Описание промышленного изобретения на имя: SOLVAY SOLEXIS S.p.A., of Italian nationality, с головным офисом в Milano, via Turati 12.
Изобретение относится к способу получения простых фторгалогенированных эфиров. Более конкретно, настоящее изобретение относится к простым фторгалогенированным эфирам, которые дегалогенированием или дегидрогалогенированием позволяют получать простые перфторвиниловые эфиры. Еще более конкретно, настоящее изобретение относится к способу получения простых перфторвиниловых эфиров, предпочтительно простого перфторметилвинилового эфира, простого перфторэтилвинилового эфира и простого перфторпропилвинилового эфира, с улучшенным выходом и селективностью, и с использованием предшественников, не принадлежащих к классу хлорфторуглеродов (CFC) и, кроме того, получаемых без дорогостоящих способов отделения от гидрированных побочных продуктов.
Хорошо известно, что CFC, из-за их влияния на озоновый слой (ODP) и из-за их сильного воздействия на окружающую среду (GWP), изгоняются или ограничиваются монреальскими протоколами и их последующими модификациями. В любом случае, в тех немногих областях, где они по-прежнему используются, необходимо предотвратить то, что CFC диспергируются в окружающей среде и, таким образом, их использование является дорогостоящим с промышленной точки зрения.
Как известно, простые перфторвиниловые эфиры представляют собой мономеры, пригодные для получения различных полимеров, от фторированных эластомеров до фторированных термочувствительных полукристаллических полимеров.
Способы получения простых перфторвиниловых эфиров известны в данной области. Патент США № 3450684 относится к простым виниловым эфирам формулы:
CF2=CFO(CF2CFX0 I-O)nICF2CF2X0 I,
где X0 I=F, Cl, CF3, H и nI может находиться в пределах от 1 до 20.
Эти соединения получают, начиная с HFPO. Способ осуществляют в несколько стадий, в соответствии со следующей схемой:
X0 ICF2CF2O-(CFX0 ICF2O)nI-1-CFX0 ICOF+HFPO ―→
X0 ICF2CF2O-(CFX0 ICF2O)nI-CF(CF3)COF
X0 ICF2CF2O-(CFX0 ICF2O)nI-CF(CF3)COF+NaOH ―→
X0 ICF2CF2O-(CFX0 ICF2O)nI-CF(CF3)COONa
212°C
X0 ICF2CF2O-(CFX0 ICF2O)nI-CF(CF3)COONa ―→
X0 ICF2CF2O-(CFX0 ICF2O)nI-CF=CF2
Выходы этого способа являются низкими.
Патент США № 3817960 относится к получению простых перфторвиниловых эфиров формулы:
CF3O(CF2O)n"CF2CF2OCF=CF2,
где n" может находиться в пределах от 1 до 5.
Синтез требует получения ацилфторида формулы:
CF3O(CF2O)n"CF2C(O)F
окислением TFE при низкой температуре, в присутствии УФ-излучения, или электрохимическим фторированием соответствующего гидрированного ацилфторида. Затем ацилфторид взаимодействует в соответствии со следующей схемой:
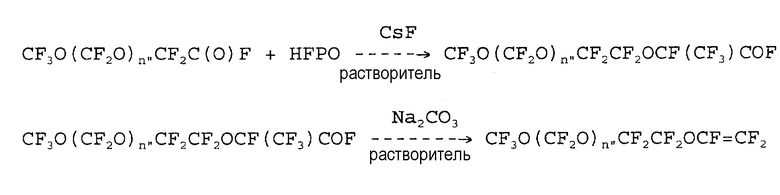
В этой схеме синтеза получение исходного ацилфторида из TFE является дорогостоящим процессом с промышленной точки зрения. Когда используется электрохимическое фторирование, выход является низким из-за образования побочных продуктов.
Патент США № 3896179 относится к отделению простых перфторвиниловых эфиров, имеющих линейную алкильную цепь, от изомерных простых перфторвиниловых эфиров с разветвленной алкильной цепью термическим разложением при температурах, находящихся в пределах 300-600°C. На самом деле разветвленные изомеры, как правило, действуют в качестве агентов переноса цепи, давая полимеры, имеющие плохие механические свойства. По этой причине, простые разветвленные виниловые эфиры являются нежелательными, в то время как простые линейные виниловые эфиры используют для получения полимеров.
Патент США № 4340750 относится к получению простых перфторвиниловых эфиров формулы:
CF2=CFOCF2R0 fX1 ,
где R0 f представляет собой C1-C20 перфторалкил, необязательно содержащий кислород, X1=H, Cl, Br, F, COOR0, CONR0R', где R0 представляет собой C1-C10 алкильную группу, и R' представляет собой H или C1-C10 алкильную группу. При получении этих соединений используется ацилфторид вместе с йодом и тетрафторэтиленом. В этом способе исключается конечная стадия щелочного пиролиза ацилфторида.
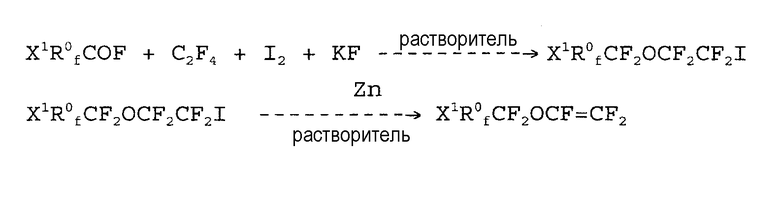
Недостатком этого способа является то, что реакция дейодофторирования (последняя стадия реакции) имеет место с низким выходом.
Патент США № 4515989 относится к получению новых соединений для синтеза простых фторвиниловых эфиров. В соответствии с патентом, синтез простого винилового эфира усовершенствуется использованием конкретного соединения, способного легко декарбоксилироваться. Для получения промежуточных соединений используют фторэпоксиды формулы:
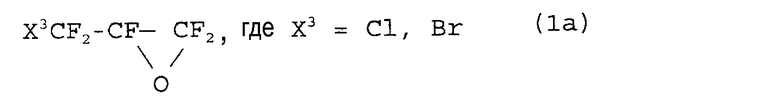
Схема реакции является следующей:
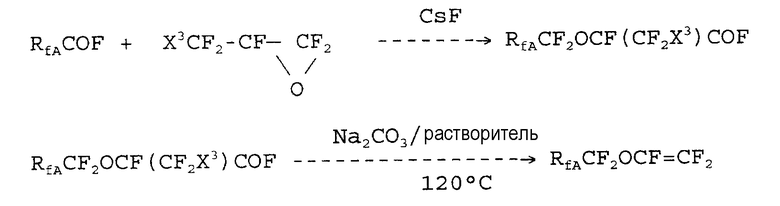
Недостатком этого способа является то, что предшественники для получения фторпероксидов (1a) трудно получать в промышленных масштабах.
Патент США № 5350497 относится к получению простых перфторалкилвиниловых эфиров фторированием с помощью фтора частично фторированных простых гидродихлорированных эфиров и последующим дехлорированием, в соответствии со следующей схемой:
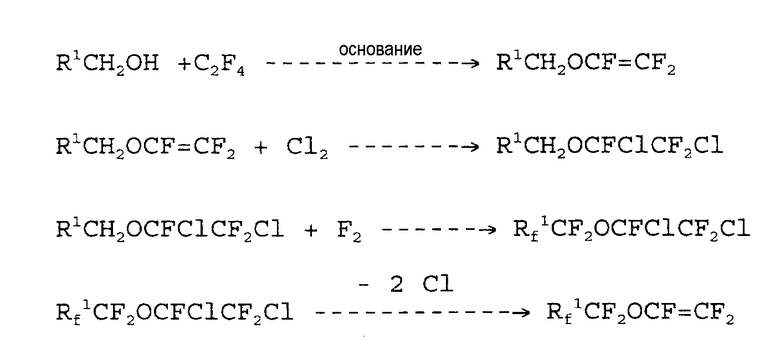
Этот способ имеет тот недостаток, что стадия фторирования с помощью фтора имеет место с невысоким выходом и используется избыток фтора для замещения всех атомов водорода.
Патент США № 6255536 описывает способ, в котором принимается во внимание синтез гидрированного предшественника, который может также быть частично галогенированным, фторирование предшественника, с получением кислотного производного, которое с помощью щелочного пиролиза разлагается до простого перфторвинилового эфира. Схема является следующей:
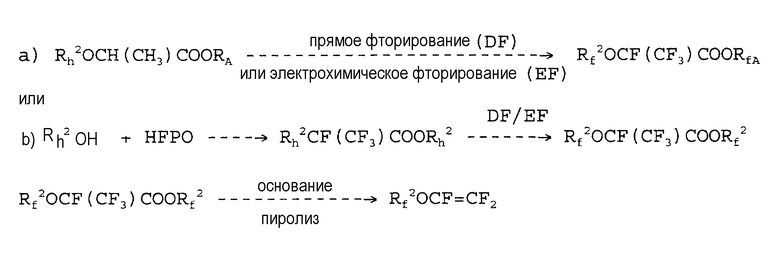
Стадия фторирования может осуществляться электрохимическим фторированием или фторированием с помощью фтора, в соответствии с патентом США № 5488142. Первое взаимодействие, как правило, имеет место при низкой селективности и образовании нежелательных побочных продуктов. При фторировании с помощью фтора, промышленно приемлемый выход и производительность не получаются. В самом деле, работа осуществляется при высоких разбавлениях гидрированного предшественника и фтора, чтобы контролировать тепло, выделяемое в ходе реакции. Кроме того, фторирование с помощью фтора требует длительных времен реакции, необходимых для получения полного фторирования соединения. Известно, что фторирование гидрированных соединений представляет собой сильно экзотермическую реакцию, которая может вызвать разрыв углерод-углерод связей с образованием нежелательных побочных продуктов. См. книгу Fluorine Chemistry; A Comprehensive Treatment, в Kirk Othmer Encyclopedia, стр. 242-259. Кроме того, для получения полного превращения, следовательно, для замещения всех атомов водорода молекулой предшественника, необходимо увеличить температуру, а следовательно, принять более жесткие условия реакции. Это обычно приводит к снижению выхода требуемого продукта, поскольку имеются вторичные реакции разложения.
Европейский патент EP 1352892 описывает способ получения простых фторированных виниловых эфиров из ацилфторидов, получаемых разложением сложных фторированных эфиров. Схема является следующей:
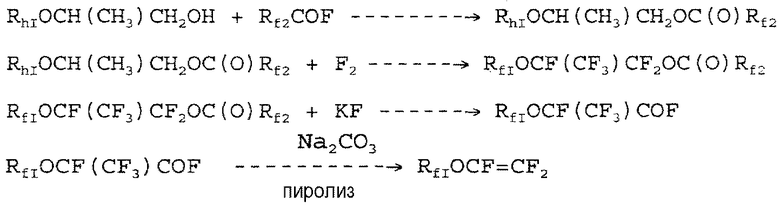
На второй стадии схемы синтеза полное фторирование частично гидрированных сложных эфиров предшественников достигается с получением соответствующих сложных перфторированных эфиров. Эта стадия полного фторирования частично фторированного сложного эфира требует, чтобы реакция осуществлялась в течение длительного времени, с несколькими добавлениями гидрированного соединения, например, бензола, чтобы способствовать полному превращению сложного эфира. Одновременно с этим должна повышаться температура реакции, например, от -10°C до комнатной температуры. Производительность этого вида фторирования является очень низкой.
Способы получения простых фторгалогенированных эфиров взаимодействием гипофторитов с галогенфторированными олефинами известны из литературы. Патент США № 4900872 описывает синтез простых фторгалогенированных эфиров взаимодействием между перфторалкил гипофторитами, разбавленными в инертном растворителе, и галогенфторированным олефином, имеющим формулу, CA1F=CA11F, где AI и AII, одинаковые или отличные друг от друга, представляют собой Cl и Br. Олефин, используемый в синтезе, описываемом в примерах этого патента, представляет собой 1,2-дихлор-1,2-дифторэтилен (CFC 1112). Синтез указанного олефина, как правило, осуществляется дегалогенированием тетрахлордифторэтана CCl2FCCl2F (CFC 112) с помощью металлического цинка в спиртовом растворителе. См., например, Houben Weyl, vol.E 10 B2, стр. 125-161. Предшественник CFC 112, используемый при этом синтезе, представляет собой хлорфторуглерод, который, как сказано, попадает под монреальские протоколы и их дополнения относительно уменьшения выбросов газов, разрушающих озоновый слой, в стратосферу. В соответствии с этими протоколами, выбросы CFC должны постепенно уменьшаться со временем, до тех пор, пока они не будут полностью устранены. В промышленности CFC 112 получают как компонент смеси различных хлорфторэтанов, симметричных и асимметричных, в основном CFC 113 (CF2Cl-CFCl2) и CFC 114 (CF2Cl-CF2Cl).
Последние соединения представляют собой больший промышленный интерес, поскольку используются в качестве хладагентов и растворителей. Способы синтеза этих смесей хлорфторэтанов освещаются, например, в Adv. Fluorine Chem. 3 (1963), "The Preparation of Organic Fluorine Compounds by Halogen Exchange" стр. 194-201, Fluorine Chem. 4 (1974), 117-139. Поскольку невозможно больше использовать соединения CFC 113 и 114, также и CFC 112, а также CFC 1112, больше недоступны в промышленности.
Ощущается необходимость в получении доступного промышленного способа синтеза простых перфторалкилвиниловых эфиров с высокими выходами и селективностью.
Неожиданно автор нашел способ, преодолевающий указанную выше техническую проблему.
Целью настоящего изобретения является способ получения простых перфторалкилвиниловых эфиров, имеющих общую формулу:
RfO-CF=CF2 (IA),
где Rf представляет собой C1-C3, предпочтительно C1-C2, перфторированный заместитель; включающий следующие стадии:
1) взаимодействия между гипофторитом формулы RfOF, где Rf является таким, как описано выше, и олефином формулы:
CY"Y=CY'Cl (II),
где Y, Y' и Y", одинаковые или отличные друг от друга, представляют собой H, Cl, Br, при условии, что Y, Y' и Y" не представляют собой одновременно водород;
2) удаления из простых фторгалогенированных эфиров формулы (III) и (III') молекул галогена (дегалогенирования)/галогенводородной кислоты (дегидрогалогенирования), где ион галогена/галогенида представляет собой Cl или Br, и получения простых виниловых эфиров формулы:
RfO-CYI=CYIIF (IV),
где YI и YII, одинаковые или отличные друг от друга, имеют значения H, Cl, Br, при условии, что как YI, так и YII не представляют собой H;
3) фторирования с помощью фтора простых виниловых эфиров (IV) и получения простых фторгалогенированных эфиров формулы:
RfO-CFYI-CF2YII (I),
где YI, YII, одинаковые или отличные друг от друга, представляют собой Cl, Br, H, при условии, что YI и YII не могут представлять собой одновременно H; Rf является таким, как указано выше;
4) удаления из простых фторгалогенированных эфиров формулы (I) молекул галогена/галогенводородной кислоты, где ион галогена/галогенида представляет собой Cl или Br, и получения простых виниловых эфиров формулы:
RfO-CF=CF2 (IA)
На стадии 1), как правило, получается смесь простых фторгалогенированных эфиров формулы:
RfO-CY"Y-CY'ClF (III),
RfO-CY'Cl-CY"YF (III'),
где Y, Y' и Y" имеют указанные выше значения; соединения (III) и (III') являются одинаковыми, когда исходный олефин (II) является симметричным.
На стадии 1) реакцию осуществляют в жидкой фазе при температурах от -130°C до 0°C, предпочтительно от -80°C до -10°C.
Перфторалкил гипофториты с количеством атомов углерода, равным или большим чем 2, известны из патента США № 4827024. Трифторметил гипофторит известен в данной области.
Необязательно могут использоваться органические растворители, инертные при условиях, используемых на стадии 1).
Эта стадия может осуществляться различными способами, например, в реактор, содержащий олефин в жидком состоянии, необязательно разбавленный растворителем, инертным в условиях реакции, вводят гипофторит, полученный в жидкой или газообразной фазе, разбавленный в соединении, инертном в условиях реакции.
Олефины формулы (II) предпочтительно выбираются из следующих: тетрахлорэтилена, трихлорэтилена, 1,2-дихлорэтилена и 1,1-дихлорэтилена.
На стадии 2) дегалогенирование (удаление хлора или брома) фторгалогенуглеродов (III) и (III') осуществляют, например, взаимодействием указанных соединений с такими переходными металлами, как цинк, медь, марганец, или с парами металлов, такими как Zn/Cu, Zn/Sn, Zn/Hg, в присутствии таких растворителей, например, как гидрированные протонные растворители, такие, например, как спирты, или таких простых гидрированных эфиров, например, как глимы, диоксан, или таких диполярных апротонных растворителей, например, как DMF, DMSO.
На стадии 2) дегидрогалогенирование (удаление HCl или HBr) фторгалогенуглеродов формулы (III) и (III') имеет место, например, при взаимодействии этих соединений с неорганическим основанием, предпочтительно NaOH или KOH, или с органическим основанием, предпочтительно первичными, вторичными или третичными алкил- или ариламинами. Как правило, используется жидкая фаза. Реакция удаления галогенводородной кислоты на стадии 2a) может необязательно осуществляться в присутствии растворителя, предпочтительно водного или спиртового. Использованием водных неорганических оснований реакция может осуществляться в присутствии такой соли четвертичного аммония или фосфония, как аммоний или фосфоний тетрабутил, предпочтительно хлорид, аммоний или фосфиний триоктилбензил, предпочтительно хлорид, и тому подобное. Альтернативно, или в смеси с солями четвертичного аммония или фосфония, могут использоваться другие соли, такие, например, как соли сульфония.
На стадии дегалогенирования или дегидрогалогенирования 2), как правило, процесс происходит при температурах в пределах 0-150°C, предпочтительно, 25-100°C.
На стадии 3) реакцию фторирования осуществляют добавлением газообразного фтора, необязательно, в присутствии инертного разбавителя, такого, например, как N2, He, и тому подобное, к соединениям формулы (IV), жидким при температуре реакции необязательным использованием растворителя или смеси инертных растворителей, находящихся в жидком состоянии в условиях, при которых осуществляют стадию 3).
На стадии 3), как правило, работа происходит при температурах в пределах между -120°C и 0°C, предпочтительно -90°C и -30°C.
Необязательные растворители, пригодные для использования на стадиях 1) и 3), выбираются из следующих: простых (пер)фторированных полиэфиров, например, (пер)фторалканов Galden®, например, с 3-10 атомами углерода, при условии, что они являются жидкими в условиях реакции, гидрофторуглеродов (HFC), гидрохлорфторуглеродов (HCFC), хлорфторуглеродов (CFC), перфтораминов, простых гидрофторированных эфиров или простых гидрофторированных полиэфиров, например, H-Galden®, или их смесей.
На стадии 4) дегалогенирование или удаление хлора или брома из простых фторгалогенированных эфиров формулы (I), например, осуществляется взаимодействием указанных соединений с такими переходными металлами, как цинк, медь, марганец, или с парами металлов, такими как Zn/Cu, Zn/Sn, Zn/Hg, в присутствии растворителей, которые могут представлять собой либо такие гидрированные протонные растворители как спирты, либо такие простые гидрированные эфиры как глимы, диоксан, или диполярных апротонных растворителей, таких как DMF, DMSO.
На стадии 4) дегидрогалогенирование или удаление HCl или HBr из простых фторгалогенированных эфиров формулы (I) имеет место, например, взаимодействием этих соединений с неорганическим основанием, предпочтительно NaOH или KOH, или органическим основанием, предпочтительно первичными, вторичными или третичными алкил- или ариламинами. Как правило, работа происходит в жидкой фазе. Реакция удаления галогенводородной кислоты на стадии 4a) может необязательно осуществляться в присутствии растворителя, предпочтительно водного или спиртового. Использованием водных неорганических оснований реакция может осуществляться в присутствии соли четвертичного аммония или фосфония, такой как аммоний или фосфоний тетрабутил, предпочтительно хлорид, аммоний или фосфиний триоктилбензил, предпочтительно хлорид, и тому подобное. Альтернативно, или в смеси с солями четвертичного аммония или фосфония, могут использоваться другие соли, такие, например, как соли сульфония. Альтернативно, или в смеси с солями четвертичного аммония или фосфония, могут использоваться другие соли, такие, например, как соли сульфония.
На стадии дегалогенирования или дегидрогалогенирования 4), как правило, работа происходит при температурах в пределах 0-150°C, предпочтительно 25-100°C.
В способе по настоящему изобретению отношение между реагентами на различных стадиях не является критичным.
В способе по настоящему изобретению давление не является критичным, и предпочтительно работа происходит при атмосферном давлении.
Способ по настоящему изобретению может осуществляться периодически, полупериодически или непрерывно.
Например, обращаясь к стадии 3), полупериодический способ может осуществляться введением газообразного фтора и соединения формулы (IV) в реактор, содержащий растворитель или смесь реакционных растворителей.
На стадии 1) может использоваться непрерывный способ, в котором газообразный гипофторит и соединение формулы (II) вводятся в реактор, до тех пор, пока не будет достигнуто стационарное состояние. На практике реагенты вводятся в реактор при известных скоростях потока непрерывной прокачкой реакционной смеси. Стационарное состояние достигается, когда концентрации реагентов и продуктов реакции в реакторе равны концентрациям реагентов и продуктов реакции на выходе из реактора.
Соединения формулы (I), получаемые с помощью способа по настоящему изобретению, представляют собой, например, следующие: CF3O-CFCl-CF2Cl, C2F5O-CFCl-CF2Cl, C3F7O-CFCl-CF2Cl.
Соединения формулы (III) и (III'), пригодные для использования на стадии 2), предпочтительно представляют собой соединения, где Rf представляет собой C1-C3, еще более предпочтительно C1-C2 перфторалкил.
Примеры этих соединений представляют собой следующие: CF3O-CHCl-CFCl2, CF3O-CCl2-CHClF, CF3O-CCl2-CCl2F, CF3O-CHCl-CHClF, CF3O-CH2-CCl2F,
C2F5O-CHCl-CFCl2, C2F5O-CCl2-CHClF, C2F5O-CCl2-CCl2F, C2F5O-CHCl-CHClF, C2F5O-CH2-CCl2F,
C3F7O-CHCl-CFCl2, C3F7O-CCl2-CHClF, C3F7O-CCl2-CCl2F, C3F7O-CHCl-CHClF, C3F7O-CH2-CCl2F.
Внезапно и неожиданно способ по настоящему изобретению позволяет получать на каждой отдельной стадии высокий выход в сочетании с высокой селективностью. Следовательно, простой перфторметилвиниловый эфир и простой перфторэтилвиниловый эфир получаются с высоким выходом и селективностью, по сравнению со способами, известными из литературы. Кроме того, способ по настоящему изобретению использует предшественники, не принадлежащие к классу хлорфторуглеродов (CFC) и получаемые без дорогостоящих процессов отделения от гидрированных побочных продуктов.
Олефины, используемые на стадии 1) способа по настоящему изобретению, являются широко доступными на рынке и экономически дешевыми.
С помощью способа по настоящему изобретению, если это желательно, может также быть получен простой перфторпропилвиниловый эфир использованием пропил гипофторита на первой стадии реакции.
Следующие далее примеры иллюстрируют, с не ограничивающими целями, настоящее изобретение.
ПРИМЕРЫ
Пример A
Синтез CF 3 OF
10 л/час газообразного фтора, 5 л/час CO и 10 л/час азота одновременно получают возможность для протекания в трубе из стали AISI 316 (внутренний диаметр 2,17 мм и длина 100 мм). Реакция запускается нагреванием зоны смешения газов при 100°C в течение нескольких минут. Все время реактор охлаждается циркуляцией воздуха, так что температура ниже чем 300°C; и температура на выходе из реактора равна 250°C. При этих условиях CO и F2 превращаются в COF2 с выходом выше чем 95% (определяют ИК-анализом вытекающей газообразной смеси).
Газообразной смеси, после охлаждения при 100°C, дают возможность для прохождения через каталитический слой, сформированный из 300 г мелкодисперсного безводного CsF, имеющего размер частиц, равный или меньший чем 0,1 мм, смешанного с 300 г иглообразной меди, имеющей диаметр 0,2 мм и длину 6-7 мм. Катализатор помещают в трубчатый реактор (внутренний диаметр 55 мм, длина 250 мм). Температура реакции между газами поддерживается при 100°C. При этих условиях COF2 превращается в CF3OF с выходом выше чем 98%, определяемым ИК-анализом вытекающей смеси.
Пример 1
Добавление CF 3 OF к трихлорэтилену
60,5 г CF3OCFCl-CF2Cl простого эфира, в качестве растворителя реакции, и 4,35 г трихлорэтилена вводят в 50 см3 стеклянный реактор, снабженный механической мешалкой. Затем реактор охлаждают до температуры -50°C криогенной баней. Через вход для барботирования в реактор вводят 21,7 г трихлорэтилена (TCE) через 5 часов с помощью насоса. Одновременно с этим вводятся, по-прежнему через вход для барботирования, 1,125 л(н.у.)/час CF3OF, разбавленного гелием при молярном отношении He/CF3OF 1,6 и вводимого при молярном отношении CF3OF/TCE, равном 1,5.
В конце реакции выпускают 97,5 г продукта. Анализом с помощью ГЖХ/МС получают превращение TCE 99,8% и селективность по двум продуктам реакции, CF3O-CHCl-CFCl2 и CF3O-CCl2-CHClF, равную 96%.
Пример 2
Дегидрохлорирование соединений CF 3 O-CHCl-CFCl 2 и CF 3 O-CCl 2 -CHClF, полученных в примере 1
50 г соединений, полученных в примере 1, и 4,1 г тетрабутиламмоний гидроксида в водном растворе, при 40% мас., вводят в 250 мл четырехгорлый реактор, снабженный магнитной мешалкой, делительной воронкой, термометром и водяным холодильником. При перемешивании добавляют 17 г NaOH в водном растворе, при 20%, сохраняя экзотермическое тепло при 34°C с помощью бани из H2O и льда. Когда добавление соды заканчивается, смесь оставляют при перемешивании при 34°C в течение дополнительных 30 минут. Ее охлаждают до 20°C: конечная смесь демонстрирует две отдельные фазы. Реакционную смесь выливают в разделительную воронку, отделяя 38,8 г органической фазы, имеющей более высокую плотность, образованную из соединения CF3OCCCl=CClF (простого хлорметилвинилового эфира, CVE), с чистотой 99% молярных. Превращение 100%, выход 92%.
Пример 3
Добавление фтора к CVE непрерывным способом
72,4 г CFCl3, в качестве растворителя, и 8 г простого хлорметилвинилового эфира вводят в такой же реактор, как используется в примере 1, охлаждаемый при температуре -70°C криогенной баней. Через вход для барботирования вводят 1,0 л(н.у.)/час F2, разбавленного азотом при молярном отношении N2/F2 1,6. Фторирование осуществляют в течение 10 минут.
В конце реакции выпускают 80,7 г продукта. Смесь анализируют ГЖХ/МС. Превращение CVE равно 31,5%, селективность для CF3O-CFCl-CF2Cl равна 79,0%.
Пример 4
Добавление фтора к CVE полупериодическим способом
В такой же реактор, как используется в примере 1, охлаждаемый при температуре -70°C криогенной баней, вводят 63,7 г CFCl3. Через вход для барботирования вводят 2,0 л(н.у.)/час F2, разбавленного азотом при молярном отношении N2/F2, равном 1,6 и 9,28 г/час CVE. Фторирование осуществляют в течение 4 часов.
В конце реакции выпускают 107,5 г смеси, которую анализируют ГЖХ/МС. Превращение CVE является количественным. Выход для CF3O-CFCl-CF2Cl равен 98,4%.
Пример 5
Дехлорирование CF 3 OCCl 2 CCl 2 F
Соединение CF3OCCl2CCl2F получают взаимодействием тетрахлорэтилена с метил гипофторитом в соответствии с J. Fluor. Chem., vol.74, 1995, 199-201.
80,0 г цинка в порошке, активированного промывкой с помощью 3н. раствора HCl, 550 мл DMF и 50 мг KI вводят, в инертной атмосфере азота, в 1-литровый трехгорлый реактор, снабженный магнитной мешалкой, делительной воронкой, термометром, соединенный через колонку Вигре и водяной холодильник с холодной ловушкой, поддерживаемой при температуре -75°C. Внутреннюю температуру поддерживают при 90°C. Затем по каплям добавляют 102,0 г CF3OCCl2CCl2F. Когда добавление заканчивается, смесь оставляют при перемешивании в течение одного часа при 90°C. 62,8 г CVE собирают в холодной ловушке. Выход CVE равен 83%.
Пример 6
Добавление CF 3 CF 2 CF 2 OF к CHCl=CHCl
Раствор, сформированный из 62 г CHCl=CHCl и 300 г CFCl3, вводят в 350 мл стеклянный реактор, снабженный механической мешалкой, охлаждаемый при температуре -90°C криогенной баней. Через вход для барботирования вводят 1,8 л(н.у.)/час CF3CF2CF2OF, полученного в соответствии с патентом США № 4900872, и 6,0 л(н.у.)/час He, в течение 5 часов и 40 минут.
Анализом ГЖХ/МС определяют превращение гипофторита 100%. Фракционной дистилляцией сырого продукта реакции получают 33,9 г продукта добавления CF3CF2CF2OCHClCHClF при выходе, вычисленном по отношению к вводимому гипофториту, 25%.
Пример 7
Добавление CF 3 CF 2 OF к CHCl=CCl 2
40 г CF2Cl-CFCl2 вводят в такой же реактор, как используют в примере 1, охлаждаемый при температуре -70°C криогенной баней. Через вход для барботирования вводят 0,76 л(н.у.)/час CF3CF2OF, полученного в соответствии с патентом США № 4900872, разбавленного азотом (молярное отношение CF3CF2OF/азот 1/10), и одновременно с этим, 4,4 г/час CHCl=CCl2.
Реакцию осуществляют в течение 3 часов. Сырой продукт (65,0 г) анализируют ГЖХ/МС. Превращение гипофторита равно 100%, и селективность по двум продуктам реакции CF3CF2O-CHCl-CFCl2 и CF3CF2O-CCl2-CHClF равна 61%.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОСТЫХ ФТОРГАЛОГЕНИРОВАННЫХ ЭФИРОВ | 2006 |
|
RU2433992C2 |
| ФТОРИРОВАННЫЕ ПРОИЗВОДНЫЕ БИСВИНИЛОКСИМЕТАНА (ВАРИАНТЫ), ПОЛИМЕРЫ И СОПОЛИМЕРЫ НА ИХ ОСНОВЕ | 1995 |
|
RU2144044C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРГАЛОГЕНЭФИРОВ | 2003 |
|
RU2329247C2 |
| ПРОСТЫЕ ФТОРВИНИЛОВЫЕ ЭФИРЫ И ПОЛУЧАЕМЫЕ ИЗ НИХ ПОЛИМЕРЫ | 2001 |
|
RU2269506C2 |
| ФТОРЭЛАСТОМЕРЫ | 2001 |
|
RU2271368C2 |
| ВУЛКАНИЗУЕМЫЙ ФТОРЭЛАСТОМЕР, СМЕСЬ, СОДЕРЖАЩАЯ ПЕРФТОРЭЛАСТОМЕР, И ПРОМЫШЛЕННОЕ ИЗДЕЛИЕ | 2002 |
|
RU2301235C2 |
| ПЕРФТОРЭЛАСТОМЕР, ВУЛКАНИЗУЕМЫЙ ПЕРОКСИДНЫМ СПОСОБОМ, СМЕСЬ, СОДЕРЖАЩАЯ ПЕРФТОРЭЛАСТОМЕР, И ПРОМЫШЛЕННОЕ ИЗДЕЛИЕ | 2002 |
|
RU2296774C2 |
| Способ получения перфтор-4-(фторсульфонил)бутилвинилового эфира | 2022 |
|
RU2800857C1 |
| ТЕТРАФТОРЭТИЛЕНОВЫЕ ТЕРМООБРАБАТЫВАЕМЫЕ СОПОЛИМЕРЫ | 1996 |
|
RU2154650C2 |
| СПОСОБ ФТОРИРОВАНИЯ ГАЛООЛЕФИНОВ | 2011 |
|
RU2596195C2 |
Изобретение относится к способу получения простых перфторалкилвиниловых эфиров, имеющих общую формулу: RfO-CF=CF2 (IA), где Rf представляет собой C1-С3, предпочтительно, C1-C2, перфторированный алкильный заместитель, включающему следующие стадии: 1) взаимодействия гипофторита формулы RfOF, где Rf является таким как выше, с олефином формулы: CY"Y=CY'Cl (II), где Y, Y' и Y", одинаковые или отличные друг от друга, представляют собой Н, Сl, Вr, при условии, что Y, Y' и Y" не представляют собой одновременно водород; 2) дегалогенирования или дегидрогалогенирования простых фторгалогенированных эфиров, полученных на стадии 1), и получения простых виниловых эфиров формулы: RfO-CYI=CYIIF (IV), где YI и YII, одинаковые или отличные друг от друга, имеют значения Н, Сl, Вr, при условии, что как YI, так и YII не одновременно представляют собой Н;
3) фторирования с помощью фтора простых виниловых эфиров (IV) и получения простых фторгалогенированных эфиров формулы: RfO-CFYI-CF2YII (I), где YI, YII, одинаковые или отличные друг от друга, представляют собой Сl, Вr, Н, при условии, что YI и YII не могут представлять собой одновременно Н, a Rf является таким как выше; 4) дегалогенирования или дегидрогалогенирования простых фторгалогенированных эфиров формулы (I) и получения простых виниловых эфиров формулы: RfO-CF=CF2 (IA). Предлагаемый способ позволяет получить целевые продукты с высокими выходами и селективностью. 14 з.п. ф-лы.
1. Способ получения простых перфторалкилвиниловых эфиров, имеющих общую формулу:
RfO-CF=CF2 (IA)
где Rf представляет собой C1-С3, предпочтительно C1-C2, перфторированный алкильный заместитель; включающий следующие стадии:
1) взаимодействия гипофторита формулы RfOF, где Rf является таким, как выше, с олефином формулы:
CY"Y=CY'Cl (II)
где Y, Y' и Y" одинаковые или отличные друг от друга представляют собой Н, Сl, Вr при условии, что Y, Y' и Y" не представляют собой одновременно водород;
2) дегалогенирования или дегидрогалогенирования простых фторгалогенированных эфиров, полученных на стадии 1), и получения простых виниловых эфиров формулы:
RfO-CYI=CYIIF (IV)
где YI и YII одинаковые или отличные друг от друга имеют значения Н, Сl, Вr при условии, что как YI, так и YII неодновременно представляют собой Н;
3) фторирования с помощью фтора простых виниловых эфиров (IV) и получения простых фторгалогенированных эфиров формулы:
RfO-CFYI-CF2YII (I)
где YI, YII одинаковые или отличные друг от друга представляют собой Cl, Br, H при условии, что YI и YII не могут представлять собой одновременно H;
Rf является таким, как выше;
4) дегалогенирования или дегидрогалогенирования простых фторгалогенированных эфиров формулы (I) и получения простых виниловых эфиров формулы:
RfO-CF=CF2 (IA).
2. Способ по п.1, в котором стадию 1) осуществляют в жидкой фазе при температурах от -130 до 0°С, предпочтительно от -80 до -10°С, необязательно в присутствии органических растворителей.
3. Способ по любому одному из пп.1 или 2, в котором олефины (II) выбираются из следующих: тетрахлорэтилена, трихлорэтилена, 1,2-дихлорэтилена и 1,1-дихлорэтилена.
4. Способ по любому одному из пп.1 или 2, в котором на стадии 2) дегалогенирование осуществляют с помощью переходных металлов, предпочтительно выбранных из цинка, меди, марганца, или с помощью пар металлов предпочтительно Zn/Cu, Zn/Sn, Zn/Hg в присутствии растворителей, выбранных из гидрированных протонных растворителей, простых гидрированных эфиров, диполярных апротонных растворителей.
5. Способ по любому одному из пп.1 или 2, в котором на стадии 2) дегидрогалогенирование осуществляют с помощью неорганического или органического основания.
6. Способ по п.5, в котором дегидрогалогенирование осуществляют в присутствии соли четвертичного аммония или фосфония, предпочтительно выбранного из аммоний тетрабутила, аммоний триоктилбензила, фосфоний тетрабутила и фосфоний триоктилбензила.
7. Способ по любому одному из пп.1 или 2, в котором на стадии 2) работа происходит при температурах в пределах 0-150°С, предпочтительно 25-100°С.
8. Способ по любому одному из пп.1 или 2, в котором на стадии 3) реакцию осуществляют добавлением газообразного фтора необязательно в присутствии инертного разбавителя, к соединениям формулы (IV), жидким при температуре реакции, необязательным использованием растворителя или смеси инертных растворителей, находящихся в жидком состоянии, при условиях, при которых осуществляют стадию 3).
9. Способ по любому одному из пп.1 или 2, в котором на стадии 3) работа происходит при температурах в пределах между -120°С и 0°С, предпочтительно -90°С и -30°С.
10. Способ по любому одному из пп.1 или 2, в котором необязательные растворители на стадиях 1) и 3) выбираются из следующих: простых (пер)фторированных полиэфиров, (пер)фторалканов, HFC, HCFC, CFC, перфтораминов, простых гидрофторированных эфиров или простых гидрофторированных полиэфиров, или их смесей.
11. Способ по любому одному из пп.1 или 2, в котором на стадии 4) дегалогенирование осуществляют с помощью переходных металлов, предпочтительно выбранных из цинка, меди, марганца, или с помощью пар металлов предпочтительно Zn/Cu, Zn/Sn, Zn/Hg в присутствии растворителей, выбранных из следующих: гидрированных протонных растворителей, простых гидрированных эфиров, диполярных апротонных растворителей.
12. Способ по любому одному из пп.1 или 2, в котором на стадии 4) дегидрогалогенирование осуществляют с помощью неорганического или органического основания.
13. Способ по п.12, в котором дегидрогалогенирование осуществляют в присутствии соли четвертичного аммония или фосфония, предпочтительно выбранной из аммоний тетрабутила, аммоний триоктилбензила, фосфоний тетрабутила и фосфоний триоктилбензила.
14. Способ по любому одному из пп.1 или 2, в котором на стадии 4) работа происходит при температурах в пределах 0-150°С, предпочтительно 25-100°С.
15. Способ по любому одному из пп.1 или 2, осуществляемый периодическим, полупериодическим или непрерывным способом.
| СПОСОБ ЭЛЕКТРОЛИТИЧЕСКОГО ОС.4ЖДЕНИЯ СПЛАВОВИНДИЯ | 0 |
|
SU201871A1 |
| US 4900872 A, 13.02.1990 | |||
| US 5350497 A, 27.09.1994 | |||
| Формирователь импульсов | 1979 |
|
SU839034A1 |
| Способ получения перфтор-2-бромэтилвинилового эфира | 1989 |
|
SU1839669A3 |

