Область техники
Настоящее изобретение относится к солям производных пиразолохинолина, обладающих ингибирующим действием в отношении фосфодиэстеразы 9 (PDE9), и их кристаллу.
Предпосылки изобретения
Известно, что циклический гуанозинмонофосфат (здесь и далее именуемый cGMP), действующий как второй посредник в клетках, играет важную роль в различных физиологических функциях, включая способности к обучению и запоминанию.
В постсинаптической части нейронных цепей головного мозга закись азота (здесь и далее именуемая NO), биосинтезируемая синтетазой оксида азота, активирует гуанилатциклазу, которая является синтетазой cGMP. Активированная гуанилатциклаза биосинтезирует cGMP из гуанозинтрифосфата. cGMP активирует cGMP-зависимую протеинкиназу (здесь и далее именуемую PKG), которая фосфорилирует различные белки, участвующие в пластичности синапсов. Известно, что активация каскада NO/cGMP/PKG участвует в индукции пластичности синапсов (долгосрочном потенцировании, здесь и далее именуемом LTP) в гиппокампе, известных как нейронный субстрат для обучаемости и запоминания (например, см. Непатентный литературный источник 1). Известно, что лекарственное средство, активирующее передачу сигнала по каскадному механизму, улучшает LTP в гиппокампе и обучаемость животных, в то время как известно, что лекарственное средство, ингибирующее каскад, характеризуется обратным действием (Непатентный литературный источник 2). Следовательно, с учетом обнаруженных фактов, ожидается, что повышение содержания cGMP в мозгу приведет к улучшению способностей к обучению и запоминанию.
cGMP метаболизируется до 5'-GMP, не обладающего свойством активации PKG с помощью фосфодиэстеразы (здесь и далее именуемой PDE). Известно, что PDE подразделяется на 11 семейств, и то, что PDE9 специфически метаболизирует cGMP, а также то, что он экспрессируется в мозгу, селезенке, тонком кишечнике и т.п. (например, см. непатентный литературный источник 3). Таким образом предполагается, что ингибирование PDE9 будет повышать содержание cGMP в головном мозге. Сообщается, что ингибитор PDE9 в действительности усиливает LTP в гиппокампе и улучшает способности к обучению и запоминанию животных при тестировании распознавания новых объектов/тестировании обучения пассивному избеганию и т.п. (Непатентный литературный источник 4). С клинической точки зрения, активность гуанилатциклазы снижается и появляется вероятность снижения уровней cGMP в верхней височной коре у пациентов с болезнью Альцгеймера (Непатентный литературный источник 5). Таким образом, существует вероятность того, что PDE9 близко соотносится с патологиями нейродегенеративных заболеваний и психиатрических заболеваний, в частности с патологиями когнитивных дисфункций и подобными в болезни Альцгеймера, такими как болезнь Александра, болезнь Альперса, болезнь Альцгеймера, боковой амиотрофический склероз (ALS; известный как болезнь Лу Герига или заболевание двигательных нейронов), атаксия-телеангиэктазия, болезнь Баттена (также известная как болезнь Шпильмейера-Фогта-Шегрена-Баттена), Деменция Бинсвангера (подкорковая атеросклеротическая энцефалопатия), биполярное расстройство, губчатая энцефалопатия крупного рогатого скота (BSE), болезнь Кэнэвэн, деменция, вызванная химиотерапией, синдром Коккейна, кортикобазальная дегенерация, болезнь Крейтцфельдта-Якоба, депрессия, синдром Дауна, дегенерация лобной и височной долей (включая лобно-височную деменцию, семантическую деменцию и прогрессивную афазию со снижениям беглости реги), Болезнь Герстманна-Штраусслера-Шейнкера, глаукома, болезнь Хантингтона (хорея), вызванное ВИЧ слабоумие, гиперкинез, болезнь Кеннеди, Корсаковский психоз (амнестический конфабуляторный синдром), Болезнь Краббе, деменция с тельцами Леви, прогрессивная логопеническая афазия, болезнь Мачадо-Джозефа (спиноцеребеллярная атаксия 3-го типа), рассеянный склероз, множественная системная атрофия (оливопонтоцеребеллярная атрофия), тяжелая миастения, болезнь Паркинсона, болезнь Пелицеуса-Мерцбахера, болезнь Пика, пресенильная деменция (мягкое когнитивное нарушение), первичный боковой склероз, первичная прогрессивная афазия, слабоумие, вызванное радиационным излучением, болезнь Рефсума (болезнь накопления фитиновой кислоты), болезнь Сандхоффа, болезнь Шильдера, шизофрения, семантическая деменция, сенильная деменция, синдром Шая-Дрейджера, спиноцеребеллярная атаксия, атрофия мышц позвоночника, синдром Стил-Ричардсона-Ольшевского (прогрессивный супрануклеарный паралич) и сосудистый амилоидоз и сосудистая деменция (мульти-инфарктная деменция).
Список использованной литературы
Непатентная литература
[Непатентный литературный источник 1] Domek-Lopacinska et al., "Cyclic GMP metabolism and its role in brain physiology", J Physiol Pharmacol., vol. 56, Suppl 2: pp. 15-34, 2005
[Непатентный литературный источник 2] Wang X., "Cyclic GMP-dependent protein kinase and cellular signaling in the nervous system", J. Neurochem., vol. 68, pp. 443-456, 1997
[Непатентный литературный источник 3] Fisher et al., "Isolation and characterization of PDE9A, a novel human cGMP-specific phosphodiesterase", J. Biol. Chem., vol. 273: pp. 15559-15564, 1998
[Непатентный литературный источник 4] van der Staay et al., "The novel selective PDE9 inhibitor BAY 73-6691 improves learning and memory in rodents", Neuropharmacology, vol. 55: pp. 908-918, 2008
[Непатентный литературный источник 5] Bonkale et al., "Reduced nitric oxide responsive soluble guanylyl cyclase activity in the superior temporal cortex of patients with Alzheimer's disease", Neurosci. Lett., vol 187, pp. 5-8, 1995
Сущность изобретения
Техническая проблема
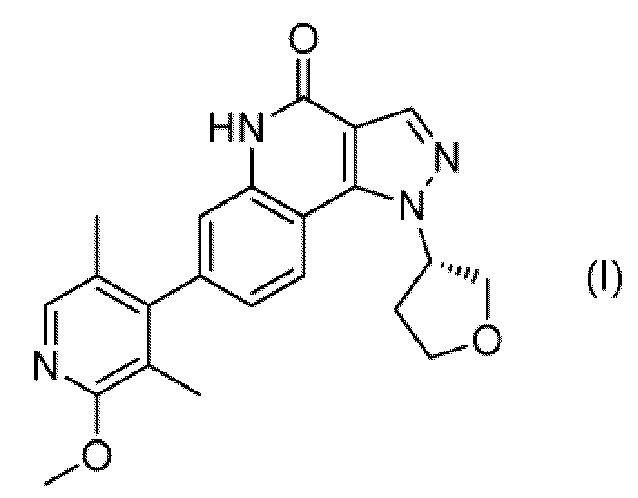
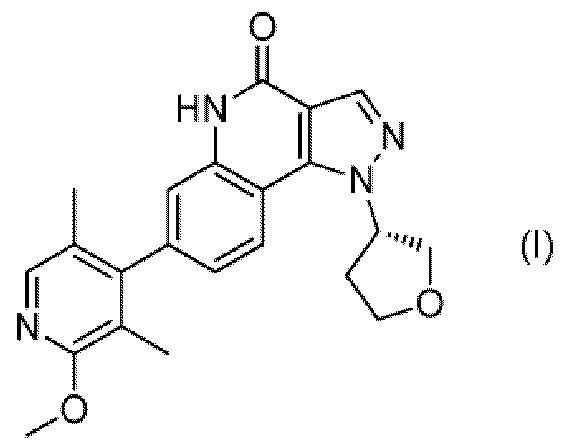
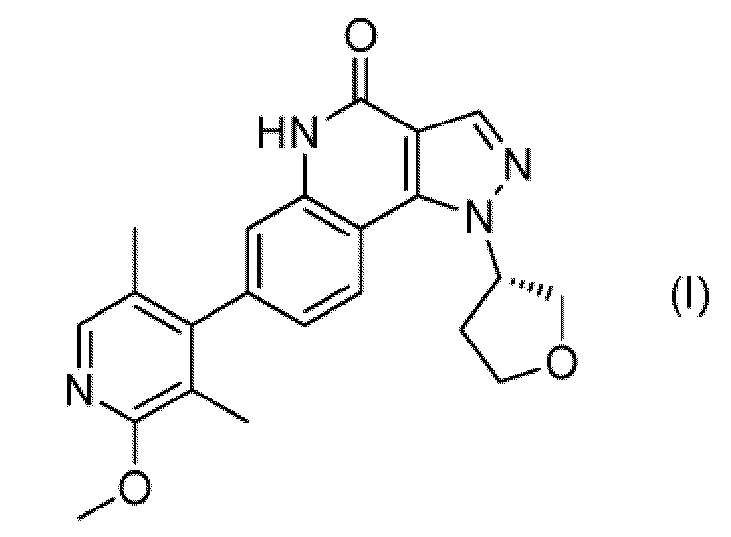
Было найдено, что соединение, представленное следующей формулой (I) ((S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он; здесь и далее именуемое соединение (I)), является новым соединением, обладающим ингибирующим действием в отношении PDE9, вследствие чего на эти изобретения была подана патентная заявка (PCT/JP 2012/075748):
Что касается соединений, применение которых в качестве фармацевтических препаратов является перспективным, то, как правило, физические свойства их солей или кристаллов этих солей имеют огромное влияние на биодоступность лекарства, чистоту лекарственной субстанции, состав фармацевтического препарата и т.п.
Задачей данного изобретения, таким образом, является предоставить соль соединения (I) или ее кристалл, применение которых в качестве лекарственной субстанции в фармацевтических препаратах является перспективным, и которые обладают улучшенными характеристиками растворимости и перорального всасывания.
Решение проблемы
Для решения обозначенных выше проблем, соединение (I) было глубоко изучено изобретателями, в результате чего были найдены соли соединения (I) или их кристаллы, что, таким образом, завершило изобретение.
Соответственно, настоящее изобретение относится к:
[1] соли (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она и кислоты, выбранной из группы, состоящей из хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, азотной кислоты, фосфорной кислоты, малоновой кислоты, малеиновой кислоты, винной кислоты, метансульфокислоты, бензолсульфокислоты и толуолсульфокислоты;
[2] мономалеату (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она;
[3] монобензолсульфонату (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она;
[4] кристаллу соли согласно [1];
[5] кристаллу мономалеата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, имеющему дифракционный пик при угле дифракции (2θ±0,2°), равном 10,1° при анализе методом рентгеновской порошковой дифракции;
[6] кристаллу монобензолсульфоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, имеющему дифракционный пик при угле дифракции (2θ±0,2°), равном 9,9° при анализе методом рентгеновской порошковой дифракции;
[7] фармацевтической композиции, включающей соль согласно [1] в качестве активного ингредиента;
[P1] соли (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она и кислоты, выбранной из группы, состоящей из неорганических кислот, органических карбоновых кислот и органических сульфокислот;
[P2] соли согласно [P1], где кислота представляет собой органическую карбоновую кислоту;
[P3] соли согласно [P2], где органическая карбоновая кислота представляет собой малоновую кислоту, малеиновую кислоту или винную кислоту;
[P4] соли согласно [P1], где кислота представляет собой органическую сульфокислоту;
[P5] соли согласно [P4], где органическая сульфокислота представляет собой метансульфокислоту, бензолсульфокислоту или толуолсульфокислоту;
[P6] соли согласно [P1], где кислота представляет собой неорганическую кислоту;
[P7] соли согласно [P6], где неорганическая кислота представляет собой хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту или фосфорную кислоту;
[Р8] мономалеату (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она;
[Р9] монобензолсульфонату (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она;
[Р10] кристаллу соли согласно [Р1];
[P11] кристаллу согласно [P10], где кислота представляет собой малоновую кислоту, малеиновую кислоту, винную кислоту, метансульфокислоту, бензолсульфокислоту, толуолсульфокислоту, хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту или фосфорную кислоту;
[Р12] кристаллу мономалеата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, имеющему дифракционный пик при угле дифракции (2θ±0,2°), равном 10,1° при анализе методом рентгеновской порошковой дифракции;
[Р12.1] кристаллу мономалеата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, имеющему дифракционные пики при углах дифракции (2θ±0,2°), равных 9,1° и 10,1° при анализе методом рентгеновской порошковой дифракции;
[Р12.2] кристаллу мономалеата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, имеющему дифракционные пики при углах дифракции (2θ±0,2°), равных 9,1°, 10,1° и 11,1° при анализе методом рентгеновской порошковой дифракции;
[Р12.3] кристаллу мономалеата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, имеющему дифракционные пики при углах дифракции (2θ±0,2°), равных 9,1°, 10,1°, 11,1°, 18,2° и 25,8° при анализе методом рентгеновской порошковой дифракции;
[Р12.4] кристаллу мономалеата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, имеющему дифракционные пики при углах дифракции (2θ±0,2°), равных 9,1°, 10,1°, 11,1°, 16,2°, 17,6°, 18,2°, 22,0°, 22,4°, 23,8° и 25,8° при анализе методом рентгеновской порошковой дифракции;
[Р12.5] кристаллу мономалеата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, имеющему пики с химическими сдвигами (м.д.), равными 13,3, 61,9, 114,3, 138,9 и 172,0 в твердотельном 13C ЯМР-спектре;
[Р13] кристаллу монобензолсульфоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, имеющему дифракционный пик при угле дифракции (2θ±0,2°), равном 9,9° при анализе методом рентгеновской порошковой дифракции;
[Р13.1] кристаллу монобензолсульфоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, имеющему дифракционные пики при углах дифракции (2θ±0,2°), равных 9,9° и 14,6° при анализе методом рентгеновской порошковой дифракции;
[Р13.2] кристаллу монобензолсульфоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, имеющему дифракционные пики при углах дифракции (2θ±0,2°), равных 9,9°, 13,7° и 14,6° при анализе методом рентгеновской порошковой дифракции;
[Р13.3] кристаллу монобензолсульфоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, имеющему дифракционные пики при углах дифракции (2θ±0,2°), равных 6,6°, 9,9°, 13,7°, 14,6° и 25,7° при анализе методом рентгеновской порошковой дифракции;
[Р13.4] кристаллу монобензолсульфоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, имеющему дифракционные пики при углах дифракции (2θ±0,2°), равных 6,6°, 9,9°, 13,7°, 14,6°, 19,0°, 19,6°, 20,5°, 21,7°, 23,5° и 25,7° при анализе методом рентгеновской порошковой дифракции;
[Р13.5] кристаллу монобензолсульфоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, имеющему пики с химическими сдвигами (м.д.), равными 16,8, 67,9, 114,0, 137,7 и 160,7 в твердотельном 13C ЯМР-спектре;
[Р14] фармацевтической композиции, включающей соль согласно [Р1] в качестве активного ингредиента;
[Р14.1] фармацевтической композиции, включающей соль согласно [Р8] или [P9] в качестве активного ингредиента;
[P14.2] фармацевтической композиции, включающей кристалл согласно [P12], [P12.1], [P12.2], [P12.3], [P12.4] или [P12.5] в качестве активного ингредиента; и
[P14.3] фармацевтической композиции, включающей кристалл согласно [P13], [P13.1], [P13.2], [P13.3], [P13.4] или [P13.5] в качестве активного ингредиента.
Полезные результаты изобретения
Соли соединения (I) и их кристаллы, предоставляемые настоящим изобретением, обладают улучшенными характеристиками растворимости и перорального всасывания, и их применение в качестве лекарственной субстанции в фармацевтических препаратах является перспективным.
Краткое описание графических материалов
На фигуре 1 представлена дифракционная картина, полученная методом рентгеновской порошковой дифракции кристалла мономалеата соединения (I), полученного в Примере 1. По оси абсцисс показан угол дифракции (2θ), и по оси ординат указана интенсивность пика.
На фигуре 2 представлена дифракционная картина, полученная методом рентгеновской порошковой дифракции кристалла монобензолсульфоната соединения (I), полученного в Примере 2. По оси абсцисс показан угол дифракции (2θ), и по оси ординат указана интенсивность пика.
На фигуре 3 представлена дифракционная картина, полученная методом рентгеновской порошковой дифракции кристалла гидрохлорида соединения (I), полученного в Примере 3. По оси абсцисс показан угол дифракции (2θ), и по оси ординат указана интенсивность пика.
На фигуре 4 представлена дифракционная картина, полученная методом рентгеновской порошковой дифракции кристалла гидробромида соединения (I), полученного в Примере 4. По оси абсцисс показан угол дифракции (2θ), и по оси ординат указана интенсивность пика.
На фигуре 5 представлена дифракционная картина, полученная методом рентгеновской порошковой дифракции кристалла п-толуолсульфоната соединения (I), полученного в Примере 5. По оси абсцисс показан угол дифракции (2θ), и по оси ординат указана интенсивность пика.
На фигуре 6 представлена дифракционная картина, полученная методом рентгеновской порошковой дифракции кристалла нитрата соединения (I), полученного в Примере 6. По оси абсцисс показан угол дифракции (2θ), и по оси ординат - интенсивность пика.
На фигуре 7 представлена дифракционная картина, полученная методом рентгеновской порошковой дифракции кристалла сульфата соединения (I), полученного в Примере 7. По оси абсцисс показан угол дифракции (2θ), и по оси ординат указана интенсивность пика.
На фигуре 8 представлена дифракционная картина, полученная методом рентгеновской порошковой дифракции кристалла метансульфоната соединения (I), полученного в Примере 8. По оси абсцисс показан угол дифракции (2θ), и по оси ординат указана интенсивность пика.
На фигуре 9 представлена дифракционная картина, полученная методом рентгеновской порошковой дифракции кристалла фосфата соединения (I), полученного в Примере 9. По оси абсцисс показан угол дифракции (2θ), и по оси ординат указана интенсивность пика.
На фигуре 10 представлена дифракционная картина, полученная методом рентгеновской порошковой дифракции кристалла L-тартрата соединения (I), полученного в Примере 10. По оси абсцисс показан угол дифракции (2θ), и по оси ординат указана интенсивность пика.
На фигуре 11 представлена дифракционная картина, полученная методом рентгеновской порошковой дифракции кристалла малоната соединения (I), полученного в Примере 11. По оси абсцисс показан угол дифракции (2θ), и по оси ординат указана интенсивность пика.
На фигуре 12 представлена кривая, показывающая результаты исследования растворимости соединения (I), мономалеата соединения (I), полученного в Примере 1, и монобензолсульфоната соединения (I), полученного в Примере 2. По оси абсцисс показано время (в минутах), и по оси ординат указана концентрация (μг/мл) в пересчете на соединение (I).
На фигуре 13 представлена кривая, показывающая изменения концентрации соединения (I) в плазме после перорального введения соединения (I) и мономалеата соединения (I), полученных в Примере 1, собакам. По оси абсцисс показано время (в часах), и по оси ординат указана концентрация соединения (μг/мл) в пересчете на соединение (I).
Описание вариантов осуществления
Соли, кристаллы и способы их получения согласно настоящему изобретению подробно проиллюстрированы ниже.
Используемый в данном контексте термин "соль" означает "соль в обычно используемом смысле" или соответствующий ей кристалл, состоящие из соединения (I), обладающего ингибирующим действием в отношении PDE9, и фармацевтически приемлемой кислоты. Термин "соль в обычно используемом смысле" относится к соединению, состоящему из положительно заряженной части основания соединения (I) и отрицательно заряженной части кислоты. Далее, "соответствующий ей кристалл" относится к кристаллическому комплексу, в котором молекулы соединения (I) и кислоты упакованы в кристаллической решетке в постоянном соотношении и в неизменной конфигурации.
В частности, соль в соответствии с настоящим изобретением представляет собой соль в обычно используемом смысле или соответствующий ей кристалл, состоящие из соединения (I) и кислоты, выбранной из группы, состоящей из органических карбоновых кислот, органических сульфокислот и неорганических кислот.
Примеры органических карбоновых кислот предпочтительно включают соль уксусной кислоты, щавелевой кислоты, малеиновой кислоты, винной кислоты, фумаровой кислоты, лимонной кислоты и малоновой кислоты, и более предпочтительно малеиновую кислоту, винную кислоту и малоновую кислоту.
Примеры органических сульфокислот предпочтительно включают метансульфокислоту, трифторметансульфокислоту, этансульфокислоту, бензолсульфокислоту, толуолсульфокислоту и камфорсульфокислоту, и более предпочтительно метансульфокислоту, бензолсульфокислоту и толуолсульфокислоту.
Примеры неорганических кислот предпочтительно включают фтористоводородную кислоту, хлористоводородную кислоту, бромистоводородную кислоту, йодистоводородную кислоту, серную кислоту, азотную кислоту, хлорную кислоту, фосфорную кислоту, угольную кислоту и бикарбонатную соль угольной кислоты, и более предпочтительно хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту и фосфорную кислоту.
Соли в соответствии с настоящим изобретением могут представлять собой сольваты. Используемый в данном контексте термин "сольват соли соединения (I)" означает твердое вещество, которое вместе образуют соль соединения (I) и молекулы растворителя. Примеры растворителей в сольватах включают растворители-кетоны, такие как ацетон, 2-бутанон и циклогексанон; растворители-сложные эфиры, такие как метилацетат и этилацетат; растворители-простые эфиры, такие как 1,2-диметоксиэтан и метил-т-бутиловый эфир; растворители-спирты, такие как метанол, этанол, 1-пропанол и изопропанол, полярные растворители, такие как N-метил-2-пирролидон, N,N-диметилформамид и диметилсульфоксид; и воду.
Используемый в данном контексте термин "кристалл" означает кристалл соли соединения (I). Соответственно, кристалл мономалеата соединения (I), например, означает кристалл соли в обычно используемом смысле, образованный соединением (I) и малеиновой кислотой, или соответствующий ей кристалл, образованный соединением (I) и малеиновой кислотой.
Примеры кристаллов, предпочтительных для данного изобретения, включают:
кристалл мономалеата соединения (I), имеющий дифракционный пик при угле дифракции (2θ±0,2°), равном 10,1° при анализе методом рентгеновской порошковой дифракции;
кристалл мономалеата соединения (I), имеющий дифракционные пики при углах дифракции (2θ±0,2°), равных 9,1° и 10,1° при анализе методом рентгеновской порошковой дифракции;
кристалл мономалеата соединения (I), имеющий дифракционные пики при углах дифракции (2θ±0,2°), равных 9,1°, 10,1° и 11,1° при анализе методом рентгеновской порошковой дифракции;
кристалл мономалеата соединения (I), имеющий дифракционные пики при углах дифракции (2θ±0,2°), равных 9,1°, 10,1°, 11,1°, 18,2° и 25,8° при анализе методом рентгеновской порошковой дифракции;
кристалл мономалеата соединения (I), имеющий дифракционные пики при углах дифракции (2θ±0,2°), равных 9,1°, 10,1°, 11,1°, 16,2°, 17,6°, 18,2°, 22,0°, 22,4°, 23,8° и 25,8° при анализе методом рентгеновской порошковой дифракции;
кристалл монобензолсульфоната соединения (I), имеющий дифракционный пик при угле дифракции (2θ±0,2°), равном 9,9° при анализе методом рентгеновской порошковой дифракции;
кристалл монобензолсульфоната соединения (I), имеющий дифракционные пики при углах дифракции (2θ±0,2°), равных 9,9° и 14,6° при анализе методом рентгеновской порошковой дифракции;
кристалл монобензолсульфоната соединения (I), имеющий дифракционные пики при углах дифракции (2θ±0,2°), равных 9,9°, 13,7° и 14,6° при анализе методом рентгеновской порошковой дифракции;
кристалл монобензолсульфоната соединения (I), имеющий дифракционные пики при углах дифракции (2θ±0,2°), равных 6,6°, 9,9°, 13,7°, 14,6° и 25,7° при анализе методом рентгеновской порошковой дифракции;
кристалл монобензолсульфоната соединения (I), имеющий дифракционные пики при углах дифракции (2θ±0,2°) равных 6,6°, 9,9°, 13,7°, 14,6°, 19,0°, 19,6°, 20,5°, 21,7°, 23,5° и 25,7° при анализе методом рентгеновской порошковой дифракции; или
кристалл мономалеата соединения (I), отличающийся тем, что он имеет пики с химическими сдвигами (м.д.), равными 13,3, 61,9, 114,3, 138,9 и 172,0 в твердотельном 13C ЯМР-спектре;
кристалл монобензолсульфоната соединения (I), отличающийся тем, что он имеет пики с химическими сдвигами (м.д.), равными 16,8, 67,9, 114,0, 137,7 и 160,7 в твердотельном 13C ЯМР-спектре.
Пики в картине порошковой рентгеновской дифракции, описанной выше, характерны для соответствующих кристаллов мономалеатов соединения (I) или кристаллов монобензолсульфонатов соединения (I).
В общем случае, в методе рентгеновской порошковой дифракции возможны ошибки в углах дифракции (2θ) в пределах ± 0,2°, поэтому необходимо учитывать, что описанные выше значения угла дифракции включают численные значения в пределах примерно ± 0,2°. Таким образом, в данное изобретение включены не только кристаллы, имеющие пики при ровно одних и тех же углах дифракции в картине порошковой рентгеновской дифракции, но также и кристаллы, имеющие пики при углах дифракции в пределах ошибки, составляющей примерно ± 0,2°.
Поэтому, фраза "имеет дифракционный пик при угле дифракции (2θ±0,2°), равном 10,1°" используется здесь, например, в значении "имеет дифракционный пик при угле дифракции (2θ), равном от 9,9° до 10,3°." То же применимо и к другим углам дифракции.
В контексте данного документа выражение "имеет пики с химическими сдвигами (м.д.), равными 13,3, 61,9, 114,3, 138,9 и 172,0" означает "имеет пики, каждый из которых в значительной степени эквивалентен пикам с химическими сдвигами (м.д.), равными 13,3, 61,9, 114,3, 138,9 и 172,0, если запись твердотельного 13C ЯМР-спектра выполняют при общепринятых условиях измерения или в значительной степени одинаковых с условиями, описываемыми в данной спецификации".
При определении того, применимо или нет выражение «имеет пики в значительной степени эквивалентные», необходимо учитывать, что вышеописанные значения химических сдвигов включают значения в пределах примерно ± 0,5 м.д., поскольку в твердотельного 13C ЯМР-спектра, как правило, возможны ошибки в значениях химических сдвигов (м.д.) в пределах ± 0,5 м.д. Таким образом, в данное изобретение включены не только кристаллы, имеющие пики при ровно одних и тех же химических сдвигах в твердотельном 13C ЯМР-спектре, но также и кристаллы, имеющие химические сдвиги в пределах ошибки, составляющей примерно ± 0,5 м.д. Поэтому, фраза "имеет пик с химическим сдвигом (м.д.), равном 13,3" используется здесь, например, в значении "имеет пик с химическим сдвигом (м.д.), равном от 12,8 до 13,8". То же применимо и к другим химическим сдвигам в твердотельных 13C ЯМР-спектрах.
Способ получения солей соединения (I) или кристаллов или подобных им продуктов, которые являются одним вариантом осуществления настоящего изобретения, будет проиллюстрирован ниже.
Получение соединения (I)
Соединение (I) согласно настоящему изобретению можно синтезировать из 3-оксотетрагидрофурана, 2-фтор-5-метилпиридина и 4-бром-2-фторбензойной кислоты в качестве исходных материалов, а именно, как описано в Сравнительном примере 1 ниже.
Способы получения солей соединения (I)
Соли соединения (I) можно получать традиционными способами получения солей. В частности, их можно получать, например, путем суспендирования или растворения соединения (I) в растворителе, при нагревании, если необходимо, а затем внесения в полученную суспензию или раствор кислоты, выбранной из группы, состоящей из органических карбоновых кислот органических сульфокислот, и неорганических кислот, и перемешивания или выдержки полученной суспензии или раствора в течение периода от нескольких минут до нескольких дней при комнатной температуре или при охлаждении на ледяной бане. Соли соединения (I) можно получать в виде кристаллов или аморфных веществ согласно способам получения. Примеры растворителей, используемых в данных способах, включают растворители-спирты, такие как этанол, 1-пропанол и изопропанол; ацетонитрил; растворители-кетоны, такие как ацетон и 2-бутанон; растворители-сложные эфиры, такие как этилацетат; растворители-насыщенные углеводороды, такие как гексан и гептан; растворители-простые эфиры, такие как метил-т-бутиловый эфир, или воду. Каждый из этих растворителей можно использовать как самостоятельно, так и в смеси из двух или более из них.
В вышеописанных способах получения соединения (I), соли соединения (I) можно получать следом за синтезом соединения (I), путем применения вышеизложенных способов.
Способы получения кристаллов солей соединения (I)
Кристалл соли соединения (I) можно получать с помощью вышеупомянутых способов получения солей соединения (I), или путем растворения соли соединения (I) при нагревании в растворителе и кристаллизации ее при охлаждении и перемешивании.
Соль соединения (I) для использования в кристаллизации может быть в любой форме: это может быть гидрат, ангидрид, аморфное вещество, кристаллическое вещество (включая те, которые состоят из множества кристаллических полиморфных форм) или их комбинация.
Примеры растворителей, используемых в кристаллизации, включают растворители-спирты, такие как метанол, этанол, изопропанол и 1-пропанол; ацетонитрил; растворители-амиды, такие как N,N-диметилформамид; растворители-сложные эфиры, такие как этилацетат; растворители-насыщенные углеводороды, такие как гексан и гептан; растворители-кетоны, такие как ацетон и 2-бутанон; растворители-простые эфиры, такие как метил-т-бутиловый эфир, или воду. Кроме того, каждый из этих растворителей можно использовать как самостоятельно, так и в смеси из двух или более из них.
Количество используемого растворителя можно подбирать подходящим образом, при условии, что нижним пределом является количество, в котором можно растворить свободную форму соединения (I) или его соль при нагревании, или полученная в нем суспензия пригодна для перемешивания; а верхним пределом является количество, в котором выход кристалла снижается незначительно.
Кристалл, полученный описанными выше способами, представляет собой кристалл одной формы. Эта форма кристалла стабильна, не подвержена переходу в другие кристаллические формы или аморфные вещества, обладает хорошими физическими свойствами и также пригодна для составления препаратов.
В ходе кристаллизации по желанию можно вносить затравочный кристалл (например, кристалл требуемой соли соединения (I)). Температура, при которой вносят затравочный кристалл, особым образом не ограничивается, но предпочтительно она составляет 0-60°C.
Как температуру, применяемую в ходе растворения при нагревании соли соединения (I), так и температуру, при которой растворяется соединение (I), можно подходящим образом выбирать в зависимости от растворителя, но предпочтительно она находится в интервале между температурой, при которой начинает кипеть раствор для перекристаллизации, и 50°C, и более предпочтительно от 65 до 55°C.
Быстрое охлаждение в ходе кристаллизации может привести к веществам с различными формами кристаллов (полиморфизму). По этой причине при охлаждении желательно регулировать скорость охлаждения соответствующим образом с учетом ее влияния на качество, размер зерна и другие характеристики кристалла. Предпочтительно, например, проводить охлаждение со скоростью охлаждения, равной от 40 до 5°С/час. Более предпочтительным является охлаждение со скоростью охлаждения равной, например, 25-5°C/час.
Кроме того, конечную температуру кристаллизации можно подходящим образом выбирать в зависимости от выхода, качества и других характеристик кристалла, но она предпочтительно равно от 30 до -25°C.
Целевой кристалл можно получать выделением кристалла, используя традиционные процедуры фильтрации, при необходимости промывкой отфильтрованного кристалла растворителем и последующей его сушкой. Растворитель для промывки кристалла можно использовать тот же, что и растворитель, используемый в ходе кристаллизации. Предпочтительно это, например, ацетон, 2-бутанон, этилацетат, метил-т-бутиловый эфир и смешанный растворитель гексан/2-бутанон (в объемном отношении 2:3).
Кристалл, выделенный в ходе процедуры фильтрации, можно сушить соответствующим образом путем выдержки на воздухе или в токе азота, или путем нагревания.
Что касается продолжительности сушки, время, требуемое для того, чтобы количество остаточного растворителя стало меньше, чем заранее установленное количество, можно выбирать соответствующим образом в зависимости от производимого количества, устройства сушильного аппарата, температуры сушки и т.п. Кроме того, сушку можно проводить в токе воздуха или при пониженном давлении. Степень разрежения давления можно определять соответствующим образом в зависимости от производимого количества, устройства сушильного аппарата, температуры сушки и т.п. После сушки полученный кристалл можно оставлять на воздухе по мере необходимости.
В вышеупомянутых способах получения соединения (I), описанные выше кристаллы можно получать вслед за синтезом соединения (I) путем дополнительного применения вышеупомянутых способов получения солей соединения (I), и если необходимо, способов получения кристаллов солей соединения (I).
Предполагается, что соли соединения (I) и их кристаллы, полученные описанными выше способами получения, повысят концентрацию cGMP в головном мозге, поскольку они обладают ингибирующим действием в отношении PDE9, как показано с помощью данных об их активности, полученных в иллюстративном фармакологическом испытании, описанном далее. Ингибирующее действие в отношении PDE9 и повышение уровней cGMP в головном мозге приводит к улучшению способности к обучению и запоминанию, и применение этих солей и кристаллов в качестве терапевтических препаратов для когнитивных дисфункций и подобных в болезни Альцгеймера является перспективным.
Соединение формулы (I) согласно данному изобретению или его фармацевтически приемлемую соль можно приготовить фармацевтическим образом, используя традиционны способ, а также можно изготовить стандартную дозу, например, пероральный препарат (таблетку, гранулу, порошок, капсулу, сироп и т.п.), препарат для инъекции (для внутривенного введения, для внутримышечного введения, для подкожного введения, для внутрибрюшинного введения и т.п.), наружный препарат (препарат для применения на кожу (мазь, пластырь и т.п.), глазные капли, капли в нос, свечи и т.п.).
При получении твердого препарата для перорального введения по мере необходимости к соединению формулы (I) или его фармацевтически приемлемой соли добавляют вспомогательное вещество, связующее, разрыхлитель, смазывающее вещество, красящее вещество и т.п., и получают таблетку, гранулу, порошок и капсулу, используя традиционные способы. На таблетку, гранулу, порошок, капсулу и т.п. при необходимости можно наносить пленку.
Примеры вспомогательного вещества включают лактозу, кукурузный крахмал и кристаллическую целлюлозу; примеры связующего включают гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу; примеры разрыхлителя включают карбоксиметилцеллюлозу кальция и кроскармеллозу натрия; примеры смазывающего вещества включают стеарат магния и стеарат кальция; примеры красящего вещества включают оксид титана; и примеры пленкообразователя включают гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу и метилцеллюлозу, но, разумеется, примеры этих добавок не ограничены данными примерами.
В таких твердых препаратах, как таблетки, капсулы, гранулы и порошки, в каждом может содержаться обычно от 0,001 до 99,5% по весу, предпочтительно от 0,01 до 90% по весу и т.п., соединения формулы (I) или его фармацевтически приемлемой соли.
При получении препарата для инъекции (для внутривенного введения, для внутримышечного введения, для подкожного введения, для внутрибрюшинного введения и т.п.) по мере необходимости к соединению формулы (I) или его фармацевтически приемлемой соли добавляют регулятор pH, буфер, суспендирующее средство, солюбилизирующее средство, антиоксидант, консервант (антисептическое средство), регулирующее тоничность средство и т.п. и препарат для инъекции можно получать традиционным способом. Составы можно подвергать лиофилизации для изготовления любых растворимых лиофилизированных препаратов.
В качестве регулятора pH и буфера можно использовать, например, органическую кислоту или неорганическую кислоту и/или их соль или подобное. Кроме того, в качестве суспендирующего средства можно использовать, например, метилцеллюлозу полисорбат 80, карбоксиметилцеллюлозу натрия или подобное. В качестве солюбилизирующего средства можно использовать, например, полисорбат 80, полиоксиэтиленсорбитанмонолаурат или подобное. В качестве антиоксиданта можно использовать, например, α-токоферол или подобное. В качестве консерванта можно использовать, например, метилпараоксибензоат, этилпарагидрооксибензоат или подобное. В качестве регулирующего тоничность средства можно использовать, например, глюкозу, натрия хлорид, маннит или подобное. Разумеется, регулятор pH, буфер, суспендирующее средство, солюбилизирующее средство, антиоксидант, консервант (антисептическое средство), регулирующее тоничность средство этим не ограничиваются.
Эти препараты для инъекций могут обычно содержать массовую долю от 0,000001 до 99,5%, предпочтительно массовую долю от 0,00001 до 90% и т.п. соли соединения (I) или ее кристалла по отношению к общей массе препаратов для инъекций.
При получении препарата для внешнего применения, исходный материал-основу добавляют к соединению формулы (I) или его фармацевтически приемлемой соли, и при необходимости к ним добавляют, например, консервант, стабилизатор, регулятор pH, антиоксидант, красящее вещество и т.п., а например, препарат для чрескожного введения (мазь, пластырь и т.п.), глазные капли, капли в нос, свечи и т.п. можно получать традиционными способами.
В качестве используемого материала-основы могут применяться различные исходные материалы, обычно используемые, например, для медицинских препаратов, квазилекарственных средств и косметических средств. Конкретные примеры материала основы включают в себя исходные материалы, такие как масла животного и растительного происхождения, минеральные масла, сложноэфирные масла, воски, эмульгаторы, высшие спирты, жирные кислоты, силиконовые масла, поверхностно-активные вещества, фосфолипиды, спирты, многоатомные спирты, водорастворимые полимеры, глинистые минералы и очищенную воду.
Эти препараты для внешнего применения каждый могут обычно содержать массовую долю от 0,000001 до 99,5%, предпочтительно массовую долю от 0,00001 до 90% и т.п. соединения формулы (I) или его фармацевтически приемлемой соли.
Дозировка соли соединения (I) или ее кристалла варьируется в зависимости от тяжести симптома, возраста, пола, веса тела, стандартной дозы, типа соли и конкретной формы заболевания и т.п. В случае применения для взрослых, обычно, перорально вводят примерно от 30 μг до 10 г, предпочтительно от 100 μг до 5 г, и более предпочтительно от 100 μг до 1 г в день, или путем инъекции, в каждом случае в виде разовой дозы или разделенных доз, вводят примерно от 30 μг до 1 г, предпочтительно от 100 μг до 500 мг, и более предпочтительно от 100 μг до 300 мг в день.
Пример
Далее настоящее изобретение будет подробно описано с примерами сравнения и примерами, но настоящее изобретение не ограничивается этими примерами сравнения и примерами.
В примерах и примерах сравнения настоящего описания используются следующие сокращения.
CDI: 1,1'-карбонилдиимидазол
DCM: дихлорметан
DMF-DMA: N,N-диметилформамид диметилацеталь
DMF: N,N-диметилформамид
DMSO: диметилсульфоксид
DTT: дитиотреитол
IPA: изопропиловый спирт
KTB: трет-бутоксид калия
MTBE: метил-т-бутиловый эфир
NBS: N-бромсукцинимид
Комплекс Pd(dppf)Cl2 и DCM: Комплекс [1,1'-бис(дифенилфосфин)ферроцен]дихлорпалладия(II) и DCM
Pd(PPh3)4: тетракис(трифенилфосфин)палладий(0)
TEA: триэтиламин
TFA: трифторуксусная кислота
THF: тетрагидрофуран
Tris: трисгидроксиметиламинометан
Химический сдвиг в спектре протонного ядерного магнитного резонанса записывали в единицах δ (м.д.) относительно тетраметилсилана, и константу связи записывали в герцах (Гц). Используются следующие сокращенные обозначения мультиплетности сигналов: s: синглет, d: дублет, t: триплет, q: квартет, m: мультиплет, brs: широкий синглет и brd: широкий дублет.
В картине порошковой рентгеновской дифракции кристаллов, изготовленных в примерах ниже, полученные кристаллы закрепляли на предметном столике прибора порошковой рентгеновской дифракции и анализировали в следующих условиях:
Условия измерения
Держатель образца: алюминий
Мишень: медь
Детектор: сцинтилляционный счетчик
Напряжение в трубке: 50 кВ
Сила тока в трубке: 300 мА
Щель: DS (отклоняющая щель) 0,5 мм (ограничивающая высоту щель 2 мм), SS (рассеивающая щель) открыта, RS (приемная щель) открыта
Скорость сканирования: 10°/мин
Время выборки: 0,02°
Диапазон сканирования: 5-35°
Гониометр: горизонтальный гониометр
Твердотельные 13C ЯМР-спектры кристаллов измеряли в следующих условиях.
Условия измерения
Используемый прибор: AVANCE400 (производства компании Bruker Corporation)
Температура при измерениях: комнатная температура (22°C)
Эталонный материал: глицин (внешний опорный сигнал: 176,03 м.д.)
Измеряемое ядро: 13C (100,6248425 МГц)
Время повторения пульса: 3 секунды
Пульсирующий режим: Измерения в режиме TOSS
Сравнительный пример 1
Синтез (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
(1) Синтез бензил 2-[дигидрофуран-3(2H)-илиден]гидразинкарбоксилата
3-Оксотетрагидрофуран (5,70 г) растворяли в метаноле (150 мл) и к раствору добавляли бензилкарбазат (10 г). Смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали. Получали 14,8 г остатка в качестве сырого продукта. Его использовали в следующей реакции без дополнительной очистки.
(2) Синтез (±)-бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилата
Бензил 2-[дигидрофуран-3(2H)-илиден]гидразинкарбоксилат (14,8 г) суспендировали в воде (96 мл). К суспензии при комнатной температуре добавляли уксусную кислоту (42,1 мл). Смесь перемешивали при комнатной температуре в течение 1 часа. Суспензия превратилась в раствор. Цианоборогидрид натрия (4,0 г) добавляли к раствору малыми порциями. Смешанный раствор перемешивали при комнатной температуре в течение двух часов. Реакционную смесь охлаждали до 0°С. Реакционную смесь нейтрализовали, добавляя 5 н. водный раствор гидроксида натрия. Смесь экстрагировали хлороформом. Органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (метанол/этилацетат, 5%). Получали соединение, указанное в заголовке (13,9 г).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 1,73-1,80 (m, 1H), 1,92-2,06 (m, 1H), 3,66-3,82 (m, 3H), 3,82-4,03 (m, 2H), 5,14 (s, 2H), 7,31-7,40 (m, 5H).
(3) Синтез (-)-бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилата и (+)-бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилата
Насыщенный водный раствор бикарбоната натрия (30 мл) добавляли в раствор (±)-бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилата (11,5 г), в MTBE (110 мл). Смесь перемешивали 10 минут при комнатной температуре и органический слой отделяли. Полученный органический слой после этого промывали насыщенным бикарбонатом натрия и соляным раствором и сушили над безводным сульфатом магния, и сиккатив удаляли фильтрацией. Фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/гексан, 25 к 50%) и целевую фракцию концентрировали. К остатку прибавляли диэтиловый эфир (30 мл) и гексан (15 мл). Выпавший твердый осадок собирали фильтрованием и сушили под пониженным давлением, получая чистый (±)-бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилат (6,17 г).
Этот продукт растворяли в этаноле и фильтровали через миллипоровый фильтр. Полученный фильтрат оптически разделяли при следующих двух типах условий.
Условия 1-го типа: CHIRALCEL™ производства компании DAICEL Corp., OD-H (20 мм Φ×250 мм длина), 20% IPA-гексан, 25 мл/мин.
Условия 2-го типа: CHIRALPAK™ производства компании DAICEL Corp., AD-H (20 мм Φ×250 мм длина), 20% IPA-гексан, 24 мл/мин. Целевую фракцию концентрировали, получая указанное в заголовке соединение с коротким временем удерживания и (-) оптическим вращением (2,60 г, >99% э.и. [OD-H, 20% IPA/гексан, время удерживания =11,2 мин]), и указанное в заголовке соединение с длинным временем удерживания и (+) оптическим вращением (2,59 г, 97,2% э.и. [OD-H, 20% IPA/гексан, время удерживания = 12,4 мин]).
(4) Синтез (S)-(тетрагидрофуран-3-ил)гидразина гидрохлорида
(-)-Бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилат (50 г) растворяли в метаноле (500 мл), и добавляли ди-т-бутилдикарбонат (92,4 г) и палладия на угле (50% влажный) (5 г). Смесь перемешивали при 25°C и 15 фунтов на кв. дюйм в течение 48 часов в атмосфере водорода. Реакционную смесь фильтровали, а затем фильтрат концентрировали при пониженном давлении. Полученный в результате остаток растворяли в диизопропиловом эфире (300 мл). После охлаждения при 0°C, к раствору добавляли хлористоводородную кислоту/диизопропиловый эфир (500 мл). Смесь перемешивали при 10°C в течение 14 часов. Выпавший твердый осадок собирали фильтрованием. Ту же операцию повторяли девять раз, исходя из (-)-бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилата (70 г), и ту же операцию производили один раз, исходя из (-)-бензил 2-(тетрагидрофуран-3-ил)гидразинкарбоксилат (50 г). Полученное твердое вещество растирали с DCM/этанолом (10/1) (1 л) в течение двух часов. Выпавший твердый осадок собирали фильтрованием. Полученное в результате твердое вещество сушили при пониженном давлении, получая указанное в заголовке соединение (235 г).
1H-ЯМР (400 МГц, DMSO-d6) δ (ppm): 1,87-2,09 (m, 2H), 3,55-3,71 (m, 2H), 3,71-3,84 (m, 3H).
Согласно результатам рентгеновской кристаллографии абсолютная конфигурация полученного указанного в заголовке соединения имеет (S)-форму.
(5) Синтез 2-фтор-3-иод-5-метилпиридина
Диизопропиламин (92 мл) добавляли к THF (1,2 л) и смесь охлаждали до -18°C в атмосфере азота. К этому раствору по каплям добавляли 2,69 M раствор (224 мл) н-бутиллития в гексане. По завершении прикапывания температуру поднимали до -5°C за 20 минут при перемешивании этой смеси. Реакционный раствор охлаждали до -73°С. К этому реакционному раствору по каплям добавляли раствор (240 мл) 2-фтор-5-метилпиридина (61 г) в THF. Реакционную смесь перемешивали при -75°C в течение трех с половиной часов. К этому реакционному раствору по каплям добавляли раствор (24 мл) иода (139 г) в THF. Реакционную смесь перемешивали при -75°C в течение 1 часа 55 минут. По завершении реакции к реакционному раствору добавляли воду (220 мл) при той же температуре. Смесь перемешивали при той же температуре в течение 5 минут. Реакционному раствору давали снова нагреться до комнатной температуры и затем добавляли воду (1,2 л). К этой смеси добавляли водный раствор (300 мл) тиосульфата натрия пентагидрата (136 г) и воду (300 мл) и смесь перемешивали 10 минут. Эту смесь экстрагировали, используя MTBE (1,2 л). Органический слой промывали насыщенным соляным раствором (500 мл). Объединенные водные слои экстрагировали, используя MTBE (1 л). Объединенные органические слои сушили над безводным сульфатом магния. Сиккатив удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. К остатку добавляли н-гептан, смесь охлаждали. Выпавший твердый осадок собирали фильтрованием. Твердое вещество промывали н-гептаном. Фильтрат охлаждали и выпавшее в осадок твердое вещество собирали фильтрованием. Процедуру повторяли 5 раз, получая указанное в заголовке соединение (109,69 г).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 2,29-2,31 (m, 3H), 7,93-8,14 (m, 2H).
ESI-MS масса/заряд 238 [M+H]+
(6) Синтез 2-фтор-4-иод-3,5-диметилпиридина
Диизопропиламин (88 мл) добавляли к THF (1,2 л) и смесь охлаждали до -18°C в атмосфере азота. К этому раствору по каплям добавляли 2,69 M раствор (215 мл) н-бутиллития в гексане. По завершении прикапывания температуру поднимали до -5°C за 30 минут при перемешивании этой смеси. Реакционный раствор охлаждали до -72°С. К этому реакционному раствору по каплям добавляли раствор (240 мл) 2-фтор-3-иод-5-метилпиридина (109,69 г) в THF. Реакционную смесь перемешивали при -74°C в течение полутора часов. К этому реакционному раствору по каплям добавляли раствор (160 мл) метилиодида (36 мл) в THF. Реакционную смесь перемешивали при температуре от -70° до -74°C в течение 2 часов. По завершении реакции к реакционному раствору добавляли воду (200 мл) при той же температуре. Смесь перемешивали при той же температуре в течение 2 минут. Реакционному раствору давали снова нагреться до комнатной температуры и затем добавляли воду (1,2 л). Эту смесь перемешивали в течение 3 минут. Затем добавляли воду (300 мл). Эту смесь экстрагировали, используя MTBE (1,2 л). Органический слой промывали насыщенным соляным раствором (500 мл). Объединенные водные слои экстрагировали, используя MTBE (1 л). Объединенные органические слои сушили над безводным сульфатом магния. Сиккатив удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. К остатку добавляли н-гептан (100 мл), и смесь охлаждали. Выпавший твердый осадок собирали фильтрованием. Твердое вещество промывали н-гептаном. Фильтрат охлаждали и выпавшее в осадок твердое вещество собирали фильтрованием. Процедуру повторяли дважды, получая указанное в заголовке соединение (86,9 г).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 2,39-2,40 (m, 6H), 7,80-7,82 (m, 1H).
ESI-MS масса/заряд 252 [M+H]+
(7) Синтез 4-иод-2-метокси-3,5-диметилпиридина
К раствору (954 мл) 2-фтор-4-иод-3,5-диметилпиридина (97,4 г) в THF при 20°C добавляли 28%-ный раствор (185 мл) метоксида натрия в метаноле. Эту смесь перемешивали при температуре от 55° до 65°C в течение 2 часов. Реакционный раствор охлаждали и затем разделяли путем внесения MTBE (1 л) и воды (1 л). Органический слой промывали насыщенным соляным раствором. Объединенные водные слои экстрагировали, используя MTBE (500 мл × 2). Объединенные органические слои сушили над безводным сульфатом магния. Сиккатив удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. К остатку добавляли н-гептан (50 мл) и смесь перемешивали при 0°С в течение 1 часа. Выпавший твердый осадок собирали фильтрованием. Твердое вещество промывали охлажденным н-гептаном (10 мл). Получали соединение, указанное в заголовке (42,6 г). Фильтрат концентрировали при пониженном давлении. К остатку добавляли н-гептан (5 мл) и смесь перемешивали при 0°С в течение 30 минут. Выпавший твердый осадок собирали фильтрованием. Твердое вещество промывали охлажденным н-гептаном (2 мл), получая указанное в заголовке соединение (20,2 г). Фильтрат концентрировали при пониженном давлении. К остатку добавляли н-гептан (5 мл) и смесь перемешивали при 0°С в течение 30 минут. Выпавший твердый осадок собирали фильтрованием. Твердое вещество промывали охлажденным н-гептаном (2 мл). Получали соединение, указанное в заголовке (10,7 г). После объединения получали соединение, указанное в заголовке (73,5 г).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 2,33-2,34 (m, 3H), 2,36-2,38 (m, 3H), 3,92 (s, 3H), 7,76 (s, 1H).
ESI-MS масса/заряд 264 [M+H]+
(8) Синтез этил 3-(4-бром-2-фторфенил)-3-оксопропаноата
CDI (8,88 г) добавляли к суспензии 4-бром-2-фторбензойной кислоты (CAS № 112704-79-7) (10 г) в DCM (97 мл) и смесь перемешивали при комнатной температуре в течение 3,5 часов. Этот раствор далее именуется "раствор 1".
К суспензии этилмалоната калия (15,5 г) в ацетонитриле (303 мл) в другой колбе прибавляли TEA (15,9 мл), а затем - хлорид магния (10,9 г), и смесь перемешивали при комнатной температуре 3 часа 10 минут. К этой реакционной смеси по каплям за 25 минут добавляли "раствор 1", приготовленный ранее, и затем реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционный раствор концентрировали до половины его объема при пониженном давлении. Полученный остаток разбавляли этилацетатом (500 мл), и после внесения 5 н. хлористоводородной кислоты (250 мл) на ледяной бане, перемешивали при комнатной температуре в течение 1 часа. Отделяли органический слой. Органический слой промывали насыщенным соляным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 5-20%), получая указанное в заголовке соединение (12,8 г).
ESI-MS масса/заряд 291 [M+H]+
(9) Синтез этил 5-(4-бром-2-фторфенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Раствор этил 3-(4-бром-2-фторфенил)-3-оксопропаноата (45 г) в DMF-DMA (165 мл) перемешивали при 50°C в течение 2 часов 15 минут. Реакционный раствор концентрировали при пониженном давлении. К остатку добавляли толуол (200 мл) и смесь концентрировали при пониженном давлении. К остатку добавляли этанол (950 мл) и смесь нагревали до 50°С. К раствору по каплям за 35 минут добавляли водный раствор (60 мл) (S)-(тетрагидрофуран-3-ил)гидразина гидрохлорида (21,6 г). Полученную реакционную смесь перемешивали при 50°C в течение 2 часов 10 минут. Реакционный раствор охлаждали до комнатной температуры и затем концентрировали до половины его объема при пониженном давлении. К остатку добавляли воду (200 мл) и этанол отгоняли при пониженном давлении. К полученному остатку добавляли этилацетат (500 мл) и органический слой отделяли. Водный слой экстрагировали этилацетатом (100 мл). Объединенные органические слои промывали насыщенным соляным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, от 10% до 15%), а затем с помощью short-path колоночной хроматографии на NH силикагеле (силикагель с покрытием из пропиламина производства компании Fuji Silysia Chemical Ltd.) (этилацетат/н-гептан, 33%), получая указанное в заголовке соединение (43,1 г).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 1,19 (t, J=7,2 Гц, 3H), 2,19-2,49 (m, 2H), 3,87-4,07 (m, 3H), 4,11-4,25 (m, 3H), 4,58-4,65 (m, 1H), 7,17-7,26 (m, 1H), 7,39-7,47 (m, 2H), 8,06 (s, 1H).
ESI-MS масса/заряд 407 [M+Na]+
(10) Синтез этил 5-[2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Смесь этил 5-(4-бром-2-фторфенил)-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата (43,1 г), бис(пинаколат)дибора (34,3 г), комплекса Pd(dppf)Cl2 с DCM (4,59 г) и ацетата калия (33,1 г) сушили при пониженном давлении, используя вакуумный насос, в течение 1 часа. Раствор сухого остатка в DMF (430 мл) перемешивали при 80°C в течение 3 часов 10 минут. Реакционному раствору давали снова нагреться до комнатной температуры и затем фильтровали через Celite™. Фильтрат концентрировали при пониженном давлении. К остатку добавляли этилацетат (430 мл) и насыщенный соляной раствор (200 мл) и смесь перемешивали 5 минут. Нерастворившиеся вещества отфильтровывали на Celite™. Органический слой отделяли от фильтрата. Водный слой снова экстрагировали этилацетатом (50 мл). Объединенные органические слои сушили над безводным сульфатом магния и фильтровали, а фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, 10-15%), получая указанное в заголовке соединение (51,9 г).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 1,16 (t, J=7,2 Гц, 3H), 1,37 (s, 12H), 2,15-2,49 (m, 2H), 3,85-4,06 (m, 3H), 4,14 (q, J=7,2 Гц, 2H), 4,20 (dd, J=15,6, 8,4 Гц, 1H), 4,57-4,66 (m, 1H), 7,30 (t, J=7,2 Гц, 0,5H), 7,35 (t, J=7,2 Гц, 0,5H), 7,63 (dd, J=5,6, 2,0 Гц, 1H), 7,70 (dd, J=7,2, 2,0 Гц, 1H), 8,06 (s, 1H).
(11) Синтез этил 5-[2-фтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата
Воду (170 мл), 4-иод-2-метокси-3,5-диметилпиридин (35,6 г), Pd(PPh3)4 (6,52 г) и карбонат цезия (110 г) добавляли к раствору этил 5-[2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата (51,9 г) в 1,4-диоксане (500 мл) и проводили реакцию в реакционной смеси при 110°C в течение 6 часов. Реакционной смеси давали остыть до комнатной температуры и органический слой отделяли. Органический слой концентрировали при пониженном давлении. Водный слой, этилацетат (700 м) и воду (100 мл) вносили в полученный остаток и органический слой отделяли. Водный слой повторно экстрагировали этилацетатом (50 мл). Объединенные органические слои последовательно промывали водой и соляным раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на NH-силикагеле (этилацетат/н-гептан, от 5% до 14%). Продукт снова очищали с помощью колоночной хроматографии на NH-силикагеле (этилацетат/н-гептан, от 2% до 10%), получая указанное в заголовке соединение (43,5 г).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 1,16 (t, J=7,2 Гц, 1,5H), 1,17 (t, J=7,2 Гц, 1,5H), 1,97 (s, 1,5H), 1,98 (s, 1,5H), 1,99 (s, 1,5H), 2,00 (s, 1,5H), 2,25-2,55 (m, 2H), 3,92-4,27 (m, 6H), 3,99 (s, 1,5H), 4,00 (s, 1,5H), 4,65-4,75 (m, 1H), 7,01 (d, J=9,2 Гц, 1H), 7,05 (d, J=7,2 Гц, 1H), 7,39 (t, J=7,2 Гц, 0,5H), 7,45 (t, J=7,2 Гц, 0,5H), 7,93 (s, 1H), 8,12 (s, 1H).
ESI-MS масса/заряд 440 [M+H]+
(12) Синтез 5-[2-фтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоновой кислоты
5 н. водный раствор гидроксида натрия (79 мл) добавляли к раствору этил 5-[2-фтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксилата (43,2 г) в этаноле (574 мл) при комнатной температуре и реакционную смесь перемешивали при 60°C два часа 10 минут. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали до половины ее объема при пониженном давлении. К остатку добавляли воду (300 мл) и этанол отгоняли при пониженном давлении. К полученному остатку добавляли MTBE (130 мл) и водный слой отделяли. Органический слой экстрагировали водой (30 мл). Объединенные водные слои подкисляли, используя 5 н. соляную кислоту (78 мл) на ледяной бане и дважды экстрагировали этилацетатом. Объединенные органические слои сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение (39,0 г).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 1,91 (s, 1,5H), 1,94 (s, 1,5H), 1,98 (s, 1,5H), 2,01 (s, 1,5H), 2,25-2,56 (m, 2H), 3,92-4,17 (m, 3H), 3,96 (s, 1,5H), 4,00 (s, 1,5H), 4,23 (dd, J=16,0, 8,0 Гц, 1H), 4,65-4,77 (m, 1H), 6,99 (brd, J=10,0 Гц, 1H), 7,03 (dr d, J=7,6 Гц, 1H), 7,38 (t, J=7,6 Гц, 0,5H), 7,44 (t, J=7,6 Гц, 0,5H), 7,90 (s, 0,5H), 7,94 (s, 0,5H), 8,14 (s, 1H).
ESI-MS масса/заряд 434 [M+Na]+
(13) Синтез 5-[2-фтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксамида
CDI (21,4 г) добавляли за один раз к раствору 5-[2-фтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоновой кислоты (38,7 г) в DMF (290 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 95 минут. В реакционную смесь добавляли 28%-ный водный аммиак (95 мл) и смесь перемешивали при комнатной температуре в течение 35 минут. В реакционную смесь снова добавляли 28%-ный водный аммиак (95 мл) и смесь перемешивали при комнатной температуре в течение 90 минут. Реакционную смесь концентрировали при пониженном давлении. Хлороформ (250 мл) и воду (80 мл) вносили в полученный остаток и органический слой отделяли. Водный слой повторно экстрагировали хлороформом (50 мл). Объединенные органические слои последовательно промывали насыщенным водным раствором хлорида аммония (60 мл × 3) и соляным раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат пропускали через подушку из силикагеля (NH-силикагель). Фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение (37,2 г).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 1,98 (brs, 6H), 2,24-2,60 (m, 2H), 3,90-4,20 (m, 3H), 3,99 (s, 3H), 4,23 (dd, J=16,0, 8,0 Гц, 1H), 4,62-4,71 (m, 1H), 5,32 (brs, 2H), 7,05 (brd, J=10,0 Гц, 1H), 7,10 (dd, J=7,6, 1,2 Гц, 1H), 7,42-7,56 (m, 1H), 7,94 (brs, 1H), 8,03 (s, 1H).
ESI-MS масса/заряд 411 [M+H]+
(14) Синтез (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Порошковый гидроксид натрия (9,43 г) добавляли за один раз к раствору 5-[2-фтор-4-(2-метокси-3,5-диметилпиридин-4-ил)фенил]-1-[(S)-тетрагидрофуран-3-ил]-1H-пиразол-4-карбоксамида (37,2 г) в DMSO (186 мл) при комнатной температуре. Реакционную смесь перемешивали при той же температуре в течение 50 минут, а затем - при 70°C в течение 45 минут. При охлаждении водой к реакционной смеси по каплям добавляли воду (600 мл), а затем по каплям добавляли уксусную кислоту (13,5 мл). Выпавший порошкообразный осадок собирали фильтрованием. Собранное целевое вещество промывали водой и МТВЕ, а затем сушили при пониженном давлении, получая указанное в заголовке соединение (34,0 г).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 1,92-1,94 (m, 3H), 1,94-1,96 (m, 3H), 2,55-2,66 (m, 1H), 2,76-2,86 (m, 1H), 4,00 (s, 3H), 4,09-4,16 (m, 1H), 4,24-4,37 (m, 2H), 4,39-4,45 (m, 1H), 5,61-5,68 (m, 1H), 7,04 (d, J=1,5 Гц, 1H), 7,08 (dd, J=1,5 Гц, 8,3Гц, 1H), 7,94 (s, 1H), 8,13 (d, J=8,3 Гц, 1H), 8,31 (s, 1H), 8,86 (s, 1H).
ESI-MS масса/заряд 391 [M+H]+
Указанное в заголовке соединение продемонстрировало оптическое вращение (-)-типа и его оптическая чистота составляла ≥99% э.и. [AD-H, 100% этанол, время удерживания: 9,7 мин].
Сравнительный пример 2
Синтез (-)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Синтез (±)-5-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
(1) Синтез этил 3-(4-бром-2-хлорфенил)-3-оксопропионата
4-Бром-2-хлорбензойную кислоту (1 г) суспендировали в DCM (10 мл). К полученной суспензии добавляли CDI (960 мг) и перемешивали при комнатной температуре в течение 4 часов. Этот раствор далее именуется "Раствор 1". Этилмалонат калия (1,1 г) суспендировали в ацетонитриле (20 мл) в другой колбе в атмосфере азота и добавляли TEA (1,5 мл). Полученный раствор охлаждали до 0°C и маленькими порциями добавляли хлорид магния (805 мг), а затем перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь охлаждали до 0°C и в нее по каплям прибавляли полученный ранее "Раствор 1". После окончания прикапывания реакционную смесь перемешивали при комнатной температуре в течение 17 часов. Реакционную смесь дополнительно перемешивали при 50°C в течение 9 часов. Реакционную смесь концентрировали при пониженном давлении и удаляли DCM. Полученный остаток охлаждали до 0°C и добавляли этилацетат (50 мл) и 2 н. хлористоводородную кислоту (20 мл) и перемешивали при комнатной температуре в течение 1 часа. Получившийся органический слой отделяли. Получившийся водный слой экстрагировали этилацетатом. Экстракт объединяли с органическим слоем и сушили безводным сульфатом магния. Сиккатив удаляли фильтрацией и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, от 0% до 10%), получая указанное в заголовке соединение (1,2 г).
ESI-MS масса/заряд 307 [M + H] +
(2) Синтез (±)-этил 5-(4-бром-2-хлорфенил)-1-(тетрагидрофуран-3-ил)-1H-пиразол-4-карбоксилата
Этил 3-(4-бром-2-хлорфенил)-3-оксопропаноат (2,00 г) растворяли в DMF-DMA (6,96 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1,5 часа. Реакционную смесь концентрировали при пониженном давлении и остаток растворяли в этаноле (40 мл). К раствору добавляли (±)-(тетрагидрофуран-3-ил)гидразина гидрохлорида (998 мг) и смесь нагревали с обратным холодильником в течение двух часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток экстрагировали этилацетатом и органический слой очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, от 10% до 30%), получая указанное в заголовке соединение (1,05 г).
ESI-MS масса/заряд 401 [M+H]+
(3) Синтез (±)-5-(4-бром-2-хлорфенил)-1-(тетрагидрофуран-3-ил)-1H-пиразол-4-карбоновой кислоты
Смесь (±)-этил 5-(4-бром-2-хлорфенил)-1-(тетрагидрофуран-3-ил)-1H-пиразол-4-карбоксилата (1,05 г) и 5 н. водного раствора гидроксида натрия (1,58 мл) перемешивали в смешанном растворителе из этанола (20 мл) и воды (5 мл) при 60°C в течение трех часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. К остатку добавляли 5 н. соляную кислоту, после чего экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния, а сиккатив отфильтровывали. Фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение (1 г).
ESI-MS масса/заряд 371 [M+H]+
(4) Синтез (±)-5-(4-бром-2-хлорфенил)-N-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-1H-пиразол-4-карбоксамида
(±)-5-(4-бром-2-хлорфенил)-1-(тетрагидрофуран-3-ил)-1H-пиразол-4-карбоновую кислоту (1 г) растворяли в DCM (20 мл) и добавляли CDI (611 мг), после чего перемешивали при комнатной температуре в течение 1 часа. В реакционную смесь добавляли 2,4-диметоксибензиламин (0,809 мл) и смесь перемешивали при комнатной температуре в течение двух часов. Насыщенный водный раствор натрия бикарбоната добавляли в реакционную смесь, а затем экстрагировали, используя DCM. Органический слой концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, от 10% до 40%), получая указанное в заголовке соединение (1,26 г).
ESI-MS масса/заряд 522 [M+H]+
(5) Синтез (±)-7-бром-5-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
(±)-5-(4-бром-2-хлорфенил)-N-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-1H-пиразол-4-карбоксамид (1,26 г) растворяли в THF (25 мл) и добавляли KTB (597 мг) при 0°C. Смесь перемешивали в течение 12 часов, при этом постепенно нагревали до комнатной температуры. Реакционную смесь охлаждали до 0°C и добавляли воду, после чего фильтровали. Остаток от фильтрации хранили отдельно. Фильтрат экстрагировали этилацетатом и органический слой концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат/н-гептан, от 10% до 70%). Полученную фракцию и остаток от фильтрации, полученный ранее, объединяли и концентрировали, получая указанное в заголовке соединение (488 мг).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 2,50-2,62 (m, 1H), 2,72-2,82 (m, 1H), 3,76 (s, 3H), 4,02 (s, 3H), 4,07-4,15 (m, 1H), 4,19-4,32 (m, 2H), 4,35-4,42 (m, 1H), 5,46-5,57 (m, 3H), 6,34 (dd, J=8,6 Гц, 2,2 Гц, 1H), 6,52 (d, J=2,2 Гц, 1H), 6,99 (d, J=8,6 Гц, 1H), 7,38 (dd, J=8,6 Гц, 1,8 Гц, 1H), 7,82 (d, J=1,8 Гц, 1H), 7,89 (d, J=8,6 Гц, 1H), 8,32 (s, 1H).
ESI-MS масса/заряд 506 [M + Na]+
(6) Синтез (±)-5-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Реакцию смеси (±)-7-бром-5-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она (300 мг), бис(пинаколат)дибора (204 мг), комплекса Pd(dppf)Cl2-DCM (13,6 мг) и ацетата калия (182 мг) проводили в смешанном растворителе из 1,4-диоксана (15 мл) и DMSO (1 мл) при 130°C в течение трех часов, используя микроволновой реактор. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток экстрагировали этилацетатом и органический слой концентрировали при пониженном давлении. Остаток наносили на подушку из силикагеля и элюировали этилацетатом, получая указанное в заголовке соединение (428 мг) в виде сырого продукта.
ESI-MS масса/заряд 532 [M+H]+
(7) Синтез 3,5-дибром-2-метоксипиридин-4-амина
Смесь 2-метоксипиридин-4-иламина (15 г) и NBS (47,3 г) перемешивали в растворителе-уксусной кислоте (150 мл) при комнатной температуре в течение трех часов. Реакционную смесь концентрировали при пониженном давлении и к остатку прибавляли 5 н. водный раствор гидроксида натрия (200 мл) при 0°С, после чего экстрагировали диэтиловым эфиром. Органический слой очищали непосредственно на подушке из силикагеля (этилацетат/н-гептан, 10%), получая указанное в заголовке соединение (32,4 г).
ESI-MS масса/заряд 283 [M+H]+
(8) Синтез 2-метокси-3,5-диметилпиридин-4-амина
Смесь 3,5-дибром-2-метоксипиридин-4-амина (16 г), триметилбороксина (19,8 мл), комплекса Pd(dppf)Cl2-DCM (4,15 г) и карбоната калия (23,5 г) нагревали с обратным холодильником в смешанном растворителе из 1,4-диоксана (320 мл) и воды (32 мл) в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. К остатку добавляли этилацетат и воду, а затем фильтровали через Celite™. Фильтрат экстрагировали этилацетатом и органический слой наносили на подушку из силикагеля (NH-силикагеля) и элюировали этилацетатом. В полученный раствор вносили NH-силикагель (30 г) и смесь концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на NH-силикагеле (этилацетат/н-гептан, от 0% до 30%), получая указанное в заголовке соединение (4,43 г).
ESI-MS масса/заряд 153 [M+H]+
(9) Синтез 4-бром-2-метокси-3,5-диметилпиридина
Смесь бромида меди(I) (12,1 г) и т-бутилнитрила (7,07 мл) перемешивали в растворителе-ацетонитриле (80 мл) при 70°C в течение 10 минут. К реакционной смеси по каплям добавляли раствор 2-метокси-3,5-диметилпиридин-4-амина (3,9 г) в ацетонитриле (40 мл) при той же температуре и смесь перемешивали при 70°C один час. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. К остатку добавляли этилацетат и насыщенный водный раствор натрия бикарбоната и смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь фильтровали через Celite™ и фильтрат экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на NH-силикагеле (н-гептан, 100%, затем на подушке из NH-силикагеля, н-гептан, 100%), получая указанное в заголовке соединение (4,3 г).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 2,28-2,29 (m, 3H), 2,29-2,31 (m, 3H), 3,93 (s, 3H), 7,77-7,84 (m, 1H).
ESI-MS масса/заряд 216 [M+H]+
(10) Синтез (±)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
Реакцию смеси (±)-5-(2,4-диметоксибензил)-1-(тетрагидрофуран-3-ил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она (219 мг), 4-бром-2-метокси-3,5-диметилпиридина (134 мг), Pd(PPh3)4 (23,8 мг) и карбоната цезия (403 мг) проводили в смешанном растворителе из 1,4-диоксана (8 мл) и воды (2 мл) при 130°C в течение 70 минут, используя микроволновой реактор. Реакционную смесь охлаждали до комнатной температуры и затем очищали, непосредственно используя колоночную хроматографию на силикагеле (этилацетат/н-гептан, от 10% до 90%). Полученный продукт присоединения растворяли в TFA (4 мл) и смесь перемешивали при 70°C в течение двух часов. Реакционную смесь охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Насыщенный водный раствор натрия бикарбоната добавляли к остатку, а затем экстрагировали, используя этилацетат. Органический слой концентрировали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле (DCM, 100%, затем - этилацетат/н-гептан, от 50% до 100%), получая указанное в заголовке соединение (78 мг).
ESI-MS масса/заряд 391 [M+H]+
(11) Синтез (+)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она и (-)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
(±)-7-(2-Метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он анализировали, используя хиральную колоночную хроматографию [колонка с хиральной фазой производства компании DAICEL Corp., AD-H (0,46 см Φ x 15 см), подвижная фаза: 100% этанол], при этом обнаружены (+)-форма при 7,8 мин и (-)-форма при 9,7 мин, что подтвердило возможность разделения оптических изомеров. (±)-7-(2-Метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она (78 мг) растворяли в смешанном растворителе из этанола (12 мл) и метанола (12 мл) и раствор фильтровали через ватный тампон. Фильтрат разделяли на оптические изомеры с помощью хиральной колоночной хроматографии [колонка с хиральной фазой: колонка AD-H, растворитель для элюирования: 100% этанол, расход: 10 мл/мин, время элюирования: 80 минут/элюирование, впрыск: 2 мл/впрыск, короткое время удерживания: (+)-форма, длительное время удерживания: (-)-форма], при этом получали 26,4 мг (+)-формы и 25,2 мг (-)-формы указанного в заголовке соединения.
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 1,92-1,94 (m, 3H), 1,94-1,96 (m, 3H), 2,55-2,66 (m, 1H), 2,76-2,86 (m, 1H), 4,00 (s, 3H), 4,09-4,16 (m, 1H), 4,24-4,37 (m, 2H), 4,39-4,45 (m, 1H), 5,61-5,68 (m, 1H), 7,04 (d, J=1,5 Гц, 1H), 7,08 (dd, J=1,5 Гц, 8,3 Гц, 1H), 7,94 (s, 1H), 8,13 (d, J=8,3 Гц, 1H), 8,31 (s, 1H), 8,86 (s, 1H).
ESI-MS масса/заряд 391 [M+H]+
(S)-7-(2-Метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-он, синтезированный согласно описанному выше Примеру сравнения 1, использовали в синтезе следующих солей.
Пример 1
Синтез мономалеата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
К (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-ону (576,32 мг) добавляли малеиновую кислоту (243,97 мг) и метил-т-бутиловый эфир (6 мл) и суспензию перемешивали при комнатной температуре в течение 2 дней. Твердое вещество отделяли фильтрованием и сушили при комнатной температуре при пониженном давлении, таким образом получая указанное в заголовке соединение (713,04 мг) в виде белого твердого вещества.
1H-ЯМР (600 МГц, DMSO-d6) δ (ppm): 1,87 (s, 3H), 1,91 (s, 3H), 2,52-2,56 (m, 2H), 3,89 (s, 3H), 3,93 (ddd, J=8, 7, 6 Гц, 1H), 4,03 (dddd, J=8, 8, 7, 2 Гц, 1H), 4,16 (ddd, J=9, 5, 3 Гц, 1H), 4,21 (dd, J=9, 6 Гц, 1H), 5,85-5,89 (m, 1H), 6,25 (s, 2H), 7,09 (dd, J=8, 1 Гц, 1H), 7,21 (d, J=1 Гц, 1H), 7,96 (s, 1H), 8,18 (s, 1H), 8,35 (d, J=8 Гц, 1H), 11,51 (s, 1H).
13C-ЯМР (100 МГц, твердое тело) δ (м.д.): 13,3, 16,1, 16,7, 29,5, 35,9, 57,2, 58,0, 61,9, 67,4, 69,7, 74,6, 111,8, 114,3, 122,5, 123,2, 125,7, 126,9, 127,9, 132,7, 133,8, 136,0, 138,9, 154,8, 156,2, 157,8, 158,9, 162,0, 163,4, 164,7, 172,0
Углы при анализе методом рентгеновской порошковой дифракции (2θ±0,2°): 9,1°, 10,1°, 11,1°, 16,2°, 17,6°, 18,2°, 22,0°, 22,4°, 23,8°, 25,8°.
Полученная методом рентгеновской порошковой дифракции дифракционная картина кристалла мономалеата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, полученного описанным выше способом, приведена на Фигуре 1.
Пример 2
Синтез монобензолсульфоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
К (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-ону (991,7 мг) добавляли 2-бутанон (10 мл) и моногидрат бензолсульфокислоты (708,0 мг) и суспензию перемешивали при комнатной температуре в течение 2 дней. Твердое вещество отделяли фильтрованием и сушили при комнатной температуре при пониженном давлении, таким образом получая указанное в заголовке соединение (1393,9 мг) в виде белого твердого вещества.
1H-ЯМР (600 МГц, DMSO-d6) δ (ppm): 1,87 (s, 3H), 1,91 (s, 3H), 2,52-2,56 (m, 2H), 3,89 (s, 3H), 3,93 (ddd, J=8, 7, 6 Гц, 1H), 4,01-4,05 (m, 1H), 4,16 (ddd, J=9, 5, 3 Гц, 1H), 4,21 (dd, J=9, 6 Гц, 1H), 5,85-5,89 (m, 1H), 7,09 (dd, J=8, 1 Гц, 1H), 7,22 (d, J=1 Гц, 1H), 7,32-7,26 (m, 3H), 7,59-7,57 (m, 2H), 7,96 (s, 1H), 8,18 (s, 1H), 8,35 (d, J=8 Гц, 1H), 11,51 (s, 1H).
13C-ЯМР (100 МГц, твердое тело) δ (м.д.): 12,8, 16,8, 31,3, 35,2, 59,1, 61,0, 61,6, 62,0, 67,9, 70,1, 70,6, 74,7, 111,6, 114,0, 117,5, 122,7, 125,4, 126,8, 128,8, 130,1, 137,7, 139,2, 146,4, 157,8, 159,6, 160,7
Углы при анализе методом рентгеновской порошковой дифракции (2θ±0,2°): 6,6°, 9,9°, 13,7°, 14,6°, 19,0°, 19,6°, 20,5°, 21,7°, 22,7°, 23,5°, 25,7°.
Полученная методом рентгеновской порошковой дифракции дифракционная картина кристалла монобензолсульфоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, полученного описанным выше способом, приведена на Фигуре 2.
Пример 3
Синтез (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она гидрохлорида
К (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-ону (984,6 мг) добавляли ацетон (20 мл) и 5 н. хлористоводородную кислоту (620 μл) и суспензию перемешивали при комнатной температуре в течение 2 дней. Твердое вещество отделяли фильтрованием и сушили при комнатной температуре при пониженном давлении, таким образом получая указанное в заголовке соединение (1100,21 мг) в виде белого твердого вещества.
Углы при анализе методом рентгеновской порошковой дифракции (2θ±0,2°): 11,2°, 12,4°, 12,7°, 17,1°, 23,5°, 26,5°, 29,4°.
Полученная методом рентгеновской порошковой дифракции дифракционная картина кристалла (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она гидрохлорида, полученного описанным выше способом, приведена на Фигуре 3.
Пример 4
Синтез (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она гидробромида
К (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-ону (614,45 мг) добавляли ацетон (6 мл) и 47%-ную бромистоводородную кислоту (220 μл) и суспензию перемешивали при комнатной температуре в течение ночи. Твердое вещество отделяли фильтрованием и сушили при комнатной температуре при пониженном давлении, таким образом получая указанное в заголовке соединение (719,23 мг) в виде белого твердого вещества.
Углы при анализе методом рентгеновской порошковой дифракции (2θ±0,2°): 5,6°, 11,1°, 12,3°, 18,5°, 19,3°, 22,9°, 23,4°, 26,3°, 29,2°.
Полученная методом рентгеновской порошковой дифракции дифракционная картина кристалла (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она гидробромида, полученного описанным выше способом, приведена на Фигуре 4.
Пример 5
Синтез п-толуолсульфоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
К (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-ону (100,6 мг) добавляли 2-бутанон (4 мл) и п-толуолсульфокислоту (65,1 мг) и суспензию перемешивали при комнатной температуре в течение ночи. Твердое вещество отделяли фильтрованием и сушили при комнатной температуре при пониженном давлении, таким образом получая указанное в заголовке соединение (153,15 мг) в виде белого твердого вещества.
Углы при анализе методом рентгеновской порошковой дифракции (2θ±0,2°): 6,5°, 9,8°, 13,9°, 14,4°, 15,3°, 18,5°, 19,3°, 20,3°, 22,8°, 23,3°, 25,4°, 28,2°.
Полученная методом рентгеновской порошковой дифракции дифракционная картина кристалла п-толуолсульфоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, полученного описанным выше способом, приведена на Фигуре 5.
Пример 6
Синтез нитрата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
К (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-ону (22,80 мг) добавляли этилацетат (300 μл) и 60%-ную азотную кислоту (5,3 μл) и суспензию перемешивали при комнатной температуре в течение ночи. Твердое вещество отделяли фильтрованием и сушили при комнатной температуре при пониженном давлении, таким образом получая указанное в заголовке соединение (14,97 мг) в виде белого твердого вещества.
Углы при анализе методом рентгеновской порошковой дифракции (2θ±0,2°): 11,1°, 11,7°, 14,8°, 15,3°, 16,4°, 19,2°, 23,6°, 24,2°, 25,8°.
Полученная методом рентгеновской порошковой дифракции дифракционная картина кристалла нитрата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, полученного описанным выше способом, приведена на Фигуре 6.
Пример 7
Синтез сульфата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
К (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-ону (23,53 мг) добавляли 2-бутанон (300 μл) и 95%-ную серную кислоту (4 μл) и суспензию перемешивали при комнатной температуре в течение ночи. Твердое вещество отделяли фильтрованием и сушили при комнатной температуре при пониженном давлении, таким образом получая указанное в заголовке соединение (27,34 мг) в виде белого твердого вещества.
Углы при анализе методом рентгеновской порошковой дифракции (2θ±0,2°): 10,7°, 14,0°, 14,5°, 16,2°, 19,1°, 20,0°, 22,8°, 23,6°, 25,3°.
Полученная методом рентгеновской порошковой дифракции дифракционная картина кристалла сульфата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, полученного описанным выше способом, приведена на Фигуре 7.
Пример 8
Синтез метансульфоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
К (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-ону (22,52 мг) добавляли 2-бутанон (300 μл) и метансульфоновую кислоту (4,5 μл) и суспензию перемешивали при комнатной температуре в течение ночи. Твердое вещество отделяли фильтрованием и сушили при комнатной температуре при пониженном давлении, таким образом получая указанное в заголовке соединение (28,69 мг) в виде белого твердого вещества.
Углы при анализе методом рентгеновской порошковой дифракции (2θ±0,2°): 12,7°, 14,8°, 17,8°, 18,7°, 23,4°, 29,8°.
Полученная методом рентгеновской порошковой дифракции дифракционная картина кристалла метансульфоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, полученного описанным выше способом, приведена на Фигуре 8.
Пример 9
Синтез фосфата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
К (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-ону (22,29 мг) добавляли 2-бутанон (300 μл) и фосфорную кислоту (4,6 μл) и суспензию перемешивали при комнатной температуре в течение ночи. После введения дополнительного количества гексана (200 μл) и перемешивания твердое вещество отделяли фильтрованием и сушили при комнатной температуре при пониженном давлении, таким образом получая указанное в заголовке соединение (23,09 мг) в виде белого твердого вещества.
Углы при анализе методом рентгеновской порошковой дифракции (2θ±0,2°): 9,5°, 11,3°, 15,2°, 16,7°, 18,4°, 23,5°, 24,0°.
Полученная методом рентгеновской порошковой дифракции дифракционная картина кристалла фосфата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, полученного описанным выше способом, приведена на Фигуре 9.
Пример 10
Синтез L-тартрата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
К (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-ону (27,42 мг) добавляли ацетон (300 μл) и L-винную кислоту (20,18 мг) и суспензию перемешивали при комнатной температуре в течение 3 дней. Твердое вещество отделяли фильтрованием и сушили при комнатной температуре при пониженном давлении, таким образом получая указанное в заголовке соединение (34,89 мг) в виде белого твердого вещества.
Углы при анализе методом рентгеновской порошковой дифракции (2θ±0,2°): 10,1°, 14,1°, 16,7°°, 17,4°, 18,2°, 20,6°, 23,4°, 24,0°, 24,3°, 26,5°.
Полученная методом рентгеновской порошковой дифракции дифракционная картина кристалла L-тартрата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, полученного описанным выше способом, приведена на Фигуре 10.
Пример 11
Синтез малоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
К (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-ону (27,70 мг) добавляли ацетон (300 μл) и малоновую кислоту (26,65 мг) и суспензию перемешивали при комнатной температуре в течение 3 дней. Твердое вещество отделяли фильтрованием и сушили при комнатной температуре при пониженном давлении, таким образом получая указанное в заголовке соединение (30,49 мг) в виде белого твердого вещества.
Углы при анализе методом рентгеновской порошковой дифракции (2θ±0,2°): 10,5°, 16,8°, 17,4°, 17,8°, 18,3°, 18,9°, 21,7°, 22,8°, 24,2°, 25,2°, 26,4°.
Полученная методом рентгеновской порошковой дифракции дифракционная картина кристалла малоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, полученного описанным выше способом, приведена на Фигуре 11.
Пример 12
Синтез мономалеата (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она
К (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-ону (500 мг) добавляли этилацетат (2 мл) и смесь перемешивали при 25°С в течение 20 минут. Раствор малеиновой кислоты (223,0 мг), полностью растворенной в этилацетате (7,5 мл), вносили в ранее полученную суспензию и смесь перемешивали при 25°С в течение 7 дней. Твердое вещество отделяли фильтрованием, кристаллы промывали этилацетатом (1 мл) и сушили при 40°С при пониженном давлении, таким образом получая указанное в заголовке соединение (366,9 мг) в виде белого твердого вещества.
Полученное соединение, указанное в заголовке, продемонстрировало такие же дифракционные пики, полученные методом порошковой рентгеновской дифракции, как и в Примере 1.
[Примеры испытаний]
Пример исследования ингибирующего действия в отношении PDE9
1) Получение человеческого рекомбинантного белка PDE9
Фрагмент hsPDE9A 1cDNA амплифицировали, основываясь на базовой последовательности (Инвентарный номер: AF048837) hsPDE9A1, зарегистрированной в базе данных GenBank, и используя следующие последовательности (Hokkaido System Science Co., Ltd.) в качестве праймеров и библиотеку cDNA человеческого гиппокампа (Clontech Laboratories, Inc.) в качестве матричной ДНК, и используя полимеразу ДНК Pfu50 (Invitrogen Corp.), и путем применения полимеразной цепной реакции (ПЦР) при следующем условии.
Праймер hPDE9-1: AGGATGGGATCCGGCTCCTCCA (SEQ №1)
Праймер hPDE9A-3: CAGGCACAGTCTCCTTCACTG (SEQ №2)
Условия ПЦР: [96°C, 5 мин] × 1 цикл, [(96°C, 10 сек), (57°C, 5 сек), (72°C, 2 мин)] × 30 циклов
Полученный фрагмент hsPDE9A 1cDNA вставляли в клонирующий вектор TOPO-TA (Invitrogen Corp.) и проверяли базовую последовательность; а затем полученное трансфицировали в вектор pcDNA 3.1/myc His-tag (Invitrogen Corp.), таким образом получая вектор для экспрессии человеческого PDE9 в клетках млекопитающих. Вектор для экспрессии человеческого PDE9 в клетках млекопитающих трансфицировали, используя кратковременную экспрессию в клетке HEK293, применяя реагент LIPOFETAMINE 2000 Reagent (Gibco). С помощью метода Вестерн-блоттинга подтвердили, что PDE9A, экспрессированный в клетке HEK293, а затем и фрагмент человеческого PDE9A 1cDNA, были трансфицированы в вектор pYNG (Katakura Industries Co., Ltd.), таким образом изготовляя вектор для экспрессии в клетках насекомого. Надосадочную жидкость гомогенизированного шелкопряда, в котором экспрессировали большое количество PDE9, очищали в Ni колонке, уравновешенной буфером А (20 ммоль/л Tris-HCl, pH: 8,0, 1 ммоль/л DTT, 10 ммоль/л имидазола). Через 1 час после начала перемешивания надосадочной жидкости в Ni колонке, проводили очистку, используя буфер B (20 ммоль/л Tris-HCl, pH: 8,0, 1 ммоль/л DTT) и элюирование проводили, используя буфер C (20 ммоль/л Tris-HCl, pH: 8,0, 1 ммоль/л DTT, 100 ммоль/л имидазола). Полученную элюированием фракцию собирали препаративно, таким образом получая раствор фермента PDE9.
2) Измерение ингибирующей активности PDE9
К раствору 100 μL буфера D (40 ммоль/л Tris-HCl, pH: 7,4, 10 ммоль/л MgCl2, 1 мМ DTT, 2 μM cGMP), содержащего [3H]-cGMP (0,5 μКи/мл), при охлаждении льдом добавляли 10 μл раствора исследуемого соединения (раствор, где соединение растворяли в DMSO и разбавляли таким образом, чтобы концентрация DMSO становилась 5%) и 90 μл раствора, приготовленного разведением раствора фермента PDE9, полученного в вышеуказанном с использованием буфера E (40 ммоль/л Tris-HCl, pH: 7,4, 10 ммоль/л MgCl2, 1 мМ DTT, 1 ммоль/л EGTA). Полученный смешанный раствор инкубировали при 30°C в течение 10 мин, а затем нагревали в течение 2 мин в кипящей воде, чтобы прекратить ферментативную реакцию, вызываемую PDE9. Затем, полученное доводили до комнатной температуры; к этому добавляли 50 μл 5'-Нуклеотидазы (Biomol GmbH, 10 единиц/мл); и полученное инкубировали при 30°C в течение 10 мин, таким образом превращая [3H]-5'-GMP, полученный в предыдущей реакции, в [3H]-гуанозин. 500 μл анионообменной смолы (Bio-Rad AG1-X2 resin, размер, меш: 200-400, H2O : смола = 2 : 1) вносили в полученную реакционную жидкость и оставляли на 10 мин, а затем центрифугировали (2000 об/мин, 10 мин); надосадочную жидкость, содержащую [3H]-гуанозин, переносили на планшет LumaPlate (PerkinElmer, Inc.) и измеряли уровень радиоактивности, используя микропланшетный сцинтилляционный и люминесцентный счетчик TopCount NXT (PerkinElmer, Inc.).
Процентную доля ингибирования исследуемым соединением рассчитывали, используя следующее выражение, принимая уровень радиоактивности контрольного образца, не содержащего исследуемое соединение, за (А), уровень радиоактивности пустого образца, не содержащего фермент, за (В), и уровень радиоактивности исследуемого соединения за (С).
Процентная доля ингибирования =100-{[(C)-(B)]/[(A)-(B)]}×100(%)
Значение IC50 для PDE9 исследуемого соединения определяли исходя из процентной доли ингибирования при различных концентрациях. Значение IC50 соединения (I), синтезированного согласно Примеру сравнения 2, относительно PDE9 составляло 0,00943 μM.
Действие на cGMP спинномозговой жидкости грызунов
Соединение вводили самцам мыши ICR (CHARLES RIVER LABORATORIES JAPAN, INC.), самцам крысы Спрег-Доули (SD) (CHARLES RIVER LABORATORIES JAPAN, INC.) или самцам крысы Лонг-Эванс (LE) (Institute for Animal Reproduction (General Incorporated Foundation)), а затем брали образцы спинномозговой жидкости под анестезией пентобарбиталом и хранили при -20°C. Измерение уровней cGMP в спинномозговой жидкости выполняли согласно процедуре ацетилирования EIA в наборе cGMP EIA (GE Healthcare) процедуре нон-ацетилирования в наборе cGMP EIA (Cayman Chemical). Результаты рассчитывали, используя следующую формулу, как увеличение (С) содержания cGMP в группе (В), где вводили исследуемое соединение, относительно содержания cGMP в группе (А), где вводили носитель (пустой образец).
Увеличение cGMP (C)=[(B)-(A)]/(A)×100(%)
В случае соединения (I), синтезированного согласно Примеру сравнения 1, увеличение cGMP составляло 274% через 1 час после введения дозы, равной 10 мг/кг крысам LE.
Действие на cGMP гиппокампа грызунов
Исследуемое соединение вводили самцам крысы SD (CHARLES RIVER LABORATORIES JAPAN, INC.) или самцам крысы LE (Institute for Animal Reproduction (foundation)). Затем проводили микроволновую обработку под анестезией пентобарбиталом и удаляли гиппокамп, сырой вес которого измеряли, затем замораживали в жидком азоте и хранили при -80°C. При измерении содержания cGMP в гиппокампе добавляли раствор 0,5 M хлорную кислоту/1 мМ EDTA таким образом, чтобы сырой вес составлял 5% (масс./об.), и смесь гомогенизировали. После гомогенизации, гомогенизированный продукт центрифугировали (10000 об/мин, 15 мин) и отбирали надосадочную жидкость. Отобранную надосадочную жидкость нейтрализовали 2 М раствором гидрокарбоната калия и центрифугировали (13000 об/мин, 10 мин). Концентрацию cGMP в надосадочной жидкости измеряли согласно процедуре нон-ацетилирования EIA набора cGMP EIA (GE Healthcare). Результаты рассчитывали, используя следующую формулу, как увеличение (С) содержания cGMP в группе (В), где вводили исследуемое соединение, относительно содержания cGMP в группе (А), где вводили носитель (пустой образец).
Увеличение cGMP (C)=[(B)-(A)]/(A)×100(%)
В случае соединения (I), синтезированного согласно Примеру сравнения 1, увеличение cGMP составляло 58% через 1 час после введения дозы, равной 10 мг/кг крысам LE.
Испытание растворимости
Каждое из соединения (I) (50 мг), соединения по Примеру 1 (30 мг) и соединения по Примеру 2 (30 мг) вносили в капсулу из гидроксипропилцеллюлозы вместе с таким же массовым количеством гидрата лактозы. 708-DS, поставленный компанией Agilent Technologies, оборудовали небольшими перемешивающими лопатками и малыми емкостями, и использовали в качестве аппарата для измерения растворимости. По одной капсуле, заполненной лекарственным средством, добавляли к 50 мл искусственного кишечного сока натощак (фосфатный буфер с рН 6,5, с добавлением 0,75 мМ лецитина и 3 мМ таурохолата натрия), который нагревали до 37°С. Лекарственное средство растворяли путем вращения перемешивающей лопатки со скоростью 50 об/мин. Из получаемого раствора отбирали пробы в ходе эксперимента и измеряли концентрации, используя ВЭЖХ. Такие испытания растворимости с использованием имитации кишечного сока натощак часто используют для оценки характеристик растворимости и перорального всасывания лекарственных средств (например, Takano et al., "Oral absorption of poorly water-soluble drugs: computer simulation of fraction absorbed in humans from a miniscale dissolution test", Pharm Res., vol. 23, pp. 1144-1156, 2006.). Результаты приведены в Таблице 1 и на графике на Фигуре 12. Концентрация соединения из Примера 2, которая достигается при его использовании, продемонстрировала значения примерно в два раза выше, а концентрация соединения из Примера 1, которая достигается при его использовании, продемонстрировала значения примерно в 4-5 раз выше относительно достигаемой концентрации соединения (I) пои его использовании.
Исследование характеристик перорального всасывания у собак
Каждое из соединения (I) (100 мг) и соединения по Примеру 1 (130 мг: мономалеат, 100 мг в пересчете на несвязанное вещество) вносили в капсулу из гидроксипропилцеллюлозы, чтобы получить образец для введения. Полученные капсулы вводили вместе с небольшим количеством воды четырем собакам бигль, и образцы крови отбирали в моменты времени, равные 0,5, 1, 2, 4, 6, 8 и 24 часам после введения. Концентрацию лекарственного средства в плазме, полученной центрифугированием, измеряли, используя ЖХ-МС/МС. Соединение (I) и соединение по Примеру 1 вводили каждому индивидууму перекрестным образом с интервалом в одну неделю (период вымывания) и сравнивали изменения в концентрации лекарственного средства в плазме. Результаты приведены в Таблице 2 и на графике на Фигуре 13. BQL означает "ниже количественно определяемого предела", и NC - не вычисляли. Величины площади под кривой концентрация лекарственного средства в плазме-время (AUC) приведены в Таблице 3. Средняя величина AUC, полученная при введении соединения (I), составляла 1,88±0.95 μг⋅ч/мл, в то время как средняя величина AUC, полученная при введении соединения по Примеру 1, составляла 4,00±0,45 μг⋅ч/мл: скорость всасывания была выше и отклонения в величинах были меньше у соединения по Примеру 1.
Таким образом, соли/кристаллы согласно настоящему изобретению продемонстрировали предпочтительные характеристики растворимости и перорального всасывания при использовании в качестве исходных материалов для фармацевтических препаратов.
(ЧАСЫ)
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРАЗОЛОХИНОЛИНОВОЕ ПРОИЗВОДНОЕ | 2012 |
|
RU2605096C2 |
| ПИРИДИНИЛПИРАЗОЛОХИНОЛИНОВЫЕ СОЕДИНЕНИЯ | 2014 |
|
RU2655172C2 |
| ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ДЕМЕНЦИИ, ПРЕДСТАВЛЯЮЩЕЕ СОБОЙ КОМБИНАЦИЮ ПРОИЗВОДНОГО ПИРАЗОЛОХИНОЛИНА И МЕМАНТИНА | 2018 |
|
RU2784428C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 7Н-ПИРРОЛО[2,3-d]ПИРИМИДИНА И ИХ СОКРИСТАЛЛОВ | 2017 |
|
RU2779212C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРА ЯНУС-КИНАЗЫ | 2017 |
|
RU2838992C2 |
| СОЛЬ ИНГИБИТОРА SYK И ЕЕ КРИСТАЛЛИЧЕСКАЯ ФОРМА | 2019 |
|
RU2818103C2 |
| СОЛИ ПРОИЗВОДНОГО ИНДАЗОЛА И ИХ КРИСТАЛЛЫ | 2017 |
|
RU2747399C2 |
| СОЛЬ ПРОИЗВОДНОГО ПИРРОЛИДИН-3-ИЛ-УКСУСНОЙ КИСЛОТЫ И ЕЕ КРИСТАЛЛЫ | 2014 |
|
RU2640047C2 |
| СОДЕРЖАЩАЯ ПРОИЗВОДНОЕ С ТРЕМЯ КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ СОЛЬ ИЛИ КРИСТАЛЛИЧЕСКАЯ ФОРМА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2020 |
|
RU2835105C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТИАЗОЛЬНОГО ПРОИЗВОДНОГО | 2016 |
|
RU2738937C2 |

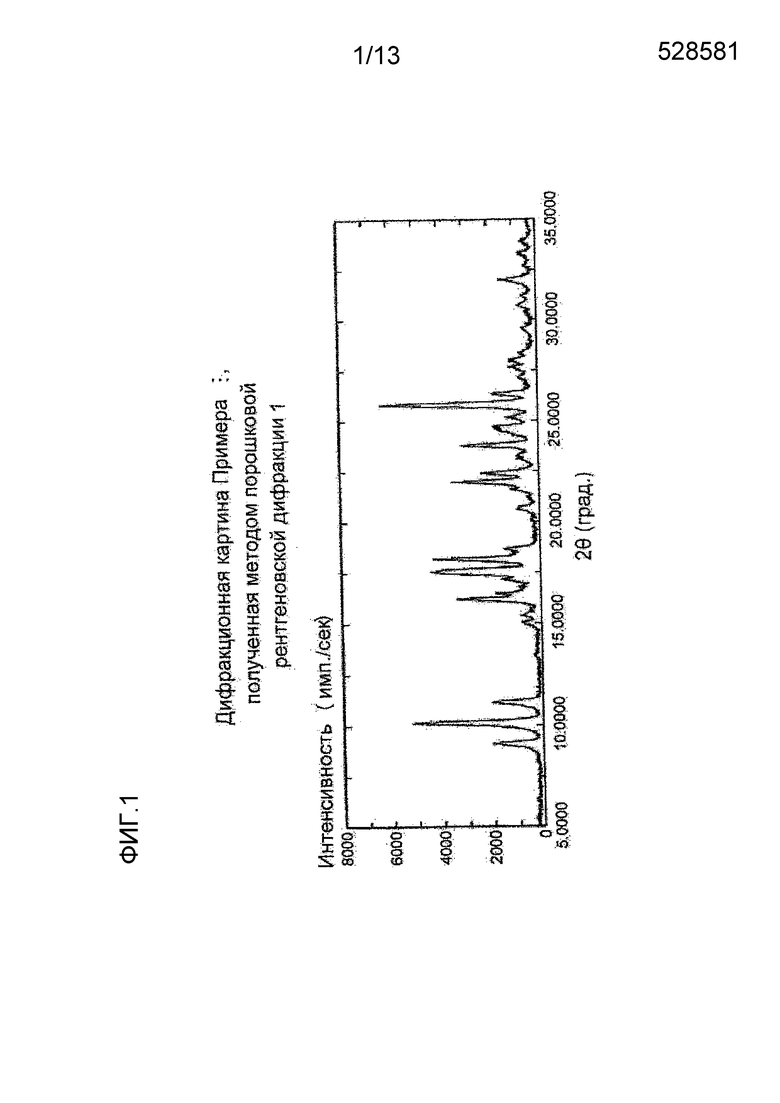
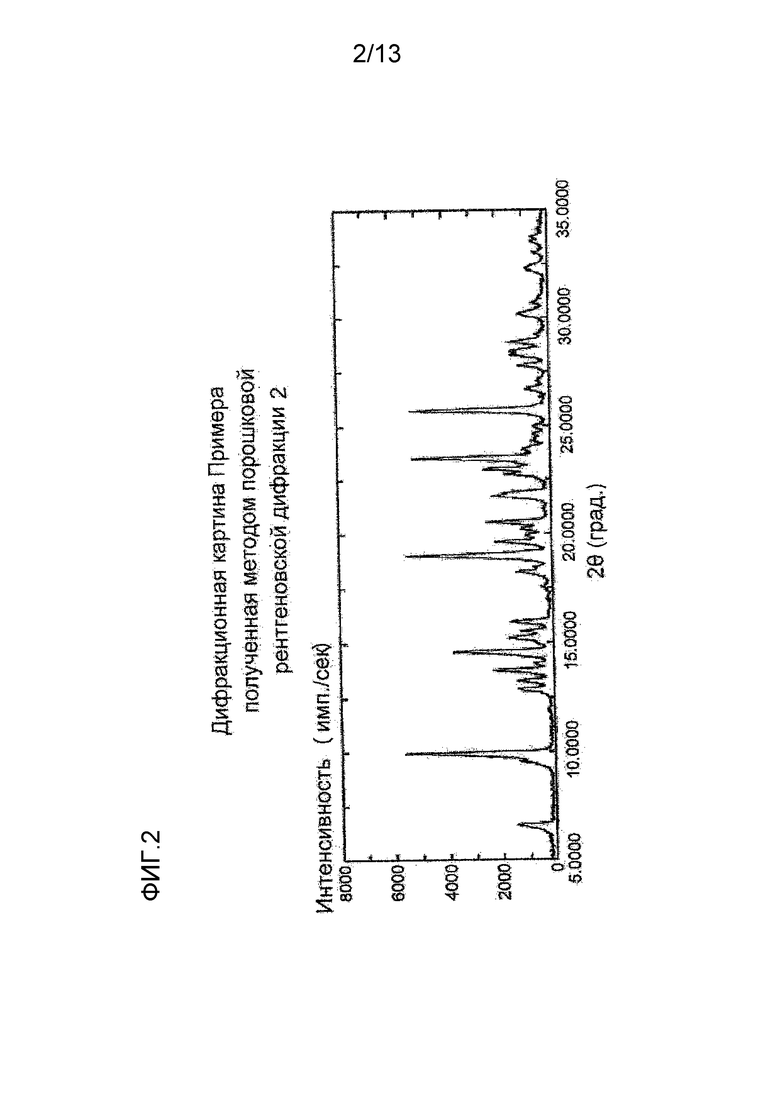
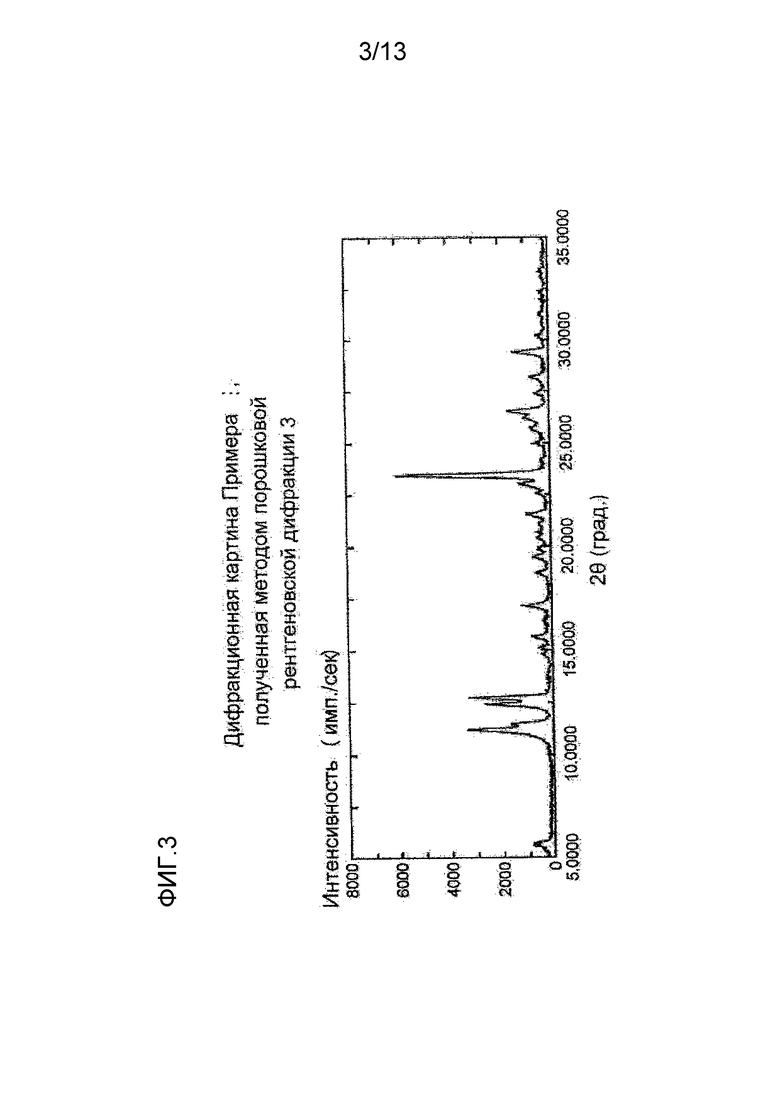
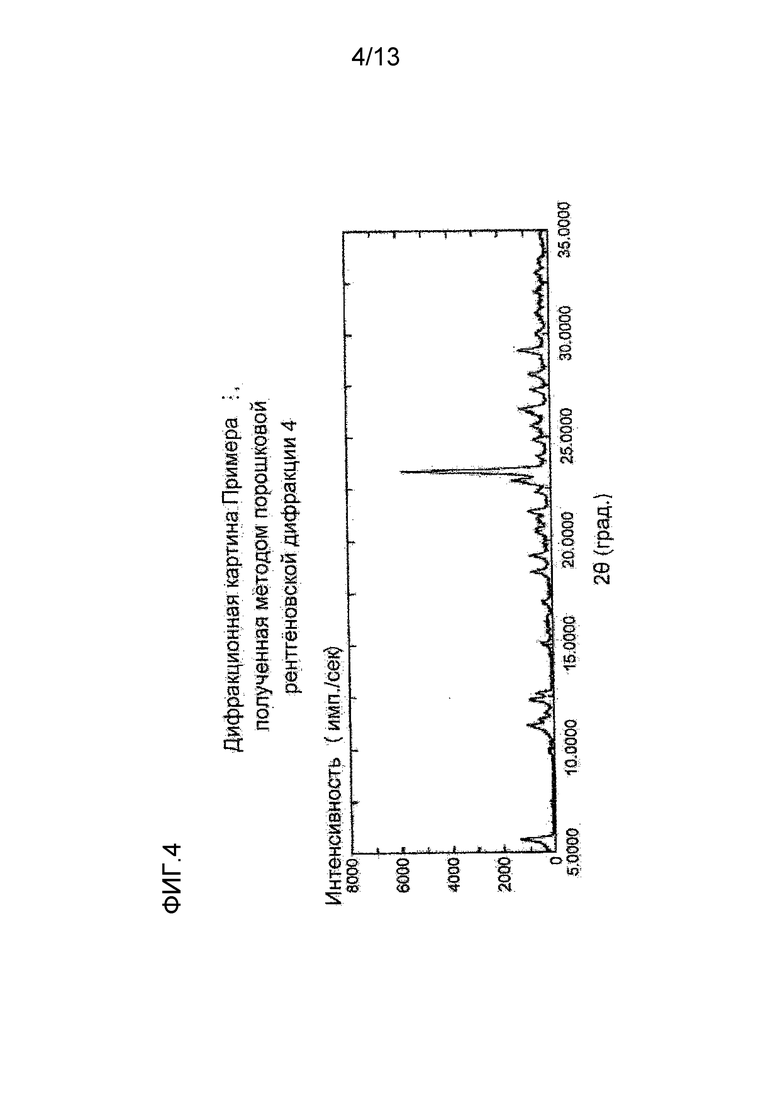
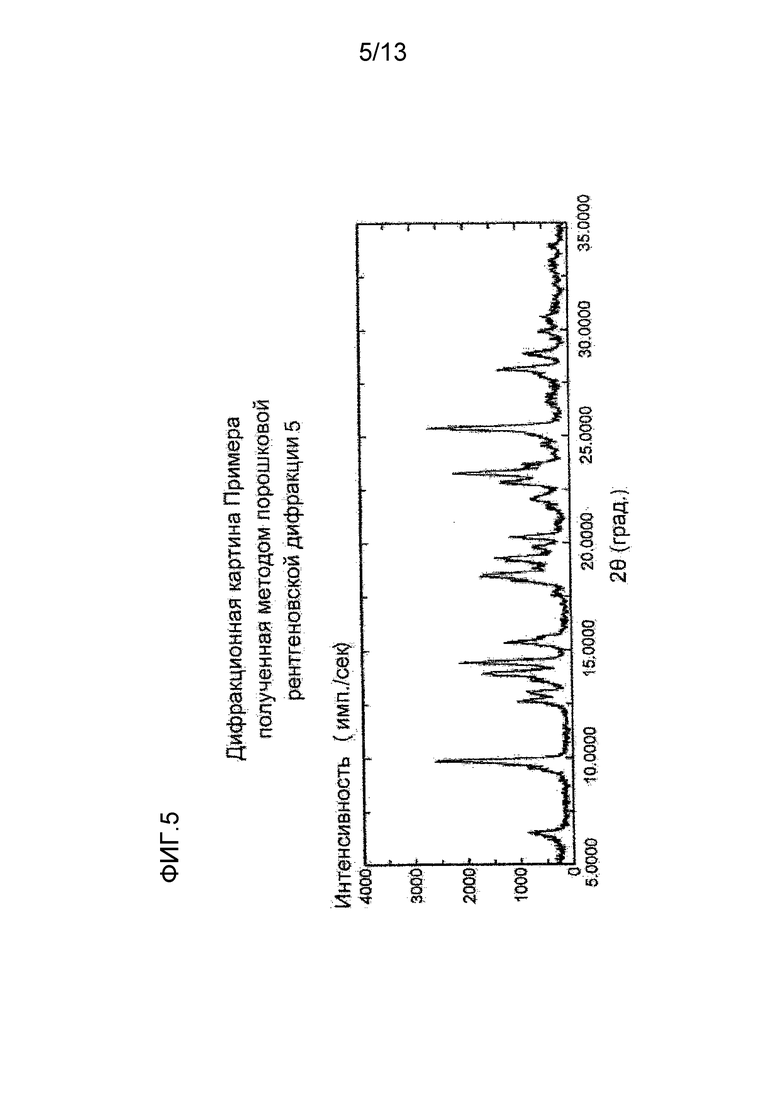
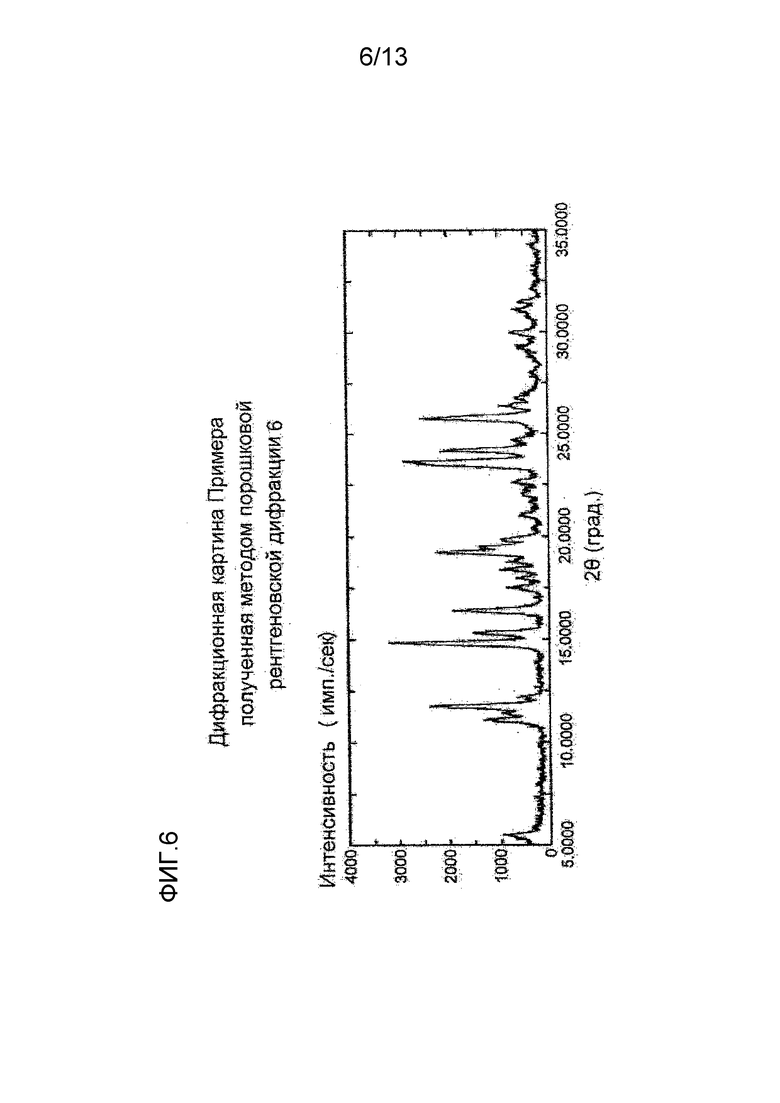
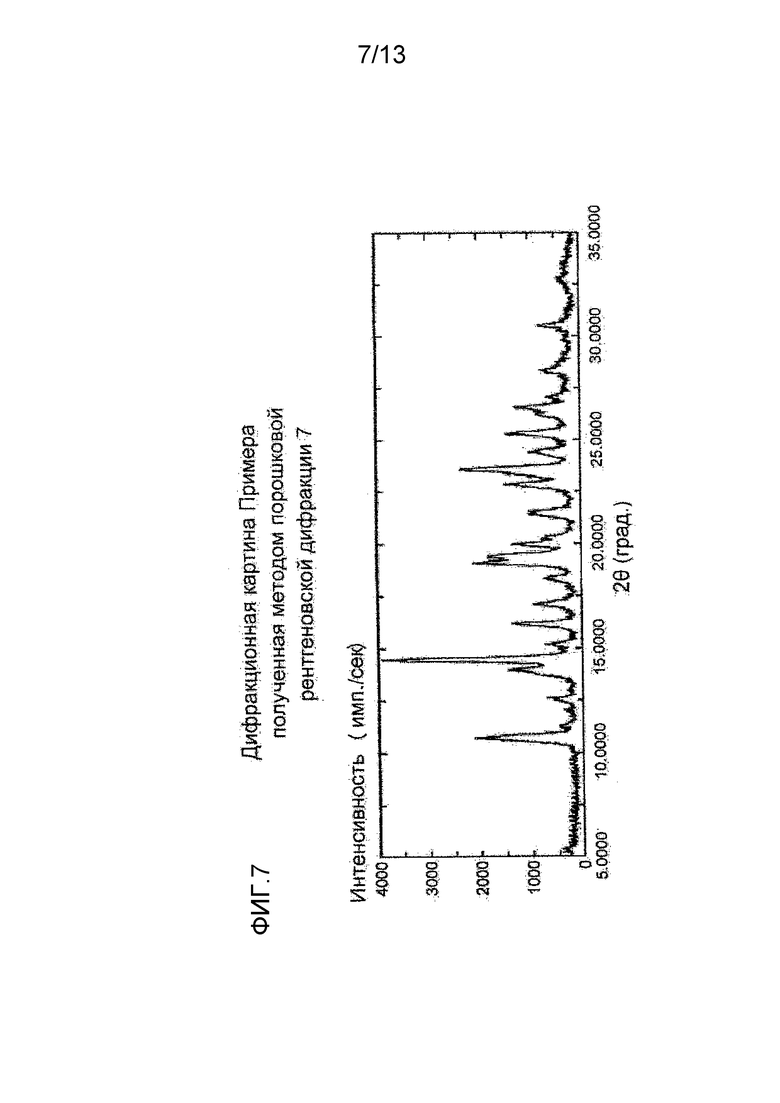
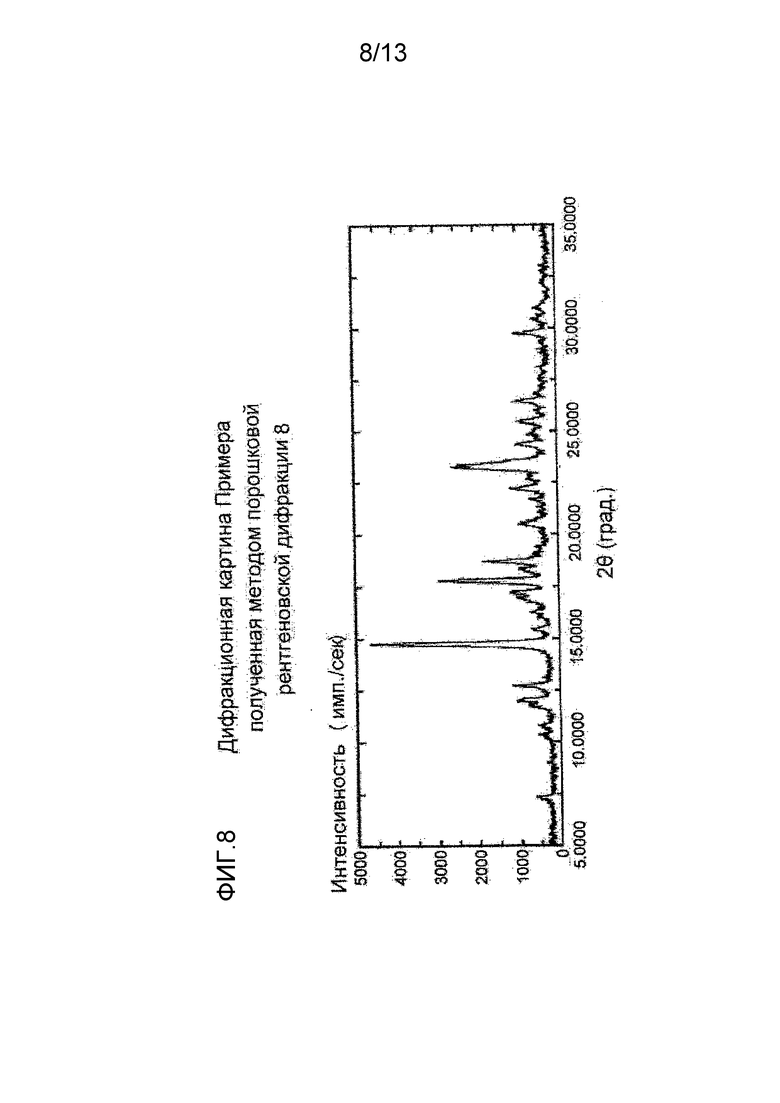
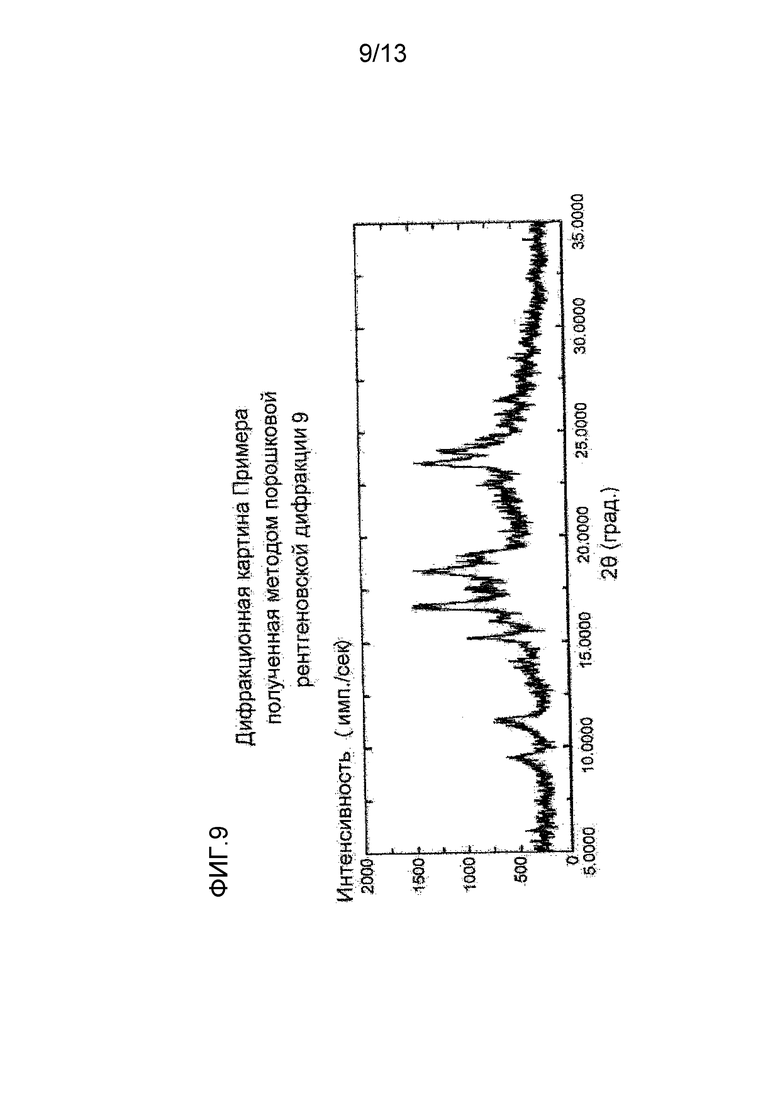
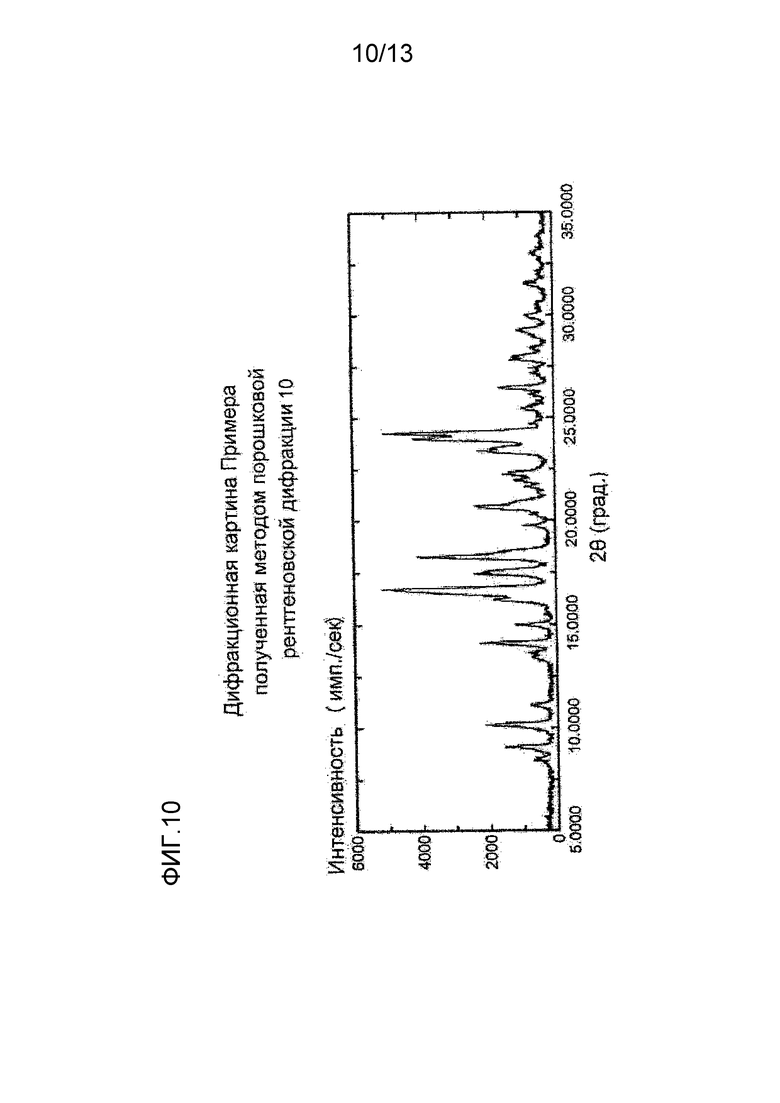
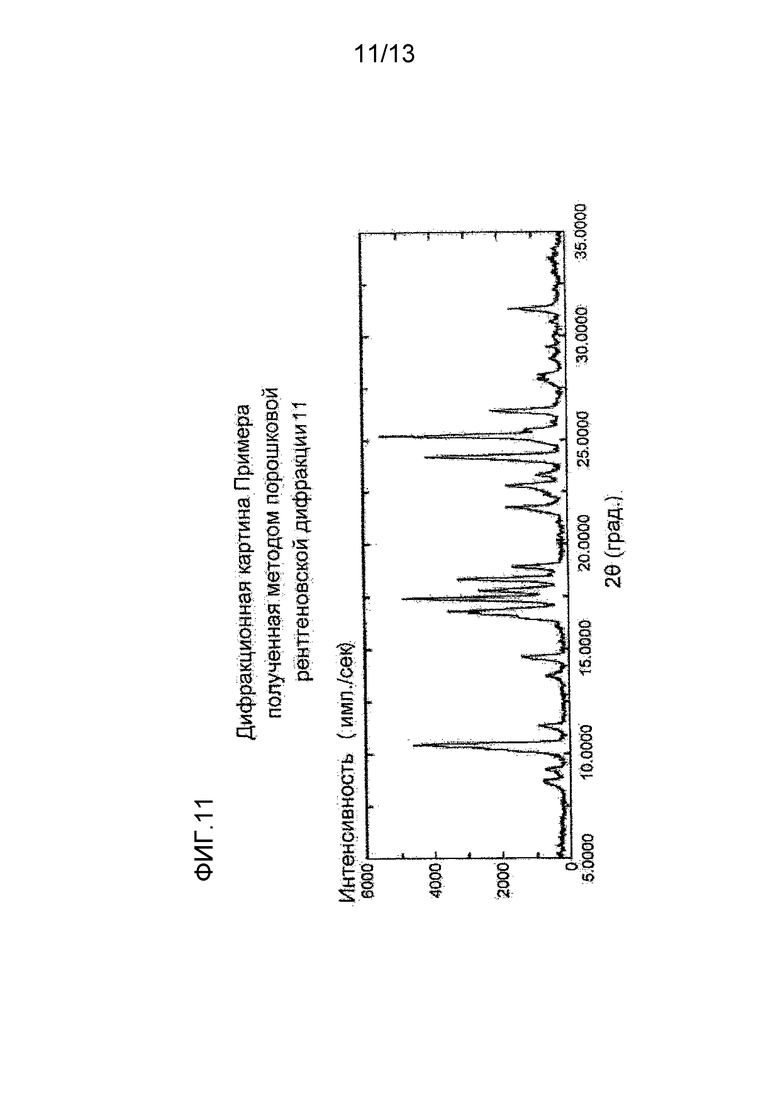
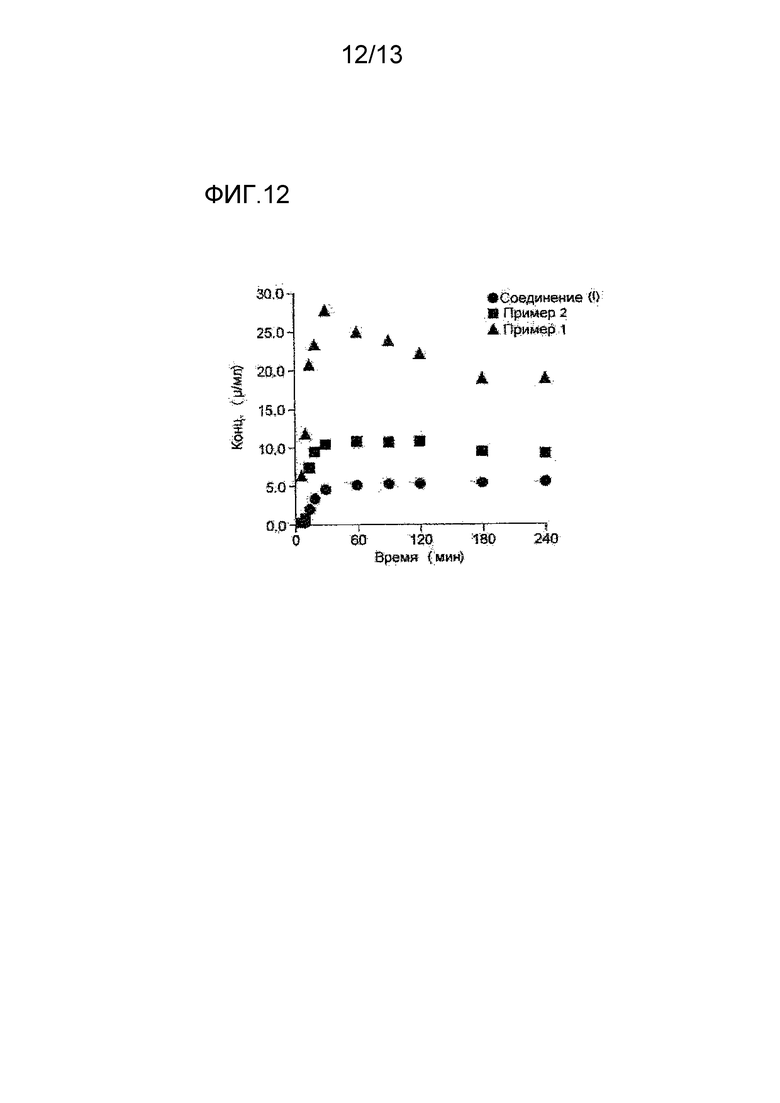
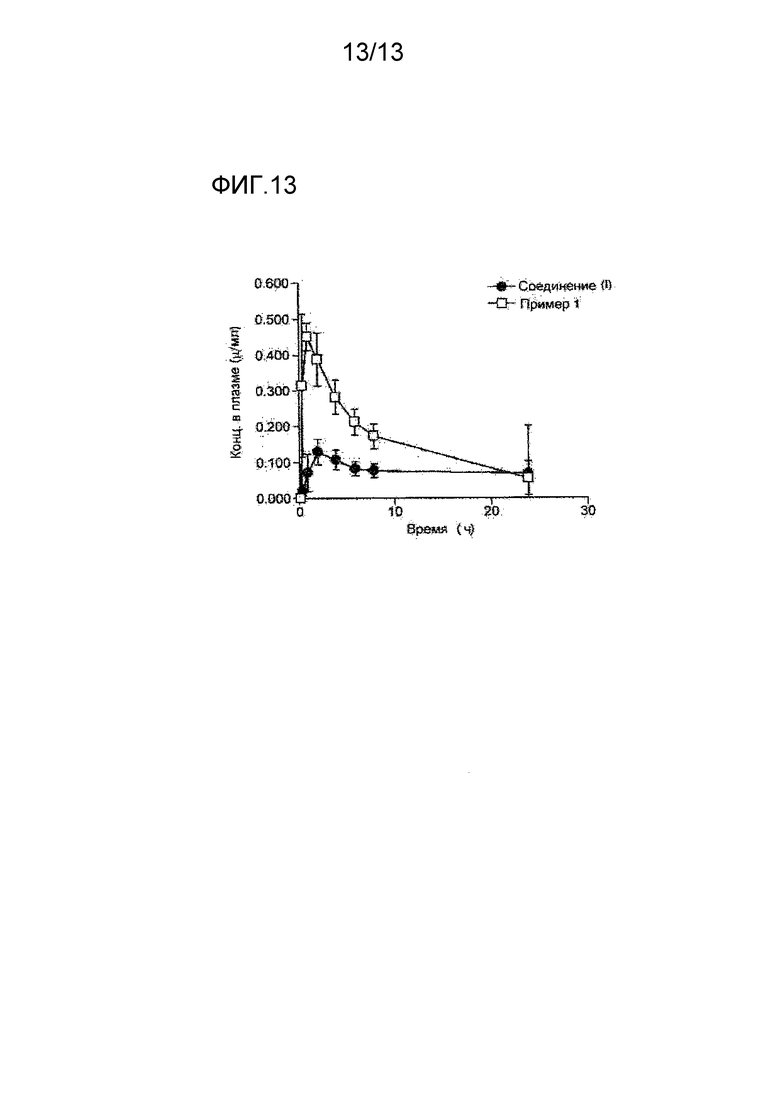
Настоящее изобретение относится к соли (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она и кислоты, выбранной из группы, состоящей из хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, азотной кислоты, фосфорной кислоты, малоновой кислоты, малеиновой кислоты, винной кислоты, метансульфокислоты, бензолсульфокислоты и толуолсульфокислоты, а также к фармацевтической композиции на основе этих соединений, которая обладает ингибирующим действием в отношении PDE9. 6 н. и 1 з.п. ф-лы, 13 ил., 3 табл., 12 пр.
1. Соль (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она и кислоты, выбранной из группы, состоящей из хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, азотной кислоты, фосфорной кислоты, малоновой кислоты, малеиновой кислоты, винной кислоты, метансульфокислоты, бензолсульфокислоты и толуолсульфокислоты.
2. Мономалеат (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она.
3. Монобензолсульфонат (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она.
4. Соль по п. 1 в кристаллической форме.
5. Мономалеат (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, в кристаллической форме имеющий дифракционный пик при угле дифракции (2θ±0,2°), равном 10,1°, при анализе методом рентгеновской порошковой дифракции.
6. Монобензолсульфоната (S)-7-(2-метокси-3,5-диметилпиридин-4-ил)-1-(тетрагидрофуран-3-ил)-1H-пиразоло[4,3-c]хинолин-4(5H)-она, в кристаллической форме имеющий дифракционный пик при угле дифракции (2θ±0,2°), равном 9,9°, при анализе методом рентгеновской порошковой дифракции.
7. Фармацевтическая композиция, обладающая ингибирующим действием в отношении PDE9, включающая соль по п. 1 в качестве активного ингредиента.
| WO 2012033144 A1, 15.03.2012 | |||
| WO 2008072779 A1, 19.06.2008 | |||
| WO 2010101230 A1, 10.09.2010 | |||
| WO 2013045400 A1, 04.04.2013 | |||
| EA 200500322 A1, 25.08.2005. |

