Настоящее изобретение относится к терапевтически активным соединениям, которые являются производными ксантина, способам получения указанных производных, фармацевтическим композициям, содержащим эти активные соединения, и применению этих соединений в терапии, особенно для лечения заболеваний, где недостаточная активация рецептора HM74A способствует развитию заболевания или где активация рецептора будет иметь полезный эффект.
Дислипидемия - общий термин, используемый для описания лиц с аберрантными липопротеиновыми профилями. Клинически, основными классами соединений, используемых для лечения пациентов с дислипидемией, имеющих поэтому риск сердечно-сосудистого заболевания, являются статины, фибраты, связывающие желчные кислоты смолы и никотиновая кислота. Никотиновая кислота (Ниацин, витамин B) использовалась клинически в течение более 40 лет у больных с различными формами дислипидемии. Первичный способ действия никотиновой кислоты осуществляется через ингибирование чувствительной к гормонам триглицеридлипазы (HSL), которое приводит к понижению плазменных уровней неэтерифицированных жирных кислот (NEFA), что, в свою очередь, изменяет жировой обмен в печени таким образом, что снижается продукция ЛПНП и ЛПОНП (липопротеинов низкой и очень низкой плотности). Уменьшенные уровни ЛПОНП, как полагают, понижают активность белка переноса сложного эфира холестерина (CETP), что приводит к увеличенному уровню ЛПВП (липопротеин высокой плотности), что может быть причиной наблюдаемых улучшений состояния сердечно-сосудистой системы. Таким образом, никотиновая кислота осуществляет очень желательное изменение липопротеиновых профилей; снижение уровней ЛПОНП и ЛПНП, увеличивая ЛПВП. Также было показано, что никотиновая кислота оказывает благоприятное действие на изменение течения заболеваний, уменьшая прогрессию и увеличивая регрессию атеросклеротических поражений и уменьшая количество случаев сердечно-сосудистых заболеваний в нескольких испытаниях.
Наблюдаемое ингибирование HSL обработкой никотиновой кислотой опосредовано уменьшением в клетках циклического аденозинмонофосфата (цАМФ), вызванным опосредованным G-белком ингибированием аденилилциклазы. Недавно связанные с G-белком рецепторы HM74 и HM74A были идентифицированы как рецепторы никотиновой кислоты (заявка PCT WO02/84298; Wise et.al. J Biol Chem., 2003, 278(11), 9869-9874). Последовательность ДНК человеческого HM74A может быть найдена в Genbank; номер доступа AY148884. Две другие публикации поддерживают это открытие (Tunaru et.al. Nature Medicine, 2003, 9(3), 352-255 and Soga et.al. Biochem Biophys Res Commun., 2003, 303(1) 364-369), однако номенклатура немного отличается. В публикации Tunaru то, что они называют человеческим HM74, фактически является HM74A, а в публикации Soga HM74b идентично HM74A. Клетки, трансфицированные таким образом, чтобы экспрессировать HM74A и/или HM74, приобретают способность вызывать Gi G-белок-опосредованный ответ при воздействии никотиновой кислоты. У мышей, у которых отсутствует гомолог HM74A (m-PUMA-G), никотиновая кислота не в состоянии уменьшить плазменные уровни NEFA.
Некоторые производные ксантина были синтезированы и раскрыты в предшествующем уровне техники. Например, в EP0389282 раскрыты производные ксантина как потенциальные медиаторы цереброваскулярных нарушений. Ряд производных ксантина был идентифицирован в качестве антагонистов рецептора аденозина Jacobson et. al., J. Med. Chem., 1993, 36, 2639-2644.
Авторы изобретения представляют группу производных ксантина, которые являются селективными агонистами рецептора никотиновой кислоты HM74A и являются, таким образом, пригодными для лечения, профилактики и супрессии заболеваний, где пониженная активация этого рецептора способствует развитию заболевания или где активация рецептора будет иметь полезный эффект.
Краткое описание изобретения
Настоящее изобретение относится к терапевтически активным производным ксантина и применению этих производных в терапии, особенно для лечения заболеваний, где пониженная активация этого рецептора HM74A способствует развитию заболевания или где активация рецептора будет иметь полезный эффект, в частности заболеваний, связанных с метаболизмом липидов, включая дислипидемию или гиперлипопротеинемию, такую как диабетическая дислипидемия и смешанная дислипидемия, сердечную недостаточность, гиперхолестеринемию, сердечно-сосудистые заболевания, включая атеросклероз, артериосклероз и гипертриглицеридемию. Как таковые соединения могут также быть использованы как терапевтические средства для лечения заболевания коронарных артерий, тромбоза, стенокардии, хронической почечной недостаточности, периферического сосудистого заболевания и инсульта, а также сердечно-сосудистых показаний, связанных с сахарным диабетом типа II, диабета типа I, резистентности к инсулину, гиперлипидемии, нервной анорексии, ожирения. Соединения могут также быть полезны для лечения воспалительных заболеваний или состояний, как изложено далее ниже.
Промежуточные соединения, композиции, методы и способы, описанные здесь, составляют дальнейшие объекты изобретения.
Подробное описание изобретения
Согласно одному аспекту изобретение относится к соединению Формулы (I)
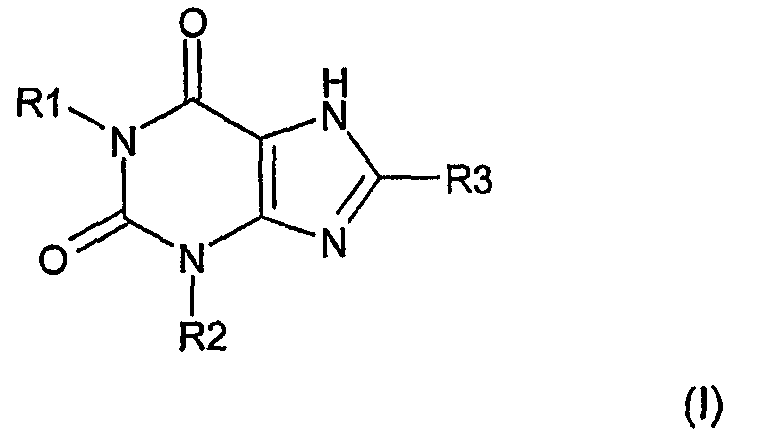
и его физиологически функциональному производному, в которой
R1 выбирают из водорода и C1-4 алкила, который может быть необязательно замещен одной или более группами, выбранными из CN и CF3;
R2 выбирают из незамещенного C3-10 алкила, C1-10 алкила, замещенного одной или более группами, выбранными из фтора и CN, C5 алкенила, неразветвленного C4 алкенила и C1-4 алкила, замещенного циклоалкилом;
и R3 выбирают из галогена и CN;
при условии, что:
(i) когда R3 обозначает Cl и R1 обозначает этил, тогда R2 отличен от пропила;
(ii) когда R3 обозначает бром и R1 обозначает пропил, тогда R2 отличен от пропила;
(iii) когда R3 обозначает Cl или бром и R1 обозначает бутил, тогда R2 отличен от бутила; и
(iv) когда R1 обозначает C1-4 алкил, CH2CN или (CH2)3CF3, тогда R2 отличен от разветвленного алкила.
Соединения полезны для лечения заболеваний, где недостаточная активация рецептора HM74A способствует развитию заболевания или где активация рецептора будет иметь полезный эффект, в частности заболеваний, связанных с метаболизмом липидов, включая дислипидемию или гиперлипопротеинемию, такую как диабетическая дислипидемия и смешанная дислипидемия, сердечную недостаточность, гиперхолестеринемию, сердечно-сосудистое заболевание, включая атеросклероз, артериосклероз и гипертриглицеридемию. Как таковые соединения могут также быть использованы как терапевтические средства для лечения заболевания коронарных артерий, тромбоза, стенокардии, хронической почечной недостаточности, периферического сосудистого заболевания и инсульта, а также сердечно-сосудистых показаний, связанных с сахарным диабетом типа II, диабета типа I, резистентности к инсулину, гиперлипидемии, нервной анорексии, ожирения. Также соединения согласно настоящему изобретению могут найти применение как агонисты или частичные агонисты HM74A (модуляторы HM74A).
В особых вариантах осуществления R1 выбирают из водорода, С1-4 алкила, CH2CN и (CH2)3CF3; в более частных случаях осуществления R1 выбирают из водорода и метила.
В некоторых вариантах осуществления R2 выбирают из незамещенного С3-10 алкила, C1-6 алкила, замещенного одним или более CN, C1-10 алкила, замещенного одним или более фтором, C5 алкенила, неразветвленного C4 алкенила и С1-4 алкила, замещенного циклоалкилом. В частности, R2 выбирают из незамещенного C3-10 алкила; (CH2)1-5CN; C2-5 алкила, замещенного одним или более фтором; C5 алкенила; и С1-4 алкила, замещенного циклоалкилом. Более предпочтительно R2 выбирают из незамещенного C4-6 н-алкила, например пентила; (CH2)1-3CN, например (CH2)CN или (CH2)3CN; C3-4 алкила, замещенного одним или более фтором, в частности, где концевой атом углерода полностью насыщен фтором, например (CH2)2-3CF3; и C5 алкенила, в частности, где имеется только одна двойная связь, например, где двойная связь расположена между четвертым и пятым атомами углерода (терминальный алкенил).
В предпочтительных вариантах осуществления R3 обозначает галоген. Более предпочтительно R3 выбирают из хлора и брома. Наиболее предпочтительно R3 обозначает хлор.
Следует понимать, что настоящее изобретение включает любую комбинацию отдельных вариантов осуществления и охватывает все комбинации частных случаев заместителей, описанных выше.
Конкретные соединения согласно настоящему изобретению включают:
(8-хлор-2,6-диоксо-1,2,6,7-тетрагидро-3H-пурин-3-ил)ацетонитрил,
3-бутил-8-хлор-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-1-метил-3-пентил-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион,
8-бром-1-метил-3-пентил-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(3,3,3-трифторпропил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-1-пропил-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-дион,
3-бутил-8-хлор-1-метил-3,7-дигидро-1H-пурин-2,6-дион,
(3-бутил-8-хлор-2,6-диоксо-2,3,6,7-тетрагидро-1H-пурин-1-ил)ацетонитрил,
8-хлор-3-(2-циклопропилэтил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-1,3-бис(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион,
4-(8-хлор-1-метил-2,6-диоксо-1,2,6,7-тетрагидро-3H-пурин-3-ил)бутаннитрил,
8-хлор-1-этил-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-дион,
1-метил-2,6-диоксо-3-пентил-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил,
8-хлор-3-пропил-1-метил-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(3-метилбутил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-пентил-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-пропил-3,7-дигидро-1H-пурин-2,6-дион,
3-бутил-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил,
8-хлор-3-(4-пентен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-гексил-3,7-дигидро-1H-пурин-2,6-дион,
4-(8-хлор-2,6-диоксо-1,2,6,7-тетрагидро-3H-пурин-3-ил)бутаннитрил,
8-хлор-3-гексил-1-метил-3,7-дигидро-1H-пурин-2,6-дион,
3-бутил-8-хлор-1-этил-3,7-дигидро-1H-пурин-2,6-дион,
[8-хлор-3-(2-циклопропилэтил)-2,6-диоксо-2,3,6,7-тетрагидро-1H-пурин-1-ил]ацетонитрил,
(8-хлор-2,6-диоксо-3-пропил-2,3,6,7-тетрагидро-1H-пурин-1-ил)ацетонитрил,
8-хлор-1-(4,4,4-трифторбутил)-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-дион,
2,2'-(8-хлор-2,6-диоксо-6,7-дигидро-1H-пурин-1,3(2H)-диил)диацетонитрил,
8-хлор-1-метил-3-(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(2-циклогексилэтил)-3,7-дигидро-1H-пурин-2,6-дион,
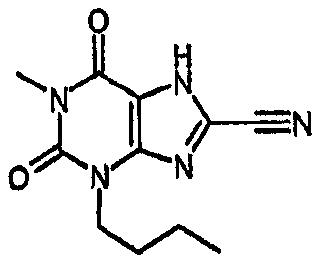
1,3-дибутил-2,6-диоксо-2,3,6,7-тетрагидро-1Н-пурин-8-карбонитрил,
1,3-дибутил-8-йод-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(4-метилпентил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(6-метилпентил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-октил-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-децил-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(циклогексилметил)-3,7-дигидро-1H-пурин-2,6-дион,
(+/-)-8-хлор-3-(3-метилпентил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(2-циклопентилэтил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(циклопропилметил)-3,7-дигидро-1H-пурин-2,6-дион,
(+/-)-8-хлор-3-(2-метилбутил)-3,7-дигидро-1H-пурин-2,6-дион,
(+/-)-8-хлор-3-(2-метилпентил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(циклобутилметил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(циклопентилметил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(3-циклопропилпропил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(2-циклобутилэтил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(4-фторбутил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(3-фторпропил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(5-фторпентил)-3,7-дигидро-1H-пурин-2,6-дион,
4-(8-хлор-1-метил-2,6-диоксо-1,2,6,7-тетрагидро-3Н-пурин-3-ил)бутаннитрил,
3-(3-бутен-1-ил)-8-хлор-3,7-дигидро-1Н-пурин-2,6-дион,
6-(8-хлор-2,6-диоксо-1,2,6,7-тетрагидро-3Н-пурин-3-ил)-2,2-диметилгексаннитрил,
8-хлор-3-(6-фторгексил)-3,7-дигидро-1Н-пурин-2,6-дион.
В настоящем описании и в формуле изобретения слова «содержит» и «включает», а также их варианты, такие как «содержат», «содержащий», «включают» и «включающий», должны пониматься включительно. То есть следует понимать, что могут быть включены другие элементы или составляющие, не упомянутые особо, если это позволяет контекст.
В настоящем описании термин «галоген» относится к фтору, хлору, брому и йоду.
В настоящем описании термин «алкил» (в значении группы или части группы) относится к прямой или разветвленной углеводородной цепи, если не указано иное, содержащей указанное число атомов углерода. Например, С3-С10алкил означает прямую или разветвленную углеводородную цепь, содержащую по меньшей мере 3 и не более 10 атомов углерода. Примеры используемых алкилов включают, но не ограничены ими, метил (Ме), этил (Et), н-пропил и изопропил. Термин «н-алкил» относится к неразветвленной углеводородной цепи.
В настоящем описании термин «циклоалкил» относится к углеводородному кольцу, содержащему от 3 до 6 атомов углерода, не содержащему гетероатомов или сопряженных двойных связей. Примеры используемых циклоалкилов включают, но не ограничены ими, циклопропил и циклогексил.
В настоящем описании термин «алкенил» относится к прямой или разветвленной углеводородной цепи, содержащей указанное число атомов углерода, которая содержит одну или более двойных связей.
В настоящем описании, когда указано, что группа является «замещенной» другой группой или имеющей «одно или более замещений», следует понимать, что замещение может иметь место в любом положении в этой группе.
В настоящем описании термин «физиологически функциональное производное» относится к любому фармацевтически приемлемому производному соединения согласно настоящему изобретению, например его амиду, и включает любую фармацевтически приемлемую соль соединения формулы (I) и любой фармацевтически приемлемый сольват соединения формулы (I), который при введении млекопитающему, такому как человек, способен приводить (прямо или косвенно) к соединению формулы (I) или его активному метаболиту или остатку. Специалисту будет понятно, что соединения формулы (I) могут быть модифицированы, чтобы получить их физиологически функциональные производные в любой из функциональных групп в соединениях, и что соединения формулы (I) могут быть, таким образом, модифицированы в более чем одном положении.
В настоящем описании термин «фармацевтически приемлемый», используемый относительно ингредиента (активного ингредиента или эксципиента), который может быть включен в фармацевтическую композицию для введения пациенту, относится к ингредиенту, являющемуся приемлемым в отношении совместимости с любым другим ингредиентом, присутствующим в фармацевтической композиции, и в отношении отсутствия вреда для реципиента.
В настоящем описании термин «сольват» относится к комплексному соединению переменной стехиометрии, образованному растворенным веществом (в настоящем изобретении соединением формулы (I), его солью или физиологически функциональным производным) и растворителем. Такие растворители для целей настоящего изобретения не должны оказывать влияния на биологическую активность растворенного вещества. Используемый растворитель может быть фармацевтически приемлемым растворителем. Примеры подходящих фармацевтически приемлемых растворителей включают воду, этанол и уксусную кислоту. Примером растворителя, который может использоваться, является вода, в случае которой сольват может быть назван гидратом рассматриваемого растворенного вещества.
Следует понимать, что для фармацевтического использования «соль или сольват», упомянутые выше, будут фармацевтически приемлемой солью или сольватом. Однако другие соли или сольваты могут найти применение, например, для получения соединения формулы (I) или для получения его фармацевтически приемлемой соли или сольвата.
Фармацевтически приемлемые соли включают описанные Berge, Bighley and Monkhouse, J. Pharm. Sci., 1977, 66, 1-19. Подходящие фармацевтически приемлемые соли включают соли щелочного металла, образованные присоединением оснований щелочного металла, таких как гидроксиды щелочного металла. Примерами подходящих солей щелочного металла является соль натрия или соль калия. Другие подходящие фармацевтически приемлемые соли включают соли щелочноземельных металлов, такие как соль кальция или соль магния, соли аммония; или соли с органическими основаниями, такими как этаноламин, триэтаноламин, этилендиамин, триэтиламин, холин и меглумин; или соли с аминокислотами, такими как аргинин, лизин и гистидин.
Соединения формулы (I) имеют потенциальную терапевтическую полезность для лечения и облегчения симптомов многих заболеваний, связанных с метаболизмом липидов, включая дислипидемию или гиперлипопротеинемию, такую как диабетическая дислипидемия и смешанная дислипидемия, сердечную недостаточность, гиперхолестеринемию, сердечно-сосудистое заболевание, включая атеросклероз, артериосклероз и гипертриглицеридемию, сахарный диабет типа II, диабет типа I, резистентность к инсулину, гиперлипидемию, нервную анорексию, ожирение. Как таковые соединения могут также использоваться как терапевтические средства для лечения заболеваний коронарных артерий, тромбоза, стенокардии, хронической почечной недостаточности, периферического сосудистого заболевания и инсульта.
Кроме того, также предполагается, что рецепторы HM74 и HM74A вовлечены в воспаление. Воспаление обозначает группу сосудистых, клеточных и неврологических реакций на травму. Воспаление может быть охарактеризовано как движение воспалительных клеток, таких как моноциты, нейтрофилы и гранулоциты, в ткани. Это обычно ассоциируется со сниженной барьерной функцией эндотелия и отеком в ткани. Воспаление относительно заболевания обычно упоминается как хроническое воспаление и может длиться всю жизнь. Такое хроническое воспаление может проявляться непосредственно через симптомы заболевания. Цель противовоспалительной терапии состоит в том, чтобы уменьшить это хроническое воспаление и обеспечить физиологический процесс лечения и восстановления ткани.
Примеры воспалительных заболеваний или состояний, для которых соединения согласно настоящему изобретению могут демонстрировать пригодность, включают воспалительные заболевания или состояния сустава, особенно артрит (например, ревматоидный артрит, остеоартрит, отторжение суставного протеза), или желудочно-кишечного тракта (например, неспецифический язвенный колит, Болезнь Крона и другие воспалительные кишечные и желудочно-кишечные заболевания, гастрит и воспаления слизистой оболочки, возникающие в результате инфекции, энтеропатия, вызванная нестероидными противовоспалительными препаратами), легкого (например, респираторный дистресс-синдром взрослых, астма, муковисцидоз или хроническое обструктивное легочное заболевание), сердца (например, миокардит), нервной ткани (например, рассеянный склероз), поджелудочной железы (например, воспаление, связанное с сахарным диабетом и его осложнениями), почки (например, гломерулонефрит), кожи (например, дерматит, псориаз, экзема, крапивница, ожоговое повреждение), глаза (например, глаукома), а также пересаженных органов (например, отторжение) и заболевания многих органов (например, системная красная волчанка, сепсис) и воспалительное осложнение вирусных или бактериальных инфекций и воспалительных состояний, связанных с атеросклерозом и после гипоксических или ишемических инсультов (с реперфузией или без), например, в мозге или при ишемической болезни сердца.
В частности, соединения по изобретению полезны для лечения и профилактики воспаления, диабета и сердечно-сосудистых заболеваний или состояний, включая атеросклероз, артериосклероз, гипертриглицеридемию и смешанную дислипидемию.
Никотиновая кислота имеет значительный профиль побочных эффектов, возможно потому, что ее вводят в больших количествах (количества граммов в сутки). Самым общим побочным эффектом является прилив крови к коже. В определенных вариантах осуществления настоящего изобретения соединения могут проявлять уменьшенные побочные эффекты по сравнению с никотиновой кислотой. HM74A был идентифицирован как рецептор с высоким сродством к никотиновой кислоте, в то время как HM74 является рецептором с более низким сродством. Соединения согласно настоящему изобретению могут найти применение в качестве селективных агонистов или частичных агонистов HM74A; в этом случае они проявляют большее сродство к HM74A, чем к HM74.
Способность соединений формулы (I) к активизации HM74A можно показать, например, используя следующий фермент и цельноклеточные тесты in vitro:
Тестирование in vitro
Для переходных трансфекций клетки HEK293T (клетки HEK293, стабильно экспрессирующие большой T-антиген SV40) выдерживали в DMEM, содержащей 10% эмбриональной телячьей сыворотки и 2 мМ глутамина. Клетки высевали в 90 мм культуральные чашки и выращивали до слияния на 60-80% (18-24 ч) до трансфекции. Человеческий HM74A (номер доступа GenBank AY148884) субклонировали в вектор экспрессии у млекопитающих (pcDNA3; Invitrogen) и трансфицировали с использованием реагента Lipofectamine. Для трансфекции 9 мкг ДНК смешивали с 30 мкл Lipofectamine в 0,6 мл Opti-MEM (Life Technologies Inc.) и инкубировали при комнатной температуре в течение 30 мин, после чего добавляли 1,6 мл Opti-MEM. Клетки подвергали воздействию смеси Lipofectamine/ДНК в течение 5 ч и добавляли 6 мл 20% (об./об.) эмбриональной телячьей сыворотки в DMEM. Клетки собирали через 48 ч после трансфекции. Обработку токсином коклюша выполняли дополнением в среду в количестве 50 нгмл-1 за 16 ч. Все исследования переходной трансфекции включали ко-трансфекцию рецептора вместе с Gi/o G белком, Go1α.
Для генерации стабильных линий клеток вышеупомянутый способ использовали для трансфекции клеток CHO-K1, высеянных в шестилуночные чашки и выращенных до 30% слияния. Эти клетки выдерживали в среде DMEM F-12 НАМ, содержащей 10% эмбриональной телячьей сыворотки и 2мМ глутамина. Через 48 ч после трансфекции среду дополняли 400 мкг/мл Geneticin (G418, Gibco) для отбора клеток, резистентных к антибиотику. Клональные линии клеток CHO-K1, стабильно экспрессирующие HM74A, подтверждали измерениями связывания [35S]-GTPγS после добавления никотиновой кислоты.
Получение мембраны P2 - Фракции частиц Р2, содержащие плазматическую мембрану, получали из клеточных паст, замороженных при -80°C после сбора. Все процедуры выполняли при 4°C. Клубок клеток ресуспендировали в 1 мл 10 мМ Nhis-HCl и 0,1мМ ЭДТА, рН 7,5 (буфер A), и гомогенизацией в течение 20 с с Ultra Turrax, с последующим проходом (5 раз) через иглу калибра 25. Клеточный лизат центрифугировали при 1000 g в течение 10 минут в микроцентрифуге для осаждения ядер и неповрежденных клеток, и фракции частиц P2 получали микроцентрифугированием при 16000 g в течение 30 мин. Фракции частиц P2 ресуспендировали в буфере A и сохраняли при -80°C до востребования.
Тесты связывания [ 35 S]-GTPγS проводили при комнатной температуре в формате с 384 лунками, основываясь на способах, описанных ранее (Wieland, T. and Jakobs, K. H. (1994) Methods Enzymol. 237, 3-13). Кратко, стандарт или тестируемые соединения разбавляли и добавляли в планшет с 384 лунками в объеме 10 мкл. Мембраны (HM74A или HM74) разбавляли в тестовом буфере (20мМ HEPES, 100мМ NaCl, 10мМ MgCl2, pH 7,4), дополненном сапонином (60 мкг/мл), шарики Leadseeker WGA (Amersham; 250 мкг/лунку) и 10 мкМ GDP, так чтобы в объеме 20 мкл, добавленном к каждой лунке, содержалось 5 мкг мембран. [35S]-GTPγS (1170 Ci/ммоль, Amersham) разбавляли (1:1500) в тестовом буфере и 20 мкл добавляли в каждую лунку. После добавления меченого лиганда планшеты запечатывали, обрабатывали импульсами и инкубировали в течение 4 часов при комнатной температуре. В конце инкубационного периода планшеты считывали на аппарате Leadseeker (VIEWLUX PLUS; Perkin-Elmer), чтобы определить уровни специфического связывания.
Тестирование in vivo
Агонисты HM74A проверяли на самцах крыс (200-250 г) Spague-Dawley, которых держали голодными в течение по меньшей мере 12 часов до исследования. Соединения вводили внутривенно (5 мл/кг) или пероральным скармливанием (10 мл/кг). Пробы крови (0,3 мл забор из вены хвоста) брали до введения и в три момента времени после введения (время в пределах от 15 минут до 8 часов после введения). Каждую пробу крови помещали в пробирки с гепарином (Becton Dickinson Microtainer, PST LH) и центрифугировали (10000 g в течение 5 минут) для получения образца плазмы. Образцы плазмы анализировали в отношении уровней неэтерифицированных жирных кислот (NEFA) с использованием коммерчески доступного набора (Randox). Ингибирование плазменных уровней NEFA относительно уровней перед введением соединений использовалось как заместитель для активности агониста HM74A.
Чтобы определить, показали ли соединения HM74A реакцию прилива крови, связанную с никотиновой кислотой, их вводили анестезированным морским свинкам. Самцов морских свинок Dunkin Hartley (300-800 г) держали голодными в течение 12 часов до анестезии смесью кетамина гидрохлорида (Vetalar, 40 мг/кг внутримышечно), ксилазина (Rompun, 8 мг/кг внутримышечно) и натрия пентобарбитона (Sagatal, 30 мг/кг внутрибрюшинно). После анестезии проводили трахеотомию и животных механически вентилировали комнатным воздухом (10-12 мл/кг, 60 вдохов/минуту). Яремную вену и сонную артерию канюлировали для внутривенного введения тестируемого соединения и отбора крови соответственно. Инфракрасный температурный зонд (Extech Instruments) помещали на 3-5 мм от края левого уха. Температуру регистрировали каждую минуту, начиная с 5 минут до введения тестируемого соединения и до 40 минут после введения тестируемого соединения. Данные автоматически собирали на компьютере Psion и затем переводили для анализа данных в крупноформатную таблицу Excel. До и в частые моменты времени после введения соединения пробы крови (0,3 мл) отбирали через каротидную артериальную канюлю и помещали в пробирки Microtainer (BD), содержащие гепарин-литий. Образцы тщательно перемешивали на барабанной мешалке для крови и затем сохраняли на льду до центрифугирования при 1200 г в течение 5 минут.
Никотиновая кислота (10 мг/кг внутривенно) приводит в среднем (± среднее стандартное отклонение) к повышению ушного температурного эквивалента до 10,42 + 1,44 (область под кривой; произвольные единицы; n=6). Для сравнения, соединение примера 30 (10 мг/кг внутривенно) приводит в среднем (± среднее стандартное отклонение) к повышению ушного температурного эквивалента до 1,52 + 0,39 (область под кривой; произвольные единицы; n=6), сокращение 85%.
Соединения согласно формуле (I) синтезировали (см. далее примеры синтеза) и проверяли в одном или более тестов, обсужденных выше. Все проиллюстрированные в примерах соединения имеют pEC50 4,9 (+/- 0,3 лог. единиц) или более и эффективность 30% или более. Некоторые конкретные соединения иллюстрируются ниже.
Общие способы очистки и анализа
Масс-спектры (МС) регистрировали на масс-спектрометре Fisons VG Platform с использованием способов положительной ионизации электрораспылением [(ES+ve с получением молекулярных ионов MH+ и М(NH4)+] или отрицательной ионизации электрораспылением [(ES-ve с получением молекулярного иона (М-Н)-].
Спектры 1Н ЯМР регистрировали, используя спектрометр Bruker DPX 400 МГц, используя тетраметилсилан в качестве внешнего стандарта.
Хроматография BiotageTM относится к очистке, выполняемой с использованием оборудования, выпускаемого Dyax Corporation (Flash 40i или Flash 150i), и картриджей, заполненных KPSil.
Масс-направленная автопрепаративная хроматография относится к способам, где материал очищали высокоэффективной жидкостной хроматографией на колонке HPLCABZ+ 5 мкм (5 см × 10 мм i.d.) с 0,1% HCO2H в воде и 95% MeCN, 5% воды (0,5% HCO2H) с использованием следующих условий градиентного элюирования: 0-1,0 минута 5% B, 1,0-8,0 минут 5→30% B, 8,0-8,9 минут 30% B, 8,9-9,0 минут 30→95% B, 9,0-9,9 минут 95% B, 9,9-10 минут 95→0% B при скорости потока 8 мл.мин-1 (система 2). Коллектор фракций Gilson 202 приводили в действие масс-спектрометром VG Platform при обнаружении заданной массы.
Препаративная ВЭЖХ относится к способам, в которых материал очищали высокоэффективной жидкостной хроматографией на колонке HPLCABZ+ 5 мкм (10 см × 21,2 мм i.d.) с 0,1% HCO2H в воде (A) и MeCN (0,5% HCO2H) (B), используя настройки условий градиентного элюирования, выраженные как градиент «от x до y» со следующей системой градиента: 0-1,45 минут x% B, 1,45-20 минут x→y% B, 20-24 минуты y→95% B, 24-30 минут 95% B, 32-34 минуты 95→x% B при скорости потока 8 мл.мин-1. Коллектор фракций Gilson 233 приводили в действие УФ (254 нм).
SPE (твердофазная экстракция) относится к использованию картриджей, выпускаемых International Sorbent Technology Ltd.
Strata Phenyl SPE относится к использованию картриджей, выпускаемых Phenomenex. Соединение загружали в картридж, предварительно обработанный MeCN и уравновешенный 5% MeCN в воде. Соединение элюировали 0,1% HCO2H в воде и MeCN (0,5% HCO2H) в подходящем градиенте на Combiflash Optix 10.
Как указано выше, соединения формулы (I) могут найти применение в медицине или ветеринарии, в частности, в качестве активаторов HM74A, для контроля дислипидемии и гиперлипопротеинемии.
Таким образом, следующий объект настоящего изобретения относится к соединению формулы (I) или его физиологически функциональному производному для применения в медицине или ветеринарии, особенно для лечения нарушений, связанных с метаболизмом липидов, включая дислипидемию или гиперлипопротеинемию, такую как диабетическая дислипидемия и смешанная дислипидемия, сердечную недостаточность, гиперхолестеринемию, сердечно-сосудистое заболевание, включая атеросклероз, артериосклероз и гипертриглицеридемию, сахарный диабет типа II, диабет типа I, резистентность к инсулину, гиперлипидемию, нервную анорексию, ожирение. Как таковые соединения также могут быть использованы для лечения заболевания коронарных артерий, тромбоза, стенокардии, хронической почечной недостаточности, периферического сосудистого заболевания и инсульта.
Следующий объект настоящего изобретения относится к соединению формулы (I) или его физиологически функциональному производному для применения для получения лекарственного средства для лечения нарушений, связанных с метаболизмом липидов, включая дислипидемию или гиперлипопротеинемию, такую как диабетическая дислипидемия и смешанная дислипидемия, сердечной недостаточности, гиперхолестеринемии, сердечно-сосудистого заболевания, включая атеросклероз, артериосклероз и гипертриглицеридемию, сахарного диабета типа II, диабета типа I, резистентности к инсулину, гиперлипидемии, нервной анорексии, ожирения. Как таковые соединения также могут быть использованы для лечения заболевания коронарных артерий, тромбоза, стенокардии, хронической почечной недостаточности, периферического сосудистого заболевания и инсульта.
Следует понимать, что указания на лечение распространяются также на профилактику, профилактику рецидива и подавление симптомов, а также лечение установленных состояний.
Согласно другому аспекту изобретение относится к применению соединения формулы (II)
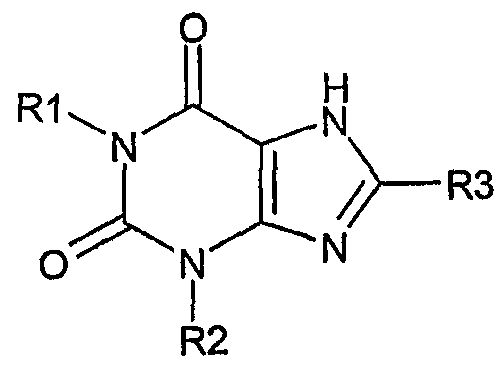
и его физиологически функционального производного, в которой:
R1 выбирают из водорода и C1-4 алкила, который может быть необязательно замещен одной или более группами, выбранными из CN и CF3;
R2 выбирают из незамещенного C2-10 алкила, C1-10 алкила, замещенного одной или более группами, выбранными из фтора и CN, C5 алкенила, неразветвленного C4 алкенила и C1-4 алкила, замещенного циклоалкилом;
и R3 выбирают из галогена и CN;
для получения лекарственного средства для лечения нарушений, связанных с метаболизмом липидов, включая дислипидемию или гиперлипопротеинемию. В частности, изобретение относится к применению соединения формулы (II) для получения лекарственного средства для лечения диабетической дислипидемии или смешанной дислипидемии, сердечной недостаточности, гиперхолестеринемии, сахарного диабета типа II, диабета типа I, резистентности к инсулину, гиперлипидемии, нервной анорексии, ожирения, заболевания коронарных артерий, тромбоза, стенокардии, хронической почечной недостаточности, инсульта и сердечно-сосудистого заболевания, включая атеросклероз, артериосклероз и гипертриглицеридемию.
В одном варианте осуществления изобретение относится к соединению формулы (II) для применения для лечения нарушений, связанных с метаболизмом липидов, включая дислипидемию или гиперлипопротеинемию. В частности, изобретение относится к применению соединения формулы (II) для получения лекарственного средства для лечения диабетической дислипидемии или смешанной дислипидемии, сердечной недостаточности, гиперхолестеринемии, сахарного диабета типа II, диабета типа I, резистентности к инсулину, гиперлипидемии, нервной анорексии, ожирения, заболевания коронарных артерий, тромбоза, стенокардии, хронической почечной недостаточности, инсульта и сердечно-сосудистого заболевания, включая атеросклероз, артериосклероз и гипертриглицеридемию.
В частных вариантах осуществления R1 выбирают из водорода, C1-4 алкила, CH2CN и (CH2)3CF3. В более частных вариантах осуществления R1 выбирают из водорода и метила.
В определенных вариантах осуществления R2 выбирают из незамещенного C3-10 алкила, C1-10 алкила, замещенного одной или более группами, выбранными из фтора и CN, C5 алкенила, неразветвленного C4 алкенила и C1-4 алкила, замещенного циклоалкилом. В частности, R2 выбирают из незамещенного C3-10 алкила, C1-6 алкила, замещенного одним или более CN, C1-10 алкила, замещенного одним или более фтором, C5 алкенила, неразветвленного C4 алкенила и С1-4 алкила, замещенного циклоалкилом. Более предпочтительно R2 выбирают из незамещенного C3-10 алкила; (CH2)1-5CN; C2-5 алкила, замещенного одним или более фтором; C5 алкенила; и С1-4 алкила, замещенного циклоалкилом. Наиболее предпочтительно R2 выбирают из незамещенного C4-6 н-алкила, например пентила; (CH2)1-3CN, например (CH2)CN или (CH2)3CN; C3-4 алкила, замещенного одним или более фтором, в частности, где концевой атом углерода полностью насыщен фтором, например (CH2)2-3CF3; и C5 алкенила, в частности, где имеется только одна двойная связь, например где двойная связь расположена между четвертым и пятым атомами углерода (терминальный алкенил).
В особых вариантах осуществления R3 обозначает галоген. Более предпочтительно R3 выбирают из хлора и брома. Наиболее предпочтительно R3 обозначает хлор.
Конкретные соединения для использования для лечения или для получения лекарственного средства для лечения заболеваний метаболизма липидов, включая дислипидемию или гиперлипопротеинемию, включают:
(8-хлор-2,6-диоксо-1,2,6,7-тетрагидро-3H-пурин-3-ил)ацетонитрил,
3-бутил-8-хлор-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-1-метил-3-пентил-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион,
8-бром-1-метил-3-пентил-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(3,3,3-трифторпропил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-1-пропил-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-дион,
3-бутил-8-хлор-1-метил-3,7-дигидро-1H-пурин-2,6-дион,
(3-бутил-8-хлор-2,6-диоксо-2,3,6,7-тетрагидро-1H-пурин-1-ил)ацетонитрил,
8-хлор-3-(2-циклопропилэтил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-1,3-бис(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион,
4-(8-хлор-1-метил-2,6-диоксо-1,2,6,7-тетрагидро-3H-пурин-3-ил)бутаннитрил,
8-хлор-1-этил-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-дион,
1-метил-2,6-диоксо-3-пентил-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил,
8-хлор-3-пропил-1-метил-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(3-метилбутил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-пентил-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-пропил-3,7-дигидро-1H-пурин-2,6-дион,
3-бутил-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил,
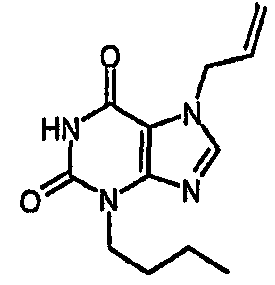
8-хлор-3-(4-пентен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-гексил-3,7-дигидро-1H-пурин-2,6-дион,
4-(8-хлор-2,6-диоксо-1,2,6,7-тетрагидро-3H-пурин-3-ил)бутаннитрил,
8-хлор-3-гексил-1-метил-3,7-дигидро-1H-пурин-2,6-дион,
3-бутил-8-хлор-1-этил-3,7-дигидро-1H-пурин-2,6-дион,
[8-хлор-3-(2-циклопропилэтил)-2,6-диоксо-2,3,6,7-тетрагидро-1H-пурин-1-ил]ацетонитрил,
(8-хлор-2,6-диоксо-3-пропил-2,3,6,7-тетрагидро-1H-пурин-1-ил)ацетонитрил,
8-хлор-1-(4,4,4-трифторбутил)-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-дион,
2,2'-(8-хлор-2,6-диоксо-6,7-дигидро-1H-пурин-1,3(2H)-диил)диацетонитрил,
8-хлор-1-метил-3-(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(2-циклогексилэтил)-3,7-дигидро-1H-пурин-2,6-дион,
1,3-дибутил-2,6-диоксо-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил,
1,3-дибутил-8-йод-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(4-метилпентил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(6-метилгептил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-октил-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-децил-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(циклогексилметил)-3,7-дигидро-1H-пурин-2,6-дион,
(+/-)-8-хлор-3-(3-метилпентил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(2-циклопентилэтил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(циклопропилметил)-3,7-дигидро-1H-пурин-2,6-дион,
(+/-)-8-хлор-3-(2-метилбутил)-3,7-дигидро-1H-пурин-2,6-дион,
(+/-)-8-хлор-3-(2-метилпентил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(циклобутилметил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(циклопентилметил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(3-циклопропилпропил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(2-циклобутилэтил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(4-фторбутил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(3-фторпропил)-3,7-дигидро-1H-пурин-2,6-дион,
8-хлор-3-(5-фторпентил)-3,7-дигидро-1H-пурин-2,6-дион,
4-(8-хлор-1-метил-2,6-диоксо-1,2,6,7-тетрагидро-3Н-пурин-3-ил)бутаннитрил,
3-(3-бутен-1-ил)-8-хлор-3,7-дигидро-1Н-пурин-2,6-дион,
6-(8-хлор-2,6-диоксо-1,2,6,7-тетрагидро-3Н-пурин-3-ил)-2,2-диметилгексаннитрил,
8-хлор-3-(6-фторгексил)-3,7-дигидро-1Н-пурин-2,6-дион,
8-хлор-3-этил-1-метил-3,7-дигидро-1Н-пурин-2,6-дион.
Следует понимать, что этот объект настоящего изобретения включает любую комбинацию частных вариантов осуществления и распространяется на все комбинации частных заместителей, описанных выше для соединений формулы (II).
Дополнительно, настоящее изобретение относится к применению соединения формулы (I) или его физиологически функционального производного для получения лекарственного средства для лечения воспалительных заболеваний или состояний суставов, в частности артрита (например, ревматоидного артрита, остеоартрита, отторжения суставного протеза), или желудочно-кишечного тракта (например, неспецифического язвенного колита, Болезни Крона и других воспалительных кишечных и желудочно-кишечных заболеваний, гастрита и воспаления слизистой оболочки, возникающего в результате инфекции, энтеропатии, вызванной нестероидными противовоспалительными препаратами), легкого (например, респираторного дистресс-синдрома взрослых, астмы, муковисцидоза или хронического обструктивного легочного заболевания), сердца (например, миокардита), нервной ткани (например, рассеянного склероза), поджелудочной железы, (например, воспаления, связанного с сахарным диабетом и его осложнениями), почки (например, гломерулонефрита), кожи (например, дерматита, псориаза, экземы, крапивницы, ожогового повреждения), глаза (например, глаукомы), а также пересаженных органов (например, отторжения) и заболеваний многих органов (например, системной красной волчанки, сепсиса) и воспалительного осложнения вирусных или бактериальных инфекций и воспалительных состояний, связанных с атеросклерозом и после гипоксических или ишемических инсультов (с реперфузией или без), например, в мозге или при ишемической болезни сердца.
В следующем или альтернативном аспекте изобретение относится к способу лечения человека или животного, страдающего состоянием, при котором недостаточная активация рецептора НМ74А способствует развитию этого состояния или где активация рецептора будет иметь полезный эффект, причем указанный способ включает введение указанному человеку или животному эффективного количества соединения формулы (I) или его физиологически приемлемой соли или сольвата.
Следует понимать, что этот объект настоящего изобретения включает любую комбинацию частных вариантов осуществления и охватывает все комбинации частных заместителей, описанных здесь выше для соединений формулы (I).
Более конкретно, настоящее изобретение относится к способу лечения нарушений, связанных с метаболизмом липидов, включая дислипидемию или гиперлипопротеинемию, такую как диабетическая дислипидемия и смешанная дислипидемия, сердечной недостаточности, гиперхолестеринемии, сердечно-сосудистого заболевания, включая атеросклероз, артериосклероз и гипертриглицеридемию, сахарного диабета типа II, диабета типа I, резистентности к инсулину, гиперлипидемии, нервной анорексии, ожирения, причем указанный способ включает введение указанному человеку или животному эффективного количества соединения формулы (I) или его физиологически приемлемой соли или сольвата. Как таковые эти соединения могут также использоваться в способах лечения заболевания коронарных артерий, тромбоза, стенокардии, хронической почечной недостаточности, периферического сосудистого заболевания и инсульта, причем указанные способы включают введение указанному человеку или животному эффективного количества соединения формулы (I).
Количество модулятора HM74A, которое требуется для достижения желательного биологического действия, будет, конечно, зависеть от множества факторов, например способа введения и точного клинического состояния реципиента. В общем, суточная доза находится в диапазоне 0,1 мг-1 г/кг, обычно 0,1-100 мг/кг. Внутривенная доза может, например, быть в диапазоне 0,01 мг-0,1 г/кг, обычно 0,01 мг-10 мг/кг, которые могут быть легко введены в виде инфузии со скоростью от 0,1 мкг до 1 мг в минуту. Инфузионные жидкости, подходящие для этой цели, могут содержать, например, от 0,01 мкг до 0,1 мг на миллилитр. Разовые дозы могут содержать, например, от 0,01 мкг до 1 г модулятора HM74A. Так, ампулы для инъекции могут содержать, например, от 0,01 мкг до 0,1 г, а перорально вводимые композиции разовой дозы, такие как таблетки или капсулы, могут содержать, например, от 0,1 мг до 1 г. Никаких токсикологических эффектов не показано/не ожидается при введении соединения по изобретению в вышеупомянутом диапазоне доз.
Соединение согласно настоящему изобретению может использоваться как соединение per se для лечения заболевания, где недостаточная активация рецептора HM74A способствует развитию заболевания или где активация рецептора будет иметь полезный эффект, примером этого является случай, где соединение по изобретению представлено с приемлемым носителем в форме фармацевтической композиции. Носитель должен, конечно, быть приемлемым в отношении совместимости с другими ингредиентами композиции и не должен быть вредным для реципиента. Носитель может быть твердым веществом или жидкостью, или и тем и другим, и может быть составлен с модулятором HM74A в виде композиции разовой дозы, например таблетки, которая может содержать от 0,05% до 95 вес.% модулятора HM74A.
Композиции включают композиции, подходящие для перорального, ректального, наружного, трансбуккального (например, подъязычного) и парентерального (например, подкожного, внутримышечного, внутрикожного или внутривенного) применения.
Изобретение также относится к способу получения такой фармацевтической композиции, который включает смешивание ингредиентов.
Композиции, подходящие для перорального введения, могут быть представлены в отдельных единицах, таких как капсулы, облатки, пастилки или таблетки, каждая из которых содержит предопределенное количество модулятора HM74A; в виде порошка или гранул; в виде раствора или суспензии в водной или неводной жидкости; или в виде эмульсии типа «масло-в-воде» или «вода-в-масле». Как правило, композиции получают однородным и непосредственным смешиванием активного модулятора HM74A с жидким или тонкодисперсным твердым носителем, или обоими, и затем, в случае необходимости, формованием продукта. Например, таблетка может быть получена прессованием или формованием порошка или гранул модулятора HM74A, необязательно с одним или более дополнительными ингредиентами. Прессованные таблетки можно получить, прессуя в подходящем механизме соединение в сыпучей форме, такой как порошок или гранулы, необязательно смешанное со связующим, лубрикантом, инертным разбавителем и/или поверхностно-активным/диспергирующим агентом(ами). Формованные таблетки можно получить, формуя в подходящем механизме измельченное в порошок соединение, увлажненное инертным жидким разбавителем.
Таблетки и капсулы для перорального введения могут содержать обычные эксципиенты, такие как связующие вещества, например сироп, гуммиарабик, желатин, сорбит, трагакант, мазь на основе камеди крахмала или поливинилпирролидона; наполнители, например лактозу, микрокристаллическую целлюлозу, сахар, кукурузный крахмал, фосфат кальция или сорбит; лубриканты, например стеарат магния, стеариновую кислоту, тальк, полиэтиленгликоль или диоксид кремния; дезинтеграторы, например картофельный крахмал, кроскармелозу натрия или натрия крахмала гликолят; или смачивающие вещества, такие как лаурилсульфат натрия. Таблетки могут быть покрыты согласно способам, известным в уровне техники. Пероральные жидкие препараты могут быть в форме, например, водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров или могут быть представлены как сухой продукт для связывания с водой или другим подходящим носителем перед использованием. Такие жидкие препараты могут содержать обычные добавки, такие как суспендирующие агенты, например сироп сорбита, метилцеллюлозу, сироп глюкозы/сахара, желатин, гидроксиметилцеллюлозу, карбоксиметилцеллюлозу, гель стеарата алюминия или гидрированные пищевые жиры; эмульгаторы, например лецитин, сорбитан моноолеат или гуммиарабик; неводные носители (которые могут включать пищевые масла), например миндальное масло, фракционированное кокосовое масло, масляные сложные эфиры, пропиленгликоль или этиловый спирт; или консерванты, например метил- или пропил-п-гидроксибензоаты или сорбиновую кислоту. Препараты могут также содержать буферные соли, ароматизирующее вещество, красители и/или подслащивающие агенты (например, маннит), в случае необходимости.
Композиции, подходящие для трансбуккального (подъязычного) введения, включают пастилки, содержащие модулятор HM74A в ароматизированной основе, обычно сахарозе и гуммиарабике или трагаканте, и пастилки, включающие модулятор HM74A в инертной основе, такой как желатин и глицерин или сахароза и гуммиарабик.
Композиции согласно настоящему изобретению, подходящие для парентерального введения, предпочтительно включают стерильные водные препараты модулятора HM74A, состав может быть изотоническим по отношению к крови предполагаемого реципиента. Эти препараты могут вводиться внутривенно, хотя введение может также быть произведено посредством подкожной, внутримышечной или внутрикожной инъекции. Такие препараты могут быть получены путем смешивания модулятора HM74A с водой и стерилизации полученного раствора и придания ему изотоничности по отношению к крови. Инъецируемые композиции согласно изобретению обычно содержат от 0,1 до 5% вес./вес. модулятора HM74A.
Таким образом, композиции согласно настоящему изобретению, подходящие для парентерального введения, включающие соединение согласно изобретению, могут быть составлены для парентерального введения болюсным вливанием или непрерывной инфузией и могут быть представлены в форме разовой дозы, например как ампулы, пузырьки, инфузии малого объема или предварительно наполненные шприцы, или в контейнерах с множеством доз с добавленным консервантом. Композиции могут находиться в таких формах, как растворы, суспензии или эмульсии в водных или неводных носителях, и могут содержать вспомогательные средства, такие как антиоксиданты, буферы, противомикробные агенты и/или регуляторы токсичности. Альтернативно, активный ингредиент может быть в порошковой форме для связывания с подходящим носителем, например стерильной апирогенной водой, перед использованием. Сухая твердая форма может быть получена путем асептического заполнения стерильного порошка в индивидуальные стерильные контейнеры или асептического заполнения стерильного раствора в каждый контейнер и сушки сублимацией.
Композиции, подходящие для ректального введения, могут быть представлены как суппозитории, содержащие разовую дозу. Они могут быть получены путем смешивания модулятора HM74A с одним или более обычных твердых носителей, например с маслом какао или глицеридами, с последующим формованием полученной смеси.
Композиции, подходящие для местного нанесения на кожу, могут иметь форму мази, крема, лосьона, пасты, геля, спрея, аэрозоля или масла. Носители, которые могут использоваться, включают вазелин, ланолин, полиэтиленгликоли, спирты и комбинации двух или более указанных носителей. Модулятор HM74A обычно присутствует в концентрации от 0,1 до 15% вес./вес. по отношению к массе композиции, например от 0,5 до 2%.
К местному нанесению в настоящем описании относят введение вдуванием и ингаляцией. Примеры различных типов препаратов для местного введения включают мази, кремы, лосьоны, порошки, пессарии, спреи, аэрозоли, капсулы или картриджи для использования в ингаляторе или инсуффляторе или капли (например, глазные капли или капли для носа).
Мази и кремы могут, например, быть составлены с водной или масляной основой с добавлением подходящего загустителя, и/или желирующих средств, и/или растворителей. Такие основы могут, таким образом, например, включать воду и/или масло, такое как вазелиновое масло или растительное масло, такое как арахисовое масло или касторовое масло, или растворитель, такой как полиэтиленгликоль. Загустители, которые могут использоваться, включают мягкий парафин, стеарат алюминия, цетостеариловый спирт, полиэтиленгликоли, микрокристаллический воск и пчелиный воск.
Лосьоны могут быть составлены с водной или масляной основой и обычно также содержат один или более эмульгаторов, стабилизирующих средств, диспергирующих агентов, суспендирующих агентов или загустителей.
Порошки для наружного применения можно сформировать при помощи любой подходящей порошковой основы, например талька, лактозы или крахмала. Капли могут быть составлены с водной или неводной основой, также включающей один или более диспергирующих агентов, солюбилизирующих агентов или суспендирующих агентов.
Композиции спрея могут быть составлены, например, в виде водных растворов или суспензий, или в виде аэрозолей, заключенных в герметичные упаковки, с использованием подходящего пропеллента, например дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, 1,1,1,2,3,3,3-гептафторпропана, 1,1,1,2-тетрафторэтана, углекислого газа или другого подходящего газа.
Капсулы и картриджи для использования в ингаляторе или инсуффляторе, например, из желатина могут содержать порошкообразную смесь соединения по изобретению и подходящей порошковой основы, такой как лактоза или крахмал.
Фармацевтические композиции согласно изобретению могут также использоваться в комбинации с другими терапевтическими средствами, например в комбинации с другими классами дислипидемических препаратов (например, статинами, фибратами, смолами, связывающими желчную кислоту, или никотиновой кислотой).
Соединения согласно настоящему изобретению могут использоваться в комбинации с одним или более другими терапевтическими средствами, например в комбинации с другими классами дислипидемических препаратов, например ингибиторами 3-гидрокси-3-метилглутарил-коэнзим А редуктазы (статины) или фибратами, или смолами, связывающими желчную кислоту, или никотиновой кислотой. Изобретение, таким образом, относится в следующем аспекте к применению такой комбинации для лечения заболеваний, где недостаточная активация рецептора HM74A способствует развитию заболевания или где активация рецептора будет иметь полезный эффект, и к применению соединения формулы (I) или (II) или его фармацевтически приемлемой соли, сольвата или физиологически функционального производного для получения лекарственного средства для комбинированной терапии нарушений, связанных с метаболизмом липидов, включая дислипидемию или гиперлипопротеинемию, такую как диабетическая дислипидемия и смешанная дислипидемия, сердечной недостаточности, гиперхолестеринемии, сердечно-сосудистого заболевания, включая атеросклероз, артериосклероз и гипертриглицеридемию, сахарного диабета типа II, диабета типа I, резистентности к инсулину, гиперлипидемии, нервной анорексии или ожирения.
Когда соединения согласно настоящему изобретению используются в комбинации с другими терапевтическими средствами, соединения могут вводиться последовательно или одновременно любым удобным путем.
Комбинации, упомянутые выше, могут быть представлены для использования в форме фармацевтической композиции, и, таким образом, фармацевтические композиции, включающие комбинацию, как определено выше, необязательно вместе с фармацевтически приемлемым носителем или эксципиентом, составляют следующий объект изобретения. Индивидуальные компоненты таких комбинаций могут вводиться последовательно или одновременно в отдельных или комбинированных фармацевтических композициях.
Следует понимать, что в случае их комбинации в той же самой композиции эти два компонента должны быть стабильными и совместимыми друг с другом и другими компонентами композиции и могут быть составлены для введения. В случае раздельной композиции они могут быть составлены в виде любой подходящей композиции, таким образом, как это известно для таких соединений в уровне техники.
В случае комбинации со вторым терапевтическим средством, активным против того же самого заболевания, доза каждого компонента может отличаться от случая, когда соединение используется индивидуально. Подходящие дозы могут быть легко определены специалистом.
Изобретение, таким образом, относится в следующем аспекте к комбинации, включающей соединение формулы (I) или (II), или его физиологически приемлемую соль, или сольват, вместе с другим терапевтически активным средством.
Комбинация, упомянутая выше, может быть представлена для использования в форме фармацевтической композиции, и, таким образом, фармацевтические композиции, включающие комбинацию, как определено выше, вместе с фармацевтически приемлемым носителем, представляют следующий аспект изобретения.
Соединения согласно настоящему изобретению имеют полезную продолжительность действия.
Соединения согласно настоящему изобретению и их соли и сольваты могут быть получены в соответствии с методологией, описанной далее, составляя следующий аспект этого изобретения.
Способ A:
Способ согласно изобретению получения соединения формулы (I) или формулы (II), в которых R1 обозначает Н или имеет то же значение, что и R2, и R3 обозначает Cl, включает:
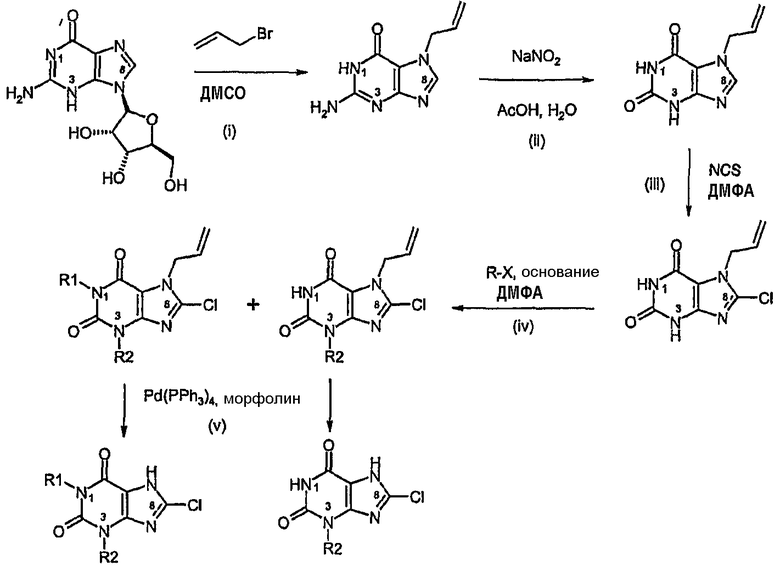
i) алкилирование гуанина аллилбромидом,
ii) диазотирование нитритом натрия с последующим гидролизом с получением ксантина,
iii) хлорирование,
iv) алкилирование в N3 и/или диалкирование в N1 и N3,
v) катализируемое палладием удаление аллильной группы.
Способ B:
Способ согласно изобретению получения соединения формулы (I) или формулы (II), в которой R3 обозначает CN, включает стадии (i) и (ii) способа А, с последующими:
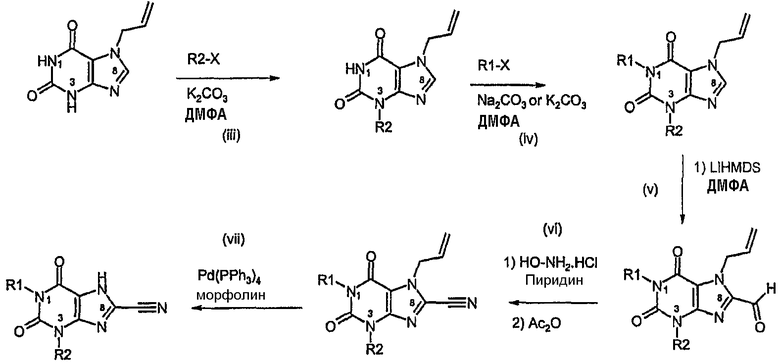
iii) алкилированием в N3,
iv) алкилированием в N1,
v) формированием альдегида в C8 путем литиирования с помощью LiHMDS и гашением ДМФА,
vi) преобразованием альдегида в нитрил,
vii) катализируемым палладием удалением аллильной группы.
Способ C:
Способ согласно изобретению получения соединения формулы (I) или формулы (II), в которой R3 обозначает Cl или Br, включает стадии (i)-(iv) способа B с последующими:
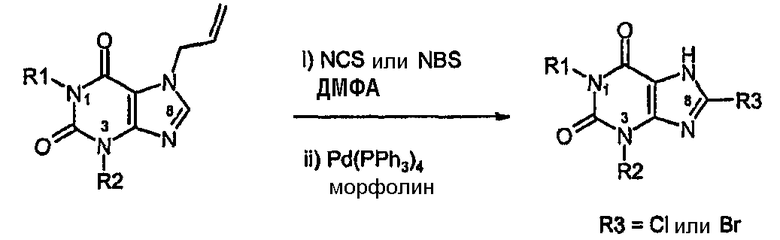
i) галогенированием в C8 с использованием NCS или NBS,
ii) катализируемым палладием удалением аллильной группы.
Способ D:
Способ согласно изобретению получения соединения формулы (I) или формулы (II), в которой R3 обозначает CN, включает стадии (i)-(iv) способа B с последующими:
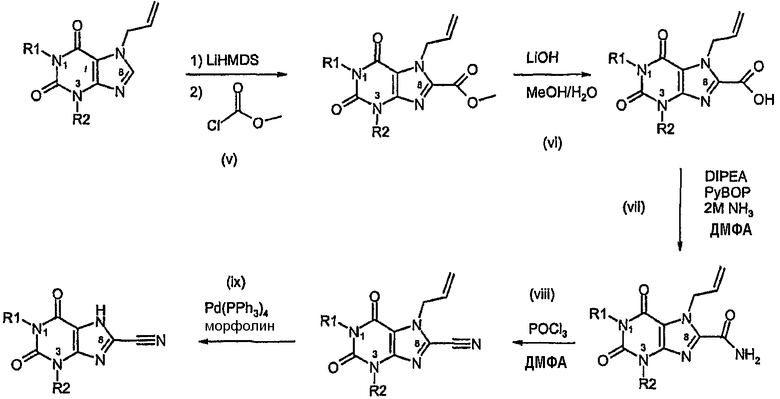
v) образованием сложного эфира,
vi) гидролизом сложного метилового эфира,
vii) преобразованием кислоты в амид,
viii) преобразованием амида в нитрил,
ix) катализируемым палладием удалением аллильной группы.
Способ Е:
Способ согласно изобретению получения соединения формулы (I) или формулы (II), в которой R3 обозначает Cl, включает:
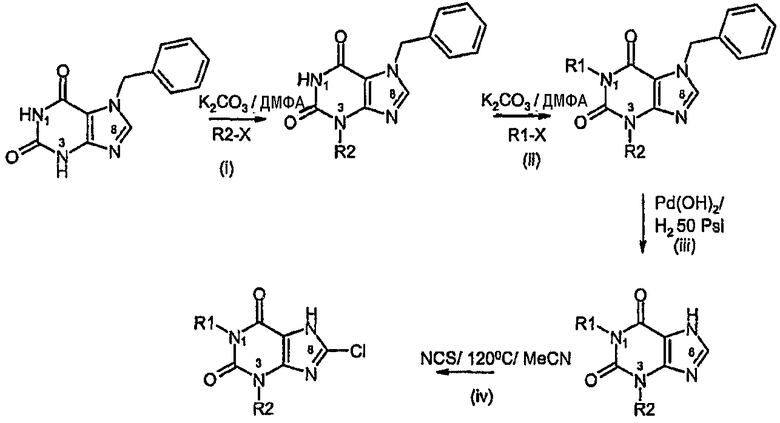
i) алкилирование в N3,
ii) алкилирование в N1,
iii) дебензилирование,
iv) хлорирование в C8.
Способ F:
Способ согласно изобретению получения соединения формулы (I) или формулы (II), в которой R1 отличается от R2 и R3 обозначает Cl, включает стадии (i)-(iv) способа А с последующими:
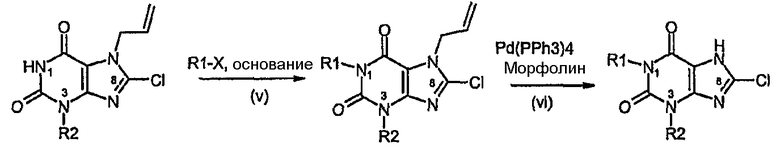
v) алкилированием в N1,
vi) катализируемым палладием удалением аллильной группы.
Способ G:
Способ согласно изобретению получения соединения формулы (I) или формулы (II), в которой R1 отличается от R2 и R3 обозначает Cl, включает стадии (i)-(v) способа F (где R2 в способе F конкретно обозначает SEM или MEM), с последующими:
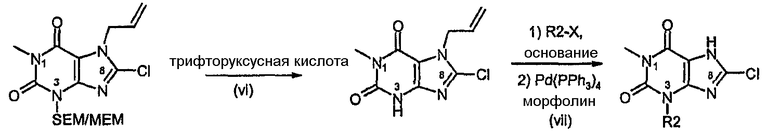
vii) расщеплением защитной группы для группы MEM или SEM,
viii) алкилированием N3 с последующим катализируемым палладием удалением аллильной группы.
Способ Н:
Способ согласно изобретению получения соединения формулы (I) или формулы (II), в которой R3 обозначает Cl, Br, I или F, включает стадии (i)-(iv) способа B с последующими:
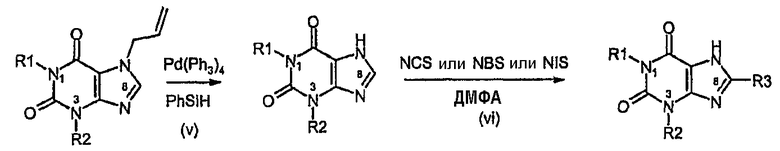
v) катализируемым палладием удалением аллильной группы,
vi) галогенированием в C8 с использованием NCS, NBS или NIS.
Способ I:
Способ согласно изобретению получения соединения формулы (I) или формулы (II), в которой R1 обозначает Н или алкил, R2 обозначает алкил и R3 обозначает Cl, включает:
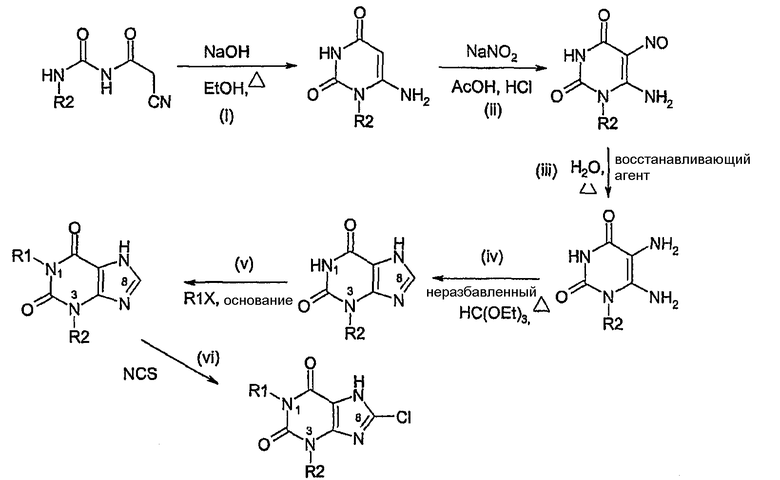
i) образование пиримидиндиона,
ii) нитрозирование,
iii) восстановление с использованием Na2S2O4 или подобного восстановителя,
iv) образование ксантина,
v) алкилирование в N1 (необязательно),
vi) галогенирование в C8 с использованием NCS.
Где желательно или необходимо, в качестве заключительной стадии в любом из вышеупомянутых способов синтеза полученное соединение формулы (I) или (II) может быть преобразовано в форму физиологически приемлемой соли, или наоборот, или одна форма соли может быть преобразована в другую физиологически приемлемую форму соли.
СОКРАЩЕНИЯ
Следующие неограничивающие примеры иллюстрирует настоящее изобретение:
Примеры синтеза
Пример 1: 8-хлор-3-(4-пентен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
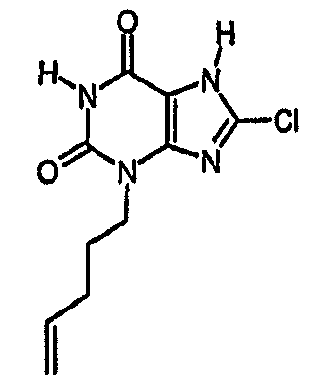
a) 2-амино-7-(2-пропен-1-ил)-1,7-дигидро-6H-пурин-6-он
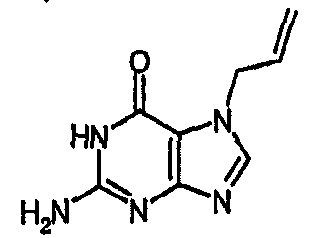
Смесь гуанозина (20 г, 0,071 моль), аллилбромида (14,7 мл, 0,169 моль) и безводного ДМСО (100 мл) перемешивали при комнатной температуре, в атмосфере азота, в течение 18 часов. Концентрированную HCl (50 мл, 37%) добавляли одной порцией и смесь перемешивали в течение 45 минут, затем выливали в MeOH (600 мл). Метанольный раствор нейтрализовали 2М раствором NaOH (водн.) и полученный белый осадок собирали фильтрованием. Твердое вещество белого цвета сушили в вакууме при 50°C в течение 18 часов с получением указанного в заголовке соединения (16 г сырого продукта, 119%). m/z 192,2 [MH+].
b) 7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
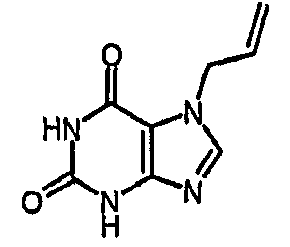
Смесь 2-амино-7-(2-пропен-1-ил)-1,7-дигидро-6H-пурин-6-она (40 г, 0,209 моль) в AcOH (900 мл) и воде (100 мл) нагревали при 55°C. Нитрит натрия (57,74 г, 0,837 моль) в воде (100 мл) добавляли по каплям. Внимание; токсичные пары. После завершения добавления (приблизительно 25 минут) реакционной смеси давали охладиться до температуры окружающей среды и затем концентрировали до приблизительно 1/3 ее первоначального объема. Добавляли воду (500 мл) и полученный осадок собирали фильтрованием. Остаток промывали водой, затем сушили при 50°C над P2O5 и в вакууме в течение 2 часов с получением указанного в заголовке соединения (17,20 г). Водную фракцию концентрировали и добавляли воду (100 мл). Полученное твердое вещество снова фильтровали и сушили. Таким образом, получали большое количество указанного в заголовке соединения (2,31 г). Комбинированный продукт (19,52 г, 49%). m/z 193,2 [MH+].
c) 8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
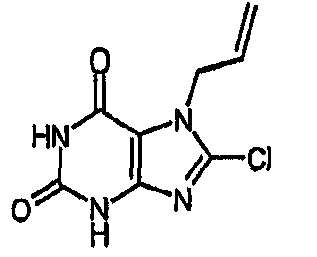
К раствору 7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона) (10,52 г, 54,7 ммоль) в безводном ДМФА (60 мл) добавляли NCS (8,04 г, 60,2 ммоль). Реакционную смесь оставляли для перемешивания в атмосфере азота при 20°C в течение 6 часов. Реакционную смесь концентрировали в вакууме с получением янтарного масла. Добавляли MeOH и оставляли на 18 часов. Полученный остаток фильтровали и сушили в вакууме с получением указанного в заголовке соединения (7,69 г, 62%). m/z 227,2 [MH+].
d) 8-хлор-3-(4-пентен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
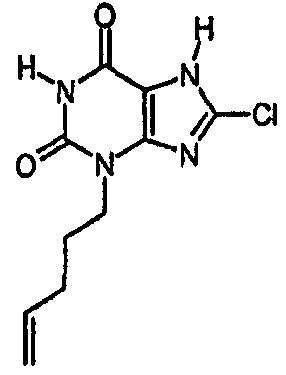
8-Хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион (0,10 г, 0,44 ммоль) растворяли в ДМФА (1,5 мл), содержащем карбонат натрия (0,12 г, 0,49 ммоль) и 5-бромпентен (0,07 г, 0,49 ммоль), и смесь перемешивали в течение 18 ч. По завершении алкилирования добавляли морфолин (0,5 мл) и тетракис(трифенилфосфин)палладий(0) (0,08 г, 0,07 ммоль) и перемешивание продолжали в течение 3,5 ч. Реакционную смесь разбавляли этилацетатом (10 мл), промывали последовательно 2N соляной кислотой (2×5 мл) и насыщенным раствором соли (3×5 мл) и органическое вещество выделяли, сушили (MgSO4) и концентрировали. Сырой продукт суспендировали в метаноле (2 мл) и очищали на картридже SPE с аминопропилом (5 г), элюируя сначала метанолом, затем 5% уксусной кислотой в метаноле, для элюирования указанного в заголовке соединения, которое выделяли в виде твердого вещества белого цвета после концентрирования (0,039 г, 35%). ЯМР; (400МГц, d6-ДМСО) 1,75 (м, 2H), 2,05 (м, 2H), 3,85 (т, 2H, J=7Гц), 4,95 (м, 1H), 5,05 (м, 1H), 5,8 (м, 1H), 11,1 (уш. с, 1H), один обмениваемый протон, не наблюдаемый при δН 13; m/z 255 [MH+].
Пример 2: 8-хлор-3-гексил-3,7-дигидро-1H-пурин-2,6-дион
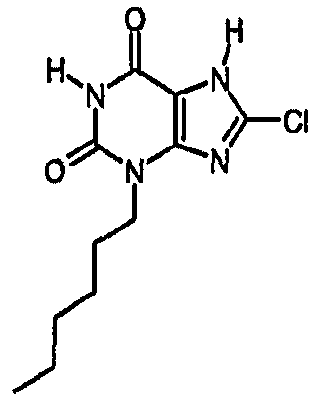
Получают подобно примеру 1, используя гексилйодид, с получением указанного в заголовке соединения.
ЯМР; δH (400МГц, d6-ДМСО) 0,85 (т, 3H, J=7Гц), 1,25 (уш.с, 6H), 1,6 (м, 2H), 3,85 (т, 2H, J=8Гц), 11,2 (уш.с, 1H), один обмениваемый протон, не наблюдаемый при δН 13; m/z 271 [MH+].
Примеры 3 и 4: (8-хлор-2,6-диоксо-1,2,6,7-тетрагидро-3H-пурин-3-ил)ацетонитрил и 2,2'-(8-хлор-2,6-диоксо-6,7-дигидро-1H-пурин-1,3(2H)-диил)диацетонитрил
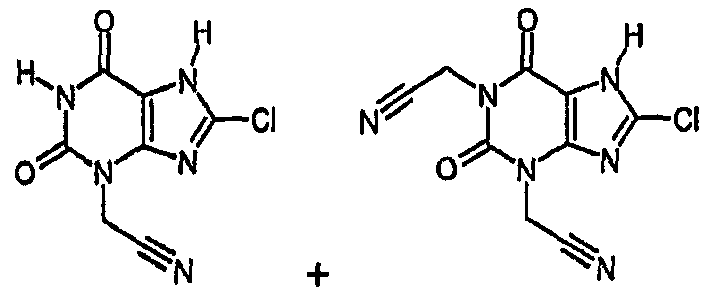
a) [8-хлор-2,6-диоксо-7-(2-пропен-1-ил)-1,2,6,7-тетрагидро-3H-пурин-3-ил]ацетонитрил и 2,2'-[8-хлор-2,6-диоксо-7-(2-пропен-1-ил)-6,7-дигидро-1H-пурин-1,3(2H)-диил]диацетонитрил
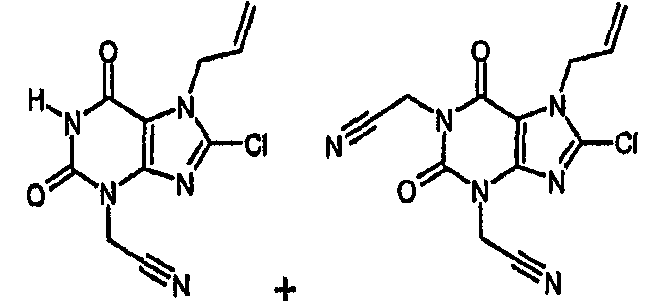
Раствор 8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (0,445 г, 2,0 ммоль) в ДМФА (8 мл) обрабатывали карбонатом натрия (0,18 г, 1,7 ммоль) и бромацетонитрилом (0,1 мл, 1,4 ммоль). Перемешиваемую смесь нагревали при 70°C в течение 3 часов, затем охлаждали до 50°C и дополнительно обрабатывали бромацетонитрилом (0,06 мл, 0,8 ммоль). Смесь выдерживали при 50°C в течение дополнительных 2 часов и затем охлаждали до температуры окружающей среды и упаривали досуха. Остаток обрабатывали 1M водной соляной кислотой (20 мл) и экстрагировали этилацетатом (2×50 мл). Органические фракции объединяли, сушили над сульфатом магния, фильтровали и упаривали. Остаток растворяли в дихлорметане (2 мл), через 20 минут полученное осажденное твердое вещество (непрореагировавший 8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион отфильтровывали и промывали дополнительным дихлорметаном). Фильтрат концентрировали в вакууме и подвергали флэш-хроматографии с использованием смеси этилацетат/циклогексан в качестве элюента в градиентном элюировании от 1:3 до 4:1. Получают два указанных в заголовке соединения:
[8-хлор-2,6-диоксо-7-(2-пропен-1-ил)-1,2,6,7-тетрагидро-3H-пурин-3-ил]ацетонитрил
Твердое вещество белого цвета (0,084 г, 16%); m/z 266 [MH+].
2,2'-[8-хлор-2,6-диоксо-7-(2-пропен-1-ил)-6,7-дигидро-1H-пурин-1,3(2H)-диил]диацетонитрил
Твердое вещество белого цвета (0,195 г, 32%); m/z 305 [MH+].
b) (8-хлор-2,6-диоксо-1,2,6,7-тетрагидро-3H-пурин-3-ил)ацетонитрил
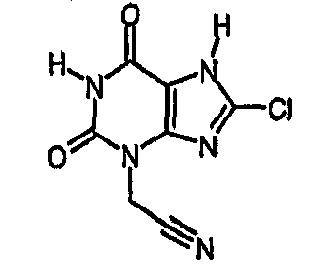
Раствор [8-хлор-2,6-диоксо-7-(2-пропен-1-ил)-1,2,6,7-тетрагидро-3H-пурин-3-ил]ацетонитрила (0,084 г, 0,32 ммоль) в ТГФ (5 мл) дегазировали последовательным применением вакуума и давления азота к реакционной смеси. Раствор затем обрабатывали морфолином (0,3 мл, 3,4 ммоль) и тетракис(трифенилфосфин)палладием(0) (0,03 г, 0,03 ммоль). Через 2 часа смесь обрабатывали 2M водной соляной кислотой (3 мл) и хлороформом (5 мл). Смесь разделяли и органическую фазу упаривали. Продукт очищали от остатка, используя масс-направленную ВЭЖХ, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (0,018 г, 25%). ЯМР δН (400МГц, d6-ДМСО) 4,95 (с, 2H), 11,49 (с, 1H), 14,63 (уш.с, 1H); m/z 226 [MH+].
c) 2,2'-(8-хлор-2,6-диоксо-6,7-дигидро-1H-пурин-1,3(2H)-диил)диацетонитрил
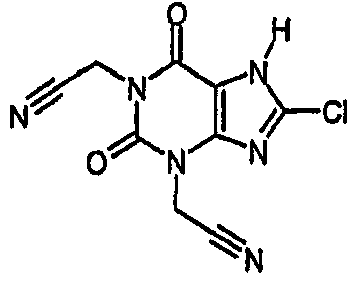
Указанное в заголовке соединение получали из 2,2'-[8-хлор-2,6-диоксо-7-(2-пропен-1-ил)-6,7-дигидро-1H-пурин-1,3(2H)-диил]диацетонитрила с использованием условий, описанных для синтеза (8-хлор-2,6-диоксо-1,2,6,7-тетрагидро-3H-пурин-3-ил)ацетонитрила.
Получают указанное в заголовке соединение в виде твердого вещества белого цвета 0,06 г (4%); ЯМР δН (400МГц, d6-ДМСО) 4,88 (с, 2H), 5,06 (с, 2H), NH не наблюдается при δН 14; m/z 282 [MNH4 +].
Пример 5: 8-хлор-3-(3,3,3-трифторпропил)-3,7-дигидро-1H-пурин-2,6-дион
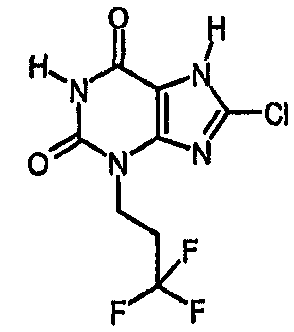
Получают подобно примеру 3, используя 3-бром-1,1,1-трифторпропан в качестве алкилирующего агента, с получением указанного в заголовке соединения.
ЯМР δН (400МГц, d6-ДМСО) 2,64-2,76 (м, 2H), 4,12 (т, 2H, J=7Гц), 11,30 (с, 1H), 14,46 (уш.с, 1H); m/z 283 [MH+].
Пример 6: 8-хлор-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-дион
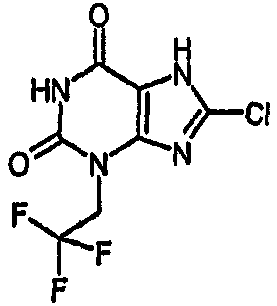
Получают подобно примеру 3, используя 2-бром-1,1,1-трифторэтан в качестве алкилирующего агента и бикарбонат натрия в качестве основания, с получением указанного в заголовке соединения.
δН (400МГц, d4-MeOD) 4,68 (кв, 2H, J=8,5Гц); m/z 267,1 [М-Н]-.
Пример 7 и 8: 8-хлор-3-(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион и 8-хлор-1,3-бис(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион
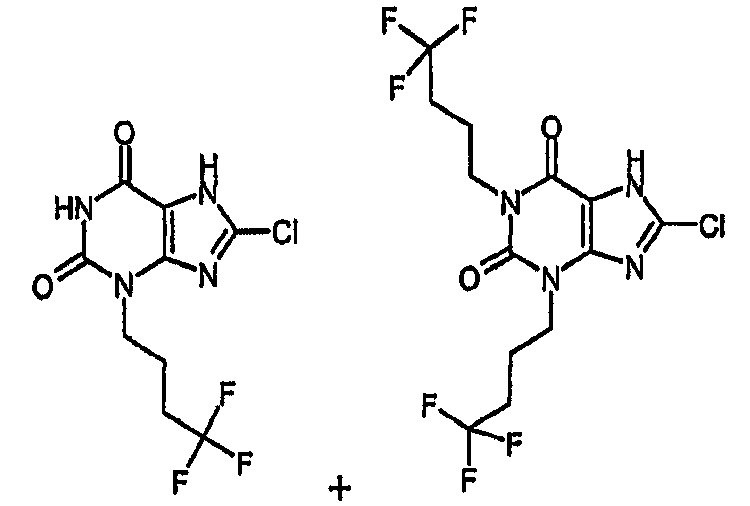
a) 8-хлор-7-(2-пропен-1-ил)-3-(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион и 8-хлор-7-(2-пропен-1-ил)-1,3-бис(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион
8-Хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион (1,5 г, 6,64 ммоль), карбонат натрия (844 мг, 7,9 ммоль) и 4-бром-1,1,1-трифторбутан (1,39 г, 7,3 ммоль) перемешивали в диметилформамиде (25 мл, сухой) в течение семи дней. Реакционную смесь распределяли между этилацетатом и водой. Органическую фазу отделяли и промывали соляной кислотой (2N), насыщенным раствором соли, сушили (MgSO4) и затем упаривали досуха. Сырой продукт растирали в простом эфире и твердое вещество собирали фильтрованием с получением 8-хлор-7-(2-пропен-1-ил)-3-(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-диона в виде твердого вещества белого цвета. (1,23 г, 57%). m/z 337 [MH+].
Восстановленный фильтрат хроматографировали на силикагеле, колонка SPE (20 г). Элюируя смесью циклогексан:этилацетат (от 10:1 до 2:1), получали 8-хлор-7-(2-пропен-1-ил)-1,3-бис(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион в виде сиропа (480 мг, 16%). m/z 447 [МН+].
b) 8-хлор-3-(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион
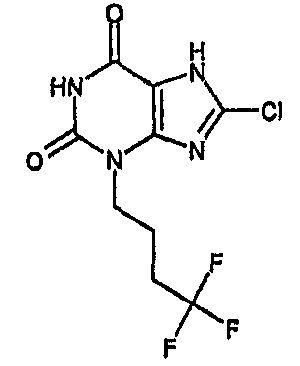
8-Хлор-7-(2-пропен-1-ил)-3-(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион (84 мг, 0,25 ммоль) и морфолин (220 мкл, 2,5 ммоль) дегазировали азотом в тетрагидрофуране (3 мл), затем добавляли тетракис(трифенилфосфин)палладий(0) (29 мг, 0,025 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Белый осадок собирали фильтрованием и промывали тетрагидрофураном и простым эфиром, получая морфолиновую соль указанного в заголовке соединения (59 мг). Ее обрабатывали 2N HCl и метанолом и растворители выпаривали досуха, после чего повторно растворяли в смеси ДМСО/MeOH и очищали препаративной ВЭЖХ, используя градиент 10-40%, с получением указанного в заголовке соединения (11 мг, 14,9%).
ЯМР δН (400МГц, d4-MeOD) 1,92-2,03 (м, 2H), 2,19-2,33 (м, 2H), 4,06 (т, 2H, J=7Гц); m/z 297 [MH+].
c) 8-хлор-1,3-бис(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион
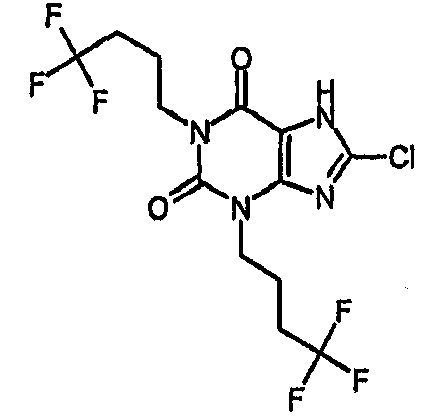
8-Хлор-7-(2-пропен-1-ил)-1,3-бис(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион (478 мг, 1,1 ммоль) и морфолин (937 мкл, 11 ммоль) дегазировали азотом в тетрагидрофуране (10 мл), затем добавляли тетракис(трифенилфосфин)палладий(0) (123 мг, 0,11 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь распределяли между дихлорметаном и 2N соляной кислотой. Органическую фазу отделяли и восстанавливали с получением сырого продукта. Его очищали SPE с аминопропилом (5 г) с последующей перекристаллизацией из ацетонитрила, получая указанное в заголовке соединение (75,5 мг, 16,9%). ЯМР. δН (400МГц, CDCl3) 1,96-2,13 (м, 4H), 2,15-2,29 (м, 4H), 4,15-4,23 (м, 4H), 12,94 (уш.с, 1H); m/z 407 [MH+].
Пример 9: 8-хлор-3-(2-циклопропилэтил)-3,7-дигидро-1H-пурин-2,6-дион
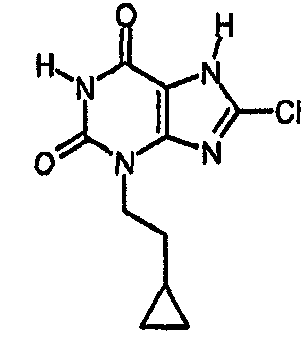
a) 8-хлор-3-(2-циклопропилэтил)-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион

8-Хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион (1,5 г, 6,64 ммоль), карбонат натрия (844 мг, 7,9 ммоль) и 2-циклопропилэтилметансульфонат (1,19 г, 7,3 ммоль) перемешивали в диметилформамиде (25 мл, сухой) в течение двух дней при 80°C. Реакционную смесь распределяли между этилацетатом и водой. Органическую фазу отделяли и промывали соляной кислотой (2N), насыщенным раствором соли, сушили (MgSO4) и затем упаривали досуха. Сырой продукт растирали в простом эфире и твердое вещество собирали фильтрованием, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,96 г, 49%). m/z 295 [MH+].
b) 8-хлор-3-(2-циклопропилэтил)-3,7-дигидро-1H-пурин-2,6-дион
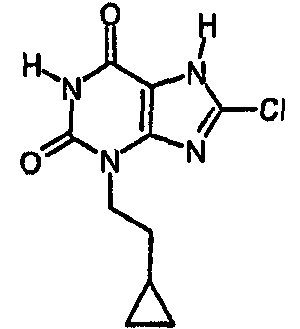
8-Хлор-3-(2-циклопропилэтил)-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион (74 мг, 0,25 ммоль) и морфолин (220 мкл, 2,5 ммоль) дегазировали азотом в тетрагидрофуране (3 мл), затем добавляли тетракис(трифенилфосфин)палладий(0) (29 мг, 0,025 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Белый осадок собирали фильтрованием и промывали тетрагидрофураном и простым эфиром, получая морфолиновую соль указанного в заголовке соединения (52 мг). Ее обрабатывали 2N HCl и метанолом и растворители выпаривали досуха перед повторным растворением в смеси ДМСО/MeOH и очисткой препаративной ВЭЖХ, используя градиент 10-40%, с получением указанного в заголовке соединения (22 мг, 34,6%).
ЯМР δН (400МГц, d4-MeOD) 0,00-0,05 (м, 2H), 0,37-0,43 (м, 2H), 0,67-0,77 (м, 1H), 1,61 (кв, 2H, J=7Гц), 4,06-4,11 (м, 2H); m/z 255 [MH+].
Пример 10: 3-бутил-8-хлор-3,7-дигидро-1H-пурин-2,6-дион
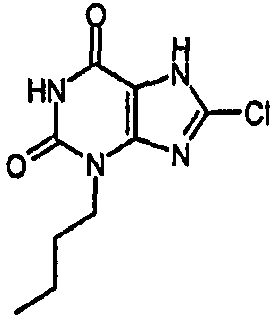
a) 3-бутил-8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
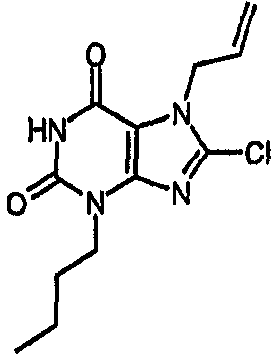
К раствору 3-бутил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (3,34 г, 13,4 ммоль) в безводном ДМФА (19 мл) добавляли NCS (1,97 г, 14,8 ммоль) и оставляли для перемешивания при комнатной температуре в атмосфере азота в течение 22 часов. Смесь концентрировали в вакууме с получением твердого вещества желтого цвета, которое фильтровали и промывали метанолом. Фильтрат концентрировали и процесс повторяли. После заключительной промывки фильтрат очищали с помощью картриджа SPE (Si, 20 г), элюируя смесью 1:1; EtOAc:циклогексан. Объединенные твердые вещества сушили в вакууме с получением указанного в заголовке соединения (2,42 г, 64%); m/z 283,3 [MH+].
b) 3-бутил-8-хлор-3,7-дигидро-1H-пурин-2,6-дион
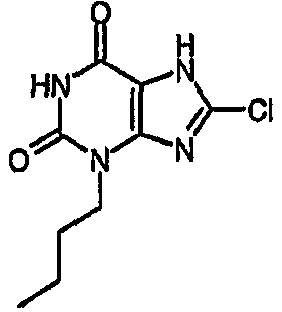
Раствор 3-бутил-8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (100 мг, 0,35 ммоль) в безводном ТГФ (4 мл) и безводном ДМСО (0,4 мл) обрабатывали Pd(PPh3)4 (61 мг, 0,053 ммоль). Смесь дегазировали под действием легкого вакуума, добавляли морфолин (308 мкл, 3,5 ммоль) и оставляли для перемешивания при комнатной температуре в атмосфере азота в течение 4 часов. Желтый раствор распределяли между 2M HCl (водн.) и EtOAc. Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали. Остаток помещали в MeOH и отправляли на SPE с аминопропилом (5 г), элюируя MeOH, затем 5% AcOH/MeOH. Фракции продукта объединяли и концентрировали в вакууме с получением указанного в заголовке соединения в виде твердого вещества белого цвета (30 мг, 35%). ЯМР; δН (400МГц, d6-ДМСО) 0,89 (т, 3H, J=7,5Гц), 1,23-1,34 (м, 2H), 1,55-1,65 (м, 2H), 3,85 (т, 2H, J=7Гц), 11,17 (с, 1H), 14,37 (уш.с, 1H); m/z 243,3 [MH+].
Пример 11: 8-хлор-3-пропил-3,7-дигидро-1H-пурин-2,6-дион

3-Пропил-3,7-дигидро-1H-пурин-2,6-дион (J. Med. Chem, 1993, 36(10), 1380-6) (0,3 г, 1,5 ммоль) и N-хлорсукцинимид (0,21 г, 1,5 ммоль) растворяли в ДМФА (5 мл) и раствор перемешивали в течение 5 ч. Раствор концентрировали и твердые остатки промывали метанолом и фильтровали, получая продукт в виде твердого вещества белого цвета (0,148 г, 42%). ЯМР; δН (400МГц, d6-ДМСО) 0,85 (т, 3H, J=7Гц), 1,65 (м, 2H), 3,8 (т, 2H, J=7Гц), 11,2 (с, 1H), один обмениваемый, не наблюдаемый при δН 13; m/z 229 [MH+].
Пример 12: 8-хлор-3-пентил-3,7-дигидро-1H-пурин-2,6-дион
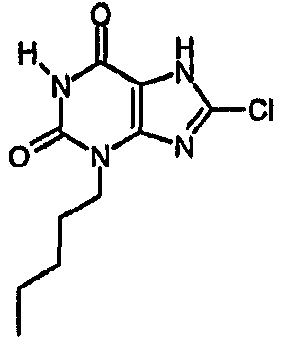
a) 8-хлор-3-пентил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
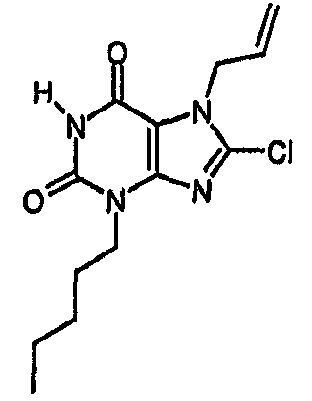
К раствору 8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (100 мг, 0,44 ммоль) в безводном ДМФА (3 мл) добавляли карбонат натрия (0,051 г, 0,484 ммоль). После 10 минут перемешивания при комнатной температуре добавляли пентилйодид (0,063 мл, 0,484 ммоль) и перемешивание продолжали в атмосфере азота при комнатной температуре в течение 18 часов. Реакционную смесь разбавляли водой (25 мл) и экстрагировали EtOAc (2×25 мл). Объединенные органические экстракты сушили (MgSO4), фильтровали и упаривали. Очисткой SPE (Si, 5 г) с элюированием смесью 4:1 EtOAc/циклогексан получали указанное в заголовке соединение в виде твердого вещества белого цвета (96 мг, 74%); m/z 297,2 [MH+].
b) 8-хлор-3-пентил-3,7-дигидро-1H-пурин-2,6-дион
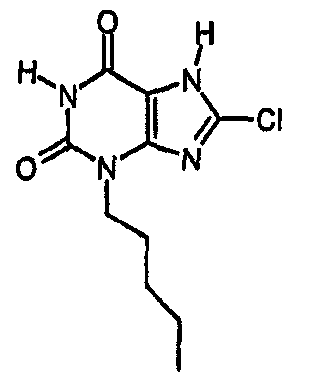
Колбу, содержащую тетракис(трифенилфосфин)палладий(0) (56 мг, 0,049 ммоль), промывали азотом, после чего добавляли раствор 8-хлор-3-пентил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (96 мг, 0,323 ммоль) в безводном ТГФ (1,5 мл), затем ДМСО (0,1 мл) и морфолин (0,28 мл, 0,049 ммоль). Полученную смесь перемешивали при комнатной температуре в атмосфере азота в течение 72 часов. Реакционную смесь растворяли в EtOAc (25 мл) и промывали 2M водной HCl (25 мл). Органический экстракт сушили (MgSO4), фильтровали и упаривали при пониженном давлении. Очищали SPE с аминопропилом (2 г) и промывали метанолом, а затем элюировали продукт 5% уксусной кислотой в метаноле. Выпариванием фракций, содержащих продукт, получали указанное в заголовке соединение в виде твердого вещества белого цвета (27 мг, 33%). ЯМР; δН (400МГц, d6-ДМСО) 0,85 (т, 3H, J=7Гц), 1,20-1,34 (м, 4H), 1,57-1,67 (м, 2H), 3,84 (т, 2H, J=7Гц), 11,19 (с, 1H), 14,38 (уш.с, 1H); m/z 257,2 [MH+].
Пример 13: 8-хлор-3-(3-метилбутил)-3,7-дигидро-1H-пурин-2,6-дион
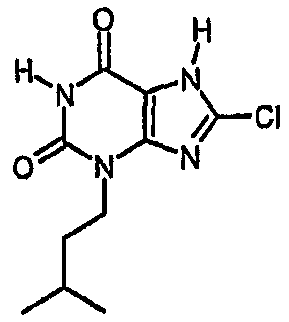
a) 8-хлор-3-(3-метилбутил)-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
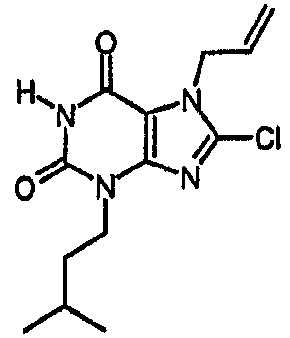
Раствор 8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (1,5 г, 6,6 ммоль) в ДМФА (40 мл) обрабатывали карбонатом натрия (0,9 г, 8,5 ммоль) и 1-бром-3-метилбутаном (1,04 г, 6,9 ммоль). Перемешиваемую смесь нагревали при 50°C в течение 18 часов, затем охлаждали и упаривали досуха. Остаток обрабатывали водой (60 мл) и экстрагировали этилацетатом (3×80 мл). Органические фракции объединяли, сушили над сульфатом магния, фильтровали и упаривали. Остаток растирали в смеси диэтилового эфира и циклогексана, получая продукт в виде твердого вещества белого цвета, которое отфильтровывали и сушили. Получали указанное в заголовке соединение в виде твердого вещества белого цвета. m/z 297
[MH+].
b) 8-хлор-3-(3-метилбутил)-3,7-дигидро-1H-пурин-2,6-дион
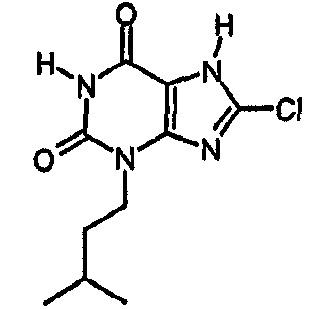
Раствор 8-хлор-3-(3-метилбутил)-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (0,074 г, 0,25 ммоль) в ТГФ (2 мл) обрабатывали морфолином (0,035 мл, 4,0 ммоль) и смесь дегазировали повторным дополнительным применением вакуума и азота к реактору. Смесь затем обрабатывали раствором тетракис(трифенилфосфин)палладия(0) (0,03 г, 0,026 ммоль) в дегазированном ТГФ (0,5 мл). Через 2 часа смесь обрабатывали 2M водной соляной кислотой (2 мл) и диэтиловым эфиром (3 мл). Осажденный продукт отфильтровывали, промывали диэтиловым эфиром и сушили. Получали указанное в заголовке соединение в виде твердого вещества белого цвета (0,036 г, 56%). ЯМР δН (400МГц, d6-ДМСО); 0,91 (д, 6H, J=6,3Гц), 1,47-1,62 (м, 3H), 3,87 (т, 2H, J=7,5Гц), 11,19 (уш.с, 1H), 14,38 (уш.с, 1H); m/z 257, 259 [MH+].
Пример 14: 4-(8-хлор-2,6-диоксо-1,2,6,7-тетрагидро-3H-пурин-3-ил)бутаннитрил
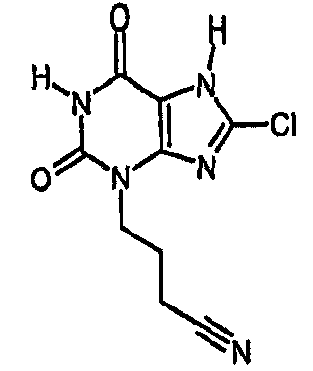
Получают, как в примере 13, с использованием 4-бромбутиронитрила в качестве алкилирующего агента. ЯМР δН (400МГц, d6-ДМСО); 1,89-2,00 (м, 2H), 2,55 (т, 2H, J=7,0Гц), 3,95 (т, 2H, J=6,5Гц), 11,25 (уш.с, 1H), 14,40 (уш.с, 1H); m/z 254 [MH+].
Пример 15: 8-хлор-3-(2-циклогексилэтил)-3,7-дигидро-1H-пурин-2,6-дион
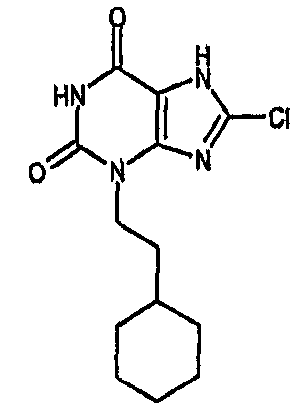
8-Хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион (100 мг, 0,442 ммоль) перемешивали с карбонатом натрия (52 мг, 0,486 ммоль) в сухом ДМФА (3 мл) в течение 30 мин. Добавляли циклогексилэтилбромид (93 мг, 0,486 ммоль) и смесь перемешивали при 37-40°C в атмосфере азота в течение 65 ч, с последующим нагреванием до 90°C в течение 18 ч. После охлаждения раствор дегазировали путем ввода и вывода азота несколько раз, добавляли тетракис(трифенилфосфин)палладий(0) (76 мг, 0,066 ммоль) и морфолин (0,385 мл, 4,42 ммоль) и смесь перемешивали в течение 18 ч. Добавляли дополнительное количество тетракис(трифенилфосфин)палладия(0) (50 мг, 0,043 ммоль) и морфолина (0,2 мл) и перемешивание продолжали еще 1 час. Добавляли этилацетат и 2M водную HCl (приблизительно 10 мл каждого) и органический слой отделяли, промывали насыщенным раствором соли и упаривали. Остаток растворяли в ТГФ и помещали на картридж SPE с 5 г аминопропила. Картридж промывали ТГФ, затем MeOH и кислотный продукт элюировали AcOH в MeOH (5% с повышением до 10%). Продукт, полученный таким образом, далее очищали автопрепаративной ВЭЖХ, получая указанное в заголовке соединение, 5,5 мг, 3%. ЯМР δН (400МГц, d6-ДМСО) 0,80-0,95 (м, 2H), 1,05-1,35 (м, 4H), 1,45-1,55 (м, 2H), 1,55-1,70 (м, 3H), 1,70-1,80 (м, 2H), 3,86 (т, 2H, J=8Гц), 11,07 (с, 1H), один обмениваемый не обнаружен. m/z 297 (MH+).
Пример 16: 3-бутил-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил
a) 3-бутил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
Перемешиваемый раствор 7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (10 г, 52 ммоль) в безводном ДМФА (100 мл) обрабатывали K2CO3 (7,91 г, 57,2 ммоль) и через 10 минут BuI (6,51 мл, 57,2 ммоль). После реакции в течение 2 дней реакционную смесь распределяли между 2M HCl (водн.) и EtOAc. Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали в вакууме с получением твердого вещества грязно-белого цвета. Его промывали горячим циклогексаном и сушили в вакууме с получением указанного в заголовке соединения (8,87 г, 68%); m/z 249,3 [MH+].
b) 3-бутил-1-метил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
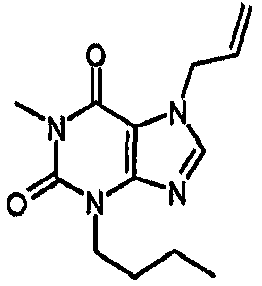
Перемешиваемый раствор 3-бутил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (1,0 г, 4,03 ммоль) в безводном ДМФА (10 мл) обрабатывали Na2CO3 (470 мг, 4,43 ммоль), затем метилйодидом (275 мкл, 4,43 ммоль). Смесь нагревали при 35°C в течение 17 часов. Добавляли K2CO3 (500 мг, 3,6 ммоль) и метилйодид (275 мкл, 4,43 ммоль) и затем перемешивали при 50°C в течение дополнительных 18 часов. Реакционной смеси давали охладиться и затем распределяли между 2M HCl (водн.) и EtOAc. Органический слой отделяли, а водный экстрагировали еще раз EtOAc. Объединенные экстракты промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали, получая желтое/коричневое масло (1,24 г). Продукт очищали SPE c силикагелем (10 г), элюируя смесью EtOAc/циклогексан. Фракции продукта объединяли и концентрировали с получением указанного в заголовке соединения в виде твердого вещества светло-желтого цвета (1,11 г, количеств.); m/z 263,3 [MH+].
c) 3-бутил-1-метил-2,6-диоксо-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбальдегид
Предварительно высушенную колбу заполняли 3-бутил-1-метил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дионом (300 мг, 1,14 ммоль) и безводным ТГФ (6 мл), охлаждали до -75°C в атмосфере азота, затем обрабатывали LiHMDS (1,37 мл 1,0M раствора в ТГФ). Полученному раствору давали нагреться до -60°C более чем за 1,5 часа, после чего добавляли безводный ДМФА (177 мкл, 2,29 ммоль). Раствору давали нагреться до -10°C более чем за 3 часа, затем реакцию гасили насыщенным раствором NH4Cl (водн.). Смесь распределяли между 1М HCl (водн.) и EtOAc. Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали с получением коричневого масла (350 мг). Продукт очищали SPE (Si, 10 г), элюируя смесью EtOAc/циклогексан, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (131 мг, 39%); ЯМР; δH (400МГц, d6-ДМСО) 0,91 (т, 3H, J=7,5Гц), 1,28-1,39 (м, 2H), 1,63-1,73 (м, 2H), 3,25 (с, 3H), 4,02 (т, 2H, J=7,5Гц), 5,03 (дд, 1H, J=17 и 1Гц), 5,17 (дд, 1H, J=10 и 1Гц), 5,31 (прибл.д, 2H, J=5,5Гц), 5,98-6,09 (м, 1H), 9,88 (с, 1H).
d) 3-бутил-1-метил-2,6-диоксо-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил
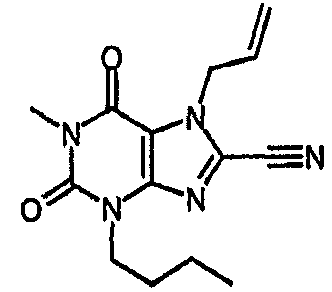
Раствор 3-бутил-1-метил-2,6-диоксо-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбальдегида в безводном пиридине (5 мл) обрабатывали гидрохлоридом гидроксиламина (63 мг, 0,91 ммоль) и нагревали при 50°C в течение 1 часа. Смеси давали охладиться, концентрировали и обрабатывали уксусным ангидридом (5 мл), затем нагревали при 100°C в течение 2,5 часов и при 125°C в течение 45 минут. Снова смеси давали охладиться, затем распределяли между водой и EtOAc. Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали с получением указанного в заголовке соединения в виде желтого остатка (230 мг сырого продукта, 114%); m/z 288,3 [MH+].
e) 3-бутил-1-метил-2,6-диоксо-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил
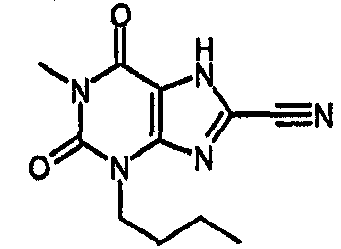
Раствор 3-бутил-1-метил-2,6-диоксо-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрила (230 мг, 0,80 ммоль) в безводном ТГФ (5 мл) и безводном ДМСО (0,5 мл) обрабатывали Pd(PPh3)4 (185 мг, 0,16 ммоль). Смесь дегазировали под действием легкого вакуума, добавляли морфолин (698 мкл) и оставляли для перемешивания при комнатной температуре в атмосфере азота в течение 2 часов. Желтый раствор распределяли между 2M HCl (водн.) и EtOAc. Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали. Остаток помещали в MeOH и отправляли на SPE с аминопропилом (5 г), элюируя MeOH, затем 5% AcOH, затем смесями 10%, 20% и 30% AcOH/MeOH. Фракции продукта объединяли и концентрировали с получением твердого вещества светло-желтого цвета (116 мг). Его промывали MeOH и указанное в заголовке соединение в виде твердого вещества белого цвета собирали фильтрованием и сушили в вакууме (55 мг, 28%). ЯМР; δH (400МГц, d6-ДМСО) 0,90 (т, 3H, J=7,5Гц), 1,25-1,35 (м, 2H), 1,59-1,68 (м, 2H), 3,24 (с, 3H), 3,96 (т, 2H, J=7Гц), NH не наблюдается при δH 15; m/z 248,2 [MH+].
Пример 17: 1-Метил-2,6-диоксо-3-пентил-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил
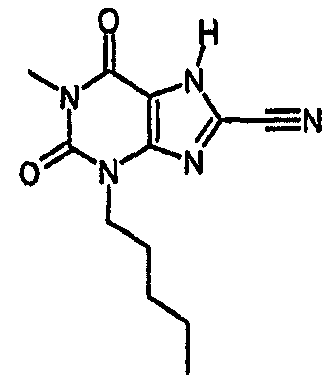
a) 3-пентил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
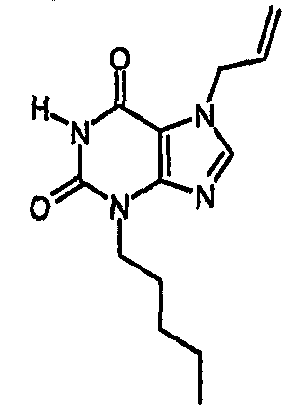
7-(2-Пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион (0,61 г, 3,2 ммоль), карбонат натрия (0,60 г, 5,7 ммоль) и пентилйодид (0,64 г, 3,2 ммоль) перемешивали в ДМФА (5 мл) при 50°C в течение 18 ч. Раствор охлаждали, разделяли между этилацетатом и насыщенным раствором соли и органические фракции выделяли, сушили (MgSO4) и концентрировали. Хроматографией на силикагеле (градиентное элюирование дихлорметан - 5:1 дихлорметан/этилацетат) получали указанное в заголовке соединение в виде твердого вещества светло-желтого цвета (0,47 г, 56%). m/z 263 [MH+].
b) 1-метил-3-пентил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
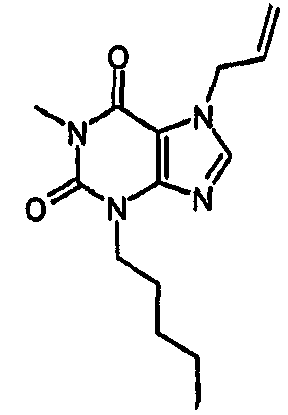
3-Пентил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион (0,20 г, 0,76 ммоль), карбонат калия (0,4 г, 2,9 ммоль) и метилйодид (0,5 мл, 4,9 ммоль) перемешивали и нагревали при 50°C в ДМФА (5 мл) в течение 3 ч. Раствору давали охладиться и разделяли его между этилацетатом и насыщенным раствором соли. Органические фракции выделяли, сушили (MgSO4) и концентрировали, получая указанное в заголовке соединение (0,21 г, 100%). m/z 277 [MH+].
c) 1-метил-2,6-диоксо-3-пентил-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбальдегид.
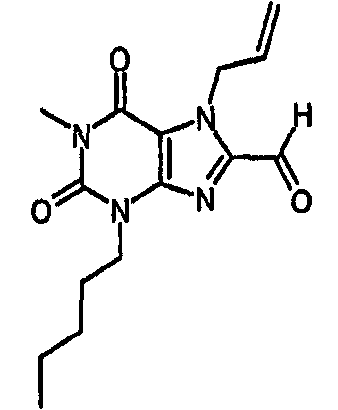
К 1-метил-3-пентил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диону (1,05 г, 3,6 ммоль) в ТГФ (15 мл) при -78°C добавляли LiHMDS (4 мл, 1M в гексане, 4 ммоль) в течение 10 мин и раствор перемешивали в течение 0,5 ч. Добавляли ДМФА (0,5 мл) и раствор перемешивали при -78°C еще 0,5 ч, затем раствору давали нагреться до температуры окружающей среды с охлаждающей ванной в течение 2 ч. Реакцию гасили 2N соляной кислотой (3 мл) и распределяли между этилацетатом и насыщенным раствором соли. Органические фракции выделяли, сушили и концентрировали. Сырой продукт хроматографировали на силикагеле (градиентное элюирование дихлорметан - 5:1 дихлорметан/этилацетат), получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,35 г, 30%). m/z 305 [MH+].
d) 1-метил-2,6-диоксо-3-пентил-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил
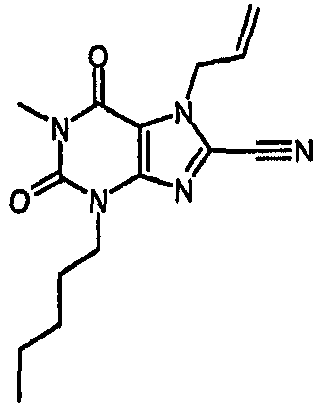
1-Метил-2,6-диоксо-3-пентил-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбальдегид (0,18 г, 0,6 ммоль) и гидрохлорид гидроксиламина (0,053 г, 0,76 ммоль) нагревали при 50°C в пиридине (5 мл) в течение 1 ч, затем охлаждали до температуры окружающей среды. Добавляли уксусный ангидрид (0,08 г, 0,78 ммоль) и раствор перемешивали в течение 18 ч. Раствор концентрировали, получая ацетат, и растворяли в уксусном ангидриде (3 мл) и нагревали при 130°C в течение 3 ч, охлаждали и концентрировали, получая сырой продукт. Хроматографией на силикагеле (элюирование дихлорметаном) получали указанное в заголовке соединение в виде прозрачного масла (0,17 г, 95%). m/z 302 [MH+].
e) 1-метил-2,6-диоксо-3-пентил-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил
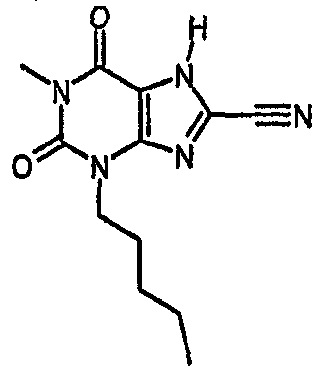
1-Метил-2,6-диоксо-3-пентил-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил (0,17 г, 0,56 ммоль) и морфолин (0,6 мл, 6,7 ммоль) растворяли в ТГФ (5 мл), содержащем ДМСО (0,5 мл). Колбу, содержащую раствор, помещали в вакуум и воздух заменяли азотом (×3). Добавляли тетракис(трифенилфосфин)палладий(0) (0,13 г, 0,11 ммоль) и раствор перемешивали в течение 2,5 ч. Раствор разделяли между этилацетатом (20 мл) и 2N соляной кислотой (10 мл) и органические фракции выделяли и промывали насыщенным раствором соли (3×10 мл). Затем органические фракции промывали 2N раствором гидроксида натрия (2×10 мл) и водную фракцию подкисляли 2N соляной кислотой и экстрагировали этилацетатом (2×10 мл). Органические фракции выделяли, сушили (MgSO4) и концентрировали, получая указанное в заголовке соединение (0,026 г, 18%). ЯМР; δH (400МГц, CDCl3) 0,92 (т, 3H, J=7Гц), 1,32-1,43 (м, 4H), 1,79 (м, 2H), 3,54 (с, 3H), 4,15 (т, 2H, J=7,5Гц), 14,35 (уш.с, 1H); m/z 262 [MH+].
Пример 18: 8-хлор-3-гексил-1-метил-3,7-дигидро-1H-пурин-2,6-дион
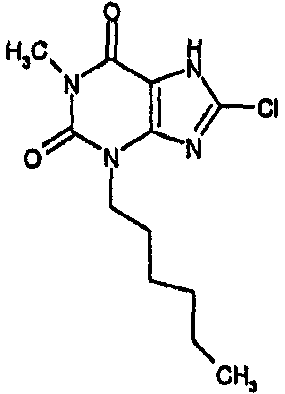
a) 8-хлор-3-({[2-(метилокси)этил]окси}метил)-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
К раствору 8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (6 г, 26,5 ммоль) в безводном ДМФА (30 мл) добавляли карбонат натрия (3,09 г, 29,15 ммоль). После 10 минут перемешивания при комнатной температуре добавляли метоксиэтоксиметилхлорид (3,03 мл, 26,5 ммоль) и перемешивание продолжали в атмосфере азота при комнатной температуре в течение 66 часов. Реакционную смесь концентрировали в вакууме и остаток растворяли в EtOAc (100 мл) и промывали насыщенным раствором соли (100 мл), водный экстракт экстрагировали DCM (100 мл) и органические экстракты сушили (MgSO4), объединяли и концентрировали в вакууме. Остаток растирали в EtOAc и отфильтровывали твердое вещество. Концентрированием фильтрата получали светло-коричневое масло, которое абсорбировали на силикагеле и очищали SPE (Si, 50 г), элюируя градиентом 1:1 EtOAc/циклогексан-EtOAc, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (2 г, 24%), m/z 315,2 [MH+].
b) 8-хлор-1-метил-3-({[2-(метилокси)этил]окси}метил)-7-(2-пропен-1-ил)-3,7-дигидро-1Н-пурин-2,6-дион
К раствору 8-хлор-3-({[2-(метилокси)этил]окси}метил)-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (2 г, 6,37 ммоль) в безводном ДМФА (15 мл) добавляли карбонат натрия (0,743 г, 7 ммоль). После 10 минут перемешивания при комнатной температуре добавляли метилйодид (0,44 мл, 7 ммоль) и перемешивание продолжали в атмосфере азота при комнатной температуре в течение 18 часов. Реакционную смесь концентрировали в вакууме и остаток растворяли в EtOAc (100 мл) и промывали насыщенным раствором соли (100 мл). Органический экстракт сушили (MgSO4), фильтровали и упаривали с получением указанного в заголовке соединения в виде желто-коричневого масла (85% чистота) (2,98 г, количеств.), m/z 329,2 [MH+].
c) 8-хлор-1-метил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
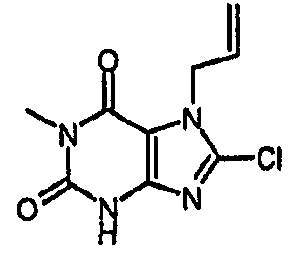
К раствору 8-хлор-1-метил-3-({[2-(метилокси)этил]окси}метил)-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (2,9 г, 6,37 ммоль) в диоксане (20 мл) и воде (20 мл) добавляли 5M HCl (20 мл). Полученную смесь нагревали при 100°C в атмосфере азота в течение 18 часов. Реакционную смесь затем концентрировали в вакууме, остаток растворяли в EtOAc (100 мл) и промывали водой. Органический экстракт сушили (MgSO4), фильтровали и упаривали. Очисткой SPE (Si, 20 г) с элюированием смесью 2:3 EtOAc/циклогексан получали указанное в заголовке соединение в виде твердого вещества белого цвета (1,04 г, 68%). m/z 241,1 [MH+].
Альтернативно, 8-хлор-1-метил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион может быть получен с защитой SEM.
a) 8-хлор-7-(2-пропен-1-ил)-3-({[2-(триметилсилил)этил]окси}метил)-3,7-дигидро-1Н-пурин-2,6-дион
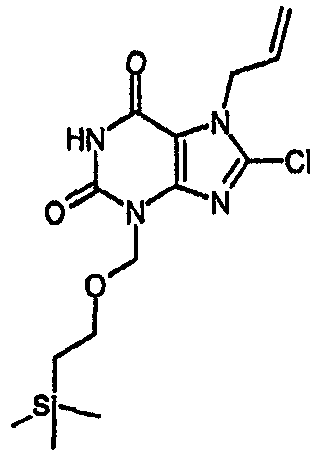
К раствору 8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (5 г, 22,1 ммоль) в ДМФА (80 мл) добавляли 2-2-(триметилсилил)этоксиметилхлорид (4,3 мл, 24,2 ммоль) и карбонат натрия (2,6 г, 24,2 ммоль). После перемешивания в течение ночи при комнатной температуре добавляли дополнительно 2-2-(триметилсилил)этоксиметилхлорид (4,3 мл, 24,2 ммоль) и карбонат натрия (1,3 г, 12,1 ммоль) и перемешивание продолжали в течение 2 часов. Реакционную смесь распределяли между 5% водным LiCl и этилацетатом. Органический экстракт отделяли, промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали. Очисткой хроматографией BiotageTM с использованием картриджа с силикагелем, элюируя смесью 1:4-1:2 этилацетат/циклогексан, получали указанное в заголовке соединение (3,14 г, 40%); m/z 374,2 [MNH4 +].
b) 8-хлор-1-метил-7-(2-пропен-1-ил)-3-({[2-(триметилсилил)этил]окси}метил)-3,7-дигидро-1H-пурин-2,6-дион
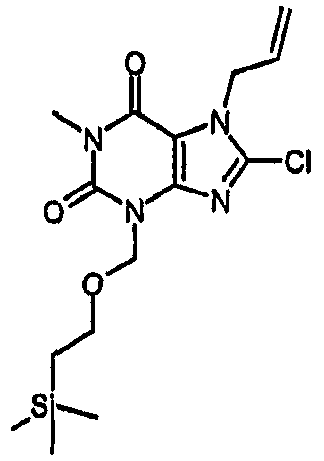
К раствору 8-хлор-7-(2-пропен-1-ил)-3-({[2-(триметилсилил)этил]окси}метил)-3,7-дигидро-1H-пурин-2,6-диона (3,14 г, 8,82 ммоль) в ДМФА (50 мл) добавляли метилйодид (0,659 мл, 10,58 ммоль) и карбонат цезия (3,45 г, 10,58 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь распределяли между водой и этилацетатом. Органический экстракт отделяли, промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали с получением указанного в заголовке соединения 2,99 г (92%); m/z 388 [MNH4 +].
c) 8-хлор-1-метил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
К раствору 8-хлор-1-метил-7-(2-пропен-1-ил)-3-({[2-(триметилсилил)этил]окси}метил)-3,7-дигидро-1H-пурин-2,6-диона (2,99 г, 8,08 ммоль) в DCM (20 мл) добавляли ТФУК (10 мл) и реакционную смесь перемешивали в течение 2,5 часов при комнатной температуре. Реакционную смесь затем концентрировали и остаток обрабатывали DCM и упаривали еще раз. Очисткой SPE (Si), элюируя смесью 1:9-4:1 этилацетат/циклогексан, получали неочищенный продукт (1,31 г), который растворяли в метаноле (20 мл) и обрабатывали насыщенным водным раствором карбоната калия (20 мл). После перемешивания в течение ночи смесь распределяли между водой, содержащей 2M HCl (1 мл), и этилацетатом. Органический экстракт отделяли, промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали с получением 0,87 г указанного в заголовке соединения (45%); m/z 241,1 [MH+].
d) 8-хлор-3-гексил-1-метил-3,7-дигидро-1H-пурин-2,6-дион
К раствору 8-хлор-1-метил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (100 мг, 0,42 ммоль) в безводном ДМФА (3 мл) добавляли карбонат натрия (58 мг, 0,54 ммоль), после 10 минут перемешивания добавляли гексилйодид (0,08 мл, 0,54 ммоль) и реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 90 часов. Добавляли Pd(PPh3)4 (73 мг, 0,063 ммоль), затем из реактора выкачивали воздух и промывали азотом (×3), добавляли морфолин (0,37 мл, 4,3 ммоль) и перемешивание при комнатной температуре в атмосфере азота продолжали в течение 4 часов. Реакционную смесь разбавляли EtOAc (25 мл) и промывали 2M водным раствором HCl (25 мл). Органический экстракт сушили (MgSO4), фильтровали и упаривали. Очисткой SPE с аминопропилом (5 г) загруженного соединения и промывкой MeOH перед элюированием продукта смесью 5% AcOH/MeOH получали указанное в заголовке соединение в виде твердого вещества белого цвета (65 мг, 54%). ЯМР; δH (400МГц, d6-ДМСО) 0,85 (т, 3H, J=7Гц), 1,23-1,33 (м, 6H), 1,58-1,68 (м, 2H), 3,22 (с, 3H), 3,91 (т, 2H, J=7,5Гц), 14,46 (уш.с, 1H); m/z 285,3 [MH+].
Пример 19: 8-хлор-1-метил-3-пропил-3,7-дигидро-1H-пурин-2,6-дион
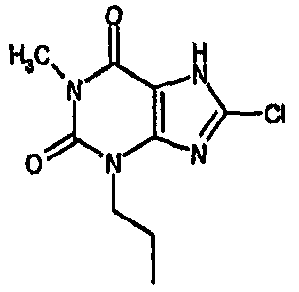
Получают подобно примеру 18, но используя пропилйодид для алкилирования по N3.
ЯМР δH (400МГц, d6-ДМСО) 0,87 (т, 3H, J=7,5Гц), 1,61-1,73 (м, 2H), 3,22 (с, 3H), 3,89 (т, 2H, J=7,5Гц), 14,45 (уш.с, 1H), m/z 243 [MH+].
Пример 20: 1,3-дибутил-2,6-диоксо-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил
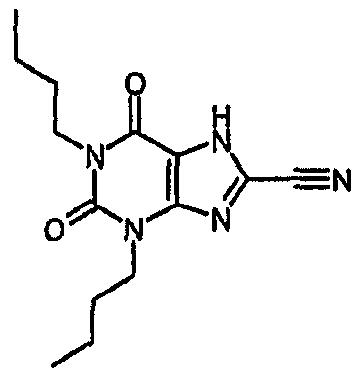
a) 1,3-дибутил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
Раствор 1,3-ди-N-бутилксантина (10 г, 38 ммоль) в безводном ДМФА (80 мл) обрабатывали K2CO3 (5,2 г, 38 ммоль), затем аллилбромидом (3,6 мл, 42 ммоль). Смесь нагревали при 55°C в атмосфере азота в течение 18 часов. После охлаждения до комнатной температуры смесь распределяли между водой и EtOAc. Несколько мл 2M HCl (водн.) добавляли, чтобы помочь разделению. Органический слой отделяли, а водный экстрагировали еще раз EtOAc. Объединенные экстракты промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали с получением указанного в заголовке соединения в виде твердого вещества грязно-белого цвета (12,23 г, 106%). m/z 305,3 [MH+].
b) метил 1,3-дибутил-2,6-диоксо-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбоксилат
Раствор 1,3-дибутил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (3,0 г, 9,9 ммоль) в безводном ТГФ (30 мл) охлаждали до -50°C и обрабатывали LiHMDS (18 мл 1,0M раствора в ТГФ, 17,8 ммоль). Через 1 час при -50°C добавляли метилхлорформиат (1,9 мл, 24,6 ммоль) и смеси давали нагреться до -30°C более чем за 2 часа, затем реакцию гасили насыщенным раствором NH4Cl (водн.). Смесь распределяли между EtOAc и 1M HCl (водн.). Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали с получением темного оранжевого масла (4,07 г). Масло помещали в смесь 15% EtOAc/циклогексан и загружали на хроматографическую колонку SiBiotageTM. Фракции продукта объединяли и концентрировали с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (1,35 г, 38%). m/z 363,2 [MH+].
c) 1,3-дибутил-2,6-диоксо-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбоновая кислота
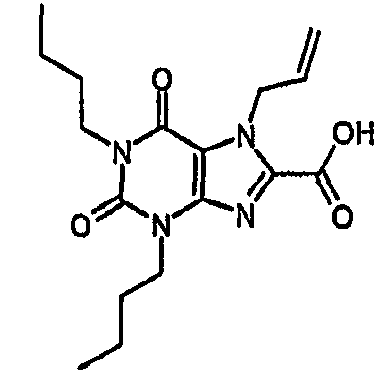
Перемешиваемый раствор метил 1,3-дибутил-2,6-диоксо-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбоксилата (1,30 г, 3,6 ммоль) в MeOH (15 мл) обрабатывали LiOH (215 мг) и водой (1,5 мл). Через 3 часа при комнатной температуре смесь разбавляли водой и рН доводили до pH5 с помощью 2M HCl (водн.). Добавляли EtOAc и затем разделяли, промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали с получением указанного в заголовке соединения в виде твердого вещества желтого цвета с чистотой 85% (1,2 г, 88%). m/z. 349,2 [MH+].
d) 1,3-дибутил-2,6-диоксо-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбоксамид
Перемешиваемый раствор 1,3-дибутил-2,6-диоксо-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбоновой кислоты (1,0 г, 2,9 ммоль) в безводном ДМФА (10 мл) последовательно обрабатывали DIPEA (1,1 мл), PyBOP и 2M NH3 (3,6 мл). Через 2 часа смесь продукта распределяли между 2M HCl (водн.) и EtOAc. Органический слой отделяли, промывали насыщенным раствором NaHCO3 (водн.), насыщенным раствором соли, затем сушили (MgSO4) и концентрировали, получая оранжевое масло (приблизительно 2 г). Продукт очищали хроматографией BiotageTM, элюируя смесями 5%→40% EtOAc/циклогексан. Соответствующие фракции объединяли и концентрировали, получая амид с чистотой 90% (790 мг, 78%). m/z. 392,3 [M+муравьиная кислота-H]-.
e) 1,3-дибутил-2,6-диоксо-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил
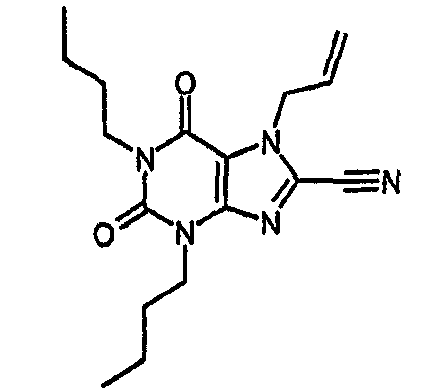
Раствор 1,3-дибутил-2,6-диоксо-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбоксамида (300 мг) в безводном ДМФА (7 мл) при 0°C обрабатывали по каплям POCl3 (237 мкл). Ванну со льдом удаляли и через 2 часа смесь распределяли между водой и Et2O. Водный слой повторно экстрагировали Et2O и объединенные экстракты отделяли, промывали водой (×2), насыщенным раствором соли, затем сушили (MgSO4) и концентрировали, получая желтое масло (312 мг). Масло помещали в циклогексан и очищали с помощью SPE (Si, 10 г), элюируя смесями EtOAc/циклогексан. Концентрированием фракций продукта получали указанное в заголовке соединение в виде бесцветного масла (150 мг, 53%). m/z 330,3 [MH+].
f) 1,3-дибутил-2,6-диоксо-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрил
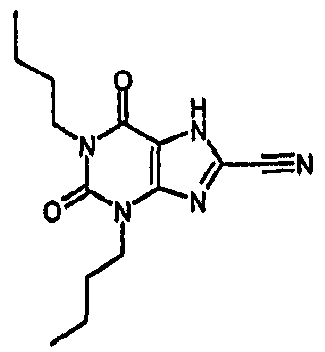
Раствор 1,3-дибутил-2,6-диоксо-7-(2-пропен-1-ил)-2,3,6,7-тетрагидро-1H-пурин-8-карбонитрила (140 мг, 0,43 ммоль) в безводном ТГФ (4 мл) и безводном ДМСО (0,4 мл) обрабатывали Pd(PPh3)4 (74 мг, 0,064 ммоль). Смесь дегазировали под действием легкого вакуума, добавляли морфолин (371 мкл) и оставляли для перемешивания при комнатной температуре в атмосфере азота в течение 4 часов. Желтый раствор распределяли между 2M HCl (водн.) и EtOAc. Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали. Остаток помещали в MeOH и отправляли на SPE с аминопропилом (5 г), элюируя MeOH, затем смесью 5%→50% AcOH/MeOH. Продукт элюировал с малым количеством примесей, которые вымывали, после концентрирования, циклогексаном с получением указанного в заголовке соединения в виде твердого вещества грязно-белого цвета (30 мг, 24%). ЯМР δН (400МГц, d6-ДМСО) 0,89 (прибл.тд, 6H, J=7 и 3Гц), 1,25-1,35 (м, 4H), 12,48-1,55 (м, 2H), 1,58-1,69 (м, 2H), 3,87 (т, 2H, J=7Гц), 3,95 (т, 2H, J=7Гц), NH не обнаружен при δН 15; m/z 290,3 [MH+].
Пример 21: 1,3-дибутил-8-йод-3,7-дигидро-1H-пурин-2,6-дион
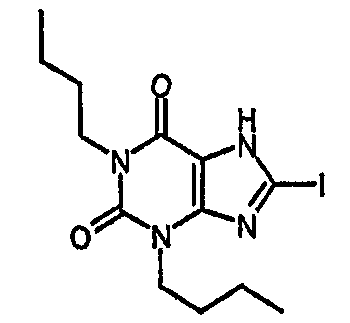
Перемешиваемый раствор 1,3-ди-N-бутилксантина (100 мг, 3,39 ммоль) в безводном ДМФА (3 мл) обрабатывали NIS (94 мг, 3,75 ммоль) и оставляли для перемешивания при комнатной температуре в атмосфере азота в течение 23 часов. Смесь распределяли между насыщенным раствором Na2SO3 (водн.) и EtOAc. Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали в вакууме. Продукт очищали на картридже SPE (Si, 5 г), элюируя смесями EtOAc/циклогексан. Фракцию продукта концентрировали с получением указанного в заголовке соединения в виде твердого вещества белого цвета (75 мг, 51%); ЯМР; δН (400МГц, d6-ДМСО) (прибл.тд, 6H, J=7,5 и 4Гц), 1,21-1,34 (м, 4H), 1,45-1,54 (м, 2H), 1,56-1,66 (м, 2H), 3,84 (т, 2H, J=7,5Гц), 3,93 (т, 2H, J=7,5Гц), 14,10 (с, 1 H); m/z 391,3 [MH+].
Пример 22: (3-бутил-8-хлор-2,6-диоксо-2,3,6,7-тетрагидро-1H-пурин-1-ил)ацетонитрил
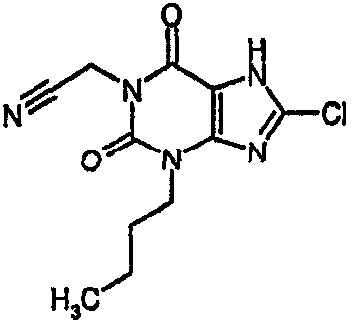
К смеси 3-бутил-8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (200 мг, 0,707 ммоль) и Cs2CO3 (254 мг, 0,778 ммоль) в безводном ДМФА (5 мл) добавляли хлорацетонитрил (0,054 мл, 0,85 ммоль). Смесь нагревали при 50°C в течение 18 часов, затем давали остыть до комнатной температуры и дегазировали под действием легкого вакуума, после чего вводили азот. Это повторяли дважды. Добавляли Pd(PPh3)4 (82 мг, 0,071 ммоль) и смесь дегазировали еще раз, после чего добавляли морфолин (0,617 мл, 7,07 ммоль) и смесь оставляли для перемешивания в течение 3 часов при комнатной температуре. Смесь распределяли между 2M HCl (водн.) и EtOAc. Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали. Остаток помещали в MeOH и отправляли на SPE с аминопропилом (5 г), элюируя MeOH, затем смесью 5-10% AcOH/MeOH. Фракцию продукта концентрировали, получая указанное в заголовке соединение, 52 мг (26%); ЯМР; δН (400МГц, d6-ДМСО) 0,90 (т, 3H, J=7,5Гц), 1,26-1,37 (м, 2H), 1,60-1,69 (м, 2H), 3,94 (т, 2H, J=7,5Гц), 4,87 (с, 2H), 14,72 (уш.с, 1H); m/z 299,2 [MNH4 +].
Пример 23: (8-хлор-2,6-диоксо-3-пропил-2,3,6,7-тетрагидро-1H-пурин-1-ил)ацетонитрил
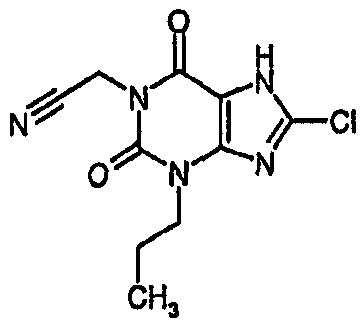
a) 8-хлор-7-(2-пропен-1-ил)-3-пропил-3,7-дигидро-1H-пурин-2,6-дион
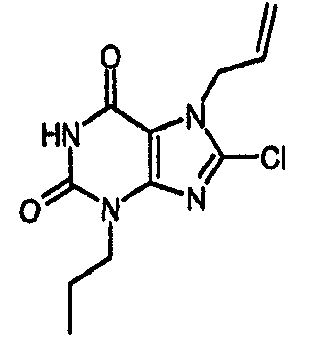
Смесь 8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (1,5 г, 6,6 ммоль), 1-йодопропана (1,2 г, 6,9 ммоль) и карбоната натрия (0,9 г, 8,5 ммоль) в ДМФА (40 мл) нагревали при 50°C в течение 18 часов. Реакционную смесь концентрировали в вакууме и остаток обрабатывали водой (60 мл) и экстрагировали этилацетатом (3×80 мл). Объединенные органические экстракты сушили (MgSO4), фильтровали и упаривали. Остаток растирали в смеси эфир/циклогексан, твердое вещество отфильтровывали и сушили, получая указанное в заголовке соединение (0,82 г, 46%); m/z 269,1 [MH+].
b) (8-хлор-2,6-диоксо-3-пропил-2,3,6,7-тетрагидро-1H-пурин-1-ил)ацетонитрил
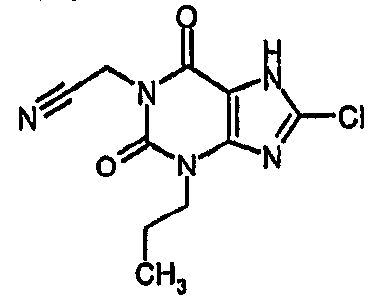
Раствор 8-хлор-7-(2-пропен-1-ил)-3-пропил-3,7-дигидро-1H-пурин-2,6-диона (0,067 г, 0,25 ммоль) в ДМФА (2 мл) обрабатывали карбонатом цезия (0,082 г, 0,25 ммоль) и бромацетонитрилом (0,044 г, 0,37 ммоль). Смесь нагревали при 80°C в течение 4 часов, затем охлаждали до температуры окружающей среды. ДМФА удаляли в вакууме и остаток обрабатывали ТГФ (2 мл). Растворитель дегазировали последовательным применением вакуума и давления азота к реакционной смеси. Смесь затем обрабатывали морфолином (0,035 мл, 0,4 ммоль) и тетракис(трифенилфосфин)палладием(0) (0,03 г, 0,026 ммоль). Через 2 часа смесь обрабатывали 2M водной соляной кислотой (2 мл) и продукт экстрагировали хлороформом (3×5 мл). Органические фракции объединяли и упаривали. Остаток подвергали очистке масс-направленной ВЭЖХ с получением указанного в заголовке соединения в виде твердого вещества белого цвета (0,022 г, 33%). ЯМР; δH (400МГц, d6-ДМСО), 0,88 (т, 3H, J=7,5Гц), 1,63-1,74 (м, 2H), 3,91 (т, 2H, J=7,5Гц), 4,87 (с, 2H), NH не наблюдается при δH 14; m/z 268 [MH+].
Пример 24: [8-хлор-3-(2-циклопропилэтил)-2,6-диоксо-2,3,6,7-тетрагидро-1H-пурин-1-ил]ацетонитрил
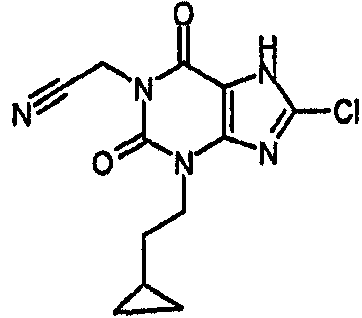
Получают, как (8-хлор-2,6-диоксо-3-пропил-2,3,6,7-тетрагидро-1H-пурин-1-ил)ацетонитрил (пример 23), используя 8-хлор-3-(2-циклопропилэтил)-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион.
ЯМР δH (400МГц, d6-ДМСО) -0,06-0,00 (м, 2H), 0,31-0,39 (м, 2H), 0,64-0,74 (м, 1H), 1,57 (кв, 2H, J=7Гц), 4,04 (т, 2H, J=7Гц), 4,87 (с, 2H), 14,68 (уш.с, 1H); m/z 294 [MH+].
Пример 25: 8-хлор-1-этил-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-дион
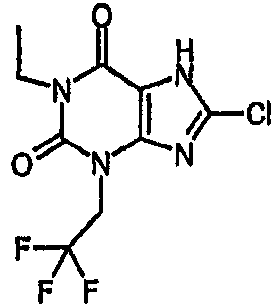
a) 8-хлор-7-(2-пропен-1-ил)-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-дион
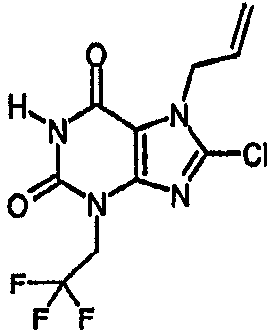
К раствору 8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (1,5 г, 6,62 ммоль) в безводном ДМФА (50 мл) добавляли бикарбонат натрия (0,98 г, 9,25 ммоль), затем 1,1,1-трифтор-2-йодэтан (1,20 г, 5,72 ммоль) и смесь нагревали при перемешивании в течение 6 ч при 50°C в атмосфере азота. Раствору давали охладиться до температуры окружающей среды в течение 10 ч, затем нагревали в течение 48 ч при 120°C. Дополнительно добавляли 1,1,1-трифтор-2-йодэтан (0,43 г, 2,05 ммоль) и смесь нагревали при 120°C еще 3 ч. Растворитель удаляли при пониженном давлении и остаток растирали в DCM, затем отфильтровывали.
Реакцию повторяли, используя 8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион (3,80 г, 16,8 ммоль), бикарбонат натрия (2,45 г, 23,1 ммоль) и 1,1,1-трифтор-2-йодэтан (4,05 г, 19,3 ммоль) в безводном ДМФА (125 мл). Смесь нагревали в течение 16 ч при 120°C, растворитель удаляли при пониженном давлении и остаток растирали в DCM, затем отфильтровывали.
Фильтраты DCM от двух фильтрований объединяли, концентрировали при пониженном давлении, затем очищали, используя хроматографию BiotageTM (элюируя смесью циклогексан/этилацетат 1:1, затем 7:3), с получением указанного в заголовке соединения в виде твердого вещества белого цвета (1,6 г, 23%). m/z 309 [MH+].
b) 8-хлор-1-этил-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-дион
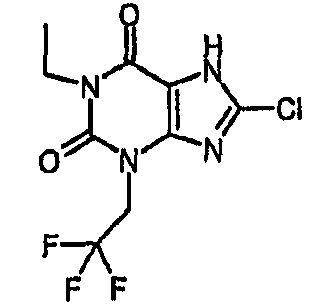
К раствору 8-хлор-7-(2-пропен-1-ил)-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-диона (0,070 г, 0,23 ммоль) в безводном ДМФА (2 мл) добавляли карбонат цезия (0,085 г, 0,26 ммоль), затем 1-йодэтан (0,061 г, 0,39 ммоль). Смесь нагревали в течение 5 ч при 80°C, затем перемешивали в течение 16 ч при температуре окружающей среды в атмосфере азота. Растворитель удаляли при пониженном давлении, используя вакуумную центрифугу, и остаток растворяли в безводном ТГФ (2,5 мл). К смеси добавляли тетракис палладия (0,030 г, 0,026 ммоль) и морфолин (0,040 г, 0,45 ммоль) и реакционную смесь дегазировали с использованием азота, затем перемешивали при температуре окружающей среды в течение 72 ч. Смесь распределяли между хлороформом и 2N водной HCl и водный слой повторно экстрагировали. Органические экстракты объединяли и выпаривали в токе азота, затем очищали, используя SPE c аминопропилом (элюируя смесью уксусная кислота:метанол:DCM, 1:2:2), с получением указанного в заголовке соединения в виде твердого вещества белого цвета с чистотой >95% (0,041 г, 60%). ЯМР δН (400МГц, d4-MeOD) 1,20 (т, 3H, J=7Гц), 4,03 (кв, 2H, J=7Гц), 4,73 (кв, 2H, J=8,5Гц), m/z 297 [MH+].
Пример 26: 8-хлор-1-пропил-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-дион
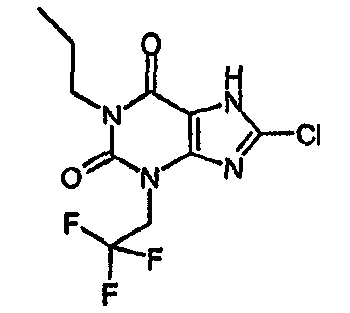
Получают подобно примеру 25, используя пропилйодид для алкилирования по N1.
ЯМР δH (400МГц, CDCl3) 0,99 (т, 3H, J=7,5Гц), 1,68-1,79 (м, 2H), 4,07 (т, 2H, J=7,5Гц), 4,77 (кв, 2H, J=8,5Гц), NH не обнаружен при δH 13; m/z 311 [MH+].
Пример 27: 8-хлор-1-(4,4,4-трифторбутил)-3-(2,2,2-трифторэтил)-3,7-дигидро-1H-пурин-2,6-дион
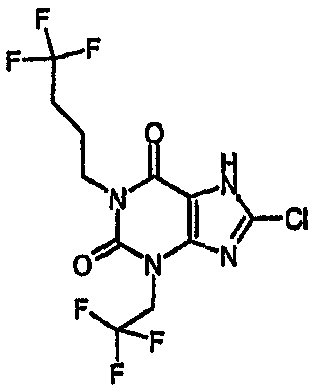
Получают подобно примеру 25, используя 4-бром-1,1,1-трифторбутан для алкилирования по N1.
ЯМР; δH (400МГц, d4-MeOD) 1,83-1,95 (м, 2H), 2,14-2,32 (м, 2H), 4,06 (т, 2H, J=7Гц), 4,74 (кв, 2H, J=8,5Гц), m/z 377 [М-Н]-.
Пример 28: 8-бром-1-метил-3-пентил-3,7-дигидро-1H-пурин-2,6-дион
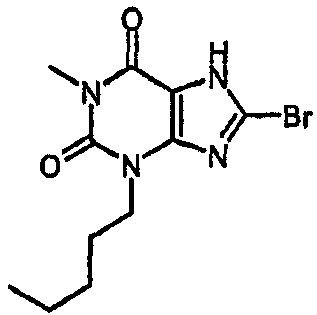
a) 1-метил-3-пентил-3,7-дигидро-1H-пурин-2,6-дион
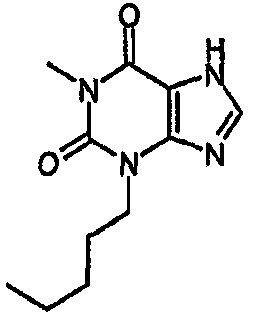
1-Метил-3-пентил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион (0,45 г, 1,63 ммоль), фенилсилан (0,25 мл, 2,03 ммоль) и тетракис(трифенилфосфин)палладий(0) (0,35 г, 0,3 ммоль) растворяли в DCM (10 мл), содержащем уксусную кислоту (6 мл). Воздух в колбе заменяли азотом, выкачивая воздух из колбы с последующим заполнением азотом (×3), и реакционную смесь нагревали при 45°C в течение 4 ч. Раствору давали охладиться, разбавляли DCM, затем промывали водой, затем насыщенным раствором бикарбоната натрия. Органические фазы выделяли, сушили и концентрировали, получая сырой продукт. Очисткой SPE (силикагель) с элюированием простым эфиром получали продукт, 0,06 г, 16%. m/z 237 [MH+].
b) 8-бром-1-метил-3-пентил-3,7-дигидро-1H-пурин-2,6-дион
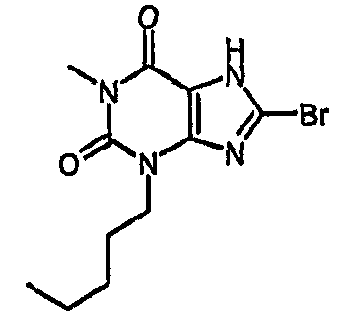
1-Метил-3-пентил-3,7-дигидро-1H-пурин-2,6-дион (0,06 г, 0,25 ммоль) растворяли в ДМФА (2 мл) и добавляли N-бромсукцинамид (0,045 г, 0,25 ммоль). Смесь перемешивали в течение 18 ч, концентрировали и сырой продукт очищали, элюируя посредством SPE с аминопропилом (5 г) сначала метанолом, затем смесью 5% уксусная кислота/метанол, для элюирования продукта. Продукт далее очищали масс-направленной auto prep, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,01 г, 12%). ЯМР δH (400МГц, d6-ДМСО) 0,86 (т, 3H, J=7Гц), 1,21-1,35 (м, 4H), 1,59-1,68 (м, 2H), 3,22 (с, 3H), 3,91 (т, 2H, J=7,5Гц), 14,39 (уш.с, 1H); m/z 315, 317 [MH+].
Пример 29: 8-хлор-1-метил-3-пентил-3,7-дигидро-1H-пурин-2,6-дион
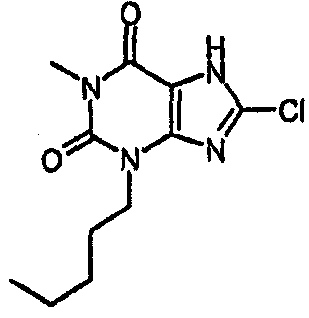
a) 8-хлор-1-метил-3-пентил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
К раствору 8-хлор-3-пентил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (3,9 г, 13,3 ммоль) в ДМФА (35 мл) добавляли карбонат цезия и смесь перемешивали в течение 10 мин, после чего добавляли йодметан (0,91 мл, 14,6 ммоль) и смесь перемешивали в течение 18 ч. Реакционную смесь распределяли между этилацетатом и 2N раствором HCl и органические фазы выделяли, сушили (MgSO4) и концентрировали. Хроматографией на силикагеле SPE, элюируя смесью циклогексан/этилацетат (5%-20%), получали продукт в виде масла, 2,78 г, 68%. m/z 311 [MH+].
b) 8-хлор-1-метил-3-пентил-3,7-дигидро-1H-пурин-2,6-дион
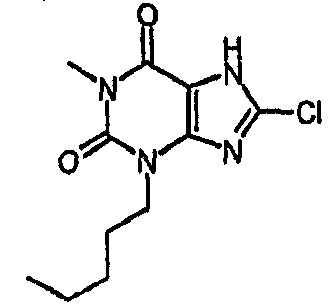
Тетракис(трифенилфосфин)палладий (1,0, 0,90 ммоль) помещали в колбу, из которой был выкачан воздух и которая затем была заполнена азотом (×3). Добавляли раствор 8-хлор-1-метил-3-пентил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (2,78 г, 8,96 ммоль) в 50 мл ТГФ и в колбе снова создавали вакуум и вводили азот. Добавляли ДМСО (4,5 мл) и морфолин (7,8 мл, 89,6 ммоль) и раствор перемешивали в течение 5 ч. Раствор распределяли между этилацетатом и 2N раствором HCl и органическую фракцию промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали. Сырой продукт очищали SPE c аминопропилом, элюируя сначала метанолом, затем метанолом, содержащим 0-15% уксусной кислоты, с получением указанного в заголовке соединения в виде твердого вещества белого цвета, 1,12 г, 46%. ЯМР δН (400МГц, d6-ДМСО) 0,86 (т, 3H, J=7Гц), 1,21-1,35 (м, 4H), 1,59-1,68 (м, 2H), 3,22 (с, 3H), 3,91 (т, 2H, J=7,5Гц), NH не обнаружен; m/z 271 [MH+].
Пример 30: 3-бутил-8-хлор-1-метил-3,7-дигидро-1H-пурин-2,6-дион
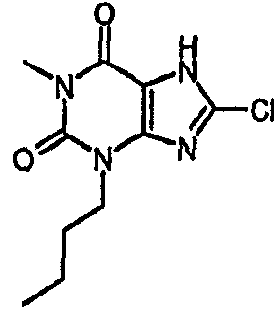
Получают подобно примеру 29, используя 3-бутил-8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион в качестве исходного материала.
ЯМР δН (400МГц, d6-ДМСО) 0,88 (т, 3H, J=7Гц), 1,25-1,35 (м, 2H), 1,6-1,66 (м, 2H), 3,22 (с, 3H), 3,91 (т, 2H, J=7,5Гц), 14,46 (уш.с, 1H); m/z 257 [MH+].
Пример 31: 4-(8-хлор-1-метил-2,6-диоксо-1,2,6,7-тетрагидро-3H-пурин-3-ил)бутаннитрил
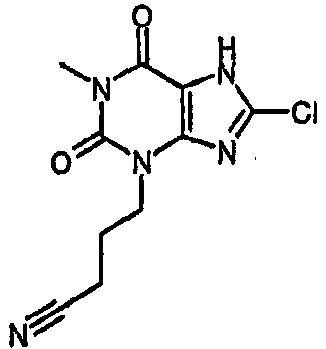
К смеси 8-хлор-1-метил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (70 мг, 0,292 ммоль) и Na2CO3 (37 мг, 0,35 ммоль) в ДМФА (3 мл) добавляли 4-бромбутиронитрил (0,035 мл, 0,35 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, затем дегазировали под действием легкого вакуума и вводили азот. Затем последовательно добавляли Pd(PPh3)4 (50 мг, 0,044 ммоль) и морфолин (0,254 мл, 2,92 ммоль). После двух часов перемешивания при комнатной температуре далее добавляли свежий Pd(PPh3)4 (50 мг, 0,044 ммоль) и перемешивание продолжали в течение ночи. Реакционную смесь распределяли между этилацетатом (20 мл) и водой (20 мл) с добавлением малого количества 2M HCl, чтобы помочь разделению. Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4) и концентрировали. Остаток помещали в MeOH и отправляли на SPE с аминопропилом (5 г), элюируя MeOH, затем смесью 3-5% AcOH/MeOH. Фракцию продукта концентрировали с получением указанного в заголовке соединения, 39,7 мг (51%); ЯМР; δН (400МГц, d6-ДМСО) 1,91-2,00 (м, 2H), 2,55 (т, 2H, J=7Гц), 3,22 (с, 3H), 4,03 (т, 2H, J=7Гц), 14,49 (уш.с, 1H); m/z 268,1 [MH+].
Пример 32: 8-хлор-1-метил-3-(4,4,4-трифторбутил)-3,7-дигидро-1H-пурин-2,6-дион
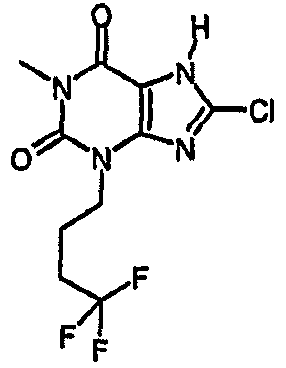
Раствор 8-хлор-1-метил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (0,048 г, 0,2 ммоль) в ТГФ (1 мл) обрабатывали карбонатом цезия (0,78 г, 0,24 ммоль) и 4-бром-1,1,1-трифторбутаном (0,044 г, 0,25 ммоль). Смесь перемешивали при температуре окружающей среды в течение 1 часа, затем нагревали при 50°C в течение 4 часов и затем охлаждали. Смесь дегазировали, поочередно применяя вакуум и давление азота к смеси, и затем обрабатывали морфолином (0,17 мл, 2 ммоль) и тетракис(трифенилфосфин)палладием(0) (0,023 г, 0,02 ммоль). Через 2 часа смесь осторожно обрабатывали 2M водной соляной кислотой (2 мл) и продукт экстрагировали хлороформом (2×4 мл). Объединенные органические фракции упаривали и продукт очищали масс-направленной ВЭЖХ с обращенными фазами, получая указанное в заголовке соединение, 6,2 мг (10%); ЯМР; δH (400МГц, d6-ДМСО); 1,84-1,92 (м, 2H), 2,28-2,35 (м, 2H), 3,22 (с, 3H), 3,99-4,03 (м, 2H) 14,31 (уш.с, 1H); m/z 311,2 [MH+].
Пример 33: 3-бутил-8-хлор-1-этил-3,7-дигидро-1H-пурин-2,6-дион
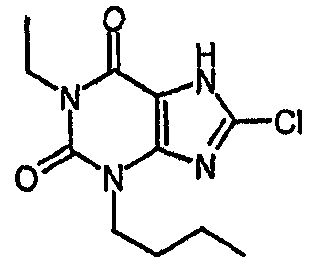
a) 3-бутил-7-(фенилметил)-3,7-дигидро-1H-пурин-2,6-дион
7-Бензил-3,7-дигидро-1H-пурин-2,6-дион (17,14 г, 70,8 ммоль) [Synthetic Communications, 20(16), 2459-2467, 1990] и карбонат калия (11,43 г, 82,8 ммоль) суспендировали в ДМФА (400 мл) при 40°C. После перемешивания в течение тридцати минут добавляли бутилйодид (8,76 мл, 77,0 ммоль) и смесь перемешивали при 40°C в течение ночи. Добавляли 50% водную уксусную кислоту (60 мл) и раствор концентрировали при пониженном давлении. Остаток суспендировали в воде (500 мл) и продукты экстрагировали хлороформом. Органические фракции собирали, концентрировали и выделяли продукт, используя флэш-хроматографию и элюируя 1% метанолом в дихлорметане, с получением продукта (9,49 г, 45%); 1Н ЯМР (400МГц; CDCl3) δ: 0,95 (3H, т), 1,34-1,41 (2H, м), 1,70-1,78 (2H, м), 4,05 (2H, т), 5,46 (2H, с), 7,31-7,40 (5H, м), 7,56 (1Н, с), 8,21 (1Н, уш.с); m/z 299 [MH+].
b) 3-бутил-1-этил-7-(фенилметил)-3,7-дигидро-1H-пурин-2,6-дион
3-Бутил-7-(фенилметил)-3,7-дигидро-1H-пурин-2,6-дион (0,429 г, 1,24 ммоль) и карбонат калия (0,256 г, 1,85 ммоль) суспендировали в ДМФА (8 мл), добавляли йодэтан (0,113 мл, 1,42 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Реакционную смесь выпаривали досуха и остаток распределяли между водой и этилацетатом. Органический слой промывали водой, затем насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении, получая указанное в заголовке соединение; 1Н ЯМР (400МГц; CDCl3) δ: 0,96 (3H, т), 1,25 (3H, т), 1,36-1,45 (2H, м), 1,72-1,76 (2H, м), 4,05-4,13 (4H, м), 5,50 (2H, с), 7,32-7,40 (5H, м), 7,52 (1H, с); m/z 327 [MH+].
c) 3-бутил-1-этил-3,7-дигидро-1H-пурин-2,6-дион
3-Бутил-1-этил-7-(фенилметил)-3,7-дигидро-1H-пурин-2,6-дион (0,353 г, 1,08 ммоль) растворяли в уксусной кислоте (30 мл), добавляли 20% гидроксид палладия на угле (0,238 г) и смесь взбалтывали в атмосфере водорода (50 psi) в течение ночи. Катализатор удаляли фильтрованием через Celite® и промывали уксусной кислотой. Фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение (0,227 г, 89%); 1H ЯМР (400МГц; CDCl3) δ: 0,97 (3H, т), 1,28 (3H, т), 1,38-1,47 (2H, м), 1,74-1,82 (2H, м), 4,12-4,17 (4H, м), 7,80 (1H, с); m/z 237 [MH+].
d) 3-бутил-8-хлор-1-этил-3,7-дигидро-1H-пурин-2,6-дион
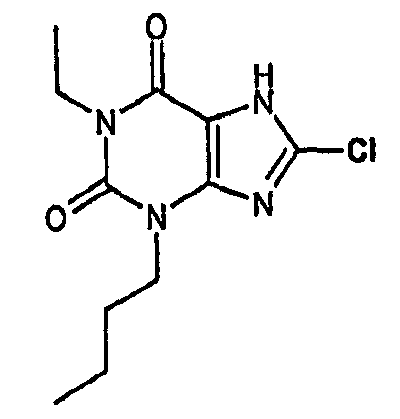
3-Бутил-1-этил-3,7-дигидро-1H-пурин-2,6-дион (100 мг, 0,42 ммоль) и NCS (56 мг, 0,42 ммоль) суспендировали в MeCN (5 мл) и нагревали при 120°C при микроволновом облучении. Реакционную смесь концентрировали при пониженном давлении и выделяли указанное в заголовке соединение, используя ВЭЖХ. [Условия ВЭЖХ, используемые для очистки: время обработки 23 минуты. Растворители: 0,1% ТФУК в MeCN и 0,1% ТФУК в воде. MeCN увеличивали от 5% до 95% линейно более чем за 15 минут. Удерживали на уровне 95% в течение 2 мин. Затем понижали до 5% линейно более чем за 1 мин, уравновешивали на уровне 5% в течение 5 минут перед следующей инъекцией.]; 1Н ЯМР (400МГц; CDCl3) δ: 0,97 (3H, т), 1,31 (3H, т), 1,38-1,45 (2H, м), 1,72-1,80 (2H, м), 4,09-4,20 (4H, м), 13,40 (1H, уш.с); m/z 271 [MH+].
Пример 34: 8-хлор-3-(4-метилпентил)-3,7-дигидро-1H-пурин-2,6-дион
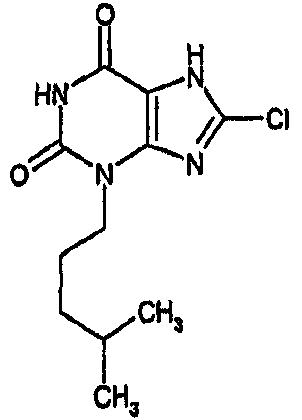
Исходя из 1-бром-4-метилпентана (81 мг).
Перекристаллизовывали из MeOH.
Выход 34,8 мг (29%), ЯМР; (400МГц, d6-ДМСО) δH 0,83 (д, 6H, J=8Гц), 1,12-1,22 (м, 2H), 1,55 (септет, 1H, J=8Гц), 1,58-1,68 (м, 2H), 3,83 (т, 2H, J=7,5Гц), 11,20 (с, 1H); m/z 271 [MH+].
Пример 35: 6-(8-хлор-2,6-диоксо-1,2,6,7-тетрагидро-3H-пурин-3-ил)-2,2-диметилгексаннитрил
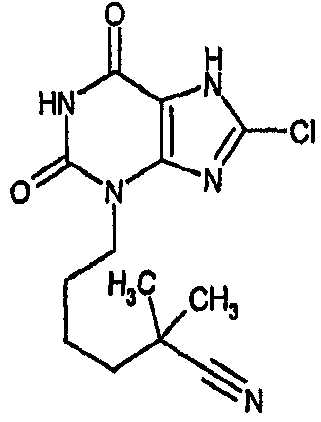
Исходя из 6-бром-2,2-диметилгексаннитрила (100 мг).
Перекристаллизовывали из MeOH.
Выход 48,5 мг (35%); ЯМР; (400МГц, d6-ДМСО) δH 1,27 (с, 6H), 1,35-1,44 (м, 2H), 1,54-1,59 (м, 2H), 1,63-1,72 (м, 2H), 3,88 (т, 2H, J=7Гц), 11,24 (с, 1H); m/z 310 [MH+].
Пример 36: 8-хлор-3-(6-метилгептил)-3,7-дигидро-1H-пурин-2,6-дион
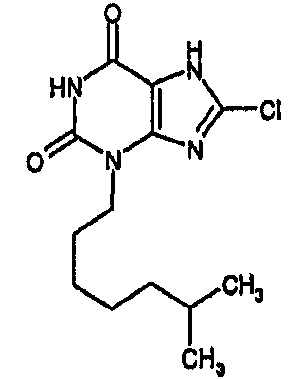
Исходя из 1-бром-6-метилгептана (95 мг).
Перекристаллизовывали из MeOH.
Выход 36 мг (27%), ЯМР; (400МГц, d6-ДМСО) δH 0,83 (д, 6H, J=7,5Гц), 1,10-1,17 (м, 2H), 1,20-1,34 (м, 4H), 1,48 (септет, 1Н, J=7,5Гц), 1,58-1,68 (м, 2H), 3,84 (т, 2H, J=8Гц), 11,22 (с, 1H); m/z 299 [MH+].
Пример 37: 8-хлор-3-октил-3,7-дигидро-1H-пурин-2,6-дион
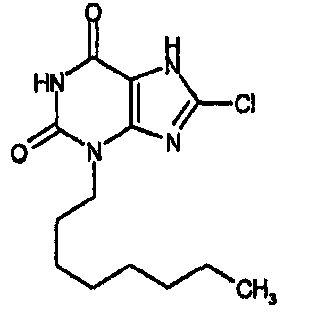
8-Хлор-3,7-дигидро-1H-пурин-2,6-дион (100 мг, 0,44 ммоль) перемешивали с карбонатом натрия (52 мг, 0,49 ммоль) в сухом ДМФА (3 мл) в течение 20 мин, затем добавляли 1-йодоктан (118 мг, 0,49 ммоль) и смесь перемешивали в атмосфере азота при 40°C в течение 65 ч. После охлаждения до комнатной температуры смесь полностью дегазировали, выкачивая воздух и снова наполняя сосуд азотом несколько раз. Добавляли тетракис(трифенилфосфин)палладий(0) (102 мг, 0,09 ммоль), смесь снова дегазировали и затем добавляли морфолин (0,385 мл, 4,4 ммоль) и перемешивание продолжали в течение 6,5 ч. Добавляли 2M HCl и EtOAc и 2-фазную систему фильтровали. Продукт присутствовал преимущественно в отфильтрованном твердом веществе, которое перекристаллизовывали из смеси ТГФ-ацетонитрил, затем MeOH, с фильтрованием, получая чистое указанное в заголовке соединение.
Выход 48 мг (36%); ЯМР; (400МГц, d6-ДМСО) δH 0,84 (т, 3H, J=7Гц), 1,18-1,30 (м, 10Н), 1,57-1,66 (м, 2H), 3,84 (т, 2H, J=7,5Гц), 11,22 (с, 1H); m/z 299 [MH+].
Пример 38: 8-хлор-3-децил-3,7-дигидро-1H-пурин-2,6-дион
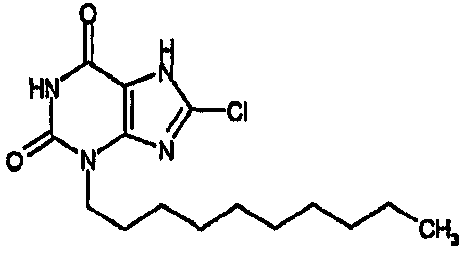
Получают согласно способу примера 37, исходя из 1-бромдекана (108 мг). Дальнейшую очистку осуществляли перекристаллизацией из MeOH с последующей масс-направленной автопрепаративной хроматографией.
Выход 2 мг (1,4%); ЯМР; (400МГц, d4-метанол) δH 0,89 (т, 3H, J=7Гц), 1,26-1,38 (м, 14H), 1,68-1,76 (м, 2H), 3,97 (т, 2H, J=7,5Гц); m/z 327 [MH+].
Пример 39: 8-хлор-3-(циклогексилметил)-3,7-дигидро-1H-пурин-2,6-дион
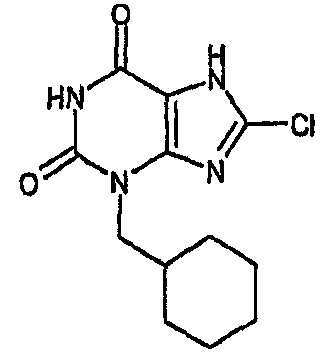
Получают подобно примеру 37, исходя из (бромметил)циклогексана (87 мг), за исключением того, что осуществляют дополнительный период нагревания при 80°C в течение 18 ч.
Перекристаллизовывают из MeOH.
Выход 31 мг (25%); ЯМР; (400МГц, d6-ДМСО) δH 0,90-1,02 (м, 2H), 1,08-1,20 (м, 3H), 1,53-1,69 (м, 5H), 1,77-1,87 (м, 1H), 3,70 (д, 2H, J=7,5Гц), 11,21 (с, 1H); m/z 283 [MH+].
Общий способ для примеров 40-46
К 8-хлор-3,7-дигидро-1H-пурин-2,6-диону (100 мг, 0,442 ммоль) в сухом ТГФ (3 мл) добавляли спирт (0,442 моль). Смесь перемешивали при 0°C и добавляли раствор дибензилазодикарбоксилата (280 мг 94% чистоты, 0,88 ммоль) в сухом ТГФ (2 мл), затем раствор трифенилфосфина (232 мг, 0,88 ммоль) в сухом ТГФ, который добавляли порциями более чем за 5 мин. После дополнительных 30 минут при 0°C перемешивание продолжали при комнатной температуре в течение 18 ч. Смесь полностью дегазировали, создавая вакуум и снова наполняя сосуд азотом несколько раз, затем добавляли тетракис(трифенилфосфин)палладий(0) (102 мг, 0,088 ммоль), затем морфолин (0,385 мл, 4,42 ммоль) и перемешивание продолжали в течение 4,5 ч. Добавляли EtOAc и 2M HCl и смесь фильтровали для удаления желтого твердого осадка. Фильтрат отделяли и органическую фазу концентрировали и повторно растворяли в смеси ТГФ и MeOH. Этот раствор отправляли на SPE с аминопропилом, элюируя смесью ТГФ-MeOH (1:1), затем MeOH и затем 5% AcOH в DCM-MeOH (1:1). Фракции продукта, полученные таким образом, концентрировали и перекристаллизовывали из MeOH с получением чистого указанного в заголовке соединения.
Пример 40: (+/-)-8-хлор-3-(3-метилпентил)-3,7-дигидро-1H-пурин-2,6-дион
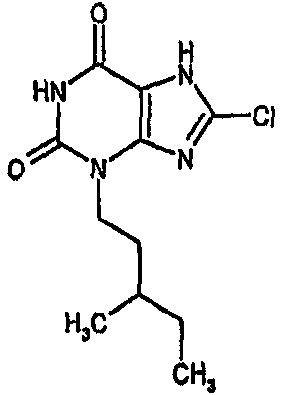
Исходя из 45 мг (+/-)-3-метил-1-пентанола.
Выход 20,2 мг (17%); ЯМР; (400МГц, d6-ДМСО) δН 0,83 (т, 3H, J=7,5Гц), 0,90 (д, 3H, J=6,5Гц), 1,12-1,21 (м, 1H), 1,30-1,48 (м, 3H), 1,58-1,68 (м, 1H), 3,87 (т, 2H, J=7,5Гц), 11,21 (с, 1H); m/z 271 [MH+].
Пример 41: 8-хлор-3-(2-циклопентилэтил)-3,7-дигидро-1H-пурин-2,6-дион
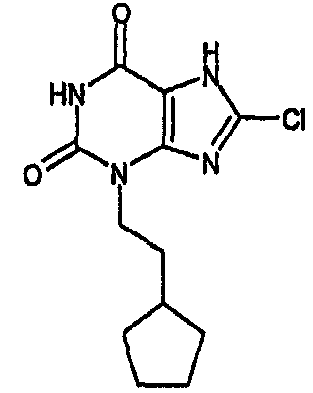
Исходя из 50 мг 2-циклопентилэтанола.
Выход 24,6 мг (20%); ЯМР; (400МГц, d6-ДМСО) δH 1,04-1,15 (м, 2H), 1,40-1,67 (м, 6H), 1,70-1,82 (м, 3H), 3,86 (т, 2H, J=7,5Гц), 11,22 (с, 1H); m/z 283 [MH+].
Пример 42: 8-хлор-3-(циклопропилметил)-3,7-дигидро-1H-пурин-2,6-дион
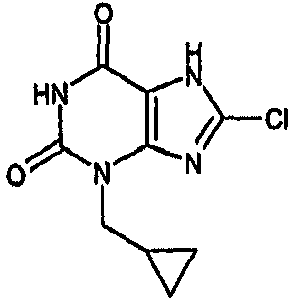
Исходя из 32 мг циклопропилметанола.
Выход 22,3 мг (21%); ЯМР; (400МГц, d6-ДМСО) δН 0,34-0,40 (м, 2H), 0,40-0,48 (м, 2H), 1,17-1,27 (м, 1H), 3,74 (д, 2H, J=7,5Гц), 11,23 (с, 1H); m/z 241 [MH+].
Пример 43: (+/-)-8-хлор-3-(2-метилбутил)-3,7-дигидро-1H-пурин-2,6-дион
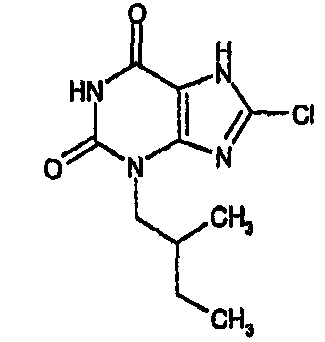
Исходя из 39 мг (+/-)-2-метил-1-бутанола.
Выход 12 мг (9,5%); ЯМР; (400МГц, d6-ДМСО) δН 0,81 (д, 3H, J=7Гц), 0,86 (т, 3H, J=7,5Гц), 1,06-1,17 (м, 1H), 1,30-1,41 (м, 1H), 1,90-2,00 (м, 1H), 3,68 (дд, 1H, J=13,5 и 8Гц), 3,75 (дд, 1H, J=13,5 и 7,5Гц), 11,22 (с, 1H); m/z 257 [MH+].
Пример 44: (+/-)-8-хлор-3-(2-метилпентил)-3,7-дигидро-1H-пурин-2,6-дион
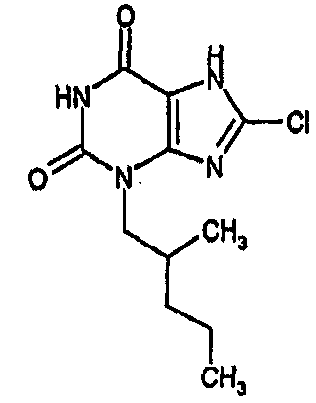
Исходя из 45 мг (+/-)-2-метил-1-пентанола.
Выход 22,4 мг (19%); ЯМР; (400МГц, d6-ДМСО) δН 0,81 (д, 3H, J=7Гц), 0,84 (т, 3H, J=7,5Гц), 1,05-1,16 (м, 1H), 1,16-1,43 (м, 3H), 1,98-2,09 (м, 1H), 3,67 (дд, 1H, J=13,5 и 8Гц), 3,74 (дд, 1H, J=13,5 и 7Гц), 11,22 (с, 1H); m/z 271 [MH+].
Пример 45: 8-хлор-3-(циклобутилметил)-3,7-дигидро-1H-пурин-2,6-дион
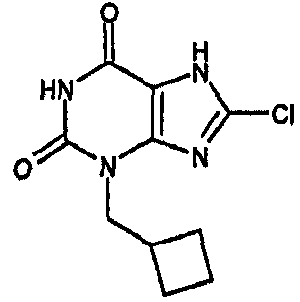
Исходя из 38 мг циклобутилметанола.
Выход 30,5 мг (27%); ЯМР; (400МГц, d6-ДМСО) δH 1,73-1,85 (м, 4H), 1,86-1,97 (м, 2H), 2,66-2,79 (м, 1H), 3,90 (д, 2H, J=7,5Гц), 11,22 (с, 1H); m/z 255 [MH+].
Пример 46: 8-хлор-3-(циклопентилметил)-3,7-дигидро-1H-пурин-2,6-дион
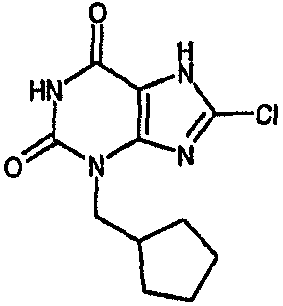
Исходя из 44 мг циклопентилметанола.
Выход 15 мг (13%); ЯМР; (400МГц, d6-ДМСО) δH 1,20-1,32 (м, 2H), 1,42-1,54 (м, 2H), 1,54-1,66 (м, 4H), 2,32-2,45 (м, 1H), 3,79 (д, 2H, J=8Гц), 11,22 (с, 1H); m/z 269 [MH+].
Пример 47: 8-хлор-3-(3-циклопропилпропил)-3,7-дигидро-1H-пурин-2,6-дион
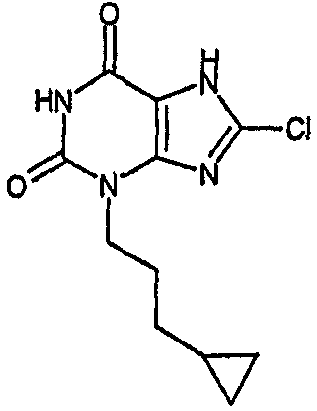
Исходя из 3-циклопропил-1-пропанола (P.J. Wagner, J. Amer. Chem. Soc., 1981, 103, 3837-3841) (44мг).
Выход 27,7 мг (23%); ЯМР; (400МГц, d6-ДМСО) δH -0,03-+0,03 (м, 2H), 0,34-0,40 (м, 2H), 0,65-0,75 (м, 1H), 1,15-1,23 (м, 2H), 1,66-1,76 (м, 2H), 3,87 (т, 2H, J=7Гц), 11,15 (с, 1H); m/z 269 [MH+].
Пример 48: 8-хлор-3-(2-циклобутилэтил)-3,7-дигидро-1H-пурин-2,6-дион

Исходя из 2-циклобутилэтанола (P. Vergnon, Eur. J. Med. Chem., 1975, 10, 65-71) (44 мг).
Выход 21,5 мг (18%); ЯМР; (400МГц, d6-ДМСО) δH 1,53-1,64 (м, 2H), 1,68-1,85 (м, 4H), 1,93-2,03 (м, 2H), 2,19-2,30 (м, 1H), 3,78 (т, 2H, J=7Гц), 11,20 (с, 1H); m/z 269 [MH+].
Пример 49: 8-хлор-3-(4-фторбутил)-3,7-дигидро-1H-пурин-2,6-дион
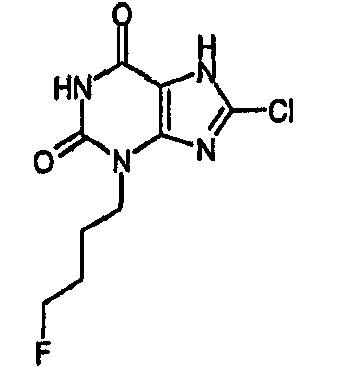
a) 8-хлор-3-(4-фторбутил)-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
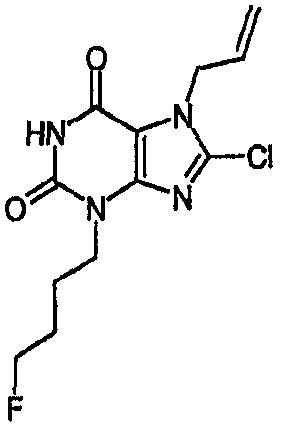
К раствору 8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (200 мг, 0,88 ммоль, 1 экв) в безводном ДМСО (1 мл) в 1,5 мл микроволновый сосуд, оборудованный мешалкой, добавляли бикарбонат натрия (113 мг, 1,07 ммоль, 1,2 экв.), затем 1-бром-4-фторбутан (114 мкл, 165 мг, 1,06 ммоль, 1,2 экв). Сосуд закрывали и нагревали при перемешивании с использованием микроволн, поддерживая температуру 120°C в течение 25 мин, с максимальной выходной мощностью 300W. Полученный темно-коричневый раствор разбавляли метанолом (1 мл) и очищали масс-направленной автопрепаративной ВЭЖХ, получая указанное в заголовке соединение в виде твердого вещества белого цвета (159 мг, 60%). m/z 301,3 [MH+].
b) 8-хлор-3-(4-фторбутил)-3,7-дигидро-1H-пурин-2,6-дион
К суспензии 8-хлор-3-(4-фторбутил)-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (100 мг, 0,33 ммоль, 1 экв) в безводном DCM (2 мл) добавляли тетракиспалладий (38 мг, 0,033 ммоль, 10 вес.%), затем уксусную кислоту (115 мкл, 121 мг, 2,01 ммоль, 6 экв.) и фенилсилан (410 мкл, 360 мг, 3,33 ммоль, 10 экв.). Полученный светло-желтый раствор перемешивали при температуре окружающей среды в течение 16 ч, получая темно-фиолетовый раствор. Растворитель удаляли в токе азота и остаток растворяли в растворе ДМСО/метанол (3 мл, 2:1) при нагревании. Желатиновой смеси давали охладиться до температуры окружающей среды, фильтровали, затем очищали масс-направленной автопрепаративной ВЭЖХ с получением указанного в заголовке соединения в виде твердого вещества белого цвета (35 мг, 43%). m/z 261,2 [MH+]; ЯМР (400МГц, MeOD), δН 4,45 (2H, дт, J=47 и 6Гц), 4,03 (2H, т, J=7Гц), 1,90-1,65 (4H, м).
Следующие соединения получали подобным способом и очищали в зависимости от ситуации препаративной или масс-направленной автопрепаративной ВЭЖХ.
Пример 50: 8-хлор-3-(3-фторпропил)-3,7-дигидро-1H-пурин-2,6-дион
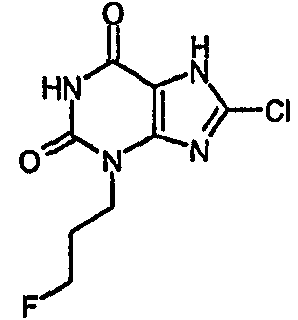
ЯМР (400МГц, MeOD), δH 4,51 (2H, дт, J=47 и 6Гц), 4,11 (2H, т, J=7Гц), 2,18-2,03 (2H, м) m/z 247 [MH+].
Пример 51: 8-хлор-3-(5-фторпентил)-3,7-дигидро-1H-пурин-2,6-дион
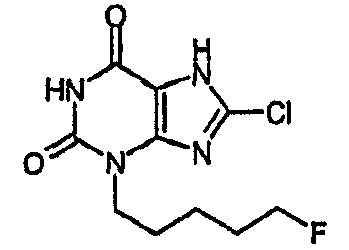
ЯМР (400МГц, MeOD), δH 4,41 (2H, дт, J=48 и 6Гц), 3,99 (2H, т, J=8Гц), 1,84-1,63 (4H, м), 1,52-1,40 (2H, м). m/z 273,29 [MH-].
Пример 52: 3-(3-бутен-1-ил)-8-хлор-3,7-дигидро-1H-пурин-2,6-дион
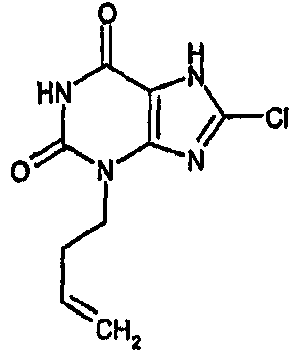
8-Хлор-3,7-дигидро-1H-пурин-2,6-дион (100 мг, 0,44 ммоль) перемешивали с карбонатом натрия (52 мг, 0,49 ммоль) в сухом ДМФА (3 мл) в течение 45 мин, затем добавляли 4-бром-1-бутен (66 мг, 0,49 ммоль) и смесь перемешивали в атмосфере азота при 40°C в течение 65 ч. После охлаждения до комнатной температуры смесь полностью дегазировали, выкачивая воздух и снова наполняя сосуд азотом несколько раз. Добавляли тетракис(трифенилфосфин)палладий(0) (102 мг, 0,09 ммоль), смесь снова дегазировали и затем добавляли морфолин (0,385 мл, 4,4 ммоль) и перемешивание продолжали в течение 6,5 ч. Добавляли 2M HCl и EtOAc и фильтровали 2-фазную систему для удаления желтого твердого осадка. Органическую фазу фильтрата отделяли и упаривали. Остаток растворяли при нагревании в ТГФ-MeOH (1:1) и загружали в картридж SPE с аминопропилом (5 г), который элюировали смесью ТГФ-MeOH (1:1), затем MeOH и затем 5% AcOH в MeOH-DCM (1:1). Фракцию продукта далее очищали масс-направленной автопрепаративной хроматографией с получением указанного в заголовке соединения.
Выход 27,5 мг (26%), ЯМР; (400МГц, d6-ДМСО) δH 2,40 (дт, 2H, J=7 и 6Гц), 3,93 (т, 2H, J=7Гц), 4,97-5,07 (м, 2H), 5,74-5,85 (м, 1H), 11,22 (с, 1H); m/z 241 [MH+].
Пример 53: 8-хлор-3-(6-фторгексил)-3,7-дигидро-1H-пурин-2,6-дион
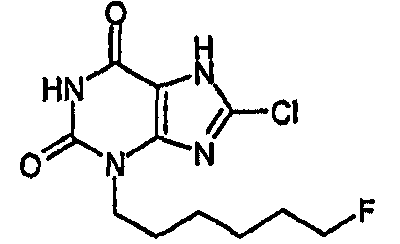
ЯМР (400МГц, MeOD), δH 4,40 (2H, дт, 48 и 6Гц), 3,98 (2H, т, 8Гц), 1,80-1,60 (4H, м), 1,52-1,35 (4H, м). m/z 287 [MH-].
Пример 54: 8-хлор-3-этил-1-метил-3,7-дигидро-1H-пурин-2,6-дион
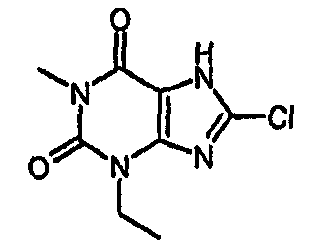
a) 8-хлор-3-({[2-(метилокси)этил]окси}метил)-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
К раствору 8-хлор-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (6 г, 26,5 ммоль) в безводном ДМФА (30 мл) добавляли карбонат натрия (3,09 г, 29,15 ммоль). После 10 минут перемешивания при комнатной температуре добавляли метоксиэтоксиметилхлорид (3,03 мл, 26,5 ммоль) и перемешивание продолжали в атмосфере азота при комнатной температуре в течение 66 часов. Реакционную смесь концентрировали в вакууме и остаток растворяли в EtOAc (100 мл), промывали насыщенным раствором соли (100 мл), водный экстракт экстрагировали DCM (100 мл) и органические экстракты сушили (MgSO4), объединяли и концентрировали в вакууме. Остаток растирали в EtOAc и отфильтровывали твердое вещество. Концентрированием фильтрата получали светло-коричневое масло, которое абсорбировали на кремнеземе и очищали SPE (Si, 50 г), элюируя градиентом 1:1 EtOAc/циклогексан-EtOAc, с получением указанного в заголовке соединения в виде твердого вещества белого цвета (2 г, 24%), m/z 315,2 [MH+].
b) 8-хлор-1-метил-3-({[2-(метилокси)этил]окси}метил)-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
К раствору 8-хлор-3-({[2-(метилокси)этил]окси}метил)-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (2 г, 6,37 ммоль) в безводном ДМФА (15 мл) добавляли карбонат натрия (0,743 г, 7 ммоль). После 10 минут перемешивания при комнатной температуре добавляли метилйодид (0,44 мл, 7 ммоль) и перемешивание продолжали в атмосфере азота при комнатной температуре в течение 18 часов. Реакционную смесь концентрировали в вакууме и остаток растворяли в EtOAc (100 мл) и промывали насыщенным раствором соли (100 мл). Органический экстракт сушили (MgSO4), фильтровали и упаривали с получением указанного в заголовке соединения в виде желтовато-коричневого масла (чистота 85%) (2,98 г, количеств.), m/z 329,2 [MH+].
c) 8-хлор-1-метил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-дион
К раствору 8-хлор-1-метил-3-({[2-(метилокси)этил]окси}метил)-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (2,9 г, 6,37 ммоль) в диоксане (20 мл) и воде (20 мл) добавляли 5M водный раствор HCl. (20 мл). Полученную смесь нагревали при 100°C в атмосфере азота в течение 18 часов. Реакционную смесь концентрировали в вакууме, остаток растворяли в EtOAc (100 мл) и промывали водой. Органический экстракт сушили (MgSO4), фильтровали и упаривали. Очисткой SPE (Si, 20г) с элюированием смесью 2:3 EtOAc/циклогексан получали указанное в заголовке соединение в виде твердого вещества белого цвета (1,04 г, 68%). m/z 241,1 [MH+].
d) 8-хлор-3-этил-1-метил-3,7-дигидро-1H-пурин-2,6-дион
К раствору 8-хлор-1-метил-7-(2-пропен-1-ил)-3,7-дигидро-1H-пурин-2,6-диона (100 мг, 0,42 ммоль) в безводном ДМФА (3 мл) добавляли карбонат натрия (58 мг, 0,54 ммоль), после 10 минут перемешивания добавляли этилйодид (0,043 мл, 0,54 ммоль) и реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 90 часов. Добавляли Pd(PPh3)4 (73 мг, 0,063 ммоль) и из реактора выкачивали воздух и промывали азотом (×3), добавляли морфолин (0,37 мл, 4,3 ммоль) и продолжали перемешивание при комнатной температуре в атмосфере азота в течение 4 часов. Реакционную смесь разбавляли EtOAc (25 мл) и промывали 2M водным раствором HCl (25 мл). Органический экстракт сушили (MgSO4), фильтровали и упаривали. Очисткой SPE с аминопропилом (5 г) загруженного соединения и промывкой MeOH перед элюированием продукта смесью 5% AcOH/MeOH получали указанное в заголовке соединение в виде твердого вещества белого цвета (67 мг, 70%). ЯМР; dH (400МГц, d6-ДМСО) 1,20 (т, 3H, J=7Гц), 3,22 (с, 3H), 3,97 (кв, 2H, J=7Гц), 14,46 (1Н, уш.с); m/z 227,2 [М-Н]-.
Все публикации, включая, но не ограничиваясь ими, патенты и заявки на патент, процитированные в настоящем описании, включены в него путем ссылки, как если бы для каждой индивидуальной публикации было специфически и индивидуально указано, что она включена в настоящее описание путем ссылки в полном объеме.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНАЦИОННОЕ ЛЕКАРСТВО | 2003 |
|
RU2328280C2 |
| АМИДОЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ КСАНТИНА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩИМ ДЕЙСТВИЕМ ФОСФОЕНОЛПИРУВАТКАРБОКСИКИНАЗЫ (ФЕПКК), СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2003 |
|
RU2295525C2 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ПУТИ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2673458C1 |
| ГИДРОКСИПУРИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2685417C1 |
| ЗАМЕЩЕННЫЕ ФЕНОКСИУКСУСНЫЕ КИСЛОТЫ, ИХ ЭФИРЫ И АМИДЫ, ВКЛЮЧАЮЩИЕ 2,6-ДИОКСО-2,3,6,7-ТЕТРАГИДРО-1Н-ПУРИН-8-ИЛОВЫЙ ФРАГМЕНТ, - АНТАГОНИСТЫ АДЕНОЗИНОВОГО A РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2477726C1 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ И КОСМЕТИКЕ | 2015 |
|
RU2712971C2 |
| АГОНИСТЫ TLR7 | 2019 |
|
RU2817014C2 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА, ИНГИБИТОР ДИПЕПТИДИЛПЕПТИДАЗЫ IV, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ И ПРИМЕНЕНИЕ НА ИХ ОСНОВЕ | 2003 |
|
RU2297418C9 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ИНГИБИТОР JAK | 2015 |
|
RU2674262C2 |
| СУЛЬФОНАМИДНОЕ ПРОИЗВОДНОЕ И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2013 |
|
RU2607081C2 |
Настоящее изобретение относится к новым производным ксантина общей формулы (I) и их фармацевтически приемлемым солям, обладающим активностью в отношении НМ74А рецептора, которые могут быть использованы в терапии для лечения диабетической дислипидемии, смешанной дислипидемии, сердечной недостаточности, гиперхолестеринемии, атеросклероза, артериосклероза, гипертриглицеридемии, сахарного диабета типа II, диабета типа I, резистентности к инсулину, гиперлипидемии, нервной анорексии, ожирения, заболевания коронарных артерий, тромбоза, стенокардии, хронической почечной недостаточности, периферического сосудистого заболевания или инсульта. В соединении формулы (I)
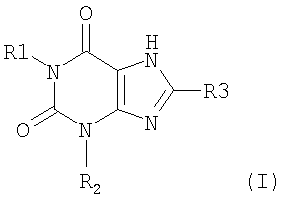
R1 представляет собой водород или метил; R2 представляет собой незамещенный н-С4-6алкил; и R3 представляет собой хлор. Изобретение также относится к фармацевтической композиции на основе новых производных ксантина. 7 н. и 9 з.п. ф-лы.
1. Соединение формулы (I)
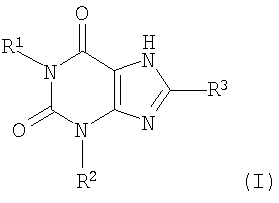
и его фармацевтически приемлемая соль, где
R1 представляет собой водород или метил;
R2 представляет собой незамещенный н-С4-6алкил; и
R3 представляет собой хлор.
2. Соединение по п.1, где R2 представляет собой н-пентил.
3. Соединение по п.1, представляющее собой 8-хлор-3-пентил-3,7-дигидро-1Н-пурин-2,6-дион или его фармацевтически приемлемую соль.
4. Соединение по п.1, представляющее собой 8-хлор-1-метил-3-пентил-3,7-дигидро-1Н-пурин-2,6-дион или его фармацевтически приемлемую соль.
5. Соединение по п.3, представляющее собой 8-хлор-3-пентил-3,7-дигидро-1Н-пурин-2,6-дион.
6. Соединение по п.4, представляющее собой 8-хлор-1-метил-3-пентил-3,7-дигидро-1 Н-пурин-2,6-дион.
7. Соединение по любому из пп.1-6, обладающее активностью в отношении НМ74А рецептора, для применения в терапии.
8. Соединение по любому из пп.1-6, обладающее активностью в отношении НМ74А рецептора, для применения для лечения диабетической дислипидемии, смешанной дислипидемии, сердечной недостаточности, гиперхолестеринемии, атеросклероза, артериосклероза, гипертриглицеридемии, сахарного диабета типа II, диабета типа I, резистентности к инсулину, гиперлипидемии, нервной анорексии, ожирения, заболевания коронарных артерий, тромбоза, стенокардии, хронической почечной недостаточности, периферического сосудистого заболевания или инсульта.
9. Соединение по любому из пп.1-6, обладающее активностью в отношении НМ74А рецептора, для применения для лечения диабетической дислипидемии, смешанной дислипидемии гиперлипопротеинемии, гиперхолестеринемии или гипертриглицеридемии.
10. Соединение по любому из пп.1-6, обладающее активностью в отношении НМ74А рецептора, для применения для лечения сахарного диабета типа II.
11. Применение соединения по любому из пп.1-6, обладающего активностью в отношении НМ74А рецептора, для получения лекарственного средства для лечения человека или животного, страдающего состоянием, при котором недостаточная активация рецептора НМ74А способствует развитию этого состояния, или где активация рецептора будет иметь полезный эффект.
12. Применение соединения по любому из пп.1-6, обладающего активностью в отношении НМ74А рецептора, для получения лекарственного средства для лечения диабетической дислипидемии, смешанной дислипидемии, сердечной недостаточности, гиперхолестеринемии, атеросклероза, артериосклероза, гипертриглицеридемии, сахарного диабета типа II, диабета типа I, резистентности к инсулину, гиперлипидемии, нервной анорексии, ожирения, заболевания коронарных артерий, тромбоза, стенокардии, хронической почечной недостаточности или инсульта.
13. Применение соединения по любому из пп.1-6, обладающего активностью в отношении НМ74А рецептора, для получения лекарственного средства для лечения диабетической дислипидемии, смешанной дислипидемии, гиперлипопротеинемии, гиперхолестеринемии или гипертриглицеридемии.
14. Применение соединения по любому из пп.1-6, обладающего активностью в отношении НМ74А рецептора, для получения лекарственного средства для лечения сахарного диабета типа II.
15. Фармацевтическая композиция, обладающая активностью в отношении НМ74А рецептора, содержащая соединение по любому из пп.1-6 в эффективном количестве и один или более физиологически приемлемых разбавителей, эксципиентов или носителей.
16. Способ получения соединения по любому из пп.1-6, включающий:
i) алкилирование в N1 или N3, или диалкирование в N1 и N3 N7 - защищенного ксантина;
ii) хлорирование в С8; и
iii) удаление защитной группы;
в любом порядке, при условии, что удаление защитной группы осуществляют после алкилирования.
| 0 |
|
SU389282A1 | |
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| ARCH, JONATHAN R | |||
| S | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| JACOBSON, KENNETH A | |||
| ET AL: "Effect of trifluoromethyl and other substituents on activity of | |||

