ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к стереоспецифическому синтезу 3-гидрокси-5β-Н-стероидных сапогенинов и их производных.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Было показано, что некоторые сапогенины и их производные (более конкретно, сапогенины, имеющие 5β-атом водорода, и еще более конкретно, соединения, имеющие 3-гидроксильную группу и 5β-атом водорода, такие как сарсасапогенин, эписарсасапогенин, смилагенин и эписмилагенин) применяются в лечении когнитивной дисфункции и других состояний. Такая активность описана, например, в WO-99/48482, WO-99/48507, WO-01/49703, WO-02/079221 и WO-01/23406, описания которых включены в настоящую работу посредством ссылки. Используемая здесь схема наименования кольцевой системы и положений атомов углерода такая, как определено в этих предшествующих публикациях.
В литературе описаны способы синтеза 3-гидроксистероидов и 3-гидроксистероидных сапогенинов. Например, синтез 3β-гидрокси-5α-Н-стероидов из соответствующих 3-кето-5β-Н-стероидов был осуществлен с боргидридом натрия в тетрагидрофуране и метаноле или с использованием алюмогидрида лития в диэтиловом эфире (Helv. Chim. Acta, 66, 192-217 (1983)).
Патент US 3875195 (1975), содержание которого включено в настоящую работу посредством ссылки, описывает каталитическое восстановление 3-кето-5β-Н-стероидов до 3β-гидрокси-5β-Н-стероидов в низшей карбоновой кислоте с никелевым катализатором Ренея и водородом под давлением. Эти авторы показывают, что восстановление по Меервейну-Пондорфу-Верлею (Meerwein-Ponndorf-Verley, MPV) дает смеси 3α- и 3β-гидроксистероидов в равных частях. Разделение таких смесей, как сообщают, является трудным.
После введения семейства высоко стерически затрудненных триалкилборгидридных восстанавливающих агентов, общеизвестных как Selectrides®, начиная с ранних 1970-х г.г. (Brown et al., J. Am. Chem. Soc. 94, 7159-7161 (1972)), появился ряд публикаций, в которых эти восстанавливающие агенты были использованы в способах синтеза некоторых стеринов. Например, в Steroids, 36, 299-303 (1980), Steroids, 45, 39-51 (1985), J. Chem. Soc. Commun. 1239-1240 (1982), Tetrahedron, 40, 851-854 (1984), Helv. Chim. Acta, 66, 192-217 (1983), патенте US 6150336 (2000), и Tetrahedron, 45, 3717-3730 (1989), описание которых включены в настоящую работу посредством ссылки, описаны стереоспецифические восстановления с помощью селектридов некоторых 3-кето-5β- и 3-кето-5α-стероидов до их соответствующих 3β-ОН, 5β-Н- и 3α-ОН, 5α-Н-стеринов.
Что касается стероидных сапогенинов, то в данной области техники описан синтез смилагенина путем восстановления смилагенона изопропоксидом алюминия в изопропиловом спирте, MPV восстановление (Marker et al., J. Amer. Chem. Soc., 62, 2525 (1940)). Маркер описал MPV восстановление сарсасапогенона с получением смеси сарсасапогенина и эписарсасапогенина (Marker and Rohrmann, J. Amer. Chem. Soc., 61, 943 (1939)). Описания этих публикаций включены в настоящую работу посредством ссылки.
В данной области техники были описаны также некоторые способы каталитической гидрогенизации (восстановления), как показано на примере получения эпитигогенина из тигогенона по Блундену (Blunden) с использованием гидрогенизации над катализатором Адамса (Adams) (оксид платины (IV)) в ледяной уксусной кислоте, содержащей 2% соляной кислоты (J. Nat. Prod. 42, 478-482 (1979); Onderstepoort J. Vet. Res., 61, 351-359 (1994)). Маркер описал гидрогенизацию сарсасапогенона с использованием катализатора Адамса в этаноле с получением эписарсасапогенина (Marker and Rohrmann, J. Amer. Chem. Soc., 61, 943 (1939)). В данной области техники было описано также восстановление с помощью боргидрида натрия, как показано на примере получения эписарсасапогенина из сарсасапогенона по Майлсу (Miles) с использованием боргидрида натрия (J. Agric. Food Chem., 41, 914-917 (1993)). В данной области техники было описано также восстановление с помощью алюмогидрида лития, как показано на примере получения эписмилагенина из смилагенона по Дьерасси (Djerassi) (J. Am. Chem. Soc., 74, 422-424, (1952)) и получения эписарсасапогенина из сарсасапогенона по Леджису (Lajis) (Steroids, 58, 387-389 (1993)). Описания этих публикаций включены в настоящую заявку посредством ссылки.
Патенты US 5703052 (1997), 5807834 (1998) и 5939398 (1999), описания которых в настоящую заявку посредством ссылки, описывают способы синтеза 3α-гидрокси-5α-Н-сапогенинов с использованием K-Selectride® при низких температурах.
В Примере 6 WO 02/079221 (опубликованной 10 октября 2002 года) описан синтез эписарсасапогенина путем восстановления сарсасапогенона с использованием три-трет-бутоксиалюмогидрида лития. Однако эта публикация не относится к предшествующему уровню техники во всех странах.
В настоящем изобретении предложен улучшенный стереоспецифический синтез 3-гидрокси-5β-водородных стероидных сапогенинов, и более предпочтительно 3β-гидрокси,5β-Н-сапогенинов, определенных и описанных в упомянутых публикациях WO 99/48482, WO 99/48507, WO 01/49703, WO 02/079221 и WO 01/23406, а также их производных, таких как, например, соответствующие сапонины и другие физиологически приемлемые формы, такие как соли и сложные эфиры, которые могут служить в качестве пролекарств.
В наиболее предпочтительном воплощении настоящего изобретения предложен эффективный стереоспецифический синтез сарсасапогенина, смилагенина, эписарсасапогенина, эписмилагенина и их пролекарств и других физиологически приемлемых форм.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В первом аспекте настоящего изобретения предложен способ стереоспецифического получения 3-гидрокси-5β-Н-стероидного сапогенина или его производного, включающий восстановление 3-кето-5β-Н-стероидного сапогенина с использованием восстанавливающего агента, содержащего стерически затрудненный органоборан или органоалюмогидрид.
3-Гидрокси-5β-Н-стероидный сапогенин, первоначально образованный с помощью стереоспецифического восстановления, затем может быть превращен в целевую производную форму, например, с использованием соответствующих методов получения производных, хорошо известных в данной области техники. Указанное превращение может происходить in situ или в другой реакционной системе и может осуществляться одновременно с восстановлением или после него.
Используемый здесь термин "стерически затрудненный органоборан" относится, в частности, к восстанавливающим агентам, содержащим триалкил- или триарилборгидрид щелочного металла, таким как, например, три-втор-бутилборгидрид лития, трисиамилборгидрид лития или трифенилборгидрид лития, или соответствующим восстанавливающим агентам, в которых литий заменен калием или натрием. Алкильные группы предпочтительно содержат от 1 до 7 атомов углерода, наиболее предпочтительно от 3 до 7 атомов углерода. Арильные группы предпочтительно содержат от 6 до 12 атомов углерода и могут быть алкил-замещенными. Такие восстанавливающие агенты иногда в совокупности называют как восстанавливающие агенты типа "Selectride", хотя следует учитывать, что используемый здесь термин "Selectride" не предназначен для ограничения изобретения восстанавливающим агентом, полученным от какого-либо конкретного производителя или из какого-либо конкретного источника, и такие восстанавливающие агенты могут быть использованы от любого производителя или из любого источника. Для более детального рассмотрения авторы изобретения ссылаются на "Reductions by the Alumino- and Borohydrides in Organic Synthesis" ("Реакции восстановления алюмо- и боргидридами в органическом синтезе"), by J. Seyden-Penne (VCH Publishers, Inc.). Предпочтительные стерически затрудненные органобораны для применения в настоящем изобретении представляют собой три-втор-бутилборгидрид лития (L-Selectride), три-втор-бутилборгидрид калия (К-Selectride), три-втор-бутилборгидрид натрия (N-Selectride), трисиамилборгидрид лития (LS-Selectride), трисиамилборгидрид калия (KS-Selectride), трифенилборгидрид калия и трифенилборгидрид лития.
Используемый здесь термин "органоалюмогидрид" относится, в частности, к любому восстанавливающему агенту, содержащему алюминий и гидридные группировки и органические группы (например, алкил или алкокси, содержащие соответственно от 1 до 7 атомов углерода), такие как бис-(2-метоксиэтокси)-алюмогидрид натрия (Red-Al®), диизобутил-алюмогидрид (DIBAL) или три-трет-бутоксиалюмогидрид лития (LTBA). Для более детального рассмотрения авторы изобретения ссылаются на "Reductions by the Alumino- and Borohydrides in Organic Synthesis", by J. Seyden-Penne (VCH Publishers, Inc.). Предпочтительные органоалюмогидриды для применения в настоящем изобретении представляют собой Red-AI, DIBAL и LTBA.
Термин "производное", используемый здесь в отношении сапогенинов, относится, в частности, к соответствующим сапонинам и другим физиологически приемлемым формам, таким как соли и сложные эфиры, которые могут служить в качестве пролекарств. Сапогенины и их производные могут легко взаимопревращаться с помощью реакций, которые хорошо известны в данной области техники. Производные формы сапогенинов могут нести производные группы в любом желаемом одном или более чем одном положении молекулы. Сапонин и сложноэфирные производные могут нести, например, производные группы в положении 3 в кольце А. Следует иметь в виду, что используемое здесь выражение "сапогенин" включает все его производные формы, если иное не следует из контекста.
В связи с третьим аспектом настоящего изобретения, описанным ниже, используемый здесь термин "производное" относится также к активированным производным сапогенинов, используемых в этой реакции.
Благодаря соответствующему выбору восстанавливающего агента, этот способ позволяет получить ряд 3α-гидрокси, 5β-Н- и 3β-гидрокси, 5β-Н-сапогенинов и их производных по существу или, по меньшей мере, преимущественно в стереоизометрически чистой форме с хорошим или превосходным общим выходом (например, выше примерно 80% конверсии) из имеющегося в продаже или легко получаемого исходного материала, обычно с исключением трудного разделения изомерной смеси.
Применение стерически затрудненных органоборановых или органоалюмогидридных восстанавливающих агентов ранее не было использовано для 3-кето-5β-Н-стероидных сапогенинов. В получении эписарсасапогенина по Майлсу (J. Agric. Food Chem., 41, 914-917 (1993)), описание которого включено в настоящую работу посредством ссылки, в качестве восстанавливающего реагента использовали боргидрид натрия, хотя более селективный реагент LTBA был известен в то время, когда эта работа появилась.
Если восстанавливающим агентом является относительно высоко стерически затрудненный органоборан (органические группы более примерно двух атомов углерода), то полученный сапогенин может представлять собой преимущественно 3β-гидрокси,5β-Н-сапогенин.
Если восстанавливающим агентом является относительно менее стерически затрудненный органоборан (органические группы до примерно двух атомов углерода), то полученный сапогенин может представлять собой преимущественно 3α-гидрокси,5β-Н-сапогенин.
Если восстанавливающим агентом является органоалюмогидрид, то полученный сапогенин может представлять собой преимущественно 3α-гидрокси, 5β-Н-сапогенин.
Выражение "3-кето, 5β-Н-сапогенин" используют здесь для удобства, чтобы сослаться на исходное вещество для восстановления, и не обязательно подразумевают насыщение или отсутствие кетогрупп в других частях молекулы, например, за пределами кольца А, при условии, что если необходимо, то любые нежелательные реакционные центры в других частях молекулы будут соответствующим образом защищены. Исходное вещество 3-кето,5β-Н-сапогенин может отличаться от целевого конечного продукта в частях молекулы, отличающихся от положения 3 в кольце А; в этом случае необходимое превращение(я) будет осуществляться способами, известными специалистам в этой области техники, как часть общего пути синтеза, ведущего к целевому конечному продукту.
Исходное вещество 3-кето,5β-Н-стероидный сапогенин может быть соответствующим образом получено путем окисления соответствующего 3-ОН-сапогенина. Например, сарсасапогенон был получен путем окисления дихроматом пиридиния, как описано Miles (J. Agric Food Chem., 41, 914-917 (1993)), окислением по Джонсу (Jones), как описано Blunden (J. Nat. Prod., 42, 478-482 (1979)) и в WO-98/07741, описания которых включены в настоящую работу посредством ссылки. Смилагенон был получен из диосгенона (который сам был получен окислением диосгенина) с использованием восстановления двойной связи α, β-ненасыщенного кетона [Marker et al., J. Am. Chem. Soc. 2525 (1940), Irismetov & Goryaev, Izv. Akad. Nauk Kaz. SSR, Ser. Khim., 2, 47-52 (1981)).
В предпочтительном примере осуществления настоящего изобретения исходное вещество 3-кето,5β-Н-стероидный сапогенин получают путем гетерогенной каталитической гидрогенизации соответствующего Δ4,3-кетостероидного сапогенина, например диосгенона. Гетерогенная каталитическая гидрогенизация превращает Δ4,3-кетостероидный сапогенин преимущественно в соответствующий 5β-Н-3-кетоновый продукт, например смилагенон, который затем восстанавливают согласно первому аспекту настоящего изобретения.
Гетерогенная каталитическая гидрогенизация может быть соответствующим образом осуществлена с использованием водорода и палладиевого катализатора в органическом растворителе. Палладиевый катализатор предпочтительно находится на носителе, таком как, например, сульфат бария, карбонат кальция, графит или углерод. Палладий предпочтительно используют в предварительно восстановленной форме.
Если исходным веществом является диосгенон, и за каталитической гидрогенизацией следует восстановление смилагенона с использованием стерически затрудненного органоборанового восстанавливающего агента, то полученный продукт представляет собой смилагенин.
Если исходным веществом является диосгенон, и за каталитической гидрогенизацией следует восстановление смилагенона с использованием органоалюмогидридного восстанавливающего агента, то полученный продукт представляет собой эписмилагенин.
Во втором аспекте настоящего изобретения предложен способ превращения 3α-гидрокси-5β-Н-стероидных сапогенинов и их производных в 3β-гидрокси-5β-Н-стероидные сапогенины и их производные, включающий приведение в контакт 3-гидрокси-активированного производного 3α-гидрокси-5β-Н-стероидного сапогенина с нуклеофильным реагентом в условиях, способствующих нуклеофильному замещению с инверсией в положении 3, с возможной последующей корректировкой 3-заместителя, при желании.
Реакция будет протекать по SN2 механизму с тем, чтобы получить в результате требуемый продукт превращения. Один протокол реакций, который может быть, в частности, упомянут, представляет собой реакцию Мицунобу (Mitsonobu) (Hughes, Organic Reactions, 42, 337-400 (1992)). Если такой протокол будут использовать для сапогенина, то 3-ОН-сапогенин превратится, через его 3-гидрокси-активированную форму, в соответствующий 3-сложный эфир с инверсией в положении 3. Используемые реагенты представляют собой диалкилазодикарбоксилат, триарилфосфин и соответствующую органическую кислоту или ее соль в соответствии с целевым сложным эфиром. Термин "алкил" предпочтительно относится к алкильным группам, содержащим от 1 до 7 атомов углерода. Термин "арил" предпочтительно относится к арильным группам, содержащим от 6 до 12 атомов углерода, и такие арильные группы могут быть алкил-замещенными.
Альтернативный протокол реакций может включать первоначальное получение активированного производного сапогенина, которое способно участвовать в нуклеофильном замещении, такое как, например, органическое сульфированное производное в 3-о-положении, такое как 3-мезилатное или 3-тозилатное производное.
Если органическая кислота, используемая в указанной реакции согласно второму аспекту настоящего изобретения, включает группу, такую как аминогруппа, участие которой иным образом будет нежелательно в этой реакции, то такая группа будет соответствующим образом защищена стандартным путем.
В общих чертах, в настоящем изобретении предложен способ синтеза полезных стероидных сапогенинов, например, смилагенина или эписмилагенина, из легко доступных веществ, например диосгенина, с использованием селективного восстановления для контроля стереохимии, как представлено для этих специфических соединений на Схеме 1 ниже:
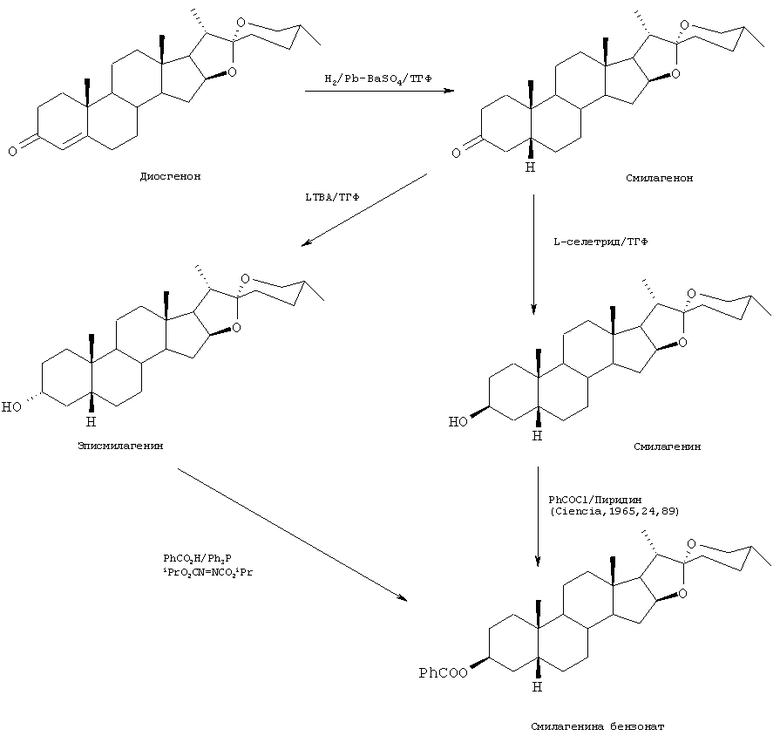
Способы по настоящему изобретению могут быть использованы для получения 3-гидрокси-5β-Н-стероидного сапогенина, такого как сарсасапогенин, смилагенин, эписарсасапогенин и эписмилагенин и их производные. Пролекарства и другие физиологически приемлемые формы сапогенинов могут быть получены из 3-ОН-соединений стандартным способом, как описано более детально ниже.
Сведения, подтверждающие возможность осуществления изобретения
Конечные продукты сапогенина
Способ по настоящему изобретению предпочтительно используют для получения конечных продуктов сапогенина, выбранных из соединений общей формулы:
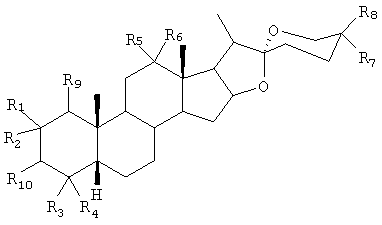
где R1, R2, R3, R4, R5, R6, R7, R8 и R9 представляют собой независимо друг от друга Н, C1-4алкил, ОН или OR (где R=С6-12арил или С1-4алкил), или R5 и R6 вместе могут представлять собой =O (карбонил) или защищенную карбонильную группу. Стереохимия в углеродном центре 3 (то есть углерод кольца А, к которому присоединена группа R10) может представлять собой либо R, либо S, и
R10 может представлять собой ОН, O-связанную сахарную группу или любую органическую сложноэфирную группу (которая включает алифатические и аминокислотные сложные эфиры).
За исключением случаев, когда в указанной формуле установлено применение условного обозначения в виде клина и штриха, и за исключением того случая, когда стереоспецифичность представляет собой признак изобретения, стереохимия в формуле точно не указана, и все стереоизомеры и изомерные смеси включены.
Используемый здесь термин "физиологически приемлемое пролекарство" означает те пролекарства соединений, которые полезны в соответствии с настоящим изобретением, и которые являются, в рамках обоснованного медицинского заключения, подходящими для применения в контакте с тканями людей и низших животных без чрезмерной токсичности, раздражения, аллергического ответа и тому подобного, соизмеримого с приемлемым соотношением риск/польза и эффективного для их подразумеваемого применения, а также цвиттер-ионные формы, где возможно, соединений по настоящему изобретению. Термин "пролекарство" означает соединения, которые быстро превращаются in vivo с получением исходного соединения указанной формулы, например, путем гидролиза в крови. Всестороннее обсуждение пролекарств предложено в следующей литературе: Design of Prodrugs (Создание пролекарств), Н. Bundgaard, Ed., Elsevier, 1985; Methods in Enzymology (Методы энзимологии), К. Widder et al., Ed., Academic Press, 42, p.309-396, 1985; Textbook of Drug Design and Development (Руководство по созданию и улучшению лекарств), Krogsgaard-Larsen and H. Bundgaard, Ed., Chapter 5; Design and Applications of Prodrugs (Создание и применение пролекарств), р. 113-193, 1991; Advanced Drug Delivery Reviews (Обзоры современных систем доставки лекарств), Н. Bundgaard, 8, р.1-38 (1992); Journal of Pharmaceutical Sciences, 77, p.285 (1988); Chem. Pharm. Bull., N. Nakeya et al., 32, p.692 (1984); Pro-drugs as Novel Delivery Systems (Пролекарства в качестве новых систем доставки), Т. Higuchi and V. Stella, Vol. 14 of A.C.S. Symposium Series, и Bioreversible Carriers in Drug Design (Биологически обратимые носители при создании лекарств), Edward В. Roche, Ed., American Pharmaceutical Association and Pergamon Press, 1978, которые включены в настоящую работу посредством ссылки.
Термин "физиологически приемлемые соли" означает относительно нетоксичные, неорганические и органические соли присоединения кислот, и соли присоединения оснований соединений по настоящему изобретению. Эти соли могут быть получены in situ во время конечного выделения и очистки соединений или путем отдельного взаимодействия очищенного соединения. Смотри, например, S.М.Berge et al., Pharmaceutical Salts, J. Pharm. Sci., 66: p.1-19 (1977), которая включена в настоящую работу посредством ссылки.
Используемый здесь термин "органический сложный эфир" относится к любому сложному эфиру, образующемуся путем взаимодействия соединения, в котором R10 представляет собой ОН, с эфир-образующей органической кислотой или ее активированным производным. Органическая кислота может представлять собой, например, алифатическую карбоновую кислоту или аминокислоту. Без ограничения, органическая сложноэфирная группа может быть выбрана, например, из групп: катилат (этоксикарбонилокси), ацетат, сукцинат, пропионат, н-бутират, изо-бутират, валерат, изовалерат, н-капроат, изо-капроат, диэтилацетат, октаноат, деканоат, лаурат, миристат, пальмитат, стеарат, бензоат, фенилацетат, фенилпропионат, циннамат, фталил, глицинат, аланинат, валинат, фенилаланинат, изолейцинат, метионинат, аргининат, аспартат, цистеинат, глутаминат, гистидинат, лизинат, пролинат, серинат, треонинат, триптофанат, тирозинат, фумарат, малеат, замещенная алифатическая группа, например, хлорацетат, метоксиацетат, защищенные аминокислотные сложноэфирные группы, например, Вос-аминоглицинат (Вос = трет-бутоксикарбонил), Вос-аминовалинат, CBZ-аминоглицинат (CBZ = бензилоксикарбонил), CBZ-аминоалинат, и замещенные ароматические сложноэфирные группы, например, п-бромбензоилокси, м-бромбензоилокси, п-метоксибензоилокси, хлорбензоат, такой как п-хлорбензоилокси, дихлорбензоат, такой как 2,4-дихлорбензоилокси, нитробензоат, такой как п-нитробензоилокси или 3,5-динитробензоилокси и т.д.
Используемый здесь термин "сахар" относится, в частности, к моно-, ди- или трисахариду и его ацилированным формам. Без ограничения, такой сахар может представлять собой, например, моноальдозу или кетозу, имеющую 5 или 6 атомов углерода, предпочтительно в цикпизованной фуранозной или пиранозной форме, или в виде α- или β-аномера, и имеющего D- или L-оптическую изомерию. Примеры подходящих сахаров включают следующие: глюкоза, манноза, фруктоза, галактоза, мальтоза, целлобиоза, сазароза, рамноза, ксилоза, арабиноза, фукоза, хиновоза, апиоза, лактоза, галактоза-глюкоза, глюкоза-арабиноза, фукоза-глюкоза, рамноза-глюкоза, глюкоза-глюкоза-глюкоза, глюкоза-рамноза, манноза-глюкоза, глюкоза-(рамноза)-глюкоза, глюкоза-(рамноза)-рамноза, глюкоза-(глюкоза)-глюкоза, галактоза-(рамноза)-галактоза и их ацилированные (например, ацетилированные) производные.
Первый аспект изобретения
Исходное вещество 3-кето,5β-Н-стероидный сапогенин для стадии, которая приводит к получению целевого сапогенина согласно первому аспекту настоящего изобретения, предпочтительно соответствует целевому сапогенину во всех положениях молекулы, за исключением группы в положении 3. Однако, при необходимости или при желании, могут быть использованы подходящие защитные группы для восстановления и последующего удаления с получением целевого сапогенина.
Используемый здесь термин "защитные группы" относится к группам, которые используют для защиты реакционноспособных функциональных групп, например гидроксильных или карбоксильных групп, если они желательны в конечном продукте, для того чтобы избежать их нежелательного участия в реакциях. Стандартные защитные группы могут быть использованы в соответствии со стандартной практикой, для примеров смотри T.W.Green и P.G.M. Wuts в "Protective Groups in Organic Chemistry" John Wiley and Sons, 1991; J.F.W McOmie в "Protective Groups in Organic Chemistry" Plenum Press, 1973.
Было обнаружено, что ряд реагентов влияет на селективность получения либо смилагенина, либо эписмилагенина, как показано в Таблице 1 ниже (проценты селективности относятся к компонентам в сыром продукте). Неожиданно авторы изобретения обнаружили, что применение К-, L- или N-Selectride® (три-втор-бутилборгидрида калия, лития или натрия) или соответствующего трифенилборгидрида приводит к образованию 3β-гидроксила, например, смилагенина, высокостереоселективным образом. Применение менее стерически затрудненного восстанавливающего агента триэтилборгидрида лития приводит к образованию 3α-гидроксила, например, эписмилагенина, высокостереоселективным образом. Неожиданно авторы также обнаружили, что применение органоалюмогидрида, такого как LTBA, также приводит к образованию 3α-гидроксила, например, эписмилагенина, высокостереоселективным образом.
Авторы изобретения обнаружили, что при стереоселективном восстановлении 3-кето, 5β-Н-стероидных сапогенинов согласно настоящему изобретению можно получить в конечном продукте молярное соотношение полученного преобладающего 3-гидроксистероида к альтернативному 3-эпимеру по меньшей мере примерно 10:1, с примерами по меньшей мере примерно 15:1.
Авторы изобретения неожиданно обнаружили, что низкие температуры (например, около -78°С) несущественны для способа по настоящему изобретению. Восстановление, как правило, можно проводить при температуре от -100°С до 25°С, предпочтительно от -40°С до 25°С, наиболее предпочтительно в интервале от примерно -10°С до 10°С, и соответственно в растворителе, выбранном из тетрагидрофура на (ТГФ), 2-метилтетрагидрофурана (МТГФ), толуола, 1,4-диоксана, трет-бутилметилового эфира и смеси этих растворителей, наиболее предпочтительно в ТГФ.
В предпочтительном примере осуществления исходное вещество 3-кето, 5β-Н-стероидный сапогенин, например, смилагенон, получают путем гетерогенной каталитической гидрогенизации соответствующего Δ4,3-кетостероидного сапогенина, например диосгенона.
Этот Δ4,3-кетостероидный сапогенин, например диосгенон, сам предпочтительно получают путем окисления соответствующего Δ5,3-гидроксистероидного сапогенина, например диосгенина, с получением αβ-ненасыщенного кетона. Следует заметить, что прямое восстановление диосгенина с использованием палладия на углероде в качестве катализатора дает преимущественно 5α-продукт тигогенин.
Маркер (Marker et al., J. Am. Chem. Soc., 62, 2525 (1940)) показал, что восстановление диосгенона до смилагенона может быть осуществлено с помощью катализатора палладий-сульфат бария в эфирном растворе в атмосфере водорода. Низкая концентрация (500 объемов; нормальные объемы для обработки находятся в пределах 5-30 объемов) и высокая загрузка катализатора (1000%; нормальные загрузки катализатора находятся в пределах 1-20%) делают описанный способ невыполнимым и неэкономичным для промышленного производства. Дополнительное соображение касается того, что эфир не подходит для промышленного производства по причинам безопасности.
Другие авторы также исследовали восстановление диосгенона до смилагенона. Дьерасси восстановил диосгенон (10 г) в этаноле (450 мл) над предварительно восстановленным 10% Pd-C (0,8 г) при атмосферном давлении. Сырой смилагенон выделяли, осаждая водой и перекристаллизовывая из смеси хлороформ/метанол с получением чистого смилагенона (7,2 г, 72%) с температурой плавления 179-183°С. Выход не изменялся при проведении реакции в присутствии гидроксида калия (3 г). Аналитически чистый образец плавился при 186-188°С (Djerassi, Yashin and Rosenkranz, J. Am. Chem. Soc., 74, 422 (1952)). Недостатком этого способа является слабое растворение из-за слабой растворимости диосгенона в этаноле.
В группе прегнанов Суворов обнаружил, что пиридин оказывает поразительный эффект на выход таких реакций гидрогенизации. Обычно при этой обработке выбранный катализатор представлял собой 10% палладий на карбонате кальция (Pd-CeCO3). Обнаружили, что в таких случаях селективность была заметно выше по сравнению с реакциями, протекающими в спиртовых растворителях, даже при добавлении каустической соды (Suvorov and Yaroslavtseva, Steroids, 1270 (1961)). Обработка, используемая в этом исследовании, включала гашение в разбавленной соляной кислоте и экстракцию продукта в хлороформе. Органический экстракт промывали разбавленной соляной кислотой, 8% водным раствором бикарбоната натрия и водой до нейтрального значения, определяемого с помощью лакмуса. Такие способы приводят к получению больших количеств водных отходов, содержащих пиридин и галогенированные растворители, которые требуют удаления, увеличивая тем самым стоимость производства.
Ирисметов продемонстрировал, что высокая селективность может быть получена при восстановлении диосгенона до смилагенона. В этой работе диосгенон (1 г) гидрогенизировали над 5% Pd-CeCO3 (1 г) в пиридине (30 мл) при атмосферном давлении. После фильтрации для удаления катализатора и выпаривания растворителя, остаток кристаллизовали из спирта с получением твердого вещества, плавящегося при 209-211°С. Выход не показан (Irismetov and Goryayev, Izv. Akad. Nauk Kaz. SSR, Ser. Khim., 2, 47 (1982)). Недостатками этой работы в том, что касается промышленного производства, являются высокие загрузки катализатора (100%) и разбавленные растворы. Пиридин является ядовитым растворителем и чаще всего используется в промышленном производстве в стехиометрических количествах в качестве кислотного поглотителя.
Патент US 736818 заявляет восстановление 3-кето-Δ4-стероидов до 5β-Н-стероидов с палладиевым катализатором в присутствии неорганического основания и в безводной среде. Предпочтительным растворителем является метанол, а предпочтительным основанием является гидроксид калия. Диосгенон не упомянут в качестве примера. Авторы настоящего изобретения обнаружили, что диосгенон плохо растворим в спиртах (в частности, в этаноле), что приводит к сильному разведению в этом способе. Такой способ требует также стадии экстрактивной обработки.
Патент US 763301 дает ссылку на полезность щелочей (то есть гидроксида натрия или калия) в повышении количества 5β-Н-продукта при восстановлении 3-кето-Δ4-стероидов. Этот патент заявляет особый пункт на применение триэтиламина в этом контексте. В качестве выбранных растворителей перечислены этанол, эфир, этилацетат и метилциклогексан, при этом 1,4-диоксан является предпочтительным растворителем.
Авторы настоящего изобретения сделали неожиданное открытие, что применение палладия на носителе, таком как сульфат бария (Pd-BaSO4) или карбонат кальция (Pd-CeCO3), в подходящем растворителе обеспечивает экономичный и масштабируемый способ. В частности, авторы настоящего изобретения обнаружили способы, которые работают при рентабельных концентрациях с использованием низких загрузок катализатора. Более того, авторы неожиданно обнаружили, что восстановленные формы этих катализаторов являются более селективными, чем невосстановленные формы, как показано в Таблице 2 ниже.
5% Pd/графит (Johnson Matthey, тип 450) и 10% Pd/C (Johnson Matthey, тип 39) также являются подходящими катализаторами для этого способа.
Подходящие растворители могут быть выбраны из тетрагидрофурана (ТГФ), 2-метилтетрагидрофурана, толуола, 1,4-диоксана, этилацетата, метил-изо-бутилкетона, наиболее предпочтительно ТГФ. Обнаружено, что эти растворители являются более предпочтительными, чем пиридин. В присутствии этих растворителей этот способ можно осуществлять при концентрации от 1 объема до 50 объемов, предпочтительно 3-30 объемов и наиболее предпочтительно при концентрации 3-10 объемов. Загрузка катализатора находится в диапазоне 1-25%, предпочтительно 1-10% и наиболее предпочтительно 1-5%.
Неожиданно авторы изобретения обнаружили, что повышение давления приводит к меньшей селективности способа. Реакцию предпочтительно проводят при давлении водорода 1-5 бар и наиболее предпочтительно при давлении водорода 1-2 бар.
Обнаружили также, что повышенная температура уменьшает селективность. Реакцию предпочтительно проводят при 15-75°С, предпочтительно при 20-50°С и наиболее предпочтительно при 20-30°С.
ТГФ обеспечивал улучшенную растворимость диосгенона при сравнении с этанолом и другими возможными эфир-замещающими растворителями, такими как диэтоксиметан и трет-бутилметиловый эфир. Это обеспечивает более высокую производительность и более экономичный способ. В этом способе предложена простая обработка по сравнению с системой этанол/водный гидроксид натрия.
Обработка включала концентрированно реакционной смеси и выделение смилагенона. Растворитель может быть использован повторно.
Ряд растворителей оказался эффективным при очистке смилагенона, включая циклогексан, 2-бутанон, ацетон, 2-пропанол и метанол; примеры этих растворителей представлены в Таблице 3 ниже.
Предпочтительный аспект изобретения состоит в том, чтобы использовать раствор смилагенона в ТГФ после гидрогенизации непосредственно на стадии восстановления согласно первому аспекту настоящего изобретения. Это устраняет необходимость обработки, выделения и сушки промежуточного смилагенона, что обеспечивает экономию времени и эксплуатационного оборудования и, следовательно, ожидаемые улучшения в стоимости производства. Авторы неожиданно обнаружили, что примеси, полученные этим способом (в основном эпитигогенин и эписмилагенин), могут быть удалены перекристаллизацией сырого смилагенина.
Второй аспект изобретения
Во втором аспекте изобретения предложено превращение 3α-гидрокси-5β-Н-стероидных сапогенинов и их производных в 3β-гидрокси-5β-Н-стероидные сапогенины и их производные, например, сложные эфиры, путем реакции стереоспецифического превращения. Например, эписарсасапогенин может быть легко превращен в новое соединение сарсасапогенина бензоат действием диизопропилазодикарбоксилата, трифенилфосфина и бензойной кислоты, согласно так называемой реакции Мицунобу (Hughes, Organic Reactions, 42, 337-400 (1992)). Поэтому сарсасапогенина бензоат и его получение представляют дополнительные признаки настоящего изобретения. Аналогично, эписмилагенин может быть превращен в известный сложный эфир смилагенина бензоат. Этот способ не ограничивается бензоатными эфирами, но может быть успешно использован для производства алифатических, например, ацетата, пропионата, н-бутирата, изо-бутирата, н-капроата, изо-капроата, пальмитата, замещенных алифатических, например, хлорацетата, метоксиацетата, защищенных аминоэфиров, например, Вос-аминоглицината, Вос-аминовалината, CBZ-аминоглицината, CBZ-аминоалината, или замещенных ароматических сложных эфиров, например, хлорбензоата, нитробензоата, дихлорбензоата и т.д.
Протокол реакций может альтернативно включать предварительное образование активированной формы 3α,5β-сапогенина, такой как метансульфонат (мезилат) или п-толуолсульфонат (тозилат). Эта активированная форма может быть затем подвергнута реакции с анионной солью карбоновой кислоты (например, солью натрия, цезия или калия), стандартным для нуклеофильного замещения путем.
Выделение полученного соединения
Соединение, полученное согласно любому аспекту настоящего изобретения, может быть выделено из реакционной смеси стандартными способами. Например, соединения могут быть выделены путем отгонки растворителя из реакционной смеси или, при необходимости, после отгонки растворителя из реакционной смеси, вливания остатка в воду с последующей экстракцией несмешивающимся с водой органическим растворителем и отгонкой растворителя из экстракта. Кроме того, продукт может быть, при желании, дополнительно очищен различными хорошо известными методами, такими как перекристаллизация, переосаждение или различные хроматографические методы, особенно колоночная хроматография или препаративная тонкослойная хроматография.
ОПИСАНИЕ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Следующие Примеры иллюстрируют, без ограничения, синтез эписарсасапогенина, смилагенина и эписмилагенина с использованием селективного восстановления для контроля стереохимии. Примеры также иллюстрируют стереоспецифическое превращение 3α-гидрокси,5β-Н-сапогенинов в 3β-гидрокси,5β-Н-сапогенины и их производные.
Пример 1
Синтез смилагенина из смилагенона с помощью L-Selectride® при -10°С
Смилагенон (657 г) растворяли в тетрагидрофуране (4000 мл), и раствор продували азотом и охлаждали, чтобы обеспечить внутреннюю температуру примерно -10°С. Добавляли L-Selectride® (2400 мл 1М в ТГФ) в течение примерно 50 минут и перемешивали в течение 90 минут. Медленно добавляли раствор лимонной кислоты (600 г) в воде (2000 мл), поддерживая температуру ниже 0°С. Смеси позволяли нагреться до температуры окружающей среды и перемешивали в течение 30 минут. Водный слой отделяли и экстрагировали дихлорметаном (2000 мл), и слои разделяли. Водный слой экстрагировали дихлорметаном (1500 мл). Объединенные органические экстракты промывали водой (4000 мл) и сушили над MgSO4. Органические экстракты упаривали досуха с получением смилагенина.
Пример 2
Синтез смилагенина из смилагенона с помощью K-Selectride® при -15°С
K-Selectride® (1600 мл; 1М в ТГФ) добавляли к раствору смилагенона (500 г) в ТГФ (3500 мл) при примерно -15°С в атмосфере азота. Реакционную смесь перемешивали при этой температуре в течение 30 минут и гасили водным раствором лимонной кислоты (393 г в 1300 мл воды), поддерживая внутреннюю температура при примерно 0°С. Смесь нагревали до температуры окружающей среды, и ТГФ упаривали при атмосферном давлении до тех пор, пока твердое вещество не выпадет в осадок. Твердое вещество отфильтровывали и сушили под вакуумом.
Твердое вещество растворяли в дихлорметане (ДХМ) (6000 мл), сушили (MgSO4) и упаривали до получения белого твердого вещества, которое перекристаллизовывали из изопропилового спирта (ИПС) (5000 мл) с получением смилагенина.
Пример 3
Синтез смилагенина из смилагенона с помощью N-Selectride® при -78°С
N-Selectride® (0,64 мл, 1М в ТГФ) добавляли к раствору смилагенона (206 мг) в ТГФ (10 мл) в течение 10 минут при -78°С. Эту смесь перемешивали и гасили 10% водным раствором лимонной кислоты (2 г в 20 мл воды), и продукт экстрагировали в ДХМ (2×50 мл), сушили (MgSO4) и упаривали до получения бесцветного масла. Масло вливали в ацетон (20 мл) и добавляли воду (50 мл). Осадок собирали фильтрацией и сушили с получением смилагенина (200 мг, 97%).
Пример 4
Синтез смилагенона из диосгенона
Диосгенон (500 г) растворяли в тетрагидрофуране (ТГФ) (2500 мл) при 40-45°С и продували азотом. Добавляли 5% Pd-BaSO4 (восстановленный) (100 г); колбу продували водородом и перемешивали в атмосфере водорода в течение примерно 6,5 часов. Колбу охлаждали до температуры окружающей среды, и катализатор удаляли фильтрацией через слой целита (50 г). Растворитель выпаривали с получением сырого смилагенона в виде твердого остатка.
Этот способ повторяли, и две порции объединяли (902,8 г) и снова суспендировали в циклогексане (2260 мл) при температуре окружающей среды в атмосфере азота в течение примерно 30 минут. Твердое вещество получали фильтрацией и сушили в вакуумной печи при примерно 40°С в течение ночи с получением очищенного смилагенона (749,1 г; 75%).
Пример 5 Синтез смилагенона из диосгенона
Диосгенон (700 г) растворяли в тетрагидрофуране (ТГФ) (4500 мл) и продували азотом. Смесь обрабатывали активированным углем (35 г) и гидрогенизировали над 5% Pd-BaSO4 (восстановленным) (35 г) при 25°С и давлении водорода 2,5 бар. Катализатор удаляли фильтрацией, и смесь концентрировали до примерно четверти объема. Воду (3000 мл) добавляли в течение примерно 30 минут, и полученное твердое вещество отфильтровывали. Твердое вещество промывали метанолом (560 мл) и сушили под вакуумом при 40-50°С с получением смилагенона (630 г, 90%).
Пример 6
Сокращение реакций гидрогенизации и восстановления
Диосгенон (500 г) растворяли в тетрагидрофуране (2500 мл) и продували азотом. Добавляли 5% Pd-BaSO4 (восстановленный) (100 г); колбу продували водородом и перемешивали в атмосфере водорода в течение примерно 5 часов. Катализатор удаляли фильтрацией через слой целита (20 г). Остаток промывали тетрагидрофураном (1000 мл), и раствор использовали непосредственно на следующей стадии.
K-Selectride® (1600 мл; 1М в тетрагидрофуране) добавляли к раствору смилагенона (500 г) в тетрагидрофуране при температуре выше примерно -15°С в атмосфере азота. Реакционную смесь перемешивали при этой температуре в течение 30 минут и гасили водным раствором лимонной кислоты (393 г в 1300 мл воды), поддерживая внутреннюю температуру при примерно 0°С. Смесь нагревали до температуры окружающей среды, и тетрагидрофуран выпаривали при атмосферном давлении до тех пор, пока твердое вещество не выпадет в осадок. Твердое вещество отфильтровывали и сушили под вакуумом.
Твердое вещество растворяли в дихлорметане (6000 мл), сушили (MgSO4) и растворитель упаривали до получения белого твердого вещества, которое перекристаллизовывали из 2-пропанола (5000 мл). Это твердое вещество затем перекристаллизовывали из ацетона (5000 мл). Твердое вещество затем перекристаллизовывали из ацетона (3500 мл). Твердое вещество сушили при 80°С в вакуумной печи с получением чистого смилагенина (154,5 г).
Т.пл. 184,7-187,0°С; [α]D 20=-73,3°; ИК νmax 3456, 2928, 1451, 1376, 1050, 979, 896 см-1; ИЭР-МС m/z 417 [М+1]+; 1H-ЯМР (CDCl3, 300 МГц): в частности, δ 4.39 (1Н, уш. к, J=8 Гц), 4.10 (1Н, уш. с), 3.46 (1Н, уш. д.д, J=11 Гц), 3.39 (1Н, т, J=11 Гц), 0.98 (3Н, с), 0.97 (3Н, д, J=7 Гц), 0.79 (3Н, д, J=7 Гц), 0.76 (3Н, с) м.д.; 13С-ЯМР (CDCl3, 126 МГц): δ 14.47, 16.43, 17.10, 20.83, 23.86, 26.48, 26.50, 27.75, 28.73, 29.89, 30.24, 31.32, 31.73, 33.46, 35.21, 35.21, 36.45, 39.78, 40.24, 40.63, 41.54, 56.41, 62.19, 66.79, 66.98, 80.87, 109.20 м.д.; С 77,94%; Н 10,75% (теоретическое значение для С27Н44O3: 77,84%; Н 10,64%).
Пример 7
Сокращение реакций гидрогенизации и восстановления
L-Selectride® (527 мл; 1М в тетрагидрофуране) добавляли к раствору смилагенона (156 г) в тетрагидрофуране (полученного гидрогенизацией диосгенона) при примерно -10°С в атмосфере азота. Реакционную смесь перемешивали при этой температуре в течение 30 минут, позволяли нагреться до температуры окружающей среды и перемешивали в течение ночи. Смесь гасили, добавляя водный раствор лимонной кислоты (311 г в 3800 мл воды) и дихлорметан (2200 мл), поддерживая внутреннюю температуру ниже 30°С. Водную фазу отделяли и снова экстрагировали дихлорметаном (400 мл). Объединенные органические экстракты промывали водным раствором лимонной кислоты (160 г в 2200 мл воды) и перегоняли до небольшого объема. Добавляли 2-пропанол (3000 мл), и смесь снова перегоняли до примерно 1/2 объема. Добавляли еще 2-пропанол (1500 мл), и смесь перегоняли до примерно 1/2 объема. Смесь нагревали до температуры образования флегмы и оставляли охлаждаться. Эту смесь затем охлаждали до 0-10°С и фильтровали. Твердое вещество сушили в вакуумной печи при 60-65°С с получением смилагенина. Выход составляет 94,0 г.
Пример 8
Восстановление смилагенона до эписмилагенина
Три-трет-бутоксиалюмогидрида лития (1М в тетрагидрофуране, 99 мл) добавляли по каплям к раствору смилагенона (32,0 г, 77,2 ммоль) в тетрагидрофуране (800 мл) с такой скоростью, чтобы поддерживалась температура 14-16°С. После того как добавление завершалось, смесь перемешивали при комнатной температуре еще 2 ч. Оставшийся восстанавливающий агент гасили, осторожно добавляя раствор хлорида аммония (30 г в 400 мл воды). Смесь фильтровали, и твердое вещество промывали дихлорметаном (300 мл). Объединенные фильтраты упаривали, и остаток распределяли между дихлорметаном (300 мл) и водой (300 мл). Водный слой дополнительно экстрагировали дихлорметаном (2×300 мл). Объединенные органические экстракты сушили (MgSO4) и упаривали с получением белого твердого вещества (25,7 г). Твердое вещество перекристаллизовывали из ацетона (1250 мл), и полученное твердое вещество (19,0 г) сушили в вакуумной печи при 40°С в течение ночи. Твердое вещество дополнительно очищали путем нагревания суспензии в ацетоне (1425 мл). Полученное твердое вещество сушили в вакуумной печи при 40°С в течение ночи. Твердое вещество окончательно очищали путем перекристаллизации из 2-пропанола (300 мл), и раствор фильтровали горячим для удаления любых неорганических веществ. Фильтрат охлаждали, твердое вещество фильтровали и сушили при 60°С в вакуумной печи в течение ночи с получением эписмилагенина (9,0 г).
Т.пл. 223-227°С; [α]D 25=-64° (с=5 г л-1, CHCl3); ИК νmax (KBr) 3392, 2937, 1451, 1369, 1051, 982, 864 см-1; ИЭР-МС m/z 417 [M+1]+; 1Н-ЯМР (CDCl3, 300 МГц): в частности, δ 4.40 (1Н, уш. к, J=8 Гц), 3.62 (1Н, септет, J=10, 10, 5, 5 Гц), 3.48 (1Н, уш. д.д, J=11 Гц), 3.37 (1Н, т, J=11 Гц), 0.97 (3Н, д, J=7 Гц), 0.95 (3Н, с), 0.79 (3Н, д, J=7 Гц), 0.75 (3Н, с) м.д.; 13С-ЯМР (CDCl3, 75 МГц) в частности: δ 14.91, 16.85, 17.55, 20.99, 23.78, 27.08, 27.49, 30.68, 31.75, 32.18, 35.09, 35.75, 35.85, 40.62, 40.91, 41.04, 41.99, 42.39, 56.74, 62.59, 67.23, 72.10, 81.30, 109.64 м.д.; С 77.77%; Н 10.59% (теоретическое значение для С27Н44О3: С 77,84%; Н 10,64%).
Пример 9
Синтез смилагенина бензоата из эписмилагенина
Раствор диизопропилазодикарбоксилата (0,81 г, 4,0 ммоль) в сухом ТГФ (2 мл) добавляли к перемешиваемому раствору эписмилагенина (0,83 г, 2,0 ммоль), трифенилфосфина (1,05 г, 4,0 ммоль) и бензойной кислоты (0,49 г, 4,0 ммоль) в сухом ТГФ (20 мл). Смесь перемешивали при комнатной температуре и контролировали с помощью ТСХ. Через 2 ч весь исходный материал был израсходован. Растворитель удаляли в вакууме, остаточный сироп растворяли в эфире (30 мл), и раствор промывали водным насыщенным раствором гидрокарбоната натрия (25 мл). Органический слой сушили над MgSO4 и пропускали через небольшой слой силикагеля, причем этот слой промывали эфиром. Объединенные промывные воды и фильтрат концентрировали в вакууме с получением смилагенина бензоата в виде белого твердого вещества.
Пример 10
Синтез сарсасапогенина бензоата из эписарсасапогенина
Раствор диизопропилазодикарбоксилата (0,81 г, 4,0 ммоль) в сухом ТГФ (2 мл) добавляли к перемешиваемому раствору эписарсасапогенина (0,83 г, 2,0 ммоль), трифенилфосфина (1,05 г, 4,0 ммоль) и бензойной кислоты (0,49 г, 4,0 ммоль) в сухом ТГФ (20 мл). Смесь перемешивали при комнатной температуре и контролировали с помощью ТСХ. Через 2 ч весь исходный материал был израсходован. Растворитель удаляли в вакууме, остаточный сироп растворяли в эфире (30 мл), и раствор промывали водным насыщенным раствором гидрокарбоната натрия (25 мл). Органический слой сушили над MgSO4 и пропускали через небольшой слой силикагеля, причем этот слой промывали эфиром. Объединенные промывные воды и фильтрат концентрировали в вакууме с получением сарсасапогенина бензоата в виде белого твердого вещества.
Т.пл. 173-175°С; 1H-ЯМР (500 МГц, CDCl3): δ 0.77 (3Н, с, 18-CH3), 1.00 (3Н, д, J=6.7 Гц, 21-CH3), 1.04 (3Н, с, 19-CH3), 1.08 (3Н, д, J=7.0 Гц, 27-CH3), 1.1-2.1 (27Н, сложный мультиплекс, алифатические), 3.31 (1Н, уш. д, J=10.9 Гц, 26-OCHH), 3.96 (1Н, уш. д.д, J=10.9, 2.0 Гц, 26-ОСНН), 4.42 (1Н, м, 16-ОСН), 5.34 (1Н, уш. с, Н-3), 7.44 (2Н, уш. т, J=7.6 Гц, ароматический Н), 7.55 (1Н, уш. т, J=7.6 Гц, ароматический Н), 8.05 (1Н, уш. д, J=7.6 Гц, ароматический Н) м.д.; 13С-ЯМР (125.6 МГц, CDCl3): δ 14.56, 16.28, 16.71, 21.17, 24.28, 25.41, 26.01, 26.19, 26.69, 27.31, 31.02, 31.33, 31.98, 35.37, 35.57, 37.92, 40.28, 40.48, 40.91, 42.36, 56.63 (С-14), 62.33 (С-17), 65.36 (С-26), 71.54 (С-3), 81.22 (С-16), 109.94 (С-22), 128.54 (ароматический С), 129.73 (ароматический С), 131.39 (ароматический С), 132.9 (ароматический С), 166.13 (карбонил) м.д.
Пример 11
Синтез эписарсасапогенина из сарсасапогенона
Раствор три-трет-бутоксиалюмогидрида лития в ТГФ (1М, 41,71 кг) добавляли (в течение примерно 2 часов) к перемешиваемому раствору сарсасапогенона (17,38 кг) в сухом ТГФ (примерно 70 кг) при температуре от -23 до -30°С в сухом азоте. Производственную линию промывали ТГФ, и смесь перемешивали при температуре от -23 до -30°С в течение примерно 3 часов. Полученный раствор осторожно гасили водным раствором сульфата натрия (5,67 кг в 28,67 кг воды). Неорганические соли удаляли фильтрацией и промывали ТГФ (184 кг). Добавляли воду (63,18 кг), и большую часть ТГФ удаляли перегонкой. Добавляли еще воду (126,44 кг), и продукт выделяли фильтрацией. Продукт промывали водой (2×17,38 кг) и ацетоном (4×13,73 кг). Продукт сушили при 35-40°С с получением эписарсасапогенина (14,48 кг).
Описанное выше в общих чертах описывает настоящее изобретение без ограничения. Вариации и модификации, как будет очевидно специалистам в этой области техники, предназначены для включения в рамки этой заявки и любых последующих патентов.
Описывается улучшенный способ стереоспецифического получения Зβ-гидрокси-5β-Н стероидного сапогенина или его производного путем восстановления 3-кето,5β-Н-стероидного сапогенина стерически затрудненным органобораном. 3β-Гидрокси,5β-Н-стероидный сапогенин или его производное могут быть получены путем восстановления 3-кето,5β-Н-стероидного сапогенина с использованием в качестве восстанавливающего агента относительно высоко стерически затрудненного органоборанового реагента или путем SN2 превращения 3α-гидрокси,5β-Н-стероидного сапогенина или его производного. Способ представляет удобный путь для получения стероидных сапогенинов, таких как сарсасапогенин, эписарсасапогенин, смилагенин, эписмилагенин и их сложные эфиры из легко доступных или легко приготавливаемых исходных веществ (например, диосгенона, предпочтительно из диосгенина). 4 и 39 з.п. ф-лы, 3 табл.
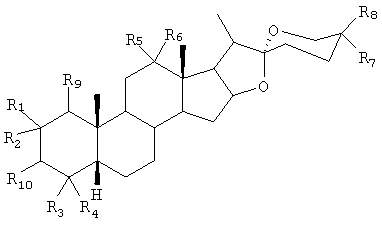
где R1, R2, R3, R4, R5, R6, R7, R8 и R9 независимо друг от друга представляют собой Н, С1-4алкил, ОН или OR (где R=C6-12арил или С1-4алкил), или R5 и R6 вместе могут представлять собой =O (карбонил) или защищенную карбонильную группу, причем стереохимия в углеродном центре 3 может быть либо R либо S, и R10 представляет собой ОН, О-связанную сахарную группу или любую органическую сложноэфирную группу.
где R1, R2, R3, R4, R5, R6, R7, R8 и R9 независимо друг от друга представляют собой Н, С1-4алкил, ОН или OR (где R=C6-12арил или С1-4алкил), или R5 и R6 вместе могут представлять собой =O (карбонил) или защищенную карбонильную группу, причем стереохимия в углеродном центре 3 может быть либо R либо S, и R10 представляет собой ОН, О-связанную сахарную группу или любую органическую сложноэфирную группу.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| US 5703052 А, 30.12.1997 | |||
| US 5939398 А, 17.08.1999. | |||

