Область техники, к которой относится изобретение
Настоящее изобретение касается кристаллических форм пиридо[3,4-d]пиримидинового производного или его сольвата, которые обладают ингибирующим действием в отношении циклин-зависимой киназы 4 и/или циклин-зависимой киназы 6 (далее в тексте именуется "CDK4/6") и которые можно применять для профилактики или лечения ревматоидного артрита, артериосклероза, фиброза легких, церебрального инфаркта и/или ракового заболевания.
Предшествующий уровень техники
Ингибиторы CDK 4/6 можно применять для лечения различных заболеваний, вызванных аномальным ростом клеток, включая рак, сердечнососудистые заболевания, заболевания почек, специфичные инфекции и аутоиммунные заболевания, и ожидается, что они будут эффективны в лечении ревматоидного артрита, артериосклероза, фиброза легких, церебрального инфаркта и рака. Причины, по которым подавление прохождения клеточного цикла и роста клеток посредством ингибирования CDK считают эффективным в лечении указанных заболеваний, основаны на описанных далее сведениях.
Ревматоидный артрит включает образование паннуса вследствие гиперпролиферации синовиальных клеток. Сообщалось, что такую гиперпролиферацию можно уменьшить путем введения животному ингибитора CDK4/6 (НПЛ 1). Комплекс CDK4-циклин D регулирует выработку MMP3 в синовиальных клетках пациентов с ревматоидным артритом. Негативная регулировка активности CDK4/6 ингибирует не только пролиферацию, но и выработку MMP3 (НПЛ 2).
Таким образом, ожидается, что ингибиторы CDK4/6 оказывают и ингибирующее действие на пролиферацию синовиальных клеток, и защитное действие на хрящи при ревматоидном артрите.
Индуцирование экспрессирования ингибирующего клеточный цикл белка p21 с помощью аденовирусного вектора было эффективно в мышиной модели фиброза легких (НПЛ 3).
Известно, что концентрация циклин D1/CDK4 повышается в крысиной модели церебрального инфаркта в связке с гибелью нейронов, вызванной локальной ишемией. Гибель нейронов уменьшается при введении флавопиридола, который представляет собой неселективный ингибитор CDK (НПЛ 4).
Механизм циклин D-CDK4/6-INK4a-Rb часто детектируется в случае человеческого рака, вызванного аномалией любых факторов, вносящих вклад в рост раковых клеток, таких как утрата функционального p16INK4a, сверхэкспрессия циклина D1, сверхэкспрессия CDK4 или утрата функционального Rb (НПЛ 5). Такая аномалия способствует переходу клеточного цикла из фазы G1 в фазу S, и данный механизм определенно играет важную роль в онкогенной трансформации или аномальном росте раковых клеток.
Ингибиторы CDK4/6 могут быть эффективны, особенно в случае опухолей, у которых наблюдается аномалия в генах, активирующих CDK4/6 киназу, таких как рак с транслокацией циклина D, рак с амплификацией или сверхэкспрессированием CDK4 или CDK6, и рак с инактивацией p16. Ингибиторы CDK4/6 могут быть эффективны в лечении рака с генетической аномалией апстрим-регулятора циклина D, количество которого увеличивается из-за дефекта апстрим-регулятора.
Многие соединения, подавляющие активность CDK4/6, были синтезированы и описаны в данной области техники, и такие соединения были клинически протестированы в лечении раковых заболеваний, таких как рак груди (НПЛ 6).
Наиболее острая и тяжелая токсичность при лучевой терапии и химиотерапии вызвана воздействием на стволовые клетки и клетки-предшественники. Ингибитор CDK4/6 вызывает временную остановку клеточного цикла у гематопоэтических стволовых клеток и клеток-предшественников, и защищает их от цитотоксичности лучевой терапии и химиотерапии. После лечения таким ингибитором, гематопоэтические стволовые клетки и клетки-предшественники (HSPC) выходят из временной спячки и затем функционируют в обычном режиме. Поэтому устойчивость к химиотерапии с применением ингибитора CDK4/6 может обеспечить серьезную защиту костного мозга (НПЛ 7).
Таким образом, ожидается, что ингибиторы CDK4/6 будут эффективны для лечения, например, ревматоидного артрита, артериосклероза, фиброза легких, церебрального инфаркта или ракового заболевания, и для защиты костного мозга, в частности для лечения ревматоидного артрита или рака и для защиты костного мозга.
Ингибиторы CDK, включая CDK4/6, описаны в ПТЛ 1 и 2.
Список процитированной литературы
Патентная литература
[ПТЛ1] WO2003/062236A
[ПТЛ2] WO2010/020675A
Непатентная литература
[НПЛ 1] Taniguchi, K et al., Nature Medicine, Vol.5, p.760-767 (1999)
[НПЛ 2] Nonomura Y et al., Arthritis & Rheumatology 2006, Jul; 54 (7): p.2074-83
[НПЛ 3] American Journal Physiology: Lung Cellular and Molecular Physiology, 2004, Vol. 286, p.L727-L733
[НПЛ 4] Proceedings of the National Academy of Sciences of the United States of America, 2000, Vol.97, p.10254-10259
[НПЛ 5] Science, Vol. 254, p.1138-1146 (1991)
[НПЛ 6] Guha M, Nature Biotechnology 2013, Mar; 31 (3): p.187
[НПЛ 7] Journal of Clinical Investigation 2010; 120 (7): p.2528-2536
Краткое описание изобретения
Проблема, на решение которой направлено изобретение
Задачей настоящего изобретения является получение новой кристаллической формы пиридо[3,4-d]пиримидинового производного или его сольвата, имеющей прекрасное ингибирующее действие в отношении CDK4/6.
Способы решения проблемы
Проведя обширные исследования, направленные на решение указанной выше задачи, авторы настоящего изобретения обнаружили, что пиридо[3,4-d]пиримидиновые производные, имеющие определенную структуру, демонстрируют прекрасное ингибирующее действие в отношении CDK4/6.
Авторы настоящего изобретения обнаружили также, что некоторые из указанных соединений могут образовывать кристаллические формы, которые химически устойчивы и могут применяться в качестве активных фармацевтических субстанций.
Таким образом, настоящее изобретение касается кристаллической формы соединения, имеющего формулу (I), или его сольвата.
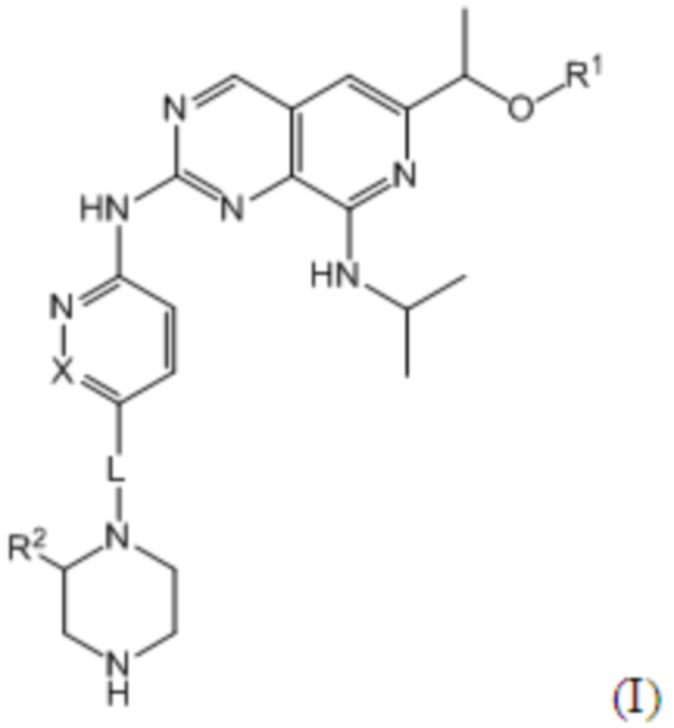
В приведенной формуле, R1 представляет собой атом водорода или C1-3 алкильную группу, R2 представляет собой атом водорода или оксо-группу (в случае оксо-группы, R2 связан с пиперазиновым кольцом двойной связью), L представляет собой простую связь или C1-3 алкиленовую группу, и X представляет собой CH или N.
Эффект изобретения
В настоящем изобретении описана кристаллическая форма пиридо[3,4-d]пиримидинового производного или его сольвата, демонстрирующая прекрасное ингибирующее действие в отношении CDK4/6, которую можно использовать в качестве профилактического или терапевтического лекарственного средства для лечения, например, ревматоидного артрита, артериосклероза, фиброза легких, церебрального инфаркта и/или рака.
Кристаллическая форма по настоящему изобретению может применяться в качестве активной фармацевтической субстанции в производстве фармацевтического продукта.
Краткое описание чертежей
Фиг. 1 представляет собой спектр порошковой рентгеновской дифракции кристаллической формы D 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она.
Фиг. 2 представляет собой спектр порошковой рентгеновской дифракции кристаллической формы A 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она.
Фиг. 3 представляет собой спектр порошковой рентгеновской дифракции кристаллической формы A 1-(6-((6-((1R)-1-метоксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридазил)пиперазина.
Фиг. 4 представляет собой спектр порошковой рентгеновской дифракции кристаллической формы B 1-(6-((6-((1R)-1-метоксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридазил)пиперазина.
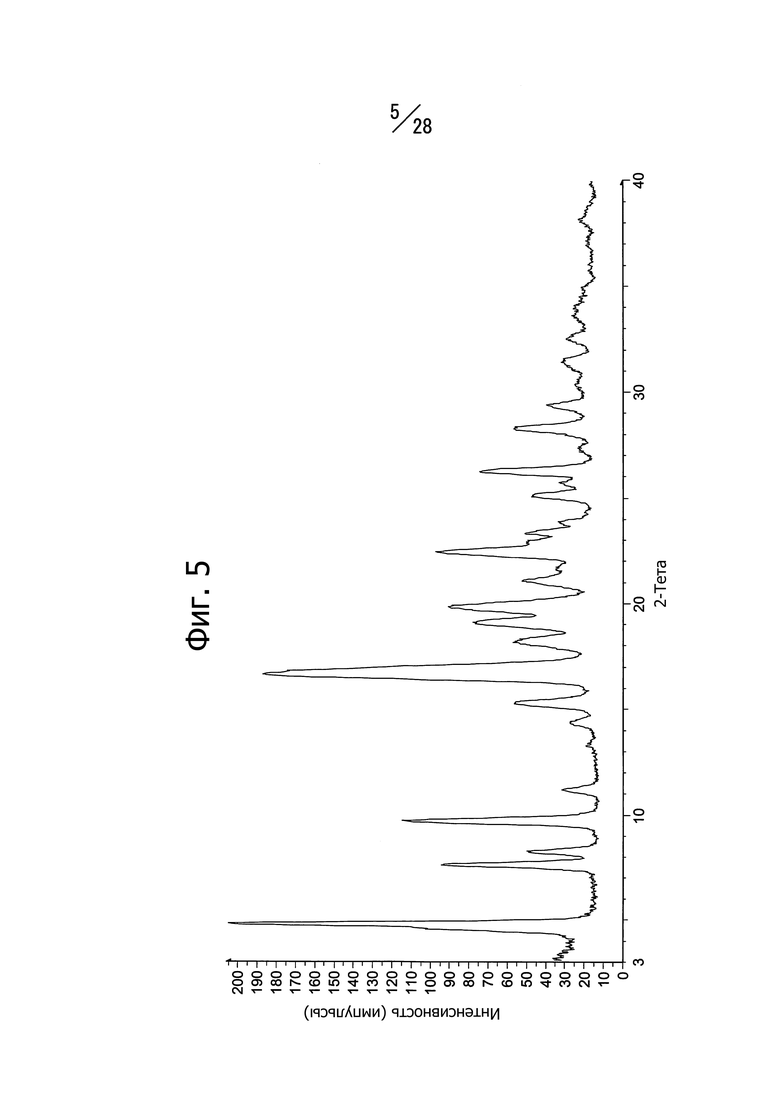
Фиг. 5 представляет собой спектр порошковой рентгеновской дифракции кристаллической формы A (R)-N8-изопропил-6-(1-метоксиэтил)-N2-(5-(пиперазин-1-илметил)пиридин-2-ил)пиридо[3,4-d]пиримидин-2,8-диамина.
Фиг. 6 представляет собой спектр порошковой рентгеновской дифракции кристаллической формы B 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она.
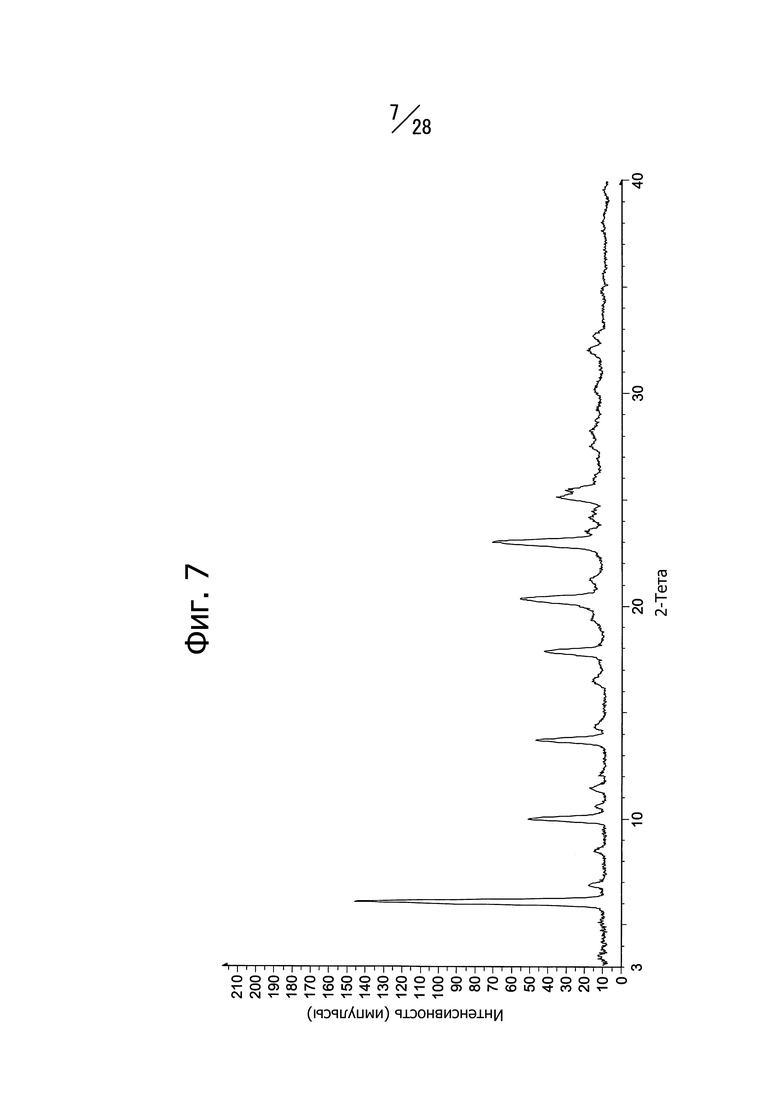
Фиг. 7 представляет собой спектр порошковой рентгеновской дифракции кристаллической формы (Кристаллическая форма C) сольвата 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она с диметилсульфоксидом.
Фиг. 8 представляет собой спектр порошковой рентгеновской дифракции кристаллической формы I 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она.
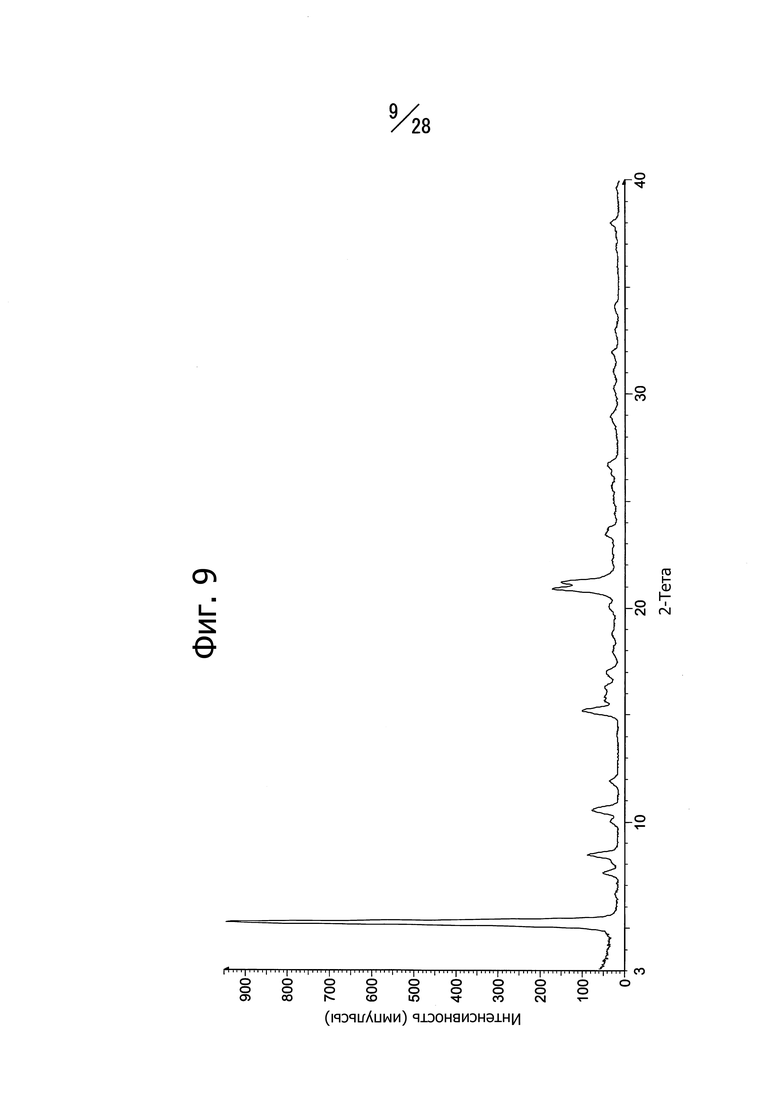
Фиг. 9 представляет собой спектр порошковой рентгеновской дифракции кристаллической формы C 1-(6-((6-((1R)-1-метоксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридазил)пиперазина.
Фиг. 10 представляет собой ИК-спектр кристаллической формы D 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она.
Фиг. 11 представляет собой ИК-спектр кристаллической формы A 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она.
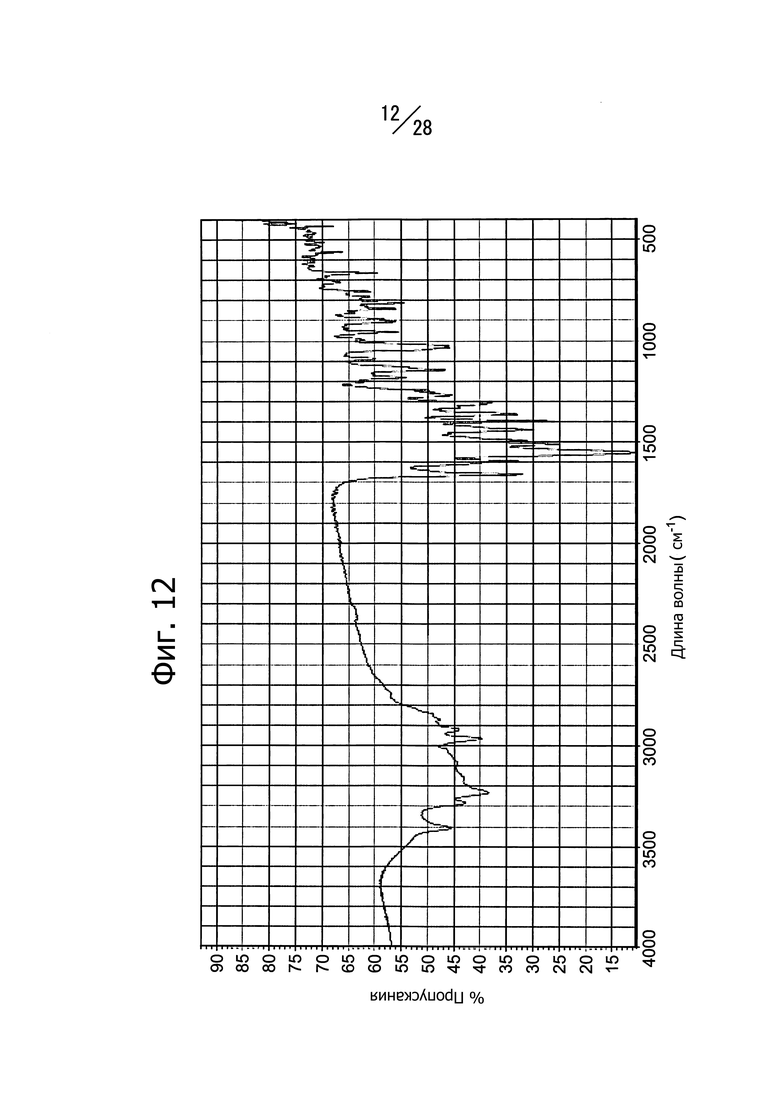
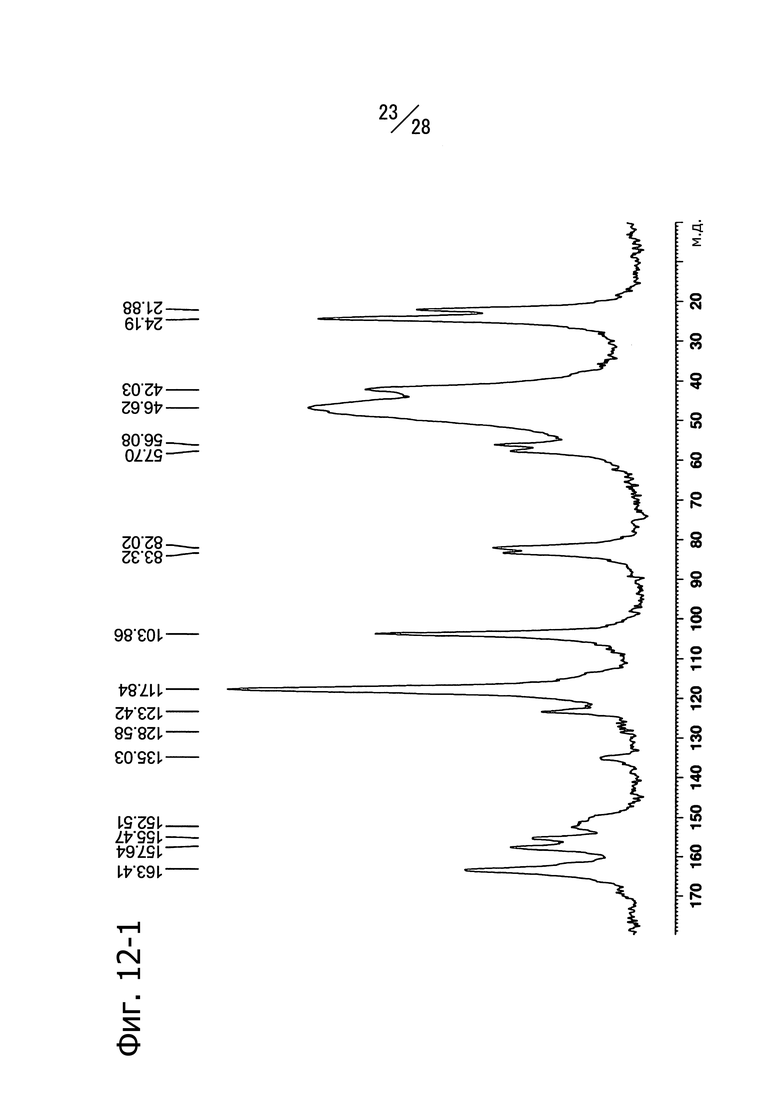
Фиг. 12 представляет собой ИК-спектр кристаллической формы (Кристаллическая форма C) сольвата 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она с диметилсульфоксидом.
Фиг. 13 представляет собой ИК-спектр кристаллической формы I 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она.
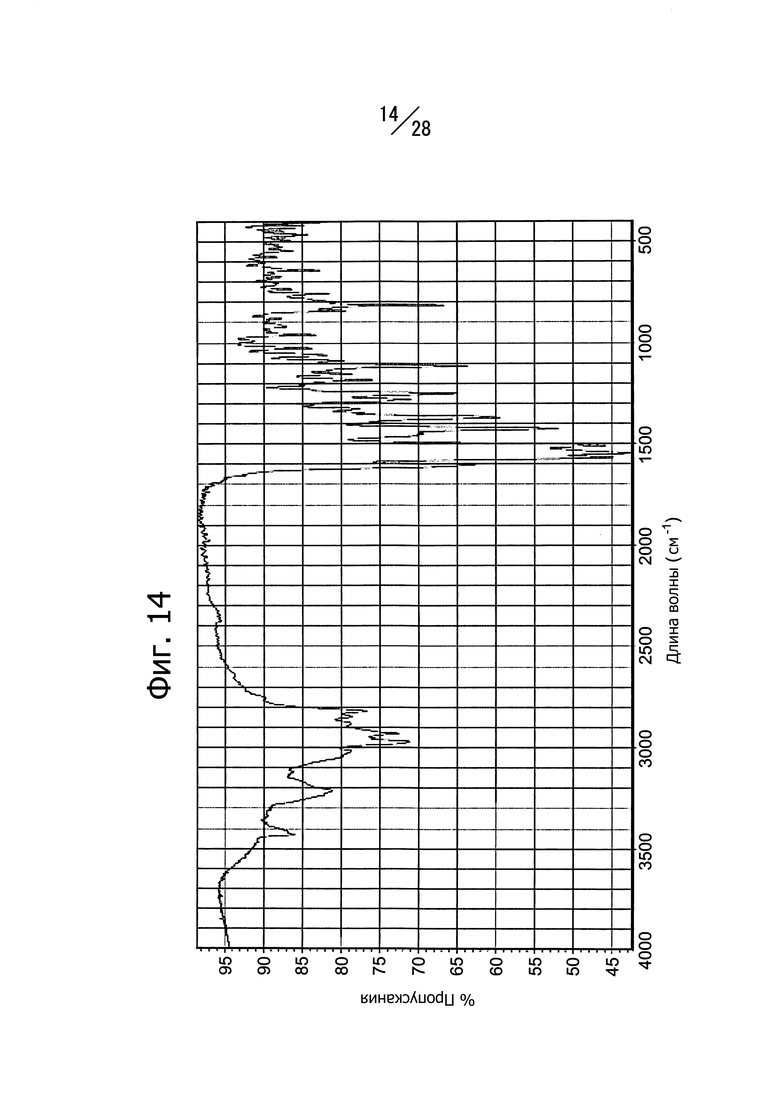
Фиг. 14 представляет собой ИК-спектр кристаллической формы A 1-(6-((6-((1R)-1-метоксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридазил)пиперазина.
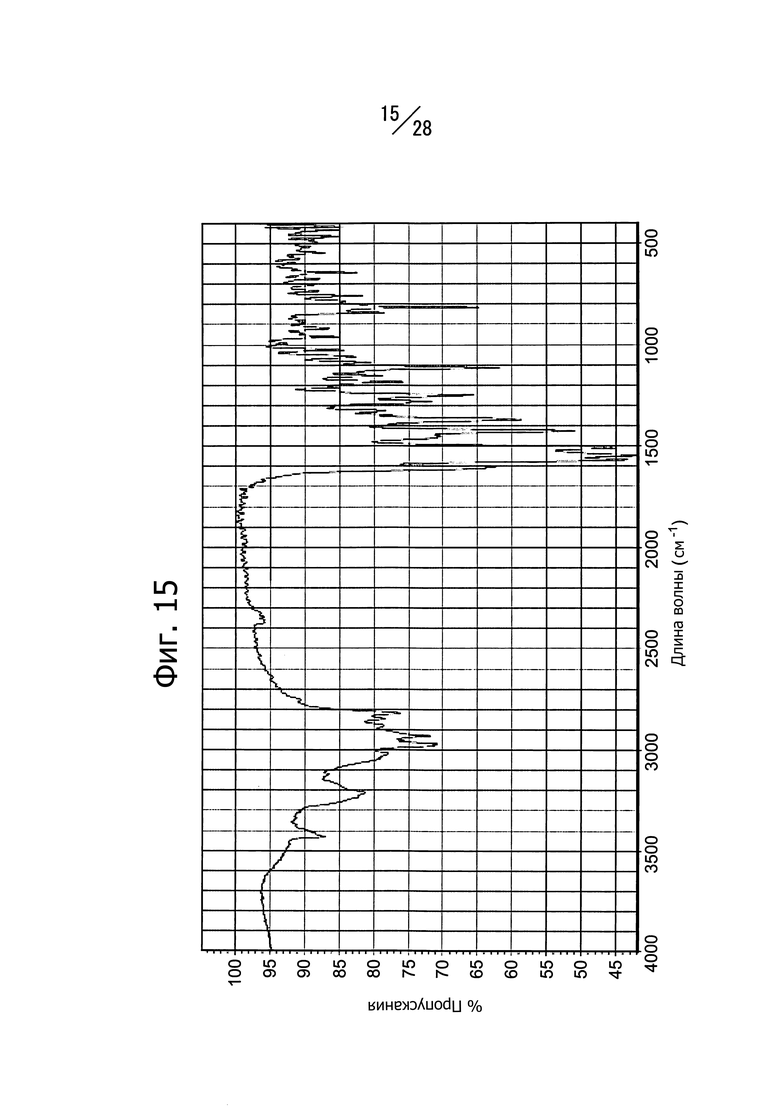
Фиг. 15 представляет собой ИК-спектр кристаллической формы C 1-(6-((6-((1R)-1-метоксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридазил)пиперазина.
Фиг. 16 представляет собой ИК-спектр кристаллической формы A (R)-N8-изопропил-6-(1-метоксиэтил)-N2-(5-(пиперазин-1-илметил)пиридин-2-ил)пиридо[3,4-d]пиримидин-2,8-диамина.
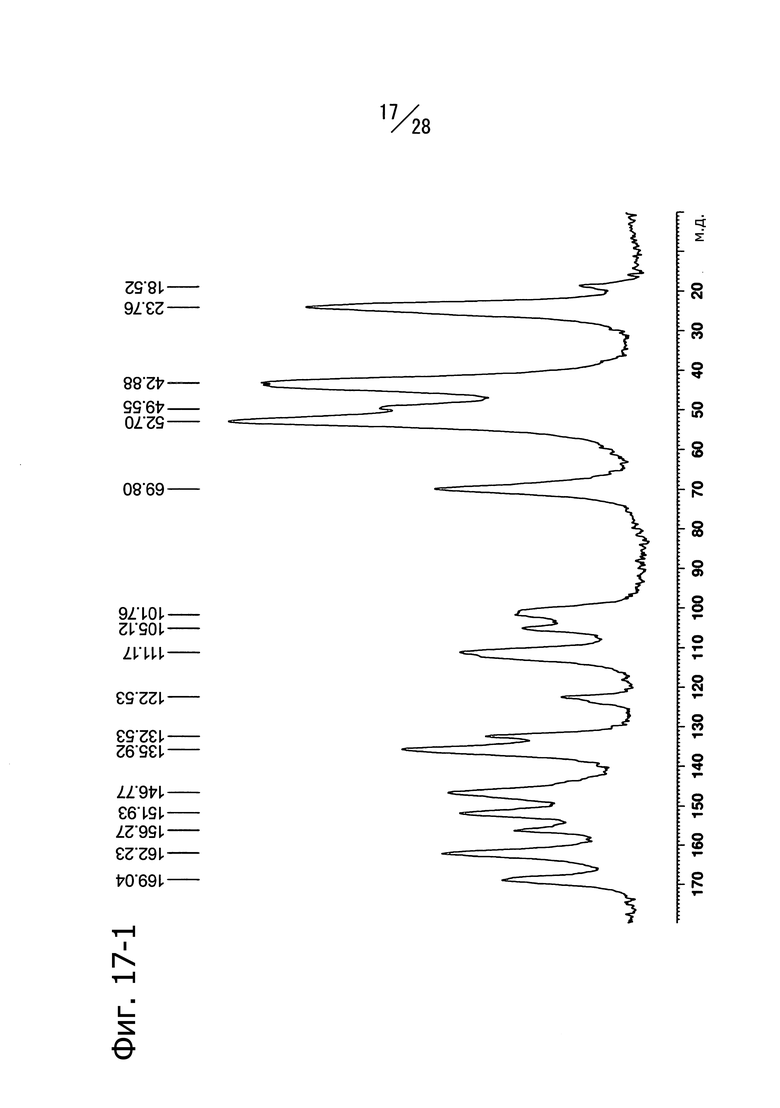
Фиг. 17-1 и 17-2 представляют собой твердотельные ЯМР-спектры (13C) кристаллической формы D 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она. Фиг. 17-1 представляет собой спектр, записанный в режиме 65000 Гц, а Фиг. 17-2 представляет собой спектр, записанный в режиме 14000 Гц.
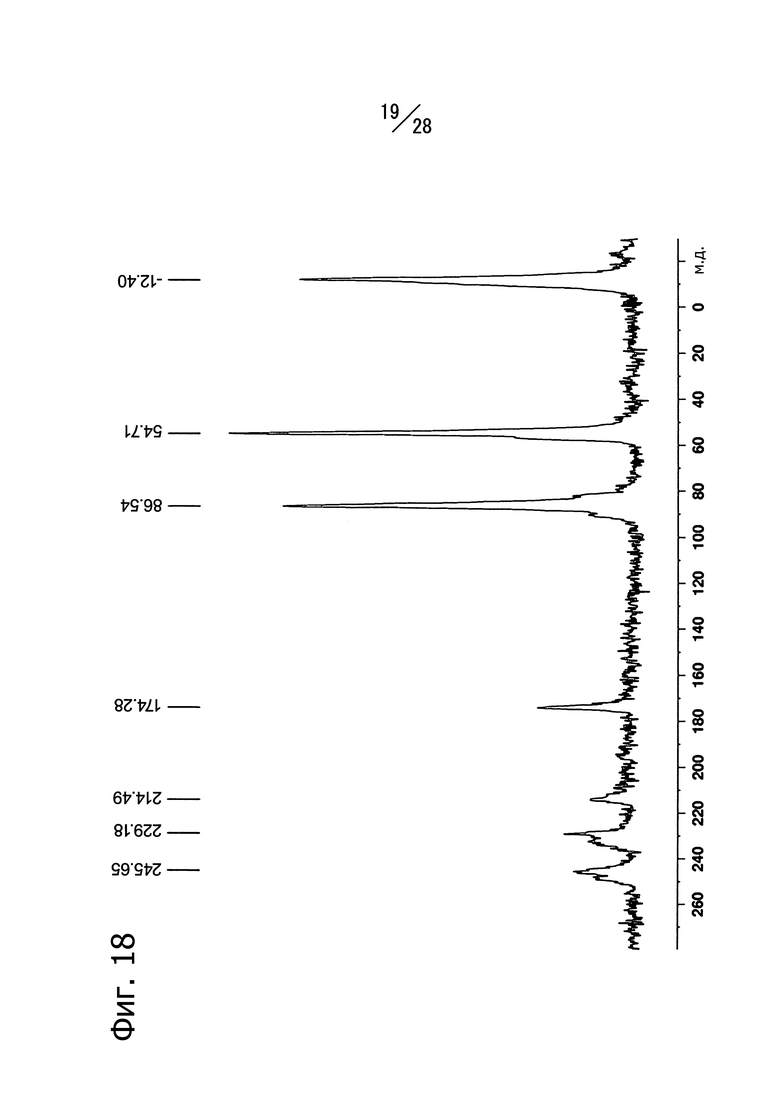
Фиг. 18 представляет собой твердотельный ЯМР-спектр (15N) кристаллической формы D 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она.
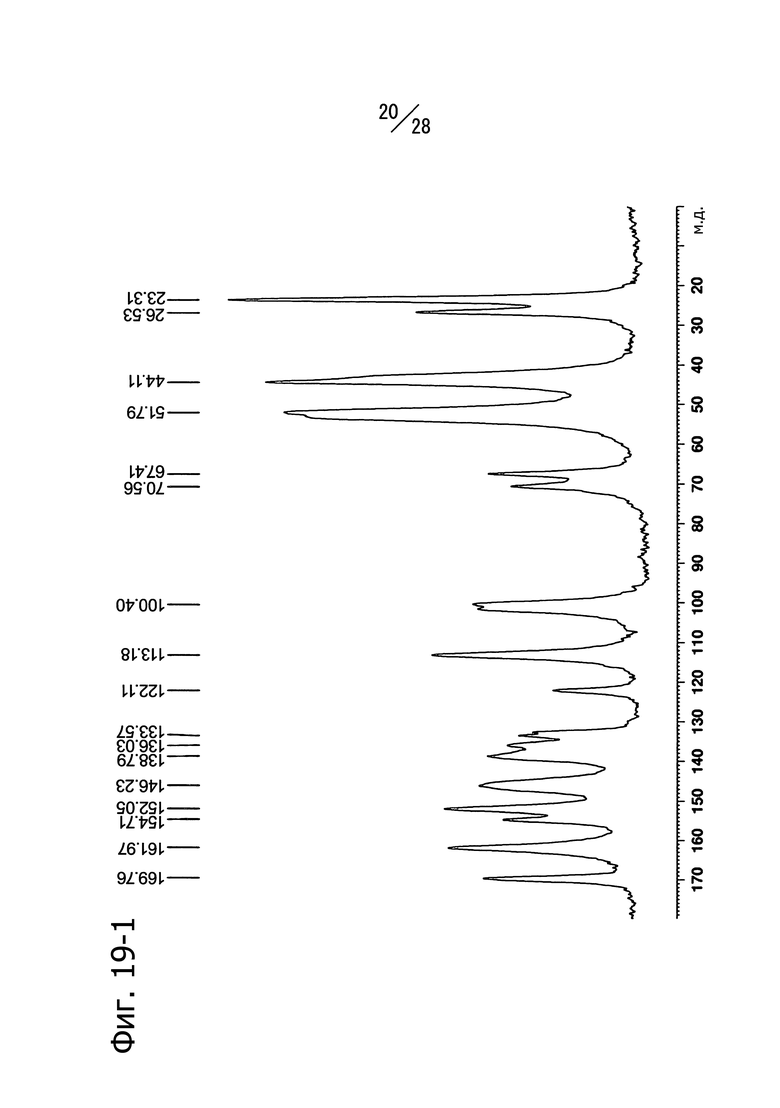
Фиг. 19-1 и 19-2 представляют собой твердотельные ЯМР-спектры (13C) кристаллической формы A 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она. Фиг. 19-1 представляет собой спектр, записанный в режиме 65000 Гц, а Фиг. 19-2 представляет собой спектр, записанный в режиме 14000 Гц.
Фиг. 20 представляет собой твердотельный ЯМР-спектр (15N) кристаллической формы A 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она.
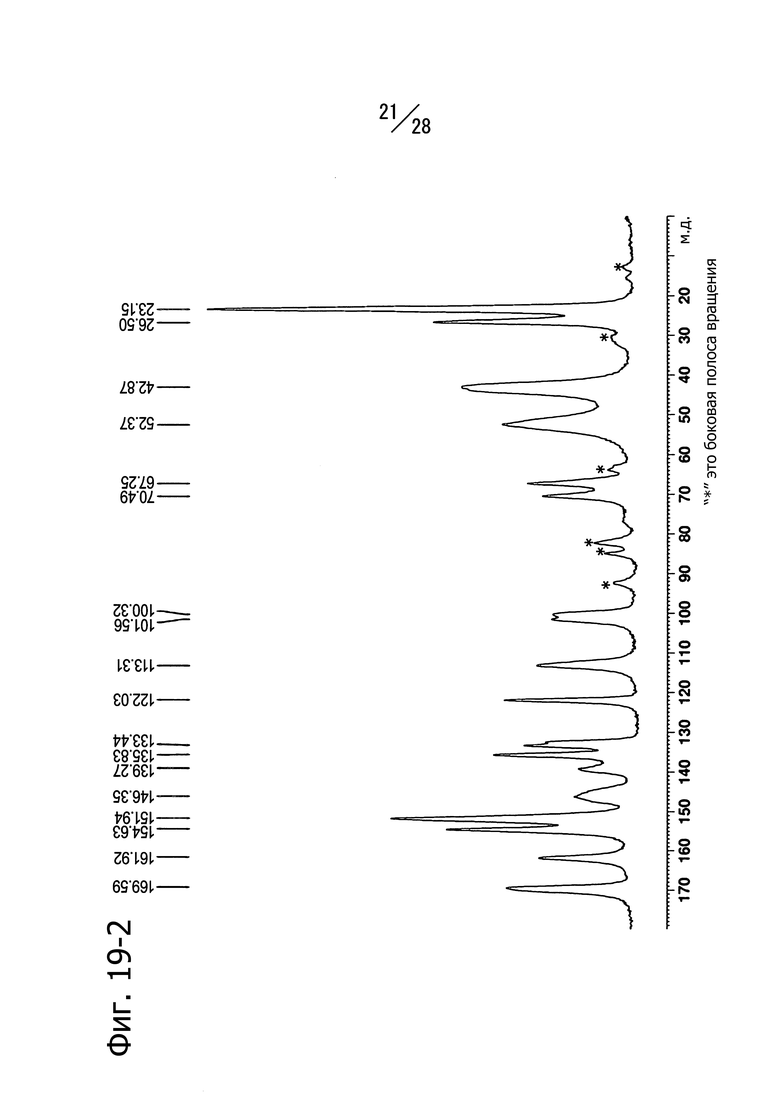
Фиг. 21-1 и 21-2 представляют собой твердотельные ЯМР-спектры (13C) кристаллической формы A 1-(6-((6-((1R)-1-метоксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридазил)пиперазина. Фиг. 21-1 представляет собой спектр, записанный в режиме 65000 Гц, а Фиг. 21-2 представляет собой спектр, записанный в режиме 14000 Гц.
Фиг. 22 представляет собой твердотельный ЯМР-спектр (15N) кристаллической формы A 1-(6-((6-((1R)-1-метоксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридазил)пиперазина.
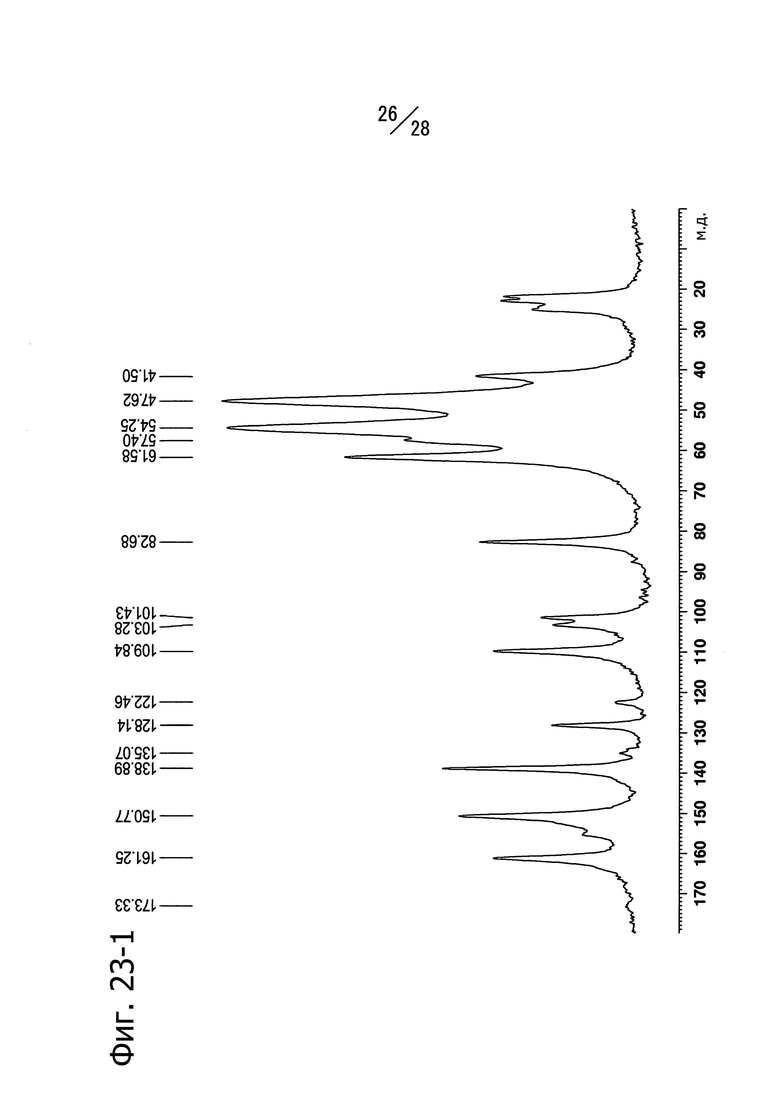
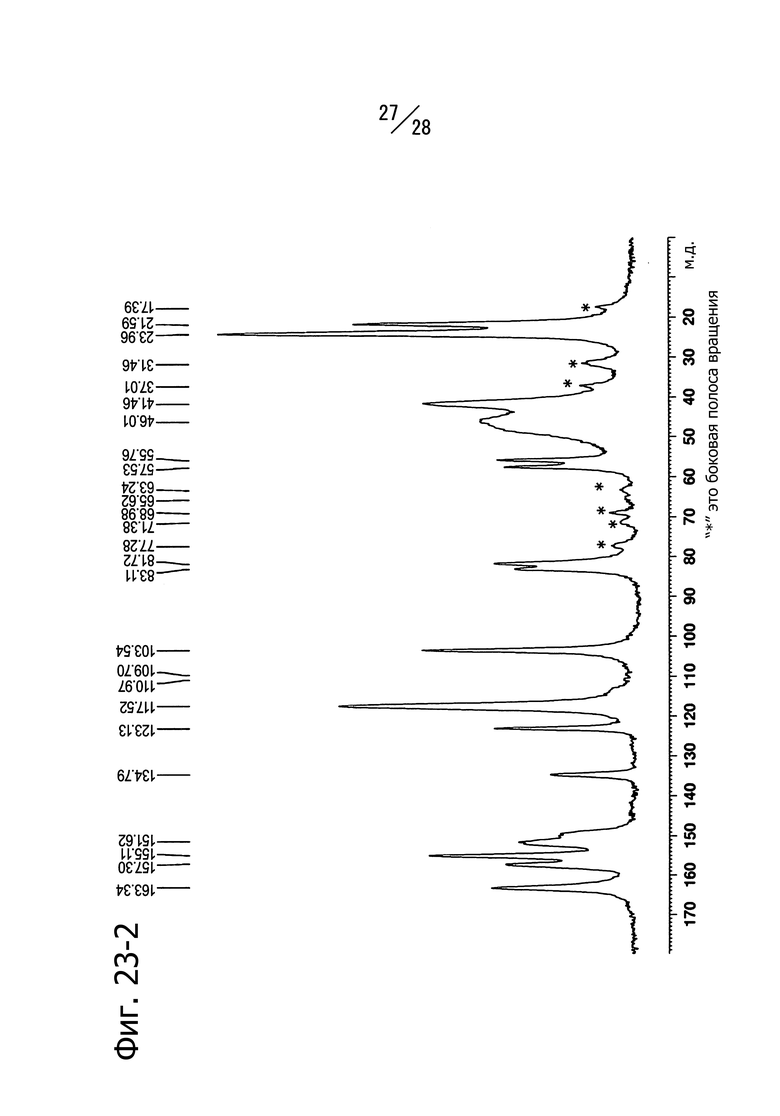
Фиг. 23-1 и 23-2 представляют собой твердотельные ЯМР-спектры (13C) кристаллической формы A (R)-N8-изопропил-6-(1-метоксиэтил)-N2-(5-(пиперазин-1-илметил)пиридин-2-ил)пиридо[3,4-d]пиримидин-2,8-диамина. Фиг. 23-1 представляет собой спектр, записанный в режиме 65000 Гц, а Фиг. 23-2 представляет собой спектр, записанный в режиме 14000 Гц.
Фиг. 24 представляет собой твердотельный ЯМР-спектр (15N) кристаллической формы A (R)-N8-изопропил-6-(1-метоксиэтил)-N2-(5-(пиперазин-1-илметил)пиридин-2-ил)пиридо[3,4-d]пиримидин-2,8-диамина.
Способы реализации изобретения
Кристаллические формы по настоящему изобретению охарактеризованы методами порошковой рентгеновской дифракции (XRD), дифференциальной сканирующей калориметрии (ДСК), ИК-Фурье спектроскопии (далее по тексту именуется “ИК-спектроскопия” или “ИК-спектры”) и/или твердотельной ЯМР спектроскопии. Спектры порошковой рентгеновской дифракции (XRD) указанных кристаллических форм являются характеристичными, и каждая из указанных кристаллических форм имеет специфичные значения дифракционных углов 2θ. Указанные кристаллические формы демонстрируют также характеристичное поведение при нагревании согласно данным дифференциальной сканирующей калориметрии (ДСК). ИК-спектры указанных кристаллических форм являются характеристичными, и каждая из указанных кристаллических форм имеет специфичный ИК-спектр с характеристичными пиками с определенными длинами волн. Твердотельные 13C ЯМР спектры указанных кристаллических форм являются характеристичными, и каждая из указанных кристаллических форм имеет специфичные значения химических сдвигов (м.д.). Твердотельные 15N ЯМР спектры указанных кристаллических форм являются характеристичными, и каждая из указанных кристаллических форм имеет специфичные значения химических сдвигов (м.д.).
Хотя настоящее изобретение охватывает любые кристаллические формы соединений, имеющих формулу (I), приведенное ниже описание фокусируется на их предпочтительных вариантах, а именно на девяти кристаллических формах следующих трех соединений:
- 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-он (далее по тексту именуется также “Соединение (a)”);
- 1-(6-((6-((1R)-1-метоксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридазил)пиперазин (далее по тексту именуется также «Соединение (b)”); и
- (R)-N8-изопропил-6-(1-метоксиэтил)-N2-(5-(пиперазин-1-илметил)пиридин-2-ил)пиридо[3,4-d]пиримидин-2,8-диамин (далее по тексту именуется также “Соединение (c)”).
Кристаллическая форма D соединения (a) имеет характеристичные пики со значениями дифракционных углов 2θ = 6.3°, 6.6°, 11.6°, 16.9° и 20.0° в спектре порошковой рентгеновской дифракции. Кристаллическая форма D соединения (a) имеет общий вид спектра порошковой рентгеновской дифракции, изображенный на Фиг. 1.
Кристаллическая форма D соединения (a) имеет эндотермический пик с экстраполированной температурой начала 277°C, согласно данным дифференциальной сканирующей калориметрии (ДСК).
Кристаллическая форма D соединения (a) имеет пики поглощения при длине волны 703 см-1, 896 см-1 и 3418 см-1 в ИК-спектре, записанном методом анализа в таблетках KBr. Кристаллическая форма D соединения (a) имеет также общий вид ИК-спектра, изображенный на Фиг. 10.
Кристаллическая форма D соединения (a) имеет пики со значениями химических сдвигов 136.0 м.д., 111.2 м.д., 105.1 м.д., 101.8 м.д., 52.7 м.д., 49.6 м.д., 42.9 м.д., 23.8 м.д. и 18.5 м.д. в твердотельном 13C-ЯМР спектре. Кристаллическая форма D соединения (a) имеет также общий вид твердотельного 13С ЯМР-спектра, изображенный на Фиг. 17-1 (6500Гц) и Фиг. 17-2 (14000Гц).
Кристаллическая форма D соединения (a) имеет пики со значениями химических сдвигов 248.6 м.д., 245.7 м.д., 229.2 м.д., 214.5 м.д., 174.3 м.д., 86.5 м.д., 54.7 м.д. и -12.4 м.д. в твердотельном 15N ЯМР-спектре. Кристаллическая форма D соединения (a) имеет также общий вид твердотельного 15N ЯМР-спектра, изображенный на Фиг. 18.
Кристаллическая форма A соединения (a) имеет характеристичные пики со значениями дифракционных углов 2θ = 5.3°, 7.3°, 10.3°, 15.1° и 17.4° в спектре порошковой рентгеновской дифракции. Кристаллическая форма A соединения (a) имеет также общий вид спектра порошковой рентгеновской дифракции, изображенный на Фиг. 2.
Кристаллическая форма A соединения (a) имеет эндотермический пик с экстраполированной температурой начала 277°C, согласно данным дифференциальной сканирующей калориметрии (ДСК).
Кристаллическая форма A соединения (a) имеет пики поглощения при длине волны 874 см-1, 1330 см-1 и 3314 см-1 в ИК-спектре, записанном методом анализа в таблетках KBr. Кристаллическая форма A соединения (a) имеет также общий вид ИК-спектра, изображенный на Фиг. 11.
Кристаллическая форма A соединения (a) имеет пики со значениями химических сдвигов 154.7 м.д., 138.8 м.д., 133.6 м.д., 113.2 м.д., 101.6 м.д., 100.4 м.д., 67.4 м.д., 51.8 м.д., 26.6 м.д. и 23.3 м.д. в твердотельном 13C-ЯМР спектре. Кристаллическая форма D соединения (a) имеет также общий вид твердотельного 13С ЯМР-спектра, изображенный на Фиг. 19-1 (6500Гц) и Фиг. 19-2 (14000Гц).
Кристаллическая форма A соединения (a) имеет пики со значениями химических сдвигов 243.6 м.д., 86.7 м.д., 56.7 м.д. и -12.4 м.д. в твердотельном 15N ЯМР-спектре. Кристаллическая форма A соединения (a) имеет также общий вид твердотельного 15N ЯМР-спектра, изображенный на Фиг. 20.
Кристаллическая форма B соединения (a) имеет характеристичные пики со значениями дифракционных углов 2θ= 5.3°, 6.0°, 6.7°, 10.4° и 20.8° в спектре порошковой рентгеновской дифракции. Кристаллическая форма B соединения (a) имеет также общий вид спектра порошковой рентгеновской дифракции, изображенный на Фиг. 6.
Кристаллическая форма B соединения (a) имеет эндотермический пик с экстраполированной температурой начала 271°C, согласно данным дифференциальной сканирующей калориметрии (ДСК).
Кристаллическая форма C соединения (a) имеет характеристичные пики со значениями дифракционных углов 2θ = 6.0°, 10.0°, 13.7°, 20.3° и 23.0° в спектре порошковой рентгеновской дифракции. Кристаллическая форма C соединения (a) имеет также общий вид спектра порошковой рентгеновской дифракции, изображенный на Фиг. 7.
Кристаллическая форма C соединения (a) имеет эндотермический пик с экстраполированной температурой начала 100°C и 278°C, согласно данным дифференциальной сканирующей калориметрии (ДСК).
Кристаллическая форма C соединения (a) имеет пики поглощения при длине волны 840 см-1, 904 см-1, 955 см-1, 1490 см-1 и 3281 см-1 в ИК-спектре, записанном методом анализа в таблетках KBr. Кристаллическая форма C соединения (a) имеет также общий вид ИК-спектра, изображенный на Фиг. 12.
Кристаллическая форма I соединения (a) имеет характеристичные пики со значениями дифракционных углов 2θ= 5.2°, 7.2°, 9.5°, 14.5°, 16.5°, 20.9°, 25.0° и 27.9° в спектре порошковой рентгеновской дифракции. Кристаллическая форма I соединения (a) имеет также общий вид спектра порошковой рентгеновской дифракции, изображенный на Фиг. 8.
Кристаллическая форма I соединения (a) имеет эндотермический пик с экстраполированной температурой начала 272°C, согласно данным дифференциальной сканирующей калориметрии (ДСК).
Кристаллическая форма I соединения (a) имеет пики поглощения при длине волны 1081 см-1 и 1260 см-1 в ИК-спектре, записанном методом анализа в таблетках KBr. Кристаллическая форма I соединения (a) имеет также общий вид ИК-спектра, изображенный на Фиг. 13.
Кристаллическая форма A соединения (b) имеет характеристичные пики со значениями дифракционных углов 2θ = 5.2°, 7.6°, 8.4°, 10.5°, 15.2°, 16.9°, 20.1°, 21.0°, 23.3° и 26.6° в спектре порошковой рентгеновской дифракции. Кристаллическая форма A соединения (b) имеет также общий вид спектра порошковой рентгеновской дифракции, изображенный на Фиг. 3.
Кристаллическая форма A соединения (b) имеет эндотермический пик с экстраполированной температурой начала 225°C, согласно данным дифференциальной сканирующей калориметрии (ДСК).
Кристаллическая форма A соединения (b) имеет пики поглощения при длине волны 703 см-1, 896 см-1 и 3418 см-1 в ИК-спектре. Кристаллическая форма A соединения (b) имеет общий вид ИК-спектра, изображенный на Фиг. 14.
Кристаллическая форма A соединения (b) имеет пики со значениями химических сдвигов 163.4 м.д., 157.6 м.д., 155.5 м.д., 117.8 м.д., 82.2 м.д., 56.1 м.д. и 42.3 м.д. в твердотельном 13C-ЯМР спектре. Кристаллическая форма A соединения (b) имеет также общий вид твердотельного 13С ЯМР-спектра, изображенный на Фиг. 21-1 (6500Гц) и Фиг. 21-2 (14000Гц).
Кристаллическая форма A соединения (b) имеет пики со значениями химических сдвигов 311.7 м.д., 232.4 м.д., 168.5 м.д., 79.5 м.д., 53.3 м.д., 32.9 м.д. и -4.3 м.д. в твердотельном 15N ЯМР-спектре. Кристаллическая форма A соединения (b) имеет также общий вид твердотельного 15N ЯМР-спектра, изображенный на Фиг. 22.
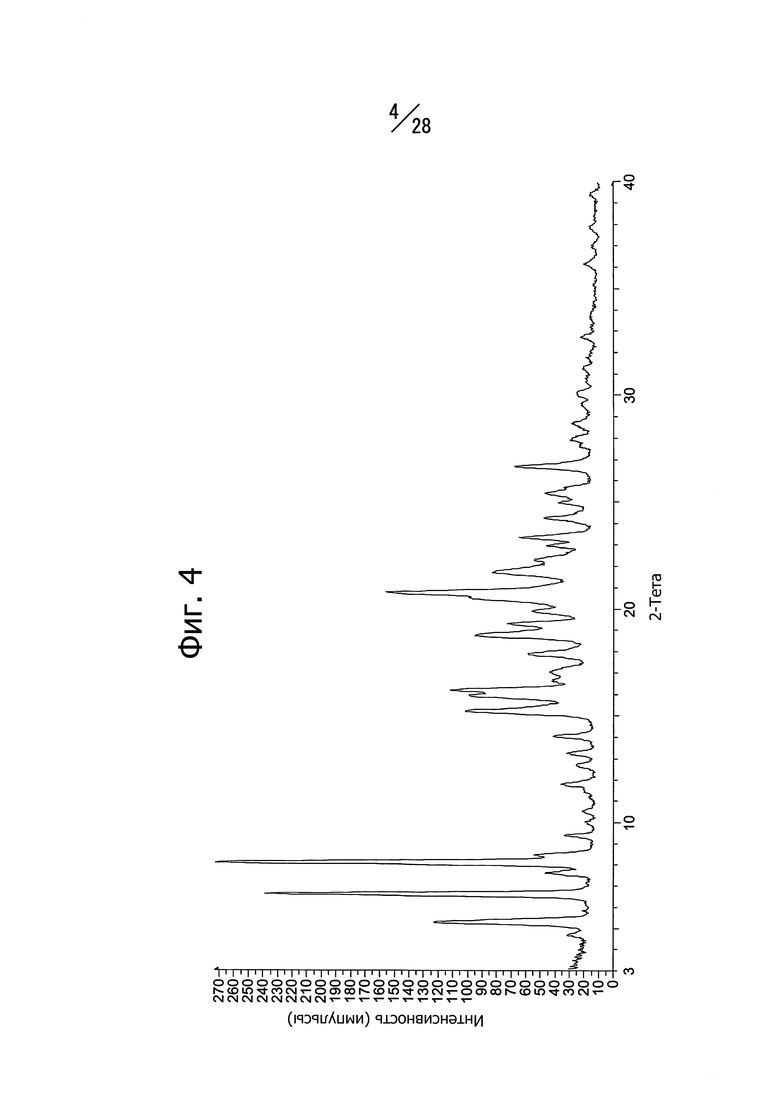
Кристаллическая форма B соединения (b) имеет в спектре порошковой рентгеновской дифракции характеристичные пики со значениями дифракционных углов 2θ = 5.2°, 6.6°, 8.1°, 15.2°, 15.9°, 16.2°, 18.8°, 20.5°, 20.8° и 21.7°. Кристаллическая форма B соединения (b) имеет общий вид спектра порошковой рентгеновской дифракции, изображенный на Фиг. 4.
Кристаллическая форма B соединения (b) имеет эндотермический пик с экстраполированной температурой начала 221°C, согласно данным дифференциальной сканирующей калориметрии (ДСК).
Кристаллическая форма C соединения (b) имеет характеристичные пики со значениями дифракционных углов 2θ = 5.2°, 7.6°, 8.4°, 10.0°, 10.5°, 11.9°, 15.2°, 17.0°, 20.9° и 21.2°в спектре порошковой рентгеновской дифракции. Кристаллическая форма C соединения (b) имеет общий вид спектра порошковой рентгеновской дифракции, изображенный на Фиг. 9. Кристаллическая форма C соединения (b) имеет также эндотермический пик с экстраполированной температурой начала 223°C, согласно данным дифференциальной сканирующей калориметрии (ДСК).
Кристаллическая форма C соединения (b) имеет пики поглощения при длине волны 1369 см-1, 1424 см-1, 1507 см-1, 1546 см-1 и 1566 см-1 в ИК-спектре, записанном методом анализа в таблетках KBr. Кристаллическая форма C соединения (b) имеет также общий вид ИК-спектра, изображенный на Фиг. 15.
Кристаллическая форма A соединения (c) имеет в спектре порошковой рентгеновской дифракции характеристичные пики со значениями дифракционных углов 2θ = 4.8°, 7.6°, 8.2°, 9.7°, 15.3°, 16.6°, 19.1°, 19.8°, 22.4° и 26.2°. Кристаллическая форма A соединения (c) имеет общий вид спектра порошковой рентгеновской дифракции, изображенный на Фиг. 5.
Кристаллическая форма A соединения (c) имеет эндотермический пик с экстраполированной температурой начала 182°C, согласно данным дифференциальной сканирующей калориметрии (ДСК).
Кристаллическая форма A соединения (c) имеет пики поглощения при длине волны 1115 см-1, 1446 см-1, 1508 см-1, 1560 см-1 и 1601 см-1 в ИК-спектре, записанном методом анализа в таблетках KBr. Кристаллическая форма A соединения (c) e имеет также общий вид ИК-спектра, изображенный на Фиг. 16.
Кристаллическая форма A соединения (c) имеет пики со значениями химических сдвигов 161.3 м.д., 150.8 м.д., 138.9 м.д., 128.1 м.д., 109.8 м.д., 82.7 м.д., 47.6 м.д., 42.5 м.д., 41.5 м.д., 24.5 м.д. и 21.7 м.д. в твердотельном 13C-ЯМР спектре. Кристаллическая форма A соединения (c) имеет также общий вид твердотельного 13С ЯМР-спектра, изображенный на Фиг. 23-1 (6500 Гц) и Фиг. 23-2 (14000Гц).
Кристаллическая форма A соединения (c) имеет пики со значениями химических сдвигов 242.8 м.д., 233.8 м.д., 219.0 м.д., 171.7 м.д., 86.9 м.д., 54.9 м.д., 11.3 м.д. и -5.5 м.д. в твердотельном 15N ЯМР-спектре. Кристаллическая форма A соединения (c) имеет также общий вид твердотельного 15N ЯМР-спектра, изображенный на Фиг. 24.
Термин “характеристичные пики” при использовании в настоящем тексте означает пики, которые видны в первую очередь и являются характерными для каждой кристаллической формы в спектре порошковой рентгеновской дифракции. Кристаллические формы, идентифицируемые по дифракционным углам, перечисленным в настоящем тексте, могут иметь другие пики, помимо характеристичных пиков.
Положение и относительная интенсивность каждого пика с дифракционным углом 2θ в спектре порошковой рентгеновской дифракции может несколько варьироваться в зависимости от условий анализа. Соответственно, идентификацию кристаллических форм следует проводить по общему сходству спектральных картин, даже если каждое значение 2θ немного отличается. Кристаллические формы в пределах погрешности измерения входят в объем настоящего изобретения. Размер погрешности измерения значения 2θ может, например, находиться в диапазоне ±0.5° или ±0.2°. Поэтому кристаллические формы, имеющие дифракционные углы, каждый из которых имеет значение в диапазоне от ±0.2° до ±0.5° от соответствующего значения, указанного выше, рассматриваются как входящие в объем притязаний кристаллической формы по настоящему изобретению.
Кристаллические формы, входящие в интервал погрешностей, возникающих из-за условий для анализа методом порошковой рентгеновской дифракции (например, погрешности прибора), также охватываются настоящим изобретением.
Термин “экстраполированная температура начала” пика в контексте дифференциальной сканирующей калориметрии (ДСК) означает температуру, при которой начинается экзотермический или эндотермический пик, т.е. температура начала экзотермического или эндотермического перехода, вычисленная экстраполированием. Экзотермический или эндотермический пик в дифференциальной сканирующей калориметрии (ДСК) может немного варьироваться в зависимости от условий анализа. Диапазон погрешностей может, например, составлять ±5°C или ±2°C. Поэтому кристаллические формы, имеющие пик в диапазоне от ±2°С до ±5°С от соответствующего значения, указанного выше, рассматриваются как входящие в объем притязаний кристаллической формы по настоящему изобретению.
В целом, химические сдвиги в твердотельном ЯМР спектре также могут иметь погрешность. Диапазон погрешности химического сдвига может составлять, например, ±0.25 м.д., обычно ±0.5 м.д. Поэтому кристаллические формы, имеющие химические сдвиги, каждый из которых находится в диапазоне от ±0.25 м.д. до ±0.5 м.д. от соответствующего значения, указанного выше, рассматриваются как входящие в объем притязаний кристаллической формы по настоящему изобретению. Различия, например, в частоте вращения образца в приборах, применяемых для съемки спектров, могут повлиять на интенсивность или наличие/отсутствие каждого пика.
В целом, пики поглощения в ИК-спектре также могут иметь погрешность. Диапазон погрешности пика поглощения может составлять, например, ±2 см-1, обычно ±5см-1. Поэтому кристаллические формы, имеющие пики поглощения, каждый из которых находится в диапазоне от ±2 см-1 до ±5 см-1 от соответствующего значения, указанного выше, рассматриваются как входящие в объем притязаний кристаллической формы по настоящему изобретению.
Кроме того, в каждом из методов – спектрометрии порошковой рентгеновской дифракции, дифференциальной сканирующей калориметрии (ДСК), ИК-спектроскопии, твердотельной 13C ЯМР спектроскопии и твердотельной 15N ЯМР спектроскопии – несмотря на то, что может иметь место различие между реальным значением для стандарта сравнения для каждой кристаллической формы (например, кристаллической формы, полученной способом, описанным в Примерах в настоящей заявке) и указанными выше значениями, такое различие принимается за погрешность измерения. А именно, кристаллические формы, имеющие дифракционные углы, эндотермические или экзотермические пики, ИК-спектры, твердотельные 13C ЯМР спектры или твердотельные 15N ЯМР спектры, которые находятся в пределах указанного выше диапазона погрешности, рассматриваются как входящие в объем притязаний кристаллической формы по настоящему изобретению.
Примеры
Далее будут описаны примеры, иллюстрирующие описание. Однако приведенные примеры не являются ограничивающими объем настоящего изобретения. Например, способы синтеза, очистки и кристаллизации соединений, описанные в приведенных ниже Примерах, являются исключительно вариантами получения кристаллической формы по настоящему изобретению, и поэтому кристаллическая форма по настоящему изобретению не ограничивается кристаллической формой, полученной способами синтеза, очистки и кристаллизации соединения, описанными в приведенных ниже примерах.
Структура кристаллических форм соединений, описанных в Примерах, и новых соединений, выделенных в ходе синтеза, была определена методами 1H-ЯМР спектрометрии или масс-спектрометрии с применением LC/MS (жидкостной хроматограф, сопряженный с масс-спектрометром).
Спектры 1H-ЯМР записывали на приборе JEOL JNM-ECZ400S (400 MГц). В качестве стандарта применяли ТМС (тетраметилсилан) (пик 0.0 м.д.) для растворителя CDCl3, или диметилсульфоксид (пик 2.49 м.д.) для ДМСО-d6. В описании 1H-ЯМР спектров (400 МГц, ДМСО-d6, CD3OD или CDCl3) приведены значения химических сдвигов (δ: м.д.) и констант спин-спинового взаимодействия (J: Гц), где с означает синглет, д – дублет, т – триплет, кв – квадруплет, ушир.с – уширенный синглет, и м означает мультиплет.
Результаты LC/MS спектрометрии приведены с указанием [M+H]+ (наблюдаемый молекулярный вес (Набл. МС), т.е. протон [H]+ добавляется к молекулярной массе [M] соединения).
Спектр рентгеновской дифракции для каждого кристаллического порошка в соответствии с Примерами записывали в следующих условиях:
Прибор: D8 DISCOVER с GADDS CS производства Bulker AXS Inc., Источник рентгеновских лучей: Cu·Kα (длина волны: 1.541838 (10-10м)), напряжение на лампе – ток лампы: 40 кВ – 40 мА, плоский графитовый монохроматор, коллиматор: φ300мкм, двумерный детектор PSPC, диапазон сканирования: от 3 до 40°.
Дифференциальную сканирующую калориметрию образцов в Примерах проводили в следующих условиях:
Прибор: DSC 8000 производство Perkin Elmer, скорость нагрева: 10°C/мин, атмосфера: азот, чашка для образца: алюминий, отбор проб: 0.1 сек, диапазон сканирования: от 25 до 300°C.
Спектр ИК-поглощения (KBr) каждого образца в Примерах записывали по методике анализа в таблетках из бромида калия в ИК-спектрометре, описанной в общих методах тестирования в Японской Фармакопее, в следующих условиях:
Прибор: AVATAR320 Nicolet iS5 производства Thermo Fisher Scientific, Диапазон сканирования: от 4000 до 400 см-1, разрешение: 4 см-1, число проходов: 16.
Твердотельный ЯМР спектр для каждого образца из Примеров записывали в следующих условиях:
Прибор: Bruker DSX300WB, регистрируемые ядра: 13C и 15N, время задержки импульса: 5 с, импульсный режим: CP/MAS.
Пример 1
В Примере 1 описана кристаллическая форма D 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил) амино)-3-пиридил)пиперазин-2-она (Соединение (a)).
Синтез соединения (a) описан ниже.
<Синтез соединения (a)-1>
Метил 5-бромо-2-метилтиопиримидин-4-карбоксилат (Соединение (a)-1) было синтезировано описанным ниже способом.
 ((a)-1)
((a)-1)
Раствор 5-бромо-2-(метилтио)пиримидин-4-карбоновой кислоты (110 г, 0.44 моль) в метаноле (1.1 л) охлаждали до 0°C при перемешивании и затем прикапывали тионилхлорид (50 мл, 0.66 моль). Полученный раствор медленно нагревали до кипения и кипятили 4 часа. Окончание реакции подтверждалось методами LC/MS и ТСХ, раствор охлаждали до комнатной температуры. Летучие компоненты удаляли при пониженном давлении, и остаток растворяли в этилацетате (1л). Полученный раствор промывали три раза 10%-ным водным раствором карбоната натрия (200мл) и два раза насыщенным раствором хлорида натрия (200 мл). Полученную органическую фазу сушили над безводным сульфатом магния. После отфильтровывания твердых компонентов фильтрат упаривали. Полученный сырой продукт очищали методом колоночной хроматографии на силикагеле, получая соединение (a)-1 (88г, 75%).
<Синтез соединения (a)-2>
Соединение (a)-2, 5-бромо-2-метилтиопиримидин-4-карбальдегид, было синтезировано описанным ниже способом.
 ((a)-2)
((a)-2)
1.7 M раствор соединения (a)-1 (25 г, 95 ммоль) в ТГФ (тетрагидрофуран) (375 мл) охлаждали при перемешивании до -78°C в токе азота. Диизобутилалюминий гидрид (84 мл, 143 ммоль) в толуоле прикапывали в полученный раствор, и полученную смесь перемешивали четыре часа при -78°C. После того как окончание реакции было подтверждено методом ТСХ, метанол прикапывали при -78°C для гашения реакционной смеси, и раствор медленно нагревали до 0°C. Полученный раствор разбавляли этилацетатом и фильтровали через слой целита при отсасывании. Фильтрат промывали насыщенным раствором хлорида натрия (200 мл) два раза, полученную органическую фазу сушили над безводным сульфатом магния, и твердую фазу отфильтровывали. Фильтрат упаривали, получая указанное в заголовке соединение (25 г, сырой продукт). Полученное сырое соединение использовали в последующей реакции без дополнительной очистки.
<Синтез соединения (a)-3>
Соединение (a)-3, (R)-3-(4-формил-2-метилтиопиримидин-5-ил)-1-метил-2-пропинил бензоат, было синтезировано описанным ниже способом.
 ((a)-3)
((a)-3)
Раствор PdCl2(PPh3)2Cl2 (7.832 г, 11.2 ммоль) и иодида меди (2.12 г, 11.2 ммоль) в 1,4-диоксане (60 мл) обескислороживали и продували аргоном. Диизопропилэтиламин (25.29 мл, 145.1 ммоль) добавляли при комнатной температуре. Полученный раствор перемешивали пять минут при комнатной температуре, и раствор смеси (26.0 г, сырой продукт) соединения (a)-2 и (5-бромо-2-метилтиопиримидин-4-ил)метоксиметанола в 1,4-диоксане (50 мл) добавляли при комнатной температуре. Затем добавляли по каплям раствор (R)-1-метилпропаргил бензоата (23.3 г, 133.9 ммоль) в 1,4-диоксане (55 мл), и полученный раствор перемешивали 16 часов при комнатной температуре. Протекание реакции отслеживали методом LC/MS. После окончания реакции, смесь разбавляли этилацетатом (400 мл), и фильтровали при отсасывании через слой целита. Целит промывали этилацетатом. Фильтрат упаривали при пониженном давлении. Полученный сырой продукт использовали в последующей реакции без дополнительной очистки.
Синтез смеси соединения (a)-2 и (5-бромо-2-метилтиопиримидин-4-ил)метоксиметанола будет описан в Сравнительном примере 1.
(Сравнительный пример 1)
Синтез смеси соединения (a)-2 и (5-бромо-2-метилтиопиримидин-4-ил)метоксиметанола
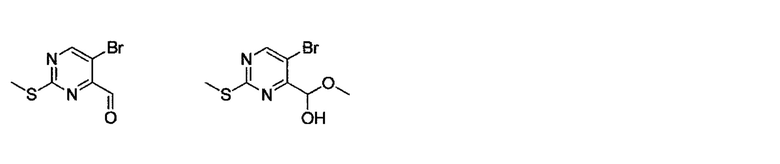
Раствор соединения (a)-2 (25 г, 95 ммоль) в ТГФ (375 мл) охлаждали при перемешивании до -78°C в токе азота. Прикапывали 1.7 M раствор диизобутилалюминий гидрида (84 мл, 143 ммоль) в толуоле, и раствор перемешивали четыре часа при -78°C. После того как окончание реакции было подтверждено методом ТСХ, прикапывали метанол при -78°C для гашения реакционной смеси. Полученный раствор медленно нагревали до 0°C. Полученный раствор разбавляли этилацетатом и фильтровали через слой целита при отсасывании. Фильтрат промывали насыщенным раствором хлорида натрия (200 мл) два раза, и полученную органическую фазу сушили над безводным сульфатом магния. Твердую фазу отфильтровывали. Фильтрат упаривали, получая смесь (25 г, сырой продукт) указанных в заголовке соединений. Полученный сырой продукт использовали в последующей реакции без дополнительной очистки.
<Синтез соединения (a)-4>
Соединение (a)-4, (R)-6-(1-(бензоилокси)этил)-2-(метилтио)пиридо[3,4-d]пиримидин-7-оксид, было синтезировано описанным ниже способом.
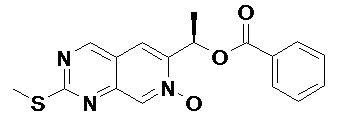 ((a)-4)
((a)-4)
Гидроксиамин моногидрохлорид (8.31 г, 119.6 ммоль) и ацетат натрия (9.81 г, 119.6 ммоль) добавляли в раствор (260 мл) соединения (a)-3 (26.0 г, 79.8 ммоль) в этаноле при комнатной температуре, и полученную смесь перемешивали 16 часов при комнатной температуре. Этанол (250 мл) добавляли в полученный раствор, добавляли карбонат калия (27.5 г, 199.4 ммоль) при комнатной температуре, и полученную смесь перемешивали три часа при 50°C. Протекание реакции отслеживали методом LC/MS. После окончания реакции, реакционную смесь фильтровали через слой целита при отсасывании. Целит промывали этилацетатом (1.0 л) и небольшим объемом метанола. Фильтрат упаривали при пониженном давлении, органический слой сушили над безводным сульфатом магния. Твердую фазу отфильтровывали. Полученный сырой продукт очищали методом колоночной хроматографии на силикагеле, получая Соединение (a)-4 (13.0 г, 48 %).
1H-ЯМР спектр соединения (a)-4 описан ниже:
1H-ЯМР (CDCl3) δ: 9.04 (1H, с), 8.79 (1H, с), 8.14 (2H, д, J = 7.5 Гц), 7.77-7.40 (4H, м), 6.66 (1H, кв, J = 6.3 Гц), 2.65 (3H, с), 1.79 (3H, д, J = 6.6 Гц).
<Синтез соединения (a)-5 >
Соединение (a)-5, (R)-1-(8-хлор-2-(метилтио)пиридо[3,4-d] пиримидин-6-ил)этил бензоат, было синтезировано описанным ниже способом.
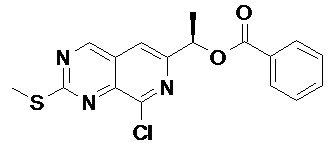 ((a)-5)
((a)-5)
Тионилхлорид (51 мл, 704 ммоль) прикапывали в раствор (130 мл) соединения (a)-4 (8.0 г, 23.5 ммоль) в дихлорметане при 0°C в токе азота. Полученный раствор перемешивали 16 часов при комнатной температуре. Протекание реакции отслеживали методом тонкослойной хроматографии (ТСХ). После окончания реакции, раствор упаривали при пониженном давлении, и полученную органическую фазу очищали методом колоночной хроматографии на оксиде алюминия, получая Соединение (a)-5 (3.2 г, 37%).
1H-ЯМР спектр соединения (a)-5 описан ниже:
1H-ЯМР (CDCl3) δ: 9.19 (1H, с), 8.16-8.12 (2H, м), 7.68 (1H, с), 7.64-7.58 (1H, м), 7.53-7.46 (2H, м), 6.27 (1H, кв, J = 6.8 Гц), 2.74 (3H, с), 1.81 (3H, д, J = 6.4 Гц).
<Соединение (a)-6>
Синтез соединения (a)-6, (R)-1-(8-(изопропиламино)-2-(метилтио)пиридо[3,4-d]пиримидин-6-ил)этил бензоата
 ((a)-6)
((a)-6)
Смесь соединения (a)-5 (3.06 г, 8.5 ммоль) и изопропиламина (18 мл) перемешивали один час при 80°C, и затем раствор охлаждали до комнатной температуры. Полученный раствор разбавляли водой и экстрагировали этилацетатом. Полученную органическую фазу промывали насыщенным раствором хлорида натрия, и затем сушили над безводным сульфатом натрия. Растворитель удаляли, и сырой продукт очищали методом колоночной хроматографии на силикагеле, получая Соединение (a)-6 (1.78 г, выход 55%).
1H-ЯМР спектр соединения (a)-6 описан ниже:
1H-ЯМР (ДМСО-d6) δ: 9.28 (1H, с), 8.08 (2H, д, J = 7.4 Гц), 7.70 (1H, т, J = 7.4 Гц), 7.57 (2H, т, J = 7.7 Гц),7.05 (1H, д, J = 8.0 Гц), 6.99 (1H, с), 5.34 (2H, с), 4.32 (1H, м), 2.66 (3H, с), 1.25 (6H, д, J = 6.5 Гц).
<Синтез соединения (a)-7>
Соединение (a)-7, (R)-1-(8-(изопропиламино)-2-(метилсульфонил)пиридо[3,4-d]пиримидин-6-ил)этил бензоат, было синтезировано описанным ниже способом.
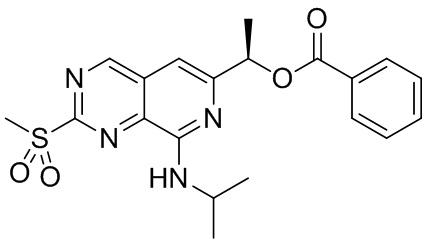 ((a)-7)
((a)-7)
Оксон (пероксимоносульфат калия) (5.72 г, 9.3 ммоль) добавляли в раствор соединения (a)-6 (1.78 г, 4.7 ммоль) в смеси тетрагидрофурана (47 мл) и воды (47 мл) при 0°C, и полученную смесь перемешивали 18 часов при комнатной температуре. Полученный раствор экстрагировали этилацетатом. Органическую фазу промывали водой, и сушили над безводным сульфатом натрия. Растворитель удаляли, и сырой продукт очищали методом колоночной хроматографии на силикагеле, получая Соединение (a)-7 (1.61 г, выход 87%).
LC/MS: (M+H)+ = 415.0.
<Синтез соединения (a)-8 >
Соединение (a)-8, трет-бутил 4-(6-нитропиридин-3-ил)-3-оксопиперазин-1-карбоксилат, было синтезировано описанным ниже способом.
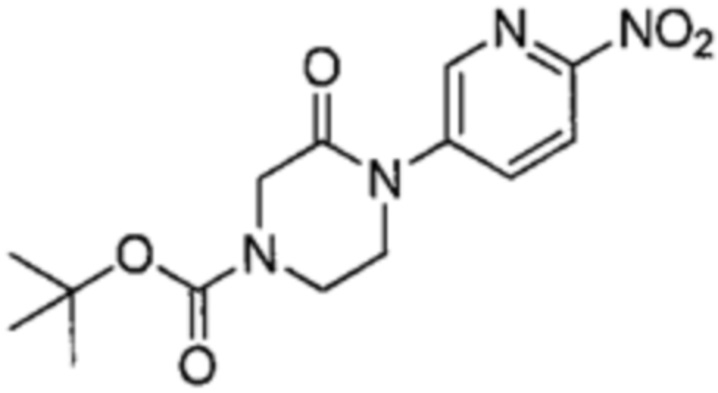 ((a)-8)
((a)-8)
2-нитро-5-бромпиридин (1.01, 5.0 моль), трет-бутил 2-оксо-4-пиперазин-карбоксилат (1.00, 5.0 моль) и карбонат цезия (3.26 г, 10.0 ммоль) суспендировали в 1,4-диоксане, и пропускали азот через суспензию в течение 30 минут. Xantphos (4,5'-бис(дифенилфосфино)-9,9'-диметилксантан) 246 мг, 0.43 ммоль) и трис(дибензилиденацетон)дипалладий (229 мг, 0.25 ммоль) добавляли в суспензию, и полученную смесь кипятили при перемешивании два часа. Полученный раствор охлаждали до комнатной температуры, и добавляли воду и этилацетат. Полученный раствор фильтровали через слой целита. Органический слой фильтрата отделяли, и водную фазу экстрагировали этилацетатом. Органические фазы объединяли и затем сушили над безводным сульфатом натрия. Твердую фазу отфильтровывали, и фильтрат упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле, получая Соединение (a)-8 (1.08 г, 67%).
1H-ЯМР (CDCl3) δ: 8.67 (1H, д, J = 2.4 Гц), 8.32 (1H, д, J = 8.8 Гц), 8.15 (1H, дд, J = 8.8, 2.4 Гц), 4.33 (2H, с), 3.93-3.83 (4H, м), 1.51 (9H, с).
<Синтез соединения (a)-9>
Соединение (a)-9, трет-бутил 4-(6-аминопиридин-3-ил)-3-оксопиперазин-1-карбоксилат, было синтезировано описанным ниже способом.
 ((a)-9)
((a)-9)
Соединение (a)-8 (1.08 г, 3.34 ммоль) растворяли в смеси этанола (45 мл) и ТГФ (22 мл). Палладий на угле (108 мг) добавляли в полученный раствор, и смесь перемешивали 24 часа в атмосфере водорода. Полученный раствор фильтровали через слой целита, и фильтрат упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле, получая Соединение (a)-9 (0.928 г, 95%).
1H-ЯМР (CDCl3) δ: 7.99 (1H, д, J = 2.4 Гц), 7.38 (1H, дд, J = 8.8, 2.4 Гц), 6.53 (1H, д, J = 8.8 Гц), 4.50 (2H, ушир.с), 4.24 (2H, с), 3.78 (2H, т, J = 5.1 Гц), 3.67 (2H, т, J = 5.4 Гц), 1.50 (9H, с).
<Синтез соединения (a)-10>
Соединение (a)-10, трет-бутил (R)-4-(6-((6-(1-(бензоилокси) этил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)пиридин-3-ил)-3-оксопиперазин-1-карбоксилат, было синтезировано описанным ниже способом.
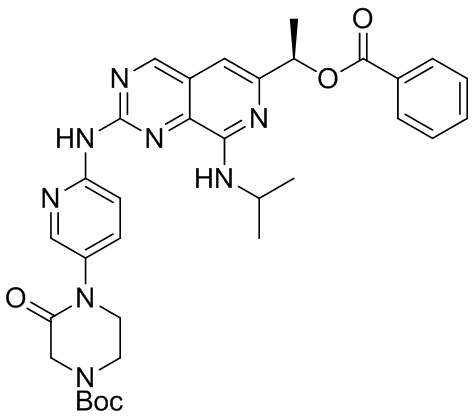 ((a)-10)
((a)-10)
Соединение (a)-7 (62 мг, 0.15 ммоль) и Соединение (a)-9 (88 мг, 0.30 ммоль) перемешивали в толуоле (0.375 мл) при 100°C в течение 6 дней. Полученный раствор очищали методом колоночной хроматографии на силикагеле, получая целевое Соединение (a)-10 (0.0092г, 10%).
1H-ЯМР (ДМСО) δ: 10.27 (1H, с), 9.27 (1H, с), 8.33 (2H, м), 8.07 (2H, м), 7.86 (1H, м), 7.70 (1H, м), 7.58 (3H, м), 7.00 (1H, с), 6.55 (1H, д), 5.98 (1H, кв), 4.27 (1H, м), 4.11 (2H, с), 3.74 (4H, м), 1.68 (3H, д), 1.45 (9H, с), 1.30 (6H, м).
<Синтез и очистка соединения (a)>
Соединение (a), 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-он, было синтезировано описанным ниже способом.
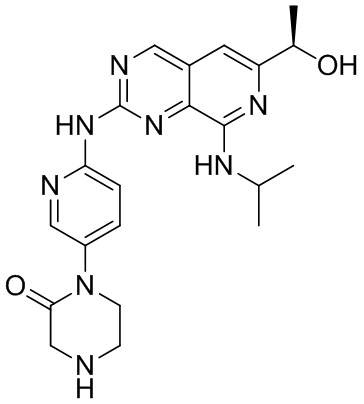 (a)
(a)
Трифторуксусную кислоту (0.15 мл) добавляли в раствор соединения (a)-10 (9.2 мг, 0.15 ммоль) в дихлорметане (0.35 мл) при комнатной температуре, и раствор перемешивали один час. После упаривания раствора досуха, добавляли тетрагидрофуран (0.15 мл) и метанол (0.15 мл), затем 4M водный раствор гидроксида лития (0.018 мл). Полученный раствор после реакции нейтрализовывали муравьиной кислотой, и продукт очищали методом колоночной хроматографии на силикагеле, получая указанное в заголовке соединение.
1H-ЯМР (ДМСО) δ: 10.16 (1H, с), 9.26 (1H, с), 8.31 (1H, м), 8.29 (1H, с), 7.81 (1H, м), 7.00 (1H, с), 6.42 (1H, м), 5.18 (1H, д), 4.63 (1H, м), 4.27 (1H, м), 3.65 (2H, м), 3.41 (2H, с), 3.05 (2H, м), 1.39 (3H, д), 1.30 (6H, м).
<Получение кристаллической формы D соединения (a)>
Раствор соединения (a), очищенного колоночной хроматографией на силикагеле, упаривали до выпадения кристаллов в осадок. Таким образом получали кристаллическую форму D соединения (a).
<Анализ кристаллической формы D соединения (a)>
На Фиг. 1 показан спектр рентгеновской дифракции полученной кристаллической формы. Наблюдаются пики при 6.3°, 6.6°, 11.6°, 16.9° и 20.0° (дифракционный угол 2θ).
Кривая теплового поведения, построенная по результатам дифференциальной сканирующей калориметрии (ДСК), имеет эндотермический пик при 277°C.
На Фиг. 10 показан спектр ИК-поглощения (ИК-спектр) полученной кристаллической формы. Наблюдаются пики поглощения при 703 см-1, 896 см-1 и 3418 см-1 (волновое число).
На Фиг. 17-1 и 17-2 показаны твердотельные 13C-ЯМР спектры полученной кристаллической формы. На Фиг.17-1 показан спектр, записанный в режиме 6500 Гц, и на Фиг.17-2 показан спектр, записанный в режиме 14000 Гц. Наблюдаются пики при 136.0 м.д., 111.2 м.д., 105.1 м.д., 101.8 м.д., 52.7 м.д., 49.6 м.д., 42.9 м.д., 23.8 м.д. и 18.5 м.д. (хим.сдвиг).
На Фиг. 18 показан твердотельный 15N-ЯМР спектр полученной кристаллической формы. Наблюдаются пики при 248.6 м.д., 245.7 м.д., 229.2 м.д., 214.5 м.д., 174.3 м.д., 86.5 м.д., 54.7 м.д. и -12.4 м.д. (хим.сдвиг).
Пример 2
В Примере 2 описана кристаллическая форма A 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она (Соединение (a)).
<Получение кристаллической формы A соединения (a)>
Кристаллическую форму A соединения (a) получали трансформацией кристаллической формы D соединения (a), полученной в Примере 1.
Кристаллическую форму D суспендировали в 5-50-кратном количестве этанола. Суспензию нагревали при перемешивании 6 часов и затем перемешивали при 0°C. Осадок отделяли фильтрованием и сушили с получением кристаллов.
Хотя нет ограничений по объему растворителя, времени нагрева, условиям перемешивания, времени фильтрования, эти параметры могут повлиять на выход кристаллов, их химическую чистоту, размер частиц и распределение частиц по размерам. Предпочтительно, чтобы эти параметры были определены соответствующим образом. Кристаллы можно отделять любым общеизвестным способом, например фильтрованием под действием силы тяжести, фильтрованием под давлением, фильтрованием при отсасывании, сушкой при нагревании или сушкой при пониженном давлении.
<Анализ кристаллической формы A соединения (a)>
На Фиг. 2 показан спектр рентгеновской дифракции полученной кристаллической формы. Наблюдаются пики при 5.3°, 7.3°, 10.3°, 15.1° и 17.4° (дифракционный угол 2θ).
Кривая теплового поведения, построенная по результатам дифференциальной сканирующей калориметрии (ДСК), имеет эндотермический пик при 277°C.
На Фиг. 11 показан спектр ИК-поглощения (ИК-спектр) полученной кристаллической формы. Наблюдаются пики поглощения при 874 см-1, 1330 см-1 и 3314 см-1 (волновое число).
На Фиг. 19-1 и 19-2 показаны твердотельные 13C-ЯМР спектры полученной кристаллической формы. На Фиг.19-1 показан спектр, записанный в режиме 6500 Гц, и на Фиг.19-2 показан спектр, записанный в режиме 14000 Гц. Наблюдаются пики при 154.7 м.д., 138.8 м.д., 133.6 м.д., 113.2 м.д., 101.6 м.д., 100.4 м.д., 67.4 м.д., 51.8 м.д., 26.6 м.д. и 23.3 м.д. (хим.сдвиг).
На Фиг. 20 показан твердотельный 15N-ЯМР спектр полученной кристаллической формы. Наблюдаются пики при 243.6 м.д., 86.7 м.д., 56.7 м.д. и -12.4 м.д. (хим.сдвиг).
Пример 3
В Примере 3 описана кристаллическая форма B 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-она (Соединение (a)).
<Получение кристаллической формы B соединения (a)>
Соединение (a) очищали аналогично Примеру 1, за исключением того, что растворителем, использующимся в колоночной хроматографии, была смесь дихлорметан/метанол = 20/1, и раствор соединения (a) упаривали до выпадения кристаллов в осадок. Таким образом получали кристаллическую форму B соединения (a).
На Фиг. 6 показан спектр рентгеновской дифракции полученной кристаллической формы. Наблюдаются пики при 5.3°, 6.0°, 6.7°, 10.4° и 20.8° (дифракционный угол 2θ).
Кривая теплового поведения, построенная по результатам дифференциальной сканирующей калориметрии (ДСК), имеет эндотермический пик при 271°C.
Пример 4
<Получение кристаллической формы C соединения (a)>
Диметилсульфоксид (5.4 мл) добавляли к кристаллической форме D (900 мг) соединения (a), и полученную смесь нагревали до 70°C. Полученный раствор охлаждали до 40°C. Добавляли ацетонитрил (6.75 мл), охлаждали до 15°C, затем перемешивали два часа. Твердую фазу отделяли фильтрованием, промывали ацетонитрилом (2.5 мл) и сушили при пониженном давлении при 40°C, получая аддукт указанного в заголовке соединения (986 мг, 92%) с диметилсульфоксидом.
<Анализ кристаллической формы C соединения (a)>
На Фиг. 7 показан спектр рентгеновской дифракции полученной кристаллической формы. Наблюдаются пики при 6.0°, 10.0°, 13.7°, 20.3° и 23.0° (дифракционный угол 2θ).
Кривая теплового поведения, построенная по результатам дифференциальной сканирующей калориметрии (ДСК), имеет эндотермический пик при 100°C и 278°C.
На Фиг. 12 показан спектр ИК-поглощения (ИК-спектр) полученной кристаллической формы. Наблюдаются пики поглощения при 840 см-1, 904 см-1, 955 см-1, 1490 см-1 и 3281 см-1 (волновое число).
Пример 5
<Получение кристаллической формы I соединения (a)>
Воду (10 мл) добавляли к кристаллической форме A (500 мг) соединения (a), смесь перемешивали четыре дня при комнатной температуре. Выпавший осадок отделяли фильтрованием и сушили при 30°C при пониженном давлении, получая указанное в заголовке соединение (432 мг, 86%).
<Анализ кристаллической формы I соединения (a)>
На Фиг. 8 показан спектр рентгеновской дифракции полученной кристаллической формы. Наблюдаются пики при 5.2°, 7.2°, 9.5°, 14.5°, 16.5°, 20.9°, 25.0° и 27.9° (дифракционный угол 2θ).
Кривая теплового поведения, построенная по результатам дифференциальной сканирующей калориметрии (ДСК), имеет эндотермический пик при 272°C.
На Фиг. 13 показан спектр ИК-поглощения (ИК-спектр) полученной кристаллической формы. Наблюдаются пики поглощения при 1081 см-1 и 1260 см-1 (волновое число).
Пример 6
В Примере 6 описана кристаллическая форма A 1-(6-((6-((1R)-1-метоксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридазил)пиперазин-1-ил(пиперазина) (Соединение (b)).
Далее описан синтез соединения (b).
<Синтез соединения (b)-1>
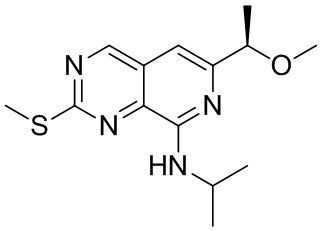
Соединение (b)-1, (R)-N-изопропил-6-(1-метоксиэтил)-2-(метилтио)пиридо[3,4-d]пиримидин-8-амин, было синтезировано описанным ниже способом.
 ((b)-1)
((b)-1)
Соединение (b)-1 было синтезировано аналогично соединениям (a)-3, (a)-4, (a)-5 и (a)-6.
1H-ЯМР (ДМСО-d6) δ: 9.27 (7H, с), 6.94 (1H, ушир.с), 6.92 (1H, с), 4.30-4.23 (1H, м), 3.29 (3H, с), 2.66 (3H, с), 1.38 (3H, д, J = 6.4 Гц), 1.32-1.25 (6H, м).
<Синтез соединения (b)-2 >
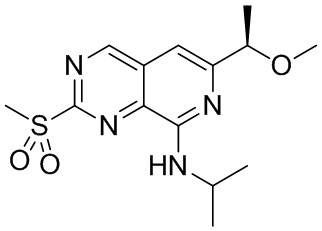
Соединение (b)-2, (R)-N-изопропил-6-(1-метоксиэтил)-2-(метилсульфонил) пиридо[3,4-d]пиримидин-8-амин, было синтезировано описанным ниже способом.
 ((b)-2)
((b)-2)
Оксон добавляли в раствор соединения (b)-1 в смеси тетрагидрофурана (ТГФ) и воды при 0°C и перемешивали 18 часов. Продукт реакции экстрагировали этилацетатом. Органический слой промывали водой и сушили над безводным сульфатом натрия. Затем удаляли растворитель, сырой продукт очищали методом колоночной хроматографии на силикагеле, получая целевое соединение (b)-2.
LC/MS: (M+H)+ = 325.10.
<Синтез соединения (b)-3 >
Соединение (b)-3, трет-бутил 4-(6-хлорпиридазин-3-ил)пиперазин-1-карбоксилат, было синтезировано описанным ниже способом.
 ((b)-3)
((b)-3)
3,6-Дихлорпиридазин (5.01 г, 33.6 ммоль) и трет-бутилпиперазин карбоксилат (6.88 г, 37.0 ммоль) растворяли в ДМФА (50 мл). Добавляли триэтиламин (11.7 мл, 50.4 ммоль), и полученную смесь перемешивали при 80°C в течение ночи. Полученный раствор охлаждали до комнатной температуры и добавляли воду. Продукт экстрагировали смесью (50 мл) дихлорметана и метанола (95:5) три раза. Объединенные органические фазы сушили над безводным сульфатом натрия, и твердую фазу отделяли фильтрованием. Фильтрат упаривали при пониженном давлении. Полученный сырой продукт промывали диэтиловым эфиром, получая целевое соединение (b)-3 (7.0 г, 70%).
<Синтез соединения (b)-4>
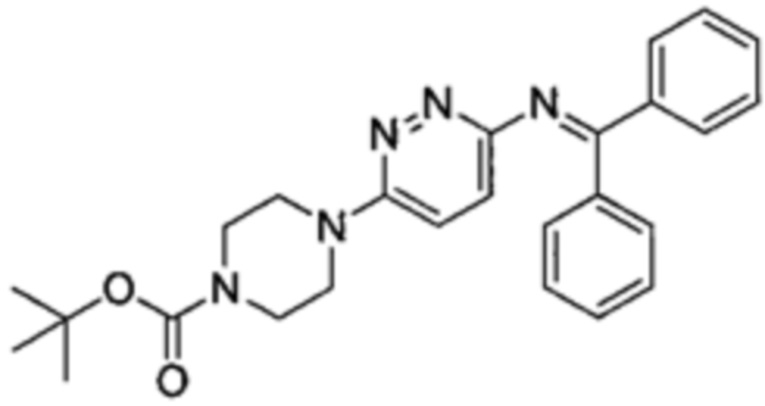
Соединение (b)-4, трет-бутил 4-(6-((дифенилметилен)амино)пиридазин-3-ил)пиперазин-1-карбоксилат, было синтезировано описанным ниже способом.
 ((b)-4)
((b)-4)
Соединение (b)-3 (59.8 мг, 0.20 ммоль), бензофенонимин (43.5 мг, 0.24 ммоль), трис(дибензилиденацетон)дипалладий (9.2 мг, 0.010 ммоль), BINAP (2,2'-бис(дифенилфосфино)-1,1'-бинафтил) (12.5 мг, 0.020 ммоль) и карбонат цезия (130.3 мг, 0.40 ммоль) суспендировали в толуоле (1.0 мл), и суспензию перемешивали при 100°C в течение ночи. Затем суспензию охлаждали до комнатной температуры, раствор фильтровали через слой целита, и целит промывали этилацетатом. Фильтрат промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. После отфильтровывания твердых компонентов, фильтрат упаривали при пониженном давлении. Остаток очищали методом колоночной хроматографии на силикагеле, получая целевое соединение (b)-4 (67 мг, 76%).
<Синтез соединения (b)-5>
Соединение (b)-5, трет-бутил 4-(6-аминопиридазин-3-ил)пиперазин-1-карбоксилат, было синтезировано описанным ниже способом.
 ((b)-5)
((b)-5)
Соединение (b)-4 (67 мг, 0.151 ммоль) растворяли в ТГФ (0.76 мл) и добавляли водный раствор лимонной кислоты (0.378 мл, 0.755 ммоль, 2 моль/л). Полученный раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь нейтрализовывали насыщенным водным раствором гидрокарбоната натрия (5 мл), и продукт экстрагировали этилацетатом (5 мл) два раза. Органические фазы объединяли и сушили над безводным сульфатом магния. После отфильтровывания твердой фазы, фильтрат упаривали при пониженном давлении. Полученный сырой продукт промывали трет-бутил-метиловым эфиром (5 мл), получая целевое соединение (b)-5 (0.30 г, 71%).
Синтез соединений, родственных примерам по настоящему изобретению, например, 6-аминопиридин-3-карбальдегида и трет-бутил 4-[(6-аминопиридин-3-ил)метил]пиперазин-1-карбоксилата, будет описан ниже в Сравнительных примерах.
<Синтез соединения (b)-6 >
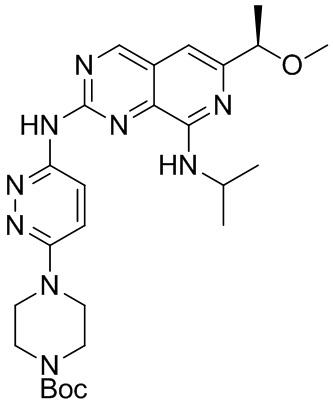
Соединение (b)-6, трет-бутил (R)-4-(6-((8-(изопропиламино)-6-(1-метоксиэтил) пиридо[3,4-d]пиримидин-2-ил)амино)пиридазин-3-ил)пиперазин-1-карбоксилат, было синтезировано описанным ниже способом.
 ((b)-6)
((b)-6)
Раствор соединения (a)-5 (708 мг, 2.2 ммоль) и Соединения (b)-5 (732 мг, 2.6 ммоль), синтезированного в Примере 1, в толуоле (5.5 мл) перемешивали три дня при 100°C. После охлаждения до комнатной температуры, раствор разбавляли этилацетатом (20 мл) и дихлорметан (100 мл). Полученный раствор промывали насыщенным раствором хлорида натрия (90 мл), затем насыщенным водным раствором гидрокарбоната натрия (10 мл). Отделяли органическую фазу, упаривали ее досуха и очищали на колонке с силикагелем, получая целевое соединение (b)-6 (510 мг, 45%).
<Синтез соединения (b)>
Соединение (b), 1-(6-((6-((1R)-1-метоксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридазил)пиперазин-1-ил(пиперазин), было синтезировано описанным ниже способом.
 (b)
(b)
Трифторуксусную кислоту (0.2 мл) добавляли в раствор соединения (b)-6 (33.2 мг, 0.063 ммоль) в дихлорметане (0.44 мл) при комнатной температуре, и полученную смесь перемешивали один час при комнатной температуре. Полученный раствор упаривали досуха, и полученный продукт очищали методом препаративной ВЭЖХ, получая целевое соединение (23.8 мг, 88%).
1H-ЯМР (ДМСО) δ: 10.24 (1H, с), 9.20 (1H, с), 8.16 (1H, д), 7.36 (1H, д), 6.86 (1H, с), 6.35 (1H, д), 6.42 (1H, м), 4.22 (2H, м), 3.43 (4H, м), 3.26 (4H, м), 2.81 (3H, м), 1.37 (3H, д), 1.26 (6H, м).
<Получение кристаллической формы A соединения (b)>
Брали соль соединения (b) с трифторуксусной кислотой (ТФУК), очищенную методом препаративной ВЭЖХ. Перемешивали эту ТФУК-соль в смеси воды и дихлорметана. Значение pH водной фазу доводили до слабощелочной (pH от 8 до 9) насыщенным водным раствором гидрокарбоната натрия и отделяли органическую фазу. Затем органическую фазу промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом натрия, растворитель удаляли, получая кристаллическую форму A.
<Анализ кристаллической формы A соединения (b)>
На Фиг. 3 показан спектр рентгеновской дифракции полученной кристаллической формы. Наблюдаются пики при 5.2°, 7.6°, 8.4°, 10.5°, 15.2°, 16.9°, 20.1°, 21.0°, 23.3° и 26.6° (дифракционный угол 2θ).
Кривая теплового поведения, построенная по результатам дифференциальной сканирующей калориметрии (ДСК), имеет эндотермический пик при 225°C.
На Фиг. 14 показан спектр ИК-поглощения (ИК-спектр) полученной кристаллической формы. Наблюдаются пики поглощения при 1369 см-1, 1424 см-1, 1508 см-1, 1545 см-1 и 1566 см-1 (волновое число).
На Фиг. 21-1 и 21-2 показаны твердотельные 13C-ЯМР спектры полученной кристаллической формы. На Фиг.21-1 показан спектр, записанный в режиме 6500 Гц, и на Фиг.21-2 показан спектр, записанный в режиме 14000 Гц. Наблюдаются пики при 163.4 м.д., 157.6 м.д., 155.5 м.д., 117.8 м.д., 82.2 м.д., 56.1 м.д. и 42.3 м.д. (хим.сдвиг).
На Фиг. 22 показан твердотельный 15N-ЯМР спектр полученной кристаллической формы. Наблюдаются пики при 311.7 м.д., 232.4 м.д., 168.5 м.д., 79.5 м.д., 53.3 м.д., 32.9 м.д. и -4.3 м.д. (хим.сдвиг).
Пример 7
В Примере 7 описана кристаллическая форма B 1-(6-((6-((1R)-1-метоксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридазил)пиперазин-1-ил(пиперазина) (Соединение (b)).
<Получение кристаллической формы B соединения (b)>
Тетрагидрофуран (0.15 мл) и метанол (0.15 мл) добавляли к ТФУК-соли, выпавшей в осадок при получении кристаллической формы A, и добавляли 4M водный раствор гидроксида лития (0.018 мл). Полученный раствор нейтрализовывали муравьиной кислотой, и продукт очищали методом колоночной хроматографии на силикагеле. Полученный раствор соединения (b) упаривали до выпадения в осадок кристаллов. Таким образом получали кристаллическую форму B соединения (b).
<Анализ кристаллической формы B соединения (b)>
На Фиг. 4 показан спектр рентгеновской дифракции полученной кристаллической формы. Наблюдаются пики при 5.2°, 6.6°, 8.1°, 15.2°, 15.9°, 16.2°, 18.8°, 20.5°, 20.8° и 21.7° (дифракционный угол 2θ).
Кривая теплового поведения, построенная по результатам дифференциальной сканирующей калориметрии (ДСК), имеет эндотермический пик при 221°C.
Пример 8
<Получение кристаллической формы C соединения (b)>
Этанол (11 мл) добавляли к кристаллической форме C (1.1 г) соединения (b), и полученную смесь перемешивали в течение ночи при комнатной температуре. Твердую фазу отделяли фильтрованием и сушили при 40°C при пониженном давлении, получая указанное в заголовке соединение (945 мг, 86%).
<Анализ кристаллической формы C соединения (b)>
На Фиг. 9 показан спектр рентгеновской дифракции полученной кристаллической формы. Наблюдаются пики при 5.2°, 7.6°, 8.4°, 10.0°, 10.5°, 11.9°, 15.2°, 17.0°, 20.9° и 21.2° (дифракционный угол 2θ).
Кривая теплового поведения, построенная по результатам дифференциальной сканирующей калориметрии (ДСК), имеет эндотермический пик при 223°C.
На Фиг. 15 показан спектр ИК-поглощения (ИК-спектр) полученной кристаллической формы. Наблюдаются пики поглощения при 1369 см-1, 1424 см-1, 1507 см-1, 1546 см-1 и 1566 см-1 (волновое число).
Пример 9
В Примере 9 описана кристаллическая форма A (R)-N8-изопропил-6-(1-метоксиэтил)-N2-(5-(пиперазин-1-илметил)пиридин-2-ил)пиридо[3,4-d]пиримидин-2,8-диамина (Соединение (c)).
Ниже описан синтез соединения (c).
<Синтез соединения (c)-1>
Соединение (c)-1, 6-аминопиридин-3-карбальдегид, было синтезировано описанным ниже способом.
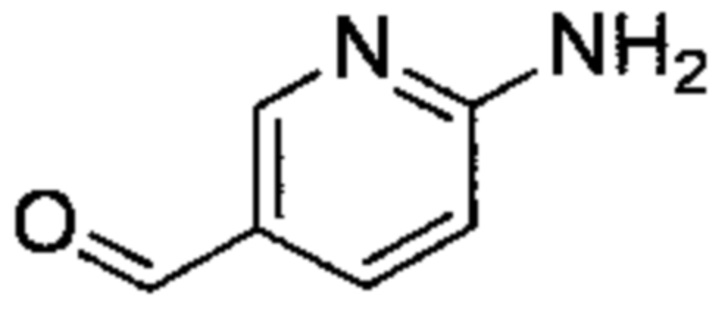 ((c)-1)
((c)-1)
6-Аминопиридин-3-карбонитрил (1.9 г, 16 ммоль) растворяли в ТГФ (160 мл), и раствор охлаждали при перемешивании до -78°C. Диизобутилалюминий гидрид (106.5 мл, 1.5M раствор в толуоле) медленно прикапывали в раствор при -78°C, нагревали при перемешивании до 20°C и перемешивали еще два часа. Добавляли в раствор ледяную воду (100 мл) для гашения реакционной смеси. Продукт экстрагировали дихлорметаном (50 мл) три раза. Затем органические фазы объединяли, раствор промывали насыщенным раствором хлорида натрия (100 мл) один раз и сушили над безводным сульфатом натрия. После отфильтровывания твердых компонентов, фильтрат упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая сырое целевое соединение (c)-1 (1.7 г). Полученный сырой продукт использовали в последующей реакции без дополнительной очистки.
<Синтез соединения (c)-2>
Соединение (c)-2, трет-бутил 4-[(6-амино пиридин-3-ил)метил]пиперазин-1-карбоксилат, было синтезировано описанным ниже способом.
 ((c)-2)
((c)-2)
6-Аминопиридин-3-карбонитрил (1.9 г, 16 ммоль) растворяли в ТГФ (160 мл), и полученную смесь охлаждали при перемешивании до -78°C. Диизобутилалюминий гидрид (106.5 мл, 1.5 M раствор в толуоле) медленно прикапывали в раствор при -78°C. Смесь нагревали при перемешивании до 20°C и перемешивали еще два часа. Добавляли в раствор ледяную воду (100 мл) для гашения реакционной смеси. Продукт экстрагировали дихлорметаном (50 мл) три раза. Затем органические фазы объединяли, раствор промывали насыщенным раствором хлорида натрия (100 мл) один раз и сушили над безводным сульфатом натрия. После отфильтровывания твердых компонентов, фильтрат упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая неочищенное целевое соединение (1.7 г).
Полученный сырой продукт (1.7 г, 13.9 ммоль) и трет-бутил пиперазин-1-карбоксилат (3.2 г, 17.2 ммоль) растворяли в дихлорметане (50 мл), и полученную смесь перемешивали восемь часов при комнатной температуре. Триацетоксиборгидрид натрия (8.84 г, 40.9 ммоль) добавляли в раствор, и полученную смесь перемешивали два часа при комнатной температуре. Протекание реакции отслеживали методом LC/MS. После окончания реакции, добавляли насыщенный водный раствор карбоната натрия (50 мл) для гашения реакционной смеси. Продукт экстрагировали этилацетатом (50 мл) три раза. Затем органические фазы объединяли, раствор промывали насыщенным раствором хлорида натрия (100 мл) один раз и сушили над безводным сульфатом натрия. После отфильтровывания твердых компонентов, фильтрат упаривали при пониженном давлении. Остаток очищали методом хроматографии на силикагеле, получая сырое целевое соединение (3.8 г, 81%).
<Синтез соединения (c)-3 >
Соединение (c)-3, трет-бутил (R)-4-((6-((8-(изопропиламино)-6-(1-метоксиэтил) пиридо[3,4-d]пиримидин-2-ил)амино) пиридин-3-ил)метил)пиперазин-1-карбоксилат, было синтезировано описанным ниже способом.
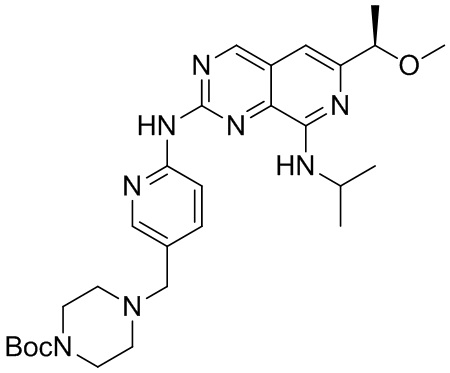 ((c)-3)
((c)-3)
Указанное в заголовке Соединение (c)-3 синтезировали аналогично соединению (b)-6, применяя соединения B-2 и C-2.
<Синтез соединения (c)>
Синтез соединения (c), (R)-N8-изопропил-6-(1-метоксиэтил)-N2-(5-(пиперазин-1-илметил)пиридин-2-ил)пиридо[3,4-d]пиримидин-2,8-диамина.
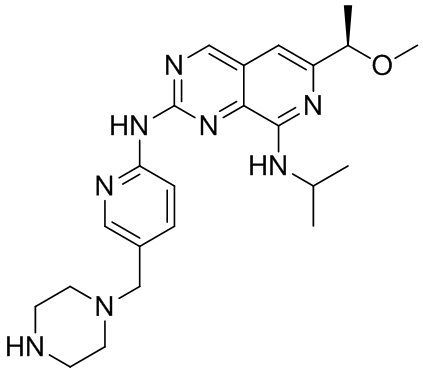 (c)
(c)
Указанное в заголовке соединение получали из Соединения (c)-3 аналогичное Соединению (a) в Примере 1.
1H-ЯМР (CDCl3) δ : 9.04 (1H, с), 8.34 (1H, д), 8.26 (1H, с), 7.74 (1H, дд), 6.84 (1H, с), 6.14 (1H, д), 4.41 (1H, м), 4.33 (1H, кв), 3.49 (2H, с), 3.41 (3H, с), 2.91 (4H, м), 2.46 (4H, ушир), 1.50 (3H, д), 1.36 (6H, м).
<Получение кристаллической формы A соединения (c)>
Раствор соединения (c), очищенного методом препаративной ВЭЖХ, упаривали до выпадения кристаллов в осадок. Таким образом получали кристаллическую форму A соединения (c).
<Анализ кристаллической формы A соединения (c)>
На Фиг. 5 показан спектр рентгеновской дифракции полученной кристаллической формы. Наблюдаются пики при 4.8°, 7.6°, 8.2°, 9.7°, 15.3°, 16.6°, 19.1°, 19.8°, 22.4° и 26.2° (дифракционный угол 2θ).
Кривая теплового поведения, построенная по результатам дифференциальной сканирующей калориметрии (ДСК), имеет эндотермический пик при 182°C.
На Фиг. 16 показан спектр ИК-поглощения (ИК-спектр) полученной кристаллической формы. Наблюдаются пики поглощения при 1115 см-1, 1446 см-1, 1508 см-1, 1560 см-1 и 1601 см-1 (волновое число).
На Фиг. 23-1 и 23-2 показаны твердотельные 13C-ЯМР спектры полученной кристаллической формы. На Фиг.23-1 показан спектр, записанный в режиме 6500 Гц, и на Фиг.23-2 показан спектр, записанный в режиме 14000 Гц. Наблюдаются пики при 161.3 м.д., 150.8 м.д., 138.9 м.д., 128.1 м.д., 109.8 м.д., 82.7 м.д., 47.6 м.д., 42.5 м.д., 41.5 м.д., 24.5 м.д. и 21.7 м.д. (хим.сдвиг).
На Фиг. 24 показан твердотельный 15N-ЯМР спектр полученной кристаллической формы. Наблюдаются пики при 242.8 м.д., 233.8 м.д., 219.0 м.д., 171.7 м.д., 86.9 м.д., 54.9 м.д., 11.3 м.д. и -5.5 м.д. (хим.сдвиг).
Пример 10
Определение ингибирующей активности в отношении человеческого CDK4/циклин D3
Оценивали ингибирующую активность Соединений (a), (b) и (c) в отношении человеческого CDK4/циклин D3.
Ингибирующую активность каждого соединения в отношении человеческого CDK4/циклин D3 определяли с помощью набора для проведения анализа (QS S Assist CDK4/Cyclin D3_FP Kit), доступного от Carna Biosciences, Inc.. Этот набор для проведения анализа определяет активность киназы по изменению флуоресцентной поляризации, вызванной связыванием киназа-фосфорилированного флуоресцентного субстрата с IMAP-связывающим реагентом, по технологии IMAP от Molecular Devices.
Для приготовления растворов образцов использовали буфер для проведения анализа (10×), входящий в состав набора, или приготовленный в лаборатории буфер для проведения анализа, имеющий тот же состав, как входящий в состав набора. Буфер для проведения анализа (10×), входящий в состав набора, разбавляли очищенной водой в десять раз, получая буфер для проведения анализа. Буфер для проведения анализа содержал 20 мM HEPES (pH 7.4), 0.01% Tween 20 и 2 мM дитиотреитол. Готовили раствор испытуемого соединения в диметилсульфоксиде (ДМСО) с концентрацией в 100 раз выше финальной концентрации, и затем разбавляли буфером для проведения анализа в 25 раз, так чтобы концентрация раствора испытуемого соединения была в четыре раза выше финальной концентрации. Раствор АТФ(5×)/субстрат/металл, входящий в набор, разбавляли буфером для проведения анализа в пять раз перед использованием. Входящий в состав набора CDK4/циклин D3 разбавляли буфером для проведения анализа до концентрации в два раза выше финальной концентрации, получая раствор фермента (финальная концентрация CDK4/циклин D3 составляла от 12.5 до 25 нг/лунку). IMAP-связывающий (5×) буфер A и IMAP-связывающий (5×) буфер B каждый разбавляли очищенной водой в пять раз, затем эти буферы смешивали так, чтобы соотношение IMAP-связывающего буфера A к IMAP-связывающему буферу B составляло 85:15, и добавляли IMAP-связывающий реагент так, чтобы имело место 400-кратное разбавление для получения детектирующего реагента.
Раствор испытуемого соединения (5 мкл) и раствор АТФ/субстрат/металл (5 мкл) помещали в 384-луночный планшет. После добавления раствора фермента или буфера для проведения анализа (10 мкл), раствор перемешивали для запуска ферментативной реакции. Общее количество раствора составляло 20 мкл/лунку, и реакционная смесь содержала 20мM HEPES (pH 7.4), 0.01% Tween 20, 2мM дитиотреитол, 100нM FITC-меченый белковый субстрат (Carna Biosciences, Inc.), 100мкM АТФ, 1мM хлорид магния, 1% ДМСО, и от 12.5 до 25 нг/лунку CDK4/циклин D3. Реакцию проводили при комнатной температуре в течение 45 минут и затем добавляли в каждую лунку реагент для выявления (60 мкл/лунку), после чего продолжали реакцию 30 минут при комнатной температуре в условиях защиты от света. Затем определяли флуоресцентную поляризацию на планшет-ридере при длине волны возбуждающего освещения 485 нм и длине волны испускаемого света 535 нм.
Добавляли раствор фермента, и определяли процент ингибирования активности фермента для испытуемого соединения, где активность фермента = 100% в случае добавления ДМСО вместо раствора испытуемого соединения, и активность фермента = 0% в случае добавления буфера для проведения анализа вместо раствора фермента и добавления ДМСО вместо раствора испытуемого соединения. Для процента ингибирования активности фермента строили кривую зависимости ответа от дозировки, для определения концентрации, на 50% ингибирующей CDK4/циклин D3.
Активность ингибирования CDK4/циклин D3 для каждого соединения была меньше 10 нM.
Пример 11
Определение ингибирующей активности в отношении человеческого CDK2/циклин A2
Оценивали ингибирующую активность Соединений A, B и C в отношении человеческого CDK2/циклин A2 с помощью набора для проведения анализа (QS S Assist CDK2/Cyclin A2_FP Kit), доступного от Carna Biosciences, Inc.. Этот набор для проведения анализа определяет активность киназы по изменению флуоресцентной поляризации, вызванной связыванием киназа-фосфорилированного флуоресцентного субстрата с IMAP-связывающим реагентом, по технологии IMAP от Molecular Devices.
Буфер для проведения анализа (10×), входящий в состав набора, разбавляли очищенной водой в десять раз, получая буфер для проведения анализа, который затем использовали для приготовления всех растворов. Буфер для проведения анализа содержал 20 мM HEPES (pH 7.4), 0.01% Tween 20 и 2 мM дитиотреитол. Каждое испытуемое соединение разбавляли диметилсульфоксидом (ДМСО) до концентрации в 100 раз выше финальной концентрации, и затем разбавляли буфером для проведения анализа в 25 раз, так чтобы концентрация испытуемого соединения была в четыре раза выше финальной концентрации, получая раствор испытуемого соединения. Раствор АТФ(5×)/субстрат/металл, входящий в набор, разбавляли буфером для проведения анализа в пять раз перед использованием. Входящий в состав набора CDK2/циклин A2 разбавляли буфером для проведения анализа до концентрации в два раза выше финальной концентрации, получая раствор фермента (финальная концентрация CDK2/циклин A2 составляла 2.5 нг/лунку). IMAP-связывающий (5×) буфер A разбавляли очищенной водой в пять раз и добавляли IMAP-связывающий реагент так, чтобы имело место 400-кратное разбавление для получения детектирующего реагента.
Раствор испытуемого соединения (5 мкл) и раствор АТФ/субстрат/металл (5 мкл) помещали в 384-луночный планшет. После добавления раствора фермента или буфера для проведения анализа (10 мкл), раствор перемешивали для запуска ферментативной реакции. Общее количество раствора составляло 20 мкл/лунку, и реакционная смесь содержала 20мM HEPES (pH 7.4), 0.01% Tween 20, 2мM дитиотреитол, 100нM FITC-меченый белковый субстрат (Carna Biosciences, Inc.), 30мкM АТФ, 5мM хлорид магния, 1% ДМСО, и 2.5 нг/лунку CDK2/циклин A2. Реакцию проводили при комнатной температуре в течение 60 минут и затем добавляли в каждую лунку реагент для выявления (60 мкл/лунку), после чего продолжали реакцию 30 минут при комнатной температуре в условиях защиты от света. Затем определяли флуоресцентную поляризацию на планшет-ридере при длине волны возбуждающего освещения 485 нм и длине волны испускаемого света 535 нм.
Добавляли раствор фермента, и определяли процент ингибирования активности фермента для испытуемого соединения, где активность фермента = 100% в случае добавления ДМСО вместо раствора испытуемого соединения, и активность фермента = 0% в случае добавления буфера для проведения анализа вместо раствора фермента и добавления ДМСО вместо раствора испытуемого соединения. Для процента ингибирования активности фермента строили кривую зависимости ответа от дозировки, для определения концентрации, на 50% ингибирующей CDK2/циклин A2.
Активность ингибирования CDK4/циклин D3 для каждого соединения (IC50) была меньше или равна 100 нM.
Пример 12
Ингибирующая активность в отношении человеческого CDK6/циклин D3
Ингибирующую активность в отношении CDK6/циклин D3 определяли методом сдвига электрофоретической подвижности. Данный метод определяет активность киназы , используя разделение белков друг от друга на основе различия электрофоретической подвижности, зависящей от молекулярного веса и электрического заряда белков. Активность киназы определяют посредством электрофоретического разделения белков, имеющих различную электроотрицательность, зависящую от степени фосфорилирования киназой.
Каждый раствор готовили с буфером для проведения анализа, содержащим 20мM HEPES (pH 7.5), 0.01% Triton X-100 и 2мM дитиотреитол. Каждое испытуемое соединение разбавляли диметилсульфоксидом (ДМСО) до концентрации в 100 раз выше финальной концентрации и затем 25-кратно разбавляли буфером для проведения анализа, так чтобы концентрация испытуемого соединения была в 4 раза выше финальной концентрации, получая раствор испытуемого соединения. Раствор АТФ/субстрат/металл готовили в концентрации в 4 раза выше финальной концентрации. Раствор фермента готовили в концентрации в 2 раза выше финальной концентрации. Финальную концентрацию фермента доводили до нужного уровня, основываясь на сигнале активности фермента и ингибирующей активности соединения, служащего положительным контролем.
Раствор испытуемого соединения (5 мкл/лунку) и раствор АТФ/субстрат/металл (5 мкл/лунку) помещали в 384-луночный планшет. После добавления раствора фермента или буфера для проведения анализа (10 мкл/лунку), смесь перемешивали для запуска ферментативной реакции. Общий объем реакционной смеси составлял 20 мкл/лунку, и реакционная смесь содержала 20мM HEPES (pH 7.5), 0.01% Triton X-100, 2мM дитиотреитол, 1000нM белковый субстрат (DYRKtide-F), 300мкM АТФ, 5мM хлорид магния, 1% ДМСО, и заданную концентрацию CDK6/циклин D3. Реакцию проводили при комнатной температуре в течение 5 часов и затем в каждую лунку добавляли останавливающий буфер (QuickScout Screening Assist MSA, Carna Biosciences, Inc.) (60 мкл/лунку) для остановки реакции. После этого разделяли белок, являющийся субстратом, и фосфорилированный белок в растворе, и количественно определяли их содержание с помощью LabChip 3000 (производство Caliper Lifesciences). Прохождение киназной реакции оценивали по соотношению продуктов (P/(P+S)), вычисленному по высоте пика (S) белка, являющегося субстратом, и высоте пика (P) фосфорилированного белка.
Добавляли раствор фермента и определяли процент ингибирования активности фермента для каждого испытуемого соединения, где активность фермента = 100% в случае добавления ДМСО вместо раствора испытуемого соединения, и активность фермента = 0% в случае добавления буфера для проведения анализа вместо раствора фермента и добавления ДМСО вместо раствора испытуемого соединения. Для процента ингибирования активности фермента строили кривую зависимости ответа от дозировки, для определения концентрации, на 50% ингибирующей CDK6/циклин D3.
Активность ингибирования CDK6/циклин D3 для каждого соединения (IC50) была меньше 10 нM.
Промышленная применимость
Кристаллическая форма соединения, имеющего формулу (I) по настоящему изобретению, можно применять в качестве действующего вещества при производстве фармацевтического средства.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ПИРИДО[3,4-d]ПИРИМИДИНА И ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 2016 |
|
RU2695337C2 |
| МНОГОКОМПОНЕНТНЫЕ КРИСТАЛЛЫ, СОДЕРЖАЩИЕ ДАЗАТИНИБ И ОПРЕДЕЛЕННЫЕ СОКРИСТАЛЛОБРАЗОВАТЕЛИ | 2013 |
|
RU2650524C2 |
| СОЛИ И ПОЛИМОРФЫ 8-ФТОР-2-{4-[(МЕТИЛАМИНО)МЕТИЛ]ФЕНИЛ}-1,3,4,5-ТЕТРАГИДРО-6Н-АЗЕПИНО[5,4,3-cd]ИНДОЛ-6-ОНА | 2011 |
|
RU2570198C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА ЯНУС-КИНАЗЫ | 2017 |
|
RU2838992C2 |
| СОЛЬ КОНДЕНСИРОВАННОГО ГЕТЕРОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО И ЕГО КРИСТАЛЛЫ | 2011 |
|
RU2557237C2 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ATR КИНАЗЫ | 2014 |
|
RU2687276C2 |
| АНТИПРОЛИФЕРАТИВНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ PAH | 2020 |
|
RU2786588C1 |
| ПИРИДО[3,4-D]ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 2017 |
|
RU2796400C2 |
| НОВЫЕ КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ-IV | 2012 |
|
RU2598072C2 |
| ИЗЕТИОНАТНАЯ СОЛЬ СЕЛЕКТИВНОГО ИНГИБИТОРА CDK4 | 2004 |
|
RU2317296C2 |














Изобретение относится к кристаллической форме соединения, имеющего формулу (I), где R1 представляет собой атом водорода или C1-3 алкильную группу, R2 представляет собой атом водорода или оксо-группу, L представляет собой простую связь или C1-3 алкиленовую группу, и X представляет собой CH или N, а именно соединение формулы (I) представляет собой 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-он, где указанная кристаллическая форма соединения имеет пики со значениями дифракционных углов 2θ = 6.3°, 6.6°, 11.6°, 16.9° и 20.0° в спектре порошковой рентгеновской дифракции или 2θ = 5.3°, 7.3°, 10.3°, 15.1° и 17.4° в спектре порошковой рентгеновской дифракции; где соединение формулы (I) представляет собой 1-(6-((6-((1R)-1-метоксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридазил)пиперазин, где указанная кристаллическая форма соединения имеет пики со значениями дифракционных углов 2θ = 5.2°, 7.6°, 8.4°, 10.5°, 15.2°, 16.9°, 20.1°, 21.0°, 23.3° и 26.6° в спектре порошковой рентгеновской дифракции или 2θ = 5.2°, 6.6°, 8.1°, 15.2°, 15.9°, 16.2°, 18.8°, 20.5°, 20.8° и 21.7° в спектре порошковой рентгеновской дифракции; где соединение формулы (I)представляет собой (R)-N8-изопропил-6-(1-метоксиэтил)-N2-(5-(пиперазин-1-илметил)пиридин-2-ил)пиридо[3,4-d]пиримидин-2,8-диамин, где указанная кристаллическая форма имеет пики со значениями дифракционных углов 2θ = 4.8°, 7.6°, 8.2°, 9.7°, 15.3°, 16.6°, 19.1°, 19.8°, 22.4° и 26.2° в спектре порошковой рентгеновской дифракции. Технический результат - кристаллические формы пиридо[3,4-d]пиримидинового производного, обладающие действием в отношении циклин-зависимой киназы 4 и/или циклин-зависимой киназы 6 (CDK4/6). 5 н. и 17 з.п. ф-лы, 28 ил., 12 пр.

1. Кристаллическая форма соединения, имеющего формулу (I):

где R1 представляет собой атом водорода или C1-3 алкильную группу, R2 представляет собой атом водорода или оксо-группу, L представляет собой простую связь или C1-3 алкиленовую группу, и X представляет собой CH или N;
где соединение представляет собой 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-он,
где указанная кристаллическая форма имеет пики со значениями дифракционных углов 2θ = 6.3°, 6.6°, 11.6°, 16.9° и 20.0° в спектре порошковой рентгеновской дифракции.
2. Кристаллическая форма по п. 1, которая имеет экстраполированную температуру начала эндотермического пика 277°C, согласно данным дифференциальной сканирующей калориметрии.
3. Кристаллическая форма по п. 1, которая имеет характеристичные пики поглощения с длиной волны 703 см-1, 896 см-1 и 3418 см-1 в ИК-спектре (метод съемки в KBr).
4. Кристаллическая форма по п. 1, которая имеет характеристичные пики 136.0 м.д., 111.2 м.д., 105.1 м.д., 101.8 м.д., 52.7 м.д., 49.6 м.д., 42.9 м.д., 23.8 м.д. и 18.5 м.д. в твердотельном ЯМР спектре (13C).
5. Кристаллическая форма по п. 1, которая имеет характеристичные пики 248.6 м.д., 245.7 м.д., 229.2 м.д., 214.5 м.д., 174.3 м.д., 86.5 м.д., 54.7 м.д. и -12.4 м.д. в твердотельном ЯМР спектре (15N).
6. Кристаллическая форма соединения, имеющего формулу (I):
где R1 представляет собой атом водорода или C1-3 алкильную группу, R2 представляет собой атом водорода или оксо-группу, L представляет собой простую связь или C1-3 алкиленовую группу, и X представляет собой CH или N;
где соединение представляет собой 1-(6-((6-((1R)-1-гидроксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридил)пиперазин-2-он,
где указанная кристаллическая форма имеет пики со значениями дифракционных углов 2θ = 5.3°, 7.3°, 10.3°, 15.1° и 17.4° в спектре порошковой рентгеновской дифракции.
7. Кристаллическая форма по п. 6, которая имеет экстраполированную температуру начала эндотермического пика 277°C, согласно данным дифференциальной сканирующей калориметрии.
8. Кристаллическая форма по п. 6, которая имеет характеристичные пики поглощения с длиной волны 874 см-1, 1330 см-1 и 3314 см-1 в ИК-спектре (метод съемки в KBr).
9. Кристаллическая форма по п. 6, которая имеет характеристичные пики 154.7 м.д., 138.8 м.д., 133.6 м.д., 113.2 м.д., 101.6 м.д., 100.4 м.д., 67.4 м.д., 51.8 м.д., 26.6 м.д. и 23.3 м.д. в твердотельном ЯМР спектре (13C).
10. Кристаллическая форма по п. 6, которая имеет характеристичные пики 243.6 м.д., 86.7 м.д., 56.7 м.д. и -12.4 м.д. в твердотельном ЯМР спектре (15N).
11. Кристаллическая форма соединения, имеющего формулу (I):
где R1 представляет собой атом водорода или C1-3 алкильную группу, R2 представляет собой атом водорода или оксо-группу, L представляет собой простую связь или C1-3 алкиленовую группу, и X представляет собой CH или N;
где соединение представляет собой 1-(6-((6-((1R)-1-метоксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридазил)пиперазин,
где указанная кристаллическая форма имеет пики со значениями дифракционных углов 2θ = 5.2°, 7.6°, 8.4°, 10.5°, 15.2°, 16.9°, 20.1°, 21.0°, 23.3° и 26.6° в спектре порошковой рентгеновской дифракции.
12. Кристаллическая форма по п. 11, которая имеет экстраполированную температуру начала эндотермического пика 225°C, согласно данным дифференциальной сканирующей калориметрии.
13. Кристаллическая форма по п. 11, которая имеет характеристичные пики поглощения с длиной волны 1369 см-1, 1424 см-1, 1508 см-1, 1545 см-1 и 1566 см-1 в ИК-спектре (метод съемки в KBr).
14. Кристаллическая форма по п. 11, которая имеет характеристичные пики 163.4 м.д., 157.6 м.д., 155.5 м.д., 117.8 м.д., 82.2 м.д., 56.1 м.д. и 42.3 м.д. в твердотельном ЯМР спектре (13C).
15. Кристаллическая форма по п. 11, которая имеет характеристичные пики 311.7 м.д., 232.4 м.д., 168.5 м.д., 79.5 м.д., 53.3 м.д., 32.9 м.д. и -4.3 м.д. в твердотельном ЯМР спектре (15N).
16. Кристаллическая форма соединения, имеющего формулу (I):
где R1 представляет собой атом водорода или C1-3 алкильную группу, R2 представляет собой атом водорода или оксо-группу, L представляет собой простую связь или C1-3 алкиленовую группу, и X представляет собой CH или N;
где соединение представляет собой 1-(6-((6-((1R)-1-метоксиэтил)-8-(изопропиламино)пиридо[3,4-d]пиримидин-2-ил)амино)-3-пиридазил)пиперазин,
где указанная кристаллическая форма имеет пики со значениями дифракционных углов 2θ = 5.2°, 6.6°, 8.1°, 15.2°, 15.9°, 16.2°, 18.8°, 20.5°, 20.8° и 21.7° в спектре порошковой рентгеновской дифракции.
17. Кристаллическая форма по п. 16, которая имеет экстраполированную температуру начала эндотермического пика 221°C, согласно данным дифференциальной сканирующей калориметрии.
18. Кристаллическая форма соединения, имеющего формулу (I):
где R1 представляет собой атом водорода или C1-3 алкильную группу, R2 представляет собой атом водорода или оксо-группу, L представляет собой простую связь или C1-3 алкиленовую группу, и X представляет собой CH или N;
где соединение представляет собой (R)-N8-изопропил-6-(1-метоксиэтил)-N2-(5-(пиперазин-1-илметил)пиридин-2-ил)пиридо[3,4-d]пиримидин-2,8-диамин,
где указанная кристаллическая форма имеет пики со значениями дифракционных углов 2θ = 4.8°, 7.6°, 8.2°, 9.7°, 15.3°, 16.6°, 19.1°, 19.8°, 22.4° и 26.2° в спектре порошковой рентгеновской дифракции.
19. Кристаллическая форма по п. 18, которая имеет экстраполированную температуру начала эндотермического пика 182°C, согласно данным дифференциальной сканирующей калориметрии.
20. Кристаллическая форма по п. 18, которая имеет характеристичные пики поглощения с длиной волны 1115 см-1, 1446 см-1, 1508 см-1, 1560 см-1 и 1601 см-1 в ИК-спектре (метод съемки в KBr).
21. Кристаллическая форма по п. 18, которая имеет характеристичные пики 161.3 м.д., 150.8 м.д., 138.9 м.д., 128.1 м.д., 109.8 м.д., 82.7 м.д., 47.6 м.д., 42.5 м.д., 41.5 м.д., 24.5 м.д. и 21.7 м.д. в твердотельном ЯМР спектре (13C).
22. Кристаллическая форма по п. 18, которая имеет характеристичные пики 242.8 м.д., 233.8 м.д., 219.0 м.д., 171.7 м.д., 86.9 м.д., 54.9 м.д., 11.3 м.д. и -5.5 м.д. в твердотельном ЯМР спектре (15N).
| Токарный резец | 1924 |
|
SU2016A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Боевая ракета | 1928 |
|
SU19094A1 |
| Innocenti, P., Woodward, H | |||
| L., Solanki, S., Naud, S., Westwood, I | |||
| M., Cronin, N | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

