Область техники
Данное изобретение относится к кристаллической форме производного 1,2-дигидропиридина [3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она], которая обладает антагонистическим действием в отношении рецептора AMPA (альфа-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты) и/или ингибирующим действием в отношении каинатного рецептора и которая может быть использована в качестве терапевтического или профилактического агента для лечения нейродегенеративных или других заболеваний, а также к способу получения этой кристаллической формы.
Уровень техники
Производные 1,2-дигидропиридина обладают антагонистическим действием в отношении рецептора AMPA и/или ингибирующим действием в отношении каинатного рецептора и полезны в качестве терапевтических или профилактических агентов против нейродегенеративных или других заболеваний. В частности, 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-он (называемый ниже соединением (1)) проявляет значительное антагонистическое действие в отношении рецептора AMPA (см. патентный документ 1).
Несмотря на то, что пример 7 в патентном документе 1 раскрывает способ получения соединения (1), там только описано, что "остаток очищен посредством колоночной хроматографии на силикагеле (этилацетат/гексан=1:2)" и в нем нет сведений о форме полученного соединения.
[патентный документ 1] WO01/96308
Раскрытие изобретения
Проблема, на решение которой направлено изобретение
Если соединение, которому свойственен кристаллический полиморфизм, используют в качестве лекарственного средства, необходимо стабильно обеспечивать соединение, обладающее однородной кристаллической формой так, чтобы можно было гарантировать однородное качество и постоянную эффективность, необходимую для лекарственного средства. Также существует потребность в кристаллической форме, способной к поддержанию постоянного качества в ходе хранения и процесса ее приготовления (такого, как смешивание и грануляция).
Так как лекарственное вещество в промышленном масштабе используется в большом количестве, требуются кристаллические формы, обладающие высоким концентрационным порогом взрываемости и минимальной энергией воспламенения, индексом взрываемости и опасности.
В целом, порошки, которые имеют тенденцию заряжаться, обладают большой адгезивностью к другим объектам; и есть опасение в отношении их адгезии к защитным материалам или коже.
Если лекарственное вещество обладает поляризуемостью, оказывается, что эффективность его получения и обрабатываемость снижаются, если соединение прилипает к вращательному лезвию на стадии перемалывания в производстве соединения, или прилипает к оборудованию для производства и собирается на нем в процессе приготовления. Если большое количество порошков, обладающих поляризуемостью, обрабатывают в промышленном масштабе, есть возможность возникновения взрыва пыли. В связи с этим требуется, чтобы в качестве лекарственного вещества использовалось соединение (кристаллическая форма), обладающее слабой поляризуемостью.
Что касается соединения, обладающего высокой фармакологической активностью (такого, как лекарственное вещество), то, для того чтобы предотвратить его воздействие на обслуживающий персонал и предупредить загрязнение предприятия, желательным является применение порошков, не имеющих тенденцию заряжаться.
По приведенным выше причинам, если активный фармацевтический ингредиент лекарственного средства получается в виде кристаллического вещества, оно предпочтительно содержит однородную кристаллическую форму, обладает постоянными предпочтительными свойствами и не содержит примеси, такие как металлы. Также существует потребность в разработке способа получения таких кристаллов в промышленном масштабе.
Соответственно, объектом данного изобретения является кристалл, содержащий однородную кристаллическую форму соединения (1), и способ его получения.
Средства для решения проблемы
В результате интенсивных и упорных исследований авторы настоящего изобретения обнаружили, что для кристаллизации соединения (1) могут быть использованы определенные растворители кристаллизации для получения соединения (1) в однородной кристаллической форме, с которым было выполнено данное изобретение.
В частности, настоящее изобретение обеспечивает, среди прочего, следующее:
(1) Кристаллический гидрат 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, имеющий пик дифракции при значении угла дифракции (2θ ± 0,2°) 8,7° при дифракции рентгеновских лучей на порошке (гидрат).
(2) Кристаллический гидрат 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, имеющий пик дифракции при значении угла дифракции (2θ ± 0,2°) 12,5° при дифракции рентгеновских лучей на порошке (гидрат).
(3) Кристаллический гидрат 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, имеющий пики дифракции при значениях угла дифракции (2θ ± 0,2°) 8,7° и 12,5° при дифракции рентгеновских лучей на порошке (гидрат).
(4) Кристаллический гидрат 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, имеющий пик поглощения при значении волнового числа 1588±1 см-1 в инфракрасном спектре поглощения (способ KBr) (гидрат).
(5) Кристаллический гидрат 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, имеющий пики поглощения при значениях волнового числа 1588±1 см-1 и 751±1 см-1 в инфракрасном спектре поглощения (способ KBr) (гидрат).
(5-2) Кристаллическое соединение по любому из пунктов (1)-(3), имеющее пик поглощения при значении волнового числа 1588±1 см-1 в инфракрасном спектре поглощения (способ KBr) (гидрат).
(5-3) Кристаллическое соединение по любому из пунктов (1)-(3), имеющее пики поглощения при значениях волнового числа 1588±1 см-1 и 751±1 см-1 в инфракрасном спектре поглощения (способ KBr) (гидрат).
(5-4) Кристаллическое соединение по любому из пунктов (1)-(5), (5-2) и (5-3), содержащее 20 част./млн или меньше палладия, предпочтительно 15 част./млн или меньше (гидрат).
(6) Кристаллический гидрат 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, имеющий пики при значениях химического сдвига, приблизительно, 146,7 част./млн и, приблизительно, 123,3 част./млн в спектре ядерного магнитного резонанса 13C в твердом состоянии (гидрат).
(7) Способ получения кристаллической формы гидрата 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она по любому из пунктов (1)-(5), (5-1), (5-2), (5-3), (5-4) и (6), включающий стадии кристаллизации 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она с помощью одного или двух растворителей кристаллизации, выбранных из группы, состоящей из спиртового растворителя, алкилкетонового растворителя и воды.
(8) Способ по п. (7), в котором растворитель кристаллизации представляет собой смешанный растворитель из ацетона и воды.
(9) Способ по п. (7), в котором растворитель кристаллизации представляет собой смешанный растворитель из ацетона и воды в объемном соотношении от 37:3 до 24:16, предпочтительно смешанный растворитель из ацетона и воды в объемном соотношении от 9:1 до 7:3 и, более предпочтительно, смешанный растворитель из ацетона и воды в объемном соотношении 8:2, образованный путем растворения кристаллов в смешанном растворителе из ацетона и воды в объемном соотношении 9:1 и последующего добавления воды к смешанному растворителю.
(10) Способ по любому из пунктов (7)-(9), в котором кристаллизацию проводят при температуре от 60 до -30°C.
(11) Способ по любому из пунктов (7)-(9), включающий стадии нагревания раствора 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, растворенного в растворителе кристаллизации при температуре 50°C или выше (предпочтительно при температуре от температуры кипения с обратным холодильником растворителя кристаллизации до 50°C, более предпочтительно при температуре от 65 до 55°C) и последующего охлаждения раствора до температуры от 10 до -20°C (предпочтительно до температуры от 10 до 5°C) при скорости охлаждения от 40 до 5°C в час (предпочтительно при скорости охлаждения от 25 до 15°C в час).
(12) Способ по любому из пунктов (7)-(11), в котором растворитель кристаллизации используется в 10-50-кратном (об./мас.) объемном отношении, в расчете на вес 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она.
Количество растворителя кристаллизации предпочтительно является 30-50-кратным (об./мас.), более предпочтительно, приблизительно, 40-кратным (об./мас.), где в качестве растворителя кристаллизации используют ацетон и воду (9:1), и, приблизительно, 45-кратным (об./мас.), где в качестве растворителя кристаллизации используют ацетон и воду (8:2).
(13) Способ по любому из пунктов (7)-(12), в котором затравочные кристаллы (небольшое количество кристаллов гидрата 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она) добавляют при температуре 60°C, или ниже (предпочтительно при температуре от 55 до 0°C, более предпочтительно от 55 до 35°C и наиболее предпочтительно, приблизительно, 40°C).
(14) Способ по любому из пунктов (7)-(13), в котором кристаллы сушат при пониженном давлении после кристаллизации.
(15) Способ по любому из пунктов (7)-(14), в котором кристаллы оставляют стоять на воздухе после кристаллизации и сушки при пониженном давлении.
(15-1) Способ по любому из пунктов (7)-(13), в котором кристаллы оставляют стоять на воздухе после кристаллизации.
(15-2) Способ по п. (14), в котором кристаллы оставляют стоять на воздухе после сушки при пониженном давлении.
(16) Безводная кристаллическая форма 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, имеющая пик дифракции при значении угла дифракции (2θ ± 0,2°) 10,3° при дифракции рентгеновских лучей на порошке (безводная форма I).
(17) Кристаллическая форма по п. (16), дополнительно имеющая пик дифракции при значении угла дифракции (2θ ± 0,2°) 19,1° при дифракции рентгеновских лучей на порошке (безводная форма I).
(18) Безводная кристаллическая форма 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, имеющая пики при значениях химического сдвига, приблизительно, 149,0 част./млн и, приблизительно, 125,6 част./млн в спектре ядерного магнитного резонанса 13C в твердом состоянии (безводная форма I).
(19) Безводная кристаллическая форма 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, имеющая пик дифракции при значении угла дифракции (2θ ± 0,2°) 16,7° при дифракции рентгеновских лучей на порошке (безводная форма V).
(20) Кристаллическая форма по п. (19), дополнительно имеющая пики дифракции при значениях угла дифракции (2θ ± 0,2°) 12,9° и 24,9° при дифракции рентгеновских лучей на порошке (безводная форма V).
(21) Безводная кристаллическая форма 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, имеющая пик поглощения при значении волнового числа 1658 ± 1 см-1 в инфракрасном спектре поглощения (способ KBr) (безводная форма V).
(22) Кристаллическая форма по п. (21), дополнительно имеющая пик поглощения при значении волнового числа 501 ± 1 см-1 в инфракрасном спектре поглощения (способ KBr) (безводная форма V).
(23) Безводная кристаллическая форма 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, имеющая пики при значениях химического сдвига, приблизительно, 145,9 част./млн и, приблизительно, 137,7 част./млн в спектре ядерного магнитного резонанса 13C в твердом состоянии (безводная форма V).
(24) Безводная кристаллическая форма 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, имеющая пики дифракции при значениях угла дифракции (2θ ± 0,2°) 23,7° и 25,0° при дифракции рентгеновских лучей на порошке (безводная форма III).
(25) Кристаллическая форма по (24), дополнительно имеющая пики дифракции при значениях угла дифракции (2θ ± 0,2°) 5,7° и 9,5° при дифракции рентгеновских лучей на порошке (безводная форма III).
(26) Лекарственное средство, содержащее кристаллическое соединение по п. (1).
(27) Фармацевтическая композиция, содержащая кристаллическое соединение по п. (1).
(28) Терапевтический или профилактический агент для лечения острого нейродегенеративного заболевания, включающий кристаллическое соединение по п. (1).
(29) Терапевтический или профилактический агент для лечения невропатии, вызванной острой фазой цереброваскулярного нарушения, черепно-мозговой травмой, повреждением спинного мозга, или гипоксией, или невропатии, вызванной гипогликемией, включающий кристаллическое соединение по п. (1).
(30) Терапевтический или профилактический агент для лечения хронического нейродегенеративного заболевания, включающий кристаллическое соединение по п. (1).
(31) Терапевтический или профилактический агент для лечения болезни Альцгеймера, болезни Паркинсона, хореи Хантингтона, амиотрофического бокового склероза или спиноцеребеллярной дегенерации, включающий кристаллическое соединение по п. (1).
(32) Терапевтический или профилактический агент для лечения эпилепсии, печеночной энцефалопатии, периферической невропатии, Паркинсонизма, спастического паралича, боли, невралгии, шизофрении, тревоги, наркотической зависимости, тошноты, рвоты, дизурии, повреждения зрения, вызванного глаукомой, повреждения слуха, вызванного антибиотиками или пищевым отравлением, включающий кристаллическое соединение по п. (1).
(33) Терапевтический или профилактический агент для лечения инфекционного энцефаломиелита, цереброваскулярной деменции, или деменции, или неврологического симптома, вызванного менингитом, включающий кристаллическое соединение по п. (1).
(34) Терапевтический или профилактический агент для лечения демиелинизирующего заболевания, включающий кристаллическое соединение по п. (1).
(35) Терапевтический или профилактический агент по п. (33), в котором инфекционный энцефаломиелит представляет собой энцефаломиелит ВИЧ.
(36) Терапевтический или профилактический агент по п. (34), в котором демиелинизирующее заболевание представляет собой энцефалит, острый спорадический энцефаломиелит, рассеянный склероз, острую полирадикулоневропатию, синдром Гийена-Барре, хроническую воспалительную демиелинизирующую полирадикулоневропатию, болезнь Макиафава-Бигнами, центральную понтомедуллярную демиелинизацию, нейромиелит зрительного нерва, болезнь Девика, болезнь Бало, ВИЧ-ассоциированную миелопатию, ВТЛЧ-ассоциированную миелопатию, прогрессирующий многоочаговый лейкоэнцефалит или вторичное демиелинизирующее заболевание.
(37) Терапевтический или профилактический агент по п. (36), в котором вторичное демиелинизирующее заболевание представляет собой красную волчанку ЦНС, нодозный полиартериит, синдром Сджогрена, саркоидоз или рассеянный васкулит головного мозга.
Результаты изобретения
В соответствии с изобретением стало возможным простое получение соединения (1) в виде однородной кристаллической формы в промышленном масштабе. Кристаллические формы в соответствии с изобретением обладают предпочтительными свойствами, такими как отсутствие поляризуемости, и пригодны для применения в качестве активного ингредиента терапевтических или профилактических агентов для лечения нейродегенеративных или других заболеваний.
Краткое описание чертежей
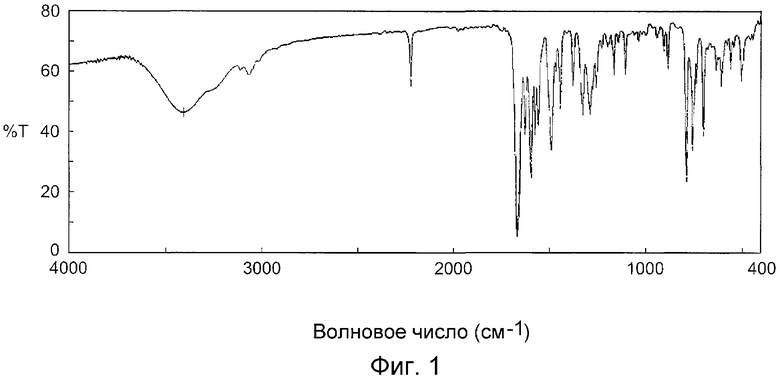
На фиг.1 показан инфракрасный спектр (способ KBr) кристаллов, полученных в примере B1.
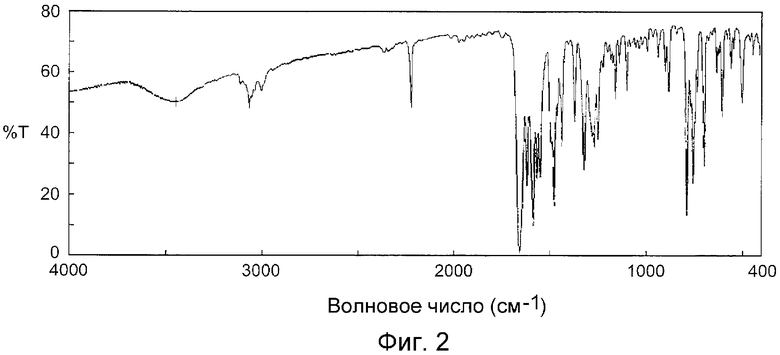
На фиг.2 показан инфракрасный спектр (способ KBr) кристаллов, полученных в примере C1.
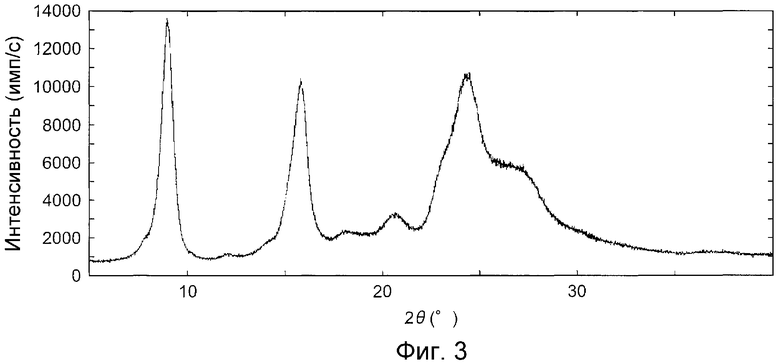
На фиг.3 показана дифракционная рентгенограмма на порошке кристаллов, полученных в контрольном примере А1.
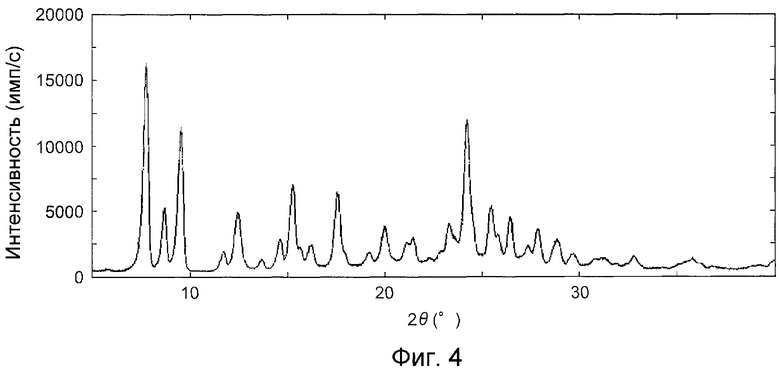
На фиг.4 показана дифракционная рентгенограмма на порошке кристаллов, полученных в примере B1.
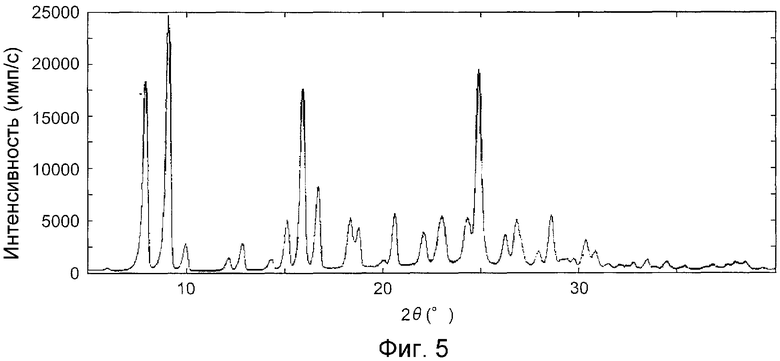
На фиг.5 показана дифракционная рентгенограмма на порошке кристаллов, полученных в примере C1.
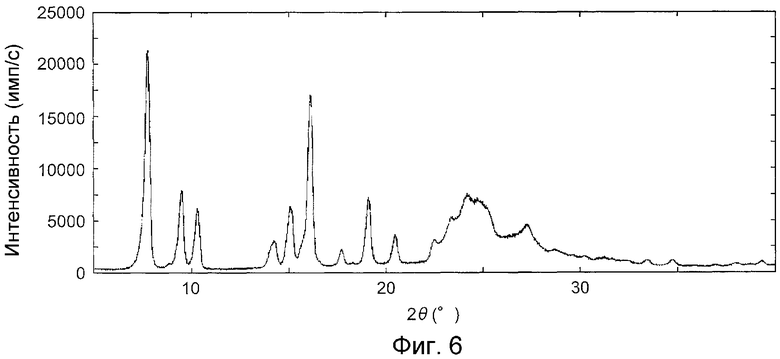
На фиг.6 показана дифракционная рентгенограмма на порошке кристаллов, полученных в примере D1.
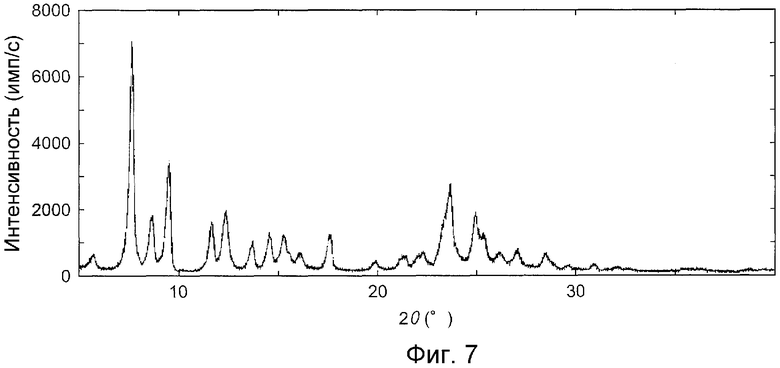
На фиг.7 показана дифракционная рентгенограмма на порошке кристаллов, как описано в примере E1.
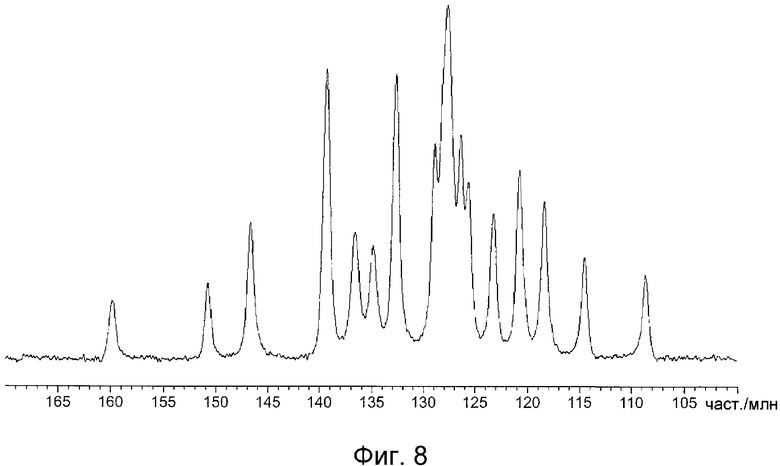
На фиг.8 показан спектр ядерного магнитного резонанса (ЯМР) 13C в твердом состоянии кристаллов, полученных в примере B1.
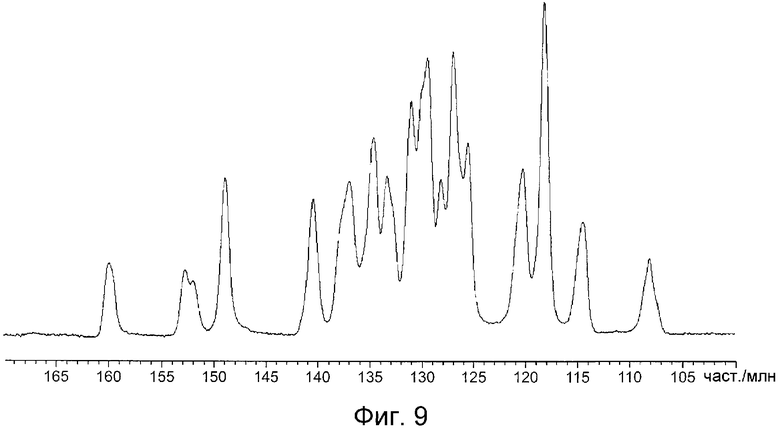
На фиг.9 показан спектр ЯМР 13C в твердом состоянии кристаллов, полученных в примере D1.
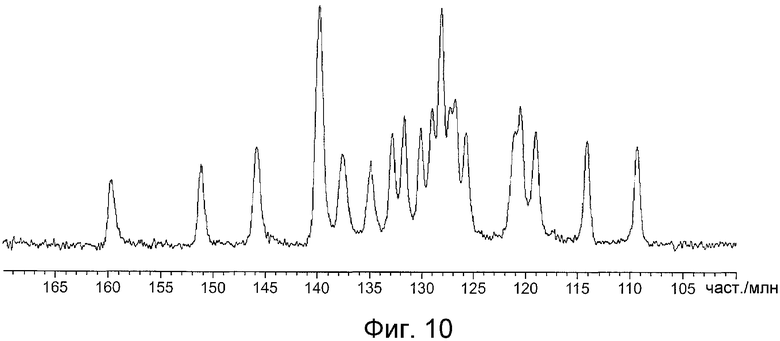
На фиг.10 показан спектр ЯМР 13C в твердом состоянии кристаллов, полученных в примере C1.
На фиг.11 показаны дифракционные рентгенограммы на порошке гидрата при различных значениях температуры.
На фиг.12 показаны дифракционные рентгенограммы на порошке гидрата при различных значениях относительной влажности.
Предпочтительный способ выполнения изобретения
Данное изобретение подробно описано ниже.
В данном описании "кристаллический гидрат 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она" является кристаллической формой 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, содержащей воду в кристалле, и само по себе количество воды, содержащейся в кристаллической форме, особым образом не ограничено; в кристаллической форме может не быть части воды. Термин также охватывает форму, в которой вода может присутствовать совместно с адгезионной водой.
Данный термин " кристаллический гидрат 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она" обозначает такую кристаллическую форму, в которой она содержит предпочтительно от 1/2 до одной молекулы воды на одну молекулу 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она в кристаллической форме, может дополнительно содержать от 0 до 1/4 молекулы адгезионной воды и может быть даже лишена от 0 до 1/2 молекулы воды в кристалле.
В частности, он обозначает следующее:
(1) Кристаллический 3/4 гидрат 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она;
(2) Кристаллический моногидрат 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она (лишенного 1/4 в кристалле);
(3) Кристаллический 1/2 гидрат 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, существующий совместно с 1/4 адгезионной воды и
(4) Кристаллический 1/2 гидрат 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она; и
(5) Кристаллический моногидрат 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она.
Кристаллические формы в соответствии с изобретением представляют собой кристаллические формы гидрата соединения (1) с характеристиками, описанными ниже. Несмотря на то, что соответствующие условия измерения для дифракционных рентгенограмм на порошке и инфракрасных спектров поглощения (способ KBr) особым образом не ограничены, измерение предпочтительно следует проводить при условиях измерения для дифракционных рентгенограмм на порошке и инфракрасных спектров поглощения (способ KBr), как будет описано ниже.
(1) Кристаллическая форма, имеющая пик дифракции при значении угла дифракции (2θ ± 0,2°) 8,7° при дифракции рентгеновских лучей на порошке;
(2) Кристаллическая форма, имеющая пик дифракции при значении угла дифракции (2θ ± 0,2°) 12,5° при дифракции рентгеновских лучей на порошке;
(3) Кристаллическая форма, имеющая пики дифракции при значениях угла дифракции (2θ ± 0,2°) 8,7° и 12,5° при дифракции рентгеновских лучей на порошке;
(4) Кристаллическая форма, имеющая пики дифракции при значениях угла дифракции (2θ ± 0,2°) показанные на фиг.4 или в таблице 5 ниже при дифракции рентгеновских лучей на порошке;
(5) Кристаллическая форма, имеющая пик поглощения при длине волны 1588±1 см-1 в инфракрасном спектре поглощения (способ KBr),
(6) Кристаллическая форма, имеющая пики поглощения при длинах волны 1588±1 см-1 и 751±1 см-1 в инфракрасном спектре поглощения (способ KBr); и
(7) Кристаллическая форма, имеющая пики поглощения при длинах волны (см-1), показанных на фиг.1 или в таблице 2 ниже в инфракрасном спектре поглощения (способ KBr).
Данные характеристические пики в дифракции рентгеновских лучей на порошке не наблюдаются в кристаллической форме, полученной способом, раскрытым в патентном документе 1 (см. контрольный пример A1, таблицу 4 и фиг.3, как описано ниже).
В отношении угла преломления (2θ) в дифракционном анализе рентгеновских лучей на порошке, в целом, погрешности значения угла преломления могут встречаться в пределах диапазона ±0,2°. В связи с этим следует понимать, что значения углов преломления могут включать числа порядка ±0,2°. Соответственно, данное изобретение охватывает не только кристаллическую форму, имеющую полностью совпадающие углы преломления пиков при дифракции рентгеновских лучей на порошке, но также и кристаллическую форму, имеющую совпадающие углы преломления пиков в пределах погрешности, приблизительно, ±0,2°.
[Гидрат]
В данном описании "имеющий пик дифракции при значении угла дифракции (2θ ± 0,2°) 8,7°" обозначает "имеющий пик дифракции при значении угла дифракции (2θ) от 8,5° до 8,9°." Термин "имеющий пик дифракции при значении угла дифракции (2θ ± 0,2°) 12,5°" обозначает "имеющий пик дифракции при значении угла дифракции (2θ) от 12,3° до 12,7°."
В данном описании "имеющий пик поглощения при значении волнового числа 1588±1 см-1" обозначает "имеющий пик поглощения при значении волнового числа от 1587 до 1589 см-1."
В данном описании "имеющий пики поглощения при значениях волнового числа 1588±1 см-1 и 751±1 см-1" обозначает "имеющий пики поглощения при значениях волнового числа от 1587 до 1589 см-1 и от 750 до 752 см-1."
В данном описании "имеющий пик при значениях химического сдвига, приблизительно, 146,7 част./млн" обозначает "имеющий пик, в основном эквивалентный 146,7 частям на миллион, при измерении спектра ЯМР 13C в твердом состоянии в нормальных условиях или в условиях, в основном идентичных описанным в данной спецификации." В данном описании "имеющий пик при значениях химического сдвига, приблизительно, 123,3 част./млн" обозначает " имеющий пик, в основном эквивалентный 123,3 частям на миллион, при измерении спектра ЯМР 13C в твердом состоянии в нормальных условиях или в условиях, в основном идентичных описанным в данной спецификации."
[Безводная форма I]
В данном описании "имеющий пик дифракции при значении угла дифракции (2θ ± 0,2°) 10,3°" обозначает "имеющий пик дифракции при значении угла дифракции (2θ) от 10,1° до 10,5°." Термин "имеющий пик дифракции при значении угла дифракции (2θ ± 0,2°) 19,1°" обозначает "имеющий пик дифракции при значении угла дифракции (2θ) от 18,9° до 19,3°."
В данном описании "имеющий пик при значениях химического сдвига, приблизительно, 149,0 част./млн" обозначает "имеющий пик, в основном эквивалентный 149,0 част./млн, при измерении спектра ЯМР 13C в твердом состоянии в нормальных условиях или в условиях, в основном идентичных описанным в данном описании." В данном описании "имеющий пик при значениях химического сдвига, приблизительно, 125,6 част./млн" обозначает "имеющий пик, в основном эквивалентный 125,6 част./млн, при измерении спектра ЯМР 13C в твердом состоянии в нормальных условиях или в условиях, в основном идентичных описанным в данном описании."
[Безводная форма V]
В данном описании "имеющий пик дифракции при значении угла дифракции (2θ ± 0,2°) 16,7°" обозначает "имеющий пик дифракции при значении угла дифракции (2θ) от 16,5° до 16,9°." Термин "имеющий пик дифракции при значении угла дифракции (2θ ± 0,2°) 12,9°" обозначает "имеющий пик дифракции при значении угла дифракции (2θ) от 12,7° до 13,1°." Термин "имеющий пик дифракции при значении угла дифракции (2θ ± 0,2°) 24,9°" обозначает "имеющий пик дифракции при значении угла дифракции (2θ) от 24,7° до 25,1°."
В данном описании "имеющий пик поглощения при значении волнового числа 1658±1 см-1" обозначает "имеющий пик поглощения при значении волнового числа от 1657 до 1659 см-1."
В данном описании "имеющий пик поглощения при значении волнового числа 501±1 см-1" обозначает "имеющий пик поглощения при значении волнового числа 500-502 см-1. "
В данном описании "имеющий пик при значениях химического сдвига, приблизительно, 145,9 част./млн" обозначает "имеющий пик, в основном эквивалентный 145,9 част./млн, при измерении спектра ЯМР 13C в твердом состоянии в нормальных условиях или в условиях, в основном идентичных описанным в данном описании." В данном описании "имеющий пик при значениях химического сдвига, приблизительно, 137,7 част./млн" обозначает "имеющий пик, в основном эквивалентный 137,7 част./млн, при измерении спектра ЯМР 13C в твердом состоянии в нормальных условиях или в условиях, в основном идентичных описанным в данном описании."
[Безводная форма III]
В данном описании "имеющий пик дифракции при значении угла дифракции (2θ ± 0,2°) 23,7°" обозначает "имеющий пик дифракции при значении угла дифракции (2θ) от 23,5° до 23,9°." Термин "имеющий пик дифракции при значении угла дифракции (2θ ± 0,2°) 25,0°" обозначает "имеющий пик дифракции при значении угла дифракции (2θ) от 24,8° до 25,2°." Термин "имеющий пик дифракции при значении угла дифракции (2θ ± 0,2°) 5,7°" обозначает "имеющий пик дифракции при значении угла дифракции (2θ) от 5,5° до 5,9°." Термин "имеющий пик дифракции при значении угла дифракции (2θ ± 0,2°) 9,5°" обозначает "имеющий пик дифракции при значении угла дифракции (2θ) от 9,3° до 9,7°."
В данном описании "алкилкетоновый растворитель" обозначает органический растворитель, представляющий собой диалкилкетон, такой как ацетон и этилметилкетон, и предпочтительно ацетон.
В данном описании "спиртовой растворитель" обозначает органический растворитель, представляющий собой C1-6 спирт, такой как метанол, этанол, 1-пропанол и 2-пропанол, и предпочтительно метанол или 1-пропанол.
В данном описании термин "при пониженном давлении" особым образом не ограничен в какой-либо степени и составляет 760 мм рт.ст. или меньше; и предпочтительно давление составляет от 760 до 0,1 мм рт.ст., более предпочтительно от 50 до 0,1 мм рт.ст. и наиболее предпочтительно от 30 до 5 мм рт.ст.
[Общий процесс получения гидрата]
Гидрат в соответствии с данным изобретением можно стабильно получать в промышленном масштабе путем приготовления соединения (1) в соответствии с примером 7 в патентном документе 1 (WO01/96308) или в примере 3 получения (описанном ниже), растворения данного соединения (1) в определенном растворителе, нагревания и кристаллизации его за счет охлаждения при перемешивании.
Соединение (1) для использования в кристаллизации может представлять собой любую форму, выбранную из гидратной, безводной, аморфной и кристаллической форм (которая включает множественные кристаллические полиморфы), и может даже представлять собой смесь указанных выше форм.
Растворители для использования в кристаллизации включают один компонент или смешанный растворитель из двух компонентов, выбранных из группы, состоящей из спиртового растворителя, алкилкетонового растворителя и воды. Растворитель предпочтительно представляет собой смешанный растворитель из ацетона и воды.
При использовании смешанного растворителя из ацетона и воды, его коэффициент смешения (объемная доля) предпочтительно составляет от 37:3 до 24:16, более предпочтительно от 9:1 до 7:3 и еще более предпочтительно, приблизительно, 8:2. Наиболее предпочтительным является смешанный растворитель, образованный путем растворения кристаллов в смешанном растворителе из ацетона и воды (9:1) с последующим добавлением воды к смешанному растворителю для приготовления раствора ацетона и воды (8:2).
Количество используемого растворителя соответственно может быть выбрано в диапазоне от нижнего предела до верхнего предела, где нижний предел представляет количество для растворения соединения (1) путем нагревания и верхний предел представляет такое количество, чтобы незначительно уменьшить выход кристаллов. Количество растворителя кристаллизации предпочтительно составляет от 10- до 50-кратного (об./мас.) как объемная доля, в расчете на массу соединения (1), более предпочтительно от 30- до 50-кратного (об./мас.). Еще более предпочтительно, данное количество является, приблизительно, 40-кратным (об./мас.) при использовании ацетона-воды (9:1); и оно является 45-кратным (об./мас.) при использовании ацетона-воды (8:2).
Температура, при которой растворяют соединение (1) путем нагревания, соответственно может быть выбрана как температура для растворения соединения (1) в зависимости от растворителя. Данная температура предпочтительно находится в диапазоне от температуры кипячения растворителя кристаллизации с обратным холодильником до 50°C, более предпочтительно от 65 до 55°C.
Любое изменение скорости охлаждения в ходе кристаллизации может приводить к получению кристаллов с различными формами (полиморфизм). В связи с этим требуется, чтобы кристаллизацию выполняли путем надлежащего контроля скорости охлаждения с учетом возможного влияния на качество и размер частиц кристаллов и т.п. Охлаждение предпочтительно выполняют при скорости от 40 до 5°C в час и более предпочтительно при скорости от 25 до 15°C в час.
Конечная температура кристаллизации также может быть подходящим образом выбрана с учетом выхода и качества кристаллов, и т.п.; и предпочтительно она составляет от 10 до -25°C.
В кристаллизации могут быть добавлены или не добавлены затравочные кристаллы, которые содержат небольшое количество кристаллов 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она. Нет особых ограничений на температуру, при которой добавляют затравочные кристаллы. Температура предпочтительно составляет 60°C или ниже, более предпочтительно от 55 до 0°C, еще более предпочтительно от 55 до 35°C и наиболее предпочтительно, приблизительно, 40°C.
Осажденные кристаллы могут быть выделены путем обычной фильтрации, в случае необходимости промыты растворителем и затем высушены для получения требуемых кристаллов. Растворитель для применения в промывке кристаллов является общим с растворителем кристаллизации, и он предпочтительно представляет собой смешанный растворитель ацетон-вода (9:1 к 1:1), более предпочтительно смешанный растворитель ацетон-вода (приблизительно 1:1).
[Способ сушки кристаллов]
Кристаллы, выделенные с помощью фильтрации, можно высушить, оставляя их в атмосфере, где является уместным, или путем нагревания.
Время, в течение которого остаток растворителя удаляют ниже заданного количества, может соответственно быть выбрано, как время сушки, в зависимости от производимого количества, сушильной установки, температуры сушки и т.п. Сушку можно выполнять либо при аэрации, либо при пониженном давлении. Уровень понижения давления может быть выбран подходящим образом, в зависимости от получаемого количества, сушильной установки, температуры сушки и т.п. В случае необходимости, полученные кристаллы после высыхания можно оставить в атмосфере.
Кристаллы, полученные с помощью описанного выше процесса, содержат однородную кристаллическую форму. Поскольку данные кристаллы наделены предпочтительными свойствами, например, что они являются устойчивыми, не имеют тенденции к легкой трансформации в другой кристалл или аморфные формы и не являются гигроскопичными, они подходят для препарата.
Использование соединения (1) в качестве терапевтического агента для лечения нейродегенеративных или других заболеваний полностью раскрыто в патентном документе 1. Кристаллические формы в соответствии с изобретением могут использоваться в качестве активного ингредиента в терапевтическом агенте для лечения нейродегенеративных или других заболеваний. Таким образом, полное раскрытие документа 1 патента включено в данное описание в качестве ссылочного материала.
Когда соединение в соответствии с данным изобретением должно быть использовано в качестве лекарственного средства, его обычно комбинируют с подходящими фармацевтическими ингредиентами для приготовления фармацевтических продуктов к применению. Несмотря на это, не следует отрицать применение кристаллической формы лекарственного вещества соединения в соответствии с изобретением в качестве лекарственного средства.
Фармацевтические ингредиенты могут включать эксципиенты, связующие вещества, смазывающие вещества, дезинтегрирующие агенты, красители, нейтрализаторы вкуса, эмульгаторы, поверхностно-активные вещества, средства для растворения, суспендирующие агенты, изотонизирующие агенты, буферные вещества, консерванты, антиоксиданты, стабилизаторы, усилители всасывания и т.п., которые обычно используют в лекарственном средстве. Если требуется, данные агенты могут быть комбинированы для применения.
Эксципиенты могут, например, включать лактозу, белый мягкий сахар, глюкозу, кукурузный крахмал, маннит, сорбит, крахмал, альфа-крахмал, декстрин, кристаллическую целлюлозу, легкий кремневый ангидрид, силикат алюминия, силикат кальция, алюмометасиликат магния, гидрофосфат кальция и т.п.
Связующие вещества могут включать, например, поливиниловый спирт, метилцеллюлозу, этилцеллюлозу, гуммиарабик, трагакант, желатин, шеллак, гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, карбоксиметилцеллюлозу натрия, поливинилпирролидон, макрогол и т.п.
Смазывающие вещества могут включать, например, стеарат магния, стеарат кальция, стеарилфумарат натрия, тальк, полиэтиленгликоль, коллоидную окись кремния и т.п.
Дезинтегрирующие агенты могут включать, например, кристаллическую целлюлозу, агар, желатин, карбонат кальция, гидрокарбонат натрия, цитрат кальция, декстрин, пектин, низкозамещенную гидроксипропилцеллюлозу, карбоксиметилцеллюлозу, карбоксиметилцеллюлозу кальция, кроскармеллозу натрия, карбоксиметилкрахмал, карбоксиметилкрахмал натрия, и т.п.
Красители могут включать полуторный оксид железа, желтый полуторный оксид железа, кармин, карамель, бета-каротин, оксид титана, тальк, рибофлавинфосфат натрия, желтый алюминиевый лак и т.п., которые были одобрены как добавки к лекарственным средствам.
Нейтрализаторы вкуса могут включать какао-порошок, ментол, ароматический порошок, мятное масло, борнеол, измельченную в порошок кору коричного дерева и т.п.
Эмульгаторы или поверхностно-активные вещества могут включать стеарилтриэтаноламин, лаурилсульфат натрия, лауриламинопропионовую кислоту, лецитин, моностеарат глицерина, эфир жирной кислоты и сахарозы, эфир глицерина и жирной кислоты и т.п.
Средства для растворения могут включать полиэтиленгликоль, пропиленгликоль, бензилбензоат, этанол, холестерин, триэтаноламин, карбонат натрия, цитрат натрия, полисорбат 80, никотинамид и т.п.
Суспендирующие агенты могут включать, в дополнение к поверхностно-активным веществам, гидрофильные полимеры, такие как поливиниловый спирт, поливинилпирролидон, метилцеллюлоза, гидроксиметилцеллюлоза, гидроксиэтилцеллюлоза и гидроксипропилцеллюлоза.
Изотонизирующие агенты могут включать глюкозу, хлорид натрия, маннит, сорбит и т.п.
Буферные вещества могут включать буферы фосфата, ацетата, карбоната, цитрата и т.п.
Консерванты могут включать метилпарабен, пропилпарабен, хлорбутанол, бензиловый спирт, фенетиловый спирт, дегидроуксусную кислоту, сорбиновую кислоту и т.п.
Антиоксиданты могут включать сульфит, аскорбиновую кислоту, альфа-токоферол и т.п.
Стабилизаторы могут включать вещества, обычно используемые в лекарственных средствах.
Усилители всасывания могут включать вещества, обычно используемые в лекарственных средствах.
Описанные выше фармацевтические продукты могут включать пероральные агенты, такие как таблетки, порошки, гранулы, капсулы, сиропы, пастилки и ингаляции; препараты для наружного применения, такие как свечи, мази, глазные мази, пластыри, глазные растворы, носовые капли, ушные капли, припарки и лосьоны; и инъекции.
Пероральные агенты для образования препаратов могут быть подходящим образом объединены со вспомогательными средствами, описанными выше. Кроме того, в случае необходимости поверхность агентов может быть покрыта.
Препараты для наружного применения могут быть подходящим образом объединены со вспомогательными средствами, в частности эксципиентами, связующими веществами, нейтрализаторами вкуса, эмульгаторами, поверхностно-активными веществами, средствами для расщепления, суспендирующими агентами, изотонизирующими агентами, консервантами, антиоксидантами, стабилизаторами или усилителями всасывания.
Инъекции для образования препаратов могут быть подходящим образом объединены со вспомогательными средствами, в частности эмульгаторами, поверхностно-активными веществами, средствами для растворения, суспендирующими агентами, изотонизирующими агентами, консервантами, антиоксидантами, стабилизаторами или усилителями всасывания.
Если соединение в соответствии с данным изобретением должно быть использовано в качестве лекарственных средств, уровень его дозировки может различаться в зависимости от симптомов, возраста или других факторов. Соединение обычно принимают в виде разовой дозированной формы или в виде раздельных введений 2-6 раз ежедневно в следующих дозах: от 0,05 до 10 мг (предпочтительно от 0,1 до 5 мг) в случае перорального агента; от 0,01 до 10 мг (предпочтительно от 0,05 до 5 мг) в случае наружного препарата; и от 0,01 до 5 мг в случае инъекции. Здесь, фактические количества, которые необходимо ввести, обозначены по отношению к пероральному агенту и инъекции, в то время как количество, которое должно быть поглощено телом, обозначено по отношению к препарату наружного применения.
Препараты для терапевтического или профилактического применения у людей, содержащие кристаллический 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-он (соединение (1)) в соответствии с изобретением, могут быть получены с помощью обычных способов, принятых в промышленной фармакологии. Конкретные примеры составов препарата показаны ниже.
Смешивали соединение в соответствии с изобретением [то есть 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-он], лактозу, низкозамещенную гидроксипропилцеллюлозу. Затем использовали поливинилпирролидон, растворенный в подходящем количестве очищенной воды, для влажной грануляции смеси. Данные грануляты сушили и затем проводили контроль размера. Низкозамещенную гидроксипропилцеллюлозу и стеарат магния смешивали с получающимися гранулятами, после чего их таблетировали. Полученные таблетки покрывали водным раствором основы для покрытия (смесь гидроксипропилметилцеллюлозы, талька, макрогола 6000, оксида титана и желтой полуторной окиси железа). Количества соответствующих материалов, используемые на каждую таблетку, показаны в таблице 1.
*2: Смесь гидроксипропилметилцеллюлозы, талька, макрогола 6000, оксида титана и желтой полуторной окиси железа
Примеры
Данное изобретение будет, в частности, подробно описано с помощью следующих примеров; однако изобретение не должно быть ограничено этими примерами.
(Пример получения 1)
Синтез 5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она
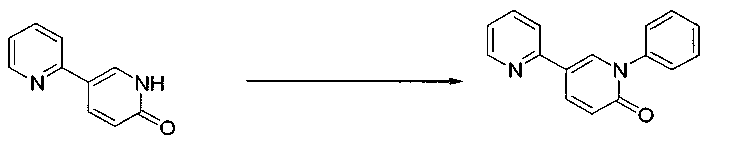
После продувки реактора азотом смесь 5-(2-пиридил)-1,2-дигидропиридин-2-она (7,33 кг: WO2004/009553), трифенилбороксина (9,0 кг), ацетата меди (безводного) (0,80 кг), воды (0,50 кг), пиридина (7,1 кг) и N,N-диметилформамида (66,7 кг) перемешивали в реакторе при внутренней температуре 28°C в течение 1 часа.
При закачке в реактор воздуха, в котором концентрация кислорода была доведена до 9% с помощью азота, при скорости 30 л/мин, реакционную смесь перемешивали при 39-40°C (внутренняя температура) в течение 16 часов для получения реакционной смеси 1A.
Воду (191 кг) и 25% водный раствор аммиака (85,8 кг) заливали в отдельный реактор и охлаждали холодной водой до 8,7°C. Затем, по истечении 3 минут, в реактор добавляли реакционную смесь 1A. Реакционную смесь перемешивали в течение 4 часов при охлаждении холодной водой. Осадок в реакционной смеси собирали путем фильтрации с центрифугой, и отфильтрованный остаток промывали 65 кг воды.
Осадок, воду (97 кг) и 25% водный раствор аммиака (43,5 кг) заливали в реактор и перемешивали в течение 1 часа, при этом температуру поддерживали с помощью теплой воды (25°C). Осадок в реакционной смеси собирали путем фильтрации с центрифугой и отфильтрованный остаток промывали 32,6 кг воды. Затем осадок сушили при пониженном давлении (60°C; 18 часов) для получения 9,6 кг 5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она.
1H ЯМР (400 МГц ДМСО-d6): δ 8,61-8,50 (м, 1H), 8,36 (д, 1H), 8,29 (дд, 1H), 7,90 (д, 1H), 7,80 (ддд, 1H), 7,56-7,45 (м, 5H), 7,27 (дд, 1H), 6,62 (д, 1H).
(Пример получения 2)
Синтез 3-бром-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она
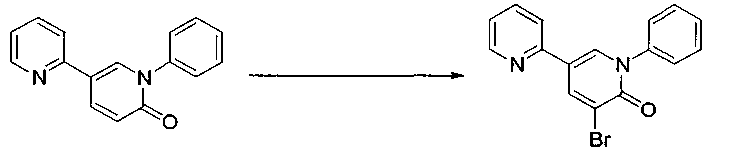
В 10-л реактор добавляли 5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-он (200 г), N-бромсукцинимид (157,7 г) и этилацетат (4 л) и реакционную смесь перемешивали под потоком азота при 30°C (внешняя температура) в течение 9 часов и 20 минут. В реакционную смесь добавляли 3% раствор гидросульфита (2 л) и толуол (2 л), и затем ее перемешивали при 55°C (внешняя температура) в течение 30 минут. После завершения реакции отделяли водный слой (нижний слой) реакционной смеси, и затем промывали органический слой водой (2 л) четыре раза. Растворитель выпаривали при перемешивании при пониженном давлении.
После этого дополнительное добавление 1,2-диметоксиэтана (4 л) и концентрирование при пониженном давлении дало необработанный продукт 3-бром-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она.
(Пример получения 3)
Синтез 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она
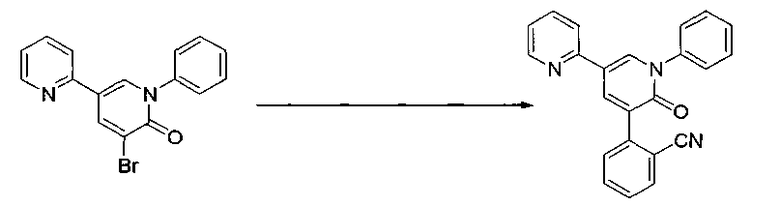
В реактор, содержащий полное количество необработанного продукта 3-бром-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, полученного в виде остатка после концентрирования в примере 2 продуцирования, добавляли 2-(1,3,2-диоксаборинан-2-ил)бензонитрил (214,9 г), ацетат палладия (3,44 г), трифенилфосфин (16,07 г), йодид меди (7,29 г), 1,2-диметоксиэтан (3,1 л) и карбонат калия (158,8 г). Перемешивание при нагревании выполняли при 70°C (внешняя температура) в атмосфере азота в течение 30 минут и затем при нагревании с обратным холодильником в течение 4 часов.
После этого в реакционную смесь добавляли этилацетат (2,5 л) при 70°C (внешняя температура), и перемешивалась смесь в течение 10 минут. Реакционную смесь фильтровали, и отфильтрованный остаток промывали этилацетатом (2,5 л). Данное полное количество фильтрата переносили в реактор, в который дополнительно добавляли 12,5% водный раствор аммиака (5 л). Перемешивание выполняли при 60°C (внешняя температура) в течение 53 минут. Отделяли нижний слой (водный слой) в реакционной смеси. В оставшийся органический слой добавляли 5% солевой раствор (2,5 л) и 25% водный раствор аммиака (2,5 л). После перемешивания отделяли нижний (водный слой). К остающемуся органическому слою дополнительно добавляли 5% солевой раствор (5 л). После перемешивания отделяли нижний слой (водный слой). Оставшийся органический слой концентрировали при пониженном давлении, и затем добавляли ацетон (4 л) с последующим концентрированием при пониженном давлении.
К данному остатку добавляли ацетон (7,2 л) и воду (0,8 л) и его растворяли путем перемешивания при 60°C (внешняя температура) в течение 1 часа и 10 минут. Затем производили охлаждение при 38°C (внешняя температура) в течение 18 минут при перемешивании. В реакционную смесь добавляли 1 г затравочных кристаллов 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она. Перемешивание проводили при 35°C (внешняя температура) в течение 30 минут. После этого реакционную смесь перемешивали при внешней температуре с понижением на 5°C каждые 30 минут и перемешивали при внешней температуре 10°C в течение 17 часов.
В реакционную смесь по каплям добавляли воду (2,29 л) при перемешивании в течение 3 часов и 10 минут. После добавления продолжали перемешивание в течение еще 1 часа и 20 минут. Реакционную смесь фильтровали, и отфильтрованный остаток промывали 2 л 50% ацетона-воды для получения 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она (526,28 г) в виде влажной массы, которая соответствовала 168,3 г в виде сухого веса.
Преобразование 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она во влажной массе в сухое вещество
Полученную влажную массу (4,378 г) взвешивали и сушили при пониженном давлении при 50°C в течение 4 часов для получения 1,4005 г высушенного порошка.
Преобразованное значение в виде сухого веса = (1,4005/4,378) x 526,28 = 168,3 г
Определение содержания по массе ацетона и воды во влажной массе 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она
Газохроматографический анализ полученной влажной массы в описанных ниже условиях позволил установить, что влажная масса, полученная в примере 3 продуцирования, содержала 168 мл ацетона и 186 мл воды.
Условия газохроматографического анализа:
Колонка: DB-WAX (30 мм Ч 0,53 мм, 1 мкм); детектор: TCD; темп. камеры: 60°C (8 мин), 60-180°C (70°C/мин), 180°C (5 мин); темп. детектора: 210°C; темп. на входе: 150°C; поток в колонке: 5,0 мл/мин; коэффициент разведения: (l:4); объем инжекции: 2 мкл
(пример 1X)
Кристаллизация 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она (гидрат)
В 10 л сосуд внесли 526,28 г 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она, полученного в виде влажной массы в примере 3 продуцирования. Из ацетона-воды, приготовленного из 5890 мл ацетона и 490 мл воды, 5,5 л добавляли в колбу и нагревали. После растворения проводили фильтрацию. При промывке 10 л сосуда и отфильтрованного остатка оставшимся полным количеством ацетона-воды весь фильтрат перенесли в 10 л сосуд.
Смесь перемешивали при внешней температуре 40°C, и после того, как внутренняя температура достигала 40°C, внешнюю температуру доводили до 35°C. Затем к смеси добавляли 842 мг гидрата 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она. После перемешивания смеси в течение 30 минут внешнюю температуру изменяли до 30°C, и затем, по истечении 30 минут, до 25°C. После этого внешнюю температуру понижали на 5°C каждые 30 минут вплоть до 15°C. После перемешивания смеси при внешней температуре 15°C в течение 30 минут внешнюю температуру дополнительно понижали до 8°C и продолжали перемешивание в течение 1 часа.
В смесь по каплям добавляли 842 мл воды при 11°C (внутренняя температура) в течение 1 часа и 10 минут. Через один час после того как было завершено добавление, внешнюю температуру изменяли до 0°C и перемешивали смесь в течение 40 минут. Затем внешнюю температуру понижали до -20°C и продолжали перемешивание в течение 15 часов.
Осадок в смеси собирали с помощью фильтрации. После того как осадок промывали 1700 мл 50% ацетона-воды, его сушили при аэрации в течение 50 минут. После этого данный осадок сушили с помощью вибрационной сушилки при 40°C при пониженном давлении в течение 11 часов и дополнительно сушили при 60°C в течение 3 часов.
После того как температура сушилки понижалась до комнатной температуры, внешний воздух впускали в сушилку при 950 гПа в течение 4 часов для получения 172,4 г 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она (кристаллическая форма гидрата).
1H ЯМР (400 МГц, ДМСО-d6): δ 8,61-8,57 (м, 1H), 8,53-8,52 (д-подобный, 1H), 8,47 (д, 1H), 8,01 (д, 1H), 7,92 (д, 1H), 7,86-7,81 (т-подобный, 1H), 7,79-7,76 (т-подобный, 1H), 7,72(д, 1H), 7,61-7,48 (м, 6H), 7,31-7,28 (м, 1H).
Остаточный палладий: 15 част./млн
(Контрольный пример А1)
Получение безводных кристаллов 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она (безводная форма II)
Продуцирование было выполнено ниже таким же способом, как процедура после исследования реакции, описанного в примере 7 в WO01/96308. Способ синтеза 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она [альтернативное название: 2-(2-оксо-1-фенил-5-(пиридин-2-ил)-1,2-дигидропиридин-3-ил)бензонитрил] описан в примере 7 в WO01/96308, а также в примере 3 продуцирования, выше.
К 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-ону (8 г) добавляли этилацетат (400 мл). Смесь нагревали при 60°C в теплой ванне. В смесь добавляли дополнительное количество ацетата (160 мл) и твердые вещества растворяли путем нагревания при 70°C в теплой ванне. После того, как в данный раствор добавляли н-гексан (80 мл), растворитель выпаривали при пониженном давлении для получения 7,7 г светло-желтого порошка.
1H ЯМР (400 МГц, ДМСО-d6): δ 8,59-8,57 (м, 1H), 8,53 (д, 1H), 8,47 (д, 1H,), 8,01 (д, 1H), 7,92 (д, 1H), 7,83 (ддд, 1H), 7,80-7,76 (м, 1H), 7,73-7,71 (д-подобный, 1H), 7,61-7,48 (м, 6H), 7,30 (дд, 1H).
(Пример B1)
Получение кристаллов 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-он гидрата (гидрат)
В 500-мл продолговатый сосуд вносили 7 г 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она. В сосуд добавляли 280 мл 90% ацетона-воды, приготовленного из 252 мл ацетона и 28 мл воды. Смесь перемешивали при нагревании в водяной бане и растворяли при кипячении с обратным холодильником (водяная баня; 65°C). После того как растворение подтвердилось, водяную баню охлаждали до 50°C. После добавления 35 мл воды в сосуд добавили 140 мг затравочных кристаллов [небольшое количество кристаллов 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она] при внутренней температуре 50°C. Для охлаждения смеси до -20°C при скорости охлаждения, приблизительно, 35°C/час использовали термостатируемую баню. После перемешивания смеси при -20°C в течение 1 часа осажденные твердые вещества собирали с помощью фильтрации и сушили при пониженном давлении (при внешней температуре 30°C в течение 1 часа и затем при 60°C в течение 2 часов). Полученные высушенные порошки (6,3 г) переносили в чашку Петри и оставляли на воздухе в течение 17 часов (влажность перед отстаиванием: 55,4%; влажность после отстаивания в течение ночи: 61,6%) для получения 6,2 г кристаллов 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она.
1H ЯМР (400 МГц, ДМСО-d6): δ 8,61-8,57 (м, 1H), 8,53 (д, 1H), 8,47 (д, 1H,), 8,01 (д, 1H), 7,92 (д, 1H), 7,83 (ддд, 1H), 7,78 (ддд, 1H), 7,73-7,71 (д-подобный, 1H), 7,61-7,48 (м, 6H), 7,30 (дд, 1H).
(Пример C1)
Получение безводных кристаллов 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она (безводная форма V)
В 500-мл сосуд вносили 9 г 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она (кристаллическая форма гидрата). В сосуд добавляли ацетон (360 мл) и смесь перемешивали при нагревании с обратным холодильником (при 70°C в водяной бане).
После растворения смесь фильтровали путем всасывания, и концентрировали фильтрат при 75°C при нормальном давлении для затвердевания. После того как твердые вещества были мелко раздроблены в ступке, к твердым веществам добавляли раствор ацетона-воды, приготовленный из 216 мл ацетона и 54 мл воды.
Смесь перемешивали при нагревании с обратным холодильником (при 75°C в водяной бане). После растворения смесь дополнительно перемешивали при нагревании с обратным холодильником в течение 2 часов и 40 минут. После этого температуру водяной бани для смеси (внешнюю температуру) понижали до значения комнатной температуры при скорости охлаждения 10°C/час, и ее перемешивали при комнатной температуре в течение 16 часов.
Осадок в реакционной смеси фильтровали путем всасывания и затем сушили при пониженном давлении (внешняя температура 20°C в течение 40 минут и затем при 60°C в течение 3 часов) для получения 7,2 г безводных кристаллов 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она.
1H ЯМР (400 МГц, ДМСО-d6): δ 8,61-8,57 (м, 1H), 8,53 (д, 1H), 8,47 (д, 1H,), 8,01 (д, 1H), 7,92 (дд, 1H), 7,83 (ддд, 1H), 7,78 (ддд, 1H), 7,72 (дд, 1H), 7,61-7,48 (м, 6H), 7,31-7,28 (м, 1H).
(Пример D1)
Получение безводных кристаллов 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она (безводная форма I)
В 1 л сосуд вносили 8 г 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она (гидрат). В сосуд добавляли этилацетат (480 мл), и смесь перемешивали при нагревании с обратным холодильником (в масляной бане) для осуществления растворения. Нагревание остановили и продолжили перемешивание, в то время как сосуд находился в масляной бане (при постепенном охлаждении). В момент, когда внутренняя температура достигла 50,9°C, в смесь добавили 0,2 г затравочных кристаллов [3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она (безводный кристалл)]. После этого перемешивание продолжали до тех пор, пока внутренняя температура не достигла 31,3°C. Смесь перемешивали в течение еще 2 часов в ледяной бане. Осажденные кристаллы собирали с помощью фильтрации и сушили при аэрации (50°C/18 часов) для получения 5,8 г безводных кристаллов 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она.
1HЯМР (400 МГц, ДМСО-d6): δ 8,58 (д, 1H), 8,53 (д, 1H), 8,47 (д, 1H,), 8,01 (д, 1H), 7,93 (д, 1H), 7,83 (ддд, 1H), 7,78 (д, 1H), 7,72 (д, 1H), 7,61-7,48 (м, 6H), 7,32-7,27 (м, 1H).
Физическая стабильность в процессе смешивания в присутствии воды или смешанного раствора вода-этанол (1:1)
(Последовательность операций)
Приблизительно 150 мг соответствующих кристаллов помещают в агатовую ступку, и операцию смешивания выполняют при комнатной температуре в течение нескольких минут, в то время как продолжается добавление по каплям воды (или смешанного растворавода-этанол (1:1)). После этого соответствующие кристаллы сушат при, приблизительно, 60°C в течение 2-3 часов.
(Результаты)
С помощью дифракционного анализа рентгеновских лучей на порошке показано, что кристаллы, полученные в контрольном примере А1, претерпели изменение кристаллической формы в ходе операции смешивания в присутствии воды или смешанного раствора вода-этанол (1:1), и та же кристаллическая форма, что и полученная в примере D1, увеличилась в количестве.
С помощью дифракционного анализа рентгеновских лучей на порошке показано, что соответствующие кристаллы, полученные в примере B1, примере C1 и примере D1, не демонстрируют изменения кристаллической формы, и они были физически устойчивыми в присутствии воды или смешанного раствора вода-этанол (1:1).
Влияние изменения температуры и влажности на Гидрат
(Аппаратура)
Система DTA рентгеновских лучей Rigaku: RINT-2000, произведенная Rigaku Corporation
(Способ осуществления)
Кристаллы, полученные в примере B1 (гидрат), измельчали в ступке и затем наносили образцы на стеклянный планшет диаметром 13 мм. Измерение выполняли при условиях, описанных ниже.
Используемые рентгеновские лучи: CuKα-луч
Напряжение на лампе: 40 кВ
Ток на лампе: 200 мА
Отклоняющая щель: 1/2 градуса
Приемная щель: 0,3 мм
Рассеивающая щель: 1/2 градуса
Скорость сканирования: 2°/мин
Шаг сканирования: 0,01°
Диапазон сканирования (2θ): от 5 до 40°
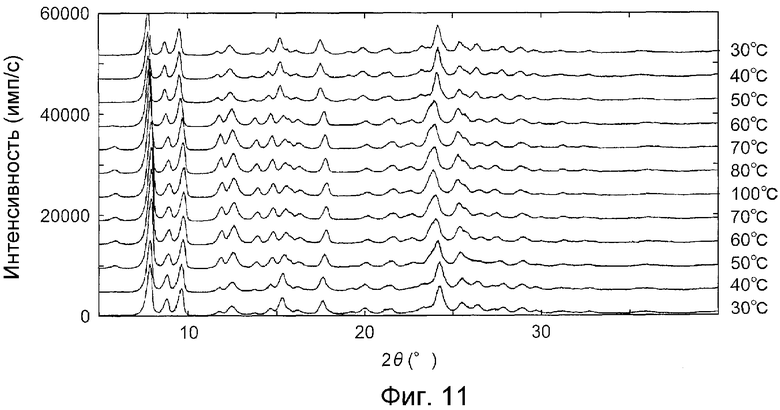
Температуру измерения последовательно меняли следующим образом, и дифракционные рентгенограммы на порошке измеряли при соответствующих значениях температуры в последовательности: 30, 40, 50, 60, 70, 80, 100, 70, 60, 50, 40 и 30°C.
(Результаты)
На фиг.11 показаны дифракционные рентгенограммы на порошке гидрата при соответствующих значениях температуры, приведенных выше. Изменения изображений дифракции рентгеновских лучей на порошке позволили обнаружить, что кристаллы из примера B1 (гидрат) трансформируются в те же кристаллы, что и кристаллы из примера E1 (безводная форма III) при, приблизительно, 60°C или выше, и снова возвращаются в гидрат, когда понижали температуру.
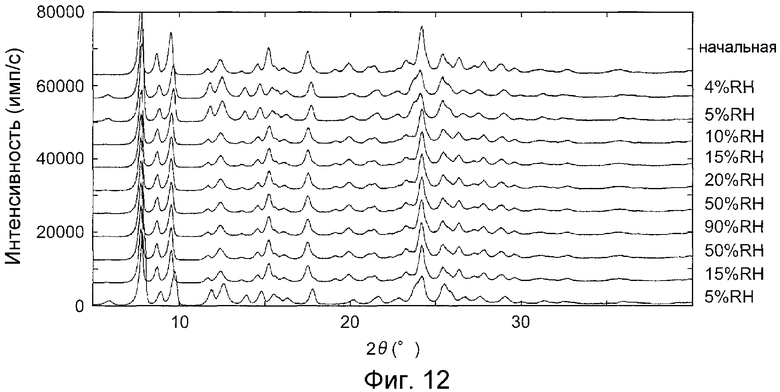
Влажность измерения последовательно меняли следующим образом, и дифракционные рентгенограммы на порошке измеряли при соответствующих значениях влажности в последовательности: 4, 5, 10, 15, 20, 50, 90, 50, 15, и 5% ОВ (относительная влажность).
(Результаты)
На фиг.12 показаны дифракционные рентгенограммы на порошке гидрата при соответствующих приведенных выше значениях влажности. Из изображений дифракции рентгеновских лучей на порошке наблюдались обратные изображения гидрата и безводной формы III при влажности больше и меньше, приблизительно, 10% ОВ. Подтверждено, что кристаллы примера B1 (гидрат) заменяются на безводную форму III при влажности, приблизительно, 10% ОВ или меньше и остаются в виде гидрата при влажности, приблизительно, 10% ОВ или больше.
Данные эксперименты, касающиеся влияния изменения температуры и влажности на гидрат, и пример 1X позволили обнаружить, что состояние осадка перед естественной сушкой представляло собой такие же кристаллы, как таковые из примера E1 (безводная форма III) или смесь безводной формы и гидрата, которая является полезным промежуточным соединением для получения гидрата.
Минимальная энергия воспламенения и нижний концентрационный предел взрываемости
(Способ осуществления)
Подходящее количество гидрата, соответствующего концентрации, равномерно поместили в кювету прибора для испытания взрывов пыли с наддувом. Воздух в 1,3 л баллоне был сжат до 50 кПа, и воздух ввели в стеклянный цилиндр путем открытия клапана под управлением соленоида для образования клубов пыли. На электрод разряда подавалась энергия по истечении 0,1 секунд после открытия клапана под управлением соленоида. Критерием воспламенения является достижение пламенем отметки воспламенения, установленной в 100 мм выше электрода разряда.
(Условия измерения нижнего концентрационного предела взрываемости)
Температура помещения, в котором производились измерения: 24°C
Влажность: 49%
Ударное давление сжатого воздуха: 50 кПа
Время запуска воспламенения: 0,1 с
Повторение теста воспламенения: 5 раз
Энергия зажигающего разряда: 10 Дж
(Условия измерения для минимальной энергии воспламенения)
Температура помещения, в котором производились измерения: 24°C
Влажность: 49%
Ударное давление сжатого воздуха: 50 кПа
Время запуска воспламенения: 0,1 с
Повторение теста воспламенения: 10 раз
(Аппаратура)
Прибор для испытания взрывов пыли с наддувом (Environmental Technology, Ltd. DES-10)
(Результаты)
Нижний концентрационный предел взрываемости: 160-170 г/м3
Минимальная энергия воспламенения: 50-100 мДж
Концентрация пыли: 1250 г/м3
Поляризуемость
(Способ осуществления)
Приблизительно 1 г соответствующих соединений взвешивают в сосуде для взвешивания (диаметр 35 мм). Палочку для перемешивания [покрытие из фторопласта (тетрафторэтиленовой смолы); 20 мм] помещают в сосуд и, после того как закрывают крышку, порошки перемешивают в течение 30 минут. Крышку открывают в то время, когда останавливают перемешивание; и статический потенциал порошка измеряют с применением измерительного прибора для статического потенциала.
(Аппаратура)
STATIRON-DZ3, произведенный Shishido Electrostatic, Ltd.
(Результаты)
Кристаллы контрольного примера A1: 70-100 В
Кристаллы примера B1: 0 В
Измерение инфракрасных спектров
Инфракрасный спектр кристаллов, полученных в примере B1, измеряли при условиях измерения, описанных ниже в соответствии со способом диска с бромидом калия для измерения инфракрасного спектра, как описано в General Tests in the Japanese Pharmacopoeia.
(Аппаратура)
FT/IR-620 произведенный JASCO Corporation
Диапазон измерений: 4000-400 см-1
Разрешение: 4 см-1
Количество интеграции: 36
Скорость сканирования: 2 мм/с
На фиг.1 показан инфракрасный спектр кристаллов, полученных в примере B1 (способ KBr), и на фиг.2 показан инфракрасный спектр кристаллов, полученных в примере C1 (способ KBr).
В таблице 2 показаны волновые числа (см-1) и значения коэффициента пропускания (%) пиков поглощения для кристаллов, полученных в примере B1. В таблице 3 показаны волновые числа (см-1) и значения коэффициента пропускания (%) пиков поглощения для кристаллов, полученных в примере C1.
Измерение дифракционной рентгенограммы на порошке
Дифракционные рентгенограммы на порошке кристаллов, полученных в соответствующих примерах, измеряли в условиях измерений, описанных ниже, в соответствии со способом измерения дифракции рентгеновских лучей, как описано в General Tests in the Japanese Pharmacopoeia.
(Аппаратура)
Система DTA рентгеновских лучей Rigaku: RINT-2000, произведенная Rigaku Corporation
(Способ осуществления)
Образец измельчали в ступке и затем наносили образцы на стеклянный планшет диаметром 13 мм. Измерение выполняли при условиях ниже.
Используемые рентгеновские лучи: CuKα-луч
Напряжение на лампе: 40 кВ
Ток на лампе: 200 мА
Отклоняющая щель: 1/2 градуса
Приемная щель: 0,3 мм
Рассеивающая щель: 1/2 градуса
Скорость сканирования: 1°/мин
Шаг сканирования: 0,01°
Диапазон сканирования (2θ): от 5 до 40°
На фиг.3 показана дифракционная рентгенограмма на порошке кристаллов, полученных в контрольном примере А1, на фиг.4 показана дифракционная рентгенограмма на порошке кристаллов, полученных в примере B1, на фиг.5 показана дифракционная рентгенограмма на порошке кристаллов, полученных в примере C1 и на фиг.6 показана дифракционная рентгенограмма на порошке кристаллов, полученных в примере D1.
В таблице 4 показаны пики и их интенсивности при углах дифракции (2θ) для кристаллов, полученных в контрольном примере А1, в таблице 5 показаны пики и их интенсивности при углах дифракции (2θ) для кристаллов, полученных в примере B1, в таблице 6 показаны пики и их интенсивности при углах дифракции (2θ) для кристаллов, полученных в примере C1 и в таблице 7 показаны пики и их интенсивности при углах дифракции (2θ) для кристаллов, полученных в примере D1.
На основании фиг.4 и таблицы 5, которые представляют дифракционную рентгенограмму на порошке кристаллов, полученных в примере B1, можно обнаружить, что дифракционная рентгенограмма на порошке кристаллов, полученных в примере B1, образует характеристический пик, имеющий угол дифракции (2θ), приблизительно, 12,5°.
Это говорит о том, что кристаллы, полученные в контрольном примере А1, не содержат ту же кристаллическую форму так же, как и кристаллы, полученные в примере B1, поскольку на фиг.3 и в таблице 4, которые представляют дифракционную рентгенограмму на порошке кристаллов, полученных в контрольном примере А1, не содержат пик, имеющий угол преломления (2θ), приблизительно, 12,5.
Пример E1 (безводная форма III)
Относительно кристаллов гидрата 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-он, дифракционную рентгенограмму на порошке измеряли при условиях, подобных описанным выше. Однако измерение проводили при скорости сканирования 2°/мин при условиях нагревания около 110°C.
На фиг.7 показана дифракционная рентгенограмма на порошке, и в таблице 8 показаны пики и интенсивность при углах преломления (2θ ± 0,2°) для кристаллов.
Измерение спектра ЯМР 13C в твердом состоянии
Спектры ЯМР 13C в твердом состоянии измеряли для кристаллов, полученных в примерах B1, C1 и D1, в следующих условиях.
Температура измерения: комнатная температура (˜22°C)
Стандартное соединение: карбонильный углерод глицина (внешний стандарт: 176,03 част./млн)
Измеряемое ядро: 13C (100,6248425 МГц)
Время повторения импульсов:
50 с для примеров C1 и D1
5 с для примера B1
Импульсный режим: измерение CP/TOSS
На фиг.8 показан спектр ЯМР 13C в твердом состоянии кристаллов, полученных в примере B1, и значения химического сдвига приведены в таблице 9. На фиг.9 показан спектр ЯМР 13C в твердом состоянии кристаллов, полученных в примере D1, и значения химического сдвига приведены в таблице 10. На фиг.10 показан спектр ЯМР 13C в твердом состоянии кристаллов, полученных в примере C1, и значения химического сдвига приведены в таблице 11.
Промышленная применимость
Кристаллические формы в соответствии с изобретением обладают предпочтительными свойствами и подходят для применения в качестве активного ингредиента терапевтических или профилактических агентов против нейродегенеративных заболеваний и т.п.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛЫ ПРОИЗВОДНЫХ 6,7-НЕНАСЫЩЕННОГО-7-КАРБАМОИЛМОРФИНАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2607084C2 |
| КРИСТАЛЛЫ ПРОИЗВОДНЫХ 6,7-НЕНАСЫЩЕННОГО-7-КАРБАМОИЛМОРФИНАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2643807C1 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА И СОЛЬ 3-(3,5-ДИХЛОР-4-ГИДРОКСИБЕНЗОИЛ)- 1,1-ДИОКСО-2,3-ДИГИДРО-1,3-БЕНЗОТИАЗОЛА | 2018 |
|
RU2772043C2 |
| СОЛЬ ПРОИЗВОДНОГО ПИРАЗОЛОХИНОЛИНА И ЕЕ КРИСТАЛЛ | 2014 |
|
RU2655171C2 |
| СТАБИЛЬНАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА ТИПИРАЦИЛА ГИДРОХЛОРИДА И СПОСОБ ЕЕ КРИСТАЛЛИЗАЦИИ | 2014 |
|
RU2640417C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ СВОБОДНОГО ОСНОВАНИЯ ПРОИЗВОДНОГО БЕНЗОФУРАНА И СПОСОБ ПОЛУЧЕНИЯ | 2018 |
|
RU2791189C2 |
| СОЛЬ МОНОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО ПИРИДИНА И ЕЕ КРИСТАЛЛ | 2015 |
|
RU2658821C1 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА ПИРИДО[3,4-D]ПИРИМИДИНОВОГО ПРОИЗВОДНОГО | 2017 |
|
RU2793759C2 |
| СОЛЬ ПРОИЗВОДНОГО ПИРРОЛИДИН-3-ИЛ-УКСУСНОЙ КИСЛОТЫ И ЕЕ КРИСТАЛЛЫ | 2014 |
|
RU2640047C2 |
| ПРОИЗВОДНОЕ ПИРИДОНА, ИМЕЮЩЕЕ ТЕТРАГИДРОПИРАНИЛМЕТИЛЬНУЮ ГРУППУ | 2015 |
|
RU2707953C2 |
Изобретение относится к кристаллическому гидрату 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она примера В1, к способу его получения, а также к безводным кристаллическим формам 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она примеров D1, C1, E1, и к фармацевтической композиции. Технический результат - простое получение 3-(2-цианофенил)-5-(2-пиридил)-1-фенил-1,2-дигидропиридин-2-она в виде однородной кристаллической формы в промышленном масштабе, обладающего антагонистическим действием в отношении рецептора АМРА и/или ингибирующим действием в отношении каинатного рецептора. 14 н. и 12 з.п. ф-лы, 11 табл.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| RU 92004391 А, 27.03.1995 | |||
| RU 2000129671 А, 20.02.2004. | |||

