Область техники
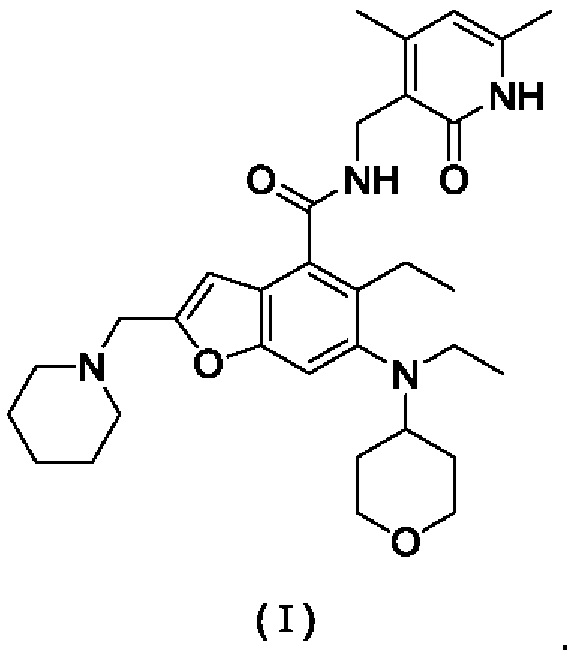
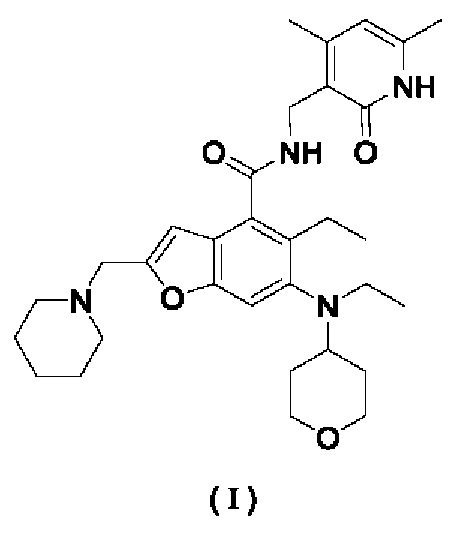
Настоящее изобретение относится к кристаллическим формам А, В, С и D N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимида, а также к способам их получения.
Предшествующий уровень техники
Лимфома - это злокачественная опухоль, происходящая из лимфоидной кроветворной системы. Она подразделяется на неходжкинскую лимфому (НХЛ) и лимфому Ходжкина (ХЛ) в зависимости от опухолевых клеток. В Азии 90% пациентов с лимфомой - это пациенты с НХЛ, имеющие лимфоциты, гистиоциты или ретикулярные клетки с различной степенью дифференциации в патологии. Согласно естественному течению НХЛ ее можно разделить на три основных клинических типа, а именно: высокоинвазивные, инвазивные и индолентные лимфомы. В соответствии с различным происхождением лимфоцитов, ее можно разделить на В-клеточную лимфому, Т-клеточную лимфому и лимфому из естественных киллеров (NK-лимфому). Основная функция В-клеток заключается в выделении различных антител для защиты организма от различных внешних вторжений.
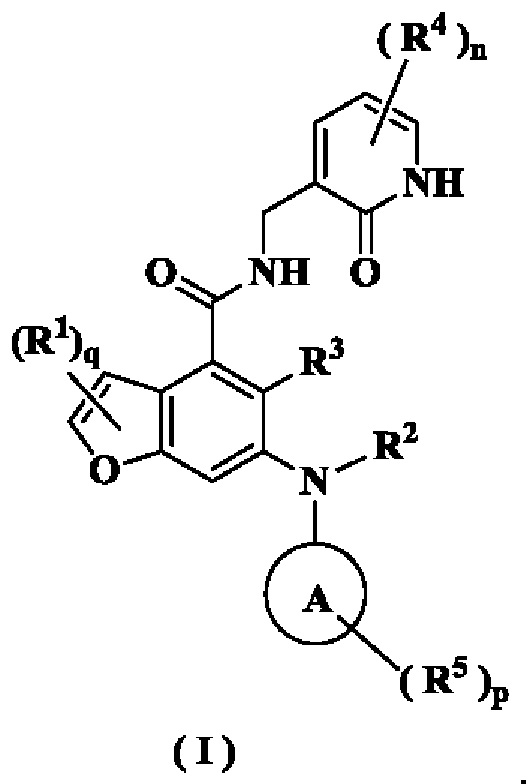
Гистоновая метилтрансфераза, кодируемая геном EZH2, является каталитическим компонентом поликомб репрессивного комплекса 2 (PRC2). Уровни EZH2 аномально повышены в раковых тканях по сравнению с нормальными тканями, a EZH2 наиболее высоко экспрессируется при запущенных формах рака или плохом прогнозе. При некоторых типах рака избыточная экспрессия EZH2 происходит одновременно с амплификацией гена EZH2. Ряд экспериментальных исследований si/shPHК (малых интерферирующих или коротких интерферирующих РНК) показывает, что снижение экспрессии EZH2 в линиях опухолевых клеток может ингибировать пролиферацию, миграцию и инвазию опухолевых клеток или ангиогенез и приводить к апоптозу. В WO 2017084494 (PCT/CN2016/104318, дата подачи 2 ноября 2016 г.) раскрыт ингибитор EZH2, имеющий следующую структуру:
Заявки на патенты, в которых раскрыт селективный ингибитор EZH2, включают WO 2012005805, WO 2012050532, WO 2012118812, WO 2012142513, WO 2012142504, WO 2013049770, WO 2013039988, WO 2013067300, WO 2015141616, WO 2011140325 и т.п.
Кристаллическая структура фармацевтически активного ингредиента часто влияет на химическую стабильность лекарственного средства. Различные условия кристаллизации и условия хранения могут привести к изменению кристаллической структуры соединения, а иногда и к сопутствующему получению других кристаллических форм. В общем, аморфный лекарственный продукт не имеет правильной кристаллической структуры и часто имеет другие недостатки, такие как плохая стабильность продукта, чрезмерно мелкие кристаллы, сложная фильтрация, легкая агломерация и плохая текучесть. Полиморфные формы лекарства имеют разные требования по хранению, производству и распространению. Следовательно, необходимо разработать кристаллическую форму соединения формулы (I) и способ ее получения, чтобы улучшить различные свойства соединения формулы (I).
Сущность изобретения
Техническая задача, решаемая настоящим изобретением, заключается в разработке кристаллических форм А, В, С и D N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимида, а также способов их получения. Кристаллические формы, полученные согласно настоящему изобретению, имеют хорошую стабильность.
Техническое решение согласно настоящему изобретению заключается в следующем:
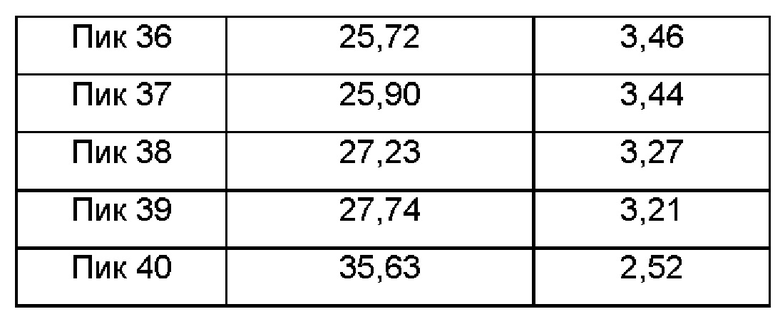
Настоящее изобретение предлагает кристаллическую форму А соединения формулы (I), отличающуюся тем, что кристаллическая форма А имеет спектр порошковой дифракции рентгеновских лучей, который получен с использованием Cu-Kα-излучения и представлен углом дифракции 26, который имеет характеристические пики при углах дифракции 2θ: 7.60, 8.51, 11.80, 12.38, 13.52, 13.73, 14.48, 15.23, 15.99, 16.10, 16.82, 16.99, 17.35, 18.24, 20.82, 21.57, 21.91, 22.57, 22.76, 22.88, 24.29, 24.47, 25.24, 25.90, 27.23 и 27.74, где диапазон погрешности угла 2θ каждого характеристического пика составляет ±0,2,
Предпочтительно имеются характеристические пики при углах дифракции 2θ: 7.60, 8.51, 10.37, 11.16, 11.80, 12.38, 12.89, 13.52, 13.73, 14.03, 14.48, 15.23, 15.99, 16.10, 16.43, 16.82, 16.99, 17.35, 18.24, 18.92, 19.17, 20.68, 20.82, 21.57, 21.91, 22.57, 22.76, 22.88, 23.53, 23.68, 24.00, 24.29, 24.47, 24.91, 25.24, 25.72, 25.90, 27.23, 27.74 и 35.63, где диапазон погрешности угла 2θ каждого характеристического пика составляет ±0,2.
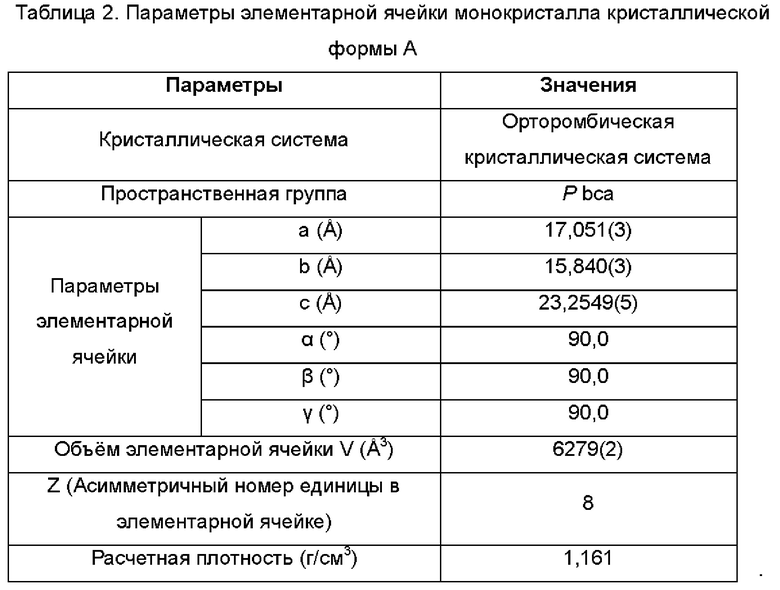
Настоящее изобретение также предоставляет данные монокристалла кристаллической формы А, характеризующиеся тем, что кристаллическая форма А представляет собой ромбическую кристаллическую систему; пространственной группой является Pbca; параметры элементарной ячейки: 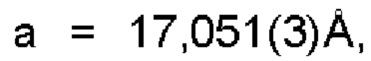
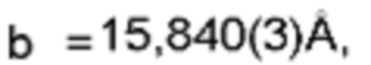
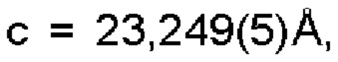 α=β=γ=90,0°; и объем элементарной ячейки составляет
α=β=γ=90,0°; и объем элементарной ячейки составляет 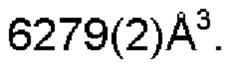
Настоящее изобретение также предлагает способ получения кристаллической формы А, отличающийся тем, что способ выбран из группы, состоящей из:
способа I, включающего растворение соединения формулы (I) в растворителе для кристаллизации, фильтрацию, промывку и сушку полученного кристалла с получением желаемой кристаллической формы А, где растворитель выбирают из группы, состоящей из амидного растворителя, смешанного растворителя из амидного растворителя и воды, смешанного растворителя из галогенуглеводорода и нитрила, причем амидный растворитель выбирают из группы, состоящей из N,N-диметилформамида и N,N-диметилацетамида, галогенуглеводородный растворитель представляет собой дихлорметан и нитрильный растворитель представляет собой ацетонитрил, и способ кристаллизации выбран из группы, состоящей из кристаллизации при комнатной температуре, кристаллизации при охлаждении, кристаллизации при испарении растворителя и кристаллизации, вызванной добавлением затравочного кристалла;
способа II, включающего растворение соединения формулы (I) в хорошем растворителе, добавление антирастворителя для кристаллизации, фильтрацию и сушку полученного кристалла с получением желаемой кристаллической формы А, где хорошим растворителем является спиртовой растворитель, причем спиртовой растворитель выбирают из группы, состоящей из метанола, этанола и изопропанола, а антирастворитель представляет собой воду, а способ кристаллизации выбирают из группы, состоящей из кристаллизации при комнатной температуре, кристаллизации при охлаждении, кристаллизации при испарении растворителя и кристаллизации, вызванной добавлением затравочного кристалла; и
способа III, включающего добавление соединения формулы (I) к растворителю, суспендирование этой смеси, фильтрацию и сушку полученного кристалла с получением желаемой кристаллической формы А, где растворитель выбирают из группы, состоящей из смешанного растворителя из амидного растворителя и воды, и смешанного растворителя из галогенуглеводорода и нитрила, причем амидный растворитель выбирают из группы, состоящей из N,N-диметилформамида и N,N-диметилацетамида, галогенуглеводородный растворитель представляет собой дихлорметан, а нитрильный растворитель представляет собой ацетонитрил.
Настоящее изобретение также предлагает способ получения монокристалла кристаллической формы А, отличающийся тем, что способ состоит из следующих стадий:
растворение соединения формулы (I) в растворителе для кристаллизации, фильтрация и сушка полученного кристалла с получением желаемого монокристалла кристалла формы А, где растворителем является смешанный растворитель из галогенуглеводорода и нитрила, галогенуглеводородным растворителем является дихлорметан и нитрильным растворителем является ацетонитрил; соотношение галогенуглеводорода к нитрилу составляет от 20:1 до 1:20 и предпочтительно - 1:10, способ кристаллизации выбирают из группы, состоящей из кристаллизации при комнатной температуре, кристаллизации при охлаждении, кристаллизации при испарении растворителя и кристаллизации, вызванной добавлением затравочного кристалла.
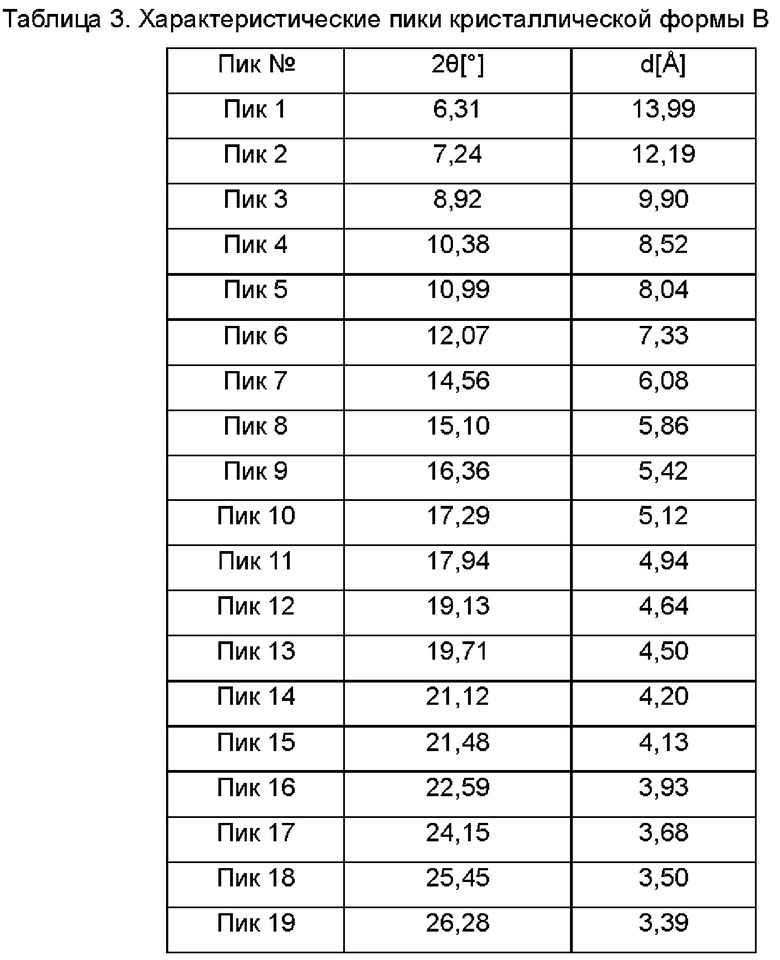
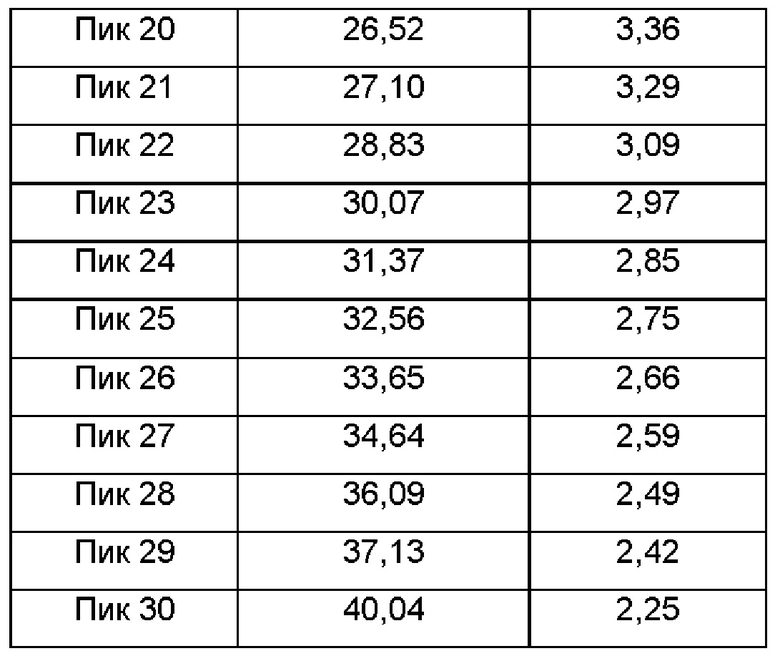
Настоящее изобретение также относится к кристаллической форме В соединения формулы (I), отличающейся тем, что кристаллическая форма В имеет спектр порошковой дифракции рентгеновских лучей, который получен с использованием Cu-Kα-излучения и представлен углом дифракции 2θ, который имеет характеристические пики при углах дифракции 2θ: 6.31, 7.24, 10.99, 12.07, 14.56, 17.94, 19.13, 19.71, 21.48, 24.15, 27.10 и 28.83, где диапазон погрешности угла 2θ каждого характеристического пика составляет ±0,2.
Предпочтительно характеристические пики имеются при углах дифракции 2θ: 6.31, 7.24, 8.92, 10.38, 10.99, 12.07, 14.56, 15.10, 16.36, 17.29, 17.94, 19.13, 19.71, 21.12, 21.48, 22.59, 24.15, 25.45, 26.28, 26.52, 27.10, 28.83, 30.07, 31.37, 32.56, 33.65, 34.64, 36.09, 37.13 и 40.04, где диапазон погрешности угла 2θ каждого характеристического пика составляет ±0,2.
Настоящее изобретение также предлагает способ получения кристаллической формы В, отличающийся тем, что способ выбран из группы, состоящей из:
способа I, включающего растворение соединения формулы (I) в растворителе для кристаллизации, фильтрацию и сушку полученного кристалла с получением желаемой кристаллической формы В, где растворитель представляет собой этанол, и способ кристаллизации выбран из группы, состоящей из кристаллизации при комнатной температуре, кристаллизации при охлаждении, кристаллизации при испарении растворителя и кристаллизации, вызванной добавлением затравочного кристалла; и
способа II, включающего растворение соединения формулы (I) в хорошем растворителе, добавление антирастворителя для кристаллизации, фильтрацию и сушку полученного кристалла с получением желаемой кристаллической формы В, где хорошим растворителем является этанол, антирастворитель представляет собой воду, а способ кристаллизации выбирают из группы, состоящей из кристаллизации при комнатной температуре, кристаллизации при охлаждении, кристаллизации при испарении растворителя и кристаллизации, вызванной добавлением затравочного кристалла.
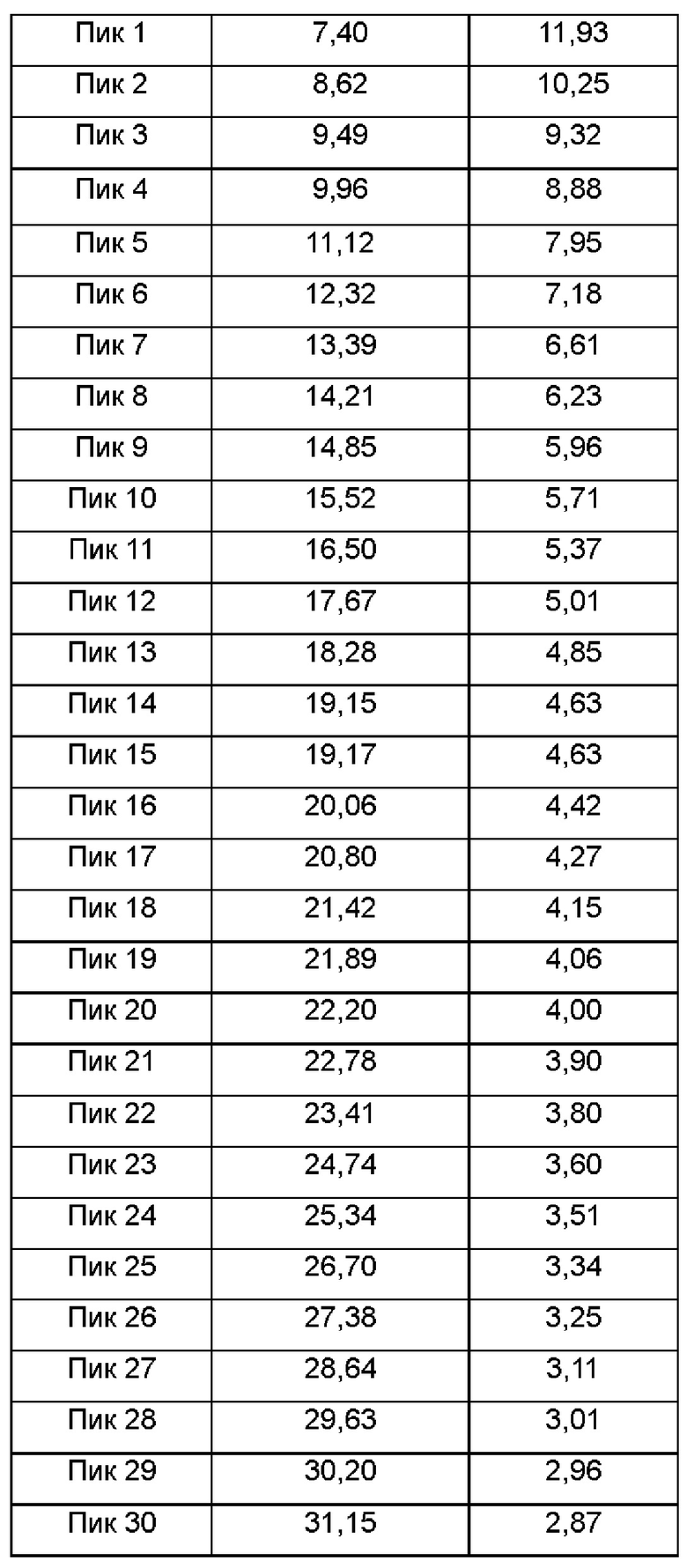
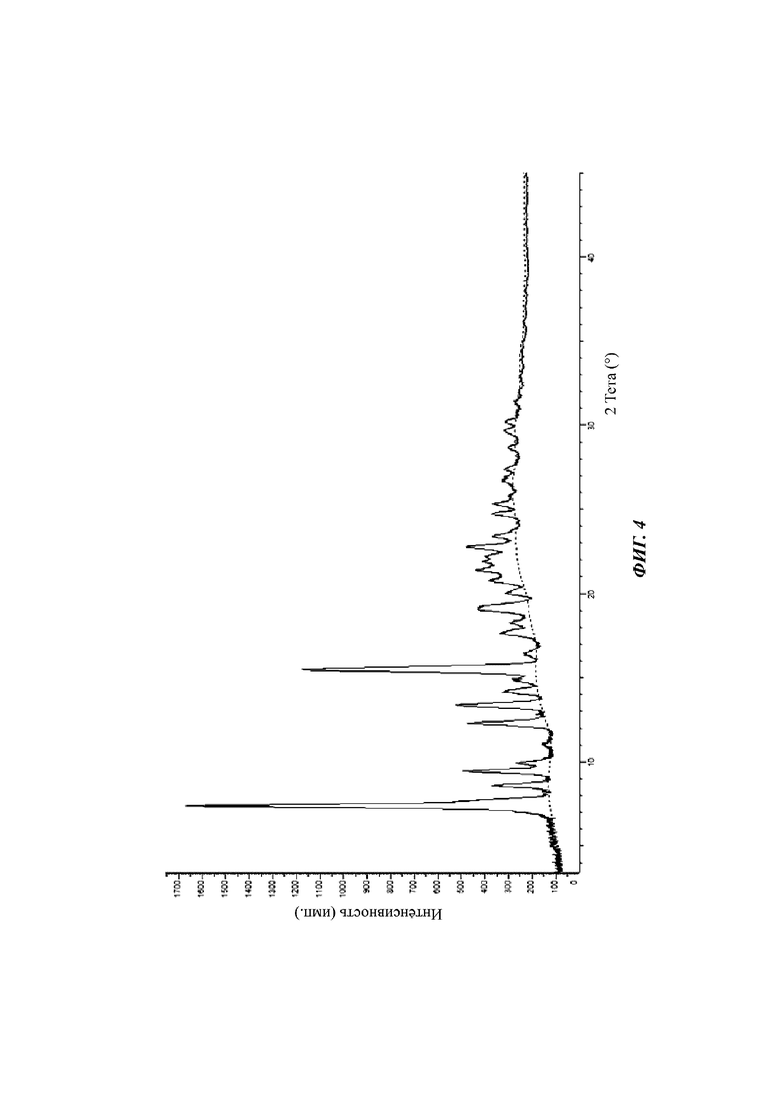
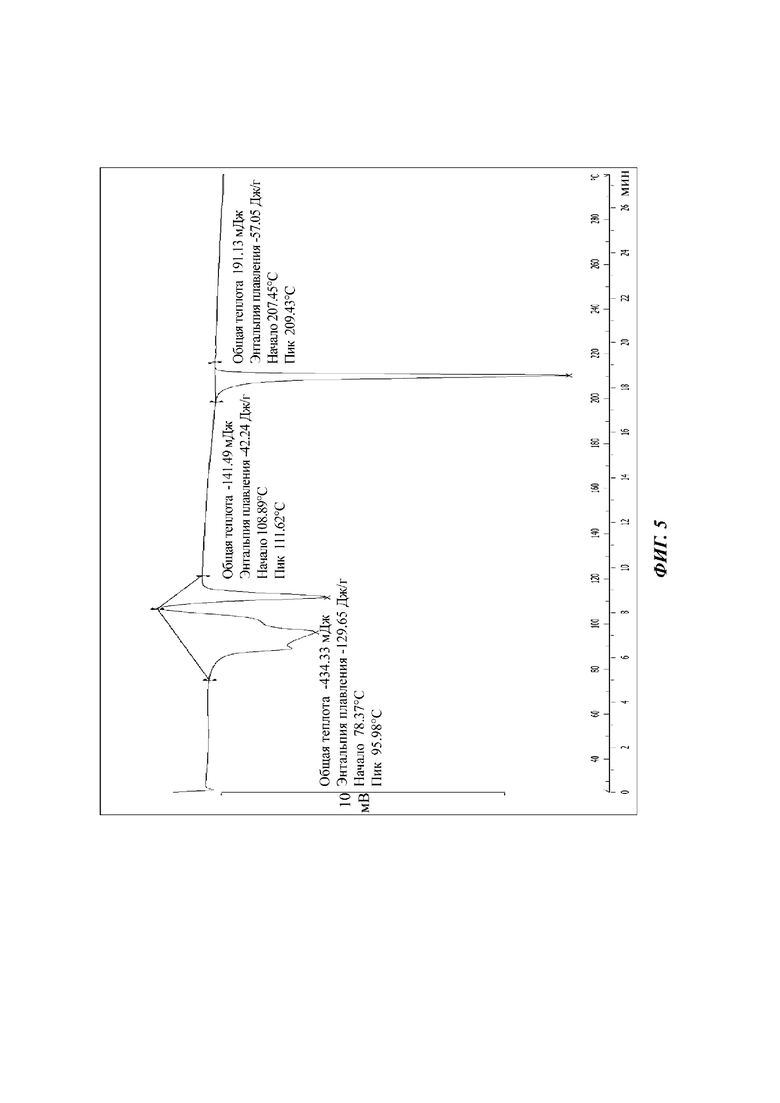
Настоящее изобретение также относится к кристаллической форме С соединения формулы (I), отличающейся тем, что кристаллическая форма С имеет спектр порошковой дифракции рентгеновских лучей, который получен с использованием Cu-Kα-излучения и представлен углом дифракции 2θ, который имеет характеристические пики при углах дифракции 2θ: 7.40, 8.62, 9.49, 12.32, 13.39, 15.52, 19.15, 19.17, 21.42 и 22.78, где диапазон погрешности угла 2θ каждого характеристического пика составляет ±0,2.
Предпочтительно характеристические пики имеются при углах дифракции 2θ: 7.40, 8.62, 9.49, 9.96, 11.12, 12.32, 13.39, 14.21, 14.85, 15.52, 16.50, 17.67, 18.28, 19.15, 19.17, 20.06, 20.80, 21.42, 21.89, 22.20, 22.78, 23.41, 24.74, 25.34, 26.70, 27.38, 28.64, 29.63, 30.20 и 31.15, где диапазон погрешности угла 2θ каждого характеристического пика составляет ±0,2.
Настоящее изобретение также предлагает способ получения кристаллической формы С, отличающийся тем, что включает следующие стадии:
растворение соединения формулы (I) в хорошем растворителе, добавление антирастворителя для кристаллизации, фильтрацию и сушку полученного кристалла с получением желаемой кристаллической формы С, где хороший растворитель представляет собой простой эфирный растворитель, причем простой эфирный растворитель представляет собой 1,4-диоксан, антирастворитель выбран из группы, состоящей из алифатического углеводородного растворителя и алициклического углеводородного растворителя, алифатический углеводородный растворитель представляет собой н-гептан и алициклический углеводородный растворитель представляет собой циклогексан, а способ кристаллизации выбирают из группы, состоящей из кристаллизации при комнатной температуре, кристаллизации при охлаждении, кристаллизации при испарении растворителя и кристаллизации, вызванной добавлением затравочного кристалла.
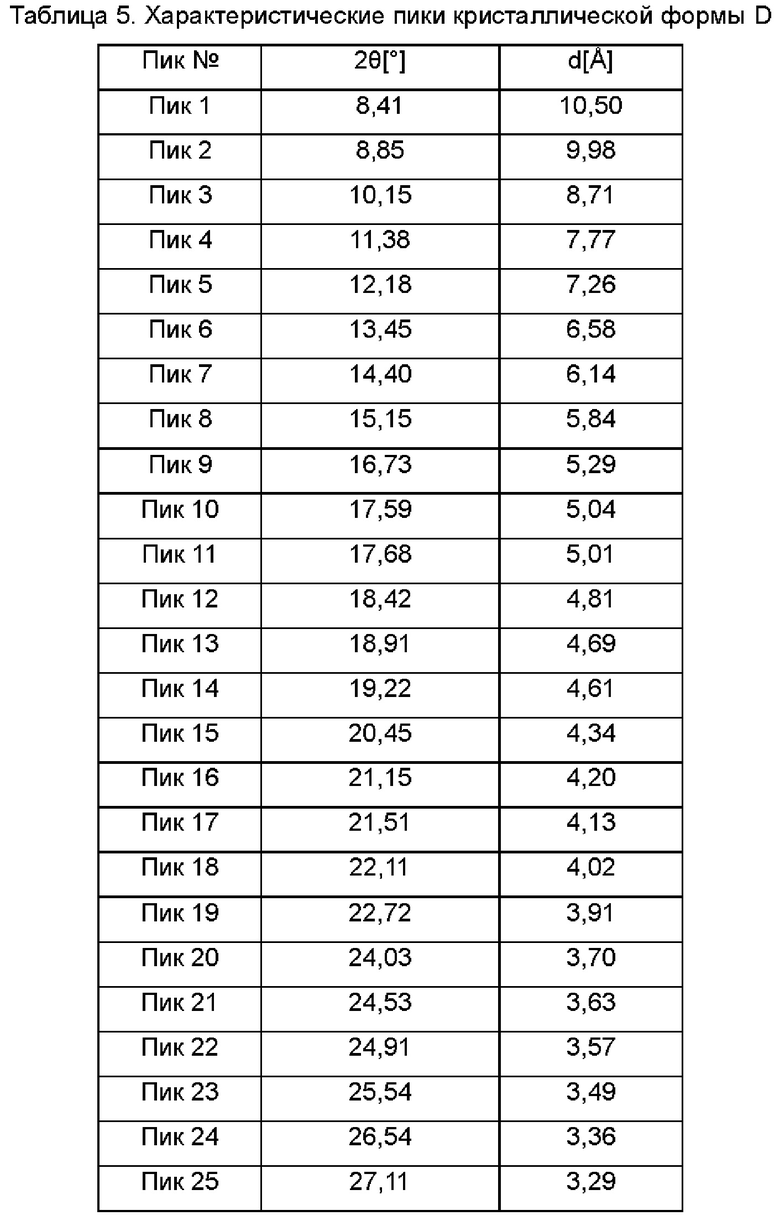
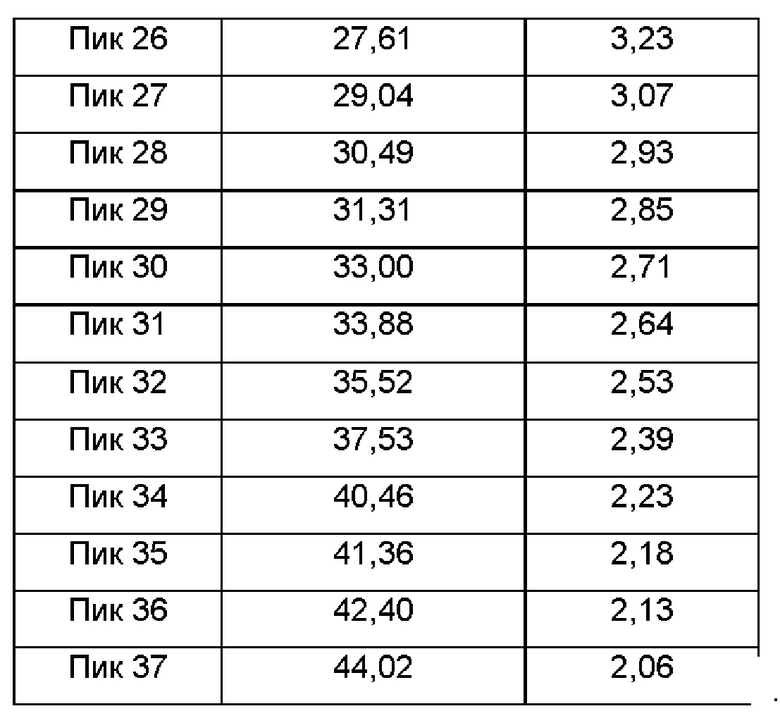
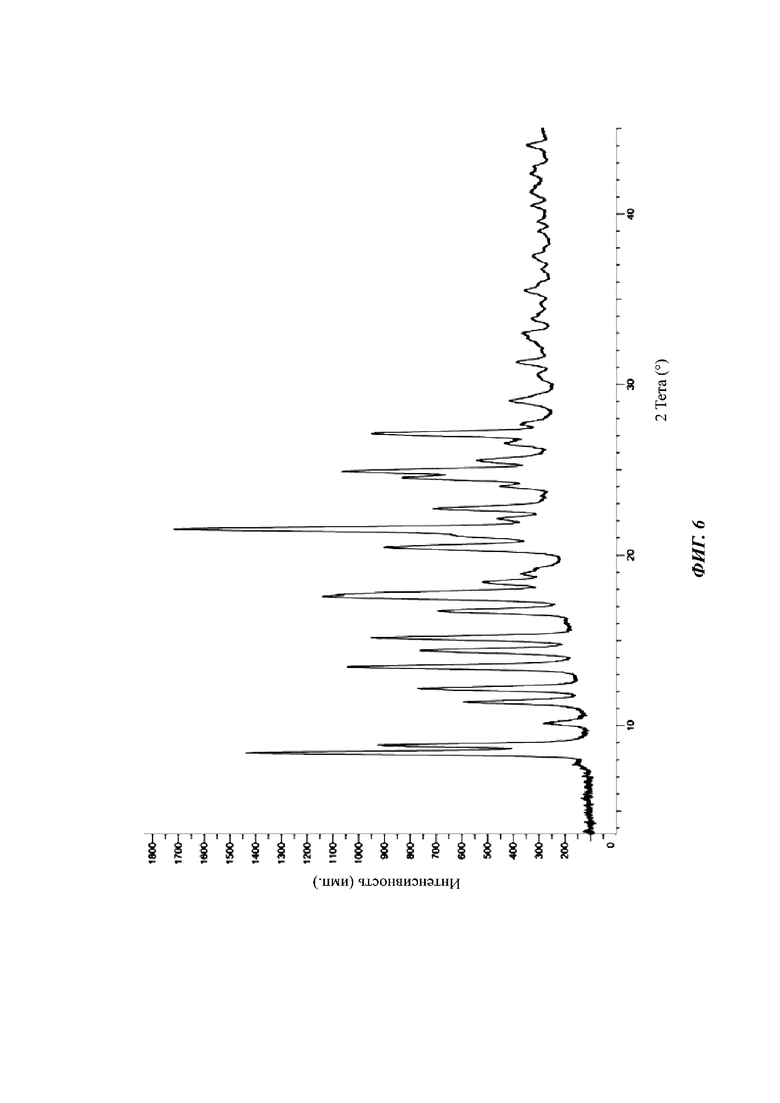
Настоящее изобретение также относится к кристаллической форме D соединения формулы (I), отличающейся тем, что кристаллическая форма D имеет спектр порошковой дифракции рентгеновских лучей, который получен с использованием Cu-Kα-излучения и представлен углом дифракции 2θ, который имеет характеристические пики при углах дифракции 2θ: 8.41, 8.85, 11.38, 12.18, 13.45, 15.15, 16.73, 17.59, 17.68, 20.45, 21.51, 22.72, 24.53, 24.91 и 27.11, где диапазон погрешности угла 2θ каждого характеристического пика составляет ±0,2.
Предпочтительно характеристические пики имеются при углах дифракции 2θ: 8.41, 8.85, 10.15, 11.38, 12.18, 13.45, 14.40, 15.15, 16.73, 17.59, 17.68, 18.42, 18.91, 19.22, 20.45, 21.15, 21.51, 22.11, 22.72, 24.03, 24.53, 24.91, 25.54, 26.54, 27.11, 27.61, 29.04, 30.49, 31.31, 33.00, 33.88, 35.52, 37.53, 40.46, 41.36, 42.40 и 44.02, где диапазон погрешности угла 2θ каждого характеристического пика составляет ±0,2.
Настоящее изобретение также предоставляет данные монокристалла кристаллической формы D, характеризующиеся тем, что кристаллическая форма D представляет собой орторомбическую кристаллическую систему; пространственной группой является Pbca; параметры элементарной ячейки: 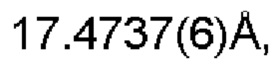
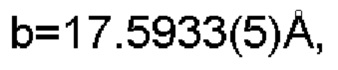
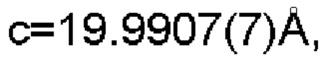 α=β=γ=90.0° и объем элементарной ячейки составляет
α=β=γ=90.0° и объем элементарной ячейки составляет 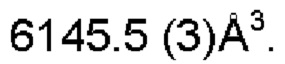
Настоящее изобретение также предлагает способ получения кристаллической формы D, отличающийся тем, что способ выбран из группы, состоящей из:
способа I, включающего растворение соединения формулы (I) в растворителе для кристаллизации, фильтрацию и сушку полученного кристалла с получением желаемой кристаллической формы D, где растворитель выбирают из группы, состоящей из спиртового растворителя, простого эфирного растворителя, смешанного растворителя из спирта и воды, смешанного растворителя из простого эфира и воды, смешанного растворителя из спирта и алифатического углеводорода, и смешанного растворителя из простого эфира и алифатического углеводорода, причем спиртовой растворитель выбирают из группы, состоящей из метанола, этанола и изопропанола, простой эфирный растворитель выбирают из группы, состоящей из тетрагидрофурана и 1,4-диоксана, а алифатическим углеводородным растворителем является н-гептан, и способ кристаллизации выбирают из группы, состоящей из кристаллизации при комнатной температуре, кристаллизации при охлаждении, кристаллизации при испарении растворителя и кристаллизации, вызванной добавлением затравочного кристалла;
способа II, включающего растворение соединения формулы (I) в хорошем растворителе, добавление антирастворителя для кристаллизации, фильтрацию и сушку полученного кристалла с получением желаемой кристаллической формы D, где хороший растворитель выбирают из группы, состоящей из спиртового растворителя и простого эфирного растворителя, спиртовой растворитель выбирают из группы, состоящей из метанола и изопропанола, простой эфирный растворитель представляет собой тетрагидрофуран, а антирастворителем является вода; или хорошим растворителем является простой эфир, простым эфирным растворителем является тетрагидрофуран, антирастворитель выбран из группы, состоящей из алифатического углеводородного растворителя и алициклического углеводородного растворителя, алифатический углеводородный растворитель представляет собой н-гептан, а алициклическим углеводородным растворителем является циклогексан, и способ кристаллизации выбран из группы, состоящей из кристаллизации при комнатной температуре, кристаллизации при охлаждении, кристаллизации при испарении растворителя и кристаллизации, вызванной добавлением затравочного кристалла;
способа III, включающего добавление соединения формулы (I) к растворителю, суспендирование этой смеси, фильтрацию и сушку полученного кристалла с получением желаемой кристаллической формы D, где растворитель выбирают из группы, состоящей из воды, сложного эфира, простого эфира, алифатического углеводорода, алициклического углеводорода, нитроалканового растворителя, арена, спирта, нитрила, галогенуглеводорода, кетона, сульфоксида, амида, смешанного растворителя из спирта и простого эфира, смешанного растворителя из спирта и воды, и смешанного растворителя из одного или более спиртов, сложноэфирный растворитель выбирают из группы, состоящей из этилацетата, изопропилацетата и бутилацетата, простой эфирный растворитель выбирают из группы, состоящей из тетрагидрофурана, 1,4-диоксана, метилового эфира пропиленгликоля и метил-трет-бутилового эфира, алифатический углеводород представляет собой н-гептан, алициклический углеводород представляет собой циклогексан, нитроалкановый растворитель представляет собой нитрометан, ареновый растворитель выбирают из группы, состоящей из ксилола и кумола, спиртовой растворитель выбирают из группы, состоящей из метанола, этанола и изопропанола, нитрильным растворителем является ацетонитрил, галогенуглеводородным растворителем является дихлорметан, кетоновым растворителем является ацетон, сульфоксидом является диметилсульфоксид, смешанный растворитель из одного или более спиртов выбирают из группы, состоящей из смешанного растворителя из метанола и этанола, смешанного растворителя из метанола и изопропанола и смешанного растворителя из этанола и изопропанола.
Настоящее изобретение также предлагает способ получения монокристалла кристаллической формы D, отличающийся тем, что способ состоит из следующих стадий:
растворение соединения формулы (I) в растворителе для кристаллизации, фильтрация и сушка полученного кристалла с получением желаемого монокристалла кристалла формы D, где растворителем является смешанный растворитель из спирта и воды, спиртовой растворитель выбирают из группы, состоящей из метанола и этанола; соотношение спирта к воде составляет от 20:1 до 1:20 и предпочтительно - 6:1, способ кристаллизации выбирают из группы, состоящей из кристаллизации при комнатной температуре, кристаллизации при охлаждении, кристаллизации при испарении растворителя и кристаллизации, вызванной добавлением затравочного кристалла.
Настоящее изобретение также относится к фармацевтической композиции кристаллической формы А, В, С или D, характеризующейся тем, что она дополнительно содержит один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
Настоящее изобретение также относится к применению кристаллической формы А, В, С или D или фармацевтической композиции для приготовления лекарственного средства для лечения заболевания, связанного с ингибитором EZH2.
В соответствии с применением настоящего изобретения заболевание, связанное с ингибитором EZH2, выбирают из группы, состоящей из лимфомы, лейкоза, рака молочной железы, рака легкого, рака предстательной железы, рака яичника, рака печени, меланомы, рабдоидной опухоли, синовиальной саркомы, мезотелиомы, рака шейки матки, рака толстой кишки, рака прямой кишки, рака желудка, рака поджелудочной железы, рака мозга, рака кожи, рака ротовой полости, рака кости, рака почки, рака мочевого пузыря, опухоли маточной трубы, опухоли яичника, опухоли брюшины, глиомы, глиобластомы, опухоли головы и шеи, и миеломы; предпочтительно из лимфомы, лейкоза, рака молочной железы, рака легкого, рака предстательной железы, рака яичника, рака печени, меланомы, рабдоидной опухоли, синовиальной саркомы и мезотелиомы; лейкоз предпочтительно представляет собой хронический миелоидный лейкоз, острый миелоидный лейкоз или лейкоз смешанного происхождения; и лимфома предпочтительно представляет собой неходжкинскую лимфому, диффузную В-крупноклеточную лимфому или фолликулярную лимфому.
Полученные кристаллические формы соединения формулы (I) определяют с помощью спектра порошковой дифракции рентгеновских лучей (XRPD) и дифференциальной сканирующей калориметрии (DSC).
Способ перекристаллизации для кристаллической формы конкретно не ограничен и может быть осуществлен посредством обычного способа перекристаллизации. Например, материал, то есть соединение формулы (I), может быть растворен в органическом растворителе с последующим добавлением антирастворителя для кристаллизации. После завершения кристаллизации желаемый кристалл может быть получен путем фильтрации и сушки.
Способ кристаллизации согласно настоящему изобретению включает кристаллизацию при комнатной температуре, кристаллизацию при охлаждении, кристаллизацию при испарении растворителя, кристаллизацию, вызванную добавлением затравочного кристалла и т.п.; и температура охлаждения составляет менее 40°С, предпочтительно от -10°С до 40°С.
Исходным материалом, используемым в способе получения кристаллической формы согласно настоящему изобретению, может быть соединение формулы (I) в любой форме, причем конкретные формы включают, но не ограничиваются этим, аморфную форму, произвольные кристаллические формы и тому подобное.
Определения
В описании и формуле изобретения настоящей заявки, если не указано иное, используемые здесь научные и технические термины имеют значения, обычно понятные специалисту в данной области. Однако, чтобы лучше понять настоящее изобретение, даны определения и пояснения некоторых соответствующих терминов. Кроме того, когда определения и пояснения терминов, представленные в настоящей заявке, не соответствуют значениям, обычно понимаемым специалистом в данной области, определения и пояснения терминов, представленные в настоящей заявке, имеют преимущественную силу.
Термин «С1-6алкил», используемый в настоящем изобретении, относится к линейному или разветвленному алкилу, содержащему от 1 до 6 атомов углерода. Его конкретные примеры включают, но не ограничиваются ими: метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, 2-метилбутил, неопентил, 1-этилпропил, н-гексил, изогексил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 2-этилбутил, 1,2-диметилпропил и тому подобное.
Термин «простой эфирный растворитель», используемый в настоящем изобретении, относится к цепочечному соединению или циклическому соединению, имеющему эфирную связь -О- и имеющему от 1 до 10 атомов углерода. Его конкретные примеры включают, но не ограничиваются ими: тетрагидрофуран, диэтиловый эфир, метиловый эфир пропиленгликоля, метил-трет-бутиловый эфир и 1,4-диоксан.
Термин «спиртовой растворитель», используемый в настоящем изобретении, относится к группе, получаемой в результате замещения одного или более атомов водорода на «С1-6алкиле» одним или более «гидрокси», где «гидрокси» и «С1-6алкил» являются такими, как определено выше. Его конкретные примеры включают, но не ограничиваются ими: метанол, этанол, изопропанол, н-пропанол, изопентанол и трифторэтанол.
Термин «сложноэфирный растворитель», используемый в настоящем изобретении, относится к комбинации низшей органической кислоты с 1-4 атомами углерода и низшего спирта с 1-6 атомами углерода. Его конкретные примеры включают, но не ограничиваются ими: этилацетат, изопропилацетат или бутилацетат.
Термин «кетонный растворитель», используемый в настоящем изобретении, относится к соединению, в котором карбонильная группа (-С(О)) связана с двумя углеводородными группами. Кетоны можно подразделить на алифатические кетоны, алициклические кетоны, ароматические кетоны, насыщенные кетоны и ненасыщенные кетоны, в зависимости от углеводородной группы в молекуле. Его конкретные примеры включают, но не ограничиваются ими: ацетон, ацетофенон, метилизобутилкетон или метилпирролидон.
Термин «нитрильный растворитель», используемый в настоящем изобретении, относится к группе, полученной в результате замещения одного или более атомов водорода на «С1-6алкиле» одним или более «циано», где «циано» и «С1-6алкил» являются такими, как определено выше. Его конкретные примеры включают, но не ограничиваются ими: ацетонитрил или пропионитрил.
Термин «алифатический углеводородный растворитель», используемый в настоящем изобретении, относится к углеводородному соединению, имеющему основные свойства алифатического соединения и имеющему от 1 до 10 атомов углерода, где атомы углерода в молекуле связаны с цепочечным углеродным скелетом, в котором два конца открыты и не образуют кольцо, например, насыщенный алифатический углеводород, включая алкановый растворитель. Его конкретные примеры включают, но не ограничиваются ими: н-бутан, н-пентан, н-гексан или н-гептан.
Термин «алициклический углеводородный растворитель», используемый в настоящем изобретении, относится к углеводородному соединению, обладающему свойствами, подобными алифатическому углеводороду, и имеющему циклический углеродный скелет с 1-8 кольцевыми атомами. Его конкретные примеры включают, но не ограничиваются ими: циклопентан и циклогексан.
Термин «амидный растворитель», используемый в настоящем изобретении, относится к соединению, содержащему карбониламино (-C(O)N) и имеющему от 1 до 10 атомов углерода. Его конкретные примеры включают, но не ограничиваются ими: N,N-диметилформамид и N,N-диметилацетамид.
Термин «ареновый растворитель», используемый в настоящем изобретении, относится к общему термину для соединения с углеродным кольцом и его производного, где молекула имеет сопряженную систему замкнутого кольца, а число π-электронов соответствует правилу Геккеля. Его конкретные примеры включают, но не ограничиваются ими: изопропилбензол и ксилол.
Термин «галогенуглеводородный растворитель», используемый в настоящем изобретении, относится к группе, полученной из «С1-6алкила», в которой один или более атомов водорода замещены одним или более «атомов галогена», где «атом галогена» и «С1-6алкил» являются такими, как определено выше. Его конкретные примеры включают, но не ограничиваются ими: метилхлорид, дихлорметан, хлороформ и четыреххлористый углерод.
Термин «нитроалкановый растворитель», используемый в настоящем изобретении, относится к группе, производной от «С1-6алкила», в которой один или более атомов водорода замещены одним или более «нитро», где «С1-6алкил» является таким, как определено выше. Его конкретные примеры включают, но не ограничиваются ими: нитрометан.
Термин «смешанный растворитель», используемый в настоящем изобретении, относится к растворителю, полученному путем смешивания одного или более различных видов органических растворителей в определенном соотношении, или к растворителю, полученному путем смешивания органического растворителя и воды в определенном соотношении. Смешанный растворитель предпочтительно представляет собой смешанный растворитель из одного или более спиртов, смешанный растворитель из спирта и простого эфира, смешанный растворитель из спирта и алифатического углеводорода, смешанный растворитель из простого эфира и алифатического углеводорода, смешанный растворитель из спирта и воды, смешанный растворитель из галогенуглеводорода и нитрила, смешанный растворитель из амида и воды или смешанный растворитель из простого эфира и воды, где спирт, простой эфир, алифатический углеводород, галогенуглеводород, амид и нитрил являются такими, как определено выше.
Термин «спектр порошковой дифракции рентгеновских лучей» или «XRPD», используемый в настоящем изобретении, относится к спектру порошковой дифракции рентгеновских лучей, который получен в соответствии с формулой Брэгга 2d sinθ=nλ (где λ - длина волны рентгеновских лучей, 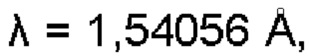 порядок дифракции n - любое положительное целое число, обычно принимающее значение дифракционного максимума первого порядка, n=1), когда рентгеновское излучение падает на определенную атомную плоскость кристалла или частичного образца кристалла, имеющего расстояние d между плоскостями, под углом скользящего падения θ (комплементарный угол падения, также называемый углом Брэгга), удовлетворяющему уравнению Брэгга.
порядок дифракции n - любое положительное целое число, обычно принимающее значение дифракционного максимума первого порядка, n=1), когда рентгеновское излучение падает на определенную атомную плоскость кристалла или частичного образца кристалла, имеющего расстояние d между плоскостями, под углом скользящего падения θ (комплементарный угол падения, также называемый углом Брэгга), удовлетворяющему уравнению Брэгга.
Термин «дифференциальная сканирующая калориметрия» или «DSC», используемый в настоящем изобретении, означает измерение разности температур и разности тепловых потоков между образцом и эталоном во время процесса нагревания или постоянной температуры образца, чтобы характеризовать все физические и химические изменения, связанные с этим тепловым воздействием, и для получения информации о фазовом изменении образца.
Термин «2θ» или «угол 2θ», используемый в настоящем изобретении, относится к углу дифракции, θ - это угол Брэгга, и единицей измерения является ° или градус. Диапазон погрешности 2θ составляет от ±0,1 до ±0,5, предпочтительно - от ±0,1 до ±0,3 и более предпочтительно ±0,2.
Термин «расстояние между плоскостями» или «межплоскостное расстояние (значение d)», используемый в настоящем изобретении, означает, что пространственная решетка характеризуется тремя непараллельными единичными векторами а, b, с, причем каждый из них соединяет две соседние точки решетки и эти три вектора делят решетку на расположенные рядом параллельные юкстагональные элементы, называемые межплоскостным расстоянием. Пространственная решетка делится в соответствии с определенными линиями элементов-параллелепипедов для получения набора линейных сеток, который называется пространственной решеткой или решеткой. Решетка отражает периодичность кристаллической структуры с геометрическими точками и линиями. Разные кристаллические плоскости имеют разные межплоскостные расстояния (то есть расстояния между двумя соседними параллельными кристаллическими плоскостями); единицей измерения является А или ангстрем.
Настоящее изобретение также относится к фармацевтической композиции, содержащей кристаллическую форму А, В, С или D соединения формулы (I) и, возможно, один или более фармацевтически приемлемых носителей и/или разбавителей. Из фармацевтической композиции может быть приготовлена любая из фармацевтически приемлемых лекарственных форм. Например, кристаллическая форма А, В, С или D соединения формулы (I) или фармацевтическая композиция согласно настоящему изобретению может быть приготовлена в виде таблетки, капсулы, пилюли, гранулы, раствора, суспензии, сиропа, инъекции (включая раствор для инъекций, стерильный порошок для инъекций и концентрированный раствор для инъекций), суппозитория, ингалятора или спрея.
Кроме того, фармацевтическую композицию согласно настоящему изобретению можно также вводить пациенту или субъекту, нуждающемуся в таком лечении, любым подходящим способом введения, таким как пероральное, парентеральное, ректальное, внутрилегочное или местное введение. Для перорального введения фармацевтическая композиция может быть приготовлена в виде пероральной композиции, например пероральной твердой композиции, такой как таблетка, капсула, пилюля, гранула и т.п.; или жидкой композиции для перорального применения, такой как пероральный раствор, пероральная суспензия, сироп и тому подобное. При приготовлении пероральной композиции фармацевтическая композиция может дополнительно содержать подходящий наполнитель, связующее, дезинтегратор, смазку и тому подобное. Для парентерального введения фармацевтическая композиция может быть приготовлена в виде препарата для инъекций, включающего раствор для инъекций, стерильный порошок для инъекций и концентрированный раствор для инъекций. При составлении композиции для инъекций фармацевтическая композиция может быть получена обычным способом современной фармацевтической промышленности. Когда готовят композицию для инъекций, в нее можно не добавлять дополнительный агент или можно добавить подходящий дополнительный агент, в зависимости от природы лекарственного средства. Для ректального введения фармацевтическая композиция может быть приготовлена в виде суппозитория и тому подобного. Для внутрилегочного введения фармацевтическая композиция может быть приготовлена в виде ингалятора или спрея и т.п. В определенных предпочтительных вариантах осуществления кристаллическая форма А, В, С или D соединения формулы (I) согласно настоящему изобретению присутствует в фармацевтической композиции или лекарственном средстве в терапевтически и/или профилактически эффективном количестве. В некоторых предпочтительных вариантах осуществления кристаллическая форма А, В, С или D соединения формулы (I) согласно настоящему изобретению присутствует в фармацевтической композиции или лекарственном средстве в стандартной дозе.
Кристаллическая форма А, В, С или D соединения формулы (I) согласно настоящему изобретению может быть использована для приготовления лекарственного средства для лечения заболевания, связанного с ингибитором EZH2. Следовательно, настоящая заявка также относится к применению кристаллической формы А, В, С или D соединения формулы (I) согласно настоящему изобретению для приготовления лекарственного средства для лечения заболевания, связанного с ингибитором EZH2. Кроме того, настоящая заявка также относится к способу ингибирования заболевания, связанного с ингибитором EZH2, включающему введение терапевтически и/или профилактически эффективного количества кристаллической формы А, В, С или D соединения формулы (I) настоящего изобретения или фармацевтической композиции согласно настоящему изобретению субъекту, нуждающемуся в этом.
В определенных предпочтительных вариантах осуществления изобретения заболевание представляет собой заболевание, связанное с ингибитором EZH2, выбранное из боли.
Полезные эффекты настоящего изобретения
По сравнению с предшествующим уровнем техники техническое решение согласно настоящему изобретению имеет следующие преимущества.
Исследования показали, что кристаллические формы А, В, С и D соединения формулы (I), полученного согласно настоящему изобретению, имеют хорошую стабильность и высокую чистоту. Получены монокристаллы кристаллических форм А и D. Кристаллические формы А, В, С и D соединения формулы (I), полученные с помощью технического решения согласно настоящему изобретению, могут соответствовать требованиям к производству, транспортировке и хранению лекарственных препаратов. Способ их получения является стабильным, воспроизводимым и контролируемым, и может быть адаптирован к промышленному производству.
Краткое описание графических материалов
На фиг. 1 показан спектр XRPD кристаллической формы А соединения формулы (I).
На фиг. 2 показан спектр XRPD кристаллической формы В соединения формулы (I).
На фиг. 3 показан спектр XRPD кристаллической формы В соединения формулы (I).
На фиг. 4 показан спектр XRPD кристаллической формы С соединения формулы (I).
На фиг. 5 показан спектр XRPD кристаллической формы С соединения формулы (I).
На фиг. 6 показан спектр XRPD кристаллической формы D соединения формулы (I).
На фиг. 7 показан спектр XRPD кристаллической формы D соединения формулы (I).
На фиг. 8 показан график 1 цикла динамической сорбции паров (DVS) кристаллической формы D соединения формулы (I).
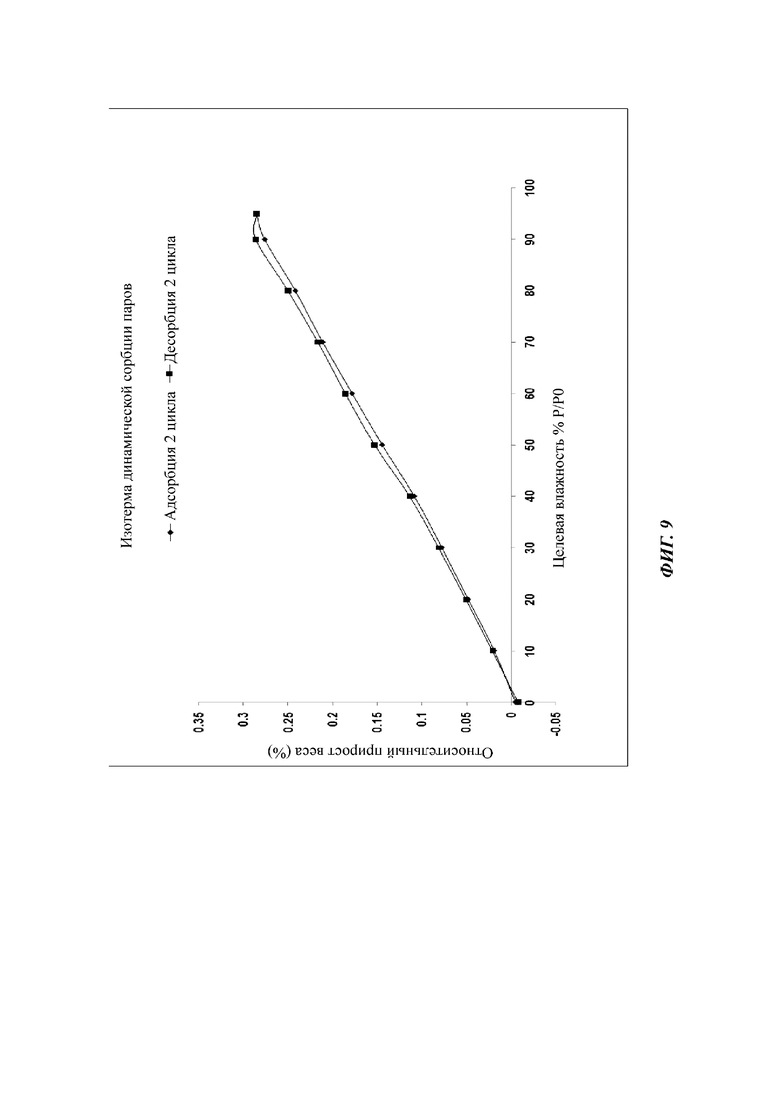
На фиг. 9 показан график 2 цикла DVS кристаллической формы D соединения формулы (I).
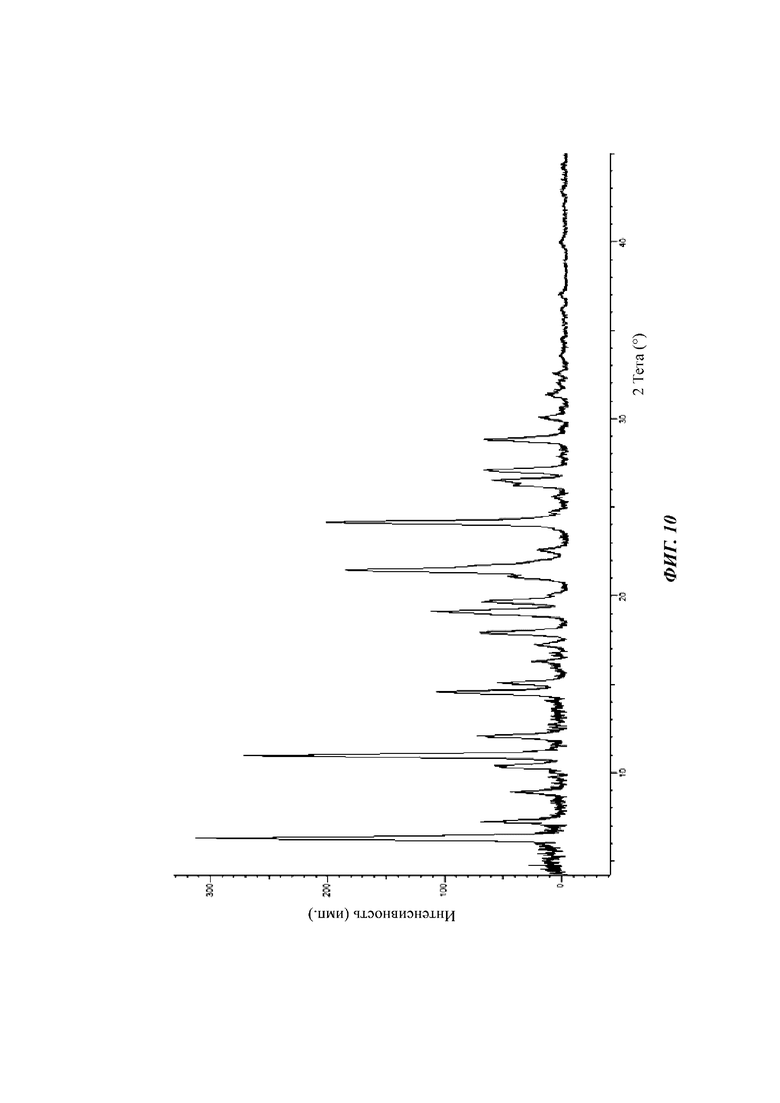
На фиг. 10 показан спектр XRPD кристаллической формы В соединения формулы (I) в день 0.
На фиг. 11 показан спектр XRPD кристаллической формы В соединения формулы (I) через 20 дней в условиях 25°С и относительной влажности 65%.
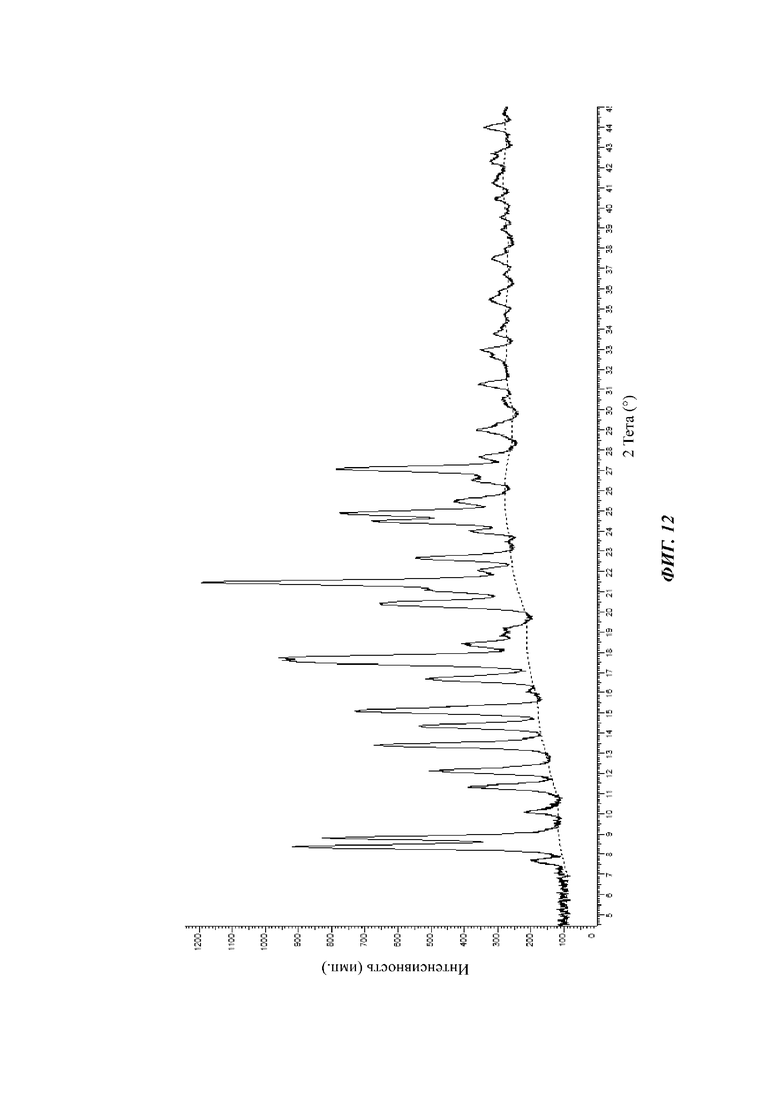
На фиг. 12 показан спектр XRPD кристаллической формы D соединения формулы (I) в день 0.
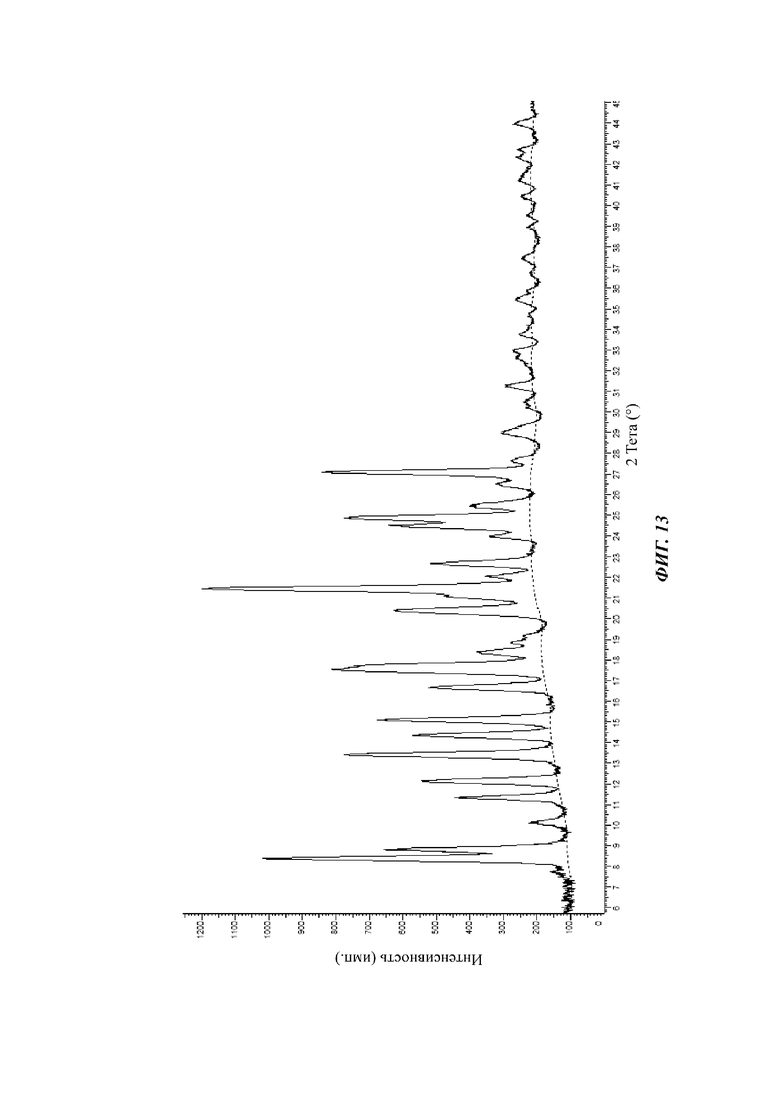
На фиг. 13 показан спектр XRPD кристаллической формы D соединения формулы (I) через 20 дней в условиях 40°С и относительной влажности 75%.
На фиг. 14 показан спектр XRPD кристаллической формы D соединения формулы (I) через 20 дней в условиях 25°С и относительной влажности 65%.
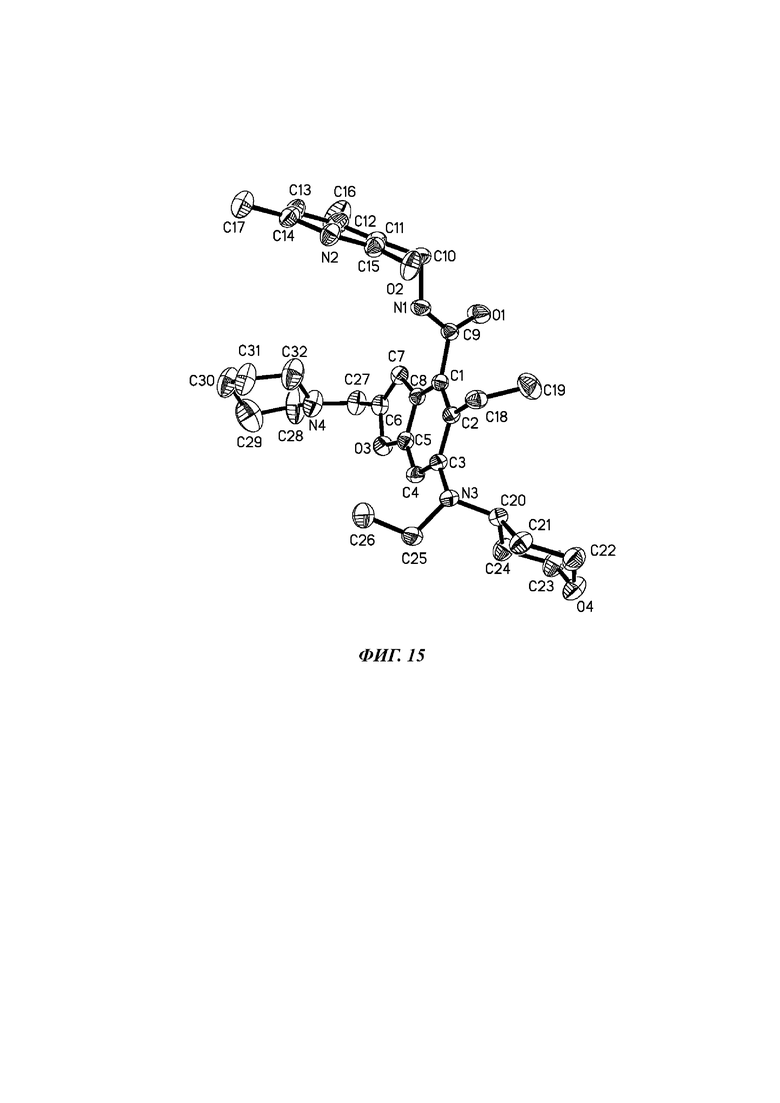
На фиг. 15 показана схема рентгеновской монокристаллической дифракционной молекулярной стереоструктуры кристаллической формы А соединения формулы (I).
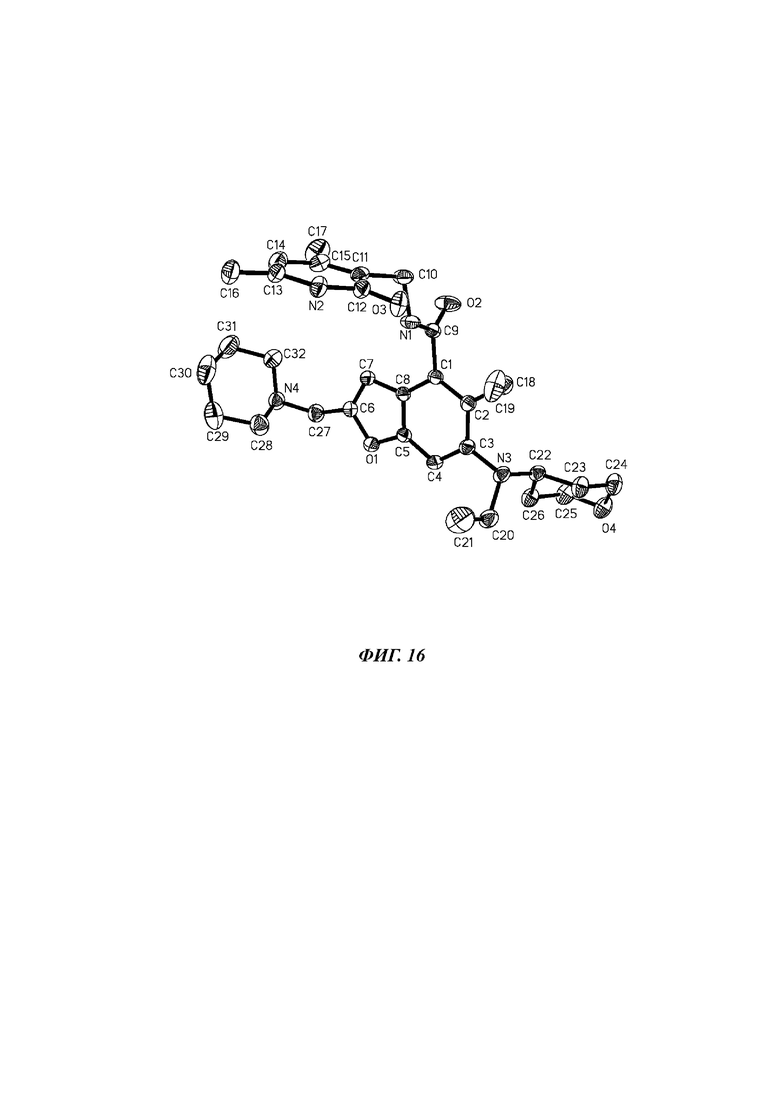
На фиг. 16 показана схема рентгеновской монокристаллической дифракционной молекулярной стереоструктуры кристаллической формы D соединения формулы (I).
На фиг. 17 показан спектр XRPD аморфной формы соединения формулы (I).
Подробное описание изобретения
Настоящее изобретение будет подробно проиллюстрировано следующими примерами. Примеры настоящего изобретения предназначены только для описания технического решения настоящего изобретения и их не следует рассматривать как ограничение сущности и объема настоящего изобретения.
Условия испытаний для приборов, используемых в экспериментах:
1. Дифференциальный сканирующий калориметр, DSC
Тип прибора: система Mettler Toledo DSC3 + STARe
Продувочный газ: азот (50 мл / мин)
Скорость нагрева: 10,0°С / мин
Диапазон температур: 20-250°С
2. Рентгеновская порошковая дифракция, XRPD
Тип прибора: порошковый дифрактометр BRUKER D8 Discover А25
Излучение: монохроматическое излучение Cu-Kα ( )
)
Режим сканирования: 9/29, диапазон сканирования: 10-48°
Напряжение: 40 кВ, Электрический ток: 40 мА
3. Динамическая сорбция паров, DVS
Тип прибора: DVS advantage
Температура: 25°С
Растворитель: вода
Изменение влажности: 0-95-0-95-0% относительной влажности, dm/dt=0,002
Сравнительный пример 1. Способ получения в примере 2 из WO 2017084494 (PCT/CN2016/104318)
Получение N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимида (соединения формулы (I))
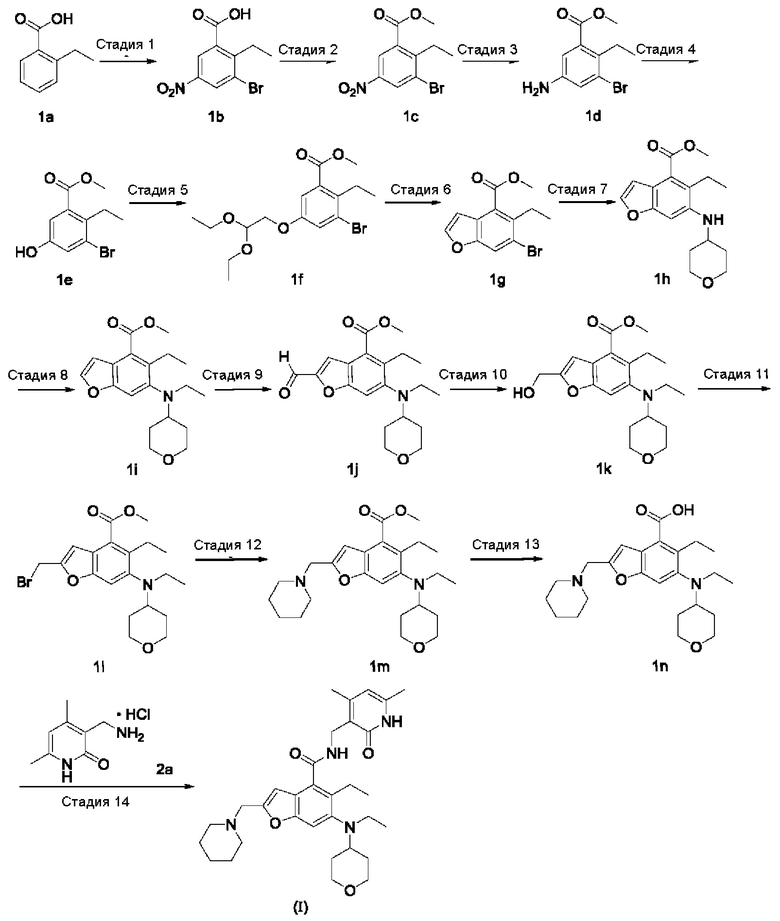
Стадия 1
3-Бром-2-этил-5-нитробензойная кислота 1b
2-Этилбензойную кислоту 1а (20,0 г, 133 ммоль, полученную в соответствии со способом, описанным в «Journal of American Chemical Society, 1991, 113 (13), 4931-6»), добавляли к 150 мл серной кислоты, затем добавляли порциями нитрат натрия (11,3 г, 133 ммоль) на ледяной бане. Реакционный раствор перемешивали в течение 3 часов, затем порциями добавляли N-бромсукцинимид (2,6 г, 14,5 ммоль). Реакционную систему перемешивали в течение 1 часа при 60°С. После завершения реакции реакционный раствор выливали в ледяную воду, хорошо перемешивали и фильтровали. Фильтрат промывали водой и концентрировали при пониженном давлении с получением сырого указанного в заголовке продукта 3-бром-2-этил-5-нитробензойной кислоты 1b (35 г) в виде белого твердого вещества, которое непосредственно использовали в следующей стадии без очистки.
Стадия 2
Метил-3-бром-2-этил-5-нитробензоат 1с
Неочищенную 3-бром-2-этил-5-нитробензойную кислоту 1b (35 г, 128 ммоль) растворяли в 200 мл N,N-диметилформамида, затем добавляли йодметан (21,8 г, 153 ммоль) и карбонат калия (35,3 г, 255 ммоль). Реакционную систему перемешивали в течение 2 часов при комнатной температуре. После завершения реакции реакционный раствор концентрировали при пониженном давлении. К реакционному раствору добавляли избыток воды и экстрагировали этилацетатом. Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого указанного в заголовке продукта метил-3-бром-2-этил-5-нитробензоата 1с (36 г) в виде желтого масла, которое использовали непосредственно в следующей стадии без очистки.
Стадия 3
Метил-5-амино-3-бром-2-этилбензоат 1d
Сырой метил-3-бром-2-этил-5-нитробензоат 1с (35,0 г, 121 ммоль) добавляли к 250 мл этанола и 150 мл воды. Реакционный раствор нагревали до 70°С, добавляли хлорид аммония (52,8 г, 969 ммоль), затем порциями добавляли порошок железа (34 г, 606 ммоль). Реакционную систему перемешивали в течение 2 часов при 70°С. После завершения реакции реакционный раствор фильтровали через целит в горячем состоянии. Осадок на фильтре промывали горячим этанолом, затем фильтрат объединяли и концентрировали при пониженном давлении. Добавляли этилацетат и насыщенный раствор бикарбоната натрия. Две фазы разделяли и водную фазу экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с использованием н-гексана и этилацетата в качестве элюента с получением указанного в заголовке продукта метил-5-амино-3-бром-2-этилбензоата 1d (22,0 г, выход 70%) в виде желтого твердого вещества.
Стадия 4
Метил-3-бром-2-этил-5-гидроксибензоат 1е
Метил-5-амино-3-бром-2-этилбензоат 1d (15,0 г, 58 ммоль) растворяли в 10 мл ацетонитрила, затем добавляли 200 мл 10%-ной серной кислоты. Реакционный раствор хорошо перемешивали и охлаждали до 3°С на ледяной бане с солью, затем по каплям добавляли 10 мл предварительно приготовленного раствора нитрита натрия (4,4 г, 64 ммоль). Реакционный раствор перемешивали в течение 4 часов при указанной температуре, по каплям добавляли 200 мл 50% серной кислоты, затем перемешивали в течение 1 часа при 90°С. После завершения реакции реакционный раствор трижды экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с использованием н-гексана и этилацетата в качестве элюента с получением указанного в заголовке продукта метил-3-бром-2-этил-5-гидроксибензоата 1е (5,5 г, выход 37%) в виде коричневого твердого вещества.
Стадия 5
Метил-3-бром-5-(2,2-диэтоксиэтокси)-2-этилбензоат 1f
Метил-3-бром-2-этил-5-гидроксибензоат 1е (35 г, 135 ммоль) растворяли в 200 мл N,N-диметилформамида, затем добавляли 2-бром-1,1-диэтоксиэтан (40 г, 202 ммоль) и карбонат калия (37 г, 269 ммоль). Реакционную систему перемешивали при 120°С в течение 12 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении для удаления N,N-диметилформамида. Реакционный раствор добавляли в воду и трижды экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с использованием н-гексана и этилацетата в качестве элюента с получением указанного в заголовке продукта метил-3-бром-5-(2,2-диэтоксиэтокси)-2-этилбензоата 1f (40 г, выход 80%) в виде светло-желтого масла.
Стадия 6
Метил-6-бром-5-этилбензофуран-4-карбоксилат 1g
Полифосфорную кислоту (30 г) добавляли к 400 мл толуола. Реакционный раствор нагревали до 100°С и добавляли 50 мл предварительно приготовленного раствора метил-3-бром-5-(2,2-диэтоксиэтокси)-2-этилбензоата 1f (40 г, 107 ммоль) в толуоле при перемешивании. Реакционный раствор перемешивали в течение 16 часов при 100°С. После завершения реакции супернатант декантировали. К остатку добавляли воду и этилацетат. Две фазы разделяли и водную фазу экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором карбоната натрия и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с использованием н-гексана и этилацетата в качестве элюента с получением указанного в заголовке продукта метил-6-бром-5-этилбензофуран-4-карбоксилата 1g (11,8 г, выход 39%) в виде желтого твердого вещества.
Стадия 7
Метил-5-этил-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 1h
Метил-6-бром-5-этилбензофуран-4-карбоксилат 1g (11,0 г, 39 ммоль), тетрагидро-2Н-пиран-4-амин (5,89 г, 58 ммоль), трис(дибензилиденацетон)дипалладий (3,6 г, 3,9 ммоль), (,9 ммоль) бис(дифенилфосфино)-1,1'-бинафталин (4,86 г, 7,8 ммоль) и карбонат цезия (38 г, 117 ммоль) растворяли в 100 мл толуола. Реакционный раствор перемешивали в течение 12 часов при 100°С. После завершения реакции реакционный раствор фильтровали через целит и осадок на фильтре промывали этилацетатом. Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с использованием н-гексана и этилацетата в качестве элюента с получением указанного в заголовке продукта метил-5-этил-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилата 1h (10,0 г, выход 85%) в виде желтого твердого вещества.
Стадия 8
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 1i
Метил-5-этил-6-((тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 1h (10,0 г, 0,033 ммоль) растворяли в 150 мл 1,2-дихлорэтана, затем добавляли ацетальдегид (7,2 г, 0,165 ммоль) и уксусную кислоту (9,9 г, 0,165 ммоль). Реакционный раствор перемешивали в течение 1 часа и добавляли триацетоксиборгидрид натрия (20,8 г, 0,1 ммоль). Реакционный раствор перемешивали в течение 12 часов при комнатной температуре. После завершения реакции реакционный раствор концентрировали при пониженном давлении, нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с использованием н-гексана и этилацетата в качестве элюента с получением указанного в заголовке продукта метил-5-этил-6-(этил(тетрагидро-2Н-пиран)-4-ил)амино)бензофуран-4-карбоксилата 1i (7,8 г, выход 71%) в виде белого твердого вещества.
MS m/z (LC-MS): 332.4 [М+1]
Стадия 9
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-формилбензофуран-4-карбоксилат 1j
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбокси-лат 1i (1,6 г, 4,8 ммоль) растворяли в 25 мл тетрагидрофурана. Реакционный раствор охлаждали до -70°С и по каплям добавляли 2,0 М диизопропиламида лития (3,6 мл, 7,3 ммоль) в атмосфере аргона. Реакционный раствор перемешивали в течение 90 минут и добавляли N.N-диметилформамид (536 мг, 7,3 ммоль). Реакционный раствор перемешивали в течение 2 часов, затем медленно нагревали до комнатной температуры. В реакционный раствор добавляли избыток хлорида аммония, хорошо перемешивали и трижды экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с использованием н-гексана и этилацетата в качестве элюента с получением указанного в заголовке продукта метил-5-этил-6-(этил-(тетрагидро-2Н-пиран-4-ил)амино)-2-формилбензофуран-4-карбоксилата 1j (1,3 г, выход 75%) в виде желтого масла.
MS m/z (ESI):360.2 [М+1]
Стадия 10
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(гидроксиметил)бензофу-ран-4-карбоксилат 1k
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-формилбензофуран-4-карбоксилат 1j (1,4 г, 3,9 ммоль) растворяли в 5 мл тетрагидрофурана и 10 мл метанола, затем добавляли боргидрид натрия (222 мг, 5,8 ммоль). Реакционный раствор перемешивали в течение 30 минут при комнатной температуре. После завершения реакции реакционный раствор концентрировали при пониженном давлении, добавляли воду и насыщенный раствор бикарбоната натрия и трижды экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с использованием н-гексана и этилацетата в качестве элюента с получением указанного в заголовке продукта метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(гидроксиметил)-бензофуран-4-карбоксилата 1k (1,4 г, выход 99%) в виде желтого масла.
Стадия 11
Метил-2-(бромметил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилат 1l
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(гидроксиметил)-бензофуран-4-карбоксилат 1k (1,0 г, 2,8 ммоль) растворяли в 30 мл тетрагидрофурана, затем по каплям добавляли трибромид фосфора (1,12 г, 4,2 ммоль). Реакционный раствор перемешивали в течение 12 часов при комнатной температуре. После завершения реакции реакционный раствор нейтрализовали насыщенным раствором бикарбоната натрия и трижды экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением сырого указанного в заголовке продукта метил-2-(бромметил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)бензофуран-4-карбоксилата 1l (1,15 г) в виде желтого масла, которое использовали непосредственно на следующей стадии без очистки.
Стадия 12
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)-бензофуран-4-карбоксилат 1m
Сырой метил-2-(бромметил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-бензофуран-4-карбоксилат 1l (1,15 г, 2,7 ммоль) растворяли в 15 мл ацетонитрила, затем по каплям добавляли 10 мл предварительно приготовленного раствора пиперидина (362 мг, 4,3 ммоль) в ацетонитриле. Реакционный раствор перемешивали в течение 30 минут при комнатной температуре. После завершения реакции реакционный раствор концентрировали при пониженном давлении и добавляли этилацетат и насыщенный раствор бикарбоната натрия. Две фазы разделяли и водную фазу экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с дихлорметаном и метанолом в качестве элюента с получением указанного в заголовке продукта метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)-бензофуран-4-карбоксилата 1m (1,2 г, выход 99%) в виде желтого масла.
MS m/z (LC-MS): 429.2[М+1]
Стадия 13
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензо-фуран-4-карбоновая кислота 1n
Метил-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил) бензофуран-4-карбоксилат 1m (1,2 г, 2,7 ммоль) растворяли в 5 мл тетрагидрофурана и 20 мл метанола, затем добавляли 5 мл 4 М раствора гидроксида натрия. Реакционный раствор перемешивали в течение 12 часов при 60°С. После завершения реакции добавляли концентрированную соляную кислоту, чтобы довести рН реакционного раствора до 4. Смесь концентрировали при пониженном давлении и остаток растворяли в смешанном растворителе дихлорметан и метанол (V:V=5:1) и фильтровали. Осадок на фильтре промывали смешанным растворителем из дихлорметана и метанола (V:V=5:1). Фильтраты объединяли и концентрировали при пониженном давлении с получением сырого указанного в заголовке продукта 5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)-бензофуран-4-карбоновой кислоты 1n (1,1 г) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии без очистки.
MS m/z (LC-MS): 415.2[М+1]
Стадия 14
N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид
(соединение формулы (I))
5-Этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)-бензофуран-4-карбоновую кислоту 1n (1,0 г, 2,4 ммоль) растворяли в 30 мл N,N-диметилформамида, затем добавляли 1-этил-3-(3-диметил-амипропил)-карбодиимид (696 мг, 3,6 ммоль), 1-гидроксибензотриазол (490 мг, 3,6 ммоль) и N,N-диизопропилэтиламин (1,56 г, 12,1 ммоль). Реакционный раствор перемешивали в течение 1 часа, затем добавляли 3-(аминометил)-4,6-диметил-пиридин-2(1Н)-она гидрохлорид 2а (593 мг, 3,0 ммоль), полученный в соответствии со способом, раскрытым в заявке на патент WO 2014097041). Реакционный раствор перемешивали в течение 12 часов при комнатной температуре. После завершения реакции в реакционный раствор добавляли избыток воды и экстрагировали смешанным растворителем из дихлорметана и метанола (V:V=8:1). Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали колоночной хроматографией на силикагеле с дихлорметаном и метанолом в качестве элюента с получением указанного в заголовке продукта N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимида (I) (750 мг, выход 57%) в виде белого твердого вещества.
MS m/z (ESI): 549.7 [М+1]
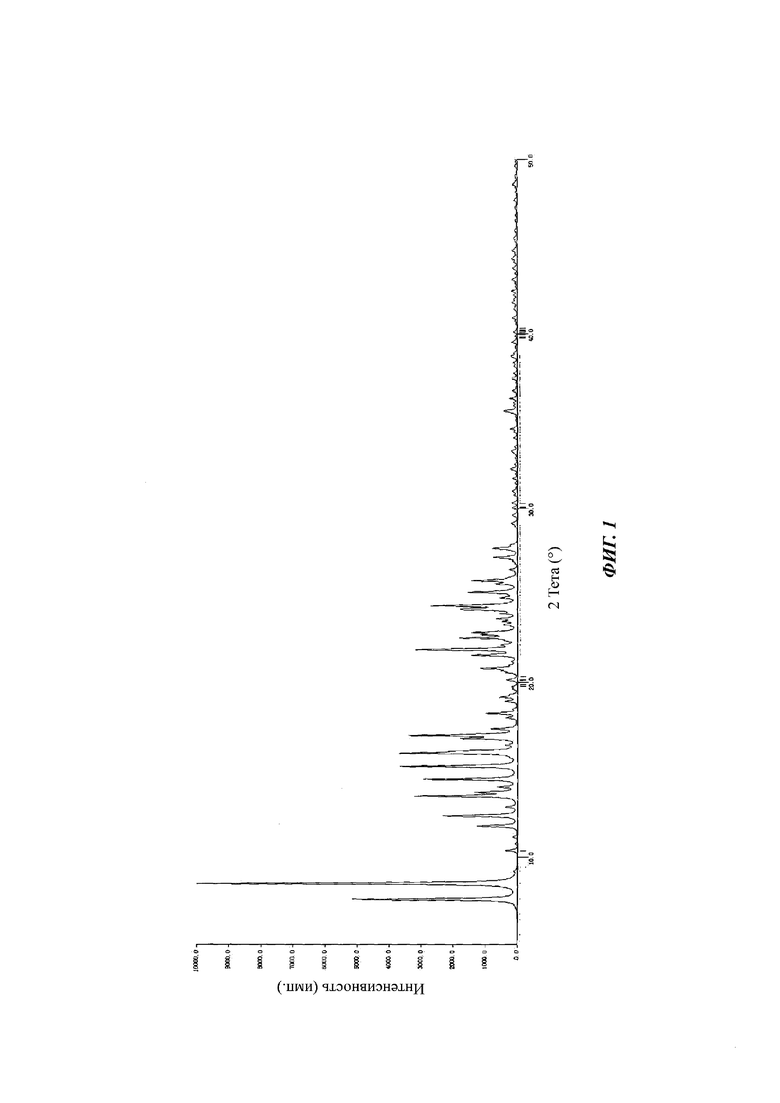
Белый твердый продукт идентифицировали по спектру XRPD как аморфную форму. Спектр XRPD аморфной формы показан на фиг. 17.
Пример 1 (Получение кристаллической формы А)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (20 мг, 0,036 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и добавляли смешанный растворитель дихлорметан-ацетонитрил (об/об, 1:10, 500 мкл). Реакционный раствор перемешивали при 25°С в течение одного часа и фильтровали. Фильтрат помещали в чистую колбу и медленно выпаривали до полного высушивания растворителя с получением твердого вещества от белого до бледно-желтого цвета. Спектр XRPD твердого образца показан на фиг. 1, а положения характеристических пиков показаны в следующей таблице:
Пример 2 (Получение монокристалла кристаллической формы А)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)-бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и растворяли в смешанном растворителе из ацетонитрила и дихлорметана (об/об, 10:1, 1 мл). Горловину колбы закрывали герметизирующей пленкой, в которой были пробиты два-три маленьких отверстия. Растворитель испаряли до получения монокристалла. Образец одномолекулярной стереоструктуры кристалла показан на фиг. 15 с помощью дифракции рентгеновских лучей на монокристалле (XRD), а параметры элементарной ячейки показаны в следующей таблице:
Пример 3 (Получение кристаллической формы В)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и растворяли в этаноле (300 мкл). Горловину колбы закрывали герметизирующей пленкой, в которой были пробиты два-три маленьких отверстия. Раствор оставляли при комнатной температуре до тех пор, пока растворитель не испарился досуха, чтобы получить твердое вещество от белого до бледно-желтого цвета. Спектр XRPD образца кристалла показан на фиг. 2. Спектр DSC показан на фиг. 3, на которой имеется один эндотермический пик и один экзотермический пик в течение периода нарастания температуры, начало экзотермического пика составляет примерно 123, 69°С и начальная температура плавления составляет около 206,31°С. Кристаллическая форма была определена как кристаллическая форма В, и положения характеристических пиков показаны в следующей таблице:
Пример 4 (Получение кристаллической формы В)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и растворяли в этаноле (300 мкл). В раствор добавляли воду (900 мкл) в качестве антирастворителя для осаждения кристаллов. Смесь фильтровали и сушили с получением бледно-желтого твердого вещества. Кристаллический образец был идентифицирован как кристаллическая форма В методом XRPD.
Пример 5 (Получение кристаллической формы С)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и растворяли в 1,4-диоксане (1 мл). К раствору добавляли н-гептан (2 мл) для осаждения кристаллов. Смесь фильтровали и сушили с получением порошка от белого до бледно-желтого цвета. Спектр XRPD образца кристалла показан на фиг. 4. Спектр DSC показан на фиг. 5, на которой имеется множество эндотермических пиков. Кристаллическая форма была определена как кристаллическая форма С, и положения характеристических пиков показаны в следующей таблице:
Пример 6 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (50 мг, 0,091 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и добавляли метанол (100 мкл). Раствор перемешивали при 25°С в течение пяти дней, фильтровали и сушили в вакууме с получением порошка от белого до бледно-желтого цвета. Спектр XRPD образца кристалла показан на фиг. 6. Спектр DSC показан на фиг. 7, где температура начала плавления составляет около 205,45°С. Кристаллическая форма была определена как кристаллическая форма D. Из графиков DVS на фиг. 8 и 9 видно, что кристаллическая форма D не имеет явной гигроскопичности. Положения характеристических пиков показаны в следующей таблице:
Пример 7 (Получение монокристалла кристаллической формы D) Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и растворяли в смешанном растворителе из метанола и чистой воды (об/об, 6:1, 1 мл). Горловину колбы закрывали герметизирующей пленкой, в которой были пробиты два-три маленьких отверстия. Растворитель выпаривали до получения монокристалла. Одномолекулярная стереоструктура образца кристалла показана на фиг. 16 методом дифракции рентгеновских лучей на монокристалле (XRD), а параметры элементарной ячейки показаны в следующей таблице:


Пример 8 (Получение монокристалла кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и растворяли в смешанном растворителе из этанола и чистой воды (по объему, 6:1, 1 мл). Горловину колбы закрывали герметизирующей пленкой, в которой были пробиты два-три маленьких отверстия. Растворитель выпаривали для получения кристалла, который идентифицировали как монокристалл кристаллической формы D с помощью дифракции рентгеновских лучей на монокристалле (XRD).
Пример 9 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и добавляли этилацетат (300 мкл). Смесь суспендировали при комнатной температуре в течение двух часов, фильтровали и сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 10 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и добавляли метиловый эфир пропиленгликоля (300 мкл). Смесь суспендировали при комнатной температуре в течение двух часов, фильтровали и сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 11 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и добавляли циклогексан (300 мкл). Смесь суспендировали при 25°С в течение двух часов, фильтровали и сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 12 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид ((30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и добавляли ксилен (300 мкл). Смесь суспендировали при 25°С в течение двух часов, фильтровали и сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 13 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и добавляли ацетонитрил (300 мкл). Смесь суспендировали при 50°С в течение двух часов, фильтровали и сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 14 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и добавляли ацетон (300 мкл). Смесь суспендировали при 50°С в течение двух часов, фильтровали и сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 15 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и добавляли нитрометан (300 мкл). Смесь суспендировали при 50°С в течение двух часов, фильтровали и сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 16 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и добавляли диметилформамид (300 мкл). Смесь суспендировали в течение двух часов, фильтровали при 50°С и сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 17 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученные в сравнительном примере 1, добавляли в реакционную колбу и растворяли в изопропаноле (1 мл). Растворитель выпаривали и полученное твердое вещество затем сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 18 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и растворяли в смешанном растворителе из метанола и воды (об/об, 19:1, 300 мкл). Растворитель выпаривали, а полученное твердое вещество затем фильтровали и сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 19 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и растворяли в диоксане (1 мл). Растворитель выпаривали, а полученное твердое вещество затем сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 20 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и растворяли в тетрагидрофуране (300 мкл). Растворитель выпаривали, а полученное твердое вещество затем фильтровали и сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 21 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и добавляли диметилсульфоксид (100 мкл). Смесь суспендировали при 50°С, фильтровали и сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 22 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и добавляли дихлорметан (100 мкл). Смесь суспендировали при 25°С, фильтровали и сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 23 (Получение кристаллической формы D)
Сырой продукт N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксимид (30 мг, 0,055 ммоль), полученный в Сравнительном примере 1, добавляли в реакционную колбу и добавляли метанол (100 мкл). Смесь суспендировали при 50°С, фильтровали и сушили с получением бледно-желтого твердого вещества, которое с помощью XRPD было идентифицировано как кристаллическая форма D.
Пример 24. Исследование долгосрочной, ускоренной стабильности кристаллических форм В и D
Образец кристаллической формы В распределяли по поверхности на воздухе для испытания стабильности образца в условиях 25°С и 65% относительной влажности (RH), и производили отбор проб в день 20. Образец кристаллической формы D распределяли по поверхности на воздухе для испытания стабильности образца в условиях 25°С и 75% относительной влажности, и 25°С и 65% относительной влажности, производили отбор проб в день 20.
Результаты испытания:
На фиг. 10 показан спектр XRPD кристаллической формы В в день 0.
На фиг. 11 показан спектр XRPD кристаллической формы В через 20 дней в условиях 25°С и 65% относительной влажности в течение 20 дней.
На фиг. 12 показан спектр XRPD кристаллической формы D в день 0.
На фиг. 13 показан спектр XRPD кристаллической формы D через 20 дней в условиях 40°С и 75% относительной влажности в течение 20 дней.
На фиг. 14 показан спектр XRPD кристаллической формы D через 20 дней в условиях 25°С и 65% относительной влажности в течение 20 дней.
Заключение испытаний:
Результаты исследования стабильности, показанные на фиг. 12, 13 и 14, показывают, что пики XRPD кристаллической формы D соединения формулы (I) существенно не изменились в условиях нахождения при 40°С и 75% относительной влажности и кристаллическая форма является стабильной. Результаты, показанные на фиг. 10 и 11, показывают, что пики XRPD кристаллической формы В соединения формулы (I) значительно изменились в условиях нахождения при 25°С и 65% относительной влажности. Можно видеть, что физическая стабильность кристаллической формы D лучше, чем кристаллической формы В в условиях нахождения при 25°С и 65% относительной влажности.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ БЕНЗОФУРАНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2016 |
|
RU2727198C2 |
| ПРИМЕНЕНИЕ ИНГИБИТОРА EZH2 В КОМБИНАЦИИ С ИНГИБИТОРОМ BTK В ПОЛУЧЕНИИ ЛЕКАРСТВЕННОГО СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ОПУХОЛИ | 2018 |
|
RU2762893C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| ПРОИЗВОДНОЕ ПИРИДОНА, ИМЕЮЩЕЕ ТЕТРАГИДРОПИРАНИЛМЕТИЛЬНУЮ ГРУППУ | 2015 |
|
RU2707953C2 |
| СОЛЕВАЯ ФОРМА ИНГИБИТОРА ГИСТОН-МЕТИЛТРАНСФЕРАЗЫ EZH2 ЧЕЛОВЕКА | 2013 |
|
RU2658911C2 |
| АРИЛ-ИЛИ ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2632193C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО БЕНЗОФУРАНА | 2018 |
|
RU2777624C2 |
| НОВЫЕ КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ-IV | 2012 |
|
RU2598072C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2629118C2 |
| ТОЗИЛАТНАЯ СОЛЬ ПРОИЗВОДНОГО 5-ПИРАЗОЛИЛ-2-ПИРИДОНА, ПОЛЕЗНАЯ В ЛЕЧЕНИИ COPD | 2010 |
|
RU2526038C2 |







Изобретение относится к кристаллической форме D соединения формулы (I), которая имеет спектр порошковой дифракции рентгеновских лучей, полученный с использованием Cu-Kα-излучения и представленный углом дифракции 2θ, который имеет характеристические пики при углах дифракции 2θ: 8,41, 8,85, 11,38, 12,18, 13,45, 15,15, 16,73, 17,59, 17,68, 20,45, 21.51, 22,72, 24,53, 24,91 и 27,11, где диапазон погрешности угла 2θ каждого характеристического пика составляет ±0,2. Кристаллическую форму D получают растворением соединения формулы (I) в растворителе для кристаллизации, фильтрацией и сушкой полученного кристалла с получением желаемой кристаллической формы D, где растворитель выбирают из группы, включающей метанол, изопропанол, простой эфирный растворитель, смешанный растворитель из метанола и воды и смешанный растворитель из этанола и воды, причем простой эфирный растворитель представляет 1,4-диоксан. Способ кристаллизации представляет собой кристаллизацию при испарении растворителя. Изобретение относится также к вариантам способа получения кристаллической формы D соединения формулы I. Кристаллическая форма D соединения формулы I по изобретению предназначена для приготовления лекарственного средства для ингибиторования EZH2. Технический результат - кристаллической формы D соединения формулы I, обладающая свойством стабильности. 4 н. и 1 з.п. ф-лы, 17 ил., 6 табл., 24 пр.
1. Кристаллическая форма D соединения формулы (I), отличающаяся тем, что кристаллическая форма D имеет спектр порошковой дифракции рентгеновских лучей, полученный с использованием Cu-Kα-излучения и представленный углом дифракции 2θ, который имеет характеристические пики при углах дифракции 2θ: 8,41, 8,85, 11,38, 12,18, 13,45, 15,15, 16,73, 17,59, 17,68, 20,45, 21.51, 22,72, 24,53, 24,91 и 27,11, где диапазон погрешности угла 2θ каждого характеристического пика составляет ±0,2
2. Кристаллическая форма D по п. 1, отличающаяся тем, что имеет характеристические пики при углах дифракции 2θ: 8,41, 8,85, 10,15, 11,38, 12,18, 13,45, 14,40, 15,15, 16,73, 17,59, 17,68, 18,42, 18,91, 19,22, 20,45, 21,15, 21,51, 22,11, 22,72, 24,03, 24,53, 24,91, 25,54, 26,54, 27,11, 27,61, 29,04, 30,49, 31,31, 33,00, 33,88, 35.52, 37,53, 40,46, 41,36, 42,40 и 44,02, где диапазон погрешности угла 2θ каждого характеристического пика составляет ±0,2.
3. Способ получения кристаллической формы D по любому из пп. 1 и 2, отличающийся тем, что способ выбирают из группы, состоящей из:
способа I, включающего растворение соединения формулы (I) в растворителе для кристаллизации, фильтрацию и сушку полученного кристалла с получением желаемой кристаллической формы D, где растворитель выбирают из группы, включающей метанол, изопропанол, простой эфирный растворитель, смешанный растворитель из метанола и воды и смешанный растворитель из этанола и воды, причем простой эфирный растворитель представляет 1,4-диоксан, и способ кристаллизации представляет собой кристаллизацию при испарении растворителя;
способа II, включающего растворение соединения формулы (I) в хорошем растворителе, добавление антирастворителя для кристаллизации, фильтрацию и сушку полученного кристалла с получением желаемой кристаллической формы D, где хороший растворитель представляет собой простой эфирный растворитель, причем простой эфирный растворитель представляет собой тетрагидрофуран, а антирастворителем является вода;
способа III, включающего добавление соединения формулы (I) к растворителю, суспендирование этой смеси, фильтрацию и сушку полученного кристалла с получением желаемой кристаллической формы D, где растворитель выбирают из группы, состоящей из сложного эфира, простого эфира, алициклического углеводорода, нитроалканового растворителя, арена, спирта, нитрила, галогенуглеводорода, кетона, сульфоксида и амида, где сложноэфирный растворитель представляет собой этилацетат, простой эфирный растворитель представляет собой метиловый эфир пропиленгликоля, алициклический углеводород представляет собой циклогексан, нитроалкановый растворитель представляет собой нитрометан, ареновый растворитель представляет собой ксилол, спиртовой растворитель представляет собой метанол, нитрильным растворителем является ацетонитрил, галогенуглеводородным растворителем является дихлорметан, кетоновым растворителем является ацетон, сульфоксидом является диметилсульфоксид, амидный растворитель представляет собой N,N-диметилформамид.
4. Фармацевтическая композиция, обладающая ингибиторной активностью в отношении EZH2, содержащая эффективное количество кристаллической формы D по любому из пп. 1 и 2, характеризующаяся тем, что дополнительно содержит один или более фармацевтически приемлемых носителей или эксципиентов.
5. Применение кристаллической формы D по любому из пп.1 и 2, или фармацевтической композиции по п.4 для приготовления лекарственного средства для ингибиторования EZH2.
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| RU 2014117632 A, 10.11.2015 | |||
| Дж | |||
| Бернштейн "Полиморфизм молекулярных кристаллов" Москва, Наука, 2007, гл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Биодоступность, с.324-330 | |||
| MINO R | |||
| CAIRA: "Crystalline Polymorphism of Organic Compounds", TOPICS IN CURRENT CHEMISTRY, 1998, vol.198, pp.163-208. | |||

