Настоящее изобретение относится к новым соединениям, представляющим собой замещенные фенокси- и бензилокси-пиперидины формулы (I), обладающим антагонистическими свойствами в отношении Р2Х7 рецептора (Р2Х7), к фармацевтическим композициям, содержащим эти соединения, к химическим способам получения этих соединений и к их применению в лечении или профилактике заболеваний, ассоциированных с активностью Р2Х7 рецептора, у животных, в частности у людей.
Р2Х7 принадлежит семейству ионотропных рецепторов Р2Х. Р2Х7 активируется внеклеточными нуклеотидами, в частности аденозинтрифосфатом (АТФ). Р2Х7 отличается от других членов семейства Р2Х специфической локализацией (в частности, в центральной нервной системе (ЦНС) и иммунокомпетентных клетках), высокой концентрацией АТФ (в миллимолярном (мМ) диапазоне), необходимой для его активации, и его способностью формировать большую пору при длительной или повторной стимуляции. Р2Х7 представляет собой лиганд-управляемый ионный канал и присутствует на клетках различных типов, главным образом на клетках, вовлеченных в воспалительный и/или иммунный процесс, конкретно на макрофагах, тучных клетках и лимфоцитах (Т и В). Активация Р2Х7 рецептора внеклеточными нуклеотидами, например АТФ, приводит к высвобождению интерлейкина-1β (IL-1β) и образованию гигантских клеток (макрофагов/микроглиальных клеток), дегрануляции (тучных клеток) и шеддингу молекул L-селектина (у лимфоцитов). Р2Х7 рецепторы также локализованы на антигенпрезентирующих клетках (АРС), кератиноцитах, ацинарных клетках слюнных желез (клетках околоушной железы), гепатоцитах, эритроцитах, эритролейкозных клетках, моноцитах, фибробластах, клетках костного мозга, нейронах и ренальных мезангиальных клетках. Известно также, что Р2Х7 рецептор является болевым сенсором в нервной системе. В экспериментах с использованием Р2Х7-дефицитных мышей продемонстрирована роль Р2Х7 в развитии боли, так как эти мыши защищены от развития как индуцированной адъювантами воспалительной боли, так и от невропатической боли, индуцированной частичным лигированием нерва. Имеется также растущее число доказательств того, что Р2Х7 или его нижележащие эффекторы, такие как интерлейкин-1β (IL-1β), вовлечены в патофизиологию некоторых неврологических расстройств, таких как болезнь Альцгеймера (J.I. Diaz-Hernandez et al, Neurobiol. Aging, 2012, 1816-1828: "In vivo P2X7 inhibition reduces Aβ plaques in AD through GSK3β"). Полагают, что P2X7 осуществляет важную функцию в нейротрансмиссии в ЦНС, оказывая активирующее воздействие на постсинаптические и/или пресинаптические нейроны и нейроглию. С использованием данных гибридизации in situ было выяснено, что мРНК Р2Х7 рецептора широко распределена по всему головному мозгу крысы. Конкретно, области с высокой экспрессией мРНК Р2Х7 были обнаружены в переднем обонятельном ядре, коре головного мозга, грушевидной коре (Pir), латеральном ядре перегородки мозга (LS), пирамидальных клетках слоев СА1, СА3, СА4 гиппокампа, мостовых ядрах, наружном клиновидном ядре и медиальном вестибулярном ядре. Сигналы гибридизации Р2Х7 также наблюдали в двигательных нейронах двигательного ядра тройничного нерва, ядра лицевого нерва, ядра подъязычного нерва и переднего рога спинного мозга.
Таким образом, существует терапевтическое обоснование для применения антагонистов Р2Х7 в лечении ряда болезненных состояний. Эти состояния включают, но не ограничиваются ими, заболевания, ассоциированные с ЦНС, такие как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, боковой амиотрофический склероз, травма спинного мозга, церебральная ишемия, травма головы, менингит, расстройства сна, расстройства настроения и тревожные расстройства, эпилепсия, ВИЧ-индуцированное нейровоспаление и поражение ЦНС, а также хроническая невропатическая и воспалительная боль. Кроме того, периферические воспалительные расстройства и аутоиммунные заболевания, включающие, но не ограничивающиеся ими, ревматоидный артрит, остеоартрит, псориаз, аллергический дерматит, астму, хроническую обструктивную болезнь легких, гиперчувствительность дыхательных путей, септический шок, бронхит, гломерулонефрит, синдром раздраженного кишечника, жировую болезнь печени, фиброз печени, повреждение кожных покровов, эмфизему легких, мышечную дистрофию, фиброз, атеросклероз, ожоговое повреждение, болезнь Крона, неспецифический язвенный колит, возрастную дегенерацию желтого пятна, рост и метастазирование злокачественных клеток, синдром Шегрена, миелобластный лейкоз, диабет, остеопороз, ишемическое заболевание сердца, все являются примерами заболеваний, при которых наблюдается вовлечение Р2Х7 рецепторов. Ввиду клинической значимости Р2Х7, идентификация соединений, которые модулируют функцию Р2Х7 рецепторов, представляет собой привлекательное направление разработки новых терапевтических агентов.
Ингибиторы Р2Х7 описаны в различных патентных заявках, таких как:
WO 2004/099146, в которой раскрыты бензамидные ингибиторы Р2Х7 рецептора и их применение в лечении воспалительных заболеваний;
WO 2009/108551, в которой раскрыты аналоги гетероариламидов и их применение для опосредуемых Р2Х7 рецептором состояний;
WO 2009/132000, в которой раскрыты замещенные хинолины и изохинолины в качестве антагонистов Р2Х7 рецептора и их применение при состояниях, опосредованных Р2Х7 рецептором;
WO 2015/119018, в которой описаны производные тиазола и оксазола в качестве антагонистов Р2Х7 рецептора и их применение при состояниях, опосредованных Р2Х7 рецептором.
Однако все еще имеется неудовлетворенная потребность в соединениях, которые способны оказывать эффективное антагонизирующее действие на Р2Х7 и которые могут быть доставлены в разные органы-мишени, являющиеся очагами развития Р2Х7-опосредуемой патологии, включая головной мозг. Такие соединения предложены в данной заявке.
Далее представлены различные воплощения данного изобретения.
Настоящее изобретение относится к тиазольным соединениям следующей формулы (I) или их фармацевтически приемлемым солям:

включая любую их стереохимически изомерную форму, где:
n равно 0 или 1;
R1 представляет собой С1-С4алкил (возможно замещенный гидроксилом или галогеном), предпочтительно метил, фторметил, дифторметил, трифторметил;
каждый из R2, R3 и R4 независимо представляет собой водород, галоген, или группы R2 и R4 вместе образуют шестичленное гетероциклическое кольцо, содержащее атом азота, при условии, что по меньшей мере один из R2, R3 и R4 не представляет собой водород;
каждый из R5 и R6 представляет собой водород или галоген при условии, что по меньшей мере один из R5 и R6 представляет собой галоген.
Используемые в вышеуказанных определениях термины.
Термины "галогено", "галоген" и "галогенид", которые могут быть использованы взаимозаменяемым образом, относятся к заместителю атому фтора, хлора, брома или йода.
Термин "стереохимически изомерные формы", использованный выше, определяет все возможные изомерные формы, которые могут быть у соединений формулы (I). Если не упомянуто или не указано иное, химическое наименование соединений означает смесь всех возможных стереохимически изомерных форм, причем указанные смеси содержат все диастереомеры и энантиомеры основной молекулярной структуры. Более конкретно, стереогенные центры могут иметь R- или S-конфигурацию; заместители на бивалентных циклических, (частично) насыщенных радикалах могут находиться либо в цис-, либо в транс-конфигурации.
Очевидно, что стереохимически изомерные формы соединений формулы (I) входят в объем настоящего изобретения.
Абсолютная стереохимическая конфигурация соединений формулы (I) и промежуточных соединений, использованных для их получения, легко может быть определена специалистами в данной области техники с использованием известных методов, таких как, например, дифракция рентгеновских лучей.
Кроме того, некоторые соединения формулы (I) и некоторые из промежуточных соединений, использованных для их получения, могут проявлять полиморфизм. Следует понимать, что настоящее изобретение охватывает любые полиморфные формы, обладающие свойствами, полезными в лечении указанных выше состояний.
Подразумевается, что упомянутые выше фармацевтически приемлемые соли представляют собой терапевтически активные нетоксичные формы солей присоединения кислоты, которые способны образовывать соединения формулы (I). Эти фармацевтически приемлемые соли присоединения кислоты легко могут быть получены путем обработки  формы такой подходящей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например соляная или бромоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобные кислоты; или органические кислоты, такие как, например, уксусная кислота, пропановая кислота, гидроксиуксусная кислота, молочная кислота, пировиноградная кислота, щавелевая кислота (т.е. этандикислота), мало новая кислота, янтарная кислота (т.е. бутандикислота), малеиновая кислота, фумаровая кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота, трифторметансуль фоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, пара-толуолсульфоновая кислота, цикламовая кислота, салициловая кислота, пара-аминосалициловая кислота, памовая кислота и подобные кислоты.
формы такой подходящей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например соляная или бромоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобные кислоты; или органические кислоты, такие как, например, уксусная кислота, пропановая кислота, гидроксиуксусная кислота, молочная кислота, пировиноградная кислота, щавелевая кислота (т.е. этандикислота), мало новая кислота, янтарная кислота (т.е. бутандикислота), малеиновая кислота, фумаровая кислота, яблочная кислота, винная кислота, лимонная кислота, метансульфоновая кислота, трифторметансуль фоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, пара-толуолсульфоновая кислота, цикламовая кислота, салициловая кислота, пара-аминосалициловая кислота, памовая кислота и подобные кислоты.
И наоборот, указанные солевые формы могут быть превращены в форму свободного основания путем обработки соответствующим основанием.
Соединения формулы (I) могут существовать как в несольватированной форме, так и в сольватированных формах. Термин "сольват" используется в данной заявке для описания ассоциации молекул, содержащей молекулу соединения по изобретению и одну или более чем одну молекулу фармацевтически приемлемого растворителя, например воды или этанола. Термин "гидрат" используется, когда указанным растворителем является вода.
Предпочтительное воплощение изобретения относится к соединениям формулы (I), как они определены выше, где:
n равно 0 или 1;
R1 представляет собой метил или дифторметил;
каждый из R2, R3 и R4 независимо представляет собой водород, фтор, хлор, или группы R2 и R4 вместе образуют шестичленное гетероциклическое кольцо, содержащее атом азота, при условии, что по меньшей мере один из R2, R3 и R4 не представляет собой водород;
каждый из R5 и R6 представляет собой водород, фтор или хлор при условии, что по меньшей мере один из R5 и R6 представляет собой галоген.
Другое воплощение изобретения относится к соединениям формулы (I), как они определены выше, где:
n равно 0 или 1;
R1 представляет собой метил или дифторметил;
каждый из R2, R3 и R4 независимо представляет собой водород, фтор или хлор при условии, что по меньшей мере один из R2, R3 и R4 не представляет собой водород;
каждый из R5 и R6 представляет собой водород, фтор или хлор при условии, что по меньшей мере один из R5 и R6 представляет собой галоген.
Другое воплощение изобретения относится к соединениям формулы (I), как они определены выше, где:
n равно 0 или 1;
R1 представляет собой метил или дифторметил;
R3 представляет собой водород, и группы R2 и R4 вместе образуют шестичленное гетероциклическое кольцо, где это шестичленное гетероциклическое кольцо вместе с фенильной группой образуют хинолиновое кольцо.
Наиболее предпочтительно, соединение формулы (I) по данному изобретению выбрано из группы, состоящей из следующих соединений:


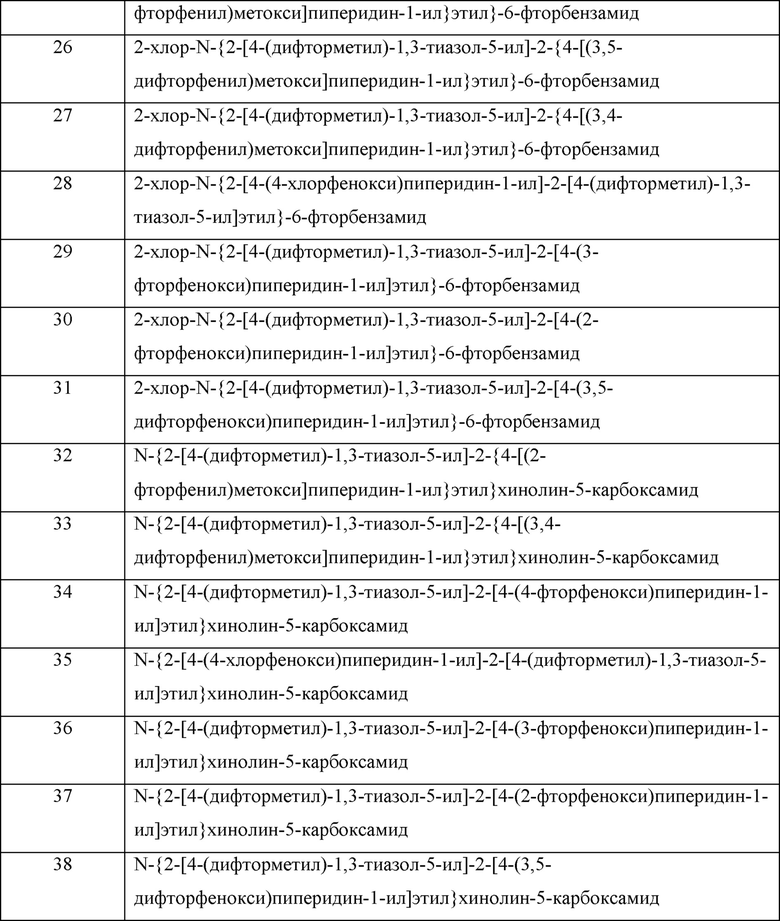
Соединения формулы (I) могут быть получены, как правило, путем осуществления взаимодействия соединения формулы (II):
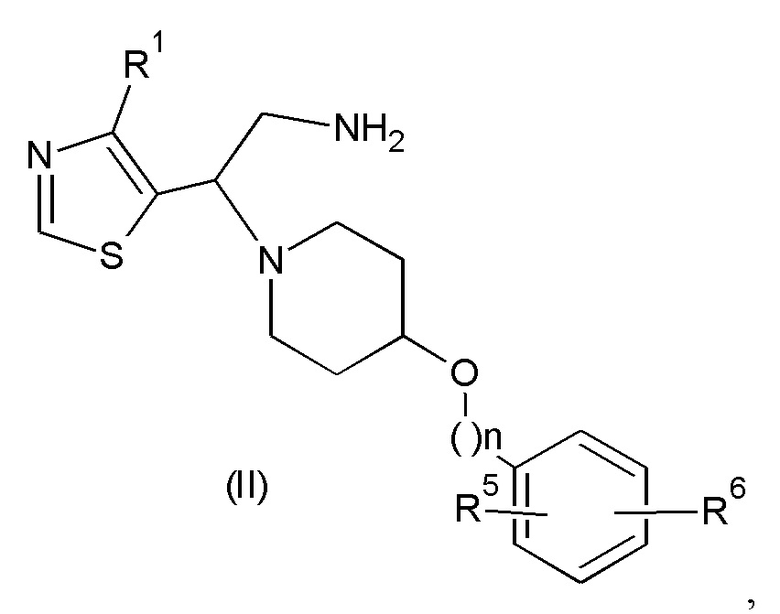
где значения n, R1, R5 и R6 такие, как определено выше, с соединением формулы (III):
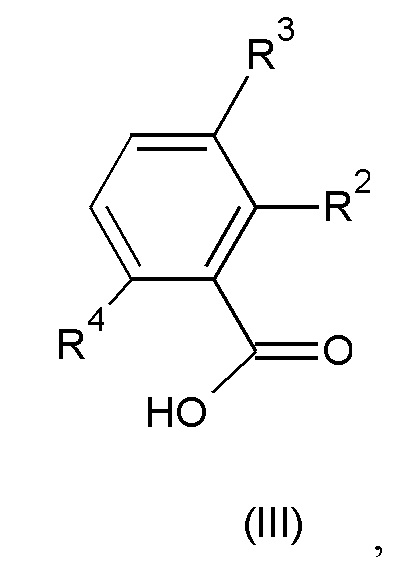
где значения R2, R3 и R4 такие, как определено выше; или
с соединением формулы (IIIa):
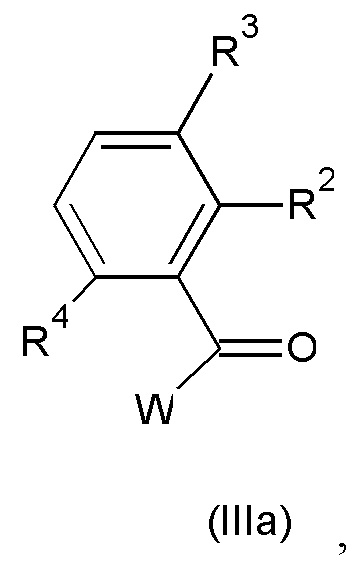
где значения R2, R3 и R4 такие, как определено выше; и W представляет собой подходящую уходящую группу;
и возможно превращения полученного соединения формулы (I) в его соль присоединения, и/или получения его стереохимически изомерных форм.
Взаимодействие соединения формулы (II) с соединением формулы (III) может быть осуществлено в по меньшей мере одном реакционно-инертном растворителе и возможно в присутствии по меньшей мере одного подходящего реагента сочетания и/или его подходящего основания. Удобно активировать карбоновую кислоту формулы (III) добавлением эффективного количества промотора реакции. Не ограничивающие примеры таких промоторов реакции включают карбонилдиимидазол, N,N'-дициклогексил-карбодиимид или 1-(3-диметиламинопропил)-3-этилкарбодиимид, гидроксибензотриазол, бензотриазолил-окситрис(диметиламино)-фосфония гексафторфосфат, тетрапирролидинофосфония гексафторфосфат, бромтрипирролидинофосфония гексафторфосфат или их функциональные производные, такие как те, которые описаны в D. Hudson, J. Org. Chem. (1988), 53, 617.
W в соединении формулы (IIIa) представляет собой подходящую уходящую группу, такую как, например, атом галогена, например фтора, хлора, брома, йода, или в некоторых случаях W может также представлять собой группу сульфонилокси, например метансульфонилокси, трифторметансульфонилокси, бензолсульфонилокси и подобные реакционноспособные уходящие группы. Взаимодействие соединения формулы (II) с соединением формулы (III) может быть осуществлено в реакционно-инертном растворителе, таком как, например, ацетонитрил, диметилацетамид, N-метил-пирролидон или DMF, и возможно в присутствии подходящего основания, такого как, например, карбонат натрия, карбонат калия или триэтиламин. Перемешивание может повышать скорость реакции. Реакцию удобно проводить при температуре в диапазоне от комнатной температуры до температуры дефлегмации реакционной смеси.
Соединения формулы (III) и формулы (IIIa) известны в данной области или могут быть получены способами, приведенными в примерах.
Соединения формулы (II) могут быть получены согласно следующей схеме:

Первичные амины (II) могут быть получены восстановлением соответствующих нитрильных производных (IV) в результате реакции образования азот-водородной связи. Не ограничивающие примеры такой реакции включают восстановление с использованием:
- водорода или источника водорода в присутствии металла, такого как никель, платина, палладий и кобальт, или его производного, такого как никель Ренея, оксид платины, оксид палладия или кобальт Ренея, в качестве катализатора;
- гидрида, такого как алюмогидрид лития, гидрид диизобутилалюминия (DIBAL), гидрид бора, или его функционального производного.
Реакция может быть осуществлена в подходящем растворителе, таком как метанол, тетрагидрофуран, уксусная кислота, диэтиловый эфир, толуол или метанольный раствор аммиака, предпочтительно при температуре от -78°С до комнатной температуры (к.т.).
Соединения формулы (IV), где R1, R5 и R6 такие, как определено для формулы (I), могут быть получены из альдегидов (VI) путем проведения реакции конденсации Штреккера с соответствующим гетероциклильным промежуточным соединением (VII) в присутствии источника цианида (V), например TMSCN или его функционального производного, в растворителе, таком как АсОН или MeCN, предпочтительно при температуре от 0°С до к.т.

Альтернативно, соединения формулы (II) также могут быть получены двухстадийным способом, описанным выше. Реакция соединений формулы (IV) с восстановителем, предпочтительно боргидридом натрия, в присутствии гексагидрата хлорида никеля(II) или гексагидрата хлорида кобальта(II) и Boc2O в растворителе, таком как МеОН, предпочтительно при температуре от 0°С до к.т., приводит к получению Вос-защищенного первичного амина формулы (VIII). После удаления защитной группы с использованием подходящей кислоты, предпочтительно TFA, получают соединения (II).
Примеры соединений формулы (VI) представлены на следующей схеме:
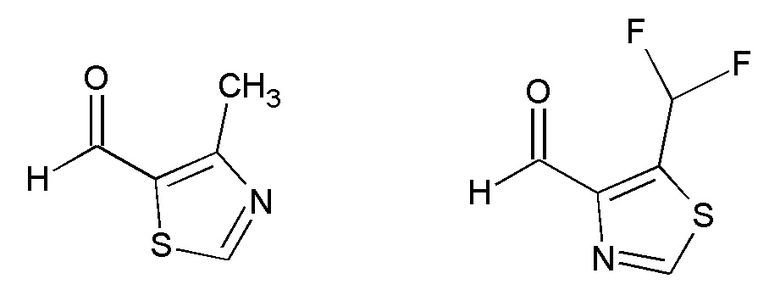
Перемешивание может повышать скорость реакции конденсации Штреккера. Исходные вещества и некоторые промежуточные соединения являются известными соединениям, и они коммерчески доступны или могут быть получены по стандартным методикам, общеизвестным в данной области техники.
Указанный способ возможно дополнительно включает в себя реакцию асимметрического синтеза с использованием хиральных вспомогательных веществ (углевода, хирального амина или циклического кетимина) и/или каталитический асимметрический синтез Штреккера (с использованием гуанидина, хирального шиффова основания или катализатора на основе 1,1'-би-2-нафтола (BINOL)).
Соединения формулы (I), полученные описанными выше способами, могут быть синтезированы в форме рацемических смесей энантиомеров, которые можно отделить друг от друга, следуя известным в данной области техники методам разделения. Те соединения формулы (I), которые получены в рацемической форме, могут быть превращены в соответствующие диастереомерные солевые формы путем взаимодействия с подходящей хиральной кислотой. Указанные диастереомерные солевые формы впоследствии разделяют, например, посредством селективной или фракционной кристаллизации, и энантиомеры высвобождают из них с использованием щелочи. Альтернативный способ разделения энантиомерных форм соединений формулы (1) включает жидкостную хроматографию с использованием хиральной неподвижной фазы. Указанные чистые стереохимически изомерные формы также могут быть получены из соответствующих чистых стереохимически изомерных форм соответствующих исходных веществ при условии, что реакция протекает стереоспецифично. Предпочтительно, в случае если желательным является конкретный стереоизомер, синтезировать указанное соединение с использованием стереоспецифических способов получения. В этих способах предпочтительно использовать энантиомерно чистые исходные вещества.
Соединения формулы (I), их фармацевтически приемлемые соли и стереоизомерные формы обладают антагонистическими свойствами в отношении Р2Х7 рецептора, что продемонстрировано в фармакологических примерах. Другими примерами известных в данной области техники реакций преобразования групп с целью превращения соединений формулы (I) в другие соединения формулы (I) являются: гидролиз сложных эфиров карбоновых кислот до соответствующей карбоновой кислоты или соответствующего спирта; гидролиз амидов до соответствующих карбоновых кислот или аминов; спирты могут быть превращены в сложные эфиры и простые эфиры; первичные амины могут быть превращены во вторичные или третичные амины; может быть проведено гидрирование по двойным связям до соответствующей одинарной связи. Исходные вещества и некоторые промежуточные соединения являются известными соединениями и имеются в продаже, или они могут быть получены в соответствии с традиционными реакционными методиками, общеизвестными в данной области техники. Соединения формулы (I), полученные описанными выше способами, могут быть синтезированы в форме рацемических смесей энантиомеров, которые можно отделить друг от друга, следуя известным в данной области техники методикам разделения. Те соединения формулы (I), которые получены в рацемической форме, могут быть превращены в соответствующие диастереомерные солевые формы путем взаимодействия с подходящей хиральной кислотой. Указанные диастереомерные солевые формы впоследствии разделяют, например, посредством селективной или фракционной кристаллизации, и энантиомеры высвобождают из них с использованием щелочи. Альтернативный способ разделения энантиомерных форм соединений формулы (I) включает жидкостную хроматографию с использованием хиральной неподвижной фазы. Указанные чистые стереохимически изомерные формы также могут быть получены из соответствующих чистых стереохимически изомерных форм соответствующих исходных веществ при условии, что реакция протекает стереоспецифично. Предпочтительно, в случае если желательным является конкретный стереоизомер, синтезировать указанное соединение с использованием стереоспецифических способов получения. В этих способах предпочтительно использовать энантиомерно чистые исходные вещества. При получении соединений формулы (I) и исходных веществ и/или промежуточных соединений, описанных в данной заявке, может быть полезным защитить определенные группы, которые являются чувствительными к реакционным условиям. Оценка полезности возможной защиты, а также выбор подходящего для защиты агента в соответствии с реакцией, проводимой при получении соединений по изобретению, и функциональной группы, подлежащей защите, находятся в пределах общеизвестных знаний специалиста. Удаление возможных защитных групп проводят в соответствии с традиционными методами. Общую информацию по использованию защитных групп в органической химии смотри в Theodora W. Greene and Peter G.M. Wuts, "Protective groups in organic synthesis", John Wiley & Sons, Inc., II Ed., 1991.
Получение солей соединений формулы (I) осуществляют известными способами. Таким образом, соединения по настоящему изобретению формулы (I) полезны в качестве лекарственного средства, особенно в лечении состояния или заболевания, опосредованного Р2Х7 рецептором, в частности лекарственного средства с антагонистической активностью в отношении Р2Х7 рецептора. Следовательно, соединения по настоящему изобретению могут быть использованы для изготовления лекарственного средства для лечения состояния или заболевания, опосредованного активностью Р2Х7 рецептора, в частности лекарственного средства с антагонистической активностью в отношении Р2Х7 рецептора.
Согласно настоящему изобретению также предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения состояний или заболеваний, выбранных из состояний или заболеваний, опосредованных Р2Х7 рецептором. В одном из воплощений согласно настоящему изобретению предложено соединение формулы (I) для применения в качестве лекарственного средства или для применения в лечении состояний или заболеваний, выбранных из состояний или заболеваний, опосредованных Р2Х7 рецептором. Кроме того, согласно настоящему изобретению также предложен способ лечения состояния, опосредованного активностью Р2Х7 рецептора, у субъекта-млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. С учетом вышеописанных механизмов действия соединения по изобретению полезны для лечения нейродегенеративных расстройств различного происхождения, таких как болезнь Альцгеймера и другие состояния с деменцией, такие как деменция, связанная с тельцами Леви, лобно-височная деменция и таупатии; амиотрофический боковой склероз, рассеянный склероз, болезнь Паркинсона и другие синдромы паркинсонизма; ВИЧ-индуцированное нейровоспаление; эссенциальный тремор; другие спиноцеребеллярные дегенерации и невропатия Шарко-Мари-Тута. Соединения по изобретению также полезны для лечения неврологических состояний, таких как эпилепсия, включая простой парциальный припадок, сложный парциальный припадок, вторично-генерализованный припадок, дополнительно включающий малый эпилептический припадок, миоклонические судороги, клонические судороги, тонические судороги, тонико-клонические судороги и атонические судороги.
Соединения по изобретению также полезны при лечении когнитивных расстройств и психиатрических расстройств. Психиатрические расстройства включают, но не ограничиваются ими, глубокую депрессию, дистимию, манию, биполярное расстройство (такое как биполярное расстройство I типа, биполярное расстройство II типа), циклотимическое расстройство, "быструю" цикличность, чрезвычайно быструю смену маниакальной и депрессивной фаз, манию, гипоманию, шизофрению, шизофреноморфные расстройства, шизоаффективные расстройства, расстройства личности, расстройства внимания с гиперактивным поведением или без него, бредовые расстройства, кратковременные психотические расстройства, индуцированные психотические расстройства, психотическое расстройство, обусловленное общим состоянием здоровья, вызванные веществами психотические расстройства или не идентифицированное психотическое расстройство, тревожные расстройства, такие как генерализованное тревожное расстройство, панические расстройства, посттравматическое стрессовое расстройство, расстройства контроля над побуждениями, фобические расстройства, диссоциативные состояния и, кроме того, зависимость от курения, лекарственную зависимость и алкоголизм, в частности биполярные расстройства, психоз, тревога и зависимость.
Соединения по настоящему изобретению полезны в предупреждении или лечении невропатической боли. Синдромы невропатической боли включают, но не ограничиваются ими: диабетическую невропатию; ишиалгию; неспецифическую боль в поясничном отделе позвоночника; боль при рассеянном склерозе; фибромиалгию; ВИЧ-обусловленную невропатию; невралгию, такую как постгерпетическая невралгия и невралгия тройничного нерва, невралгия Мортона, каузалгия; и боль, обусловленную физической травмой, ампутацией, фантомную боль, раковым заболеванием, токсинами или хроническими воспалительными состояниями; центральную боль, такую как боль, наблюдаемую при таламических синдромах; смешанные центральные и периферические формы боли, такие как комплексные региональные болевые синдромы (CRPS), также называемые симпатическими рефлекторными дистрофиями.
Соединения по изобретению также полезны в лечении хронической боли. Хроническая боль включает, но не ограничивается ими, хроническую боль, вызванную воспалением или связанным с воспалением состоянием, остеоартритом, ревматоидным артритом, острым повреждением или травмой, боль в верхнем отделе позвоночника и боль в поясничном отделе позвоночника (являющуюся результатом системного, регионарного или первичного заболевания позвоночника, такого как радикулопатия), боль в костях (обусловленную остеоартритом, остеопорозом, костным метастазированием или неизвестными причинами), тазовую боль, боль, ассоциированную с травмой спинного мозга, кардиальную боль в грудной клетке, некардиальную боль в грудной клетке, центральную постинсультную боль, миофасциальный болевой синдром, боль при серповидноклеточной анемии, боль при раковом заболевании, боль при болезни Фабри, боль при синдроме приобретенного иммунодефицита (СПИД), боль в старческом возрасте или боль, причиной которой являются головная боль, синдром височно-нижнечелюстного сустава, подагра, фиброз или компрессионные синдромы верхней апертуры грудной клетки, в частности, ревматоидный артрит и остеоартрит.
Соединения по изобретению также полезны при лечении острой боли, вызванной острым повреждением, болезнью, спортивно-медицинскими повреждениями, синдромом запястного канала, ожогами, растяжениями и напряжениями в скелетно-мышечной системе, мышечно-сухожильным растяжением, шейно-плечевыми болевыми синдромами, диспепсией, язвой желудка, язвой двенадцатиперстной кишки, дисменореей, эндометриозом или хирургическим вмешательством (таком как, при операции на открытом сердце или коронарном шунтировании), в лечении послеоперационной боли, боли при камнях в почках, боли в желчном пузыре, боли при камнях в желчном пузыре, родовой боли или зубной боли.
Соединения по изобретению также полезны в лечении головных болей, таких как мигрень, головная боль напряжения, трансформированная мигрень или развивающаяся головная боль, кластерная головная боль, а также расстройств, связанных с вторичной головной болью, таких как расстройства, вызванные инфекциями, метаболическими расстройствами или другими системными болезнями, и других острых головных болей, пароксизмальной гемикрании и тому подобных, являющихся результатом ухудшения вышеупомянутых первичных и вторичных головных болей.
Соединения по изобретению также полезны в лечении таких заболеваний, как головокружение, шум в ушах, мышечный спазм и другие расстройства, включая, но не ограничиваясь ими, сердечно-сосудистые заболевания (такие как сердечная аритмия, инфаркт миокарда или стенокардия, гипертензия, сердечная ишемия, церебральная ишемия), эндокринные нарушения (такие как акромегалия или несахарный диабет), заболевания, при которых патофизиология расстройства включает избыточную секрецию или гиперсекрецию или другую неподходящую клеточную секрецию эндогенного вещества (такого как катехоламин, гормон или фактор роста).
Соединения по изобретению также полезны в селективном лечении заболевания печени, такого как воспалительные заболевания печени, например хронический вирусный гепатит В, хронический вирусный гепатит С, алкогольное поражение печени, первичный билиарный цирроз, аутоиммунный гепатит, фиброз печени, неалкогольный стеатогепатит и отторжение трансплантата печени.
Соединения по изобретению ингибируют воспалительные процессы, затрагивающие все системы организма. Поэтому они полезны в лечении воспалительных процессов скелетно-мышечной системы, для которых ниже приведен список примеров, не являющийся исчерпывающим списком всех целевых расстройств: артритоподобные состояния, такие как, анкилозирующий спондилит, артрит шейного отдела позвоночника, фибромиалгия, подагра, ювенильный ревматоидный артрит, пояснично-крестцовый артрит, остеоартрит, остеопороз, псориатический артрит, ревматическая болезнь; расстройства, затрагивающих кожу и родственные ткани: экзема, псориаз, дерматит и воспалительные состояния, такие как солнечный ожог; расстройства дыхательной системы: астма, аллергический ринит и респираторный дистресс-синдром, легочные расстройства, в которые вовлечено такое воспаление, как астма и бронхит; хроническая обструктивная болезнь легких; расстройства иммунной и эндокринной систем: узелковый периартрит, тиреоидит, апластическая анемия, склеродермия, тяжелая миастения, рассеянный склероз и другие демиелинизирующие заболевания, энцефаломиелит, саркоидоз, почечный синдром, синдром Бехчета, полимиозит, гингивит.
Соединения по изобретению также полезны в лечении расстройств желудочно-кишечного тракта (ЖКТ), таких как воспалительные заболевания кишечника (IBD), включающие, но не ограничивающиеся ими, неспецифический язвенный колит, болезнь Крона, илеит, проктит, глютеновую болезнь, энтеропатии, микроскопичесий или коллагенозный колит, эозинофильный гастроэнтерит или паучит, возникающий после проктоколэктомии и постиленатального анастомоза, и синдром раздраженного кишечника, включающий любые расстройства, ассоциированные с абдоминальной болью и/или абдоминальным дискомфортом, такие как пилороспазм, нервная диспепсия, синдром раздраженной толстой кишки, спастический колит, спастический кишечник, невроз кишечника, функциональный колит, слизистый колит, колит, вызванный приемом слабительных средств, и функциональная диспепсия; а также для лечения атрофического гастрита, гастрита различной этиологии (gastritis varialoforme), неспецифического язвенного колита, пептической язвы, изжоги и другого поражения ЖКТ, например, в результате инфекции Helicobacter pylori, гастроэзофагеальной рефлюксной болезни, гастропареза, такого как диабетический гастропарез; и других функциональных расстройств кишечника, таких как неязвенная диспепсия (NUD); рвота, диарея и висцеральное воспаление внутренних органов.
Соединения по изобретению также полезны в лечении расстройств мочеполовых путей, таких как гиперактивность мочевого пузыря, простатит (хронический бактериальный и хронический небактериальный простатит), простатодиния, интерстициальный цистит, недержание мочи и доброкачественная гиперплазия предстательной железы, аднексит, пельвиоперитонит, бартолинит и вагинит, в частности гиперактивность мочевого пузыря и недержание мочи.
Соединения по изобретению также полезны в лечении офтальмологических заболеваний, таких как ретинит, ретинопатия, увеит и острое повреждение ткани глаза, возрастная макулярная дегенерация или глаукома, конъюнктивит.
Соединения по изобретению также полезны в лечении расстройств приема пищи, таких как нервная анорексия, включая подтипы ограничительного типа и типа с компульсивным перееданием/очищением кишечника (при помощи слабительных средств); нервная булимия, включая подтипы булимии с очищением кишечника и булимии без очищения кишечника; ожирение; компульсивные расстройства приема пищи; компульсивное переедание; и не классифицированное расстройство приема пищи.
Соединения по изобретению также полезны в лечении аллергического дерматита, гипервосприимчивости дыхательных путей, хронической обструктивной болезни легких (COPD), бронхита, септического шока, синдрома Шегрена, гломерулонефрита, атеросклероза, роста и метастазирования клеток злокачественного образования, миелобластного лейкоза, диабета, менингита, остеопороза, ожогового повреждения, ишемической болезни сердца, инсульта, заболевания периферических кровеносных сосудов, варикозного расширения вен, глаукомы.
Термин "подвергание лечению" и "лечение", использованный в данном описании, относится к терапевтическому, паллиативному и профилактическому лечению, включая реверсирование, облегчение, подавление развития или предупреждение заболевания, расстройства или состояния, к которому применяется такой термин, или одного или более симптомов такого заболевания, расстройства или состояния.
Дополнительно согласно настоящему изобретению предложены фармацевтические композиции, содержащие по меньшей мере один фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы (I).
Чтобы приготовить фармацевтические композиции по настоящему изобретению, эффективное количество конкретного соединения, в форме основания или соли присоединения кислоты, в качестве активного ингредиента объединяют в однородной смеси по меньшей мере с одним фармацевтически приемлемым носителем, который может иметь самые разнообразные формы в зависимости от формы препарата, необходимого для введения. Эти фармацевтические композиции желательно находятся в стандартной лекарственной форме, предпочтительно, для перорального введения, ректального введения, чрескожного введения или парентеральной инъекции.
Например, при изготовлении композиций в пероральной лекарственной форме можно использовать любой из обычных жидких фармацевтических носителей, таких как, например, вода, гликоли, масла, спирты и тому подобное, в случае пероральных препаратов в жидкой форме, таких как суспензии, сиропы, эликсиры и растворы; или можно использовать твердые фармацевтические носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие вещества, разрыхлители и тому подобное, в случае порошков, пилюль, капсул и таблеток. Ввиду легкости их введения таблетки и капсулы представляют собой наиболее удобную пероральную стандартную лекарственную форму, и, разумеется, в этом случае используют твердые фармацевтические носители. В случае композиций для парентеральных инъекций фармацевтический носитель будет преимущественно содержать стерильную воду, хотя для улучшения растворимости активного ингредиента в состав могут быть включены другие ингредиенты.
Растворы для инъекций могут быть приготовлены, например, с использованием фармацевтически приемлемого носителя, содержащего физиологический раствор, раствор глюкозы или смесь их обоих. Суспензии для инъекций также могут быть приготовлены с использованием подходящих жидких носителей, суспендирующих агентов и тому подобного. Фармацевтический носитель в композициях, подходящих для чрескожного введения, возможно может содержать агент, усиливающий проникновение через кожу, и/или пригодный увлажняющий агент, возможно в комбинации с небольшими количествами пригодных добавок, которые не оказывают существенного вредного воздействия на кожу. Указанные добавки могут быть выбраны с целью облегчения введения активного ингредиента в кожу и/или быть полезными при изготовлении требуемых композиций. Эти композиции для местного применения можно вводить различными способами, например, в виде трансдермального пластыря, препарата для капельного нанесения ("spot-on") или мази. Разумеется, соли присоединения соединений формулы (1), благодаря своей повышенной растворимости в воде по сравнению с соответствующей основной формой являются более подходящими для приготовления водных композиций.
Особенно предпочтительным является приготовление фармацевтических композиций по изобретению в стандартной лекарственной форме для удобства введения и единообразия дозировки.
Термин "стандартная лекарственная форма", используемый в данном описании, относится к физически дискретным единицам, подходящим в качестве однократных дозировок, причем каждая единица содержит предварительно заданное количество активного ингредиента, рассчитанное исходя из оказания желаемого терапевтического эффекта, совместно с необходимым фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (в том числе таблетки с насечкой для их деления, или таблетки, покрытые оболочкой), капсулы, пилюли, пакетики с порошком, облатки, растворы или суспензии для инъекций, отмеренные дозы, соответствующие чайной ложке, отмеренные дозы, соответствующие столовой ложке, и т.п. и их изолированные кратные количества.
Для перорального введения фармацевтические композиции по настоящему изобретению могут иметь форму твердых лекарственных форм, например таблеток (как в проглатываемых, так и в жевательных формах), капсул или желатиновых капсул, приготовленных традиционными способами, вместе с фармацевтически приемлемыми эксципиентами и носителями, такими как связующие вещества (например, прежелатинизированный кукурузный крахмал, поливинилпирролидон, гидроксипропилметилцеллюлоза и т.п.), наполнители (например, лактоза, микрокристаллическая целлюлоза, фосфат кальция и т.п.), смазывающие вещества (например, стеарат магния, тальк, диоксид кремния и т.п.), разрыхлители (например, картофельный крахмал, натрия крахмала гликолят и т.п.), увлажняющие агенты (например, лаурилсульфат натрия) и т.п. Такие таблетки также могут быть покрыты оболочкой способами, общеизвестными в данной области техники.
Жидкие препараты для перорального введения могут иметь форму, например, растворов, сиропов или суспензий, или они могут быть приготовлены в виде сухого продукта для смешивания с водой и/или другим подходящим жидким носителем перед использованием. Такие жидкие препараты могут быть приготовлены традиционными методами, возможно вместе с другими фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сироп сорбита, метилцеллюлоза, гидроксипропилметилцеллюлоза или гидрогенизированные пищевые жиры), эмульгирующие агенты (например, лецитин или аравийская камедь), неводные носители (например, миндальное масло, сложные эфиры жирных кислот или этиловый спирт), подсластители, корригенты, агенты, маскирующие вкус и запах, и консерванты (например, метил- или пропил-пара-гидроксибензоаты или сорбиновая кислота).
Фармацевтически приемлемые подсластители, пригодные для использования фармацевтических композициях по изобретению, предпочтительно включают по меньшей мере один подсластитель с интенсивным вкусом, такой как аспартам, ацесульфам калия, цикламат натрия, алитам, дигидрохалконовый подсластитель, монеллин, стевиозид-сукралоза (4,1',6'-трихлор-4,1',6'-тридезоксигалактосахароза) или, предпочтительно, сахарин, натриевая или кальциевая соль сахарина, и возможно по меньшей мере один объемный подсластитель, такой как сорбит, маннит, фруктоза, сахароза, мальтоза, изомальт, глюкоза, гидрогенизированный глюкозный сироп, ксилит, карамель или мед. Подсластители с интенсивным вкусом обычно используют в низких концентрациях. Например, в случае натриевой соли сахарина указанная концентрация может колебаться в диапазоне от примерно 0,04% до 0,1% (масса/объем) от конечной композиции. Объемный подсластитель можно эффективно использовать в более высоких концентрациях, изменяющихся в диапазоне от примерно 10% до примерно 35%, предпочтительно от примерно 10% до примерно 15% (масса/объем). Фармацевтически приемлемые корригенты, которые могут маскировать ингредиенты с горьким вкусом в композициях с низкими дозами, предпочтительно включают фруктовые корригенты, такие как вишневые, малиновые, черносмородиновые или клубничные корригенты. Сочетание двух корригентов может обеспечивать очень хорошие результаты. В композициях с высокими дозами могут потребоваться более сильные фармацевтически приемлемые корригенты, такие как карамель-шоколад (Caramel Chocolate), освежающая мята (Mint Cool), "фантазия" (Fantasy) и т.п.
Каждый корригент может присутствовать в конечной композиции в концентрации в диапазоне от примерно 0,05% до 1% (масса/объем). Предпочтительно использовать сочетания указанных сильных корригентов. Предпочтительно использовать корригент, который не подвергается какому-либо изменению или не теряет свой вкус и/или цвет в условиях данной композиции.
Соединения формулы (I) могут быть приготовлены в виде композиции для парентерального введения путем инъекции, обычно внутривенной, внутримышечной или подкожной инъекции, например, болюсной инъекции или непрерывной внутривенной инфузии. Композиции для инъекций могут быть представлены в стандартной лекарственной форме, например в ампулах или многодозовых контейнерах, содержащей добавленный в нее консервант. Они могут находиться в таких формах, как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать технологические агенты, такие как агенты, обеспечивающие изотоничность, суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, активный ингредиент может присутствовать в порошковой форме для смешивания с подходящим носителем, например стерильной апирогенной водой, перед использованием.
Соединения формулы (I) также могут быть приготовлены в виде ректальных композиций, таких как суппозитории или удерживающие клизмы, содержащие, например, традиционные суппозиторные основы, такие как масло какао и/или другие глицериды.
Специалисты в области лечения заболеваний, связанных с регуляцией лиганд-управляемых ионных каналов, без труда определят терапевтически эффективное количество соединения формулы (I) на основании представленных ниже результатов тестирования. Обычно предполагается, что терапевтически эффективная доза будет составлять от примерно 0,001 мг/кг до примерно 50 мг/кг массы тела, более предпочтительно от примерно 0,01 мг/кг до примерно 10 мг/кг массы тела подлежащего лечению пациента. Может оказаться приемлемым введение терапевтически эффективной дозы в форме двух или более субдоз через подходящие интервалы времени в течение суток. Указанные субдозы могут быть приготовлены в виде стандартных лекарственных форм, при этом каждая из них содержит, например, от примерно 0,1 мг до примерно 1000 мг, более конкретно от примерно 1 до примерно 500 мг активного ингредиента на одну стандартную лекарственную форму.
В настоящем документе "терапевтически эффективное количество" соединения представляет собой количество соединения, в результате введения которого индивидууму или животному уровень такого соединения у индивидуума или животного оказывается достаточно высоким, чтобы вызывать заметный антагонистический ответ в отношении Р2Х7 рецепторов.
Точная дозировка и частота введения зависят от конкретного используемого соединения формулы (I), конкретного подвергаемого лечению состояния, степени тяжести подвергаемого лечению состояния, возраста, массы и общего физического состояния конкретного пациента, а также другого препарата, который этот пациент может принимать, что хорошо известно специалистам в данной области техники. Кроме того, указанное "терапевтически эффективное количество" можно уменьшать или увеличивать в зависимости от реакции подвергаемого лечению пациента и/или в зависимости от оценки врача, назначающего соединения по настоящему изобретению. Поэтому упомянутые выше диапазоны эффективных количеств, принимаемых в течение одних суток, представляют собой всего лишь рекомендации.
Номенклатура и структуры
В общем, использованная в этой заявке номенклатура основана на ChemSketch™ (ACDLabs) и создана в соответствии с систематической номенклатурой IUPAC. Химические структуры, показанные в данном документе, были получены с использованием ISIS®, версия 2.2. Любая свободная валентность, показанная на атоме углерода, кислорода, серы или азота в приведенных в данном документе структурах, указывает на присутствие атома водорода, если не указано иное. Если азотсодержащее гетероарильное кольцо показано со свободной валентностью на атоме азота, и на этом гетероарильном кольце показаны переменные, такие как R1, R2, R3 и т.д., то такие переменные могут быть связаны с атомом азота, имеющим свободную валентность, или могут быть присоединены к нему. Если в структуре имеется хиральный центр, но для этого хирального центра не показана никакая конкретная стереохимия, то данной структурой охвачены оба энантиомера, ассоциированные с данным хиральным центром. Если структура, показанная в данном описании, может существовать в нескольких таутомерных формах, то все такие таутомеры охвачены данной структурой. Подразумевается, что атомы, представленные в структуре, приведенной в данном документе, охватывают все существующие в природе изотопы таких атомов. Так, например, подразумевается, что атомы водорода, представленные в данном описании, включают дейтерий и тритий, и подразумевается, что атомы углерода включают изотопы С13 и С14.
Сокращения
В описании схем и примеров могут быть использованы следующие сокращения:
АсОН: уксусная кислота;
безв.: безводный;
AcONa: ацетат натрия;
Вос: трет-бутил-карбонат;
Вос2О: ди-трет-бутилдикарбонат;
КХ: колоночная хроматография;
DAST: трифторид диэтиламиносеры;
DCM: дихлорметан;
DEA: диэтиламин;
DIAD: диизопропилазодикарбоксилат;
DIBAL: гидрид диизобутилалюминия;
DIPEA: диизопропилэтиленамин;
DMAP: диметиламино пиридин;
DMF: диметилформамид;
DMSO: диметилсульфоксид;
Et2O: диэтиловый эфир;
EtOAc: этил ацетат;
EtOH: этанол;
ЭРИ: электрораспылительная ионизация;
HBTU: гексафторфосфат N,N,N',N'-тетраметил-O-(1Н-бензотриазол-1-ил)-урония;
ч: час;
М: молярный;
MeCN: ацетонитрил;
МеОН: метанол;
мин: минута(ы);
Ni Ренея: никель Ренея;
ЯМР: ядерный магнитный резонанс;
к.т. комнатная температура;
TFA: трифторуксусная кислота;
THF: тетрагидрофуран;
ТСХ: тонкослойная хроматография;
TMSCN: триметилсилилцианид;
УЭЖХ-МС: ультраэффективная жидкостная хроматография-масс-спектрометрия;
XPhos: 4,5-бис(дифенилфосфино)-9,9-диметилксантен.
Экспериментальная часть
Следующие далее примеры иллюстрируют настоящее изобретение. Если конкретно не указано иное, все показатели (в частности процентные содержания и количества) относятся к массе.
Примеры синтеза
А. Синтез промежуточных соединений 1b-1d
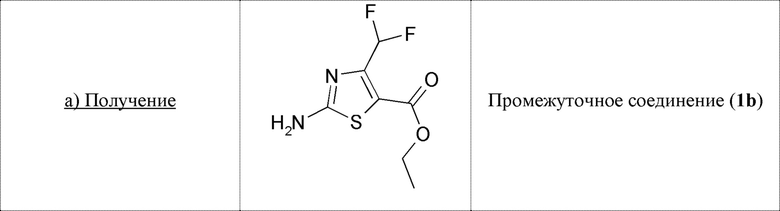
Сульфурилхлорид (1,23 мл, 15,2 ммоль, 1,01 экв.) добавляли по каплям при 0°С к этил-4,4-дифторацетоацетату (2,5 г, 15,0 ммоль, 1 экв.) в атмосфере азота и перемешивали в течение ночи при комнатной температуре. Реакционную смесь разбавляли EtOAc (20 мл) и вливали в смесь лед/вода (20 мл). Органический слой сушили над безв. Na2SO4, фильтровали и упаривали с получением 3,2 г неочищенного вещества в 2-хлор-4,4-дифторацетоацетате в виде желтого масла. Это неочищенное вещество растворяли в этаноле (10 мл), обрабатывали тиомочевиной (3,2 г, 30 ммоль, 2 экв.) и нагревали в микроволновом реакторе в течение 1 ч при 100°С. Затем растворитель удаляли в вакууме, и остаток распределяли в нас. NaHCO3 (10 мл) и EtOAc (10 мл). Органический слой промывали рассолом (20 мл), сушили над безв. Na2SO4, фильтровали и упаривали. Неочищенное вещество обрабатывали диэтиловым эфиром, фильтровали и сушили в вакууме с получением 1,37 г (выход 41%) промежуточного соединения 1b в виде желтого твердого вещества.

Промежуточное соединение 1b (1,37 г, 6,16 ммоль, 1 экв.) растворяли в диоксане (35 мл), добавляли изоамилнитрит (2,24 мл, 16,64 ммоль, 2,7 экв.), и эту реакционную смесь нагревали в течение 1 часа при 80°С. Растворитель удаляли выпариванием при пониженном давлении, и остаток очищали флэш-хроматографией на силикагеле (EtOAc/петролейный эфир 10/90) с получением промежуточного соединения 1с (1,02 г, выход 80%) в виде желтого твердого вещества.

Промежуточное соединение 1с (0,758 г, 3,66 ммоль, 1 экв.) растворяли в сухом DCM (18,5 мл) в атмосфере аргона и охлаждали до -75°С. Добавляли по каплям 1М диизобутилалюминия гидрид в DCM (4,1 мл, 4,1 ммоль, 1,12 экв.), и реакционную смесь перемешивали при -70°С. Через 1,5 ч добавляли по каплям 1М гидрид диизобутилалюминия в DCM (2,5 мл, 2,5 ммоль, 0,68 экв.), и реакционную смесь перемешивали дополнительно в течение 1 ч при -70°. Реакционную смесь нагревали до 0°С и обрабатывали водой (0,264 мл), 15% NaOH (0,264 мл) и водой (0,66 мл) в этом порядке. Затем смесь перемешивали в течение 5 минут при 0°С, затем в течение 30 минут при комнатной температуре. Последовательно добавляли воду (0,24 мл) и 15% NaOH (0,130 мл), и реакционную смесь перемешивали при комнатной температуре до образования осадка. Смесь фильтровали, и затем растворитель выпаривали. Остаток очищали флэш-хроматографией на силикагеле (DCM/петролейный эфир 80/20→100% DCM) с получением желтого масла (0,34 мг, выход 40%), содержащего промежуточное соединение 1d (чистота примерно 70%), которое использовали как есть.
В. Синтез промежуточных соединений: α-аминонитрилов
Исходные вещества
Все замещенные производные 4-фенилокси- или 4-бензилокси-пиперидина и 4-метил-1,3-тиазол-5-карбальдегида, использованные в качестве исходных веществ, были приобретены у поставщиков химических реагентов:


Общая методика
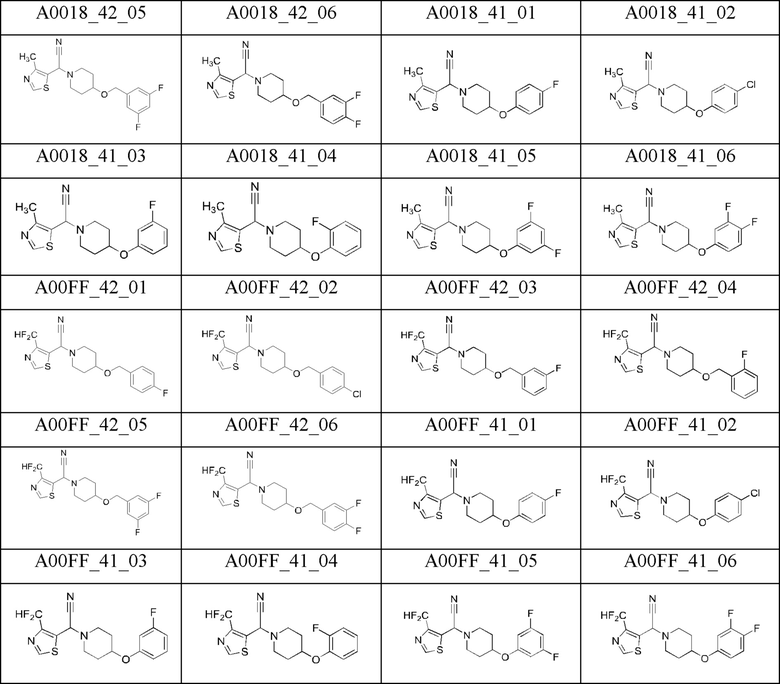
Гидрохлорид производного 4-замещенного пиперидина (1 экв.) суспендировали в 2-3 мл DCM и добавляли TEA (1,1-2 экв.). Смесь перемешивали в течение нескольких минут, растворитель выпаривали на роторном испарителе, и остаток сушили в вакууме в течение 15 минут при 40°С. Не содержащий гидрохлорида амин (1 экв.), тиазолил-альдегид (180-300 мг, 1,2-1,5 экв.) и AcONa (3,5 экв.) растворяли в ледяной АсОН (5-8 мл). Смесь перемешивали при комнатной температуре в атмосфере аргона в течение 3 ч и затем охлаждали до 0°С. Добавляли по каплям TMSCN (3-12 экв.), и смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1-3 суток. В то же время, если это необходимо согласно результатам анализа методом ЖХ-МС, добавляли TMSCN (3-6 экв.) (вплоть до 12 экв. TMSCN), и реакционную смесь перемешивали в течение 24 ч. Затем растворитель выпаривали на роторном испарителе при 40-45°С. К остатку добавляли насыщенный раствор NaHCO3 (20-50 мл). При необходимости, добавляли твердый NaHCO3 и воду для увеличения рН до 8. Смесь экстрагировали DCM (5 мл × 3-5). Объединенные органические фазы сушили (безв. Na2SO4) и упаривали. Неочищенное вещество очищали флэш-хроматографией (SiO2) с использованием смеси гексан/ацетон (0->30%) с получением чистого а-аминонитрила (выход 28-68%).
Используя эту методику, были получены промежуточные соединения А0018_42_01 (выход 54%), А0018_42_02 (выход 55%), А0018_42_03 (выход 55%), А0018_42_04 (выход 42%), А0018_42_05 (выход 63%), А0018_42_06 (выход 50%), А0018_41_01 (выход 45%), А0018_41_02 (выход 60%), А0018_41_03 (выход 46%), А0018_41_04 (выход 68%), А0018_41_05 (выход 65%), А0018_41_06 (выход 43%), A00FF_42_04 (выход 30%), A00FF_42_05 (выход 56%), A00FF_42_06 (выход 36%), A00FF_41_01 (выход 31%), A00FF_41_02 (выход 57%), A00FF_41_03 (выход 53%), A00FF_41_04 (выход 53%), A00FF_41_05 (выход 28%), начиная с 4-метил-1,3-тиазол-5-карбальдегида или, соответственно, 4-дифторметил-1,3-тиазол-5-карбальдегида и 4-(4-фторбензилоксипиперидина), 4-(4-хлорбензилоксипиперидина), 4-(3-фторбензилоксипиперидина), 4-(2-фторбензилоксипиперидина), 4-(3,5-дифторбензилоксипиперидина), 4-(3,4-дифторбензилоксипиперидина), 4-(4-фторфенилоксипиперидина), 4-(4-хлорфенилоксипиперидина), 4-(3-фторфенилоксипиперидина), 4-(2-фторфенилоксипиперидина), 4-(3,5-дифторфенилоксипиперидина) или 4-(3,4-дифторфенилоксипиперидина).

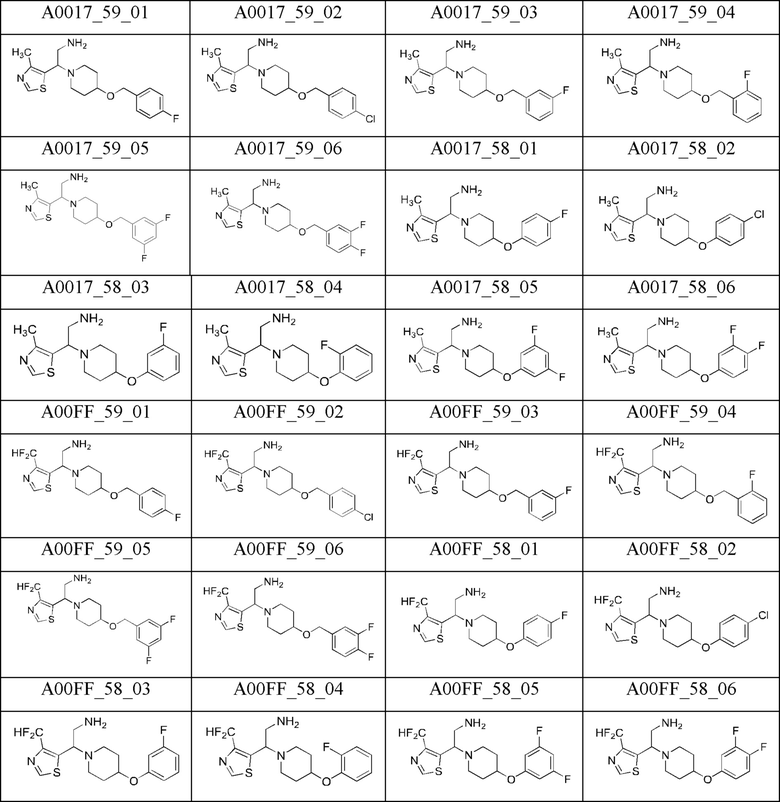
С. Получение диаминов (Общая методика)
Цианидное производное (100-270 мг, 1 экв.) растворяли в сухом DCM в атмосфере аргона и охлаждали в бане лед-соль. Медленно добавляли 1М раствор DIBAL (3 экв.) в DCM (порциями, 1 экв. каждые 30 минут), и смесь перемешивали в течение в течение еще одного часа. В этот раствор (при 0°С) добавляли по каплям воду (1 мл), и смесь перемешивали до окончания образования осадка. DCM удаляли в вакууме, и остаток суспендировали в AcOEt. Полученное твердое вещество отфильтровывали и промывали 4-6 раз AcOEt. Объединенные органические слои сушили над Na2SO4, упаривали до образования маслянистого остатка и сушили не менее 1 ч в вакууме при 38-40°С. Неочищенный продукт (выход 68-97%) использовали без дополнительной очистки на следующей стадии синтеза.
С использованием этой методики:
промежуточное соединение А0017_59_01 (выход 94%) было получено из А0018_42_01;
промежуточное соединение А0017_59_02 (выход 83%) было получено из А0018_42_02;
промежуточное соединение А0017_59_03 (выход 87%) было получено из А0018_42_03;
промежуточное соединение А0017_59_04 (выход 97%) было получено из А0018_42_04;
промежуточное соединение А0017_59_05 (выход 65%) было получено из А0018_42_05;
промежуточное соединение А0017_59_06 (выход 88%) было получено из А0018_42_06;
промежуточное соединение А0017_58_01 (выход 62%) было получено из А0018_41_01;
промежуточное соединение А0017_58_02 (выход 95%) было получено из А0018_41_02;
промежуточное соединение А0017_58_03 (выход 67%) было получено из А0018_41_03;
промежуточное соединение А0017_58_04 (выход 89%) было получено из А0018_41_04;
промежуточное соединение А0017_58_05 (выход 80%) было получено из А0018_41_05;
промежуточное соединение А0017_58_06 (выход 95%) было получено из А0018_41_06;
промежуточное соединение A00FF_59_04 (выход 87%) было получено из A00FF_42_04;
промежуточное соединение A00FF_59_05 (выход 81%) было получено из A00FF_42_05;
промежуточное соединение A00FF_59_06 (выход 80%) было получено из A00FF_42_06;
промежуточное соединение A00FF_58_01 (выход 89%) было получено из A00FF_41_01;
промежуточное соединение A00FF_58_02 (выход 94%) было получено из A00FF_41_02;
промежуточное соединение A00FF_58_03 (выход 88%) было получено из A00FF_41_03;
промежуточное соединение A00FF_58_04 (выход 92%) было получено из A00FF_41_04
промежуточное соединение A00FF_58_05 (выход 93%) было получено из A00FF_41_05.
D. Общая методика синтеза конечных соединений
Получение Соединений 1-38
Смесь карбоновой кислоты (25-115 мг, 1 экв.), HATU (1,1 экв.) или EDCl (1 экв.)/HOBt (1 экв.) и DIPEA (2-3 экв.) в безводном DMF или DCM (1-2 мл) перемешивали в течение 10-30 минут в атмосфере аргона. Затем добавляли раствор неочищенного амина (1 экв.) в безводном DCM или DMF (1-3 мл), и эту реакционную смесь перемешивали в течение ночи. Добавляли воду и насыщенный раствор NaHCO3, и продукт экстрагировали 4-6 раз DCM. Объединенные органические слои сушили над Na2SO4 и упаривали. Продукт очищали методом FCC (SiO2, DCM->AcOEt->0-10 МеОН/AcOEt) или методом SFC (5-10% MeOH/sc(сверхкритический) СО2), и фракции с целевым продуктом упаривали и сушили в глубоком вакууме в течение 16-72 ч. Выходя 8-71%.
С использованием этой методики были получены следующие соединения:
Соединение 1 (выход 19%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и А0017_59_01 в DMF+DCM, очистка методом SFC;
Соединение 2 (выход 19%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и А0017_59_02 в DMF, очистка методом SFC;
Соединение 3 (выход 23%) было получено из 2-хлор-6-фторбензойной кислоты, EDCl/HOBt и А0017_59_03 в DCM, очистка методом SFC;
Соединение 4 (выход 30%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и А0017_59_04 в DMF, очистка методом SFC;
Соединение 5 (выход 16%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и А0017_59_05 в DMF, очистка методом FCC, затем методом SFC;
Соединение 6 (выход 12-18%) было получено из 2-хлор-6-фторбензойной кислоты, EDCl/HOBt или HATU и А0017_59_06 в DCM или DMF+DCM, очистка методом SFC;
Соединение 7 (выход 16%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и А0017_58_01 в DMF+DCM, очистка методом SFC;
Соединение 8 (выход 13-16%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и А0017_58_02 в DMF+DCM or DMF, очистка методом FCC preceded by SFC;
Соединение 9 (выход 48%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и А0017_58_03 в DMF, очистка методом FCC;
Соединение 10 (выход 12%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и А0017_58_04 в DMF, очистка методом FCC;
Соединение 11 (выход 8%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и А0017_58_05 в DCM, очистка методом SFC с предшествующей очисткой методом FCC;
Соединение 12 (выход 29%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и А0017_58_06 в DMF+DCM, очистка методом SFC;
Соединение 13 (выход 28%) было получено из хинолин-5-карбоновой кислоты, HATU и А0017_59_01 в DCM, очистка методом FCC;
Соединение 14 (выход 25%) было получено из хинолин-5-карбоновой кислоты, HATU и А0017_59_02 в DMF, очистка методом FCC;
Соединение 15 (выход 32%) было получено из хинолин-5-карбоновой кислоты, HATU и А0017_59_03 в DMF+DCM, очистка методом FCC;
Соединение 16 (выход 31%) было получено из хинолин-5-карбоновой кислоты, HATU и А0017_59_04 в DMF+DCM, очистка методом FCC;
Соединение 17 (выход 29%) было получено из хинолин-5-карбоновой кислоты, HATU и А0017_59_05 в DMF, очистка методом FCC;
Соединение 18 (выход 20%) было получено из хинолин-5-карбоновой кислоты, EDCl/HOBt и А0017_59_06 в DCM, очистка методом FCC;
Соединение 19 (выход 28%) было получено из хинолин-5-карбоновой кислоты, HATU и А0017_58_01 в DMF, очистка методом FCC;
Соединение 20 (выход 27%) было получено из хинолин-5-карбоновой кислоты, HATU и А0017_58_02 в DMF, очистка методом FCC;
Соединение 21 (выход 71%) было получено из хинолин-5-карбоновой кислоты, HATU и А0017_58_03 в DMF, очистка методом FCC;
Соединение 22 (выход 25%) было получено из хинолин-5-карбоновой кислоты, HATU и А0017_58_04 в DMF, очистка методом FCC;
Соединение 23 (выход 15%) было получено из хинолин-5-карбоновой кислоты, EDCl/HOBt и А0017_58_05 в DCM, очистка методом FCC;
Соединение 24 (выход 17-27%) было получено из хинолин-5-карбоновой кислоты, EDCl/HOBt или HATU и А0017_58_06 в DCM или DMF+DCM, очистка методом FCC;
Соединение 25 (выход 30%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и A00FF_59_04 в DMF+DCM, очистка методом SFC;
Соединение 26 (выход 19%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и A00FF_59_05 в DMF+DCM, очистка двумя отдельными FCC;
Соединение 27 (выход 26%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и A00FF_59_06 в DMF+DCM, очистка методом SFC;
Соединение 28 (выход 19%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и A00FF_58_02 в DMF+DCM, очистка методом SFC;
Соединение 29 (выход 22%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и A00FF_58_03 в DMF+DCM, очистка методом SFC;
Соединение 30 (выход 30%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и A00FF_58_04 в DMF+DCM, очистка методом SFC;
Соединение 31 (выход 29%) было получено из 2-хлор-6-фторбензойной кислоты, HATU и A00FF_58_05 в DMF+DCM, очистка методом SFC;
Соединение 32 (выход 30%) было получено из хинолин-5-карбоновой кислоты, HATU и A00FF_59_04 в DMF+DCM, очистка методом SFC;
Соединение 33 (выход 31%) было получено из хинолин-5-карбоновой кислоты, HATU и A00FF_59_05 в DMF+DCM, очистка методом SFC;
Соединение 34 (выход 34%) было получено из хинолин-5-карбоновой кислоты, HATU и A00FF_58_01 в DMF+DCM, очистка методом SFC preceded by FCC;
Соединение 35 (выход 19%) было получено из хинолин-5-карбоновой кислоты, HATU и A00FF_58_02 в DMF+DCM, очистка методом FCC preceded by SFC;
Соединение 36 (выход 22%) было получено из хинолин-5-карбоновой кислоты, HATU и A00FF_58_03 в DMF+DCM, очистка методом SFC;
Соединение 37 (выход 22%) было получено из хинолин-5-карбоновой кислоты, HATU и A00FF_58_04 в DMF+DCM, очистка методом SFC
Соединение 38 (выход 35%) было получено из хинолин-5-карбоновой кислоты, HATU и A00FF_58_05 в DMF+DCM, очистка методом SFC.
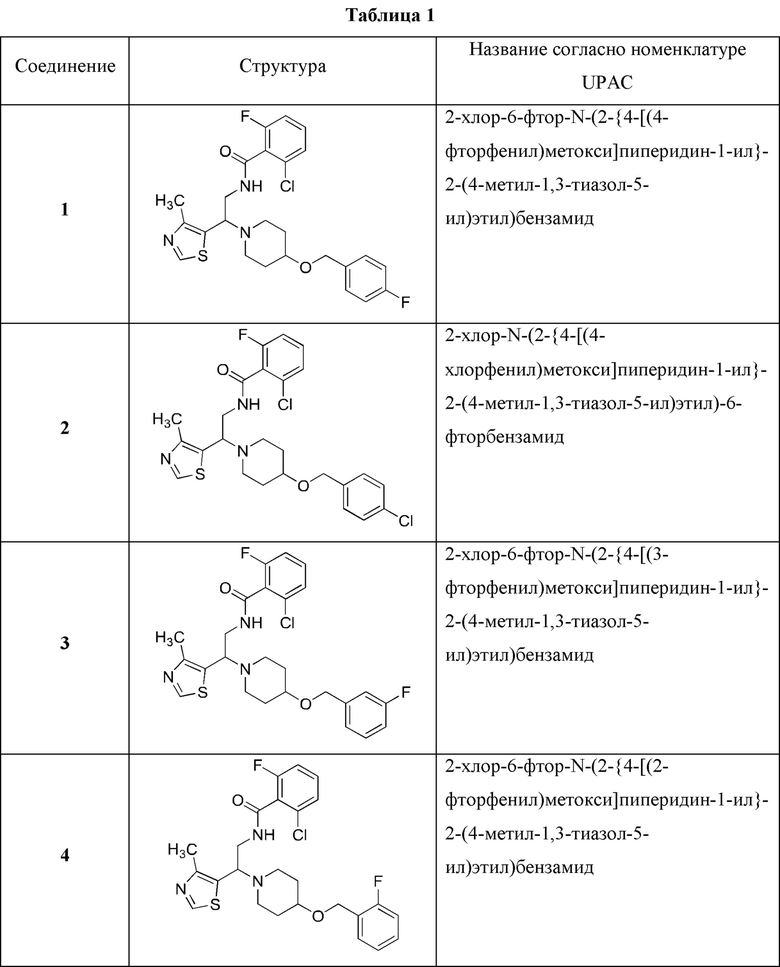
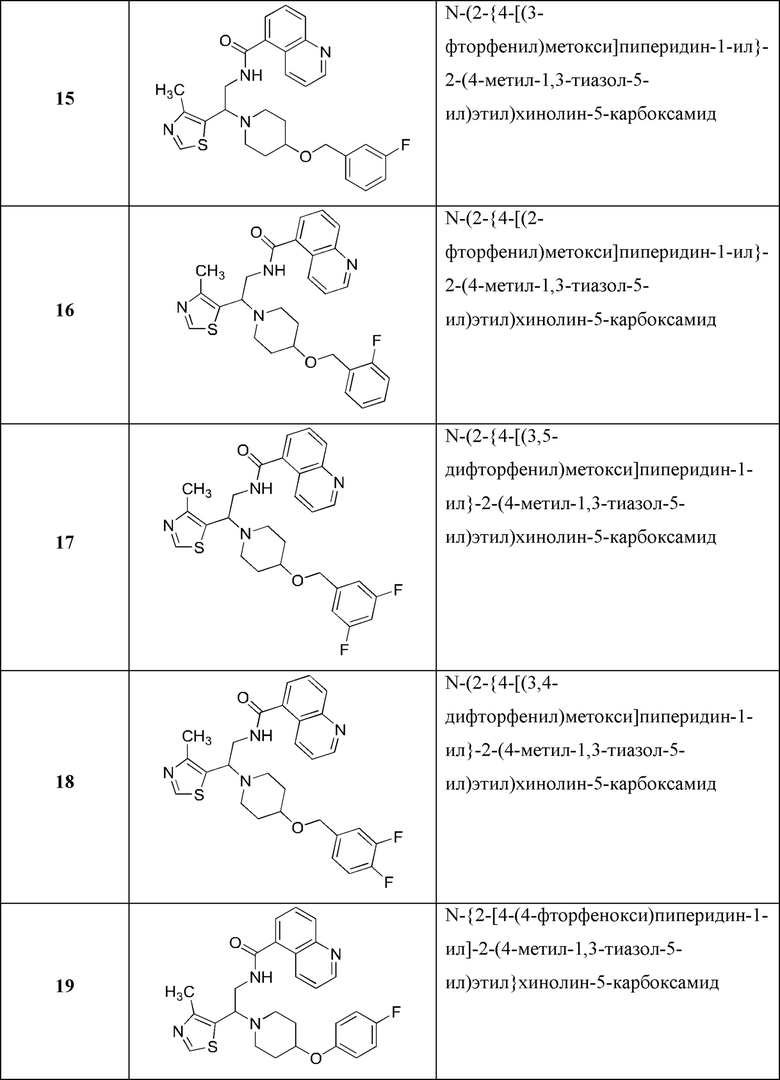
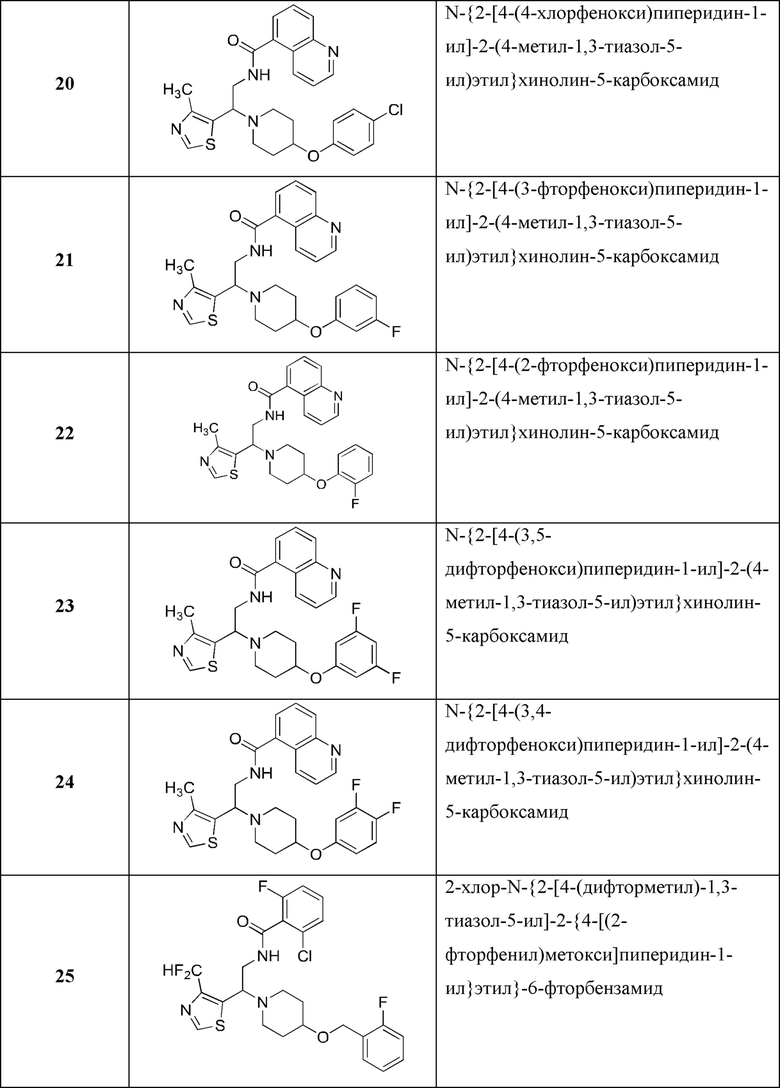
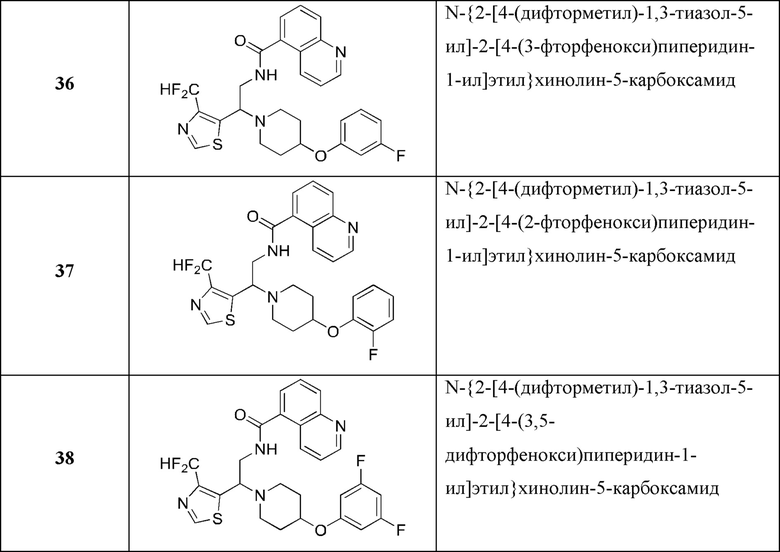
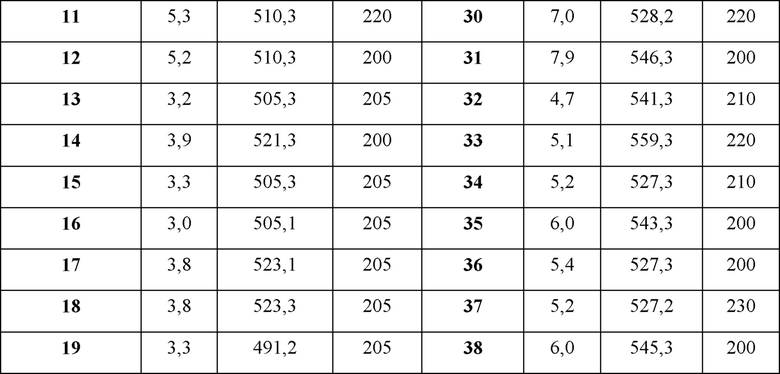
В Таблице 1 приведен список конечных соединений, которые были получены и протестированы согласно экспериментальной методике, описанной для Примера 1.




Система очистки
Флэш-хроматография (FCC)
Разделения методом FCC осуществляли на Interchim puriFlash®430, Interchim puriFlash®450 или Interchim puriFlash® 4250-250, оснащенном УФ-детектором. Тип силикагелевых колонок: Interchim puriFlash® SiHP (высококачественный диоксид кремния) 50 мкм, 4-25 г.
Сверхкритическая флюидная хроматография (SFC):
Разделения методом FCC осуществляли на системе Waters Prep 100q SFC System, оснащенной фотодиодным детектором и детектором MS QDa. Тип силикагелевой колонки: Viridis Prep Silica 2-ЕР (2-этилпиридин) OBD, 19×100 мм, 5 мкм. Использованный метод: растворитель (А): CO2, растворитель (В): метанол; условия градиента: от 5%-10% В за 8 минут; ABPR (автоматический регулятор обратного давления) 120 бар (12000 кПа); Т=40°С.
Аналитическая часть
Общая методика ЖХ-МС
Измерения методом ВЭЖХ осуществляли с использованием модуля Dionex Ultimate 3000, включающего в себя насос для четырехкомпонентных смесей с дегазатором, автосамплер, термостат для колонки (установленный на 25°С), детектор на диодной матрице (DAD) (обычно используемая длина волны 200 нм) и колонку Kinetex ХВ С18 4,6×50 мм, 2,6 мкм. Скорость потока элюата составляла 0,5 мл/мин. Использовали две подвижные фазы: подвижная фаза А: 0,1%-ный раствор муравьиной кислоты в воде (MiliQ); подвижная фаза В: 0,1%-ный раствор муравьиной кислоты в ацетонитриле (J.T. Baker для ВЭЖХ), и их использовали для выполнения условий градиента от 20% В до 80% за 6,7 мин, выдержка при 80% В в течение 1,3 мин, условий градиента от 80% В до 95% за 0,3 мин, выдержка при 95% В, и условия градиента до 20% В за 0,5 мин и выдержка при этих условиях в течение 2 минут для того, чтобы заново уравновесить колонку. Использовали впрыскиваемый объем 1,0 мкл. Поток из колонки отводили в МС спектрометр. МС детектор (НСТ Bruker) был скомпонован с источником электрораспылительной ионизации. Масс-спектры получали путем сканирования от 100 до 1000 Да. Напряжение на капиллярной игле составляло 4 кВ в режиме положительной ионизации, и температура источника поддерживалась при 365°С. В качестве газа-распылителя использовали азот; скорость потока составляла 9,0 л/мин. Сбор данных осуществляли с использованием программы Data Analysis Bruker Program.

Определение характеристик ЯМР
Спектры 1H-ЯМР и 13С-ЯМР регистрировали на спектрометре Bruker Avance III HD 400 МГц с использованием CDCl3 или CD3OD в качестве растворителя. Химические сдвиги (δ) приведены в частях на миллион (млн-1) относительно остаточного сигнала пика не полностью дейтерированных растворителей для 1H-ЯМР, заданного как 7,26 млн-1 для CHCl3 и 3,31 млн-1 для CHD2OD, или относительно сигнала пика дейтерированных растворителей для 13С-ЯМР, заданного как 77,16 млн-1 для CHCl3 и 49,00 млн-1 для CD3OD.






Фармакологические примеры
В анализе с использованием автоматизированного метода фиксации потенциала "пэтч-кламп" было обнаружено, что соединения по изобретению активны в отношении канала Р2Х7 человека.
С целью проведения прямого мониторинга блокирования канала Р2Х7 был разработан и осуществлен электрофизиологический анализ на автоматизированном приборе для электрофизиологических исследований QPatch16X.
Клетки HEK-293, экспрессирующие каналы Р2Х7, культивировали в модифицированной минимальной поддерживающей среде Игла (ЕМЕМ).
За 72 часа до начала эксперимента во флаконы Т225 высевали 5 миллионов клеток. Непосредственно перед экспериментом клетки дважды промывали, открепляли от стенок флаконов, используя смесь трипсина и EDTA (этилендиаминтетрауксусная кислота), ресуспендировали в растворе для суспендирования и помещали в QPatch16X.
В день эксперимента из растворов соединений (20 мМ в 100%-ном DMSO), которые хранили при -20°С, готовили разведенные растворы (первое разведение 1:20 в 100% DMSO для получения 1 мМ исходного раствора, затем следует получение 1 мкМ раствора во внеклеточном растворе + последовательное разведение 1:10).
Стандартные эксперименты с фиксацией потенциала на цельных клетках проводили при комнатной температуре. Для этих экспериментов использовали многоканальную технологию, и данные регистрировали при 2 кГц.
Внутриклеточный раствор содержал (в мМ): 135 CsF, 10 NaCl, 1 EGTA (этиленгликольтетрауксусная кислота), 10 HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота) (рН 7,2, доведенный добавлением CsOH), а внеклеточный раствор содержал (в мМ): 145 NaCl, 4 KCl, 0,5 MgCl2, 1 CaCl2, 10 HEPES, 10 Glc (глюкоза) (рН 7,2, доведенный добавлением NaOH).
После установления герметичного контакта и перехода к конфигурации "цельная клетка" потенциал клеток фиксировали при -80 мВ. Ток через канал P2XR7 вызывали, применяя только 100 мкМ BzATP (4 раза), а затем в присутствии возрастающих концентраций исследуемых соединений (1, 10, 100 и 1000 нМ).
Периоды предварительного инкубирования 5-8 соответствуют возрастающим концентрациям интересующего соединения (1, 10, 100 и 1000 нМ), как иллюстрируется на Фиг. 1.
Измеряли максимальный входящий ток, вызванный BzATP, в отсутствие или в присутствии возрастающих концентраций исследуемых соединений, и выполняли нормирование. Потенциальный модулирующий эффект измеряли в % от контроля и в виде величины IC50, определенной посредством выравнивания кривых зависимости доза-ответ с использованием следующего уравнения:
Y=100/(1+10^((LogIC50-Х)*коэффициент Хилла)),
где:
X: логарифм концентрации;
Y: нормированный ответ, от 100% до 0%, уменьшающийся по мере увеличения X;
LogIC50: в тех же логарифмических единицах, что и X;
коэффициент Хилла: угловой коэффициент или HS, безразмерная величина.
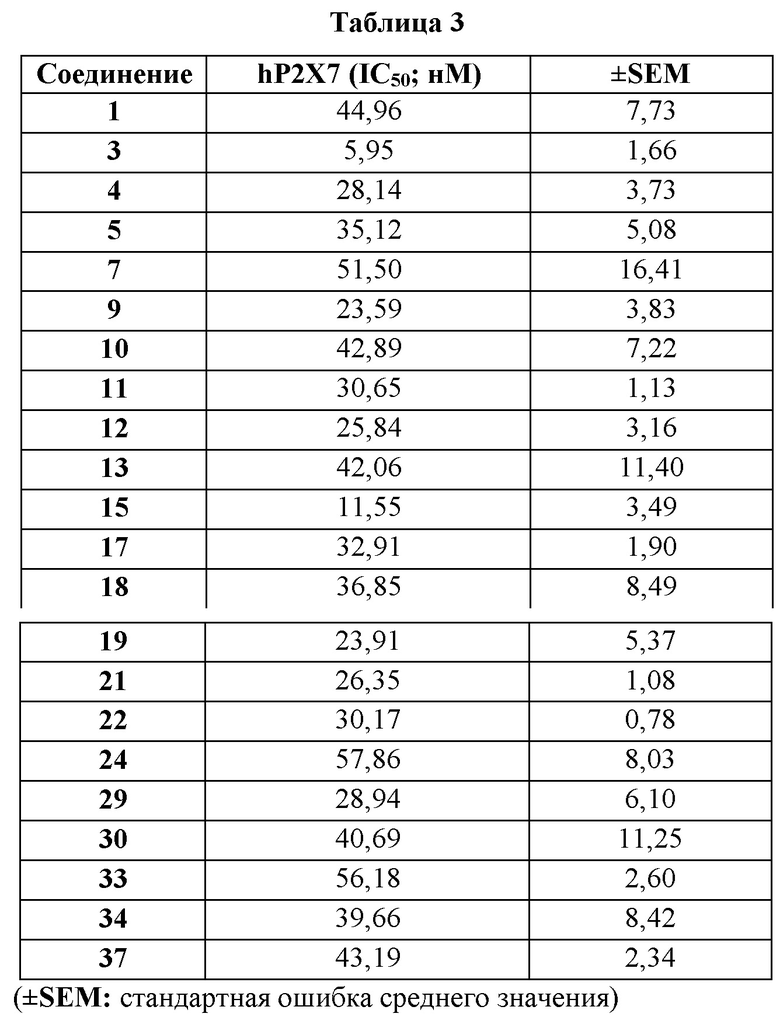
Используя набор для определения содержания кальция с использованием Fluo-8 без стадии отмывки Screen Quest™, было обнаружено, что соединения по изобретению являются ингибиторами Р2Х7 крысы.
Поступление Са2+ измеряли в клетках HEK-293, стабильно трансфицированных рецептором, используя набор для определения содержания кальция с использованием Fluo-8 без стадии отмывки Screen Quest™ (AAt Bioquest®, №по каталогу 36316). Кратко, сразу после попадания внутрь клетки липофильные блокирующие группы Fluo-8 отщепляются под действием неспецифических клеточных эстераз с образованием в результате отрицательно заряженного флуоресцентного красителя, который остается внутри клеток. Его флуоресценция усиливается при связывании с ионами кальция. Когда клетки HEK-293/Р2Х7 стимулируют Bz-ATP, Са2+ поступает в клетки и сигнал флуоресценции от Fluo-8 NW (без стадии отмывки) возрастает. Этот краситель имеет спектр поглощения, сочетаемый с возбуждением при 488 нм под действием в качестве источников возбуждения аргоновых лазеров, и длина волны его находится в диапазоне 515-575 нм.
Для рутинного тестирования соединений клетки HEK-293, стабильно трансфицированные P2X7R крысы, высевали в течение ночи в ростовой среде в количестве 10000, 15000 или 20000 клеток на лунку в 384-луночном планшете согласно уровню ответной реакции после оттаивания. Через 24 часа среду удаляли, и клетки предварительно нагружали при к.т.в течение 1 часа 20 мкл на лунку Fluo-8 NW приготовленного в буфере Тироде (Tyrode) с содержанием 0,3 мМ Са2+/в отсутствие Mg2+.
Соединения по изобретению тестировали в 8 концентрациях (в 4 повторах для каждой концентрации): 10; 3,16; 1; 0,316; 0,1; 0,0316; 0,01; и 0,00316 мкМ, в одном и том же планшете.
Соединения тестировали в системе FLIPRTETRA согласно следующему методу:
• первое впрыскивание в FLIPRTETRA 10 мкл 3х тестируемого соединения (в буфере Тироде, содержащем 0,3 мМ Са2+ /не содержащем Mg2+ + DMSO 0,5% конечная концентрация)
• инкубирование 5 минут
• второе впрыскивание в FLIPRTETRA 15 мкл 3х BzATP при примерно EC80 (в буфере Тироде, содержащем 0,3 мМ Са2+ /не содержащем Mg2+ + BSA 0,0003% конечная концентрация)
• считывание флуоресценции в течение 3 минут.
Между одним планшетом и следующим наконечники интенсивно промывали водой, затем 100%-ным DMSO и в конце водой во избежание переноса остатка предыдущей пробы в наконечниках.
Эффект тестируемых соединений измеряли в виде процента ингибирования относительно референсного антагониста, и значения IC50 вычисляли соответственно.
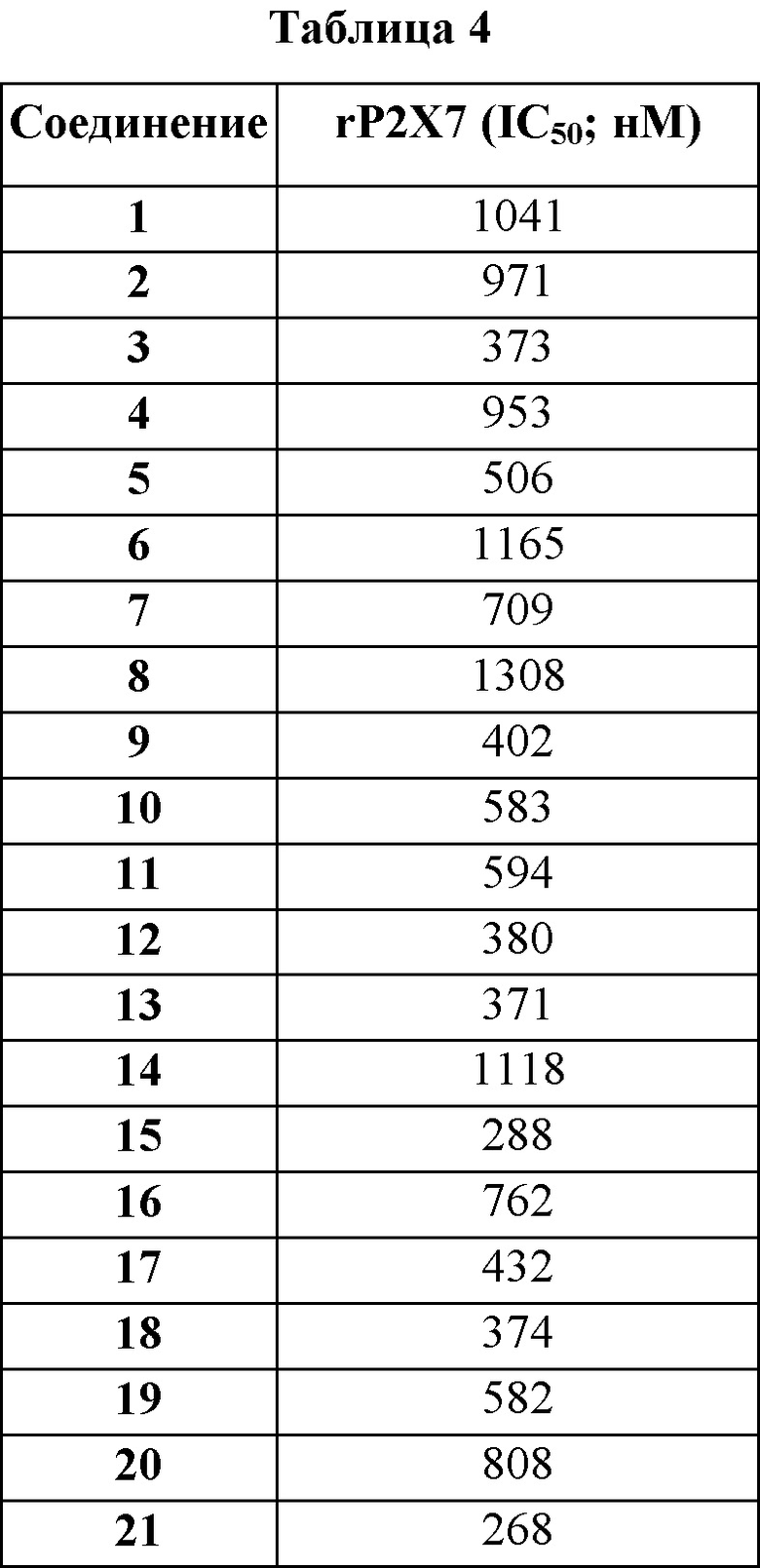
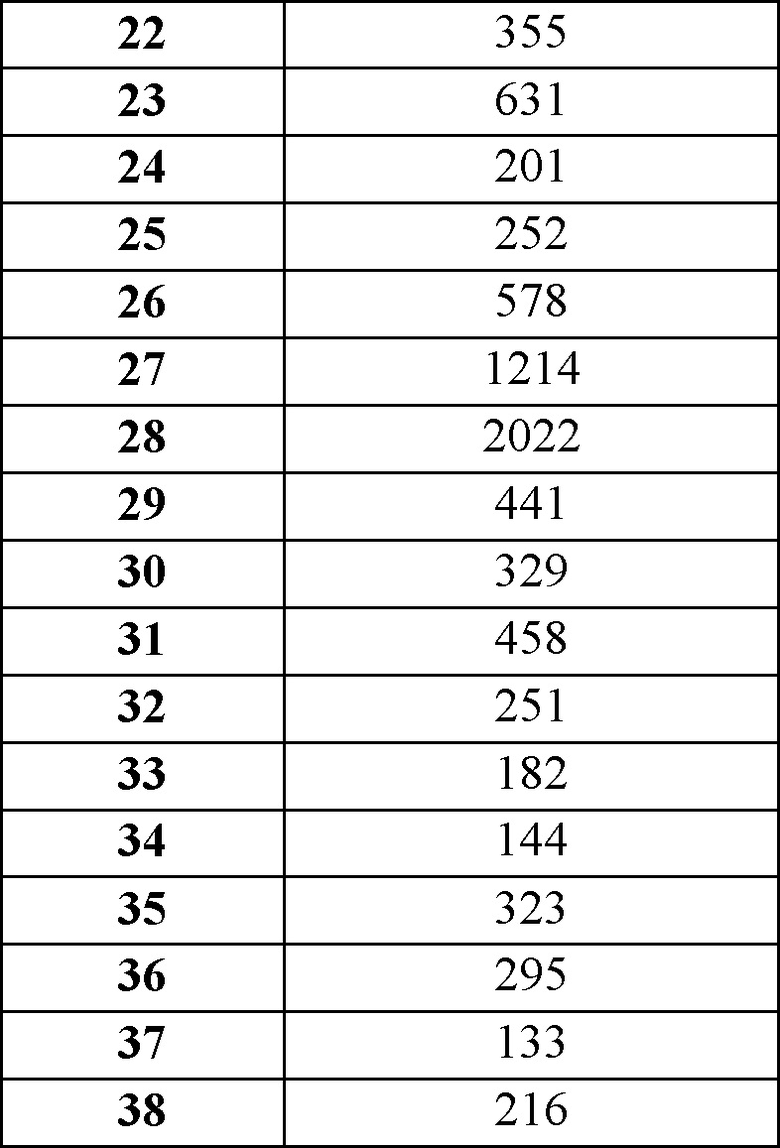
Было установлено, что соединения по настоящему изобретению неожиданно являются более сильнодействующими, чем очень близкое соединение Примера из WO 2015/118019, как указано в Таблице 5.
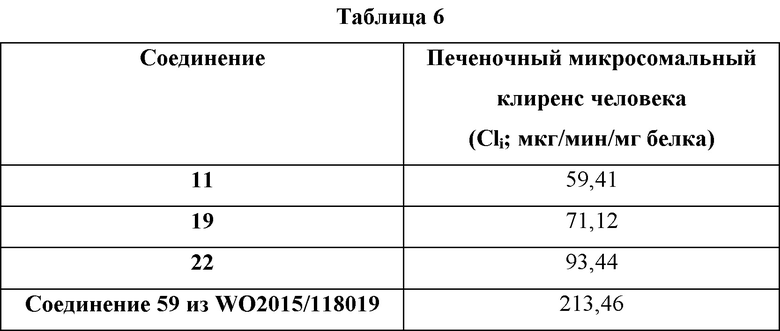
Оценка in vitro тестируемых соединений в отношении метаболической стабильности с использованием печени человека
Микросомы
Тест-система с использованием микросом печени человека (мыши)
Концентрация тестируемого соединения: 1 мкМ
Точки времени: 0, 5, 10, 30 и 60 минут
Конечная концентрация белка: 1 мг/мл
Число повторов: два
Калий-фосфатный буфер рН 7,4 100 мМ
Конечная точка: % остатка тестируемого соединения, период полувыведения, Clint (внутренний клиренс)
Биоанализ методом ЖХ-МС/МС
Приготовление и разведение тестируемого соединения:
Выполняли приготовление 10 мМ исходного раствора тестируемого соединения в DMSO и разводили его смесью вода:ацетонитрил (1:1) до концентрации 1 мМ. Рабочую концентрацию 100 мкМ получали дополнительным разведением смесью вода:ацетонитрил (1:1).
Приготовление калий-фосфатного буфера рН 7.4:
100 мл воды качества Milli Q будут нужно добавить к K2HPO4 (1,398 г) и KH2PO4 (0,27 г) до достижения конечного значения рН 7,4.
Методика анализа:
Смесь для предварительного инкубирования: 2,5 мкл тестируемого соединения +75 мкл микросом печени в количестве 3,33 мг/мл +85 мкл 100 мМ калий-фосфатного буфера (Предварительно инкубировать в течение 10 мин при 37°С).
60 мин без кофактора: 32,5 мкл смеси для предварительного инкубирования +17,5 мкл 100 мМ калий-фосфатного буфера (инкубировать в течение 60 мин при 37°С)
Проба 0 мин: 16,25 мкл смеси для предварительного инкубирования +200 мкл ацетонитрила, содержащего внутренний стандарт, +8,75 мкл смеси для инкубирования с кофактором, содержащей 62 мкл кофактора (2,5 мМ) + остаток смеси для инкубирования (инкубированной в течение 60 мин при 37°С).
Приготовление пробы: 25 мкл смеси для инкубирования + 200 мкл ацетонитрила, содержащего внутренний стандарт, + вортекс в течение 5 мин при 1200 об/мин + центрифугирование в течение 10 мин при 4000 об/мин. Развести надосадочную жидкость в 2 раза водой и впрыскивать на ЖХ-МС/МС.
Биоанализ: метод ЖХ-МС/МС с использованием типичных для ЖХ условий градиента; подробности будут представлены в отчете.
Вычисление: % остатка тестируемого вещества = [площадь пика в момент времени в мин/площадь пика при 0 мин]*100; Ке1 угловой коэффициент, полученный из графика зависимости log % остатка от времени (мин); период полувыведения: t1/2 (мин)=0,693/Ke1.
Внутренний клиренс (мкл/мин/мг)=ABS (Ke1/концентрация белка)*1000.
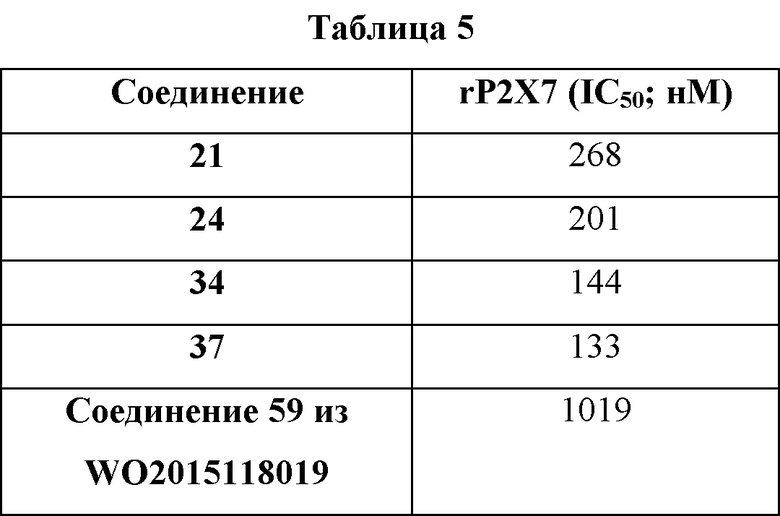
Было обнаружено, что проиллюстрированные примерами соединения по настоящему изобретению неожиданно являются в 2-4 раза более стабильными в тестах на стабильность с использованием микросом печени человека, чем очень похожее соединение, проиллюстрированного примером в WO 2015118019, как указано в Таблице 6.
| название | год | авторы | номер документа |
|---|---|---|---|
| Замещенные тиазолы или оксазолы в качестве антагонистов Р2Х7 рецепторов | 2015 |
|
RU2701858C1 |
| N-(2-ГИДРОКСИЭТИЛ)-N-МЕТИЛ-4-(ХИНОЛИН-8-ИЛ(1-(ТИАЗОЛ-4-ИЛМЕТИЛ)ПИПЕРИДИН-4-ИЛИДЕН)МЕТИЛ)БЕНЗАМИД, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ БОЛИ, ТРЕВОГИ И ДЕПРЕССИИ | 2007 |
|
RU2454414C2 |
| Гетероциклические антагонисты Р2Х7 | 2018 |
|
RU2768865C2 |
| НОВЫЕ 5,7-ДИЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ [1,3]ТИАЗОЛО[4,5-d]ПИРИМИДИН-2-(3Н)-АМИНА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2007 |
|
RU2437889C2 |
| НОВЫЕ 5,7-ДИЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ [1,3]ТИАЗОЛО[4,5-d]ПИРИМИДИН-2(3Н)-ОНА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2007 |
|
RU2441012C2 |
| МОНОГИДРАТ МЕЗИЛАТА N-[5-(АМИНОСУЛЬФОНИЛ)-4-МЕТИЛ-1,3-ТИАЗОЛ-2-ИЛ]-N-МЕТИЛ-2-[4-(2-ПИРИДИНИЛ)ФЕНИЛ]АЦЕТАМИДА | 2012 |
|
RU2669388C1 |
| НОВАЯ КРИСТАЛЛИЧЕСКАЯ ФОРМА 2-{ 3-[2-(1-{ [3,5-БИС(ДИФТОРМЕТИЛ)-1Н-ПИРАЗОЛ-1-ИЛ]АЦЕТИЛ} ПИПЕРИДИН-4-ИЛ)-1,3-ТИАЗОЛ-4-ИЛ]-4,5-ДИГИДРО-1,2-ОКСАЗОЛ-5-ИЛ} -3-ХЛОРФЕНИЛМЕТАНСУЛЬФОНАТА | 2014 |
|
RU2691948C2 |
| (2S,4R)-5-(5'-ХЛОР-2'-ФТОРБИФЕНИЛ-4-ИЛ)-4-(ЭТОКСИОКСАЛИЛАМИНО)-2-ГИДРОКСИМЕТИЛ-2-МЕТИЛПЕНТАНОВАЯ КИСЛОТА | 2016 |
|
RU2726623C2 |
| МОНОГИДРАТ МЕЗИЛАТА N-[5-(АМИНОСУЛЬФОНИЛ)-4-МЕТИЛ-1,3-ТИАЗОЛ-2-ИЛ]-N-МЕТИЛ-2-[4-(2-ПИРИДИНИЛ)ФЕНИЛ]АЦЕТАМИДА | 2012 |
|
RU2620604C2 |
| ПРОИЗВОДНЫЕ N-ФЕНИЛ(ПИПЕРИДИН-2-ИЛ)МЕТИЛ-БЕНЗАМИДА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2004 |
|
RU2351588C2 |

Изобретение относится к конкретным соединениям, представляющим собой замещенные фенокси- и бензилоксипиперидины, соответствующие структурной формуле (I). Также изобретение относится к способу получения соединений по изобретению и фармацевтической композиции, на основе таких соединений. Соединения по изобретению предназначены к применению в лечении или профилактике заболеваний, ассоциированных с активностью Р2Х7 рецептора. 3 н. и 2 з.п. ф-лы, 1 ил., 6 табл.

1. Соединение, выбранное из группы, состоящей из:
2-хлор-N-(2-{4-[(4-хлорфенил)метокси]пиперидин-1-ил}-2-(4-метил-1,3-тиазол-5-ил)этил)-6–фторбензамида,
2-хлор-6-фтор-N-(2-{4-[(3-фторфенил)метокси]пиперидин-1-ил}-2-(4-метил-1,3-тиазол-5-ил)этил)бензамида,
2-хлор-6-фтор-N-(2-{4-[(2-фторфенил)метокси]пиперидин-1-ил}-2-(4-метил-1,3-тиазол-5-ил)этил)бензамида,
2-хлор-N-(2-{4-[(3,5-дифторфенил)метокси]пиперидин-1-ил}-2-(4-метил-1,3-тиазол-5-ил)этил)-6-фторбензамида,
2-хлор-6-фтор-N-{2-[4-(4-фторфенокси)пиперидин-1-ил]-2-(4-метил-1,3-тиазол-5-ил) этил}бензамида,
2-хлор-6-фтор-N-{2-[4-(3-фторфенокси)пиперидин-1-ил]-2-(4-метил-1,3-тиазол-5-ил)этил}бензамида,
2-хлор-6-фтор-N-{2-[4-(2-фторфенокси)пиперидин-1-ил]-2-(4-метил-1,3-тиазол-5-ил)этил}бензамида,
2-хлор-N-{2-[4-(3,5-дифторфенокси)пиперидин-1-ил]-2-(4-метил-1,3-тиазол-5-ил)этил}-6-фторбензамида,
2-хлор-N-{2-[4-(3,4-дифторфенокси)пиперидин-1-ил]-2-(4-метил-1,3-тиазол-5-ил)этил}-6-фторбензамида,
N-(2-{4-[(4-фторфенил)метокси]пиперидин-1-ил}-2-(4-метил-1,3-тиазол-5-ил)этил)хинолин-5-карбоксамида,
N-(2-{4-[(3-фторфенил)метокси]пиперидин-1-ил}-2-(4-метил-1,3-тиазол-5-ил)этил)хинолин-5-карбоксамида,
N-(2-{4-[(2-фторфенил)метокси]пиперидин-1-ил}-2-(4-метил-1,3-тиазол-5-ил)этил)хинолин-5-карбоксамида,
N-(2-{4-[(3,5-дифторфенил)метокси]пиперидин-1-ил}-2-(4-метил-1,3-тиазол-5-ил)этил) хинолин-5-карбоксамида,
N-(2-{4-[(3,4-дифторфенил)метокси]пиперидин-1-ил}-2-(4-метил-1,3-тиазол-5-ил) этил)хинолин-5-карбоксамида,
N-{2-[4-(4-фторфенокси)пиперидин-1-ил]-2-(4-метил-1,3-тиазол-5-ил)этил}хинолин-5-карбоксамида,
N-{2-[4-(4-хлорфенокси)пиперидин-1-ил]-2-(4-метил-1,3-тиазол-5-ил)этил}хинолин-5-карбоксамида,
N-{2-[4-(3-фторфенокси)пиперидин-1-ил]-2-(4-метил-1,3-тиазол-5-ил)этил}хинолин-5-карбоксамида,
N-{2-[4-(2-фторфенокси)пиперидин-1-ил]-2-(4-метил-1,3-тиазол-5-ил)этил}хинолин-5-карбоксамида,
N-{2-[4-(3,5-дифторфенокси)пиперидин-1-ил]-2-(4-метил-1,3-тиазол-5-ил)этил}хинолин-5-карбоксамида,
N-{2-[4-(3,4-дифторфенокси)пиперидин-1-ил]-2-(4-метил-1,3-тиазол-5-ил)этил}хинолин-5-карбоксамида,
2-хлор-N-{2-[4-(дифторметил)-1,3-тиазол-5-ил]-2-{4-[(2-фторфенил)метокси]пиперидин-1-ил}этил}-6-фторбензамида,
2-хлор-N-{2-[4-(дифторметил)-1,3-тиазол-5-ил]-2-{4-[(3,5-дифторфенил)метокси]пиперидин-1-ил}этил}-6-фторбензамида,
2-хлор-N-{2-[4-(дифторметил)-1,3-тиазол-5-ил]-2-[4-(3-фторфенокси)пиперидин-1-ил]этил-6-фторбензамида
2-хлор-N-{2-[4-(дифторметил)-1,3-тиазол-5-ил]-2-[4-(2-фторфенокси)пиперидин-1-ил]этил}-6-фторбензамида,
2-хлор-N-{2-[4-(дифторметил)-1,3-тиазол-5-ил]-2-[4-(3,5-дифторфенокси)пиперидин-1-ил]этил}-6-фторбензамида,
N-{2-[4-(дифторметил)-1,3-тиазол-5-ил]-2-{4-[(2-фторфенил)метокси]пиперидин-1-ил}этил}хинолин-5-карбоксамида,
N-{2-[4-(дифторметил)-1,3-тиазол-5-ил]-2-{4-[(3,4-дифторфенил)метокси]пиперидин-1-ил}этил}хинолин-5-карбоксамида,
N-{2-[4-(дифторметил)-1,3-тиазол-5-ил]-2-[4-(4-фторфенокси)пиперидин-1-ил]этил}хинолин-5-карбоксамида,
N-{2-[4-(4-хлорфенокси)пиперидин-1-ил]-2-[4-(дифторметил-1,3-тиазол-5-ил]этил}хинолин-5-карбоксамида,
N-{2-[4-(дифторметил)-1,3-тиазол-5-ил]-2-[4-(3-фторфенокси)пиперидин-1-ил]этил}хинолин-5-карбоксамида,
N-{2-[4-(дифторметил)-1,3-тиазол-5-ил]-2-[4-(2-фторфенокси)пиперидин-1-ил]этил}хинолин-5-карбоксамида,
N-{2-[4-(дифторметил)-1,3-тиазол-5-ил]-2-[4-(3,5-дифторфенокси)пиперидин-1-ил]этил}хинолин-5-карбоксамида, или
его фармацевтически приемлемая соль.
2. Способ получения соединения, как оно определено в п.1, включающий стадии:
1) взаимодействия соединения формулы (II):
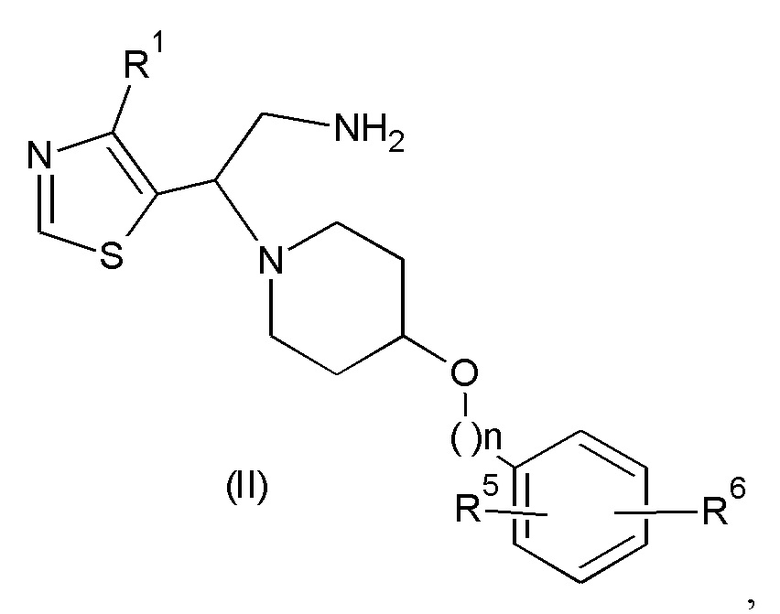
где n равно 0 или 1;
R1 представляет собой С1-С4алкил, возможно замещенный галогеном;
каждый из R5 и R6 представляет собой водород или галоген, при условии, что по меньшей мере один из R5 и R6 представляет собой галоген,
с соединением формулы (III):
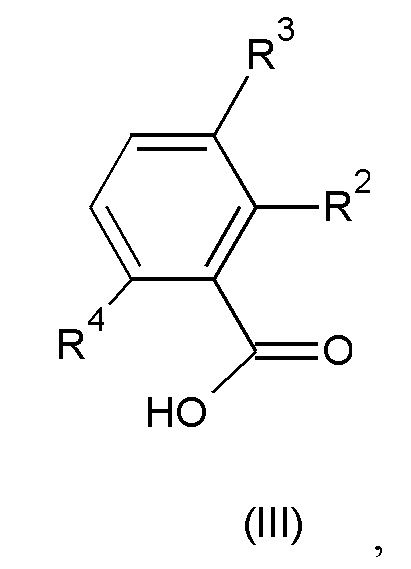
где каждый из R2, R3 и R4 независимо представляет собой водород или галоген, при условии, что по меньшей мере один из R2, R3 и R4 не представляет собой водород, или группы R2 и R4 вместе образуют шестичленное гетероциклическое кольцо, содержащее атом азота; или
с соединением формулы (IIIa):
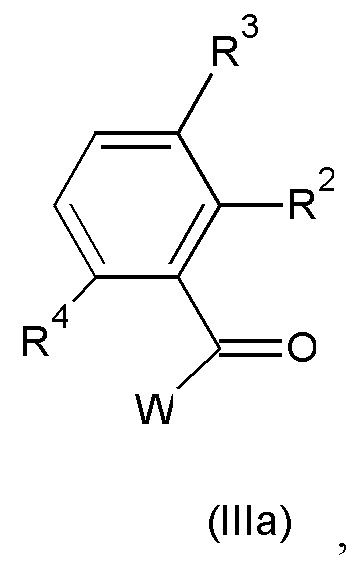
где значения R2, R3 и R4 такие, как определено выше; и W представляет собой подходящую уходящую группу;
и возможно превращения полученного соединения в его соль.
3. Фармацевтическая композиция, обладающая антагонистической активностью в отношении Р2Х7 рецептора, которая содержит терапевтически эффективное количество соединения по п.1 или его фармацевтически приемлемой соли и фармацевтически приемлемый разбавитель и/или носитель.
4. Соединение по п.1 или его фармацевтически приемлемая соль для применения в качестве лекарственного средства, обладающего антагонистической активностью в отношении Р2Х7 рецептора.
5. Соединение по п.1 для применения в предупреждении и/или лечении состояний или заболеваний, опосредованных активностью Р2Х7 рецептора, которые выбраны из группы, состоящей из нейродегенеративных, когнитивных, психиатрических расстройств, невропатической боли, хронической боли, воспалительных процессов скелетно-мышечной системы, расстройств желудочно-кишечного тракта, расстройств урогенитального тракта, офтальмических заболеваний.
| WO 2015118019 A1, 13.08.2015 | |||
| WO 2009132000 A1, 29.10.2009 | |||
| WO 2014091415 A1, 19.06.2014 | |||
| ПРОИЗВОДНЫЕ ИНДОЛА ИЛИ БЕНЗИМИДАЗОЛА ДЛЯ МОДУЛЯЦИИ IkB-КИНАЗЫ | 2003 |
|
RU2318820C2 |

