Область техники
Настоящее изобретение относится к области медицины. В частности, настоящее изобретение относится к новому дейтерированному производному хенодезоксихолевой кислоты, а также фармацевтической композиции, содержащей данное соединение.
Уровень техники
Фарнезоидный X рецептор (FXR) является членом семейства ядерных рецепторов, экспрессия которого в основном происходит в органах пищеварительной системы, например, в печени, тонком кишечнике и др., и участвует в таких связях, как метаболизм желчных кислот и метаболизм холестерина. Желчные кислоты выполняют различные физиологические функции и играют важную роль в таких процессах, как абсорбция жира, транспорт, распределение и динамическая балансировка уровня холестерина. Фарнезоидный X рецептор действует как рецептор желчных кислот, таких как хенодезоксихолевая кислота, и поддерживает баланс желчных кислот в организме путем регулирования экспрессии генов, участвующих в метаболизме желчных кислот. Также фарнезоидный X рецептор также играет важную роль в динамической балансировке глюкозы и чувствительности к инсулину в организме. Таким образом, ожидается, что агонисты фарнезоидного X рецептора будут использоваться при разработке лекарственных препаратов для лечения неалкогольного стеатогепатита, неалкогольной жировой болезни печени, желчных камней, первичного билиарного цирроза, цирроза, фиброза печени, диабета, гиперхолестеринемии, атеросклероза, ожирения, гипертриглицеридемии и др.
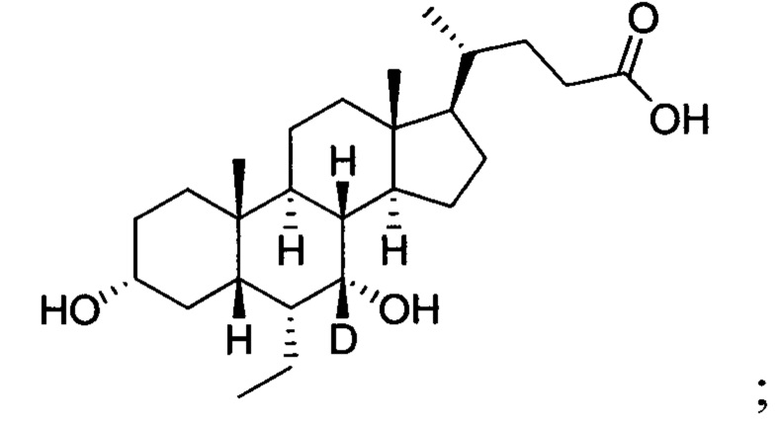
Дезоксихолевая кислота и ее производные представляют собой класс агонистов фарнезоидных X рецепторов. Ряд производных хенодезоксихолевой кислоты представлен в патентах WO 2010059859 и WO 2005082925, в которых соединение обетихолевой кислоты является селективным агонистом фарнезоидного X рецептора, химическим названием которого является 3α, 7α-дигидрокси-6α-этил-5β-холан-24-оваю кислота, и которые могут быть использованы для лечения неалкогольного стеатогепатита и неалкогольных жировых заболеваний печени. В настоящее время, обетихолевая кислота находится на III этапе клинического исследования.
Хотя обетихолевая кислота показывает лучший клинический эффект в снижении воспаления печени и уровня фиброза, а также способствует снижению веса и повышению чувствительности к инсулину и т.д., также существуют другие побочные эффекты, такие как зуд и повышение уровня липопротеинов низкой плотности. Таким образом, селективный поиск агонистов фарнезоидного X рецептора, которые также будут обладать высокой активностью и быть безопасными, является очень затруднительным.
Соответственно, в данной области техники существует потребность в разработке соединений, обладающих хорошим эффектом активации фарнезоидных X рецепторов или лучшими фармакодинамическими/фармакокинетическими свойствами.
Сущность изобретения
Задачей настоящего изобретения является получение нового класса соединений, обладающих способностью активации фарнезоидного X рецептора и лучшими фармакодинамическими/ фармакокинетическими свойствами, а также их использование.
В первом аспекте настоящего изобретения представлено дейтерированное производное хенодезоксихолевой кислоты, представленное формулой (I) или его кристаллической формой, фармацевтически приемлемой солью, гидратами или сольватами:
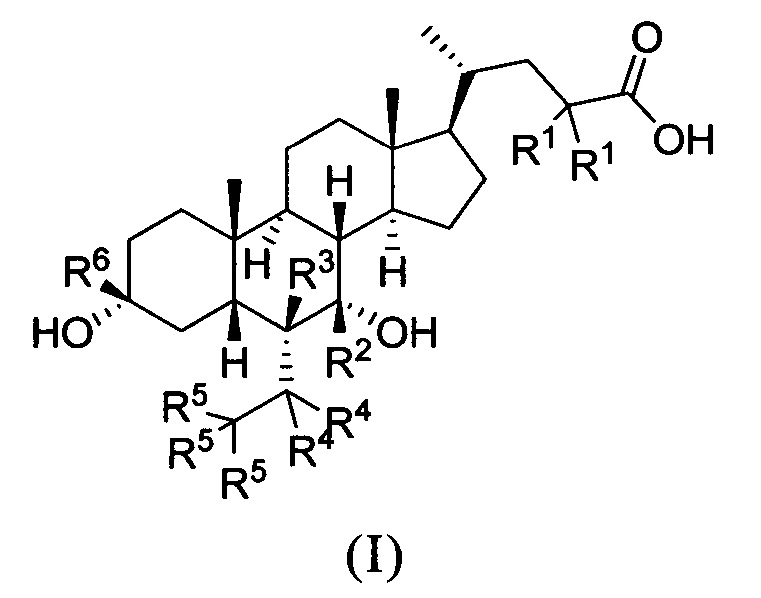
где:
R1, R2, R3, R4, R5 и R6 независимо друг от друга представляют собой водород или дейтерий;
с оговоркой, что по меньшей мере один из R1, R2, R3, R4, R5 или R6 представляет собой дейтерий.
В другом предпочтительном варианте осуществления изобретения, содержание изотопов дейтерия в позиции, замещаемой дейтерием, по меньшей мере превышает естественное содержание изотопов дейтерия (около 0,015%), предпочтительнее превышает более чем на 30%, предпочтительнее превышает более чем на 50%, предпочтительнее превышает более чем на 75%, предпочтительнее превышает более чем на 95%, предпочтительнее превышает более чем на 99%.
В другом предпочтительном варианте осуществления изобретения, соединение формулы (I) содержит по меньшей мере один атом дейтерия, предпочтительнее два атома дейтерия, предпочтительнее три атома дейтерия, предпочтительнее пять атомов дейтерия, предпочтительнее шесть атомов дейтерия.
В другом предпочтительном варианте осуществления изобретения, R1 представляет собой водород или дейтерий.
В другом предпочтительном варианте осуществления изобретения, R2 представляет собой водород или дейтерий.
В другом предпочтительном варианте осуществления изобретения, R3 представляет собой водород или дейтерий.
В другом предпочтительном варианте осуществления изобретения, R4 и R5 выбираются по отдельности из водорода или дейтерия.
В другом предпочтительном варианте осуществления изобретения, R6 представляет собой водород или дейтерий.
В другом предпочтительном варианте осуществления изобретения, R1 представляет собой дейтерий.
В другом предпочтительном варианте осуществления изобретения, R2 представляет собой дейтерий.
В другом предпочтительном варианте осуществления изобретения, R3 представляет собой дейтерий.
В другом предпочтительном варианте осуществления изобретения, R4 представляет собой дейтерий и/или R5 представляет собой дейтерий.
В другом предпочтительном варианте осуществления изобретения, R2 представляет собой дейтерий и/или R1 представляет собой дейтерий.
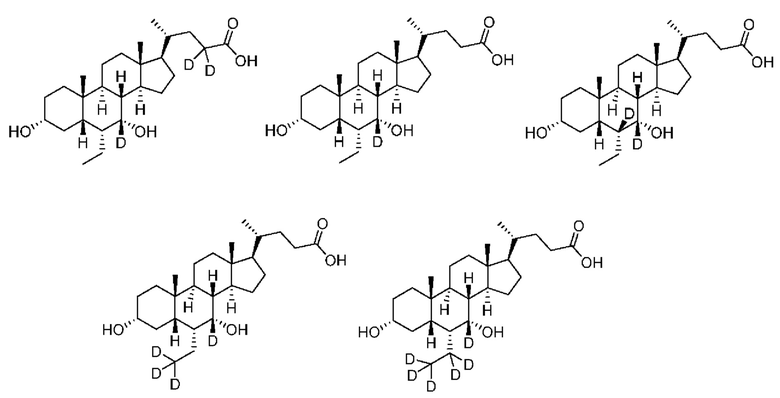
В другом предпочтительном варианте осуществления изобретения, соединение является одним из следующих соединений или его фармацевтически приемлемой солью:
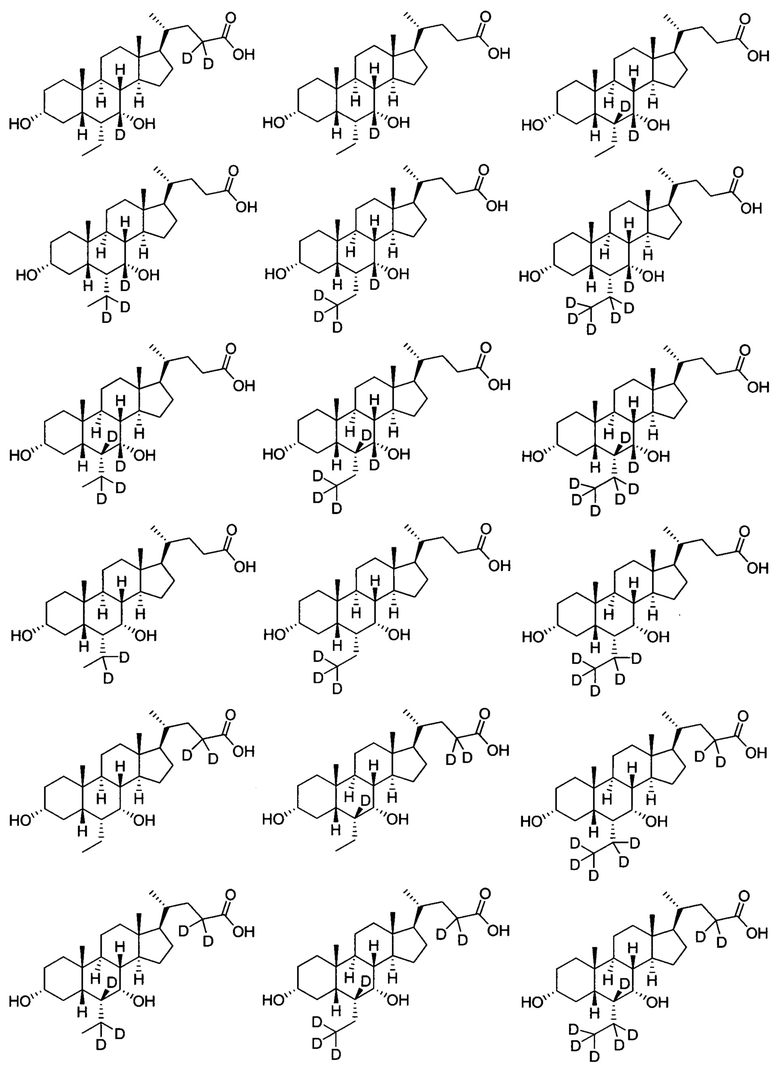
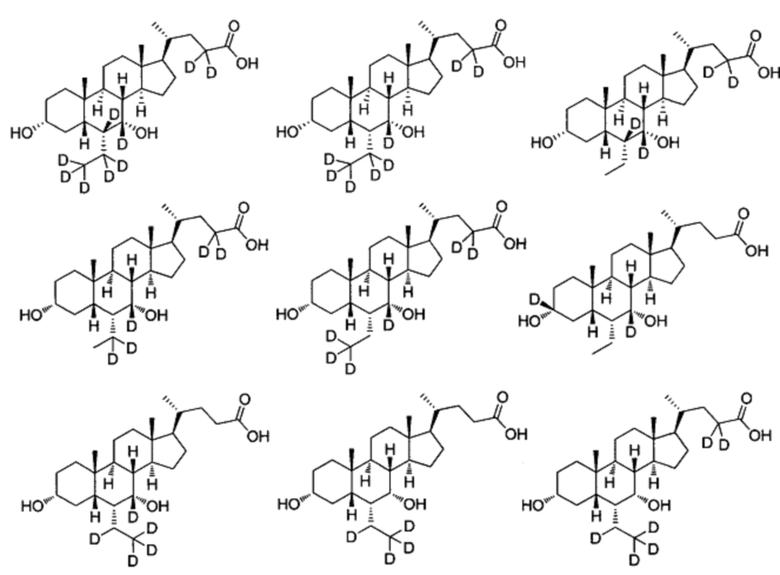
В другом предпочтительном варианте осуществления изобретения, соединение является одним из следующих соединений или его фармацевтически приемлемой солью:
3α, 7α-дигидрокси-6α-этил-7-d-5β-холан-24-овая кислота;
 3α, 7α-дигидрокси-6α-этил-6, 7-d2-5β-холан-24-овая кислота;
3α, 7α-дигидрокси-6α-этил-6, 7-d2-5β-холан-24-овая кислота;
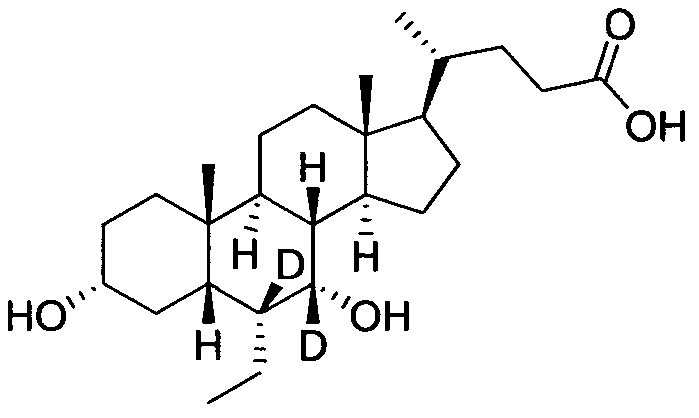
3α, 7α-дигидрокси-6α-этил-7, 23, 23-d3-5β-холан-24-овая кислота;
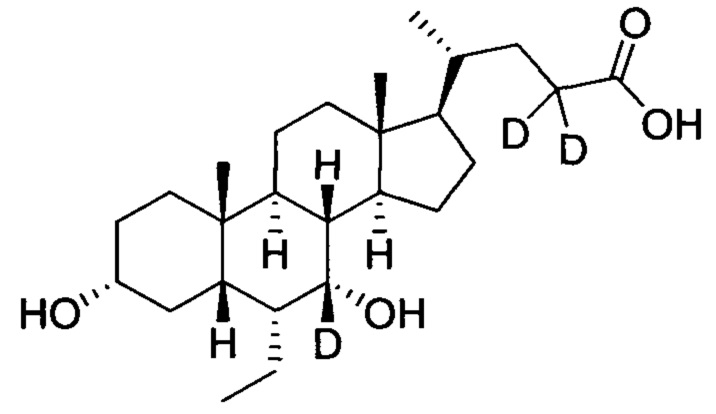
3α, 7α-дигидрокси-6α-(этил-d5)-7-d-5β-холан-24-овая кислота;
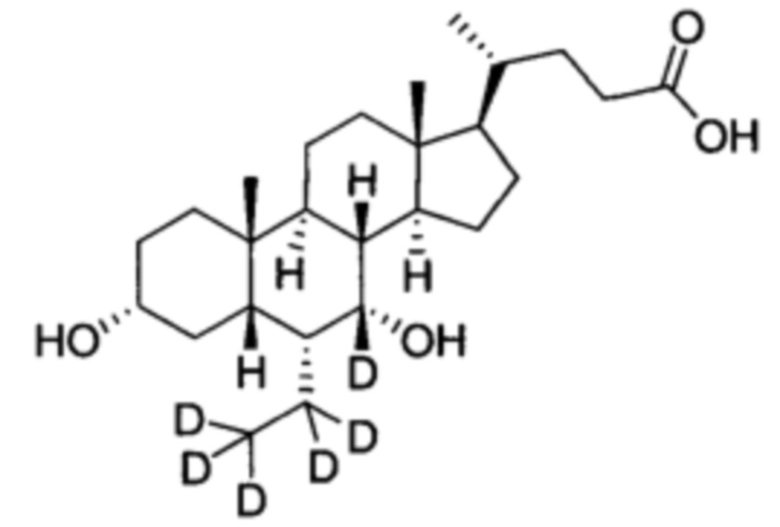
3α, 7α-дигидрокси-6α-(этил-d5)-5β-холан-24-овая кислота;
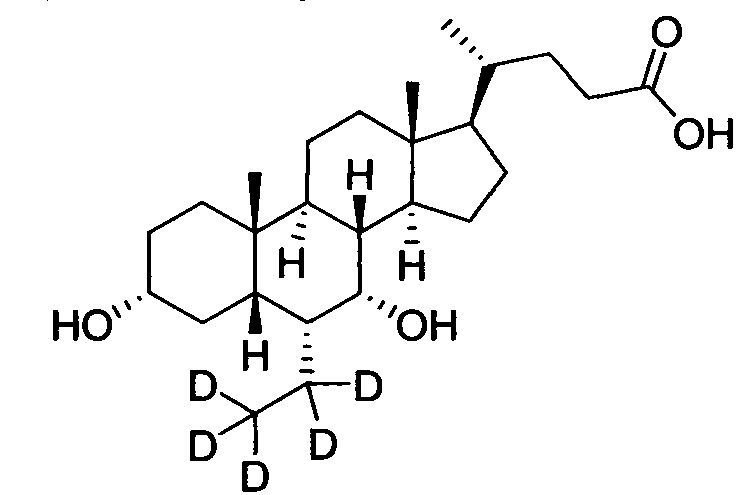
3α, 7α-дигидрокси-6α-этил-23, 23-d2-5β-холан-24-овая кислота;
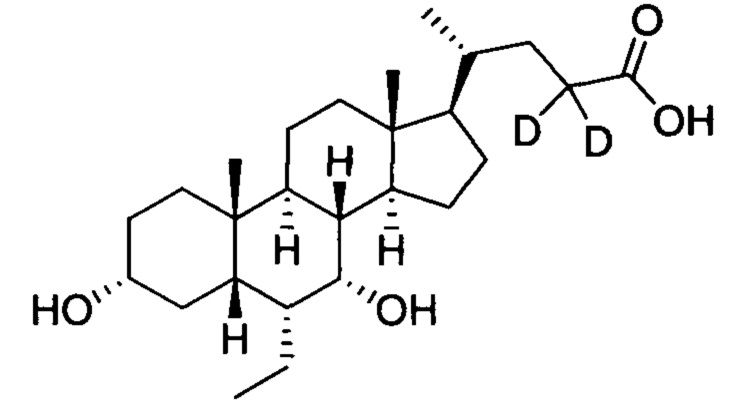
3α, 7α-дигидрокси-6α-(этил-d5)-23, 23-d2-5β-холан-24-овая кислота;
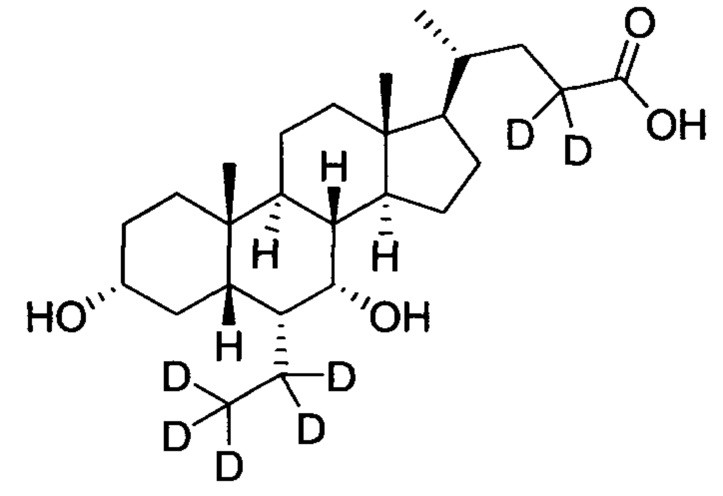
3α, 7α-дигидрокси-6α-(этил-d5)-6, 23, 23-d3-5β-холан-24-овая кислота;
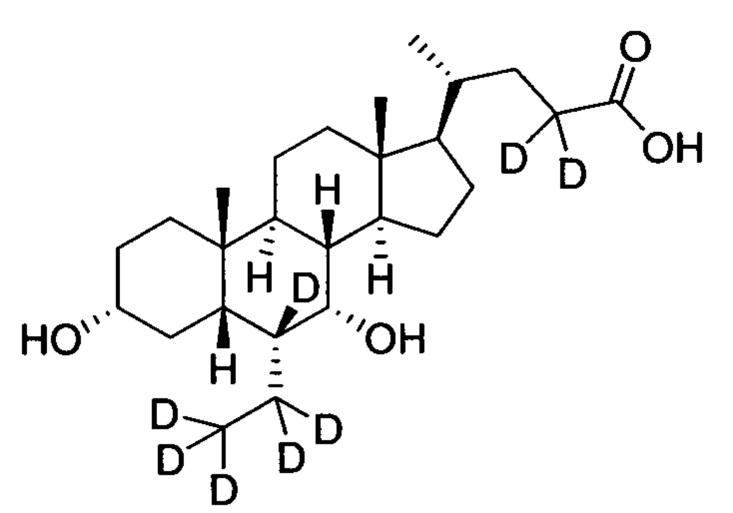
3α, 7α-дигидрокси-6α-(этил-(d5)-7, 23, 23-d3-5β-холан-24-овая кислота;
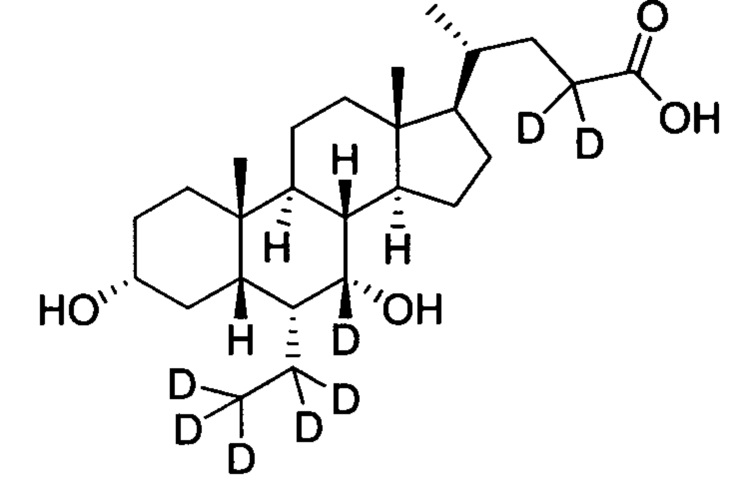
3α, 7α-дигидрокси-6α-этил-6, 7, 23, 23-d4-5β-холан-24-овая кислота;
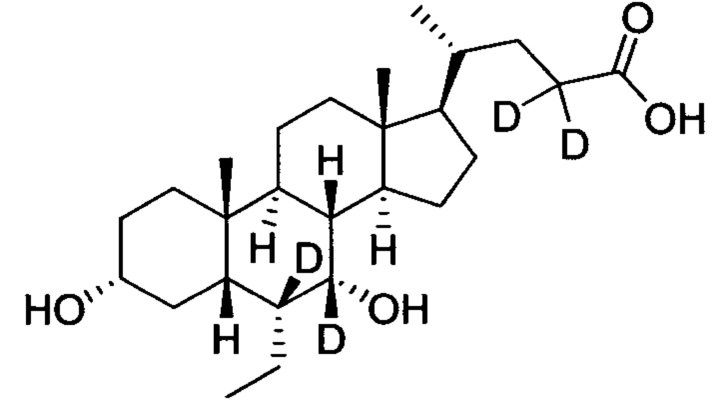
3α, 7α-дигидрокси-6α-(этил-d5)-6, 7, 23, 23-d4-5β-холан-24-овая кислота;
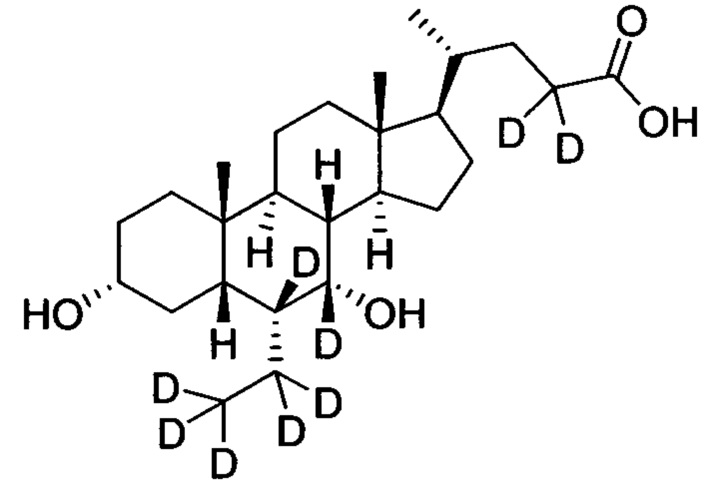
В другом предпочтительном варианте осуществления изобретения, соединение представляет собой
 и имеет следующие характеристики: расчетное значение MS: 421; измеренное значение MS: 422 (М+Н)+, 444 (M+Na)+.
и имеет следующие характеристики: расчетное значение MS: 421; измеренное значение MS: 422 (М+Н)+, 444 (M+Na)+.
В другом предпочтительном варианте осуществления изобретения, соединение представляет собой
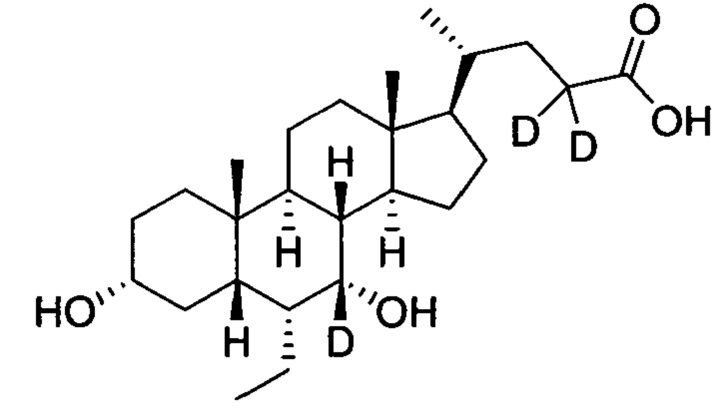 ; и имеет следующие характеристики: расчетное значение MS: 423; измеренное значение MS: 424 (М+Н)+, 446(M+Na)+.
; и имеет следующие характеристики: расчетное значение MS: 423; измеренное значение MS: 424 (М+Н)+, 446(M+Na)+.
В другом предпочтительном варианте осуществления изобретения, соединение представляет собой
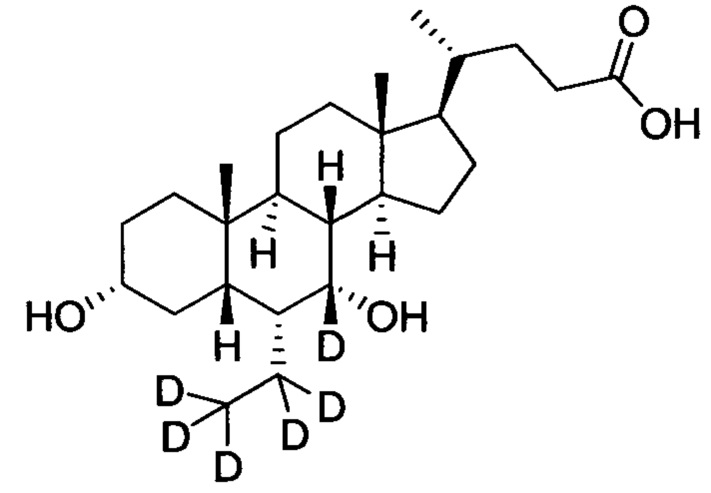 ; и имеет следующие характеристики: расчетное значение MS: 426; измеренное значение MS: 427 (М+Н)+, 449 (M+Na)+.
; и имеет следующие характеристики: расчетное значение MS: 426; измеренное значение MS: 427 (М+Н)+, 449 (M+Na)+.
В другом предпочтительном варианте осуществления изобретения, соединение представляет собой
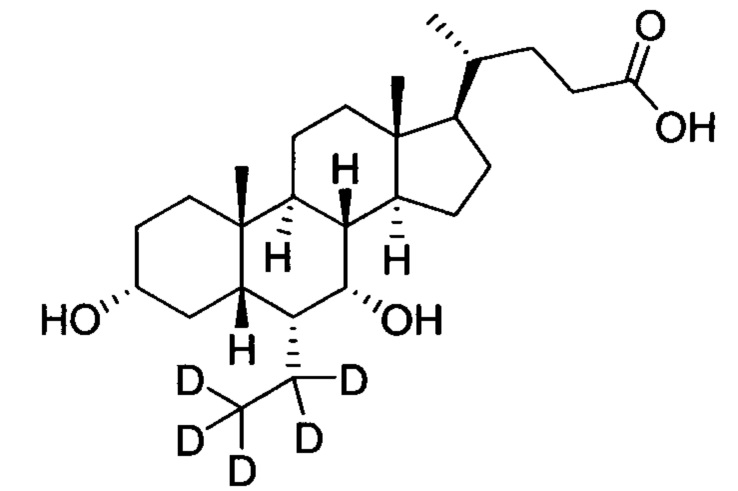 ; и имеет следующие характеристики: расчетное значение MS: 425; измеренное значение MS: 426 (М+Н)+, 448 (M+Na)+.
; и имеет следующие характеристики: расчетное значение MS: 425; измеренное значение MS: 426 (М+Н)+, 448 (M+Na)+.
В другом предпочтительном варианте осуществления изобретения, соединение представляет собой
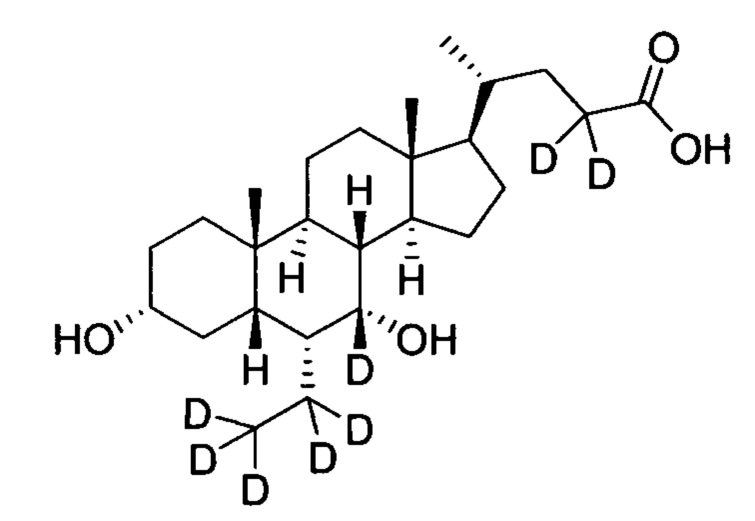 ; и имеет следующие характеристики: расчетное значение MS: 428; измеренное значение MS: 429 (М+Н)+, 451 (M+Na)+.
; и имеет следующие характеристики: расчетное значение MS: 428; измеренное значение MS: 429 (М+Н)+, 451 (M+Na)+.
В другом предпочтительном варианте осуществления изобретения, соединение представляет собой
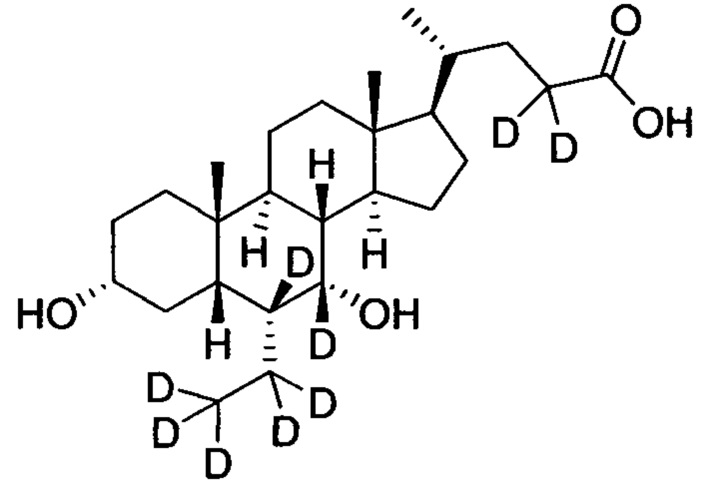 ; и имеет следующие характеристики: расчетное значение MS: 429; измеренное значение MS: 430 (М+Н)+, 452 (M+Na)+.
; и имеет следующие характеристики: расчетное значение MS: 429; измеренное значение MS: 430 (М+Н)+, 452 (M+Na)+.
В другом предпочтительном варианте осуществления изобретения, соединение представляет собой
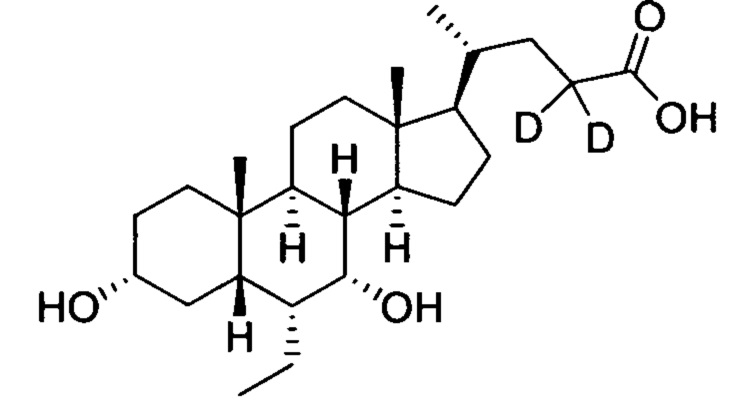 ; и имеет следующие характеристики: расчетное значение MS: 422; измеренное значение MS: 423 (М+Н)+, 445 (M+Na)+.
; и имеет следующие характеристики: расчетное значение MS: 422; измеренное значение MS: 423 (М+Н)+, 445 (M+Na)+.
В другом предпочтительном варианте осуществления изобретения, соединение представляет собой
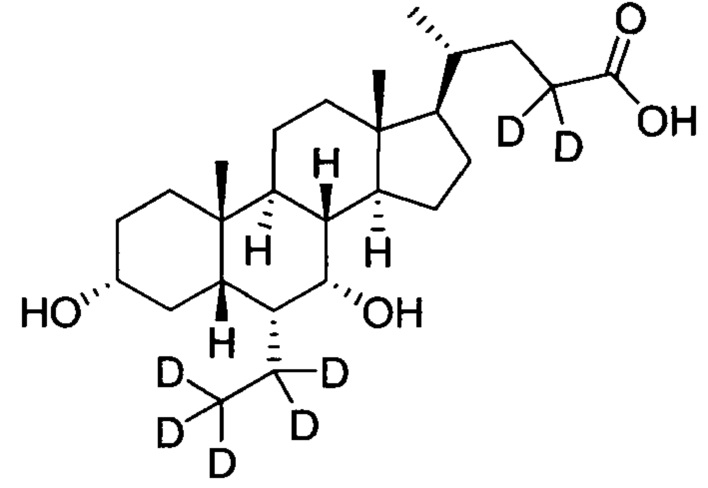 ; и имеет следующие характеристики: расчетное значение MS: 427; измеренное значение MS: 428 (М+Н)+, 450 (M+Na)+.
; и имеет следующие характеристики: расчетное значение MS: 427; измеренное значение MS: 428 (М+Н)+, 450 (M+Na)+.
В другом предпочтительном варианте осуществления изобретения, соединение не содержит какого-либо недейтерированного соединения.
В другом предпочтительном варианте осуществления изобретения, недейтерированным соединением является обербиновая кислота, то есть 3α, 7α-дигидрокси-6α-этил-5β-холан-24-овая кислота.
В другом предпочтительном варианте осуществления изобретения, соединение приготавливают способом, описанном в примерах 1-4.
Во втором аспекте настоящего изобретения представлен способ приготовления фармацевтической композиции, содержащий следующий этап: смешивание соединений первого аспекта настоящего изобретения или их кристаллических форм, фармацевтически приемлемых солей, гидратов или сольватов и фармацевтически приемлемого носителя для получения фармацевтической композиции.
В третьем аспекте настоящего изобретения представлена фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и соединения первого аспекта настоящего изобретения или их кристаллическую форму, фармацевтически приемлемые соли, гидраты или сольваты.
В другом предпочтительном варианте осуществления изобретения, фармацевтическая композиция представлена в виде инъекций, капсул, таблеток, пилюлей, порошков или гранул.
В другом предпочтительном варианте осуществления изобретения, фармацевтическая композиция содержит другие терапевтические средства, а другие терапевтические средства в свою очередь представляют собой лекарства для лечения раковых заболеваний, сердечно-сосудистых заболеваний, воспалений, инфекций, иммунных заболеваний, нарушений обмена веществ или нарушений, связанных с трансплантацией органов.
В другом предпочтительном варианте осуществления изобретения, раковые заболевания включают в себя (без ограничений): рак легких, рак молочной железы, рак предстательной железы, рак пищевода, рак прямой кишки, рак толстой кишки, рак крови (или злокачественное заболевание крови), рак костей, рак почки, рак желудка, рак печени или колоректальный рак.
В другом предпочтительном варианте осуществления изобретения, под раком подразумевается рак печени.
Более предпочтительно, чтобы дополнительное терапевтическое средство включало в себя (без ограничений): сорафениб, регорафениб, дуонафениб, цисплатин, доксорубицин, гемцитабин, ФОЛФОКС, децитабин, капецитабин, статины (ловастатин, симвастатин, правастатин, мевастатин, флувастатин, аторвастатин, церивастатин, розувататин и др.), розиглитазон, пиоглитазон, метформин, акарбозу, воглибозу, сульфонилмочевины (глипизид, гликлазид, глимепирид и др.), гипогликемические ингибиторы дипептидилпептидазы-4 (DPP-4) (такие как ситаглиптин, вилдаглиптин, алоглиптин, тражента и др.), гипогликемические ингибиторы натрийзависимых переносчиков глюкозы (SGLT2) (такие как дапаглифлозин, канаглифлозин и др.), агонисты рецептора глюкагонаподобного пептида-1 (GLP-1) (такие как экзенатид, лираглутид, ликсисататид, ликсисенатид и др.), интерферон, пегилированный интерферон, препараты для лечения гепатита С (такие как Софосбувир, телапревир, Боцепревир, АСН-3102, Даклатасвир, Делеобувир, Ледипасвир и др.), препараты для лечения гепатита В (такие как ламивудин, адефовир дипивоксил, телбивудин, энтекавир, тенофовир дизопроксил, клевудин и др.).
В четвертом аспекте настоящего изобретения представлено применение соединений первого аспекта настоящего изобретения или их кристаллической формы, фармацевтически приемлемых солей, гидратов или сольватов или фармацевтической композиции третьего аспекта настоящего изобретения при приготовлении фармацевтических композиций агониста фарнезоидного X рецептора (FXR) и/или агониста рецептора желчной кислоты, сопряженного с G-белком (GPBAR или TGR5).
В другом предпочтительном варианте осуществления изобретения представлено использование фармацевтической композиции при приготовлении лекарственных средств для лечения и профилактики следующих заболеваний: неалкогольного стеатогепатита, неалкогольной жировой болезни печени, желчных камней, первичного билиарного цирроза, цирроза, фиброза печени, диабета, атеросклероза, ожирения.
В пятом аспекте настоящего изобретения представлен способ лечения с применением агониста фарнезоидного X рецептора (FXR) и/или агониста рецептора желчной кислоты, сопряженного с G-белком (GPBAR или TGR5), или терапевтический способ лечения заболеваний (таких как рак, неалкогольный стеатогепатит, неалкогольная жировая болезнь печени, желчные камни, первичный билиарный цирроз, цирроз, фиброз печени, диабет, атеросклероз, ожирение), включающий в себя следующие этапы: введение соединений первого аспекта настоящего изобретения или их кристаллических форм, фармацевтически приемлемых солей, гидратов или сольватов, или введения фармацевтической композиции третьего аспекта настоящего изобретения нуждающемуся субъекту.
Следует понимать, что в настоящем изобретении, каждая из технических особенностей, описанных выше и ниже (например, в Примерах), может быть объединена с любой другой, тем самым образуя новые или предпочтительные технические решения, которые не требуют повторного определения в настоящем документе.
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
В результате исследований, изобретатель неожиданно обнаружил, что дейтерированные производные хенодезоксихолевой кислоты или их фармацевтически приемлемые соли обладают явно лучшими фармакокинетическими и/или фармакодинамическими свойствами при сравнении с недейтерированным соединением, и поэтому более пригодны для использования в качестве соединений агониста фарнезоида X рецептора (FXR) и/или агониста рецептора желчной кислоты, сопряженного с G-белком (GPBAR или TGR5), к тому же, они более подходят для использования при приготовлении лекарственных средств для лечения рака и заболеваний, связанных с фарнезоидным X рецептором (FXR) и/или рецептором желчной кислоты, сопряженным с G-белком (GPBAR или TGR5). Настоящее изобретение выполнено основываясь на этом.
ОПРЕДЕЛЕНИЯ
Используемый в данном документе термин «галоген» относится к F, Cl, Br и I. Предпочтительнее, если атом галогена выбирается из F, Cl и Br.
Используемый в данном документе термин «лучшие фармакокинетические и/или фармакодинамические свойства» относится к лекарственным средствам с большим периодом полувыведения (t1/2) или к лекарственным средствам с более сильным воздействием на организм (AUC) или к лекарственным средствам с более высокой максимальной концентрацией (Cmax) или к лекарственным средствам с более длительным периодом выведения из организма.
Используемый в данном документе термин «дейтерированный» означает, что один или несколько атомов водорода в соединении или группе замещены дейтерием.
Используемый в данном документе термин «недейтерированное соединение» означает соединение, в котором атомное отношение дейтерия не превышает естественное содержание изотопов дейтерия (0,015%).
В другом предпочтительном варианте осуществления изобретения, содержание изотопов дейтерия в позиции, замещаемой дейтерием, превышает естественное содержание изотопов дейтерия (0,015%), предпочтительнее превышает более чем на 50%, предпочтительнее превышает более чем на 75%, предпочтительнее превышает более чем на 95%, предпочтительнее превышает более чем на 97%, предпочтительнее превышает более чем на 99%, предпочтительнее превышает более чем на 99,5%.
В другом предпочтительном варианте осуществления изобретения, соединение формулы (I) содержит по меньшей мере один атом дейтерия, предпочтительнее два атома дейтерия, три атома дейтерия, предпочтительнее четыре атома дейтерия, предпочтительнее шесть атомов дейтерия.
Предпочтительнее, что бы в соединении с формулой (I), О представлял собой 16O.
В другом предпочтительном варианте осуществления изобретения, в соединении, содержание изотопа 16O в позиции атома кислорода составляет ≥95%, предпочтительнее ≥99%.
АКТИВНЫЕ КОМПОНЕНТЫ
Используемый в данном документе термин «соединение настоящего изобретения» относится к соединению формулы (I). Термин также охватывает кристаллические формы, фармацевтически приемлемые соли, гидраты или сольваты соединения формулы (I).
Используемый в данном документе термин «фармацевтически приемлемые соли» относится к солям, подходящим к использованию в фармацевтической композиции, которая получается путем смешивания соединения настоящего изобретения с кислотой или основой. Фармацевтически приемлемые соли включают в себя неорганические и органические соли. Предпочтительным типом солей являются соли, полученные из соединений настоящего изобретения и кислоты. Кислоты, подходящие для образования солей, включают в себя, без ограничений, аминокислоты, такие как пролин, фенилаланин, аспарагиновая кислота, глутаминовая кислота и т.д. Другими предпочтительными типами солей являются соли, полученные путем смешивания соединений настоящего изобретения и основ, к примеру, соли щелочных металлов (например, соли натрия или калия), соли щелочноземельных металлов (например, соли кальция или магния), соли аммония (например, соли низшего аммониевого алкила или другие фармацевтически приемлемые соли аминов), соль метиламина, соль этиламина, соль пропиламина, соль диметиламина, соли триметиламина, соли диэтиламина, соли триэтиламина, соли трет-бутиламина, соли этилендиамина, соли гидроксиэтиламина, соли бигидроксиэтиламина, соли тригидроксиэтиламина и соли аминов, полученные с помощью морфолина, пиперазина и лизина.
Термин «сольват» означает смешивание соединения настоящего изобретения и молекул растворителя для получения комплексного удельного отношения. «Гидрат» означает смешивание соединения настоящего изобретения с водой для получения комплексного соединения.
Помимо этого, соединения настоящего изобретения дополнительно содержат хиральные энантиомеры или рацематы производной хенодезоксихолевой кислоты формулы (I).
Кроме того, соединения настоящего изобретения дополнительно включают в себя глюкуронидные конъюгаты (глюкурониды) и тауриновые конъюгаты производной хенодезоксихолевой кислоты формулы (I).
Кроме того, соединения настоящего изобретения дополнительно включают в себя депо-формы производной хенодезоксихолевой кислоты формулы (I). Термин «депо-форма» включает в себя соединения, которые сами по себе могут быть биологически активными или неактивными и при введении соответствующим способом в организм человека, посредством метаболических или химических реакций превращаются в класс соединений формулы (I), соль или раствор соединения формулы (I). Депо-формы включают в себя, без ограничений, сложные эфиры карбоновой кислоты, сложные эфиры угольной кислоты, сложные эфиры фосфорной кислоты, сложные эфиры азотной кислоты, сложные эфиры серной кислоты, сложные эфиры сульфонов, сложный эфир сульфоксидов, аминосоединения, карбаматы, азосоединения, фосфорамиды, глюкозиды, простые эфиры, ацетали и др.
СПОСОБ ПРИГОТОВЛЕНИЯ
Далее более конкретно описано приготовление соединений формулы (I), но эти конкретные способы получения не возлагают никаких ограничений на настоящее изобретение. Соединения настоящего изобретения также могут быть легко приготовлены путем необязательного сочетания различных синтетических способов, описанных здесь или известных в данной области техники, такое сочетание может быть легко выполнено специалистом в области техники настоящего изобретения.
Известны способы приготовления дейтерированного производного хенодезоксихолевой кислоты и его физиологически совместимых солей, используемых в настоящем изобретении. Приготовление соответствующего дейтерированного производного хенодезоксихолевой кислоты может быть совершено с использованием соответствующего дейтерированного исходного соединения и синтезирования по тому же методу. Например, соединение формулы (I) может быть приготовлено в соответствии со способом, описанным в WO 02072598, за исключением того, что вместо дейтерированного материала используется недейтерированный материал.
Как правило, в процессе приготовление, каждая реакция обычно проводится в инертном растворителе при температуре от комнатной до температуры флегмы (например, от 0°С до 200°С, предпочтительнее от 0°С до 100°С). Время реакции обычно составляет от 0,1 до 60 часов, предпочтительнее от 0,5 до 48 часов.
Следующие типовые способы приготовления 1 и 2 могут быть использованы при синтезе соединений настоящего изобретения с формулой (I).
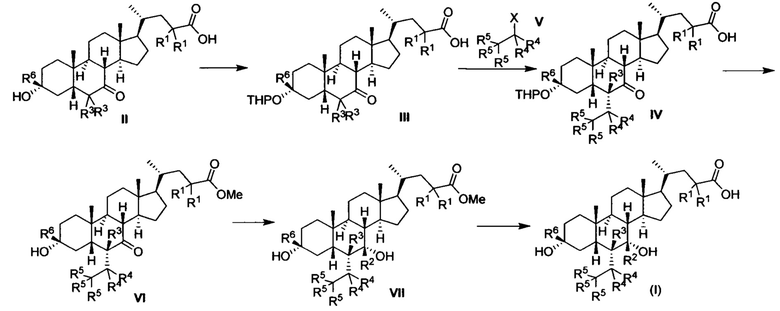
Синтетический способ 1
Где R1, R2, R3, R4, R5 и R6 соответствуют определениям выше, а X представляет собой галоген.
Как показано в синтетическом способе 1, соединение III получается посредством защиты гидроксила соединения II с помощью ТНР. Соединение III и соединение V подвергаются реакции замещения для получения соединение IV с помощью щелочи. Соединение IV подвергается снятию защитных групп и этерификации в кислоте и метаноле для получения соединения VI. Соединение VI восстанавливается для получения соединения VII. Наконец, соединение VII настоящего изобретения получается посредством гидролиза соединения VII. Вышеуказанная реакция проводится в инертном растворителе, таком как дихлорметан, ацетонитрил, н-гексан, толуол, тетрагидрофуран, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, уксусная кислота, бутанол, пропанол и др. при температуре от 0 до 200°С.
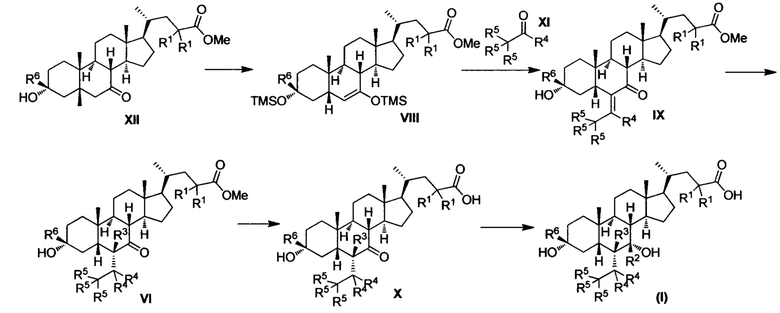
Синтетический способ 2
Как показано в Синтетическом способе 2, соединение VIII получается посредством защиты гидроксила сложного метилового эфира соединения XII с помощью TMS. Соединение IX получается с помощью проведения реакции альдольной конденсации и реакции отщепления соединения VIII и альдегидного соединения XI. Соединение IX восстанавливается для получения соединения VI; соединение VI гидролизуется для получением соединения X и, наконец, соединение X восстанавливается для получения соединения I.
Упомянутая реакция проводится в инертном растворителе, таком как дихлорметан, ацетонитрил, н-гексан, толуол, тетрагидрофуран, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, уксусная кислота, бутанол, пропанол и др. при температуре от -100°С до 200°С.
ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ВВЕДЕНИЕ
Соединения настоящего изобретения обладают отменной способностью активации фарнезоидного X рецептора (FXR) и/или рецептора холевой кислоты, сопряженного с G-белком (GPBAR или TGR5). Следовательно, соединение настоящего изобретения и кристаллические формы, фармацевтически приемлемые неорганические или органические соли, гидраты или сольваты, а также фармацевтическая композиция, содержащая соединение настоящего 25 изобретения в качестве основного активного компонента, могут быть использованы для лечения, профилактики и облегчение заболеваний, вызванных фарнезоидным X рецептором (FXR) и/или рецептором холевой кислоты, сопряженным с G-белком (GPBAR или TGR5). В соответствии с известным уровнем техники, использование соединений настоящего изобретения полезно при лечении следующих заболеваний: рака, неалкогольного стеатогепатита, неалкогольной жировой болезни печени, желчных камней, первичного билиарного цирроза, цирроза, фиброза печени, диабета, атеросклероза, ожирения и др.
Фармацевтическая композиция изобретения содержит соединение настоящего изобретения или его фармацевтически приемлемые соли в безопасном и эффективном диапазоне доз а также фармацевтически приемлемые эксципиенты или носители. «Безопасное и эффективное количество» означает: количество соединения, достаточное для значительного улучшения состояния, но которое не вызовет серьезных побочных эффектов. Как правило, фармацевтическая композиция содержит 0,5-2000 мг кристаллических форм настоящего изобретения на одну дозу, предпочтительнее 1-500 мг кристаллических форм настоящего изобретения на одну дозу, предпочтительнее, чтобы «одна доза» представляла собой одну капсулу или таблетку.
«Фармацевтически приемлемый носитель» означает один или несколько совместимых твердых или жидких наполнителей или желеобразные материалы, которые подходят для применения человеком, и которые также должны обладать достаточной чистотой и достаточно низкой токсичностью. «Совместимый» в данном документе означает, что каждый компонент в составе композиции и соединение настоящего изобретения могут быть хорошо смешаны между собой без существенного снижения эффективности соединений. Некоторые примеры фармацевтически приемлемых носителей включают в себя целлюлозу и ее производные (например, карбоксиметилцеллюлоза натрия, натрий-этил целлюлоза, ацетат целлюлозы и др.), желатин, тальк, твердые смазочные вещества (например, стеариновая кислота, стеарат магния), сульфат кальция, растительные жиры (например, соевое масло, кунжутное масло, арахисовое масло, оливковое масло и др.), полиолы (например, пропилен-гликоль, глицерин, маннитол, сорбитол и др.), эмульгаторы (например, Tween®), смачивающий реагент (например, додецилсульфат натрия), красящие добавки, ароматизаторы, стабилизаторы, антиоксиданты, консерванты, апирогенная вода и др.
Какое-либо особое ограничение способа введения соединения или фармацевтических композиций настоящего изобретения отсутствует, а типовой способ введения включает в себя (без ограничений): пероральный, введение в двенадцатиперстную кишку, ректальный, парентеральный (внутривенный, внутримышечный или подкожный) и местное применение.
Твердая лекарственная форма для перорального введения включает в себя капсулы, таблетки, пилюли, порошки и гранулы. В данных твердых лекарственных формах активные соединения смешиваются по меньшей мере с одним традиционным инертным эксципиентом (или носителем), например, цитратом натрия или CaHPO4, или смешивается с любым из следующих компонентов: (а) наполнители или компатибилизатор, например, крахмал, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; (б) связующие вещества, например, гидроксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и аравийская камедь; (в) увлажнитель, например, глицерин; (г) распадающиеся вещества, например, агар, карбонат кальция, картофельный крахмал или тапиоковый крахмал, альгиновая кислота, определенные композитные силикаты и карбонат натрия; (д) замедляющие растворение реагенты, например, парафин; (е) ускорители впитывания, например, четвертичные аммониевые соединения; (ж) смачивающие реагенты, например, цетиловый спирт и глицерилмоностеарат; (з) адсорбенты, например, каолин; и (и) смазочные вещества, например, тальк, стеарат кальция, стеарат магния, твердый полиэтиленгликоль, лаурилсульфат натрия или их смеси. Лекарственные формы в капсулах, таблетках и пилюлях могут также содержать буферные агенты.
Твердые лекарственные формы, такие как таблетки, сахарные пустышки, капсулы, пилюли и гранулы, могут быть подготовлены с использованием материалов покрытия, таких как кишечнорастворимые оболочки и любых других материалов, известных в данной области техники. Они могут содержать непрозрачное вещество. Активные компоненты в композициях могут высвобождаться с задержкой в заданной части желудочно-кишечного тракта. Примеры интегрированных компонентов включают в себя полимеры и воски. При необходимости, активные соединения и один или несколько вышеуказанных эксципиентов могут образовывать микрокапсулы.
Жидкие лекарственные формы для перорального введения включают в себя фармацевтически приемлемые эмульсии, растворы, взвеси, сиропы или вытяжки. Кроме активных соединений, жидкие лекарственные формы могут содержать какие-либо обычные традиционные инертные разбавители, известные в данной области техники, например, воду или иные растворители, сжижающие реагенты и эмульгаторы, например, этанол, изопропанол, угольноэтиловый эфир, этилацетат, пропилен-гликоль, 1,3-бутандиол, диметилформамид, а также масло, в частности, хлопковое масло, арахисовое масло, масло из кукурузных зерен, оливковое масло, касторовое масло и кунжутное масло или их сочетания.
Кроме данных инертных разбавителей, композиция также может содержать присадки, например, смачивающие реагенты, эмульгаторы и суспендирующее вещество, подсластители, ароматизаторы и отдушки.
В дополнение к активным соединениям, взвесь может содержать суспендирующее вещество, например, этоксилированный изооктадеканол, полиоксиэтиленовый сорбитол и сложные эфиры сорбитана, микрокристаллическую целлюлозу, метанол, алюминий и агар или их сочетания.
Композиции для парэнтеральных инъекций могут содержать физиологически приемлемые стерильные водные или безводные растворы, дисперсии, взвеси или эмульсии, а также стерильные порошки, которые можно повторно растворить в стерильных инъекционных растворах или дисперсиях. Подходящие водные и безводные носители, разбавители, растворители или эксципиенты включают в себя воду, этанол, полиолы и их подходящие смеси.
Дозированные формы для местного применения соединений по изобретению включают в себя мази, порошки, пластыри, аэрозоль и ингаляторы.
Активные ингредиенты смешиваются с физиологически приемлемыми носителями и любыми консервантами, буферными агентами или пропел лентами, в стерильных условиях, если это необходимо.
Соединения настоящего изобретения могут вводиться отдельно или в сочетании с любыми другими фармацевтически приемлемыми соединениями.
Когда используются фармацевтические композиции, к млекопитающим (например, человек) применяется безопасное и эффективное количество соединения настоящего изобретения, при этом доза введения - это фармацевтически эффективная доза. Для человека весом 60 кг суточная доза обычно составляет 0,5-2000 мг, предпочтительнее 1-500 мг.
Конечно, конкретная доза должна также зависеть от различных факторов, например, способ введения, состояние здоровья пациента, которые связаны с навыками опытного терапевта.
Соединения настоящего изобретения обладают рядом преимуществ по сравнению с дейтерированными соединениями известного уровня техники. Основными преимуществами настоящего изобретения являются:
(1) Соединения настоящего изобретения обладают отменной способностью активации в отношении фарнезоидного X рецептора (FXR) и/или рецептора желчной кислоты, сопряженного с G-белком (GPBAR или TGR5).
(2) Метаболизм дейтерированных соединений в организме изменяется по технологии дейтерирования, что придает соединению лучшие фармакокинетические характеристики. В этом случае, доза может быть изменена и превращена в препарат длительного действия для улучшения применимости.
(3) Водород в соединениях был замещен дейтерием, концентрация лекарственного средства соединения в организме животных может быть усилена изотопным эффектом дейтерия, что повышает эффективность лекарственного средства.
(4) Водород в соединениях был замещен дейтерием, так как некоторые метаболиты являются подавленными, безопасность соединения может быть улучшена.
Настоящее изобретение будет дополнительно проиллюстрировано ниже со ссылкой на конкретные примеры. Следует понимать, что данные примеры предназначены только для иллюстрации изобретения, а не для ограничения объема изобретения. Экспериментальные способы, не требующие особых условий, описанные в следующих примерах, обычно выполняются в обычных условиях или в соответствии с инструкциями производителя. Если не указано иное, части и процентные доли рассчитываются по весу.
Пример 1 Синтез 3α, 7α-дигидрокси-6α-этил-7-d-5β-холан-24-овой кислоты (соединение 1)
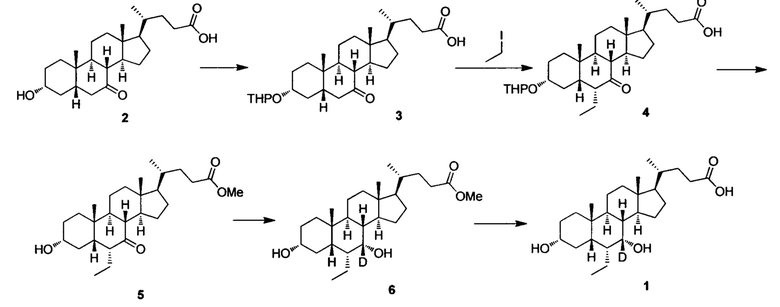
1. Синтез 3α-тетрагидропиранилокси-7-кето-5β-холан-24-овой кислоты (соединение 3)
3α-гидрокси-7-кето-5β-холан-24-овая кислота (10,0 г, 25,6 ммоль) растворяют в диоксане (150 мл) в колбе. Моногидрат n-толуолсульфоновой кислоты (0,49 г, 2,56 ммоль) и 3,4-дигидро-2Н-пиран (4,31 г, 51,2 ммоль) добавляются по очереди. После перемешивания при комнатной температуре в течение 1 часа, капельно добавляется раствор аммиака в метаноле для доведения рН до 8-9. После удаления летучих компонентов посредством выпаривания, остаток экстрагируется с помощью этилового эфира уксусной кислоты. Органическая фаза затем промывается насыщенным раствором бикарбоната натрия, водой и насыщенным рассолом и подвергается сушке с помощью Na2SO4. После фильтрации, фильтрат конденсируется с использованием роторного испарителя. Полученный неочищенный продукт очищается посредством колоночной хроматографии на силикагеле (соотношение этилового эфира уксусной кислоты к петролейному эфиру = 1/3), для получения белого с металлическим оттенком твердого целевого соединения (9,72 г, 80%).
2. Синтез 3α-тетрагидропиранилокси-6α-этил-7-кето-5β-холан-24-овой кислоты (соединение 4)
В колбу добавляется диизопропиламин (5,8 г, 57,6 ммоль) в безводном тетрагидрофуране (400 мл) и охлаждается до -78°С. При -60°, по очереди, капельно, добавляются н-бутиллитий (23,1 мл, 2,5 М в гексане) и гексаметилфосфорный триамид (НМРА, 10,3 г, 57,6 ммоль). После перемешивания в течение 1 часа при -70°С, капельно добавляется в предварительно охлажденный (-78°С) раствор 3α-тетрагидропиранилокси-7-кето-5β-холан-24-овой кислоты (соединение 3, 9,1 г, 19,2 ммоль) в безводном тетрагидрофуране (200 мл), а затем перемешивается еще 30 мин. Затем, йодэтан (29,9 г, 192 ммоль) в безводном тетрагидрофуране (1000 мл) и реакционная смесь медленно добавляются и перемешиваются при комнатной температуре в течение ночи. После удаления летучих компонентов с помощью вакуума, остаток доводится до рН 2-3 с использованием 10% раствора соляной кислоты и экстрагируется с помощью этилового эфира уксусной кислоты. Объединенная органическая фракция затем промывается 5% раствором гипосульфита натрия, водой и рассолом, сушится с использованием Na2SO4 и фильтруется. Фильтрат выпаривается для получения целевого продукта, который использовался непосредственно на следующем этапе без дополнительной очистки.
3. Синтез Метил 3α-гирокси-6α-этил-7-кето-5β-холан-24-оата (соединение 5)
Неочищенное соединение 3α-тетрагидропиранилокси-6α-этил-7-кето-5β-холан-24-овой кислоты, полученное на предыдущем этапе, растворяется в растворе соляной кислоты в метаноле (2 н., 120 мл) и перемешивается при температуре флегмы в течение 16 часов. После удаления летучих компонентов с помощью вакуума, остаток экстрагируется с помощью этилового эфира уксусной кислоты. Объединенная органическая фракция затем промывается водой, насыщенным раствором NaHCO3 и рассолом, сушится с использованием Na2SO4 и фильтруется. После выпаривания фильтрата, остаток очищается посредством колоночной хроматографии на силикагеле (20-40% раствор этилового эфира уксусной кислоты/гексана) для получения целевого соединения 5 (1,8 г, выход 21,7% из соединения 3).
4. Синтез Метил 3α, 7α-дигирокси-6α-этил-7-d-5β-холан-24-оата (соединение 6)
Метил 3α-гирокси-6α-этил-7-кето-5β-холан-24-оат (1,5 г, 3,5 ммоль) растворяется в метаноле (6 мл) в колбе и перемешивается. Добавляется бородеутерид натрия (NaBD4, 0,3 г, 7 ммоль, Sigma-Aldrich) и смесь перемешивается при комнатной температуре еще в течении 3 часов. Добавляется вода для гашения реакции. Смесь концентрируется с помощью высокого вакуума и экстрагируется с помощью этилового эфира уксусной кислоты. Объединенная органическая фракция затем промывается водой и рассолом, сушится с использованием Na2SO4 и фильтруется. Фильтрат выпаривается для получения целевого соединения (1,3 г, 85%).
5. Синтез 3α, 7α-дигидрокси-6α-этил-7-d-5β-холан-24-овой кислоты (соединение 1)
В колбу добавляется метил 3α, 7α-дигирокси-6α-этил-7-d-5β-холан-24-оат (1,2 г, 2,8 ммоль), раствор гидроксида натрия в воде (10%, 2,24 г, 5,6 ммоль) и раствор безводного тетрагидрофурана/МеОН/водяной смеси (1/3/2, 20 мл). Смесь перемешивается при 40°С в течение 6 часов. Добавляется 3 н. раствор соляной кислоты для доведения рН до 2-3. Смесь экстрагируется с помощью этилового эфира уксусной кислоты. Объединенная органическая фракция затем промывается водой и рассолом, сушится с использованием Na2SO4 и фильтруется. Фильтрат концентрируется и остаток очищается посредством колоночной хроматографии на силикагеле (5% раствор метанола/дихлорметана) для получения целевого соединения (0,87 г, 75%). 1Н ЯМР (400 МГц, CDCl3+CD3OD) δ: 3,46 (m, 1Н), 2,35-0,74 (m, 27Н), 0,95 (d, 3Н), 0,89-0,92 (m, 6Н), 0,68 (s, 3Н). Масс-спектрометрия с электрораспылением (м/з): 422 (М+Н)+, 444 (M+Na)+.
Альтернативный способ приготовления Соединения 1
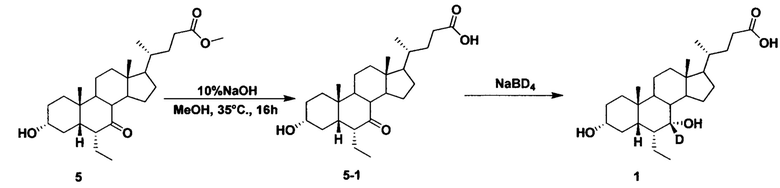
В колбу поочередно добавляется Метил 3α-гирокси-6α-этил-7-кето-5β-холан-24-оат (2,0 г, 4,6 ммоль), раствор гидроксида натрия в воде (10%, 4,0 мл) и раствор метанола/водной смеси (3/1, 20 мл). Смесь перемешивается при 35°С в течение 16 часов. После концентрирования, в остаток добавляется вода (10 мл), рН доводится до 2-3 с помощью 1 н. раствора соляной кислоты. Осадок фильтруется и промывается очищенной водой с последующим высушиванием для получения соединения 5-1 (1,7 г, 88%).
Соединение 5-1 (1,0 г, 2,4 ммоль), раствор гидроксида натрия (50%, 0,5 мл) и вода (8,0 мл) добавляются в колбу. Бородеутерид натрия (103 мг, 2,4 ммоль) добавляется порциями и смесь перемешивается при 100°С в течение ночи. Смесь охлаждается до комнатной температуры и добавляется 1 н. раствора соляной кислоты для доведения рН до 2-3. Осадок фильтруется, промывается водой и сушится для получения целевого соединения (520 мг, 51%). ЯМР (400 МГц, DMSO-d6) δ: 11,95 (brs, 1Н), 4,23-4,01 (m, 2H), 3,16-3,11 (m, 1H), 2,28-2,20 (m, 1H), 2,15-2,07 (m, 1H), 1,93-0,83 (m, 34H), 0,61 (s, 3H).
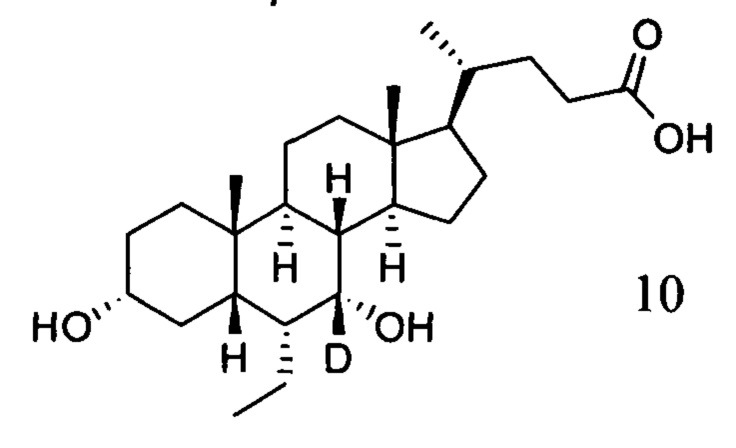
Пример 2 Синтез 3α, 7α-дигидрокси-6α-(этил-d5)-7-d-5β-холан-24-овой кислоты (соединение 10)
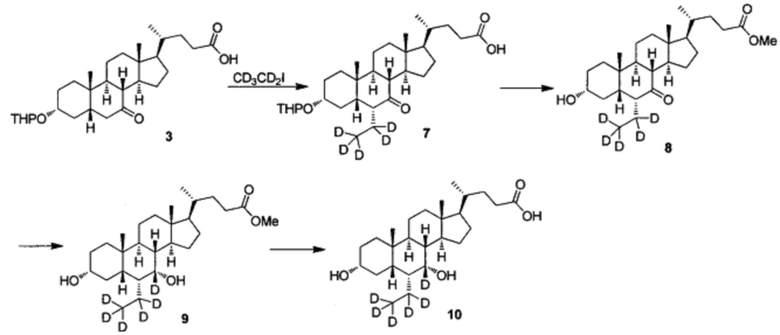
1. Синтез 3α-тетрагидропиранилокси-6α-(этил-d5)-7-кето-5β-холан-24-овой кислоты (соединение 7)
В колбу добавляется диизопропиламин (2,3 г, 23 ммоль) в безводном тетрагидрофуране (200 мл) и охлаждается до -78°С. Температура поддерживается при -60°С, затем, по очереди, капельно, добавляются н-бутиллитий (9,2 мл, 2,5 М в гексане) и гексаметилфосфорный триамид (НМРА, 4,2 г, 23 ммоль). После добавления, смесь перемешивается в течение 1 часа при -70°С. Предварительно охлажденный (-78°С) раствор 3α-тетрагидропиранилокси-7-кето-5β-холан-24-оата (Соединение 3, 3,6 г, 7,6 ммоль) в безводном тетрагидрофуране (100 мл) добавляется капельно. После перемешивания в течение еще 30 мин, медленно добавляется йодэтан-d5 (6,2 г, 38 ммоль) в безводном тетрагидрофуране (200 мл) и смесь перемешивается при комнатной температуре в течение ночи. После удаления летучих компонентов с помощью концентрирования под вакуумом, остаток доводится до рН 2-3 с использованием 10% раствора соляной кислоты и экстрагируется с помощью этилового эфира уксусной кислоты. Объединенная органическая фракция затем промывается 5% раствором гипосульфита натрия, водой и рассолом, сушится с помощью Na2SO4 и концентрируется для получения целевого продукта, который непосредственно использовался на следующем этапе без дополнительной очистки.
2. Синтез Метил 3α-гирокси-6α-(этил-d5)-7-кето-5β-холан-24-оата (соединение 8)
3-тетрагидропиранилокси-6α-(этил-d5)-7-кето-5β-холан-24-овая кислота, полученная на предыдущем этапе, растворяется в растворе соляной кислоты в метаноле (2 н., 30 мл) и перемешивается при температуре флегмы в течение 16 часов. После удаления летучих компонентов с помощью концентрирования под вакуумом, остаток экстрагируется с помощью этилового эфира уксусной кислоты. Объединенная органическая фракция затем промывается водой, насыщенным раствором NaHCO3 и рассолом, сушится с использованием Na2SO4 и концентрируется, а остаток очищается посредством колоночной хроматографии на силикагеле (20-40% раствор этилового эфира уксусной кислоты/гексана) для получения твердого вещества (0,6 г, выход 18%).
3. Синтез Метил 3α, 7α-дигирокси-6α-(этил-d5)-7-d-5β-холан-24-оата (соединение 9)
Метил 3α-гидрокси-6α-(этил-d5)-7-кето-5β-холан-24-оат (0,3 г, 0,68 ммоль) растворяется в метаноле (3 мл), добавляется в колбу и перемешивается. Добавляется бородеутерид натрия (60 мг, 1,4 ммоль) и смесь перемешивается при комнатной температуре еще в течении 3 часов. Добавляется вода для гашения реакции. Смесь концентрируется с помощью высокого вакуума и экстрагируется с помощью этилового эфира уксусной кислоты. Объединенная органическая фракция затем промывается водой и рассолом, сушится с использованием Na2SO4 и фильтруется. Фильтрат концентрируется для получения целевого соединения в виде белого твердого вещества (0,25 г, 82%).
4. Синтез 3α, 7α-дигидрокси-6α-(этил-d5)-7-d-5β-холан-24-овой кислоты (соединение 10)
В колбу по очереди добавляются метил 3α, 7α-дигирокси-6α-(этил-d5)-7-d-5β-холан-24-оат (0,24 г, 0,54 ммоль), раствор гидроксида натрия (10%, 0,44 г, 1,1 ммоль) и безводный тетрагидрофуран/МеОН/водяная смесь (1/3/2, 5 мл). Смесь перемешивается при 40°С в течение 6 часов. 3 н. раствор соляной кислоты добавляется для доведения рН до 2-3, а затем смесь экстрагируется с помощью этилового эфира уксусной кислоты. Объединенная органическая фракция затем промывается водой и рассолом, сушится с использованием Na2SO4, фильтруется и концентрируется для получения твердого неочищенного продукта, очищается посредством колоночной хроматографии на силикагеле (5% раствор метанола/дихлорметана) для получения целевого соединения (0,18 г, 78%). 1Н ЯМР (400 МГц, CDCl3+CD3OD) δ: 3,47 (m, 1H), 2,36-0,74 (m, 25Н), 0,95 (d, 3Н), 0,91 (s, 3Н), 0,66 (s, 3Н). Масс-спектрометрия с электрораспылением (м/з): 427 (М+Н)+, 449 (M+Na)+.
Пример 3 Синтез 3α, 7α-дигидрокси-6α-(этил-d5)-5β-холан-24-овой кислоты (соединение 12)
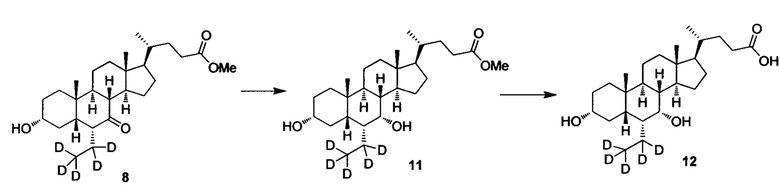
1. Синтез 3α, 7α-дигидрокси-6α-(этил-d5)-5β-холан-24-овой кислоты (соединение 11)
Затем в колбу поочередно добавляются метил 3α-гирокси-6α-(этил-d5)-7-кето-5β-холан-24-оат (0,3 г, 0,68 ммоль) и метанол (3 мл) и перемешиваются. Добавляется бородеутерид натрия (NaBH4, 60 мг, 1,4 ммоль) и смесь перемешивается при комнатной температуре еще в течении 3 часов. Добавляется вода для гашения реакции. Смесь концентрируется с помощью высокого вакуума и экстрагируется с помощью этилового эфира уксусной кислоты. Объединенная органическая фракция затем промывается водой и рассолом, сушится с использованием Na2SO4 и фильтруется. Фильтрат выпаривается для получения целевого соединения в виде белого твердого вещества (0,24 г, 81%).
2. Синтез 3α, 7α-дигидрокси-6α-(этил-d5)-5β-холан-24-овой кислоты (соединение 12)
В колбу по очереди добавляются 3α, 7α-дигидрокси-6α-(этил-d5)-5β-холан-24-овая кислота (0,24 г, 0,54 ммоль), раствор гидроксида натрия (10%, 0,44 г, 1,1 ммоль) и безводный тетрагидрофуран/МеОН/водяная смесь (1/3/2, 5 мл). Смесь перемешивается при 40°С в течение 6 часов. Добавляется 3 н. водный раствор соляной кислоты для доведения рН до 2-3. Смесь экстрагируется с помощью этилового эфира уксусной кислоты. Объединенная органическая фракция затем промывается водой и рассолом, сушится с использованием Na2SO4 и фильтруется. Фильтрат концентрируется для получения неочищенного продукта, который потом очищается посредством колоночной хроматографии на силикагеле (5% раствор метанола/дихлорметана) для получения целевого соединения (0,16 г, 72%). 1Н ЯМР (400 МГц, DMSO-d6) δ: 11,97 (brs, 1H), 4,32 (d, J=4,0 Hz, 1H), 4,07 (d, J=4,0 Hz, 1H), 3,50 (s, 1H),3,14-3,13 (m, 1H), 2,27-2,20 (m, 1H), 2,15-2,07 (m, 1H), 1,93-0,84 (m, 29H), 0,61 (s, 3H). Масс-спектрометрия с электрораспылением (м/з): 426 (М+Н)+, 448 (M+Na)+.
Пример 4 Синтез 3α, 7α-дигидрокси-6α-(этил-d3)-7-d-5β-холан-24-овой кислоты (соединение 18)
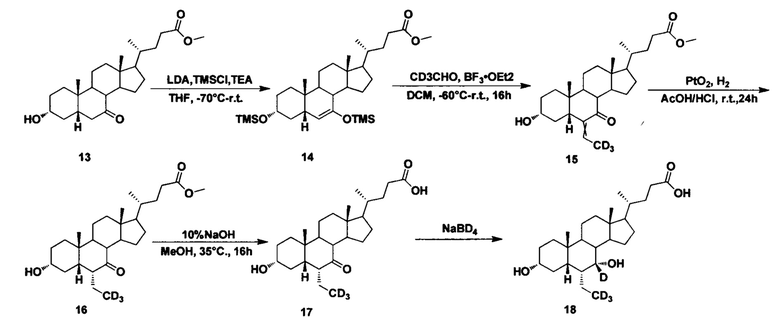
1. Синтез Метил 3α, 7-бис (триметилсилилокси)-5β-холан-6-ен-24-оата (соединение 14)
В четырехгорлую колбу добавляются диизопропиламид лития (68 мл, 135,9 ммоль, 2 M в безводном тетрагидрофуране/гептане/этилбензоле) и безводный тетрагидрофуран (50 мл). Во время перемешивания при 70°С, под защитой азота добавляется триметилхлорсилан (12,1 г, 111,1 ммоль) и смесь перемешивается в течение 30 мин. Раствор метил 2,3α-гидрокси, 7-оксо-холан-24-оата (соединение 13 (10 г) в безводном тетрагидрофуране (50 мл)) добавляется капельно по прошествии 30 мин, затем смесь перемешивается в течение еще 1 часа при 70°С. Триэтиламин (35,2 г, 348 ммоль) добавляется при температуре около -70°С. Смесь перемешивается при -70°С в течение еще 1 часа, а затем нагревается до комнатной температуры и перемешивается в течение ночи. Смесь охлаждается льдом и гасится медленным капельным добавлением насыщенного раствора NaHCO3. Смесь экстрагируется с помощью этилацетата, а водная фаза экстрагируется с помощью этилового эфира уксусной кислоты. Объединенная органическая фракция затем промывается насыщенным водным раствором NaHCO3 и рассолом, сушится с использованием Na2SO4 и концентрируется, а неочищенный продукт очищается посредством колоночной хроматографии на силикагеле (ЕА/РЕ=2%) для получения целевого соединения (12,9 г, выход 95%).
2. Синтез Метил 3а-гидрокси-6-(этилиден-d3)-7-кето-5β-холан-24-оата (соединение 15)
В четырехгорлую колбу добавляются метил 3α, 7-бис (триметилсилилокси)-5β-холан-6-ен-24-оат (11,0 г, 18,2 ммоль) и дихлорметан (60 мл). К перемешиваемой смеси добавляется (метил-d3) альдегид (2,1 мл, 36,4 ммоль) при -40°С. Во время перемешивания при температуре около -60°С в течение 10 мин, капельно добавляется раствор BF3.OEt2 (10 мл) в 20 мл дихлорметана. После перемешивания в течение еще 3 часов при -60°С, смесь естественным образом нагревается до комнатной температуры и перемешивается в течение ночи. Смесь охлаждается льдом и медленно гасится насыщенным водным раствором NaHCO3, перемешивается и экстрагируется. Водная фаза промывается дихлорметаном (60 мл), а в объединенную органическую фракцию добавляется 3 н. раствор соляной кислоты и смесь перемешивается в течение 1 часа попутно находясь в водяной бане. Реакция гасится насыщенным водным раствором NaHCO3, повторно экстрагируется, а водный слой промывается дихлорметаном. Объединенные органические слои сушатся с использованием Na2SO4 и концентрируются, а неочищенный продукт очищается посредством колоночной хроматографии на силикагеле (ЕА/РЕ=25-35%) для получения целевого соединения (6,1 г, выход 70%).
3. Синтез Метил 3α-гидрокси-6α-(этил-d3)-7-кето-5β-холан-24-оата (соединение 16)
Добавляются метил 3α-гидрокси-6-(этилиден-d3)-7-кето-5β-холан-24-оат (0,18 г, 0,42 ммоль), уксусная кислота (10 мл), концентрированный раствор соляной кислоты (0,5 мл) и оксид платины (IV) (20 мг). Смесь наводороживается и в течение 12 часов реагирует находясь в водородной атмосфере при комнатной температуре. Смесь фильтруется и концентрируется для получения целевого соединения (0,17 г, 94%).
4. Синтез 3α-гидрокси-6α-(этил-d3)-7-кето-5β-холан-24-кислоты (соединение 17)
В реакционную колбу добавляются метил 3α-гидрокси-6α-(этил-d3)-7-кето-5β-холан-24-оат (0,17 г, 0,39 ммоль), водный раствор гидроксида натрия (10%, 8,0 мл) и метанол/вода (4,5/1, 11 мл). Смесь перемешивается в течение 16 часов при 35°С, а затем концентрируется. После добавления воды добавляется 1 н. уксусной кислоты для доведения рН до 2-3 и фильтруется. Осадок промывается чистой водой и сушится для получения целевого соединения (0,14 г, 79%).
5. Синтез 3α, 7α-дигидрокси-6α-(этил-d3)-7-d-5β-холан-24-кислоты (соединение 18)
Поочередно добавляются 3α-гидрокси-6α-(этил-d3)-7-кето-5β-холан-24-кислота (65 мг, 0,15 ммоль), водный раствор гидроксида натрия (50%, 200 мг) и вода (3,0 мл). Во время перемешивания добавляется бородеутерид натрия (13 мг, 0,3 ммоль) и смесь перемешивается в течение 16 часов при 100°С. Смесь охлаждается до комнатной температуры и добавляется 1 н. раствора соляной кислоты для доведения рН до 2-3. Осадок фильтруется, промывается очищенной водой и сушится для получения целевого соединения (45 мг, 69%). 1Н ЯМР (400 МГц, DMSO-d6) δ: 11,95 (brs, 1H), 4,31 (s, J=4,0 Hz, 1Н), 4,04 (d, J=8,0 Hz, 1H), 3,14-3,13 (m, 1H), 2,27-2,20 (m, 1H), 2,15-2,07 (m, 1H), 1,93-0,84 (m, 31H), 0,61 (s, 3Н). Масс-спектрометрия с электрораспылением (м/з): 425 (М+Н)+, 447 (M+Na)+.
Пример 5 Синтез 3α, 7α-дигидрокси-6α-(этил-d3)-5β-холан-24-овой кислоты (соединение 19)
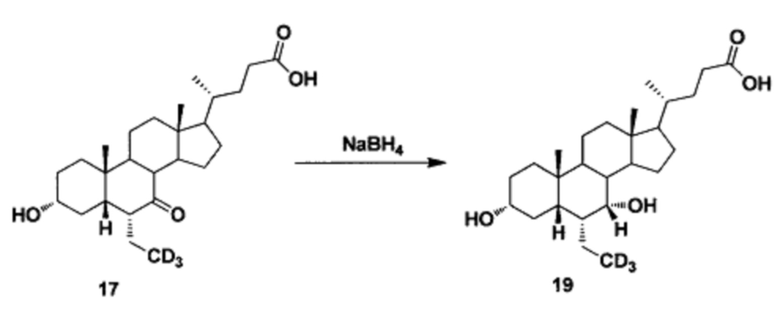
В реакционную колбу поочередно добавляются 3α-гидрокси-6α-(этил-d3)-7-кето-5β-холан-24-кислота (65 мг, 0,15 ммоль), водный раствор гидроксида натрия (50%, 200 мг) и вода (3,0 мл). Во время перемешивания добавляется борогидрид натрия (13 мг, 0,3 ммоль) и смесь перемешивается в течение 16 часов при 100°С. Смесь охлаждается до комнатной температуры и добавляется 1 н. раствора соляной кислоты для доведения рН до 2-3. Осадок фильтруется, промывается очищенной водой и сушится для получения целевого соединения (51 мг, 78%). 1Н ЯМР (400 МГц, DMSO-d6) δ: 11,97 (brs, 1Н), 4,32 (d, J=4,0 Hz, 1H), 4,07 (d, J=4,0 Hz, 1H), 3,50 (s, 1H), 3,14-3,13 (m, 1H), 2,27-2,20 (m, 1H), 2,15-2,07 (m, 1H), 1,93-0,84 (m, 31H), 0,61 (s, 3Н). Масс-спектрометрия с электрораспылением (м/з): 424 (М+Н)+, 446(M+Na)+.
Пример 6 Синтез 3α, 7α-дигидрокси-6α-(этил-d4)-7-d-5β-холан-24-овой кислоты (соединение 23)
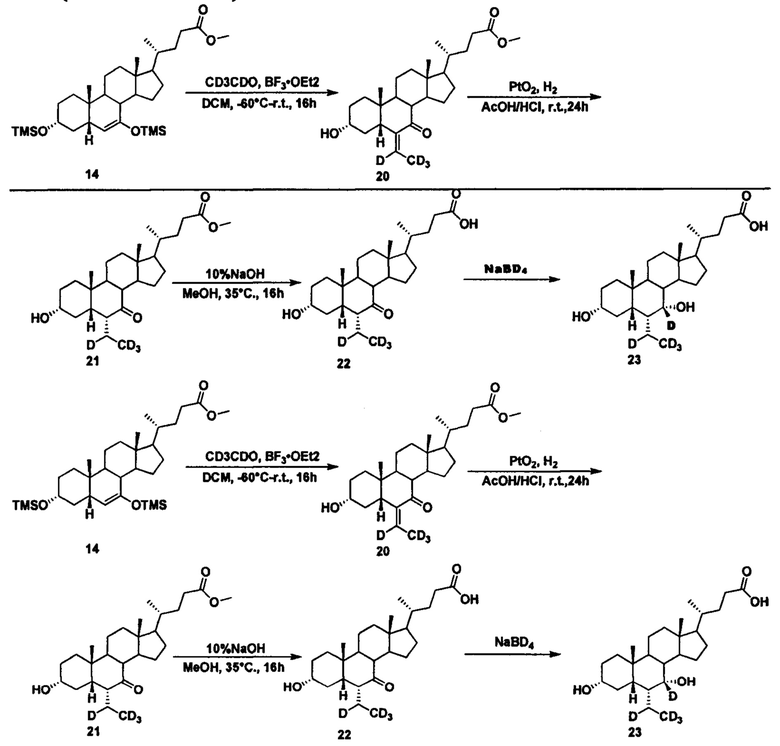
1. Синтез метил 3α-гидрокси-6-(этилиден-d4)-7-кето-5β-холан-24-оата (соединение 20)
В четырехгорлую колбу поочередно добавляются метил 3α, 7-бис(триметилсилилокси)-5β-холан-6-ен-24-оат (11,0 г, 18,2 ммоль), дихлорметан (60 мл) и ацетальдегид-d4 (2,1 мл, 36,4 ммоль) при -40°C. После перемешивания в течение 10 мин при -60°C, капельно, медленно добавляется раствор BF3.OEt2 (10 мл) в дихлорметане (20 мл). Смесь перемешивается в течение еще 3 часов при -60°C, естественным образом нагревается до комнатной температуры и перемешивается в течение ночи. Пока смесь находится в водяной бане, медленно добавляется насыщенный раствор NaHCO3 и перемешивается, а затем смесь экстрагируется и водный слой промывается дихлорметаном. В объединенную органическую фракцию добавляется 3 н. уксусной кислоты и смесь перемешивается в течение 1 часа попутно находясь в водяной бане. Смесь гасится насыщенным водным раствором NaHCO3, повторно экстрагируется, а водный слой промывается дихлорметаном. Объединенный органический слой сушится с помощью безводного Na2SO4 и концентрируется. Неочищенный продукт очищается посредством колоночной хроматографии на силикагеле (процент этилового эфира уксусной кислоты/петролейного эфира = 25-35%), для получения целевого соединения (5,2 г, 59%).
2. Синтез метил 3α-гидрокси-6α-(этил-d4)-7-кето-5β-холан-24-оата (соединение 21)
К раствору добавляются метил 3α-гидрокси-6-(этилиден-d4)-7-кето-5β-холан-24-оата (0,18 г, 0,42 ммоль) в уксусной кислоте (10 мл), концентрированная уксусная кислота (0,5 мл) и оксид платины (IV) (20 мг). Смесь наводороживается и в течение 12 часов перемешивается в водородной атмосфере при комнатной температуре. Смесь фильтруется и концентрируется для получения целевого соединения (0,16 г, 88%).
3. Синтез 3α-гидрокси-6α-(этил-d4)-7-кето-5β-холан-24-кислоты (соединение 22)
Добавляются метил 3α-гидрокси-6α-(этил-d4)-7-кето-5β-холан-24-оат (0,16 г, 0,36 ммоль), водный раствор гидроксида натрия (10%, 8,0 мл) и метанол/вода (4,5/1, 11 мл). Смесь перемешивается в течение 16 часов при 35°С. Смесь концентрируется и в нее добавляется вода. Добавляется 1 н. уксусной кислоты для доведения рН до 2-3. Осадок фильтруется, промывается очищенной водой и сушится для получения целевого соединения (0,12 г, 73%).
4. Синтез 3α,7α-дигидрокси-6α-(этил-d4)-7-d-5β-холан-24-кислоты (соединение 23)
В колбу добавляются 3α-гидрокси-6α-(этил-d4)-7-кето-5β-холан-24-кислота (60 мг, 0,14 ммоль), водный раствор гидроксида натрия (50%, 200 мг) и вода (3,0 мл). Во время перемешивания добавляется бородеутерид натрия (13 мг, 0,3 ммоль) и смесь перемешивается в течение 16 часов при 100°С. Смесь охлаждается до комнатной температуры и добавляется 1 н. раствора соляной кислоты для доведения рН до 2-3. Осадок фильтруется, промывается очищенной водой и сушится для получения целевого соединения (40 мг, 67%). 1Н ЯМР (400 МГц, DMSO-d6) δ: 11,97 (brs, 1Н), 4,32 (s, 1Н), 4,07 (d, J=8,0 Hz, 1Н), 3,16-3,11 (m, 1H), 2,28-2,19 (m, 1H), 2,15-2,07 (m, 1H), 1,93-0,84 (m, 30Н), 0,61 (s, 3Н). Масс-спектрометрия с электрораспылением (м/з): 426 (М+Н)+, 448 (M+Na)+.
Пример 7. Синтез 3α, 7α-дигидрокси-6α-(этил-d4)-5β-холан-24-кислоты (соединение 24)
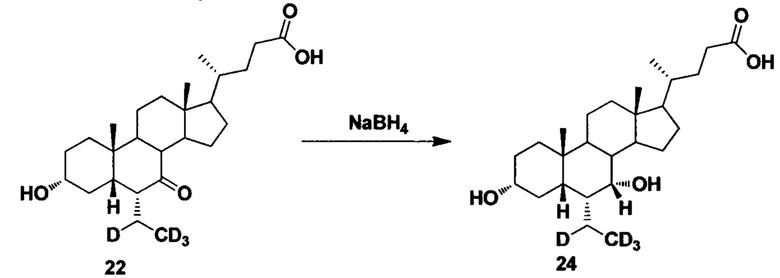
В колбу добавляются 3α-гидрокси-6α-(этил-d4)-7-кето-5β-холан-24-кислота (30 мг, 0,07 ммоль), водный раствор гидроксида натрия (50%, 50 мг) и вода (2,0 мл). Во время перемешивания добавляется борогидрид натрия (10 мг, 0,15 ммоль) и смесь перемешивается в течение 16 часов при 100°С. Смесь охлаждается до комнатной температуры и добавляется 1 н. раствора соляной кислоты для доведения рН до 2-3. Осадок фильтруется, промывается очищенной водой и сушится для получения целевого соединения (22 мг, 70%). 1Н ЯМР (400 МГц, DMSO-d6) δ: 11,97 (brs, 1Н), 4,32 (d, J=4,0 Hz, 1H), 4,07 (d, J=4,0 Hz, 1H), 3,50 (s, 1H), 3,14-3,13 (m, 1H), 2,27-2,20 (m, 1H), 2,15-2,07 (m, 1H), 1,93-0,84 (m, 30Н), 0,61 (s, 3Н). Масс-спектрометрия с электрораспылением (м/з): 425 (М+Н)+, 447 (M+Na)+.
Пример 8. Синтез 3α, 7α-дигидрокси-6α-этил-7, 23, 23-d3-5β-холан-24-кислоты (соединение 25)
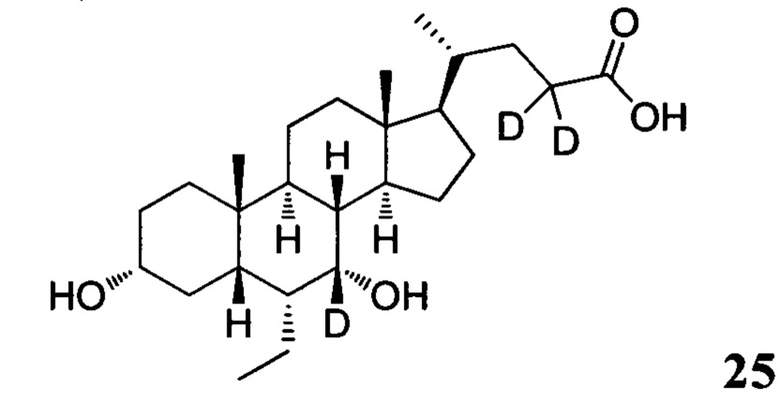
3α, 7α-дигидрокси-6α-этил-7-d-5β-холан-24-овая кислота (0,2 г) растворяется в дейтероксиде натрия в оксиде дейтерия. Смесь перемешивается в течение 24 часов при комнатной температуре. После удаления растворителей с помощью высокого вакуума, остаток растворяется в дейтероксиде натрия в оксиде дейтерия. Смесь перемешивается еще в течение 24 часов при комнатной температуре. 3 н. раствор соляной кислоты добавляется для доведения рН до 2-3, а затем смесь экстрагируется с помощью этилацетата. Объединенная органическая фаза затем промывается очищенной водой и рассолом, сушится с использованием Na2SO4 и фильтруется. Фильтрат концентрируется и остаток очищается посредством колоночной хроматографии на силикагеле (5% раствор метанола/дихлорметана) для получения целевого соединения. Масс-спектрометрия с электрораспылением (м/з): 424 (М+Н)+, 446 (M+Na)+.
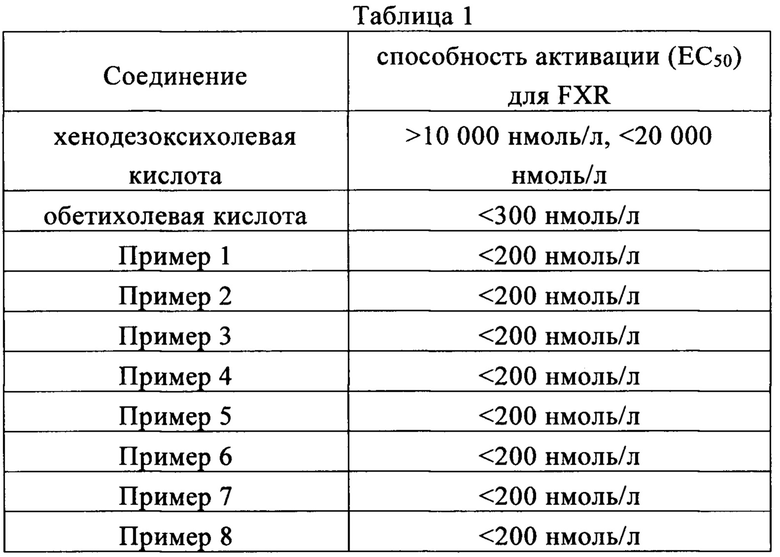
Пример 9: Фармакокинетическая оценка на крысах
Самцам крыс линии Спрег-Доули, 7-8 недельного возраста, масса тела которых составляла около 210 г, 6 крыс на группу, посредством дуоденального введения было введено 1 мкмоль/мин/кг дозы (а) контрольной группе: обетихолевой кислоты или (б) опытной группе: соединения, приготовленные в примерах с 1 по 8. Введение проводилось в течение 1 часа при скорости введения 2,5 мл/ч. Сравнивались фармакокинетика плазмы и кинетика выделения желчи.
Крыс кормили стандартным кормом и водой. Крыс подвергли 16 часам голодания перед началом теста. Препарат был растворен в физиологическом растворе. Кровь бралась из левой бедренной вены. Взятия крови проводилось за 0,5 часа до введения, через 0,5 часа, 1 час, 1,5 часа, 2 часа, 2,5 часа, 3 часа и 3,5 часа после введения. Желчь собиралась во время и через 2,5 часа после введения с интервалом в 15 мин.
Перед началом анализа, плазма и желчь хранились при -70°С. Концентрация соединений изобретения в плазме и желчи определялась с помощью жидкостной хроматографии с масс-спектрометрией/масс-спектрометрии. Фармакокинетические параметры рассчитывались на основе плазменной концентрации у каждого животного в разные моменты времени и концентрации препарата в желчи.
В результате было обнаружено, что соединение настоящего изобретения способствует более высокому содержанию вещества в плазме крови и лучшей секреции желчи в организме животного по сравнению с контрольным соединением Аобеликовой кислоты, поэтому имеет лучшие фармакодинамические свойства и терапевтический эффект.
Пример 10: Фармакодинамическая оценка in vitro соединений настоящего изобретения на фарнезоидный X рецептор (FXR)
Активация фарнезоидного X рецептора (FXR) соединениями настоящего изобретения определяется с помощью Анализа рекрутинга коактиватора, то есть с помощью AlphaScreen. В специфической экспериментальной программе фармакодинамической оценки in vitro использовались литературные материалы Журнала фармакологии и экспериментальной терапии 350:56-68 за июль 2014 года.
Результаты испытаний показаны в Таблице 1. Видно, что соединения настоящего изобретения обладают отменной способностью активации в отношении фарнезоидного X рецептора (FXR).
Пример 11. Фармацевтическая композиция
Вышеуказанные вещества смешиваются обычными способами и затем покрываются обычными желатиновыми капсулами для получения 1000 капсул.
Все литературные материалы, упомянутые в настоящей заявке, включены в данный документ путем отсылки, как если бы каждый из них был включен отдельно путем отсылки. Кроме того, следует понимать, что после прочтения вышеуказанного материала, специалисты в данной области техники могут внести различные изменения и модификации в настоящее изобретение. Данные эквиваленты также входят в область, определяемую прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДУЛЯТОРЫ ROR-ГАММА | 2012 |
|
RU2658013C2 |
| АГЕНТЫ ДЛЯ КРОВЯНОГО ДЕПО ДЛЯ ДИАГНОСТИКИ С ПОМОЩЬЮ ЯДЕРНОГО МАГНИТНОГО РЕЗОНАНСА | 1999 |
|
RU2250765C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГАММА-АМИДОВ ГЛУТАМОВОЙ КИСЛОТЫ | 1999 |
|
RU2395490C2 |
| ЛЕЧЕНИЕ ЗАБОЛЕВАНИЯ ДЫХАТЕЛЬНОЙ СИСТЕМЫ | 2013 |
|
RU2693382C2 |
| ДВОЙНЫЕ МЕТАЛЛЦИАНИДНЫЕ КАТАЛИЗАТОРЫ ДЛЯ ПОЛУЧЕНИЯ ПОЛИЭФИРПОЛИОЛОВ | 2000 |
|
RU2235589C2 |
| КАТАЛИЗАТОРЫ НА ОСНОВЕ ДВОЙНЫХ МЕТАЛЛОЦИАНИДОВ ДЛЯ ПОЛУЧЕНИЯ ПОЛИЭФИРПОЛИОЛОВ | 2000 |
|
RU2254164C2 |
| Средства для ингибирования фермента тирозил-ДНК-фосфодиэстеразы 1 на основе желчных кислот | 2018 |
|
RU2689335C1 |
| ПРОИЗВОДНЫЕ 19-НОРПРЕГНЕНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОЕДИНЕНИЯ | 1997 |
|
RU2166509C2 |
| Малотоксичные 1,3,4-оксадиазольные производные дезоксихолевой кислоты с простатопротекторным и противовоспалительным действием | 2023 |
|
RU2819604C1 |
| СТЕРОИДНОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2000 |
|
RU2240325C2 |
Изобретение относится к дейтерированному производному хенодезоксихолевой кислоты, представленному формулой (I), или его фармацевтически приемлемой соли. В формуле (I) R1, R3, R4, R5 и R6 независимо друг от друга представляют собой водород или дейтерий; R2 представляет собой дейтерий. Изобретение также относится к фармацевтической композиции, используемой в качестве агониста фарнезоидного Х рецептора (FXR), и к применению соединения для получения фармацевтической композиции. Технический результат: получены новые дейтерированные производные хенодезоксихолевой кислоты, представленные формулой (I), обладающие способностью активации фарнезоидного Х рецептора. 3 н. и 4 з.п. ф-лы, 1 табл., 11 пр.
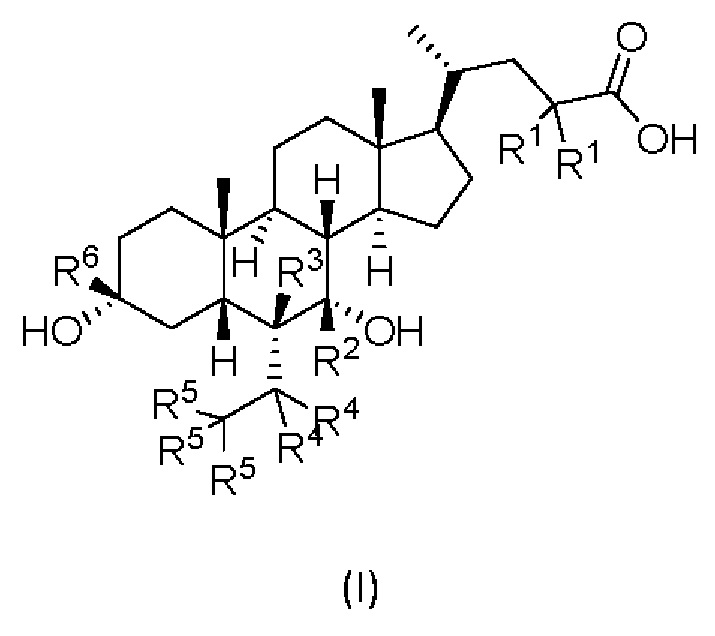
1. Дейтерированное производное хенодезоксихолевой кислоты, представленное формулой (I) или его фармацевтически приемлемая соль
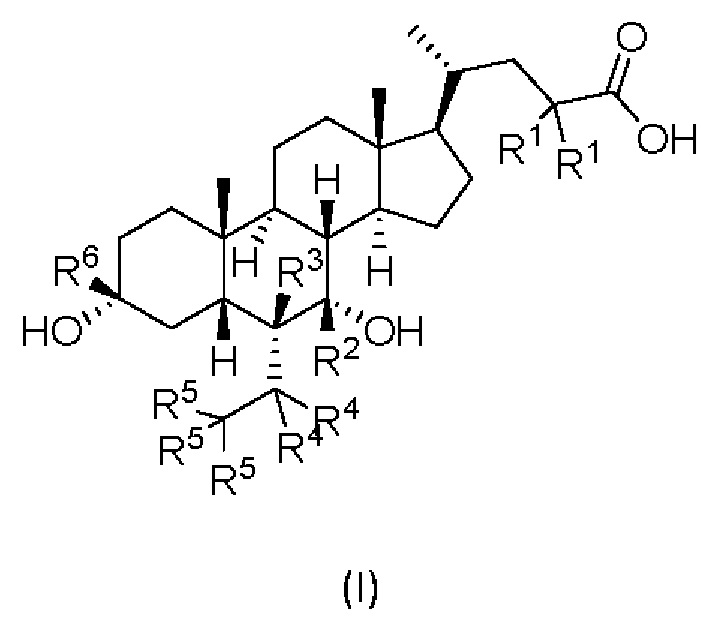
где R1, R3, R4, R5 и R6 независимо друг от друга представляют собой водород или дейтерий;
R2 представляет собой дейтерий.
2. Соединение по п. 1, где R1 представляет собой дейтерий.
3. Соединение по п. 1, где соединение является соединением, выбранным из следующей группы соединений или их фармацевтически приемлемых солей
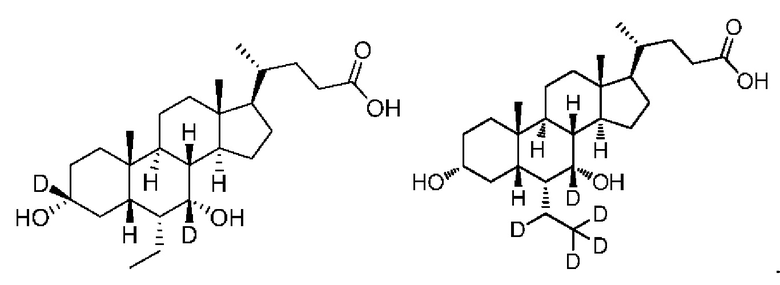
4. Фармацевтическая композиция, используемая в качестве агониста фарнезоидного X рецептора (FXR), содержащая безопасное и эффективное количество соединения по любому из пп. 1-3 или фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
5. Фармацевтическая композиция по п. 4, где фармацевтическая композиция представлена в форме раствора для инъекций, капсул, таблеток, пилюлей, порошка или гранул.
6. Применение соединения по п. 1 или его фармацевтически приемлемой соли для получения фармацевтической композиции, используемой в качестве агониста фарнезоидного X рецептора (FXR).
7. Применение по п. 6, отличающееся тем, что упомянутая фармацевтическая композиция используется при приготовлении лекарственных средств для лечения и профилактики заболеваний, выбранных из группы, состоящей из неалкогольного стеатогепатита, неалкогольной жировой болезни печени, желчекаменной болезни, первичного билиарного цирроза, цирроза, фиброза печени, диабета, атеросклероза, ожирения.
| СОВМЕЩЕННОЕ АНТЕННОЕ УСТРОЙСТВО | 1991 |
|
RU2072598C1 |
| WO 2005082925 A2, 09.09.2005 | |||
| WO 2013192097 A1, 27.12.2013 | |||
| WO 2015061421 A1, 30.04.2015 | |||
| Приспособление для ограничения силы тормозного тока при электродинамическом торможении моторных повозок электрических железных дорог | 1928 |
|
SU20140A1 |

