор охлаждают до 50oC. Добавляют 3 л изопропилового эфира и затем реакционную смесь охлаждают и при 30oC осаждают. После перемешивания при 15oC в течение 2-х часов продукт фильтруют. Осадок на фильтре промывают 2 л изопропилового эфира и затем высушивают в сушильном шкафу с получением 1,219 г белого твердого вещества (85,3%, чистота по ВЭЖХ 99,6%).
1H ЯМР (CD3OD) □ 7,41-7,24 (m, 7H), 7,15 (m, 1H), 5.67 (dd, 5.67), 4.29 (m, 1H), 3.82 (m, 2H), 3.42 (m, 1H), 2.91 (m, 1H), 2.76-2.72 (m, 1H), 2.59-2.53 (m, 1H), 2.42 (m, 1H), 2.11 (m, 1H), 1.72-1.65 (m, 1H).
ХИАД/МС (M+1)+ = 409
ВЭЖХ tr = 9,96 мин (Inertsil C8, 4,6 х 150 мм, 225 нм, 1 мл/мин, 30:70 MeCN:(0,2% TEA + 0,1% H3PO4 в воде)т
Описывается способ получения соединений пирролидинил гидроксамовой кислоты, которые используются в качестве анестезирующих противовоспалительных или нейропротекторных средств. Описываются также новые промежуточные соединения, которые полезны для получения целевых продуктов. 9 с. и 9 з.п. ф-лы.
2. Композиция, содержащая 1-(2-гидрокси-2-фенил-этил)-пирролидин-3-иловый эфир бензойной кислоты структуры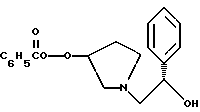
и его региоизомер структуры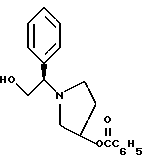
3. 1-(2-Хлор-2-фенил-этил)-пирролидин-3-иловый эфир бензойной кислоты структуры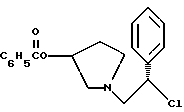
4. 1-(2-Бензилоксиамино-2-фенил-этил)-пирролидин-3-иловый эфир бензойной кислоты структуры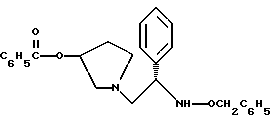
5. Оксалат 1-(2-бензилоксиамино-2-фенил-этил)-пирролидин-3-илового эфира бензойной кислоты структуры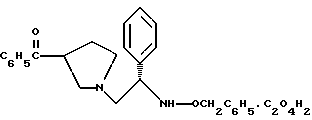
6. N-бензилокси-2-(3,4-дихлор-фенил)-N-[2-(3-гидрокси-пирролидин-1-ил)-1-фенил-этил]-ацетамида бензойной кислоты эфир структуры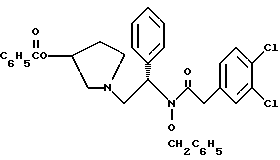
7. N-бензилокси-2-(3,4-дихлор-фенил)-N-[2-(3-гидрокси-пирролидин-1-ил)-1-фенил-этил]-ацетамид структуры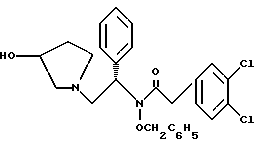
8. Способ получения соединения структуры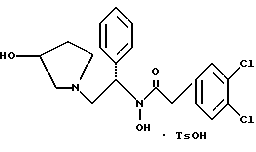
где TsOH - пара-толуолсульфоновая кислота, при котором соединение структуры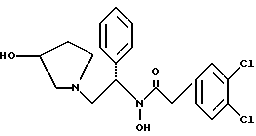
подвергают взаимодействию с пара-толуолсульфоновой кислотой.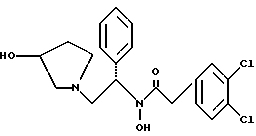
удалением бензильной группы из соединения структуры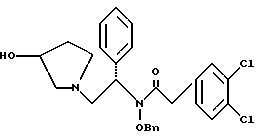
где Bn обозначает бензил, в присутствии катализатора палладий на углероде.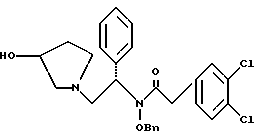
где Bn обозначает бензил, замещением Bz-группы, где Bz обозначает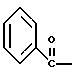
на водород в соединении структуры: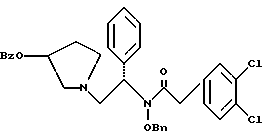
11. Способ по п.10, который далее включает способ получения соединения формулы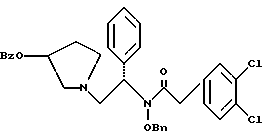
где Bz обозначает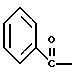
Bn обозначает бензил,
взаимодействием соединения формулы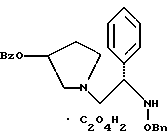
с 3,4-дихлорфенилацетил хлоридом в присутствии основания.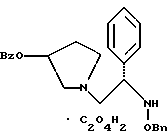
где Bz обозначает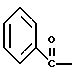
Bn обозначает бензил,
взаимодействием соединения формулы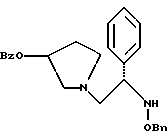
с щавелевой кислотой.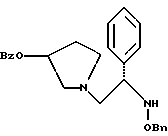
где Bz обозначает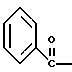
Bn обозначает бензил,
взаимодействием соединения формулы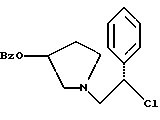
с NH2OBn в присутствии основания.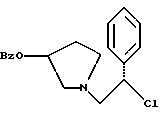
где Bz обозначает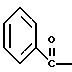
взаимодействием смеси соединений формул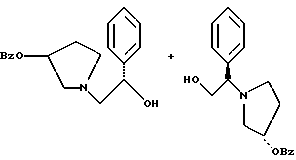
с метансульфонилхлоридом в присутствии основания.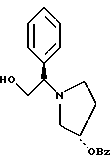
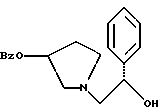
где Bz обозначает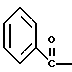
взаимодействием соединения формулы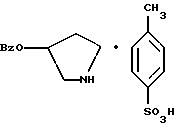
с соединением формулы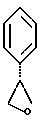
в присутствии основания.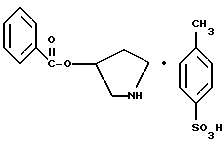
взаимодействием соединения формулы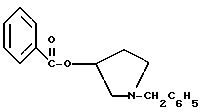
с водородом в присутствии пара-толуолсульфоновой кислоты.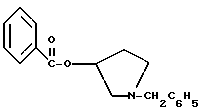
взаимодействием соединения формулы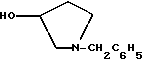
с бензоилхлоридом.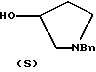
где Bn обозначает бензил,
подвергают взаимодействию с бензоилхлоридом с использованием метиленхлорида в качестве растворителя с получением соединения формулы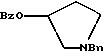
Bz обозначает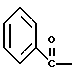
Bn обозначает бензил,
полученное соединение подвергают взаимодействию с пара-толуолсульфоновой кислотой с использованием катализатора палладий на углероде, ТГФ (тетрагидрофуран) в качестве растворителя, водорода и температуры 50oC с получением соединения формулы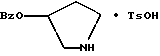
где Bz обозначает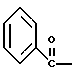
TsOH обозначает пара-толуолсульфоновую кислоту,
полученное соединение подвергают взаимодействию с соединением формулы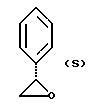
с использованием толуола в качестве растворителя, гидроксида натрия и температуры 100oC с получением смеси соединений формул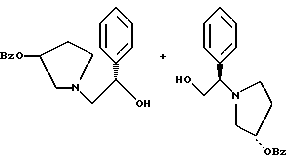
где Bz обозначает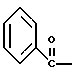
полученную смесь соединений подвергают взаимодействию с метансульфонилхлоридом с использованием дихлорэтана в качестве растворителя и триэтиламина с получением соединения формулы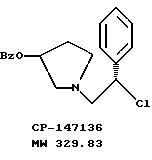
где Bz обозначает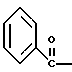
полученное соединение подвергают взаимодействию с гидрохлоридом O-бензилгидроксиламина с использованием изопропанола в качестве растворителя и триэтиламина с получением соединения формулы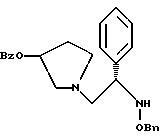
где Bz обозначает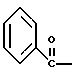
Bn обозначает бензил,
полученное соединение подвергают взаимодействию с щавелевой кислотой с использованием изопропанола в качестве растворителя с получением соединения формулы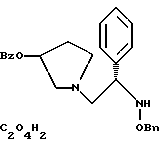
где Bz обозначает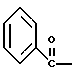
Bn обозначает бензил,
полученное соединение подвергают взаимодействию с соединением формулы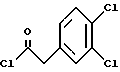
с использованием метиленхлорида в качестве растворителя и суспензии бикарбоната натрия в воде с получением соединения формулы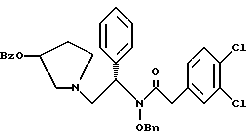
где Bz обозначает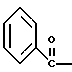
Bn обозначает бензил,
полученное соединение подвергают взаимодействию с гидроксидом лития, растворенным в воде, с использованием ТГФ и метанола в качестве растворителей с получением соединения формулы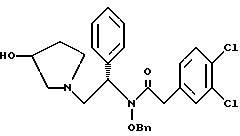
где Bn обозначает бензил,
полученное соединение подвергают взаимодействию с тремя эквивалентами концентрированной соляной кислоты с использованием катализатора 5% палладий на углероде и метанола в качестве растворителя с получением соединения формулы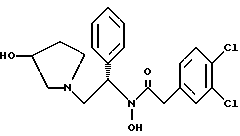
полученное соединение подвергают взаимодействию с пара-толуолсульфоновой кислотой с использованием метиленхлорида и этилацетата в качестве растворителей с получением соединения формулы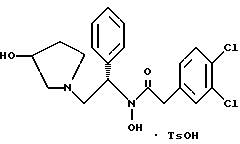
где TsOH обозначает пара-толуолсульфоновую кислоту.
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| УСТРОЙСТВО для НАГРЕВА ДЕТАЛЕЙ В ЭЛЕКТРОЛИТЕ | 0 |
|
SU254545A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| DE 4034785 A1, 29.02.1996 | |||
| Способ получения производных 2-аминоэтанола | 1989 |
|
SU1727531A3 |

