Область техники
Настоящее изобретение относится к фармацевтическим средствам, включающим ингибитор дипептидилпептидазы IV (DPPIV) и бигуанидный агент, которые усиливают действия или эффекты активного циркулирующего глюкагон-подобного пептида-1 (GLP-1) и/или активного циркулирующего глюкагон-подобного пептида-2 (GLP-2).
Уровень техники
Глюкагонподобный пептид-1 (GLP-1) является гормоном, о котором известно, что он секретируется в ответ на потребление пищи из L клеток в дистальной части тонкой кишки. Он усиливает секрецию инсулина из панкреатических β клеток глюкозазависимым образом. GLP-1 разрушается и быстро инактивируется дипептидилпептидазой IV (DPPIV). Таким образом, ингибиторы DPPIV могут использоваться в качестве профилактических и/или терапевтических средств при таких заболеваниях, как диабет (особенно диабет типа II) и тучность или ожирение, с которыми связаны уровни GLP-1. Ингибиторы DPPIV были разработаны в ходе клинических испытаний и описаны в патентных документах 1, 2 и 3.
Метформин, бигуанидный агент, обычно использовался в качестве профилактического и/или терапевтического средства при диабете.
В последние годы было подряд сообщено о новых открытиях: уровни GLP-1 увеличиваются у тучных недиабетических пациентов после введения метформина (непатентный документ 1); и сочетание метформина и GLP-1 является эффективным для лечения диабета типа II (непатентный документ 2). Однако, также если уровень GLP-1 кратковременно повышается под действием метформина, GLP-1 быстро разрушается и инактивируется под действием DPPIV, как описывается выше. Следовательно, повышенный уровень GLP-1 не поддерживается в течение длительного периода времени, и, таким образом, эффект GLP-1 сильно снижается. Это является проблемой, которую предстоит решить.
В непатентных документах 3 и 4 предположена возможность комбинированного использования ингибитора DPPIV и метформина. В патентных документах 4-8 описано комбинированное использование ингибитора DPPIV и бигуанидного агента. Однако в этих документах не описаны конкретные результаты испытаний по комбинированному использованию данных средств. Иными словами, нет никакого комбинационного лекарства, которое содержит ингибитор DPPIV и метформин и о котором известно, что оно усиливает эффекты GLP-1.
Сообщалось, что глюкагон-подобный пептид-2 (GLP-2) является гормоном, секретируемым в ответ на потребление пищи из L клеток в дистальной части тонкой кишки как GLP-1, и что он может использоваться для профилактики и/или лечения желудочно-кишечных заболеваний (непатентные документы 5-9). Однако, как и GLP-1, GLP-2 быстро разрушается и инактивируется под действием DPPIV. Соответственно, существовала потребность в разработке средств, подавляющих разрушение GLP-2, и, следовательно, усиления эффектов GLP-2. Однако нет никаких сообщений, описывающих увеличение уровня GLP-2 после введения метформина или усиление эффектов GLP-2 путем комбинированного использования ингибитора DPPIV и метформина.
[Патентный документ 1]
Патент США № 6166063
[Патентный документ 2]
Патент США № 6011155
[Патентный документ 3]
Патент США № 6548481
[Патентный документ 4]
WO 01/52825
[Патентный документ 5]
WO 01/97808
[Патентный документ 6]
Патентная заявка США № 2002/0161001
[Патентный документ 7]
Патентная заявка США № 2002/0198205
[Патентный документ 8]
Патентная заявка США № 2003/0105077
[Непатентный документ 1]
Edoardo Mannucci, and eight other authors, "Diabetes Care", 24(3): 489-494 (2001) Mar.
[Непатентный документ 2]
Mette Zander, and four other authors, "Diabetes Care", 24(4): 720-725 (2001) Apr.
[Непатентный документ 3]
Simon A. Hinke, and five other authors, "Biochemical and Biophysical Research Communications", 291(5): 1302-1308 (2002) Mar.
[Непатентный документ 4]
Simon A. Hinke, and nine other authors, "Diabetes Care", 25(8): 1490-1491 (2002) Aug.
[Непатентный документ 5]
Robin P. Boushey, and two other authors, "American Journal of Physiology", 277(8): E937-E947 (1999)
[Непатентный документ 6]
D.L. Sigalet, "Current Opinion in Investigational Drugs", 2(4): 505-509 (2001) Apr.
[Непатентный документ 7]
Daniel J. Drucker, "Gut", 50(3): 428-435 (2002)
[Непатентный документ 8]
Daniel J. Drucker "Gastroenterology", 122(2): 531-544 (2002) Feb.
[Непатентный документ 9]
Robin P. Boushey, and two other authors, "Cancer Research", 61:687-693 (2001) Jan.
Описание изобретения
Целью настоящего изобретения является разработка фармацевтических средств, которые усиливают фармакологические эффекты активного циркулирующего GLP-1 и/или активного циркулирующего GLP-2 путем подавления разрушения GLP-1 и/или GLP-2, когда уровни повышены бигуанидным агентом.
Авторы настоящего изобретения провели обширные исследования с учетом вышеуказанных предпосылок и обнаружили, что комбинированное использование ингибитора DPPIV и бигуанидного агента усиливает фармакологические действия активного циркулирующего GLP- 1 и/или активного циркулирующего GLP-2. Это является следствием того, что ингибитор DPPIV подавляет разрушение активного циркулирующего GLP-1 и/или активного циркулирующего GLP-2, когда их уровни увеличиваются бигуанидным агентом. Таким образом было создано настоящее изобретение.
В частности, настоящее изобретение включает:
<1> фармацевтическое средство, содержащее в сочетании ингибитор дипептидилпептидазы IV и бигуанидный агент;
<2> фармацевтическое средство по <1>, которое усиливает эффекты активного циркулирующего глюкагон-подобного пептида-1 (GLP-1) и/или активного циркулирующего глюкагон-подобного пептида-2 (GLP-2);
<3> фармацевтическое средство, которое усиливает эффекты активного циркулирующего GLP-2;
<4> фармацевтическое средство, содержащее в сочетании ингибитор дипептидилпептидазы IV и фармацевтическое средство по <3>;
<5> фармацевтическое средство по <1> или <4>, в котором ингибитор дипептидилпептидазы IV является любым соединением, выбранным из:
(S)-1-((3-гидрокси-1-адамантил)амино)ацетил-2-цианпирролидина;
(S)-1-(2-((5-цианпиридин-2-ил)амино)этиламиноацетил)-2-цианпирролидина;
изолейцин-тиазолидида; изолейцин-пирролидида; и валин-пирролидида;
или его солью или гидратом;
<6> фармацевтическое средство по <1> или <4>, в котором ингибитор дипептидилпептидазы IV является соединением, представленным следующей формулой, или его солью или гидратом,
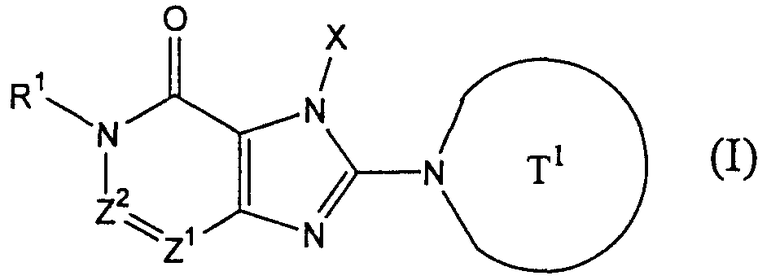
(в которой
Т1 представляет собой моноциклическую или бициклическую 4-12-членную гетероциклическую группу, содержащую один или два атома азота в кольце, которая может иметь один или несколько заместителей;
Х представляет собой С1-6 алкильную группу, которая может иметь один или несколько заместителей, С2-6 алкенильную группу, которая может иметь один или несколько заместителей, С2-6 алкинильную группу, которая может иметь один или несколько заместителей, С6-10 арильную группу, которая может иметь один или несколько заместителей, 5-10-членную гетероарильную группу, которая может иметь один или несколько заместителей, С6-10 арил С1-6 алкильную группу, которая может иметь один или несколько заместителей, или 5-10-членную гетероарил С1-6 алкильную группу, которая может иметь один или несколько заместителей;
Z1 и Z2,каждый, независимо, представляют собой атом азота или группу, представленную формулой -CR2=;
R1 и R2, каждый, независимо, представляют собой группу формулы -А0-А1-А2
(в которой А0 представляет собой одинарную связь или С1-6 алкиленовую группу, которая может иметь 1-3 заместителя, выбранных из группы В, состоящей из заместителей, описанных ниже;
А1 представляет собой одинарную связь, атом кислорода, атом серы, сульфинильную группу, сульфонильную группу, карбонильную группу, группу, представленную формулой -О-СО-, группу, представленную формулой -СО-О-, группу, представленную формулой -NRA-, группу, представленную формулой -CO-NRА, группу, представленную формулой -NRA-CO-, группу, представленную формулой -SO2-NRA-, или группу, представленную формулой -NRA-SO2-;
А2 и RA, каждый, независимо, представляют собой атом водорода, атом галогена, циан группу, С1-6алкильную группу, С3-8циклоалкильную группу, С2-6алкенильную группу, С2-6алкинильную группу, С6-10арильную группу, 5-10-членную гетероарильную группу, 4-8-членную гетероциклическую группу, 5-10-членную гетероарил С1-6алкильную группу, С6-10арил С1-6алкильную группу, или С2-7алкилкарбонильную группу;
однако А2 и RA, каждый, независимо, могут иметь 1-3 заместителя, выбранных из группы заместителей В, описанных ниже;
когда Z2 представляет собой группу, представленную формулой -CR2=;
R1 и R2 могут в сочетании образовывать 5-7-членное кольцо,
за исключением случаев, в которых: [1] R1 представляет собой атом водорода; Z1 представляет собой атом азота; и Z2 представляет собой -СН=; и [2] Z1 представляет собой атом азота; и Z2 представляет собой -С(ОН)=;
<Группы заместителей В>
Группа заместителей В представляет собой группу, состоящую из гидроксильной группы, меркаптогруппы, циангруппы, нитрогруппы, атома галогена, трифторметильной группы, С1-6алкильной группы, которая может иметь один или несколько заместителей, С3-8циклоалкильной группы, С2-6алкенильной группы, С2-6алкинильной группы, С6-10арильной группы, 5-10-членной гетероарильной группы, 4-8-членной гетероциклической группы, С1-6 алкоксигруппы, С1-6 алкилтиогруппы, группы, представленной формулой -SO2-NRB1-RB2, группы, представленной формулой NRB1-СО-RB2, группы, представленной формулой -NRB1-RB2 (в которой RB1 и RB2, каждый, независимо, представляют собой атом водорода или С1-6 алкильную группу), группы, представленной формулой -СО-RB3 (в которой RB3 представляет собой 4-8-членную гетероциклическую группу), группы, представленной формулой -СО-RB4-RB5, и группы, представленной формулой -СН2-СО-RB4-RB5 (в которой RВ4 представляет собой одинарную связь, атом кислорода или группу, представленную формулой -NRB6; RВ5 и RВ6, каждый, независимо, представляет собой атом водорода, С1-6 алкильную группу, С3-8 циклоалкильную группу, С2-6 алкенильную группу, С2-6 алкинильную группу, С6-10 арильную группу, 5-10-членную гетероарильную группу, 4-8-членную гетероциклическую С1-6 алкильную группу, С6-10 арил С1-6 алкильную группу, или 5-10-членную гетероарил С1-6 алкильную группу));
<7> фармацевтическое средство по <6>, в котором Т1 представляет собой пиперазин-1-ильную группу или 3-аминопиперидин-1-ильную группу;
<8> фармацевтическое средство по <6>, в котором Т1 представляет собой пиперазин-1-ильную группу:
<9> фармацевтическое средство по любому из <6>-<8>, в котором Х представляет собой 3-метил-2-бутен-1-ильную группу, 2-бутинильную группу, бензильную группу или 2-хлорфенильную группу;
<10> фармацевтическое средство по любому из п<6>-<8>, в котором Х представляет собой 3-метил-2-бутен-1-ильную группу или 2-бутин-1-ильную группу;
<11> фармацевтическое средство по любому из <6>-<8>, в котором Х представляет собой 2-бутин-1-ильную группу;
<12> фармацевтическое средство по любому из <6>-<11>, в котором Z1 представляет собой атом азота; и
Z2 представляет собой группу, представленную формулой -CR2=
(в которой R2 имеет значения, определенные в <6>);
<13> фармацевтическое средство по любому из <6>-<11>, в котором Z2 представляет собой атом азота; и
Z1 представляет собой группу, представленную формулой -CR2=
(в которой R2 имеет значения, определенные в <6>);
<14> фармацевтическое средство по любому из <6>-<13>, в котором R1 представляет собой либо метильную группу, цианбензильную группу, фторцианбензильную группу, фенетильную группу, 2-метоксиэтильную группу либо 4-метоксикарбонилпиридин-2-ильную группу;
<15> фармацевтическое средство по любому из <6>-<13>, в котором R1 представляет собой либо метильную группу, либо цианбензильную группу;
<16> фармацевтическое средство по любому из <6>-<15>, в котором R2 представляет собой или атом водорода, циангруппу, метоксигруппу, карбамоилфенилоксигруппу, или группу, представленную формулой:
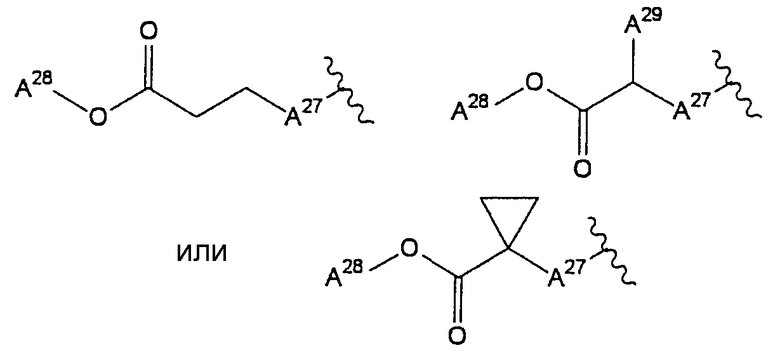
(в которой
А27 представляет собой атом кислорода, атом серы, или -NH-;
А28 и А29, каждый, независимо, представляют собой атом водорода или С1-6 алкильную группу);
<17> фармацевтическое средство по любому из <6>-<15>, в котором R2 представляет собой атом водорода, циангруппу, или 2-карбамоилфенилоксигруппу;
<18> фармацевтическое средство по <6>, в котором соединение представленное формулой (I), является соединением, выбранным из:
(1) 7-(2-бутинил)-2-циан-1-метил-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она;
(2) 3-(2-бутинил)-5-метил-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она;
(3) 2-(3-аминопиперидин-1-ил)-3-(2-бутинил)-5-метил-3,5-дигидроимидазо[4,5-d]пиридазин-4-она;
(4) 2-[7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]бензамида;
(5) 7-(2-бутинил)-1-(2-цианбензил)-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-карбонитрила; и
(6) 2-[3-(2-бутинил)-4-оксо-2-(пиперазин-1-ил)-3,4-дигидро-имидазо[4,5-d]пиридазин-5-илметил]бензонитрила;
или их соли или гидрата;
<19> фармацевтическое средство по <1> или <4>, в котором ингибитором дипептидилпептидазы IV является соединение, представленное следующей формулой, или его солью или гидратом,
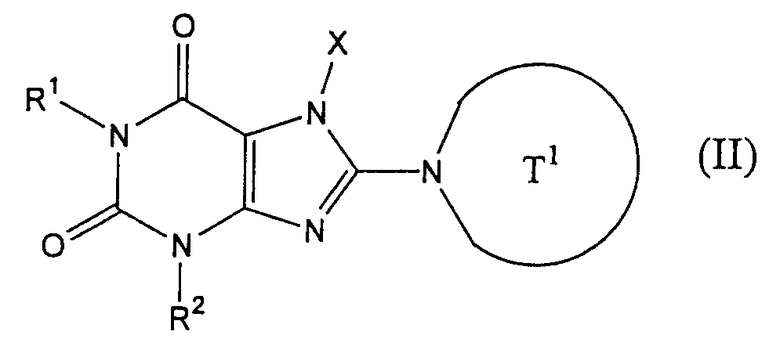
(в которой T1, X, R1 и R2 имеют значения, определенные в <6>);
<20> фармацевтическое средство по <19>, в котором T1 представляет собой пиперазин-1-ильную группу;
<21> фармацевтическое средство по <19> или <20>, в котором Х представляет собой 2-бутинильную группу или 2-хлорфенильную группу;
<22> фармацевтическое средство по <19> или <20>, в котором Х представляет собой 2-бутинильную группу;
<23> фармацевтическое средство по любому из <19>-<22>, в котором R1 представляет собой атом водорода, метильную группу, 2-пропинильную группу, 2-бутинильную группу, цианметильную группу, фенетильную группу, феноксиэтильную группу или группу, представленную формулой:
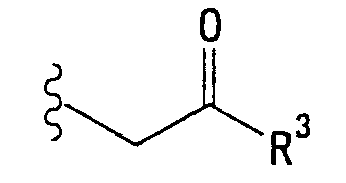
(в которой R3 представляет собой гидроксильную группу, С1-6 алкоксигруппу или фенильную группу);
<24> фармацевтическое средство по любому из <19>-<23>, в котором R2 представляет собой атом водорода, С1-6 алкильную группу, этоксиэтильную группу, тетрагидрофуранилметильную группу или группу, представленную формулой:
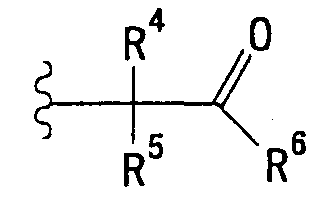
(в которой R4 и R5 являются идентичными или отличными друг от друга и независимо представляют собой атом водорода, метильную группу или фенильную группу; и
R6 представляет собой гидроксильную группу, С1-6 алкоксигруппу или фенильную группу)
или группу, представленную формулой:
<25> фармацевтическое средство по <19>, в котором соединение, представленное формулой (II), является соединением, выбранным из:
(1) 7-(2-бутинил)-1,3-диметил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона;
(2) 7-(2-бутинил)-3-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона;
(3) метил [7-(2-бутинил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-ил]ацетата;
(4) 7-(2-бутинил)-3-метил-8-(пиперазин-1-ил)-1-(2-пропинил)-3,7-дигидропурин-2,6-диона;
(5) 1,7-бис(2-бутинил)-3-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона;
(6) [7-(2-бутинил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-ил]ацетонитрила;
(7) 7-(2-бутинил)-3-метил-1-[(2-оксо-2-фенил)этил]-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона;
(8) 7-(2-бутинил)-3-этил-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона;
(9) метил [7-(2-бутинил)-1-метил-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетата;
(10) 7-(2-бутинил)-3-(2-тетрагидрофуранил)метил-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона;
(11) метил [7-(2-бутинил)-1-метил-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]фенилацетата;
(12) 7-(2-бутинил)-3-пропил-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона;
(13) 7-(2-бутинил)-3-(2-оксо-2-фенэтил)-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона;
(14) этил 2-[7-(2-бутинил)-1-метил-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]пропионата;
(15) 7-(2-бутинил)-3-(2-этоксиэтил)-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона;
(16) 7-(2-бутинил)-3-изопропил-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона;
(17) 7-(2-бутинил)-3-(3,3-диметил-2-оксобутил)-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона;
(18) 7-(2-бутинил)-1-метил-3-(2-оксопирролидин-3-ил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона;
(19) 7-(2-бутинил)-3-(2-этоксиэтил)-1-(2-оксо-2-фенилэтил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона;
(20) метил [7-(2-бутинил)-2,6-диоксо-1-(2-оксо-2-фенилэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетата;
(21) этил [7-(2-бутинил)-2,6-диоксо-1-(2-фенэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетата;
(22) [7-(2-бутинил)-2,6-диоксо-1-(2-фенэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетата;
(23) 7-(2-бутинил)-3-[2-оксо-2-(пирролидин-1-ил)этил]-1-(2-фенэтил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона;
(24) 2-[7-(2-бутинил)-2,6-диоксо-1-(2-фенэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]-N-метилацетамида;
(25) 2-[7-(2-бутинил)-2,6-диоксо-1-(2-фенэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]-N-циклопропилацетамида;
(26) 2-[7-(2-бутинил)-2,6-диоксо-1-(2-фенэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]-N-фенилацетамида; и
(27) 2-[7-(2-бутинил)-2,6-диоксо-1-(2-фенэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]-N-(2-пропинил)ацетамида;
или его соли или гидрата;
<26> фармацевтическое средство по <1>, в котором бигуанидным агентом является метформин;
<27> фармацевтическое средство по <1> или <2>, который является профилактическим или терапевтическим средством при заболевании, связанном с активным циркулирующим GLP-1 и/или активным циркулирующим GLP-2;
<28> фармацевтическое средство по <27>, в котором заболеванием является, по крайней мере, любое, выбранное из группы, состоящей из диабета, ожирения, гиперлипидемии и желудочно-кишечных заболеваний;
<29> фармацевтическое средство по <3> или <4>, которое является профилактическим или терапевтическим средством при заболевании, связанном с активным циркулирующим GLP-2;
<30> фармацевтическое средство по <29>, в котором заболеванием является желудочно-кишечное заболевание;
<31> способ профилактики или лечения заболевания, связанного с активным циркулирующим GLP-1 и/или активным циркулирующим GLP-2, которое содержит введение фармацевтического средства по <1> или <2> в эффективном количестве;
<32> применение фармацевтического средства по <1> или <2> для получения профилактического или терапевтического средства при заболевании, связанном с активным циркулирующим GLP-1 и/или активным циркулирующим GLP-2;
<33> способ профилактики или лечения заболевания, связанного с активным циркулирующим GLP-2, который включает введение фармацевтического средства по <3> или <4> в эффективном количестве;
<34> применение фармацевтического средства по <3> или <4> для получения профилактического или терапевтического средства при заболевании, связанном с активным циркулирующим GLP-2;
<35> способ усиления эффектов активного циркулирующего GLP-1 и/или активного циркулирующего GLP-2, который включает применение фармацевтического средства по <1> или <2>;
и
<36> способ усиления эффектов активного циркулирующего GLP-2, который включает применение фармацевтического средства по <3> или <4>.
Настоящее изобретение также включает:
<37> средство для усиления эффектов активного циркулирующего глюкагон-подобного пептида-1 (GLP-1) и/или активного циркулирующего глюкагон-подобного пептида-2 (GLP-2), которое содержит в сочетании ингибитор дипептидилпептидазы IV и бигуанидный агент;
<38> средство для усиления эффектов активного циркулирующего глюкагон-подобного пептида-2 (GLP-2), которое содержит в качестве активного ингредиента бигуанидный агент;
<39> средство для усиления эффектов активного циркулирующего глюкагон-подобного пептида-2 (GLP-2), которое содержит в сочетании ингибитор дипептидилпептидазы IV и бигуанидный агент;
<40> профилактическое или терапевтическое средство при диабете, ожирении, гиперлипидемии или желудочно-кишечных заболеваниях, которое усиливает эффекты активного циркулирующего глюкагон-подобного пептида-1 (GLP-1) и которое содержит ингибитор дипептидилпептидазы IV и бигуанидный агент в качестве активных ингредиентов;
<41> профилактическое или терапевтическое средство при желудочно-кишечных заболеваниях, которое усиливает эффекты активного циркулирующего глюкагон-подобного пептида-2 (GLP-2) и которое содержит ингибитор дипептидилпептидазы IV и бигуанидный агент в качестве активных ингредиентов; и
<42> профилактическое или терапевтическое средство при диабете, ожирении, гиперлипидемии или желудочно-кишечных заболеваниях, которое содержит ингибитор дипептидилпептидазы IV и бигуанидный агент в качестве активных ингредиентов.
В разделах с <37> по <42>, предпочтительно, чтобы ингибитором дипептидилпептидазы IV был указанный в любом из <5>-<25>, перечисленных выше, а бигуанидным агентом является средство, определенный выше в <26>.
Наилучший способ осуществления изобретения
В настоящем описании структурная формула соединения иногда представляет собой какой-то определенный изомер для удобства описания. Однако соединения по настоящему изобретению могут включать все возможные изомеры, такие как структурно возможные геометрические изомеры, оптические изомеры, образующиеся ввиду наличия асимметрических атомов углерода, стереоизомеры, таутомеры и смеси изомеров, и не ограничиваются формулами, используемыми для удобства описания, и могут быть любым из двух изомеров или смесью обоих изомеров. Таким образом, соединения по настоящему изобретению могут быть или оптически активными соединениями, имеющими асимметрический атом углерода в их молекулах, или их рацематами, и не ограничиваются каким-либо одним из них, а включают и оба. Кроме того, соединения по настоящему изобретению могут проявлять кристаллический полиморфизм, и аналогичным образом не ограничиваются какой-либо одной из форм, и могут быть в любой из форм их кристаллов или существовать в виде смеси двух или более кристаллических форм. Соединения по настоящему изобретению также включают как безводные, так и гидратированные формы. Вещества, продуцируемые вследствие in vivo метаболизма соединений по изобретению, также охватываются объемом пунктов формулы изобретения.
Далее определены термины и символы, используемые в данном описании, а настоящее изобретение описано более подробно.
Используемое здесь выражение "С1-6алкильная группа" относится к линейной или разветвленной алкильной группе, содержащей 1-6 атомов углерода, которая является одновалентной группой, полученной при удалении любого из атомов водорода из алифатических углеводородов, содержащих 1-6 углеродов, и включает, в частности, например, метильную группу, этильную группу, 1-пропильную, 2-пропильную, 2-метил-1-пропильную, 2-метил-2-пропильную, 1-бутильную, 2-бутильную, 1-пентильную, 2-пентильную, 3-пентильную, 2-метил-1-бутильную, 3-метил-1-бутильную группу, 2-метил-2-бутильную, 3-метил-2-бутильную, 2,2-диметил-1-пропильную, 1-гексильную, 2-гексильную, 3-гексильную группу, 2-метил-1-пентильную группу, 3-метил-1-пентильную, 4-метил-1-пентильную, 2-метил-2-пентильную, 3-метил-2-пентильную, 4-метил-2-пентильную, 2-метил-3-пентильную, 3-метил-3-пентильную, 2,3-диметил-1-бутильную, 3,3-диметил-1-бутильную группу, 2,2-диметил-1-бутильную группу, 2-этил-1-бутильную группу, 3,3-диметил-2-бутильную группу и 2,3-диметил-2-бутильную группу.
Используемое здесь выражение "С2-6 алкенильная группа" относится к линейной или разветвленной алкенильной группе, содержащей 2-6 атомов углерода, и включает, в частности, например, винильную группу, аллильную группу, 1-пропенильную, 2-пропенильную, 1-бутенильную, 2-бутенильную, 3-бутенильную группу, пентенильную группу и гексенильную группу.
Используемое здесь выражение "С2-6 алкинильная группа" относится к линейной или разветвленной алкинильной группе, содержащей 2-6 атомов углерода, и включает, в частности, например, этинильную группу, 1-пропинильную, 2-пропинильную, бутинильную группу, пентинильную группу и гексинильную группу.
Используемое здесь выражение "С3-8 циклоалкильная группа" относится к циклической алифатической углеводородной группе, содержащей 3-8 атомов углерода, и включает, в частности, например, циклопропильную группу, циклобутильную, циклопентильную группу, циклогексильную группу, циклогептильную группу и циклооктильную группу.
Используемое здесь выражение "С1-6 алкиленовая группа" относится к дивалентной группе, полученной при удалении еще одного произвольного атома водорода из "С1-6 алкильной группы", определенной выше, и включает, в частности, например, метиленовую группу, 1,2-этиленовую группу, 1,1-этиленовую группу, 1,3-пропиленовую группу, тетраметиленовую, пентаметиленовую группу и гексаметиленовую группу.
Используемое здесь выражение "С3-8 циклоалкиленовая группа" относится к дивалентной группе, полученной при удалении еще одного произвольного атома водорода из "С3-8 циклоалкильной группы", определенной выше.
Используемое здесь выражение "С1-6 алкоксигруппа" относится к оксигруппе, связанной с "С1-6 алкильной группой", определенной выше, и включает, в частности, например, метоксигруппу, этоксигруппу, 1-пропилоксигруппу, 2-пропилокси, 2-метил-1-пропилокси, 2-метил-2-пропилокси, 1-бутилокси, 2-бутилокси, 1-пентилокси, 2-пентилокси, 3-пентилокси, 2-метил-1-бутилокси, 3-метил-1-бутилоксигруппу, 2-метил-2-бутилокси, 3-метил-2-бутилокси, 2,2-диметил-1-пропилокси, 1-гексилокси, 2-гексилокси, 3-гексилокси группу, 2-метил-1-пентилокси группу, 3-метил-1-пентилокси, 4-метил-1-пентилокси, 2-метил-2-пентилокси, 3-метил-2-пентилокси, 4-метил-2-пентилокси, 2-метил-3-пентилокси, 3-метил-3-пентилокси, 2,3-диметил-1-бутилокси, 3,3-диметил-1-бутилоксигруппу, 2,2-диметил-1-бутилоксигруппу, 2-этил-1-бутилоксигруппу, 3,3-диметил-2-бутилоксигруппу, и 2,3-диметил-2-бутилоксигруппу.
Используемое здесь выражение "С1-6 алкилтиогруппа" относится к тиогруппе, связанной с "С1-6 алкильной группой", определенной выше, и включает, в частности, например, метилтиогруппу, этилтиогруппу, 1-пропилтиогруппу, 2-пропилтио, бутилтиогруппу и пентилтиогруппу.
Используемое здесь выражение "С2-7 алкоксикарбонильная группа" относится к карбонильной группе, связанной с "С1-6 алкоксигруппой", определенной выше, и включает, в частности, например, метоксикарбонильную группу, этоксикарбонильную группу, 1-пропилоксикарбонильную группу и 2-пропилоксикарбонильную группу.
Используемое здесь выражение "С2-7 алкилкарбонильная группа" относится к карбонильной группе, связанной с "С1-6 алкильной группой", определенной выше, и включает, в частности, например, метилкарбонильную группу, этилкарбонильную группу, 1-пропилкарбонильную группу и 2-пропилкарбонильную группу.
Используемый здесь термин "атом галогена" относится к атому фтора, атому хлора, атому брома или атому йода.
Используемое здесь выражение "С6-10 арильная группа" относится к ароматической циклической углеводородной группе, содержащей 6-10 атомов углерода, и включает, в частности, например, фенильную группу, 1-нафтильную группу и 2-нафтильную группу.
Используемый здесь термин "гетероатом" относится к атому серы, атому кислорода или атому азота.
Используемое здесь выражение "5-10-членное гетероарильное кольцо" относится к ароматическому 5-10-членному кольцу, содержащему один или несколько гетероатомов, и, в частности, включает, например пиридиновое кольцо, тиофеновое кольцо, фурановое кольцо, пиррольное кольцо, оксазольное кольцо, изоксазольное кольцо, тиазольное кольцо, тиадиазольное кольцо, изотиазольное, имидазольное кольцо, триазольное, пиразольное кольцо, фуразановое, оксадиазольное, пиридазиновое, пиримидиновое, пиразиновое, триазиновое, индольное, изоиндольное, индазольное, хроменовое кольцо, хинолиновое, изохинолиновое, циннолиновое, хиназолиновое, хиноксалиновое кольцо, нафтиридиновое, фталазиновое, пуриновое, птеридиновое, тиенофурановое, имидазотиазольное, бензофурановое, бензотиофеновое, бензоксазольное, бензотиазольное, бензотиадиазольное, бензимидазольное, имидазопиридиновое, пирролпиридиновое, пирролпиримидиновое кольцо и пиридопиримидиновое кольцо. Предпочтительные "5-10-членные гетероарильные кольца" включают пиридиновое кольцо, тиофеновое, фурановое, пиррольное, имидазольное, 1,2,4-триазольное, тиазольное, тиадиазольное, пиразольное, фуразановое, тиадиазольное, пиридазиновое, пиримидиновое, пиразиновое, изохинолиновое, бензоксазольное, бензотиазольное и бензимидазольное кольца. Наиболее предпочтительным примером является пиридиновое кольцо.
Используемое здесь выражение "5-10-членная гетероарильная группа" относится к одновалентной или дивалентной группе, полученной удалением любого одного или двух атомов водорода из "5-10-членной гетероарильной группы", описанной выше.
Используемое здесь выражение "4-8-членное гетероциклическое кольцо" относится к неароматическому кольцу, в котором:
(i) число атомов, составляющих кольцо, равняется 4-8;
(ii) атомы, составляющие кольцо, включают 1-2 гетероатома;
(iii) кольцо может содержать 1-2 двойные связи;
(iv) кольцо может содержать 1-3 карбонильные группы; и
(v) кольцо является моноциклическим.
В частности, 4-8-членное гетероциклическое кольцо включает, например, азетидиновое кольцо, пирролидиновое, пиперидиновое, азепановое, азокановое, тетрагидрофурановое кольцо, тетрагидропирановое, морфолиновое, тиоморфолиновое, пиперазиновое, тиазолидиновое, диоксановое, имидазолиновое, тиазолиновое кольцо и кольцо, представленное одной из формул:
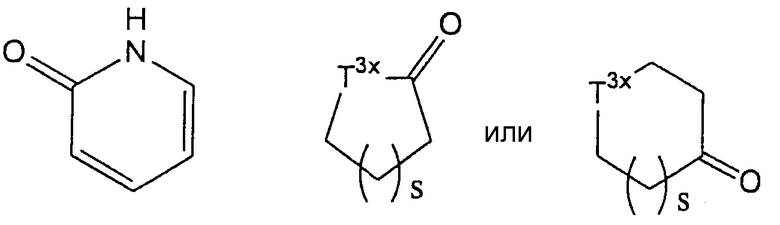
(где S представляет собой целое число от 1 до 3; T3x представляет собой метиленовую группу, атом кислорода или группу, представленную формулой -NT4х-, в которой T4х представляет собой атом водорода или С1-6 алкильную группу. Предпочтительно, "4-8-членные гетероциклические кольца" включают пирролидиновое кольцо, пиперидиновое, азепановое, морфолиновое, тиоморфолиновое, пиперазиновое, дигидрофуран-2-оновое и тиазолидиновое кольца.
Используемое здесь выражение "4-8-членная гетероциклическая группа" относится к одновалентной или дивалентной группе, полученной удалением любого одного или двух атомов водорода из "4-8-членной гетероциклической группы", описанной выше. Предпочтительно, "4-8-членные гетероциклические группы" включают пиперидин-1-ильную группу, пирролидин-1-ильную группу и морфолин-4-ильную группу.
Используемое здесь выражение "С6-10 арил С1-6 алкильная группа" относится к группе, полученной замещением "С6-10 арильной группой", определенной выше, произвольного атома водорода в "С1-6 алкильной группе", определенной выше, и, в частности, включает, например, бензильную группу, фенетильную группу и 3-фенил-1-пропильную группу.
Используемое здесь выражение "5-10-членная гетероарил С1-6 алкильная группа" относится к группе, полученной замещением "5-10-членной гетероарильной группой", определенной выше, произвольного атома водорода в "С1-6 алкильной группе", определенной выше, и, в частности, включает, например, 2-пиридилдметильную и 2-тиенилметильную группу.
Используемое здесь выражение "4-8-членная гетероциклическая С1-6 алкильная группа" относится к группе, полученной замещением "4-8-членной гетероциклической группой", определенной выше, произвольного атома водорода в "С1-6 алкильной группе", определенной выше.
Используемое здесь выражение "моноциклическая или бициклическая 4-12-членная гетероциклическая группа, содержащая один или два атома азота в кольце, которое может иметь один или несколько заместителей", относится к неароматической циклической группе, которая может иметь один или несколько заместителей. В неароматических циклических группах:
(i) число атомов, составляющих кольцо циклической группы, равняется 4-12;
(ii) атомы, составляющие кольцо циклической группы, включают один или два атома азота; и
(iii) группа является моноциклической или бициклической структурой.
В частности, группа представлена формулой:
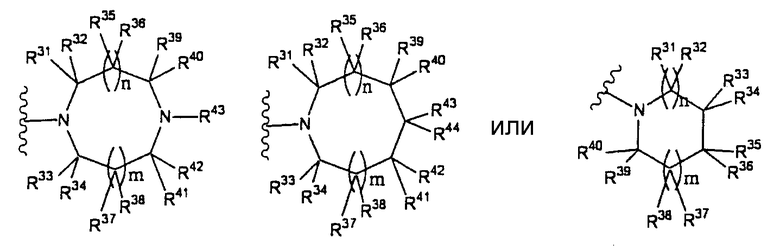
(где n и m, каждый, независимо, представляют собой 0 или 1; R31-R44 независимо представляют собой атом водорода или заместитель, выбранный из заместителей, на которые дается ссылка в выражении "которое может иметь один или несколько заместителей" (группа заместитель S определена ниже); любые два из R31-R44 могут в сочетании образовывать С1-6 алкиленовую группу).
Используемое здесь выражение "которая может иметь один или несколько заместителей" означает, что группа может иметь один или несколько заместителей в любом сочетании в способных к замещению положениях. В частности, такие заместители включают, например, заместитель, выбранный из группы заместителя S, определенной ниже.
<Группа заместитель S>
Данная группа состоит из:
(1) атома галогена,
(2) гидроксильной группы,
(3) меркаптогруппы,
(4) нитрогруппы,
(5) циангруппы,
(6) формильной группы,
(7) карбоксильной группы,
(8) трифторметильной группы,
(9) трифторметокси группы,
(10) аминогруппы,
(11) оксогруппы,
(12) иминогруппы, и
(13) группы, представленной формулой -T1x-T2x (где T1x представляет собой одинарную связь, С1-6 алкиленовую группу, атом кислорода, группу, представленную формулой -СО-, группу, представленную формулой -S-, группу, представленную формулой -S(O)-, группу, представленную формулой -S(O)2-, группу, представленную формулой -O-CO-, группу, представленную формулой -CO-O-, группу, представленную формулой -NRT-, группу, представленную формулой -CO-NRT-, группу, представленную формулой -NRT-CO-, группу, представленную формулой -SO2-NRT-, группу, представленную формулой -NRT-SO2, группу, представленную формулой -NH-CO-NRT-, или группу, представленную формулой -NH-CS-NRT-;
T2x представляет собой атом водорода, С1-6 алкильную группу, С3-8 циклоалкильную группу, С2-6 алкенильную группу, С2-6 алкинильную группу, фенильную группу, 1-нафтильную группу, 2-нафтильную группу, 5-10-членный гетероарильную группу или 4-8-членную гетероциклическую группу;
RT представляет собой атом водорода, С1-6 алкильную группу, С3-8 циклоалкильную группу, С2-6 алкенильную группу или С2-6 алкинильную группу;
при условии, что Т2х и RT, каждый, может, независимо, иметь 1-3 заместителя, выбранных из группы заместителя Т, определенной ниже).
<Группа заместитель Т>
Данная группа состоит из гидроксила, циан, атома галогена, С1-6 алкила, С3-8 циклоалкила, С2-6 алкенила, С2-6 алкинила, фенила, 1-нафтила, 2-нафтила, 5-10-членного гетероарила, 4-8-членного гетероциклического кольца, С1-6 алкокси, С1-6 алкилтио, С2-7 алкоксикарбонильной группы и др.
<Группа заместитель S>, предпочтительно, состоит из
(1) атома галогена,
(2) гидроксильной группы,
(3) циангруппы,
(4) карбоксильной группы,
(5) трифторметильной группы,
(6) трифторметоксигруппы,
(7) аминогруппы,
(8) С1-6 алкильной группы,
(9) С3-8 циклоалкильной группы,
(10) С2-6 алкенильной группы,
(11) С2-6 алкинильной группы,
(12) фенильной группы и
(13) С1-6 алкоксигруппы.
Используемое здесь выражение "С1-6 алкильная группа, которая может иметь один или несколько заместителей" в группе заместителе В, определенном выше, относится к "С1-6 алкильной группе", которая может иметь одну или несколько групп, выбранных из заместителей, на которую дается ссылка в выражении "которая может иметь один или несколько заместителей" в замещаемых положениях. Предпочтительно, "С1-6 алкильная группа, которая может иметь один или несколько заместителей", относится к С1-6 алкильной группе, которая может иметь один или два заместителя, выбранных из группы, состоящей из циангруппы, карбоксильной группы, С2-7 алкоксикарбонильной группы, группы, представленной формулой -NR3TCOR4T, группы, представленной формулой -CONR3TR4T (где R3T и R4T, каждый, независимо, представляет собой атом водорода или С1-6 алкильную группу), и С1-6 алкоксигруппы.
В формуле (I), указанной выше, фраза "когда Z2 представляет собой группу формулы -CR2=, R1 и R2 могут в сочетании образовывать 5-7-членное кольцо" обозначает, что соединения, представленные формулой (I), приведенной выше, включают соединения (III), представленные формулой:
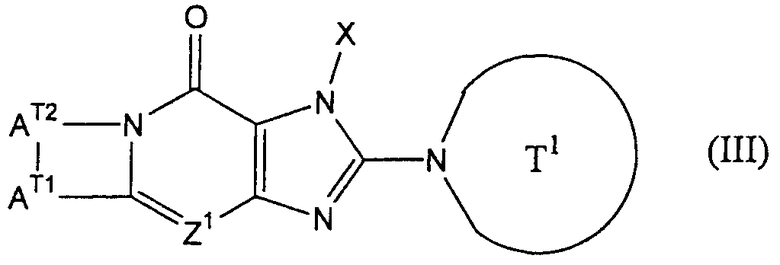
(где Z1, Х и Т1 имеют значения, определенные выше; АТ1 представляет собой атом кислорода, атом серы, сульфинильную группу, сульфонильную группу, карбонильную группу, метиленовую группу, которая может иметь один или несколько заместителей, или атом азота, который может иметь один или несколько заместителей; АТ2 представляет собой С2-6 алкиленовую группу, которая может иметь один или несколько заместителей). В формуле (III), показанной выше, АТ1, предпочтительно, представляет собой атом кислорода, а АТ2, предпочтительно, представляет собой С2-4 алкиленовую группу.
Используемое здесь выражение "цианбензильная группа" относится к бензильной группе, имеющей одну циангруппу, и, в частности, включает, например, 2-цианбензильную группу, 3-цианбензильную группу и 4-цианбензильную группу.
Используемое здесь выражение "фторцианбензильная группа" относится к бензильной группе, имеющей один атом фтора и одну циангруппу, и, в частности, включает, например, 2-циан-4-фторбензильную группу и 2-циан-6-фторбензильную группу.
Используемое здесь выражение "карбамоилфенилокси группа" относится к фенилокси группе, имеющей группу, представленную формулой -CONH2, и, в частности, включает например 2-карбамоилфенилоксигруппу, 3-карбамоилфенилокси группу и 4-карбамоилфенилоксигруппу.
Используемые термины "фенилокси" и "фенокси" являются эквивалентными.
В данном контексте нет никаких ограничений в отношении типа "солей", коль скоро соли являются фармацевтически приемлемыми и происходят из любого соединения по настоящему изобретению. Такие соли включают, например, соли неорганических кислот, соли органических кислот, соли неорганических оснований, соли органических оснований и кислые или основные соли аминокислот.
Примеры предпочтительных солей неорганических кислот включают гидрохлорид, гидробромид, сульфат, нитрат и фосфат. Примеры предпочтительных солей органических кислот включают ацетат, сукцинат, фумарат, малеат, тартрат, цитрат, лактат, стеарат, бензоат, метансульфонат и п-толуолсульфонат.
Примеры предпочтительных солей неорганических оснований включают соли щелочных металлов, такие как натриевые соли и калиевые соли; соли щелочно-земельных металлов, такие как кальциевые соли и магниевые соли; соли алюминия; и аммониевые соли. Примеры предпочтительных солей органических оснований включают диэтиламиновые соли, диэтаноламиновые соли, соли меглумина и соли N,N'-дибензилэтилендиамина.
Примеры предпочтительных кислых солей аминокислот включают аспартат и глутамат. Примеры предпочтительных основных солей аминокислот включают аргининовые соли, лизиновые соли и орнитиновые соли.
Используемое здесь выражение "усиливающие эффекты активного циркулирующего GLP-1 и/или активность циркулирующего GLP-2" означает, что эффекты активного циркулирующего GLP-1 и/или активного циркулирующего GLP-2 усиливаются благодаря увеличенным уровням данных пептидов в крови, что является результатом их усиленной секреции или подавленного разрушения.
Используемое здесь выражение "усиливающие эффекты активного циркулирующего GLP-2" означает, что эффект активного циркулирующего GLP-2 усиливаются благодаря увеличенным уровням данного пептида в крови, что является результатом его усиленной секреции или подавленного разрушения.
Эффекты активного циркулирующего GLP-1 включают усиление секреции инсулина глюкозазависимым образом; усиление биосинтеза инсулина; подавление секреции глюкагона; промотирование обновления β клеток; активирование гликогенсинтазы в печени; подавление потребления пищи; подавление прироста массы, подавление опорожнения желудка; и подавление секреции желудочной кислоты.
Эффекты активного циркулирующего GLP-2 включают промотирование роста эпителиальных клеток кишечника; промотирование роста эпителиальных клеток в желудочно-кишечном тракте; подавление апоптоза эпителиальных клеток в желудочно-кишечном тракте; поддержание функции желудочно-кишечного барьера; усиление абсорбции глюкозы; подавление секреции желудочной кислоты; и усиление тока крови в желудочно-кишечном тракте.
Выражение "усиление эффекта(ов)" означает усиление эффектов, описанных выше.
Используемый здесь термин "бигуанидный агент" относится, например, к фенформину, метформину и буформину, которые являются средствами, которые оказывают следующие действия: подавление глюконеогенеза и гликогенолиза в печени; усиление чувствительности скелетных мышц к инсулину; подавление абсорбции глюкозы в кишечном тракте; и снижение массы путем подавления потребления пищи. Предпочтительным бигуанидным агентом является метформин.
В данном описании "заболевание, которое связано с активным циркулирующим GLP-1 и/или активным циркулирующим GLP-2" включает, например, диабет, ожирение, гиперлипидемию, гипертензию, артериосклероз и желудочно-кишечные заболевания.
В данном описании "заболевание, которое связано с активным циркулирующим GLP-2", включает, например, желудочно-кишечные заболевания.
Используемое здесь выражение "и/или" означает как "и", так и "или".
(S)-1-((3-Гидрокси-1-адамантил)амино)ацетил-2-цианпирролидин может быть получен по способу, описанному в патенте США № 6166063.
(S)-1-(2-((5-Цианпиридин-2-ил)амино)этил-аминоацетил)-2-цианпирролидин может быть получен по способу, описанному в патенте США № 6011155.
Изолейцинтиазолидид, изолейцинпирролидид и валинпирролидид могут быть получены согласно методу, описанному в патенте США № 6548481,
Соединение, представленное формулой (II), приведенной здесь, может быть получено с помощью метода, описанного ниже в разделе [Типичные методы синтеза], или любого из методов, описанных в публикации патентной заявки США № 2002/0161001; публикации патентной заявки США № 2003/0105077 и публикации патентной заявки США № 2002/0198205.
[Типичные методы синтеза]
Представительные способы получения соединений по настоящему изобретению, представленных формулами (I) и (II), приведенными выше, описаны ниже.
Каждый символ в способах получения описан ниже.
R31-R42, n, m, R1, R2, X, А0, А1, А2, RA и Т1 имеют те же значения, что определены выше.
U1, U3 и Hal, каждый, независимо, представляют собой удаляемую группу, такую как атом хлора, атом брома, атом йода, метансульфонилоксигруппу или п-толуолсульфонилоксигруппу.
Rp1, Rp2 и Rp3, каждый, независимо, представляет собой -NH-защитную группу, такую как пивалилоксиметильная группа и триметилсилилэтоксиметильная группа.
Rp4 представляет собой группу, защищающую гидроксильную группу, такую как трет-бутилдиметилсилильная группа и трет-бутилдифенилсилильная группа.
Rp5 представляет собой NH-защитную группу, такую как N,N-диметилсульфамоил, тритил, бензил и трет-бутоксикарбонил.
U2 и U4, каждый, независимо, представляют собой атом хлора, атом брома, атом йода, метансульфонилокси группу, п-толуолсульфонилокси группу, группу, представленную формулой -В(ОН)2, 4,4,5,5-тетраметил-1,3,2-диоксаборан-2-ильную группу, или группу, представленную формулой -Sn(Rz)3 (где Rz представляет собой С1-6 алкильную группу).
Rх2 представляет собой группу, представленную формулой -О-А2, группу, представленную формулой -S-A2, группу, представленную формулой -N(RA)A2, или 4-8-членную гетероциклическую группу, которая может иметь один или несколько заместителей (например, 1-пирролидинил, 1-морфолинил, 1-пиперазинил или 1-пиперидил) и др.
Rх3 представляет собой группу, представленную формулой -А0-А1-А2, такую как циангруппа, С1-6 алкильная группа, которая может иметь один или несколько заместителей, С3-8 циклоалкильная группа, которая может иметь один или несколько заместителей, С2-6 алкенильная группа, которая может иметь один или несколько заместителей, С2-6 алкинильная группа, которая может иметь один или несколько заместителей, С6-10 арильная группа, которая может иметь один или несколько заместителей.
A2COOR представляет собой С1-6 алкильную группу, С3-8 циклоалкильную группу, С2-6 алкенильную группу, С2-6 алкинильную группу, С6-10 арильную группу, 5-10-членную гетероарильную группу, 4-8-членную гетероциклическую группу, 5-10-членную гетероарил С1-6 алкильную группу, или С6-10 арил С1-6 алкильную группу, каждая из которых содержит сложноэфирную группу.
A2COOН представляет собой С1-6 алкильную группу, С3-8 циклоалкильную группу, С2-6 алкенильную группу, С2-6 алкинильную группу, С6-10 арильную группу, 5-10-членную гетероарильную группу, 4-8-членную гетероциклическую группу, 5-10-членную гетероарил С1-6 алкильную группу, или С6-10 арил С1-6 алкильную группу, каждая из которых содержит карбоновую кислоту.
A2NO2 представляет собой С1-6 алкильную группу, С3-8 циклоалкильную группу, С2-6 алкенильную группу, С2-6 алкинильную группу, С6-10 арильную группу, 5-10-членную гетероарильную группу, 4-8-членную гетероциклическую группу, 5-10-членную гетероарил С1-6 алкильную группу, или С6-10 арил С1-6 алкильную группу, каждая из которых содержит нитрогруппу.
A2NH2 представляет собой С1-6 алкильную группу, С3-8 циклоалкильную группу, С2-6 алкенильную группу, С2-6 алкинильную группу, С6-10 арильную группу, 5-10-членную гетероарильную группу, 4-8-членную гетероциклическую группу, 5-10-членную гетероарил С1-6 алкильную группу, или С6-10 арил С1-6 алкильную группу, каждая из которых содержит аминогруппу.
A2СN представляет собой С1-6 алкильную группу, С3-8 циклоалкильную группу, С2-6 алкенильную группу, С2-6 алкинильную группу, С6-10 арильную группу, 5-10-членную гетероарильную группу, 4-8-членную гетероциклическую группу, 5-10-членную гетероарил С1-6 алкильную группу, или С6-10 арил С1-6 алкильную группу, каждая из которых содержит нитрильную группу.
AСОNH2 представляет собой С1-6 алкильную группу, С3-8 циклоалкильную группу, С2-6 алкенильную группу, С2-6 алкинильную группу, С6-10 арильную группу, 5-10-членную гетероарильную группу, 4-8-членную гетероциклическую группу, 5-10-членную гетероарил С1-6 алкильную группу, или С6-10 арил С1-6 алкильную группу, каждая из которых содержит карбоксильную амидную группу.
М представляет собой -MgCl, -MgBr, -Sn(Rz)3 (где Rz имеет значения, определенные выше) и др.
Термин "комнатная температура" относится к температуре от около 20 до около 30°С.
Т1а определен как группа, представленная символом Т1, или представляет собой группу формулы:
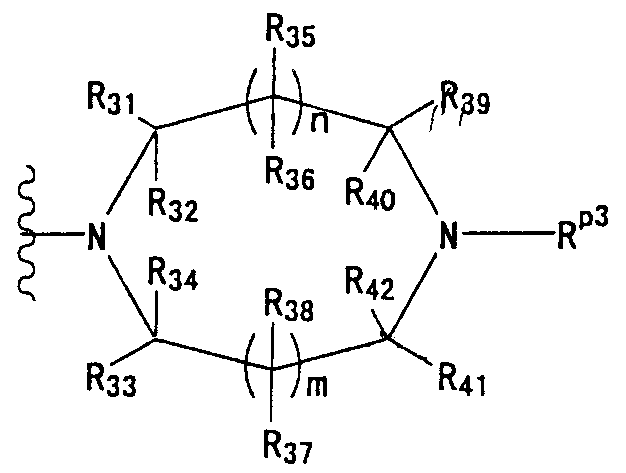
группу, представленную формулой:
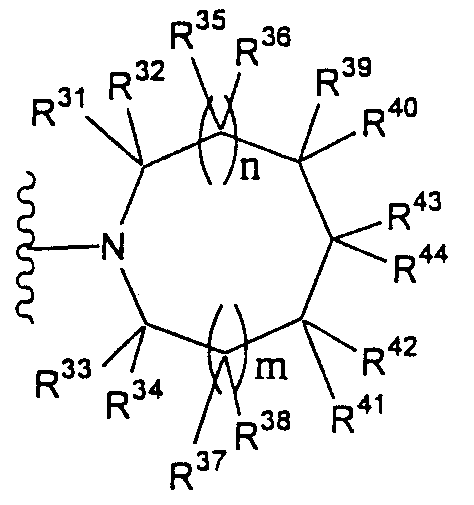
(в которой R31-R44 имеют значения, определенные выше, за исключением того, что любой из R31-R44 представляет собой -NH-Rр3), или
группу, представленную формулой:
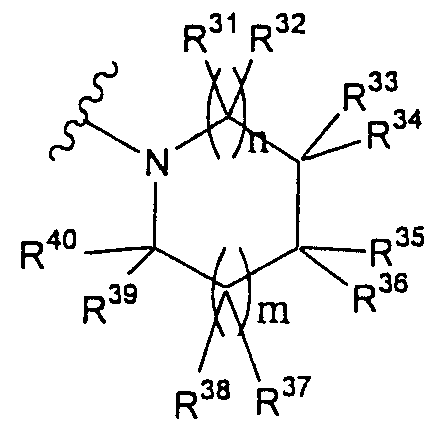
(в которой R31-R40 имеют значения, определенные выше, за исключением того, что любой из R31-R40 представляет собой -NH-Rр3).
s представляет собой 1-4.
R51-R50, каждый, независимо, представляют собой атом водорода, С1-6 алкильную группу или С6-10 арильную группу.
В примерах реакций, представленных на следующих реакционных схемах, если не указано иное, количества используемых реагентов, катализаторов и других (эквивалент, массовый % и массовое соотношение) представлены в соотношении к основному соединению в каждой реакционной схеме. Основное соединение представляет собой соединение, представленное химической формулой в реакционной схеме и имеющее скелет соединений по настоящему изобретению.
Способ получения А
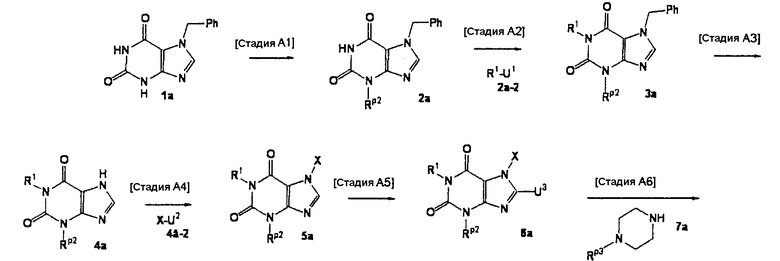
[Стадия А1]
На данной стадии -NH-защитный реагент подвергают взаимодействию с соединением (1а) [CAS No. 56160-64-6], что давало соединение (2а). Условия реакции подбирают в зависимости от типа используемого -NH-защитного реагента. Реакцию можно проводить в условиях, которые обычно используются для введения реагента, использующего защитную группу.
-NH-защитным реагентом может быть реагент, который обычно используется для введения -NH-защитной группы. В частности, такие -NH-защитные реагенты включают, например, хлорметилпивалат. Предпочтительно, использовать 1-2 эквивалента защитного реагента. Растворители реакции включают ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран и диметоксиэтан. Предпочтительно, используется N,N-диметилформамид.
Реакцию можно проводить в присутствии основания. Примеры оснований, используемых в реакции, включают карбонат цезия, карбонат лития, карбонат натрия, карбонат калия и гидрид натрия. Предпочтительно, используется гидрид натрия. В данном случае основание, предпочтительно, используется в количестве 1-5 эквивалентов. Реакцию можно проводить при температуре в интервале от 0°С до 150°С. Предпочтительно, температурой реакции является комнатная температура.
[Стадия А2]
На данной стадии соединение (2а) подвергают взаимодействию с соединением (2а-2), что давало соединение (3а).
Соединением (2а-2) может быть любое соединение, которое является элетрофильным реагентом, таким как алкилгалогенид. Конкретные примеры включают такие алкилгалогениды, как йодметан, йодэтан, йодпропан и бензилбромид; алкенилгалогениды, такие как аллилбромид и 1-бром-3-метил-2-бутен; и алкинилгалогениды, такие как пропаргилбромид и 1-бром-2-бутин. Предпочтительно, используются один-два эквивалента электрофильного реагента.
Растворители реакции включают, например, диметилсульфоксид, N,N-диметилфоромамид, N-метилпирролидон, диоксан, тетрагидрофуран и толуол.
Реакцию можно проводить в присутствии или в отсутствии основания. Примеры оснований, используемых в реакции, включают гидроксид лития, гидроксид натрия, гидроксид калия, карбонат лития, карбонат натрия, карбонат калия, карбонат цезия, гидрид лития, гидрид натрия, гидрид калия, бутиллитий, метиллитий, бис(триметилсилил)амид лития, бис(триметилсилил)амид натрия и бис(триметилсилил)амид калия. В данном случае, предпочтительно, используют один-два эквивалента основания. Реакцию можно проводить при температуре от 0°С до 150°С.
[Стадия А3]
На данной стадии бензильная группа в 7 положении удаляется из соединения (3а), что давало соединение (4а).
В частности, соединение (4а) может быть получено из соединения (3а), например, с помощью каталитического восстановления в атмосфере водорода в присутствии катализатора на основе металла, но условия реакции не ограничиваются этим.
Конкретные растворители реакции включают, например, метанол, этанол, пропанол, уксусную кислоту, диметилсульфоксид, N,N-диметилформамид, N-метилпирролидон, диоксан, тетрагидрофуран и толуол. Примеры катализаторов на основе металла включают палладий на угле, окись платины и никель Ренея. Катализатор на основе металла, предпочтительно, используется в количестве 0,5-50 мас.%. Предпочтительно, давление водорода составляет 1-5 атм. Реакцию можно проводить при температуре в интервале от 0°С до 150°С.
[Стадия А4]
На данной стадии соединение (4а) подвергают взаимодействию с соединением (4а-2), что давало соединение (5а).
Конкретными примерами соединения (4а-2) являются алкилгалогениды, такие как йодметан, йодэтан, йодпропан и бензилбромид; алкенилгалогениды, такие как аллилбромид и 1-бром-3-метил-2-бутен; или алкинилгалогениды, такие как пропаргилбромид и 1-бром-2-бутин. Данные галогениды, предпочтительно, используются в количестве от одного до двух эквивалентов.
Растворители реакции включают диметилсульфоксид, N,N-диметилформамид, N-метилпирролидон, диоксан, тетрагидрофуран и толуол.
Реакцию можно проводить в присутствии или отсутствии основания. Примеры используемых в реакции оснований включают гидроксид лития, гидроксид натрия, гидроксид калия, карбонат лития, карбонат натрия, карбонат калия, карбонат цезия, гидрид лития, гидрид натрия, гидрид калия, бутиллитий, метиллитий, бис(триметилсилил)амид лития, бис(триметилсилил)амид натрия и бис(триметилсилил)амид калия. В данном случае, предпочтительно, используют от 1 до 4 эквивалентов основания. Реакцию можно проводить при температуре в интервале от 0°С до 150°С.
Соединение (5а) может быть получено при взаимодействии соединения (4а) с соединением (4а-2) в присутствии медного катализатора и основания. В данном случае, предпочтительно, использовать 0,1-2 эквивалента медного катализатора и 1-10 эквивалентов основания.
В данной реакции соединением (4а-2) может быть арилбороновая кислота, гетероарилбороновая кислота или такая кислота, в которой Х представляет собой С6-10 арильную группу, которая может иметь один или несколько заместителей, или 5-10-членную гетероарильную группу, которая может иметь один или несколько заместителей, и U2 представляет собой -B(OH)2 или аналогичный. Предпочтительно, используют от одного до трех эквивалентов соединения (4а-2).
В данном случае реакционные растворители включают дихлорметан, хлороформ, 1,4-диоксан, тетрагидрофуран, толуол, пиридин, N,N-диметилформамид и N-метилпирролидон.
Основания включают триэтиламин, диизопропилэтиламин, пиридин и N,N-диметиламинопиридин. Медные катализаторы включают ацетат меди (II), трифторацетат меди (II) и хлорид меди (II) и йодид меди (II). Реакцию можно проводить при температуре в интервале от 0°С до 150°С.
[Стадия А5]
На данной стадии соединение (5а) подвергают взаимодействию с галогенирующим средством, что давало соединение (6а).
Конкретные примеры галогенирующих средств включают, например, N-хлорсукцинимид, N-бромсукцинимид и N-йодсукцинимид. Галогенирующее средство, предпочтительно, используется в количестве от 1 до 4 эквивалентов.
Растворители реакции включают ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран и диметоксиэтан. Реакцию можно проводить при температуре в интервале от 0°С до 150°С.
[Стадия А6]
На данной стадии соединение (6а) подвергают взаимодействию с соединением (7а), что давало соединение (8а). В данном случае, предпочтительно, используется 1-4 эквивалента соединения (7а).
Реакцию можно проводить, например, в растворителе, таком как тетрагидрофуран, ацетонитрил, N,N-диметилформамид, N-метилпирролидон, метанол, этанол, 1,4-диоксан, толуол и ксилол, или в отсутствии растворителя. Реакцию можно проводить при температуре в интервале от 0°С до 200°С, в присутствии или отсутствии основания. Примеры оснований включают триэтиламин, карбонат калия и 1,8-диазабицикло[5,4,0]ундецен. В данном случае, предпочтительно, используется 1-4 эквивалента основания.
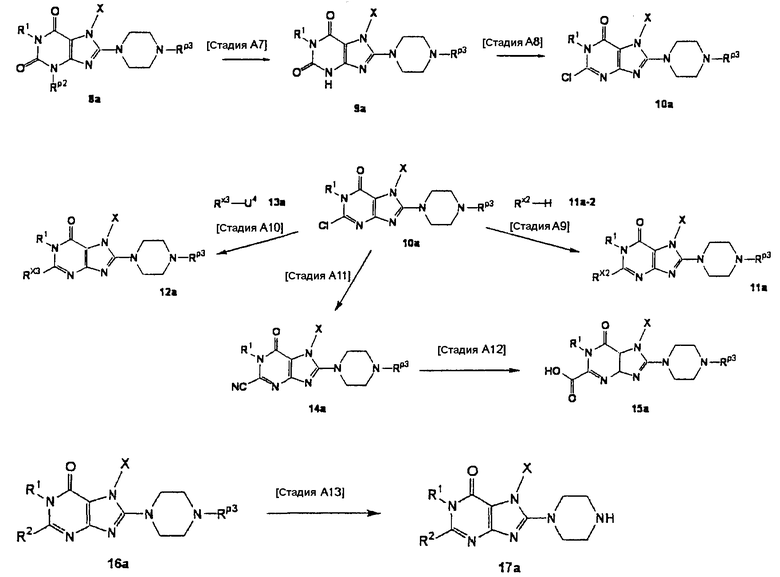
[Стадия А7]
На данной стадии удаляется -NH-защитная группа в 3 положении соединения (8а), что давало соединение (9а). Условия реакции выбираются в зависимости от типа -NH-защитной группы, подлежащей удалению. Реакция удаления защиты может проводиться в условиях, которые обычно используются для защитной группы.
Например, когда Rp2 представляет собой пивалилоксиметильную группу, реакцию можно проводить в метаноле или в смешанном растворе метанола и тетрагидрофурана с использованием основания, такого как метилат натрия, гидрид натрия или 1,8-диазабицикло[5,4,0]-7-ундецен, при температуре 0°С-150°С. В данном случае, предпочтительно, используется 0,1-2 эквивалента основания.
Альтернативно, когда Rp2 представляет собой триметилсилилэтоксиметильную группу, реакцию можно проводить в растворителе, таком как ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран или диметоксиэтан, с использованием фторидного реагента, такого как тетрабутиламмонийфторид или фторид цезия, при температуре 0°С-150°С. В данном случае, предпочтительно, используется 1-5 эквивалентов фторидного реагента.
[Стадия А8]
На данной стадии соединение (9а) хлорируется, что давало соединение (10а).
Нет никаких конкретных ограничений в отношении условий реакции, и реакцию можно проводить в стандартных условиях хлорирования. Например, реакцию можно проводить при температуре 0°С-150°С в растворителе, таком как хлорокись фосфора. В данном случае, предпочтительно, использовать 10-200-кратное количество галогенирующего средства по массе.
Альтернативно, когда Rp3 представляет собой трет-бутоксикарбонильную группу или такую, которая удаляется в описанных выше условиях с использованием хлорокиси фосфора или аналогичного, защитная группа должна снова вводиться.
Нет никаких конкретных ограничений в отношении условий реакции защиты. В случае трет-бутоксикарбонильной группы реакцию можно проводить с использованием -NH-защитного реагента, такого как ди-трет-бутилдикарбонат, в растворителе, таком как ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран или диметоксиэтан, в присутствии основания, такого как гидроксид лития, гидроксид натрия, гидроксид калия, карбонат лития, карбонат натрия, карбонат калия, карбонат цезия, бикарбонат калия, бикарбонат натрия или триэтиламин, при 0°С-150°С.
[Стадия А9]
На данной стадии соединение (10а) подвергают взаимодействию с соединением (11а-2), что давало соединение (11а).
Соединение (11а-2) включает производные спиртов или производные фенолов, представленные формулой А2-ОН, производные аминов, представленные формулой A2(RA)NH, или тому подобные, и производные тиолов, представленные формулой A2-SH. В данном случае соединение (11а-2), предпочтительно, используется в количестве от 1 до 10 эквивалентов или в 5-100-кратном количестве по массе.
Растворители реакции включают ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран, диметоксиэтан, метанол и этанол.
Реакцию можно проводить в присутствии или отсутствии основания. Основания, используемые для реакции, включают гидроксид лития, гидроксид натрия, гидроксид калия, карбонат лития, карбонат натрия, карбонат калия, карбонат цезия, гидрид лития, гидрид натрия, гидрид калия, бутиллитий, метиллитий, бис(триметилсилил)амид лития, бис(триметилсилил)амид натрия, бис(триметилсилил)амид калия и триэтиламин. В данном случае, предпочтительно, используется от 1 до 10 эквивалентов основания. Реакцию можно проводить при температуре в интервале от 0°С до 150°С.
[Стадия А10]
На данной стадии соединение (10а) подвергают взаимодействию с соединением (13а) в присутствии катализатора на основе металла, что давало соединение (12а). В данном случае, предпочтительно, используется от 1 до 50 эквивалентов соединения (13а).
Растворители реакции включают ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран, диметоксиэтан, метанол и этанол.
Катализаторы на основе металла включают палладиевый катализатор и медный катализатор. Палладиевые катализаторы включают тетракистрифенилфосфинпалладий, ацетат палладия и дибензилиденацетонпалладий. Медный катализатор включает йодид меди. Предпочтительно, использовать 0,01-2 эквивалента катализатора на основе металла.
Реакцию можно проводить в присутствии фосфорорганического лиганда. Когда реакция осуществляется в присутствии фосфорорганического лиганда, примеры лигандов включают о-толилфосфин и дифенилфосфиноферроцен. В данном случае, предпочтительно, использовать 1-5 эквивалентов фосфорорганического лиганда по отношению к металлическому катализатору.
Реакцию можно проводить в присутствии или отсутствии основания. Основания, используемые для реакции, включают гидроксид лития, гидроксид натрия, гидроксид калия, карбонат лития, карбонат натрия, карбонат калия, карбонат цезия, гидрид лития, гидрид натрия, гидрид калия, фосфат калия, бис(триметилсилил)амид лития, бис(триметилсилил)амид натрия, бис(триметилсилил)амид калия и триэтиламин. Реакцию можно проводить при температуре в интервале от 0°С до 150°С.
[Стадия А11]
На данной стадии соединение (10а) подвергают взаимодействию с реагентом цианидирования, что давало соединение (14а).
В частности, реагенты цианидирования включают, например, цианид натрия и цианид калия. Он, предпочтительно, используется в количестве от 1 до 20 эквивалентов.
Растворители для реакции включают, например, ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран, диметоксиэтан, метанол и этанол. Реакцию можно проводить при температуре в интервале от 0°С до 150°С.
[Стадия А12]
На данной стадии циангруппа соединения (14а) гидролизуется, что давало соединение (15а). Нет никаких особых ограничений в отношении условий реакции, и реакцию можно проводить в условиях, используемых обычно для превращения циангруппы в карбамоильную группу с помощью гидролиза.
Растворители реакции включают N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран, диметоксиэтан, метанол, этанол и смешанный растворитель из тетрагидрофурана и метанола.
Реакцию можно проводить в присутствии или отсутствии основания. Когда используется основание, реакцию можно проводить с использованием водного раствора основания, такого как гидроксид калия, гидроксид натрия, гидроксид лития или аммиак. Реакцию можно проводить путем давления водного раствора перекиси водорода (предпочтительно, водного раствора 30% перекиси водорода).
Реакцию можно проводить при температуре в интервале от 0°С до 150°С.
[Стадия А13]
На данной стадии Rр3 соединения (16а) удаляется, что давало соединение (17а). Соединения (11а), (12а), (14а), (15а) и другие могут использоваться в качестве соединения (16а).
Реакция удаления защиты для Rр3 может осуществляться в стандартных условиях удаления -NH-защитной группы.
Например, когда Rр3 представляет собой трет-бутоксикарбонильную группу, реакцию можно проводить в присутствии кислоты, такой как безводный метанольный раствор хлорида водорода, безводный этанольный раствор хлорида водорода, безводный диоксановый раствор хлорида водорода, трифторуксусная кислота или муравьиная кислота.
Альтернативный способ получения соединения (10а) описан ниже.
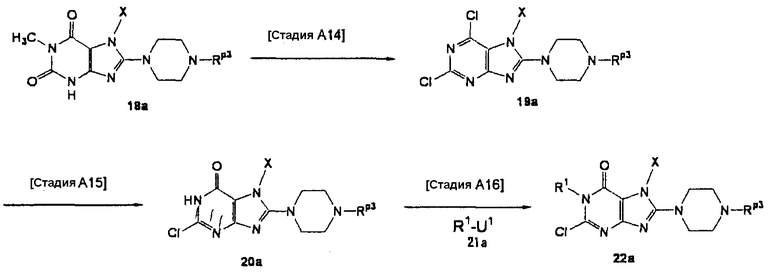
[Стадия А14]
На данной стадии соединение (18а) хлорируется, что давало соединение (19а). Нет никаких конкретных ограничений в отношении условий реакции, и реакцию можно проводить в стандартных условиях хлорирования. Например, реакцию можно проводить в растворителе, таком как хлорокись фосфора, при температуре в интервале от 0°С до 150°С. Предпочтительно, используют 10-200-кратное количество по массе реагента хлорирования.
Когда Rp3 представляет собой трет-бутоксикарбонильную группу или такую, которая удаляется в описанных выше условиях с использованием хлорокиси фосфора или аналогичного, защитная группа должна снова вводиться.
Нет никаких конкретных ограничений в отношении условий реакции защиты, и когда Rp3 представляет собой трет-бутоксикарбонильную группу, реакцию можно проводить с использованием -NH-защитного реагента, такого как ди-трет-бутилдикарбонат, в растворителе, таком как ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран и диметоксиэтан, в присутствии основания, такого как гидроксид лития, гидроксид натрия, гидроксид калия, карбонат лития, карбонат натрия, карбонат калия, карбонат цезия, бикарбонат калия, бикарбонат натрия или триэтиламин, при температуре в интервале от 0°С до 150°С.
[Стадия А15]
На данной стадии соединение (19а) частично гидролизуется, что давало соединение (20а). Реакция осуществляется в присутствии основания, такого как ацетат натрия, карбонат калия или гидроокись натрия. Предпочтительно, используется от одного до десяти эквивалентов основания. Растворители для реакции включают диметилсульфоксид, N-метилпирролидон, тетрагидрофуран, воду и их смеси. Реакцию можно проводить при температуре в интервале от 0°С до 100°С.
[Стадия А16]
На данной стадии соединение (20а) подвергают взаимодействию с соединением (21а), что давало соединение (22а). Реакцию можно проводить в тех же условиях, что используются на [Стадии А2] способа получения А.
Альтернативный способ получения соединения (19а) описывается ниже.
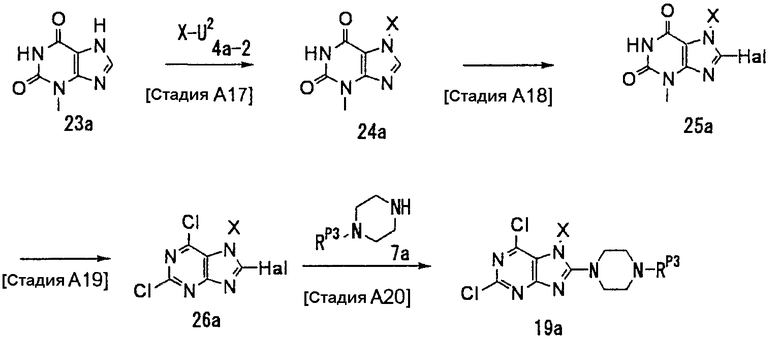
[Стадия А17]
На данной стадии осуществляется реакция замещения с использованием соединения (23а) {CAS No. 1076-22-8] и соединения (4а-2), что давало соединение (24а).
Реакцию можно проводить в тех же условиях, что используются на [Стадии А4] способа получения А.
[Стадия А18]
На данной стадии соединение (24а) подвергается взаимодействию с галогенирующим средством, что давало соединение (25а).
Реакцию можно проводить в тех же условиях, что используются на [Стадии А5] способа получения А.
[Стадия А19]
На данной стадии соединение (25а) хлорируется, что давало соединение (26а).
Нет никаких особых ограничений относительно условий реакции, и соединение (25а) может подвергаться взаимодействию с хлорокисью фосфора, пентахлоридом фосфора или их смесью в растворителе или в отсутствии растворителя при температуре от 0°С до 150°С. Растворители включают, например, толуол, ацетонитрил и дихлорэтан.
[Стадия А20]
На данной стадии соединение (26а) подвергается взаимодействию с соединением (7а), что давало соединение (19а).
Реакцию можно проводить в тех же условиях, что используются на [Стадии А6] способа получения А.
Способ получения В
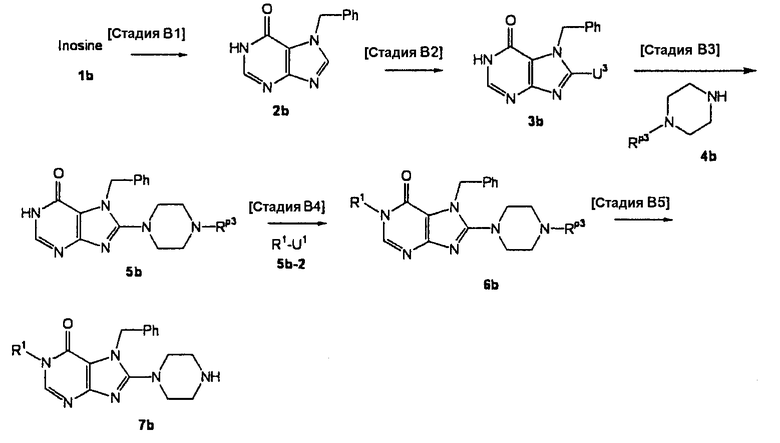
[Стадия В1]
На данной стадии соединение (1b) бензилируется, а сахарная цепь расщепляется, что давало соединение (2b).
Нет никаких особых ограничений в отношении условий реакции. Соединение (2b) может быть получено при взаимодействии соединения (1b) с бензилбромидом в растворителе, таком как ацетонитрил, N,N-диметилформамид, N-метилпирролидон, диметилсульфоксид, 1,4-диоксан, тетрагидрофуран, диметоксиэтан, метанол или этанол, при температуре от 0°С до 150°С, добавлением 3-10 эквивалентов соляной кислоты и инкубированием смеси при температуре от 0°С до 150°С для расщепления сахарной части. Предпочтительно, использовать 1-3 эквивалента бензилбромида.
[Стадия В2]
На данной стадии соединение (2b) подвергают взаимодействию с галогенирующим средством, что давало соединение (3b). Реакция галогенирования может проводиться в тех же условиях, что используются на [Стадии А5] способа получения А.
[Стадия В3]
На данной стадии соединение (3b) подвергают взаимодействию с соединением (4b), что давало соединение (5b). Реакцию можно проводить в тех же условиях, что используются на [Стадии А6] способа получения А.
[Стадия В4]
На данной стадии соединение (5b) подвергают взаимодействию с соединением (5b-2), что давало соединение (6b). Реакцию можно проводить в тех же условиях, что используются на [Стадии А2] способа получения А.
[Стадия В5]
На данной стадии Rр3 соединения (6b) удаляется, что давало соединение (7b). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения В-2
Соединение (9b), представленное формулой:
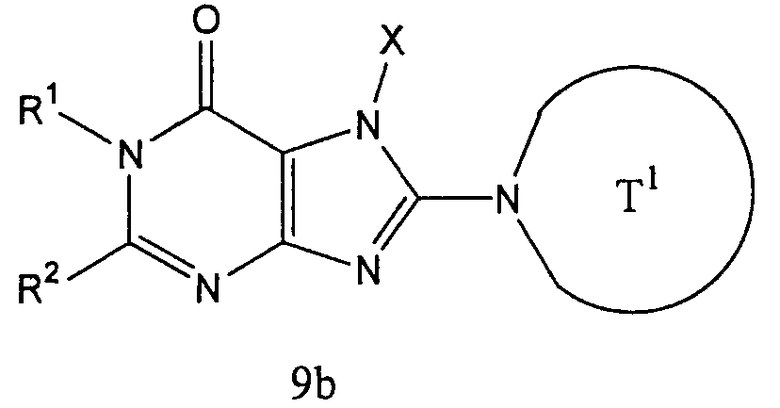
может получаться с использованием соединения (8b), представленного формулой Н-Т1а, вместо соединения (7а) на [Стадии А6] способа получения А, описанного выше, в тех же условиях, что используются на [Стадии А6], и затем соответствующим образом с применением [Стадии А7]-[Стадии А13], описанной выше.
Соединение (10b), представленное формулой:
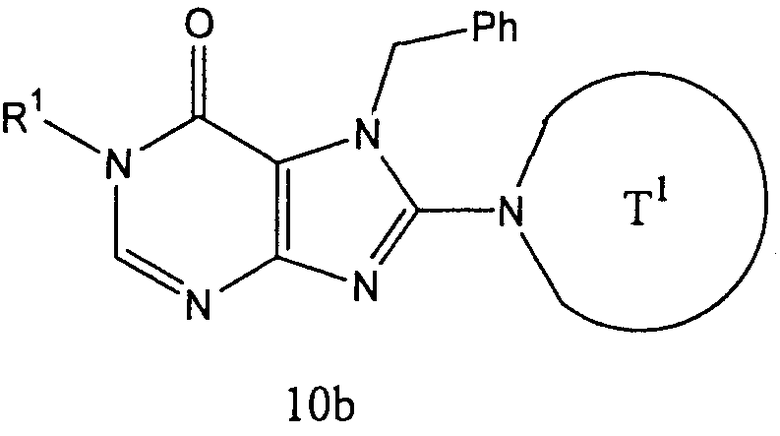
может получаться с использованием соединения (8b), представленного формулой Н-Т1а, вместо соединения (3b) на [Стадии В3] способа получения В, описанного выше, в тех же условиях, что используются на [Стадии В3], и затем соответствующим образом с применением [Стадии В4]-[Стадии В6], описанной выше. Предпочтительные примеры соединения (8b) включают трет-бутиловый эфир пиперидин-3-ил-карбаминовой кислоты.
Способ получения С
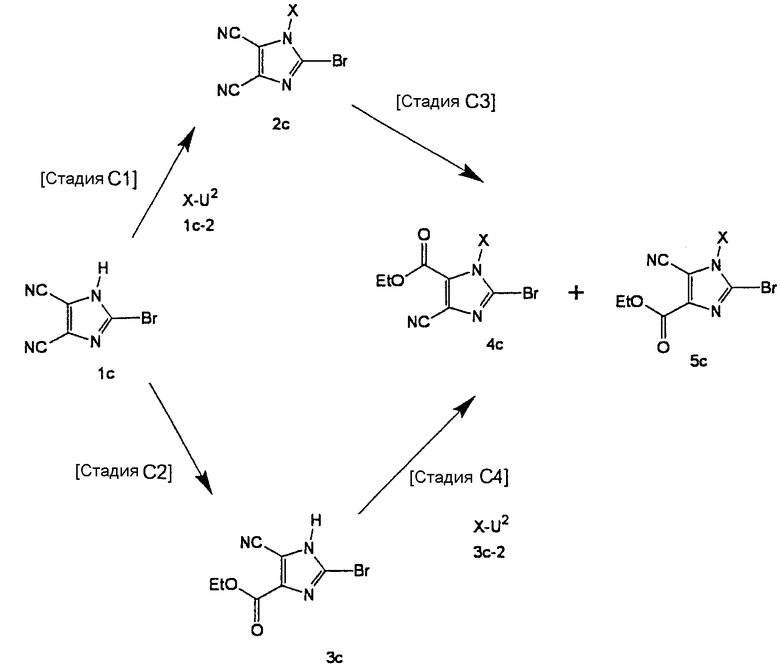
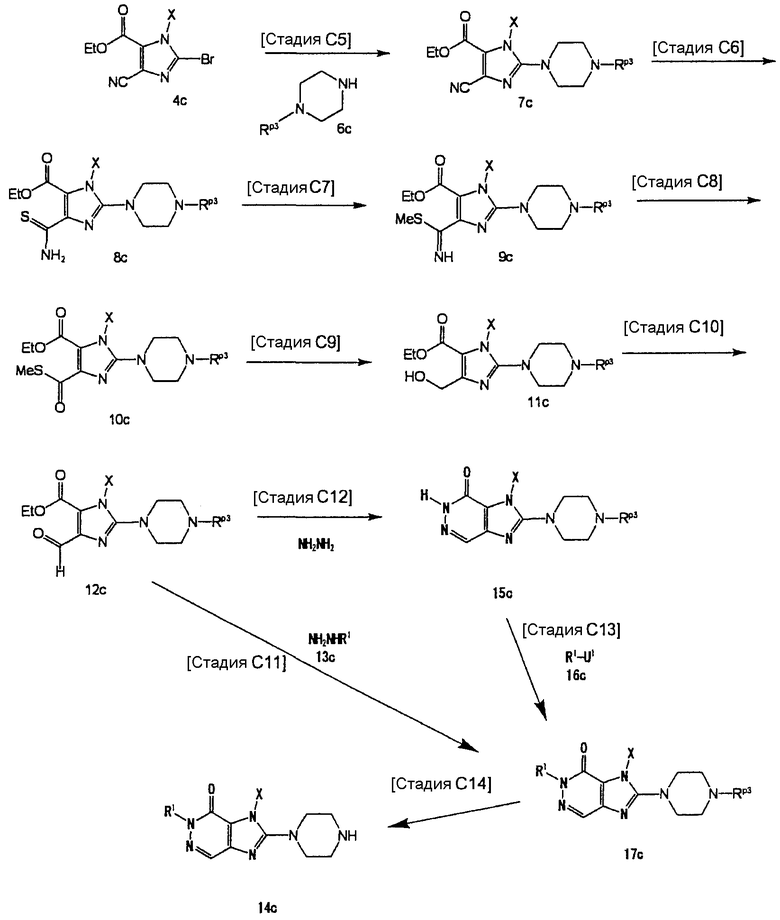
[Стадия С1]
На данной стадии соединение (1с) подвергают взаимодействию с соединением (1с-2), что давало соединение (2с). Реакцию можно проводить в тех же условиях, что используются на [Стадии А4] способа получения А.
[Стадия С2]
На данной стадии соединение (1с) подвергают взаимодействию с этанолом, что давало соединение (3с).
Соединение (3с) может быть получено, например, с помощью нагревания этанольного раствора соединения (2с) при кипении с обратным холодильником в присутствии кислоты, такой как серная кислота или соляная кислота. Однако условия реакции не ограничиваются этим. В данной реакции, предпочтительно, использовать один-два эквивалента кислоты.
[Стадия С3]
На данной стадии соединение (2с) подвергают взаимодействию с этанолом, что давали соединения (4с) и (5с). Реакцию можно проводить в тех же условиях, что используются на [Стадии С2] способа получения С.
[Стадия С4]
На данной стадии соединение (3с) подвергают взаимодействию с соединением (3с-2), что давало соединения (4с) и (5с). Реакцию можно проводить в тех же условиях, что используются на [Стадии А4] способа получения А.
[Стадия С5]
На данной стадии соединение (4с) подвергают взаимодействию с соединением (6с), что давало соединение (7с). Реакцию можно проводить в тех же условиях, что используются на [Стадии А6] способа получения А.
[Стадия С6]
На данной стадии соединение (7с) тиоамидируется, что давало соединение (8с). Растворители для реакции включают метанол, этанол, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран и диметоксиэтан. Реагенты тиоамидирования включают сульфид аммония, сульфид натрия и сульфид водорода. Предпочтительно, использовать 2-10 эквивалентов реагента тиоамидирования. Когда в качестве тиоамидирующего средства используется сульфид водорода, реакция осуществляется в присутствии основания, такого как триэтиламин или N,N-диизопропилэтиламин. Реакцию можно проводить при температуре в интервале от 0°С до 150°С.
[Стадия С7]
На данной стадии соединение (8с) подвергают взаимодействию с метилирующим реагентом, что давало соединение (9с). Метилирующие реагенты включают тетрафторборат триметилоксония, метилсульфат, метилйодид и триметилфосфит. Предпочтительно, использовать 1,0-1,5 эквивалента метилирующего реагента.
Когда в качестве метилирующего реагента используется тетрафторборат триметилоксония, соединение (9с) может получаться путем осуществления реакции в галогенированном растворителе, таком как дихлорметан, при температуре в интервале от 0°С до 50°С.
Когда в качестве метилирующего реагента используется метилсульфат, метилйодид или триметилфосфит, соединение (9с) может быть получено осуществлением реакции в присутствии основания, такого как карбонат калия, триэтиламин или N,N-диизопропилэтиламин. В данном случае, предпочтительно, использовать 1,0-1,5 эквивалента основания. Растворители для реакции включают ацетон, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран и диметоксиэтан. Реакцию можно проводить при температуре в интервале от 0°С до 100°С.
[Стадия С8]
На данной стадии соединение (9с) гидролизуется, что давало соединение (10с).
Нет никаких особых ограничений в отношении условий реакции гидролиза. Реакцию можно проводить в смешанном растворителе этанола и воды в присутствии кислоты, такой как серная кислота, соляная кислота или п-толуолсульфоновая кислота, при температуре в интервале от 0°С до 80°С. В данном случае, предпочтительно, использовать 5-50 эквивалентов кислоты.
Когда Rр3 является группой, такой как трет-бутоксикарбонильная группа, которая удаляется в описанных выше условиях, следует снова вводить защитную группу. Нет никаких особых ограничений в отношении условий реакции для введения данной защитной группы. Когда Rр3 представляет собой трет-бутоксикарбонильную группу, реакцию можно проводить с использованием реагента, такого как трет-бутилдикарбонат, в растворителе, таком как дихлорметан, хлороформ, N,N-диметилформамид, или тетрагидрофуран, в присутствии основания, такого как пиридин, 4-аминопиридин, триэтиламин и N,N-диизопропилэтиламин, при температуре в интервале от 0°С до 80°С. В данном случае, предпочтительно, использовать 2-3 эквивалента основания.
[Стадия С9]
На данной стадии соединение (10с) подвергают взаимодействию с восстанавливающим реагентом, что давало соединение (11с).
Нет никаких особых ограничений в отношении условий реакции восстановления. Реакцию можно проводить взаимодействием соединения (10с) с водородом в присутствии никеля Ренея в растворителе, таком как бензол, этанол, 2-пропанол или ацетон, при температуре в интервале от 0°С до 50°С, или альтернативно взаимодействием соединения (10с) с восстанавливающим средством, таким как боргидрид натрия, в растворителе, таком как метанол, этанол или 2-метил-2-пропанол, или в смешанном растворителе из воды и тетрагидрофурана, при температуре в интервале от 0°С до 50°С, или альтернативно взаимодействием соединения (10с) с восстанавливающим средством, таким как боргидрид натрия, в присутствии 1 - 5 эквивалентов ртутной соли, такой как ацетат ртути, в растворителе, таком как метанол, этанол или 2-метил-2-пропанол, при температуре в интервале от 0°С до 50°С. Предпочтительно, использовать два-три эквивалента восстанавливающего средства.
[Стадия С10]
На данной стадии соединение (11с) подвергают взаимодействию окисления, что давало соединение (12с).
Когда в реакции окисления используется такой окислитель, как диоксид марганца, хлорформиат пиридиния или дихромат пиридиния, соединение (12с) может получаться путем осуществления реакции в растворителе, таком как дихлорметан или хлороформ, при температуре в интервале от 20°С до 80°С. Альтернативно, соединение (12с) может также получаться с помощью осуществления реакции в стандартных условиях окисления первичного спирта в альдегид, такого как окисление Сверна. Предпочтительно, использовать 5-20 эквивалентов окислителя.
[Стадия С11]
На данной стадии соединение (12с) подвергают взаимодействию с соединением (13с), что давало соединение (17с). В данном случае, предпочтительно, использовать 2-10 эквивалентов соединения (13с).
Соединение (17с) может быть получено, например, с помощью объединения соединений (12с) и (13с) в растворителе, таком как метанол, этанол, 1-метил-2-пирролидон, 1,4-диоксан, тетрагидрофуран или диметоксиэтан, или в отсутствии растворителя и реакции смеси при температуре 20°С-150°С. Однако условия реакции не ограничиваются указанными.
[Стадия С12]
На данной стадии соединение (12с) подвергают взаимодействию с гидразином, что давало соединение (15с). Реакцию можно проводить в тех же условиях, что используются на [Стадии С11] способа получения С. Предпочтительно, использовать 2-10 эквивалентов гидразина.
[Стадия С13]
На данной стадии осуществляется реакция замещения с использованием соединения (15с) и соединения (16с), что давало соединение (17с). Реакцию можно проводить в тех же условиях, что используются на [Стадии А2] способа получения А. Предпочтительно, использовать 1-3 эквивалента соединения (16с).
[Стадия С14]
На данной стадии удаляется группа Rр3 соединения (17с), что давало соединение (14с). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
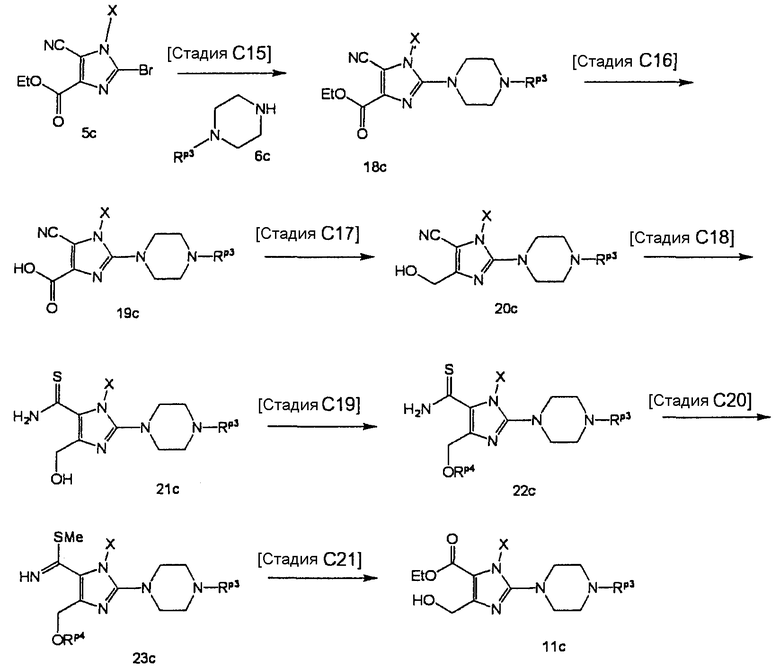
[Стадия С15]
На данной стадии соединения (5с) подвергают взаимодействию с соединением (6с), что давало соединение (18с). Реакцию можно проводить в тех же условиях, что используются на [Стадии А6] способа получения А.
[Стадия С16]
На данной стадии соединение (18с) гидролизуется, что давало соединение (19с).
Нет никаких особых ограничений в отношении условий реакции гидролиза. Например, соединение (19с) может быть получено с помощью инкубирования соединения (18с) в присутствии основания при температуре в интервале от 0°С до 100°С.
Растворители для реакции включают метанол, этанол, тетрагидрофуран, воду или их смеси. Основания включают гидроксид лития, гидроксид натрия и гидроксид калия. Предпочтительно, использовать 1-2 эквивалента основания.
[Стадия С17]
На данной стадии соединение (19с) подвергают взаимодействию с восстанавливающим средством, что давало соединение (20с). Восстановление может достигаться в стандартных условиях восстановления карбоновой кислоты в метиловый спирт.
Восстанавливающие средства включают борановые производные, такие как комплекс боран-тетрагидрофуран и комплекс боран-метилсульфид, и боргидрид натрия. Предпочтительно, использовать 5-30 эквивалентов восстанавливающего средства.
Когда в качестве восстанавлитвающего средства используется борановое производное, соединение (20с) может быть получено осуществлением реакции с использованием растворителя, такого как 1,4-диоксан, тетрагидрофуран или диметоксиэтан, при температуре в интервале от -78°С до 35°С.
Альтернативно, когда в качестве восстанавливающего средства используется боргидрид натрия, сначала соединение (19с) подвергают взаимодействию с активатором, таким как изобутилхлорформиат, при температуре в интервале от -78°С до 20°С, затем подвергают взаимодействию с восстанавливающим средством, таким как боргидрид натрия, при температуре в интервале от -78°С до 35°С с получением соединения (20с). Растворители для реакции включают 1,4-диоксан, тетрагидрофуран и диметоксиэтан.
[Стадия С18]
На данной стадии соединение (20с) тиоамидируется, что давало соединение (21с). Реакцию можно проводить в тех же условиях, что используются на [Стадии С6] способа получения С.
[Стадия С19]
На данной стадии соединение (21с) подвергают взаимодействию с силилирующим средством в присутствии основания, что давало соединение (22с).
Растворители для реакции включают дихлорметан, N,N-диметилформамид, 1,4-диоксан, тетрагидрофуран и диметоксиэтан. Основания включают имидазол, пиридин, 4-диметиламинопиридин, триэтиламин и N,N-диизопропилэтиламин. Силилирующие средства включают трет-бутилдиметилхлорсилан и трет-бутилхлордифенилсилан. Предпочтительно, использовать 1,0-1,5 эквивалента основания и 1,0-1,5 эквивалента силилирующего средства. Реакцию можно проводить при температуре в интервале от 0°С до 80°С.
[Стадия С20]
На данной стадии соединение (22с) метилируется, что давало соединение (23с).
Реакцию можно проводить в тех же условиях, что используются на [Стадии С7] способа получения С.
[Стадия С21]
На данной стадии соединение (23с) гидролизуется, что давало соединение (24с).
Нет никаких особых ограничений по условиям реакции гидролиза. Соединение (24с) может быть получено осуществлением реакции в смешанном растворителе из этанола и воды в присутствии кислоты, такой как серная кислота, соляная или п-толуолсульфоновая кислота, при температуре в интервале от 50°С до 100°С.
Когда такая реакция приводит в результате к удалению группы -Rр3, -NH-группу повторно защищают с помощью реакции защиты. В частности, например, когда Rр3 представляет собой трет-бутоксикарбонильную группу, реакцию можно проводить с использованием такого реагента, как трет-бутилдикарбонат, в растворителе, таком как дихлорметан, хлороформ, N,N-диметилформамид или тетрагидрофуран, в присутствии основания, такого как пиридин, 4-аминопиридин, триэтиламин или N,N-диизопропилэтиламин, при температуре в интервале от 0°С до 80°С. Однако реакция не ограничивается указанным.
Способ получения D
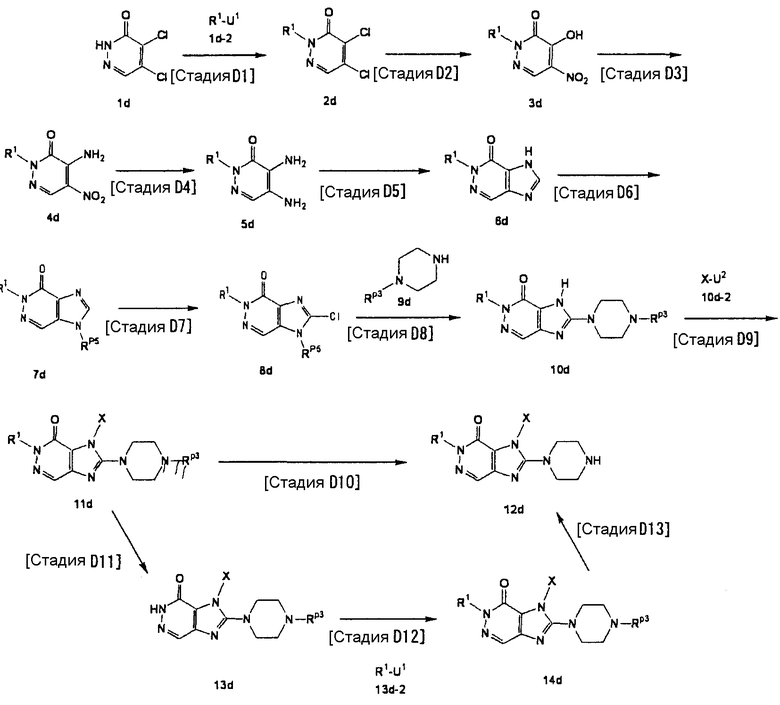
[Стадия D1]
На данной стадии соединение (1d) вводится в реакцию с соединением (1d-2), что давало соединение (2d).
В частности, соединение (1d-2) включает, например, алкилгалогениды, такие как йодметан, йодэтан, йодпропан, бензилбромид, 2-бромацетофенон, хлорметилбензиловый эфир и бромацетонитрил; алкенилгалогениды, такие как аллилбромид и 1-бром-3-метил-2-бутен; и алкинилгалогениды, такие как пропаргилбромид и 1-бром-2-бутин. Предпочтительно, использовать 1-1,5 эквивалента соединения (1d-2).
Растворители для реакции включают N,N-диметилформамид, N-метилпирролидон, тетрагидрофуран, 1,2-диметоксиэтан, 1,4-диоксан и дихлорметан. Реакцию можно проводить в присутствии или отсутствии основания. Основания, используемые в реакции, включают 1,8-диазабицикло[5,4,0]ундецен, триэтиламин, N,N-диизопропилэтиламин и гидрид натрия. В данном случае, предпочтительно, использовать 1-1,5 эквивалента основания. Реакцию можно проводить при температуре в интервале от 0°С до 150°С.
[Стадия D2]
На данной стадии соединение (2d) вводится в реакцию с нитрильной солью, что давало соединение (3d).
Растворители для реакции включают смешанный растворитель из воды и растворителя, выбранного из N,N-диметилформамида, N-метилпирролидона, тетрагидрофурана, 1,2-диметоксиэтана и 1,4-диоксана. Нитритные соли включают нитрит натрия и нитрит калия. Предпочтительно, использовать 3-5 эквивалентов нитрита. Реакцию можно проводить при температуре в интервале от 20°С до 120°С.
[Стадия D3]
На данной стадии соединение (3d) вводится в реакцию с аммиаком, что давало соединение (4d). Предпочтительно, использовать 10-20 эквивалентов аммиака.
Реакцию можно проводить в растворителе, таком как метанол, этанол или 1,4-диоксан, при температуре в интервале от 20°С до 200°С.
[Стадия D4]
На данной стадии соединение (4d) подвергается каталитическому восстановлению в атмосфере водорода или в присутствии 2-3 эквивалентов гидразина с использованием катализатора на основе металла, что давало соединение (5d).
Растворители для реакции включают метанол, этанол, N,N-диметилформамид, тетрагидрофуран, 1,2-диметоксиэтан, 1,4-диоксан, воду или смешанный растворитель из указанных. Катализаторы на основе металла включают палладий на угле, окись платины и никель Ренея. Предпочтительно, использовать металлический катализатор в количестве 0,5-10% по массе. Реакцию можно проводить при температуре в интервале от 0°С до 150°С.
[Стадия D5]
На данной стадии соединение (5d) подвергают взаимодействию с ортоформиатным эфиром, что давало соединение (6d).
Реакция осуществляется в присутствии ангидрида карбоновой кислоты, такого как уксусный ангидрид. Ортоформиатные эфиры включают метилортоформиат и этилортоформиат. Предпочтительно, использовать 1-20-кратное количество ортоформиатного эфира по массе и 3-10 эквивалентов ангидрида карбоновой кислоты. Реакцию можно проводить при температуре в интервале от 20°С до 200°С.
[Стадия D6]
На данной стадии NH группа в 1-положении соединения (6d) подвергается защите, что давало соединение (7d).
Защитные реагенты включают N,N-диметилсульфамоилхлорид, тритилхлорид, ди-трет-бутилдикарбонат и бензилбромид. Предпочтительно, использовать 1-1,5 эквивалента защитного реагента. Растворители для реакции включают дихлорметан, хлороформ, четыреххлористый углерод, толуол, N,N-диметилформамид и тетрагидрофуран. Основания включают пиридин, 4-диметиламинопиридин, 1,8-диазабицикло[5,4,0]ундецен, триэтиламин и N,N-диизопропилэтиламин. В типичных случаях, предпочтительно, использовать 1,2 эквивалента основания. Однако, когда защитным реагентом является ди-трет-бутилдикарбонат, предпочтительно используют 0,005-0,1 эквивалента 4-диметиламинопиридина. Реакцию можно проводить при температуре в интервале от 20°С до 200°С.
[Стадия D7]
На данной стадии соединение (7d) хлорируется, что давало соединение (8d).
Нет никаких особых ограничений по условиям реакции. Например, реакция осуществляется следующим образом. Соединение (7d) подвергают взаимодействию с основанием при температуре в интервале от -100°С до 20°С, а затем вводится в реакцию с ними хлорирующий средство. Данная реакция дает соединение (8d). Соединение (8d) может быть также получено при взаимодействии соединения (7d) с основанием в присутствии реагента хлорирования. Растворители для реакции включают, например, диэтиловый эфир, тетрагидрофуран 1,2-диметоксиэтан и 1,4-диоксан. Основания включают н-бутиллитий, трет-бутиллитий, диизопропиламид лития, бис(триметилсилил)амид лития и диизопропиламид магния. Предпочтительно, использовать 1-1,5 эквивалента основания. Хлорирующие реагенты включают гексахлорэтан и N-хлорсукцинимид. Предпочтительно, использовать 1-3 эквивалента хлорирующего реагента.
[Стадия D8]
На данной стадии соединение (8d) подвергают взаимодействию с соединением (9d), что давало соединение (10d). Реакцию можно проводить в тех же условиях, что используются на [Стадии А6] способа получения А.
[Стадия D9]
На данной стадии осуществляется реакция замещения с использованием соединения (10d) и соединения (10d-2), что давало соединение (11d). Реакцию можно проводить в тех же условиях, что используются на [Стадии А4] способа получения А.
[Стадия D10]
На данной стадии Rp3 соединения (11d) удаляется, что давало соединение (12d). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
[Стадия D11]
На данной стадии группа в 5 положении соединения (11d) получается деалкилированием, что давало соединение (13d). Нет никаких особых ограничений в отношении условий реакции деалкилирования. Например, такую реакцию можно проводить следующим образом:
когда R1 представляет собой бензилоксиметильную группу, соединение (11d) подвергают взаимодействию с 3-10 эквивалентами трибромида бора, трихлорида бора или аналогичного в растворителе, таком как дихлорметан, при температуре в интервале от -100°С до 20°С. Данная реакция дает соединение (13d).
Когда такая реакция приводит в результате к удалению группы Rp3, -NH- снова защищается с помощью реакции защиты. В частности, например, когда Rp3 представляет собой трет-бутоксикарбонильную группу, реакцию можно проводить с использованием такого реагента, как трет-бутилдикарбонат, в растворителе, таком как дихлорметан, хлороформ, N,N-диметилформамид или тетрагидрофуран, в присутствии основания, такого как пиридин, 4-аминопиридин, триэтиламин или N,N-диизопропилэтиламин, при температуре в интервале от 0°С до 80°С. Однако реакция не ограничивается указанным.
[Стадия D12]
На данной стадии соединение (13d) подвергают взаимодействию с соединением (13d-2), что давало соединение (14d). Реакцию можно проводить в тех же условиях, что используются на [Стадии D1] способа получения D.
[Стадия D13]
В данном случае, Rp3 соединения (14d) удаляется, что давало соединение (12d). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
Альтернативный способ получения соединения (11d) описывается ниже.
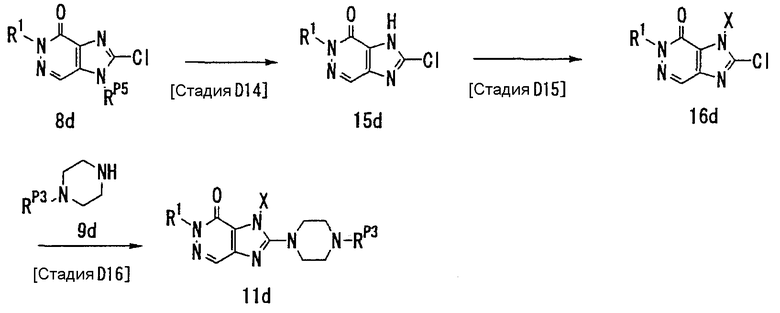
[Стадия D14]
На данной стадии соединение (8d) подвергают взаимодействию удаления защиты, что давало соединение (15d).
Удаление защиты может достигаться в стандартных условиях реакции в зависимости от типа защитной группы. Например, в случае трет-бутоксикарбонильной группы удаление защиты может достигаться осуществлением реакции с использованием такого основания, как гидроксид натрия, карбонат калия и аммиак, в тетрагидрофуране, N,N-диметилформамиде, метаноле, этаноле, воде или в смешанном растворителе из указанных при температуре в интервале от 0°С до 100°С. Когда после хлорирования на предыдущей стадии добавляются растворитель и основание, удаление защиты может достигаться без выделения соединения (8d).
[Стадия D15]
В данном случае X вводится в соединение (15d), что давало соединение (16d). Реакцию можно проводить с использованием X-U2 в тех же условиях, что используются на [Стадии А4] способа получения А.
Спирт (X-OH) может вводиться с использованием реакции Мицунобу. В частности, соединение (16d) может получаться при взаимодействии спирта (Х-ОН) с диалкиловым эфиром азодикарбоновой кислоты и трифенилфосфином в растворителе, таком как тетрагидрофуран, при температуре в интервале от -70°С до 50°С.
[Стадия D16]
На данной стадии соединение (16d) подвергают взаимодействию с соединением (9d), что давало соединение (11d). Реакцию можно проводить в тех же условиях, что используются на [Стадии А6] способа получения А.
Способ получения Е
Соединение (1е), представленное формулой:
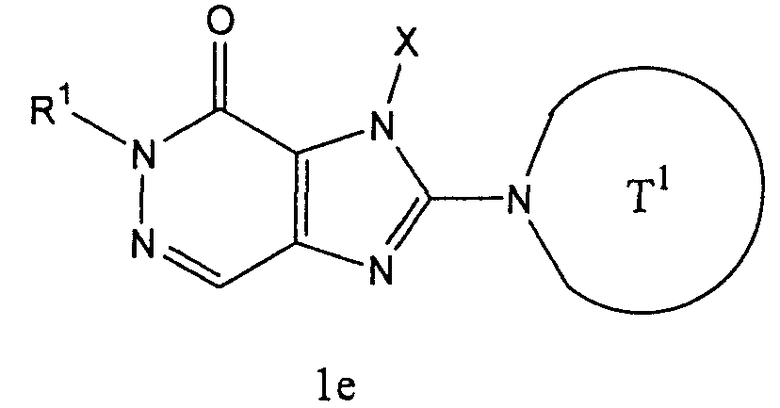
может получаться с использованием соединения (8b), представленного формулой Н-Т1а, вместо соединения (6с), используемого на [Стадии С5] или [Стадии С15] способа получения С, описанного выше, в тех же условиях, что используются на [Стадии С5], и затем соответствующим образом применением [Стадии С6]-[Стадии С21], описанных выше.
Соединение (1е), представленное формулой:
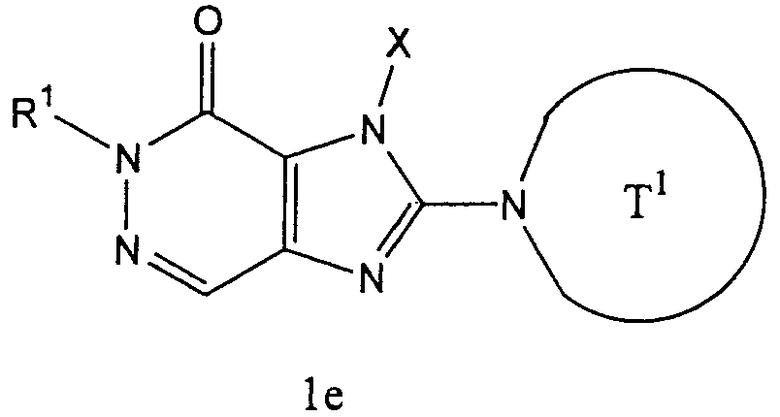
может получаться с использованием соединения (8b), представленного формулой Н-Т1а, вместо соединения (9d) на [Стадии D8] способа получения D, описанного выше, в тех же условиях, что используются на [Стадии D8], и затем соответствующим образом применением [Стадии D9]-[Стадии D13], описанных выше.
Способ получения F
[Стадия F1]
На данной стадии сложноэфирная группа соединения (1f) гидролизуется, что давало соединение (2f). Реакцию можно проводить в тех же условиях, что используются на [Стадии С16] способа получения С.
[Стадия F2]
На данной стадии Rp3 соединения (2f) удаляется, что давало соединение (3f). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения G
[Стадия G1]
На данной стадии нитрогруппа соединения (1g) восстанавливается, что давало соединение (2g).
Растворители для реакции включают метанол, этанол, тетрагидрофуран, воду или их смеси. Восстанавливающие средства включают железо, олово и цинк. Катализаторы включают соляную кислоту и аммониевую соль, такую как хлорид аммония. Реакцию можно проводить при температуре в интервале от 20°С до 120°С.
[Стадия G2]
На данной стадии Rp3 соединения (2g) удаляется, что давало соединение (3g). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения Н
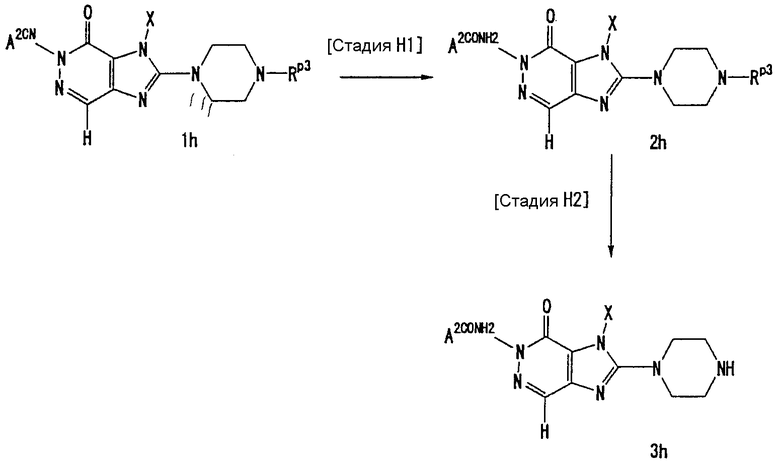
[Стадия Н1]
На данной стадии нитрильная группа соединения (1h) гидролизуется, что давало соединение (2h).
Нет никаких особых ограничений по условиям реакции. Например, реакция осуществляется следующим образом. Соединение (2h) может быть получено с помощью реакции соединения (1h) с перекисью водорода в присутствии основания при температуре в интервале от -20°С до 50°С. Растворители включают метанол, этанол, тетрагидрофуран, воду или их смесь. Основания включают аммиак и алкиламины, такие как триэтиламин.
[Стадия Н2]
На данной стадии Rp3 соединения (2h) удаляется, что давало соединение (3h). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения I
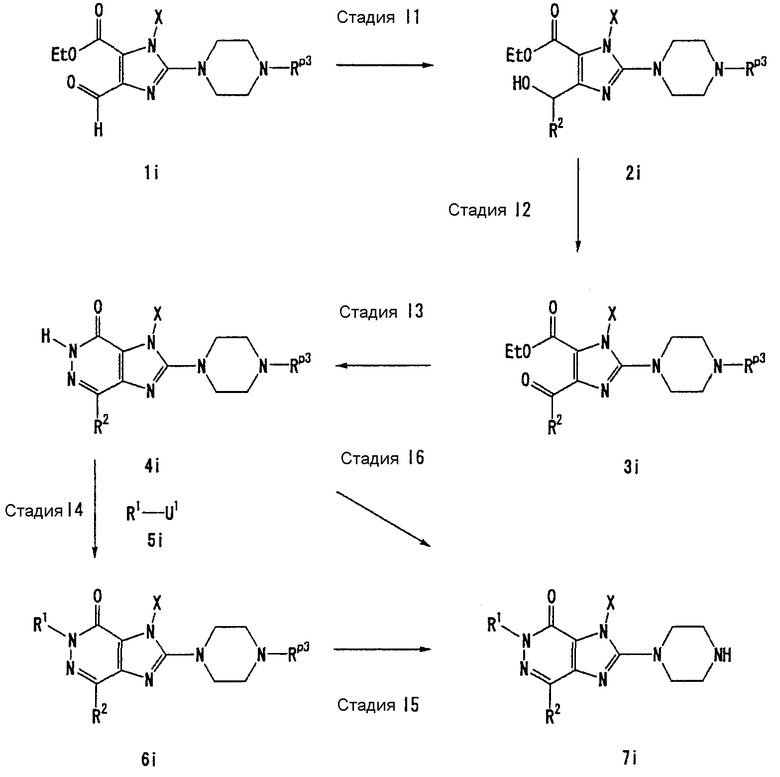
[Стадия I1]
На данной стадии соединение (1i) подвергают взаимодействию с алкилметаллическим средством или арилметаллическим средством, что давало соединение (2i).
Нет никаких особых ограничений по условиям реакции. Например, реакция осуществляется следующим образом. Соединение (1i) может подвергаться реакции со средством, таким как алкиллитий, ариллитий, алкильный реагент Гриньяра или арильный реагент Гриньяра, в растворителе, таком как диэтиловый эфир или тетрагидрофуран, при температуре в интервале от -100°С до 100°С. Альтернативно, данное соединение может подвергаться реакции с алкилцинком или арилцинком в растворителе, таком как N,N-диметилформамид или 1-метил-2-пирролидон, при температуре в интервале от 0°С до 50°С.
[Стадия I2]
На данной стадии соединение (2i) окисляется, что давало соединение (3i). В качестве окислителя может использоваться типичный реагент, который обычно используется при окислении спирта. В частности, например, в качестве окислителя может применяться двуокись марганца, в растворителе, таком как дихлорметан или хлороформ, при температуре в интервале от 20°С до 100°С. Альтернативно, в качестве окислителя может использоваться трехокись серы - пиридин в растворителе, таком как диметилсульфоксид, при температуре в интервале от 20°С до 100°С. Альтернативно, может использоваться перйодинан Десс-Мартина в растворителе, таком как дихлорметан или хлороформ, при температуре в интервале от -50°С до 50°С.
[Стадия I3]
На данной стадии соединение (3i) подвергают взаимодействию с гидразином, что давало соединение (4i). Реакцию можно проводить в тех же условиях, что используются на [Стадии С12] способа получения С.
[Стадия I4]
На данной стадии осуществляется реакция замещения с использованием соединения (4i) и соединения (5i), что давало соединение (6i). Реакцию можно проводить в тех же условиях, что используются на [Стадии А2] способа получения А.
[Стадия I5]
На данной стадии Rp3 соединения (6i) удаляется, что давало соединение (7i). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
[Стадия I6]
На данной стадии Rp3 соединения (4i) удаляется, что давало соединение (7i), когда R1 соединения (7i) представляет собой Н.
Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения J
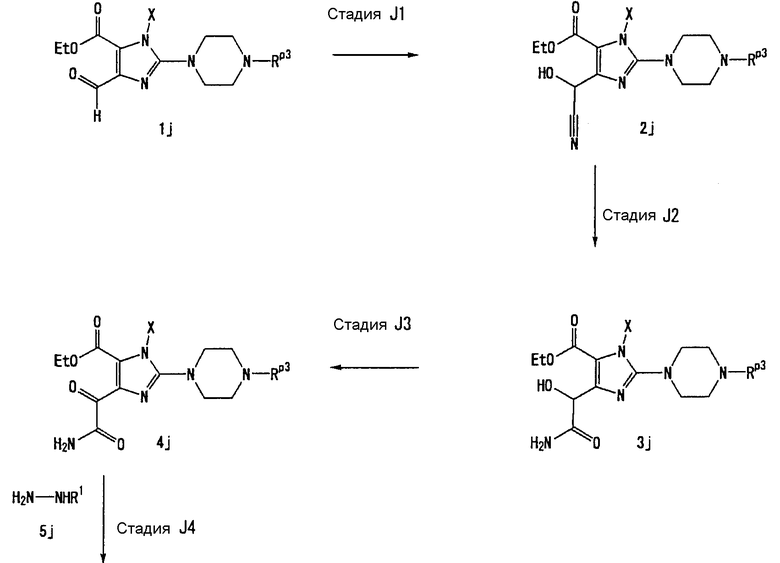
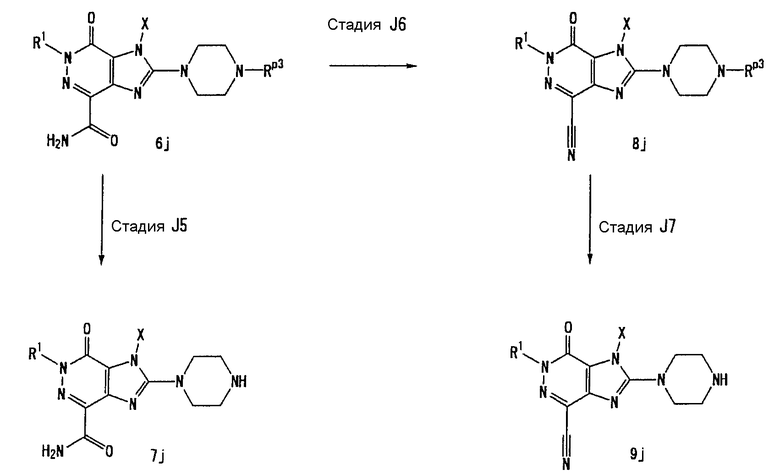
[Стадия J1]
На данной стадии соединение (1j) подвергают взаимодействию со средством цианидирования в присутствии катализатора, что давало соединение (2j).
Средства цианидирования включают цианид натрия и цианид калия. Катализаторы включают уксусную кислоту. Растворители включают, например, ацетонитрил. Реакцию можно проводить при температуре в интервале от 0°С до 100°С.
[Стадия J2]
На данной стадии нитрильная группа соединения (2j) гидролизуется, что давало соединение (3j). Реакцию можно проводить в тех же условиях, что используются на [Стадии Н1] способа получения Н.
[Стадия J3]
На данной стадии гидроксильная группа соединения (3j) окисляется, что давало соединение (4j). Реакцию можно проводить в тех же условиях, что используются на [Стадии I2] способа получения I.
[Стадия J4]
На данной стадии соединение (4j) подвергают взаимодействию с соединением (5j), что давало соединение (6j). Реакцию можно проводить в тех же условиях, что используются на [Стадии С11] способа получения С.
[Стадия J5]
На данной стадии Rp3 соединения (6j) удаляется, что давало соединение (7j). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
[Стадия J6]
На данной стадии карбамоильная группа соединения (6j) дегидратируется в присутствии основания, что давало соединение (8j).
Дегидратирующие средства включают, например, хлорокись фосфора. Основания включают алкиламины, такие как триэтиламин. Растворители включают дихлорметан и хлороформ. Альтернативно, реакцию можно проводить в отсутствии растворителя. Реакцию можно проводить при температуре в интервале от 0°С до 100°С.
[Стадия J7]
На данной стадии Rp3 соединения (8j) удаляется, что давало соединение (9j). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения К
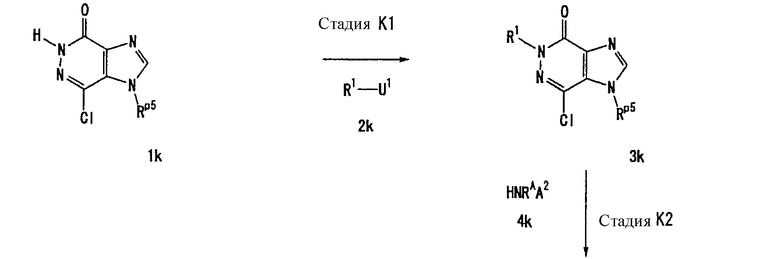
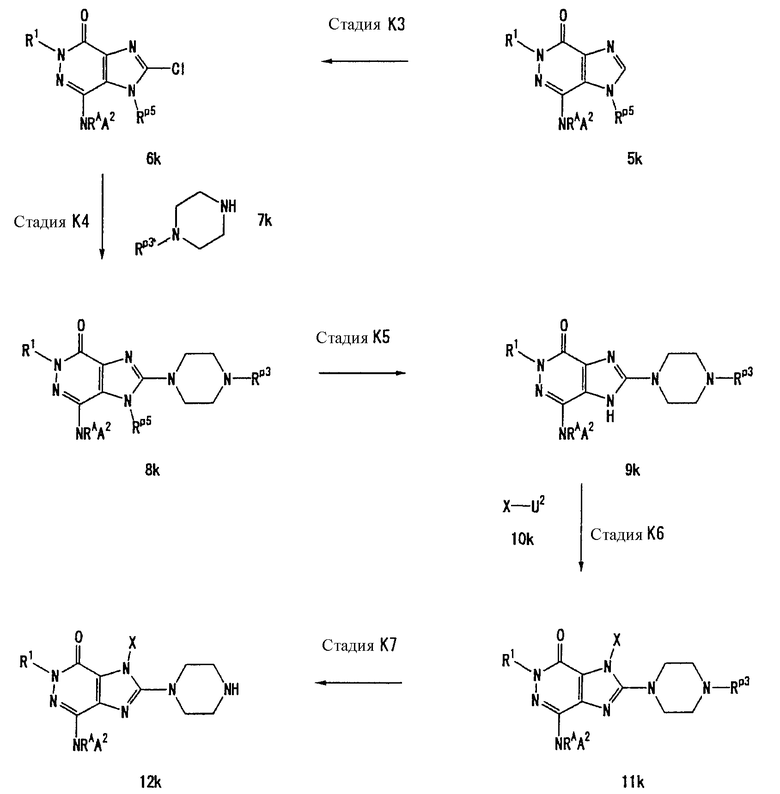
[Стадия К1]
На данной стадии осуществляется реакция замещения с использованием соединения (1k) и соединения (2k), что давало соединение (3k). Реакцию можно проводить в тех же условиях, что используются на [Стадии А2] способа получения А.
[Стадия К2]
На данной стадии осуществляется реакция замещения с использованием соединения (3k) и соединения (4k), что давало соединение (5k).
Соединение (5k) может получаться, например, с помощью реакции смеси соединений (3k) и (4k) в растворителе, таком как метанол, этанол, 1-метил-2-пирролидон, 1,4-диоксан, тетрагидрофуран или диметоксиэтан, или в отсутствии растворителя, при температуре в интервале от 20°С до 200°С. Однако условия реакции не ограничиваются указанными.
[Стадия К3]
На данной стадии соединение (5k) хлорируется, что давало соединение (6k). Реакцию можно проводить в тех же условиях, что используются на [Стадии D7] способа получения D.
[Стадия К4]
На данной стадии соединение (6k) подвергают взаимодействию с соединением (7k), что давало соединение (8k). Реакцию можно проводить в тех же условиях, что используются на [Стадии А6] способа получения А.
[Стадия К5]
На данной стадии Rp5 соединения (8k) удаляется, что давало соединение (9k).
Реакция удаления защиты для Rp5 может проводиться в стандартных условиях удаления -NH-защитной группы.
Например, когда Rp5 представляет собой бензильную группу, реакцию можно проводить с использованием металла, такого как литий или натрий, в жидком аммиаке при температуре в интервале от -78°С до -30°С.
[Стадия К6]
На данной стадии осуществляется реакция замещения с использованием соединения (9k) и соединения (10k), что давало соединение (11k). Реакцию можно проводить в тех же условиях, что используются на [Стадии А4] способа получения А.
[Стадия К7]
На данной стадии Rp3 соединения (11k) удаляется, что давало соединение (12k). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения L
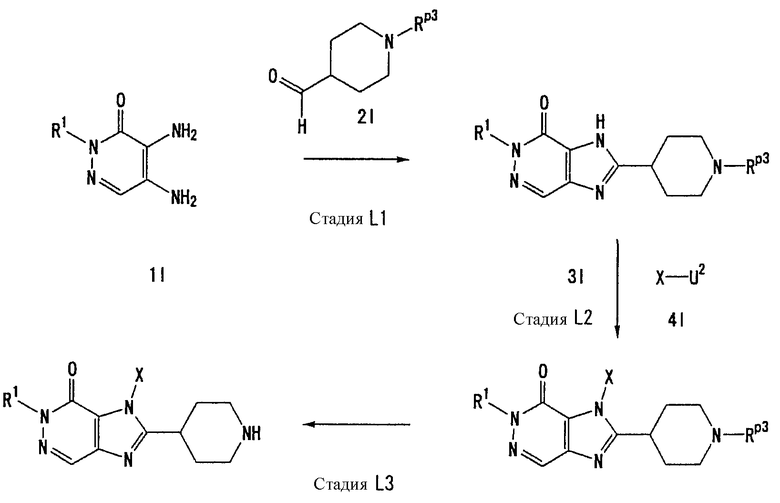
[Стадия L1]
На данной стадии соединение (1l) подвергают взаимодействию с соединением (2l) в присутствии окислителя, что давало соединение (3l). Окислители включают соли, такие как хлорид железа (III). Растворители включают метанол, этанол и воду. Реакцию можно проводить при температуре в интервале от 20°С до 100°С.
Когда такая реакция приводит в результате к удалению группы -Rр3, -NH-группу снова защищают с помощью реакции защиты. В частности, например, когда Rр3 представляет собой трет-бутоксикарбонильную группу, реакцию можно проводить с использованием такого реагента, как ди-трет-бутилдикарбонат, в растворителе, таком как дихлорметан, хлороформ, N,N-диметилформамид или тетрагидрофуран, в присутствии основания, такого как пиридин, 4-аминопиридин, триэтиламин или N,N-диизопропилэтиламин, при температуре в интервале от 0°С до 80°С. Однако реакция не ограничивается указанным.
[Стадия L2]
На данной стадии соединение (3l) подвергают взаимодействию с соединением (4l), что давало соединение (5l). Реакцию можно проводить в тех же условиях, что используются на [Стадии А4] способа получения А.
[Стадия L3]
На данной стадии Rp3 соединения (5l) удаляется, что давало соединение (6l). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения M
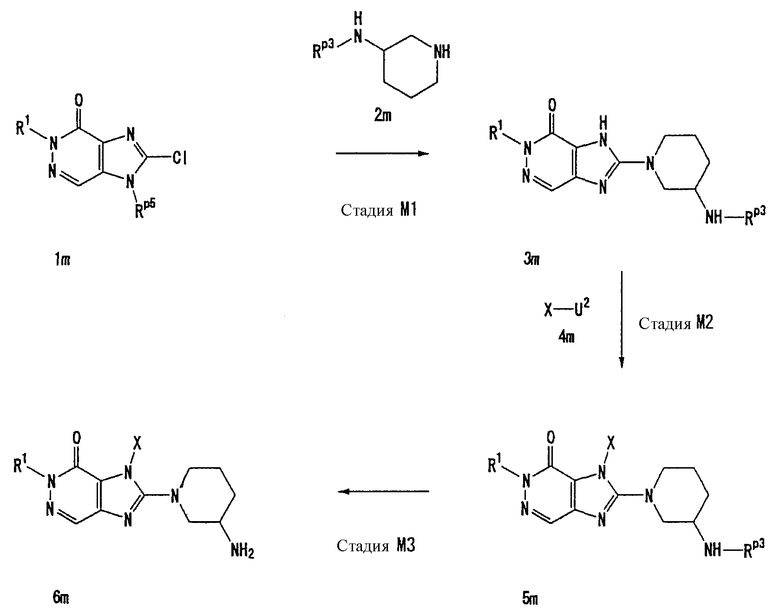
[Стадия M1]
На данной стадии соединение (1m) подвергают взаимодействию с соединением (2m), что давало соединение (3m). Реакцию можно проводить в тех же условиях, что используются на [Стадии А6] способа получения А.
[Стадия M2]
На данной стадии соединение (3m) подвергают взаимодействию с соединением (4m), что давало соединение (5m). Реакцию можно проводить в тех же условиях, что используются на [Стадии А4] способа получения А.
[Стадия M3]
На данной стадии Rp3 соединения (5m) удаляется, что давало соединение (6m). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения N
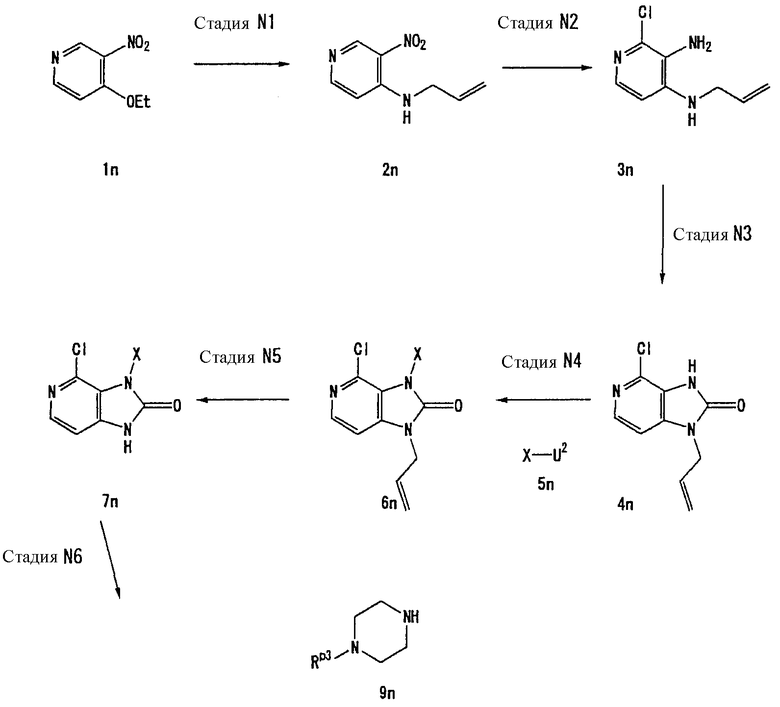
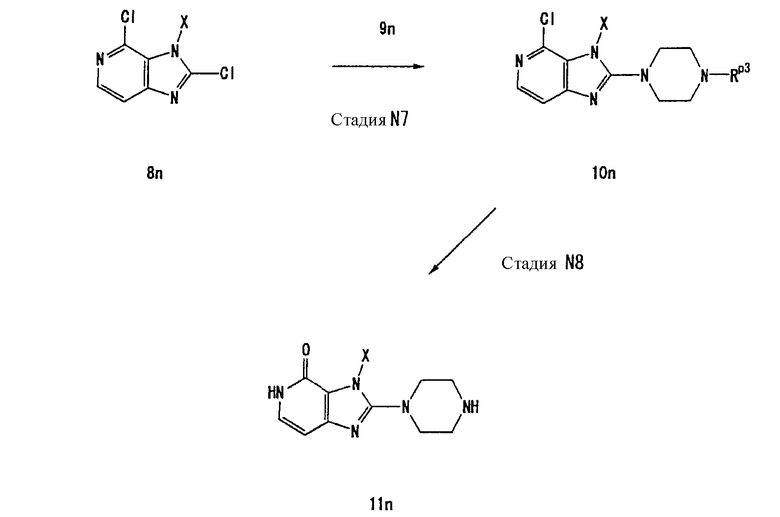
[Стадия N1]
На данной стадии соединение (1n) подвергают взаимодействию с аллиламином, что давало соединение (2n). Реакцию можно проводить при температуре в интервале от 20°С до 150°С. Растворители для реакции включают метанол, этанол, воду и смешанные растворители из указанных.
[Стадия N2]
На данной стадии соединение (2n) восстанавливается, при этом хлорируясь, что давало соединение (3n). Восстанавливающие средства включают соли олова, такие как хлорид олова. Растворители включают концентрированную соляную кислоту. Реакцию можно проводить при температуре в интервале от 20°С до 150°С.
[Стадия N3]
На данной стадии соединение (3n) подвергают взаимодействию с N,N'-дисукцинимидилкарбонатом, что давало соединение (4n).
Реакцию можно проводить с использованием растворителя, такого как ацетонитрил или тетрагидрофуран. Реакцию можно проводить при температуре в интервале от 20°С до 100°С.
[Стадия N4]
На данной стадии соединение (4n) подвергают взаимодействию с соединением (5n), что давало соединение (6n).
Реакцию можно проводить в тех же условиях, что используются на [Стадии А4] способа получения А.
[Стадия N5]
На данной стадии аллильная группа удаляется из соединения (6n), что давало соединение (7n).
Соединение (7n) может получаться, например, с помощью реакции соединения (6n) с осмиевой кислотой и перйодатом натрия в растворителе, таком как тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан или вода, при температуре в интервале от 20°С до 100°С. Однако условия реакции не ограничиваются данным примером.
[Стадия N6]
На данной стадии соединение (7n) хлорируется, что давало соединение (8n).
Нет никаких особых ограничений в отношении условий реакции. Реакцию можно проводить в стандартных реакционных условиях, используемых для хлорирования. Соединение (8n) может быть получено, например, с использованием такого реагента, как пентахлорид фосфора, в растворителе, таком как хлорокись фосфора, при температуре 0°С-150°С.
[Стадия N7]
На данной стадии соединение (8n) подвергают взаимодействию с соединением (9n), что давало соединение (10n). Реакцию можно проводить в тех же условиях, что используются на [Стадии А6] способа получения А.
[Стадия N8]
На данной стадии группа Rp3 соединения (10n) удаляется, что давало соединение (11n). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения О
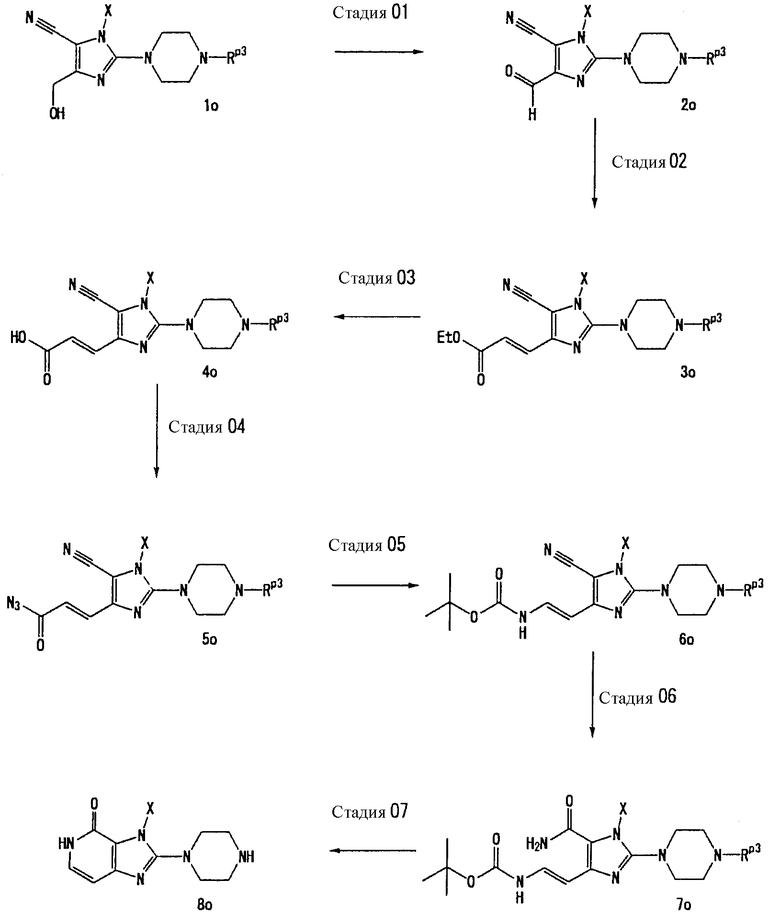
[Стадия О1]
На данной стадии гидроксильная группа соединения (1о) окисляется, что давало соединение (2о). Реакцию можно проводить в тех же условиях, что используются на [Стадии I2] способа получения I.
[Стадия О2]
На данной стадии соединение (2о) подвергают взаимодействию с этилдиэтилфосфоноацетатом в присутствии основания, что давало соединение (3о).
Основания включают гидрид натрия и диизопропиламид лития. Растворители включают, например, тетрагидрофуран и N,N-диформамид. Реакцию можно проводить при температуре в интервале от 0°С до 100°С.
[Стадия О3]
На данной стадии сложный эфир соединения (3о) гидролизуется, что давало соединение (4о). Реакцию можно проводить в тех же условиях, что используются на [Стадии С16] способа получения С.
[Стадия О4]
На данной стадии соединение (4о) подвергают взаимодействию с дифенилфосфорилазидом в присутствии основания, что давало соединение (5о).
Растворители для реакции включают толуол, трет-бутанол, тетрагидрофуран и дихлорметан. Основания включают третичные амины, такие как триэтиламин и диизопропилэтиламин. Реакцию можно проводить при температуре в интервале от -50°С до 50°С.
[Стадия О5]
На данной стадии соединение (5о) подвергается перегруппировке, что давало соединение (6о).
Реакцию можно проводить в трет-бутаноле при температуре в интервале от 50°С до 100°С.
[Стадия О6]
На данной стадии нитрильная группа соединения (6о) гидролизуется, что давало соединение (7о). Реакцию можно проводить в тех же условиях, что используются на [Стадии Н1] способа получения Н.
[Стадия О7]
На данной стадии соединение (7о) подвергают взаимодействию с кислотой, что давало соединение (8о).
Кислоты включают соляную кислоту, серную кислоту и трифторуксусную кислоту. Растворители включают метанол, этанол, 1,4-диоксан, воду и их смеси. Реакцию можно проводить при температуре в интервале от 0°С до 50°С.
Способ получения Р
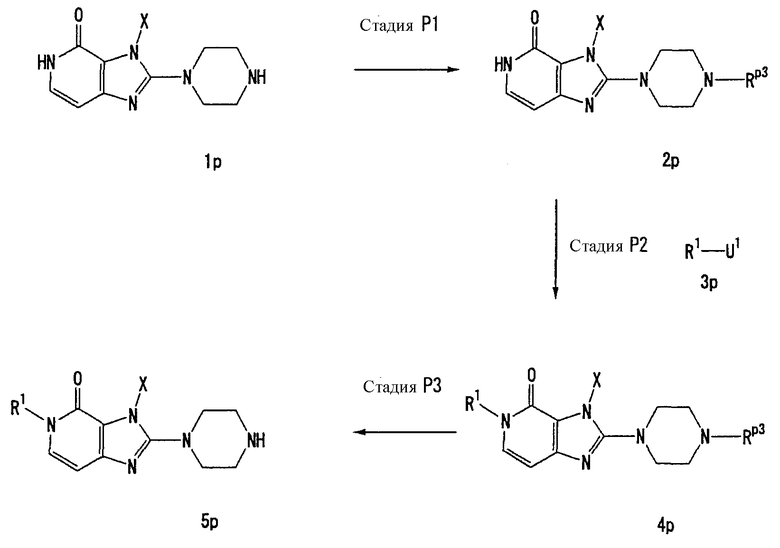
[Стадия Р1]
На данной стадии соединение (1р) подвергается защите, что давало соединение (2р).
В качестве реагента, защищающего NH группу, может использоваться типичный реагент, защищающий NH группу, который обычно используется при защите NH груп. Например, когда Rp3 представляет собой трет-бутоксикарбонильную группу, реакцию можно проводить при температуре в интервале от 0°С до 80°С с использованием такого реагента, как ди-трет-бутилдикарбонат, в растворителе, таком как дихлорметан, хлороформ, N,N-диметилформамид и тетрагидрофуран, в присутствии основания, такого как пиридин, 4-аминопиридин, триэтиламин и N,N-диизопропилэтиламин.
[Стадия Р2]
На данной стадии соединение (2р) подвергают взаимодействию с соединением (3р), что давало соединение (4р). Реакцию можно проводить в тех же условиях, что используются на [Стадии А2] способа получения А.
[Стадия Р3]
На данной стадии группа Rp3 соединения (4р) удаляется, что давало соединение (5р). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения Q
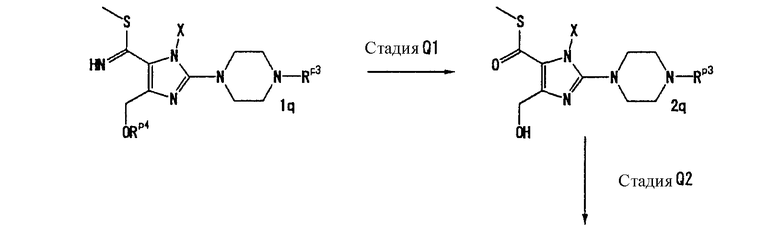
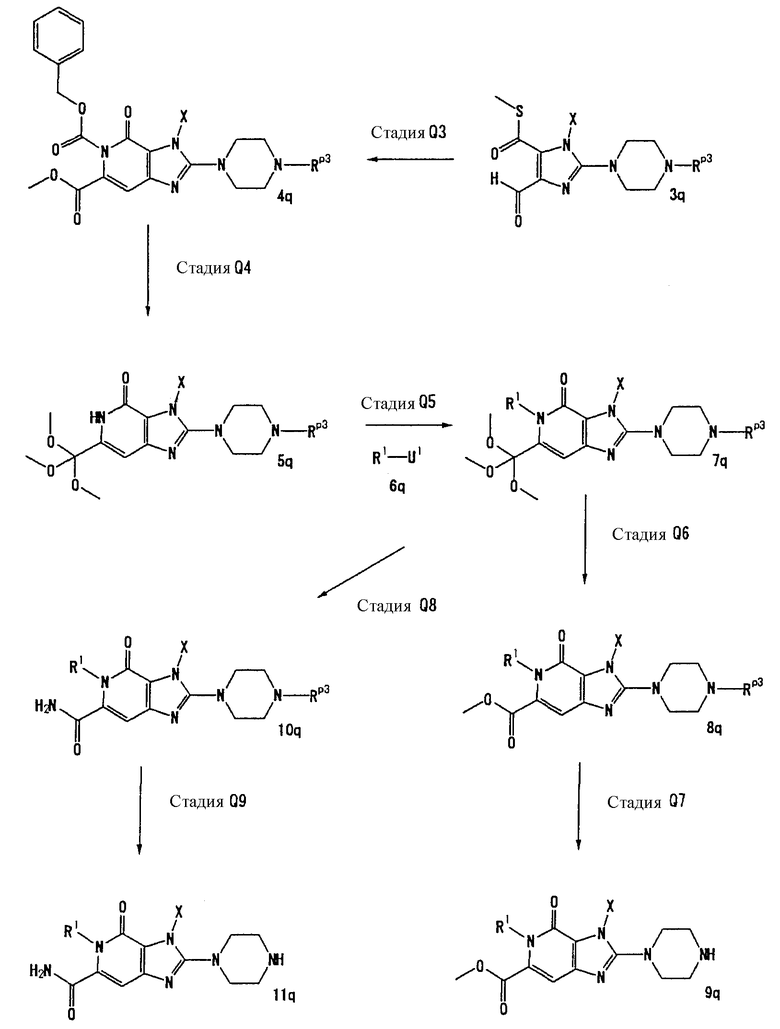
[Стадия Q1]
На данной стадии соединение (1q) гидролизуется, что давало соединение (2q).
Реакционные растворители включают тетрагидрофуран, метанол и этанол. Кислоты включают неорганические кислоты, такие как соляная кислота и серная кислота. Реакцию можно проводить при температуре в интервале от 0°С до 100°С.
[Стадия Q2]
На данной стадии гидроксильная группа соединения (2q) окисляется, что давало соединение (3q). Реакцию можно проводить в тех же условиях, что используются на [Стадии I2] способа получения I.
[Стадия Q3]
На данной стадии соединение (3q) подвергают взаимодействию с метилбензилоксикарбониламино(диметоксифосфорил)ацетатом в присутствии основания, что давало соединение (4q).
Основания включают гидрид натрия, трет-бутоксид калия и 8-диазабицикло[5,4,0]-7-ундецен. Растворители включают дихлорметан, тетрагидрофуран и N,N-диметилформамид. Реакцию можно проводить при температуре в интервале от 0°С до 100°С.
[Стадия Q4]
На данной стадии соединение (4q) подвергают взаимодействию с метоксидом натрия, что давало соединение (5q).
В качестве растворителя может использоваться метанол. Реакцию можно проводить при температуре в интервале от 0°С до 80°С.
[Стадия Q5]
На данной стадии соединение (5q) подвергают взаимодействию с соединением (6q), что давало соединение (7q). Реакцию можно проводить в тех же условиях, что используются на [Стадии А2] способа получения А.
[Стадия Q6]
На данной стадии соединение (7q) подвергают взаимодействию с кислотой, что давало соединение (8q). Реакцию можно проводить в тех же условиях, что используются на [Стадии О7] способа получения О.
[Стадия Q7]
На данной стадии группа Rp3 соединения (8q) удаляется, что давало соединение (9q). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения A.
[Стадия Q8]
На данной стадии соединение (7q) подвергают взаимодействию с аммиаком, что давало соединение (10q).
Реакционные растворители включают метанол, этанол и воду. Реакцию можно проводить при температуре в интервале от 20°С до 150°С.
[Стадия Q9]
На данной стадии группа Rp3 соединения (10q) удаляется, что давало соединение (11q). Реакцию можно проводить в тех же условиях, что используются на [Стадии А13] способа получения A.
Способ получения R
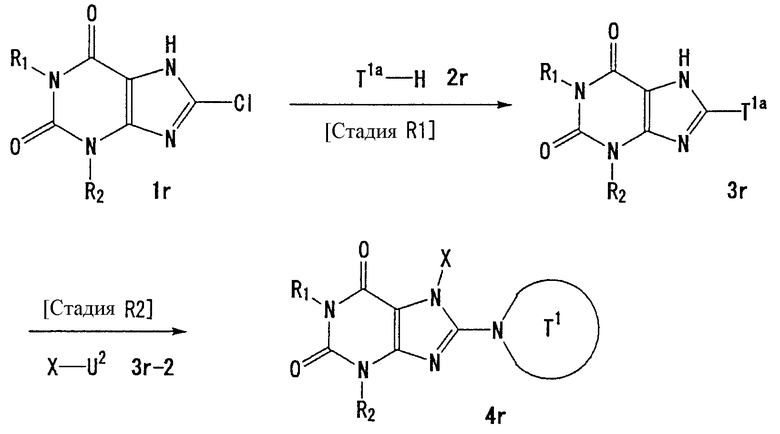
[Стадия R1]
На данной стадии соединение (1r) подвергают взаимодействию с соединением (2r), что давало соединение (3r).
Реакцию можно проводить в тех же условиях, что используются на [Стадии А6] способа получения А.
[Стадия R2]
На данной стадии вводится заместитель в аминогруппу в положении 7 соединения (3r) с помощью реакции замещения между соединением (3r) и соединением (3r-2), и Rp3 затем удаляется, что давало соединение (4r).
Реакция замещения проводится в тех же условиях, что используются на [Стадии А4] способа получения А.
Реакция удаления защиты для Rp3 осуществляется в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения S
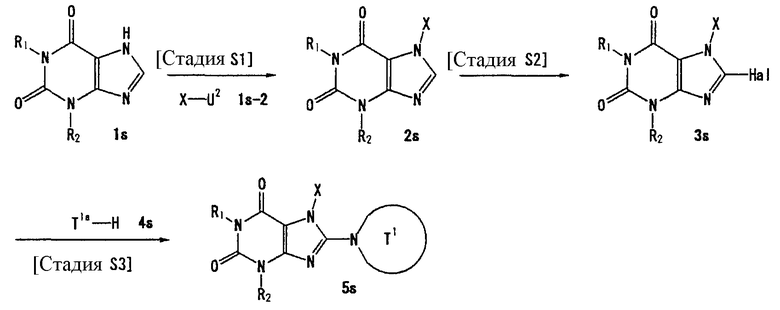
[Стадия S1]
На данной стадии вводится заместитель в аминогруппу в положении 7 соединения (1s) с помощью реакции замещения между соединением (1s) и соединением (1s-2), что давало соединение (2s).
Реакция замещения проводится в тех же условиях, что используются на [Стадии А4] способа получения А.
[Стадия S2]
На данной стадии соединение (2s) подвергают взаимодействию с галогенирующим средством, что давало соединение (3s).
Реакция галогенирования проводится в тех же условиях, что используются на [Стадии А5] способа получения А.
[Стадия S3]
На данной стадии соединение (3s) подвергают взаимодействию с соединением (4s), и Rp3 затем удаляется, что давало соединение (5s).
Реакция сочетания проводится в тех же условиях, что используются на [Стадии А6] способа получения А.
Реакция удаления защиты для Rp3 может осуществляться в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения Т
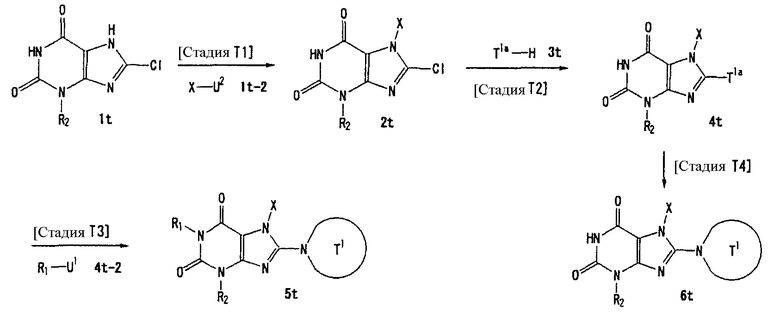
[Стадия Т1]
На данной стадии вводится заместитель в аминогруппу в положении 7 соединения (1t) с помощью реакции замещения между соединением (1t) и соединением (1t-2), что давало соединение (2t).
Реакция замещения проводится в тех же условиях, что используются на [Стадии А4] способа получения А.
[Стадия T2]
На данной стадии соединение (2t) подвергают взаимодействию с соединением (3t), что давало соединение (4t).
Реакция проводится в тех же условиях, что используются на [Стадии А6] способа получения А.
[Стадия T3]
На данной стадии соединение (4t) алкилируется в 1 положении, и Rp3 затем удаляется, что давало соединение (5t).
Реакция алкилирования проводится в тех же условиях, что используются на [Стадии А2] способа получения А.
Реакция удаления защиты для Rp3 проводится в тех же условиях, что используются на [Стадии А13] способа получения А.
[Стадия T4]
На данной стадии Rp3 удаляется из соединения (4t), что давало соединение (6t).
Реакция удаления защиты для Rp3 может осуществляться в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения U
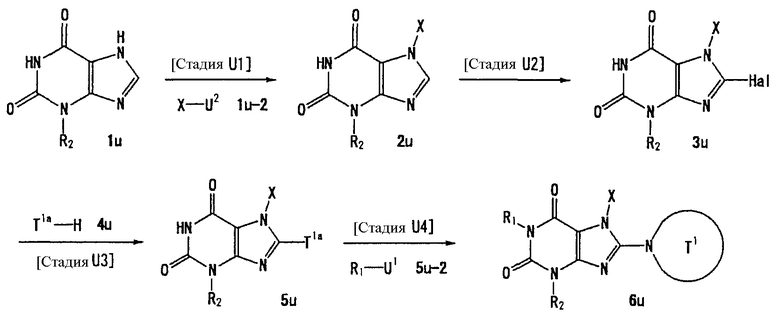
[Стадия U1]
На данной стадии вводится заместитель в аминогруппу в 7 положении соединения (1u) с помощью реакции замещения между соединением (1u) и соединением (1u-2), что давало соединение (2u).
Реакция замещения проводится в тех же условиях, что используются на [Стадии А4] способа получения А.
[Стадия U2]
На данной стадии соединение (2u) подвергают взаимодействию с галогенирующим средством, что давало соединение (3u).
Реакция галогенирования проводится в тех же условиях, что используются на [Стадии А5] способа получения А.
[Стадия U3]
На данной стадии соединение (3u) подвергают взаимодействию с соединением (4u), что давало соединение (5u).
Реакция проводится в тех же условиях, что используются на [Стадии А6] способа получения А.
[Стадия U4]
На данной стадии соединение (5u) алкилируется в 1 положении, и затем Rp3 удаляется, что давало соединение (6u).
Реакция алкилирования проводится в тех же условиях, что используются на [Стадии А2] способа получения А.
Реакция удаления защиты для Rp3 проводится в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения V
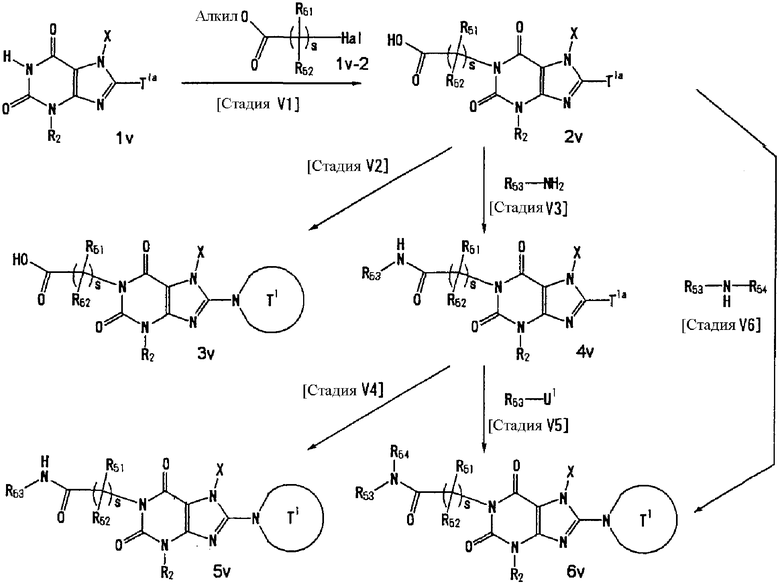
(где каждый символ является таким, как определен выше; и "Алкил" представляет собой С1-6 алкильную группу.)
[Стадия V1]
На данной стадии соединение (1v) алкилируется в 1 положении и гидролизуется, что давало соединение (2v).
Нет никаких особых ограничений в отношении условий реакции алкилирования. Например, алкилированное соединение может быть получено с помощью инкубирования соединения, представленного формулой (1v-2), такого как метилбромацетат или этилбромацетат; в присутствии основания, такого как гидроокись лития, гидроокись натрия, гидроокись калия, карбонат лития, карбонат натрия, карбонат калия, карбонат цезия, гидрид лития, гидрид натрия, гидрид калия, бутиллитий, метиллитий, бис-триметилсилиламид лития, бис-триметилсилиламид натрия или бис-триметилсилиламид калия; в растворителе, таком как диметилсульфоксид, N,N-диметилформамид, N-метилпирролидон, диоксан, тетрагидрофуран или толуол; при температуре в интервале от 0°С до 150°С.
Нет никаких особых ограничений в отношении условий реакции гидролиза. Например, реакцию можно проводить с использованием водного раствора гидроксида лития, гидроксида натрия или гидроксида калия; в растворителе, таком как метанол, этанол, пропанол, диметилсульфоксид, N,N-диметилформамид, N-метилпирролидон, диоксан или тетрагидрофуран; при температуре в интервале от 0°С до 150°С.
[Стадия V2]
На данной стадии группа Rp3 удаляется из соединения (2v), что давало соединение (3v). Реакция удаления защиты для Rp3 проводится в тех же условиях, что используются на [Стадии А13] способа получения A.
[Стадия V3]
На данной стадии соединение (2v) амидируется, что давало соединение (4v).
Нет никаких особых ограничений в отношении условий реакции амидирования. Например, реакцию можно проводить с использованием ацилирующего средства, такого как этилхлорформиат или изобутилхлорформиат; в присутствии органического основания, такого как триэтиламин или N,N-диизопропилэтиламин; в растворителе, таком как ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран или диметоксиэтан; с соответствующим амином при температуре в интервале от 0°С до 150°С.
[Стадия V4]
На данной стадии группа Rp3 удаляется из соединения (4v), что давало соединение (5v).
Реакция удаления защиты для Rp3 проводится в тех же условиях, что используются на [Стадии А13] способа получения A.
[Стадия V5]
На данной стадии соединение (5v) алкилируется, а затем Rp3 удаляется, что давало соединение (6v).
Реакция алкилирования проводится в тех же условиях, что используются на [Стадии А2] способа получения А.
Реакция удаления защиты для Rp3 проводится в тех же условиях, что используются на [Стадии А13] способа получени A.
[Стадия V6]
На данной стадии соединение (2v) амидируется, а затем Rp3 удаляется, что давало соединение (6v).
Нет никаких особых ограничений в отношении условий реакции амидирования. Например, амидирование может проводиться с использованием конденсирующего средства, такого как 1,1'-карбонилдиимидазол или диэтилцианфосфонат; в растворителе, таком как диметилсульфоксид, N,N-диметилформамид или тетрагидрофуран. Если требуется, можно добавлять в реакцию органическое основание, такое как триэтиламин. Реакцию можно проводить при температуре в интервале примерно от температуры охлаждения льдом до комнатной температуры.
Реакция удаления защиты для Rp3 проводится в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения W
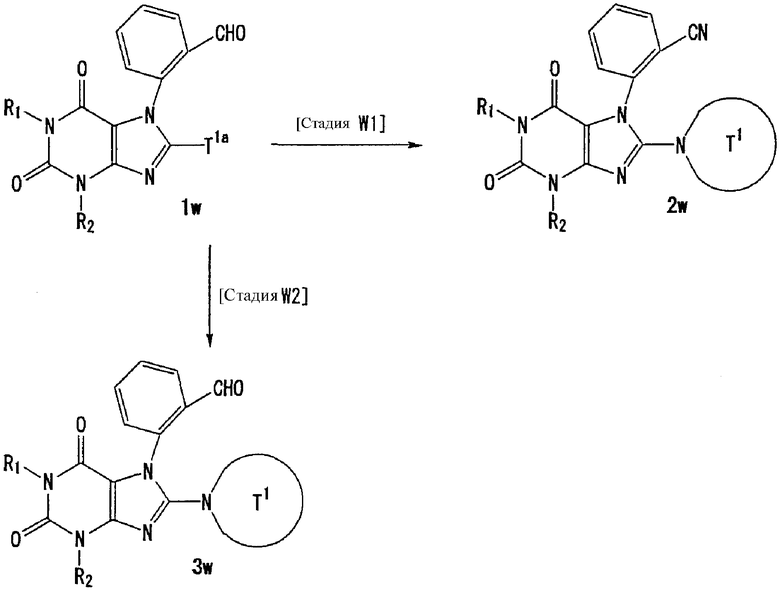
[Стадия W1]
На данной стадии соединение (1w) гидрокси-иминируется и полученная гидроксильная группа обрабатывается с помощью сульфонилирования с последующим удалением Rp3, что давало соединение (2w).
Нет никаких особых ограничений в отношении условий реакции гидрокси-иминирования. Например, реакция гидрокси-иминирования может осуществляться с использованием такого реагента, как гидроксиламингидрохлорид; в присутствии основания, такого как ацетат калия или ацетат натрия; в растворителе, таком как вода, метанол, этанол, пропанол, диметилсульфоксид, N,N-диметилформамид, N-метилпирролидон, диоксан, тетрагидрофуран или толуол.
Нет никаких особых ограничений в отношении условий реакции сульфонилирования. Например, реакция сульфонилирования может проводиться с использованием метансульфонилхлорида, тозилхлорида, 4-нитробензолсульфонилхлорида или аналогичного; в присутствии основания, такого как триэтиламин, диизопропилэтиламин, пиридин или N,N-диметиламинопиридин; в растворителе, таком как дихлорметан, хлороформ, диоксан, тетрагидрофуран, толуол или пиридин; при температуре в интервале от 0°С до 150°С.
Реакция удаления защиты для Rp3 проводится в тех же условиях, что используются на [Стадии А13] способа получения А.
[Стадия W2]
На данной стадии Rp3 удаляется из соединения (1w), что давало соединение (3w).
Реакция удаления защиты для Rp3 проводится в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения Х
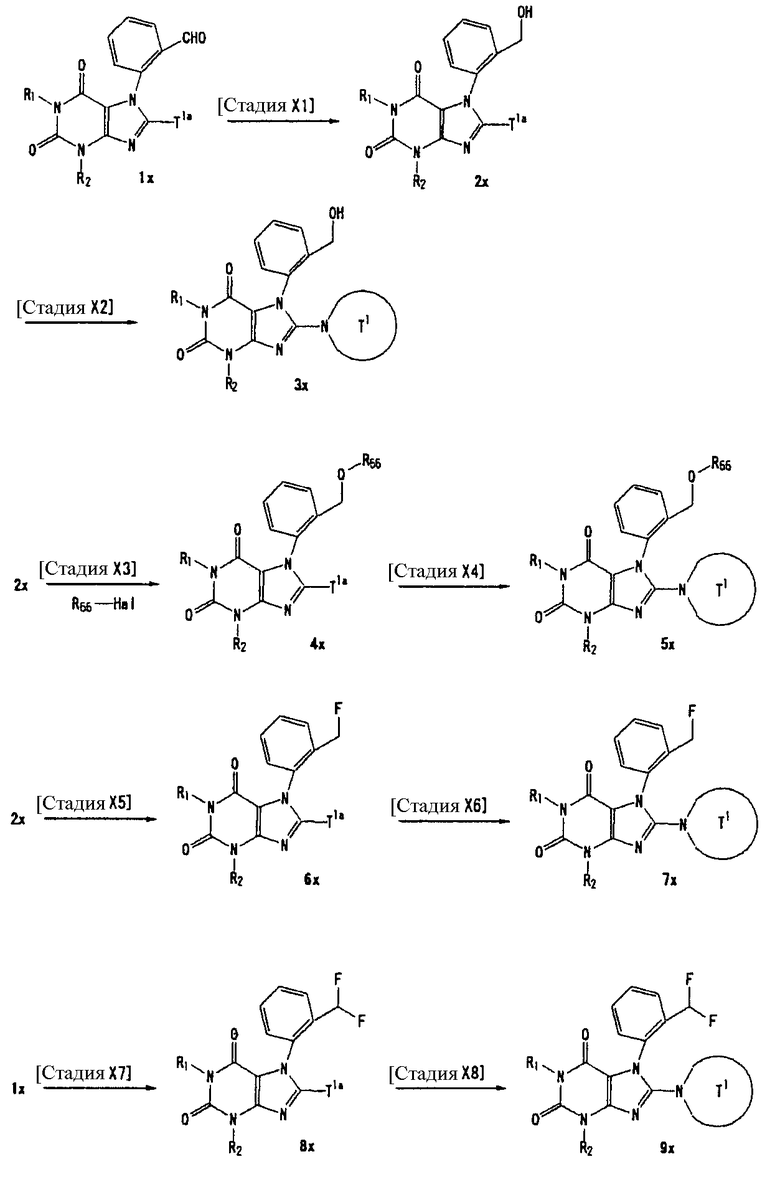
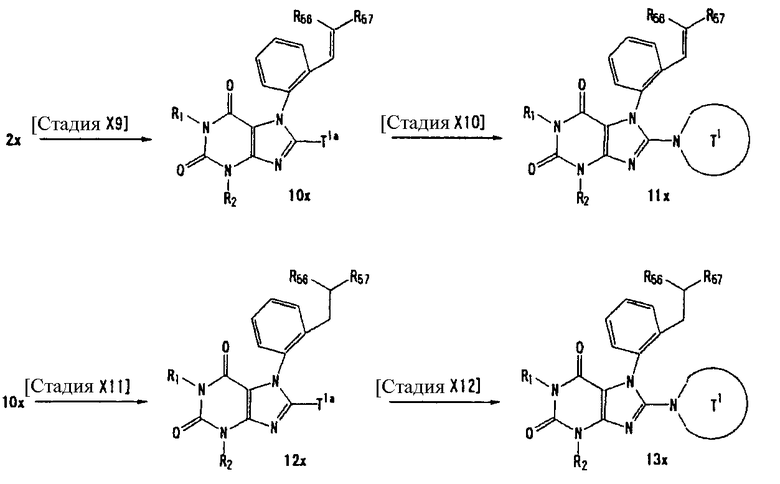
[Стадия Х1]
На данной стадии соединение (1х) восстанавливается, что давало соединение (2х).
Нет никаких особых ограничений в отношении условий реакции. Например, реакцию можно проводить с использованием восстанавливающего средства, такого как боргидрид лития, боргидрид натрия или боргидрид калия; в растворителе, таком как метанол, этанол, ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран, диметоксиэтан, или в смешанном равстворе данных растворителей; при температуре в интервале от 0°С до 150°С.
[Стадия Х3]
На данной стадии соединение (2х) алкилируется, что давало соединение (4х).
Нет никаких особых ограничений в отношении условий реакции алкилирования. Например, реакция алкилирования может осуществляться с использованием галогенированного алкила; в присутствии основания, такого как гидрид лития, гидрид натрия, гидрид калия, гидроокись лития, гидроокись натрия или гидроокись калия; в растворителе, таком как метанол, этанол, ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран или диметоксиэтан.
[Стадия Х5]
На данной стадии соединение (2х) фторируется, что давало соединение (6х).
Нет никаких особых ограничений в отношении условий реакции. Например, реакцию можно проводить с использованием фторирующего средства, такого как трифторид трис-диметиламиносульфата; в растворителе, таком как дихлорметан, 1,2-дихлорэтан, ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран или диметоксиэтан; при температуре в интервале от -78°С до 150°С.
[Стадия Х7]
На данной стадии соединение (1х) фторируется, что давало соединение (8х).
Нет никаких особых ограничений в отношении условий реакции. Например, реакцию можно проводить с использованием фторирующего средства, такого как трифторид трис-диметиламиносульфата; в растворителе, таком как дихлорметан, 1,2-дихлорэтан, ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран или диметоксиэтан; при температуре в интервале от -78°С до 150°С.
[Стадия Х9]
На данной стадии соединение (2х) подвергают взаимодействию Виттиг-Хорнер-Эммонс'а, что давало соединение (10х).
Нет никаких особых ограничений по условиям реакции. Например, реакцию можно проводить с использованием реагента, такого как фосфониевая соль или фосфонатный эфир; в присутствии основания, такого как гидрид лития, гидрид натрия, гидрид калия, трет-бутоксид калия или бутиллитий; в растворителе, таком как дихлорметан, 1,2-дихлорэтан, ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран или диметоксиэтан; при температуре в интервале от -78°С до 150°С.
[Стадия Х11]
На данной стадии соединение (10х) восстанавливается, что давало соединение (12х).
Нет никаких особых ограничений по условиям реакции восстановления. Например, реакцию можно проводить в присутствии катализатора на основе металла, такого как палладий на угле, окись платины или никель Ренея; в растворителе, таком как метанол, этанол, пропанол, диметилсульфоксид, N,N-диметилформамид, N-метилпирролидон, диоксан, тетрагидрофуран или толуол; в атмосфере водорода при температуре в интервале от 0°С до 150°С.
[Стадия Х2], [Стадия Х4], [Стадия Х6], [Стадия Х8], [Стадия Х10] и [Стадия Х12]
Rp3 удаляется из соединений (2х), (4х), (6х), (8х), (10х) и (12х), что давало соответственно соединения (3х), (5х), (7х), (9х), (11х) и (13х).
Реакция удаления защиты для Rp3 проводится в тех же условиях, что используются на [Стадии А13] способа получения А.
Способ получения Y
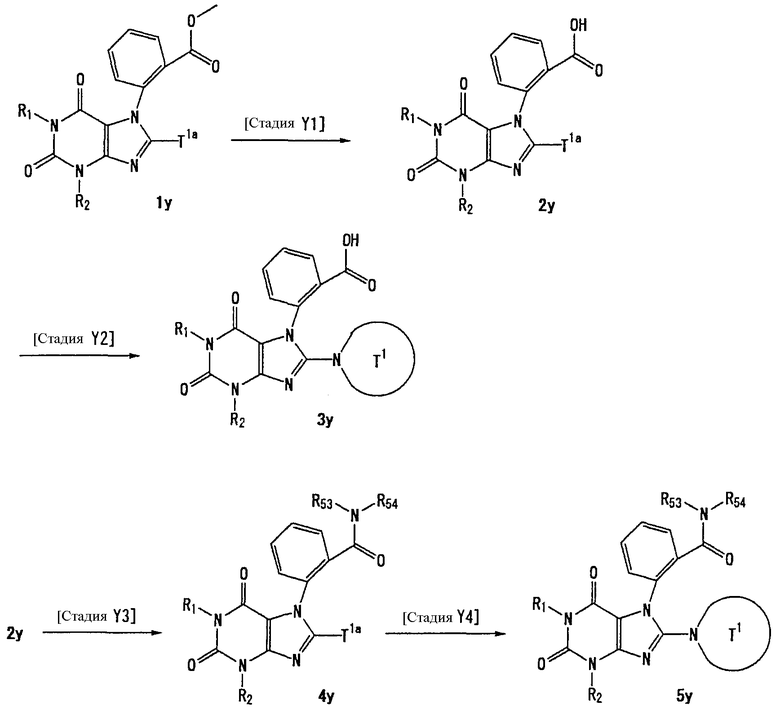
[Стадия Y1]
На данной стадии соединение (1y) гидролизуется, что давало соединение (2y).
Нет никаких особых ограничений в отношении условий реакции гидролиза. Например, гидролиз может проводиться с использованием водного раствора гидроксида лития, гидроксида натрия или гидроксида калия; в растворителе, таком как метанол, этанол, ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран или диметоксиэтан; при температуре в интервале от 0°С до 150°С.
[Стадия Y3]
На данной стадии соединение (2y) амидируется, что давало соединение (4y).
Реакция амидирования проводится в тех же условиях, что используются на [Стадии V6] способа получения V.
[Стадия Y2] и [Стадия Y4]
На данной стадии группа Rp3 удаляется из соединений (2у) и (4у), что давало соответственно соединения (3у) и (5у).
Реакция удаления защиты для Rp3 проводится в тех же условиях, что используются на [Стадии А13] способа получения A.
Способ получения Z
Является альтернативным по отношению к способу получения соединения (2u), описанному в способе получения U.
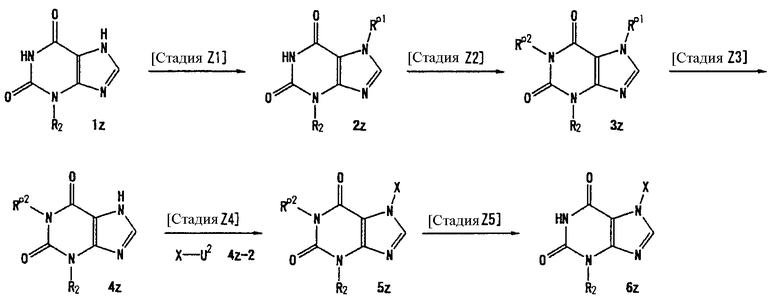
[Стадия Z1]
На данной стадии соединение (1z) защищается по аминогруппе 7 положения, что давало соединение (2z).
Нет никаких особых ограничений в отношении типов групп, используемых для защиты аминогруппы, условий реакции и других переменных. Например, когда защитной группой является бензильная группа, реакцию можно проводить с использованием алкилирующего средства, такого как бензилбромид; в присутствии основания, такого как карбонат цезия, карбонат лития, карбонат натрия или карбонат калия; в растворителе, таком как ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран или диметоксиэтан; при температуре в интервале от 0°С до 150°С.
[Стадия Z2]
На данной стадии соединение (2z) защищается в 1 положении, что давало соединение (3z).
Нет никаких особых ограничений в отношении типов групп, используемых для защиты аминогруппы, условий реакции и других переменных. Например, когда защитной группой является пивалилоксиметильная группа, реакцию можно проводить с использованием алкилирующего средства, такого как хлорметилпивалат; в присутствии основания, такого как карбонат цезия, карбонат лития, карбонат натрия или карбонат калия; в растворителе, таком как ацетонитрил, N,N-диметилформамид, N-метилпирролидон, 1,4-диоксан, тетрагидрофуран или диметоксиэтан; при температуре в интервале от 0°С до 150°С.
[Стадия Z3]
На данной стадии соединение (3z) подвергают взаимодействию удаления защиты по аминогруппе 7-положения, что давало соединение (4z).
Условия реакции варьируют в зависимости от типов используемых защитных груп. Например, когда защитной группой является бензильная группа, реакцию можно проводить в присутствии катализатора на основе металла, такого как палладий на угле, окись платины или никель Ренея; в растворителе, таком как метанол, этанол, пропанол, диметилсульфоксид, N,N-диметилформамид, N-метилпирролидон, диоксан, тетрагидрофуран или толуол; в атмосфере водорода при температуре в интервале от 0°С до 150°С.
[Стадия Z4]
На данной стадии вводится заместитель в аминогруппу в 7 положении соединения (4z) с помощью реакции замещения между соединением (4z) и соединением (4z-2), что давало соединение (5z).
Реакция защиты проводится в тех же условиях, что используются на [Стадии А4] способа получени A.
[Стадия Z5]
На данной стадии защитная группа в 1 положении удаляется из соединения (5z), что давало соединение (6z)(=2u).
Условия реакции варьируют в зависимости от типов используемых защитных групп. Например, когда защитной группой является пивалилоксиметильная группа, в реакции может использоваться основание, такое как метоксид натрия, гидрид натрия или диазабициклоундец-7-ен; в растворителе, таком как метанол или смешанный растворитель из метанола и тетрагидрофурана; при температуре в интервале от 0°С до 150°С.
Способ получения АА
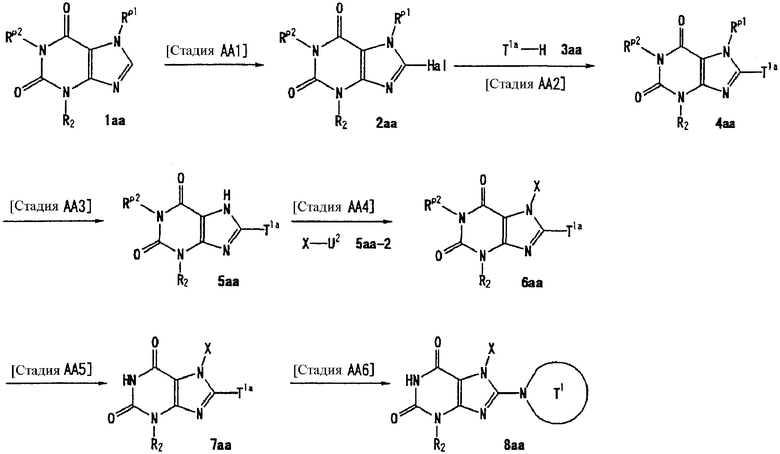
[Стадия АА1]
На данной стадии соединение (1аа) подвергают взаимодействию с галогенирующим средством, что давало соединение (2аа).
Реакция галогенирования проводится в тех же условиях, что используются на [Стадии А5] способа получения А.
[Стадия АА2]
На данной стадии соединение (2аа) подвергают взаимодействию с соединением (3аа), что давало соединение (4аа).
Реакция проводится в тех же условиях, что используются на [Стадии А6] способа получения А.
[Стадия АА3]
На данной стадии защитная группа по аминогруппе 7 положения удаляется из соединения (4аа), что давало соединение (5аа).
Реакция удаления защиты проводится в тех же условиях, что используются на [Стадии Z3] способа получения Z.
[Стадия АА4]
На данной стадии вводится заместитель в аминогруппу в 7 положении соединения (5аа) с помощью реакции замещения между соединением (5аа) и соединением (5аа-2), что давало соединение (6аа).
Реакция замещения проводится в тех же условиях, что используются на [Стадии А4] способа получения A.
[Стадия АА5]
На данной стадии защитная группа в 1 положении удаляется из соединения (6аа), что давало соединение (7аа).
Реакция удаления защиты проводится в тех же условиях, что используются на [Стадии Z5] способа получения Z.
[Стадия АА6]
На данной стадии группа Rp3 удаляется из соединения (7аа), что давало соединение (8аа).
Реакция удаления защиты для Rp3 проводится в тех же условиях, что используются на [Стадии А13] способа получения A.
Способ получения ВВ
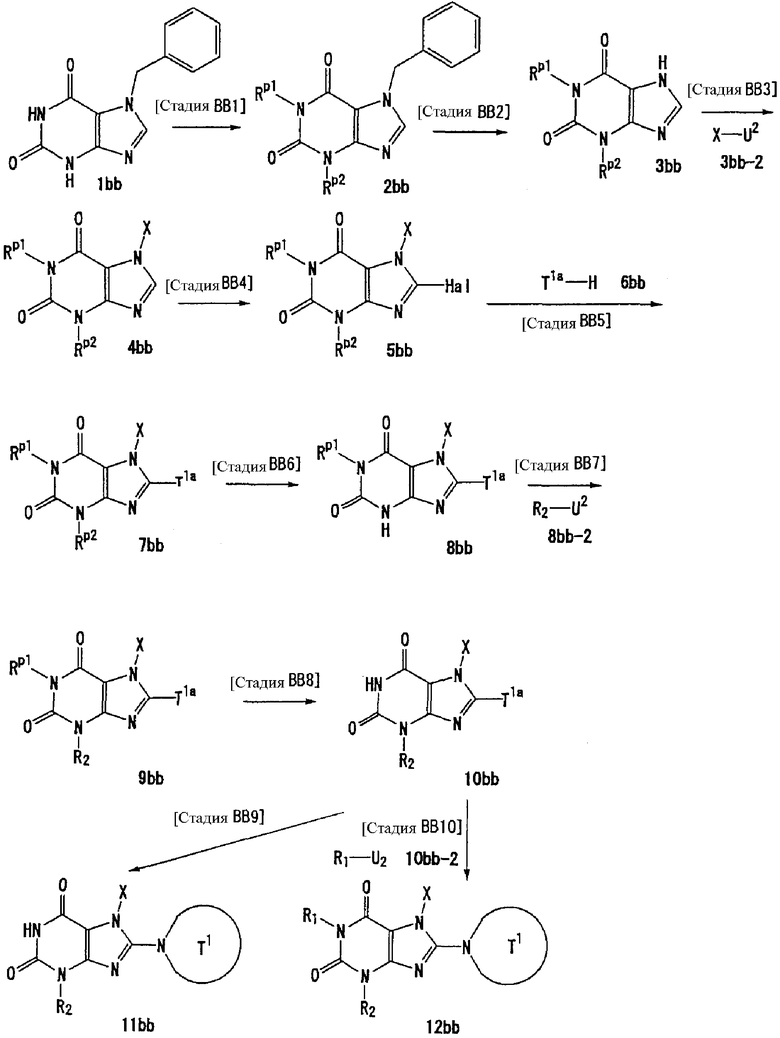
[Стадия ВВ1]
На данной стадии соединение (1bb) защищается по группам в 1 и 3 положениях, что давало соединение (2bb).
Реакция проводится в тех же условиях, что используются на [Стадии Z2] способа получения Z.
[Стадия BB2]
На данной стадии защитная группа аминогруппы в 7 положении удаляется из соединения (2bb), что давало соединение (3bb).
Реакция удаления защиты проводится в тех же условиях, что используются на [Стадии Z3] способа получения Z.
[Стадия BB3]
На данной стадии вводится заместитель в аминогруппу в 7 положении соединения (3bb) с помощью реакции замещения между соединением (3bb) и соединением (3bb-2), что давало соединение (4bb).
Реакция замещения проводится в тех же условиях, что используются на [Стадии А4] способа получения A.
[Стадия ВВ4]
На данной стадии соединение (4bb) подвергают взаимодействию с галогенирующим средством, что давало соединение (5bb).
Реакция галогенирования проводится в тех же условиях, что используются на [Стадии А5] способа получения A.
[Стадия ВВ5]
На данной стадии соединение (5bb) подвергают взаимодействию с соединением (6bb), что давало соединение (7bb).
Реакция проводится в тех же условиях, что используются на [Стадии А6] способа получения А.
[Стадия BB6]
На данной стадии защитная группа в 3 положении удаляется из соединения (7bb), что давало соединение (8bb).
Реакция удаления защиты проводится в тех же условиях, что используются на [Стадии Z5] способа получения Z.
[Стадия BB7]
На данной стадии вводится заместитель в группу в 3-положении соединения (8bb) с помощью реакции замещения между соединением (8bb) и соединением (8bb-2), что давало соединение (9bb).
Реакция замещения проводится в тех же условиях, что используются на [Стадии А4] способа получения A.
[Стадия ВВ8]
На данной стадии защитная группа в 1 положении удаляется из соединения (9bb), что давало соединение (10bb).
Реакция удаления защиты проводится в тех же условиях, что используются на [Стадии Z5] способа получения Z.
[Стадия BB9]
На данной стадии группа Rp3 удаляется из соединения (10bb), что давало соединение (11bb).
Реакция удаления защиты для Rp3 проводится в тех же условиях, что используются на [Стадии А13] способа получения A.
[Стадия BB10]
На данной стадии вводится заместитель в группу в 3 положении соединения (10bb) с помощью реакции замещения между соединением (10bb) и соединением (10bb-2), и затем Rp3 удаляется, что давало соединение (12bb).
Реакция замещения проводится в тех же условиях, что используются на [Стадии А4] способа получения A.
Реакция удаления защиты для Rp3 проводится в тех же условиях, что используются на [Стадии А13] способа получения A.
Способ получения CC
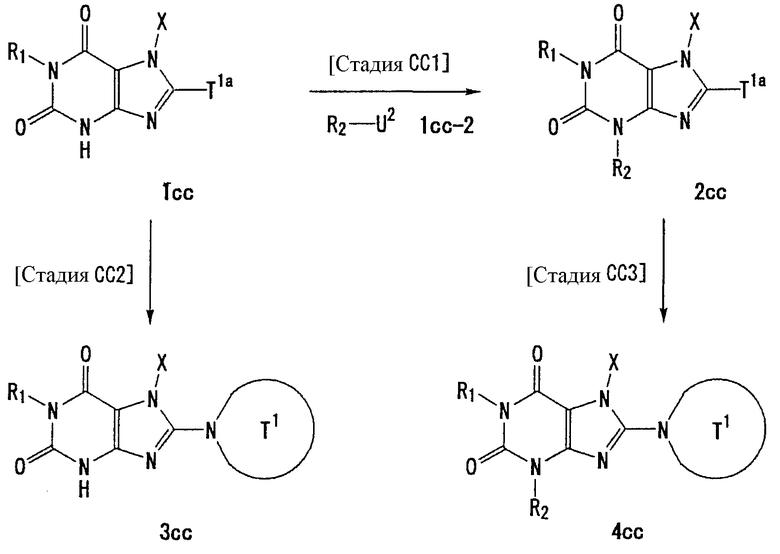
[Стадия CC1]
На данной стадии вводится заместитель в группу в 3 положении соединения (1сс) с помощью реакции замещения между соединением (1сс) и соединением (1сс-2), что давало соединение (2сс).
Реакция замещения проводится в тех же условиях, что используются на [Стадии А4] способа получения A.
[Стадия СС2] [Стадия СС3]
На данных стадиях группа Rp3 удаляется из соединений (1сс) и (2сс), что давало соответственно соединения (3сс) и (4сс).
Реакция удаления защиты для Rp3 проводится в тех же условиях, что используются на [Стадии А13] способа получения A.
Способы, описанные выше, являются представительными способами получения соединений (I) и (II) по настоящему изобретению. Исходными соединениями и различными реагентами, используемыми в способах получения соединений по настоящему изобретению, могут быть соли, гидраты или сольваты в зависимости от типа используемых исходных материалов и растворителей и не ограничиваются, при условии, что они не ингибируют реакции. Типы используемых растворителей зависят от типов используемых исходных соединений и реагентов и не ограничиваются, при условии, что они растворяют материалы до некоторой степени и не тормозят реакции. Когда соединения (I) и (II) по настоящему изобретению получают в свободных формах, такие соединения могут быть преобразованы в соли или гидраты, которые являются возможными формами соединений (I) и (II), описанными выше, в соответствии с обычным методом.
Когда соединения (I) и (II) по настоящему изобретению получаются в виде солей или гидратов, такие продукты могут быть преобразованы в свободные формы соединений (I) и (II), описанных выше, обычными методами.
Кроме того, различные изомеры соединений (I) и (II) по настоящему изобретению (например, геометрические изомеры, энантиомеры на основе асимметрического углерода, ротамеры, стереоизомеры и таутомеры) могут быть очищены и выделены обычными методами выделения, включающих перекристаллизацию, метод диастереомерной соли, метод разделения, основанный на ферментах, и различные хроматографические методы (например, тонкослойную хроматографию, колоночную хроматографию и газовую хроматографию).
Фармецевтические средства по настоящему изобретению могут быть получены с помощью объединения активных ингредиентов, т.е. ингибитора DPPIV бигуанидного агента или фармацевтического средства, который способен усиливать эффекты активного циркулирующего GLP-2. Активные ингредиенты, описанные выше, могут входить в состав препарата отдельно или в сочетанях, и они могут быть смешаны с фармацевтически приемлемыми носителями, эксципиентами, связующими и тому подобное. Стандартные лекарственные формы фармацевтических средств, описанных выше, включают пероральные препараты, например гранулы, микрогранулы, порошки, таблетки, таблетки с покрытиями, капсулы и сиропы; и непероральные препараты, например инъекционные препараты (внутривенные инъекции, подкожные инъекции, внутримышечные инъекции и др.), суппозитории и препараты для наружного применения (трансдермальные терапевтические препараты, мази и др.).
Такие готовые формы препаратов могут быть получены с использованием типичных эксципиентов, связующих, дезинтегрирующих средств, смазочных средств, окрашивающих средств, вкусовых или ароматизирующих средств; и, если требуется, стабилизаторов, эмульгаторов, абсорбирующих средств, детергентов, регуляторов рН, консервирующих средств, антиоксидантов и др., и материалов, используемых в качестве ингредиентов фармацевтических препаратов согласно обычным методам. Данные материалы включают, например, (1) животные и растительные масла, такие как масло соевых бобов, говяжий жир, и синтетические глицериды; (2) углеводороды, такие как жидкий парафин, сквалан и твердый парафин; (3) сложноэфирные масла, такие как октилдодецилмиристат и изопропилмиристат; (4) высшие спирты, такие как цетостеариловый спирт и бегениловый спирт; (5) силиконовые смолы; (6) силиконовые масла; (7) детергенты, такие как полиоксиэтиленовый эфир жирной кислоты, сорбитановый эфир жирной кислоты, глицериновый эфир жирной кислоты, полиоксиэтиленсорбитановый эфир жирной кислоты, полиоксиэтиленовое гидрированное касторовое масло и полиоксиэтилен-полиоксипропиленовый блок-сополимер; (8) водорастворимые полимеры, такие как гидроксиэтилцеллюлоза, полиакриловая кислота, карбоксивиниловый полимер, полиэтиленгликоль, поливинилпирролидон и метилцеллюлоза; (9) низшие спирты, такие как этанол и изопропанол; (10) полиатомные спирты, такие как глицерин, пропиленгликоль, дипропиленгликоль и сорбит; (11) сахара, такие как глюкоза и сахароза; (12) неорганический порошок, такой как безводная кремниевая кислота, алюмосиликат магния и силикат алюминия; и (13) чистую воду.
Эксципиенты включают, например, лактозу, кукурузный крахмал, белый сахар, глюкозу, маннит, сорбит, кристаллическую целлюлозу и двуокись кремния. Связующие включают, например, поливиниловый спирт, поливиниловый эфир, метилцеллюлозу, этилцеллюлозу, аравийскую камедь, трагакант, желатин, шеллак, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, поливинилпирролидон, полипропиленгликоль-полиоксиэтиленовый блок-сополимер, меглумин, цитрат кальция, декстрин и пектин. Дезинтегрирующие средства включают, например, крахмал, агар, желатиновый порошок, кристаллическую целлюлозу, карбонат кальция, бикарбонат натрия, цитрат кальция, декстрин, пектин и кальциевую карбоксиметилцеллюлозу. Смазочные средства включают, например, стеарат магния, тальк, полиэтиленгликоль, двуокись кремния и гидрированное растительное масло. Окрашивающие средства включают средства, которые являются фармацевтически приемлемыми. Вкусовые или ароматизирующие средства включают порошок какао, перечномятную камфору, ароматический порошок, перечномятное масло, Борнео камфору и циннамоновый порошок. Антиоксиданты включают те, которые являются фармацевтически приемлемыми, такие как аскорбиновая кислота и α-токоферол.
Оральный препарат может быть получен путем смешения активных ингредиентов с эксципиентом; и, если требуется, связующим, дезинтегрирующим средством, смазочным средством, окрашивающим средством, вкусовым средством или аналогичным; и формулирования или преобразования смеси в порошок, микрогранулы, гранулы, таблетки, таблетки с покрытием, капсулы или т.п.; в соответствии с обычными методами. Таблетки и гранулы могут быть покрыты сахаром или желатином, или, если необходимо, любыми другими соответствующими покрытиями. Растворы, такие как сиропы или инъецируемые препараты, подлежащие введению, могут быть введены в состав препарата смешением соединения по настоящему изобретению с регулятором рН, солюбилизирующим средством, изотонизирующим средством или тому подобное; и, если требуется, со вспомогательными солюбилизирующим средством, стабилизатором, буфером, суспендирующим средством, антиоксидантом или тому подобное согласно обычным методам. Раствор может высушиваться замораживанием. Примерами предпочтительных суспендирующих средств являютс метилцеллюлоза, Полисорбат 80, гидроксиэтилцеллюлоза, аравийская камедь, порошкообразный трагакант, натриевая карбоксиметилцеллюлоза и полиоксиэтиленсорбитан-моно-лаурат. Примерами предпочтительных вспомогательных солюбилизирующих средств являются полиоксиэтиленовое гидрированное касторовое масло, Полисорбат 80, никотинамид и полиоксиэтиленсорбитан-моно-лаурат. Примерами предпочтительных стабилизаторов являются сульфит натрия, метасульфит натрия и простой эфир. Примерами предпочтительных консервирующих средств являются метил-пара-оксибензоат, этил-пара-оксибензоат, сорбиновая кислота, фенол, крезол и хлоркрезол. Нет никаких ограничений в отношении способов получения наружных препаратов, и такие препараты могут быть получены обычными методами. Различные материалы, обычно используемые для получения фармацевтических препаратов, квази-лекарств, косметических препаратов, и другие, включающие животные и растительные масла, минеральные масла, сложноэфирные масла, воски, высшие спирты, жирные кислоты, силиконовые масла, детергенты, фосфолипиды, спирты, моногоатомные спирты, водорастворимые полимеры, глиняные минералы и чистая вода могут включаться в качестве материалов основы. Далее, наружные препараты по настоящему изобретению могут содержать регуляторы рН, антиоксиданты, хелатирующие средства, антибактериальные/противогрибковые средства, окрашивающие средства (красители) или ароматизирующие средства, по необходимости. В дополнение к изложенному, наружные препараты по настоящему изобретению могут также содержать средства, которые вызывают дифференциацию, промотируют ток крови, активируют клетки, и антимикробные средства, противовоспалительные средства, витамины, аминокислоты, увлажнители, кератолитические средства и другие, если необходимо.
Нет никаких ограниченний в отношении методов введения для фармацевтических средств согласно настоящему изобретению. Ингибитор DPPIV и или бигуанидный агент, или фармацевтическое средство, который усиливает эффекты активного циркулирующего GLP-2, могут использоваться во время введения в сочетании. Например, методы введения включают (1) введение препарата, полученного конъюгированием ингибитора DPPIV и или бигуанидного агента, или фармацевтического средства, который усиливает эффекты активного циркулирующего GLP-2; (2) одновременное введение двух типов препаратов, которые получают с помощью раздельного формулирования ингибитора DPPIV и или бигуанидного агента, или фармацевтического средства, который усиливает эффекты активного циркулирующего GLP-2; и (3) раздельное введение двух типов препаратов, которые получают раздельным формулированием ингибитора DPPIV и или бигуанидного агента, или фармацевтического средства, который усиливает эффекты активного циркулирующего GLP-2, в различные точки времени (например, введение их по порядку, сначала ингибитора DPPIV, а затем или бигуанидного агента, или фармацевтического средства, который усиливает эффекты активного циркулирующего GLP-2, или в обратном порядке).
Доза фармацевтических средств согласно настоящему изобретению может быть выбрана на основе стандартной дозы каждого средства. Доза может быть соответствующим образом выбрана на основе возраста пациента, массы, пола, тяжести симптомов, дозировочной формы и типа болезни. Когда ингибитором DPPIV, подлежащим введению орально или парентерально является (S)-1-((3-гидрокси-1-адамантил)амино)ацетил-2-цианпирролидин или (S)-1-(2-((5-цианпиридин-2-ил)амино)этиламиноацетил)-2-цианпирролидин, доза может типично выбираться в интервале от 0,1 до 250 мг/взрослого/день, предпочтительно, 1-100 мг/взрослого/день. Когда ингибитором DPPIV, подлежащим введению орально или парентерально, является изолейцинтиазолидид, изолейцинпирролидид или валинпирролидид, доза может типично выбираться в интервале 0,01-2,0 мг/кг/день, предпочтительно, 0,01-1,0 мг/кг/день. Когда ингибитором DPPIV является соединение, представленное формулой (I) или (II), или его соль или гидрат, и его предназначают вводить орально взрослому, доза может типично выбираться в интервале 0,03-1000 мг/день, предпочтительно, 0,1-500 мг/день, более предпочтительно, 0,1-100 мг/день. Когда ингибитором DPPIV является соединение, представленное формулой (I) или (II), или его соль или гидрат, и его предполагают вводить парентерально взрослому, доза типично может выбираться в интервале примерно 1-3000 мкг/кг/день, предпочтительно, около 3-1000 мкг/кг/день. Когда ингибитор DPPIV предполагается использовать в сочетании с еще одним средством, например бигуанидным агентом, доза типично варьирует от 10 до 2500 мг/взрослому/день и, предпочтительно, составляет в интервале от 100 до 1000 мг/взрослому/день.
В настоящем изобретении как ингибитор DPPIV, так и бигуанидный агент может вводиться однократно или несколько раз в ежедневной дозе, описанной выше.
Соотношение доз между соответствующими средствами в фармацевтических средствах согласно настоящему изобретению может выбираться соответствующим образом на основе возраста пациента, массы, пола, тяжести симптомов, дозировочной формы и типа заболевания. Например, соотношение масса:массовая доза между ингибитором DPPIV и бигуанидным агентом может типично находиться в интервале от 1:1 до 1:2500, предпочтительно, 1:10-1:250.
(S)-1-((3-гидрокси-1-адамантил)амино)ацетил-, указанный здесь, может вводиться в дозе, выбранной в интервале 3-1000 мкг/кг. Когда ингибитор DPPIV используется в сочетании с еще одним средством, например бигуанидным агентом, доза типично варьирует от 10 до 2500 мг/взрослому/день и, предпочтительно, составляет от 100 до 1000 мг/взрослому/день.
Соединения по настоящему изобретению, представленные формулами (I) и (II), показанными выше, могут получаться по способам, описанным ниже в примерах. Однако соединения по настоящему изобретению ни при каких обстоятельствах не должны рассматриваться как ограниченные конкретными примерами, описанными ниже.
[Примеры получения]
Пример получения 1
трет-Бутил 4-[1-(2-бутинил)-6-метил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилат
(а) трет-Бутил 5-метил-4-оксо-4,5-дигидроимидазо[4,5-d]пиридазин-1-карбоксилат
Смесь, состоящую из 1,0 г 5-метил-3,5-дигидроимидазо[4,5-d]пиридазин-4-она, 16 мг 4-диметиламинопиридина, 1,6 г ди-трет-бутил дикарбоната и 5 мл тетрагидрофурана, перемешивали при комнатной температуре в течение ночи. Затем к раствору добавляли 0,5 мл раствора тетрагидрофурана, содержащего 300 мг ди-трет-бутил дикарбоната, и полученную смесь перемешивали при комнатной температуре в течение 3 часов. К реакционной смеси добавляли 5 мл трет-бутил метилового эфира, и смесь охлаждали льдом. Полученные в результате кристаллы собирали с помощью фильтрования, что давало 1,63 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,72 (с, 9H) 3,93 (с, 3H) 8,38 (с, 1H) 8,54 (с, 1H).
(b) 2-Хлор-5-метил-1,5-дигидроимидазо[4,5-d]пиридазин-4-он
8,4 мл гексаметилдисилазида лития (1,0 М раствор тетрагидрофурана) по каплям, в течение 1 часа, в атмосфере азота при 0°С добавляли к 300 мл раствору тетрагидрофурана, содержащего 1,68 г трет-бутил 5-метил-4-оксо-4,5-дигидроимидазо[4,5-d]пиридазин-1-карбоксилата и 4,15 г гексахлорэтана. Полученную смесь перемешивали в течение 30 минут. К раствору добавляли 2 н аммиачную воду, и смесь перемешивали в течение 3 часов. Затем реакционный раствор концентрировали до 50 мл и промывали 20 мл трет-бутил метилового эфира. Раствор подкисляли концентрированной соляной кислотой. Полученный осадок собирали с помощью фильтрования и промывали последовательно 10 мл воды и 10 мл трет-бутил метилового эфира. Таким образом получали 1,03 г указанного в заголовке соединения.
1H-ЯМР (ДМСО-d6)
δ 1,45 (с, 9H) 3,72 (с, 3H) 8,33 (с, 1H).
(c) 3-(2-Бутинил)-2-хлор-5-метил-3,5-дигидроимидазо[4,5-d]пиридазин-4-он
7,72 г 2-хлор-5-метил-1,5-дигидроимидазо[4,5-d]пиридазин-4-она суспендировали в 400 мл тетрагидрофурана в атмосфере азота, затем туда добавляли 14,22 г трифенилфосфина и 3,85 г 2-бутин-1-ола. Полученную смесь охлаждали до 0°С. Затем, по каплям, добавляли 100 мл раствора тетрагидрофурана, содержащего 12,55 г ди-трет-бутилового эфира азодикарбоновой кислоты, и реакционную смесь перемешивали в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении. К остатку добавляли 50 мл дихлорметана и 50 мл трифторуксусной кислоты, и смесь перемешивали в течение 15 часов. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток растворяли в 400 мл этилацетата и промывали 200 мл 5 н водного раствора гидроксида натрия. Водный слой экстрагировали 100 мл этилацетата. Органические слои объединяли вместе, сушили над сульфатом магния и концентрировали при пониженном давлении. Получившийся в результате остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 8,78 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (4:1).
1H-ЯМР (CDCl3)
δ 1,82 (т, J=2,3 Гц, 3H) 3,87 (с, 3H) 5,32 (кв, J=2,3 Гц, 2H) 8,19 (с, 1H).
(d) трет-Бутил 4-[1-(2-бутинил)-6-метил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилат
5 мл 1-метил-2-пирролидона в атмосфере азота добавляли к смеси, состоящей из 1,183 г 3-(2-бутинил)-2-хлор-5-метил-3,5-дигидроимидазо[4,5-d]пиридазин-4-она, 0,829 г карбоната калия и 1,395 г трет-бутил пиперазин-1-карбоксилата. Полученную смесь перемешивали при 130°С в течение 6 часов. Реакционную смесь охлаждали и в нее добавляли 50 мл воды. Затем реакционную смесь экстрагировали 100 мл этилацетата. Органический слой дважды промывали 50 мл воды и затем 50 мл насыщенного водного раствора хлорида натрия. Органический слой сушили над сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 1,916 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (1:4).
1H-ЯМР (CDCl3)
δ 1,52 (с, 9H) 1,83 (т, J=2,3 Гц, 3H) 3,38-3,42 (м, 4H) 3,61-3,64 (м, 4H) 3,85 (с, 3H) 5,09 (кв, J=2,3 Гц, 2H) 8,13 (с, 1H).
Пример получения 2
трет-Бутил 4-[7-(2-бутинил)-2,6-дихлор-7Н-пурин-8-ил]пиперазин-1-карбоксилат
(а) 7-(2-Бутинил)-3-метил-3,7-дигидропурин-2,6-дион
55,3 мл 1-бром-2-бутина и 84,9 г безводного карбоната калия добавляли к смеси 100 г 3-метил ксантина [CAS №1076-22-8] и 1000 мл N,N-диметилформамида. Полученную смесь перемешивали при комнатной температуре в течение 18 часов. К реакционному раствору добавляли 1000 мл воды, и смесь перемешивали при комнатной температуре в течение 1 часа. Полученный белый остаток собирали с помощью фильтрования. Белое твердое вещество промывали водой и затем трет-бутил метиловым эфиром. Таким образом получали 112 г указанного в заголовке соединения.
1H-ЯМР (ДМСО-d6)
δ 1,82 (т, J=2,2 Гц, 3H) 3,34 (с, 3H) 5,06 (кв, J=2,2 Гц, 2H) 8,12 (с, 1H) 11,16 (ушир.с, 1H).
(b) 7-(2-Бутинил)-8-хлор-3-метил-3,7-дигидропурин-2,6-дион
112 г 7-(2-бутинил)-3-метил-3,7 дигидропурин-2,6-диона растворяли в 2200 мл N,N-диметилформамида и добавляли 75,3 г N-хлорсукцинимида. Полученную смесь перемешивали при комнатной температуре в течение 5 часов. К реакционному раствору добавляли 2200 мл воды, и смесь перемешивали при комнатной температуре в течение 1,5 часов. Белый осадок собирали с помощью фильтрования, и белое твердое вещество промывали водой и затем трет-бутил метиловым эфиром. Таким образом получали 117 г указанного в заголовке соединения.
1H-ЯМР (ДМСО-d6)
δ 1,78 (т, J=2,0 Гц,3H) 3,30 (с, 3H) 5,06 (кв, J=2,0 Гц, 2H) 11,34 (ушир.с, 1H).
(с) 7-(2-Бутинил)-2,6,8-трихлор-7Н-пурин
Смесь 2,52 г 7-(2-бутинил)-8-хлор-3-метил-3,7-дигидропурин-2,6-диона и 100 мл хлорокиси фосфора перемешивали при 120°C в течение 14 часов. После того, как реакционную смесь охлаждали, в раствор добавляли 4,15 г пентахлорида фосфора. Полученную смесь перемешивали при 120°С в течение 24 часов. После того, как реакционный раствор охлаждали до комнатной температуры, растворитель выпаривали при пониженном давлении. Остаток растворяли в тетрагидрофуране. Раствор вливали в насыщенный раствор бикарбоната натрия, и смесь экстрагировали этилацетатом. Полученный в результате органический слой промывали водой, затем насыщенным соляным раствором и затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат:гексан=1:3), что давало 2,40 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,82 (т, J=2,4 Гц, 3H) 5,21 (кв, J=2,4 Гц, 2H).
(d) трет-Бутил 4-[7-(2-бутинил)-2,6-дихлор-7Н-пурин-8-ил]пиперазин-1-карбоксилат
Смесь 2,4 г 7-(2-бутинил)-2,6,8-трихлор-7Н-пурина, 1,46 г бикарбоната натрия, 2,43 г трет-бутил пиперазин-1-карбоксилата и 45 мл ацетонитрила перемешивали при комнатной температуре в течение 2 часов и 20 минут. Затем добавляли 0,73 г бикарбоната натрия и 1,21 г трет-бутил пиперазин-1-карбоксилата, и получившуюся в результате смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь экстрагировали смесью этилацетат-вода, и органический слой промывали 1 н соляной кислотой, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Остаток растирали с диэтиловым эфиром. Кристаллы собирали с помощью фильтрования и промывали диэтиловым эфиром. Таким образом получали 3,0 г указанного в заголовке соединения в виде белого твердого вещества.
1H-ЯМР (ДМСО-d6)
δ 1,42 (с, 9H) 1,83 (т, J=2 Гц, 3H) 3,48-3,55 (м, 4H) 3,57-3,63 (м, 4H) 4,89 (кв, J=2 Гц, 2H).
[Примеры]
Пример1
Трифторацетат этил [7-(2-хлорфенил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]ацетата
(а) [7-Бензил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионат
8,66 г 7-бензилксантина растворяли в 300 мл N,N-диметилформамида и туда добавляли 1,57 г гидрида натрия и 7,7 мл хлорметил пивалата. Полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционный раствор разбавляли этилацетатом и промывали водой и 1 н соляной кислотой. Органический слой сушили над безводным сульфатом магния и затем фильтровали. Растворитель выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 2,66 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (1:1).
1H-ЯМР (CDCl3)
δ 1,18 (с, 9H) 5,45 (с, 2H) 6,06 (с, 2H) 7,34-7,39 (м, 5H) 7,58 (с, 1H) 8,18 (с, 1H).
(b) [7-Бензил-1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионат
2,66 г [7-бензил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионата растворяли в 30 мл N,N-диметилформамида и добавляли 1,6 г карбоната калия и 1 мл метил йодида. Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли этилацетатом и промывали водой и 1 н соляной кислотой. Органический слой сушили над безводным сульфатом магния и затем фильтровали. Растворитель выпаривали при пониженном давлении. Остаток растирали с толуолом. Таким образом получали 2,16 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,18 (с, 9H) 3,41 (с, 3H) 5,49 (с, 2H) 6,11 (с, 2H) 7,26-7,39 (м, 5H) 7,57 (с, 1H).
(с) [1-Метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионат
2,349 г [7-бензил-1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионата растворяли в 100 мл уксусной кислоты и добавляли 1 г 10% палладия на угле. Реакционную смесь перемешивали в атмосфере азота при комнатной температуре в течение ночи. Реакционную смесь фильтровали и концентрировали, что давало 1,871 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,19 (с, 9H) 3,48 (с, 3H) 6,17 (с, 2H) 7,83 (с, 1H).
(d) [7-(2-Хлорфенил)-1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионат
1,60 г [1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионата, 1,83 г 2-хлорфенилборной кислоты и 1,5 г ацетата меди (II) суспендировали в 30 мл N,N-диметилформамида и добавляли 3 мл пиридина. Смесь перемешивали при комнатной температуре в течение трех дней. Реакционную смесь фильтровали через короткую колонку, заполненную силикагелем, и фильтрат разбавляли этилацетатом. Органический слой промывали 1 н соляной кислотой, водой, насыщенным соляным раствором и сушили над безводным сульфатом магния, затем фильтровали. Фильтрат концентрировали. Остаток суспендировали в простом эфире, и суспензию фильтровали. Фильтрат очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 724 мг указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (3:2).
(е) трет-Бутил 4-[7-(2-хлорфенил)-3-(2,2-диметилпропионилоксиметил)-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
724 мг [7-(2-хлорфенил)-1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионата суспендировали в 15 мл N,N-диметилформамида и добавляли 760 мг N-хлорсукцинимида. Реакционный раствор перемешивали в течение ночи и затем разбавляли этилацетатом. Раствор промывали водой и 1 н соляной кислотой и сушили над безводным сульфатом магния, затем фильтровали. Фильтрат концентрировали. Таким образом получали 764 мг [8-хлор-7-(2-хлорфенил)-1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионата. Данное соединение перемешивали с 4 г трет-бутил пиперазин-1-карбоксилата. Смесь нагревали при 150°С и перемешивали в течение 3 часов. К реакционной смеси добавляли этилацетат и воду, и смесь разделяли. Органический слой промывали 1 н соляной кислотой и сушили над безводным сульфатом магния, затем фильтровали. Фильтрат концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 724 мг указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (3:2).
(f) трет-Бутил 4-[7-(2-хлорфенил)-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
трет-Бутил 4-[7-(2-хлорфенил)-3-(2,2-диметилпропионилоксиметил)-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат растворяли в смеси 10 мл метанола и 20 мл тетрагидрофурана и добавляли 200 мг гидрида натрия. Полученную смесь перемешивали при комнатной температуре в течение ночи. К реакционному раствору добавляли 1 н соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния, затем фильтровали. Фильтрат концентрировали. Остаток суспендировали в эфире и смесь фильтровали. Таким образом получали 450 мг указанного в заголовке соединения.
1H-ЯМР (ДМСО-d6)
δ 1,35 (с, 9H) 3,04 (с, 3H) 3,06-3,12 (м, 4H) 3,17-3,22 (м, 4H) 7,48 (дт, J=1,6, 7,6 Гц, 1H) 7,53 (дт, J=2,0, 7,6 Гц, 1H) 7,63 (дд, J=2,0, 8,0 Гц, 1H) 7,65 (дд, J=1,6, 8,0 Гц, 1H).
(g) трет-Бутил 4-[2-хлор-7-(2-хлорфенил)-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат (g-1) и трет-бутил 4-[2,6-дихлор-7-(2-хлорфенил)-7Н-пурин-8-ил]пиперазин-1-карбоксилат (g-2)
78 мг трет-бутил 4-[7-(2-хлорфенил)-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 3 мл хлорокиси фосфора, и смесь перемешивали при 120°С в течение ночи. Реакционный раствор концентрировали, и остаток растворяли в 1 мл тетрагидрофурана. Данный раствор вливали в суспензию, состоящую из 50 мг ди-трет-бутил дикарбоната, 1 мл тетрагидрофурана и 0,5 мл воды, содержащей 100 мг бикарбоната натрия. Полученную смесь перемешивали при комнатной температуре в течение трех часов. Реакционную смесь разбавляли этилацетатом и промывали водой. Органический слой сушили над безводным сульфатом магния, затем фильтровали. Фильтрат концентрировали и остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 16 мг трет-бутил 4-[2,6-дихлор-7-(2-хлорфенил)-7Н-пурин-8-ил]пиперазин-1-карбоксилата из фракции, элюированной смесью гексан-этилацетат (3:2) и 10 мг трет-бутил 4-[2-хлор-7-(2-хлорфенил)-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата из фракции, элюированной смесью гексан-этилацетат (1:9).
(h) Трифторацетат этил [7-(2-хлорфенил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]ацетата
10 мг трет-бутил 4-[2-хлор-7-(2-хлорфенил)-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата и 10 мг этил гликолята растворяли в 0,2 мл N-метилпирролидона и добавляли 10 мг гидрида натрия. Смесь перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор растворяли в этилацетате и смесь промывали 1 н соляной кислотой. Таким образом получали 24 мг трет-бутил 4-[7-(2-хлорфенил)-2-этоксикарбонилметокси-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата. 8 мг данного соединения растворяли в трифторуксусной кислоте и смесь концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 2,11 мг указанного в заголовке соединения.
MS m/e (ESI) 447 (MH+-CF3COOH).
Пример 4
Трифторацетат метил 2-[7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]фенилацетата
(а) [7-(2-Бутинил)-1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионат
1,871 г [1-метил-2,6-диоксо-1,2,6,7-тетрадигидропурин-3-ил]метил-2,2-диметилпропионата растворяли в 30 мл N,N-диметилформамида и добавляли 1,5 г карбоната калия и 0,7 мл 2-бутинил бромида. Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли этилацетатом и промывали водой и 1 н соляной кислотой. Органический слой сушили над безводным сульфатом магния, затем фильтровали. Растворитель выпаривали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 2,12 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (3:2).
(b) 7-(2-Бутинил)-1-метил-3,7-дигидропурин-2,6-дион
Указанное в заголовке соединение получали с помощью обработки [7-(2-бутинил)-1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионата способом, аналогичным примененному в примере (1f).
1H-ЯМР (CDCl3)
δ 1,91 (т, J=2,4 Гц, 3H) 3,39 (с, 3H) 5,10 (с, 2H) 7,93 (с, 1H) 10,62 (с, 1H).
(c) трет-Бутил 4-[7-(2-бутинил)-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
Указанное в заголовке соединение получали с помощью обработки [7-(2-бутинил)-1-метил-3,7-дигидропурин-2,6-диона способом, аналогичным примененному в примере (1е).
1H-ЯМР (CDCl3)
δ 1,48 (с, 9H) 1,83 (т, J=2,4 Гц, 3H) 3,37 (с, 3H) 3,37-3,39 (м, 4H) 3,58-3,60 (м, 4H) 4,87 (с, 2H) 9,68 (с, 1H).
(d) Трифторацетат метил 2-[7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]фенилацетата
8 мг трет-бутил 4-[7-(2-бутинил)-1-метил-2,6-диоксо-2,3,6,7-тетрадигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата и 10 мг метил 2-бромфенилацетата растворяли в 0,2 мл N,N-диметилформамида и добавляли 10 мг карбоната калия. Смесь перемешивали при 50°С в течение ночи. К реакционной смеси добавляли этилацетат, и смесь промывали водой и 1 н соляной кислотой. Органический слой концентрировали. Остаток растворяли в трифторуксусной кислоте и смесь концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 1,07 мг целевого соединения указанного в заголовке.
MS m/e (ESI) 451 (MH+-CF3COOH).
Пример 7
Трифторацетат 7-(2-бутинил)-2-циклопентилокси-1-метил-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
Указанное в заголовке соединение получали по способу примера 4 с использованием бромциклопентана вместо метил 2-бромфенилацетата в Примере (4d).
MS m/e (ESI) 371 (MH+-CF3COOH).
Пример 9
Этил 2-[7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]пропионат
Трифторацетат указанного в заголовке соединения получали по способу примера 4 с использованием 2-бромпропионата вместо метил 2-бромфенилацетата в Примере (4d). Соединение очищали с помощью хроматографии с использованием NH-силикагеля (силикагель, поверхность которого модифицирована аминогруппами: Fuji Silysia Chemical Ltd. NH-DM 2035). Таким образом, указанное в заголовке соединение получали из фракции, элюированной смесью этилацетат-метанол (20:1).
MS m/e (ESI) 404 (MH+).
Пример 11
Трифторацетат 7-(2-бутинил)-2-метокси-1-метил-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
(а) трет-Бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат (а-1) и трет-бутил 4-[7-(2-бутинил)-2,6-дихлор-7Н-пурин-8-ил]пиперазин-1-карбоксилат (а-2)
5,127 г трет-бутил 4-[7-(2-бутинил)-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 75 мл хлорокиси фосфора и затем смесь перемешивали при 120°С в течение ночи. Реакционный раствор концентрировали и остаток растворяли в 50 мл тетрагидрофурана. Данный раствор вливали в суспензию, состоящую из 7 г ди-трет-бутил дикарбоната, 50 мл тетрагидрофурана, 100 мл бикарбоната натрия и 200 мл воды, и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь вливали в этилацетат и смесь промывали водой. Органический слой сушили над безводным сульфатом магния, затем фильтровали. Фильтрат концентрировали, и остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 1,348 г трет-бутил 4-[7-(2-бутинил)-2,6-дихлор-7Н-пурин-8-ил]пиперазин-1-карбоксилата [1H-ЯМР (CDCl3) δ 1,50 (с, 9H) 1,87 (т, J=2,4 Гц, 3H) 3,64 (м, 8H) 4,81 (кв, J=2,4 Гц, 2H)] из фракции, элюированной смесью гексан-этилацетат (1:1) и 1,238 г трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата [1H-ЯМР (CDCl3) δ 1,49 (с, 9H) 1,83 (т, J=2,4 Гц, 3H) 3,42-3,44 (м, 4H) 3,59-3,62 (м, 4H) 3,73 (с, 3H) 4,93 (кв, J=2,4 Гц, 2H)] из фракции, элюированной смесью гексан-этилацетат (1:9).
(b) Трифторацетат 7-(2-бутинил)-2-метокси-1-метил-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
8 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,2 мл метанола и туда добавляли 10 мг гидрида натрия. Смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли 1 н соляной кислоты, и смесь экстрагировали этилацетатом. Органический слой концентрировали и остаток растворяли в трифторуксусной кислоте. Смесь концентрировали и остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 1,72 мг соединения указанного в заголовке.
MS m/e (ESI) 317 (MH+-CF3COOH).
Пример 12
7-(2-Бутинил)-2-этокси-1-метил-8-(пиперазин-1-ил)-1,7-дигидропурин-6-он
Трифторацетат указанного в заголовке соединения получали по способу примера 11 с использованием этанола вместо метанола в Примере (11b). Данное соединение очищали с помощью хроматографии с использованием NH-силикагеля. Таким образом, соединение, указанное в заголовке, получали из фракции, элюированной смесью этилацетат-метанол (20:1).
1H-ЯМР (CDCl3)
δ 1,42 (т, J=7,2 Гц, 3H) 1,82 (т, J=2,4 Гц, 3H) 3,02-3,06 (м, 4H) 3,40-3,42 (м, 4H) 3,46 (с, 3H) 4,51 (кв, J=7,2 Гц, 2H) 4,90 (кв, J=2,4 Гц, 2H).
МС m/e (ESI) 331 (MH+).
Пример 13
Этил [7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]ацетат
Пример 14
[7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]уксусная кислота
Трифторацетат этил [7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]ацетата и трифторацетат [7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]уксусной кислоты [MS m/e (ESI) 361 (MH+-CF3COOH)] получали с помощью обработки трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата с использованием этил 2-гидроксиацетата вместо этанола по способу примера 11. Трифторацетат этил [7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]ацетата очищали с помощью хроматографии с использованием NH-силикагеля. Таким образом, этил [7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]ацетат [1H-ЯМР (CDCl3) δ 1,29 (т, J=7,2 Гц, 3H) 1,83 (т, J=2,4 Гц, 3H) 3,02-3,06 (м, 4H) 3,38-3,41 (м, 4H) 3,55 (с, 3H) 4,22 (кв, J=7,2 Гц, 2H) 4,90 (кв, J=2,4 Гц, 2H) 5,03 (с, 2H). МС m/e (ESI) 389 (MH+)] получали из фракции, элюированной смесью этилацетат-метанол (20:1).
Пример 16
Этил 1-[7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]циклопропанкарбоксилат
Трифторацетат указанного в заголовке соединения получали по способу примера 13 с использованием этил 1-гидроксициклопропанкарбоксилата вместо этил 2-гидроксиацетата в Примере 13. Соединение очищали с помощью хроматографии с использованием NH-силикагеля. Таким образом, соединение, указанное в заголовке, получали из фракции, элюированной смесью этилацетат-метанол (20:1).
1H-ЯМР (CDCl3)
δ 1,19 (т, J=7,2 Гц, 3H) 1,39-1,42 (м, 2H) 1,67-1,71 (м, 2H) 1,83 (т, J=2,4 Гц, 3H) 3,02-3,05 (м, 4H) 3,37-3,40 (м, 4H) 3,49 (с, 3H) 4,14 (кв, J=7,2 Гц, 2H) 4,90 (кв, J=2,4 Гц, 2H).
МС m/e (ESI) 415 (MH+).
Пример 20
Трифторацетат 7-(2-бутинил)-1-метил-2-фенокси-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
Указанное в заголовке соединение получали по способу примера 13 с использованием фенола вместо этил 2-гидроксиацетата в примере 13.
MS m/e (ESI) 379 (MH+-CF3COOH).
Пример 22
Трифторацетат 7-(2-бутинил)-1,2-диметил-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
8 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата и 2 мг тетракис(трифенилфосфин)палладия растворяли в 0,2 мл диоксана и туда добавляли 0,2 мл хлорида метилцинка (1,5 М раствор тетрагидрофурана). Смесь перемешивали при 50°С в течение 0,5 часа. Реакционный раствор концентрировали и остаток растворяли в трифторуксусной кислоте. Смесь концентрировали, и остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 4,56 мг указанного в заголовке соединения.
MS m/e (ESI) 301 (MH+-CF3COOH).
Пример 29
Трифторацетат 7-(2-бутинил)-1-метил-2-диметиламино-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
8 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,2 мл водного раствора 40% диметиламина, и смесь перемешивали при 80°С в течение 5 часов. Реакционный раствор концентрировали, и остаток растворяли в трифторуксусной кислоте. Смесь концентрировали, и остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 6,95 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,82 (т, J=2,4 Гц, 3H) 2,83 (с, 6H) 3,02-3,05 (м, 4H) 3,39-3,42 (м, 4H) 3,56 (с, 3H) 4,90 (д, J=2,4 Гц, 2H)
МС m/e (ESI) 330 (MH+-CF3COOH).
Пример 41
Трифторацетат 7-(2-бутинил)-2-(2-этоксиэтиламино)-1-метил-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
10 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,15 мл 1-метил-2-пирролидона и туда добавляли 20 мкл 2-этоксиэтиламина. После того, как смесь нагревали при 80°С в течение 12 часов, реакционный раствор концентрировали в обильном токе азота. Полученный остаток растворяли в 0,40 мл трифторуксусной кислоты и смесь концентрировали в обильном токе газообразного азота. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 6,95 мг указанного в заголовке соединения.
MS m/e (ESI) 374 (MH+-CF3COOH).
Пример 53
Трифторацетат (S)-1-[7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-ил]пирролидин-2-карбоновой кислоты
Получали 4,07 мг указанного в заголовке соединения по способу примера 41 с использованием L-пролин трет-бутилового эфира вместо 2-этоксиэтиламина в Примере 41.
MS m/e (ESI) 400 (MH+-CF3COOH).
Пример 63
Трифторацетат (R)-1-[7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-ил]пирролидин-2-карбоновой кислоты
6 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,15 мл 1-метил-2-пирролидона и туда добавляли 15 мг гидрохлорида D-пролин метилового эфира и 50 мкл триэтиламина. После того, как реакционную смесь нагревали при 80°С в течение 12 часов, реакционный раствор концентрировали в обильном токе газообразного азота. Остаток растворяли в растворе, состоящем из 0,20 мл этанола и 0,20 мл 5 н водного раствора гидроксида натрия. Смесь перемешивали при комнатной температуре в течение 5 часов и затем концентрировали в обильном токе газообразного азота. Остаток растворяли в 0,40 мл трифторуксусной кислоты, и смесь концентрировали в обильном токе газообразного азота. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 3,42 мг указанного в заголовке соединения.
MS m/e (ESI) 400 (MH+-CF3COOH).
Пример 64
Трифторацетат 2-[7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-иламино]пропионовой кислоты
1,12 мг указанного в заголовке соединения получали по способу примера 63 с использованием гидрохлорида DL-аланин метилового эфира вместо гидрохлорида D-пролин метилового эфира в Примере 63.
MS m/e (ESI) 374 (MH+-CF3COOH).
Пример 68
Трифторацетат метил [7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илсульфанил]ацетата
6 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,15 мл 1-метил-2-пирролидона и туда добавляли 20 мкл метил меркаптоацетата и 6 мг карбоната калия. Смесь перемешивали при комнатной температуре в течение 5 часов. К реакционному раствору добавляли насыщенный водный раствор хлорида аммония и смесь экстрагировали этилацетатом. Органический слой концентрировали и остаток растворяли в 0,40 мл трифторуксусной кислоты. Раствор концентрировали в обильном токе газообразного азота. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 4,83 мг указанного в заголовке соединения.
MS m/e (ESI) 391 (MH+-CF3COOH).
Пример 73
Трифторацетат 7-(2-бутинил)-1-метил-8-(пиперазин-1-ил)-2-(пиридин-2-илсульфанил)-1,7-дигидропурин-6-она
4,66 мг указанного в заголовке соединения получали по способу примера 68 с использованием 2-меркаптопиридина вместо метилмеркаптоацетата в Примере 68.
MS m/e (ESI) 396 (MH+-CF3COOH).
Пример 76
Трифторацетат 7-(2-бутинил)-2-изопропилсульфанил-1-метил-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
6 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,15 мл 1-метил-2-пирролидона и добавляли 15 мг натриевой соли пропан-2-тиола. Смесь перемешивали при комнатной температуре в течение 5 часов. К реакционному раствору добавляли насыщенный водный раствор хлорида аммония, и смесь экстрагировали этилацетатом. Органический слой концентрировали, и остаток растворяли в 0,40 мл трифторуксусной кислоты. Раствор концентрировали в обильном токе газообразного азота. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 4,56 мг указанного в заголовке соединения.
MS m/e (ESI) 361 (MH+-CF3COOH).
Пример 79
Трифторацетат [7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илсульфанил]уксусной кислоты
6 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,15 мл N-метилпирролидона и к ним добавляли 20 мкл метил меркаптоацетата и 6 мг карбоната калия. После перемешивания смеси при комнатной температуре в течение 5 часов к реакционному раствору добавляли насыщенный водный раствор хлорида аммония. Смесь экстрагировали этилацетатом. Органический слой концентрировали. Полученный остаток растворяли в растворе, состоящем из 0,20 мл этанола и 0,20 мл 5 н водного раствора гидроксида натрия. Смесь перемешивали при комнатной температуре в течение ночи и затем концентрировали в обильном токе газообразного азота. Остаток растворяли в 0,40 мл трифторуксусной кислоты и раствор концентрировали в обильном токе газообразного азота. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 0,96 мг трифторацетата 7-(2-бутинил)-2-меркапто-1-метил-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она [MS m/e(ESI)319(MH+-CF3COOH)] и 0,61 мг трифторацетата [7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илсульфанил]уксусной кислоты.
[MS m/e (ESI) 377 (MH+-CF3COOH)].
Пример 82
Трифторацетат 7-(2-бутинил)-2-циан-1-метил-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
8 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,2 мл N-метилпирролидона и добавляли 10 мг цианида натрия. Смесь перемешивали при 50°С в течение 1 часа. К реакционной смеси добавляли воду и смесь экстрагировали этилацетатом. Органический слой концентрировали, что давало 14 мг трет-бутил 4-[7-(2-бутинил)-2-циан-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата. 5 мг данного соединения растворяли в трифторуксусной кислоте и раствор концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 4,12 мг указанного в заголовке соединения.
MS m/e (ESI) 312 (MH+-CF3COOH).
Пример 83
7-(2-Бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-карбоксамид
(а) трет-Бутил 4-[7-(2-бутинил)-2-карбамоил-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
176 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 2 мл N-метилпирролидона и добавляли 100 мг цианида натрия. Смесь перемешивали при 50°С в течение 0,5 часа. К реакционной смеси добавляли воду, и смесь экстрагировали этилацетатом. Органический слой концентрировали, что давало 170 мг трет-бутил 4-[7-(2-бутинил)-2-циан-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата. 98 мг данного соединения растворяли в смеси 3 мл тетрагидрофурана и 2 мл метанола и добавляли 0,5 мл водного раствора 20% аммиака и 0,5 мл водного раствора 30% перекиси водорода. Смесь перемешивали при комнатной температуре в течение ночи. К реакционному раствору добавляли этилацетат и смесь промывали водой. Органический слой сушили над безводным сульфатом магния, затем фильтровали. Растворитель выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 77 мг указанного в заголовке соединения из фракции, элюированной смесью этилацетат-метанол.
1H-ЯМР (CDCl3)
δ 1,49 (с, 9H) 1,83 (т, J=1,2 Гц, 3H) 3,42-3,49 (м, 4H) 3,58-3,65 (м, 4H) 3,95 (с, 3H) 5,01 (д, J=2,4 Гц, 2H) 5,54 (ушир. 1H) 7,61 (ушир. 1H).
(b) 7-(2-Бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-карбоксамид
77 мг трет-бутил 4-[7-(2-бутинил)-2-карбамоил-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 1 мл трифторуксусной кислоты, и раствор концентрировали. Остаток очищали с помощью хроматографии с использованием NH-силикагеля. Таким образом получали 49 мг указанного в заголовке соединения из фракции, элюированной смесью этилацетат-метанол (5:1).
1H-ЯМР (CDCl3)
δ 1,83 (т, J=2,4 Гц, 3H) 3,05-3,07 (м, 4H) 3,45-3,48 (м, 4H) 3,94 (с, 3H) 4,98 (с, 2H) 5,57 (ушир. 1H) 7,65 (ушир. 1H).
Пример 86
Гидрохлорид 7-(2-бутинил)-2-метокси-1-(2-фенилэтил)-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
(а) [7-Бензил-2,6-диоксо-1-(2-фенилэтил)-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионат
Смесь, состоящую из 500 мг [7-бензил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионата, 0,38 мл 2-бромэтилбензола, 390 мг безводного карбоната калия и 5 мл N,N-диметилформамида, перемешивали в масляной бане при 50°С в течение 2 часов. Реакционную смесь экстрагировали этилацетатом и водой, и органический слой промывали водой и затем насыщенным соляным раствором. Органическую жидкость сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Остаток подвергали кристаллизации смесью этилацетат-гексан, что давало 540 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,19 (с, 9H) 2,92-2,98 (м, 2H) 4,19-4,25 (м, 2H) 5,48 (с, 2H) 6,11 (с, 2H) 7,17-7,40 (м, 10H) 7,54 (с, 1H).
(b) [7-(2-Бутинил)-8-хлор-2,6-диоксо-1-(2-фенилэтил)-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионат
Смесь, состоящую из 540 мг [7-Бензил-2,6-диоксо-1-(2-фенилэтил)-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионата, 50 мг 10% палладия на угле и 8 мл уксусной кислоты, перемешивали в атмосфере водорода при комнатной температуре в течение ночи. Реакционную смесь фильтровали и затем концентрировали при пониженном давлении, что давало 410 мг остатка.
Весь остаток объединяли с 0,15 мл 1-бром-2-бутина, 300 мг безводного карбоната калия и 5 мл N,N-диметилформамида. Смесь перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор экстрагировали этилацетатом и водой. Органический слой промывали водой и затем насыщенным соляным раствором. Органическую жидкость сушили над безводным сульфатом магния и концентрировали при пониженном давлении, что давало 470 мг остатка.
Весь остаток объединяли со 180 мг N-хлорсукцинимида и 5 мл N,N-диметилформамида. Смесь перемешивали при комнатной температуре в течение 2 часов. После добавления к реакционному раствору 0,5 мл водного раствора 1М тиосульфата натрия, смесь экстрагировали этилацетатом и водой. Органический слой промывали водой и затем насыщенным соляным раствором. Органическую жидкость сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. С помощью кристаллизации смесью этилацетат-гексан получали 380 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,21 (с, 9H) 1,83 (т, J=2 Гц, 3H) 2,92-2,98 (м, 2H) 4,19-4,25 (м, 2H) 5,11 (кв, J=2 Гц, 2H) 6,05 (с, 2H) 7,18-7,32 (м, 5H).
(с) трет-бутил 4-[7-(2-бутинил)-2,6-диоксо-1-(2-фенилэтил)-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
Смесь, состоящую из 380 мг [7-(2-бутинил)-8-хлор-2,6-диоксо-1-(2-фенилэтил)-1,2,6,7-тетрагидропурин-3-ил]метил-2,2-диметилпропионата, 460 мг трет-бутил пиперазин-1-карбоксилата и 0,5 мл N-метилпирролидона, перемешивали в масляной бане при 150°С в течение 15 минут. Реакционную смесь экстрагировали этилацетатом и водой, и органический слой промывали водой и затем насыщенным соляным раствором. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток растворяли в смеси этилацетат/гексан (1/1). Раствор фильтровали через небольшое количество силикагеля и затем промывали смесью этилацетат/гексан (1/1). Фильтрат объединяли с промывающим раствором. Перемешанный раствор концентрировали при пониженном давлении, что давало 570 мг остатка.
Весь остаток объединяли с 5 мл тетрагидрофурана и 2,5 мл метанола. К смеси добавляли 33 мг гидрида натрия, и получившуюся в результате смесь перемешивали при комнатной температуре в течение 30 минут. К реакционному раствору добавляли 1 мл 1 н соляной кислоты и смесь экстрагировали этилацетатом и водой, далее промывали водой и затем насыщенным соляным раствором. Органическую жидкость сушили над безводным сульфатом магния и концентрировали при пониженном давлении, что давало 350 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,50 (с, 9H) 1,85 (т, J=2 Гц, 3H) 2,91-2,98 (м, 2H) 3,37 (ушир.с, 4H) 3,56-3,62 (м, 4H) 4,15-4,22 (м, 2H) 4,87 (кв, J=2 Гц, 2H) 7,18-7,35 (м, 5H).
(d) трет-Бутил 4-[7-(2-бутинил)-2-хлор-6-оксо-1-(2-фенилэтил)-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
Смесь, состоящую из 290 мг трет-бутил 4-[7-(2-бутинил)-2,6-диоксо-1-(2-фенилэтил)-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата и 4 мл хлорокиси фосфора, нагревали и перемешивали в масляной бане при 120°С в течение 8 часов. Реакционный раствор концентрировали при пониженном давлении, и остаток растворяли в 5 мл тетрагидрофурана. Данный раствор, по каплям, добавляли к смеси, состоящей из 250 мг ди-трет-бутил дикарбоната, 10 мл насыщенного раствора бикарбоната натрия и 10 мл тетрагидрофурана, в то время, как смесь перемешивали и охлаждали льдом. Смесь инкубировалась при комнатной температуре в течение 4 часов и затем экстрагировали этилацетатом. Органический слой промывали водой, затем насыщенным соляным раствором, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием 30-50% смеси этилацетат/гексан. Затем материал подвергали дальнейшей очистке с помощью колоночной хроматографии с обратной фазой с использованием 50-100% смеси метанол/вода, что давало 60 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,49 (с, 9H) 1,84 (т, J=2 Гц, 3H) 3,10-3,16 (м, 2H) 3,40-3,46 (м, 2H) 3,57-3,63 (м, 4H) 4,42-4,49 (м, 4H) 4,94 (кв, J=2 Гц, 2H) 7,21-7,34 (м, 5H).
(е) Гидрохлорид 7-(2-бутинил)-2-метокси-1-(2-фенилэтил)-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
10 мг гидрида натрия (60%; масляный) добавляли к смеси, состоящей из 7 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-6-оксо-1-(2-фенилэтил)-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата и 0,5 мл метанола. Смесь перемешивали при комнатной температуре в течение 20 минут. К реакционному раствору добавляли воду. Смесь экстрагировали этилацетатом. Органический слой промывали водой и затем насыщенным соляным раствором и концентрировали. К остатку добавляли 0,5 мл трифторуксусной кислоты. Смесь перемешивали при комнатной температуре в течение 30 минут и затем концентрировали. Остаток очищали с помощью колоночной хроматографии с обратной фазой с использованием 20-80% смеси метанол/вода (содержащей 0,1% концентрированной соляной кислоты), что давало 4,3 мг указанного в заголовке соединения.
1H-ЯМР (ДМСО-d6)
δ 1,80 (ушир.с, 3H) 2,85 (т, J=7 Гц, 2H) 3,28 (ушир.с, 4H) 3,48-3,54 (м, 4H) 3,83 (с, 3H) 4,15 (т, J=7 Гц, 2H) 4,97 (ушир.с, 2H) 7,16-7,24 (м, 3H) 7,29 (т, J=8 Гц, 2H) 9,08 (ушир.с, 2H).
Пример 88
Гидрохлорид [7-(2-бутинил)-6-оксо-1-(2-фенилэтил)-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илсульфанил]ацетата
Указанное в заголовке соединение синтезировали по способу примера 86 с использованием метилтиогликолята вместо метанола и использованием карбоната калия в качестве основания в Примере 86(е).
1H-ЯМР (ДМСО-d6)
δ 1,80 (с, 3H) 2,96 (т, J=8 Гц, 2H) 3,29 (ушир.с, 4H) 3,50-3,56 (м, 4H) 3,68 (с, 3H) 4,16 (с, 2H) 4,23 (т, J=8 Гц, 2H) 4,99 (с, 2H) 7,24-7,38 (м, 5H) 8,96 (ушир.с, 2H).
Пример 95
Трифторацетат 7-(2-бутинил)-2-хлор-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
(а) трет-Бутил 4-[7-(2-бутинил)-2-хлор-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
Смесь, состоящую из 1,0 г трет-бутил 4-[7-(2-бутинил)-2,6-дихлор-7Н-пурин-8-ил]пиперазин-1-карбоксилата, 580 мг ацетата натрия и 10 мл диметилсульфоксида, перемешивали в масляной бане при 80°С в течение 24 часов. Реакционный раствор экстрагировали этилацетатом и водой. Органический слой промывали водой и затем насыщенным соляным раствором, затем сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием 50-70% смеси этилацетат/гексан и кристаллизовали смесью этилацетат/гексан, что давало 800 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,49 (с, 9H) 1,83 (т, J=2 Гц, 3H) 3,44 (ушир.с, 4H) 3,56-3,63 (м, 4H) 4,94 (кв, J=2 Гц, 2H).
(b) Трифторацетат 7-(2-бутинил)-2-хлор-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
8 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в трифторуксусной кислоте и раствор концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 3,45 мг указанного в заголовке соединения.
MS m/e (ESI) 307 (MH+-CF3COOH).
Пример 96
Гидрохлорид 2-[7-(2-бутинил)-2-диметиламино-6-оксо-8-(пиперазин-1-ил)-6,7-дигидропурин-1-илметил]бензонитрила
(а) трет-Бутил 4-[7-(2-бутинил)-2-хлор-1-(2-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
Смесь, состоящую из 100 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата, 60 мг 2-цианбензилбромида, 68 мг безводного карбоната калия и 1 мл N,N-диметилформамида, перемешивали при комнатной температуре в течение 4 часов. К реакционному раствору добавляли смесь этилацетат/гексан (1/1) и вода. Нерастворимый материал удаляли с помощью фильтрования. Фильтрат экстрагировали этилацетатом. Органический слой промывали водой и затем насыщенным соляным раствором, сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием 30-50% смеси этилацетат/гексан, что давало 50 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,49 (с, 9H) 1,83 (т, J=2 Гц, 3H) 3,43-3,49 (м, 4H) 3,58-3,64 (м, 4H) 4,95 (кв, J=2 Гц, 2H) 5,72 (с, 2H) 7,06 (д, J=8 Гц, 1H) 7,39 (т, J=8 Гц, 1H) 7,51 (т, J=8 Гц, 1H) 7,71 (д, J=8 Гц, 1H).
(b) трет-Бутил 4-[7-(2-бутинил)-1-(2-цианбензил)-2-диметиламино-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
Смесь, состоящую из 8 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-(2-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата, 20 мкл водного раствора 50% диметиламина и 0,2 мл N,N-диметилформамида, перемешивали при комнатной температуре в течение 2 часов. Реакционный раствор экстрагировали этилацетатом и водой. Органический слой промывали водой, затем насыщенным соляным раствором и затем концентрировали. Остаток отделяли с помощью тонкослойной хроматографии на силикагеле с использованием 70% смеси этилацетат/гексан, что давало 6,5 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,50 (с, 9H) 1,81 (т, J=2 Гц, 3H) 2,73 (с, 6H) 3,38-3,45 (м, 4H) 3,56-3,64 (м, 4H) 4,91, (кв, J=2 Гц, 2H) 5,55 (с, 2H) 7,07 (д, J=8 Гц, 1H) 7,32 (т, J=8 Гц, 1H) 7,46, (т, J=8 Гц, 1H) 7,65 (д, J=8 Гц, 1H).
(с) Гидрохлорид 2-[7-(2-бутинил)-2-диметиламино-6-оксо-8-(пиперазин-1-ил)-6,7-дигидропурин-1-илметил]бензонитрила
6,5 мг трет-бутил 4-[7-(2-бутинил)-1-(2-цианбензил)-2-диметиламино-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,5 мл трифторуксусной кислоты, и смесь оставляли стоять при комнатной температуре в течение 20 минут. Реакционный раствор концентрировали, и остаток очищали с помощью колоночной хроматографии с обратной фазой с использованием 20-80% смеси метанол/вода (содержащей 0,1% концентрированной соляной кислоты), что давало 6,4 мг указанного в заголовке соединения.
1H-ЯМР (ДМСО-d6)
δ 1,76 (с, 3H) 2,69 (с, 6H) 3,28 (ушир.с, 4H) 3,51 (ушир.с, 4H) 4,91 (с, 2H) 5,40 (с, 2H) 7,04 (д, J=8 Гц, 1H) 7,43 (т, J=8 Гц, 1H) 7,60 (т, J=8 Гц, 1H) 7,83 (д, J=8 Гц, 1H) 8,90 (ушир.с, 2H).
Пример 98
Гидрохлорид 2-[7-(2-бутинил)-2-метокси-6-оксо-8-(пиперазин-1-ил)-6,7-дигидропурин-1-илметил]бензонитрила
Указанное в заголовке соединение синтезировали по способу примера 96 с использованием метанола вместо диметиламина и использованием карбоната калия в качестве основания в Примере 96(b).
1H-ЯМР (ДМСО-d6)
δ 1,79 (с, 3H) 3,28 (ушир.с, 4H) 3,48-3,56 (м, 4H) 3,91 (с, 3H) 4,97 (с, 2H) 5,32 (с, 2H) 7,19 (д, J=8 Гц, 1H) 7,48 (т, J=8 Гц, 1H) 7,63 (т, J=8 Гц, 1H) 7,87 (д, J=8 Гц, 1H) 9,05 (ушир.с, 2H).
Пример 109
Трифторацетат 7-бензил-1-метил-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
(а) 7-Бензил-1,7-дигидропурин-6-он
18,23 г инозина растворяли в 90 мл диметилсульфоксида и добавляли 16 мл бензилбромида. Смесь перемешивали при комнатной температуре в течение ночи. Реакционный раствор вливали в 3 л этилацетата. Полученный супернатант (всплывающий слой) удаляли и осажденное масло растворяли в 10% соляной кислоте (135 мл). Раствор нагревался при 70°С с перемешиванием в течение 4 часов. Раствор охлаждали до комнатной температуры и затем нейтрализовали до рН 7 с использованием 5 н водного раствора гидроксида натрия. Осажденное твердое вещество собирали фильтрованием и сушили, что давало 12,748 г указанного в заголовке соединения.
(b) трет-Бутил 4-(7-бензил-6-оксо-6,7-дигидро-1Н-пурин-8-ил)пиперазин-1-карбоксилат
12,748 г 7-бензил-1,7-дигидропурин-6-она растворяли в 150 мл N,N-диметилформамида и туда добавляли 7,9 г N-хлорсукцинимида. Реакционный раствор перемешивали в течение ночи и затем разбавляли этилацетатом. Раствор промывали водой и 1 н соляной кислотой, и сушили над безводным сульфатом магния. Раствор фильтровали и фильтрат концентрировали, что давало 6,103 г 7-бензил-8-хлор-1,7-дигидропурин-6-она. Данное соединение объединяли с 20 г трет-бутил пиперазин-1-карбоксилата и смесь нагревали при 150°С. После перемешивания в течение 1 часа реакционную смесь объединяли с этилацетатом и водой и распределяли. Органический слой промывали 1 н соляной кислотой и сушили над безводным сульфатом магния. После фильтрования фильтрат концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 1,539 г указанного в заголовке соединения из фракции, элюированной смесью этилацетат-метанол (10:1).
1H-ЯМР (CDCl3)
δ 1,39 (с, 9H) 3,07-3,10 (м, 4H) 3,35-3,39 (м, 4H) 5,44 (с, 2H) 7,16-7,18 (м, 2H) 7,22-7,32 (м, 3H) 7,91 (с, 1H) 12,18 (с, 1H).
(с) Трифторацетат 7-бензил-1-метил-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
15 мг Трет-бутил 4-(7-бензил-6-оксо-6,7-дигидро-1Н-пурин-8-ил)пиперазин-1-карбоксилата растворяли в 1 мл N,N-диметилформамида и туда добавляли 10 мг гидрида натрия и 10 мкл метилйодида. Смесь перемешивали при комнатной температуре в течение 3 дней, затем добавляли этилацетат и воду и слои разделяли. Органический слой концентрировали и остаток растворяли в трифторуксусной кислоте. Раствор концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусную кислоту)), что давало 4,31 мг указанного в заголовке соединения.
MS m/e (ESI) 325 (MH+-CF3COOH).
Пример 115
Трифторацетат 3-(2-бутинил)-5-метил-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
(а) Этил 2-бром-3-(2-бутинил)-5-циан-3Н-имидазол-4-карбоксилат
4,56 мл серной кислоты добавляли к 170 мл этанола, содержащего 16,80 г 2-бром-1Н-имидазол-4,5-дикарбонитрила [CAS №50847-09-1], и смесь нагревали с обратным холодильником в течение 48 часов. Раствор охлаждали и затем к нему добавляли 500 мл этилацетата и 200 мл воды. Органический слой сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток растворяли в N,N-диметилформамиде и туда добавляли 14,1 г карбоната калия и 8,6 мл 2-бутинилбромида. Смесь перемешивали при комнатной температуре в течение 18 часов. К раствору добавляли 500 мл этилацетата, и смесь три раза промывали 300 мл воды и затем 300 мл насыщенного раствора хлорида натрия. Затем раствор сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 4,09 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (9:1).
1H-ЯМР (CDCl3)
δ 1,43 (т, J=7,2 Гц, 3H) 1,81 (с, 3H) 4,47 (кв, J=7,2 Гц, 2H) 5,16 (с, 2H).
(b) трет-Бутил 4-[1-(2-бутинил)-4-циан-5-этоксикарбоксил-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
4,09 г этил 2-бром-3-(2-бутинил)-5-циан-3Н-имидазол-4-карбоксилата объединяли с 7,70 г трет-бутил пиперазин-1-карбоксилата, и смесь нагревали до 150°С с перемешиванием в течение 50 минут. Реакционная смесь растворяли в толуоле. Смесь очищалась с помощью колоночной хроматографии на силикагеле. Таким образом получали 4,47 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (2:1).
1H-ЯМР (CDCl3)
δ 1,43 (т, J=7,2 Гц, 3H) 1,47 (с, 9H) 1,82 (т, J=2,3 Гц, 3H) 3,08-3,13 (м, 4H) 3,57-3,61 (м, 4H) 4,44 (кв, J=7,2 Гц, 2H) 4,89 (кв, J=2,3 Гц, 2H).
(с) трет-Бутил 4-[1-(2-бутинил)-5-этоксикарбонил-4-тиокарбамоил-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
5 мл водного раствора 50% сульфида аммония добавляли к 20 мл раствора этанола, содержащего 0,80 г трет-бутил 4-[1-(2-бутинил)-4-циан-5-этоксикарбонил-1Н-имидазол-2-ил]пиперазин-1-карбоксилата, и смесь нагревали при 60°С в течение 14 часов. К смеси добавляли 100 мл этилацетата и 50 мл воды, и органический слой последовательно промывали 50 мл воды и 50 мл насыщенного раствора хлорида натрия. Реакционный раствор сушили над безводным сульфатом магния и затем фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 0,58 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (3:2).
1H-ЯМР (CDCl3)
δ 1,43 (т, J=7,2 Гц, 3H) 1,48 (с, 9H) 1,82 (т, J=2,3 Гц, 3H) 3,12-3,16 (м, 4H) 3,54-3,59 (м, 4H) 4,44 (кв, J=7,2 Гц, 2H) 4,89 (кв, J=2,3 Гц, 2H) 7,41 (ушир.с, 1H) 8,88 (ушир.с, 1H).
(d) трет-Бутил 4-[1-(2-бутинил)-5-этоксикарбонил-4-метилсульфанилкарбонимидоил-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
0,235 г триметилоксонийтетрафторбората добавляли к 20-мл раствора дихлорметана, содержащего 0,58 г трет-бутил 4-[1-(2-бутинил)-5-этоксикарбонил-4-тиокарбамоил-1Н-имидазол-2-ил]пиперазин-1-карбоксилата, и смесь перемешивали при комнатной температуре в течение 18 часов. К раствору добавляли 50 мл дихлорметана, и смесь промывали 20 мл насыщенного раствора бикарбоната натрия. Смесь сушили над безводным сульфатом магния и концентрировали при пониженном давлении, что давало 0,55 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,41 (т, J=7,2 Гц, 3H) 1,47 (с, 9H) 1,81 (т, J=2,3 Гц, 3H) 2,39 (с, 3H) 3,12-3,16 (м, 4H) 3,56-3,59 (м, 4H) 4,42 (кв, J=7,2 Гц, 2H) 4,80 (кв, J=2,3 Гц, 2H).
(е) трет-Бутил 4-[1-(2-бутинил)-5-этоксикарбонил-4-метилсульфанилкарбонил-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
5 мл 2 н водного раствора соляной кислоты добавляли в 30-мл раствор этанола, содержащий 0,55 г трет-бутил 4-[1-(2-бутинил)-5-этоксикарбонил-4-метилсульфанилкарбонимидоил-1Н-имидазол-2-ил]пиперазин-1-карбоксилата, и смесь нагревали при 60°С в течение 5 часов. После того, как реакционный раствор концентрировали при пониженном давлении, к нему добавляли 25 мл этилацетата и 1 н раствор гидроксида натрия. Водный слой экстрагировали 25 мл этилацетата и органические слои объединяли вместе. Смесь промывали 10 мл насыщенного раствора хлорида натрия, содержащего 1 мл 1 н раствора гидроксида натрия, и сушили над безводным сульфатом магния. Раствор фильтровали и фильтрат концентрировали при пониженном давлении. Остаток растворяли в 10 мл дихлорметана и туда добавляли 0,10 мл триэтиламина и 0,256 г ди-трет-бутил дикарбоната. Смесь перемешивали при комнатной температуре в течение 15 часов и затем к ней добавляли 25 мл этилацетата. Смесь последовательно промывали 10 мл 0,1 н соляной кислоты, 10 мл насыщенного раствора бикарбоната натрия и 10 мл насыщенного раствора хлорида натрия и затем сушили над безводным сульфатом магния. Раствор концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 0,15 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (4:1).
1H-ЯМР (CDCl3)
δ 1,43 (т, J=7,1 Гц, 3H) 1,48 (с, 9H) 1,81 (т, J=2,3 Гц, 3H) 2,40 (с, 3H) 3,16-3,20 (м, 4H) 3,55-3,59 (м, 4H) 4,35 (кв, J=7,1 Гц, 2H) 4,80 (кв, J=2,3 Гц, 2H).
(f) трет-Бутил 4-[1-(2-бутинил)-5-этоксикарбонил-4-гидроксиметил-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
0,187 г меркурий(II)ацетата и 0,090 г боргидрида натрия при 0°С добавляли к 8 мл этанольного раствора, содержащего 0,265 г трет-бутил 4-[1-(2-бутинил)-5-этоксикарбонил-4-метилсульфанилкарбонил-1Н-имидазол-2-ил]пиперазин-1-карбоксилата, и смесь перемешивали при комнатной температуре в течение 4 часов. После добавления к раствору 0,187 г меркурий(II)ацетата и 0,090 г боргидрида натрия смесь перемешивали при комнатной температуре в течение 15 часов. К раствору добавляли 100 мл этилацетата и 50 мл 0,5 н соляной кислоты и органический слой последовательно промывали 50 мл воды и 50 мл насыщенного раствора хлорида натрия. Смесь сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. 0,172 г исходного материала собирали из фракции, элюированной смесью гексан-этилацетат (4:1). Затем получали 0,061 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (1:4).
1H-ЯМР (CDCl3)
δ 1,42 (т, J=7,1 Гц, 3H) 1,48 (с, 9H) 1,81 (т, J=2,3 Гц, 3H) 3,17-3,21 (м, 4H) 3,41 (т, J=4,8 Гц, 1H) 3,56-3,60 (м, 4H) 4,36 (кв, J=7,1 Гц, 2H) 4,75 (д, J=4,8 Гц, 2H) 4,81 (кв, J=2,3 Гц, 2H).
(g) трет-Бутил 4-[1-(2-бутинил)-5-этоксикарбонил-4-формил-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
0,120 г диоксида марганца добавляли к 2-мл дихлорметановому раствору, содержащему 0,061 г трет-бутил 4-[1-(2-бутинил)-5-этоксикарбонил-4-гидроксиметил-1Н-имидазол-2-ил]пиперазин-1-карбоксилата, и смесь перемешивали при комнатной температуре в течение 15 часов. Реакционный раствор фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 0,055 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (7:3).
1H-ЯМР (CDCl3)
δ 1,42 (т, J=7,1 Гц, 3H) 1,48 (с, 9H) 1,82 (т, J=2,3 Гц, 3H) 3,23-3,26 (м, 4H) 3,55-3,59 (м, 4H) 4,45 (кв, J=7,1 Гц, 2H) 4,89 (кв, J=2,3 Гц, 2H) 10,36 (с, 1H).
(h) трет-Бутил 4-[1-(2-бутинил)-6-метил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилат
0,05 мл метилгидразина добавляли к 2,5-мл этанольного раствора, содержащего 0,055 г трет-бутил 4-[1-(2-бутинил)-5-этоксикарбонил-4-формил-1Н-имидазол-2-ил]пиперазин-1-карбоксилата. Смесь перемешивали при 80°С в течение 15 часов и затем нагревали при 130°С в течение 14 часов. Реакционный раствор концентрировали при пониженном давлении. Затем остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 0,035 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (1:1).
1H-ЯМР (CDCl3)
δ 1,52 (с, 9H) 1,83 (т, J=2,3 Гц, 3H) 3,38-3,42 (м, 4H) 3,61-3,64 (м, 4H) 3,85 (с, 3H) 5,09 (кв, J=2,3 Гц, 2H) 8,13 (с, 1H).
МС m/e (ESI) 387,4 (MH+).
(i) Трифторацетат 3-(2-бутинил)-5-метил-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
0,4 мл трифторуксусной кислоты добавляли к 0,4-мл раствору дихлорметана, содержащего 0,0351 г трет-бутил 4-[1-(2-бутинил)-6-метил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилата и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 0,0295 г указанного в заголовке соединения.
1H-ЯМР (CD3OD)
δ 1,83 (т, J=2,3 Гц, 3H) 3,45-3,49 (м, 4H) 3,65-3,69 (м, 4H) 3,83 (с, 3H) 5,15 (кв, J=2,3 Гц, 2H) 8,20 (с, 1H).
МС m/e (ESI) 287,09 (MH+-CF3COOH).
Пример 116
Трифторацетат 5-бензилоксиметил-3-(2-бутинил)-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
(а) Диметиламид 5-бензилоксиметил-4-оксо-4,5-дигидроимидазо[4,5-d]пиридазин-1-сульфокислоты
2,08 г триэтиламина, 2,80 г N,N-диметилсульфамоилхлорида и 0,22 г 4-диметиламинопиридина добавляли к 50 мл раствора дихлорметана, содержащего 3,04 г 5-бензилоксиметилимидазо[4,5-d]пиридазин-4-она [CAS №82137-50-6] (R. Paul Gagnier, Michael J. Halat, and Brian A. Otter Journal of Heterocyclic Chemistry, 21, p481, 1984), и смесь нагревали с обратным холодильником в течение 4 часов. К раствору добавляли 250 мл этилацетата, и смесь последовательно промывали 50 мл водного раствора 1 н соляной кислоты, 50 мл насыщенного раствора бикарбоната натрия и 50 мл насыщенного раствора хлорида натрия. Смесь сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 2,86 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (2:3).
1H-ЯМР (CDCl3)
δ 2,98 (с, 6H) 4,77 (с, 2H) 5,74 (с, 2H) 7,30-7,39 (м, 5H) 8,21 (с, 1H) 8,46 (с, 1H).
(b) Диметиламид 5-бензилоксиметил-2-хлор-4-оксо-4,5-дигидроимидазо[4,5-d]пиридазин-1-сульфокислоты
5,3 мл н-бутиллития (2,0М раствор циклогексана) в атмосфере азота при -78°С добавляли к 150 мл раствора тетрагидрофурана, содержащего 3,34 г диметиламида 5-бензилоксиметил-4-оксо-4,5-дигидроимидазо[4,5-d]пиридазин-1-сульфокислоты, и смесь перемешивали при -78°С в течение 1 часа. Затем к данному раствору добавляли 20 мл раствора тетрагидрофурана, содержащего 3,26 г гексахлорэтана. Смесь оставляли для нагрева до комнатной температуры. К раствору добавляли 25 мл 5% водного раствора хлорида аммония и смесь экстрагировали 50 мл этилацетата. Органический слой последовательно промывали 25 мл воды и 25 мл насыщенного раствора хлорида натрия и затем сушили над безводным сульфатом магния. Органическая жидкость концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 2,31 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (2:3).
1H-ЯМР (CDCl3)
δ 3,12 (с, 6H) 4,77 (с, 2H) 5,70 (с, 2H) 7,30-7,39 (м, 5H) 8,48 (с, 1H).
(с) трет-Бутил 4-(6-бензилоксиметил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил)пиперазин-1-карбоксилат
Смесь, состоящую из 2,31 г диметиламида 5-бензилоксиметил-2-хлор-4-оксо-4,5-дигидроимидазо[4,5-d]пиридазин-1-сульфокислоты и 4,49 г трет-бутил пиперазин-1-карбоксилата, нагревали при 150°С в атмосфере азота в течение 2,5 часов. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 1,94 г указанного в заголовке соединения из фракции, элюированной этилацетатом.
1H-ЯМР (CDCl3)
δ 3,54-3,58 (м, 4H) 3,71-3,75 (м, 4H) 4,68 (с, 2H) 5,65 (с, 2H) 7,25-7,35 (м, 5H) 8,21 (с, 1H) 12,58 (ушир.с, 1H).
(d) трет-Бутил 4-[6-бензилоксиметил-1-(2-бутинил)-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилат
0,74 г карбоната калия и 0,078 г 2-бутинилбромида добавляли к 20-мл раствора N,N-диметилформамида, содержащего 0,216 г трет-бутил 4-(6-бензилоксиметил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил)пиперазин-1-карбоксилата, и смесь перемешивали при комнатной температуре в течение 16 часов. Затем к раствору добавляли 50 мл этилацетата. Органический слой трижды промывали 20 мл воды и затем 10 мл насыщенного раствора хлорида натрия. Раствор сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 0,139 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (3:2).
1H-ЯМР (CDCl3)
δ 1,50 (с, 9H) 1,86 (т, J=2,3 Гц, 3H) 3,38-3,44 (м, 4H) 3,61-3,66 (м, 4H) 4,72 (с, 2H) 5,10 (кв, J=2,3 Гц, 2H) 5,65 (с, 2H) 7,25-7,38 (м, 5H) 8,18 (с, 1H).
(е) Трифторацетат 5-бензилоксиметил-3-(2-бутинил)-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
Получали 0,0043 г указанного в заголовке соединения с помощью обработки 0,0073 г трет-бутил 4-[6-бензилоксиметил-1-(2-бутинил)-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилата и очистки продукта по способу примера 115(i).
1H-ЯМР (CD3OD)
δ 1,83 (т, J=2,3 Гц, 2H) 3,45-3,49 (м, 4H) 3,65-3,69 (м, 4H) 4,69 (с, 2H) 5,15 (кв, J=2,3 Гц, 2H) 5,64 (с, 2H) 7,17-7,32 (м, 5H) 8,20 (с, 1H).
МС m/e (ESI) 393,28 (MH+-CF3COOH).
Пример 117
Трифторацетат 3-(2-бутинил)-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
8 мл раствора дихлорметана, содержащего 0,123 г трет-бутил 4-[6-бензилоксиметил-1-(2-бутинил)-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилата, в атмосфере азота, охлаждали до -78°С и к нему добавляли 1,9 мл треххлористого бора (1,0М раствор дихлорметана). Смесь перемешивали при -78°С в течение 5 часов и к ней добавляли 10 мл смеси растворителей 1:1 дихлорметан-метанол. Смесь перемешивали при -78°С в течение 2 часов и затем ей давали возможность нагреться до комнатной температуры. Растворитель концентрировали при пониженном давлении и в смесь добавляли 10 мл метанола. После этого раствор снова концентрировали при пониженном давлении. Остаток растворяли в 3 мл пиридина, и смесь нагревали с обратным холодильником в течение 2 часов. 0,3 мл данного раствора концентрировали при пониженном давлении. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 0,005 г соединения, указанного в заголовке.
1H-ЯМР (CD3OD)
δ 1,83 (т, J=2,3 Гц, 3H) 3,45-3,49 (м, 4H) 3,65-3,69 (м, 4H) 5,16 (кв, J=2,3 Гц, 2H) 8,21 (с, 1H).
МС m/e (ESI) 273,16 (MH+-CF3COOH).
Пример 118
Гидрохлорид 2-[7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]бензамида
(а) трет-Бутил 4-[7-(2-бутинил)-2-(2-карбамоилфенокси)-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
200 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 2,0 мл 1-метил-2-пирролидона и к ним добавляли 85 мг салициламида и 129 мг карбоната калия. Смесь перемешивали при 100°С в течение 2 часов. После охлаждения реакционной смеси до комнатной температуры к ней добавляли 5,0 мл воды. После перемешивания смеси при комнатной температуре в течение 1 часа белый осадок собирали с помощью фильтрования. Полученное белое твердое вещество промывали водой и простым эфиром, что давало 221 мг указанного в заголовке соединения (89%).
1H-ЯМР (ДМСО-d6)
δ 1,43 (с, 9H) 1,79 (т, J=2,5 Гц, 3H) 3,23-3,27 (м, 4H) 3,36 (с, 3H) 3,48-3,52 (м, 4H) 4,95 (кв, 2,5 Гц, 2H) 6,59 (тд, J=8,0, 1,0 Гц, 1H) 6,63 (дд, J=8,0, 1,0 Гц, 1H) 7,14 (ддд, J=8,0, 7,5, 2,0 Гц, 1H) 7,80 (дд, J=7,5, 2,0 Гц, 1H).
МС m/e (ESI) 522 (MH+).
(b) Гидрохлорид 2-[7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]бензамида
210 мг трет-бутил 4-[7-(2-бутинил)-2-(2-карбамоилфенокси)-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата объединяли с 3,5 мл метанола и 2,1 мл 4 н раствора соляной кислоты-этилацетата. После перемешивания при комнатной температуре в течение 4 часов реакционный раствор концентрировали в обильной струе газообразного азота. Полученный в результате остаток промывали этанолом и этилацетатом, что давало 177 мг указанного в заголовке соединения (96%).
1H-ЯМР (ДМСО-d6)
δ 1,82 (т, J=2,3 Гц, 3H) 3,28-3,32 (м, 4H) 3,48 (с, 3H) 3,54-3,58 (м, 4H) 5,04 (кв, 2,3 Гц, 2H) 6,96 (ушир.т, J=7,0 Гц, 1H) 6,99 (ушир.д, J=8,0 Гц, 1H) 7,46 (ддд, J=8,0, 7,0, 1,5 Гц, 1H) 7,93 (ушир.д, J=8,0 Гц, 1H)
МС m/e (ESI) 422 (MH+-HCl).
Пример 119
3-(2-Бутинил)-5-метил-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-он
(а) 5-Метил-1-тритил-1,5-дигидроимидазо[4,5-d]пиридазин-4-он
78,8 г 5-метил-1,5-дигидроимидазо[4,5-d]пиридазин-4-она [CAS №76756-58-6] (Shih-Fong Chen and Raymond P. Panzica, Journal of Organic Chemistry 46, p2467, 1981) суспендировали в 2,5 л дихлорметана при комнатной температуре и к нему добавляли 78,8 г триэтиламина. К смеси добавляли 176 г трифенилхлорметана, после чего она перемешивали в течение 3 часов. К смеси добавляли 7,5 л этилацетата. После последовательного промывания 3-мя литрами воды и 3 л насыщенного раствора хлорида натрия смесь сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 136,5 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (от 20:80 до 0:100).
1H-ЯМР (CDCl3)
δ 3,79 (с, 3H) 6,92 (с, 1H) 7,07-7,13 (м, 6H) 7,32-7,40 (м, 9H) 7,87 (с, 1H).
(b) 2-Хлор-5-метил-1-тритил-1,5-дигидроимидазо[4,5-d]пиридазин-4-он
220 мл гексаметилдисилазида лития (1,0 М раствор тетрагидрофурана) при -75°С в атмосфере азота добавляли к 4 л раствора тетрагидрофурана, содержащего 68,3 г 5-метил-1-тритил-1,5-дигидроимидазо[4,5-d]пиридазин-4-она, и смесь перемешивали при -75°С в течение 1 часа. Затем к раствору добавляли 200 мл тетрагидрофуранового раствора, содержащего 82,3 г гексахлорэтана. Смеси давали возможность нагреться до -20°С. Добавляли 5 л 5% водного раствора хлорида аммония, и смесь экстрагировали 4 л этилацетата. Органический слой последовательно промывали 5 л воды и 5 л насыщенного раствора хлорида натрия. Раствор сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток суспендировали в 150 мл трет-бутилметилового эфира и затем собирали с помощью фильтрования. Твердое вещество дважды промывали 100 мл трет-бутилметилового эфира, что давало 69,7 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 3,78 (с, 3H) 5,81 (с, 1H) 7,25-7,27 (м, 6H) 7,28-7,38 (м, 9H).
(с) трет-Бутил 4-(6-метил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил)пиперазин-1-карбоксилат
69,7 г 2-Хлор-5-метил-1-тритил-1,5-дигидроимидазо[4,5-d]пиридазин-4-она объединяли с 153,4 г трет-бутилпиперазин-1-карбоксилата, и смесь перемешивали и нагревали до 100°С в атмосфере азота. Когда реакционная смесь становилась легко мешаемой (перемешиваемой), температура доводилась до 150°С. Смесь выдерживалась при данной температуре в течение 1 часа. Реакционному раствору давали возможность охладиться и затем его суспендировали в 250 мл трет-бутилметилового эфира. Суспендированный материал собирали с помощью фильтрования. Твердое вещество дважды промывалось 200 мл трет-бутилметилового эфира и три раза 200 мл воды. Твердое вещество снова дважды промывалось 200 мл трет-бутилметилового эфира и сушили, что давало 50,3 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,50 (с, 9H) 3,56-3,62 (м, 4H) 3,73-3,80 (м, 4H) 3,87 (с, 3H) 8,16 (с, 1H) 12,65 (ушир.с, 1H).
(d) трет-Бутил 4-[1-(2-бутинил)-6-метил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилат
43,9 г карбоната калия и 27,8 мл 2-бутинилбромида при 15°С в атмосфере азота последовательно добавляли к 5,5 л раствора N,N-диметилформамида, содержащего 88,4 г трет-бутил 4-(6-метил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил)пиперазин-1-карбоксилата. Реакционный раствор перемешивали при комнатной температуре в течение 22 часов и затем вливали в 10 л воды. Смесь экстрагировали 5 л этилацетата. Органический слой последовательно промывали два раза 5 л воды и 5 л насыщенного раствора хлорида натрия. Водный слой дважды экстрагировали 3 л этилацетата. Органические слои объединяли вместе и затем сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 54,3 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (от 3:2 до 3:7).
1H-ЯМР (CDCl3)
δ 1,52 (с, 9H) 1,83 (т, J=2,3 Гц, 3H) 3,38-3,42 (м, 4H) 3,61-3,64 (м, 4H) 3,85 (с, 3H) 5,09 (кв, J=2,3 Гц, 2H) 8,13 (с, 1H).
(е) 3-(2-Бутинил)-5-метил-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-он
200 мл трифторуксусной кислоты добавляли к 200 мл раствора дихлорметана, содержащего 54,3 г трет-бутил 4-[1-(2-бутинил)-6-метил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилата, и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали при пониженном давлении и остаток растворяли в 500 мл этилацетата. Постепенно добавляли 1 л 10% водного раствора бикарбоната натрия. Затем к раствору добавляли 1 л этилацетата и 500 мл 5 н водного раствора гидроксида натрия. Органический слой отделяли. Затем водный слой пять раз экстрагировали 1 л дихлорметана. Органические слои объединяли вместе, промывали 500 мл водного раствора 2 н гидроксида натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток перекристаллизовывали из этилацетата, что давало 30,5 г кристаллического соединения, указанного в заголовке.
1H-ЯМР (CDCl3)
δ 1,84 (т, J=2,3 Гц, 3H) 3,05-3,09 (м, 4H) 3,38-3,44 (м, 4H) 3,85 (с, 3H) 5,06 (кв, J=2,3 Гц, 2H) 8,13 (с, 3H).
Пример 119-2
Толуол-4-сульфонат 3-(2-бутинил)-5-метил-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
98,7 мг 3-(2-Бутинил)-5-метил-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она растворяли в 1 мл этанола и затем, при одновременном перемешивании раствора, к нему добавляли 1 мл раствора этанола, содержащего 101 мг моногидрата п-толуолсульфокислоты. Смесь охлаждали льдом в течение 2 часов с одновременным перемешиванием. Осадок собирали с помощью фильтрования и затем сушили при пониженном давлении при 50°С в течение 1 часа, что давало 153,2 мг указанного в заголовке соединения.
1H-ЯМР (ДМСО-d6)
δ 1,79 (т, J=2 Гц, 3H) 2,27 (с, 3H) 3,25-3,35 (м, 4H) 3,50-3,54 (м, 4H) 3,70 (с, 3H) 5,13 (д, J=2 Гц, 2H) 7,10 (д, J=8 Гц, 2H) 7,47 (д, J=8 Гц, 2H) 8,25 (с, 1H) 8,79 (ушир.с, 2H).
Далее, 107,95 мг указанного в заголовке соединения перекристаллизовывали из ацетона, что давало 84,9 мг кристаллического продукта.
Пример 120
Трифторацетат 2-(3-аминопиридин-1-ил)-3-(2-бутинил)-5-метил-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
(а) 9Н-Флуорен-9-илметил-3-трет-бутоксикарбониламинопиперидин-1-карбоксилат
1,84 г диизопропилэтиламина и 4,71 г дифенилфосфорилазида добавляли к 10 мл раствора трет-бутанола, содержащего 5,01 г 9Н-флуорен-9-илметил-3-карбоксипиперидин-1-карбоксилата, и смесь нагревали при 60°С в атмосфере азота в течение 18 часов. Реакционный раствор охлаждали и к нему добавляли 150 мл этилацетата. Органический слой промывали последовательно 100 мл 5% водного раствора серной кислоты, 100 мл 5% водного раствора бикарбоната натрия, 100 мл воды и 100 мл насыщенного раствора хлорида натрия и затем сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 1,88 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (4:1).
1H-ЯМР (CDCl3)
δ 1,45 (с, 9H) 1,45-1,72 (м, 3H) 1,82-1,87 (ушир.с, 1H) 3,09-3,30 (ушир.с, 2H) 3,58 (ушир.с, 2H) 3,82-3,98 (ушир.с, 1H) 4,24 (т, J=7,2 Гц, 1H) 4,27-4,48 (ушир.с, 2H) 4,52-4,59 (ушир.с, 1H) 7,32 (дд, J=10,3, 10,0 Гц, 2H) 7,39 (т, J=10,0 Гц, 2H) 7,59 (д, J=10,0 Гц, 2H) 7,75 (д, J=10,3 Гц, 2H).
(b) трет-бутил пиперидин-3-илкарбамат
25 мл диэтиламина добавляли к 250 мл раствора этанола, содержащего 1,88 г 9Н-флуорен-9-илметил-3-трет-бутоксикарбониламинопиперидин-1-карбоксилата, и смесь перемешивали при комнатной температуре в течение 18 часов. После того, как раствор концентрировали при пониженном давлении, остаток растворяли в смеси, состоящей из 150 мл толуола и 100 мл 10% водного раствора лимонной кислоты. Водный слой подщелачивали 5 н водным раствором гидроксида натрия и затем дважды экстрагировали 100 мл дихлорметана. Органические слои объединяли вместе, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, что давало 0,79 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,45 (с, 9H) 1,41-1,53 (м, 2H) 1,65-1,72 (м, 1H) 1,79-1,86 (м, 1H) 2,48-2,56 (м, 1H) 2,64-2,70 (м, 1H) 2,78-2,86 (м, 1H) 3,06 (дд, J=12,0,4,0 Гц, 1H) 3,48-3,62 (ушир.с, 1H) 4,71-4,88 (ушир.с, 1H).
(с) Трифторацетат 2-(3-аминопиперидин-1-ил)-3-(2-бутинил)-5-метил-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
0,020 г 2-хлор-5-метил-1-тритил-1,5-дигидроимидазо[4,5-d]пиридазин-4-она и 0,040 г трет-бутил пиперидин-3-илкарбамата объединяли вместе, и смесь нагревали в атмосфере азота при 150°С в течение 1 часа. Реакционную смесь очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 0,016 г трет-бутил [1-(6-метил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил)пиперидин-3-ил]карбамата из фракции, элюированной этилацетатом. 0,0080 г данного соединения растворяли в 0,6 мл N,N-диметилформамида и затем к нему добавляли 0,0038 г карбоната калия и 0,003 мл 2-бутинилбромида. Смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь распределяли между 1 мл этилацетата и 1 мл воды, и органический слой концентрировали. Остаток растворяли в 0,5 мл дихлорметана и затем к нему добавляли 0,5 мл трифторуксусной кислоты. Через 1 час реакционный раствор концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 0,0046 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,74-1,80 (ушир.с, 1H) 1,82 (ушир.с, 3H) 1,96-2,19 (ушир.м, 3H) 3,43-3,79 (ушир.м, 5H) 3,86 (с, 3H) 5,05 (ушир.д, J=16,0 Гц, 1H) 5,23 (ушир.д, J=16,0 Гц, 1H) 8,15 (с, 1H).
Пример 122
2-[7-(2-Бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]бензамид
53,0 г трет-бутил 4-[7-(2-бутинил)-2-(2-карбамоилфенокси)-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 160 мл трифторуксусной кислоты, и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору, по каплям, добавляли 1250 мл 2М водного раствора гидроксида натрия и смесь перемешивали при комнатной температуре в течение одного часа и 50 минут. Полученный в результате белый осадок собирали с помощью фильтрования. Белое твердое вещество промывали водой и затем этанолом и сушили при 60°С в течение ночи, что давало 42,8 г указанного в заголовке соединения.
1H-ЯМР (ДМСО-d6)
δ 1,78 (т, J=2,4 Гц, 3H) 2,82-2,86 (м, 4H) 3,18-3,22 (м, 4H) 3,36 (с, 3H) 4,91 (кв, 2,4 Гц, 2H) 6,58 (тд, J=8,4, 1,2 Гц, 1H) 6,63 (дд, J=8,0, 0,8 Гц, 1H) 7,14 (ддд, J=8,0, 7,2, 2,0 Гц, 1H) 7,80 (дд, J=7,6, 2,0 Гц, 1H).
МС m/e (ESI) 422 (MH+).
Пример 126
Трифторацетат 3-[7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илсульфанил]пропионовой кислоты
7 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,15 мл 1-метил-2-пирролидона и затем к ним добавляли 20 мкл 3-меркаптопропионовой кислоты и 6 мг карбоната калия. Смесь перемешивали при комнатной температуре в течение 5 часов. К реакционному раствору добавляли насыщенный раствор хлорида аммония и смесь экстрагировали этилацетатом. Органический слой концентрировали и остаток растворяли в 0,40 мл трифторуксусной кислоты. Раствор концентрировали в обильном токе газообразного азота. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 4,60 мг указанного в заголовке соединения.
MS m/e (ESI) 391 (MH+-CF3COOH).
Пример 129
Трифторацетат 7-(2-бутинил)-1-метил-8-(пиперазин-1-ил)-2-пропилсульфанил-1,7-дигидропурин-6-она
Получали 4,61 мг указанного в заголовке соединения по способу примера 126 с использованием пропан-1-тиола вместо 3-меркаптопропионовой кислоты.
MS m/e (ESI) 361 (MH+-CF3COOH).
Пример 142
Трифторацетат 7-(2-бутинил)-1-метил-8-(пиперазин-1-ил)-2-(тиазол-2-илсульфанил)-1,7-дигидропурин-6-она
Получали 3,86 мг указанного в заголовке соединения по способу примера 126 с использованием тиазол-2-тиола вместо 3-меркаптопропионовой кислоты.
MS m/e (ESI) 402 (MH+-CF3COOH).
Пример 146
Трифторацетат 7-(2-бутинил)-1-метил-8-(пиперазин-1-ил)-2-[1-(тиофен-2-ил)этилсульфанил]-1,7-дигидропурин-6-она
Получали 0,51 мг указанного в заголовке соединения по способу примера 126 с использованием 1-(тиофен-2-ил)этантиола вместо 3-меркаптопропионовой кислоты.
MS m/e (ESI) 429 (MH+-CF3COOH).
Пример 147
Трифторацетат 7-(2-бутинил)-1-метил-2-(1-метил-1Н-имидазол-2-илсульфанил)-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
5 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,15 мл 1-метил-2-пирролидона и затем туда добавляли 10 мг 1-метил-1Н-имидазол-2-тиола и 8 мг карбоната калия. Смесь перемешивали при комнатной температуре в течение 5 часов. К реакционному раствору добавляли насыщенный раствор хлорида аммония и смесь экстрагировали этилацетатом. Органический слой концентрировали, и остаток растворяли в 0,40 мл трифторуксусной кислоты. Раствор концентрировали в обильной струе газообразного азота. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 3,75 мг указанного в заголовке соединения.
MS m/e (ESI) 399 (MH+-CF3COOH).
Пример 159
Трифторацетат 7-(2-бутинил)-1-метил-2-(4-метилтиазол-2-илсульфанил)-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она
Получали 4,01 мг указанного в заголовке соединения по способу примера 147 с использованием 4-метилтиазол-2-тиола вместо 1-метил-1Н-имидазол-2-тиола.
MS m/e (ESI) 416 (MH+-CF3COOH).
Пример 229
Гидрохлорид 7-(2-бутинил)-1-(2-цианбензил)-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-карбонитрила
(а) трет-Бутил 4-[7-(2-бутинил)-2-циан-1-(2-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
Смесь, содержащую 8 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-(2-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата, полученного в примере 96(а), 10 мг цианида натрия и 0,3 мл N,N-диметилформамида, перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь экстрагировали смесью этилацетат-вода и органический слой промывали водой, а затем насыщенным солевым раствором. Органический слой концентрировали. Остаток очищали с помощью тонкослойной хроматографии (50% этилацетат-гексан), что давало 6,1 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,50 (с, 9H) 1,83 (с, 3H) 3,50 (с, 4H) 3,58-3,64 (м, 4H) 4,99 (с, 2H) 5,74 (с, 2H) 7,02 (д, J=8 Гц, 1H) 7,44 (т, J=8 Гц, 1H) 7,55 (т, J=8 Гц, 1H) 7,74 (д, J=8 Гц, 1H).
(b) Гидрохлорид 7-(2-бутинил)-1-(2-цианбензил)-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-карбонитрила
Смесь, содержащую 6,1 мг трет-бутил 4-[7-(2-бутинил)-2-циан-1-(2-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата и 0,2 мл трифторуксусной кислоты, перемешивали при комнатной температуре в течение 20 минут. Реакционный раствор концентрировали и остаток очищали с помощью колоночной хроматографии с обратной фазой с использованием в качестве растворителя 20%-60%-ной смеси метанол-вода (0,1% концентрированной соляной кислоты), что давало 5,0 мг указанного в заголовке соединения.
1H-ЯМР (ДМСО-d6)
δ 1,80 (с, 3H) 3,30 (с, 4H) 3,60-3,70 (м, 4H) 5,09 (с, 2H) 5,60 (с, 2H) 7,27 (д, J=8 Гц, 1H) 7,54 (т, J=8 Гц, 1H) 7,68 (т, J=8 Гц, 1H) 7,94 (д, J=8 Гц, 1H) 9,36 (ушир.с, 2H).
Пример 230
Трифторацетат амида 3-[7-(2-бутинил)-1-(2-цианбензил)-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]пиридин-2-карбоновой кислоты
7 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-(2-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,2 мл 1-метил-2-пирролидона и затем туда добавляли 8 мг 3-гидроксипиридин-2-карбоксиламида и 8 мг карбоната калия. Смесь перемешивали при 100°С в течение 2 часов. К реакционной смеси добавляли 1 н соляной кислоты и смесь экстрагировали этилацетатом. Органический слой концентрировали и остаток растворяли в трифторуксусной кислоте. Раствор концентрировали и остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 2,93 мг указанного в заголовке соединения.
MS m/e (ESI) 524 (MH+-CF3COOH).
Пример 234
Трифторацетат 2-[7-(2-бутинил)-1-(2-цианбензил)-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]бензамида
Получали 3,74 мг указанного в заголовке соединения по способу примера 230 с использованием салициламида вместо 3-гидроксипиридин-2-карбоксиламида.
MS m/e (ESI) 523 (MH+-CF3COOH).
Пример 235
Трифторацетат 2-[7-(2-бутинил)-1-(4-цианбензил)-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]бензамида
(а) трет-Бутил 4-[7-(2-бутинил)-2-хлор-1-(4-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
100 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 1,2 мл N,N-диметилформамида и затем туда добавляли 97 мг 4-цианбензилбромида и 68 мг карбоната калия. Смесь перемешивали при комнатной температуре в течение 4 часов. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония и смесь экстрагировали этилацетатом. Органический слой концентрировали и остаток очищали с помощью хроматографии на силикагеле, что давало 71 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,49 (с, 9H) 1,84 (т, J=2,5 Гц, 3H) 3,43-3,47 (м, 4H) 3,59-3,63 (м, 4H) 4,94 (кв, 2,5 Гц, 2H) 5,53 (с, 2H) 7,42 (д, J=8,0 Гц, 2H) 7,62 (д, J=8,0 Гц, 2H).
(b) Трифторацетат 2-[7-(2-бутинил)-1-(4-цианбензил)-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]бензамида
12 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-(4-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,3 мл 1-метил-2-пирролидона и затем туда добавляли 10 мг салициламида и 10 мг карбоната калия. Смесь перемешивали при 100°С в течение 12 часов. К реакционному раствору добавляли 1 н соляной кислоты и смесь экстрагировали этилацетатом. Органический слой концентрировали и остаток растворяли в трифторуксусной кислоте. Раствор концентрировали и остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 6,69 мг указанного в заголовке соединения.
MS m/e (ESI) 523 (MH+-CF3COOH).
Пример 238
Трифторацетат 2-[7-(2-бутинил)-1-(3-цианбензил)-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]бензамида
(а) трет-Бутил 4-[7-(2-бутинил)-2-хлор-1-(3-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
100 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 1,2 мл N,N-диметилформамида и затем туда добавляли 97 мг 3-цианбензилбромида и 68 мг карбоната калия. Смесь перемешивали при комнатной температуре в течение 12 часов. Затем к реакционному раствору добавляли насыщенный раствор хлорида аммония и смесь экстрагировали этилацетатом. Органический слой концентрировали и остаток очищали с помощью хроматографии на силикагеле, что давало 71 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,49 (с, 9H) 1,84 (т, J=2,5 Гц, 3H) 3,43-3,47 (м, 4H) 3,59-3,63 (м, 4H) 4,94 (кв, 2,5 Гц, 2H) 5,53 (с, 2H) 7,42 (д, J=8,0 Гц, 2H) 7,62 (д, J=8,0 Гц, 2H).
(b) Трифторацетат 2-[7-(2-бутинил)-1-(3-цианбензил)-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]бензамида
12 мг трет-бутил 4-[7-(2-бутинил)-2-хлор-1-(3-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,3 мл 1-метил-2-пирролидона, и затем туда добавляли 10 мг салициламида и 10 мг карбоната калия. Смесь перемешивали при 100°С в течение 5 часов. К реакционному раствору добавляли 1 н соляной кислоты и смесь экстрагировали этилацетатом. Органический слой концентрировали и остаток растворяли в трифторуксусной кислоте. Раствор концентрировали и остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 8,76 мг указанного в заголовке соединения.
MS m/e (ESI) 523 (MH+-CF3COOH).
Пример 242
Гидрохлорид 8-(3-аминопиперидин-1-ил)-7-(2-бутинил)-1-(2-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-2-карбонитрила
(а) Бензил-3-трет-бутоксикарбониламинопиперидин-1-карбоксилат
88 г бензилхлорформиата (бензиловый эфир хлормуравьиной кислоты) (30% раствор толуола) по каплям добавляли к смеси, состоящей из 24,3 г этилпиперидин-3-карбоксилата, 26 мл триэтиламина и 300 мл этилацетата, на протяжении 30 минут, с одновременным охлаждением смеси льдом. Реакционную смесь фильтровали для удаления нерастворимого материала. Фильтрат снова фильтровали через небольшое количество силикагеля. Фильтрат концентрировали.
К остатку добавляли 200 мл этанола и 40 мл 5М водного раствора гидроксида натрия. Смесь перемешивали при комнатной температуре в течение ночи. Реакционный раствор концентрировали и к остатку добавляли 200 мл воды. Смесь экстрагировали трет-бутилметиловым эфиром. К водному слою добавляли 5М водный раствор соляной кислоты и смесь экстрагировали этилацетатом. Органический слой промывали водой и затем насыщенным соляным раствором. Органический слой сушили над безводным сульфатом магния и затем концентрировали, что давало маслянистый остаток (30,9 г).
Смесь, состоящую из 30 г данного остатка, 24,5 мл дифенилфосфорилазида, 15,9 мл триэтиламина и 250 мл трет-бутанола, перемешивали при комнатной температуре в течение 1,5 часов. Смесь далее перемешивали в масляной бане при 100°С в течение 20 часов. Реакционный раствор концентрировали и остаток экстрагировали смесью этилацетат-вода. Органический слой промывали разбавленным водным раствором бикарбоната натрия и затем насыщенным соляным раствором. Органический слой сушили над безводным сульфатом магния и затем концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием 10%-20% смеси этилацетат-гексан, с последующей перекристаллизацией из смеси этилацетат-гексан, что давало 21,4 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,43 (с, 9H) 1,48-1,92 (м, 4H) 3,20-3,80 (м, 5H) 4,58 (ушир.с, 1H) 5,13 (с, 2H) 7,26-7,40 (м, 5H).
(b) трет-Бутилпиперидин-3-илкарбамат
Смесь, состоящую из 10 г бензил-3-трет-бутоксикарбониламинопиперидин-1-карбоксилата, 500 мг 10% палладия на угле и 100 мл этанола, перемешивали при комнатной температуре в атмосфере водорода в течение ночи. Катализатор удаляли с помощью фильтрования. Фильтрат концентрировали и сушили, что давало 6,0 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,44 (с, 9H) 1,47-1,80 (м, 4H) 2,45-2,60 (м, 1H) 2,60-2,75 (м, 1H) 2,75-2,90 (м, 1H) 3,05 (дд, J=3 Гц, 12 Гц, 1H) 3,57 (ушир.с, 1H) 4,83 (ушир.с, 1H).
(с) трет-Бутил [1-[7-(2-бутинил)-2,6-дихлор-7Н-пурин-8-ил]пиперидин-3-ил]карбамат
Смесь, состоящую из 1,25 г 7-(2-бутинил)-2,6,8-трихлор-7Н-пурина, 1,0 г трет-бутил пиперидин-3-илкарбамата и 10 мл ацетонитрила, перемешивали при комнатной температуре в течение 10 минут. В течение 10 минут по каплям добавляли 0,63 мл триэтиламина и затем смесь непрерывно перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор распределяли между этилацетатом и водой и органический слой промывали насыщенным соляным раствором. Органический слой сушили над безводным сульфатом магния и затем концентрировали. Остаток кристаллизовался смесью трет-бутил метиловый эфир-гексан, что давало 1,79 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,43 (с, 9H) 1,60-2,02 (м, 4H) 1,83 (т, J=2 Гц, 3H) 3,32-3,41 (м, 1H) 3,42-3,52 (м, 1H) 3,67-3,76 (м, 1H) 3,80-3,91 (м, 1H) 4,76-4,90 (м, 3H).
(d) трет-бутил [1-[7-(2-бутинил)-2-хлор-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперидин-3-ил]карбамат
Смесь, состоящую из 1,79 г трет-бутил [1-[7-(2-бутинил)-2,6-дихлор-7Н-пурин-8-ил]пиперидин-3-ил]карбамата, 1,0 г ацетата натрия и 18 мл диметилсульфоксида, перемешивали в масляной бане при 120°С в течение 3 часов. Смесь удаляли из масляной бани и к реакционному раствору добавляли 18 мл воды. Смесь охлаждали до комнатной температуры. Кристаллы собирали с помощью фильтрования и промывали водой и затем трет-бутилметиловым эфиром. Затем кристаллы сушили, что давало 1,59 г указанного в заголовке соединения.
1H-ЯМР (ДМСО-d6)
δ 1,39 (с, 9H) 1,34-1,88 (м, 4H) 1,78 (с, 3H) 2,81 (т, J=11 Гц, 1H) 2,95 (т, J=11 Гц, 1H) 3,48-3,60 (м, 2H) 3,64 (д, J=6 Гц, 1H) 4,90 (с, 2H) 6,94 (д, J=8 Гц, 1H).
(е) трет-Бутил [1-[7-(2-бутинил)-2-хлор-1-(2-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперидин-3-ил]карбамат
Смесь, состоящую из 100 мг трет-бутил [1-[7-(2-бутинил)-2-хлор-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперидин-3-ил]карбамата, 66 мг безводного карбоната калия, 70 мг 2-цианбензилбромида и 1 мл N,N-диметилформамида, перемешивали при комнатной температуре в течение 5 часов. Реакционный раствор распределяли между этилацетатом и водой, и органический слой промывали водой и затем насыщенным соляным раствором. Органический слой сушили над безводным сульфатом магния и затем концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием 50% смеси этилацетат-гексан, что давало 44,7 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,44 (с, 9H) 1,59-1,81 (м, 2H) 1,83 (т, J=2 Гц, 3H) 1,86-1,94 (м, 2H) 3,20-3,50 (м, 3H) 3,66 (д, J=7 Гц, 1H) 3,86 (ушир.с, 1H) 4,88-5,06 (м, 3H) 5,72 (с, 2H) 7,06 (д, J=8 Гц, 1H) 7,38 (т, J=8 Гц, 1H) 7,51 (т, J=8 Гц, 1H) 7,70 (д, J=8 Гц, 1H).
(f) трет-Бутил [1-[7-(2-бутинил)-2-циан-1-(2-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперидин-3-ил]карбамат
Смесь, состоящую из 15 мг трет-бутил [1-[7-(2-бутинил)-2-хлор-1-(2-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперидин-3-ил]карбамата, 20 мг цианида натрия и 0,2 мл N,N-диметилформамида, перемешивали при комнатной температуре в течение 3 часов. Реакционный раствор распределяли между этилацетатом и водой, и органический слой промывали водой и затем насыщенным соляным раствором. Затем органический слой концентрировали и остаток очищали с помощью тонкослойной хроматографии с использованием в качестве растворителя 50% смеси этилацетат-гексан (проводили три раза), что давало 10,3 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,44 (с, 9H) 1,52-1,98 (м, 4H) 1,81 (т, J=2 Гц 3H) 3,24 (дд, J=7 Гц, 12 Гц, 1H) 3,30-3,40 (м, 1H) 3,46-3,56 (м, 1H), 3,72 (д, J=12 Гц, 1H) 3,86 (ушир.с, 1H) 4,86-5,10 (м, 3H) 5,73 (с, 2H) 7,00 (д, J=8 Гц, 1H) 7,42 (т, J=8 Гц, 1H) 7,54 (дт, J=2 Гц, 8 Гц, 1H) 7,73 (дд, J=2 Гц, 8 Гц, 1H).
(g) Гидрохлорид 8-(3-аминопиперидин-1-ил)-7-(2-бутинил)-1-(2-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-2-карбонитрила
Смесь, состоящую из 10,3 мг трет-бутил [1-[7-(2-бутинил)-2-циан-1-(2-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперидин-3-ил]карбамата и 0,2 мл трифторуксусной кислоты, перемешивали в течение 20 минут. Реакционный раствор концентрировали, и остаток очищали с помощью обратной фазы колоночной хроматографии с использованием в качестве растворителя 20%-80% смеси метанол/вода (содержащей 0,1% концентрированной соляной кислоты), что давало 8,0 мг указанного в заголовке соединения.
1H-ЯМР (ДМСО-d6)
δ 1,60-1,74 (м, 2H) 1,79 (т, J=2 Гц, 3H) 1,88-2,03 (м, 2H) 3,14-3,28 (м, 2H) 3,42 (ушир.с, 1H) 3,52-3,82 (м, 2H) 4,98-5,12 (м, 2H) 5,58 (с, 2H) 7,26 (д, J=8 Гц, 1H) 7,53 (т, J=8 Гц, 1H) 7,66 (т, J=8 Гц, 1H) 7,93 (д, J=8 Гц, 1H) 8,16 (ушир.с, 3H).
Пример 243
Гидрохлорид 2-[8-(3-аминопиперидин-1-ил)-7-(2-бутинил)-2-метокси-6-оксо-6,7-дигидропурин-1-илметил]бензонитрила
Смесь, состоящую из 15 мг трет-бутил [1-[7-(2-бутинил)-2-хлор-1-(2-цианбензил)-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперидин-3-ил]карбамата, 20 мг безводного карбоната калия и 0,2 мл метанола, перемешивали в течение 3 часов. Последующие шаги выполнялись в соответствии с аналогичной процедурой, примененной в Примере 242(f) и (g). Таким образом, было синтезировано указанное в заголовке соединение.
1H-ЯМР (ДМСО-d6)
δ 1,58-1,72 (м, 2H) 1,84-1,94 (м, 1H) 1,96-2,04 (м, 1H) 3,08-3,20 (м, 2H) 3,36-3,70 (м, 3H) 3,90 (с, 3H) 4,90-5,02 (м, 2H) 5,32 (с, 2H) 7,20 (д, J=8 Гц, 1H) 7,47 (т, J=8 Гц, 1H) 7,63 (т, J=8 Гц, 1H) 7,87 (д, J=8 Гц, 1H) 8,12 (ушир.с, 3H).
Пример 248
Трифторацетат 2-[8-(3-аминопиперидин-1-ил)-7-(2-бутинил)-1-метил-6-оксо-6,7-дигидро-1Н-пурин-2-илокси]бензамида
(а) трет-бутил [1-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперидин-3-ил]карбамат
700 мг трет-бутил [1-[7-(2-бутинил)-2-хлор-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперидин-3-ил]карбамата растворяли в 7,0 мл диметилсульфоксида и затем туда добавляли 114 мкл метилйодида и 299 мг карбоната калия. Смесь перемешивали при комнатной температуре в течение 30 минут и к реакционному раствору добавляли 40 мл воды. Смесь перемешивали при комнатной температуре в течение 30 минут и белый осадок собирали с помощью фильтрования. Полученное белое твердое вещество промывали водой и затем гексаном, что давало 540 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,44 (с, 9H) 1,72-1,94 (м, 4H) 1,81 (т, J=2,4 Гц, 3H) 3,16-3,92 (м, 5H) 3,72 (с, 3H) 4,91 (дд, J=17,6, 2,4 Гц, 1H) 5,01 (д, J=17,6 Гц, 1H).
(b) Трифторацетат 2-[8-(3-аминопиперидин-1-ил)-7-(2-бутинил)-1-метил-6-оксо-6,7-дигидро-1Н-пурин-2-илокси]бензамида
10 мг трет-бутил [1-[7-(2-бутинил)-2-хлор-1-метил-6-оксо-6,7-дигидро-1Н-пурин-8-ил]пиперидин-3-ил]карбамата растворяли в 0,3 мл 1-метил-2-пирролидона и затем туда добавляли 10 мг салициламида и 10 мг карбоната калия. Смесь перемешивали при 100°С в течение 2 часов. К реакционному раствору добавляли 1 н соляную кислоту и смесь экстрагировали этилацетатом. Органический слой концентрировали и остаток растворяли в трифторуксусной кислоте. Раствор концентрировали, и остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 5,54 мг указанного в заголовке соединения.
MS m/e (ESI) 436(MH+-CF3COOH).
Пример 258
Трифторацетат 3-(2-бутинил)-2-(пиперазин-1-ил)-5-(2-пропинил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
(а) трет-Бутил 4-[1-(2-бутинил)-7-оксо-6,7дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилат
0,299 г триэтиламина, 0,023 г 4-диметиламинопиридина и 0,645 г ди-трет-бутилдикарбоната при комнатной температуре добавляли к 20 мл раствора N,N-диметилформамида, содержащего 0,448 г трифторацетата 3-(2-бутинил)-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она, и смесь перемешивали в течение 5 часов. Затем к данному раствору добавляли 2 мл 5 н водного раствора гидроксида натрия, и смесь перемешивали в течение 1 часа. Реакционный раствор вливали в смесь 200 мл этилацетата и 100 мл насыщенного водного раствора хлорида аммония. Органический слой промывали два раза 100 мл воды и затем 100 мл насыщенного водного раствора хлорида натрия. Органическая жидкость сушили над сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 0,298 г указанного в заголовке соединения из фракции, элюированной этилацетатом.
1H-ЯМР (CDCl3)
δ 1,50 (с, 9H) 1,84 (т, J=2,3 Гц, 3H) 3,41 (м, 4H) 3,63 (м, 4H) 5,06 (кв, J=2,3 Гц, 2H) 8,17 (с, 1H) 9,92 (ушир.с, 1H).
(b) Трифторацетат 3-(2-бутинил)-2-(пиперазин-1-ил)-5-(2-пропинил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
0,005 г карбоната калия и 0,003 мл 3-бром-1-пропина добавляли к 0,5 мл раствора N,N-диметилформамида, содержащего 0,010 г трет-бутил 4-[1-(2-бутинил)-7-оксо-6,7дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилата, и смесь перемешивали при комнатной температуре в течение 10 часов. К реакционному раствору добавляли 1 мл этилацетата и 1 мл воды и слои разделяли. Органический слой концентрировали и получившийся в результате остаток растворяли в смеси, состоящей из 0,5 мл дихлорметана и 0,5 мл трифторуксусной кислоты. Смесь перемешивали в течение 1 часа и затем концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 0,011 г указанного в заголовке соединения.
MS m/e (ESI) 311,29 (MH+-CF3COOH).
Пример 266
Трифторацетат 3-(2-бутинил)-5-[2-(3-метоксифенил)-2-оксоэтил]-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
Указанное в заголовке соединение получали с использованием трет-бутил 4-[1-(2-бутинил)-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилата и 2-бром-3'-метокси-ацетофенона по способу, представленному в Примере 258(b).
MS m/e (ESI) 421,33 (MH+-CF3COOH).
Пример 267
Трифторацетат 2-[3-(2-бутинил)-4-оксо-2-(пиперазин-1-ил)-3,4-дигидроимидазо[4,5-d]пиридазин-5-илметил]бензонитрила
Указанное в заголовке соединение получали с использованием трет-бутил 4-[1-(2-бутинил)-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилата и 2-бромметилбензонитрила по способу, представленному в Примере 258(b).
1H-ЯМР (CD3OD)
δ 1,81 (т, J=2,5 Гц, 3H) 3,45-3,49 (м, 4H) 3,66-3,70 (м, 4H) 5,15 (кв, J=2,5 Гц, 2H) 5,62 (с, 2H) 7,34 (дд, J=7,6,1,5 Гц, 1H) 7,45 (тд, J=7,6,1,5 Гц, 1H) 7,59 (тд, J=7,6,1,7 Гц, 1H) 7,75 (дд, J=7,6,1,7 Гц, 1H) 8,25 (с, 1H).
МС m/e (ESI) 388,32 (MH+-CF3COOH).
Пример 297
Трифторацетат 2-[3-(2-бутинил)-4-оксо-2-(пиперазин-1-ил)-3,4-дигидроимидазо[4,5-d]пиридазин-5-илметил]-3-фторбензонитрила
Указанное в заголовке соединение получали с использованием трет-бутил 4-[1-(2-бутинил)-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилата и 2-бромметил-3-фторбензонитрила по способу, представленному в Примере 258(b).
MS m/e (ESI) 406,25 (MH+-CF3COOH).
Пример 308
Трифторацетат 3-бензил-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
(а) трет-Бутил 4-(1-бензил-6-бензилоксиметил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали с использованием трет-бутил 4-(6-бензилоксиметил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил)пиперазин-1-карбоксилата и бензилбромида по способу, представленному в Примере 116(d).
1H-ЯМР (CDCl3)
δ 1,48 (с, 9H) 3,13-3,18 (м, 4H) 3,50-3,54 (м, 4H) 4,72 (с, 2H) 5,61 (с, 2H) 5,65 (с, 2H) 7,20-7,35 (м, 10H) 8,22 (с, 1H).
(b) Трифторацетат 3-бензил-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
Указанное в заголовке соединение получали с помощью обработки трет-бутил 4-(1-бензил-6-бензилоксиметил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил)пиперазин-1-карбоксилата по способу, представленному в Примере 117.
1H-ЯМР (CD3OD)
δ 3,31-3,37 (м, 4H) 3,40-3,46 (м, 4H) 5,68 (с, 2H) 7,22-7,36 (м, 5H) 8,25 (с, 1H).
МС m/e (ESI) 311,24 (MH+-CF3COOH).
Пример 309
Трифторацетат 3-бензил-5-метил-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
(а) трет-Бутил 4-(1-бензил-7-оксо-6,7дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали с использованием трифторацетата 3-бензил-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она по способу, представленному в Примере 258(а).
1H-ЯМР (CDCl3)
δ 1,47 (с, 9H) 3,12-3,16 (м, 4H) 3,47-3,52 (м, 4H) 5,58 (с, 2H) 7,20-7,34 (м, 5H) 8,20 (с, 1H) 10,04 (ушир.с, 1H).
(b) Трифторацетат 3-бензил-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
Указанное в заголовке соединение получали с использованием трет-бутил 4-(1-бензил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил)пиперазин-1-карбоксилата и метилйодида по способу, представленному в Примере 258(b).
1H-ЯМР (CD3OD)
δ 3,29-3,35 (м, 4H) 3,36-3,41 (м, 4H) 3,83 (с, 3H) 5,68 (с, 2H) 7,21-7,34 (м, 5H) 8,20 (с, 1H).
МС m/e (ESI) 325,01 (MH+-CF3COOH).
Пример 311
Трифторацетат 3-бензил-5-(2-фенилэтил)-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она
Указанное в заголовке соединение получали с использованием трет-бутил 4-[1-бензил-7-оксо-6,7-дигидро-1Н-имидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилата и (2-бромэтил)бензола по способу, представленному в Примере 258(b).
1H-ЯМР (CDCl3)
δ 3,11 (т, J=8,1 Гц, 2H) 3,24-3,29 (м, 4H) 3,37-3,42 (м, 4H) 4,46 (т, J=8,1 Гц,2H) 5,58 (с, 2H) 7,09-7,34 (м, 10H) 8,20 (с, 1H)
МС m/e (ESI) 415,54 (MH+-CF3COOH).
Пример 332
Трифторацетат 1-(2-бутинил)-6-метил-7-оксо-2-(пиперазин-1-ил)-6,7-дигидроимидазо[4,5-d]пиридазин-4-карбоксамида
(а) трет-бутил 4-[1-(2-бутинил)-4-(циангидроксиметил)-5-метоксикарбонил-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
0,200 г цианида натрия и 0,010 мл уксусной кислоты добавляли в 15 мл ацетонитрильного раствора трет-бутил-4-[1-(2-бутинил)-5-метоксикарбонил-4-формил-1Н-имидазол-2-ил]пиперазин-1-карбоксилата, и смесь перемешивали при комнатной температуре в течение 16 часов. К раствору добавляли 100 мл этилацетата, и смесь промывали два раза 50 мл воды и затем 50 мл насыщенного раствора хлорида натрия. Органический слой сушили над сульфатом магния, и растворитель концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 0,274 г указанного в заголовке соединения из фракции, элюированной смесью этилацетат-гексан (2:3).
1H-ЯМР (CDCl3)
δ 1,49 (с, 9H) 1,83 (т, J=2,5 Гц, 3H) 3,19-3,23 (м, 4H) 3,56-3,60 (м, 4H) 3,95 (с, 3H) 4,68 (д, J=9,0 Гц, 1H) 4,82 (кв, J=2,5 Гц, 2H) 5,72 (д, J=9,0 Гц, 1H).
(b) трет-Бутил 4-[1-(2-бутинил)-4-(карбамоилгидроксиметил)-5-метоксикарбонил-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
3,2 мл 30% водного раствора перекиси водорода и 3,2 мл 28% водного раствора аммиака при 5°С добавляли к 8 мл раствора метанола, содержащего 0,274 г трет-бутил 4-[1-(2-бутинил)-4-(циангидроксиметил)-5-метоксикарбонил-1Н-имидазол-2-ил]пиперазин-1-карбоксилата, и смесь перемешивали в течение 15 часов. К раствору добавляли 100 мл насыщенного раствора бисульфита натрия и смесь дважды экстрагировали 100 мл этилацетата. Органические слои объединяли вместе. Объединенные органические слои сушили над сульфатом магния и концентрировались при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 0,039 г указанного в заголовке соединения из фракции, элюированной смесью метанол-этилацетат (1:9).
1H-ЯМР (CDCl3)
δ 1,48 (с, 9H) 1,83 (т, J=2,5 Гц, 3H) 3,13-3,25 (м, 4H) 3,54-3,57 (м, 4H) 3,91 (с, 3H) 4,33-4,37 (ушир.с, 1H) 4,77 (кв, J=2,5 Гц, 2H) 5,54 (с, 1H) 5,63 (с, 1H) 6,82 (с, 1H).
(с) трет-Бутил 4-[4-аминооксалил-1-(2-бутинил)-5-метоксикарбонил-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
0,051 мл триэтиламина и 1 мл раствора диметилсульфоксида, содержащего 0,058 г серного ангидрида пиридина (серная трехокись пиридина), при 0°С добавляли к 2 мл раствора дихлорметана, содержащего 0,038 г трет-бутил 4-[1-(2-бутинил)-4-(карбамоилгидроксиметил)-5-метоксикарбонил-1Н-имидазол-2-ил]пиперазин-1-карбоксилата, и смесь перемешивали при комнатной температуре в течение 15 часов. Затем добавляли 0,102 мл триэтиламина и 1 мл раствора диметилсульфоксида, содержащего 0,116 г серного ангидрида пиридина, и смесь перемешивали при комнатной температуре в течение 8 часов. К раствору добавляли 50 мл этилацетата и органический слой последовательно промывали 20 мл водного раствора 1% серной кислоты, 20 мл насыщенного раствора бикарбоната натрия и 20 мл насыщенного раствора хлорида натрия. Органический слой сушили над сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 0,021 г указанного в заголовке соединения из фракции, элюированной смесью этилацетат-гексан (2:1).
1H-ЯМР (CDCl3)
δ 1,48 (с, 9H) 1,82 (т, J=2,5 Гц, 3H) 3,19-3,23 (м, 4H) 3,56-3,59 (м, 4H) 3,84 (с, 3H) 4,84 (кв, J=2,5 Гц, 2H) 5,62 (ушир.с, 1H) 7,02 (ушир.с, 1H).
(d) трет-бутил 4-[1-(2-бутинил)-4-карбамоил-6-метил-7-оксо-6,7-дигидро-1Н-дигидроимидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилат
Указанное в заголовке соединение получали с использованием трет-бутил 4-[4-аминооксалил-1-(2-бутинил)-5-метоксикарбонил-1Н-имидазол-2-ил]пиперазин-1-карбоксилата по способу, представленному в Примере 115(h).
1H-ЯМР (CDCl3)
δ 1,50 (с, 9H) 1,84 (т, J=2,3 Гц, 3H) 3,46-3,50 (м, 4H) 3,63-3,66 (м, 4H) 3,99 (с, 3H) 5,12 (кв, J=2,3 Гц, 2H) 6,16 (с, 1H) 8,85 (с, 1H).
(е) Трифторацетат 1-(2-бутинил)-6-метил-7-оксо-2-(пиперазин-1-ил)-6,7-дигидроимидазо[4,5-d]пиридазин-4-карбоксамида
Указанное в заголовке соединение получали с использованием трет-бутил 4-[1-(2-бутинил)-4-карбамоил-6-метил-7-оксо-6,7-дигидро-1Н-дигидроимидазо[4,5-d]пиридазин-2-ил]пиперазин-1-карбоксилата по способу, представленному в Примере 115(i).
MS m/e (ESI) 330,18 (MH+-CF3COOH).
Пример 338
Трифторацетат 3-(2-бутинил)-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-с]пиридин-4-она
(а) 2-бром-1-(2-бутинил)-1Н-имидазол-4,5-дикарбонитрил
69,8 г карбоната калия и 50 мл раствора N,N-диметилформамида, содержащего 74 мл 1-бром-2-бутина, добавляли к 520 мл раствора N,N-диметилформамида, содержащего 90,6 г 2-бром-1Н-имидазол-4,5-дикарбонитрила [CAS № 50847-09-1], и смесь нагревали при 50°С в течение 8 часов. К раствору добавляли 1 л этилацетата и 500 мл воды, и органический слой дважды промывали 500 мл воды и затем 500 мл насыщенного раствора хлорида натрия. Органический слой сушили над сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 48,0 г указанного в заголовке соединения из фракции, элюированной смесью этилацетат-гексан (1:4).
1H-ЯМР (CDCl3)
δ 1,87 (т, J=2,3 Гц, 3H) 4,85 (кв, J=2,3 Гц, 2H).
(b) Этил 2-бром-1-(2-бутинил)-5-циан-1Н-имидазол-4-карбоксилат
25 мл концентрированной серной кислоты добавляли к 500 мл раствора этанола, содержащего 48,0 г 2-бром-1-(2-бутинил)-1Н-имидазол-4,5-дикарбонитрила, и смесь нагревали с обратным холодильником в течение 110 часов. Реакционный раствор охлаждали до комнатной температуры и затем концентрировали при пониженном давлении. Остаток растворяли в смеси, состоящей из 500 мл этилацетата и 500 мл воды, и рН раствора доводили до 8 с использованием гидроксида калия. Водный слой экстрагировали 500 мл этилацетата, и органические слои объединяли вместе. Органический слой сушили над сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 21,7 г указанного в заголовке соединения из фракции, элюированной смесью этилацетат-гексан (1:3).
1H-ЯМР (CDCl3)
δ 1,43 (т, J=7,0 Гц, 3H) 1,87 (т, J=2,3 Гц, 3H) 4,46 (кв, J=7,0 Гц, 2H) 4,85 (кв, J=2,3 Гц, 2H).
(с) трет-Бутил 4-[1-(2-бутинил)-5-циан-4-этоксикарбонил-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
25,1 г указанного в заголовке соединения получали с использованием 21,7 г этил 2-бром-1-(2-бутинил)-5-циан-1Н-имидазол-4-карбоксилата по способу, представленному в Примере 115(b).
1H-ЯМР (CDCl3)
δ 1,43 (т, J=7,0 Гц, 3H) 1,49 (с, 9H) 1,87 (т, J=2,3 Гц, 3H) 3,22-3,26 (м, 4H) 3,56-3,61 (м, 4H) 4,44 (кв, J=7,0 Гц, 2H) 4,68 (кв, J=2,3 Гц, 2H).
(d) трет-Бутил 4-[1-(2-бутинил)-4-карбокси-5-циан-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
16 мл 5 н водного раствора гидроксида натрия добавляли к 500 мл раствора этанола, содержащего 25,1 г трет-бутил 4-[1-(2-бутинил)-5-циан-4-этоксикарбонил-1Н-имидазол-2-ил]пиперазин-1-карбоксилата, и смесь перемешивали при комнатной температуре в течение 2 часов. Затем растворитель концентрировали при пониженном давлении. Остаток растворяли в смеси, состоящей из 1 л этилацетата и 500 мл воды. К раствору добавляли 50 мл 2 н соляной кислоты. Органический слой промывали 200 мл насыщенного раствора хлорида натрия и сушили над сульфатом магния. Органическая жидкость концентрировали при пониженном давлении, что давало 23,2 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ 1,49 (с, 9H) 1,87 (т, J=2,3 Гц, 3H) 3,22-3,26 (м, 4H) 3,56-3,61 (м, 4H) 4,68 (кв, J=2,3 Гц, 2H).
(е) трет-Бутил 4-[1-(2-бутинил)-5-циан-4-гидроксиметил-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
6,9 г триэтиламина и затем 100 мл раствора тетрагидрофурана, содержащего 10,19 г изобутилхлорформиата по каплям добавляли к 600 мл тетрагидрофурана, содержащего 22,9 г трет-бутил 4-[1-(2-бутинил)-4-карбокси-5-циан-1Н-имидазол-2-ил]пиперазин-1-карбоксилата при -10°С. После удаления осадка с помощью фильтрования раствор снова охлаждали до -10°С. К раствору по каплям добавляли 100 мл водного раствора, содержащего 9,45 г боргидрида натрия. Через 1 час к раствору добавляли 500 мл этилацетата и 500 мл воды. рН раствора доводилось до 5 с использованием 1 н соляной кислоты, а затем доводилось до 10 с использованием насыщенного раствора бикарбоната натрия. Органический слой последовательно промывали 500 мл воды и 500 мл насыщенного раствора хлорида натрия. Органический слой сушили над сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 19,1 г указанного в заголовке соединения из фракции, элюированной смесью этилацетат-гексан (4:1).
1H-ЯМР (CDCl3)
δ 1,48 (с, 9H) 1,84 (т, J=2,3 Гц, 3H) 2,26 (т, J=6,3 Гц, 1H) 3,13-3,17 (м, 4H) 3,53-3,57 (м, 4H) 4,58 (кв, J=2,3 Гц, 2H) 4,64 (д, J=6,3 Гц, 2H).
(f) трет-Бутил 4-[1-(2-бутинил)-5-циан-4-формил-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
3,28 г двуокиси магния добавляли к 5 мл раствора дихлорметана, содержащего 1,35 г трет-бутил 4-[1-(2-бутинил)-5-циан-4-гидроксометил-1Н-имидазол-2-ил]пиперазин-1-карбоксилата. Реакционный раствор перемешивали при комнатной температуре в течение 15 часов, затем перемешивали и нагревали с обратным холодильником в течение 5 часов. Раствор фильтровали и затем концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 1,11 г указанного в заголовке соединения из фракции, элюированной смесью этилацетат-гексан (2:3).
1H-ЯМР (CDCl3)
δ 1,50 (с, 9H) 1,88 (т, J=2,3 Гц, 3H) 3,24-3,28 (м, 4H) 3,59-3,63 (м, 4H) 4,70 (кв, J=2,3 Гц, 2H) 9,87 (с, 1H).
(g) трет-Бутил 4-[1-(2-бутинил)-5-циан-4-(2-этоксикарбонилвинил)-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
0,038 г гидрида натрия при 5°С в атмосфере азота добавляли к 5 мл раствора тетрагидрофурана, содержащего 0,243 г диэтилфосфонацетата этила. Добавляли 0,310 г трет-бутил 4-[1-(2-бутинил)-5-циан-4-формил-1Н-имидазол-2-ил]пиперазин-1-карбоксилата, растворенного в 5 мл тетрагидрофурана, и смесь перемешивали в течение 30 минут. К раствору добавляли 50 мл этилацетата и 25 мл 0,1 н гидроксида натрия. Органический слой сушили над сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 0,380 г указанного в заголовке соединения из фракции, элюированной смесью этилацетат-гексан (3:7).
1H-ЯМР (CDCl3)
δ 1,33 (т, J=7,4 Гц, 3H) 1,50 (с, 9H) 1,86 (т, J=2,3 Гц, 3H) 3,19-3,23 (м, 4H) 3,55-3,59 (м, 4H) 4,25 (кв, J=7,4 Гц, 2H) 4,59 (кв, J=2,3 Гц, 2H) 6,70 (д, J=15,8 Гц, 1H) 7,50 (д, J=15,8 Гц, 1H).
(h) трет-бутил 4-[1-(2-бутинил)-5-циан-4-(2-карбоксивинил)-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
Указанное в заголовке соединение получали с использованием трет-бутил 4-[1-(2-бутинил)-5-циан-4-(2-этоксикарбонилвинил)-1Н-имидазол-2-ил]пиперазин-1-карбоксилата по способу, представленному в Примере 338(d).
1H-ЯМР (CDCl3)
δ 1,50 (с, 9H) 1,86 (т, J=2,3 Гц, 3H) 3,19-3,23 (м, 4H) 3,55-3,59 (м, 4H) 4,59 (кв, J=2,3 Гц, 2H) 6,70 (д, J=15,8 Гц, 1H) 7,50 (д, J=15,8 Гц, 1H).
(i) трет-Бутил 4-[1-(2-бутинил)-5-циан-4-(2-азидкарбонилвинил)-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
Смесь, состоящую из 0,200 г трет-бутил 4-[1-(2-бутинил)-5-циан-4-(2-карбоксивинил)-1Н-имидазол-2-ил]пиперазин-1-карбоксилата, 0,073 мл триэтиламина и 2 мл раствора трет-бутанола, содержащего 0,108 мл дифенилфосфорилазида, нагревали при 50°С в атмосфере азота в течение 4 часов. К раствору добавляли 50 мл этилацетата и смесь промывали 20 мл воды. Органический слой сушили над сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 0,178 г указанного в заголовке соединения из фракции, элюированной смесью этилацетат-гексан (2:3).
1H-ЯМР (CDCl3)
δ 1,48 (с, 9H) 1,86 (т, J=2,2 Гц, 3H) 3,19-3,23 (м, 4H) 3,55-3,59 (м, 4H) 4,59 (кв, J=2,2 Гц, 2H) 6,67 (д, J=15,4 Гц, 1H) 7,56 (д, J=15,4 Гц, 1H).
(j) трет-Бутил 4-[4-(2-трет-бутоксикарбониламиновинил)-1-(2-бутинил)-5-циан-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
10 мл раствора трет-бутанола, содержащего 0,178 г трет-бутил 4-[1-(2-бутинил)-5-циан-4-(2-азидкарбонилвинил)-1Н-имидазол-2-ил]пиперазин-1-карбоксилата, нагревали с обратным холодильником в атмосфере азота в течение 15 часов. Растворитель концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 0,169 г указанного в заголовке соединения из фракции, элюированной смесью этилацетат-гексан (9:11).
1H-ЯМР (CDCl3)
δ 1,48 (с, 9H) 1,84 (т, J=2,2 Гц, 3H) 3,16-3,19 (м, 4H) 3,54-3,58 (м, 4H) 4,51 (кв, J=2,2 Гц, 2H) 5,83 (д, J=15,0 Гц, 1H) 6,43-6,53 (м, 1H) 7,55-7,66 (м, 1H).
(k) трет-Бутил 4-[4-(2-трет-бутоксикарбониламиновинил)-1-(2-бутинил)-5-карбамоил-1Н-имидазол-2-ил]пиперазин-1-карбоксилат
Указанное в заголовке соединение получали с использованием трет-бутил 4-[4-(2-трет-бутоксикарбониламиновинил)-1-(2-бутинил)-5-циан-1Н-имидазол-2-ил]пиперазин-1-карбоксилата по способу, представленному в Примере 332(b).
1H-ЯМР (CDCl3)
δ 1,48 (с, 9H) 1,84 (т, J=2,2 Гц, 3H) 3,21-3,25 (м, 4H) 3,54-3,58 (м, 4H) 4,68 (кв, J=2,2 Гц, 2H) 5,90 (ушир.с, 1H) 6,36 (ушир.д, J=14,8 Гц, 1H) 6,92 (ушир.д, J=8,4 Гц, 1H) 7,45 (ушир.с, 1H) 7,52 (м, 1H).
(l) Трифторацетат 3-(2-бутинил)-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-с]пиридин-4-она
0,1 мл 5 н соляной кислоты добавляли к 0,3 мл раствора этанола, содержащего 0,0075 г трет-бутил 4-[4-(2-трет-бутоксикарбониламиновинил)-1-(2-бутинил)-5-карбамоил-1Н-имидазол-2-ил]пиперазин-1-карбоксилата, и смесь перемешивали при комнатной температуре в течение 15 часов. Растворитель концентрировали при пониженном давлении. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой (с использованием подвижной фазы ацетонитрил-вода (содержащей 0,1% трифторуксусной кислоты)), что давало 0,0043 г указанного в заголовке соединения.
1H-ЯМР (CD3OD)
δ 1,81 (т, J=2,4 Гц, 3H) 3,45-3,48 (м, 4H) 3,62-3,65 (м, 4H) 5,15 (кв, J=2,4 Гц, 2H) 6,60 (д, J=7,1 Гц, 1H) 7,18 (д, J=7,1 Гц, 1H).
МС m/e (ESI) 272,32 (MH+-CF3COOH).
Пример 339
Трифторацетат 3-(2-бутинил)-5-(2-фенилэтил)-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-с]пиридин-4-она
(а) трет-Бутил 4-[3-(2-бутинил)-4-оксо-4,5-дигидро-3Н-имидазо[4,5-с]пиридин-2-ил]пиперазин-1-карбоксилат
Указанное в заголовке соединение получали с использованием трифторацетата 3-(2-бутинил)-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-с]пиридин-4-она по способу, представленному в Примере 258(a).
1H-ЯМР (CDCl3)
δ 1,49 (с, 9H) 1,83 (т, J=2,3 Гц, 3H) 3,35-3,39 (м, 4H) 3,60-3,64 (м, 4H) 5,07 (кв, J=2,3 Гц, 2H) 6,55 (д, J=7,1 Гц, 1H) 6,97 (д, J=7,1 Гц, 1H).
(b) Трифторацетат 3-(2-бутинил)-5-(2-фенилэтил)-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-с]пиридин-4-она
Указанное в заголовке соединение получали с использованием трет-бутил 4-[3-(2-бутинил)-4-оксо-4,5-дигидро-3Н-имидазо[4,5-с]пиридин-2-ил]пиперазин-1-карбоксилата и (2-бромэтил)бензола по способу, представленному в Примере 258(b).
1H-ЯМР (CD3OD)
δ 1,83 (т, J=2,4 Гц, 3H) 3,05 (т, J=7,3 Гц, 2H) 3,45-3,48 (м, 4H) 3,62-3,65 (м, 4H) 4,26 (т, J=7,3 Гц, 2H) 5,18 (кв, J=2,4 Гц, 2H) 6,46 (д, J=7,3 Гц, 1H) 7,15 (д, J=7,3 Гц, 1H) 7,16-7,30 (м, 5H).
МС m/e (ESI) 376,36 (MH+-CF3COOH).
Пример 340
Трифторацетат 3-(2-бутинил)-5-(2-феноксиэтил)-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-с]пиридин-4-она
Указанное в заголовке соединение получали с использованием трет-бутил 4-[3-(2-бутинил)-4-оксо-4,5-дигидро-3Н-имидазо[4,5-с]пиридин-2-ил]пиперазин-1-карбоксилата и (2-бромэтил)фенилового эфира по способу, представленному в Примере 258(b).
1H-ЯМР (CD3OD)
δ 1,80 (т, J=2,4 Гц, 3H) 3,45-3,48 (м, 4H) 3,62-3,65 (м, 4H) 4,30 (т, J=5,5 Гц, 2H) 4,44 (т, J=5,5 Гц, 2H) 5,16 (кв, J=2,4 Гц, 2H) 6,59 (д, J=6,1 Гц, 1H) 6,87-6,91 (м, 3H) 7,20-7,24 (м, 2H) 7,50 (д, J=6,1 Гц, 1H).
МС m/e (ESI) 392,34 (MH+-CF3COOH).
Пример 341
Трифторацетат 3-(2-бутинил)-5-(2-оксо-2-фенилэтил)-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-с]пиридин-4-она
Указанное в заголовке соединение получали с использованием трет-бутил 4-[3-(2-бутинил)-4-оксо-4,5-дигидро-3Н-имидазо[4,5-с]пиридин-2-ил]пиперазин-1-карбоксилата и 2-бромацетофенона по способу, представленному в Примере 258(b).
1H-ЯМР (CD3OD)
δ 1,79 (т, J=2,3 Гц, 3H) 3,46-3,50 (м, 4H) 3,64-3,68 (м, 4H) 5,16 (кв, J=2,3 Гц, 2H) 5,61 (с, 2H) 6,65 (д, J=7,3 Гц, 1H) 7,37 (д, J=7,3 Гц, 1H) 7,57 (т, J=8,0 Гц, 2H) 7,69 (т, J=8,0 Гц, 1H) 8,10 (д, J=8,0 Гц, 2H).
МС m/e (ESI) 392,34 (MH+-CF3COOH).
Пример 353
7-(2-Бутинил)-1,3-диметил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-дион
(а) трет-бутил 4-[7-(2-бутинил)-1,3-диметил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
4,9 г 8-хлортеофиллина и 5 г карбоната калия растворяли в 100 мл N,N-диметилформамида и затем добавляли 2,4 мл 1-бром-2-бутина. Полученную смесь перемешивали при комнатной температуре в течение ночи и затем разбавляли этилацетатом и промывали водой. Полученное нерастворимое белое твердое вещество собирали с помощью фильтрования и промывали этилацетатом, что давало 3,8 г 7-(2-Бутинил)-8-хлор-1,3-диметил-3,7-дигидропурин-2,6-диона. Затем 1,8 г полученного 7-(2-бутинил)-8-хлор-1,3-диметил-3,7-дигидропурин-2,6-диона объединяли с 3,7 г трет-бутил 1-пиперазин-карбоксилата, и смесь перемешивали при 150°С в течение 1 часа. После охлаждения до комнатной температуры смесь экстрагировали этилацетатом. Органический слой промывали водой и затем насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Растворитель отгоняли при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 1,6 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (1:4).
1H-ЯМР (CDCl3)
δ: 1,49 (с, 9H) 1,82 (т, J=2,4 Гц, 3H) 3,33-3,36 (м, 4H) 3,40 (с, 3H) 3,52 (с, 3H) 3,58-3,61 (м, 4H) 4,88 (кв, J=2,4 Гц, 2H).
(b) 7-(2-Бутинил)-1,3-диметил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-дион
2,5 г трет-бутил 4-[7-(2-бутинил)-1,3-диметил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 15 мл трифторуксусной кислоты. Смесь перемешивали при комнатной температуре в течение 30 минут. Растворитель отгоняли при пониженном давлении. Затем остаток очищали с помощью колоночной хроматографии с использованием NH силикагеля (силикагель, поверхность которого модифицирована аминогруппами: Fuji Silysia Chemical Ltd. NH-DM 2035). Таким образом получали 1,6 г указанного в заголовке соединения из фракции, элюированной этилацетатом.
1H-ЯМР (CDCl3)
δ: 1,82 (т, J=2,4 Гц, 3H) 3,13-3,16 (м, 4H) 3,40 (с, 3H) 3,46-3,48 (м, 4H), 3,52 (с, 3H) 4,87 (кв, J=2,4 Гц, 2H).
Пример 354
7-(2-Бутинил)-3-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-дион
(а) трет-Бутил 4-[7-(2-бутинил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
1,1 г 3-метилксантина растворяли в 15 мл N,N-диметилформамида и затем добавляли 1,0 г карбоната калия и 0,64 мл 1-бром-2-бутина. Полученную смесь перемешивали при комнатной температуре в течение ночи и затем разбавляли этилацетатом и промывали водой. Полученное в результате нерастворимое белое твердое вещество собирали с помощью фильтрования и промывали этилацетатом, что давало 1,3 г 7-(2-бутинил)-3-метил-3,7-дигидропурин-2,6-диона. Далее, 1,3 г полученного 7-(2-Бутинил)-3-метил-3,7-дигидропурин-2,6-диона растворяли в 15 мл N,N-диметилформамида и затем к смеси, одновременно охлаждаемой на льду, добавляли 0,89 г N-хлорсукцинимида. Данную смесь перемешивали при комнатной температуре в течение 3 часов и затем разбавляли этилацетатом и промывали водой. Полученное в результате нерастворимое белое твердое вещество собирали с помощью фильтрования и промывали этилацетатом, что давало 1,1 г 7-(2-бутинил)-8-хлор-3-метил-3,7-дигидропурин-2,6-диона. Затем 1,4 г полученного 7-(2-Бутинил)-3-метил-3,7-дигидропурин-2,6-диона объединяли с 2,8 г трет-бутил 1-пиперазин-карбоксилата и смесь перемешивали при 150°С в течение 1 часа. Данную смесь затем охлаждали до комнатной температуры и экстрагировали этилацетатом. Органический слой промывали водой и затем насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Растворитель отгоняли при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 1,1 г указанного в заголовке соединения из фракции, элюированной смесью гексан-этилацетат (1:4).
1H-ЯМР (CDCl3)
δ: 1,49 (с, 9H) 1,82 (т, J=2,4 Гц, 3H) 3,35-3,37 (м, 4H) 3,47 (с, 3H) 3,58-3,61 (м, 4H) 4,85 (кв, J=2,4 Гц, 2H) 7,73 (с, 1H).
(b) 7-(2-Бутинил)-3-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-дион
Указанное в заголовке соединение получали с использованием трет-бутил 4-[7-(2-бутинил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата по способу, представленному в Примере 353(b).
1H-ЯМР (CDCl3)
δ: 1,82 (т, J=2,4 Гц, 3H) 3,02-3,05 (м, 4H) 3,37-3,39 (м, 4H) 3,48 (с, 3H) 4,85 (кв, J=2,4 Гц, 2H).
Пример 355
Трифторацетат метил [7-(2-бутинил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-ил]ацетата
15 мг трет-бутил 4-[7-(2-бутинил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата и 7 мг карбоната калия растворяли в 1 мл N,N-диметилформамида и затем добавляли 10 мкл метилбромацетата. Полученную смесь перемешивали при комнатной температуре в течение ночи и затем разбавляли этилацетатом и промывали водой. Растворитель отгоняли и остаток затем растворяли в 0,5 мл трифторуксусной кислоты. Смесь перемешивали при комнатной температуре в течение 30 минут. Растворитель отгоняли, и половина остатка очищалась с помощью ВЭЖХ (высокоэффективной жидкостной хроматографии) с обратно-фазной колонкой с использованием в качестве растворителя для элюирования смеси вода-ацетонитрил-трифторуксусная кислота. Таким образом получали 6,9 мг указанного в заголовке соединения.
MS m/e (ESI) 375 (MH+-CF3COOH).
Пример 356
Трифторацетат 7-(2-бутинил)-1-(2-этоксиэтил)-3-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием (2-бромэтил)этилового эфира с помощью той же процедуры, описанной в примере 355.
MS m/e (ESI) 375(MH+-CF3COOH).
Пример 357
Трифторацетат 7-(2-бутинил)-3-метил-8-(пиперазин-1-ил)-1-(2-пропинил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием пропаргилбромида с помощью той же процедуры, описанной в примере 355.
MS m/e(ESI)341(MH+-CF3COOH)
Пример 358
Трифторацетат 1,7-бис(2-бутинил)-3-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием 1-бром-2-бутина с помощью той же процедуры, описанной в примере 355.
MS m/e (ESI) 355 (MH+-CF3COOH).
Пример 359
Трифторацетат [7-(2-бутинил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-ил]ацетонитрила
Указанное в заголовке соединение получали с использованием бромацетонитрила с помощью той же процедуры, описанной в примере 355.
MS m/e (ESI) 342 (MH+-CF3COOH).
Пример 360
Трифторацетат 7-(2-бутинил)-1-этил-3-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием этилйодида с помощью той же процедуры, описанной в примере 355.
MS m/e (ESI) 331 (MH+-CF3COOH).
Пример 361
Трифторацетат 7-(2-бутинил)-3-метил-1-[(2-оксо-2-фенил)этил]-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием 2-бромацетофенона с помощью той же процедуры, описанной в примере 355.
MS m/e (ESI) 421 (MH+-CF3COOH).
Пример 362
Трифторацетат 7-(2-бутинил)-1-[2-(4-хлорфенил)-2-оксоэтил]-3-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием 2-бром-4'-хлорацетофенона с помощью той же процедуры, описанной в примере 355.
MS m/e (ESI) 455 (MH+-CF3COOH).
Пример 363
Трифторацетат 7-(2-бутинил)-3-метил-1-(2-феноксиэтил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием 2-феноксиэтилбромида с помощью той же процедуры, описанной в примере 355.
MS m/e (ESI) 423 (MH+-CF3COOH).
Пример 364
Трифторацетат 2-[7-(2-бутинил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-илметил]бензонитрила
Указанное в заголовке соединение получали с использованием 2-цианбензилбромида с помощью той же процедуры, описанной в примере 355.
MS m/e (ESI) 418 (MH+-CF3COOH).
Пример 365
Трифторацетат метил 4-[7-(2-бутинил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-илметил]бензоата
Указанное в заголовке соединение получали с использованием метил 4-(бромметил)бензоата с помощью той же процедуры, описанной в примере 355.
MS m/e (ESI) 451 (MH+-CF3COOH).
Пример 366
Трифторацетат метил 3-[7-(2-бутинил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-илметил]бензоата
Указанное в заголовке соединение получали с использованием метил 3-(бромметил)бензоата с помощью той же процедуры, описанной в примере 355.
MS m/e (ESI) 451 (MH+-CF3COOH).
Пример 367
Трифторацетат 7-(2-бутинил)-3-метил-1-(2-фенилэтил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием 2-(бромэтил)бензола с помощью той же процедуры, описанной в примере 355.
MS m/e (ESI) 407 (MH+-CF3COOH).
Пример 368
Трифторацетат 2-[7-(2-бутинил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-илметил]-N-фенилацетамида
25 мг трет-бутил 4-[1-карбоксиметил-3-метил-7-(2-бутинил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 1 мл тетрагидрофурана. Затем к смеси добавляли 5 мкл анилина, 9 мг 1,1-карбонилдиимидазола и 8 мкл триэтиламина. Полученную смесь перемешивали при 60°С в течение 5 часов. Раствор разбавляли этилацетатом и промывали водой и затем сушили над безводным сульфатом магния. Растворитель отгоняли, и остаток затем растворяли в 0,5 мл трифторуксусной кислоты. Смесь перемешивали при комнатной температуре в течение 30 минут. Растворитель отгоняли, и половина остатка очищалась с помощью ВЭЖХ с обратно-фазной колонкой с использованием в качестве растворителя для элюирования смеси вода-ацетонитрил-трифторуксусная кислота. Таким образом получали 2,74 мг указанного в заголовке соединения.
MS m/e (ESI) 436 (MH+-CF3COOH).
Пример 369
Трифторацетат 7-(2-метоксифенил)-1,3-диметил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
(а) трет-бутил-4-(1,3-диметил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил)пиперазин-1-карбоксилат
3,5 г 8-хлортеофиллина и 11,69 г трет-бутилпиперазин-1-карбоксилата смешивали и перемешивали при 110°С на протяжении ночи. Затем смесь разбавляли этилацетатом, а затем водой. Получающееся нерастворимое белое твердое вещество собирали фильтрованием и промывали этилацетатом, что давало 3,65 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,48 (с, 9H) 3,38 (с, 3H) 3,54-3,57 (м, 7H) 3,66-3,69 (м, 4H) 11,58 (с, 1H).
(b) Трифторацетат 7-(2-метоксифенил)-1,3-диметил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
11 мг трет-бутил-4-(1,3-диметил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил)пиперазин-1-карбоксилата, 15 мг 2-метоксифенилбороновой кислоты и 10 мг ацетата меди (II) суспендировали в 0,5 мл безводного тетрагидрофурана, а затем добавляли 0,1 мл пиридина. Получающуюся смесь перемешивали при комнатной температуре в течение пяти дней. Реакционный раствор фильтровали через короткую колонку, заполненную NH силикагелем, и фильтрат концентрировали. Остаток растворяли в 0,5 мл трифторуксусной кислоты, и смесь перемешивали при комнатной температуре в течение 30 минут. После того, как растворитель концентрировали, получающийся остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой. Таким образом получали 3,53 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 3,05-3,20 (м, 4H) 3,29 (с, 3H) 3,50-3,51 (м, 7H) 3,81 (с, 3H) 7,04-7,07 (м, 2H) 7,26-7,30 (м, 1H) 7,47 (дт, J=2,0, 8,0 Гц, 1H).
МС m/e (ESI) 371 (MH+-CF3COOH).
Пример 370
Трифторацетат 7-(2-цианфенил)-1,3-диметил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
(а) трет-бутил 4-[7-(2-формилфенил)-1,3-диметил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
226 мг трет-бутил 4-(1,3-диметил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил)пиперазин-1-карбоксилата, 200 мг 2-формилфенилбороновой кислоты и 200 мг ацетата меди (II) суспендировали в 5 мл безводного тетрагидрофурана, а затем добавляли 0,2 мл пиридина. Получающуюся смесь перемешивали при комнатной температуре в течение пяти дней. Реакционный раствор фильтровали через короткую колонку, заполненную силикагелем, и фильтрат концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом получали 51 мг указанного в заголовке соединения из фракции, элюированной смесью 1:1 гексан-этилацетата.
1H-ЯМР (CDCl3)
δ: 1,42 (с, 9H) 3,10-3,14 (м, 4H) 3,25-3,34 (м, 7H) 3,60 (с, 3H) 7,53 (дд, J=1,2, 8,0 Гц, 1H) 7,63-7,67 (м, 1H) 7,73-7,78 (м, 1H) 8,02-8,04 (м, 1H) 9,86 (с, 1H).
(b) Трифторацетат 7-(2-цианфенил)-1,3-диметил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
13 мг трет-бутил 4-[7-(2-формилфенил)-1,3-диметил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата и 10 мг гидрохлорида гидроксиламина растворяли в смеси, содержащей 1 мл этанола и 0,2 мл воды. К смеси добавляли приблизительно 10 мг ацетата калия. Получающуюся смесь перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор разбавляли этилацетатом, а затем промывали водным раствором бикарбоната натрия. Органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, что давало трет-бутил 4-[7-[2-(гидроксииминометил)фенил]-1,3-диметил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат. Данное соединение растворяли в 0,5 мл дихлорметана, а затем добавляли приблизительно 0,05 мл триэтиламина и 0,05 мл метансульфонилхлорида. Получающуюся смесь перемешивали при комнатной температуре в течение 0,5 часа. Смесь отгоняли, и остаток растворяли в трифторуксусной кислоте. Раствор концентрировали, и остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой, что давало 4,14 мг указанного в заголовке соединения.
МС m/e (ESI) 366 (MH+ -CF3COOH).
Пример 371
Трифторацетат 7-(2-винилфенил)-1,3-диметил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
9 мг третичного бутоксида калия растворяли в 1 мл тетрагидрофурана, а затем добавляли 31 мг метилтрифенилфосфонийбромида. Получающуюся смесь перемешивали при комнатной температуре в течение 30 минут. К смеси, которую затем перемешивали при комнатной температуре в течение 1 часа, добавляли 1 мл тетрагидрофуранового раствора, содержащего 20 мг трет-бутил 4-[7-(2-формилфенил)-1,3-диметил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата. Реакционный раствор разбавляли этилацетатом, а затем водой. Органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, что давало 40 мг трет-бутил 4-[7-(2-винилфенил)-1,3-диметил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата. 12 мг данного соединения растворяли в трифторуксусной кислоте. Раствор концентрировали, и остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой, что давало 4,38 мг указанного в заголовке соединения.
МС m/e (ESI) 367 (MH+ -CF3COOH).
Пример 372
7-(2-Хлорфенил)-1,3-диметил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-дион
(а) 7-(2-Хлорфенил)-1,3-диметил-3,7-дигидропурин-2,6-дион
510 мг теофиллина, 1 г 2-хлорфенилбороновой кислоты и 220 мг ацетата меди (II) суспендировали в 10 мл N,N-диметилформамида, а затем добавляли 1 мл пиридина. Получающуюся смесь перемешивали при комнатной температуре на протяжении ночи. Реакционный раствор разбавляли этилацетатом и промывали 30% аммиачной водой. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток растирали с простым эфиром, что давало 147 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 3,72 (с, 3H) 3,68 (с, 3H) 7,43-7,51 (м, 3H) 7,57-7,60 (м, 1H) 7,68 (с, 1H).
(b) 8-Хлор-7-(2-хлорфенил)-1,3-диметил-3,7-дигидропурин-2,6-дион
138 мг 7-(2-хлорфенил)-1,3-диметил-3,7-дигидропурин-2,6-диона и 78 мг N-хлорсукцинимида суспендировали в 1 мл N,N-диметилфоромамида. Получающуюся смесь перемешивали при комнатной температуре в течение двух часов. Реакционный раствор разбавляли этилацетатом и промывали водой. Органический слой сушили над безводным сульфатом магния, а затем фильтровали. Фильтрат концентрировали при пониженном давлении, что давало 151 мг указанного в заголовке соединения.
(с) трет-Бутил 4-[7-(2-хлорфенил)-1,3-диметил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
142 мг 8-Хлор-7-(2-хлорфенил)-1,3-диметил-3,7-дигидропурин-2,6-диона объединяли с 500 мг трет-бутилпиперазин-1-карбоксилата. Смесь перемешивали при 150°С в течение 4 часов, а затем разбавляли этилацетатом и промывали водой. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом из фракции, элюированной смесью 2:3 гексан-этилацетат, получали 143 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,43 (с, 9H) 3,21-3,23 (м, 4H) 3,30 (с, 3H) 3,31-3,35 (м, 4H) 3,58 (с, 3H) 7,42-7,51 (м, 3H) 7,55-7,57 (м, 1H).
(d)7-(2-Хлорфенил)-1,3-диметил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-дион
102 мг трет-бутил 4-[7-(2-хлорфенил)-1,3-диметил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 5 мл трифторуксусной кислоты. Получающуюся смесь перемешивали при комнатной температуре в течение 30 минут. Растворитель отгоняли, и остаток очищали с помощью колоночной хроматографии с использованием NH-силикагеля. Затем из фракции, элюированной смесью 9:1 этилацетата и метанола, получали 109 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 2,77 (дт, J=1,6, 4,8 Гц, 4H) 3,24 (т, J=5,2 Гц, 4H) 3,30 (с, 3H) 3,58 (с, 3H) 7,41-7,44 (м, 2H) 7,48-7,51 (м, 1H) 7,55-7,56 (м, 1H).
Пример 373
Трифторацетат 7-(2-хлорфенил)-3-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
(а) 7-Бензил-3-метил-3,7-дигидропурин-2,6-дион
2,882 г 3-метилксантина суспендировали в 40 мл N,N-диметилформамида, а затем добавляли 3 г карбоната калия и 2,5 мл бензилбромида. Получающуюся смесь перемешивали при комнатной температуре на протяжении ночи. Реакционный раствор разбавляли этилацетатом и промывали 1 н соляной кислотой. Выпавшие в осадок кристаллы собирали фильтрованием и промывали этилацетатом. Таким образом получали 3,18 г указанного в заголовке соединения.
1H-ЯМР (d6-ДМСО)
δ: 3,32 (с, 3H) 5,42 (с, 2H) 7,27-7,35 (м, 5H) 8,21 (с, 1H) 11,13 (с, 1H).
(b) 7-Бензил-3-метил-2,6-диоксо-2,3,6,7-тетрагидропурин-1-илметил 2,2-диметилпропионат
3,18 г 7-бензил-3-метил-3,7-дигидропурин-2,6-диона суспендировали в 40 мл N,N-диметилформамида. К смеси добавляли 2,6 г карбоната калия и 2,15 мл хлорметилпивалата. Получающуюся смесь перемешивали при 40°С на протяжении ночи. Реакционный раствор разбавляли этилацетатом и промывали 1 н соляной кислотой. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом, из фракции, элюированной 1:3 смесью гексана и этилацетата, получали 4,26 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ:1,19 (с, 9H) 3,58 (с, 3H) 5,48 (с, 2H) 6,04 (с, 2H) 7,32-7,39 (м, 5H) 7,58 (с, 1H).
(с) 3-Метил-2,6-диоксо-2,3,6,7-тетрагидропурин-1-илметил 2,2-диметилпропионат
4,26 г 7-Бензил-3-метил-2,6-диоксо-2,3,6,7-тетрагидропурин-1-илметил 2,2-диметилпропионата растворяли в 100 мл уксусной кислоты и затем добавляли 1,5 г 10% палладия на угле. Получающуюся смесь перемешивали в атмосфере водорода при комнатной температуре на протяжении ночи. Реакционный раствор фильтровали через целит, и фильтрат концентрировали, что давало 2,98 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,19 (с, 9H) 3,66 (с, 3H) 6,12 (с, 2H) 7,86 (с, 1H).
(d) 7-(2-Хлорфенил)-3-Метил-2,6-диоксо-2,3,6,7-тетрагидропурин-1-илметил 2,2-диметилпропионат
Указанное в заголовке соединение получали с использованием 3-метил-2,6-диоксо-2,3,6,7-тетрагидропурин-1-илметил 2,2-диметилпропионата с помощью той же процедуры, описанной в примере 372-(а).
(е) 8-Хлор-7-(2-хлорфенил)-3-метил-3,7-дигидропурин-2,6-дион
144 мг 7-(2-хлорфенил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидропурин-1-илметил 2,2-диметилпропионата растворяли в смеси, содержащей 2 мл метанола и 1 мл тетрагидрофурана, а затем добавляли 20 мг гидрида натрия. Получающуюся смесь перемешивали при комнатной температуре на протяжении ночи. Реакционный раствор разбавляли этилацетатом и промывали 1 н соляной кислотой. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток растирали со смесью этилацетата и диэтилового эфира, что давало 72 мг 7-(2-хлорфенил)-3-метил-3,7-дигидропурин-2,6-диона. Данное соединение растворяли в 1 мл N,N-диметилформамида, а затем добавляли 35 мг N-хлорсукцинимида. Получающуюся смесь перемешивали при комнатной температуре на протяжении ночи, и реакционный раствор разбавляли этилацетатом и промывали 1 н соляной кислотой. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, что давало 58 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 3,59 (с, 3H) 7,42 (дд, J=1,6, 7,6 Гц, 1H) 7,47 (дт, J=1,6, 9,2 Гц, 1H) 7,54 (дт, J=1,6, 7,2 Гц, 1H) 7,61 (дт, J=1,6, 7,6 Гц, 1H) 7,93 (ушир. 1H).
(f) трет-Бутил 4-[7-(2-хлорфенил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
58 мг 8-хлор-7-(2-хлорфенил)-3-метил-3,7-дигидропурин-2,6-диона объединяли со 150 мг 1-(третрет-бутоксикарбонил)пиперазина, и смесь перемешивали при 150°С в течение 4 часов. Реакционный раствор разбавляли этилацетатом и промывали водой. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом из фракции, элюированной этилацетатом, получали 44 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,41 (с, 9H) 3,17-3,24 (м, 4H) 3,25-3,41 (м, 4H) 3,53 (с, 3H) 7,41-7,51 (м, 3H) 7,55 (дд, J=2,0, 7,6 Гц, 1H) 7,66 (ушир. 1H).
(g) Трифторацетат 7-(2-хлорфенил)-3-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
8 мг трет-бутил 4-[7-(2-[хлорфенил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в трифторуксусной кислоте и затем раствор концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой, что давало 3,86 мг указанного в заголовке соединения.
MS m/e (ESI) 361 (MH+-CF3COOH).
1H-ЯМР (CDCl3)
δ: 2,76 -2,79 (м, 4H) 3,23-3,26 (м, 4H) 3,53 (с, 3H) 7,40-7,43 (м, 2H) 7,48-7,53 (м, 2H).
Пример 374
Трифторацетат метил [7-(2-хлорфенил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-ил]ацетата
18 мг трет-бутил 4-[7-(2-[хлорфенил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 1 мл N,N-диметилформамида и затем добавляли 0,1 мл метилбромацетата и 10 мг карбоната калия. Получающуюся смесь перемешивали при комнатной температуре в течение 3 дней. Реакционный раствор разбавляли этилацетатом и промывали водой. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток растворяли в трифторуксусной кислоте и раствор концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой, что давало 8,79 мг указанного в заголовке соединения.
MS m/e (ESI) 433 (MH+-CF3COOH).
Пример 375
Трифторацетат [7-(2-хлорфенил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-ил]ацетонитрила
Пример 376
Трифторацетат [7-(2-хлорфенил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-ил]ацетамида
18 мг трет-бутил 4-[7-(2-[хлорфенил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 1 мл N,N-диметилформамида и затем добавляли 0,1 мл бромацетонитрила и 10 мг карбоната калия. Получающуюся смесь перемешивали при комнатной температуре в течение 3 дней. Реакционный раствор разбавляли этилацетатом и промывали водой. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток растворяли в 1 мл ацетонитрила и затем добавляли 0,05 мл триметилсилилйодида. Получающуюся смесь перемешивали при комнатной температуре в течение 1 часа. Затем к смеси добавляли метанол. Реакционный раствор концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой, что давало 7,43 мг трифторацетата [7-(2-хлорфенил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-ил]ацетонитрила. [MS m/e (ESI) 400 (MH+-CF3COOH)] и 3,71 мг трифторацетата [7-(2-хлорфенил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-ил]ацетамида. [MS m/e (ESI) 418 (MH+-CF3COOH)].
Пример 377
Трифторацетат 7-(2-хлорфенил)-3-метил-1-(2-фенэтил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием 2-фенэтилбромида с помощью той же самой процедуры, описанной в примере 374.
MS m/e (ESI) 465 (MH+-CF3COOH).
Пример 378
Трифторацетат [7-(2-хлорфенил)-3-метил-1-(2-оксо-2-фенилэтил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием фенацилбромида с помощью той же самой процедуры, описанной в примере 374.
MS m/e (ESI) 479 (MH+-CF3COOH).
Пример 379
Трифторацетат 7-(2-метоксифенил)-3-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием 2-метоксифенилбороновой кислоты с помощью той же самой процедуры, описанной в примере 373.
MS m/e (ESI) 476 (MH+-CF3COOH).
Пример 380
Трифторацетат [7-(2-метоксифенил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-ил]ацетонитрила
Пример 381
Трифторацетат 2-[7-(2-метоксифенил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-ил]ацетамида
Трифторацетат [7-(2-метоксифенил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-ил]ацетонитрила [MS m/e (ESI) 396 (MH+-CF3COOH)] и трифторацетат 2-[7-(2-метоксифенил)-3-метил-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-ил]ацетамида [MS m/e (ESI) 414 (MH+-CF3COOH)] получали с использованием трет-бутил 4-[7-(2-метоксифенил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата с помощью тех же самых процедур, что использовались в Примерах 375 и 376.
Пример 382
Трифторацетат 7-(2-метоксифенил)-3-метил-1-(2-оксо-2-фенилэтил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием трет-бутил 4-[7-(2-метоксифенил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата и 2-бромацетофенона с помощью той же самой процедуры, описанной в примере 374.
MS m/e (ESI) 475 (MH+-CF3COOH).
Пример 383
Трифторацетат [7-(2-метоксифенил)-3-метил-1-(2-фенилэтил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием трет-бутил 4-[7-(2-метоксифенил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата и (2-бромэтил)бензола с помощью той же самой процедуры, описанной в примере 374.
MS m/e (ESI) 461 (MH+-CF3COOH).
Пример 384
7-(2-Винилфенил)-3-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-дион
(а) трет-Бутил 4-[7-бензил-1-(2,2-диметилпропионилоксиметил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
Указанное в заголовке соединение получали с использованием 7-бензил-3-метил-2,6-диоксо-2,3,6,7-тетрагидропурин-1-илметил 2,2-диметилпропионата с помощью той же самой процедуры, описанной в примере 373-(е) и (f).
(b) трет-Бутил 4-[1-(2,2-диметилпропионилоксиметил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
2,227 г трет-бутил 4-[7-бензил-1-(2,2-диметилпропионилоксиметил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 100 мл уксусной кислоты и затем добавляли 1 г 10% палладия на угле. Получающуюся смесь перемешивали в атмосфере водорода при комнатной температуре на протяжении ночи. Реакционный раствор фильтровали. Фильтрат концентрировали, что давало 1,89 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,09 (с, 9H) 1,41 (с, 9H) 3,36 (с, 3H) 3,37-3,42 (м, 4H) 3,45-3,50 (м, 4H) 5,82 (с, 2H).
(с) трет-Бутил 4-[1-(2,2-диметилпропионилоксиметил)-7-(2-винилфенил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
Указанное в заголовке соединение получали с использованием трет-бутил 4-[1-(2,2-диметилпропионилоксиметил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата с помощью той же процедуры, описанной в примерах 370 и 371.
1H-ЯМР (CDCl3)
δ: 1,15 (с, 9H) 1,58 (с, 9H) 3,18 (ушир. 4H) 3,30 (ушир. 4H) 3,58 (с, 3H) 5,32 (д, J=11,2 Гц, 1H) 5,75 (д, J=17,2 Гц, 1H) 6,39 (дд, J=10,8, 17,2 Гц, 1H) 7,34 (дд, J=1,2, 7,6 Гц, 1H) 7,40 (дт, J=1,6, 7,2 Гц, 1H) 7,46 (дт, J=1,6, 7,6 Гц, 1H) 7,69 (дд, J=1,6, 8,0 Гц, 1H).
(d) 7-(2-Винилфенил)-3-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-дион
187 мг трет-бутил 4-[1-(2,2-диметилпропионилоксиметил)-7-(2-винилфенил)-3-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 3 мл метанола, а затем добавляли 14 мг гидрида натрия. Получающуюся смесь перемешивали при комнатной температуре на протяжении ночи. Реакционный раствор нейтрализовали 1 н соляной кислотой, а затем экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель отгоняли. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом, из фракции, элюированной 3:2 смесью гексан-этилацетат, получали 108 мг трет-бутил 4-[3-метил-2,6-диоксо-7-(2-винилфенил)-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата. Данное соединение растворяли в 2 мл трифторуксусной кислоты и затем раствор концентрировали. Остаток очищали с использованием NH-силикагеля. Таким образом из фракции, элюированной смесью 15:1 этилацетата и метанола получали 84 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 2,73 (т, J=5,2 Гц, 4H) 3,19 (т, J=5,2 Гц, 4H) 3,54 (с, 3H) 5,32 (дд, J=1,2, 10,8 Гц, 1H) 5,74 (д, J=0,8, 17,6 Гц, 1H) 6,41 (дд, J=10,8, 17,2 Гц, 1H) 7,33 (дд, J=1,2, 6,0 Гц, 1H) 7,38 (дт, J=1,6, 7,6 Гц, 1H) 7,45 (дт, J=1,6, 7,6 Гц, 1H) 7,68 (дд, J=1,6, 8,0 Гц, 1H).
Пример 385
Трифторацетат 7-(2-хлорфенил)-3-этил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
(а) Гидрохлорид 2-амино-7-бензил-1,7-дигидропурин-6-она
100 г гуанозина суспендировали в 500 мл диметилсульфоксида. К суспрензии при комнатной температуре по каплям добавляли 100 мл бензилбромида. Получающуюся реакционная смесь перемешивали при комнатной температуре в течение 4 часов. Затем к реакционной смеси добавляли 250 мл концентрированной соляной кислоты, и получающуюся смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь выливали в 3 л метанола, и смесь перемешивали на протяжении ночи. Выпавшие в осадок кристаллы собирали фильтрованием, а затем промывали метанолом. Кристаллы сушили на воздухе при 60°С в течение 24 часов, что давало 82,5 г указанного в заголовке соединения.
1H-ЯМР (d6-ДМСО)
δ: 5,23 (с, 2H) 7,32-7,42 (м, 5H) 8,92 (с, 1H).
(b) 7-бензил-3,7-дигидропурин-2,6-дион
Белая суспензия, состоящая из 12,88 г гидрохлорида 2-амино-7-бензил-1,7-дигидропурин-6-она, 320 мл уксусной кислоты и 32 мл воды перемешивали при 110°С в течение 10 минут, а затем при 50°С в течение 10 минут. Затем в реакционную смесь при 50°С медленно по каплям добавляли 32 мл водного раствора, содержащего 12,88 г нитрита натрия. Получающуюся реакционная смесь перемешивали при 50°С в течение 15 часов. Получающуюся светло-коричневая суспензия фильтровали, что давало 4,27 г указанного в заголовке соединения.
1H-ЯМР (d6-ДМСО)
δ: 5,39 (с, 2H) 7,27-7,35 (м, 5H) 8,11 (с, 1H) 10,86 (с, 1H) 11,57 (с, 1H).
(с) [7-Бензил-3-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидропурин-1-ил]метил 2,2-диметилпропионат
9,54 г 7-бензил-ксантина растворяли в 250 мл N,N-диметилформамида, а затем добавляли 17 г карбоната калия и 14,2 мл хлорметилпивалата. Получающуюся смесь перемешивали при 50°С на протяжении ночи. Реакционный раствор разбавляли этилацетатом и промывали водой и 1 н соляной кислотой. Органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель отгоняли. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом из фракции, элюированной смесью 3:2 гесан-этилацетат, получали 12,8 г указанного в заголовке соединения.
(d) [3-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидропурин-1-ил]метил 2,2-диметилпропионат
Указанное в заголовке соединение получали с использованием [7-бензил-3-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидропурин-1-ил]метил 2,2-диметилпропионата с помощью той же процедуры, описанной в примере 384-(b).
(е) [7-(2-Хлорфенил)-3-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидропурин-1-ил]метил 2,2-диметилпропионат
Указанное в заголовке соединение получали с использованием [3-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидропурин-1-ил]метил 2,2-диметилпропионата с помощью той же процедуры, описанной в примере 373-(d).
1H-ЯМР (CDCl3)
δ: 1,16 (с, 9H) 1,22 (с, 9H) 5,99 (с, 2H) 6,19 (с, 2H) 7,42-7,52 (м, 3H) 7,58-7,61 (м, 1H) 7,73 (с, 1H).
(f) трет-бутил 4[7-(2-хлорфенил)-1,3-бис-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
Указанное в заголовке соединение получали с использованием [7-(2-хлорфенил)-3-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидропурин-1-ил]метил 2,2-диметилпропионата с помощью той же процедуры, описанной в примере 373-(е) и (f).
1H-ЯМР (CDCl3)
δ: 1,16 (с, 9H) 1,23 (с, 9H) 1,44 (с, 9H) 3,20-3,35 (м, 4H) 3,32-3,37 (м, 4H) 5,92 (с, 2H) 6,09 (с, 2H) 7,41-7,49 (м, 2H) 7,52-7,57 (м, 2H).
(g) трет-бутил 4-[7-(2-хлорфенил)-1-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
2,227 г трет-бутил 4-[7-(2-хлорфенил)-1,3-бис-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворялось в смеси, содержащей 10 мл тетрагидрофурана и 20 мл метанола, а затем добавляли 0,518 мл 1,8-диазабицикло[5,4,0]ундец-7-ена. Получающуюся смесь перемешивали при комнатной температуре на протяжении ночи. К реакционному раствору добавляли 1 н соляной кислоты. Получающееся выпавшее в осадок твердое вещество собирали фильтрованием и сушили, что давало 1,025 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,16 (с, 9H) 1,44 (с, 9H) 3,22-3,24 (м, 4H) 3,33-3,35 (м, 4H) 5,90 (с, 2H) 7,43-7,47 (м, 2H) 7,51-7,57 (м, 2H) 8,71 (ушир.с, 1H).
(h) Трифторацетат 7-(2-хлорфенил)-3-этил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
8 мг трет-бутил 4-[7-(2-хлорфенил)-1-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,3 мл N,N-диметилформамида, а затем добавляли 0,05 мл йодэтана и 20 мг карбоната калия. Реакционную смесь перемешивали при 50°С на протяжении ночи. К реакционному раствору добавляли этилацетат, и смесь промывали водой. Органический слой концентрировали. Остаток растворяли в метаноле и затем добавляли 5 мг гидрида натрия. Смесь перемешивали при комнатной температуре в течение 3 часов. Реакционный раствор нейтрализовали 1 н соляной кислотой, а затем экстрагировали этилацетатом. Растворитель концентрировали. Остаток растворяли в трифторуксусной кислоте, и затем раствор концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой, что давало 4,49 мг указанного в заголовке соединения.
MS m/e (ESI) 375 (MH+-CF3COOH).
Пример 386
Трифторацетат 7-(2-хлорфенил)-3-(2-оксо-2-фенэтил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием фенацилбромида с помощью той же самой процедуры, описанной в примере 385-(h).
MS m/e (ESI) 465 (MH+-CF3COOH).
Пример 387
Трифторацетат 7-(2-хлорфенил)-3-(2-оксотетрагидрофуран-3-ил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Пример 388
Трифторацетат 2-[7-(2-хлорфенил)-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидрогидропурин-3-ил]-4-гидроксибутановой кислоты
Трифторацетат 7-(2-хлорфенил)-3-(2-оксотетрагидрофуран-3-ил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона [MS m/e (ESI) 431 (MH+-CF3COOH)] и трифторацетат 2-[7-(2-хлорфенил)-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]-4-гидроксибутановой кислоты [MS m/e (ESI) 449 (MH+-CF3COOH)] получались с использованием α-бром-γ-бутиролактона с помощью той же самой процедуры, описанной в примере 385-(h).
Пример 389
Трифторацетат 2-[7-(2-хлорфенил)-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидрогидропурин-3-ил]ацетамида
Указанное в заголовке соединение получали с использованием 2-бромацетамида с помощью той же самой процедуры, описанной в примере 385-(h).
1H-ЯМР (d6-ДМСО)
δ: 2,97-3,04 (м, 4H) 3,22-3,34 (м, 4H) 4,43 (с, 2H) 7,18 (ушир.с, 1H) 7,49-7,59 (м, 2H) 7,62 (с, 1H) 7,66-7,71 (м, 2H) 10,90 (с, 1H).
МС m/e (ESI) 404 (MH+-CF3COOH).
Пример 390
Трифторацетат [7-(2-хлорфенил)-1-метил-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]уксусной кислоты
(а) трет-Бутил 4-[7-(2-хлорфенил)-3-карбоксиметил-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
87 мг трет-бутил 4-[7-(2-хлорфенил)-3-метоксикарбонилметил-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 2 мл метанола, а затем добавляли 0,2 мл 5 н водного раствора гидроксида натрия. Получающуюся смесь перемешивали при комнатной температуре в течение двух часов, а затем нейтрализовали 1 н соляной кислотой. Смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель отгоняли, что давало указанное в заголовке соединение.
(b) Трифторацетат [7-(2-хлорфенил)-1-метил-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]уксусной кислоты
26 мг трет-бутил 4-[7-(2-хлорфенил)-3-карбоксиметил-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в трифторуксусной кислоте, и смесь концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой, что давало 10,73 мг указанного в заголовке соединения.
1H-ЯМР (d6-ДМСО)
δ: 3,15-3,18 (м, 4H) 3,26 (с, 3H) 3,46-3,49 (м, 4H) 4,80 (с, 2H) 7,50-7,59 (м, 2H) 7,63-7,68 (м, 2H).
МС m/e (ESI) 419 (MH+-CF3COOH).
Пример 391
Трифторацетат 2-[7-(2-хлорфенил)-1-метил-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетамида
(а) трет-Бутил 4-[7-(2-хлорфенил)-3-ацетамид-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
53 мг трет-бутил 4-[7-(2-хлорфенил)-3-карбоксиметил-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 1 мл тетрагидрофурана, а затем добавляли 0,03 мл триэтиламина и 0,015 мл этилхлоркарбоната. Получающуюся смесь перемешивали при комнатной температуре в течение 15 минут, а затем добавляли 0,1 мл водного раствора 30% аммиака. Реакционный раствор разбавляли этилацетатом и промывали водой и 1 н соляной кислотой. Органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель отгоняли, что давало 53 мг указанного в заголовке соединения.
(b) Трифторацетат 2-[7-(2-хлорфенил)-1-метил-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетамида
53 мг трет-бутил 4-[7-(2-хлорфенил)-3-ацетамид-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в трифторуксусной кислоте, и раствор концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой, что давало 23,31 мг указанного в заголовке соединения.
1H-ЯМР (d6-ДМСО)
δ: 3,15-3,18 (м, 4H) 3,26 (с, 3H) 3,45-3,48 (м, 4H) 4,76 (с, 2H) 7,50-7,59 (м, 2H) 7,62-7,68 (м, 2H).
МС m/e (ESI) 418 (MH+-CF3COOH).
Пример 392
Трифторацетат [7-(2-хлорфенил)-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]уксусной кислоты
Указанное в заголовке соединение получали с использованием трет-бутил 4-[7-(2-хлорфенил)-3-метоксикарбонилметил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата с помощью той же самой процедуры, описанной в примере 390-(а) и (b).
MS m/e (ESI) 405 (MH+-CF3COOH).
Пример 393
Трифторацетат [7-(2-хлорфенил)-2,6-диоксо-1-фенэтил-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]уксусной кислоты
Указанное в заголовке соединение получали с использованием трет-бутил 4-[7-(2-хлорфенил)-3-метоксикарбонилметил-2,6-диоксо-1-фенэтил-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата с помощью той же самой процедуры, описанной в примере 390-(а) и (b).
MS m/e (ESI) 509 (MH+-CF3COOH).
Пример 394
Трифторацетат 2-[7-(2-хлорфенил)-2,6-диоксо-1-фенэтил-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетамида
Указанное в заголовке соединение получали с использованием трет-бутил 4-[7-(2-хлорфенил)-3-карбоксиметил-2,6-диоксо-1-фенэтил-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата с помощью той же самой процедуры, описанной в примере 391-(а) и (b).
MS m/e (ESI) 508 (MH+-CF3COOH).
Пример 395
Трифторацетат [7-(2-хлорфенил)-1-метил-8-(пиперазин-1-ил)]-3,7-дигидропурин-2,6-диона
(а) [7-Бензил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил 2,2-диметилпропионат
8,66 г 7-бензилксантина растворяли в 300 мл N,N-диметилформамида, а затем добавляли 1,57 г гидрида натрия и 7,7 мл хлорметилпивалата. Получающуюся смесь перемешивали при комнатной температуре на протяжении ночи. Реакционный раствор разбавляли этилацетатом, а затем промывали водой и 1 н соляной кислотой. Органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель отгоняли. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом из фракции, элюированной 1:1 смесью гексан-этилацетат, получали 2,66 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,18 (с, 9H) 5,45 (с, 2H) 6,06 (с, 2H) 7,34-7,39 (м, 5H) 7,58 (с, 1H) 8,18 (с, 1H).
(b) [7-Бензил-1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил 2,2-диметилпропионат
2,66 г [7-бензил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил] метил 2,2-диметилпропионата растворяли в 30 мл N,N-диметилфоромамида, а затем добавляли 1,6 г карбоната калия и 1 мл йодметана. Получающуюся смесь перемешивали при комнатной температуре на протяжении ночи. Реакционный раствор разбавляли этилацетатом и промывали водой и 1 н соляной кислотой. Органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель отгоняли. Остаток растирали с толуолом, что давало 2,16 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,18 (с, 9H) 3,41 (с, 3H) 5,49 (с, 2H) 6,11 (с, 2H) 7,26-7,39 (м, 5H) 7,57 (с, 1H).
(с) [1-Метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил 2,2-диметилпропионат
2,16 г указанного в заголовке соединения получали с использованием [7-бензил-1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил] метил 2,2-диметилпропионата с помощью той же самой процедуры, что описана в Примере 385-(d).
1H-ЯМР (CDCl3)
δ: 1,19 (с, 9H) 3,48 (с, 3H) 6,17 (с, 2H) 7,83 (с, 1H).
(d) [7-(2-Хлорфенил)-1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил 2,2-диметилпропионат
Указанное в заголовке соединение получали с использованием [1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил] метил 2,2-диметилпропионата с помощью той же самой процедуры, что описана в Примере 385-(е).
(е) трет-Бутил 4-[7-(2-хлорфенил)-3-(2,2-диметилпропионилоксиметил)-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
Указанное в заголовке соединение получали с использованием [7-(2-хлорфенил)-1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил] метил 2,2-диметилпропионата с помощью той же самой процедуры, что описана в Примере 385-(f).
(f) трет-Бутил 4-[7-(2-хлорфенил)-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
Указанное в заголовке соединение получали с использованием трет-бутил 4-[7-(2-хлорфенил)-3-(2,2-диметил-пропионилоксиметил)-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата с помощью той же самой процедуры, что описана в Примере 373-(е).
1H-ЯМР (d6-ДМСО)
δ: 1,35 (с, 9H) 3,04 (с, 3H) 3,06-3,12 (м, 4H) 3,17-3,22 (м, 4H) 7,48 (дт, J=1,6, 7,6 Гц, 1H) 7,53 (дт, J=2,0, 7,6 Гц, 1H) 7,63 (дд, J=2,0, 8,0 Гц, 1H) 7,65 (дд, J=1,6, 8,0 Гц, 1H).
(g) Трифторацетат 7-(2-хлорфенил)-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием трет-бутил 4-[7-(2-хлорфенил)-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилатата с помощью той же самой процедуры, что описана в Примере 391-(b).
1H-ЯМР (d6-ДМСО)
δ: 2,95-3,03 (м, 4H) 3,14 (с, 3H) 3,23-3,34 (м, 4H) 7,49-7,62 (м, 2H) 7,66-7,71 (м, 2H) 10,90 (с, 1H).
МС m/e (ESI) 361 (MH+-CF3COOH).
Пример 396
Трифторацетат [7-(2-бутинил)-3-этил-1-метил-8-(пиперазин-1-ил)]-3,7-дигидропурин-2,6-диона
(а) [7-(2-бутинил)-1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил 2,2-диметилпропионат
1,871 г [1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил 2,2-диметилпропионата растворяли в 30 мл N,N-диметилформамида, а затем добавляли 1,5 г карбоната калия и 0,7 мл 2-бутинилбромида. Получающуюся смесь перемешивали при комнатной температуре на протяжении ночи. Реакционный раствор разбавляли этилацетатом, а затем промывали водой и 1 н соляной кислотой. Органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель отгоняли. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом из фракции, элюированной смесью 3:2 гесан-этилацетат, получали 2,12 г указанного в заголовке соединения.
(b) 7-(2-Бутинил)-1-метил-3,7-дигидропурин-2,6-дион
Указанное в заголовке соединение получали с использованием [7-(2-бутинил)-1-метил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил 2,2-диметилпропионата с помощью той же процедуры, что описана в Примере 395-(f).
1H-ЯМР (CDCl3)
δ: 1,91 (т, J=2,4 Гц, 3H) 3,39 (с, 3H) 5,10 (с, 2H) 7,93 (с, 1H) 10,62 (с, 1H).
(c) трет-бутил 4-[7-(2-бутинил)-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
Указанное в заголовке соединение получали с использованием [7-(2-бутинил)-1-метил-3,7-дигидропурин-2,6-диона с помощью той же самой процедуры, что описана в Примере 395-(е).
1H-ЯМР (CDCl3)
δ: 1,48 (с, 9H) 1,83 (т, J=2,4 Гц, 3H) 3,37 (с, 3H) 3,37-3,39 (м, 4H) 3,58-3,60 (м, 4H) 4,87 (с, 2H) 9,68 (с, 1H).
(d) Трифторацетат 7-(2-бутинил)-3-этил-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием трет-бутил 4-[7-(2-бутинил)-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата с помощью той же самой процедуры, что описана в Примере 385-(h).
MS m/e (ESI) 331 (MH+-CF3COOH).
Пример 397
Трифторацетат 7-(2-бутинил)-3-бензил-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием бензилбромида с помощью той же самой процедуры, что описана в Примере 396-(d).
1H-ЯМР (CDCl3)
δ: 1,83 (т, J=2,4 Гц, 3H) 3,03-3,06 (м, 4H) 3,38 (с, 3H) 3,38-3,41 (м, 4H) 4,84 (кв, J=2,4 Гц, 2H) 5,21 (с, 2H) 7,26-7,30 (м, 3H) 7,52-7,54 (м, 2H).
МС m/e (ESI) 393 (MH+-CF3COOH).
Пример 398
Трифторацетат метил [7-(2-бутинил)-1-метил-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетата
Указанное в заголовке соединение получали с использованием метилбромацетата с помощью той же самой процедуры, что описана в Примере 396-(d).
1H-ЯМР (CDCl3)
δ: 1,84 (т, J=2,4 Гц, 3H) 3,00-3,03 (м, 4H) 3,34-3,36 (м, 4H) 3,40 (с, 3H) 3,79 (с, 3H) 4,78 (с, 2H) 4,84 (кв, J=2,4 Гц, 2H).
МС m/e (ESI) 375 (MH+-CF3COOH).
Пример 399
Трифторацетат 7-(2-бутинил)-3-циклобутил-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
8 мг трет-бутил 4-[7-(2-бутинил)-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,4 мл N,N-диметилформамида, а затем добавляли 10 мг карбоната калия и 0,01 мл циклобутилбромида. Получающуюся смесь перемешивали при 50°С на протяжении ночи. Реакционный раствор разбавляли этилацетатом. Органический слой концентрировали. Остаток растворяли в трифторуксусной кислоте, и раствор концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой, что давало 3,72 мг указанного в заголовке соединения.
MS m/e (ESI) 357 (MH+-CF3COOH).
Пример 400
Трифторацетат 7-(2-бутинил)-3-(2-тетрагидрофуранил)метил-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием 2-бромметил-тетрагидрофурана с помощью той же самой процедуры, что описана в Примере 399.
1H-ЯМР (CDCl3)
δ: 1,70-1,77 (м, 1H) 1,84 (т, J=2,4 Гц, 3H) 1,88-1,93 (м, 1H) 1,97-2,06 (м, 2H) 3,01-3,04 (м, 4H) 3,34-3,36 (м, 4H) 3,39 (с, 3H) 3,77 (дд, J=8,4, 14,0 Гц, 1H) 3,92-3,97 (м, 2H) 4,19 (дд, J=8,4, 13,6 Гц, 1H) 4,45-4,50 (м, 1H) 4,83 (кв, J=2,4 Гц, 2H).
МС m/e (ESI) 387 (MH+-CF3COOH).
Пример 401
Трифторацетат 2-[7-(2-бутинил)-1-метил-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетамида
Указанное в заголовке соединение получали с использованием 2-бромацетамида с помощью той же самой процедуры, что описана в Примере 399.
1H-ЯМР (CDCl3)
δ: 1,68 (т, J=2,4 Гц, 3H) 3,15-3,19 (м, 4H) 3,23 (с, 3H) 3,46-3,51 (м, 4H) 4,55 (с, 2H) 4,71 (кв, J=2,4 Гц, 2H) 6,00 (ушир. 1H) 6,91 (ушир. 1H).
МС m/e (ESI) 360 (MH+-CF3COOH).
Пример 402
Трифторацетат метил [7-(2-бутинил)-1-метил-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]фенилацетата
Указанное в заголовке соединение получали с использованием метил 2-бромфенил-ацетата с помощью той же самой процедуры, что описана в Примере 399.
1H-ЯМР (CDCl3)
δ: 1,83 (т, J=2,4 Гц, 3H) 3,02-3,05 (м, 4H) 3,36-3,38 (м, 4H) 3,37 (с, 3H) 3,80 (с, 3H) 4,82 (кв, J=2,4 Гц, 2H) 6,50 (с, 1H) 7,30-7,32 (м, 3H) 7,65-7,67 (м, 2H).
МС m/e (ESI) 451 (MH+-CF3COOH).
Пример 403
Трифторацетат 7-(2-бутинил)-3-пропил-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием йодпропана с помощью той же самой процедуры, что описана в Примере 399.
MS m/e (ESI) 345 (MH+-CF3COOH).
Пример 404
Трифторацетат 7-(2-бутинил)-3-(2-оксо-2-фенэтил)-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием фенацилбромида с помощью той же самой процедуры, что описана в Примере 399.
1H-ЯМР (CDCl3)
δ: 1,85 (т, J=2,4 Гц, 3H) 2,96-2,99 (м, 4H) 3,28-3,31 (м, 4H) 3,41 (с, 3H) 4,85 (кв, J=2,4 Гц, 2H) 5,48 (с, 2H) 7,50-7,54 (м, 2H) 7,61-7,65 (м, 1H) 8,02-8,05 (м, 2H).
МС m/e (ESI) 421 (MH+-CF3COOH).
Пример 405
Трифторацетат этил 2-[7-(2-бутинил)-1-метил-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]пропионата
Указанное в заголовке соединение получали с использованием этил 2-бромпропионата с помощью той же самой процедуры, что описана в Примере 399.
1H-ЯМР (CDCl3)
δ: 1,23 (т, J=7,2 Гц, 3H) 1,70 (д, J=7,2 Гц, 3H) 1,84 (т, J=2,4 Гц, 3H) 3,00-3,03 (м, 4H) 3,33-3,37 (м, 4H) 3,38 (с, 3H) 4,15-4,25 (м, 2H) 4,85 (кв, J=2,4 Гц, 2H) 5,43 (кв, J=7,2 Гц, 1H).
МС m/e (ESI) 403 (MH+-CF3COOH).
Пример 406
Трифторацетат 7-(2-бутинил)-3-(2-оксо-тетрагидрофуран-3-ил)-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием α-бром-γ-бутиролактона с помощью той же самой процедуры, что описана в Примере 399.
1H-ЯМР (CDCl3)
δ: 1,84 (т, J=2,4 Гц, 3H) 2,59-2,68 (м, 1H) 2,69-2,91 (м, 1H) 3,01-3,03 (м, 4H) 3,34-3,37 (м, 5H) 3,38 (с, 3H) 4,39-4,45 (м, 1H) 4,68 (дт, J=2,8, 9,2 Гц, 2H) 4,84 (ушир. 2H).
МС m/e (ESI) 387 (MH+-CF3COOH).
Пример 407
Трифторацетат 7-(2-бутинил)-3-(2-этоксиэтил)-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием 2-этоксиэтилбромида с помощью той же самой процедуры, что описана в Примере 399.
1H-ЯМР (CDCl3)
δ: 1,16 (т, J=7,2 Гц, 3H) 1,83 (т, J=2,4 Гц, 3H) 3,01-3,06 (м, 4H) 3,33-3,46 (м, 4H) 3,39 (с, 3H) 3,58 (кв, J=7,2 Гц, 2H) 3,77 (т, J=6,0 Гц, 2H) 4,26 (т, J=6,0 Гц, 2H) 4,85 (кв, J=2,4 Гц, 2H).
МС m/e (ESI) 375 (MH+-CF3COOH).
Пример 408
Трифторацетат 7-(2-бутинил)-3-изопропил-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием 2-йодпропана с помощью той же самой процедуры, что описана в Примере 399.
MS m/e (ESI) 345 (MH+-CF3COOH).
Пример 409
Трифторацетат 7-(2-бутинил)-3-(3,3-диметил-2-оксобутил)-1-метил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием 1-бромпинаколона с помощью той же самой процедуры, что описана в Примере 399.
MS m/e (ESI) 401 (MH+-CF3COOH).
Пример 410
Гидрохлорид 7-(2-бутинил)-1-метил-3-(2-оксопирролидин-3-ил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием 3-бром-2-оксопирролидина с помощью той же самой процедуры, что описана в Примере 399.
1H-ЯМР (d6-ДМСО)
δ: 1,80 (т, J=2 Гц, 3H) 2,32-2,48 (м, 2H) 3,17 (с, 3H) 3,20-3,55 (м, 10H) 4,96 (кв, J=2 Гц, 2H) 5,14 (т, J=10 Гц) 7,94 (ушир.с, 1H) 9,04 (ушир.с, 2H).
Пример 411
Трифторацетат 7-(2-бутинил)-3-(2-этоксиэтил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
(а) трет-Бутил 4-[7-(2-бутинил)-1,3-бис-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
Смесь, содержащую 1,0 г [3-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидропурин-1-ил]метил 2,2-диметилпропионата, 0,28 мл 1-бром-2-бутина, 0,73 г безводного карбоната калия и 15 мл N,N-диметилформамида, перемешивали при комнатной температуре в течение двух часов. Реакционный раствор экстрагировали этилацетатом-водой. Органический слой промывали водой и насыщенным раствором хлорида натрия, а затем сушили над безводным сульфатом магния. Жидкость концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием смеси 20-30% этилацетат/гексан, что давало 1,06 г [7-(2-бутинил)-3-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидропурин-1-ил]метил 2,2-диметилпропионата.
Все количество данного соединения объединяли с 390 мг N-хлорсукцинимида и 5 мл N,N-диметилформамида. Смесь перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор экстрагировали этилацетатом и водой. Органический слой промывали водой и насыщенным раствором хлорида натрия, а затем сушили над безводным сульфатом магния. Жидкость концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием смеси 20-30% этилацетат/гексан, что давало 1,18 г [7-(2-бутинил)-8-хлор-3-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидропурин-1-ил]метил 2,2-диметилпропионата.
Все количество соединения объединяли с 1,4 г трет-бутилпиперазин-1-карбоксилата, и смесь перемешивали при 150°С на масляной бане, при этом ее перемешивали в течение 30 минут. Реакционный раствор очищали с помощью колоночной хроматографии с использованием смеси 20-30% этилацетат/гексан, что давало 1,34 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,18 (с, 18H) 1,49 (с, 9H) 1,84 (т, J=2 Гц, 3H) 3,36 (т, J=5 Гц, 4H) 3,58 (т, J=5 Гц) 4,86 (кв, J=2 Гц, 2H) 6,02 (с, 2H), 6,03 (с, 2H).
(b) трет-Бутил 4-[7-(2-бутинил)-1-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
0,63 г трет-бутил 4-[7-(2-бутинил)-1,3-бис-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в смешанном растворителе из 4 мл тетрагидрофурана и 2 мл метанола, а затем добавляли 0,18 мл диазабицикло[5,4,0]ундецена. Получающуюся смесь перемешивали при комнатной температуре на протяжении ночи. Реакционный раствор концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом из фракции, элюированной гексаном-этилацетатом (1:5), получали 0,29 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,19 (с, 9H) 1,48 (с, 9H) 1,83 (т, J=2,4 Гц, 3H) 3,37-3,39 (м, 4H) 3,58-3,60 (м, 4H) 4,86 (кв, J=2,4 Гц, 2H) 6,00 (с, 2H) 9,08 (с, 1H).
(с) Трифторацетат 7-(2-бутинил)-3-(2-этоксиэтил)-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
50 мг трет-бутил 4-[7-(2-бутинил)-1-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата и 15 мг карбоната калия растворяли в 1,2 мл N,N-диметилформамида, а затем добавляли 12 мкл 2-бромэтилэтилового эфира. Получающуюся смесь перемешивали при 60°С в течение двух часов, а затем разбавляли этилацетатом и промывали водой. Жидкость сушили над безводным сульфатом магния. Органический слой концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом из фракции, элюированной смесью гексан-этилацетат (2:1), получали трет-бутил 4-[7-(2-бутинил)-1-(2,2-диметилпропионилоксиметил)-3-(2-этоксиэтил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат. Затем получающийся трет-бутил 4-[7-(2-бутинил)-1-(2,2-диметилпропионилоксиметил)-3-(2-этоксиэтил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат растворяли в смешанном растворителе из 1,0 мл тетрагидрофурана и 0,5 мл метанола, а затем добавляли 5 мг гидрида натрия. Получающуюся смесь перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор нейтрализовали 2 н соляной кислотой и экстрагировали этилацетатом. Затем органический слой сушили над безводным сульфатом магния. Растворитель отгоняли, что давало трет-бутил 4-[7-(2-бутинил)-3-(2-этоксиэтил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат. 1/4 эквивалента получающегося трет-бутил 4-[7-(2-бутинил)-3-(2-этоксиэтил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 0,5 мл трифторуксусной кислоты, и смесь перемешивали при комнатной температуре в течение 30 минут. Растворитель отгоняли. Затем половина аликвоты остатка очищалась с помощью ВЭЖХ с использованием колонки с обратной фазой с водой-ацетонитрилом-трифторуксусной кислотой в качестве растворителя для элюирования, что давало 3,2 мг указанного в заголовке соединения.
MS m/e (ESI) 361 (MH+-CF3COOH).
Пример 412
Трифторацетат метил [7-(2-бутинил)-3-(2-этоксиэтил)-2,6-диоксо-8-(пиперазин-1-ил)-2,3,6,7-тетрагидропурин-1-ил]ацетата
1/4 эквивалента трет-бутил 4-[7-(2-бутинил)-3-(2-этоксиэтил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата, полученного в Примере 411-(с), и 7 мг карбоната калия растворяли в 0,8 мл N,N-диметилформамида, а затем добавляли 10 мкл метилбромацетата. Получающуюся смесь перемешивали при комнатной температуре на протяжении ночи, а затем разбавляли этилацетатом и промывали водой. Жидкость сушили над безводным сульфатом магния. Органический слой концентрировали, и затем остаток растворяли в 0,5 мл трифторуксусной кислоты. Смесь перемешивали при комнатной температуре в течение 30 минут. Растворитель отгоняли, и половина аликвоты остатка очищалась с помощью ВЭЖХ с использованием колонки с обратной фазой с водой-ацетонитрилом-трифторуксусной кислотой в качестве растворителя для элюирования, что давало 3,2 мг указанного в заголовке соединения.
MS m/e (ESI) 433 (MH+-CF3COOH).
Пример 413
Трифторацетат 7-(2-бутинил)-3-(2-этоксиэтил)-1-(2-оксо-2-фенилэтил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Указанное в заголовке соединение получали с использованием 2-бромацетофенона с помощью той же процедуры, что описана в примере 412,
MS m/e (ESI) 479 (MH+-CF3COOH).
Пример 414
Трифторацетат метил [7-(2-бутинил)-1-(2-этоксиэтил)-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетата
(а) трет-Бутил 4-[7-(2-бутинил)-3-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
1,1 г трет-бутил 4-[7-(2-бутинил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата и 0,43 г карбоната калия растворяли в 15 мл N,N-диметилформамида. Затем к смеси на льду добавляли 0,60 мл хлорметилпивалата. Получающуюся смесь перемешивали при комнатной температуре на протяжении ночи, а затем разбавляли этилацетатом и промывали водой. Получающееся нерастворимое белое твердое вещество собирали фильтрованием и промывали смешанным растворителем из гексана и этилацетата (1:1), что давало 0,57 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,18 (с, 9H) 1,49 (с, 9H) 1,83 (т, J=2,4 Гц, 3H) 3,33-3,36 (м, 4H) 3,57-3,59 (м, 4H) 4,84 (кв, J=2,4 Гц, 2H) 5,99 (с, 2H) 7,72 (с, 1H).
(b) Трифторацетат метил[7-(2-бутинил)-1-(2-этоксиэтил)-2,6-диоксо-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетата
40 мг трет-бутил 4-[7-(2-бутинил)-3-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата и 17 мг карбоната калия растворяли в 1,5 мл N,N-диметилформамида, а затем добавляли 14 мкл 2-бромэтилэтилового эфира. Получающуюся смесь перемешивали при 60°С в течение 5 часов, а затем разбавляли этилацетатом и промывали водой. Жидкость сушили над безводным сульфатом магния. Растворитель отгоняли, и остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом из фракции, элюированной смесью гексан-этилацетат (1:1), получали трет-бутил 4-[7-(2-бутинил)-3-(2,2-диметилпропионилоксиметил)-1-(2-этоксиэтил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат. Затем получающийся трет-бутил 4-[7-(2-бутинил)-3-(2,2-диметилпропионилоксиметил)-1-(2-этоксиэтил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат растворяли в смешанном растворителе из 1,0 мл тетрагидрофурана и 0,5 мл метанола, а затем добавляли 5 мг гидрида натрия. Получающуюся смесь перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор нейтрализовали 2 н соляной кислотой и экстрагировали этилацетатом. Затем органический слой сушили над безводным сульфатом магния и растворитель отгоняли. Получающийся остаток растворяли в 1 мл N,N-диметилформамида, а затем добавляли 10 мг карбоната калия и 10 мкл метилбромацетата. Получающуюся смесь перемешивали при комнатной температуре в течение 2 часов, а затем разбавляли этилацетатом и промывали водой. Органический слой концентрировали, и остаток растворяли в 0,5 мл трифторуксусной кислоты. Получающуюся смесь перемешивали при комнатной температуре в течение 30 минут. Растворитель отгоняли, и затем половину аликвоты остатка очищали с помощью ВЭЖХ с использованием колонки с обратной фазой с водой-ацетонитрилом-трифторуксусной кислотой в качестве растворителя для элюирования, что давало 6,2 мг указанного в заголовке соединения.
MS m/e (ESI) 433(MH+-CF3COOH).
Пример 415
Трифторацетат метил [7-(2-бутинил)-2,6-диоксо-1-(2-оксо-2-фенилэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетата
Указанное в заголовке соединение получали с использованием 2-бромацетофенона с помощью процедуры, описанной в примере 414.
MS m/e (ESI) 479(MH+-CF3COOH).
Пример 416
Гидрохлорид этил [7-(2-бутинил)-2,6-диоксо-1-(2-фенэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетата
(а) Этил (7-бензил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил)ацетат
Смесь, содержащую 3,0 г 7-бензил-3,7-дигидропурин-2,6-диона, 2,0 г безводного карбоната калия и 60 мл N,N-диметилформамида, перемешивали при 40°С на масляной бане, а затем добавляли 1,5 г этилбромацетата. Получающуюся смесь перемешивали в течение 4 часов при 40°С. Реакционный раствор разбавляли этилацетатом и водой и экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором хлорида натрия, а затем сушили над безводным сульфатом магния. Жидкость концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием 20-40% смеси (20% 2-пропанол/этилацетат)/гексан, что давало 1,3 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,28 (т, J=7 Гц, 3H) 4,23 (кв, J=7 Гц, 2H) 4,78 (с, 2H) 5,04 (с, 2H) 7,31-7,39 (м, 5H) 7,51 (с, 1H) 8,01 (ушир.с, 1H).
(b) Этил [7-бензил-1-(2-фенилэтил)-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]ацетат
Смесь, содержащую 300 мг этил (7-бензил-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил)ацетата, 250 мг безводного карбоната калия, 0,25 мл 2-бромэтилбензола и 5 мл N,N-диметилформамида, перемешивали при 50°С на масляной бане в течение двух часов. Реакционный раствор разбавляли этилацетатом и водой и экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором хлорида натрия, а затем сушили над безводным сульфатом магния. Жидкость концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием смеси 10-20% (20% 2-пропанол/этилацетат)/гексан, что давало 366 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,29 (т, J=7 Гц, 3H) 2,95 (т, J=8 Гц, 2H) 4,22 (т, J=8 Гц, 2H) 4,24 (кв, J=7 Гц, 2H) 4,83 (с, 2H) 5,48 (с, 2H) 7,17-7,39 (м, 10H) 7,49 (с, 1H).
(с) Этил [7-(2-бутинил)-8-хлор-1-(2-фенилэтил)-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]ацетат
Каталитическое количество 10% палладия на угле добавляли к смеси, содержащей 366 мг этил [7-бензил-1-(2-фенилэтил)-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]ацетата и 10 мл уксусной кислоты. Получающуюся смесь перемешивали в атмосфере водорода при комнатной температуре на протяжении ночи. После того, как катализатор удаляли фильтрованием, жидкость концентрировали при пониженном давлении, что давало 320 мг остатка. Все количество сконцентрированного остатка объединяли с 260 мг безводного карбоната калия, 0,1 мл 1-бром-2-бутина и 5 мл N,N-диметилформамида. Получающуюся смесь перемешивали при комнатной температуре в течение двух часов. Реакционный раствор разбавляли этилацетатом и водой и экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором хлорида натрия, а затем сушили над безводным сульфатом магния. Жидкость концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием смеси 20-30% этилацетат/гексан, что давало 290 мг масляного материала. Все количество масляного материала объединяли с 3 мл N,N-диметилформамида и 120 мг N-хлорсукцинимида. Получающуюся смесь перемешивали при комнатной температуре в течение одного часа. Реакционный раствор экстрагировали этилацетатом и водой. Органический слой промывали водой и насыщенным раствором хлорида натрия, а затем сушили над безводным сульфатом магния. Жидкость концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с использованием смеси 20-30% этилацетат/гексан, что давало 273 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,31 (т, J=7 Гц, 3H) 1,82 (т, J=2 Гц, 3H) 2,94 (т, J=8 Гц, 2H) 4,21 (т, J=8 Гц, 2H) 4,25 (кв, J=7 Гц, 2H) 4,78 (с, 2H) 5,09 (кв, J=2 Гц, 2H) 7,19-7,24 (м, 1H), 7,26-7,33 (м, 4H).
(d) трет-Бутил 4-[7-(2-бутинил)-3-этоксикарбонилметил-1-(2-фенилэтил))-2,6-диоксо-1,2,6,7-тетрагидропурин-8-ил]пиперазин-1-карбоксилат
Смесь, содержащую 273 мг этил [7-(2-бутинил)-8-хлор-1-(2-фенилэтил)-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]ацетата и 360 мг трет-бутил пиперазин-1-карбоксилата, нагревали при 150°С на масляной бане в течение 30 минут. Реакционный раствор очищали с помощью колоночной хроматографии на силикагеле с использованием смеси 20-30% этилацетат-гексан, что давало 320 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,30 (т, J=7 Гц, 3H) 1,49 (с, 9H) 1,84 (т, J=2 Гц, 3H) 2,93 (т, J=8 Гц, 2H) 3,33 (т, J=5 Гц, 4H) 3,57 (т, J=5 Гц, 4H) 4,19 (т, J=8 Гц, 2H) 4,25 (кв, J=7 Гц, 2H) 4,76 (с, 2H) 4,86 (кв, J=2 Гц, 2H) 7,19 (т, J=7 Гц, 1H) 7,25-7,34 (м, 4H).
(е) Гидрохлорид этил [7-(2-бутинил)-2,6-диоксо-1-(2-фенэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетата
Смесь, содержащую 27 мг трет-бутил 4-[7-(2-бутинил)-3-этоксикарбонилметил-1-(2-фенилэтил))-2,6-диоксо-1,2,6,7-тетрагидропурин-8-ил]пиперазин-1-карбоксилата и 0,25 мл трифторуксусной кислоты, перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор концентрировали, и остаток очищали с помощью колоночной хроматографии с обратной фазой с использованием смеси 20-80% метанол/вода (содержащей 0,1% концентрированной соляной кислоты), что давало 17 мг указанного в заголовке соединения.
1H-ЯМР (d6-ДМСО)
δ: 1,22 (т, J=7 Гц, 3H) 1,82 (т, J=2 Гц, 3H) 2,80 (т, J=8 Гц, 2H) 3,22-3,28 (м, 4H) 3,46-3,51 (м, 4H) 4,05 (т, J=8 Гц, 2H) 4,17 (кв, J=7 Гц, 2H) 4,69 (с, 2H) 4,96 (кв, J=2 Гц, 2H) 7,19-7,24 (м, 3H) 7,30 (т, J=7 Гц, 2H).
Пример 417
Гидрохлорид [7-(2-бутинил)-2,6-диоксо-1-(2-фенэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]уксусной кислоты
(f) трет-бутил 4-[7-(2-бутинил)-3-карбоксиметил-1-(2-фенилэтил))-2,6-диоксо-1,2,6,7-тетрагидропурин-8-ил]пиперазин-1-карбоксилат
Смесь, содержащую 190 мг трет-бутил 4-[7-(2-бутинил)-3-этоксикарбонилметил-1-(2-фенилэтил)-2,6-диоксо-1,2,6,7-тетрагидропурин-8-ил]пиперазин-1-карбоксилата, 3 мл этанола и 0,5 мл 1 н водного раствора гидроксида натрия, перемешивали на масляной бане при 50°С в течение двух часов. К реакционному раствору добавляли 0,55 мл водного раствора 1 н соляной кислоты, а затем смесь экстрагировали этилацетатом и водой. Органический слой промывали водой и насыщенным раствором хлорида натрия и сушили над безводным сульфатом магния. Жидкость концентрировали при пониженном давлении, и к жидкости для кристаллизации добавляли этилацетат-гексан. Таким образом получали 166 мг указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,49 (с, 9H) 1,84 (т, J=2 Гц, 3H) 2,93 (т, J=8 Гц, 2H) 3,34 (т, J=5 Гц, 4H) 3,58 (т, J=5 Гц, 4H) 4,19 (т, J=8 Гц, 2H) 4,82 (с, 2H) 4,85 (кв, J=2 Гц, 2H) 7,19 (т, J=7 Гц, 1H) 7,24-7,33 (м, 4H).
(g) Гидрохлорид [7-(2-бутинил)-2,6-диоксо-1-(2-фенэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]уксусной кислоты
2,2 мг указанного в заголовке соединения получали с использованием 22 мг трет-бутил 4-[7-(2-бутинил)-3-карбоксиметил-1-(2-фенилэтил))-2,6-диоксо-1,2,6,7-тетрагидропурин-8-ил]пиперазин-1-карбоксилата с помощью процедуры, описанной в примере 416-(е).
1H-ЯМР (d6-ДМСО)
δ: 1,82 (т, J=2 Гц, 3H) 2,80 (т, J=8 Гц, 2H) 3,23-3,28 (м, 4H) 3,46-3,53 (м, 4H) 4,05 (т, J=8 Гц, 2H) 4,59 (с, 2H) 4,96 (кв, J=2 Гц, 2H) 7,19-7,25 (м, 3H) 7,30 (т, J=7 Гц, 2H).
Пример 418
Гидрохлорид 7-(2-бутинил)-3-[2-оксо-2-(пирролидин-1-ил)этил]-1-(2-фенэтил-8-(пиперазин-1-ил)-3,7-дигидропурин-2,6-диона
Смесь, содержащую 20 мг трет-бутил 4-[7-(2-бутинил)-3-карбоксиметил-1-(2-фенилэтил)-2,6-диоксо-1,2,6,7-тетрагидропурин-8-ил]пиперазин-1-карбоксилата, 8 мкл диэтилцианфосфата, 10 мкл триэтиламина, 20 мкл пирролидина и 0,3 мл N,N-диметилформамида, оставляли стоять при комнатной температуре в течение 3 дней. Реакционный раствор разбавляли этилацетатом и водой и экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором хлористого натрия и затем концентрировали. К остатку добавляли 0,5 мл трифторуксусной кислоты, и получающуюся смесь инкубировали при комнатной температуре в течение 30 минут. Реакционный раствор концентрировали, и остаток очищали с помощью колоночной хроматографии с обратной фазой с использованием смеси 20-80% метанол/вода (содержащей 0,1% концентрированной соляной кислоты), что давало 3,2 мг указанного в заголовке соединения.
1H-ЯМР (d6-ДМСО)
δ: 1,76-1,84 (м, 5H) 1,95 (квинтет, J=7 Гц, 2H), 2,79 (т, J=8 Гц, 2H) 3,22-3,34 (м, 6H) 3,45-3,52 (м, 4H) 3,55 (т, J=7 Гц, 2H) 4,03 (т, J=8 Гц, 2H) 4,68 (с, 2H) 4,96 (кв, J=2 Гц, 2H) 7,18-7,26 (м, 3H) 7,31 (т, J=8 Гц, 2H).
Пример 419
Гидрохлорид 2-[7-(2-бутинил)-2,6-диоксо-1-(2-фенэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]-N-метилацетамида
Указанное в заголовке соединение синтезировали с использованием водного раствора метиламина с помощью процедуры, описанной в примере 418.
1H-ЯМР (d6-ДМСО)
δ: 1,82 (т, J=2 Гц, 3H) 2,61 (д, J=5 Гц, 3H) 2,79 (т, J=8 Гц, 2H) 3,20-3,28 (м, 4H) 3,44-3,52 (м, 4H) 4,03 (т, J=8 Гц, 2H) 4,48 (с, 2H) 4,96 (кв, J=2 Гц, 2H) 7,19-7,26 (м, 3H) 7,31 (т, J=7 Гц, 2H) 8,09 (ушир.д, J=5 Гц, 1H).
Пример 420
Гидрохлорид 2-[7-(2-бутинил)-2,6-диоксо-1-(2-фенэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]-N-циклопропилацетамида
Указанное в заголовке соединение синтезировали с использованием циклопропиламина с помощью процедуры, описанной в примере 418.
1H-ЯМР (d6-ДМСО)
δ: 0,39-0,44 (м, 2H) 0,60-0,66 (м, 2H) 1,82 (т, J=2 Гц, 3H) 2,60-2,68 (м, 1H) 2,79 (т, J=8 Гц, 2H) 3,20-3,30 (м, 4H) 3,44-3,54 (м, 4H) 4,03 (т, J=8 Гц, 2H) 4,44 (с, 2H) 4,96 (кв, J=2 Гц, 2H) 7,19-7,27 (м, 3H) 7,31 (т, J=8 Гц, 2H) 8,27 (д, J=4 Гц, 1H).
Пример 421
Гидрохлорид 2-[7-(2-бутинил)-2,6-диоксо-1-(2-фенэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]-N-фенилацетамида
Указанное в заголовке соединение синтезировали с использованием анилина с помощью процедуры, описанной в примере 418.
1H-ЯМР (d6-ДМСО)
δ: 1,83 (т, J=2 Гц, 3H) 2,81 (т, J=8 Гц, 2H) 3,20-3,30 (м, 4H) 3,44-3,54 (м, 4H) 4,05 (т, J=8 Гц, 2H) 4,74 (с, 2H), 4,98 (кв, J=2 Гц, 2H) 7,06 (т, J=8 Гц, 1H) 7,18-7,35 (м, 7H) 7,56 (д, J=8 Гц, 2H) 9,01 (ушир.с, 2H) 10,39 (с, 1H).
Пример 422
Гидрохлорид 2-[7-(2-бутинил)-2,6-диоксо-1-(2-фенэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]-N-(2-пропинил)ацетамида
Указанное в заголовке соединение синтезировали с использованием пропаргиламина с помощью процедуры, описанной в примере 418.
1H-ЯМР (d6-ДМСО)
δ: 1,81 (т, J=3 Гц) 2,80 (т, J=8 Гц, 2H) 3,18 (т, J=2 Гц 1H), 3,22-3,32 (м, 4H) 3,44-3,54 (м, 4H) 3,90 (дд, J=2 Гц, 5 Гц, 2H) 4,03 (т, J=8 Гц, 2H) 4,51 (с, 2H) 4,96 (кв, J=2 Гц, 2H) 7,16-7,34 (м, 5H) 8,66 (т, J=5 Гц, 1H) 8,96 (ушир.с, 2H).
Пример 423
Гидрохлорид этил [7-(2-бутинил)-2,6-диоксо-1-(2-феноксиэтил)-8-(пиперазин-1-ил)-1,2,6,7-тетрагидропурин-3-ил]ацетата
Указанное в заголовке соединение синтезировали с использованием 2-бромэтилфенилового эфира с помощью процедуры, описанной в примере 416.
1H-ЯМР (d6-ДМСО)
δ:1,20 (т, J=7 Гц, 3H) 1,81 (с, 3H) 3,22-3,28 (м, 4H) 3,46-3,53 (м, 4H) 4,06-4,19 (м, 4H) 4,25 (т, J=6 Гц, 2H) 4,69 (с, 2H) 4,97 (с, 2H) 6,88-6,96 (м, 3H) 7,26 (т, J=7 Гц, 2H) 8,96 (ушир.с, 2H).
Пример 424
Трифторацетат этил [1-метил-2,6-диоксо-8-(пиперазин-1-ил)-7-(2-винилфенил)-1,2,6,7-тетрагидропурин-3-ил]ацетата
(а) [1-(2,2-Диметилпропионилоксиметил)-7-(2-формилфенил)-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил 2,2-диметилпропионат
10,2 г [3-(2,2-диметилпропионилоксиметил)-2,6-диоксо-2,3,6,7-тетрагидропурин-1-ил]метил 2,2-диметилпропионата, 8,04 г 2-формилфенилбороновой кислоты и 7,30 г ацетата меди (II) суспендировали в 50 мл N,N-диметилформамида, а затем добавляли 4,34 мл пиридина. Смесь перемешивали при комнатной температуре в течение 37 часов. Реакционный раствор разбавляли этилацетатом и промывали водой. Органический слой сушили над безводным сульфатом магния и фильтровался. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Из фракции, элюированной гексаном-этилацетатом (1:2), получали 4,12 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,16 (с, 9H) 1,23 (с, 9H) 5,95 (с, 2H) 6,20 (с, 2H) 7,46-7,48 (м, 1H) 7,42-7,78 (м, 2H) 7,75 (с, 1H) 8,03-8,06 (м, 1H) 9,92 (с, 1H).
(b) [8-Хлор-1-(2,2-диметилпропионилоксиметил)-7-(2-формилфенил)-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил 2,2-диметилпропионат
2,50 г [1-(2,2-диметилпропионилоксиметил)-7-(2-формилфенил)-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил 2,2-диметилпропионата и 896 мг N-хлорсукцинимида растворяли в 25 мл N,N-диметилформамида. Получающуюся смесь перемешивали при комнатной температуре в течение 8 часов. Реакционный раствор разбавляли этилацетатом и промывали водой. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом из фракции, элюированной гексаном-этилацетатом (2:1), получали 2,0 г указанного в заголовке соединения.
1H-ЯМР (CDCl3)
δ: 1,15 (с, 9H) 1,24 (с, 9H) 5,91 (с, 2H) 6,14 (с, 2H) 7,49-7,51 (м, 1H) 7,81-7,83 (м, 2H) 8,03-8,06 (м, 1H) 9,92 (с, 1H).
(с) трет-Бутил 4-[1,3-бис-(2,2-диметилпропионилоксиметил)-7-(2-формилфенил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
2,0 г [8-хлор-1-(2,2-диметилпропионилоксиметил)-7-(2-формилфенил)-2,6-диоксо-1,2,6,7-тетрагидропурин-3-ил]метил 2,2-диметилпропионата объединяли с 2,15 г трет-бутилпиперазин-1-карбоксилата. Получающуюся смесь перемешивали при 150°С в течение 70 минут. Реакционную смесь разбавляли хлороформом, а затем очищали с помощью колоночной хроматографии на силикагеле. Таким образом из фракции, элюированной гексаном-этилацетатом (1:1), получали 1,94 г указанного в заголовке соединения.
(d) трет-Бутил 4-[1,3-бис(2,2-диметилпропионилоксиметил)-2,6-диоксо-7-(2-винилфенил)-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
3,52 г метилтрифенилфосфонийбромида разбавляли в 20 мл тетрагидрофурана, а затем добавляли 948 мг третичного бутоксида калия. Получающуюся смесь перемешивали при комнатной температуре в течение 1 часа. К реакционной смеси при комнатной температуре добавляли 20 мл раствора тетрагидрофурана, содержащего 1,94 г трет-бутил 4-[1,3-бис(2,2-диметилпропионилоксиметил)-7-(2-формилфенил)-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата. Смесь перемешивали при комнатной температуре в течение 3 часов и 50 минут. Реакционный раствор разбавляли этилацетатом, а затем промывали водой. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом из фракции, элюированной гексаном-этилацетатом (2:1), получали 704 мг указанного в заголовке соединения.
(е) трет-Бутил 4-[1-(2,2-диметилпропионилоксиметил)-2,6-диоксо-7-(2-винилфенил)-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
704 мг трет-бутил 4-[1,3-бис(2,2-диметилпропионилоксиметил)-2,6-диоксо-7-(2-винилфенил)-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в смешанном растворителе из 7 мл тетрагидрофурана и 14 мл метанола, а затем добавляли 51 мг гидрида натрия. Получающуюся смесь перемешивали при комнатной температуре в течение 17 минут. Реакционный раствор разбавляли хлороформом и промывали насыщенным раствором хлорида натрия. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом из фракции, элюированной гексаном-этилацетатом (2:3), получали 510 мг указанного в заголовке соединения.
(f) трет-Бутил 4-[1-(2,2-диметилпропионилоксиметил)-3-этоксикарбонилметил-2,6-диоксо-7-(2-винилфенил)-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
80 мг трет-бутил 4-[1-(2,2-диметилпропионилоксиметил)-2,6-диоксо-7-(2-винилфенил)-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 2 мл N,N-диметилформамида, а затем добавляли 19 мкл этилбромацетата и 22 мг карбоната калия. Получающуюся смесь перемешивали при комнатной температуре в течение 14 часов. Реакционный раствор разбавляли этилацетатом и промывали водой. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, что давало 89 мг указанного в заголовке соединения.
(g) трет-Бутил 4-[3-этоксикарбонилметил-2,6-диоксо-7-(2-винилфенил)-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
89 мг трет-бутил 4-[1-(2,2-диметилпропионилоксиметил)-3-этоксикарбонилметил-2,6-диоксо-7-(2-винилфенил)-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в смешанном растворителе, содержащем 1 мл тетрагидрофурана и 2 мл метанола, а затем добавляли 7 мг гидрида натрия. Получающуюся смесь перемешивали при комнатной температуре в течение 3,5 часов. Реакционный раствор разбавляли этилацетатом и промывали водой. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле. Таким образом из фракции, элюированной гексаном-этилацетатом (1:2), получали 60 мг указанного в заголовке соединения.
(h) трет-Бутил 4-[3-этоксикарбонилметил-1-метил-2,6-диоксо-7-(2-винилфенил)-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилат
60 мг трет-бутил 4-[3-этоксикарбонилметил-2,6-диоксо-7-(2-винилфенил)-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 2 мл N,N-диметилформамида, а затем добавляли 17 мкл метилйодида и 17 мг карбоната калия. Получающуюся смесь перемешивали при комнатной температуре в течение 13 часов. Реакционный раствор разбавляли этилацетатом и промывали водой. Органический слой сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, что давало 48 мг указанного в заголовке соединения.
(i) Трифторацетат этил [1-метил-2,6-диоксо-8-(пиперазин-1-ил)-7-(2-винилфенил)-1,2,6,7-тетрагидропурин-3-ил]ацетата
8 мг трет-бутил 4-[3-этоксикарбонилметил-1-метил-2,6-диоксо-7-(2-винилфенил)-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в трифторуксусной кислоте, и затем раствор концентрировали. Остаток очищали с помощью высокоэффективной жидкостной хроматографии с обратной фазой, что давало 2,68 мг указанного в заголовке соединения.
MS m/e (ESI) 439 (MH+-CF3COOH).
Пример 425
Трифторацетат [1-метил-2,6-диоксо-8-(пиперазин-1-ил)-7-(2-винилфенил)-1,2,6,7-тетрагидропурин-3-ил]уксусной кислоты
40 мг трет-бутил 4-[3-этоксикарбонилметил-1-метил-2,6-диоксо-7-(2-винилфенил)-2,3,6,7-тетрагидро-1Н-пурин-8-ил]пиперазин-1-карбоксилата растворяли в 4 мл тетрагидрофурана, а затем добавляли 1 мл 2 н гидроксида натрия. Получающуюся смесь перемешивали при 90°С в течение 4 часов. Реакционный раствор концентрировали при пониженном давлении, а затем обрабатывали с помощью азеотропной перегонки с использованием толуола. Остаток растворяли в трифторуксусной кислоте, и раствор концентрировали. Остаток очищали с помощью высоко эффективной жидкостной хроматографии с обратной фазой, что давало 29,5 мг указанного в заголовке соединения.
MS m/e (ESI) 411 (MH+-CF3COOH).
Следующие формулы представляют собой соединения, которые, что было подтверждено, синтезированы в соответствии с общими методами синтеза, описанными выше, и теми же методами, описанными выше в Примерах получения и Примерах.
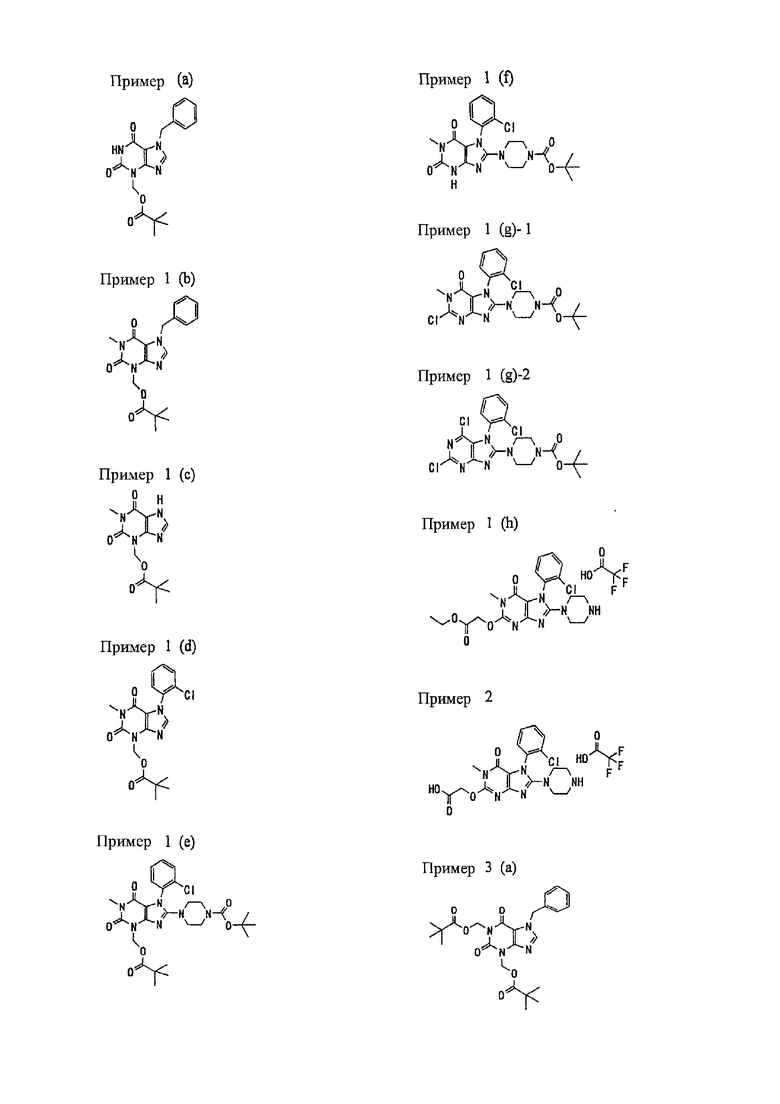
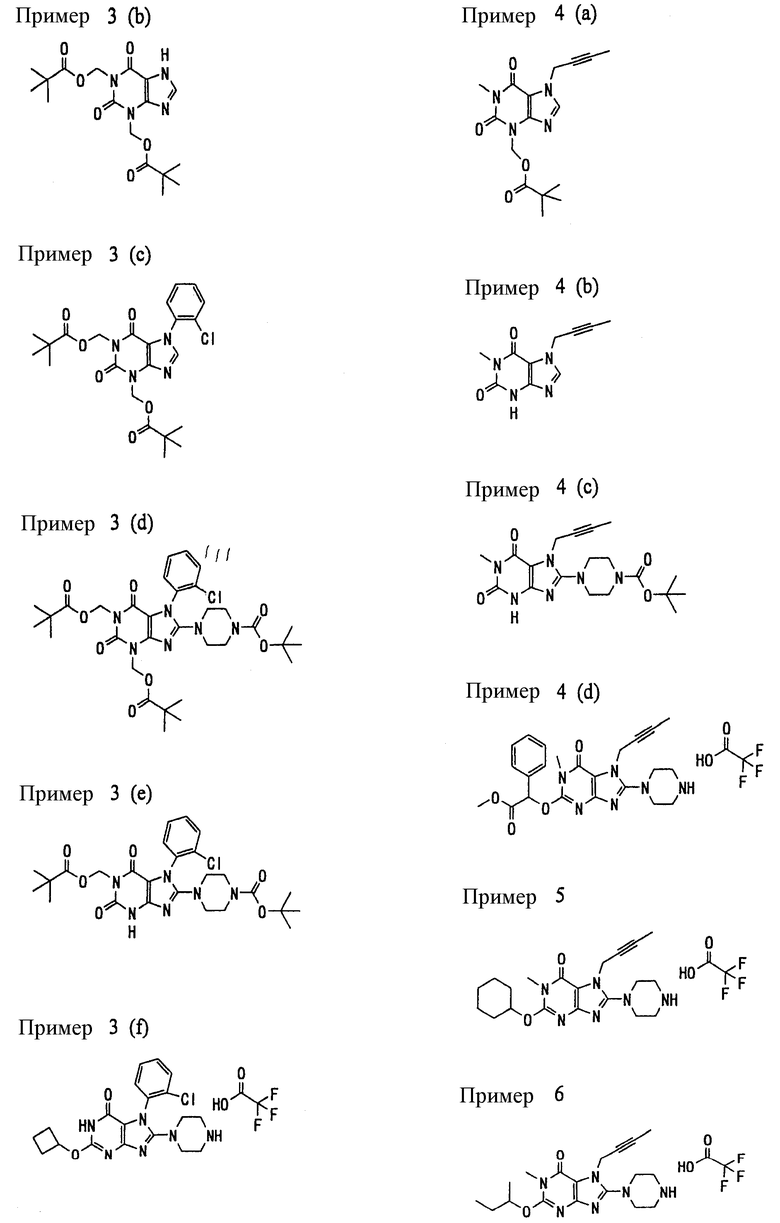
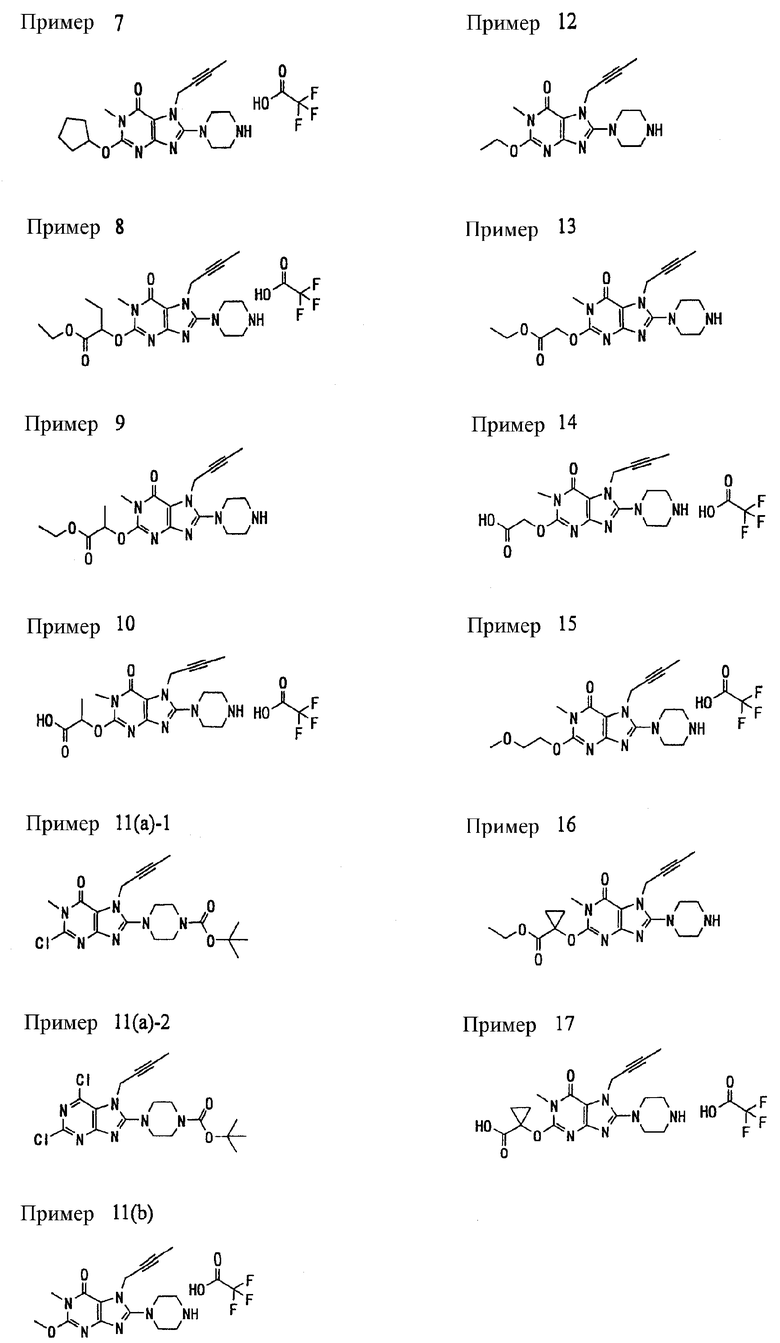
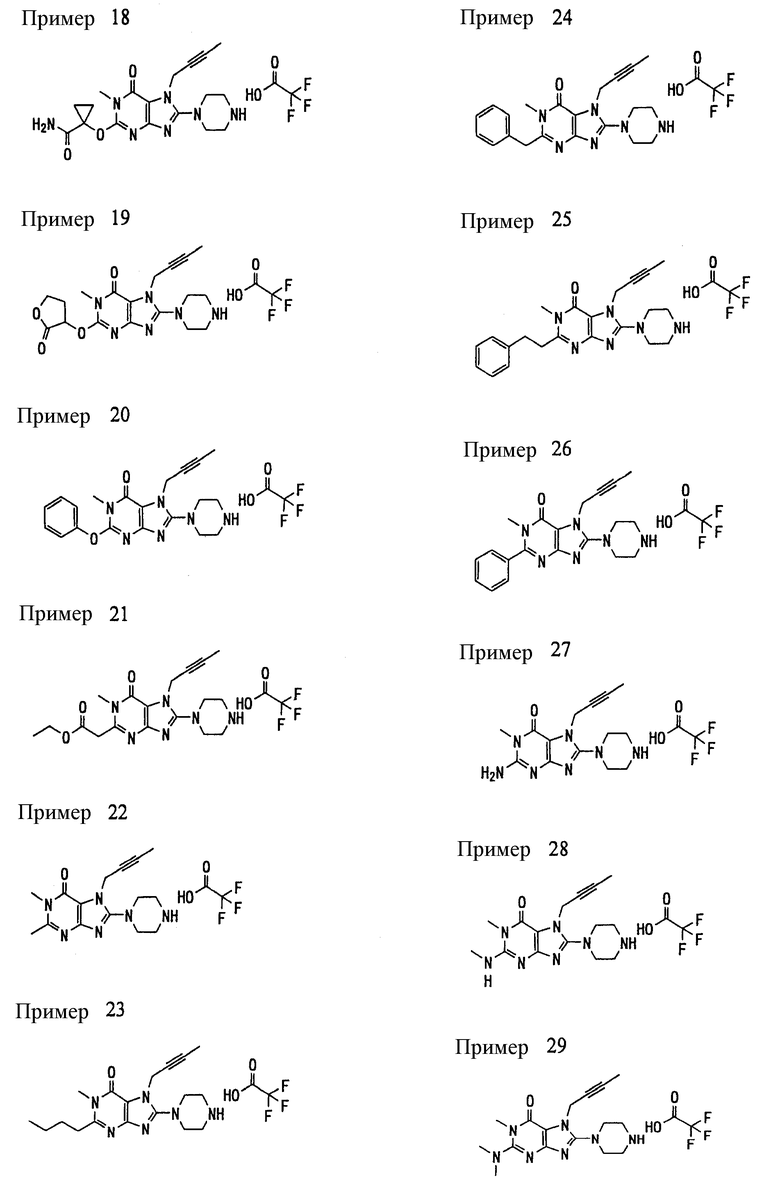
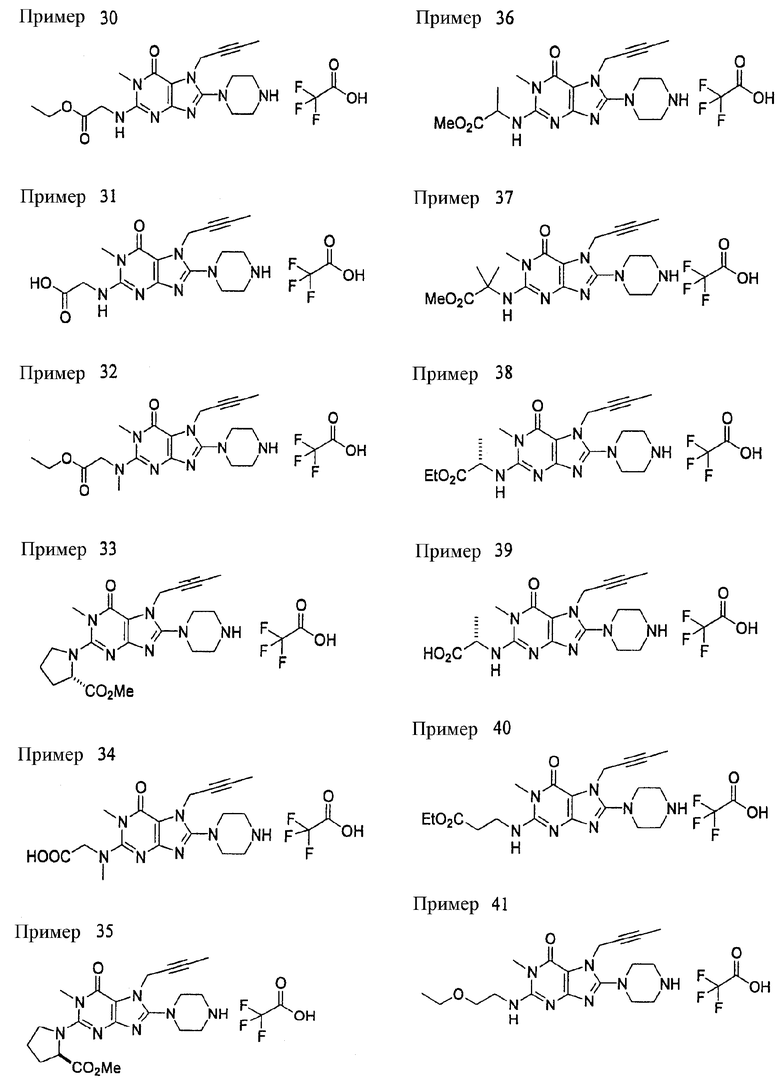
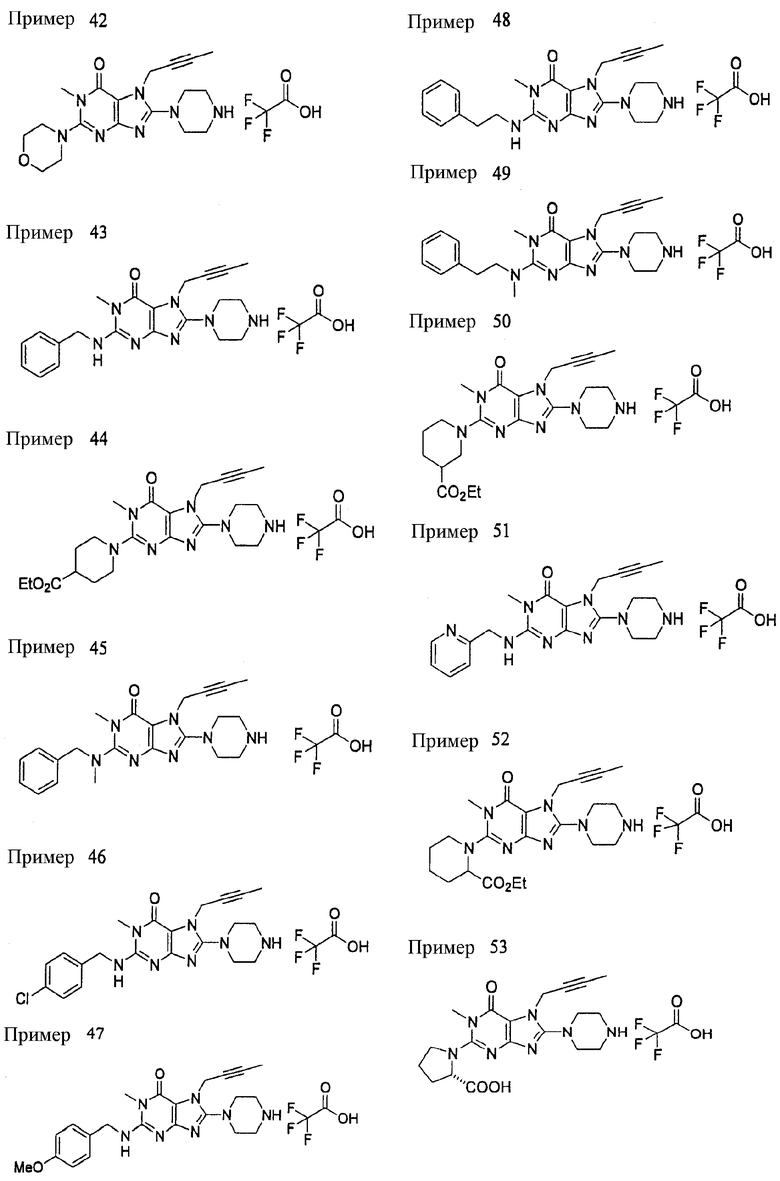
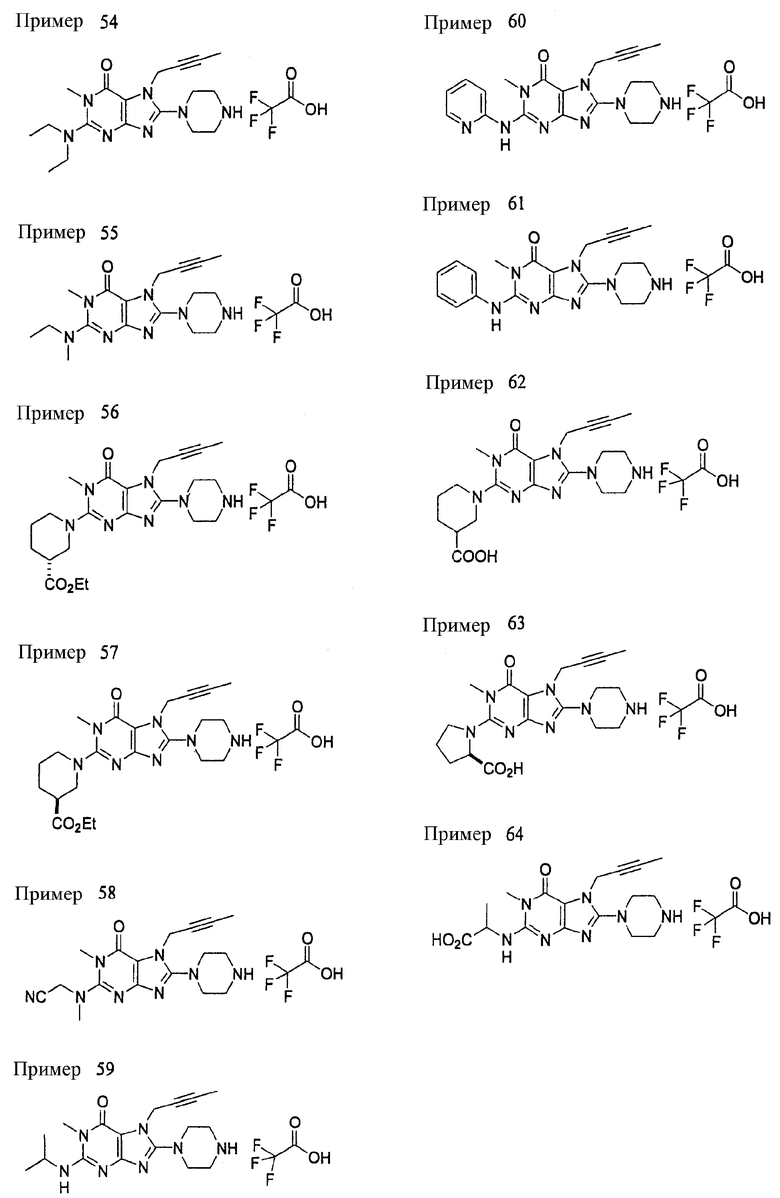
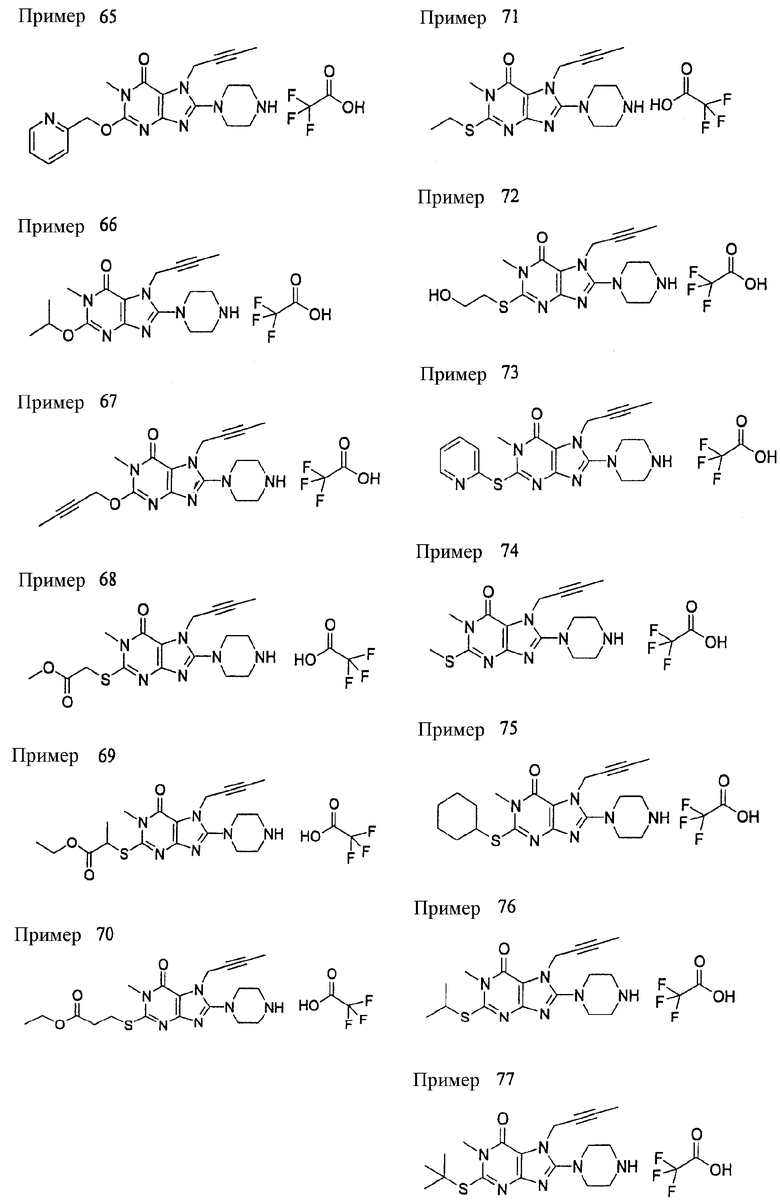
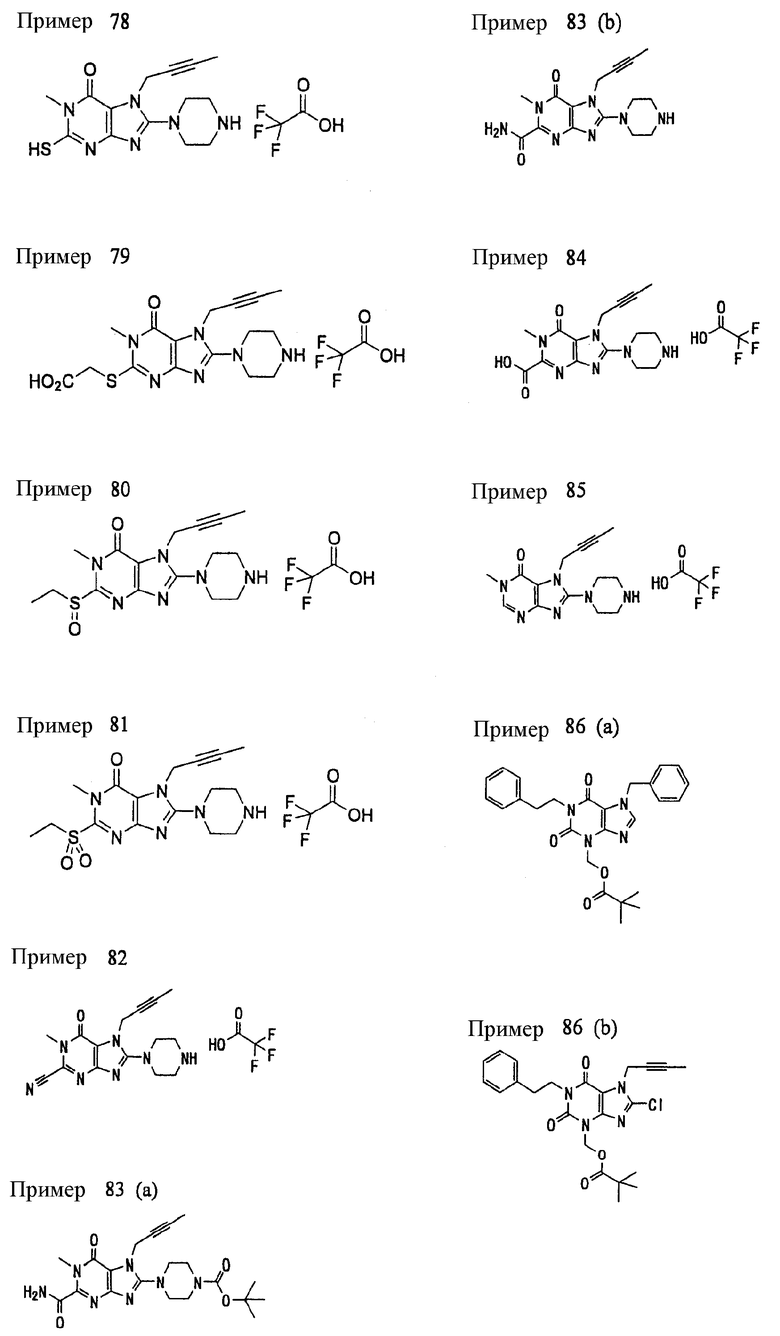
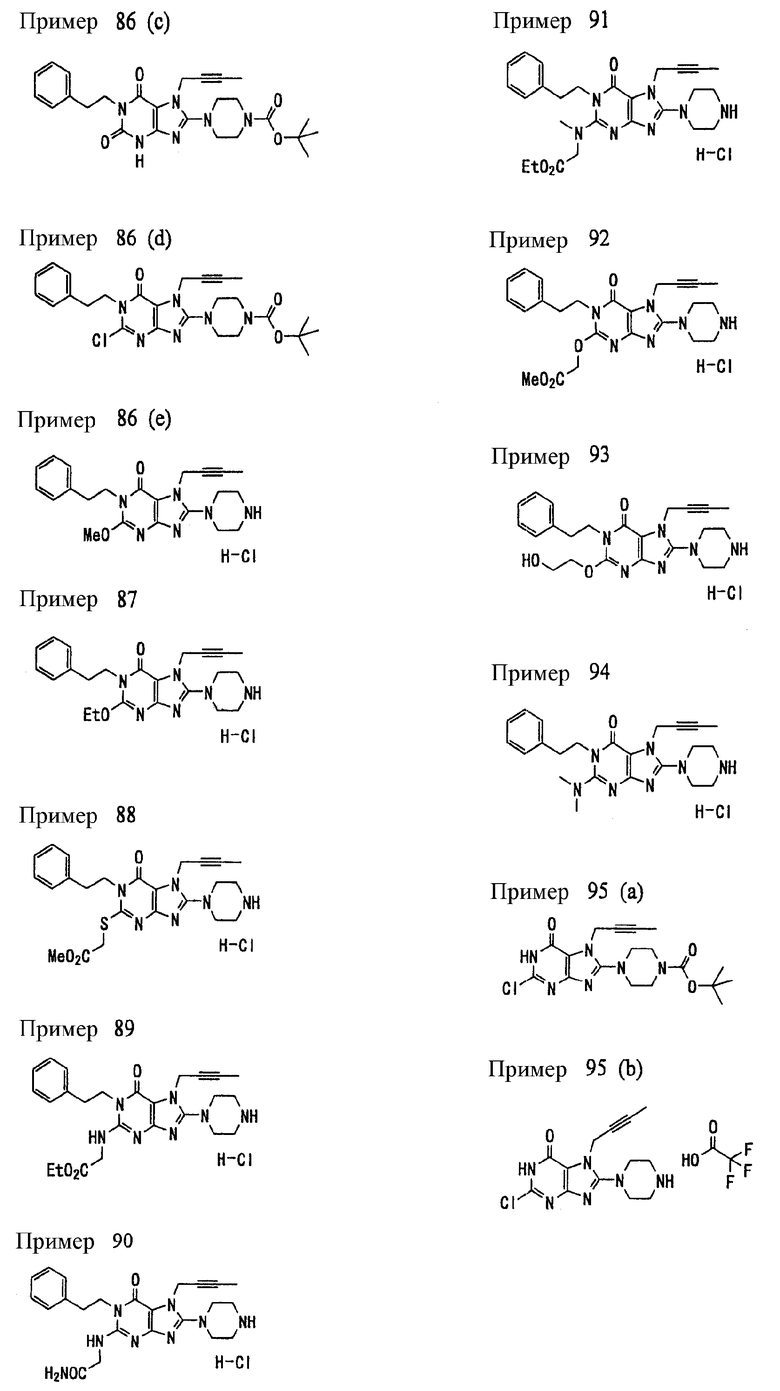
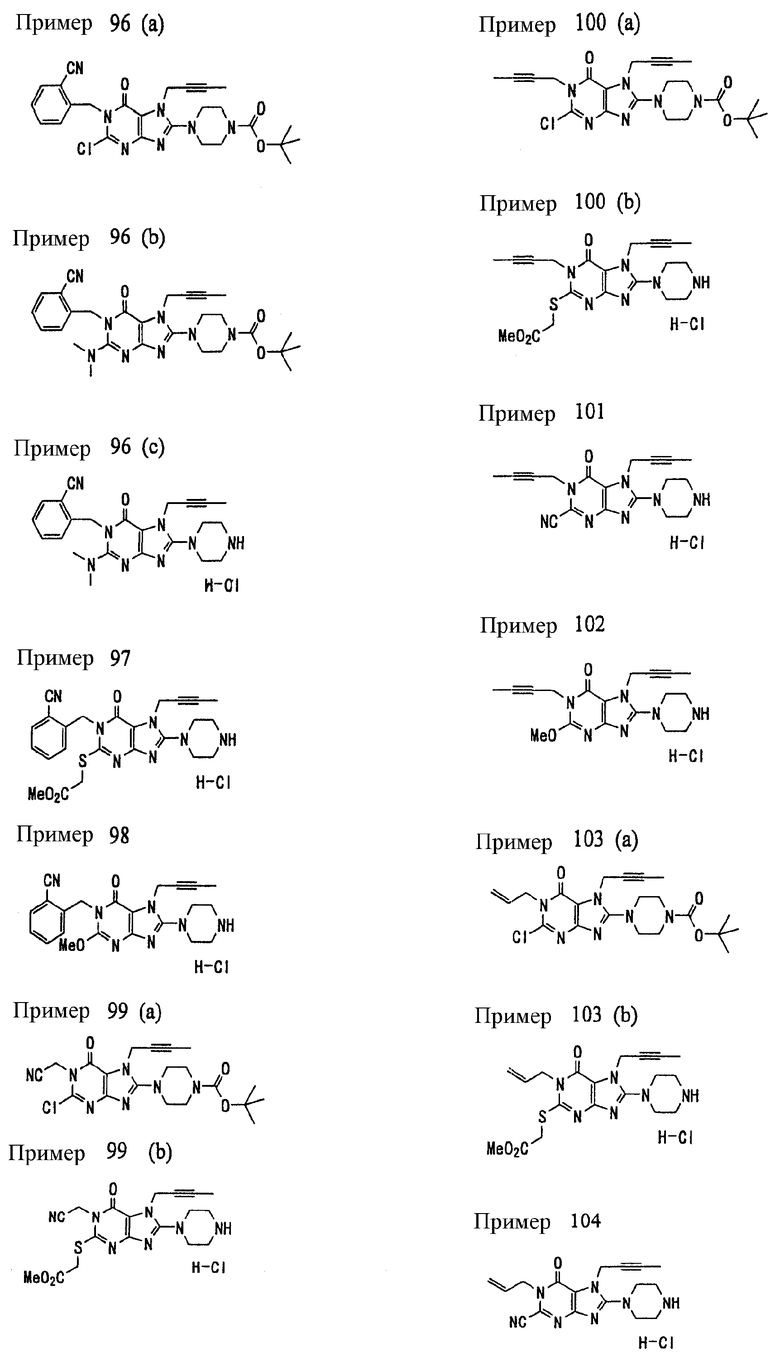
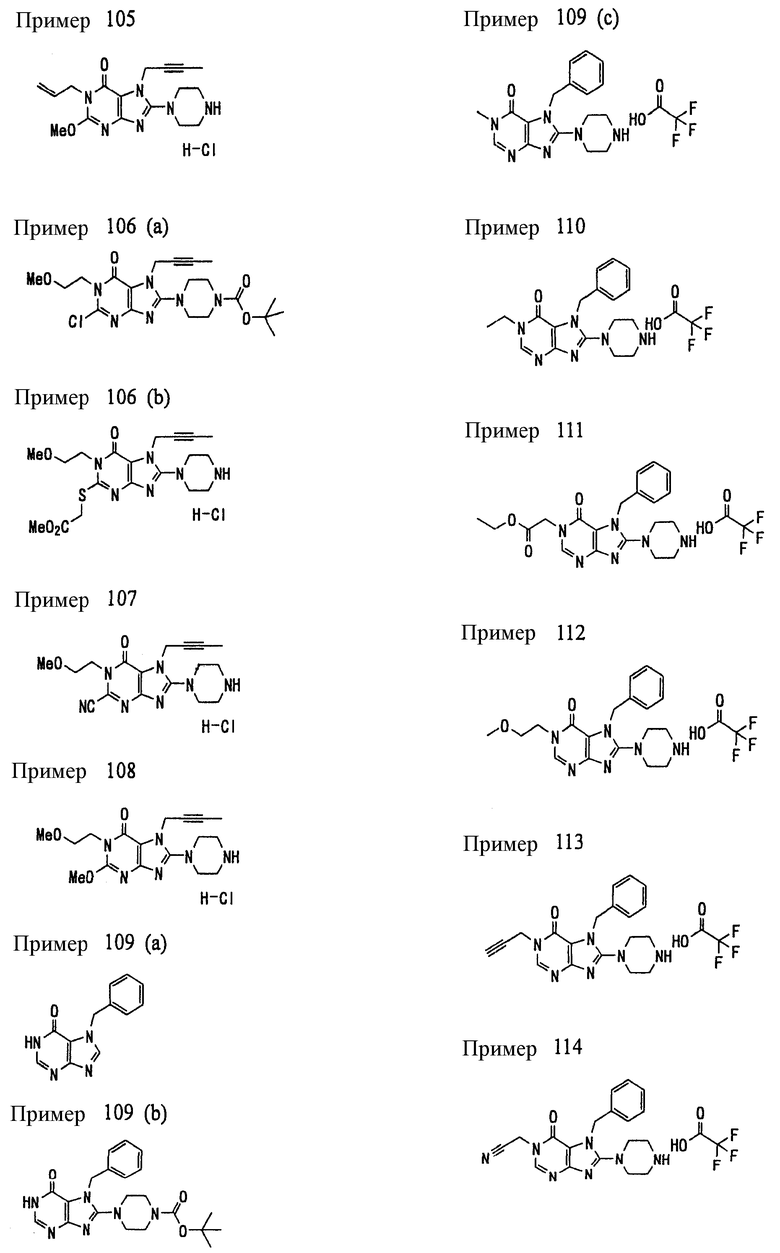
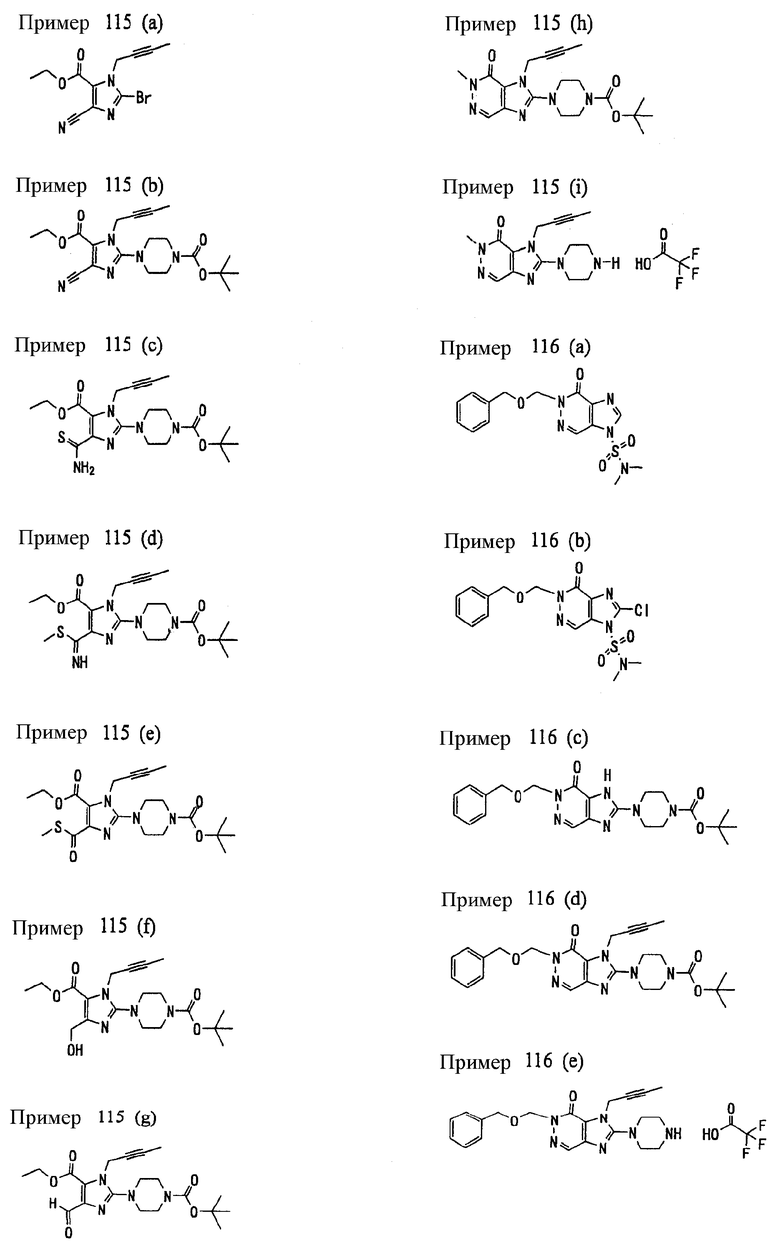
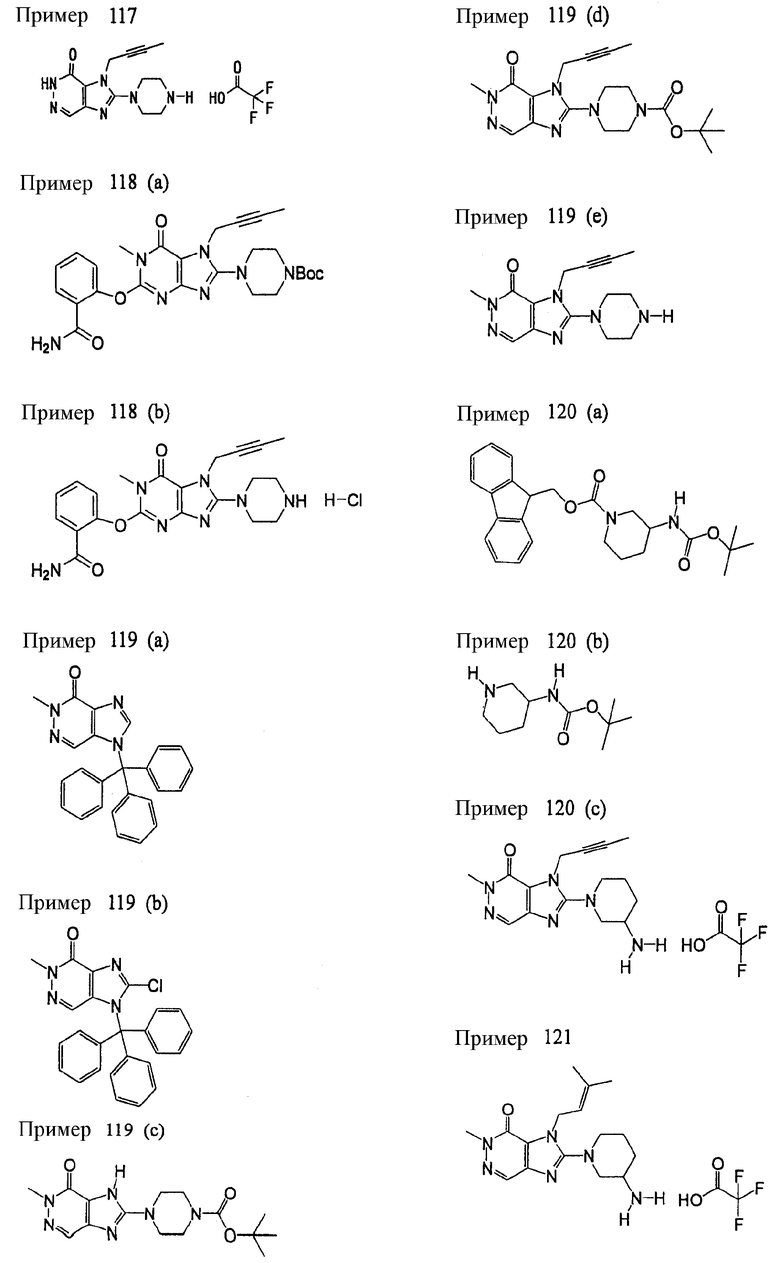
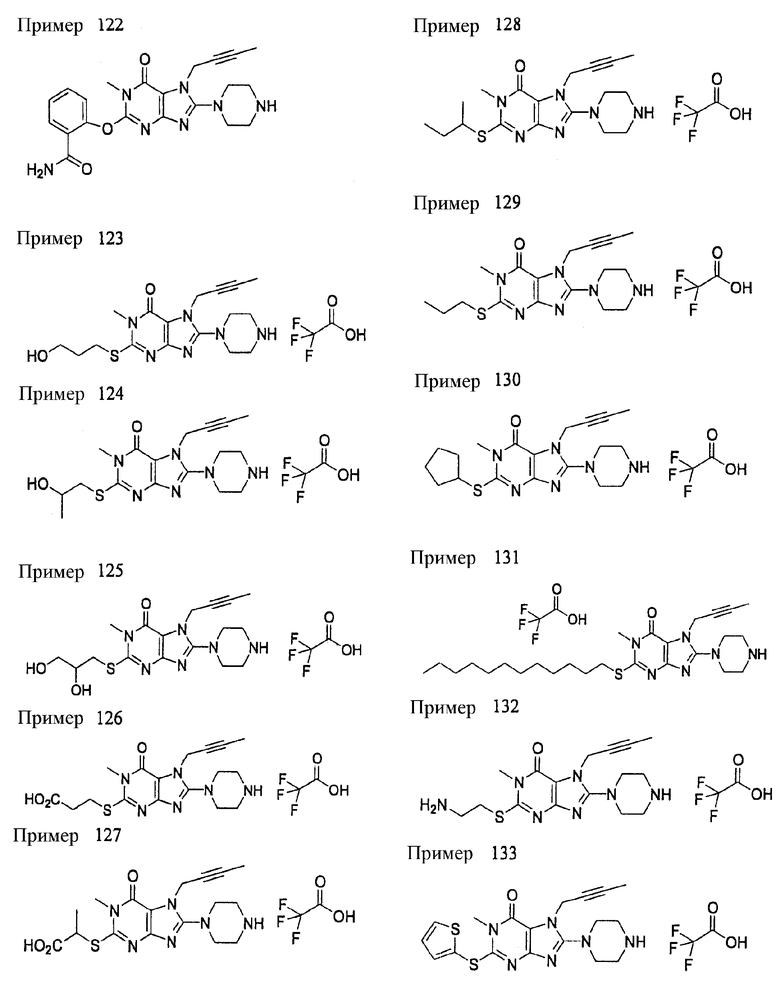
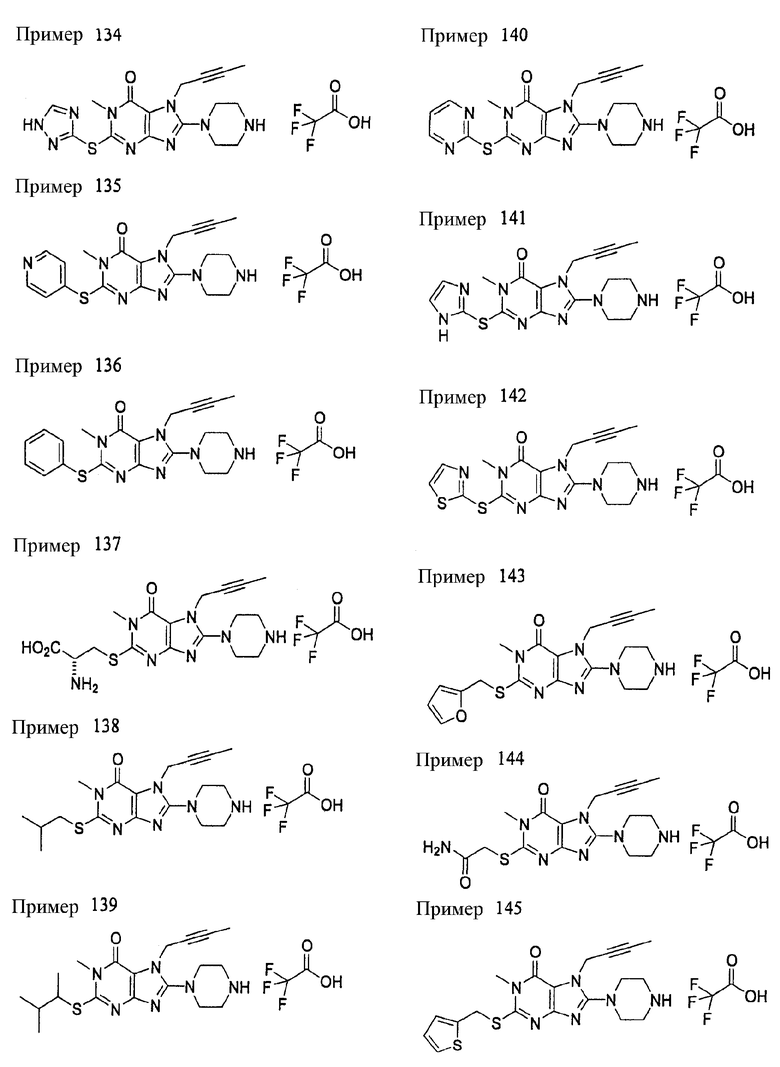
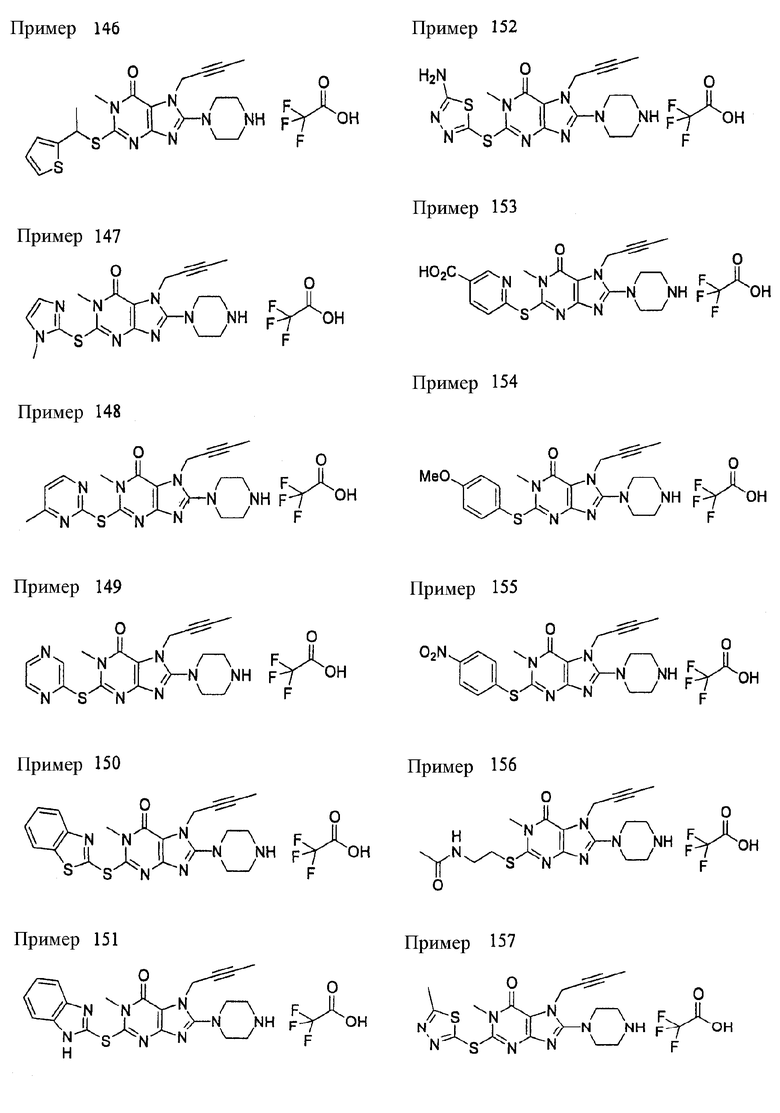
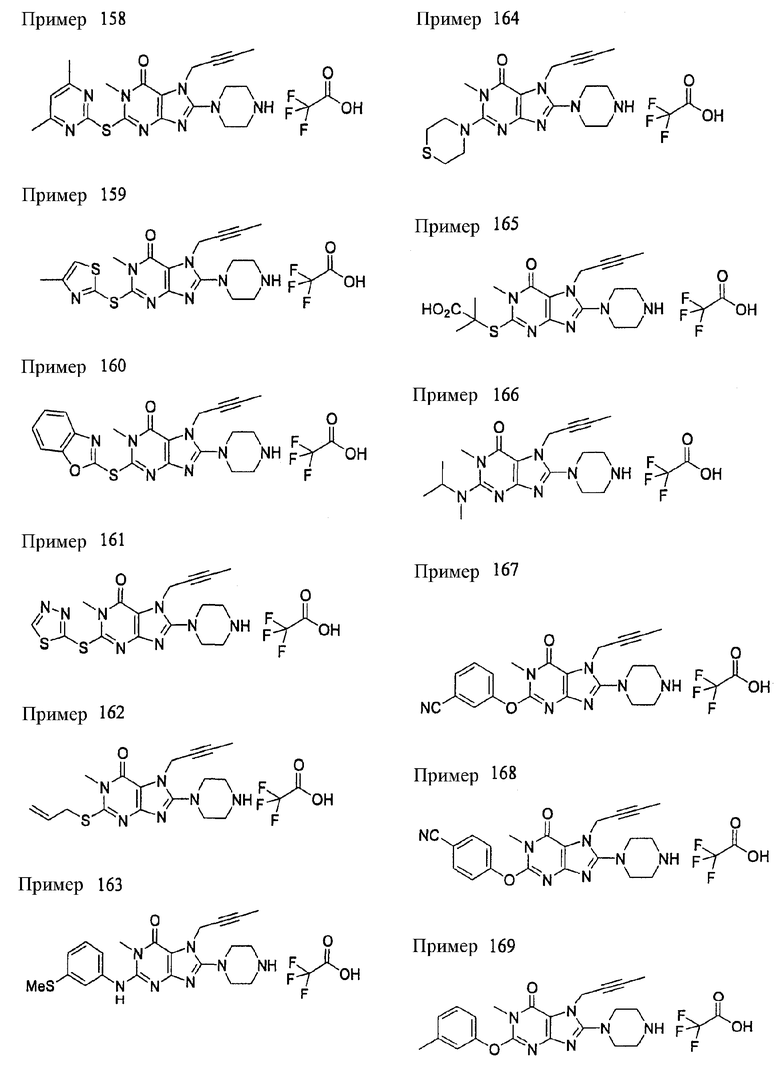
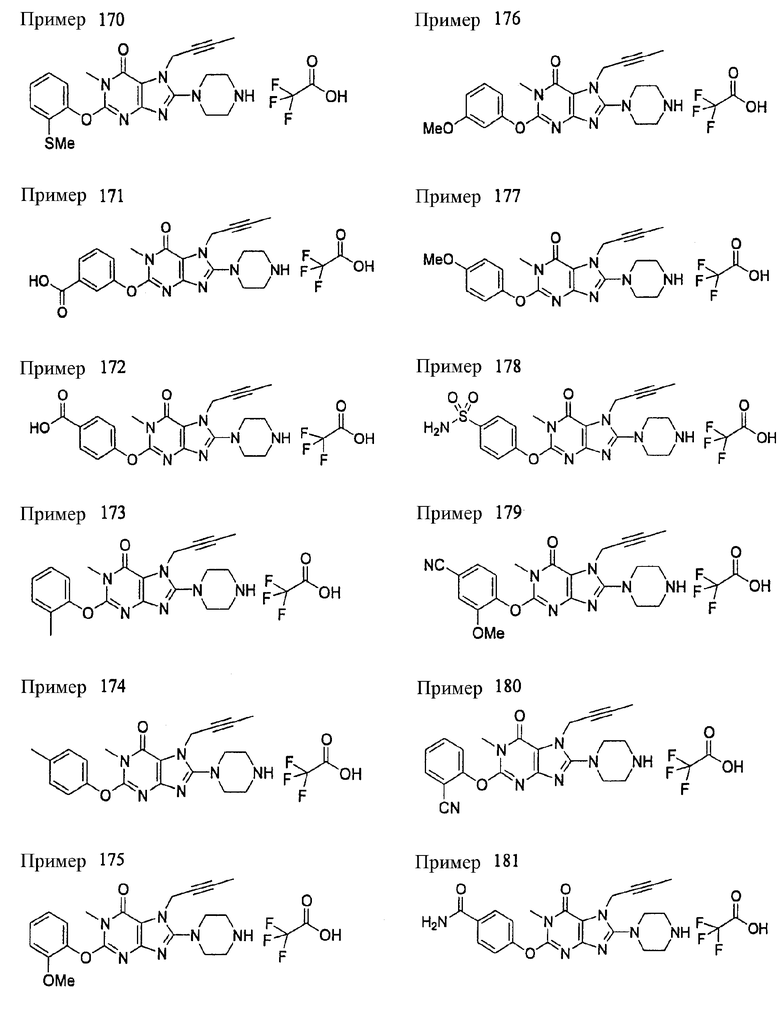
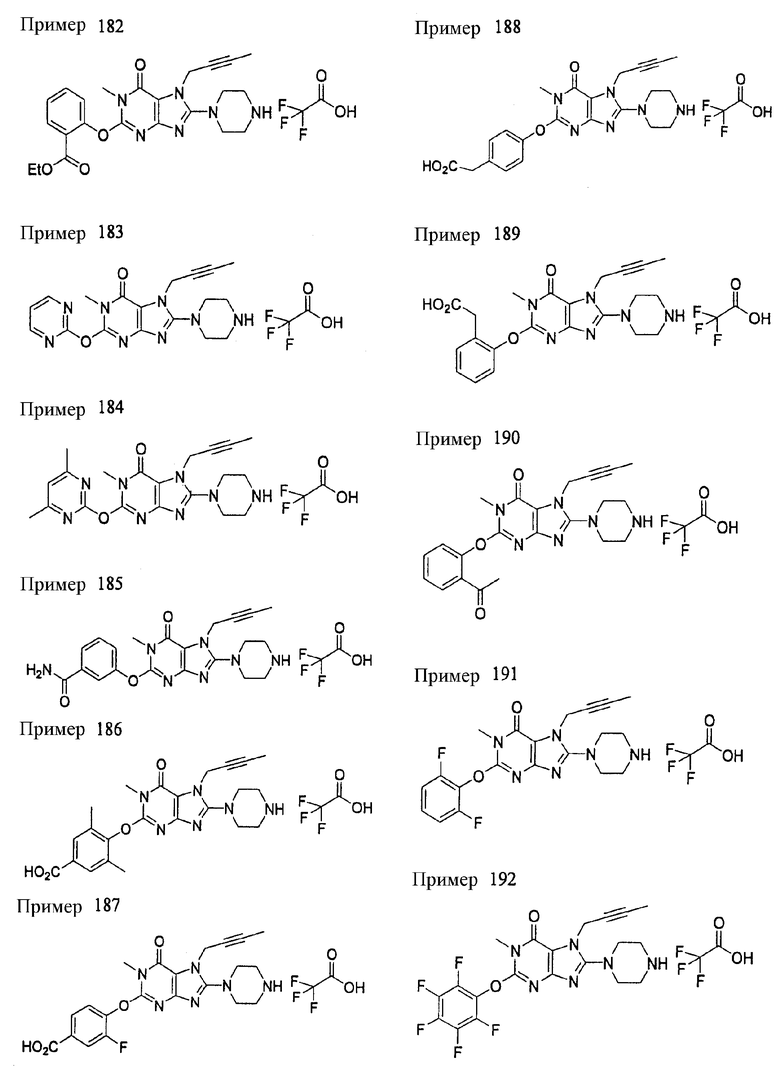
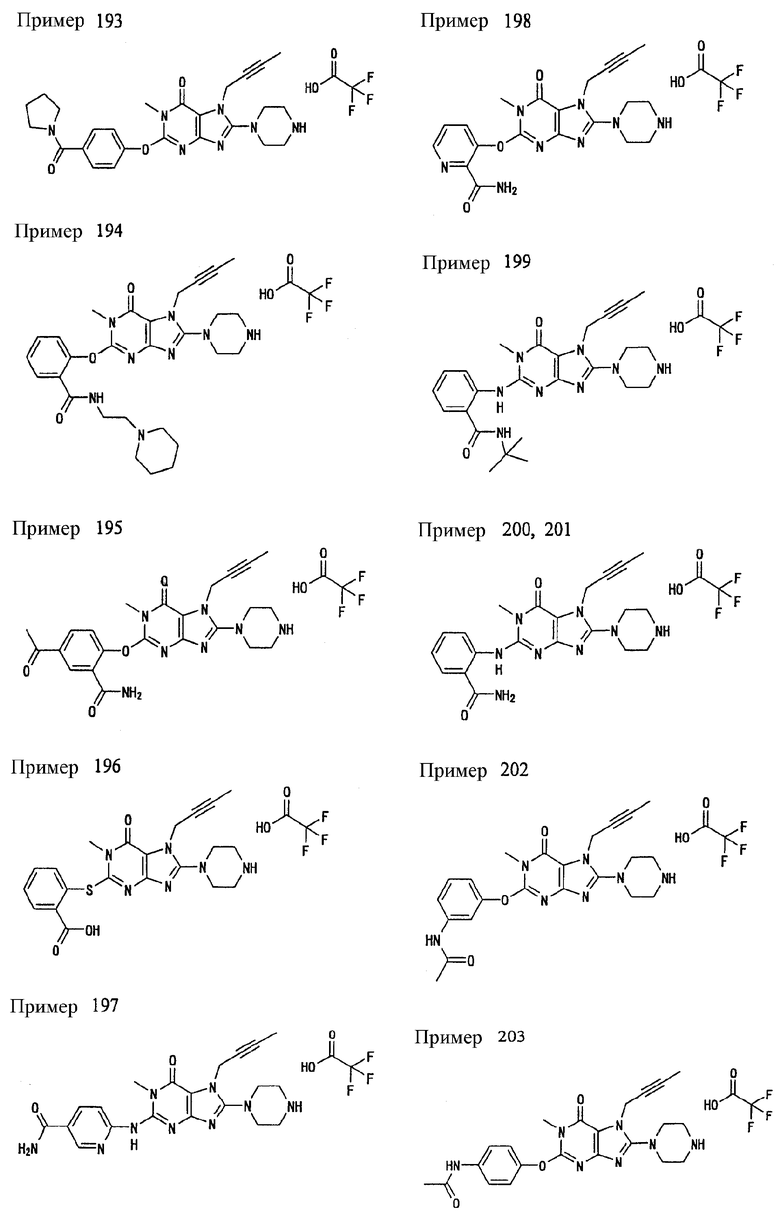
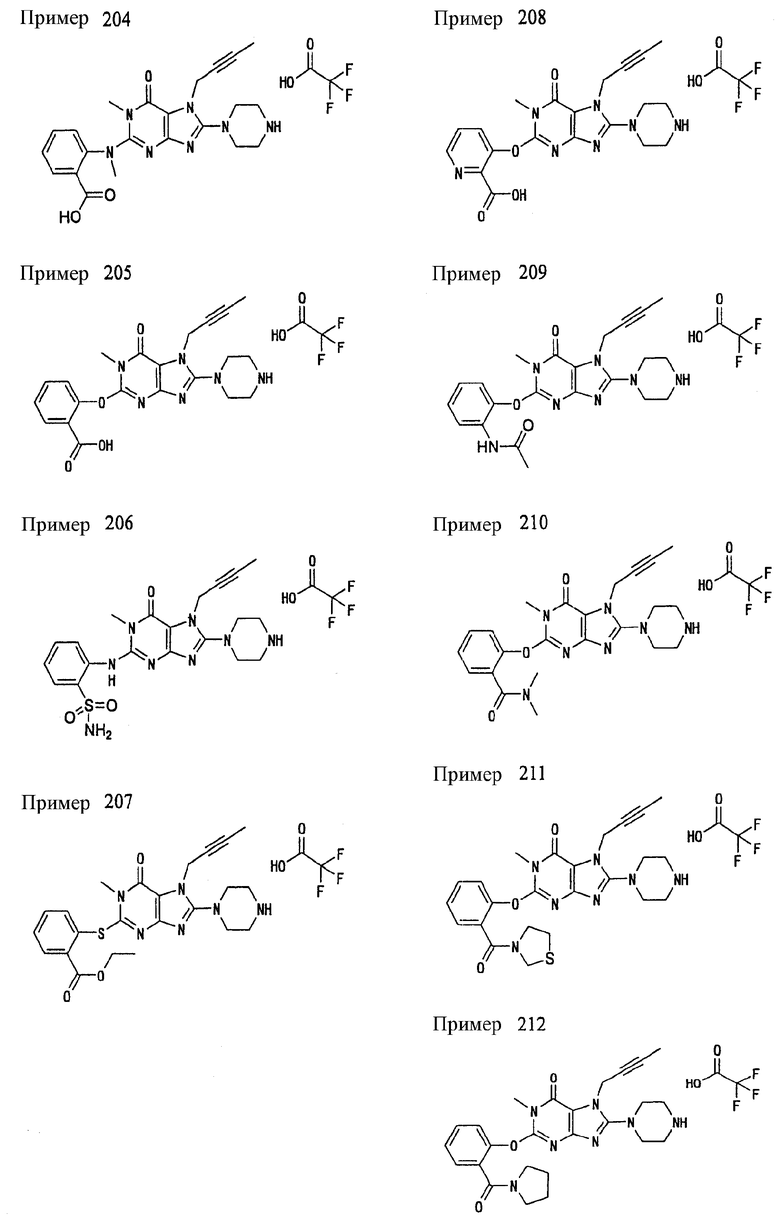
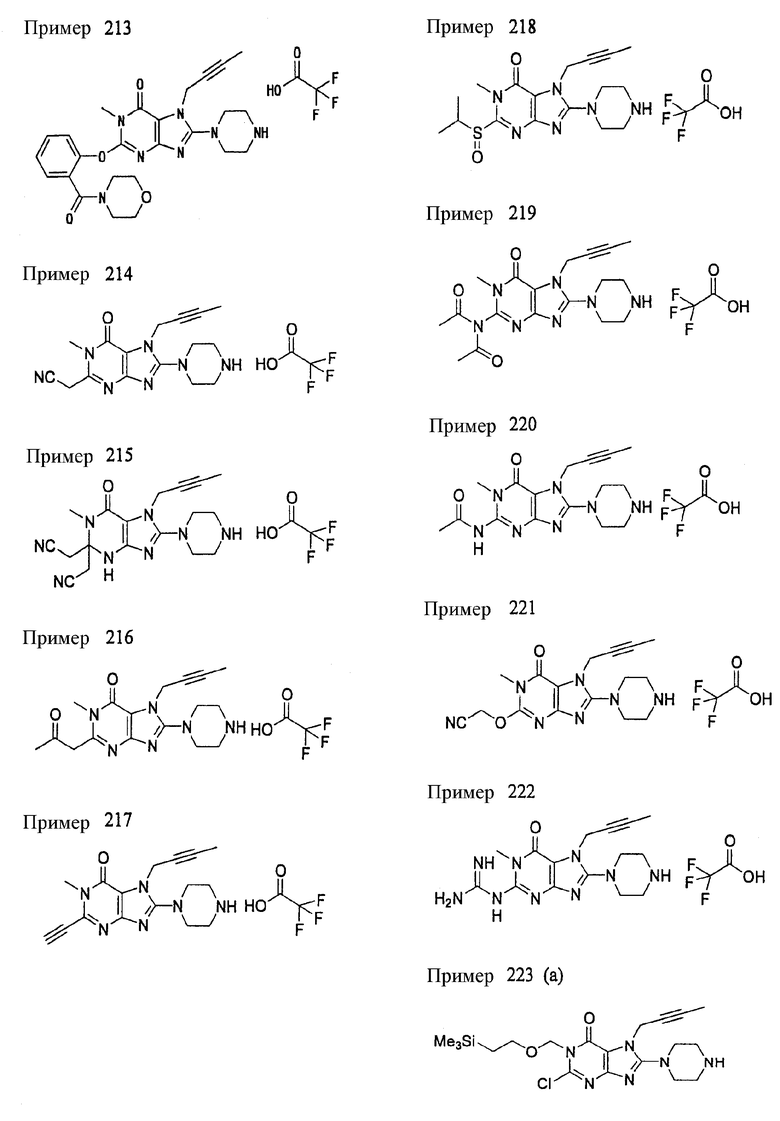
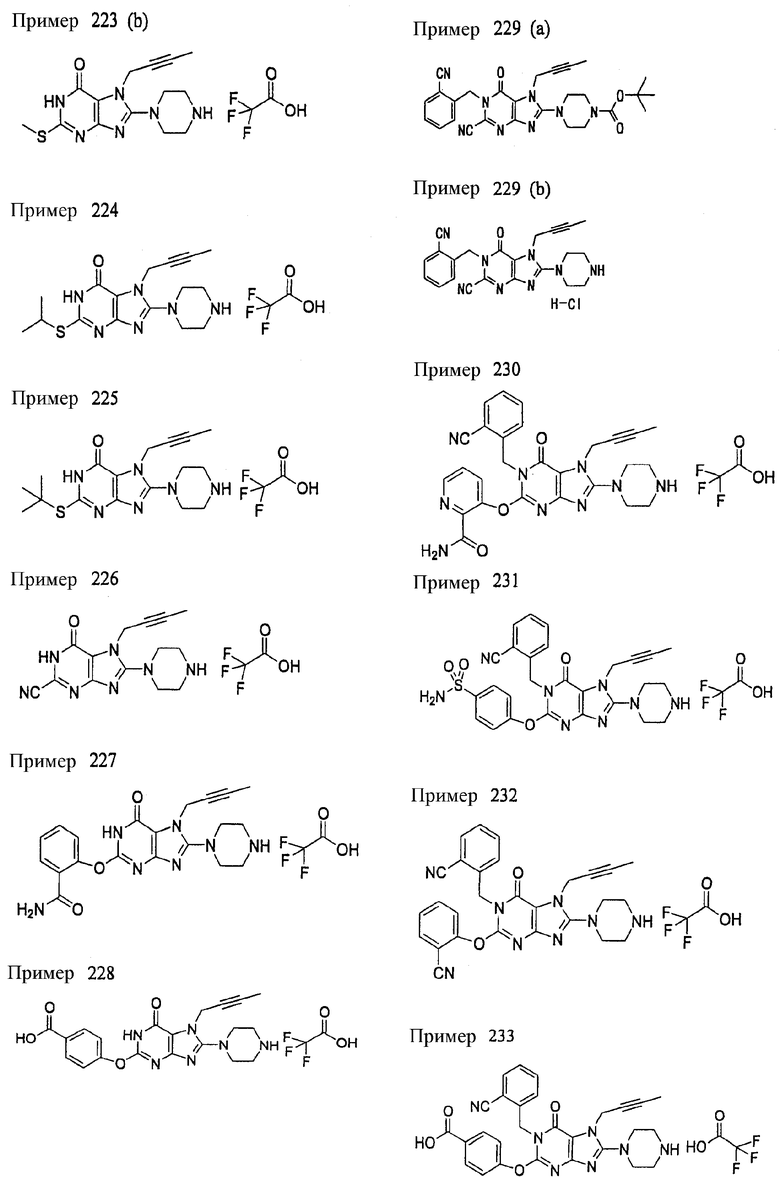
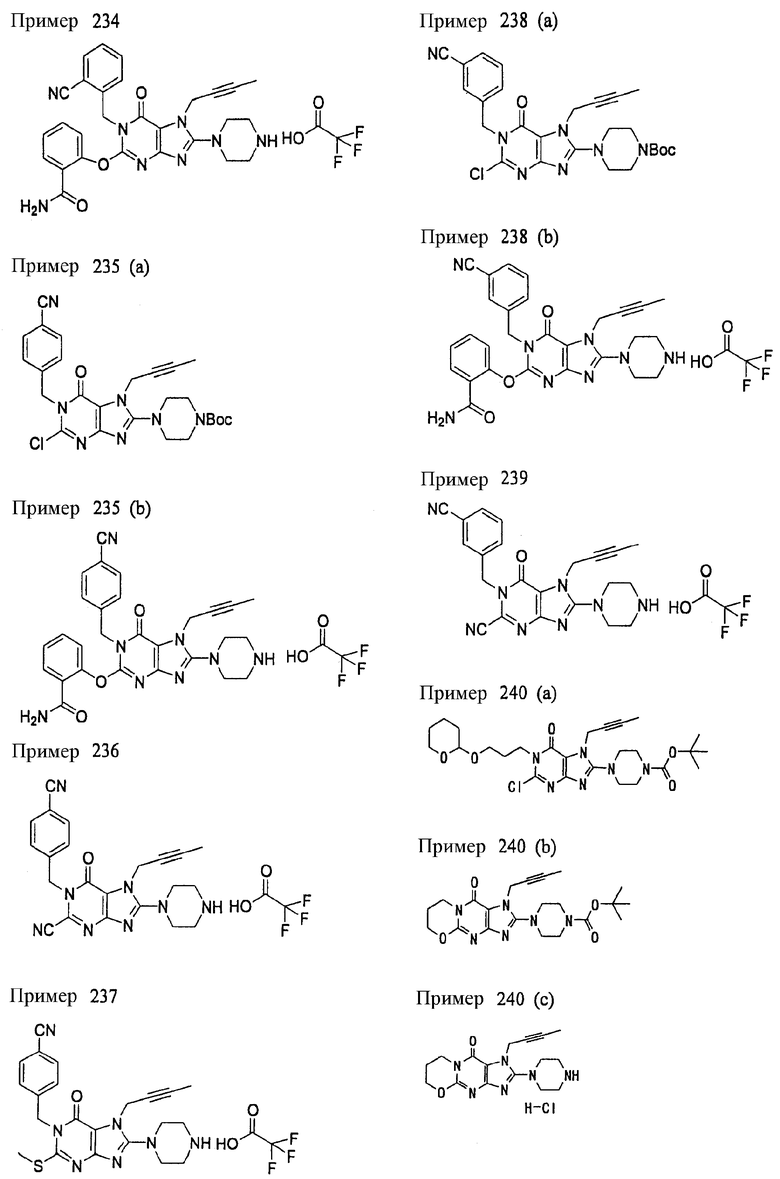
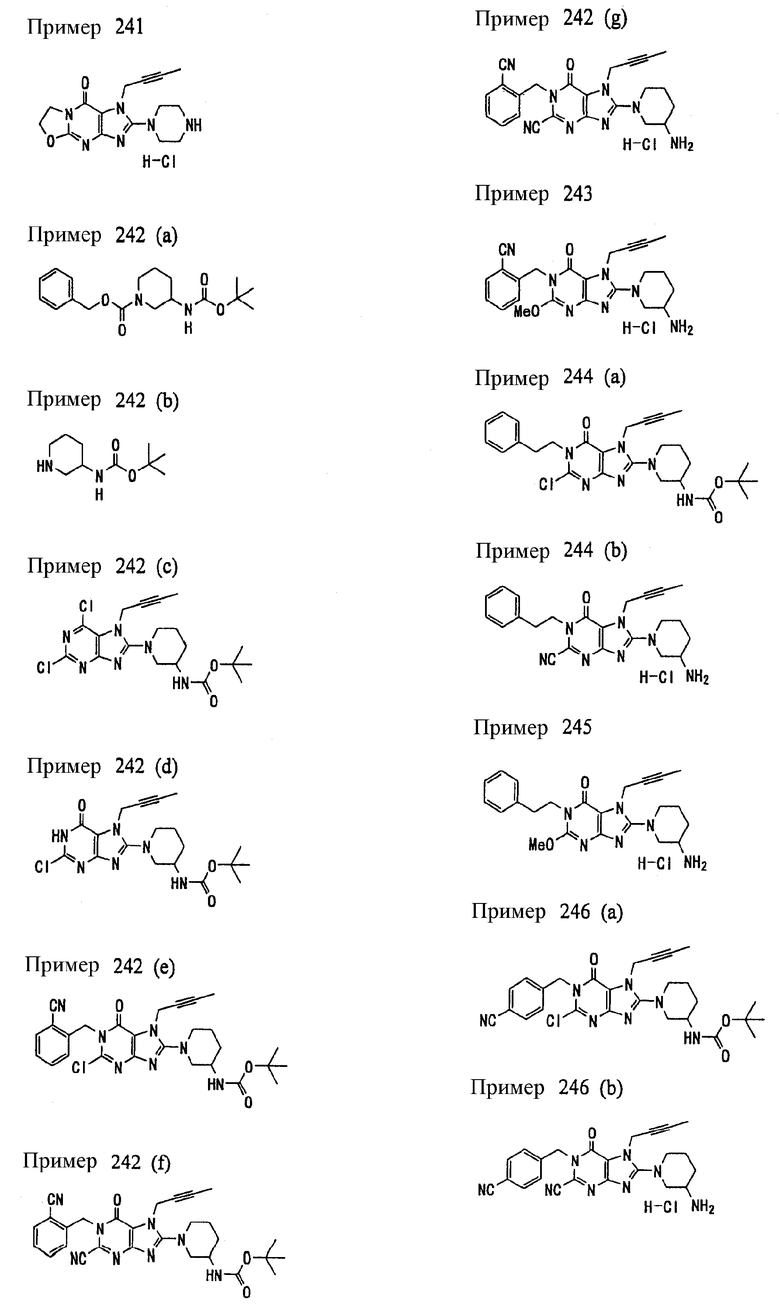
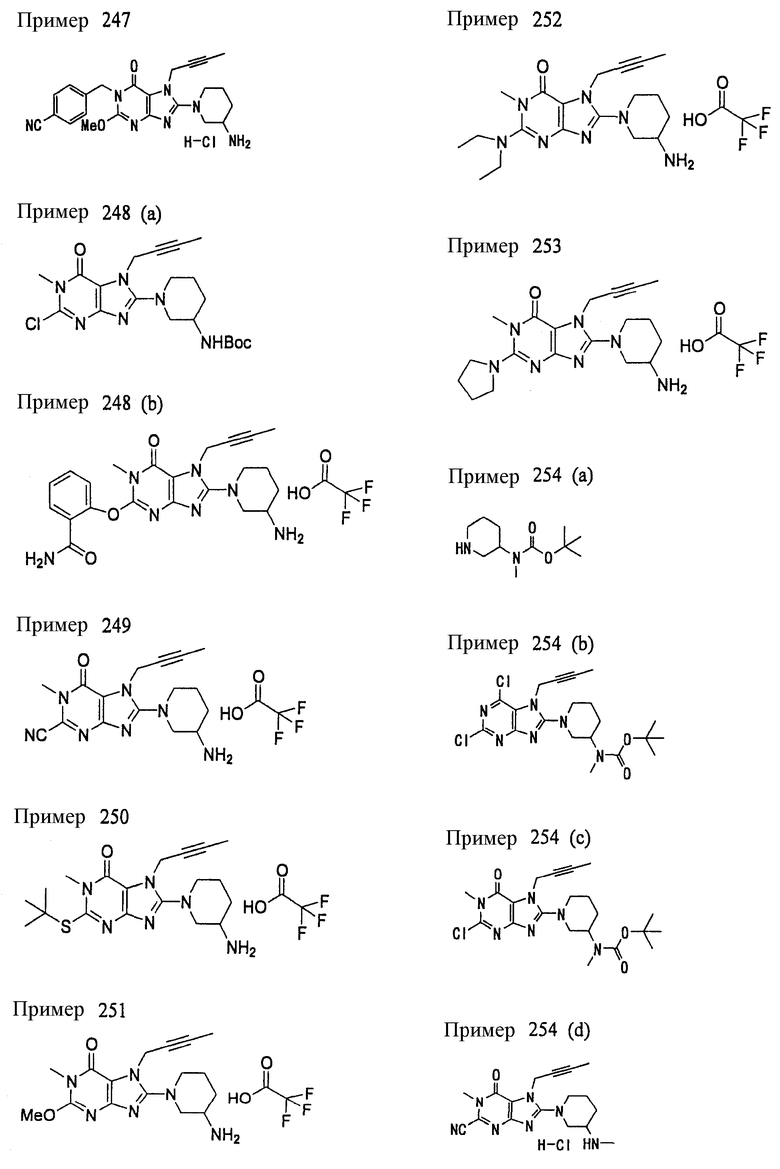
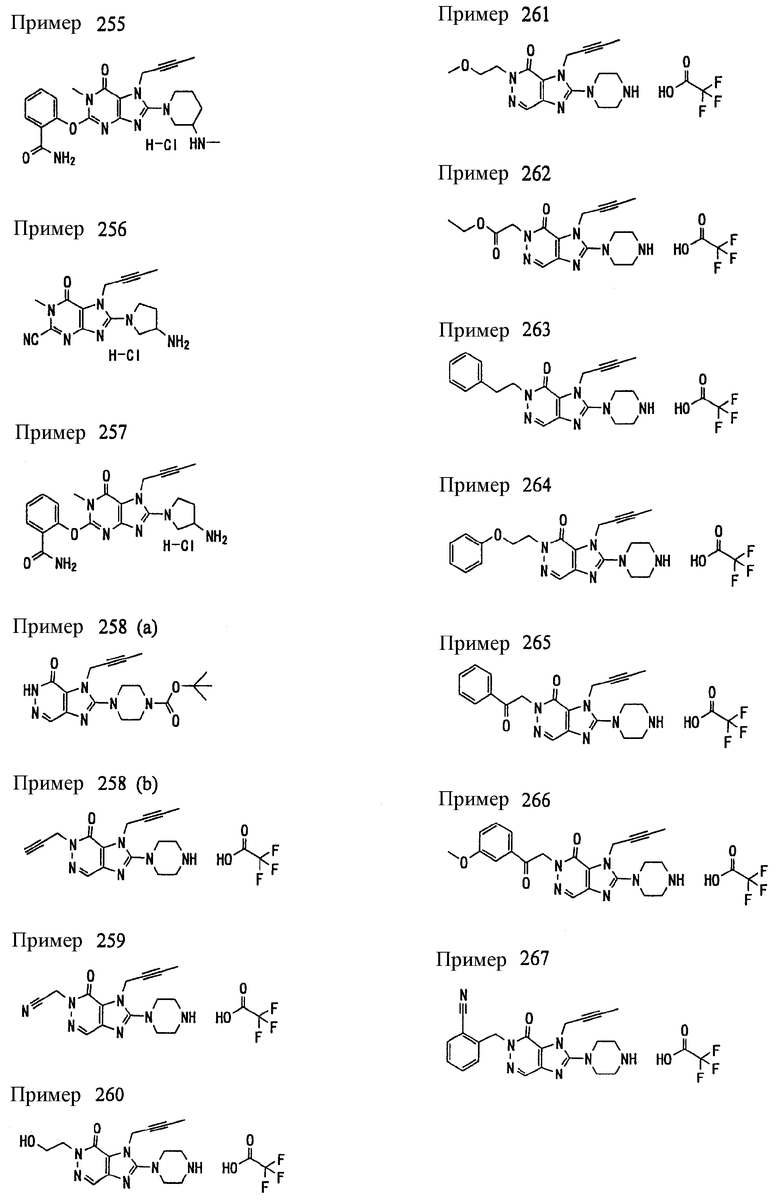
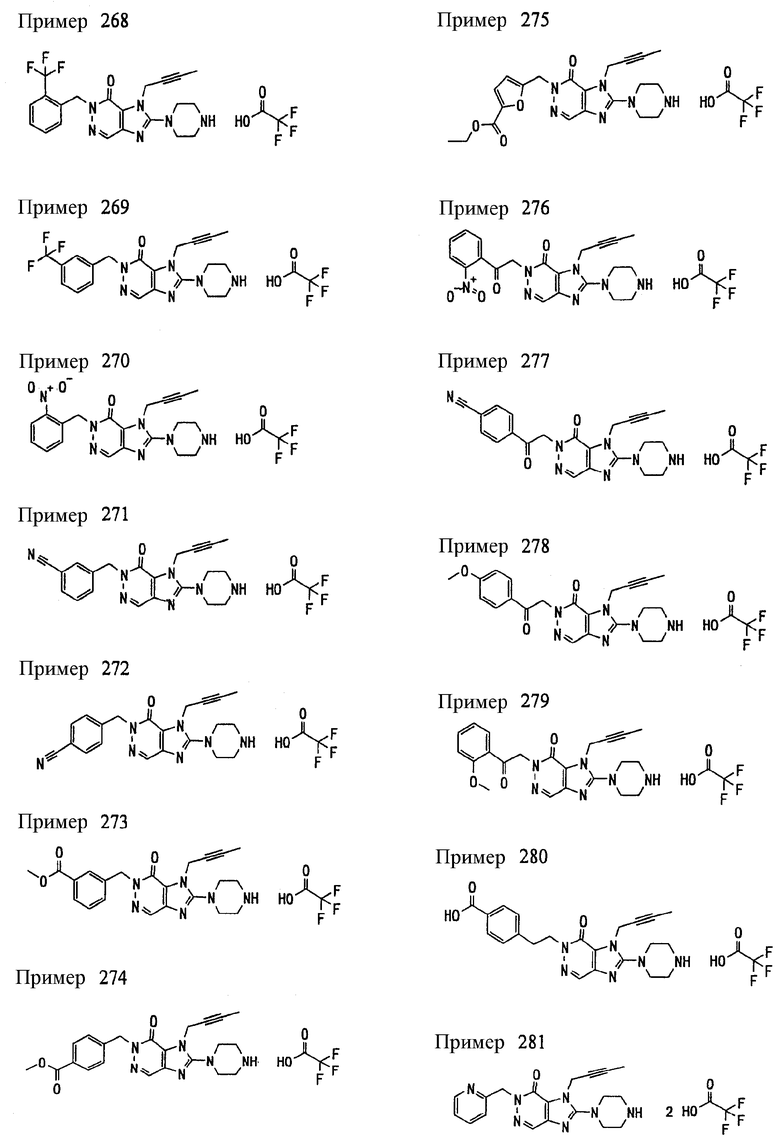
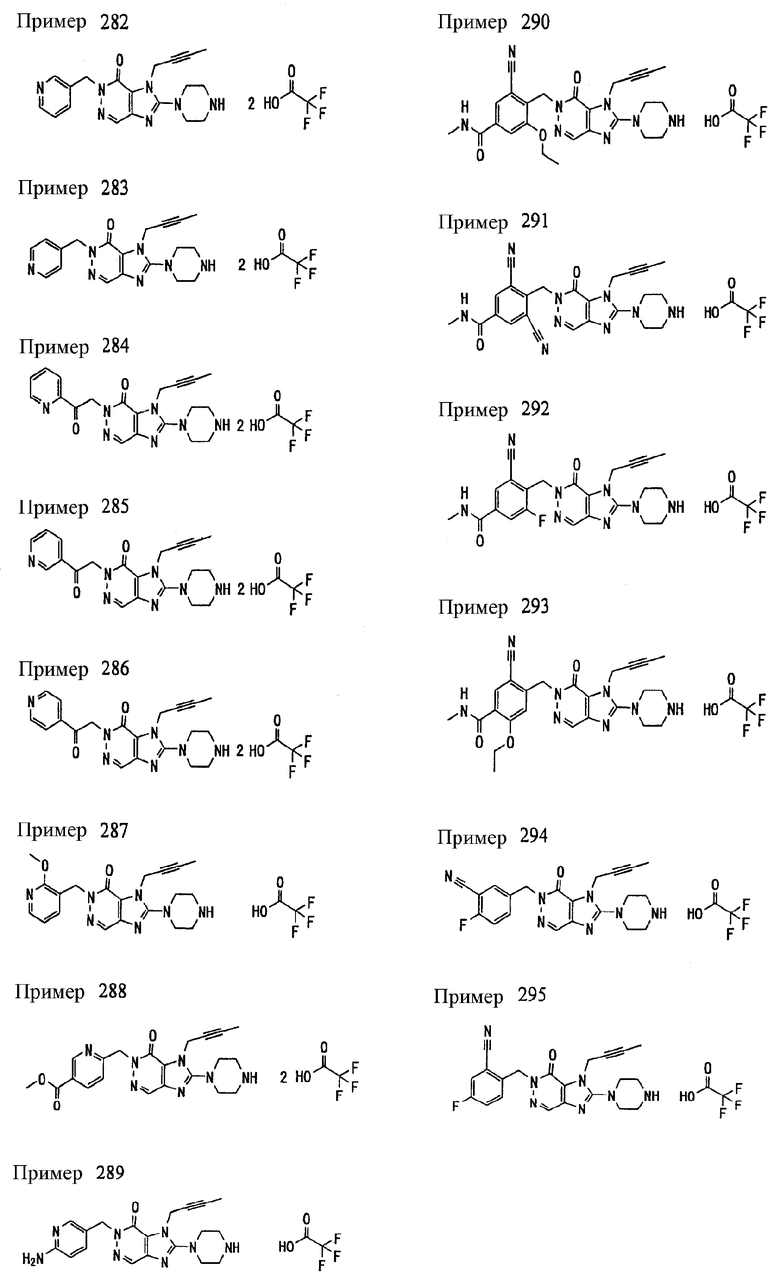
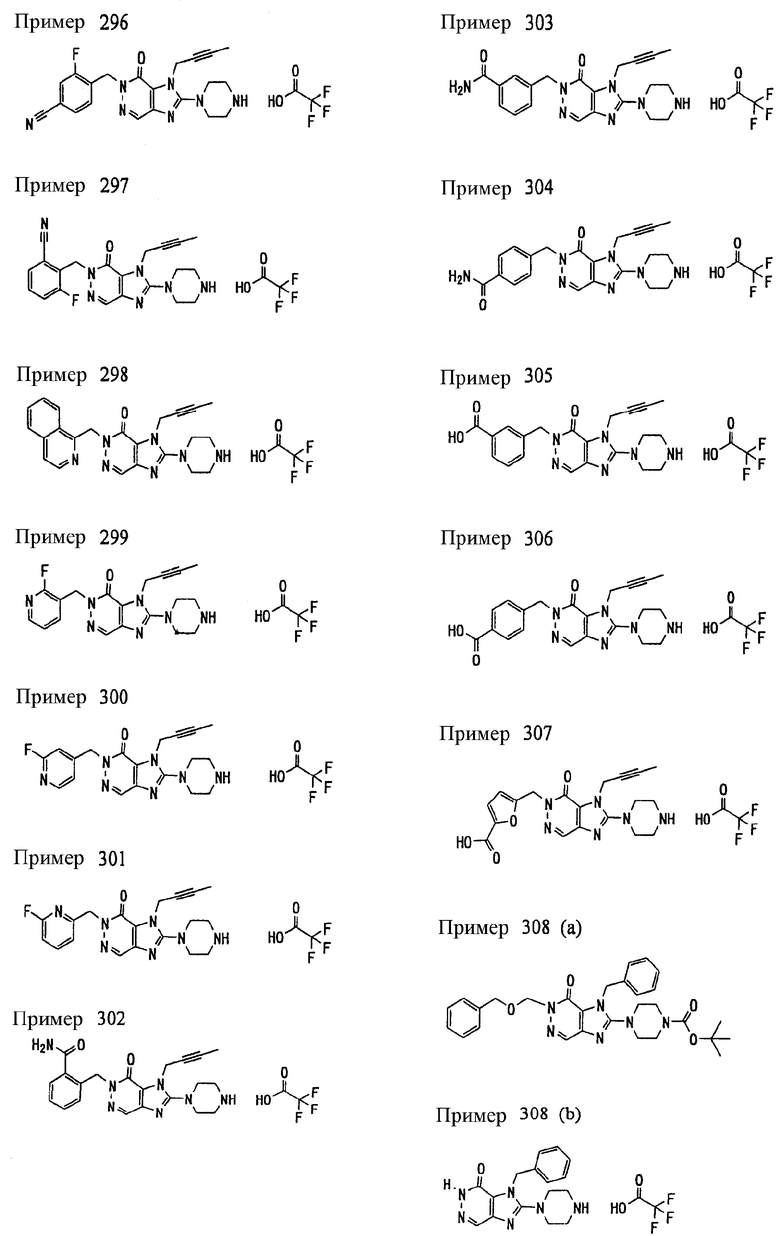
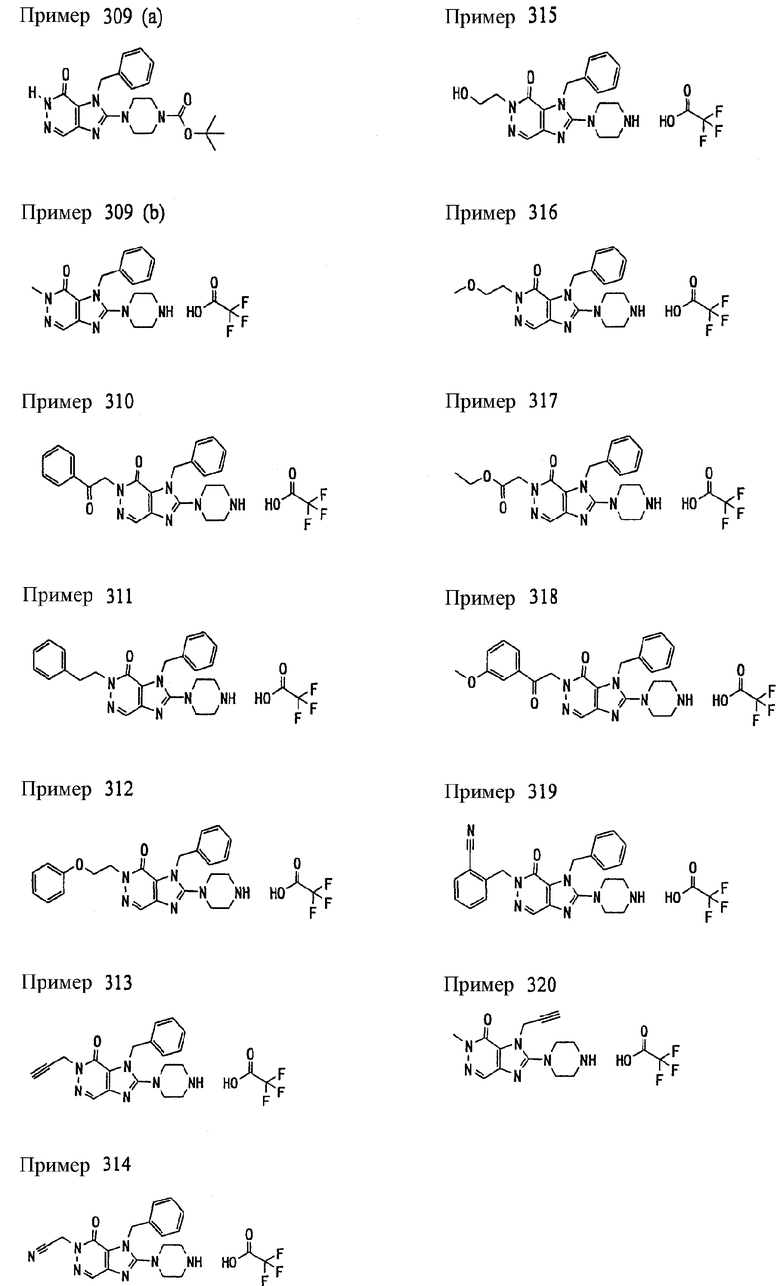
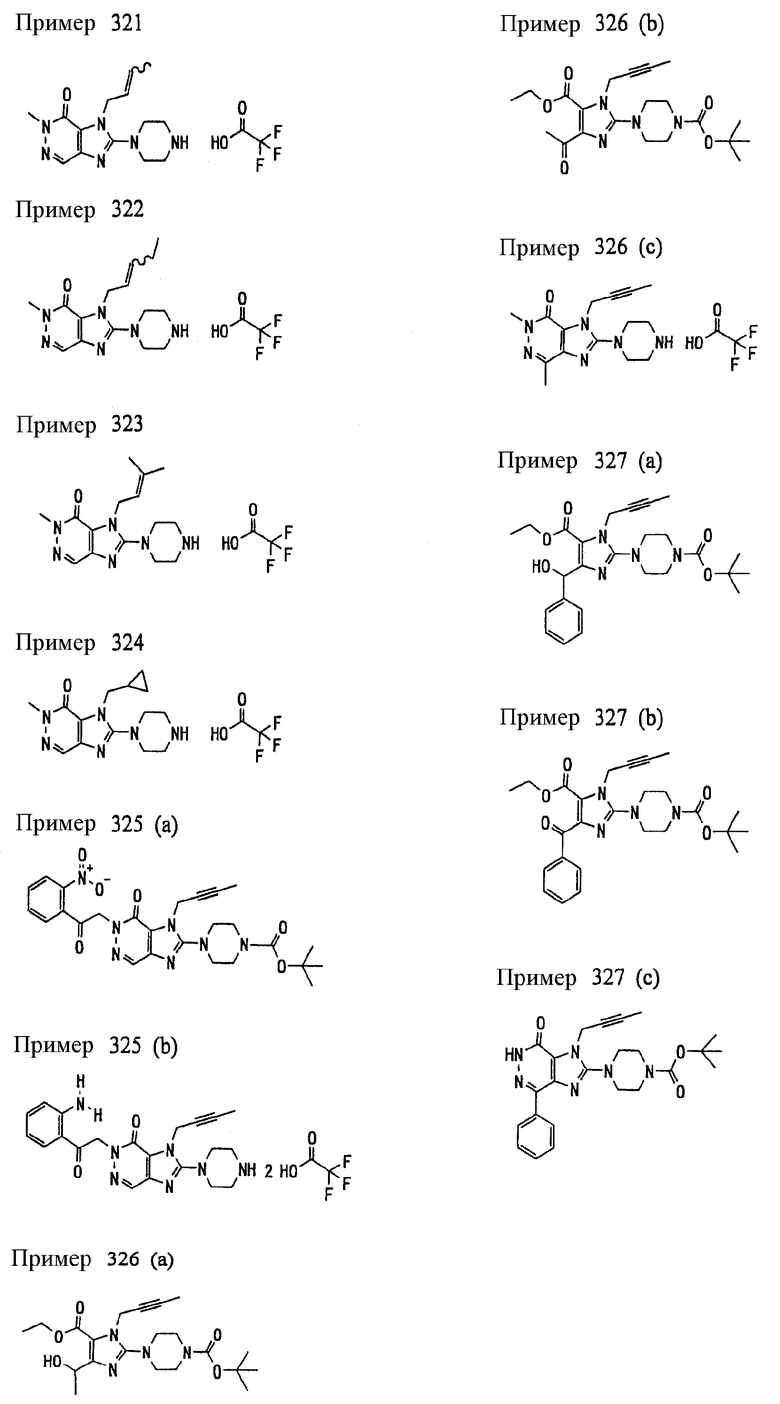
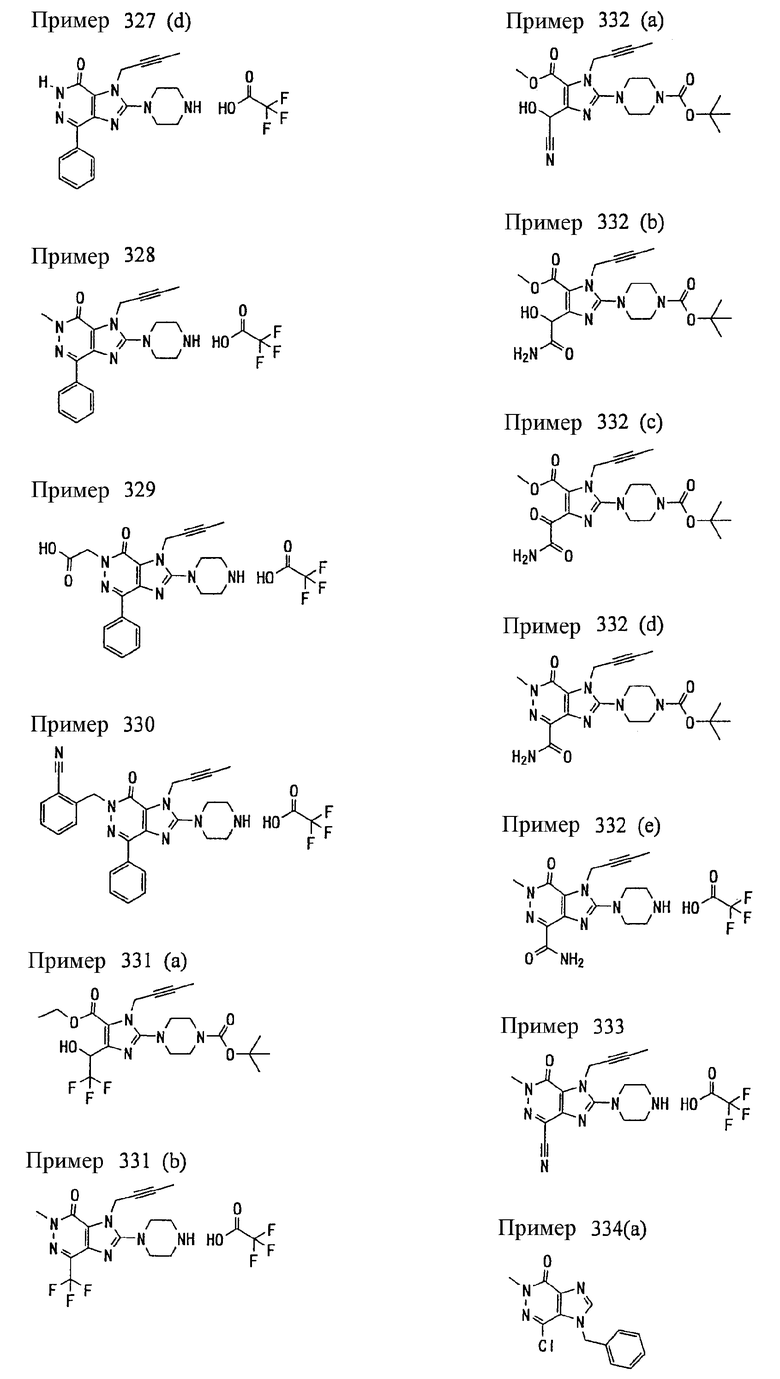
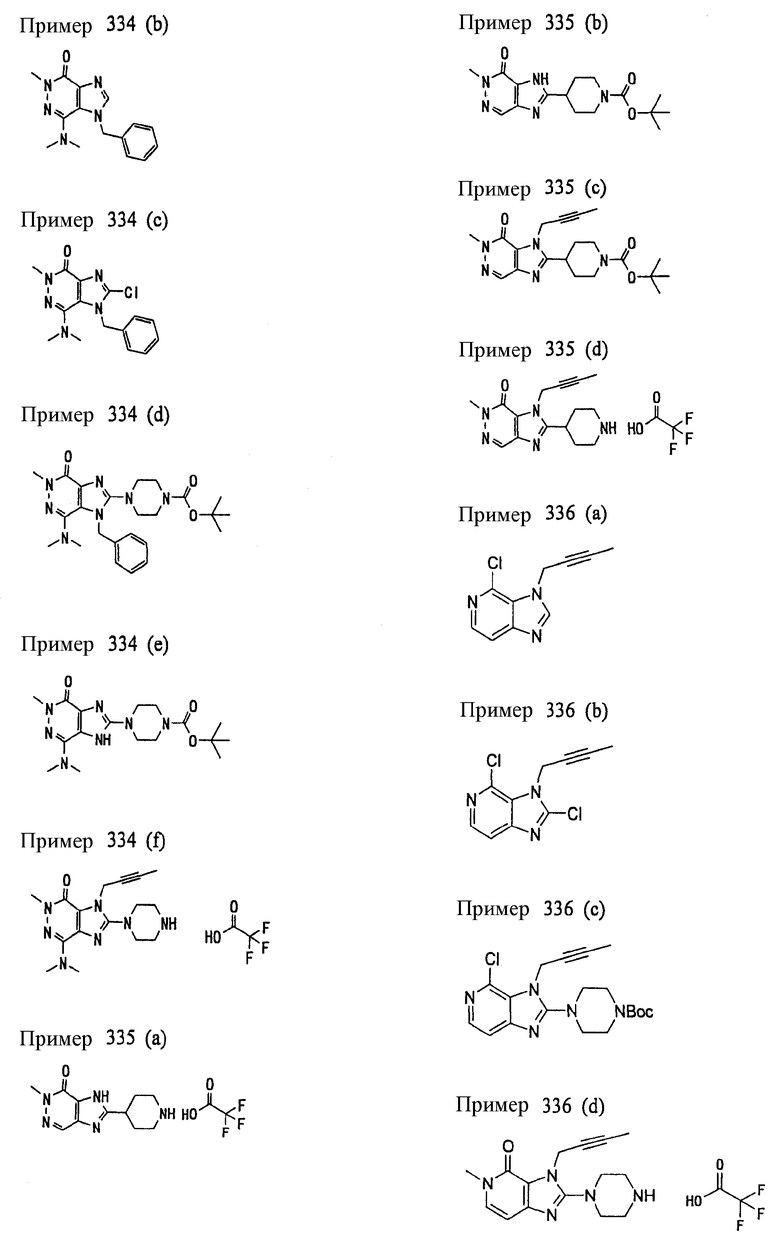
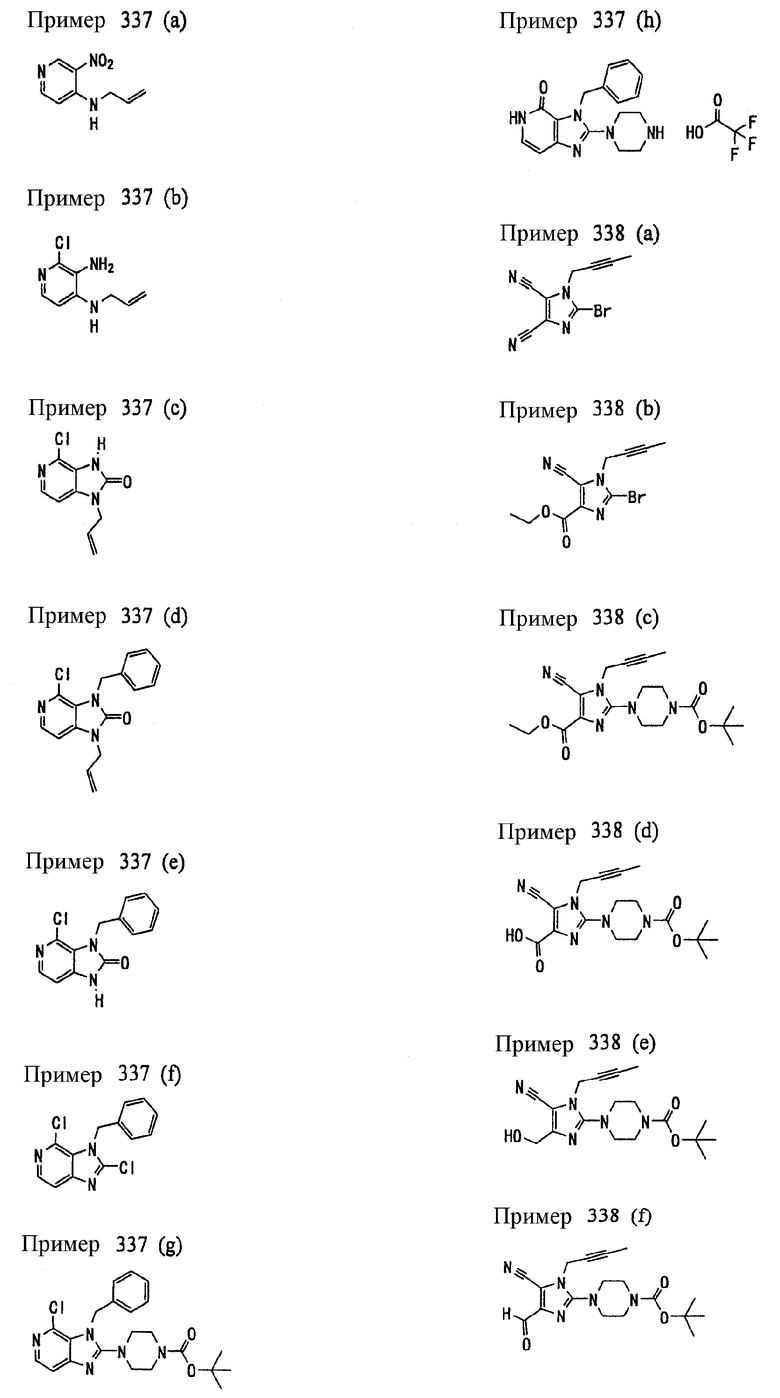
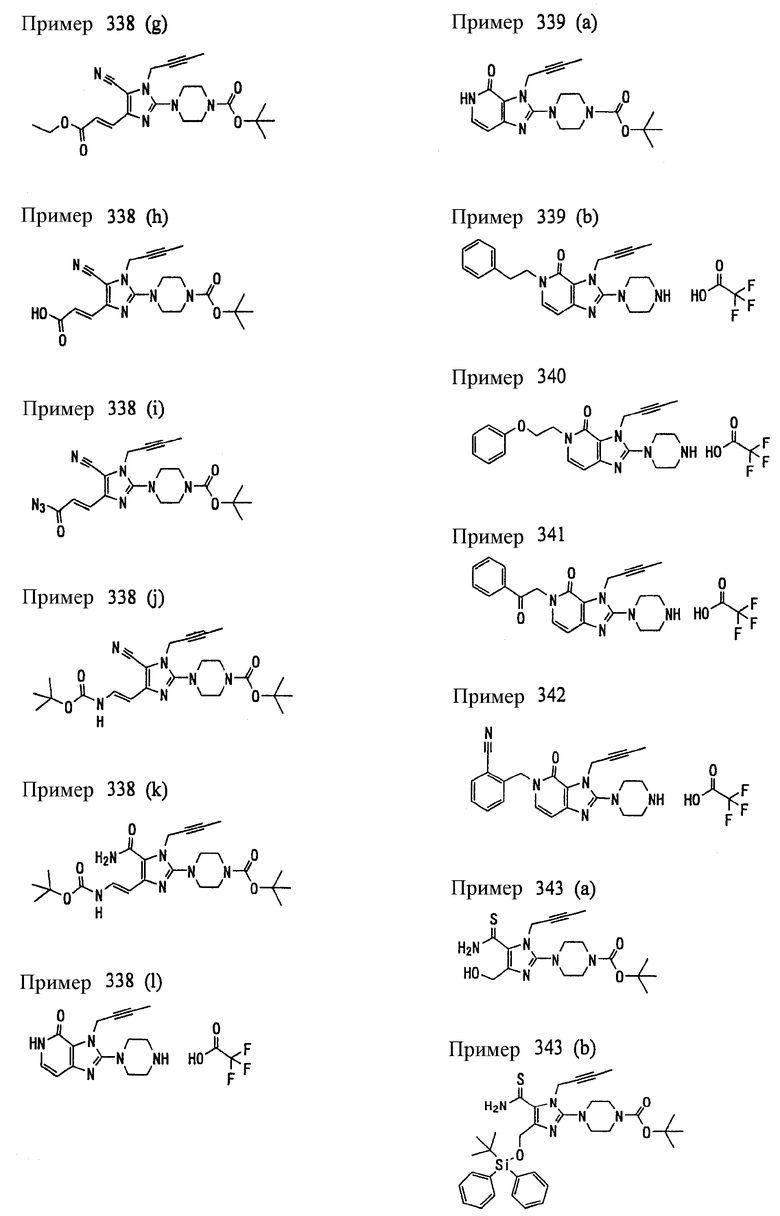
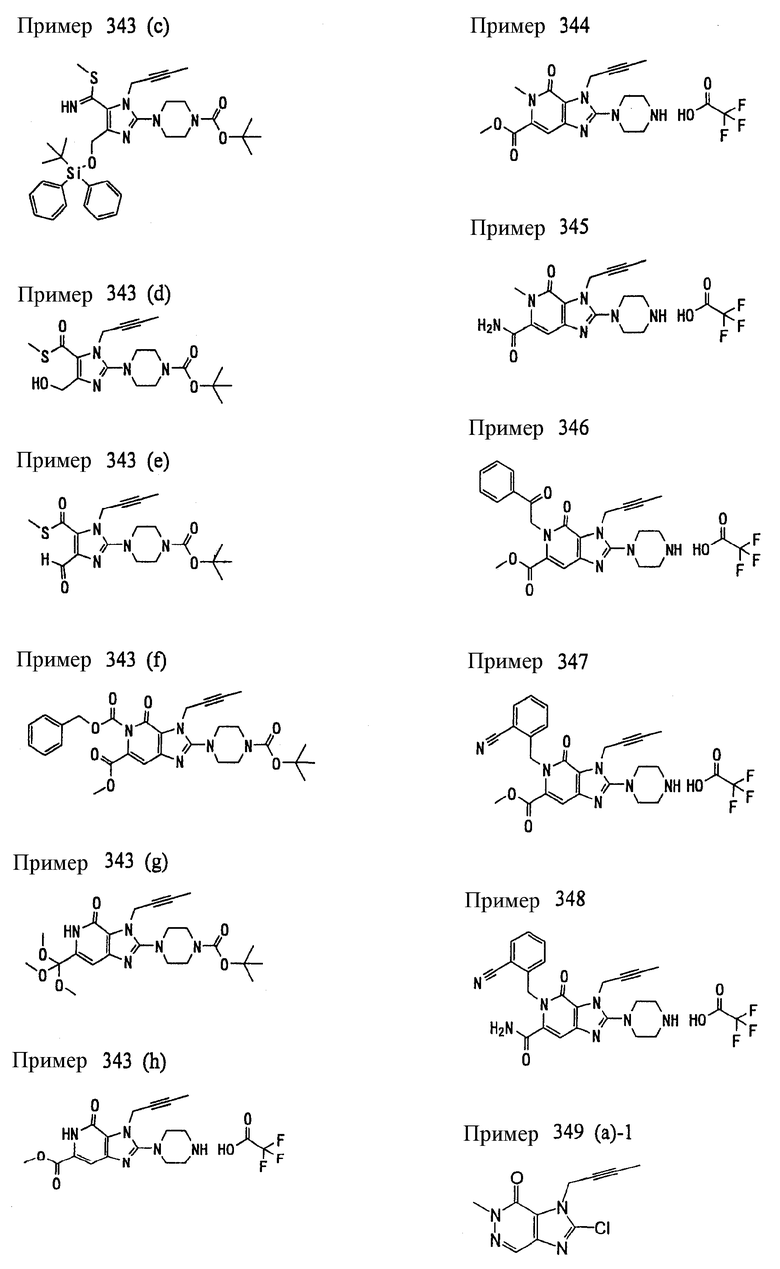
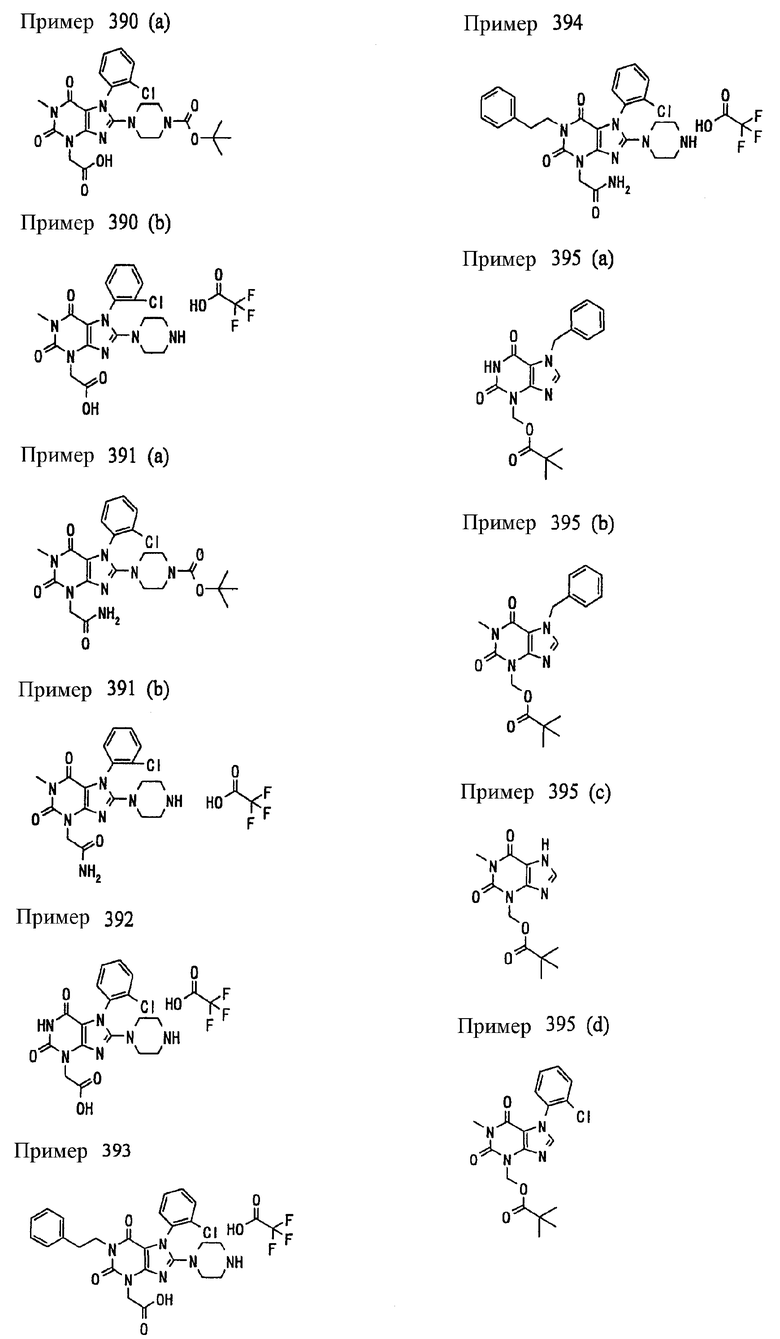
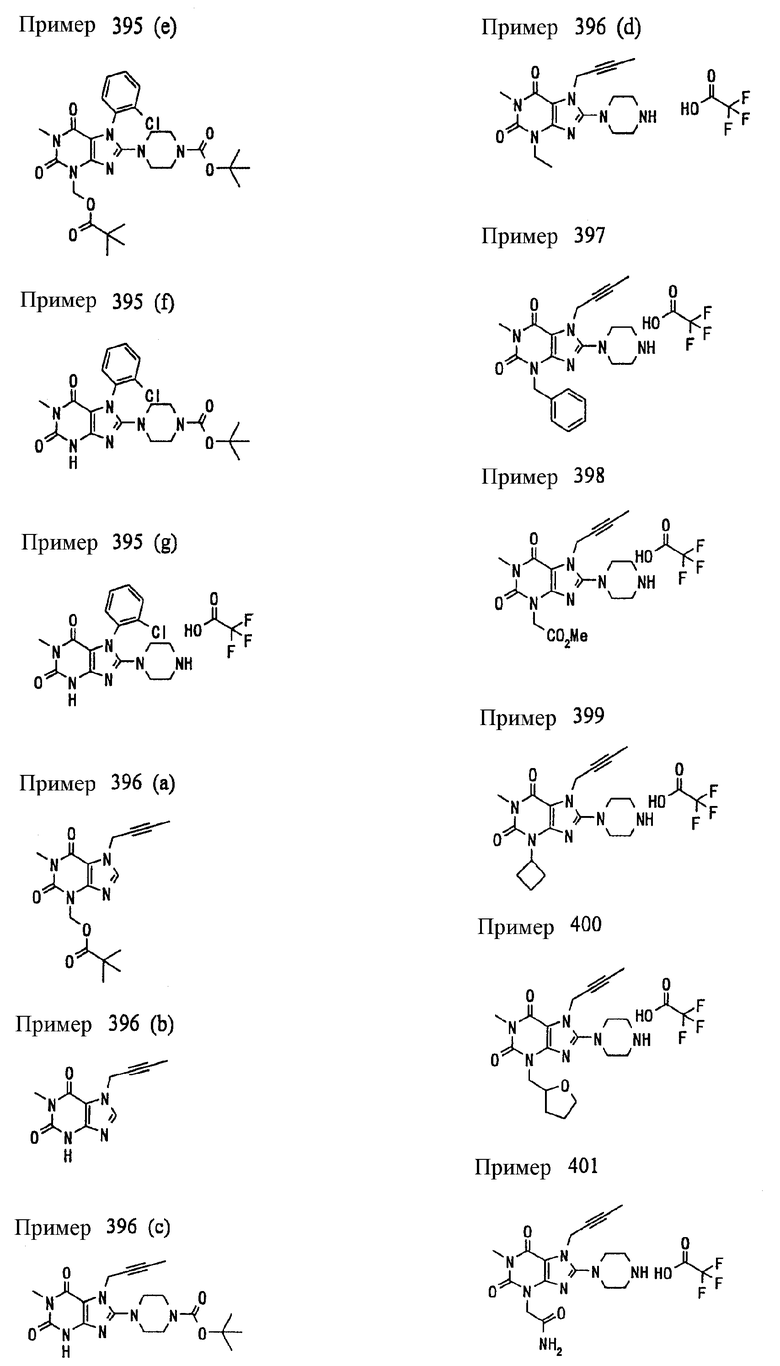
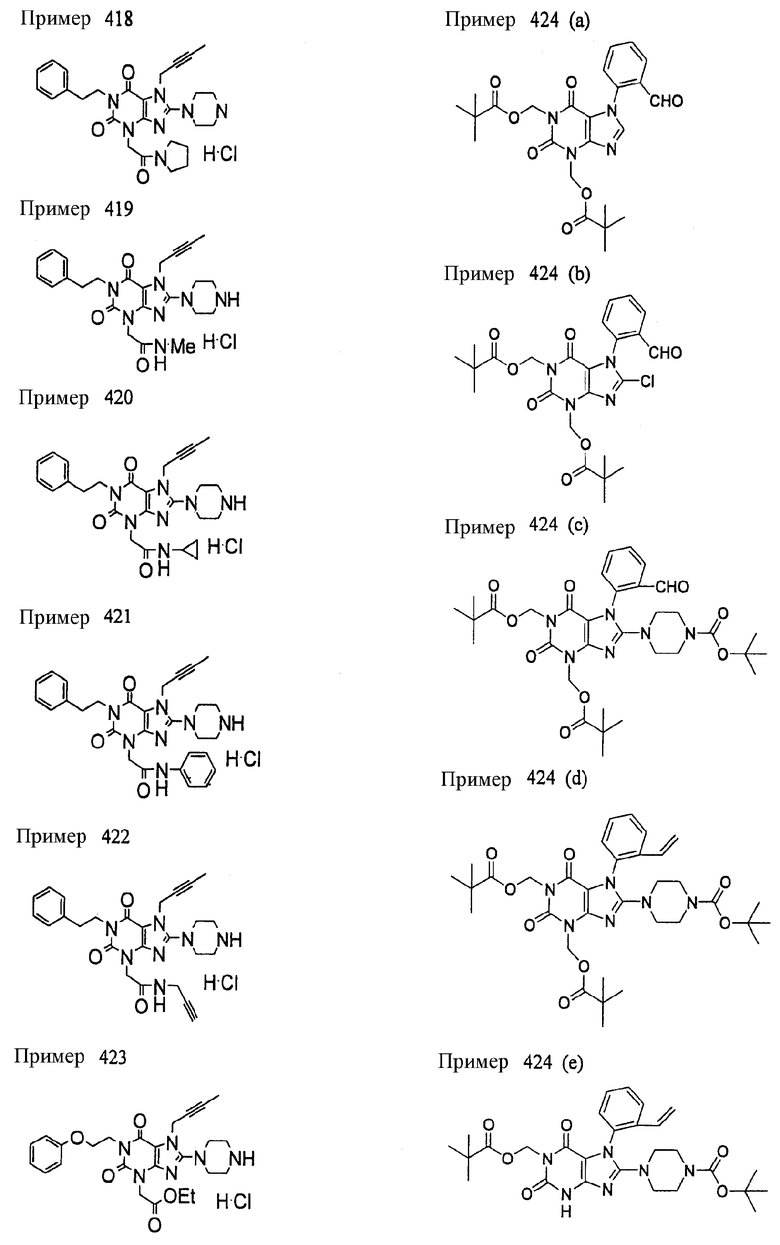
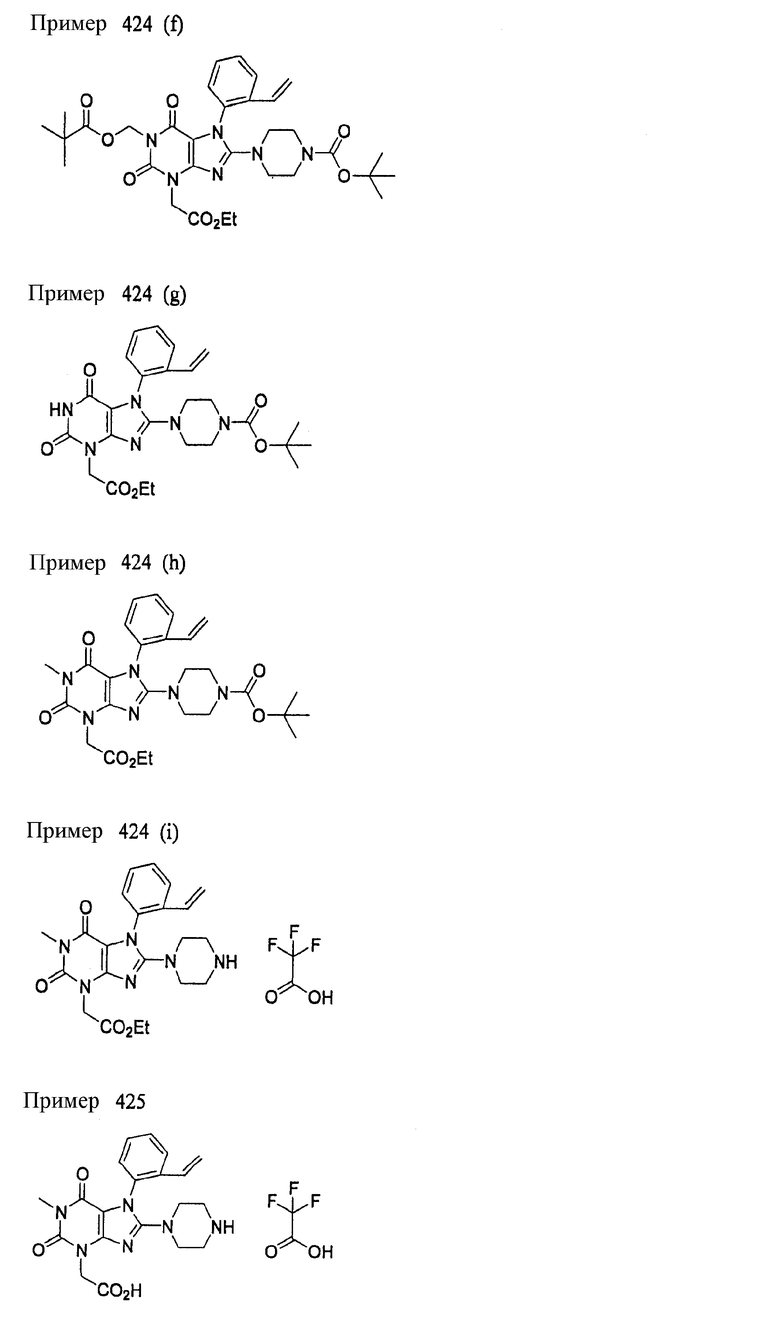
[Пример анализа 1]
Анализ соединений, представленных формулой (I), на DPPIV-ингибирующую активность
DPPIV, полученную из свиных почек, растворяли в реакционном буфере (50мМ Трис-HCl (рН 7,4)/0,1% BSA) в конечной концентрации 10 мU/мл. 110 мкл аликвота данного раствора фермента добавляли к реакционной системе, а затем добавляли 15 мкл средства. Реакционный раствор инкубировался при комнатной температуре в течение 20 минут. К реакционной системе (в конечной концентрации 0,33 мМ) добавляли 25 мкл раствора, содержащего 2 мМ Gly-Pro-п-нитроанилида для начала ферментной реакции. Время реакции составляло 20 минут. Для прекращения реакции добавляли 25 мкл 1 н раствора фосфата. Спектральная поглощательная способность образца измерялась при 405 нм. Определялась степень ингибирования ферментной реакции, и IC50 вычислялась на основании поглощательной способности.
[Пример анализа 2]
Влияние метформина, буформина и фенформина на уровень GLP-1 у крыс с дефицитом DPPIV
Животные: самцы крыс Фишер с дефицитом DPPIV (закупаемые у Charles River Japan, Inc.)
Методы:
[Получение и введение испытуемых соединений]
Каждое испытуемое соединение суспендировали в растворе 0,5% метилцеллюлозы в дозах, показанных в Таблице 2, а затем вводилось орально в объеме 5 мл/кг. Группе контрольного носителя вводили орально раствор 0,5% метилцеллюлозы в объеме 5 мл/кг.
[Сбор крови и анализ GLP-1]
Неанестезированную крысу слегка разрезали по хвостовой вене бритвенным лезвием и брали кровь непосредственно до и через 1, 3 и 5 часов после введения испытуемого соединения или раствора 0,5% метилцеллюлозы. 250 мкл крови собирали у крыс с использованием гепаринизированного капилляра и переносили в центрифужную пробирку. Супернатант, полученный с помощью центрифугирования (при 10000 g при 4°С в течение 2 минут), анализировали на уровень GLP-1 с использованием Активного GLP-1 набора ЭЛИЗА (Linco).
Результаты:
Результат представлен в виде "средней величины ± стандартная ошибка". Соответствующие величины оценивались и сравнивались с помощью теста Даннетт'а и показаны в Таблице 2.
(% от показателя до введения)
***: Р < 0,001 против контрольной группы носителя
Группа крыс с дефицитом DPPIV, которой вводили метформин в дозе 300 мг/кг, имела значительно повышенный уровень активного GLP-1 в плазме через пять часов после введения. Группа крыс с дефицитом DPPIV, которой вводили буформин в дозе 100 мг/кг, имела значительно повышенный уровень активного GLP-1 в плазме через один и пять часов после введения. Далее, группа крыс с дефицитом DPPIV, которой вводили фенформин в дозе 100 мг/кг, имела значительно повышенный уровень активного GLP-1 в плазме через 1, 3 и 5 часов после введения.
[Пример анализа 3]
Влияние метформина и ингибитора DPPIV (валинпирролидид (Val-Pyr)), используемых по одному или в сочетании на уровень GLP-1 у нормальных крыс
Животные: DPPIV-интактные самцы нормальных крыс Фишер (закупаемые у CLEA Japan, Inc.)
Методы:
[Получение и введение испытуемых соединений]
Каждое испытуемое соединение суспендировали в растворе 0,5% метилцеллюлозы в дозах, показанных в Таблице 3, а затем вводилось орально в объеме 5 мл/кг. Группе контрольного носителя вводили орально раствор 0,5% метилцеллюлозы в объеме 5 мл/кг.
[Сбор крови и анализ GLP-1]
Неанестезированную крысу слегка разрезали по хвостовой вене бритвенным лезвием и брали кровь непосредственно до и через 1, 3 и 5 часов после введения испытуемого соединения или раствора 0,5% метилцеллюлозы. 250 мкл крови собирали у крыс с использованием гепаринизированного капилляра и переносили в центрифужную пробирку. Супернатант, полученный с помощью центрифугирования (при 10000 g при 4°С в течение 2 минут), анализировался на уровень GLP-1 с использованием Активного GLP-1 набора ЭЛИЗА (Linco).
Результаты:
Результат представлен в виде "средней величины ± стандартная ошибка". Соответствующие величины оценивались и сравнивались с помощью теста Даннетт'а и показаны в Таблице 3.
(% от показателя до введения)
+
валинпирролидид
+
30
***: Р < 0,001 против контрольной группы носителя
Когда метформин или ингибитор DPPIV дается по одному, нет никакого увеличения уровня активного GLP-1, Однако уровень активного GLP-1 значительно повышался через 1, 3 и 5 часов после введения в группе, которой вводился метформин и ингибитор DPPIV в сочетании. Данный результат говорит о том, что уровень активного GLP-1 повышался вследствие усиления секреции GLP-1 метформином и подавления разрушения GLP-1 ингибитором DPPIV.
[Пример анализа 4]
Влияние метформина и ингибитора DPPIV, используемых по одному (Примеры 82, 119, 120, 122, 229 и 267) или в сочетании, на уровень GLP-1 у нормальных крыс
Животные: DPPIV-интактные самцы нормальных крыс Фишер (закупаемых у CLEA Japan, Inc.)
Методы:
[Получение и введение испытуемых соединений]
Каждое испытуемое соединение суспендировали в растворе 0,5% метилцеллюлозы в дозах, показанных в Таблицах 4-6, а затем вводилось орально в объеме 5 мл/кг. Группе контрольного носителя вводили орально раствор 0,5% метилцеллюлозы в объеме 5 мл/кг.
[Сбор крови и анализ GLP-1]
Неанестезированную крысу слегка разрезали по хвостовой вене бритвенным лезвием и брали кровь непосредственно до и через 3 часа после введения испытуемого соединения или раствора 0,5% метилцеллюлозы. 250 мкл крови собирали у крыс с использованием гепаринизированного капилляра и переносили в центрифужную пробирку. Супернатант, полученный с помощью центрифугирования (при 10000 g при 4°С в течение 2 минут), анализировался на уровень GLP-1 с использованием Активного GLP-1 набора ЭЛИЗА (Linco).
Результаты:
Результат представлен в виде "средней величины ± стандартная ошибка". Соответствующие величины оценивались и сравнивались с помощью теста Даннетт'а и показаны в Таблицах 4-6.
(% от показателя до введения)
(% от показателя до введения)
(% от показателя до введения)
Когда метформин или ингибитор DPPIV давался по одному, не было никакого увеличения уровня активного GLP-1. Однако уровень активного GLP-1 значительно повышался через 3 часа после введения в группе, которая получала метформин и ингибитор DPPIV в сочетании. Данный результат говорит о том, что уровень активного GLP-1 повышался вследствие усиления секреции GLP-1 метформином и подавления разрушения GLP-1 ингибитором DPPIV.
[Пример анализа 5]
Влияние метформина и ингибитора DPPIV (валинпирролидид (Val-Pyr)), используемых по одному или в сочетании, на переносимость глюкозы, инсулин и на уровень GLP-1, потребление корма и массы тела у крыс Zucker fα/fα
Животные: крысы Zucker fα/fα (закупаемые у Charles River Japan, Inc.)
Методы:
[Получение и введение испытуемых соединений]
Каждое испытуемое соединение растворяли в дистиллированной воде в дозах, показанных в Таблицах, приведенных ниже, а затем вводили орально в объеме 5 мл/кг. Группе контрольного носителя вводили орально дистиллированную воду в объеме 5 мл/кг. Каждое испытуемое соединение или дистиллированную воду давали орально в указанной выше дозе два раза в день ежедневно (в 10:00 до полудня и 4:00 после полудня) в течение 14 дней. Крысы испытывались на переносимость глюкозы в первый день серии опытов с введением. Во время опыта дистиллированную воду и испытуемые соединения давали за 0,5 часа до глюкозной нагрузки.
[Процедура сбора крови и определения уровней глюкозы в крови и GLP-1]
Для опыта на переносимость глюкозы неанестезированную крысу слегка разрезали по хвостовой вене бритвенным лезвием и брали кровь непосредственно до введения испытуемого соединения или дистиллированной воды и непосредственно до и через 0,5, 1, 2 и 3 часа после нагрузки глюкозы. 250 мкл крови собирали у крыс с использованием гепаринизированного капилляра и переносилось в центрифужную пробирку. Супернатант, полученный с помощью центрифугирования (при 10000 g при 4°С в течение 2 минут), анализировался на уровень GLP-1 с использованием Активного GLP-1 набора ЭЛИЗА (Linco). В то же самое время собирали 10 мкл крови и смешивали со 140 мкл 0,6 М раствора перхлорной кислоты. Смесь центрифугировали (при 3000 g при 4°С в течение 10 минут), и получающийся супернатант анализировали на глюкозу с использованием Теста на глюкозу Wako II (Wako Pure Chemical Industries, Inc.). Уровень глюкозы в крови определялся во время измерения через 3 часа после нагрузки глюкозы.
[Определение потребления корма и массы тела]
Потребление корма и масса тела определялись в 4:00 после полудня через 14 дней серии опытов по введению. Общее потребление корма и масса тела определялись на протяжении 14 дней для каждой экспериментальной группы.
Результаты:
Результат представлен в виде "средней величины ± стандартная ошибка". Соответствующие величины оценивались и сравнивались с помощью теста Даннетт'а и показаны в Таблицах 7-10.
Доза
(мг/кг)
+
валинпирролидид (30)
Доза
(мг/кг)
+
валинпирролидид (30)
Доза
(мг/кг)
+
Валинпирролидид (30)
+
валинпирролидид
+
30
В течение опыта на переносимость глюкозы уровень активного GLP-1 значительно повышался в группе, которой вводился ингибитор DPPIV, не было никакого значительного увеличения уровня активного GLP-1 в группе, которой вводился метформин. Однако уровень активного GLP-1 повышался синергистически в группе, которой вводился как метформин, так и ингибитор DPPIV. Данный результат говорит о том, что уровень активного GLP-1 повышался вследствие усиленной секреции GLP-1, вызванной метформином, и подавленного разрушения GLP-1 благодаря ингибитору DPPIV, как описывалось выше.
Испытание на переносимость глюкозы показало, что переносимость глюкозы улучшалась в каждой группе, которой вводили по одному или метформин, или ингибитор DPPIV. Однако переносимость глюкозы улучшалась синергистически в группе, которой вводили метформин и ингибитор DPPIV в сочетании, по сравнению с группами, которым вводилось любое соединение по одному.
В течение опыта на переносимость глюкозы уровень инсулина значительно увеличивался зависимым от глюкозы образом в группе, которой вводили только ингибитор DPPIV, при этом не было никакого значительного увеличения уровня инсулина в группах, которым вводили или один метформин, или метформин и ингибитор DPPIV в сочетании. Данный результат говорит о том, что эффект, наблюдаемый в группе, которой вводился метформин, основан на экстра-панкреатическом действии данного средства, в то время как данный эффект в группе, которой вводили ингибитор DPPIV, основан на зависимом от глюкозы увеличении уровня инсулина благодаря увеличению уровня активного GLP-1. С другой стороны, он говорит о том, что группа, которой вводили метформин и ингибитор DPPIV в сочетании, имела синергистически улучшенную переносимость глюкозы благодаря усиленной восприимчивости к инсулину, на основе экстра-панкреатического действия метформина и синергистического увеличения уровня активного GLP-1, являющегося результатом комбинированного введения.
Кроме того, снижение потребления корма и подавление прироста массы наблюдались только в группе, которой вводили метформин и ингибитор DPPIV в сочетании в течение 14 дней. Можно сделать вывод, что синергистическое увеличение уровня GLP-1 вследствие комбинированного использования метформина и ингибитора DPPIV приводило к уменьшению потребления корма через гипоталамус, которое в свою очередь приводило в результате к подавлению прироста массы.
В дополнение к сказанному, синергистическое уменьшение уровней глюкозы в крови и инсулина в течение голодания были обнаружены в группе, которой вводили метформин и ингибитор DPPIV в сочетании в течение 14 дней. Возможно, что это является результатом усиленного метаболизма глюкозы вследствие синергистического улучшения переносимости глюкозы и подавленного прироста массы в группе, которой вводили метформин и ингибитор DPPIV в сочетании. Это говорит о том, что комбинированное использование метформина и ингибитора DPPIV является эффективным для лечения диабета типа II.
[Пример анализа 6]
Влияние метформина на уровень GLP-2 у крыс с дефицитом DPPIV
Животные: самцы крыс Фишер с дефицитом DPPIV (закупаемые у Charles River Japan, Inc.)
Методы:
[Получение и введение испытуемых соединений]
Каждое испытуемое соединение суспендировали в растворе 0,5% метилцеллюлозы в дозах, показанных в Таблице 11, а затем вводилось орально в объеме 5 мл/кг. Группе контрольного носителя вводили орально водный раствор 0,5% метилцеллюлозы в объеме 5 мл/кг.
[Сбор крови и определение уровня GLP-2]
Неанестезированную крысу слегка разрезали по хвостовой вене бритвенным лезвием и спускали кровь непосредственно до и через 1, 3 и 5 часов после введения испытуемого соединения или раствора 0,5% метилцеллюлозы. 250 мкл крови собирали у крыс с использованием гепаринизированного капилляра и переносили в центрифужную пробирку. Супернатант, полученный с помощью центрифугирования (при 10000 g при 4°С в течение 2 минут), анализировался на уровень GLP-2 с использованием GLP-2 набора ЭЛИЗА (Yanaihara Institute Inc.).
Результаты:
Результат представлен в виде "средней величины ± стандартная ошибка". Соответствующие величины оценивались и сравнивались с помощью t-теста и показаны в Таблице 11.
В группе, которой вводили метформин уровень GLP-2 в плазме, значительно повышался через 1, 3 и 5 часов после введения у крыс с дефицитом DPPIV. Данный результат говорит о том, что комбинированное использование метформина и ингибитора DPPIV смогло синергистически усилить действие GLP-2, таким образом, может быть эффективным для лечения желудочно-кишечных заболеваний.
[Пример анализа 7]
Влияние метформина и ингибитора DPPIV (валинпирролидид (Val-Pyr)), используемых по одному или в сочетании на атрофию тонкой кишки, вызываемую 5-фторурацилом (5-FU)
Животные: мыши BALB/c AnCrj (закупаемые у Cyarles River Japan, Inc.)
Методы:
[Получение и введение испытуемых соединений]
5-FU (закупаемый у Sigma) суспендировали в растворе 0,5% метилцеллюлозы и затем вводили орально в объеме 10 мл/кг/день (от 8 до 9 до полудня в течение 3 дней (60 мг/кг). Каждое испытуемое соединение суспендировали в растворе 0,5% метилцеллюлозы в дозах, показанных в Таблице 12, и затем вводили орально дважды в день, в объеме 10 мл/кг (8-9 час до полудня и в 3-4 после полудня). Группе контрольного носителя вводили орально раствор 0,5% метилцеллюлозы в объеме 10 мл/кг. Группа, которая не получала 5-FU, определяется как нормальная контрольная группа.
[Сбор проб тонкой кишки]
Мышей держали в голоде в течение 18 часов после введения после полудня на третий день введения ряда соединений. На следующий день мышей убивали цервикальной дислокацией, а затем весь тонкий кишечник удаляли с помощью скальпеля и измеряли влажную массу.
Результаты:
Результат представлен в виде "средней величины ± стандартная ошибка". Соответствующие величины оценивались и сравнивались с помощью теста Tukey и показаны в Таблице 12.
+
валинпирролидид
+
30
5-FU значительно снижал влажную массу тонкого кишечника мыши. Введение или метформина, или ингибитора DPPIV по одному не приводило в результате ни к какому изменению влажной массы тонкого кишечника в группе мышей, обработанных 5-FU. В противоположность этому, комбинированное введение метформина и ингибитора DPPIV приводило в результате к значительному увеличению влажной массы тонкого кишечника. Данное увеличение могло быть вызвано усилением активности GLP-2, являющимся результатом комбинированного использования метформина и ингибитора DPPIV. Это говорит о том, что комбинированное использование метформина и ингибитора DPPIV может использоваться для лечения желудочно-кишечных заболеваний, так как увеличение уровня GLP-2 приводит к подавлению апоптоза и усилению роста эпительальных клеток тонкого кишечника.
Промышленная применимость
Фармацевтические средства, включающие ингибитор DPPIV и бигуанидный агент согласно настоящему изобретению, усиливают действие активного циркулирующего GLP-1 и/или активного циркулирующего GLP-2 и могут использоваться в качестве профилактических и/или терапевтических средств от диабета, ожирения, гиперлипидемии, желудочно-кишечных заболеваний и аналогичных. Кроме того, если фармацевтические средства согласно настоящему изобретению используются в сочетании, соответствующие средства могут вводиться в меньших дозах по сравнению со случаями, когда каждое средство дается индивидуально, что может снижать риск пагубных побочных эффектов бигуанидных агентов (например, симптомов желудочно-кишечной системы, таких как диаррея).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА, ИНГИБИТОР ДИПЕПТИДИЛПЕПТИДАЗЫ IV, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ И ПРИМЕНЕНИЕ НА ИХ ОСНОВЕ | 2003 |
|
RU2297418C9 |
| СОЕДИНЕНИЯ С КОНДЕНСИРОВАННЫМ 1,3-ДИГИДРОИМИДАЗОЛЬНЫМ ЦИКЛОМ | 2003 |
|
RU2328498C9 |
| АМИДОЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КСАНТИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩИМ ДЕЙСТВИЕМ ФОСФОЕНОЛПИРУВАТКАРБОКСИКИНАЗЫ (ФЕПКК), СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2003 |
|
RU2295525C2 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ 637: ПИРИДОПИРИМИДИНДИОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2008 |
|
RU2479584C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2685417C1 |
| НОВЫЕ СОЕДИНЕНИЯ | 2005 |
|
RU2395511C2 |
| СОЕДИНЕНИЯ ТИЕНОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ | 2004 |
|
RU2331648C2 |
| ПРОИЗВОДНЫЕ МОРФОЛИНОПУРИНА, ОБЛАДАЮЩИЕ PI3K И/ИЛИ mTOR ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ | 2009 |
|
RU2490269C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ТРОМБОТИЧЕСКИХ ЗАБОЛЕВАНИЙ | 1995 |
|
RU2156763C2 |
Данное изобретение относится к области фармакологии, а именно к фармацевтическим средствам, содержащим в сочетании ингибитор дипептидилпептидазы IV (DPPIV) и бигуанидный агент. Предложено также применение фармацевтического средства для получения профилактического или терапевтического средства и к способу усиления эффектов активного циркулирующего GLP-1 и/или активного циркулирующего GLP-2. Также изобретение касается способа профилактики или лечения заболевания, связанного с активным циркулирующим GLP-1 и/или активным циркулирующим GLP-2, в частности диабета, ожирения, гиперлипидемии, желудочно-кишечных заболеваний. Сочетание ингибитора DPPIV формулы (I) и бигуанидного агента, а именно фенформина, метформина или буформина, усиливает действие активного циркулирующего глюкагонподобного пептида-1 (GLP-1) и/или активного циркулирующего глюкагонподобного пептида-2 (GLP-2) и, следовательно, подавляет разрушение GLP-1 и GLP-2, когда их уровни увеличиваются бигуанидным агентом. 4 н. и 13 з.п. ф-лы, 12 табл.
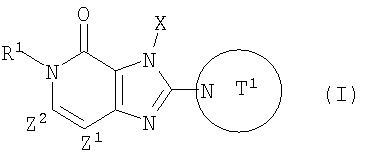
в которой Т1 представляет собой моноциклическую или бициклическую 4-12-членную гетероциклическую группу, содержащую один или два атома азота в кольце, которая может иметь один или несколько заместителей; X представляет собой С1-6алкильную группу, которая может иметь один или несколько заместителей, С2-6 алкенильную группу, которая может иметь один или несколько заместителей, С2-6 алкинильную группу, которая может иметь один или несколько заместителей, С6-10 арильную группу, которая может иметь один или несколько заместителей, 5-10-членную гетероарильную группу, которая может иметь один или несколько заместителей, С6-10арил C1-6алкильную группу, которая может иметь один или несколько заместителей, или 5-10-членную гетероарил С1-6 алкильную группу, которая может иметь один или несколько заместителей;
Z1 и Z2 каждый, независимо, представляют собой атом азота или группу, представленную формулой -CR2=;
R1 и R2 каждый, независимо, представляют собой группу формулы - A0-А1-А2,
в которой А0 представляет собой одинарную связь или С1-6алкиленовую группу, которая может иметь 1-3 заместителя, выбранных из группы В, состоящей из заместителей, описанных ниже;
А1 представляет собой одинарную связь, атом кислорода, атом серы, сульфинильную группу, сульфонильную группу, карбонильную группу, группу, представленную формулой -O-СО-, группу, представленную формулой -СО-O-, группу, представленную формулой -NRA-, группу, представленную формулой -CO-NRA, группу, представленную формулой - NRA-CO, группу, представленную формулой -SO2-NRA-, или группу, представленную формулой -NRA-SO2-;
А2 и RA каждый, независимо, представляют собой атом водорода, атом галогена, циано группу, C1-6алкильную группу, С3-8циклоалкильную группу, С2-6алкенильную группу, С2-6алкинильную группу, С6-10арильную группу, 5-10-членную гетероарильную группу, 4-8-членную гетероциклическую группу, 5-10-членную гетероарил C1-6алкильную группу, С6-10арил С1-6алкильную группу, или С2-7алкилкарбонильную группу;
однако, А2 и RA каждый, независимо, могут иметь 1-3 заместителя, выбранные из заместителей группы В, описанных ниже;
когда Z2 представляет собой группу, представленную формулой - CR2=;
R1 и R2 могут в сочетании образовывать 5-7-членное кольцо; за исключением случаев, когда: [1] R1 представляет собой атом водорода; Z1 представляет собой атом азота; и Z2 представляет собой -СН=; и [2] Z1 представляет собой атом азота; и Z2 представляет собой - С(ОН)=;
<Группы заместителей В>
Группа заместителей В представляет собой группу, состоящую из гидроксильной группы, меркаптогруппы, цианогруппы, нитрогруппы, атома галогена, трифторметильной группы, C1-6алкильной группы, которая может иметь один или несколько заместителей, С3-8циклоалкильной группы, С2-6алкенильной группы, С2-6алкинильной группы, С6-10арильной группы, 5-10-членной гетероарильной группы, 4-8-членной гетероциклической группы, C1-6алкоксигруппы, C1-6алкилтиогруппы, группы, представленной формулой -SO2-NRB1-RB2, группы, представленной формулой NRB1-CO-RB2, группы, представленной формулой -NRB1-RB2, (в которой RB1 и RB2 каждый, независимо, представляет собой атом водорода или C1-6алкильную группу), группу, представленную формулой -CO-RB3, (в которой RB3 представляет собой 4-8-членную гетероциклическую группу), группу, представленную формулой -CO-RB4-RB5, и группу представленную формулой -CH2-CO-RB4-RB5 (в которой RB4 представляет собой одинарную связь, атом кислорода или группу, представленную формулой -NRB6; RB5 и RB6 каждый, независимо, представляют собой атом водорода, C1-6алкильную группу, С3-8циклоалкильную группу, С2-6алкенильную группу, С2-6алкинильную группу, С6-10арильную группу, 5-10-членную гетероарильную группу, 4-8-членную гетероциклическую C1-6алкильную группу, С6-10арил С1-6алкильную группу, или 5-10-членную гетероарил C1-6алкильную группу).
Z2 представляет собой группу, представленную формулой -CR2=
(в которой R2 имеет значения, определенные в п.1).
Z1 представляет собой группу, представленную формулой -CR2=
(в которой R2 имеет значения, определенные в п.1).
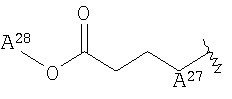
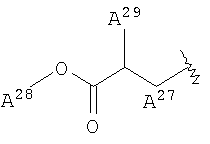
или 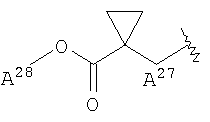
в которой А27 представляет собой атом кислорода, атом серы, или -NH-;
А28 и А29 каждый, независимо, представляет собой атом водорода или С1-6алкильную группу.
(1) 7-(2-бутинил)-2-циано-1-метил-8-(пиперазин-1-ил)-1,7-дигидропурин-6-она;
(2) 3-(2-бутинил)-5-метил-2-(пиперазин-1-ил)-3,5-дигидроимидазо[4,5-d]пиридазин-4-она;
(3) 2-(3-аминопиперидин-1-ил)-3-(2-бутинил)-5-метил-3,5-дигидроимидазо[4,5-d]пиридазин-4-она;
(4) 2-[7-(2-бутинил)-1-метил-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-илокси]бензамида;
(5) 7-(2-бутинил)-1-(2-цианбензил)-6-оксо-8-(пиперазин-1-ил)-6,7-дигидро-1Н-пурин-2-карбонитрила; и
(6) 2-[3-(2-бутинил)-4-оксо-2-(пиперазин-1-ил)-3,4-дигидроимидазо[4,5-d]пиридазин-5-илметил]бензонитрила,
или их фармацевтически приемлемые соли, или гидрат.
| Способ регулирования режима работы конденсатора | 1982 |
|
SU1052825A1 |
| WO 02002560 А2, 10.01 | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| ЭТАНОЛАММОНИЕВАЯ СОЛЬ ПОЛИ-5-ВИНИЛТЕТРАЗОЛА В КАЧЕСТВЕ ПЛЕНКООБРАЗУЮЩЕГО КОМПОНЕНТА ДЛЯ КОСМЕТИЧЕСКИХ ПРЕПАРАТОВ | 1992 |
|
RU2068420C1 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Устройство для формирования потока сыпучей массы | 1983 |
|
SU1097808A1 |
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| HINKE S.A | |||
| et al | |||
| On combination therapy of diabetes with metformin and dipeptidyl peptidase IV inhibitors, Diabetes Care | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| DRUCKER D.J | |||
| Biological actions | |||

