Область техники
Настоящее изобретение относится к новым хинуклидиновым производным и их применению в качестве фармацевтических средств. Вследствие их фармакологического профиля соединения по настоящему изобретению могут быть полезны для лечения таких разнообразных заболеваний или расстройств, как связанные с холинэргической системой центральной нервной системы (ЦНС), периферической нервной системы (ПНС), заболеваний или расстройств, связанных с сокращением гладкой мускулатуры, эндокринных заболеваний или расстройств, заболеваний или расстройств, связанных с нейродегенерацией, заболеваний или расстройств, связанных с воспалением, болью, и синдрома отмены, вызванного прекращением употребления химических веществ.
Предшествующий уровень техники
Эндогенный холинэргический нейротрансмиттер, ацетилхолин, осуществляет свое биологическое действие посредством двух типов холинэргических рецепторов: мускариновых ацетилхолинорецепторов (mAChR) и никотиновых ацетилхолинорецепторов (nAChR).
Хорошо установлено, что мускариновые ацетилхолиновые рецепторы важны в отношении памяти и сознания, и множество исследований, направленных на разработку агентов для лечения расстройств, связанных с памятью, сфокусировано на синтезе модуляторов мускариновых ацетилхолиновых рецепторов.
Действительно, некоторые расстройства ЦНС могут быть связаны с холинэргической недостаточностью, дофаминэргической недостаточностью, адренэргической недостаточностью или серотонэргической недостаточностью.
Brown et al. [Brown et al.: Quinuclidine Inhibitors of 2,3-Oxidosqualene Cyclase-Lanosterol Synthase: Optimization from Lipid Profiles; J Med. Chem. 1999, 42, 1306-1311] описывает синтез 3-замещенных хинуклидиновых производных, полезных в качестве ингибиторов биосинтеза холестерина. О действии на никотиновые и/или моноаминовые рецепторы не сообщается.
Сущность изобретения
Настоящее изобретение посвящено предоставлению новых хинуклидиновых производных, которые являются модуляторами никотиновых и/или моноаминовых рецепторов, причем эти модуляторы полезны для лечения заболеваний или расстройств, связанных с холинэргическими рецепторами и, в частности, с никотиновым ацетилхолиновым рецептором, моноаминовым рецептором, в частности с серотониновым рецептором (5-HTR), дофаминовым рецептором (DAR) и норэпинефриновым рецептором (NER), и биогенными аминовыми транспортерами для серотонина (5-НТ), дофамина (DA) и норэпинефрина (NE).
Вследствие их фармакологического профиля соединения по настоящему изобретению могут быть полезны для лечения таких разнообразных заболеваний или расстройств, как связанные с холинэргической системой центральной нервной системы (ЦНС), периферической нервной системы (ПНС), заболеваний или расстройств, связанных с сокращением гладкой мускулатуры, эндокринных заболеваний или расстройств, заболеваний или расстройств, связанных с нейродегенерацией, заболеваний или расстройств, связанных с воспалением, болью, и синдрома отмены, вызванного прекращением употребления химических веществ.
Соединения по настоящему изобретению могут также быть полезны в качестве диагностических средств или агентов мониторинга при различных способах диагностики и, в частности, для рецепторной визуализации (нейровизуализации) in vivo, и их можно использовать в меченой или немеченой форме.
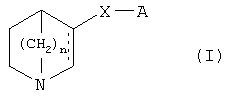
Соответственно, в первом аспекте изобретения предложены хинуклидиновые производные, представленные формулой 1

их энантиомер, или смесь их энантиомеров, или их фармацевтически приемлемая соль присоединения, или их ониевая соль, где
 представляет собой возможную двойную связь;
представляет собой возможную двойную связь;
n представляет собой 1, 2 или 3;
X представляет собой линкер, выбранный из -O-, -O-CH2-, -O-СН2-СН2-, -S-, -SO-, -SO2-, -СН2-, -S-CH2-CH2-, -СН2-, -С(=СН2)-, -NH-, -N(алкил)-, -С(=O)-, -C(=S)-,


А представляет собой моноциклическую или полициклическую, карбоциклическую или гетероциклическую группу, возможно, замещенную один или более раз заместителями, выбранными из группы, состоящей из алкила, циклоалкила, циклоалкил-алкила, алкокси, гидрокси-алкокси, алкокси-алкила, алкокси-алкокси, циклоалкокси, циклоалкокси-алкила, циклоалкокси-алкокси, галогено, CF3, CN, NO2, NH2, карбокси, карбамоила, амидо, сульфамоила и фенила, или другой моноциклической или полициклической, карбоциклической или гетероциклической группой, причем эта дополнительная моноциклическая или полициклическая, карбоциклическая или гетероциклическая группа может, возможно, быть замещена один или более раз заместителями, выбранными из группы, состоящей из алкила, циклоалкила, циклоалкил-алкила, алкокси, гидрокси-алкокси, алкокси-алкила, алкокси-алкокси, циклоалкокси, циклоалкокси-алкила, циклоалкокси-алкокси, галогено, CF3, CN, NO2, NH2, карбокси, карбамоила, амидо, сульфамоила и фенила;
при условии, что, однако,
если Х представляет собой О или S;
тогда А не является фенилом или фенилом, замещенным чем-либо другим, отличным от фенильной группы (то есть бифенильной группой).
В еще одном аспекте изобретения предложены фармацевтические композиции, содержащие терапевтически эффективное количество хинуклидинового производного по настоящему изобретению.
В третьем аспекте данное изобретение относится к применению хинуклидинового производного по настоящему изобретению или его фармацевтически приемлемой соли присоединения для производства фармацевтической композиции/лекарственного средства для лечения, предупреждения или облегчения заболевания, или расстройства, или состояния млекопитающего, включая человека, причем это заболевание, расстройство или состояние чувствительно к действию модулятора никотинового ацетилхолинового рецептора.
В дополнительном аспекте изобретения предложен способ лечения или облегчения заболевания или расстройства организма животного, включая человека, которое чувствительно к действию модулятора никотинового ацетилхолинового рецептора, причем этот способ включает в себя стадию введения в такой организм нуждающегося в этом животного, включая человека, терапевтически эффективного количества хинуклидинового производного по настоящему изобретению.
Другие объекты изобретения будут очевидны специалисту в данной области техники из последующего детального описания и примеров.
Детальное описание изобретения
Хинуклидиновые производные
В первом аспекте в настоящем изобретении предложены новые хинуклидиновые производные, представленные формулой 1
их энантиомер, или смесь их энантиомеров, или их фармацевтически приемлемая соль присоединения, или их ониевая соль, где
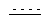 представляет собой возможную двойную связь;
представляет собой возможную двойную связь;
n представляет собой 1, 2 или 3;
X представляет собой линкер, выбранный из -O-, -O-CH2-, -O-CH2-CH2-, -S-, -SO-, -SO2-, -СН2-, -S-CH2-CH2-, -СН2-, -С(=СН2)-, -NH-, -N(алкил)-, -С(=O)-, -C(=S)-,
А представляет собой моноциклическую или полициклическую, карбоциклическую или гетероциклическую группу, возможно, замещенную один или более раз заместителями, выбранными из группы, состоящей из алкила, циклоалкила, циклоалкил-алкила, алкокси, гидрокси-алкокси, алкокси-алкила, алкокси-алкокси, циклоалкокси, циклоалкокси-алкила, циклоалкокси-алкокси, галогено, CF3, CN, NO2, NH2, карбокси, карбамоила, амидо, сульфамоила и фенила, или другой моноциклической или полициклической, карбоциклической или гетероциклической группой, причем эта дополнительная моноциклическая или полициклическая, карбоциклическая или гетероциклическая группа может, возможно, быть замещена один или более раз заместителями, выбранными из группы, состоящей из алкила, циклоалкила, циклоалкил-алкила, алкокси, гидрокси-алкокси, алкокси-алкила, алкокси-алкокси, циклоалкокси, циклоалкокси-алкила, циклоалкокси-алкокси, галогено, CF3, CN, NO2, NH2, карбокси, карбамоила, амидо, сульфамоила и фенила,
при условии, что, однако,
если Х представляет собой О или S;
тогда А не является фенилом или фенилом, замещенным чем-либо другим, отличным от фенильной группы (то есть если Х представляет собой О или S, и А представляет собой фенильную группу, тогда эта фенильная группа должна быть только бифенильной группой).
В предпочтительном воплощении хинуклидиновое производное по настоящему изобретению представляет собой соединение формулы I, где  представляет собой одинарную (ковалентную) связь.
представляет собой одинарную (ковалентную) связь.
В другом предпочтительном воплощении хинуклидиновое производное по настоящему изобретению представляет собой соединение формулы I, где n представляет собой 1, 2 или 3.
В третьем предпочтительном воплощении хинуклидиновое производное по настоящему изобретению представляет собой соединение формулы I, где Х представляет собой линкер, выбранный из -O-, -O-CH2-, -O-CH2-CH2-, -S- и -СН2-. В более предпочтительном воплощении Х представляет собой линкер, выбранный из -O-, -O-СН2- и -O-CH2-CH2-.
В четвертом предпочтительном воплощении хинуклидиновое производное по настоящему изобретению представляет собой соединение формулы I, где А представляет собой моноциклическую или полициклическую карбоциклическую группу, выбранную из фенила; инданила, в частности 4-инданила и 5-инданила; инденила, в частности 1-инденила, 2-инденила и 3-инденила; нафтила, в частности 1-нафтила и 2-нафтила; 5,6,7,8-тетрагидро-нафтила, в частности 5,6,7,8-тетрагидро-1-нафтила и 5,6,7,8-тетрагидро-2-нафтила; азуленила, в частности 1-азуленила, 2-азуленила и 3-азуленила; и флуоренила, в частности 1-флуоренила, 2-флуоренила, 3-флуоренила и 4-флуоренила; и антраценила, в частности 1-антраценила и 2-антраценила; причем данная карбоциклическая группа, возможно, замещена один или два раза заместителями, выбранными из группы, состоящей из алкила, циклоалкила, циклоалкил-алкила, алкокси, гидрокси-алкокси, алкокси-алкила, алкокси-алкокси, циклоалкокси, циклоалкокси-алкила, циклоалкокси-алкокси, галогено, CF3, CN, NO2, NH2, карбокси, карбамоила, амидо, сульфамоила и фенила.
В пятом предпочтительном воплощении хинуклидиновое производное по настоящему изобретению представляет собой соединение формулы I, где А представляет собой ароматическую моноциклическую или полициклическую карбоциклическую группу, выбранную из фенила; инденила, в частности 1-инденила, 2-инденила и 3-инденила; нафтила, в частности 1-нафтила и 2-нафтила; азуленила, в частности 1-азуленила, 2-азуленила и 3-азуленила; и антраценила, в частности 1-антраценила и 2-антраценила, причем данная ароматическая карбоциклическая группа, возможно, замещена один или два раза заместителями, выбранными из группы, состоящей из алкила, циклоалкила, циклоалкил-алкила, алкокси, гидрокси-алкокси, алкокси-алкила, алкокси-алкокси, циклоалкокси, циклоалкокси-алкила, циклоалкокси-алкокси, галогено, CF3, CN, NO2, NH2, карбокси, карбамоила, амидо, сульфамоила и фенила.
В наиболее предпочтительном воплощении хинуклидиновое производное по изобретению формулы I представляет собой
(±)-3-(2-фенилфенилокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(3-фенилфенилокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(4-фенилфенилокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(4-фенилфенил-метокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(нафталин-2-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(5,6,7,8-тетрагидро-2-нафтилокси)-1-аза-бицикло[2.2.2]октан или
(±)-3-(5-инданилокси)-1-аза-бицикло[2.2.2]октан;
или его энантиомер, или его фармацевтически приемлемую соль присоединения, или его ониевую соль.
В шестом предпочтительном воплощении хинуклидиновое производное по изобретению представляет собой соединение формулы I, где А представляет собой моноциклическую или полициклическую гетероциклическую группу, выбранную из пиридила, в частности пирид-2-ила, пирид-3-ила и пирид-4-ила; тиенила, в частности тиен-2-ила и тиен-3-ила; фуранила, в частности фуран-2-ила и фуран-3-ила; пиридазинила, в частности пиридазин-3-ила и пиридазин-4-ила; тиазолила, в частности тиазол-2-ила, тиазол-4-ила и тиазол-5-ила; тиадиазолила, в частности 1,3,4-тиадиазол-2-ила, 1,3,4-тиадиазол-5-ила, 1,2,4-тиадиазол-3-ила и 1,2,4-тиадиазол-6-ила; хинолинила, в частности хинолин-2-ила, хинолин-3-ила, хинолин-4-ила, хинолин-5-ила и хинолин-6-ила; хиноксалинила, в частности хиноксалин-2-ила и хиноксалин-3-ила; бензимидазолила, в частности бензимидазол-2-ила; бензоксазолила, в частности бензоксазол-2-ила, бензтиазолила, в частности бензтиазол-2-ила, причем моноциклическая или полициклическая гетероциклическая группа, возможно, замещена один или более раз заместителями, выбранными из группы, состоящей из алкила, циклоалкила, циклоалкил-алкила, алкокси, гидрокси-алкокси, алкокси-алкила, алкокси-алкокси, циклоалкокси, циклоалкокси-алкила, циклоалкокси-алкокси, галогено, CF3, CN, NO2, NH2, карбокси, карбамоила, амидо, сульфамоила и фенила, или другой моноциклической или полициклической, карбоциклической или гетероциклической группой, причем эта дополнительная моноциклическая или полициклическая, карбоциклическая или гетероциклическая группа может, возможно, быть замещена один или более раз заместителями, выбранными из группы, состоящей из алкила, циклоалкила, циклоалкил-алкила, алкокси, гидрокси-алкокси, алкокси-алкила, алкокси-алкокси, циклоалкокси, циклоалкокси-алкила, циклоалкокси-алкокси, галогено, CF3, CN, NO2, NH2, карбокси, карбамоила, амидо, сульфамоила и фенила.
В седьмом предпочтительном воплощении хинуклидиновое производное по изобретению представляет собой соединение формулы I, где А представляет собой моноциклическую гетероциклическую группу, выбранную из пиридила, в частности пирид-2-ила, пирид-3-ила и пирид-4-ила; тиенила, в частности тиен-2-ила и тиен-3-ила; фуранила, в частности фуран-2-ила и фуран-3-ила; пиридазинила, в частности пиридазин-3-ила и пиридазин-4-ила; тиазолила, в частности тиазол-2-ила, тиазол-4-ила и тиазол-5-ила; тиадиазолила, в частности 1,3,4-тиадиазол-2-ила, 1,3,4-тиадиазол-5-ила, 1,2,4-тиадиазол-3-ила и 1,2,4-тиадиазол-5-ила; причем данная моноциклическая гетероциклическая группа, возможно, замещена один или более раз заместителями, выбранными из группы, состоящей из алкила, циклоалкила, алкокси, циклоалкокси, галогено, CF3, CN, NO2, NH2, фенила, 2-тиенила, 3-тиенила, 2-фуранила, 3-фуранила и 3-пиридинила, причем данные фенильные, 2-тиенильные, 3-тиенильные, 2-фуранильные, 3-фуранильные и 3-пиридинильные группы могут, возможно, быть замещены один или два раза заместителями, выбранными из группы, состоящей из алкила, циклоалкила, алкокси, галогено, CF3, CN, NO2, NH2 и фенила.
В наиболее предпочтительном воплощении хинуклидиновое производное по изобретению формулы I представляет собой
(±)-3-(3,4,5-трихлор-тиен-2-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(5-бром-тиазол-2-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(5-фенил-тиазол-2-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-[5-(2,4-дифтор-фенил)-тиазол-2-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[5-(3-тиенил)-тиазол-2-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[5-(2-тиенил)-тиазол-2-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[5-(3-фуранил)-тиазол-2-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[5-(3-пиридил)-тиазол-2-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-(6-хлор-пиридазин-3-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(6-бром-пиридазин-3-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(6-фенил-пиридазин-3-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-[6-(3-тиенил)пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[6-(2-тиенил)пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[6-(2-фуранил)-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[6-(3-фуранил)-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[6-(3-пиридил)-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-(5-фенил-1,3,4-тиадиазол-2-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(5-фенил-1,2,4-тиадиазол-3-илокси)-1-аза-бицикло[2.2.2]октан или
(±)-3-[5-(2-тиенил)-1,3,4-тиадиазол-2-илокси]-1-аза-бицикло[2.2.2]октан;
или его энантиомер, или его фармацевтически приемлемую соль присоединения, или его ониевую соль.
В восьмом предпочтительном воплощении хинуклидиновое производное по изобретению представляет собой соединение формулы I, где А представляет собой полициклическую гетероциклическую группу, выбранную из индолила, в частности индол-2-ила и индол-3-ила; изоиндолила, в частности изоиндол-2-ила; хинолинила, в частности хинолин-2-ила, хинолин-3-ила, хинолин-4-ила, хинолин-5-ила и хинолин-6-ила; хиноксалинила, в частности хиноксалин-2-ила и хиноксалин-3-ила; бензимидазолила, в частности бензимидазол-2-ила; бензоксазолила, в частности бензоксазол-2-ила, бензтиазолила, в частности бензтиазол-2-ила; бензизотиазолила, в частности бензизотиазол-3-ила; бензтриазолила, в частности 1,2,3-бензтриазол-1-ила; имидазо[1,2-b]пиридазинила, в частности имидазо[1,2-b]пиридазин-6-ила; дибензофуранила, в частности дибензофуран-2-ила; причем данная моноциклическая или полициклическая гетероциклическая группа, возможно, замещена один или более раз заместителями, выбранными из группы, состоящей из алкила, циклоалкила, алкокси, циклоалкокси, галогено, CF3, CN, NO2, NH2 и фенила, причем данная фенильная группа может, возможно, быть замещена один или более раз заместителями, выбранными из группы, состоящей из алкила, циклоалкила, алкокси, галогено, CF3, CN, NO2, NH2 и фенила.
В наиболее предпочтительном воплощении хинуклидиновое производное по изобретению формулы I представляет собой
(±)-3-[(1,3-дион)-2-изоиндолил-метокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[(1,3-дион)-2-изоиндолил-этокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-(2-хинолинилокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(2-хинолинилокси)-1 -аза-бицикло[2.2.2]октана метилиум йодид;
(±)-3-(6-хинолинилокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(2-хиноксалинилокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(2-хиноксалинилокси)-1 -аза-бицикло[2.2.2]октана метилиум йодид;
(±)-3-(3-хлор-2-хиноксалинилокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(3-метокси-2-хиноксалинилокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(бензоксазол-2-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(бензотиазол-2-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(6-хлор-бензотиазол-2-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(1,2-бензоизотиазол-3-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(1,2-бензоизотиазол-3-илокси)-1 -аза-бицикло[2.2.2]октан;
(±)-3-(1-метил-бензоимидазол-2-илокси)-1-аза-бицикло[2.2.2]октан; или
(±)-3-(бензотриазол-1-илокси)-1-азабицикло[2.2.2]октан;
или его энантиомер, или его фармацевтически приемлемую соль присоединения, или его ониевую соль.
В другом предпочтительном воплощении хинуклидиновое производное по изобретению представляет собой соединение формулы II

где
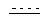 представляет собой возможную двойную связь;
представляет собой возможную двойную связь;
n представляет собой 1, 2 или 3;
X представляет собой линкер, выбранный из -O-, -O-СН2-, -O-СН2-СН2-, -S-, -SO-, -SO2-, -СН2-, -S-CH2-CH2-, -СН2-, -С(=СН2)-, -NH-, -N(алкил)-, -С(=O)-, -C(=S)-,
Y представляет собой О, S, SO2 или NR', где R' представляет собой водород или алкил.
В более предпочтительном воплощении данного аспекта хинуклидиновое производное по изобретению представляет собой соединение формулы II, где представляет собой одинарную (ковалентную) связь.
В другом предпочтительном воплощении данного аспекта хинуклидиновое производное по изобретению представляет собой соединение формулы II, где n представляет собой 1, 2 или 3.
В третьем предпочтительном воплощении данного аспекта хинуклидиновое производное по изобретению представляет собой соединение формулы II, где Х представляет собой линкер, выбранный из -O-, -О-СН2-, -O-СН2-СН2-, -S- и -СН2-.
В четвертом предпочтительном воплощении данного аспекта хинуклидиновое производное по изобретению представляет собой соединение формулы II, где Y представляет собой О, S, SO2 или NR', где R' представляет собой водород или алкил.
В наиболее предпочтительном воплощении хинуклидиновое производное по изобретению формулы II представляет собой
(±)-3-(дибензофуран-2-илокси)-1-азабицикло[2.2.2]октан;
или его энантиомер, или его фармацевтически приемлемую соль присоединения, или его ониевую соль.
В еще одном предпочтительном воплощении хинуклидиновое производное по изобретению представляет собой соединение формулы III

где
представляет собой возможную двойную связь;
n представляет собой 1, 2 или 3;
X представляет собой линкер, выбранный из -O-, -O-CH2-, -O-CH2-CH2-, -S-, -SO-, -SO2-, -CH2-, -S-CH2-CH2-, -CH2-, -C(=CH2)-, -NH-, -N(алкил)-, -С(=O)-, -C(=S)-,
В представляет собой моноциклическую или полициклическую, карбоциклическую или гетероциклическую группу, возможно, замещенную один или более раз заместителями, выбранными из группы, состоящей из алкила, циклоалкила, циклоалкил-алкила, алкокси, гидрокси-алкокси, алкокси-алкила, алкокси-алкокси, циклоалкокси, циклоалкокси-алкила, циклоалкокси-алкокси, галогено, CF3, CN, NO2, NH2, карбокси, карбамоила, амидо, сульфамоила и фенила, или другой моноциклической или полициклической, карбоциклической или гетероциклической группой, причем эта дополнительная моноциклическая или полициклическая, карбоциклическая или гетероциклическая группа может, возможно, быть замещена один или более раз заместителями, выбранными из группы, состоящей из алкила, циклоалкила, циклоалкил-алкила, алкокси, гидрокси-алкокси, алкокси-алкила, алкокси-алкокси, циклоалкокси, циклоалкокси-алкила, циклоалкокси-алкокси, галогено, CF3, CN, NO2, NH2, карбокси, карбамоила, амидо, сульфамоила и фенила.
В более предпочтительном воплощении данного аспекта хинуклидиновое производное по настоящему изобретению представляет собой соединение формулы III, где представляет собой одинарную (ковалентную) связь.
В другом предпочтительном воплощении данного аспекта хинуклидиновое производное по изобретению представляет собой соединение формулы III, где n представляет собой 1, 2 или 3.
В третьем предпочтительном воплощении данного аспекта хинуклидиновое производное по изобретению представляет собой соединение формулы III, где Х представляет собой линкер, выбранный из -O-, -O-CH2-, -O-СН2-СН2-, -S- и -СН2-.
В четвертом предпочтительном воплощении данного аспекта хинуклидиновое производное по изобретению представляет собой соединение формулы III, где В представляет собой моноциклическую или полициклическую, карбоциклическую или гетероциклическую группу, возможно, замещенную один или более раз заместителями, выбранными из группы, состоящей из алкила, циклоалкила, циклоалкил-алкила, алкокси, гидрокси-алкокси, алкокси-алкила, алкокси-алкокси, циклоалкокси, циклоалкокси-алкила, циклоалкокси-алкокси, галогено, CF3, CN, NO2, NH2, карбокси, карбамоила, амидо, сульфамоила и фенила, или другой моноциклической или полициклической, карбоциклической или гетероциклической группой, причем эта дополнительная моноциклическая или полициклическая, карбоциклическая или гетероциклическая группа может, возможно, быть замещена один или более раз заместителями, выбранными из группы, состоящей из алкила, циклоалкила, циклоалкил-алкила, алкокси, гидрокси-алкокси, алкокси-алкила, алкокси-алкокси, циклоалкокси, циклоалкокси-алкила, циклоалкокси-алкокси, галогено, CF3, CN, NO2, NH2, карбокси, карбамоила, амидо, сульфамоила и фенила.
В пятом предпочтительном воплощении данного аспекта хинуклидиновое производное по изобретению представляет собой соединение формулы III, где В представляет собой фенильную группу, причем данный фенил, возможно, замещен один или два раза заместителями, выбранными из группы, состоящей из алкила, циклоалкила, алкокси, циклоалкокси, галогено, CF3, CN, NO2, NH2 и фенила.
В наиболее предпочтительном воплощении хинуклидиновое производное по изобретению формулы III представляет собой
(±)-3-(2-фенил-имидазо[1,2-b]пиридазин-6-илокси)-1-аза-бицикло[2.2.2]октан;
или его энантиомер, или его фармацевтически приемлемую соль присоединения, или его ониевую соль.
Считается, что любая комбинация двух или более чем двух воплощений, описанных здесь, находится в рамках данного изобретения.
Определение заместителей
В контексте данного изобретения галогено представляет собой фторо, хлоро, бромо или иодо. Таким образом, тригалометильная группа представляет собой, например, трифторметильную группу, трихлорметильную группу и подобные тригалоидзамещенные метильные группы.
В контексте данного изобретения алкильная группа обозначает унивалентную, насыщенную, прямую или разветвленную углеводородную цепь. Углеводородная цепь предпочтительно содержит от одного до восемнадцати атомов углерода (С1-18алкил), более предпочтительно от одного до шести атомов углерода (C1-6-алкил; низший алкил), включая пентил, изопентил, неопентил, третичный пентил, гексил и изогексил. В предпочтительном воплощении алкил представляет собой С1-4-алкильную группу, включая бутил, изобутил, вторичный бутил и третичный бутил. В другом предпочтительном воплощении настоящего изобретения алкил представляет собой С1-3-алкильную группу, которая, в частности, может представлять собой метил, этил, пропил или изопропил.
В контексте данного изобретения циклоалкильная группа обозначает циклическую алкильную группу, предпочтительно содержащую от трех до семи атомов углерода (С3-7-циклоалкил), включая циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
В контексте данного изобретения циклоалкил-алкильная группа обозначает циклоалкильную группу, как определено выше, причем данная циклоалкильная группа замещена по алкильной группе, так же как определено выше. Примеры предпочтительных циклоалкил-алкильных групп по изобретению включают в себя циклопропилметил и циклопропилэтил.
В контексте данного изобретения алкокси-группа обозначает "алкил-O-" группу, где алкил такой, как определено выше. Примеры предпочтительных алкокси-группп по настоящему изобретению включают метокси и этокси.
В контексте данного изобретения гидрокси-алкокси группа обозначает алкокси-группу, как определено выше, причем данная алкокси-группа замещена одной или более чем одной гидрокси-группами. Предпочтительные гидрокси-алкокси группы по настоящему изобретению включают в себя 2-гидрокси-этокси, 3-гидрокси-пропокси, 4-гидрокси-бутокси, 5-гидрокси-пентокси и 6-гидрокси-гексокси.
В контексте данного изобретения циклоалкокси группа обозначает "циклоалкил-O-" группу, где циклоалкил такой, как определено выше.
В контексте данного изобретения алкокси-алкильная группа обозначает "алкил-O-алкил-" группу, где алкил такой, как определено выше. Примеры предпочтительных алкокси-алкильных групп по настоящему изобретению включают в себя метокси-метил, метокси-этил, этокси-метил и этокси-этил.
В контексте данного изобретения алкокси-алкокси группа обозначает "алкил-О-алкил-O-" группу, где алкил такой, как определено выше. Примеры предпочтительных алкокси-алкокси групп по настоящему изобретению включают в себя метокси-метокси, метокси-этокси, этокси-метокси и этокси-этокси.
В контексте данного изобретения циклоалкокси-алкильная группа обозначает "циклоалкил-O-алкил" группу, где циклоалкил и алкил такие, как определено выше.
В контексте данного изобретения циклоалкокси-алкоси группа обозначает "циклоалкил-О-алкил-O-" группу, где циклоалкил и алкил такие, как определено выше.
В контексте данного изобретения моно- или полициклическая карбоциклическая группа представляет собой моно- или полициклическую карбоциклическую группу, содержащую углерод только в качестве атома в составе кольца. Кольцевая структура может, в частности, быть ароматической (то есть арильная группа), или насыщенной, или частично насыщенной.
Предпочтительные моно- или полициклические карбоциклические группы по настоящему изобретению включают фенил; инданил, в частности 4-инданил и 5-инданил; инденил, в частности 1-инденил, 2-инденил и 3-инденил; нафтил, в частности 1-нафтил и 2-нафтил; 5,6,7,8-тетрадигро-нафтил, в частности 5,6,7,8-тетрадигро-1-нафтил и 5,6,7,8-тетрадигро-2-нафтил; азуленил, в частности 1-азуленил, 2-азуленил и 3-азуленил; флуоренил, в частности 1-флуоренил, 2-флуоренил, 3-флуоренил и 4-флуоренил; и антраценил, в частности 1-антраценил и 2-антраценил.
Моно- или полициклическая карбоциклическая группа может, в частности, быть ароматической группой (арильной). Предпочтительные арильные группы по настоящему изобретению включают фенил; инденил, в частности 1-инденил, 2-инденил и 3-инденил; нафтил, в частности 1-нафтил и 2-нафтил; азуленил, в частности 1-азуленил, 2-азуленил и 3-азуленил; и антраценил, в частности 1-антраценил и 2-антраценил.
В контексте данного изобретения моно- или полициклическая гетероциклическая группа представляет собой моно- или полициклическое соединение, которое содержит один или более чем один гетероатом в своей кольцевой структуре. Термин полигетероциклические группы включает в себя бензоконденсированные пяти- и шестичленные гетероциклические кольца, содержащие один или более чем один гетероатом. Предпочтительные гетероатомы включают азот (N), кислород (О) и серу (S). Одна или более из этих кольцевых структур может, в частности, быть ароматической (то есть представлять собой гетероарил).
Предпочтительные моноциклические гетероциклические группы по настоящему изобретению включают в себя пиридил, в частности пирид-2-ил, пирид-3-ил и пирид-4-ил; тиенил, в частности тиен-2-ил и тиен-3-ил; фуранил, в частности фуран-2-ил и фуран-3-ил; пиридазинил, в частности пиридазин-3-ил и пиридазин-4-ил; тиазолил, в частности тиазол-2-ил, тиазол-4-ил и тиазол-5-ил; и тиадиазолил, в частности 1,3,4-тиадиазол-2-ил, 1,3,4-тиадиазол-5-ил, 1,2,4-тиадиазол-3-ил и 1,2,4-тиадиазол-5-ил.
Предпочтительные полициклические гетероциклические группы по настоящему изобретению включают индолил, в частности индол-2-ил и индол-3-ил; изоиндолил, в частности изоиндол-2-ил; хинолинил, в частности хинолин-2-ил, хинолин-3-ил, хинолин-4-ил, хинолин-5-ил и хинолин-6-ил; хиноксалинил, в частности хиноксалин-2-ил и хиноксалин-3-ил; бензимидазолил, в частности бензимидазол-2-ил; бензоксазолил, в частности бензоксазол-2-ил; бензтиазолил, в частности бензтиазол-2-ил; бензизотиазолил, в частности бензизотиазол-3-ил; бензтриазолил, в частности 1,2,3-бензтриазол-1-ил; имидазо[1,2-b]пиридазинил, в частности имидазо[1,2-b]пиридазин-6-ил; и дибензофуранил, в частности дибензофуран-2-ил.
Фармацевтически приемлемые соли
Хинуклидиновое производное по настоящему изобретению может быть предложено в любой форме, подходящей для планируемого введения. Подходящие формы включают в себя фармацевтически (то есть физиологически) приемлемые соли и пре- или пролекарственные формы хинуклидинового производного по настоящему изобретению.
Примеры фармацевтически приемлемых солей присоединения включают, без ограничения, нетоксичные соли присоединения неорганических и органических кислот, как, например, гидрохлорид, полученный из соляной кислоты, гидробромид, полученный из бромистоводородной кислоты, нитрат, полученный из азотной кислоты, перхлорат, полученный из перхлорной кислоты, фосфат, полученный из фосфорной кислоты, сульфат, полученный из серной кислоты, формиат, полученный из муравьиной кислоты, ацетат, полученный из уксусной кислоты, аконат, полученный из аконитовой кислоты, аскорбат, полученный из аскорбиновой кислоты, бензолсульфонат, полученный из бензолсульфоновой кислоты, бензоат, полученный из бензойной кислоты, циннамат, полученный из коричной кислоты, цитрат, полученный из лимонной кислоты, эмбонат, полученный из эмбоновой кислоты, энантат, полученный из энантовой кислоты, фумарат, полученный из фумаровой кислоты, глутамат, полученный из глутаминовой кислоты, гликолят, полученный из гликолевой кислоты, лактат, полученный из молочной кислоты, малеат, полученный из малеиновой кислоты, малонат, полученный из малоновой кислоты, манделат, полученный из миндальной кислоты, метансульфонат, полученный из метансульфоновой кислоты, нафталин-2-сульфонат, полученный из нафталин-2-сульфоновой кислоты, фталат, полученный из фталевой кислоты, салицилат, полученный из салициловой кислоты, сорбат, полученный из сорбиновой кислоты, стеарат, полученный из стеариновой кислоты, сукцинат, полученный из янтарной кислоты, тартрат, полученный из винной кислоты, толуол-лара-сульфонат, полученный из пара-толуолсульфоновой кислоты, и тому подобное. Такие соли могут быть получены с помощью процедур, хорошо известных и описанных в данной области техники.
Другие кислоты, такие как щавелевая кислота, которые не могут считаться фармацевтически приемлемыми, могут быть полезны при получении солей, полезных в качестве промежуточных соединений при получении химического соединения по настоящему изобретению и его фармацевтически приемлемой соли присоединения кислоты.
Примеры фармацевтически приемлемых катионных солей химического соединения по настоящему изобретению включают, без ограничения, соль натрия, калия, кальция, магния, цинка, алюминия, лития, холина, лизина и аммония и тому подобное химического соединения по настоящему изобретению, содержащего анионную группу. Такие катионные соли могут быть получены с помощью процедур, хорошо известных и описанных в данной области техники.
В контексте данного изобретения "ониевые соли" N-содержащих соединений также рассматриваются как фармацевтически приемлемые соли (азониевые соли). Предпочтительные азониевые соли включают алкилониевые соли, в частности метил- и этилониевые соли; циклоалкилониевые соли, в частности циклопропилониевые соли; и циклоалкилалкилониевые соли, в частности циклопропилметилониевые соли.
Стерические изомеры
Хинуклидиновые производные по настоящему изобретению могут существовать в (+) и (-) формах, а также в рацемических формах (±). Рацематы этих изомеров и отдельные изомеры сами по себе входят в рамки настоящего изобретения.
Рацемические формы могут быть разделены на оптические антиподы с помощью известных способов и методик. Один путь разделения диастереомерных солей заключается в применении оптически активной кислоты и выделении оптически активного аминного соединения путем обработки основанием. Другой способ разделения рацематов на оптические антиподы основан на хроматографии на оптически активном матриксе. Рацемические соединения по настоящему изобретению могут быть, таким образом, разделены на их оптические антиподы, например, путем фракционированной кристаллизации d- или I- (тартраты, манделаты или камфорсульфонаты) солей.
Хинуклидиновые производные по настоящему изобретению могут также быть разделены путем образования диастереомерных амидов путем взаимодействия химических соединений по настоящему изобретению с оптически активной активированной карбоновой кислотой, такой как, например, кислота, полученная из (+) или (-) фенилаланина, (+) или (-) фенилглицина, (+) или (-) камфановой кислоты или путем образования диастереомерных карбаматов путем взаимодействия химического соединения по настоящему изобретению с оптически активным хлорформиатом или тому подобным.
Дополнительные способы разделения оптических изомеров известны из области техники. Такие способы включают в себя те, что описаны Jagues J, Collet A & Wilen S в "Enantiomers. Racemates and Resolutions". John Wiley and Sons, New York (1981).
Оптически активные соединения также могут быть получены из оптически активных исходных материалов.
Способы получения
Хинуклидиновые производные по настоящему изобретению могут быть получены с помощью традиционных способов химического синтеза, например тех, что описаны в рабочих примерах. Исходные материалы для способов, описанных в настоящей заявке, известны или легко могут быть приготовлены с помощью традиционных способов из коммерчески доступных химических веществ.
Также одно соединение по настоящему изобретению может быть превращено в другое соединение по настоящему изобретению с использованием общепринятых способов.
Конечные продукты реакций, описанных здесь, могут быть выделены с помощью общепринятых методик, например путем экстракции, кристаллизации, перегонки, хроматографии и тому подобного.
Биологическая активность
Настоящее изобретение относится к новым хинуклидиновым производным, которые, как обнаружено, являются холинэргическими лигандами никотиновых ацетилхолиновых рецепторов (nAChR) и модуляторами моноаминовых рецепторов, в частности транспортеров биогенных аминов, таких как серотониновый рецептор (5-HTR), дофаминовый рецептор (DAR) и норэпинефриновый рецептор (NER), и транспортеров биогенных аминов для серотонина (5-НТ), дофамина (DA) и норэпинефрина (NE). Также предпочтительные хинуклидиновые производные по настоящему изобретению демонстрируют избирательную α7 активность, как показано в рабочих примерах. Соединения по настоящему изобретению могут, в частности, быть агонистами, частичными агонистами, антагонистами и аллостерическими модуляторами рецептора.
Вследствие своего фармакологического профиля хинуклидиновые производные по настоящему изобретению могут быть полезны для лечения разнообразных заболеваний или состояний, таких как заболевания, связанные с ЦНС, заболевания, связанные с ПНС, заболевания, связанные с сокращением гладкой мускулатуры, эндокринные расстройства, заболевания, связанные с нейродегенерацией, заболевания, связанные с воспалением, болью, и синдром отмены, вызванный прекращением употребления химических веществ.
В предпочтительном воплощении хинуклидиновые производные по настоящему изобретению используют для лечения заболеваний, расстройств или состояний, связанных с центральной нервной системой. Такие заболевания или расстройства включают тревогу, когнитивные расстройства, дефицит обучения, дефицит и дисфункцию памяти, болезнь Альцгеймера, дефицит внимания, синдром дефицита внимания с гиперактивностью (СДВГ), болезнь Паркинсона, болезнь Гентингтона, боковой амиотрофический склероз, синдром Жиля де ла Туретта, психоз, депрессию, манию, маниакальную депрессию, шизофрению, обсессивно-компульсивные расстройства (ОКР), панические расстройства, расстройства питания, такие как нервная анорексия, булимия и ожирение, нарколепсию, ноцицепцию, СПИД-деменцию, старческую деменцию, периферическую невропатию, аутизм, дислексию, позднюю дискинезию, гиперкинезию, эпилепсию, булимию, пост-травматический синдром, социальную фобию, расстройства сна, псевдодеменцию, синдром Ганзера, предменструальный синдром, синдром поздней лютеальной фазы, синдром хронической усталости, мутизм, трихотилломанию и нарушение суточного ритма организма.
В предпочтительном воплощении заболевания расстройства или состояния, связанные с центральной нервной системой, при которых используются хинуклидиновые производные по настоящему изобретению, представляют собой когнитивные расстройства, психоз, шизофрению и/или депрессию.
В другом предпочтительном воплощении хинуклидиновые производные по настоящему изобретению могут быть полезны для лечения заболеваний, расстройств или состояний, связанных с сокращениями гладкой мускулатуры, включая конвульсивные расстройства, стенокардию, преждевременные роды, судороги, диарею, астму, эпилепсию, позднюю дискинезию, гиперкинезию, преждевременную эякуляцию и затрудненную эрекцию.
В еще одном предпочтительном воплощении хинуклидиновые производные по настоящему изобретению могут быть полезны для лечения эндокринных расстройств, таких как тиреотоксикоз, феохромоцитома, гипертензия и аритмии.
В еще одном предпочтительном воплощении хинуклидиновые производные по настоящему изобретению могут быть полезны для лечения нейродегенеративных расстройств, включая временную аноксию и индуцированную нейродегенерацию.
В еще одном предпочтительном воплощении хинуклидиновые производные по настоящему изобретению могут быть полезны для лечения воспалительных заболеваний, расстройств или состояний, включая воспалительные кожные расстройства, такие как акне и розовые угри, болезнь Крона, воспалительное заболевание кишечника, язвенный колит и диарея.
В еще одном предпочтительном воплощении хинуклидиновые производные по настоящему изобретению могут быть полезны для лечения слабой, умеренной или даже сильной боли острого, хронического или рецидивирующего характера, а также боли, вызванной мигренью, послеоперационной боли и фантомной боли в ампутированной конечности. Боль, в частности, может представлять собой невропатическую боль, хроническую головную боль, центральную боль, боль, связанную с диабетической невропатией, с посттерапевтической невралгией или с повреждением периферических нервов.
Наконец, хинуклидиновые производные по настоящему изобретению могут быть полезны для лечения синдрома отмены, вызванного прекращением применения веществ, вызывающих привыкание. Такие вещества, вызывающие привыкание, включают никотин-содержащие продукты, такие как табак, опиоиды, такие как героин, кокаин и морфин, бензодиазепины и бензодиазепиноподобные лекарства и алкоголь. Отмена веществ, вызывающих привыкание, в общем, влечет за собой травматическое переживание, характеризующееся тревогой и фрустрацией, гневом, тревогой, трудностями концентрации, беспокойством, сниженным сердечным ритмом и повышенным аппетитом и увеличением массы.
В данном контексте "лечение" подразумевает лечение, предупреждение, профилактику и облегчение синдрома отмены и абстиненции, а также лечение, приводящее к добровольному сокращению приема веществ, вызывающих привыкание.
В другом аспекте хинуклидиновые производные по настоящему изобретению используют в качестве диагностических агентов, например, для идентификации и локализации никотиновых рецепторов в различных тканях.
Фармацевтические композиции
В другом аспекте изобретения предложены новые фармацевтические композиции, содержащие терапевтически эффективное количество хинуклидиновых производных по настоящему изобретению.
Хотя химическое соединение по настоящему изобретению для применения в терапии можно вводить в форме исходного химического соединения, предпочтительно вводить активный ингредиент, возможно, в форме физиологически приемлемой соли, в фармацевтической композиции вместе с одним или более чем одним адъювантом, эксципиентом, носителем, буфером, разбавителем и/или другим обычным фармацевтическим вспомогательным веществом.
В предпочтительном воплощении в изобретении предложены фармацевтические композиции, содержащие хинуклидиновое производное вместе с одним или более чем одним фармацевтически приемлемым носителем для него и, возможно, другими терапевтическими и/или профилактическими ингредиентами, известными и используемыми в данной области техники. Носитель(и) должен быть «приемлемым» в смысле совместимости с другими ингредиентами препарата и не быть вредным для реципиента.
Фармацевтическую композицию по настоящему изобретению можно вводить любым удобным путем, который подходит для желаемой терапии. Предпочтительные пути введения включают в себя пероральное введение, в частности в таблетках, в капсулах, в драже, в порошке или в жидкой форме, и парентеральное введение, в частности кожное, подкожное, внутримышечное, или внутривенную инъекцию. Фармацевтическую композицию по настоящему изобретению может производить специалист в данной области путем применения стандартных способов и общепринятых методик, подходящих для желаемого препарата. При желании могут быть использованы композиции, адаптированные для замедленного высвобождения активного ингредиента.
Дополнительные детали по методикам приготовления и введения можно найти в последнем издании Remington's Pharmaceutical Sciences (Maack Publishing Co., Easton, PA).
Действительная дозировка зависит от природы и тяжести заболевания, которое лечат, находится на усмотрении лечащего врача и может варьировать путем титрования дозировки в отношении конкретных примеров данного изобретения, для получения желаемого терапевтического эффекта. Однако в настоящем изобретении предполагается, что фармацевтические композиции, содержащие от примерно 0,1 до примерно 500 мг активного ингредиента на индивидуальную дозу, предпочтительно от примерно 1 до примерно 100 мг, наиболее предпочтительно от примерно 1 до примерно 10 мг, подходят для терапевтического лечения.
Активный ингредиент можно вводить в виде одной или нескольких доз в сутки. Удовлетворительный результат может, в некоторых случаях, быть получен при такой низкой дозировке, как 0,1 мкг/кг в/в (внутривенно) и 1 мкг/кг п.о. (перорально). Верхняя граница диапазона дозировок, как считается в настоящем изобретении, составляет примерно 10 мг/кг в/в и 100 мг/кг п.о. Предпочтительные дозировки составляют от примерно 0,1 мкг/кг до примерно 10 мг/кг/сутки в/в и от примерно 1 мкг/кг до примерно 100 мг/кг/сутки п.о.
Способы терапии
Вследствие своего фармакологического профиля соединения по настоящему изобретению могут быть полезны для лечения таких разнообразных заболеваний или состояний, как заболевания, связанные с центральной нервной системой, заболевания, связанные с периферической нервной системой, заболевания, связанные с сокращением гладкой мускулатуры, эндокринные заболевания, заболевания, связанные с нейродегенерацией, заболевания, связанные с воспалением, болью, и синдром отмены, вызванный прекращением употребления химических веществ.
В другом аспекте изобретения предложены способы лечения, предупреждения или облегчения заболеваний, или расстройств, или состояний организма животного, включая человека, причем это заболевание или расстройство чувствительно к действию модулятора моноаминового рецептора, причем данный способ включает в себя стадию введения в такой организм нуждающегося в этом животного, включая человека, терапевтически эффективного количества хинуклидинового производного по настоящему изобретению.
В контексте данного изобретения термин "лечение" включает в себя лечение, предупреждение, профилактику или облегчение, а термин "заболевание" включает в себя болезнь, заболевание, расстройство или состояние, связанное с рассматриваемым заболеванием.
В настоящее время предполагается, что подходящая дозировка лежит в диапазоне от примерно 0,1 до примерно 500 миллиграмм активного вещества в сутки, более предпочтительно от примерно 10 до примерно 70 миллиграмм активного вещества в сутки, вводимого один или два раза в сутки, в зависимости, как правило, от точного способа введения, формы введения, симптома, в отношении которого направлено введение, вовлеченного субъекта и массы тела вовлеченного субъекта и, дополнительно, от предпочтения и опыта лечащего врача или ответственного ветеринара.
ПРИМЕРЫ
Изобретение далее проиллюстрировано со ссылкой на следующие примеры, которые не подразумевают какого-либо ограничения объема заявленного изобретения.
Общие замечания: все реакции, в которые вовлечены чувствительные к воздуху реагенты или промежуточные вещества, осуществляли в атмосфере азота и в безводных растворителях. В качестве осушителя в процедурах обработки использовали сульфат магния; растворители выпаривали при пониженном давлении.
Способ А
Соль фумаровой кислоты и (±)-3-(нафталин-2-илокси)-1-аза-бицикло[2.2.2]октана
(Соединение А1)
К смеси 2-нафтола (5,0 г, 34,5 ммоль), (±)-3-хинуклидинола (2,94 г, 23,1 ммоль), трифенилфосфина (9,0 г, 34,5 ммоль) и тетрагидрофурана (100 мл) добавляли: диэтилазодикарбоксилат (5,4 мл, 34,5 ммоль) при комнатной температуре в течение 30 минут. Реакционной смеси давали перемешаться в течение 20 часов при 50°С. Добавляли водный гидроксид натрия (100 мл, 1 М).
Смесь экстрагировали дихлорметаном (3×100 мл). Хроматография на силикагеле с дихлорметаном, метанолом и концентрированным аммиаком (89:10:1) дала соединение, указанное в заголовке. Соответствующую соль получали путем добавления смеси диэтилового эфира и метанола (9:1), насыщенной при помощи фумаровой кислоты. Выход 3,7 г (43%). Т. пл. 140,9-141,6°С.
Соль фумаровой кислоты и (±)-3-(4-фенилфенилокси)-1-аза-бицикло[2.2.2]октана (Соединение А2)
Получали в соответствии со способом А из 4-фенилфенола. Т. пл. 173,5-185,1°С.
Свободное основание (±)-3-(3-фенилфенилокси)-1-аза-бицикло[2.2.2]октана (Соединение A3)
Получали в соответствии со способом А из 3-фенилфенола. Продукт выделяли в виде масла.
Соль фумаровой кислоты и (±)-3-(2-фенилфенилокси)-1-аза-бицикло[2.2.2]октана (Соединение А4)
Получали в соответствии со способом А из 2-фенилфенола. Т. пл. 125,4°С.
Соль фумаровой кислоты и (±)-3-(6-хинолинокси)-1-аза-бицикло[2.2.2]октана (Соединение А5)
Получали в соответствии со способом А из 6-гидроксихинолина. Т. пл. 146,0-147,0°С.
Соль фумаровой кислоты и (±)-3-(5-инданилокси)-1-аза-бицикло[2.2.2]октана (Соединение А6)
Получали в соответствии со способом А из 5-инданола. Т. пл. 149,3-150,5°С.
Соль фумаровой кислоты и (±)-3-(5,6,7,8-тетрагидро-2-нафтилокси)-1-аза-бицикло[2.2.2]октана (Соединение А7)
Получали в соответствии со способом А из 5,6,7,8-терагидро-2-нафтола. Т.пл. 109,7-111,3°С.
Способ В
Соль фумаровой кислоты и (±)-2-(1-аза-бицикло[2.2.2]окт-3-илокси)-хинолина (Соединение В1)
Смесь (±)-3-хинуклидинола (2,0 г, 15,7 ммоль), 2-хлорхинолина (2,6 г, 15,7 ммоль) и ДМФ (диметилсульфоксид) (30 мл) перемешивали при комнатной температуре. Гидрид натрия (0,94 г, 23,6 ммоль, 60% в масле) добавляли небольшими порциями. Реакционную смесь перемешивали в течение 1,5 часов при 50°С. Добавляли водный гидроксид натрия (50 мл, 1 М), за чем следовала экстракция диэтиловым эфиром (3×50 мл). Объединенные эфирные фазы промывали водным гидроксидом натрия (2×50 мл, 1М). Соответствующую соль получали путем добавления смеси диэтилового эфира и метанола (9:1), насыщенной при помощи фумаровой кислоты. Выход 4,62 г (79%). Т. пл. 160,0-160,5°С.
Соль фумаровой кислоты и (±)-3-(6-хлор-бензотиазол-2-илокси)-1-аза-бицикло[2.2.2]октана (Соединение В2)
Получали в соответствии с процедурой В из 2,6-дихлорбензотиазола. Т. пл. 203-205°С.
Соль фумаровой кислоты и (±)-3-(бензотиазол-2-илокси)-1-аза-бицикло[2.2.2]октана (Соединение В3)
Получали в соответствии с процедурой В из 2-хлорбензотиазола. Т. пл. 173,7-174,2°С.
Соль фумаровой кислоты и (±)-2-(1-аза-бицикло[2.2.2]окт-3-илокси)-3-хлорхиноксалина (Соединение В4)
Получали в соответствии с процедурой В из 2,3-дихлорхиноксалина. Т. пл. 120,8-122,1°С.
Соль фумаровой кислоты и (±)-3-(1-метил-бензоимидазол-2-илокси)-1-аза-бицикло[2.2.2]октана (Соединение В5)
Получали в соответствии с процедурой В из 2-хлор-1-метилбензоимидазола. Т. пл. 184,9-185,9°С.
Соль фумаровой кислоты и (±)-3-(бензоксазол-2-илокси)-1-аза-бицикло[2.2.2]октана (Соединение В6)
Получали в соответствии с процедурой В из 2-хлорбензоксазола. Т. пл. 187,2-188,8°С.
Соль фумаровой кислоты и (±)-2-(1-аза-бицикло[2.2.2]окт-3-илокси)-хиноксалина (Соединение В7)
Получали в соответствии с процедурой В из 2-хлорхиноксалина. Т. пл. 127,7-128,5°С.
Соль фумаровой кислоты и (±)-3-(6-фенилпиридазин-3-илокси)-1-аза-бицикло[2.2.2]октана (Соединение В8)
Получали в соответствии с процедурой В из 3-хлор-6-фенилпиридазина. Т. пл. 168,5-172,0°С.
Соль фумаровой кислоты и (±)-3-(5-фенил-1.3.4-тиадиазол-2-илокси)-1-аза-бицикло[2.2.2]октана (Соединение В9)
Получали в соответствии с процедурой В из 2-хлор-5-фенил-1,3,4-тиадиазола. Т. пл. 168,5-172,0°С.
Соль фумаровой кислоты и (±)-3-(5-бром-тиазол-2-илокси)-1-аза-бицикло[2.2.2]октана (Соединение В10)
Получали в соответствии с процедурой В из 2,5-дибромтиазола, используя в качестве температуры реакции 0°С. Т. пл. 157,8-162,1°С.
Соль фумаровой кислоты и (±)-3-(1,2-бензоизотиазол-3-илокси)-1-аза-бицикло[2.2.2]октана (Соединение В11)
Получали в соответствии с процедурой В из 3-хлор-1,2-бензоизотиазола. Т. пл. 172,3-173,6°С.
Соль фумаровой кислоты и (±)-3-(5-фенил-1,2,4-тиадиазол-3-илокси)-1-аза-бицикло[2.2.2]октана (Соединение В12)
Получали в соответствии с процедурой В из 3-хлор-5-фенил-1,2,4-тиадиазола. Т. пл. 155,0-159,3°С.
Соль фумаровой кислоты и (±)-3-(6-бром-пиридазин-3-илокси)-1-аза-бицикло[2.2.2]октана (Соединение В13)
Получали в соответствии с процедурой В из 3,6-дибромпиридазина. Т. пл. 152,8°С.
Соль фумаровой кислоты и (±)-3-(6-хлор-пиридазин-3-илокси)-1-аза-бицикло[2.2.2]октана (Соединение В14)
Получали в соответствии с процедурой В из 3,6-дихлорпиридазина. Т. пл. 164-164,5°С.
Соль фумаровой кислоты и (±)-3-(3,4,5-трихлор-тиофенил-2-илокси)-1-аза-бицикло[2.2.2]октана (Соединение В15)
Получали в соответствии с процедурой В, используя условия: трет-бутоксид калия, макроциклический эфир (18:6), из тетрахлортиофена. Т. пл. 188-189,4°С.
Соль фумаровой кислоты и (±)-3-(3-метокси-2-хиноксалинилокси)-1-аза-бицикло[2.2.2]октана (Соединение В16)
Смесь (±)-2-(1-аза-бицикло[2.2.2]окт-3-илокси)-3-хлорхиноксалина (Соединение В4; 1,38 г, 4,76 ммоль), карбоната цезия (1,55 г, 4,76 ммоль) и метанола (15 мл) перемешивали в течение 3 часов при 45°С. Добавляли водный гидроксид натрия (50 мл, 1М), за чем следовала экстракция диэтиловым эфиром (3×50 мл). Соответствующую соль получали путем добавления смеси диэтилового эфира и метанола (9:1), насыщенной при помощи фумаровой кислоты. Выход 0,51 г, 27%. Т. пл. 168,5-170,0°С.
Соль фумаровой кислоты и (±)-3-[5-(3-тиенил)-1,3,4-тиадиазол-2-илокси1-1-аза-бицикло[2.2.2]октана (Соединение В17)
Получали в соответствии с процедурой В из 2-хлор-5-(3-тиенил)-1,3,4-тиадиазола. Т. пл. 186-188°С.
Соль фумаровой кислоты и (±)-3-[(1,3-дион)-2-изоиндолил-метиленокси]-1-азабицикло[2.2.2]октана (Соединение В18)
Получали в соответствии с процедурой В из N-(2-бромметил)-фталимида. Т. пл. 212-213°С.
Свободное основание (±)-3-[(1,3-дион)-2-изоиндолил-этиленокси]-1-азабицикло[2.2.2]октана (Соединение В19)
Получали в соответствии с процедурой В из N-(2-бромэтил)-фталимида. Выделяли как свободное основание, масло.
Соль фумаровой кислоты и (±)-3-(бензотриазол-1-илокси)-1-азабицикло[2.2.2]октана (Соединение В20)
Получали в соответствии с процедурой В из 1-(хлорметил)-1Н-бензотриазола. Т. пл. 163,3-164,5°С.
Способ С
Соль метилиум йодид (±)-2-(1-аза-бицикло[2.2.2]окт-3-илокси)-хиноксалина (Соединение C1)
Смесь (±)-3-(хиноксалин-2-илокси)-1-аза-бицикло[2.2.2]октана (1,27 г, 5,0 ммоль), дихлорметана (10 мл) добавляли при -70°С: метилиодид (0,31 г, 5,0 ммоль), растворенный в дихлорметане (1,5 мл), добавляли в течение 10 минут. Реакционную смесь перемешивали при -70°С в течение 40 минут. Реакционной смеси давали перемешиваться при комнатной температуре в течение 3 часов.
Осадок выделяли путем фильтрования. Т. пл. 229-230°С.
Метилиум йодид (±)-2-(1-аза-бицикло[2.2.2]окт-3-илокси)-хинолина (Соединение С2)
Получали в соответствии со способом С из (±)-2-(1-аза-бицикло[2.2.2]окт-3-илокси)-хинолина. Т. пл. 156,6-175,2°С.
Метилиум йодид (±)-3-(1,2-бензоизотиазол-3-илокси)-(1-аза-бицикло[2.2.2]октана (Соединение С3)
Получали в соответствии со способом С из (±)-3-(1,2-бензоизотиазол-3-илокси)-1-аза-бицикло[2.2.2]октана. Т. пл. 180,1-186,4°С.
Способ D
Соль фумаровой кислоты и (+)-3-(5-фенил-тиазол-2-илокси)-1-аза-бицикло[2.2.2]октана (Соединение D1)
Смесь (±)-3-(5-бром-тиазол-2-илокси)-1-аза-бицикло[2.2.2]октана (1,25 г, 4,32 ммоль), фенилбороновой кислоты (0,791 г, 6,48 ммоль), Pd(PPh3)4 (0,150 г, 0,13 ммоль), водного карбоната калия (6,5 мл, 2 М), 1,3-пропандиола (0,97 мл, 13,0 ммоль) и 1,2-диметоксиэтана (30 мл) перемешивали с обратным холодильником в течение 15 часов. Добавляли водный гидроксид натрия (50 мл, 1М), смесь экстрагировали этилацетатом (3×50 мл). Хроматография на силикагеле дихлорметаном, метанолом и конц. аммиаком (89:10:1) дала соединение, указанное в заголовке. Выход 3,7 г (43%). Соответствующую соль получали путем добавления смеси диэтилового эфира и метанола (9:1), насыщенной при помощи фумаровой кислоты. Т. пл. 170,9-172,2°С. Соль фумаровой кислоты и (±)-3-[5-(2,4-дифторфенил)-тиазол-2-илокси]-1-аза-бицикло[2.2.2]октана (Соединение D2)
Получали в соответствии со способом D. Т. пл. 84,3-86,3°С.
Соль фумаровой кислоты и (±)-3-[5-(3-тиенил)-тиазол-2-илокси]-1-аза-бицикло[2.2.2]октана (Соединение D3)
Получали в соответствии со способом D. Т. пл. 68,5-74,3°С.
Соль фумаровой кислоты и (±)-3-[5-(2-тиенил)-тиазол-2-илокси]-1-аза-бицикло[2.2.2]октана (Соединение D4)
Получали в соответствии со способом D. Т. пл. 152,6-154,9°С.
Соль фумаровой кислоты и (±)-3-[5-(3-фуранил)-тиазол-2-илокси]-1-аза-бицикло[2.2.2]октана (Соединение D5)
Получали в соответствии со способом D. Т. пл. 127,6-136,2°С.
Соль фумаровой кислоты и (±)-3-[5-(3-пиридил)-тиазол-2-илокси]-1-аза-бицикло[2.2.2]октана (Соединение D6)
Получали в соответствии со способом D. Т. пл. 82,7-86,0°С.
Соль фумаровой кислоты и (±)-3-[6-(3-тиенил)-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октана (Соединение D7)
Получали в соответствии со способом D из (±)-3-(6-бром-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октана. Т. пл. 197,9°С.
Соль фумаровой кислоты и (±)-3-[6-(2-тиенил)-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октана (Соединение D8)
Получали в соответствии со способом D из (±)-3-(6-бром-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октана. Т. пл. 180,3-191,1°С.
Соль фумаровой кислоты и (±)-3-[6-(2-фуранил)-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октана (Соединение D9)
Получали в соответствии со способом D из (±)-3-(6-бром-пиридазин-3-илокси)-1-аза-бицикло[2.2.2]октана. Т. пл. 175,8-178,2°С.
Соль фумаровой кислоты и (±)-3-[6-(3-фуранил)-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октана (Соединение D10)
Получали в соответствии со способом D из (±)-3-(6-бром-пиридазин-3-илокси)-1-аза-бицикло[2.2.2]октана. Т. пл. 224,8-225,4°С.
Соль фумаровой кислоты и (±)-3-[6-(3-пиридил)-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октана (Соединение D11)
Получали в соответствии со способом D из (±)-3-(6-бром-пиридазин-3-илокси)-1-аза-бицикло[2.2.2]октана. Т. пл. 137,2-143,2°С.
Способ Е
Соль фумаровой кислоты и (±)-3-(4-фенилфенил-метиленокси)-1-аза-бицикло[2.2.2]октана (Соединение Е1)
Смесь (±)-3-хинуклидинола (2,0 г, 15,7 ммоль), 4-фенилбензилхлорида (3,2 г, 15,7 ммоль), гидрида натрия, 60% с маслом (1,26 г, 31,4 ммоль) и ДМФ (30 мл) при 50°С в течение 4,5 часов. Добавляли водный гидроксид натрия (100 мл, 1М). Смесь экстрагировали диэтиловым эфиром (3×50 мл). Хроматография на силикагеле дихлорметаном, метанолом и конц. аммиаком (89:10:1) дала соединение, указанное в заголовке. Выход 2,0 г (29%).
Соответствующую соль получали путем добавления смеси диэтилового эфира и метанола (9:1), насыщенной при помощи фумаровой кислоты. Т. пл. 159,8-160,4°С.
Соединение может также быть названо (±)-3-(бифенил-4-ил-метокси)-хинуклидин.
Способ F
Соль фумаровой кислоты и (±)-3-(2-фенил-имидазо[1,2-b]пиридазин-6-илокси)-1-азабицикло[2.2.2]октана (Соединение F1)
К смеси 6-хлор-2-фенил-имидазо[1,2-b]пиридазина (приготовленного в соответствии с J.Heterocycl. Chem. 39, 737, 2002) (3,6 г, 15,7 ммоль), (±)-3-хинуклидинола (2,0 г, 15,7 ммоль) в ДМФ (30 мл) в течение 20 минут добавляли гидрид натрия (1,26 г, 31,4 ммоль), при комнатной температуре, за чем следовало перемешивание при 50°С в течение 4 часов. Добавляли водный гидроксид натрия (100 мл, 1М). Смесь экстрагировали диэтиловым эфиром (3×100 мл). Хроматография на силикагеле дихлорметаном, метанолом и конц. аммиаком (89:10:1) дала соединение, указанное в заголовке. Выход 2,9 г (57%).
Соответствующую соль получали путем добавления смеси диэтилового эфира и метанола (9:1), насыщенной при помощи фумаровой кислоты. Т. пл. 211-216°С.
Способ G
Соль фумаровой кислоты и (±)-3-(дибензофуран-2-илокси)-1-азабицикло[2.2.2]октана (Соединение G1)
К смеси (±)-3-хинуклидинола (3,0 г, 23,6 ммоль), 2-гидроксидибензофурана (4,3 г, 23,6 ммоль), трифенилфосфина (9,29 г, 35,4 ммоль) и ТГФ (тетрагидрофурана) добавляли диэтилазодикарбоксилат (6,3 мл, 35,4 ммоль) в течение периода времени 40 минут при комнатной температуре. Смесь перемешивали при 50°С в течение 7 суток. Добавляли водный гидроксид натрия (100 мл, 1М). Смесь экстрагировали дихлорметаном (3×100 мл). Хроматография на силикагеле дихлорметаном, метанолом и конц. аммиаком (89:10:1) дала соединение, указанное в заголовке. Выход 2,0 г (29%).
Соответствующую соль фумаровой кислоты получали путем добавления смеси диэтилового эфира и метанола (9:1), насыщенной при помощи фумаровой кислоты. Т. пл. 131,3-133,8°С.
Соединение может также быть названо (±)-3-(дибензофуран-2-илокси)-хинуклидин.
Биологическая активность
In vitro подавление связывания 3H-α-бунгаротоксина в головном мозгу крысы
В данном примере определяли сродство соединений по изобретению для связывания α7-подтипа никотиновых рецепторов.
α-Бунгаротоксин представляет собой пептид, выделенный из яда змеи семейства Elapidae Bungarus multicinctus. Он обладает высоким сродством к нейрональным и нейромышечным никотиновым рецепторам, где он действует как сильный антагонист.
3H-α-Бунгаротоксин метит никотиновые ацетилхолиновые рецепторы, образованные изоформой α7 субъединицы, найденной в головном мозгу, и α1 изоформой в нервномышечном соединении.
Приготовление ткани
Приготовления осуществляли при 0-4°С. Кору головного мозга от самцов крыс Wistar (150-250 г) гомогенизировали в течение 10 секунд в 15 мл 20 мМ буфера Hepes, содержащего 118 мМ NaCl, 4,8 мМ KCl, 1,2 мМ MgSO4, 2,5 мМ CaCl2 (pH 7,5), используя гомогенизатор Ultra-Turrax. Тканевую суспензию подвергали центрифугированию при 27000 × g в течение 10 минут. Супернатант отбрасывали и осадок промывали дважды путем центрифугирования при 27000 × g в течение 10 минут в 20 мл свежего буфера и конечный осадок затем ресуспендировали в свежем буфере, содержащем 0,01% БСА (бычьего сывороточного альбумина) (35 мл на г исходной ткани) и использовали для анализа связывания.
Анализ
Аликвоты по 500 мкл гомогената добавляли к 25 мкл тестируемого раствора и 25 мкл 3H-α-бунгаротоксина (конечная концентрация 2 нМ), смешивали и инкубировали в течение 2 часов при 37°С. Неспецифическое связывание определяли, используя (-)-никотин (конечная концентрация 1 мМ). После инкубации к образцам добавляли 5 мл ледяного буфера Hepes, содержащего 0,05% ПЭИ (полиэтиленимина) и выливали непосредственно на стекловолоконные фильтры Whatman GF/C (предварительно замоченные в 0,1% ПЭИ в течение по меньшей мере 6 часов) под подсосом и немедленно промывали 2×5 мл ледяного буфера.
Радиоактивность на фильтрах определяли с помощью общепринятого жидкостно-сцинтилляционного измерения активности. Специфическое связывание получают как общее связывание минус неспецифическое связывание.
Тестируемая величина дана как IC50 (концентрация тестируемого вещества, которая подавляет специфическое связывание 3H-α-бунгаротоксина на 50%).
Результаты этих экспериментов представлены в таблице 1 ниже.
Примеры фармацевтической композиции
а)
Соединение формулы (I) смешивают с лактозой, кроскармеллозой и поливинилпирролидоном в течение приблизительно 10 минут. Затем добавляют стеарат магния, полученную смесь перемешивают в течение приблизительно 2 минут и прессуют в форме таблетки.
(б)
Соединение формулы (I) смешивают с лактозой и кроскармеллозой в течение приблизительно 10 минут. Затем добавляют стеарат магния, полученную смесь перемешивают в течение приблизительно 2 минут и ею заполняют желатиновые капсулы.
Настоящее изобретение относится к новым хинуклидиновым производным и их применению в качестве фармацевтических средств. Вследствие своего фармакологического профиля соединения по настоящему изобретению могут быть полезны для лечения таких разнообразных заболеваний или расстройств, как связанные с холинэргической системой центральной нервной системы (ЦНС), периферической нервной системы (ПНС), заболеваний или расстройств, связанных с сокращением гладкой мускулатуры, эндокринных заболеваний или расстройств, заболеваний или расстройств, связанных с нейродегенерацией, заболеваний или расстройств, связанных с воспалением, болью, и синдрома отмены, вызванного прекращением употребления химических веществ. 3 н. и 8 з.п. ф-лы, 1 табл.
или его фармацевтически приемлемая соль присоединения, где
 представляет собой одинарную (ковалентную) связь;
представляет собой одинарную (ковалентную) связь;
n представляет собой 2;
Х представляет собой линкер, выбранный из -О- и -О-СН2-; и
А представляет собой моноциклическую или полициклическую карбоциклическую группу, выбранную из инданила, нафтила и 5,6,7,8-тетрагидро-нафтила; или
моноциклическую гетероциклическую группу, выбранную из пиридазинила и тиазолила, которая возможно замещена фенилом, дифторзамещенным фенилом, пиридилом, фуранилом, тиенилом или галогеном; или
полициклическую гетероциклическую группу, выбранную из изоиндолила, хинолинила и хиноксалинила, которая возможно замещена галогеном или низшим алкокси; или
полициклическую гетероциклическую группу, выбранную из бензоксазолила, бензтиазолила, бензизотиазолила, бензтриазолила и имидазо[1,2-b]пиридазинила, которая возможно замещена фенилом.
(±)-3-(5,6,7,8-тетрагидро-2-нафтилокси)-1-аза-бицикло[2.2.2]октан или
(±)-3-(5-инданилокси)-1-аза-бицикло[2.2.2]октан;
или его фармацевтически приемлемую соль присоединения.
(±)-3-(5-фенил-тиазол-2-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-[5-(2,4-дифторфенил)-тиазол-2-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[5-(3-тиенил)-тиазол-2-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[5-(2-тиенил)-тиазол-2-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3 - [5 -(3 -фуранил)-тиазол-2-илокси] -1 -аза-бицикло [2.2.2] октан;
(±)-3-[5-(3-пиридил)-тиазол-2-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-(6-хлор-пиридазин-3-илокси)-1 -аза-бицикло[2.2.2]октан;
(±)-3-(6-бром-пиридазин-3-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(6-фенил-пиридазин-3-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-[6-(3-тиенил)-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[6-(2-тиенил)-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[6-(2-фуранил)-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[6-(3-фуранил)-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октан;
(±)-3-[6-(3-пиридил)-пиридазин-3-илокси]-1-аза-бицикло[2.2.2]октан;
или его фармацевтически приемлемую соль присоединения.
полициклическую гетероциклическую группу, выбранную из изоиндолила, хинолинила и хиноксалинила, которая возможно замещена галогеном или низшим алкокси; или
полициклическую гетероциклическую группу, выбранную из
бензоксазолила, бензтиазолила, бензизотиазолила, бензтриазолила и имидазо[1,2-b]пиридазинила, которая возможно замещена фенилом.
(±)-3-(2-хинолинилокси)-1-аза-бицикло[2.2.2]октанаметилиум йодид;
(±)-3-(6-хинолинилокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(2-хиноксалинилокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(2-хиноксалинилокси)-1-аза-бицикло[2.2.2]октана метилиум йодид;
(±)-3-(3-хлор-2-хиноксалинилокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(3-метокси-2-хиноксалинилокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(бензоксазол-2-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(бензтиазол-2-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(6-хлор-бензтиазол-2-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(1,2-бензизотиазол-3-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(1,2-бензизотиазол-3-илокси)-1-аза-бицикло[2.2.2]октан;
(±)-3-(бензтриазол-1 -илокси)-1-аза-бицикло[2.2.2]октан; или
(±)-3-(2-фенил-имидазо[1,2-b]пиридазин-6-илокси)-1-азабицикло[2.2.2]октан;
или его фармацевтически приемлемую соль присоединения.
Приоритет установлен по наиболее поздней дате от 02.10.2002.
| ПРОИЗВОДНЫЕ 1,2,5-ТИАДИАЗОЛА, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2158262C2 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| US 5852037 A1, 22.12.1998 | |||
| US 5763457 A, 09.06.1998 | |||
| US 5998404 A, 07.12.1999 | |||
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| US 5589477 A, 31.12.1996 | |||
| WO 9931097 A1, 24.06.1999 | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |

