ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к замещенному арилметиламиновому соединению, его получению, лекарственному препарату, включающему такое соединение, его использованию и его интермедиатам.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Воспалительные процессы с вовлечением тучных клеток, в частности астма, становятся все более серьезной проблемой здравоохранения. Астма часто характеризуется прогрессивным развитием гиперреактивной реакции трахеи и бронхов как на иммуноспецифические аллергены, так и на общие химические или физические стимулы, которая приводит к формированию хронического воспаления. Лейкоциты, содержащие рецепторы IgE, особенно тучные клетки и базофилы, присутствуют в эпителии и выстилающей гладкой мышечной ткани бронхов. Такие лейкоциты первоначально активируются посредством связывания специфических вдыхаемых антигенов с рецепторами IgE, а затем высвобождают ряд химических медиаторов. Например, дегрануляция тучных клеток приводит к выделению протеогликанов, пероксидазы, арилсульфатазы В, химазы и триптазы, что вызывает сужение бронхиол.
Триптаза хранится в секреторных гранулах тучных клеток и является основной секреторной протеазой тучных клеток человека. Триптаза вовлечена в разнообразные биологические процессы, в том числе деградацию сосудорасширяющих и бронхорелаксирующих нейропептидов (Caughey et al., J. Pharmacol. Exp. Ther., 1988, 244, стр. 133-137; Franconi et al., J. Pharmacol. Exp. Ther., 1988, 248, стр. 947-951; Tam et al., Am. J. Respir. Cell Mol. Biol., 1990, 3, стр. 27-32) и модуляцию бронхиального ответа на гистамин (Sekizawa et al., J. Clin. Invest., 1989, 83, стр. 175-179).
Поэтому ингибиторы триптазы могут быть полезны в качестве противовоспалительных агентов (K Rice, P.A. Sprengler, Current Opinion in Drug Discovery and Development, 1999, 2(5), стр. 463-474), особенно при лечении хронической астмы (M.Q. Zhang, H. Timmerman, Mediators Inflamm., 1997, 112, стр. 311-317), а также могут быть полезны при лечении или профилактике аллергического ринита (S. J. Wilson et al., Clin. Exp. Allergy, 1998, 28, стр. 220-227), синдрома хронической раздражимости кишечника (S.C. Bischoff et al., Histopathology, 1996, 28, стр. 1-13), псориаза (A. Naukkarinen et al., Arch. Dermatol. Res., 1993, 285, стр. 341-346), конъюктивита (A.A.Irani et al., J. Allergy Clin. Immunol., 1990, 86, стр. 34-40), атопического дерматита (A. Jarvikallio et al., Br. J. Dermatol., 1997, 136, стр. 871-877), ревматоидного артрита (L.C. Tetlow et al., Ann. Rheum. Dis., 1998, 54, стр. 549-555), остеоартрита (M.G. Buckley et al., J. Pathol., 1998, 186, стр. 67-74), подагрического артрита, ревматоидного спондилита и болезней разрушения суставных хрящей.
Кроме того, было показано, что триптаза является эффективным митогеном для фибробластов, что указывает на ее участие в легочном фиброзе при астме и интерстициальных легочных процессах (Ruoss et al., J. Clin. Invest., 1991, 88, стр. 493-499).
Поэтому ингибиторы триптазы могут быть полезны при лечении или профилактике фиброзных заболеваний (J.A. Cairns and A.F. Walls, J. Clin. Invest., 1997, 99, стр. 1313-1321), например фиброза, склеродермии, легочного фиброза, цирроза печени, фиброза миокарда, нейрофибром и гипертрофических рубцов.
Кроме того, ингибиторы триптазы могут быть полезны при лечении или профилактике инфаркта миокарда, инсульта, стенокардии и других последствий разрыва атеросклеротических бляшек (M. Jeziorska et al., J. Pathol., 1997, 182, стр. 115-122).
Было также обнаружено, что триптаза активирует простромелизин, который, в свою очередь, активирует коллагеназу, тем самым, инициируя разрушение хряща и периодонтальной соединительной ткани соответственно.
Поэтому ингибиторы триптазы могут быть полезны при лечении или профилактике артрита, периодонтальных заболеваний, диабетической ретинопатии и роста опухолей (W.J. Beil et al., Exp. Hematol., (1998) 26, стр. 158-169). Ингибиторы триптазы также могут быть полезны при лечении анафилакции (L.B. Schwarz et al., J. Clin. Invest., 1995, 96, стр. 2702-2710), рассеянного склероза (M. Steinhoff et al., Nat. Med. (N. Y.), 2000, 6(2), стр. 151-158), пептических язв и синцитиальных вирусных инфекций.
Замещенные арилметиламины, представленные соединениями формулы (A), их получение, фармакологические препараты, содержащие такие соединения, и их фармацевтическое использование при лечении болезненных состояний, которые могут корректироваться ингибированием триптазы, изложены в находящейся на рассмотрении патентной заявке США № 09/843126. В общую информацию о соединениях формулы (А) в патентной заявке США № 09/843126 включено и соединение настоящего изобретения формулы I. Вместе с тем в патентной заявке США с № 09/843126 конкретно не раскрывается информация о соединении формулы I.
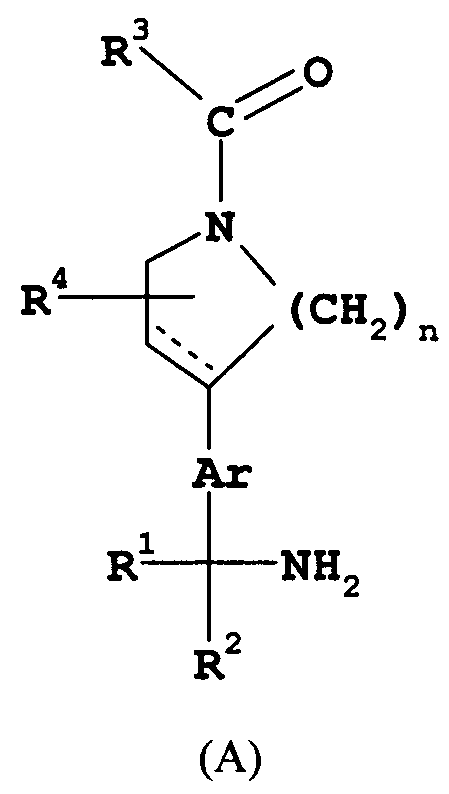
Соответственно, существует потребность в новом и полезном соединении, которое обладает особенно ценными фармацевтическими свойствами в своей способности ингибировать триптазу. Такое соединение должно с легкостью найти применение в лечении пациентов, страдающих болезнями, которые можно облегчить введением ингибитора триптазы, например воспалительные состояния с участием тучных клеток, воспаление и заболевания или расстройства, связанные с деградацией сосудорасширяющих и бронхорелаксирующих нейропептидов.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению формулы I

или пролекарству, фармацевтически приемлемой соли или сольвату указанного соединения.
Кроме того, настоящее изобретение относится к фармацевтической композиции, включающей фармацевтически эффективное количество соединения формулы I и фармацевтически приемлемый носитель.
Кроме того, настоящее изобретение относится к применению соединения формулы I в качестве ингибитора триптазы, предусматривающему введение соединения в состав, включающий триптазу. Настоящее изобретение также относится к применению соединения формулы I для лечения пациентов, страдающих или подверженных физиологическим заболеваниям, при которых необходимо повышение уровня ингибитора триптазы, при этом такое лечение предусматривает введение пациенту терапевтически эффективного количества соединения по пункту 1.
Настоящее изобретение также относится к способу получения соединения формулы I.
Краткое описание чертежей
Аспекты, особенности и преимущества настоящего изобретения будут более понятны из последующих подробных описаний, рассматриваемых в совокупности с сопроводительными чертежами, которые приводятся исключительно для иллюстрации и не носят ограничительного характера для настоящего изобретения, к ним относятся следующие.
Фиг.1: уровни соединения I в плазме, бронхоальвеолярном лаваже (БАЛ) и легких, измеренные через 2 часа после введения соединения I в дозе 1 мг/кг перорально. Значения представляют собой среднее ± стандартная ошибка для 6-8 животных.
Фиг.2: уровни соединения I в плазме и легких, измеренные через 24 часа после введения. Значения представляют собой среднее ± стандартная ошибка для 3-4 животных.
ПОЛНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
Используемые выше и во всем тексте настоящего описания и прилагаемой формулы изобретения термины, если не указано иначе, имеют следующие принятые значения.
Использованный в настоящем описании термин «соединение настоящего изобретения» и эквивалентные ему выражения охватывают соединение формулы I, как это описано выше, и включает пролекарство, фармацевтически приемлемую соль и сольват, например гидрат. Точно так же ссылка на интермедиаты, независимо от того, включены ли они сами в формулу изобретения, охватывают соли и сольваты в случаях, где это допускается контекстом. Для полной ясности конкретные случаи, где это допускается контекстом, иногда указываются в тексте, но такие случаи носят чисто иллюстративный характер и не предполагают исключения других случаев, где контекстом допускается подобное толкование.
Термин «лечение» или «лечить», используемый в настоящем документе, включает профилактическую терапию, а также лечение диагностированного состояния больного.
«Пациент» означает человека или другое млекопитающее.
«Эффективное количество» призвано описывать количество соединения, эффективного в создании желательного терапевтического эффекта.
«Пролекарство» означает соединение, которое пригодно для введения пациенту, не обладает нежелательной токсичностью, не вызывает раздражения, аллергической реакции и т.п. и в ходе метаболических процессов в организме (например, гидролиза) может превращаться в соединение настоящего изобретения. Подробное обсуждение пролекарств приводится в T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems, Vol. 14 of the A. C. S. Symposium Series, а также в Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, обе работы в силу ссылки включены в настоящий документ.
Конкретные или предпочтительные варианты осуществления
Кроме того, настоящее изобретение относится к применению соединения формулы I для лечения пациентов, страдающих физиологическими заболеваниями, которые можно облегчить введением пациенту терапевтически эффективного количества соединения формулы I. Конкретные варианты осуществления для физиологических заболеваний, которые можно лечить с помощью соединения настоящего изобретения, включают, но, конечно же, не ограничиваются перечисленным, воспалительные заболевания, например воспаление суставов, артрит, ревматоидный артрит, ревматоидный спондилит, подагрический артрит, травматический артрит, краснушный артрит, псориатический артрит и другие хронические воспалительные заболевания суставов. Другие варианты осуществления для физиологических заболеваний, которые можно лечить с использованием настоящего изобретения, включают такие физиологические заболевания, как хроническое обструктивное заболевание легких (ХОЗЛ), обострение ХОЗЛ, разрушение суставного хряща, глазной конъюктивит, весенний конъюктивит, синдром повышенной раздражимости кишечника, астма, аллергический ринит, интерстициальные легочные процессы, фиброз, склеродермия, легочный фиброз, цирроз печени, фиброз миокарда, нейрофибромы, гипертрофические рубцы, различные дерматологические заболевания, например атопический дерматит и псориаз, инфаркт миокарда, инсульт, стенокардия и другие последствия разрыва атеросклеротических бляшек, а также периодонтальное заболевание, диабетическая ретинопатия, рост опухолей, анафилаксия, рассеянный склероз, пептические язвы и синцитиальные вирусные инфекции.
В одном из конкретных вариантов осуществлений настоящее изобретение относится к применению соединения формулы I для лечения пациентов, страдающих астмой, которое предусматривает введение пациенту физиологически эффективного количества вещества.
В другом конкретном варианте осуществления настоящее изобретение относится к применению соединения формулы I для лечения пациентов, страдающих ХОЗЛ, которое предусматривает введение пациенту физиологически эффективного количества вещества.
В другом конкретном варианте осуществления настоящее изобретение относится к применению соединения формулы I для лечения пациентов, страдающих обострением ХОЗЛ, которое предусматривает введение пациенту физиологически эффективного количества вещества.
В другом конкретном варианте осуществления настоящее изобретение относится к применению соединения формулы I для лечения пациентов, страдающих аллергическим ринитом, которое предусматривает введение пациенту физиологически эффективного количества вещества.
В другом конкретном варианте осуществления настоящее изобретение относится к применению соединения формулы I для лечения пациентов, страдающих воспалением суставов, которое предусматривает введение пациенту физиологически эффективного количества вещества.
В другом конкретном варианте осуществления настоящее изобретение относится к применению соединения формулы I для лечения пациентов, страдающих синдромом повышенной раздражимости кишечника, которое предусматривает введение пациенту физиологически эффективного количества вещества.
Кроме того, настоящее изобретение распространяется на фармацевтическую композицию, включающую соединение формулы I, второе соединение, выбранное из группы, включающей бета-адренергический агонист, антихолинергическое средство, противовоспалительный кортикостероид и противовоспалительный агент, а также их фармацевтически приемлемый носитель. В такой композиции соединение формулы I и второе соединение присутствуют в количествах, которые обеспечивают терапевтически эффективную активность, то есть аддитивный или синергетический эффект. К конкретным воспалительным заболеваниям или нарушениям, которые можно лечить такой фармацевтической композицией, среди прочих, относится астма.
Кроме того, настоящее изобретение относится к способу лечения пациентов, страдающих воспалительными нарушениями, который предусматривает введение пациенту соединения формулы I и второго соединения, выбранного из группы, включающей бета-адренергический агонист, антихолинергическое средство, противовоспалительный кортикостероид и противовоспалительный агент. В таком способе соединение формулы I и второе соединение присутствуют в количествах, которые обеспечивают терапевтически эффективную активность, то есть аддитивный или синергический эффект. В таком способе настоящего изобретения соединение настоящего изобретения может вводиться пациенту перед вторым соединением, а второе соединение может вводиться пациенту перед соединением настоящего изобретения или соединение настоящего изобретения и второе соединение могут вводиться одновременно. Конкретные примеры адренергических агонистов, антихолинергических средств, противовоспалительных кортикостероидов и противовоспалительных агентов, применяющихся в соответствии с данным способом, описаны ниже.
Фармакологические препараты
Как отмечалось выше, соединение настоящего изобретения проявляет полезную фармакологическую активность и соответственно может включаться в состав фармацевтических композиций и использоваться в лечении пациентов, страдающих определенными медицинскими нарушениями. Таким образом, в соответствии с дополнительным аспектом настоящее изобретение предусматривает фармацевтические композиции, состоящие из соединения изобретения и его фармацевтически приемлемого носителя. Термин «фармацевтически приемлемый», который используется в настоящем документе, предпочтительно означает утвержденный директивным органом правительства, в частности федерального правительства или правительства штата, или внесенный в Фармакопею США или другую общепринятую фармакопею для использования на животных и, в особенности, на человеке. Пригодные фармацевтические носители описаны в книге E.W. Martin "Remington's Pharmaceutical Sciences".
В соответствии с настоящим изобретением фармацевтические композиции могут готовиться по общепринятым методикам с использованием одного или нескольких фармацевтически приемлемых вспомогательных веществ или наполнителей. К вспомогательным веществам, среди прочего, относятся разбавители, наполнители, связующие, дезинтегрирующие вещества, облегчающие скольжение вещества, смазывающие вещества, поверхностно-активные вещества, стерильные водные среды и различные нетоксичные органические растворители. Составы могут иметь форму таблеток, капсул, пилюль, составов с замедленным высвобождением, гранул, порошков, водных растворов или суспензий, инъекционных растворов, эликсиров или сиропов и могут содержать один или несколько агентов, выбираемых из группы, содержащей подсластители, отдушки, красители или стабилизаторы, с тем чтобы получать фармацевтически приемлемые препараты. Выбор носителя и содержание активного вещества в носителе обычно определяется в соответствии с растворимостью и химическими свойствами активного соединения, конкретным способом введения и положениями, которые должны соблюдаться в фармацевтической практике. Например, для приготовления таблеток могут использоваться наполнители, такие как лактоза, микрокристаллическая целлюлоза, желатинированный крахмал, немодифицированный крахмал, силикативированная микрокристаллическая целлюлоза, маннитол, сорбитол, ксилитол, декстраты, фруктоза, цитрат натрия, карбонат кальция, дикальцийфосфат дигидрат, безводный дикальцийфосфат, сульфат кальция, наряду со связующими, такими как поливинилпирролидон, гидроксипропилметилцеллюлоза, этилцеллюлоза, гидроксиэтилцеллюлоза, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, желатинированный крахмал, крахмал, полиэтиленгликоли, полиэтиленоксид, поликарбофилы, желатин и камедь, и дезинтегрирующие агенты, такие как натриевая соль кроскармелозы, натриевая соль гликолята крахмала, кросповидон, крахмал, микрокристаллическая целлюлоза, альгиновая кислота и некоторые сложные силикаты в сочетании со смазывающими веществами, такими как стеарат магния, стеарат кальция, стеариновая кислота, гидрогенизированное растительное масло, минеральное масло, полиэтиленгликоли, эфиры глицерина и жирных кислот, лаурилсульфат натрия и облегчающие скольжение вещества, такие как двуокись кремния, тальк, крахмал, наряду с некоторыми поверхностно-активными веществами, такими как лаурилсульфат натрия, эфиры сорбитана, полиоксиэтиленовые эфиры жирных кислот, полоксамер, полиоксиэтиленовый эфир, докузат натрия, полиэтоксилированное касторовое масло и бензалконий хлорид. Для приготовления капсулы удобно использовать наполнители, такие как лактоза, микрокристаллическая целлюлоза, прежелатинированный крахмал, немодифицированный крахмал, силикативированная микрокристаллическая целлюлоза в чистом виде или смесь двух или более наполнителей с добавлением или без добавления связующих, которые приведены выше, наряду с подходящим поверхностно-активным агентом (агентами), дезинтегрирующими веществами, облегчающими скольжение веществами, смазывающими веществами и пр., как это указано выше. При использовании водных суспензий они могут содержать эмульгаторы или вещества, которые способствуют образованию суспензий. Могут также использоваться разбавители, например сахароза, этанол, полиэтиленгликоль, пропиленгликоль, глицерин и хлороформ или их смеси. Такими фармацевтически приемлемыми носителями могут также быть стерилизованная вода и масла, в том числе нефтяного, животного, растительного или синтетического происхождения, например арахисовое масло, соевое масло, минеральное масло, кунжутное масло и подобные им масла. При внутривенном введении лекарственного препарата предпочтительным носителем является вода. Физиологические растворы и водные растворы декстрозы и глицерина также могут использоваться в качестве жидких носителей, особенной для инъекционных растворов. К подходящим фармацевтическим наполнителям относятся маннитол, альбумин человеческой сыворотки, крахмал, глюкоза, лактоза, желатин, солод, рис, мука, мел, силикагель, карбонат магния, стеарат магния, стеарат натрия, глицеринмоностеарат, тальк, хлорид натрия, сухое снятое молоко, глицерин, пропилен, гликоль, вода, этанол и подобные им вещества. Такие препараты могут иметь форму растворов, суспензий, таблеток, пилюль, капсул, порошков, лекарственных форм с длительным высвобождением вещества и им подобных.
Естественно, фармацевтическая композиция настоящего изобретения будет содержать терапевтически эффективное количество действующего вещества наряду с соответствующим количеством носителя, с тем чтобы обеспечить форму для надлежащего введения пациенту. Несмотря на то что внутривенная инъекция является очень эффективной формой введения, могут использоваться и другие способы, например инъекция или пероральное, назальное или парентеральное введение, которые обсуждаются ниже.
Методы лечения
Соединение формулы I обладает способностью ингибировать триптазу в соответствии с тестами, описанными в литературе и изложенными ниже в настоящем описании, и результаты таких тестов предположительно коррелируют с фармакологической активностью в организме человека и других млекопитающих. Таким образом, еще в одном варианте осуществления настоящее изобретение относится к применению соединения формулы I или содержащей его композиции для лечения пациентов, страдающих или подверженных болезненным состояниям, которые можно облегчить посредством введения ингибитора триптазы. Например, соединение формулы I полезно для лечения воспалительных заболеваний, таких как воспаление суставов, в том числе артрит, ревматоидный артрит и другие виды артрита, такие как ревматоидный спондилит, подагрический артрит, травматический артрит, краснушный артрит, псориатический артрит, остеоартрит или иное хроническое воспалительное заболевание суставов или заболевания разрушения суставного хряща, глазной конъюктивит, весенний конъюктивит, синдром повышенной раздражимости кишечника, астма, аллергический ринит, интерстициальные легочные процессы, фиброз, склеродермия, легочный фиброз, цирроз печени, фиброз миокарда, нейрофибромы, гипертрофические рубцы, различные дерматологические заболевания, например атопический дерматит и псориаз, инфаркт миокарда, инсульт, стенокардия и другие последствия разрыва атеросклеротических бляшек, а также периодонтальное заболевание, диабетическая ретинопатия, рост опухолей, анафилаксия, рассеянный склероз, пептические язвы или синцитиальные вирусные инфекции.
Согласно другим вариантам осуществления изобретения в нем приводится способ лечения пациента, человека или животного, страдающего или подверженного заболеваниям, которые можно облегчить введением ингибитора триптазы, например, заболевания, которые были описаны выше в настоящем документе, и такое лечение предусматривает введение пациенту эффективного количества соединения изобретения или препарата, содержащего соединение изобретения.
Комбинированная терапия
Как пояснялось выше, в сочетании с соединением формулы I могут использоваться другие фармацевтически активные вещества в зависимости от болезни, лечение которой проводится. Например, при лечении астмы могут включаться бета-адренергетические агонисты, такие как альбутерол, тербуталин, формотерол, фенотерол или преналин, а также антихолинергические средства, такие как ипратропий бромид, противовоспалительные кортикостероиды, такие как беклометазон дипропионат, триамцинолон ацетонид, флюнизолид или дексаметазон и противовоспалительные агенты, такие как натрия кромогликат и недокромил-натрий. Таким образом, настоящее изобретение распространяется на фармацевтическую композицию, включающую соединение формулы I и второе соединение, которое выбирается из группы, включающей бета-адренергический агонист, антихолинергическое средство, противовоспалительный кортикостероид и противовоспалительный агент, а также их фармацевтически приемлемый носитель. Конкретные фармацевтические носители, применяющиеся для данной фармацевтической композиции, описаны в настоящем описании.
Кроме того, настоящее изобретение относится к способу лечения пациентов, страдающих от астмы, который предусматривает введение пациенту соединения настоящего изобретения и второго соединения, выбранного из группы, включающей бета-адренергический агонист, антихолинергическое средство, противовоспалительный кортикостероид и противовоспалительный агент. В таком комбинированном методе соединение настоящего изобретения может вводиться перед вторым соединением, соединение настоящего изобретения может вводиться после второго соединения или соединение настоящего изобретения и второе соединение могут вводиться одновременно.
Способы доставки
В соответствии с настоящим изобретением соединение формулы I или фармацевтическая композиция, включающая данное соединение, могут вводиться пациенту парентерально, через слизистую оболочку, например, перорально, назально, пульмонально или ректально, или же чрескожным образом.
Пероральная доставка
В настоящем описании предлагаются для использования пероральные твердые дозированные формы, общее описание которых приводится в Remington's Pharmaceutical Sciences, 18th Ed.1990 (Mack Publishing Co. Easton PA 18042), глава 89, который в силу ссылки на него включается в настоящее описание. К твердым дозированным формам относятся таблетки, капсулы, пилюли, пастилки или лепешки, облатки или пеллеты. Для составления таких препаратов может использоваться липосомальное или протеиноидное инкапсулирование (как, например, протеиноидные микросферы, представленные в патенте U.S. Patent № 4925673). Может использоваться липосомальное инкапсулирование, и могут быть получены производные липосом и различных полимеров (например, патент U.S. Patent No. 5013556). Описание возможных твердых дозированных форм для терапевтического применения приводится в Marshall, K. In: Modern Pharmaceutics Edited by G.S. Banker and C.T. Rhodes, глава 10, 1979, которая в качестве ссылки включается в настоящее описание. В общем случае состав будет включать соединение настоящего изобретения и инертные ингредиенты, которые обеспечивают защиту от среды желудка и высвобождение биологически активного материала, то есть соединения настоящего изобретения, в кишечнике.
Кроме того, конкретно предлагаются пероральные дозированные формы соединения настоящего изобретения. Такое соединение может быть химически модифицировано, с тем чтобы пероральная доставка была бы более эффективной. В общем случае предложенная химическая модификация включает присоединение по крайней мере одного функционального фрагмента к самой молекуле компонента, причем такой функциональный фрагмент обеспечивает (а) ингибирование протеолиза и (b) усвоение в кровоток из желудка или кишечника. Кроме того, желательно повысить общую стабильность соединения настоящего изобретения и увеличить время циркуляции в организме. К примерам таких функциональных фрагментов относятся полиэтиленгликоль, сополимеры этиленгликоля и пропиленгликоля, карбоксиметилцеллюлоза, декстран, поливиниловый спирт, поливинилпирролидон и полипролин. Abuchowski and Davis, 1981, "Soluble Polymer-Enzyme Adducts" In: Enzymes as Drugs, Hocenberg and Roberts, eds., Wiley-Interscience, New York, NY, pp. 367-383; Newmark et al., 1982, J. Appl. Biochem. 4:185-189. К другим полимерам, которые могут быть использованы, относятся поли-1,3-диоксолан и поли-1,3,6-триоксокан. Как отмечалось выше, предпочтительными для фармацевтического применения являются функциональные фрагменты, содержащие полиэтиленгликоль.
Для соединения настоящего изобретения местом высвобождения может быть желудок, тонкий кишечник (двенадцатиперстная кишка, тощая кишка или подвздошная кишка) или толстый кишечник. Специалистам в данной области известны доступные составы, которые не будут растворяться в желудке, но будут высвобождать вещество в двенадцатиперстной кишке или в другой области кишечника. Предпочтительно высвобождение позволит избежать отрицательных воздействий среды желудка либо посредством защиты соединения настоящего изобретения, либо за счет высвобождения вещества вне среды желудка, например в кишечнике.
Чтобы обеспечить абсолютную устойчивость в желудке, необходимо покрытие, непроницаемое по крайней мере до рН 5,0. Примерами наиболее распространенных инертных ингредиентов, которые используются в качестве энтеральных покрытий, являются тримеллитат ацетилцеллюлозы (ТАЦ), гидроксипропилметилцеллюлозы фталат (ГПМЦФ), ГПМЦФ 50, ГПМЦФ 55, поливинилацетат фталат (ПВАФ), Eudragit L30D, Aquateric, фталат ацетилцеллюлозы (ФАЦ), Eudragit L, Eudragit S и шеллак. Такие покрытия могут использоваться в форме смешанных пленок.
Покрытия или смесь покрытий могут также использоваться на таблетках, которые не предназначены для защиты от среды желудка. К ним могут относиться сахарные покрытия или покрытия, которые облегчают проглатывание таблетки. Капсулы могут состоять из твердой оболочки (например, желатин) для доставки сухого терапевтического средства, то есть порошка; для жидких форм может использоваться мягкая желатиновая оболочка. Материалом оболочки для облаток может быть густой крахмал или другая съедобная бумага. Для приготовления пилюль, лепешек, формованных таблеток или таблетированных порошков могут использоваться методики концентрации влаги.
Терапевтическое средство может входить в состав препарата в виде агрегатов мелких частиц в форме гранул или пеллет с размером частиц около 1 мм. Состав материала для введения в виде капсул может также представлять собой порошок, слегка прессованные вкладки или даже таблетки. Терапевтическое средство может быть приготовлено посредством прессования.
Могут включаться красители и отдушки. Например, может готовиться лекарственная форма соединения настоящего изобретения (например, инкапсулирование в липосомах или микросферах), а затем вноситься в пищевой продукт, например охлажденный напиток, содержащий красители и отдушки.
Можно разбавлять или увеличивать объем терапевтического средства за счет инертного материала. К таким разбавителям могут относиться углеводы, особенно маннитол, альфа-лактоза, безводная лактоза, целлюлоза, сахароза, модифицированные декстраны и крахмал. Некоторые неорганические соли могут также использоваться в качестве наполнителей, в том числе трифосфат кальция, карбонат магния и хлорид натрия. Ряд промышленно выпускаемых разбавителей включает Fast-Flo, Emdex, STA-Rx 1500, Emcompress и Avicell.
Дезинтегрирующие вещества могут включаться в состав терапевтического средства в виде твердой дозированной формы. К материалам, используемым в качестве дезинтегрирующих средств, среди прочих, относятся промышленно выпускаемые дезинтегрирующие средства на основе крахмала, например Explotab. Могут применяться натриевая соль гликолята крахмала, амберлит, натриевая соль карбоксиметилцеллюлозы, ультрамилопектин, альгинат натрия, желатин, апельсиновая цедра, кислая карбоксиметилцеллюлоза, природная губка и бентонит. Другой формой дезинтегрирующих веществ являются нерастворимые катионобменные смолы. Камеди в порошке могут использоваться в качестве дезинтегрирующих веществ и связующих, и к ним могут относиться такие порошковые камеди как агар, карайя или трагакант. В качестве дезинтегрирующих веществ также полезно применять альгиновую кислоту и ее натриевую соль.
Связующие могут использоваться для формования терапевтического средства в твердую таблетку, и к таким связующим относятся материалы из природных продуктов, например камедь, трагакант, крахмал и желатин. Среди других связующих - метилцеллюлоза (МЦ), этилцеллюлоза (ЭЦ) и карбоксиметилцеллюлоза (КМЦ). Для гранулирования терапевтического средства могут использоваться поливинилпирролидон (ПВП) и гидроксипропилметилцеллюлоза (ГПМЦ) в спиртовых растворах.
В состав терапевтического средства может включаться снижающий трение агент, с тем чтобы предотвратить слипание в процессе приготовления состава. Смазывающие вещества могут использоваться в качестве прослойки между терапевтическим средством и стенкой матрицы, и к ним, среди прочих, могут относиться стеариновая кислота, в том числе ее магниевые и кальциевые соли, политетрафторэтилен (ПТФЭ), жидкий парафин, растительные масла и воски. Могут также применяться растворимые смазывающие вещества, например лаурилсульфат натрия, лаурилсульфат магния, полиэтиленгликоль различного молекулярного веса, Carbowax 4000 и 6000.
Могут добавляться облегчающие скольжение вещества, которые улучшают текучесть лекарства при приготовлении состава и способствуют реорганизации при формовании. К облегчающим скольжение веществам относятся крахмал, тальк, пирогенный кремнезем и гидратированный алюмосиликат.
Чтобы способствовать растворению терапевтического средства в водной среде, в качестве смачивателя может добавляться поверхностно-активное вещество. К поверхностно-активным веществам могут относиться анионные детергенты, например лаурилсульфат натрия, диоктилсульфосукцинат натрия и диоктилсульфонат натрия. Возможно применение катионных детергентов, которые могут включать бензалконий хлорид и бензетомий хлорид. Перечень возможных неионогенных детергентов, которые могут входить в состав препарата в качестве поверхностно-активных веществ, включает лауромакрогол 400, полиоксилстеарат 40, полиоксиэтиленовое производное гидрогенизированного касторового масла 10, 50 и 60, глицерилмоностеарат, полисорбат 40, 60, 65 и 80, сахарный эфир жирных кислот, метилцеллюлоза и карбоксиметилцеллюлоза. Такие поверхностно-активные вещества могут присутствовать в составе препарата соединения настоящего изобретения как в чистом виде, так и в смеси в различных соотношениях.
К добавкам, которые в состоянии усилить усвоение соединения настоящего изобретения, относятся, например, жирные кислоты: олеиновая кислота, линолевая кислота и линоленовая кислота. Желательным может быть препарат для перорального применения с регулируемым высвобождением действующего вещества. Лекарство может включаться в инертную матрицу, которая обеспечивает высвобождение либо за счет диффузии, либо посредством механизмов выщелачивания, например, камеди. В состав препарата также могут входить медленно распадающиеся матрицы. Ряд энтеральных покрытий также обладают замедленным эффектом высвобождения.
Другой формой регулируемого высвобождения данного терапевтического средства является метод, основанный на терапевтической системе OROS (Alza Corp.), то есть препарат заключается в полупроницаемую мембрану, которая позволяет воде проникать внутрь и за счет осмотических эффектов выталкивать препарат через единственный тонкий просвет.
Для приготовления препарата могут использоваться другие покрытия. К ним относятся разнообразные сахара, которые могут наноситься в ванне для покрытий. Терапевтический препарат может также вводиться в таблетке с пленочным покрытием, и применяемые для этого материалы подразделяются на 2 группы. К первой относятся неэнтерические материалы, и она включает метилцеллюлозу, этилцеллюлозу, гидроксиэтилцеллюлозу, метилгидроксиэтилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, натриевую соль карбоксиметилцеллюлозы, провидон и полиэтиленгликоли. Вторую группу образуют энтерические материалы, которые обычно представлены эфирами фталевой кислоты.
Для обеспечения оптимального пленочного покрытия может использоваться смесь материалов. Пленочное покрытие может наноситься в ванне для покрытий или в псевдоожиженном слое, или покрытием путем прессования.
Пульмональная доставка
В настоящем документе также предлагается пульмональная доставка соединения настоящего изобретения как индивидуально, так и в виде фармацевтической композиции. Соединение доставляется в легкие млекопитающих при вдыхании и через выстилающий легочный эпителий переносится в кровоток. Другие сообщения об этом включают Adjei et al., 1990, Pharmaceutical Research, 7:565-569; Adjei et al., 1990, International Journal of Pharmaceutics, 63:135-144 (лейпролид ацетат); Braquet et al., 1989, Journal of Cardiovascular Pharmacology, 13 (suppl. 5):143-146 (эндотелин-1); Hubbard et al., 1989, Annals of Internal Medicine, Vol. III, pp. 206-212 (a1- антитрипсин); Smith et al., 1989, J.Clin. Invest. 84:1145-1146 (a-1-протеиназа); Oswein et al., 1990, "Aerosolization of Proteins", Proceedings of Symposium on Respiratory Drug Delivery II, Keystone, Colorado, March (рекомбинантный гормон роста человека); Debs et al., 1988, J. Immunol. 140:3482-3488 (интерферон-гамма и фактор некроза опухоли альфа) и Platz et al., U.S. Patent No. 5284656 (колониестимулирующий фактор гранулоцитов). Метод и состав для пульмональной доставки препаратов системного воздействия описан в патенте U.S. Patent № 5451569, выданном 19 сентября 1995 года Wong et al.
Для практического использования в настоящем изобретении предлагается широкий круг механических устройств, предназначенных для пульмональной доставки терапевтических препаратов, в том числе, среди прочих, аэрозольных аппаратов, дозирующих ингаляторов и порошковых ингаляторов, все они известны специалистам в данной области.
Некоторые конкретные примеры промышленно выпускаемых устройств, пригодных для практического применения в настоящем изобретении, включают аэрозольный аппарат Ultravent, производимый Mallinckrodt, Inc., St. Louis, Missouri; аэрозольный аппарат Acorn II, выпускаемый Marquest Medical Products, Englewood, Colorado; дозирующий ингалятор Ventolin, который производится Glaxo Inc., Research Triangle Park, North Carolina, а также порошковый ингалятор Spinhaler, выпускаемый Fisons Corp., Bedford, Massachusetts, и многие другие.
Все подобные устройства требуют использования препаратов, пригодных для дозирования соединения настоящего изобретения. Каждый препарат, как правило, пригоден для использования с определенным типом используемого устройства, и может предусматривать применение соответствующего пропеллента в дополнение к обычным разбавителям, вспомогательным веществам и/или носителям, пригодным для проведения лечения. Также предлагается использовать липосомы, микрокапсулы или микросферы, комплексы включения или другие виды носителей. Химически модифицированное соединение настоящего изобретения может также входить в состав различных препаратов в зависимости от типа химической модификации или типа используемого устройства.
В состав препаратов, пригодных для использования в аэрозольном аппарате, как в струевом, так и в ультразвуковом, будет обычно входить соединение настоящего изобретения, растворенное в воде с концентрацией примерно от 0,1 до 25 мг соединения на 1 мл раствора. Препарат может также включать буфер и простой сахар (например, для стабилизации и регулирования осмотического давления). Состав в аэрозольном аппарате может также содержать поверхностно-активное вещество для уменьшения или предотвращения поверхностно-стимулированной агрегации соединения, вызванного распылением раствора при образовании аэрозоля.
Составы препарата для использования в дозирующем устройстве для ингаляции будут обычно содержать мелко измельченный порошок, содержащий соединение изобретения, взвешенный в пропелленте с помощью поверхностно-активного вещества. В качестве пропеллента может использоваться любой традиционный материал, который применяется для этих целей, например хлорфторуглеводород, гидрохлорфторуглеводород, гидрофторуглеводород или углеводород, в том числе трихлорфторметан, дихлордифторметан, дихлортетрафторэтанол и 1,1,1,2-тетрафторэтан или их комбинации. К пригодным поверхностно-активным веществам относятся сорбитантриолеат и соевый лецитин. Олеиновая кислота может также быть пригодна для использования в качестве поверхностно-активного вещества.
Препараты для дозирования с помощью порошкового устройства для ингаляции будут содержать мелко измельченный порошок, содержащий соединение настоящего изобретения, и могут также включать наполнитель, например лактозу, сорбитол, сахарозу или маннитол, в количествах, которые способствуют распылению порошка из устройства, например от 50 до 90 вес.% препарата. Соединение настоящего изобретения должно преимущественным образом готовиться в измельченной форме со средним размером частиц менее 10 мм (или микрон), предпочтительнее всего от 0,5 до 5 мм, для наиболее эффективной доставки в периферические легкие.
Назальная доставка
Также предлагается назальная доставка соединения настоящего изобретения. Назальная доставка обеспечивает попадание соединения в кровоток непосредственно после введения терапевтического препарата в нос без необходимости депонирования продукта в легких. Препараты для назальной доставки включают составы с декстраном или циклодекстраном.
Чрескожная доставка
В настоящем изобретении находит применение множество разнообразных методов чрескожного применения того или иного препарата, например через трансдермальный пластырь. Трансдермальные пластыри описываются, например, в U.S. Patent № 5407713, выданный 18 апреля 1995 года Rolando et al.; U.S. Patent № 5352456, выданный 4 октября 1994 года Fallon et al.; U.S. Patent № 5332213, выданный 9 августа 1994 года D'Angelo et al.; U.S. Patent № 5336168, выданный 9 августа 1994 года Sibalis; U.S. Patent № 5290561, выданный 1 марта 1994 года Farhadieh et al.; U.S. Patent № 5254346, выданный 19 октября 1993 года Tucker et al.; U.S. Patent № 5164189, выданный 17 ноября 1992 года Berger et al.; U.S. Patent № 5163899, выданный 17 ноября 1992 года Sibalis; U.S. Patent №№ 5088977 и 5087240, выданные 18 февраля 1992 года Sibalis; U.S. Patent № 5008110, выданный 16 апреля 1991 года Benecke et al., и U.S. Patent № 4921475, выданный 1 мая 1990 года Sibalis, раскрытие информации по каждому из которых в силу ссылки на них полностью включается в настоящий документ.
Можно с легкостью установить, что чрескожный путь введения можно сделать более эффективным за счет использования агента, усиливающего проникновение через кожу, например агентов, описанных в U.S. Patent № 5164189 (выше), U.S. Patent № 5008110 (выше) и U.S. Patent № 4879119, выданный 7 ноября 1989 года Aruga et al., раскрытие информации по каждому из которых в силу ссылки на них полностью включается в настоящий документ.
Местное применение
Для местного применения могут использоваться гели (на водной или спиртовой основе), кремы или мази, содержащие соединения изобретения. Соединения изобретения могут также входить в состав геля или матричной основы для применения в пластыре, которые будут обеспечивать регулируемое высвобождение соединения через трансдермальный барьер.
Ректальное введение
Твердые составы для ректального введения включают суппозитории, приготовленные в соответствии с известными методами и содержащие соединение изобретения.
Дозировка
Процент активного компонента в составах изобретения может различаться, при этом необходимо, чтобы он содержал такую долю, которая обеспечивала бы соответствующую дозировку. Понятно, что несколько единичных дозированных форм могут вводиться примерно одновременно. Применяемая доза будет определяться врачом и зависит от желаемого терапевтического эффекта, способа введения и продолжительности лечения, а также состояния пациента. Для взрослых дозы обычно порядка 0,001-50, предпочтительно порядка 0,001-5 мг/кг веса тела при введении посредством ингаляций, порядка 0,01-100, предпочтительно от 0,1 до 70, более точно от 0,5 до 10 мг/кг веса тела в день при пероральном применении и порядка 0,001-10, предпочтительно от 0,01 до 1 мг/кг веса тела в день при внутривенном введении. В каждом конкретном случае дозы будут определяться в соответствии с факторами, характерными для проходящего лечения, например возраста, веса, общего состояния здоровья и других особенностей, которые могут влиять на эффективность лекарственного препарата.
Кроме того, в соответствии с изобретением соединение может вводиться так часто, как это необходимо для достижения желаемого терапевтического эффекта. Некоторые пациенты могут проявлять быструю ответную реакцию на более высокую или более низкую дозу, и для них могут оказаться достаточными гораздо более слабые дозы. Для других пациентов могут понадобиться долгосрочные курсы лечения с частотой от 1 до 4 доз в день в соответствии с физиологическими потребностями каждого конкретного пациента. Вообще, действующее вещество может применяться перорально от 1 до 4 раз в день. Конечно, для некоторых пациентов придется установить не более одной или двух доз в день.
Естественно, пациент, для которого введение соединения настоящего изобретения оказывается эффективной терапевтической схемой, предпочтительно является человеком, но может быть и любым животным. Таким образом, как может с легкостью установить любой специалист в данной области, способы и фармацевтические композиции настоящего изобретения особенно подходят для введения любому животному, в частности млекопитающему, в том числе, но не ограничено, домашним животным, например, относящимся к семейству кошачьих или псовых, сельскохозяйственным животным, например, среди прочих, коровам, лошадям, козам, овцам и свиньям, диким животным (в условиях дикой природы или в зоологическом саду), подопытным животным, например мышам, крысам, кроликам, козам, овцам, свиньям, собакам, кошкам и пр., птицам, например курам, индейкам, певчим птицам, то есть для применения в ветеринарной медицине.
Подробная информация о приготовлении
Соединение формулы I может быть получено посредством применения или адаптации известных методов, под которыми подразумеваются методы, использовавшиеся прежде либо описанные в литературе, например изложенные в работе R.C. Larock in Comprehensive Organic Transformations, VCH publishers, 1989, или как это описано в настоящем документе.
В описанных ниже реакциях может возникать необходимость защитить реакционные функциональные группы, например аминогруппы, чтобы избежать их нежелательного участия в реакциях. Могут использоваться традиционные защитные группы в соответствии с принятой практикой, примеры см. в T.W. Greene and P.G.M. Wuts in "Protective Groups in Organic Chemistry" John Wiley and Sons, 1991.
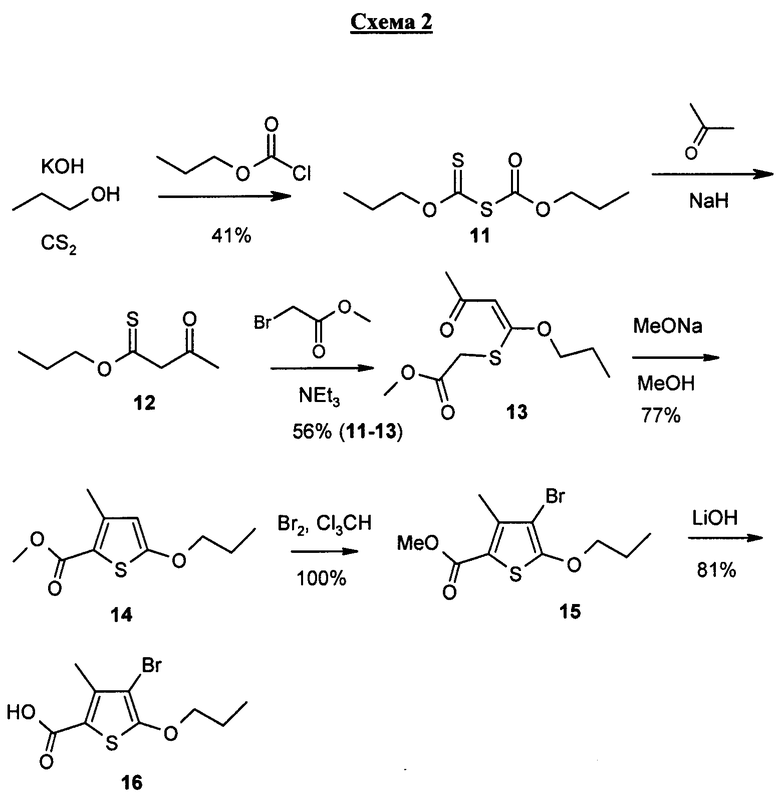
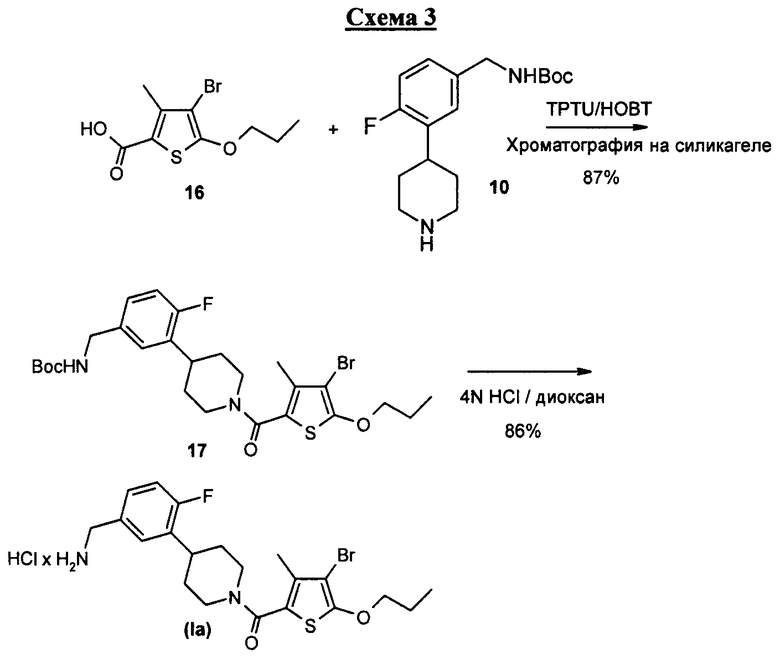
В частности, соединение формулы I может быть получено, как это показано на схемах 1-3.
Например, соединение настоящего изобретения представляет собой ахиральное соединение, получение которого включает конвергентный синтез. Ниже на схеме 1 приведены методы, конечной стадией которых является получение амина 10. Следующая схема 2 содержит методы, которые приводят к получению кислоты 16. На схеме 3 ниже показаны методы, в результате которых получается соединение формулы I, причем синтез соединения формулы I включает двустадийную последовательность. Получение 10, 16 и соединения формулы I настоящего изобретения поочередно обсуждается ниже.
Соединение 2 превращается в соединение 3 посредством защиты аминогруппы агентом для защиты аминов, например 1,2-бис(хлордиметилсилил)этаном, в присутствии третичного амина, например триэтиламина, в подходящем инертном растворителе, например дихлорметане, с тем чтобы получить защищенное соединение 3.
Соединение 3 превращается в соединение 5 алкилированием соединения 4 с использованием соединения 3 в условиях проведения реакций алкилирования, включающих сильное основание, например н-бутиллитий, в подходящем апротонном растворителе, например тетрагидрофуране, с тем чтобы получить гидроксильное производное, соединение 5.
Соединение 5 превращается в соединение 6 посредством снятия защиты его аминогруппы агентом для снятия защиты, например сильной неорганической кислотой, такой как фосфорная кислота, в присутствии инертного растворителя, например гептана, с тем чтобы получить соединение 6 без защиты.
Соединение 6 превращается в соединение 7 посредством дегидратирования с применением сильной неорганической кислоты, например фосфорной кислоты, и последующей нейтрализации продукта с помощью сильного неорганического основания, например водного гидроксида натрия, с тем чтобы получить дегидратированное соединение 7.
Соединение 7 превращается в соединение 8 посредством введения защиты аминогруппы с помощью агента для защиты аминов, например BOC ангидрида, в смешанной водно-органической системе растворителей, в которой органический растворитель представляет собой полярный органический растворитель, например метанол, с использованием сильного неорганического основания, например водного гидроксида натрия, с тем чтобы получить защищенное BOC соединение 8.
Соединение 8 превращается в соединение 9 посредством гидрогенизации с помощью восстановителя, например гидроксида палладия на углеродной подложке (20%), в системе смешанных растворителей, например метанольная уксусная кислота, с тем чтобы получить пиперидиновую соль со снятой защитой, соединение 9.
Соединение 9 превращается в свою свободную основную форму, соединение 10, путем нейтрализации с применением сильного неорганического основания, например водного гидроксида натрия, с тем чтобы получить конечное соединение 10.
Сероуглерод превращается в соединение 11 при ацилировании с использованием ацилирующего агента, пропилхлорформиата, в присутствии сильного неорганического основания, например гидроокиси калия, в н-пропаноле, с тем чтобы получить соединение 11.
Соединение 11 превращается в соединение 12 при алкилировании ацетона соединением 11 в присутствии сильного основания, например гидрида натрия, с тем чтобы получить алкилированное соединение 12.
Соединение 12 превращается в соединение 13 при алкилировании соединения 12 альфа-бромметилацетатом в присутствии третичного амина, например триэтиламина, с тем чтобы получить алкилированное соединение 13.
Соединение 13 превращается в соединение 14 циклизацией в сильно основной среде, например в метилате натрия, в присутствии протонного органического растворителя, например метанола, с тем чтобы получить циклическое соединение 14.
Соединение 14 превращается в соединение 15 бромированием в инертном органическом растворителе, например трихлорметане или в смеси трет-бутилметиловый эфир/гептан, с тем чтобы получить бромированное соединение 15.
Соединение 15 превращается в соединение 16 при гидролизе с использованием сильного неорганического основания, например гидроксида лития.
Соединение 16 превращается в соединение 17 в реакции сочетания с соединением 10 с применением агента реакции сочетания, например TPTU/HOBT или EDC, в присутствии инертного растворителя, например дихлорметана, и третичного амина, например диизопропилэтиламина, в безводной среде, с тем чтобы получить продукт сочетания, соединение 17.
Соединение 17 превращается в соединение I при снятии защиты в сильно кислой среде, например в соляной кислоте, в присутствии полярного органического растворителя, например диоксана, с тем чтобы получить соединение I без защиты.

Соединение настоящего изобретения является основанием, такое соединение пригодно для применения в форме свободного основания или в форме его фармацевтически приемлемой кислотно-аддитивной соли.
Кислотно-аддитивные соли могут быть более удобной формой для использования, и на практике использование солевой формы по существу означает применение формы свободного основания. К кислотам, которые могут использоваться для приготовления кислотно-аддитивных солей, относятся предпочтительно такие, которые при смешении со свободным основанием приводят к фармацевтически приемлемым солям, то есть солям, анионы которых являются нетоксичными для пациента в фармакологических дозах солей, так что полезные эффекты ингибирования, присущие свободному основанию, не искажаются побочными эффектами, приписываемыми анионам. Хотя фармацевтически приемлемые соли указанных основных соединений являются предпочтительными, все кислотно-аддитивные соли представляют собой полезные источники формы свободного основания, даже если конкретная соль, сама по себе, нужна только лишь как промежуточный продукт, например, если соль получается исключительно в целях очистки и идентификации или если она используется как интермедиат при получении фармацевтически приемлемой цели с помощью ионообменных процессов. Фармацевтически приемлемые соли, которые относятся к сфере настоящего изобретения, включают полученные из минеральных и органических кислот, и к ним относятся гидрогалогениды, например гидрохлорид и гидробромид, сульфаты, фосфаты, нитраты, сульфаматы, ацетаты, цитраты, лактаты, тартраты, малонаты, оксалаты, салицилаты, пропионаты, сукцинаты, фумараты, малеаты, метилен-бис-бета-гидроксинафтоаты, бензоаты, тозилаты, гентизинаты, изетионаты, ди-п-толуоилтартраты, метансульфонаты, этансульфонаты, бензолсульфонаты, п-толуолсульфонаты, циклогексилсульфаматы и хинаты. Более конкретным примером соли соединения формулы является гидрохлоридная соль.
Кроме возможности использовать сами соли соединения изобретения в качестве активных соединений, они также пригодны для целей очистки соединения, например, за счет использования разницы в растворимости между солями и исходным соединением, побочными продуктами и/или исходным материалом с помощью методик, известных специалистам в области.
Согласно еще одной особенности изобретения кислотно-аддитивные соли соединения настоящего изобретения могут быть получены при реакции свободного основания с подходящей кислотой с применением или адаптацией известных методов. Например, соли соединения настоящего изобретения, образованные при добавлении кислот, могут быть получены либо растворением свободного основания в воде или водном спиртовом растворе, либо в других подходящих растворителях, содержащих подходящую кислоту, и выделением соли посредством испарения раствора или же реакцией свободного основания и кислоты в органическом растворителе, в этом случае соль отделяется непосредственно или может быть получена концентрированием раствора.
Кислотно-аддитивные соли соединения настоящего изобретения могут извлекаться из солей с применением или адаптацией известных методов. Например, исходное соединение настоящего изобретения может быть получено из его солей, образованных при добавлении кислот, обработкой щелочами, например водным раствором бикарбоната натрия или водным раствором аммиака.
Исходные материалы и интермедиаты могут быть получены с применением или адаптацией известных методов, например методов, описанных в справочных примерах, или их в очевидных химических эквивалентах.
Настоящее изобретение также относится к некоторым интермедиатам из приведенных выше схем, и описанные в настоящем документе процессы их получения сами по себе являются дополнительными признаками настоящего изобретения.
Примеры
Для более глубокого понимания настоящего изобретения можно сослаться на следующие предпочтительные, но не единственные примеры, которые приводятся в качестве иллюстрации изобретения. Следующие примеры приводятся в целях более полной иллюстрации предпочтительных осуществлений изобретения. Вместе с тем его ни в коем случае не следует рассматривать как ограничение широкого охвата настоящего изобретения.
В спектрах ядерного магнитного резонанса (ЯМР), приведенных ниже, величины химического сдвига выражены в м.д. относительно тетраметилсилана. Сокращения имеют следующие значения: ш = широкий, дд = дублет дублетов, с = синглет; м = мультиплет.
ПРИМЕР 1
Стадия 1: получение 1-(3-бром-4-фторбензил)-2,2,5,5-тетраметил[1,2,5] азадисилолидина (соединение 3)
110 г (0,46 моль) 3-бром-4-фторбензиламингидрохлорида (2) суспендируют в 900 мл метиленхлорида в 2-литровой трехгорлой круглодонной колбе, снабженной азотной рубашкой, термопарным температурным датчиком с тефлоновым покрытием и механической мешалкой, и охлаждают до ˜5°C на ледяной бане. Добавляется всего 144 г (1,42 моль, 3,1 экв. д=0,7), что приводит к образованию густой суспензии. Затем по каплям приливают раствор 1,2-бис(хлордиметилсилил)этана (100 г, 0,46 моль) в 250 мл метиленхлорида в течение более 1,5 часов, поддерживая при этом температуру реакционной смеси между 5 и 8°C. Смесь перемешивают в течение 30 мин и затем подогревают до комнатной температуры. Осадок триэтиламингидрохлорида отфильтровывают. Фильтрат концентрируют в вакууме (40°C, <50 мбар), добавляют 1 л пентана и фильтрованием отделяют дополнительный осадок Et3N × HCl, который при этом образуется. Процесс повторяют еще раз с 1 л пентана. Фильтрат концентрируют в вакууме (40°C, <5 мбар) до бесцветной жидкости, которая кристаллизуется при охлаждении с образованием 159 г (˜100%) соединения 3 в виде белого кристаллического порошка.
Завершение реакции необходимо отслеживать с помощью 1H ЯМР, с тем чтобы убедиться в практическом отсутствии любых остатков исходного материала 2 и триэтиламингидрохлорида, поскольку они приводят к расходованию н-бутиллития на следующей стадии, тем самым снижая выход реакции.
Стадия 2: получение 1-бензил-4-[2-фтор-5-(2,2,5,5-тетраметил[1,2,5]азадисилолидин-1-илметил)фенил] пиперидин-4-ола (соединение 5)
Раствор соединения 3 (159 г, 0,4574 моль) в 1,5 л безводного ТГФ помещают в 5-литровую трехгорлую круглодонную колбу, снабженную азотной рубашкой, термопарным температурным датчиком с тефлоновым покрытием и механической мешалкой, и охлаждают до -75°C. К этому перемешиваемому раствору по каплям приливают 2,5 M раствор н-бутиллития (192 мл, 0,48 моль, 1,05 экв.) в течение примерно 1 часа, поддерживая при этом температуру реакционной смеси в интервале между -72 и -75°C. Через 30 мин по каплям добавляют раствор 1-бензил-4-пиперидона (соединение 4; 88,2 г, 0,47 моль, 1,02 экв.) в безводном ТГФ (350 мл + 50 мл для промывания) в течение более 1 часа, поддерживая при этом температуру реакции ниже -70°C. После перемешивания в течение 20 мин при температуре от -70 до -75°C к реакционной смеси приливают 200 мл метанола (цвет меняется с оранжевого на желтый), затем реакционную смесь оставляют нагреваться до комнатной температуры. Реакционную смесь концентрируют в вакууме (50°C), чтобы получить 295 г коричневой маслянистой жидкости, соединение 5.
Стадия 3: получение 4-(5-аминометил-2-фторфенил)-1-бензилпиперидин-4-ола (фосфатная соль) (соединение 6)
Неочищенное соединение 5 (˜295 г) разбавляют 1 л метиленхлорида и переносят в 3-литровую трехгорлую круглодонную колбу, снабженную термопарным температурным датчиком с тефлоновым покрытием и механической мешалкой. К этому раствору медленно приливают 53 г 85% H3PO4 (1 экв.) при перемешивании (экзотермическая реакция) и смесь концентрируют в вакууме (40°C). Добавляют 1 л метиленхлорида и смесь концентрируют в вакууме (40°C, 1 мбар) до желтой пены, которую затем размешивают в 1 л гептана. Образуется осадок, который отделяют фильтрованием. Коричневый осадок сушат, с тем чтобы получить 190 г соединения 6. МС: обнаружен m/z 315 (M+H).
Стадия 4: получение 3-(1-бензил-1,2,3,6-тетрагидропиридин-4-ил)-4-фторбензиламина (соединение 7)
Все 190 г высушенного соединения 6 переносят в 2-литровую трехгорлую круглодонную колбу, снабженную термопарным температурным датчиком с тефлоновым покрытием и механической мешалкой. Добавляется всего 600 мл 85% H3PO4. Полученную суспензию постепенно нагревают до 100°C при перемешивании. Момент завершения реакции отслеживают по ВЭЖХ (обычно от 2 до 3 часов). После окончания реакции реакционную смесь охлаждают до комнатной температуры и разбавляют 800 мл воды. Водный слой промывают эфиром (2 × 200 мл). Водный слой затем нейтрализуют 50% водн. NaOH до pH > 9, поддерживая при этом температуру ниже 30°C. Такой водный раствор представляет собой высококонцентрированную буферную систему, рН которой остается неизменным примерно 7-8, до момента достижения точки нейтрализации. Подтверждение завершения нейтрализации получают добавлением нескольких капель основания к небольшому образцу супернатанта, чтобы убедиться в прекращении выпадения осадка. Значительное количество соли выпадает в осадок. Смесь фильтруют и осадок промывают ДХМ (дихлорметан) (примерно 3 л) и 2 л воды. Органический слой (нижний) отделяют и промывают водой (2 × 1 л). Органический раствор концентрируют в вакууме (40°C), чтобы получить 107 г коричневой маслянистой жидкости, соединение 7. МС: обнаружен m/z 297 (M+H).
Стадия 5: получение трет-бутилового эфира [3-(1-бензил-1,2,3,6-тетрагидропиридин-4-ил)-4-фторбензил]карбаминовой кислоты (соединение 8)
В 2-литровую трехгорлую круглодонную колбу, снабженную термопарным температурным датчиком с тефлоновым покрытием и механической мешалкой, помещают 50 г соединения 7 и раствор метанола (800 мл) и воды (400 мл). Добавляют 36,8 г BOC ангидрида и 2 мл 50% NaOH и смесь перемешивают при комнатной температуре. Продукт выпадает в осадок из раствора после 2 часов перемешивания. Если продукт реакции с BOC не выпадает в осадок, декантируют верхний водный слой и к маслянистой жидкости приливают н-гептан для кристаллизации продукта. Полученную смесь перемешивают в течение ночи при комнатной температуре. Осадок отделяют фильтрованием и размешивают в 1,05 л смеси MeOH/вода (2:1 по объему) в течение 4 часов, затем отделяют фильтрованием и сушат в течение 4 дней, чтобы получить 40 г (общий выход составляет примерно от 35 до 48% по соединению 3) соединения 8 в виде светло-желтого порошка. Чистота 96,3% по ВЭЖХ. МС: m/z 397 (M+H), 398 (M+2H).
Стадия 6: получение ацетатной соли трет-бутилового эфира 6-(4-фтор-3-пиперидин-4-илбензил)карбаминовой кислоты (соединение 9)
64 г (0,16 моль) соединения 8, 6,4 г гидроксида палладия/C 20%, 19,5 г (2 экв.) ледяной уксусной кислоты и 250 мл метанола загружают в 1-литровый сосуд для гидрогенизации. Реакционный сосуд продувают (N2/вакуум, 3 раза), затем заполняют H2 до 40 фунтов на кв. дюйм и встряхивают в течение ночи при комнатной температуре. Катализатор отделяют фильтрованием и фильтрат концентрируют в вакууме (40°C) до получения маслянистой жидкости. Добавляют 200 мл изопропилового эфира и перемешивают в течение ночи. Белый осадок, который при этом выпадает, отделяют фильтрованием, промывают изопропиловым эфиром и сушат, с тем чтобы получить 53,5 г белого порошка с чистотой по ВЭЖХ 95,4%. Порошок размешивают в 500 мл МТБЭ в течение 5 часов, затем отделяют фильтрованием и снова размешивают в 500 мл МТБЭ в течение ночи. Порошок отделяют фильтрованием и сушат, с тем чтобы получить 51,3 г (86,3%) белого порошка ацетата соединения 9. Элементный анализ: расчет для C17H25FN2O2: C 61,94; H 7,93; N 7,6. Найдено: C 62,0; H 8,17; N 7,49. По Карлу Фишеру: 0,34% воды. ВЭЖХ: Rt 9,14 мин, чистота 97% по площади под кривой. МС: m/z 309 (M+H), 310 (M+2H).
Стадия 7: получение трет-бутилового эфира (4-фтор-3-пиперидин-4-илбензил) карбаминовой кислоты (соединение 10)
Ацетат соединения 9 (51 г) растворяют в 400 мл воды и рН корректирут до 5 добавлением 2N HCl. Водный раствор промывают эфиром (2 × 200 мл). Водный слой нейтрализуют до pH > 12 50% водн. NaOH и экстрагируют эфиром (2 × 300 мл). Органический слой промывают водой и сушат над Na2SO4, а затем концентрируют до маслянистой жидкости. К маслянистой жидкости приливают 200 мл н-пентана и перемешивают в течение 3 часов. Твердый продукт отделяют фильтрованием, промывают н-пентаном и сушат при комнатной температуре под стандартным вакуумом в течение 24 часов, чтобы получить 42 г соединения 10 (98%) в виде белого порошка. Масс-спектроскопия с инонизацией электрораспылением, m/z 309 (M + H). Элементный анализ: расчет для C17H25FN2O2: C 66,21; H 8,17; N 9,08. Найдено, без поправки на воду: C 64,30; H 8,64; N 8,77. По Карлу Фишеру: 2,57% воды. ВЭЖХ: Rt 9,16 мин, чистота 98,1% по площади под кривой.
Примечание. Если чистота ацетата соединения 9 по ВЭЖХ меньше 95%, вместо чистого н-пентана для кристаллизации продукта (свободное основание) из маслянистой жидкости можно использовать смесь н-пентана и эфира (до 1:1 по объему). Продукт очень хорошо растворим в эфире, так что если используется большее количество эфира, еще больше продукта будет потеряно, что тем самым приведет к снижению выхода.
ПРИМЕР 2
Стадия 1: получение бис(пропокситиокарбонил)сульфида (соединение 11)
84,2 г порошка гидроксида калия (1,27 моль) добавляют к 530 мл н-пропанола, помещенного в трехгорлую круглодонную колбу, снабженную механической мешалкой и охлаждающей баней, при комнатной температуре. Затем к раствору по каплям в течение более 1 часа приливают 80 мл сероуглерода (1,33 моль) через добавочную воронку с выравниванием давления. Перемешивание продолжается в течение 3 часов. Добавляют 200 мл воды. Через добавочную воронку с выравниванием давления по каплям приливают 73,5 г (0,6 моль) чистого пропилхлорформиата. Полученную смесь перемешивают в течение ночи при комнатной температуре. Затем смесь разбавляют 400 мл гептана. Водный слой дважды экстрагируют 100 мл гептана. Объединенный органический слой промывают 100 мл воды, 100 мл соляного раствора, а затем сушат над карбонатом калия и упаривают. Остаточный пропанол удаляют перегонкой под высоким вакуумом, чтобы получить прозрачную желтую жидкость. 10 г прозрачной желтой жидкости растворяют в 10 г гептана и очищают хроматографией на силикагеле, элюируя гептаном, чтобы получить 8,35 г соединения 11 (выход 41%).
Стадия 2: получение O-пропилового эфира 3-оксотиомасляной кислоты (соединение 12)
1,82 г (45 ммоль) гидрида натрия суспендируют в 40 мл толуола. К полученной суспензии при перемешивании приливают раствор 1,74 г (30 ммоль) ацетона и 4,76 г (20 ммоль) соединения 11 в 10 мл толуола при 40°C. Реакцию инициируют внесением небольшого количества гидрида калия. При этом выделяются пузырьки, и цвет реакционной смеси меняется с желтого на оранжевый. Реакционную смесь перемешивают при 40°C в течение одного часа, а затем охлаждают до 0°C на водяной бане со льдом. Реакционную смесь затем выливают в стакан, содержащий 11 мл 4N HCl, лед и 150 мл эфира. Органический слой отделяют и концентрируют. Сырой продукт очищают хроматографией на силикагеле, элюируя 5% этилацетатом в гептане для получения О-пропилового эфира 3-оксотиомасляной кислоты (соединение 12).
Стадия 3: получение метилового эфира (3-оксо-1-пропоксибут-1-енилсульфанил) уксусной кислоты (соединение 13)
Соединение 12 затем растворяют в 40 мл ДМФ и охлаждают до 0°C. К полученному раствору добавляют 4,6 г альфа-бромметилацетата в 5 мл ДМФ и 5,2 мл триэтиламина. Происходит мгновенное образование белого осадка. Полученная суспензия перемешивается при 0°C в течение 2 часов, а затем выливается в смесь эфир/лед-вода. Органический слой отделяют и концентрируют. Сырой продукт очищают хроматографией на силикагеле, а затем элюируют 3% метанолом в ДХМ, с тем чтобы получить 2,61 г соединения 13 (выход 56% по двум стадиям).
Стадия 4: получение метилового эфира 3-метил-5-пропокситиофен-2-карбоновой кислоты (соединение 14)
Смесь 2,61 г (11,24 ммоль) соединения 13 и 2 мл 0,5 M смеси метилат натрия/метанол в 40 мл метанола нагревают при 70-73°C в течение 40 мин. Смесь затем выливают в эфир/лед-вода. Органический слой отделяют и концентрируют. Сырой продукт очищают хроматографией на силикагеле, элюируя ДХМ, чтобы получить 1,85 г (выход 76,8%) соединения 14.
Стадия 5: получение метилового эфира 4-бром-3-метил-5-пропокситиофен-2-карбоновой кислоты (соединение 15)
К раствору 1,22 г (5,7 ммоль) соединения 14 в 20 мл трихлорметана приливают 11 мл 0,55 M смеси бром/трихлорметан при 0°C и перемешивают в течение 10 мин. Смесь гасят водным раствором сульфита натрия, а затем экстрагируют ДХМ. Органический слой отделяют и концентрируют. Сырой продукт очищают, пропуская через слой карбонат калия-силикагель, и смывают ДХМ, чтобы получить 1,67 г (выход 100%) соединения 15.
Стадия 6: получение 4-бром-3-метил-5-пропокситиофен-2-карбоновой кислоты (соединение 16)
К раствору 1,67 г соединения 15 (5,70 ммоль) в 27 мл диоксана добавляют 10 мл 2 M LiOH/вода и 9 мл воды. Полученную смесь перемешивают в течение 4 часов при комнатной температуре. Смесь затем разбавляют 10 мл воды и дважды экстрагируют 10 мл эфира. Водный раствор охлаждают на водяной бане со льдом и подкисляют 4 M HCl. Белый порошок собирают фильтрованием с отсасыванием, чтобы получить 1,28 г (выход 80,5%) соединения 16.
ПРИМЕР 3
Стадия 1: получение трет-бутилового эфира {3-[1-(4-бром-3-метил-5-пропокситиофен-2-карбонил)пиперидин-4-ил]-4-фторбензил}карбаминовой кислоты (соединение 17)
К раствору 170 мг соединения 16 (0,61 ммоль) в 25 мл ДХМ в токе N2 добавляют 170 мг TPTU (0,61 ммоль) и 80 мг 1-гидрокси-1H-бензотриазола (HOBt) (0,61 ммоль). Перемешивание продолжается в течение 3 минут. Затем к смеси приливают раствор 200 мг (0,65 моль) соединения 10 в 5 мл ДХМ и 0,3 мл диизопропилэтиламина (1,2 ммоль). Полученную смесь перемешивают в течение 24 часов при комнатной температуре. Смесь затем промывают 20 мл воды, сушат над безводным сульфатом натрия и концентрируют. Сырой маслянистый продукт очищают хроматографией на силикагеле, элюируя 3% метанолом в ДХМ, чтобы получить 0,3 г (выход 86,5%) соединения 17.
1H ЯМР (CDCl3 ,ТМС): δ 7,14-7,04 (м, 2H), 6,95 (дд, H), 4,82 (уш. с, H), 4,38 (уш. д, 2H), 4,23 (д, 2H), 4,05 (т, 2H), 3,17-2,95 (м, 3H), 2,21 (с, 3H), 1,92-1,59 (м, 6H), 1,44 (с, 9H), 1,05 (т, 3H). МС (ИЭР+): 569 (M++1).
Стадия 2: получение гидрохлорида [4-(5-аминометил-2-фторфенил)пиперидин-1-ил]-(4-бром-3-метил-5-пропокситиофен-2-ил)метанон (соединение I)
Раствор 0,25 г соединения 17 (0,44 ммоль) в 8 мл 4 M HCl/диоксан перемешивают в токе азота при комнатной температуре в течение 3 часов. Раствор разбавляют 30 мл эфира и перемешивают в течение 5 мин. Жидкость над осадком декантируют. Осадок промывают 30 мл эфира и вновь декантируют жидкость. Затем осадок растворяют в 3% метаноле в ДХМ и очищают хроматографией на силикагеле, элюируя от 3 до 10% метанола в ДХМ. Соединение I, 0,19 г (выход 86,4%), получают в виде аморфного вещества, удаляя упариванием растворитель из объединенных чистых фракций продукта. Продукт представляет собой аморфное стекло.
1H ЯМР (CDCl3, ТМС): δ 8,62 (уш. с, 3H), 7,53-7,43 (м, H), 7,36-7,26 (м, H), 6,97 (дд, H), 4,22 (уш. д, 2H), 4,18-3,99 (м, 4H), 3,16-2,90 (м, 3H), 2,17 (с, 3H), 2,00-1,60 (м, 6H), 1,02 (т, 3H). МС (ИЭР+): 469 (M++1). 100% чистота по ЖХ/МС (УФ 220 нм и полный счет ионов).
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
Свойства соединения настоящего изобретения также продемонстрированы на примерах: 1) его ингибирующей способности в отношении бета-триптазы (значения IC50 и Ki) и 2) его активности, измеренной в модели гиперреактивности дыхательных путей на морских свинках (пероральная средняя эффективная доза ED50).
ПОРЯДОК ПРОВЕДЕНИЯ ИСПЫТАНИЙ «IN VITRO»
Поскольку все воздействия триптазы, как это описано в разделе о предпосылках изобретения, зависят от ее каталитической активности, соединения, которые ингибируют ее каталитическую активность будут потенциально ингибировать воздействие триптазы. Ингибирование такой каталитической активности может измеряться с помощью ферментного анализа и клеточного анализа «in vitro».
Ингибирование активности триптазы подтверждается применением либо триптазы, выделенной из легких человека, либо рекомбинантной человеческой бета-триптазы, экспрессированной в дрожжевых клетках. По существу эквивалентные результаты получаются при использовании изолированного нативного фермента или экспрессированного фермента. В процессе анализа используется 96-ячеечная микропланшетка (Costar 3590) с L-пироглютамил-L-пролил-L-аргинин-п-нитроанилидом (S2366: Quadratech) в качестве субстрата (в основном в соответствии с описанием в McEuen et. al. Biochem Pharm, 1996, 52, стр. 331-340). Анализ производится при комнатной температуре с использованием 0,5 mM субстрата (2 × Km), и данные микропланшетки получают с помощью считывателя микропланшеток (считыватель Beckman Biomek Plate) на длине волны 405 нм.
Материалы и методы для первичного скрининга триптазы (хромогенный анализ)
Аналитический буфер
50 mM Tris (pH 8,2), 100 mM NaCl, 0,05% Tween 20, 50 мкг/мл гепарина.
Субстрат
S2366 (исходные растворы 2,5 mM).
Фермент
Исходные растворы очищенной рекомбинантной бета-триптазы 310 мкг/мл.
Протокол (одноточечное определение)
Добавить 60 мкл разбавленного субстрата (конечная концентрация 500 мкМ в аналитическом буфере) в каждую ячейку.
Добавить соединение (по две ячейки), конечная концентрация 20 мкМ, объем 20 мкл.
Добавить фермент в конечной концентрации 50 нг/мл, объем 20 мкл.
Общий объем для каждой ячейки составляет 100 мкл.
Встряхивть в течение непродолжительного времени и инкубировать при комнатной температуре в темноте в течение 30 мин.
Считывать поглощение на 405 нм.
Для каждой из ячеек предусмотрены следующие контрольные пробы:
общие: 60 мкл субстрата, 20 мкл буфера (с конечной концентрацией 0,2% ДМСО), 20 мкл фермента,
неспецифические: 60 мкл субстрата, 40 мкл буфера (с 0,2% ДМСО),
общие: 60 мкл субстрата, 20 мкл буфера (без ДМСО), 20 мкл фермента,
неспецифические: 60 мкл субстрата, 40 мкл буфера (без ДМСО).
Протокол (определение IC50 и Ki)
Протокол по существу такой же, как и описанный выше, за исключением того, что соединение добавляется в две ячейки со следующими конечными концентрациями: 0,01, 0,03, 0,1, 0,3, 1, 3, 10 мкМ (все разбавления производятся вручную). Для каждого анализа, как для точечного, так и для определения IC50, используется эталонное соединение, чтобы получить IC50 для сравнения. По значению IC50 можно рассчитать Ki с помощью следующей формулы: Ki = IC50/(1 + [субстрат]/Km).
Ингибирующая способность бета-триптазы для соединения формулы I определяется значениями IC50 и Ki для 76 нМ и 15 нМ соответственно.
ПОРЯДОК ПРОВЕДЕНИЯ ИСПЫТАНИЙ «IN VIVO»
Протокол анализа
Сенсибилизация и применение лекарственного препарата. Самцы морской свинки Хартли (225-250 г) сенсибилизируются яичным альбумином (0,5 мл 1% раствора, внутрибрюшинно и подкожно). На 4 день животные получали повторную дозу (внутрибрюшинно) 0,5 мл 1% яичного альбумина. На 21 день животные получали пероральную дозу (2 мл/кг) либо носителя (0,5% метилцеллюлоза/0,2% Tween 80), либо тестового соединения за 2 часа до антигенной стимуляции. За тридцать минут до антигенной стимуляции животным также вводили мепирамин (30 мг/кг, внутрибрюшинно), чтобы предотвратить анафилактический коллапс. Затем животных в течение 5 минут подвергали воздействию аэрозоля физиологического раствора (контрольные животные) или 1% яичного альбумина с помощью аэрозольного аппарата deVilbiss Ultraneb.
Оценка гиперчувствительности дыхательных путей: в течение восемнадцати - двадцати четырех часов после стимуляции животных анестезировали с применением сочетания кетамина (133 мг/кг) и ксилазина (24 мг/кг), вводимых внутримышечно, проводили хирургическую подготовку и фиксировали в общем плетизмографе для определения функции легких. Животных подключали к аппаратам искусственной вентиляции легких Ugo-Basile, обеспечивающим дыхательный объем 1 мл/100 г с частотой 50 вдохов/минуту через трахеальную канюлю. В яремную вену также вводилась канюля для гистаминовой стимуляции. Заполненная водой пищеводная канюля помещалась таким образом, чтобы обеспечить возможность регистрации транспульмонального давления. Транспульмональное давление определяется по разности между трахеальной и пищеводной канюлями с помощью датчика дифференциального давления. Объем, поток воздуха и транспульмональное давление отслеживались с помощью системы пульмонального анализа (программное обеспечение Buxco XA) и использовались для расчета легочной резистентности (см H2O/мл·сек) и динамической растяжимости (мл/см H2O). Резистентность дыхательных путей и динамическая растяжимость рассчитываются для каждого вдоха. Гистамин вводится внутривенно, и рассчитывается реактивность по отношению к нарастающим концентрациям (0,3-20 мкг/кг). ED50 оцениваются по значениям площади под кривой, полученным для отдельных кривых «доза-эффект» гистамина.
Уровни препарата в плазме и в легких
В сателлитных группах определялись уровни концентрации соединения в плазме и в легких. Для определения уровней препарата использовалось от трех до четырех морских свинок из каждой группы лечения экспериментальным препаратом. В заданный момент времени (2 или 24 часа после введения) животных умерщвляли и посредством пункции сердца отбирали 1 мл образца крови, собирая их в шприцы, покрытые гепарином, которые содержали 20 мкл (на 1 мл крови) 5 mM раствора гидралазина. Плазма отделялась от клеточного компонента крови центрифугированием и хранилась при -20°C до проведения анализа. Образцы легких препарируются с освобождением от соединительных тканей, промакиваются досуха, взвешиваются и хранятся в 20 мл флаконах, содержащих 5 мл 5 mM раствора гидралазина в физиологическом растворе. Образцы замороженной плазмы затем переносятся на сухой лед к образцам экспериментальной группы фармакокинетики для определения уровней концентрации соединения.
Результаты
Эффективность соединения I определяют по гиперчувствительности дыхательных путей к гистамину у сенсибилизированных морских свинок, пероральным путем. Соединение I не оказывает воздействия на базальную резистентность дыхательных путей или базальную динамическую растяжимость легких.
Сенсибилизация и разовая стимуляция яичным альбумином приводит к повышению бронхиальной реактивности к гистамину, которая регистрируется как сдвиг влево на кривой «доза-эффект» спазмогена и как заметное увеличение площади под кривой для резистентности дыхательных путей и растяжимости легких. Абсолютные значения отражают увеличение резистентности дыхательных путей и снижение растяжимости легких.
При пероральном введении за 2 часа до стимуляции яичным альбумином соединение I обеспечивает заметную защиту от антиген-стимулированной гиперчувствительности верхних дыхательных путей с ED50 = 0,1 мг/кг, как показывают оценки по резистентности дыхательных путей и динамической растяжимости легких.
В отдельной группе животных уровни соединения I определяются в бронхоальвеолярном лаваже (БАЛ), легких и плазме через 2 часа после введения соединения (1 мг/кг, перорально). Регистрируется заметное количество соединения I в органах-мишенях, например в легких и БАЛ (без учета фактора разбавления для БАЛ). В плазме обнаруживаются гораздо более низкие уровни соединения.
В сателлитной группе животных через 24 часа после введения также измерялись уровни соединения I как в легких, так и в плазме. В то время как в плазме не удалось обнаружить соединения I, в легких морских свинок через 24 часа после введения регистрировались зависимые от дозы его заметные количества. Было также показано, что соединение I обладает продолжительным действием со средней ED50 0,4 мг/кг при введении за 24 часа до стимуляции антигеном. Такая продолжительность действия находится в согласии с его протяженным длительным воздействием.
Данные по пероральному введению соединения настоящего изобретения в модели гиперчувствительности дыхательных путей на морских свинках четко демонстрируют, что соединение проявляет ингибирующую активность в отношении триптазы. Следовательно, соединение настоящего изобретения с легкостью находит применение в качестве фармацевтического препарата для лечения широкого круга заболеваний, связанных с триптазой, и естественно в методах лечения таких заболеваний у пациента.
Настоящее изобретение не должно ограничиваться в объеме теми конкретными осуществлениями, которые описаны в данном документе. В самом деле, различные модификации изобретения, кроме уже описанных в настоящем описании, будут очевидны для специалистов из приведенного выше описания и приложенных чертежей. Предполагается, что такие модификации попадают в сферу охвата прилагаемой формулы изобретения.
В настоящем описании цитируются различные публикации, которые в силу ссылки на них полностью включаются в представленный текст.


Описывается соединение формулы (I) или фармацевтически приемлемая соль. Соединение обладает ингибирующим действием в отношении бета-триптазы. Описываются способ лечения пациентов, страдающих или подверженных физиологическим заболеваниям, при которых необходимо повышение уровня ингибитора триптазы, и фармацевтическая композиция на основе соединения формулы (I). 3 н. и 10 з.п. ф-лы, 2 ил.


или фармацевтически приемлемая соль указанного соединения.
| 0 |
|
SU190101A1 |

