Настоящее изобретение относится к способу получения фторированных виниловых эфиров.
Более конкретно, настоящее изобретение относится к способу получения фторгалогенэфиров, из которых в процессе дегалогенирования образуются фторированные виниловые эфиры. Способ по изобретению позволяет получать фторгалогенэфиры с улучшенной селективностью.
Как известно, фторированные виниловые эфиры представляют собой класс ценных мономеров для получения разнообразных полимеров, от фторированных эластомеров до пригодных для тепловой обработки полукристаллических фторированных полимеров.
Из уровня техники известны способы получения фторгалогенэфиров, основанные на взаимодействии гипофторитов с олефинами. В наиболее известных способах получения гипофторитов используются катализаторы на основе фторидов металлов.
В патенте US 4827024 описано получение гипофторитов в непрерывном режиме путем фторирования, при взаимодействии эквимолярных количеств фтора и галогенированных карбонильных соединений, содержащих по меньшей мере два атома углерода, в присутствии катализаторов, состоящих из CsF, индивидуально или в смеси с такими металлами, как, например, медь. Как правило, эти металлы, помимо их использования в качестве подложки для катализатора (CsF), также облегчают теплообмен, т.е. рассеивают тепло, выделяющееся в процессе синтеза гипофторитов.
Металлическая подложка в описанном выше методе из уровня техники должна выполнять две основные функции: 1) обеспечивать доступность катализатора для реагентов, 2) облегчать теплообмен, поддерживая под контролем в заданном интервале температуру каталитического слоя. Последней, но не менее важной характеристикой подложки является абсолютная инертность к действию реагентов и продуктов реакции.
В патентах US 4816599, US 4801409 и US 4962282 гипофториты предпочтительно получают путем воздействия избытка фтора на ацилфториды с целью их полного превращения гипофториты, таким образом, чтобы концентрация ацилфторидов на каталитическом слое оставалась очень низкой, поскольку известно, что некоторые ацилфториды разлагаются в присутствии CsF. Смотри, например, Carl G. Ktrespan in Journal of Fluorine Chemistry, 16 (1980) 385-390.
В ходе проведенных Заявителем исследований известных из уровня техники способов получения гипофторитов с использованием описанных выше катализаторов было показано, что при использовании указанных каталитических систем как в периодическом, так и в непрерывном режиме, их каталитическая активность со временем быстро снижается. Заявитель, в частности, обнаружил, что активность значительно снижается, вплоть до полной дезактивации катализатора, если в реакции образования гипофторитов используют катализатор в сочетании с избытком фтора по сравнению со стехиометрическим количеством, причем такие условия в способах уровня техники указываются как предпочтительные.
Таким образом, из уровня техники следует, что синтез гипофторитов следует проводить в избытке фтора, чтобы насколько возможно избежать описанных выше неудобств. При работе в указанных условиях известный из уровня техники катализатор очень быстро, за два-три дня, дезактивируется. При таких коротких сроках действия практически невозможно создать промышленную установку с непрерывным режимом работы.
Кроме того, при синтезе гипофторитов в периодическом режиме последовательное неоднократное использование каталитического слоя без подложки приводит к очень низким выходам в реакции получения гипофторитов, и наблюдается очень быстрая дезактивация.
Из уровня техники известны способы получения фторированных виниловых эфиров. В патенте US 4900872 описано получение перфторвинилэфирных предшественников путем непрерывного взаимодействия перфторалкильных гипофторитов, растворенных в инертном растворителе, и олефина формулы CAIF=CA'IF, где А и А', одинаковые или различные, представляют собой Cl и Br. В этом патенте указано, что указанные гипофториты могут подаваться непосредственно из реактора, в котором происходит их синтез в газовой фазе путем взаимодействия фтора с ацилфторидом в присутствии катализатора. Полученные соединения превращают в перфторвиниловые эфиры путем дегалогенирования с участием цинка. Этот способ имеет такие же недостатки, которые были описаны выше в связи с получением гипофторитов. В частности, причиной недостатков указанных способов является необходимость синтеза и немедленного использования гипофторитов, которые, как известно, являются нестабильными соединениями, в особенности, когда число атомов углерода в перфторалкильной цепи гипофторита равно или превышает 2. Кроме того, известно, что в синтезе гипофторитов необходимо использовать катализатор, недостатки которого описаны выше.
Таким образом возникла необходимость разработки доступного способа получения фторгалогенэфиров, лишенного недостатков, присущих способам, известным из уровня техники.
Заявитель неожиданно обнаружил, что путем использования описанного далее способа можно решить указанную техническую задачу и таким образом разработать доступный непрерывный или периодический промышленный процесс очень высокой селективностью.
Объектом настоящего изобретения является способ получения (пер)фторгалогенэфиров общей формулы (I):
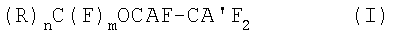
где:
А и А', одинаковые или различные, представляют собой Cl или Br или одно из А и А' представляет собой водород, а другое - галоген, выбранный из Cl, Br;
R может иметь следующие значения: F или фторированный, предпочтительно перфторированный, заместитель, выбранный из следующих групп: линейный или разветвленный С1-С20алкил, более предпочтительно C1-С10; С3-С7циклоалкил; ароматическая группа, С6-С10арилалкил, алкиларил; С5-С10гетероцикл или алкилгетероцикл;
если R представляет собой фторированный или перфторированный алкил, циклоалкил, арилалкил, алкиларил, он может необязательно содержать в цепи один или более атомов кислорода;
если R представляет собой фторированный заместитель, он может необязательно содержать один или более атомов Н и/или один или более атомов галогена, отличных от F;
n представляет собой целое число, равное 1 или 2; m равно 3-n;
путем взаимодействия карбонильных соединений формулы (II):
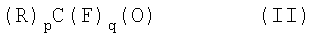
где:
р представляет собой целое число, равное 1 или 2;
q представляет собой целое число, равное 0 или 1,
при условии, что если р=2, то q=0; если р=1, то q=1;
R такое, как указано выше;
в жидкой фазе с фтором и олефиновыми соединениями формулы (III):

где А и А' такие, как указано выше,
при температуре от -120°С до -20°С, предпочтительно от -100°С до -40°С, необязательно в присутствии растворителя, инертного в условиях проведения взаимодействия.
Фтор, используемый в реакции, может быть необязательно разбавлен инертным газом, таким как, например, азот или гелий.
Соединения формулы (II), которые можно использовать, представляют собой ацилфториды, например, COF2, CF3COF, C2F5COF, С3F7COF, C7F15COF, CF3CF(OCF3)CF2CF2COF, CF3О(CF2)2COF; кетоны, такие как гексафторацетон, перфтордиизопропилкетон, и т.д. Ацилфториды являются предпочтительными.
Соединения формулы (III) представляют собой, например, 1,2-дихлор-1,2-дифторэтилен (CFC 1112), 1,2-дибром-1,2-дифторэтилен, предпочтительно CFC 1112.
Процесс в соответствии с настоящим изобретением проводят в отдельном реакторе, и взаимодействие можно осуществлять в полунепрерывном или непрерывном режиме.
Полунепрерывный процесс можно осуществлять, например, путем подачи газообразного фтора в реактор, содержащий карбонильные соединения формулы (II) и олефиновые соединения формулы (III). Молярное соотношение (II)/(III) может изменяться в широком интервале, например, от 0.05 до 10. Подачу фтора продолжают до полного превращения олефина. Наступление этого момента можно определить по прекращению выделения тепла в процессе реакции. Фактически, при проведении взаимодействия соединений (III) и (II), например, при -100°С, имеет место экзотермическая реакция, поскольку соединения взаимодействуют с элементарным фтором, и температура повышается примерно на 5°-15°С. Следовательно, это взаимодействие прекращается, если, например, соединение (III) было полностью израсходовано. В этот момент температура в реакторе возвращается к начальному значению.
В непрерывном процессе газообразный фтор и соединения (II), (III) подают в реактор до тех пор, пока не достигается устойчивое состояние. На практике реагенты подают в реактор с установленными скоростями потока, а реакционная смесь непрерывно выводится. Устойчивое состояние наступает тогда, когда концентрация трех реагентов и продуктов реакции в реакторе равна концентрации реагентов и продуктов реакции, вытекающих из реактора.
Молярные соотношения реагентов в способе по настоящему изобретению не имеют особых ограничений, например, молярное соотношение (II)/(III) может находиться в интервале от 0.05 до 10, a F2/(III) - от 0.05 до 10.
В способе по настоящему изобретению в качестве растворителей можно использовать соединения, которые в приведенном выше температурном интервале являются жидкими и инертными. Например, можно использовать соединения, выбранные из (пер)фторуглеродов, (пер)фторэфиров, (пер)фторполиэфиров, перфтораминов, или соответствующие смеси. Специалист в данной области может выбрать для использования в качестве растворителей соединения из приведенных выше классов на основании их физических свойств.
Заявитель неожиданно обнаружил, что взаимодействие между соединением формулы (II), олефином формулы (III) и элементарным фтором в приведенном выше температурном интервале непосредственно приводит к получению фторгалогенэфиров формулы (I) с улучшенной селективностью. Этот результат является удивительным и неожиданным. Кроме того, в способе по изобретению не используется катализатор, в отличие от известных из уровня техники способов получения фторгалогенэфиров из гипофторитных предшественников. Таким образом, отсутствие катализатора в способе по изобретению заметно упрощает проведение процесса, в особенности, в промышленном масштабе. Действительно, затраты на регенерацию катализатора, его замену и в целом на эксплуатацию части установки, содержащей каталитический реактор, являются весьма обременительными.
Заявитель обнаружил, что при использовании известного из уровня техники способа получения фторгалогенэфиров путем взаимодействия между олефином и гипофторитом, полученным из соответствующего ацилфторида, большое количество гипофторита разлагается. Скорость разложения гипофторита возрастает с увеличением его молекулярной массы; на практике это происходит, если гипофторит иной, чем метилгипофторит. Смотри сравнительные Примеры. Способ по изобретению имеет улучшенную селективность, даже если фторгалогенэфиры получают исходя из соединений формулы (II), независимо от числа атомов углерода в составе R. В Примерах изобретения, если соединение (II) представляет собой ацетилфторид или пропионилфторид, селективность по фторгалогенэфиру по существу одного порядка. Кроме того, по сравнению со способами, известными из уровня техники, в которых для образования гипофторитов используют катализатор, способ по изобретению имеет более высокую производительность, поскольку нет необходимости останавливать установку для регенерации или замены катализатора. Таким образом из установки для осуществления способа по изобретению исключаются части, относящиеся к получению катализатора, эксплуатации каталитической системы и регенерации катализатора.
Заявитель обнаружил, что при осуществлении способа по настоящему изобретению образуется незначительное количество продуктов разложения соединения (II). Смотри Примеры.
Более того, способ по настоящему изобретению также позволяет работать при низкой степени превращения соединения (II) с высокой селективностью по фторгалогенэфиру.
Соединения формулы (II), в отличие от гипофторитов, которые являются нестабильными соединениями, не разлагаются при взаимодействии с реакционной средой, и их можно выделить, например, при помощи перегонки. При работе с гипофторитами это невозможно по причине опасности, возникающей при использовании этих соединений как в процессе реакции, так и при их выделении. Хорошо известно, что при использовании гипофторитов им дают возможность полностью вступить во взаимодействие, чтобы они не накапливались в реакционной среде.
Следующие примеры иллюстрируют изобретение, но не предназначены для его ограничения.
ПРИМЕР 1
Синтез CF3-CF2-CF2OCFCl-CF2Cl
57 г CFCl=CFCl (CFC 1112) и 15 г CF3-CF2COF (перфторпропионил фторид, PFPF) вводят в 50 см3 стеклянный реактор, снабженный механической мешалкой, и выдерживают раствор при температуре -100°С.
Через клапан для барботажа подают фтор, разбавленный азотом (молярное соотношение фтор/азот 1:5), в течение 6.5 часов. Во время подачи фтора отмечается умеренная экзотермическая реакция.
Газ, поступающий из реактора, пропускают через сепаратор, содержащий фторированный растворитель, в котором поддерживают температуру -80°С.
По окончании реакции извлекают растворы из реактора и сепаратора и анализируют их при помощи газовой хроматографии.
Материальный баланс реакции составляет 93.6% в расчете на раствор, извлеченный из реактора, и на соединения в составе фторированной жидкости в сепараторе, при этом степени превращения олефина CFC 1112 и PFPF составляют 100% и 61.25%, соответственно.
Основными продуктами взаимодействия PFPF являются:
A) CF3-CF2-CF2OCFCl-CF2Cl (пропильный аддукт)
B) CF3-CF2-CF2O(CFCl)3-CF2Cl
Селективность составляет 73% для А) и 17% для В) в расчете на PFPF.
Параллельно основной реакции также протекает реакция фторирования CFC 1112 до CF2Cl-CF2Cl (CF 114) и фтордимеризации до CF2Cl-(CFCl)3-CF2Cl (димер CFC 1112); селективность по отношению к CFC 1112 составляет 40% для каждого соединения.
Молярный баланс олефина составляет примерно 98%. Раствор, извлеченный из реактора, перегоняют. Структуру соединения
А) подтверждают при помощи 19F-ЯМР анализа.
ПРИМЕР 2 (сравнительный)
Синтез CF3-CF2-CF2O-CFCl-CF2Cl в соответствии с методом, известным из уровня техники
a) Синтез гипофторита CF3-CF2-CF2OF согласно патенту US 4827024.
2.7 г Nл/час фтора, разбавленного азотом (молярное соотношение фтор/азот 1/10), и 2.3 Nл/час CF3-CF2-COF (PFPF) подают в 500 см3 металлический трубчатый реактор, заполненный катализатором CsF, смешанным с медной проволокой для рассеяния тепла, выделяющегося при реакции. PFPF превращается в гипофторит CF3-CF2-CF2OF с выходом 99.5%.
b) Синтез CF3-CF2-CF2O-CFCl-CF2Cl путем взаимодействия гипофторита и CFC 1112 в соответствии с патентом US 4900872.
Гипофторит, полученный на стадии а), подают в реактор CSTR-типа (корпусной реактор непрерывного действия с мешалкой), содержащий 121.2 г CFCl=CFCl (CFC 1112) и 452 г CF2Cl-CF3 (CFC 115) в качестве реакционного растворителя, и выдерживают при температуре -90°С. Реакцию проводят в течение 4 часов, затем реактор разгружают и анализируют раствор при помощи газовой хроматографии.
Массовый баланс реакции составляет 84.3%, и селективность по пропильному аддукту относительно PFPF составляет 48.1%. Раствор перегоняют, и выход выделенного соединения соответствует аналитическим расчетам.
Количество молей образовавшегося COF2, определенное при помощи ацидиметрического титрования соединения в выделившемся газе (отходящем газе), что дает индекс разложения гипофторита путем β-расщепления, составляет 36% от исходного PFPF.
ПРИМЕР 3 (сравнительный)
Синтез CF3-CF2O-CFCl-CF2Cl (этильный аддукт)
a) Синтез гипофторита
Проводят процесс аналогично стадии а) Примера 2 (сравнительного), но подают 2.3 Nл/час CF3-COF (PFAF) в каталитический реактор. Выход гипофторита CF3-CF2OF равен 99.6%.
b) Синтез CF3-CF2O-CFCl-CF2Cl
Проводят процесс аналогично стадии b) Примера 2 (сравнительного), но без использования растворителя, путем подачи в CSTR реактор 300 г CFC 1112. Материальный баланс реакции составляет 90%.
Раствор, извлеченный из реактора, анализируют при помощи газовой хроматографии, и выход этильного аддукта CF3-CF2O-CFCl-CF2Cl, в расчете на загрузку гипофторита составляет 75%. Остальные 25% гипофторита разлагаются с образованием COF2.
Для выделения этильного аддукта раствор перегоняют. Выделенное количество соответствует выходу, предварительно рассчитанному при помощи газовой хроматографии.
ПРИМЕР 4
Синтез CF3-CF2O-CFCl-CF2Cl
49 г CF3-COF (PFAF) и 24.4 г CFCl=CFCl (CFC 1112) вводят в устройство, описанное в Примере 1. Реакционную смесь выдерживают при температуре -87°С и подают 1.5 Nл/час фтора, разбавленного азотом (молярное соотношение фтор/азот 1/5), в течение 129 минут.
Массовый баланс реакции составляет 90.5% при степени превращения PFAF 10%. Анализ при помощи газовой хроматографии показывает, что основным продуктом является CF3-CF2O-CFCl-CF2Cl (этильный аддукт) и селективность по отношению к PFAF составляет 80%. Остальные 20% составляют побочные продукты.
ПРИМЕР 3
Синтез CF3-(CF2)6-CF2O-CFCl-CF2Cl
19.65 г CF3-OCFCl-CF2Cl в качестве растворителя, 23.07 г CFCl=CFCl (CFC 1112) и 7.57 г CF3-(CF2)6-COF вводят в устройство, описанное в Примере 1. Реакционную смесь выдерживают при температуре -55°С и подают 1.5 Nл/час фтора, разбавленного азотом (молярное соотношение фтор/азот 1/5), в течение 110 минут.
Массовый баланс реакции составляет 92.2%, степень превращения ацилфторида составляет 25%.
Излеченный раствор анализируют при помощи газовой хроматографии, и основным продуктом является эфир CF3-(CF2)6-CF2O-CFCl-CF2Cl, который образуется в селективностью 90% по отношению к исходному ацилфториду. Остальные 10% составляют побочные продукты.
Раствор обрабатывают водой и перегоняют отделенную органическую фазу. 19F ЯМР и GC MS анализы подтверждают структуру указанного выше соединения.
ПРИМЕР 6
Синтез CF3CF(OCF3)CF2CF2CF2O-CFCl-CF2Cl
Пример 1 повторяют, вводя в реактор 16.58 г CF3CF(OCF3)CF2CF2COF и 48.85 г CFCl=CFCl (CFC 1112). Реакционную смесь выдерживают при температуре -81°С и подают 1.5 Nл/час фтора, разбавленного азотом (молярное соотношение фтор/азот 1/5). Процесс проводят в течение 264 минут и материальный баланс составляет 93.5% со степенью превращения исходного ацилфторида, равной 41.8%. Извлеченный раствор анализируют при помощи газовой хроматографии и основными соединениям являются следующие перфторгалогенэфиры:
-CF3CF(OCF3)CF2CF2CF2O-CFCl-CF2Cl,
-CF3CF(OCF3)CF2CF2CF2O-(CFCl)3-CF2Cl,
которые образуются с селективностью по отношению к исходному ацилфториду, равной 85% и 7.4%, соответственно.
Раствор обрабатывают водой и из отделенной органической фазы отгоняют эфир CF3CF2CF(OCF3)CF2CF2O-CFCl-CF2Cl, структуру которого подтверждают при помощи 19F ЯМР анализа.
Полученное соединение подвергают дехлорированию цинком в диметилформамиде при 75°С с получением соответствующего винилового эфира CF3CF2CF(OCF3)CF2CF2O-CF=CF2.
ПРИМЕР 7
Синтез CF3O-CFCl-CF2Cl
В устройство, описанное в Примере 1, в котором поддерживают температуру -100°С, вводят 19.2 г CF2Cl-CF3.
Затем подают 1.5 Nл/час фтора, разбавленного азотом (молярное соотношение фтор/азот 1/2.5), 1.5 Nл/час CFCl=CFCl и 1,5 Nл/час COF2.
Реакция продолжается в течение 2 часов, в конце реактор разгружают. Реакционную смесь анализируют при помощи газовой хроматографии.
Обнаружено, что степень превращения CFC 1112 составляет 69.0%, а COF2 - 57.0%.
Реакционную смесь перегоняют. Выделяют 12.0 г соединения формулы CF3O-CFCl-CF2Cl (19F ЯМР).
Селективность по отношению к COF2 составляет 66.7%, а по отношению к CFC 1112 - 44.6%.
Обнаружено, что одновременно с синтезом приведенного выше соединения, в реакторе также идут реакции фторирования CFC 1112 с образованием CFC 114 и фтордимеризации до CF2Cl-CFCl-CFCl-CF2Cl.
Селективность, рассчитанная по отношению к CFC 1112, для продукта CFC 114 составляет 35.1%, а для димера CFC 1112 - 7.3%.
Молярный баланс олефина составляет 99%.
ПРИМЕР 8
Синтез (CF3)2-CF-O-CFCl-CF2Cl
В устройство Примера 1, в котором поддерживают температуру -80°С, загружают 16.8 г CF3С(O)CF3. Затем подают 1.7 Nл/час фтора, разбавленного азотом (молярное соотношение фтор/азот 1/2.5), и 1.5 Nл/час диCFCl=CFCl.
Реакция продолжается в течение 4 часов, и в конце реактор разгружают. Реакционную смесь анализируют при помощи газовой хроматографии.
Обнаружено, что степень превращения как CFC 1112,так и CF3С(O)CF3 составляет 100%.
Реакционную смесь перегоняют. Выделяют 19.5 г соединения формулы (CF3)2-CF-O-CFCl-CF2Cl (19F ЯМР).
Селективность по отношению к CF3С(O)CF3 составляет 57.0%, а по отношению к CFC 1112 - 20.9%.
Обнаружено, что одновременно с синтезом приведенного выше соединения, в реакторе также идут реакции фторирования CFC 1112 с образованием CFC 114 и фтордимеризации до CF2Cl-CFCl-CFCl-CF2Cl.
Селективность, рассчитанная по отношению к CFC 1112, для продукта CFC 114 составляет 62.7%, а для димера CFC 1112 - 1.6%.
Молярный баланс составляет 99% для CFC 1112 и 90% для CF3С(O)CF3.
ПРИМЕР 9
Синтез CF3-CF2-CF2-O-CHF-CF2Cl
В устройство Примера 1, в котором поддерживают температуру -90°С, загружают 70.0 г CF3CF2COF и 11.3 г диCHF=CFCl (CFC 1122а). Затем подают 1.5 Nл/час фтора, разбавленного азотом (молярное соотношение фтор/азот 1/4).
Реакция продолжается в течение 1.5 часов, и в конце реактор разгружают. Реакционную смесь анализируют при помощи газовой хроматографии.
Обнаружено, что степень превращения CFC 1122а составляет 100%, а CF3CF2COF - 20.5%.
Выделяют 1.2 г CF3-CF2-CF2-O-CHF-CF2Cl. Селективность по отношению к CF3CF2COF составляет 4.4%, а по отношению к CFC 1122а - 3.4%.
Обнаружено, что одновременно с синтезом приведенного выше соединения также идут реакции фторирования CFC 1122а с образованием CFC 124а и фтордимеризации до CF2Cl-CFH-CFCl-CF2H и его изомеров CF2Cl-CFH-CFH-CF2Cl и CF2H-CFCl-CFCl-CF2H.
Селективность, рассчитанная по отношению к CFC 1122а, для CFC 124а составляет 39.4%, а для димеров CFC 1122а - 13.6%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОСТЫХ ФТОРГАЛОГЕНИРОВАННЫХ ЭФИРОВ | 2006 |
|
RU2433992C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОСТЫХ ФТОРГАЛОГЕНИРОВАННЫХ ЭФИРОВ | 2006 |
|
RU2425022C2 |
| ФТОРИРОВАННЫЕ ПРОИЗВОДНЫЕ БИСВИНИЛОКСИМЕТАНА (ВАРИАНТЫ), ПОЛИМЕРЫ И СОПОЛИМЕРЫ НА ИХ ОСНОВЕ | 1995 |
|
RU2144044C1 |
| ПРОСТЫЕ ФТОРВИНИЛОВЫЕ ЭФИРЫ И ПОЛУЧАЕМЫЕ ИЗ НИХ ПОЛИМЕРЫ | 2001 |
|
RU2269506C2 |
| ФТОРЭЛАСТОМЕРЫ | 2001 |
|
RU2271368C2 |
| СПОСОБ ОКИСЛЕНИЯ ТЕТРАФТОРЭТИЛЕНА ДО ПОЛУЧЕНИЯ ПЕРОКСИДНЫХ ПРОСТЫХ ПЕРФТОРПОЛИЭФИРОВ | 1997 |
|
RU2194725C2 |
| ФТОРЭЛАСТОМЕРЫ | 2005 |
|
RU2383555C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕРФТОРПОЛИЭФИРОВ | 1994 |
|
RU2120450C1 |
| ПЕРФТОРЭЛАСТОМЕРЫ | 2005 |
|
RU2378294C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГАЛОИДИРОВАННЫХ 1,3-ДИОКСОЛАНОВ И ГАЛОИДИРОВАННЫЕ 1,3-ДИОКСОЛАНЫ | 1991 |
|
RU2039055C1 |
Изобретение относится к способу получения (пер)фторгалогенэфиров общей формулы (I):
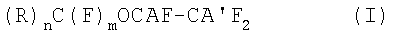 ,
,
которые используют при получении фторированных виниловых эфиров, которые представляют собой класс ценных мономеров для получения разнообразных полимеров. При этом в формуле (I) А и А' одинаковые или различные, представляют собой Cl или Br или одно из А и А' представляет собой водород, а другое - галоген, выбранный из Cl, Br; R=F или фторированный, предпочтительно перфторированный, заместитель, выбранный из следующих групп: линейный или разветвленный С1-С20алкил; С3-С7циклоалкил; С6-С10ароматическая группа, С6-С10арилС1-С20алкил, C1-С20алкилС6-С10арил, или С1-С20алкилС5-С10гетероцикл: если R представляет собой фторированный или перфторированный алкил, циклоалкил, арилалкил, алкиларил, он может необязательно содержать в цепи один или более атомов кислорода; если R представляет собой фторированный заместитель, он может необязательно содержать один или более атомов Н и/или один или более атомов галогена, отличных от F; n представляет собой целое число, равное 1 или 2; m равно 3-n. Способ включает взаимодействие карбонильных соединений формулы (II):
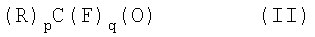 ,
,
где р представляет собой целое число, равное 1 или 2; q представляет собой целое число, равное 0 или 1, при условии, что если р=2, то q=0; если р=1, то q=1; в жидкой фазе с элементарным фтором и олефиновыми соединениями формулы (III):
 .
.
Способ позволяет получить фторгалогенэфиры с улучшенной селективностью. 7 з.п. ф-лы.
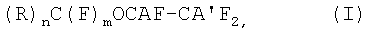
где А и А', одинаковые или различные, представляют собой Cl или Br или одно из А и А' представляет собой водород, а другое - галоген, выбранный из Cl, Br;
R - F или фторированный, предпочтительно перфторированный, заместитель, выбранный из следующих групп: линейный или разветвленный C1-С20алкил; С3-С7циклоалкил; С6-С10ароматическая группа, С6-С10арилС1-С20алкил, С1-С20алкилС6-С10арил или С1-С20алкилС5-С10гетероцикл:
если R представляет собой фторированный или перфторированный алкил, циклоалкил, арилалкил, алкиларил, он может необязательно содержать в цепи один или более атомов кислорода;
если R представляет собой фторированный заместитель, он может необязательно содержать один или более атомов Н и/или один или более атомов галогена, отличных от F;
n представляет собой целое число, равное 1 или 2;
m равно 3-n,
путем взаимодействия карбонильных соединений формулы (II)
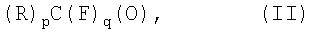
где р представляет собой целое число, равное 1 или 2;
q представляет собой целое число, равное 0 или 1, при условии, что если р=2, то q=0; если р=1, то q=1;
R такое, как указано выше,
в жидкой фазе с элементарным фтором и олефиновыми соединениями формулы (III):

где А и А' такие, как указано выше,
при температуре от -120 до -20°С, предпочтительно от -100 до -40°С, необязательно в присутствии растворителя, инертного в условиях проведения взаимодействия.
| Способ получения этил-бета-хлорпропилового эфира | 1958 |
|
SU121785A1 |
| US 4900872 A, 13.02.1990 | |||
| US 4872024 A, 02.05.1989 | |||
| СПОСОБ ЭЛЕКТРОЛИТИЧЕСКОГО ОС.4ЖДЕНИЯ СПЛАВОВИНДИЯ | 0 |
|
SU201871A1 |
| Синтезы фторорганических соединений | |||
| М.: Химия, 1973, стр.26-28. | |||

